
Home » Uncategorized (Page 113)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AMBROXOL

Ambroxol is a secretolytic agent used in the treatment of respiratory diseases associated with viscid or excessive mucus. It is the active ingredient of Mucosolvan, Mucobrox, Mucol, Lasolvan, Mucoangin, Surbronc, Ambolar, and Lysopain. The substance is a mucoactive drug with several properties including secretolytic and secretomotoric actions that restore the physiological clearance mechanisms of the respiratory tract, which play an important role in the body’s natural defence mechanisms. It stimulates synthesisand release of surfactant by type II pneumocytes. Surfactant acts as an anti-glue factor by reducing the adhesion of mucus to thebronchial wall, in improving its transport and in providing protection against infection and irritating agents.[1][2] Ambroxol is often administered as an active ingredient in cough syrup.
Ambroxol is indicated as “secretolytic therapy in bronchopulmonary diseases associated with abnormal mucus secretion and impaired mucus transport. It promotes mucus clearance, facilitates expectoration and eases productive cough, allowing patients to breathe freely and deeply”.[3]

Ambroxolhydrochloridetablets in Japan
There are many different formulations developed since the first marketing authorisation in 1978. Ambroxol is available as syrup, tablets, pastilles, dry powder sachets, inhalation solution, drops and ampules as well aseffervescent tablets.
Ambroxol also provides pain relief in acute sore throat. Pain in sore throat is the hallmark of acutepharyngitis.[4] Sore throat is usually caused by a viral infection. The infection is self limited and the patient recovers normally after a few days. What is most bothering for the patient is the continuous pain in the throat maximized when the patient is swallowing. The main goal of treatment is thus to reduce pain. The main property of Ambroxol for treating sore throat is the local anaesthetic effect, described first in the late 1970s,[5][6] but explained and confirmed in more recent work.
Ambroxol is a potent inhibitor of the neuronal Na+ channels.[7] This property led to the development of alozenge containing 20 mg of ambroxol. Many state-of-the-art clinical studies[4] have demonstrated the efficacy of Ambroxol in relieving pain in acute sore throat, with a rapid onset of action, with its effect lasting at least three hours. Ambroxol is also anti-inflammatory, reducing redness in a sore throat.
Ambroxol has recently been shown to increase activity of the lysosomal enzyme glucocerebrosidase. Because of this it may be a useful therapeutic agent for both Gaucher disease and Parkinson’s disease.
Ambroxol is a secretolytic agent used in the treatment of respiratory diseases associated with viscid or excessive mucus. It is the active ingredient of Mucosolvan, Lasolvan or Mucoangin. The substance is a mucoactive drug with several properties including secretolytic and secretomotoric actions that restore the physiological clearance mechanisms of the respiratory tract which play an important role in the body’s natural defence mechanisms. It stimulates synthesis and release of surfactant by type II pneumocytes. Surfactants acts as an anti-glue factor by reducing the adhesion of mucus to the bronchial wall, in improving its transport and in providing protection against infection and irritating agents.
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | R02AD05 R05CB06 R07AA03 |
C 13 H 18 Br 2 N 2 O | 378.11 g / mol | 18683-91-5 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| trans-4-(2-Amino-3,5-dibrombenzylamino)-cyclohexanol | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Identifiers | |
| ATC code | R05CB06 |
| PubChem | CID 2132 |
| ChemSpider | 10276826 |
| UNII | 200168S0CL |
| KEGG | D07442 |
| ChEMBL | CHEMBL153479 |
| Chemical data | |
| Formula | C13H18Br2N2O |
| Mol. mass | 378.10 |
Synthesis pathway
| Синтез a) |
|---|
  |
Synthesis

