
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

Cefuroxime Axetil
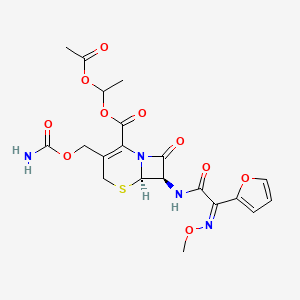
Cefuroxime Axetil (1-(acetyloxy) ethyl ester of cefuroxime, is (RS)-1-hydroxyethyl (6R,7R)-7-[2-(2-furyl)glyoxyl-amido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylate, 7 2 -(Z)-(O-methyl-oxime), 1-acetate 3-carbamate.
Its molecular formula is C 20 H 22 N 4 O 10S, and it has a molecular weight of 510.48.
Cefuroxime Axetil is used orally for the treatment of patients with mild-to-moderate infections, caused by susceptible strains of the designated microorganisms.
Cefuroxime axetil is a second generation oral cephalosporin antibiotic. It was discovered by Glaxo now GlaxoSmithKline and introduced in 1987 as Zinnat.[1] It was approved by FDA on Dec 28, 1987.[2] It is available by GSK as Ceftin in US[3] and Ceftum in India.[4]
It is an acetoxyethyl ester prodrug of cefuroxime which is effective orally.[5] The activity depends on in vivo hydrolysis and release of cefuroxime.
Cefuroxime is chemically (6R, 7R)-3-carbamoyloxymethyl-7-[(Z)-2-(fur-2-yl)-2-methoxy-iminoacetamido] ceph-3-em-4-carboxylic acid and has the structural Formula II:

Cefuroxime axetil having the structural Formula I:

is the 1-acetoxyethyl ester of cefuroxime, a cephalosporin antibiotic with a broad spectrum of activity against gram-positive and gram negative micro-organisms.
This compound as well as many other esters of cefuroxime, are disclosed and claimed in U.S. Pat. No. 4,267,320. According to this patent, the presence of an appropriate esterifying group, such as the 1-acetoxyethyl group of cefuroxime axetil, enhances absorption of cefuroxime from the gastrointestinal tract, whereupon the esterifying group is hydrolyzed by enzymes present in the human body.
Because of the presence of an asymmetric carbon atom at the 1-position of the 1-acetoxyethyl group, cefuroxime axetil can be produced as R and S diastereoisomers or as a racemic mixture of the R and S diastereoisomers. U.S. Pat. No. 4,267,320 discloses conventional methods for preparing a mixture of the R and S isomers in the crystalline form, as well as for separating the individual R and S diastereoisomers.
The difference in the activity of different polymorphic forms of a given drug has drawn the attention of many workers in recent years to undertake the study on polymorphism. Cefuroxime axetil is the classical example of amorphous form exhibiting higher bioavailability than the crystalline form.
U.S. Pat. No. 4,562,181 and the related U.S. Pat. Nos. 4,820,833; 4,994,567 and 5,013,833, disclose that cefuroxime axetil in amorphous form, essentially free from crystalline material and having a purity of at least 95% aside from residual solvents, has a higher bioavailability than the crystalline form while also having adequate chemical stability.
These patents disclose that highly pure cefuroxime axetil can be recovered in substantially amorphous form from a solution containing cefuroxime axetil by spray drying, roller drying, or solvent precipitation. In each case, crystalline cefuroxime axetil is dissolved in an organic solvent and the cefuroxime axetil is recovered from the solution in a highly pure, substantially amorphous form.
Another U.S. Pat. No. 5,063,224 discloses that crystalline R-cefuroxime axetil which is substantially free of S-isomer is readily absorbed from the stomach and gastrointestinal tract of animals and is therefore ideally suited to oral therapy of bacterial infections.
According to this patent, such selective administration of R-cefuroxime axetil results in surprisingly greater bioavailability ability of cefuroxime, and thus dramatically reduces the amount of unabsorbable cefuroxime remaining in the gut lumen, thereby diminishing adverse side effects attributable to cefuroxime.
