
Home » Uncategorized (Page 112)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tioconazole UK-20349 an antifungal agent

Tioconazole;UK-20349;Trosyd;Trosyl;Vagistat-1
l-[2-(2-chloro-3-thienyl)methoxy]-2-(2,4- dichlorophenyl)ethyl]-lH-imidazole,
1-[2-(2-Chloro-3-thienylmethoxy)-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole
65899-73-2
Launched – 1983, Bristol-Myers Squibb
Tioconazole is an antifungal medication of the imidazole class used to treat infections caused by a fungus or yeast. It is marketed under the brand names Trosyd and Gyno-Trosyd (Pfizer). Tioconazole ointments serve to treat women’s vaginal yeast infections.[1]They are available in one day doses, as opposed to the 7-day treatments more common in use in the past.
Tioconazole topical (skin) preparations are also available for ringworm, jock itch, athlete’s foot, and tinea versicolor or “sun fungus”.

Side effects
Side effects (for the women’s formulas) may include temporary burning/irritation of the vaginal area, moderate drowsiness, headachesimilar to a sinus headache, hives, and upper respiratory infection. These side effects may be only temporary, and do not normally interfere with the patient’s comfort enough to outweigh the end result.
|
|
| Systematic (IUPAC) name | |
|---|---|
| (RS)-1-[2-[(2-Chloro-3-thienyl)methoxy]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole | |
| Clinical data | |
| Trade names | Vagistat-1 |
| AHFS/Drugs.com | monograph |
| Legal status | |
| Routes | Topical |
| Identifiers | |
| CAS number | 65899-73-2 |
| ATC code | D01AC07 G01AF08 |
| PubChem | CID 5482 |
| DrugBank | DB01007 |
| KEGG | D00890 |
| Synonyms | Thioconazole |
| Chemical data | |
| Formula | C16H13Cl3N2OS |
| Mol. mass | 387.711 g/mol |
http://www.google.com/patents/EP0934279A1?cl=en
Imidazole derivatives, in particular, l-[2-(2-chloro-3-thienyl)methoxy]-2-(2,4- dichlorophenyl)ethyl]-lH-imidazole, commonly referred to as tioconazole, are known for their antifungal therapeutic properties. US 4,062,966 discloses a process for the preparation of l-aryl-2-(l -imidazolyl) alkyl ethers and thioethers which employs arylation of an appropriate 1 -aryl-2-(l -imidazolyl)alkanol or alkane thiol having the formula