| melting point | 233-234.5 | Kack, J., Koss, F.W., Schraven, E. and Beisenherz, G.; US. Patent 3,536,713; October 27, 1970; assigned to Boehringer lngelheim G.m.b.H. |
Kack, J., Koss, F.W., Schraven, E. and Beisenherz, G.; US. Patent 3,536,713; October 27,
1970; assigned to Boehringer lngelheim G.m.b.H.
Trade Names
| Page | Trade name | Manufacturer |
|---|---|---|
| Germany | Ambrobeta | betapharm |
| AmbroGEKSAL | Hexal | |
| Mucosal | Boehringer Ingelheim | |
| various generic drugs | ||
| France | Muksol | Leurquin Milan |
| Surbronk | Boehringer Ingelheim | |
| Italy | Ambrotus | Epifarma |
| ATUS | Metapharma | |
| Mukoarikodil | Menarini | |
| Mucosal | Boehringer Ingelheim, 1982 | |
| Viskomucil | Institute of Organic Chem. | |
| Japan | Mucosal | Teijin |
| Ukraine | AMBROKSOL | Ltd. “Pilot Plant” HNTSLS “m. Kharkiv, Ukraine |
| Ambroxol hydrochloride | CJC BHFZ, m. Kyiv, Ukraine | |
| AMBROBENE | ratiopharm GmbH, Germany | |
| LAZOLVAN® | Boehringer Ingelheim International GmbH, Germany | |
| various generic drugs | ||
Formulations
-
ampoule 15 mg;
-
Capsules of 45 mg, 75 mg;
-
drops 7.5 mg, 30 mg,
-
dry syrup 1.5%, 3%;
-
Effervescent tablets 30 mg, 60 mg;
-
coated tablets 30 mg, 60 mg;
-
granules 1.5%, 3%;
-
inhalation solution of 7.5 mg;
-
inaektsiya 1.000 mg;
-
solution of 0.3%, 0.75%;
-
Syrup 0.3%;
-
Tablets of 15 mg, 30 mg, 60 mg (hydrochloride)
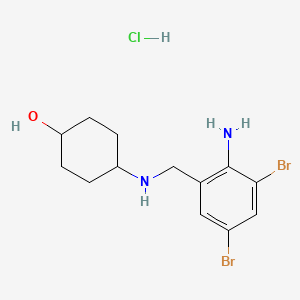
4-[(2-amino-3,5-dibromobenzyl)amino]cyclohexanol hydrochloride(1:1)
References
- Sanderson RJ et al. (1976), “Morphological and physical basis for lung surfactant action”, Respir Phys 27 (3): 379–92, doi:10.1016/0034-5687(76)90066-9,PMID 989610
- Kido H et al. (Nov 2004), “Secretory leukoprotease inhibitor and pulmonary surfactant serve as principal defenses against influenza A virus infection in the airway and chemical agents up-regulating their levels may have therapeutic potential.”, Biol Chem 385 (11): 1029–34, doi:10.1515/bc.2004.133, PMID 15576322
- Malerba M, Ragnoli B. (August 2008), “Ambroxol in the 21st century: pharmacological and clinical update”, Expert Opin Drug Metab Toxicol. 4 (8): 1119–29,doi:10.1517/17425255.4.8.1119, PMID 18680446
- de Mey C. et al. (2008), “Efficacy and safety of ambroxol lozenges in the treatment of acute uncomplicated sore throat”, Arzneimittelforschung 58 (11): 557–68,doi:10.1055/s-0031-1296557, PMID 19137906
- Püschmann S, Engelhorn R. (1978), “Pharmakologische Untersuchungen des Bromhexin-Metaboliten Ambroxol (Pharmacological study on the bromhexine-metabolite ambroxol)”, Arzneimittelforschung 28 (5a): 889–98, PMID 581987
- Klier KF, Papendick U. (1977), “Die lokalanaesthetische Wirkung von NA-872-haltigen Augentropfen (The local anesthetic effect of NA872-containing eyedrops)”, Med Monatsschr. 31 (12): 575–8, PMID 593223
- Weiser T. (2006), “Comparison of the effects of four Na+ channel analgesics on TTX-resistant Na+ currents in rat sensory neurons and recombinant Nav1.2 channels”,Neurosci Lett. 395 (3): 179–84, doi:10.1016/j.neulet.2005.10.058, PMID 16293367
- [1] Drugs.com, Ambroxol, accessed 21 January 2014
- http://drugsynthesis.blogspot.co.uk/2011/11/laboratory-synthesis-of-ambroxol_30.html
- DE 1 593 579 (Thomae; appl. 10.5.1966).
- DOS 2 218 647 (Thomae; appl. 18.4.1972).
- DOS 2 223 193 (Thomae; appl. 12.5.1972).
- Keck, J.: Justus Liebigs Ann. Chem. (JLACBF) 707, 107 (1967).
Links
-
GB 1 178 034 (Boehringer Ingelheim; appl. 10.5.1967; D-prior. 10.5.1966).
-
U.S. 3 536 713 (Boehringer Ingelheim; 27.10.1970; appl. 10.5.1967; S-prior. 10.5.1966).
-
DE 1 593 579 (Thomae; appl. 10.5.1966).
-
DOS 2 218 647 (Thomae; appl. 18.4.1972).
-
DOS 2 223 193 (Thomae; appl. 12.5.1972).
-
Keck, J .: Justus Liebigs Ann. Chem. (JLACBF) 707, 107 (1967).
ORGANIC SPECTROSCOPY
Telmapitant……Tachykinin NK1 Antagonists
, click to zoom.")
Telmapitant
TELMAPITANT; Telmapitant (USAN); Telmapitant [USAN]; 552292-58-7; HJ5FE4153B; D10391
(5R,8S)-8-[[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy]methyl]-8-phenyl-1,3,9-triazaspiro[4.5]decane-2,4-dione
1,3,7-Triazaspiro[4.5]decane-2,4-dione, 8-[[(1R)-1-[3,5-bis(trifluoromethyl)phenyl]ethoxy]methyl]-8-phenyl-, (5R,8S)-
(5R,8S)-8-(((1R)-1-(3,5-Bis(Trifluoromethyl)phenyl)ethoxy)methyl)-8-phenyl-1,3,7- triazaspiro(4.5)decane-2,4-dione
1,3,7-Triazaspiro(4.5)decane-2,4-dione,
8-(((1R)-1-(3,5-bis(trifluoromethyl)phenyl)ethoxy)methyl)-8-phenyl-, (5R,8S)-
Molecular Formula: C24H23F6N3O3
Molecular Weight: 515.448139
cas 552292-58-7
Merck & Co. (innovator)
Treatment of Nausea and Vomiting,
SYNTHESIS
……………………………………….
US7902366
http://www.google.com/patents/US7902366
Example 43a Example 43b