British Patent Specification No. 2,145,409 discloses a process for obtaining pure crystalline cefuroxime axetil and is said to be an improvement over British Patent Specification No. 1,571,683. Sodium cefuroxime is used as the starting material in the disclosed specification, which in turn, is prepared from either 3-hydroxy cefuroxime or cefuroxime.
Said process involves an additional step of preparing sodium cefuroxime, and therefore is not economical from commercial point of view.
CEFTIN (cefuroxime axetil) Tablets and CEFTIN (cefuroxime axetil) for Oral Suspension contain cefuroxime as cefuroxime axetil. CEFTIN (cefuroxime axetil) is a semisynthetic, broad-spectrum cephalosporin antibiotic for oral administration.
Chemically, cefuroxime axetil, the 1-(acetyloxy) ethyl ester of cefuroxime, is (RS)-1-hydroxyethyl (6R,7R)-7-[2-(2-furyl)glyoxyl-amido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylate, 72-(Z)-(O-methyl-oxime), 1-acetate 3-carbamate. Its molecular formula is C20H22N4O10S, and it has a molecular weight of 510.48.
Cefuroxime axetil is in the amorphous form and has the following structural formula:
 |
CEFTIN (cefuroxime axetil) Tablets are film-coated and contain the equivalent of 250 or 500 mg of cefuroxime as cefuroxime axetil. CEFTIN (cefuroxime axetil) Tablets contain the inactive ingredients colloidal silicon dioxide, croscarmellose sodium, hydrogenated vegetable oil, hypromellose, methylparaben, microcrystalline cellulose, propylene glycol, propylparaben, sodium benzoate, sodium lauryl sulfate, and titanium dioxide.
CEFTIN (cefuroxime axetil) for Oral Suspension, when reconstituted with water, provides the equivalent of 125 mg or 250 mg of cefuroxime (as cefuroxime axetil) per 5 mL of suspension. CEFTIN (cefuroxime axetil) for Oral Suspension contains the inactive ingredients acesulfame potassium, aspartame, povidone K30, stearic acid, sucrose, tutti-frutti flavoring, and xanthan gum.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-Acetoxyethyl (6R,7R)-3-[(carbamoyloxy)methyl]-7-{[(2Z)-2-(2-furyl)-2-(methoxyimino)acetyl]amino}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate | |
| Clinical data | |
| Identifiers | |
| PubChem | CID 6321416 |
| ChemSpider | 4882027 |
| ChEMBL | CHEMBL1095930 |
| Synonyms | Cefuroxime 1-acetoxyethyl ester |
| Chemical data | |
| Formula | C20H22N4O10S |
| Mol. mass | 510.475 g/mol |

dsc

http://www.google.com/patents/US5013833


http://www.google.com/patents/US6833452
EXAMPLE 1
Dicyclohexylamine (17.2 g) in N,N-dimethylacetamide (50 ml) was added to a solution of cefuroxime acid (42.4 g) in N,N-dimethylacetamide (300 ml) at about −10° C. (R,S)1-Acetoxethylbromide (33.4 g) in N,N-dimethylacetamide (50 ml) was added to the above solution and the reaction mixture was stirred for 45 minutes at about −3 to 0° C. Potassium carbonate (1.1 g) was added to the reaction mixture and it was further stirred at that temperature for about 4 hours. The reaction mixture was worked up by pouring into it ethyl acetate (1.0 It), water (1.2 It) and dilute hydrochloric acid (3.5% w/w, 200 ml). The organic layer was separated and the aqueous layer was again extracted with ethyl acetate. The combined organic extracts were washed with water, dilute sodium bicarbonate solution (1%), sodium chloride solution and evaporated in vacuo to give a residue. Methanol was added to the residue and the crude product was precipitated by adding water.
The resulting precipitate was filtered off and recrystallized from the mixture of ethylacetate, methanol and hexane. The precipitated product was filtered, washed and dried to give pure crystalline cefuroxime axetil (42.5 g).
Assay (by HPLC on anhydrous basis)-98.2% w/w; Diastereoisomer ratio-0.53; Total related substances-0.48% w/w.