wherein Rl to R4 are each H or C,^ alkyl, Ar is phenyl, or substituted phenyl wherein said substitutents are halogen, C,^ alkyl, C,_6 alkoxy, thienyl, or halothienyl, and, Z is oxygen or sulfur. In accordance with US’966, the reaction comprises converting the alcohol or thiol in a suitable solvent to its alkali metal derivative by treatment with a strong base, such as an alkali metal amide or hydride, and reacting with the appropriate aralkyl halide ofthe formula
X-(CH2)η-Y
where n is 1 or 2, Y is an aromatic heterocyclic group or substituted heterocyclic group, wherein substitutents are halogen, C,.6 alkyl, or C,.6 alkoxy atoms, thienyl or halothienyl group, and X is a halogen, preferably chlorine. Tetrahydrofuran (THF) is the preferred solvent taught in US ‘966. Reaction temperatures may range from about 0 °C to reflux temperature ofthe solvent and reaction times range from about 1 hour to about 24 hours. The product is isolated with water, extracted with ether, and may be purified as the free base or converted to a salt, e.g. the hydrochloride, and purified by recrystallization. A disadvantage ofthe process disclosed in US ‘966 is that THF is a peroxide generator which presents the potential for an explosion. From a commercial viewpoint, peroxide generators are not preferred due to the dangers associated therewith.
GB 1 522 848 discloses a process for the preparation of imidazoles useful as antifungal agents involving a labor intensive, multi-sequence reaction of an imidazole ether with a reactive ester. Like US ‘966, THF is employed presenting similar concerns in the synthesis ofthe desired imidazole product.
According to the Pharmaceutical Manufacturing Encyclopedia, tioconazole is prepared by dissolving l-(2,4-dichlorophenyl)-2-(l- imidazolyl)ethanol in THF and sodium hydride and heating to about 70 βC. The resulting mixture is then contacted with 2-chloro-3- chloromethylthiophene and heated to reflux (about 67 CC). The resulting product is filtered, saturated with hydrogen chloride, triturated and recrystallized to obtain the purified tioconazole hydrochloride product having a melting point of about 170 βC. This salt must then be freebased to form the product used in pharmaceutical formulations. This route, like those discussed above, also presents the dangers of a potential explosion. There is thus a continuing need for a commercially viable, synthetic route for the production of imidazoles, in particular tioconazole.
…………………….
see US 4062966
http://www.google.com/patents/US4062966
………………………….
References
- Tioconazole, Mayo Clinic
-
References1:
Gymer, G.E.; DE 2619381 .
References2:Hillier, K.; Blancafort, P.; Castaner, J.; Serradell, M.N.; Tioconazole. Drugs Fut 1980, 5, 10, 509.
- Growth quantification and rapid drug susceptibility testing of uropathogenic Candida albicans by isothermal microcalorimetry
28th Congr Eur Assoc Urol (March 15-19, Milan) 2013, Abst 618 - Difference in percutaneous absorption and intracutaneous distribution in guinea pigs among topical antifungal drugs (tioconazole solution, tioconazole cream, miconazole nitrate solution and bifonazole solution)
Biol Pharm Bull 2004, 27(9): 1428 - A randomized comparison of the nail surface remainder of three nail lacquers containing amorolfine 5%, ciclopirox 8%, or tioconazole 28% in healthy volunteers
63rd Annu Meet Am Acad Dermatol (AAD) (February 18-22, New Orleans) 2005, Abst P1805
Literature References:
Antimycotic imidazole derivative. Prepn: G. E. Gymer, BE 841309; idem, (1976, 1977 both to Pfizer).
Antifungal spectrum: S. Jevons, Antimicrob. Agents Chemother. 15, 597 (1979); F. C. Odds, J. Antimicrob. Chemother. 6,749 (1980).
Pharmacology: M. S. Marriott et al., Dermatologica 166, Suppl. 1, 1 (l983).
Clinical trial in dermatomycosis: Y. M. Clayton et al., Clin. Exp. Dermatol. 7, 543 (1982). Series of articles on pharmacology and clinical efficacy in gynecological use:Gynak. Rundsch. 23, Suppl. 1, 1-60 (l983).
MHRA’s Guidance for Software as a Medical Device (including Apps)
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
The British MHRA (Medicines and Healthcare Products Regulatory Agency) has published a guidance for developers of “software as a medical device” = “stand-alone software”. The text also expressly addresses “apps”. Get the details here.
Whereas in the pharmaceutical business software plays a role in the manufacture of products, it can also act as two parts in the medical device business – one in the manufacture of a device and one as a medical device as such – i.e. software as a medical device. The British Health Authority – MHRA – has published a current guidance on software as a medical device, also called stand-alone software, intended for developers of such software. This guidance also addresses the increasingly encountered topic “apps”. The text doesn’t cover software that is part of a medical device, e.g. software that controls a CT scanner.
The guidance itself is very short and divided into 6 main chapters:
- Introduction
- Key points and existing…
View original post 306 more words
Questions and Answers on the Topic “Pharmaceutical Water”
DRUG REGULATORY AFFAIRS INTERNATIONAL

In the following News, you will find questions on pharmaceutical water preparation and distribution frequently asked during our courses, as well as their respective answers. Read more here.
During our courses and conferences participants quite frequently raise questions on pharmaceutical water preparation and distribution. Therefore following you will find some of these questions and their respective answers.
Question 1: Which concentrations of ozone are required in water systems?
The technical literature delivers different information about the ozone concentrations in water systems: e.g. ISPE Baseline Water and Steam: 0.02 ppm – 0.2 ppm; Collentro, Pharmaceutical Water: 0.2 ppm – 0.5 ppm and W.Setz, Ciba-Geigy 1990: max 0.04 ppm, for sanitisation 0.05 ppm.
The indications provided by the ISPE Baseline refer to the concentration required to prevent microbial growth. One can thus assume that a concentration of 20 ppb ozone can prevent any growth.
If systemic protection is desired i.e. the…
View original post 894 more words
EU Commission publishes long-awaited EU GMP Guide Chapters 3 and 5
DRUG REGULATORY AFFAIRS INTERNATIONAL
The EU Commission has published the long-awaited, revised chapters 3 and 5 of the EU GMP Guide. The change focuses on the prevention of cross-contamination as well as on the statement concerning the need for dedicated facilities. Continue reading.
The EU Commission had published its first draft of the chapter 3 “Premises and Equipment” and 5 “Production” for comments in early 2013 (see news from 04/12/2013). The content concerns the measures for avoiding cross-contamination and the regulation relative to which products have to be produced in dedicated facilities.
The mention of specific products for which a dedication is required – as provided in the currently valid version of chapter 3 – is missing in the now published version. The quality risk management approach is maintained. Also remaining are the exceptions where dedication is required – which are:
- The risk cannot be adequately controlled by operational and/ or technical measures…
View original post 214 more words
AMRI Introduces Protein Expression & Purification Solutions

|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


| Albany Molecular Research Inc. (AMRI) 26 Corporate Circle Albany, NY 12203 |



21′α-Cyanoanhydrovinblastine

Some derivatives ) are known as being intermediates in the preparation of anti-tumor medicaments such as vinblastine, vincristine and vinorelbine.