Step 1:

To a suspension of lactol Compound 3 (60 g, 93.0 mmol, 1 equiv.) and Wittig Reagent (93.5 g, 200.0 mmol, 2.15 equiv.) in toluene (800 ml) stirred at −78° C. under N2, a solution of KHMDS (0.5M in toluene, 558 ml, 280.0 mmol, 3 equiv.) was added dropwise at −78° C. The cooling bath was removed and the yellow mixture was warmed to RT to form a red solution. The mixture was allowed to stir at 23° C. for further 1 h before being quenched with saturated NH4Cl solution. EtOAc was added and layers were separated. The separated aqueous layer was extracted with EtOAc (2×500 ml). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [5% EtOAc-hexane to 10% EtOAc-hexane] gave alkene Compound 42 as white solid (40.5 g, 68%), Electrospray MS [M+1]+ 638.1. Continuous elution gave an impure cyclized product Compound 43.
Step 2:

A suspension of alkene Compound 42 (40.5 g, 64 mmol, 1 equiv.) and PtO2 (1.44 g, 6.4 mmol, 0.1 equiv.) in EtOH (400 ml) were stirred under a H2 balloon at 23° C. for 24 h. Another batch of PtO2 (1.44 g, 6.4 mmol, 0.1 equiv) was added and the mixture was stirred for another 24 h at 23° C. The catalyst was filtered via a pad of Celite. This solution of alkane Compound 44 was used in the next step without further purification.
Step 3:

p-TsOH.H2O (2.42 g, 13.0 mmol) was added to the ethanolic solution of alkane Compound 44 from above and the solution was heated to reflux for 4 h. The solution was cooled to RT and neutralized with Et3N. Solvents were removed in vacuum and EtOAc was added. Saturated NaHCO3 solution was added and layers were separated. The separated aqueous layer was extracted with EtOAc (300 ml×2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [10% ether-hexane] gave enamide Compound 45 (first batch) as yellow oil. Some intermediate and starting material were recovered as yellow oil by continuous elution with [50% EtOAc-hexane]. The yellow oil was dissolved in toluene and 10 mol % p-TsOH was added. The mixture was heated to reflux for 2 h and cooled to RT. Work up was as above and the combined enamide Compound 45 (25 g, 70%), Electrospray MS [M+1]+ 564.1, was obtained as yellow oil.
Step 4:

BH3.Me2S (13.6 ml, 133 mmo, 3.02 equiv) was added to a solution of enamide Compound 45 (25 g, 44.0 mmol,1 equiv.) in THF at 23° C. under N2. The mixture was stirred at 23° C. for 18 h and then cooled over an ice-water bath. A solution of NaOH (500 ml, 2N) was added slowly followed by a solution of H202 (500 ml, 30% aqueous). The mixture was allowed to stir from 0° C. to 23° C. for 18 h. Layers were separated and the separated aqueous layer was extracted with Et2O (500 ml×2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [hexane-EtOAc, 3:1 (v/v)] gave alcohol Compound 46 as colorless oil (19 g, 74%), Electrospray MS [M+1]+ 582.1.
Step 5:

Oxalyl chloride (5.7 ml, 65.3 mmol, 2 equiv.) was added to a solution of DMSO (9.3 ml, 131.0 mmol, 4 equiv.) in CH2Cl2 (300 ml) at −78° C. under N2. The mixture was stirred at −78° C. for 15 min before a solution of alcohol Compound 46 (19 g, 32.7 mmol. 1 equiv.) in CH2Cl2 (50 ml) was added. The mixture was stirred at −78° C. for a further 1 h and Et3N (32 ml, 228.9 mmol, 7 equiv.) was added. The cooling bath was removed and the mixture was warmed to RT before it was quenched with saturated NaHCO3 solution. Layers were separated and the aqueous was extracted with CH2Cl2 (300 ml×2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [hexane-ether, 4:1 (v/v)] gave ketone Compound 47 as colorless oil (15 g, 80%), Electrospray MS [M+1]+ 580.1.
Step 6:

EtOH (150 ml) was added to Cbz-ketone Compound 47 (15 g, 25.88 mmol, 1 equiv.), followed by NH4(CO3)2 (9.95 g, 103.5 mmol, 4 equiv.) and a solution of KCN (3.4 g, 51.77 mmol, 2 equiv.). The resulting mixture was heated at 58° C. under N2 for 72 h. TLC (1:1 EtOAc:hexane) revealed complete consumption of the starting material. The reaction mixture was cooled to RT and poured into sat. aq. NaHCO3 (200 ml) and extracted with EtOAc (3×200 ml). The combined organic layers were dried over MgSO4 and concentrated in vacuo to afford crude Cbz-hydantoin Compound 48 (16.5 g, 98%), Electrospray MS [M+1]+650.1. The crude material was used in the next reaction without further purification.
Step 7:
The crude Cbz-hydantoin Compound 48 (16.5 g, 25.4 mmol, 1 equiv.) was dissolved in MeOH (220 ml) and 20% Pd(OH)2—C (3.6 g) was added. The reaction mixture was shaken in a parr shaker under H2 atmosphere at 40 psi for 18 h. TLC (1:1 EtOAc:hexane) revealed complete consumption of the starting material. The reaction mixture was filtered through a pad of celite and the celite was washed with MeOH. The resulting solution was concentrated in vacuo. The crude product was purified by column chromatography on a Biotage (3:2, EtOAc:hex). Two major spots were collected. The less-polar spot corresponds to the isomer Example 43a (3 g, overall 20% over two steps), Electrospray MS [M+1]+ 516.1. The more polar spot corresponds to the isomer Example 43b (4.5 g, overall 30% over two steps), Electrospray MS [M+1]+ 516.1.
………………………………..
http://www.google.com/patents/WO2003051840A1?cl=en
Example 43a Example 43b

Step 1 :
Compound 3

To a suspension of lactol Compound 3 (60g, 93.0mmol, lequiv.) and Wittig Reagent (93. δg, 200.0mmol, 2.1 δequiv.) in toluene (800ml) stirred at -78°C under δ N2, a solution of KHMDS (O.δM in toluene, δδδml, 280.0mmol, 3equiv.) was added dropwise at -78°C. The cooling bath was removed and the yellow mixture was warmed to RT to form a red solution. The mixture was allowed to stir at 23°C for further 1 h before being quenched with saturated NH CI solution. EtOAc was added and layers were separated. The separated aqueous layer was extracted with EtOAc 0 (2 x δOOml). The combined organic layers were dried (MgSO ) and filtered.
Removal of solvents in vacuum followed by Biotage column chromatography [δ% EtOAc-hexane to 10% EtOAc-hexane] gave alkene Compound 42 as white solid (40. δg, 68%), Electrospray MS [M+1]+ 638.1. Continuous elution gave an impure cyclized product Compound 43. δ Step 2:
Compound 42