…………………………………
http://www.google.com/patents/EP1409492B1?cl=en
-
The present invention relates to an improved method for synthesis of cefuroxime axetil of formula (I) in high purity substantially free of the corresponding 2-cephem(Δ2)-ester of formule (II) and other impurities. The compound produced is valuable as a prodrug ester of the corresponding cephalosporin- 4-carboxylic acid derivative i. e. cefuroxime, particularly suitable for oral administration in various animal species and in man for treatment of infections caused by gram-positive and gram-negative bacteria.


BACKGROUND OF THE INVENTION
-
[0002]One of the ways to improve the absorption of cephalosporin antibiotics which are poorly absorbed through the digestive tract is to prepare and administer the corresponding ester derivatives at the 4-carboxylic acid position. The esters are then readily and completely hydrolysed in vivoby enzymes present in the body to regenerate the active cephalosporin derivative having the free carboxylic acid at the 4-position.
-
[0003]Among the various ester groups that can be prepared and administered only a selected few are biologically acceptable, in addition to possessing high antibacterial activity and broad antibacterial spectrum. Clinical studies on many such potential “prodrug esters” such as cefcanel daloxate (Kyoto), cefdaloxime pentexil tosilate (Hoechst Marion Roussel) and ceftrazonal bopentil (Roche), to name a few have been discontinued, while ceftizoxime alapivoxil ((Kyoto) in under Phase III clinical studies. The cephalosporin prodrug esters which have been successfully commercialised and marketed include cefcapene pivoxil (Flomox® , Shionogi), cefditoren pivoxil (Spectracef®, Meiji Seika), cefetamet pivoxil (Globocef®, Roche), cefotiam hexetil (Taketiam®, Takeda), cefpodoxime proxetil (Vantin®, Sankyo), cefteram pivoxil (Tomiron®, Toyama) and cefuroxime axetil (Ceftin® and Zinnat®, Glaxo Wellcome).
-
[0004]Typically, such (3,7)-substituted-3-cephem-4-carboxylic acid esters represented by formula (I A) are synthesised by reacting the corresponding (3,7)-substituted-3- cephem-4-carboxylic acid derivative of formula (III A), with the desired haloester compound of formula (IV A) in a suitable organic solvent. The synthesis is summarised in Scheme-I, wherein in compounds of formula (I A), (II A), (III A) and (IV A) the groups R1 and R2 at the 3- and 7-positions of the β-lactam ring are substituents useful in cephalosporin chemistry ; R3 is the addendum which forms the ester function and X is halogen.

-
[0005]However, the esterification reaction which essentially involves conversion of a polar acid or salt derivative to a neutral ester product invariably produces the corresponding (3,7)-substituted-2-cephem (Δ2)-4-carboxylic acid ester derivative of formula (II A) in varying amounts, arising out of isomerisation of the double bond from the 3-4 position to the 2-3 position as well as other unidentified impurities.
-
[0006]It has been suggested [D. H. Bentley, et. al., Tetrahedron Lett., 1976, 41, 3739] that the isomerisation results from the ability of the 4-carboxylate anion of the starting carboxylic acid to abstract a proton from the 2-position of the 3-cephem-4-carboxylic acid ester formed, followed by reprotonation at 4-position to give the said Δ2-ester. It has also been suggested [R. B. Morin, et. al., J. Am. Chem. Soc., 1969, 91, 1401 ; R. B. Woodward, et. al., J. Am. Chem. Soc., 1966, 88, 852] that the equilibrium position for isomerisation is largely determined by the size of the ester addendum attached at the 4-carboxylic acid position.