R=CH3, vinblastine
R=CHO, vincristine
n=2, anhydrovinblastine
n=1, vinorelbine
The remarkable anti-tumor properties of these complex natural molecules, extracted from the Madagascar periwinkle, Carantheus roseus, are known and they are already used in anti-cancer treatment. Vinblastine and vincristine are “spindle poisons” which oppose the formation of the mitotic spindle during cellular division, thus preventing cellular proliferation.
Vincristine and vinblastine are active agents in the treatment of leukemia, lymphosarcoma and solid tumors. Vinblastine is also used in the treatment of Hodgkin’s disease.
Vinorelbine is currently used in the treatment of the most widespread form of cancer of the lungs, that is lung cancer of non-small cells. It is also used in the treatment of metastasic cancers of the breast.
The methods currently used for preparing vinblastine and vincristine involve extraction of these molecules from plants. The plants have to be crushed and dried before these substances can be extracted. The extraction process is long and costly, given that the extract obtained is very complex, containing at least 200 different alkaloids. The yields are also very low; 5 to 10 g of vinoblastine are obtained per ton of dried plant material, and 0.5 to 1 g of vincristine per ton of dried plant material.
Many research groups have thus tried to achieve synthesis of these molecules by using more efficient procedures which enable better yields and which make use of derivatives with interesting anti-tumor properties but which are endowed with lower levels of toxicity.
 just an animation
just an animation
The patent FI 882 755, filed by the HUATAN-MAKI Oy Company, relates to the formation of vinblastine and vincristine by irradiation of catharanthine and of vindoline with UV radiation in an acidic aqueous solution, under an atmosphere of oxygen or an inert gas. The yields obtained in these reactions are extremely low.
Furthermore, other processes are known which make use of anhydrovinblastine which is an intermediate in the synthesis of vinblastine, vincristine and also of vinorelbine.
Anhydrovinblastine is thus a key chemical intermediate which enables access to all alkaloids of the vinblastine type. This intermediate is synthesised by coupling catharanthine and vindoline.

The latter two alkaloids are also extracted from the Madagascar periwinkle but, in contrast to vincristine and vinblastine, they represent the main constituents of the extract obtained. In fact, 400 g of catharanthine per ton of dried plant material and 800 g of vindoline per ton of dried plant material are obtained.
The preparation of anhydrovinblastine by coupling catharanthine and vindoline is therefore a favoured route for synthesising this intermediate product.
There are several methods for preparing anhydrovinblastine from catharanthine and vindoline.
The patent FR 2 296 418 filed by ANVAR describes a process during the course of which the N-oxide of catharanthine is coupled to vindoline in the presence of trifluoroacetic anhydride.
When this process is performed at ambient temperature only the inactive 16′-R epimer of anhydrovinblastine is obtained. The naturally occurring active 16′-S epimer is obtained as the major product when this reaction is performed at a temperature which is at least 50° C. lower and under an inert gas. Nevertheless, even at low temperature, 10% of the 16′-R epimer of anhydrovinblastine is still produced.

This process has several disadvantages. The operating conditions are extremely restrictive due to the use of anhydrous solvents, the low temperature and the atmosphere of inert gas. The product obtained has to be subjected to a purification procedure due to the presence of 10% of the 16′-R epimer of anhydrovinblastine. The yield of isolated anhydrovinblastine is low, of the order of 35%.
A second process, suggested by VUKOVIC et al. in the review “Tetrahedron” (1998, volume 44, pages 325-331) describes a coupling reaction between catharanthine and vindoline initiated by ferric ions. Catharanthine is also oxidised in this reaction. The yield of anhydrovinblastine is of the order of 69% when the reaction is performed under an atmosphere of inert gas. However, this process has the major disadvantage that it leads to many secondary products. These are impurities resulting from further oxidation of the dimeric alkaloids formed, whatever the chosen operating conditions. This makes the purification stage difficult and delicate.
An improved process was suggested in the patent U.S. Pat. No. 5,037,977 and this increases the yield of anhydrovinblastine to 89%. However, this improvement is described only for very small amounts of reagents and its extension to the industrial scale seems to be difficult. In any case, these processes based on ferric ions lead in all cases to many secondary products due to the fact that these ions are responsible for parasitic reactions.
A third process described by GUNIC et al. in “Journal of the Chemical Society Chemical Communications” (1993), volume 19, pages 1496-1497, and by Tabakovic et al. in “Journal of Organic Chemistry” (1997), volume 62, pages 947-953, describes a coupling reaction between catharanthine and vindoline as a result of anodic oxidation of catharanthine. However, this process also suffers from disadvantages which, on the one hand, are due to the requirement for an inert atmosphere and, on the other hand, are connected with the nature of the electrochemical process itself, involving wear of the electrodes, difficulty in controlling the reproducibility and the cost of electrolytes. And, as in all the preceding methods, the anhydrovinblastine is contaminated with about 10% of the 16′-R epimer of anhydrovinblastine.
http://www.google.com/patents/US6365735
EXAMPLE 11 Preparation of 21′α-Cyanoanhydrovinblastine
0.537 mmol of catharanthine hydrochloride (200 mg), 0.537 mmol of vindoline (245 mg) and 0.054 mmol of dimethyl viologen (14 mg) and 0.028 mmol of triphenylpyrilium hydrogen sulfate (11 mg) are added to 50 ml of 0.1 N sulfuric acid. The entire mixture is irradiated with light of wavelength λ>400 nm in a Pyrex irradiation flask, under an atmosphere of oxygen. The reaction is terminated after 2 h 30 min of irradiation.
The aqueous phase is then saturated with lithium tetrafluoroborate and then extracted with dichloromethane. A solution of 15 ml of dichloromethane containing 100 μl (1.34 mmol, 2 eq.) of trimethylsilyl cyanide, TMSCN, is then added to the reaction medium. The organic phase is washed with a solution of 0.1 M sodium carbonate, dried and evaporated under reduced pressure at 20° C.