A suspension of alkene Compound 42 (40. δg, 64mmol, lequiv.) and PtO2 (1.44g, 6.4mmol, 0.1 equiv.) in EtOH (400ml) were stirred under a H2 balloon at 23°C for 24 h. Another batch of PtO2 (1.44g, 6.4mmol, 0.1 equiv) was added and the 0 mixture was stirred for another 24 h at 23°C. The catalyst was filtered via a pad of Celite. This solution of alkane Compound 44 was used in the next step without further purification. Step 3:
Compound 44

p-TsOH.H2O (2.42g, 13.0mmol) was added to the ethanolic solution of alkane
Compound 44 from above and the solution was heated to reflux for 4 h. The solution was cooled to RT and neutralized with Et3N. Solvents were removed in vacuum and EtOAc was added. Saturated NaHCO3 solution was added and layers
5 were separated. The separated aqueous layer was extracted with EtOAc (300ml x
2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [10% ether- hexane] gave enamide Compound 45 (first batch) as yellow oil. Some intermediate and starting material were recovered as yellow oil by continuous elution with 0 [50%EtOAc-hexane]. The yellow oil was dissolved in toluene and 10mol% p-TsOH was added. The mixture was heated to reflux for 2 h and cooled to RT. Work up was as above and the combined enamide Compound 45 (2δg, 70%), Electrospray
MS [M+1]+ 664.1 , was obtained as yellow oil.
Step 4:

BH3.Me2S (13.6ml, 133mmo, 3.02 equiv) was added to a solution of enamide Compound 45T25g, 44.0mmol, lequiv.) in THF at 23°C under N2. The mixture was stirred at 23°C for 18 h and then cooled over an ice-water bath. A solution of NaOH (600ml, 2N) was added slowly followed by a solution of H O2 (600ml, 30% 0 aqueous). The mixture was allowed to stir from 0°C to 23°C for 18 h. Layers were separated and the separated aqueous layer was extracted with Et.20 (600ml x 2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [hexane-EtOAc, 3:1 (v/v)] gave alcohol Compound 46 as colorless oil (19g, 74%), Electrospray MS [M+1]+ δ 582.1. Step 5:
Compound 46

Oxalyl chloride (δ.7ml, 6δ.3mmol, 2equiv.) was added to a solution of DMSO (9.3ml, 131.0mmol, 4equiv.) in CH2CI2 (300ml) at -78°C under N2. The mixture was 0 stirred at -78°C for 1 δ min before a solution of alcohol Compound 46 (19g, 32.7mmol. lequiv.) in CH2CI2 (50ml) was added. The mixture was stirred at -78°C for a further 1 h and Et3N (32ml, 228.9mmol, 7equiv.) was added. The cooling bath was removed and the mixture was warmed to RT before it was quenched with saturated NaHCO3 solution. Layers were separated and the aqueous was extracted with CH2CI2 (300ml x 2). The combined organic layers were dried (MgSO4) and filtered. Removal of solvents in vacuum followed by Biotage column chromatography [hexane-ether, 4:1 (v/v)] gave ketone Compound 47 as colorless oil (1δg, 80%), Electrospray MS [M+1]+ 680.1.