-
[0007]The 2-cephem-4-carboxylic acid esters of formula (II A) are not only unreactive as antibacterial agents but are undesired by-products. Pharmacopoeias of many countries are very stringent about the presence of the 2-cephem analogues in the finished sample of (3,7)-substituted-3-cephem-4-carboxylic acid esters and set limits for the permissible amounts of these isomers. Due to the structural similarity of the 2-cephem and 3-cephem analogues it is very difficult to separate the two isomers by conventional methods, such as chromatography as well as by fractional crystallisation. In addition to this removal of other unidentified impurities formed in the reaction, entails utilisation of tedious purification methods, thus overall resulting in,
- a) considerable loss in yield, increasing the cost of manufacture and
- b) a product of quality not conforming to and not easily amenable for upgradation to pharmacopoeial standards.
-
[0008]Several methods are reported in the prior art for synthesis of cefuroxime axetil of formula (I) and various (3,7)-substituted-3-cephem-4-carboxylic acid esters of formula (I A), with attempts to minimise the unwanted Δ2-isomers formed in such reactions as well as conversion of the Δ2-isomer thus formed back to the desired Δ3– isomer. The prior art methods can be summarised as follows:
- i) US Patent No, 4 267 320 (Gregson et. al.) describes a method for synthesis of cefuroxime axetil comprising reaction of cefuroxime acid or its alkali metal salts or onium salts with (R,S)-1-acetoxyethyl bromide in an inert organic solvent selected from N,N-dimethylacetamide, N,N-dimethylformamide, dimethyl sulfoxide, acetone, acetonitrile and hexamethylphosphoric triamide at a temperature in the range of -50 to +1150° C. The patent mentions that when alkali metal salts, specially potassium salt of cefuroxime acid are employed the reaction can be carried out in a nitrile solvent in the presence of a crown ether. When cefuroxime acid is employed the reaction is carried out in the presence of a weak inorganic base such as sodium carbonate or potassium carbonate, which is added prior to the addition of the haloester. The patent further mentions that the use of potassium carbonate in conjunction with the haloester, specially the bromo or iodo ester is preferred since it helps to minimise the formation of the Δ2-isomer. Ideally, substantially equivalent amounts of cefuroxime acid and the base is employed.
The US Patent No. 4 267 320 also describes methods, wherein the said esterification is carried out in the presence of an acid binding agent, which serve to bind hydrogen halide liberated in the reaction, thereby controlling the formation of the Δ2-isomer. The acid binding agents that are utilised include a tertiary amine base such as triethylamine or N, N-dimethylamine ; an inorganic base such as calcium carbonate or sodium bicarbonate and an oxirane compound such as ethylene oxide or propylene oxide.
However, from the examples provided in the above patent the yield of cefuroxime axetil and other (3,7)-substituted-3-cephem-4-carboxylic acid esters obtained is found to be only of about 50%, implying formation of substantial amounts of impurities in the reaction. Indeed, when cefuroxime acid is reacted with (R,S)-1-acetoxyethyl bromide in the presence of 0.55 molar equivalents of sodium carbonate or potassium carbonate in N,N-dimethylacetamide as solvent, as per the process disclosed in this patent, it is found that substantial amounts of the Δ2-isomer in a proportion ranging from 10-22% is formed, in addition to other unknown impurities. Also, substantial amounts of the starting cefuroxime acid remains unreacted even after 5 hrs of reaction. Isolation of the product generally affords a gummy material, which resists purification even after repeated crystallisations.
Moreover, the use of the acid binding agents mentioned in the above patent, specially tertiary amines and inorganic bases lead to cleavage of the β-lactam ring and also promote the undesired Δ2-isomerisation, thereby enhancing the level of impurities formed in the reaction. - ii) GB Patent No. 2 218 094 describes a method by which the Δ2-isomers formed during esterification can be converted back to the desired Δ3-isomers. The method comprises of oxidation of the dihydrothiazine ring in the mixture of Δ2– and Δ3– cephalosporin acid esters to the corresponding sulfoxide derivatives with suitable oxidising agents, whereby the Δ2-isomer gets isomerised to the corresponding Δ3-isomer during oxidation and the Δ3– cephalosporin acid ester sulfoxide is isolated. The sulfide group is regenerated back by reduction of the sulfoxide function with suitable reducing agents.
Typically, the oxidation is carried out using m-chloroperbenzoic acid and the reduction achieved by use of an alkali metal halide in presence of acetyl chloride in presence of an inert organic solvent or by use of a phosphorous trihalide.