The only product in the residue (403 mg, 0.509 mmol, 95%) is recrystallised from absolute isopropanol. 340 mg of white crystals of 21′α-cyanoanhydrovinblastine (0.430 mmol; yield: 80%) are recovered.

C47H55N5O8
M.pt. 212° C. (iPrOH) IR film 3450, 2950, 2220, 1740, 1610 cm−1; MS M/z (relative intensity) 818 (MH+, 3), 122 (100), 108 (21);
NMR 1H (500 MHz, CDCl3) 9.78 (s, 1H, OH), 8.04 (s, 1H, Na′H), 7.51 (1H, H-9′), 7.16 (1H, H-11′), 7.13 (1H, H-12′), 7.12 (1H, H-10′), 6.63 (s, 1H, H-9), 6.13 (s, 1H, H-12), 5.85 (m, 1H, H-14), 5.47 (s, 1H, Hα-17), 5.54 (m, 1H, H-15′), 5.30 (m 1H, H-15), 4.18 (1H, H62-2), 3.60 (s, 3H, C16′—COOCH3), 3.38 (1H, H62-3), 3.35 (1H, Hβ-3′), 3.31 (1H, Hβ-5), 3.25 (1H, Hβ-6′), 3.24 (m, 1H, Hβ-5′), 3.15 (1H, Hβ-17′), 3.14 (m, 1H, Hα-5′), 3.12 (1H, Hα-6′), 2.82 (1H, Hα-3), 2.72 (s, 3H, NaCH3), 2.66 (s, 1H, Hα-21), 2.62 (1H, Hα-3′), 2.46 (1H, Hα-5), 2.40 (1H, Hα-17′), 2.20 (1H, Hβ-5), 2.11 (s, 3H, CH3—COO), 2.11 (1H, H-19′), 2.03 (1H, H-19′), 1.80 (1H, Hα-6), 1.80 (1H, H-19), 1.35 (1H, H-19), 1.21 (m, 1H, H-14′), 1.04 (3H, H-18′), 0.81 (3H, H-18).
NMR 13C (125 MHz, CDCl3) 174.69 (C16′—COOCH3), 171.74 (C16—COOCH3), 171.03130.01 (C15), 129.34 (C8′), 129.16 (C15′), 124.63 (C14), 123.48 (C9), 123.24 (C8), 122.49 (C11′), 121.00 (C10), 119.21 (C10′), 119.21 (CN), 118.35 (C9′), 115.65 (C7′), 110.64 (C11—OCH3), 55.40 (C16′), 53.30 (C7), 52.46 (C16′—COOCH3), 52.30 (C16—COOCH3), 52.26 (C5′), 50.50 (C5), 50.41 (C5), 44.86 (C6), 44.48 (C3′), 42.76 (C20), 38.32 (Na—CH3), 34.00 (C17′), 33.28 (C14′), 30.92 (C19), 28.63 (C8′), 25.92 (C19′), 21.19 (CH3—COO), 11.86 (C18′), 8.50 (C18).

| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4737586 | Apr 29, 1986 | Apr 12, 1988 | Agence Nationale De Valorisation De La Recherche | Process for the preparation of bis-indolic compounds |
| US5037977 | Aug 8, 1989 | Aug 6, 1991 | Mitsui Petrochemical Industries Ltd. | Reacting catharanthine with vindoline in presence of ferric ions, inactivating iron with ligand, reducing |
| DE3801450A1 | Jan 20, 1988 | Aug 18, 1988 | Univ British Columbia | Verfahren fuer die synthese von vinblastin und vincristin |
| DE3826412A1 | Aug 3, 1988 | Feb 16, 1989 | Univ British Columbia | Verfahren fuer die synthese von vinblastin und vincristin |
| WO1989012056A1 | Jun 9, 1989 | Dec 14, 1989 | Huhtamaeki Oy | Process for the preparation of dimeric catharanthus alkaloids |
| Reference | ||
|---|---|---|
| 1 | E. Gunic et al., “Electrochemical Synthesis of Anhydrovinblastine“, J. Chem. Soc., Chem. Commun., 1993, pp. 1496-1497. | |
| 2 | I. Tabakovic et al., “Anodic Fragmentation of Catharanthine and Coupling with Vindoline. Formation of Anhydrovinblastine“, J. Org. Chem., 1997, vol. 62, pp 947-953. | |
| 3 | J. Vucovik et al., “Production of 3′,4′-anhydrovinblastine: a Unique Chemical Synthesis“, Pergamon Journals Ltd., 1988, vol. 44, pp. 325-331. | |
| 4 | Richard J. Sundberg et al.; “Mechanistic aspects of the formation of anhydrovinblastine by Potier-Polonovski oxidative coupling of catharanthine and vindoline. Spectroscopic observation and chemical reactions of intermediates” Tetrahedron., vol. 48, No. 2,-Jan. 10, 1992; pp. 277-296, XP002083507 Oxford GB-the whole document. | |
| 5 | Richard J. Sundberg et al.; “Oxidative fragmentation of catharanthine by dichlorodicyanoquinone“; Journal of Organic Chemistry,-Mar. 1, 1991; pp. 1689-1692, XP002083508 Easton US -the whole document. | |
| 6 | Richard J. Sundberg et al.; “Photoactivated C16-C21 fragmentation of catharanthine” Tetrahedron Letters, vol. 32, No. 26, Jun. 24, 1992, pp. 3035-3038 XP002083509 Oxford GB-the whole document. | |
| 7 | Richard J. Sundberg et al.; “Mechanistic aspects of the formation of anhydrovinblastine by Potier-Polonovski oxidative coupling of catharanthine and vindoline. Spectroscopic observation and chemical reactions of intermediates” Tetrahedron., vol. 48, No. 2,—Jan. 10, 1992; pp. 277-296, XP002083507 Oxford GB—the whole document. | |
| 8 | Richard J. Sundberg et al.; “Oxidative fragmentation of catharanthine by dichlorodicyanoquinone“; Journal of Organic Chemistry,—Mar. 1, 1991; pp. 1689-1692, XP002083508 Easton US —the whole document. | |
| 9 | Richard J. Sundberg et al.; “Photoactivated C16-C21 fragmentation of catharanthine” Tetrahedron Letters, vol. 32, No. 26, Jun. 24, 1992, pp. 3035-3038 XP002083509 Oxford GB—the whole document. | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7235564 * | Dec 3, 2004 | Jun 26, 2007 | Amr Technology, Inc. | 11′-substituted; potent inhibitors of cellular mitosis and proliferation |
| US7238704 * | Dec 3, 2004 | Jul 3, 2007 | Amr Technology, Inc. | For use as inhibitors of cellular mitosis and proliferation |
| US7745619 | Oct 31, 2007 | Jun 29, 2010 | Albany Molecular Research, Inc. | alkaloids; anticarcinogenic, antiproliferative agent; inhibitor of cellular mitosis and cell proliferation; binding to tubulin leads to cell cycle arrest in M phase and subsequently to apoptosis; antiallergen, antiinflammatory, antidiabetic, autoimmune diseases; asthma, arthritis, Alzheimer’ disease |
| US7842802 | Dec 10, 2008 | Nov 30, 2010 | Albany Molecular Research, Inc. | Vinorelbine derivatives |
| US8048872 | Apr 29, 2008 | Nov 1, 2011 | Stat of Oregon Acting by and Through The Oregon State Board of Higher Education on Behalf of the University of Oregon | Treatment of hyperproliferative diseases with vinca alkaloid N-oxide and analogs |
| US8053428 | Apr 6, 2007 | Nov 8, 2011 | Albany Molecular Research, Inc. | Vinorelbine derivatives |
| WO2005055939A2* | Dec 3, 2004 | Jun 23, 2005 | Amr Technology Inc | Vinca derivatives |



Acebutolol……..For the management of hypertension and ventricular premature beats in adults.

Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | C07AB04 C07BB04 |
C 18 H 28 N 2 O 4 | 336.43 g / mol | 37517-30-9 |
| (R) be the bases | C07AB04 C07BB04 |
C 18 H 28 N 2 O 4 | 336.43 g / mol | 68107-81-3 |
| (S) be the bases | C07AB04 C07BB04 |
C 18 H 28 N 2 O 4 | 336.43 g / mol | 68107-82-4 |
| (RS) -monogidrohlorid | C07AB04 C07BB04 |
C 18 H 28 N 2 O 4 · HCl | 372.89 g / mol | 34381-68-5 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (RS)-N-{3-acetyl-4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl}butanamide | |
| Clinical data | |
| Trade names | Sectral |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a687003 |
| Licence data | US FDA:link |
| Pregnancy cat. | C (AU) B (US) |
| Legal status | ℞ Prescription only |
| Routes | oral, iv |
| Pharmacokinetic data | |
| Bioavailability | 40% (range 35 to 50%) |
| Metabolism | Hepatic |
| Half-life | 3-4 hours (parent drug) 8-13 hours (active metabolite) |
| Excretion | Renal: 30% Biliary: 60% |
| Identifiers | |
| CAS number | 37517-30-9 |
| ATC code | C07AB04 |
| PubChem | CID 1978 |
| DrugBank | DB01193 |
| ChemSpider | 1901 |
| UNII | 67P356D8GH |
| KEGG | D02338 |
| ChEBI | CHEBI:2379 |
| ChEMBL | CHEMBL642 |
| Chemical data | |
| Formula | C18H28N2O4 |
| Mol. mass | 336.426 g/mol |
| Physical data | |
| Melt. point | 121 °C (250 °F) |
Application
-
antagonist of β-adrenergic
-
β-blocker
Classes of substances
-
Acetophenones
-
1-aryloxy-3-amino-2-propanol
-
Butyric acid anilides
-

-
Synthesis pathway
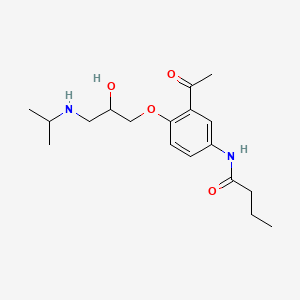

| Synthesis a) |
|---|
   |
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| Germany | Printemps | Bayer |
| Sali-Printemps | – “- | |
| Tredalat | – “- | |
| France | Sektral | Sanofi-Aventis |
| United Kingdom | Sekadreks | Aventis |
| Sektral | Aventis | |
| Italy | Atsekor | SPA |
| AlOl | SIT | |
| Printemps | Bayropharm | |
| Sektral | Rhône-Poulenc Rorer | |
| Japan | Atsetanol | Sanofi-Aventis Chugai |
| Sektral | Organon | |
| USA | – “- | Wyeth-Ayerst |
| Ukraine | No | No |
Formulations
-
ampoule 25 mg;
-
Capsules 100 mg, 200 mg;
-
Tablets of 200 mg, 400 mg, 500 mg (as hydrochloride)
Pharmacology
Acebutolol is a cardioselective beta blocker with ISA (intrinsic sympathomimetic activity; see article on pindolol). It is therefore more suitable than non cardioselective beta blockers, if a patient with asthma or chronic obstructive pulmonary disease (COPD) needs treatment with a beta blocker. (For these reasons, it may be a beta-blocker of choice in inclusion in Polypill strategies). In doses lower than 800mg daily its constricting effects on the bronchial system and smooth muscle vessels are only 10% to 30% of those observed under propranolol treatment, but there is experimental evidence that the cardioselective properties diminish at doses of 800mg/day or more.
The drug has lipophilic properties, and therefore crosses the blood–brain barrier. Acebutolol has no negative impact on serum lipids (cholesterol and triglycerides). No HDL decrease has been observed. In this regard, it is unlike many other beta blockers which have this unfavourable property.
The drug works in hypertensive patients with high, normal, or low renin plasma concentrations, although acebutolol may be more efficient in patients with high or normal renin plasma concentrations. In clinically relevant concentrations, a membrane-stabilizing effect does not appear to play an important role.
Pharmacokinetics
Acebutolol is well absorbed from the GI tract, but undergoes substantial first-pass-metabolization, leading to a bioavailability of only 35% to 50%. Peak plasma levels of acebutolol are reached within 2 to 2.5 hours after oral dosing. Peak levels of the main active metabolite, diacetolol, are reached after 4 hours. Acebutolol has a half-life of 3 to 4 hours, and diacetolol a half-life of 8 to 13 hours.
Acebutolol undergoes extensive hepatic metabolization resulting in the desbutyl amine acetolol which is readily converted into diacetolol. Diacetolol is as active as acebutolol (equipotency) and appears to have the same pharmacologic profile. Geriatric patients tend to have higher peak plasma levels of both acebutolol and diacetolol and a slightly prolonged excretion. Excretion is substantially prolonged in patients with renal impairment, and so a dose reduction may be needed. Liver cirrhosis does not seem to alter the pharmacokinetic profile of the parent drug and metabolite.
Indications
- hypertension
- ventricular and atrial cardiac arrhythmia
- acute myocardial infarction in high-risk patients
- Smith-Magenis syndrome
Contraindications
- Stable or Unstable Angina (due to its partial agonist or ISA activity)
Contraindications and Precautions
Acebutolol may not be suitable in patients with Asthma bronchiale or COPD due to its bronchoconstricting (β2 antagonistic) effects.
Side effects
The development of anti-nuclear antibodies (ANA) has been found in 10 to 30% of patients under treatment with acebutolol. A systemic disease with arthralgic pain and myalgias has been observed in 1%. A lupus erythematosus-like syndrome with skin rash and multiforme organ involvement is even less frequent. The incidence of both ANA and symptomatic disease under acebutolol is higher than under Propranolol. Female patients are more likely to develop these symptoms than male patients. Some few cases of hepatotoxicity with increased liver enzymes (ALT, AST) have been seen. Altogether, 5 to 6% of all patients treated have to discontinue acebutolol due to intolerable side effects. When possible, the treatment should be discontinued gradually in order to avoid a withdrawal syndrome with increased frequency of angina and even precipitation of myocardial infarction.
Dosage
The daily dose is 200mg – 1,200mg in a single dose or in 2 divided doses as dictated by the severity of the condition to be treated. Treatment should be initiated with low doses, and the dose should be increased cautiously according to the response of the patient. Acebutolol is particularly suitable for antihypertensive combination treatment with diuretics, if acebutolol alone proves insufficient. In some countries injectable forms for i.v.-injection with 25mg acebutolol exist, but these are only for cases of emergency under strict clinical monitoring. The initial dose is 12.5 to 25mg, but additional doses may be increased to 75 to 100mg, if needed. If further treatment is required, it should be oral.