EtOH (150ml) was added to Cbz-ketone Compound 47 (15g, 2δ.88mmol, lequiv.), followed by NH (CO )2 (9.9δg, 103.5mmol, 4equiv.) and a solution of KCN (3.4g, 61.77mmoI, 2equiv.). The resulting mixture was heated at 68°C under N2 for 72 h. TLC (1 :1 EtOAc:hexane) revealed complete consumption of the starting
1δ material. The reaction mixture was cooled to RT and poured into sat. aq. NaHCO3 (200 ml) and extracted with EtOAc (3 x 200ml). The combined organic layers were dried over MgSO4 and concentrated in vacuo to afford crude Cbz-hydantoin Compound 48 (16.δg, 98%), Electrospray MS [M+1]+ 650.1. The crude material was used in the next reaction without further purification.
20 Step 7:
The crude Cbz-hydantoin Compound 48 (16.5g, 2δ.4mmol, lequiv.) was dissolved in MeOH (220ml) and 20% Pd(OH)2-C (3.6g) was added. The reaction mixture was shaken in a parr shaker under H2 atmosphere at 40 psi for 18 h. TLC (1 :1 EtOAc:hexane) revealed complete consumption of the starting material. The
26 reaction mixture was filtered through a pad of celite and the celite was washed with MeOH. The resulting solution was concentrated in vacuo. The crude product was purified by column chromatography on a Biotage (3:2, EtOAc:hex). Two major spots were collected. The less-polar spot corresponds to the isomer Example 43a (3 g, overall 20% over two steps), Electrospray MS [M+1]+ 616.1. The more polar spot
30 corresponds to the isomer Example 43b (4.6 g, overall 30% over two steps), Electrospray MS [M+1]+ 616.1.
|
4-29-2011
|
NK1 ANTAGONISTS
|
|
|
3-9-2011
|
NK1 antagonists
|
| English translation of Knabe, J., et al., “Racemates and Enantiomers of . . . ,” Pharmazie 52(12):912-919 (1997). | ||
| 2 | English translation of Schult, Karl E., et al., “Hydantoin-Derivate as Potential . . . ,” Eur. J. Med. Chem.-Chimica Therapeutics 13(1):25-31 (1978). | |
| 3 | English translation of Schult, Karl E., et al., “Hydantoin-Derivate as Potential . . . ,” Eur. J. Med. Chem.—Chimica Therapeutics 13(1):25-31 (1978). | |
| 4 | Knabe, J., et al., “Racemates and Enantiomers of Basic Substituted 5-Phenylhydantoins . . . ,” Pharmazie 52(12): 912-919 (1997). | |
| 5 | Oh, Chang-Hyun et al., “Synthesis of New Hydantoin-3-Acetic Acid Derivatives . . . ,” Bull. Korean Chem. Soc. 9(4):231-235 (1988). | |
| 6 | Shulte, Karl E., et al., “Hydantoin-Derivate als . . . ,” Eur. J. Med. Chem.-Chimica Therapeutica 13(1):25-31 (1978). | |
| 7 | Shulte, Karl E., et al., “Hydantoin-Derivate als . . . ,” Eur. J. Med. Chem.—Chimica Therapeutica 13(1):25-31 (1978). | |
| 8 | Wu, X. et al., “Generation of Cyclopenta [c] piperidines and Pyrrolo [3,4-c]piperidines- . . . ,” Tetrahedron 56(34): 6279-6290 (2000). | |
| 9 | * | Xiujuan Wu et al 2000. , Stereoselective transformation of 2H-1,4-Oxazin-2-ones into 2,(2),5,5-tri- and tetrasubstituted Analogues. . . |
| US6436928 * | Dec 14, 2000 | Aug 20, 2002 | Schering Corporation | Selective neurokinin antagonists |
| US6635639 * | Feb 13, 2002 | Oct 21, 2003 | Nps Allelix Corp. | Use of N-alkylamino-heterocylic compounds for the treatment of migraine |
| US7041682 * | Jul 2, 2003 | May 9, 2006 | Schering Corporation | Antiemetics, antidepressants, anxiolytic agents, antitussive agents |
| US7122677 * | Nov 12, 2002 | Oct 17, 2006 | Scherig Corporation | NK1 antagonists |
| US20060094720 * | Dec 15, 2005 | May 4, 2006 | Neng-Yang Shih | NK1 antagonists |
| US20060223804 * | Jun 30, 2005 | Oct 5, 2006 | Schering Corporation | NK1 antagonists |
| EP0790248A1 | Jan 20, 1997 | Aug 20, 1997 | Pfizer Limited | 3-Aza-piperidone- (tetrahydropyrimidin-2-one) and 3-oxa-piperidone (1,3 oxazin-2-one) derivatives, their preparation and their use as tachykinin/neurokinin antagonists |
key
Telmapitant, Merck, Tachykinin NK1 Antagonists
ORGANIC SPECTROSCOPY
Aldoxorubicin…CytRx is pouring money into R&D of cancer-fighting drugs

Aldoxorubicin, DOXO-EMCH
N’-[1-[4(S)-(3-Amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranosyloxy)-2(S),5,12-trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydronaphthacen-2-yl]-2-hydroxyethylidene]-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanohydrazide
1H-Pyrrole-1-hexanoic acid, 2,5-dihydro-2,5-dioxo-, (2E)-2-[1-[(2S,4S)-4-[(3-amino-2,3,6-trideoxy-α-L-lyxo– hexopyranosyl)oxy]-1,2,3,4,6,11-hexahydro-2,5,12- trihydroxy-7-methoxy-6,11-dioxo-2-naphthacenyl]-2- hydroxyethylidene]hydrazide
CytRx is pouring money into R&D of cancer-fighting drugs see article
Los Angeles Times
s most promising cancer-fighting drug, aldoxorubicin, is “sort of like a guided … Phase 3 clinical trial of a second-line treatment for soft-tissue sarcoma.