Although, this method provides the desired Δ3-isomers in good purity, it cannot be considered as an industrially feasible method since it involves a two step process of oxidation and reduction, isolation of the intermediate products at each stage and necessary purifications, all resulting in considerable loss of the desired product and increase in the cost of manufacture. Moreover, the use of acetyl halide and phosphorous trihalide in the reduction step cannot be applied to cephalosporin derivatives that are sensitive to these reagents.
A similar method has been reported by Kaiser et. al. in J. Org. Chem., 1970, 35, 2430. - (iii)Mobasherry et. al. in J. Org. Chem., 1986, 51, 4723 describe preparation of certain Δ3-cephalosporin-4-carboxylic acid esters by reaction of the corresponding 3-cephem-4-carboxylic acids (in turn prepared form the corresponding carboxylic acid alkali metal salts) with an haloester in presence of 1.1 eq of sodium carbonate in the presence 1.2-1.5 eq of an alkyl halide and in presence of a solvent comprising of a mixture of N,N-dimethylformamide and dioxane. The authors claim that the method provides of Δ3– cephalosporin-4-carboxylic acid esters unaccompanied by the corresponding Δ2-isomer.
However, the method involves an additional step in that the starting 3-cephem-4-carboxylic acid ester derivatives are obtained from the corresponding alkali metal salts prior to reaction. In addition, longer reaction times of about 24 hrs coupled with the fact that it utilises dioxane, a potent carcinogen, not recommended by International Conference on Harmonisation (ICH) on industrial scale renders the method unattractive commercially.
Moreover, on duplication of the method exactly as described in the article it is found that about 3-4% of the corresponding Δ2-isomer is indeed formed in the reaction in addition to other unidentified impurities. Also, substantial amounts of the starting cephalosporin carboxylic acid is recovered unreacted. - (iv)Shigeto et. al. in Chem. Pharm. Bull., 1995, 43(11), 1998 have carried out the esterification of certain 7-substituted-3-cephem-4-carboxylic acid derivatives with 1-iodoethyl isopropyl carbonate in a solvent system containing a mixture of N, N-dimethylformamide and dioxane in a 3:5 ratio. A conversion to the corresponding 3-cephem- 4-carboxylate ester was achieved in only 34%, out of which the Δ2-isomer amounted to about 8%.
Esterification of 7-formamido-3-(N,N-dimethylcarbamoyloxy)methyl-3-cephem-4-carboxylic acid sodium salt with a suitable haloester in presence of solvents such as N, N-dimethylacetamide and N, N-dimethylformamide, with formation of about 0.8 to 3.0% of the Δ2-isomer is also reported in the above article by Shigeto et. al. The 7-formamido group was cleaved under acidic conditions to give the corresponding 7-amino derivative contaminated with only about 0.4% of the corresponding Δ2-isomer. The minimisation of the percentage of Δ2-isomer is attributed to the relative unstability of 7-amino-2-cephem-4-carboxylic acid esters in acidic conditions, facilitating isomerisation of the 2-cephem intermediate to the 3-cephem derivative.
However, the method does not have a general application, especially for synthesis of commercially valuable cephalosporin derivatives containing hydroxyimino or alkoxyimino substituents in the 7-amino side chain addendum, since these oxyimino functions exhibit a tendency to isomerise from the stable (Z)-configuration to the relatively undesirable(E)-configuration under acidic conditions. This would render separation of the two isomers cumbersome. Moreover, longer reaction times of about 18-20 hrs to effect the isomerisation of the double bond from the 2- position to the 3-position and use of toxic dioxane as solvent impose further limitations on the method.
(v) Demuth et. al. in J. Antibiotics, 1991, 44, 200 have utilised the N, N-dimethylformamide-dioxane system in the coupling of 1-iodocephem-4-nitrobenzyl ester with naldixic acid sodium salt and recommend use of dioxane since it reduces the basicity of the quinolone carboxylate and lowers the polarity of the reaction medium.