Sectral (acebutolol HCl) is a selective, hydrophilic beta-adrenoreceptor blocking agent with mild intrinsic sympathomimetic activity for use in treating patients with hypertension and ventricular arrhythmias. It is marketed incapsule form for oral administration. Sectral (acebutolol) capsules are provided in two dosage strengths which contain 200 or 400 mg of acebutolol as the hydrochloride salt. The inactive ingredients present are D&C Red 22, FD&C Blue 1, FD&C Yellow 6, gelatin, povidone, starch, stearic acid, and titanium dioxide. The 200 mg dosage strength also contains D&C Red 28 and the 400 mg dosage strength also contains FD&C Red 40. Acebutolol HCl has the following structural formula:
 Acebutolol HCl is a white or slightly off-white powder freely soluble in water, and less soluble in alcohol. Chemically it is defined as the hydrochloride salt of (±)N-[3-Acetyl-4-[2- hydroxy-3-[(1-methylethyl)amino]propoxy]phenyl] butanamide.
Acebutolol HCl is a white or slightly off-white powder freely soluble in water, and less soluble in alcohol. Chemically it is defined as the hydrochloride salt of (±)N-[3-Acetyl-4-[2- hydroxy-3-[(1-methylethyl)amino]propoxy]phenyl] butanamide.
External links
EXAMPLE 4 Crude 5-butyramido-2′-(2,3-epoxypropoxy)acetophenone (16 g), isopropylamine (20 g.) and ethanol (100 ml.) were heated together under reflux for 4 hours. The reaction mixture was concentrated under reduced pressure and theresidual oil was dissolved in N hydrochloric acid. The acid solution was extracted with ethyl acetate, theethyl acetate layers being discarded. The acidic solution was brought to pH 11 with 2N aqueous sodium hydroxide solution and then extracted with chloroform. The dried chloroform extracts were concentrated under reduced pressure to give an oil which was crystallised from a mixture of ethanol and diethyl ether to give 5′-butyramido-2- (2-hydroxy-3-isopropylaminopropoxy)acetophenone (3 g.), m.p. 119l23C.
Similarly prepared was cyclohexylamino-2-hydroxypropoxy)acetophenone, m.p. 112113C.
Crude 5-butyramido-2-(2,3-epoxypropoxy)acetophenone used as startingmaterial was prepared as follows:
p-Butyramidophenol (58 g.; prepared according to Fierz-David and Kuster, loc.cit.), acetyl chloride (25.4 g.) and benzene (500 ml.) were heated together under reflux until a solution formed (12 hours). This solution was cooled and treated with water. The benzene layer was separated and the aqueous layer was again extracted with benzene.
The combined benzene extracts were dried and evaporated to dryness under reduced pressure to give pbutyramidophenyl acetate (38 g.) as an off-white solid, mp. 102-l03C. A mixture of p-butyramidophenyl acetate (38 g.), aluminium chloride (80 g.) and 1,l,2,2-tetrachloroethane (250 ml.) was heated at 140C. for 3 hours. The reaction mixture was cooled and treated with iced water. The tetrachloroethane layer was separated and the aqueous layer was extracted with chloroform. The combined organic layers were extracted with 2N aqueous sodium hydroxide and the alkaline solution was acidified to pH 5 with concentrated hydrochloric acid. The acidified solution was extracted with chloroform and the chloroform extract was dried and concentrated under reduced pressure to give 5′-butyramido-2-hydroxyacetophenone 15.6 g.), m.p. 114l17C. A solution of 5-butyramido-2′- hydroxyacetophenone (15.6 g.) in ethanol (100 ml.) was added to an ethanolic solution of sodium ethoxide which was prepared from sodium (1.62 g.) and ethanol (100 ml.). The resulting solution’was evaporated to dryness under reduced pressure and dimethylformamide (100 ml.) was added to the solid’residue. Ap-
proximately ml. of dimethylformamide was removed by distillation under reduced pressure. Epichlorohydrin ml.) was added and the solution was heated at 100C. for 4 hours. The solution was concentrated under reduced pressure to give a residual oil which was treated with water to’give a solid. The solid was dissolved in ethanol and the resulting solution was treated with charcoal, filtered and concentrated under reduced pressure to give crude 5-butyramido- 2-(2,3-epoxypropoxy)acetophenone (16 g.), m.p. 1101 16C.
The crude compound may be purified by recrystallisation from ethyl acetate, after, treatment with decolourizing charcoal, to give pure 5′-butyramido-2′-(2,3- epoxypropoxy)acetophenone, m.p. 136138C.
Links
-
GB 1247384 (May & Baker; appl. 22.12.1967).
-
DAS 1,815,808 (May & Baker; appl. 19.12.1968; GB -prior. 22.12.1967, 5/14/1968, 2.8.1968).
-
US 3,726,919 (May & Baker; 10/4/1973; GB -prior. 22.12.1967, 05.14.1968, 2.8.1968).
-
US 3,857,952 (May & Baker; 31.12.1974; GB -prior. 22.12.1967, 14.05.1968, 2.8.1968).