Aldoxorubicin
http://www.ama-assn.org/resources/doc/usan/aldoxorubicin.pdf
 in phase 3 Cytrx Corporation
in phase 3 Cytrx Corporation
(E)-N’-(1-((2S,4S)-4-(((2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl)-2-hydroxyethylidene)-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanehydrazide hydrochloride
1H-Pyrrole-1-hexanoic acid, 2,5-dihydro-2,5-dioxo-, (2E)-2-[1-[(2S,4S)-4-[(3-amino-
2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-1,2,3,4,6,11-hexahydro-2,5,12-trihydroxy-
7-methoxy-6,11-dioxo-2-naphthacenyl]-2-hydroxyethylidene]hydrazide
N’-[(1E)-1-{(2S,4S)-4-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-2,5,12-
trihydroxy-7-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-2-yl}-2-
hydroxyethylidene]-6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanohydrazide
MOLECULAR FORMULA C37H42N4O13
MOLECULAR WEIGHT 750.7
SPONSOR CytRx Corp.
CODE DESIGNATION
- Aldoxorubicin
- INNO 206
- INNO-206
- UNII-C28MV4IM0B
CAS REGISTRY NUMBER 1361644-26-9
CAS: 151038-96-9 (INNO-206); 480998-12-7 (INNO-206 HCl salt), 1361644-26-9
| QC data: | |
| Safety Data Sheet (MSDS): |

hydrochloride
CAS: 151038-96-9
Chemical Formula: C37H42N4O13
Exact Mass: 750.27484
Molecular Weight: 750.75
| Certificate of Analysis: | |
| QC data: | |
| Safety Data Sheet (MSDS): |

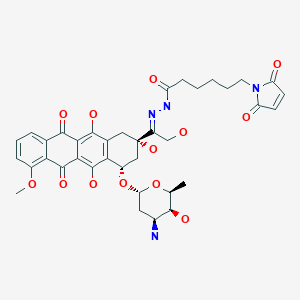
| In vitro protocol: | Clin Cancer Res. 2012 Jul 15;18(14):3856-67 |
| In vivo protocol: | Clin Cancer Res. 2012 Jul 15;18(14):3856-67.Invest New Drugs. 2010 Feb;28(1):14-9.Invest New Drugs. 2012 Aug;30(4):1743-9.Int J Cancer. 2007 Feb 15;120(4):927-34. |
| Clinical study: | Expert Opin Investig Drugs. 2007 Jun;16(6):855-66. |