However, low yields of about 35% and use of toxic dioxane makes the method of little industrial application. - (vi) Wang et. al. in US Patent No. 5 498 787 claim a method for preparation of certain (3,7)-substituted-3-cephem-4-carboxylic acid prodrug esters, unaccompanied by the analogous 2-cephem esters comprising reaction of the corresponding (3,7)-substituted-3-cephem-4-carboxylic acid alkali metal salts with suitable haloesters in the presence of catalytic amounts of a quaternary ammonium or quarternary phosphonium salt. Among the prodrug esters covered in this patent is cefuroxime axetil.
US Patent No. 5 498 787 claims that among the quarternary ammonium salts, such salts with acid counter ion, specially tetrabutyl ammonium sulfate (TBA+HSO4 –) is the most preferred. When the molar ratio of TBA+HSO4 –/cefuroxime sodium was above 0.40 no Δ2-isomer was detected, when the said molar ratio was below 0.40 and near about 0.20 the molar ratio of Δ2/Δ3 isomers formed was about 2.0%. When no TBA+HSO4 – was added the molar ratio of Δ2/Δ3 isomers formed was about 10.0%. Examples 1 and 2 of this patent illustrate the esterification of cefuroxime sodium in presence of TBA+HSO4 – and indicate that the Δ2-isomer was not detected after 3-12 hours of reaction. The same patent also establishes the superiority of TBA+HSO4 – over other salts, specially tetrabutyl ammonium iodide (TBA+I–) since use of the latter salt resulted in considerable isomerisation of the double bond giving the undesired Δ2-isomer in predominant amounts.
The present inventors have, however, found that when cefuroxime sodium is reacted with (R,S)- 1-acetoxyethyl bromide in the presence of tetrabutylammonium sulfate (TBA+HSO4 –) as per the method covered in US Patent No. 5,498 787 the same did not necessarily result in the production of the desired Δ3isomer free of the undesired Δ2 isomer and other impurities. Also, such process had limitations in that the reaction could not be completed at times even at the end of 5.0hrs. Moreover, the separation of the impurities; from the product proved cumbersome and could not be removed from the product even after successive crystallisations. - (vii) H. W. Lee et. al., Syntheic Communications, 1998, 28(23), 4345-4354 have demonstrated a method essentially similar to that claimed in US Patent No. 5 498 787 . The method of preparation of various esters of cefotaxime consists of reacting cefotaxime sodium with the requisite haloester compound in a suitable solvent and in presence of quarternary ammonium salts as phase transfer catalysts. It is claimed that when no quarternary ammonium salts are added the molar ratio (%) of Δ2/Δ3 isomers formed is about 10%. The formation of Δ2– isomer is minimised when quarternary ammonium salts are added and particularly when the molar ratio of TBA+HSO4 –/cefotaxime sodium employed is 0.80 the formation of the Δ2– isomer is completely inhibited.
However, this method requires long hours (~18-24 hrs) and is carried out at higher temperatures (40-45° C) and as such may not be suitable for cephalosporin derivatives that are sensitive to heat. - (viii)H. W. Lee et. al. in Synthetic Communications, 1999, 29(11), 1873-1887 demonstrate a method for preparation of number of (3,7)-substituted-3-cephem-4-carboxylic acid esters comprising reacting the corresponding (3,7)-substituted-3-cephem-4-carboxylic acid derivatives with a base selected form cesium carbonate or cesium bicarbonate either used alone or in combination with potassium carbonate, sodium carbonate, potassium bicarbonate and sodium bicarbonate. The authors established that the formation of Δ2– isomers could be minimised by utilisation of a solvent combination ofN, N-dimethyl formamide and dioxane. The use of the latter mentioned solvent i. e. dioxane was expected to lower polarity of the reaction medium and thereby reduce the basicity of the transient 3-cephem-4-carboxylate anion formed in the reaction and thus preventing the isomerisation of the double bond from the 3-4 position to the 2-3 position.
The formation of the Δ2– isomer was found to be dependent on the amount of dioxane in the solvent mixture, the more the proportion of dioxane lesser the degree of isomerisation.