 |
CLINICAL TRIALS………… JAPAN
DRUG REGULATORY AFFAIRS INTERNATIONAL

Clinical trials
Clinical trials, also known as clinical studies, are scientific studies of drugs, medical devices or other treatments in humans. These studies are most often conducted for the following reasons:
- To verify the safety and effectiveness of potential drugs, medical devices or other treatments,
- To compare trial-treatments against existing treatments,
- To determine better ways to use treatments to make them more effective, easier to use, or to decrease side effects,
- To determine how best to use a treatment in a specific population. For example, in children or in a particular ethnic group.
Clinical trials for new drug development
There are three main stages in clinical trials for drug development: Phases I, II and III. Phase I clinical trials are conducted with 50~200 healthy participants and involve incremental dose increases of investigational drug within a predefined dose range to evaluate tolerability and safety, monitor food and drug interaction, as well…
View original post 2,862 more words
The Procedure for Manufacturing Drugs in Mie Prefecture, Japan.
DRUG REGULATORY AFFAIRS INTERNATIONAL
The Procedure for Manufacturing Drugs in Mie Prefecture, Japan.
The following details the necessary procedure for the commencement of manufacture (or importing) of drugs in Mie Prefecture, Japan.
Note: The procedures described below are applicable in Mie Prefecture, Japan, as of April 2002. Due to future amendments and the disparities of laws in different prefectures, it is necessary to be informed as to the correct application procedures directly by the relevant prefecture.
1. For Manufacture (or Importing) of Drugs
Approval for the manufacture (importing) of each item, and a manufacturing (importing) license are required for the manufacture (or importing) of drugs.
| Drug Manufacture | Approval | The quality, effectiveness and safety of the drug under application must pass the examination. However, drugs listed on the Pharmacopoeia of Japan do not require approval. |
|---|---|---|
| License | The structural conditions (building and facilities) and human resource requirements (e.g. Administrators) of the drug manufacturing facilities must pass… |
View original post 2,249 more words
Bacteria responsible for gum disease facilitates development and progression of rheumatoid arthritis
10 AUG 2012
Gum disease 4 times as common in rheumatoid arthritis patients
Might be potential trigger and/or help sustain inflammation suggest researchers
Gum disease is not only four times as common among patients with the autoimmune disease rheumatoid arthritis as it is among their healthy peers, but it also tends to be more severe, indicates a small study published online in the Annals of the Rheumatic Diseases.
The researchers base their findings on 91 adults with confirmed rheumatoid arthritis (RA) and a comparison group of 93 healthy people, matched for age and sex.
All participants were non-smokers, as smoking is a known risk factor for rheumatoid arthritis. And it is strongly associated with the production of antibodies, indicative of a systemic reaction to a person’s own proteins (ACPAs), and which often predates the development of rheumatoid arthritis by several years.
And none had been treated with arthritis drugs known…
View original post 332 more words