However, yields of representative esters obtained by the method are in the range of 45-85 %, implying that the reaction is accompanied by formation of substantial amounts of impurities and that the isomerisation is dependent on the nature of the substituent at 3α-position of the cephalosporin nucleus as well as on the nature of the haloester employed. Moreover, the method utilises dioxane, not desirable for reasons mentioned herein earlier and expensive cesium salts. This method, therefore, also has limited application. - (ix) Y.S. Cho et. al., in Korean J. Med. Chem., 1995, 5(1), 60-63 describe synthesis of several cephalosporin prodrug esters and their efficacy on oral administration. The esters were synthesised by reacting the corresponding cephalosporin-4-carboxylic acid derivative with the respective haloester derivative in presence of cesium carbonate and N, N-dimethylacetamide. The yields of the ester derivatives obtained are in the range of only 25-56%, indicating formation of substantial amounts of impurities in the reaction.
- i) US Patent No, 4 267 320 (Gregson et. al.) describes a method for synthesis of cefuroxime axetil comprising reaction of cefuroxime acid or its alkali metal salts or onium salts with (R,S)-1-acetoxyethyl bromide in an inert organic solvent selected from N,N-dimethylacetamide, N,N-dimethylformamide, dimethyl sulfoxide, acetone, acetonitrile and hexamethylphosphoric triamide at a temperature in the range of -50 to +1150° C. The patent mentions that when alkali metal salts, specially potassium salt of cefuroxime acid are employed the reaction can be carried out in a nitrile solvent in the presence of a crown ether. When cefuroxime acid is employed the reaction is carried out in the presence of a weak inorganic base such as sodium carbonate or potassium carbonate, which is added prior to the addition of the haloester. The patent further mentions that the use of potassium carbonate in conjunction with the haloester, specially the bromo or iodo ester is preferred since it helps to minimise the formation of the Δ2-isomer. Ideally, substantially equivalent amounts of cefuroxime acid and the base is employed.

Example – 1
- Preparation of (R, S -1-Acetoxyethyl-3-carbamoyloxymethyl-7-[(Z)-2-(fur-2-yl)-2-methoxyiminoacetamido]ceph-3-em-4-carboxylate (Cefuroxime axetil, I) :
Without use of GrouplI
- /
II metal phosphate and C1-4 alcohol
-
[0045](R, S)-1-Acetoxyethyl bromide (1.6gms; 0.0094moles) was added to a mixture of cefuroxime acid (2gms; 0.0047moles) and potassium carbonate (0.326gms; 0.00235moles) in N,N-dimethylacetamide (10 ml) at 5°C and stirred at 0 to 20° C for 180 minutes Ethyl acetate was added to the reaction mixture, followed by 3% aqueous sodium bicarbonate solution (15ml). The organic layer containing the title product, Δ2 isomer (8.51%) and unidentified impurities (X1-1.86% and X2 – 3.54%) was separated and washed with 10% aqueous NaCl solution. The organic solvent was evaporated off under vacuum to give 1.08gms (44.90%) of the title compound as a gummy solid.
-
[0046]HPLC analysis : Purity (compound I) – 89.11% ; Impurities : Δ2 isomer (II) – 8.51%, X1 – 1.86% and X2 – 3.54%
………………………………..


References
- “Our history – About GSK”. GlaxoSmithKline.
- http://www.drugs.com/monograph/cefuroxime-axetil.html
- https://www.gsksource.com/gskprm/en/US/adirect/gskprm?cmd=ProductsByName#C
- “Our products”. GlaxoSmithKline.
- Walter Sneader. Drug Discovery: A History. John Wiley, Chichester, UK. ISBN 0-471-89979-8.
Literature References:
Prepn: M. C. Cook et al., DE 2439880; eidem, US 3974153 (1973, 1976 both to Glaxo).
Prepn of the 1-acetoxyethyl ester: M. Gregson, B. Sykes, DE 2706413; eidem, US 4267320 (1977, 1981 both to Glaxo).
In vitro studies: C. H. O’Callaghan et al., Antimicrob. Agents Chemother. 9, 511 (1976); R. N. Jones et al., ibid. 12, 47 (1977).
In vitro antibacterial activity, human pharmacokinetics: C. H. O’Callaghan et al., J. Antibiot. 29, 29 (1976).
Pharmacology: H. Freiesleben et al., Proc. 10th Int. Congr. Chemother., Zürich, 1977 (Am. Soc. for Microbiol., Washington, 1978) II, pp 873-874.
Pharmacokinetics: P. E. Gower, ibid. 877-878; J. Kosmidis et al., ibid. 875-876.
Clinical studies: P. F. Wood et al., ibid. 1042-1044; R. Norrby et al., J. Antimicrob. Chemother. 3, 355 (1977).
Review of antibacterial activity, pharmacology and therapeutic efficacy: R. N. Brogden et al., Drugs 17, 233-266 (1979).
Comprehensive description: T. J. Wozniak, J. R. Hicks, Anal. Profiles Drug Subs. 20, 209-236 (1991).
|
5-18-2005
|
Intermediates in cephalosporin production
|
|
|
12-22-2004
|
Process for the preparation of highly pure crystalline (R,S)-cefuroxime axetil
|

| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US5847118 * | Jul 25, 1997 | Dec 8, 1998 | Apotex, Inc. | Methods for the manufacture of amorphous cefuroxime axetil |
| US6060599 * | Jun 17, 1998 | May 9, 2000 | Ranbaxy Laboratories Limited | Process for the preparation of cefuroxime axetil in an amorphous form |
| US6107290 * | Sep 16, 1999 | Aug 22, 2000 | Hammi Pharm Co., Ltd. | Non-crystalline cefuroxime axetil solid dispersant, process for preparing same and composition for oral administration thereof |
| US6323193 | Aug 21, 2000 | Nov 27, 2001 | Ranbaxy Laboratories Limited | Bioavailable oral dosage form of cefuroxime axetil |
| US6384213 | May 19, 2000 | May 7, 2002 | Ranbaxy Laboratories Limited | Process for preparing a pure, pharmacopoeial grade amorphous form of cefuroxime axetil |
| US6534494 | Jan 27, 1999 | Mar 18, 2003 | Ranbaxy Laboratories Limited | Process for the preparation of cefuroxime axetil in an amorphous form |
| US6833452 | Jul 16, 2001 | Dec 21, 2004 | Ranbaxy Laboratories Limited | Process for the preparation of highly pure crystalline (R,S)—cefuroxime axetil |
| US6911441 * | Dec 16, 2002 | Jun 28, 2005 | Akzo Nobel N.V. | Prolonged release pharmaceutical composition |
| US7507813 | Jul 22, 2005 | Mar 24, 2009 | Nanomaterials Technology Pte Ltd. | Amorphous cefuroxime axetil and preparation process therefore |
| CN1909889B | Jan 10, 2005 | Jun 2, 2010 | 韩美药品株式会社 | Cefuroxime axetil granule and process for the preparation thereof |
| EP1619198A1 * | Jul 14, 2005 | Jan 25, 2006 | Nanomaterials Technology Pte Ltd | Amorphous cefuroxime axetil and preparation process therefore |
| WO1999065919A1 * | Jan 27, 1999 | Dec 23, 1999 | Ranbaxy Lab Ltd | Process for the preparation of cefuroxime axetil in an amorphous form |
| WO2001010410A1 * | Jul 25, 2000 | Feb 15, 2001 | Hanmi Pharm Ind Co Ltd | Non-crystalline cefuroxime axetil solid dispersant, process for preparing same and composition for oral administration thereof |
| WO2003014126A1 * | Aug 1, 2002 | Feb 20, 2003 | Marco Alpegiani | Process for the preparation of highly pure cefuroxime axetil |
| WO2005065658A1 * | Jan 10, 2005 | Jul 21, 2005 | Hee Chul Chang | Cefuroxime axetil granule and process for the preparation thereof |