Aldoxorubicin (INNO-206): Aldoxorubicin, also known as INNO-206, is the 6-maleimidocaproyl hydrazone derivative prodrug of the anthracycline antibiotic doxorubicin (DOXO-EMCH) with antineoplastic activity. Following intravenous administration, doxorubicin prodrug INNO-206 binds selectively to the cysteine-34 position of albumin via its maleimide moiety. Doxorubicin is released from the albumin carrier after cleavage of the acid-sensitive hydrazone linker within the acidic environment of tumors and, once located intracellularly, intercalates DNA, inhibits DNA synthesis, and induces apoptosis. Albumin tends to accumulate in solid tumors as a result of high metabolic turnover, rapid angiogenesis, hyervasculature, and impaired lymphatic drainage. Because of passive accumulation within tumors, this agent may improve the therapeutic effects of doxorubicin while minimizing systemic toxicity.
“Aldoxorubicin has demonstrated effectiveness against a range of tumors in both human and animal studies, thus we are optimistic in regard to a potential treatment for Kaposi’s sarcoma. The current standard-of-care for severe dermatological and systemic KS is liposomal doxorubicin (Doxil®). However, many patients exhibit minimal to no clinical response to this agent, and that drug has significant toxicity and manufacturing issues,” said CytRx President and CEO Steven A. Kriegsman. “In addition to obtaining valuable information related to Kaposi’s sarcoma, this trial represents another opportunity to validate the value and viability of our linker technology platform.” The company expects to announce Phase-2 study results in the second quarter of 2015.
Kaposi’s sarcoma is an orphan indication, meaning that only a small portion of the population has been diagnosed with the disease (fewer than 200,000 individuals in the country), and in turn, little research and drug development is being conducted to treat and cure it. The FDA’s Orphan Drug Act may grant orphan drug designation to a drug such as aldoxorubicin that treats a rare disease like Kaposi’s sarcoma, offering market exclusivity for seven years, fast-track status in some cases, tax credits, and grant monies to accelerate research
The widely used chemotherapeutic agent doxorubicin is delivered systemically and is highly toxic, which limits its dose to a level below its maximum therapeutic benefit. Doxorubicin also is associated with many side effects, especially the potential for damage to heart muscle at cumulative doses greater than 450 mg/m2. Aldoxorubicin combines doxorubicin with a novel single-molecule linker that binds directly and specifically to circulating albumin, the most plentiful protein in the bloodstream. Protein-hungry tumors concentrate albumin, thus increasing the delivery of the linker molecule with the attached doxorubicin to tumor sites. In the acidic environment of the tumor, but not the neutral environment of healthy tissues, doxorubicin is released. This allows for greater doses (3 1/2 to 4 times) of doxorubicin to be administered while reducing its toxic side effects. In studies thus far there has been no evidence of clinically significant effects of aldoxorubicin on heart muscle, even at cumulative doses of drug well in excess of 2,000 mg/m2.
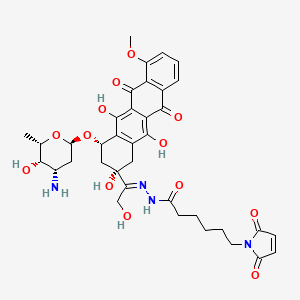
INNO-206 is an anthracycline in early clinical trials at CytRx Oncology for the treatment of breast cancer, HIV-related Kaposi’s sarcoma, glioblastoma multiforme, stomach cancer and pancreatic cancer. In 2014, a pivotal global phase 3 clinical trial was initiated as second-line treatment in patients with metastatic, locally advanced or unresectable soft tissue sarcomas. The drug candidate was originally developed at Bristol-Myers Squibb, and was subsequently licensed to KTB Tumorforschungs. In August 2006, Innovive Pharmaceuticals (acquired by CytRx in 2008) licensed the patent rights from KTB for the worldwide development and commercialization of the drug candidate. No recent development has been reported for research that had been ongoing for the treatment of small cell lung cancer (SCLC).
INNO-206 is a doxorubicin prodrug. Specifically, it is the 6-maleimidocaproyl hydrazone of doxorubicin. After administration, the drug candidate rapidly binds endogenous circulating albumin through the acid sensitive EMCH linker. Circulating albumin preferentially accumulates in tumors, bypassing uptake by other non-specific sites including the heart, bone marrow and the gastrointestinal tract. Once inside the acidic environment of the tumor cell, the EMCH linker is cleaved and free doxorubicin is released at the tumor site. Like other anthracyclines, doxorubicin inhibits DNA and RNA synthesis by intercalating between base pairs of the DNA/RNA strand, thus preventing the replication of rapidly-growing cancer cells. It also creates iron-mediated free oxygen radicals that damage the DNA and cell membranes. In 2011, orphan drug designation was assigned in the U.S. for the treatment of pancreatic cancer and for the treatment of soft tissue sarcoma.
CytRx Corporation (NASDAQ:CYTR) has announced it has initiated a pivotal global Phase 3 clinical trial to evaluate the efficacy and safety of aldoxorubicin as a second-line treatment for patients with soft tissue sarcoma (STS) under a Special Protocol Assessment with the FDA. Aldoxorubicin combines the chemotherapeutic agent doxorubicin with a novel linker-molecule that binds specifically to albumin in the blood to allow for delivery of higher amounts of doxorubicin (3.5 to 4 times) without several of the major treatment-limiting toxicities seen with administration of doxorubicin alone.
According to a news from Medicalnewstoday.com; CytRx holds the exclusive worldwide rights to INNO-206. The Company has previously announced plans to initiate Phase 2 proof-of-concept clinical trials in patients with pancreatic cancer, gastric cancer and soft tissue sarcomas, upon the completion of optimizing the formulation of INNO-206. Based on the multiple myeloma interim results, the Company is exploring the possibility of rapidly including multiple myeloma in its INNO-206 clinical development plans.
According to CytRx’s website, In preclinical models, INNO-206 was superior to doxorubicin with regard to ability to increase dosing, antitumor efficacy and safety. A Phase I study of INNO-206 that demonstrated safety and objective clinical responses in a variety of tumor types was completed in the beginning of 2006 and presented at the March 2006 Krebskongress meeting in Berlin. In this study, doses were administered at up to 4 times the standard dosing of doxorubicin without an increase in observed side effects over historically seen levels. Objective clinical responses were seen in patients with sarcoma, breast, and lung cancers.
INNO-206 – Mechanism of action:
According to CytRx’s website, the proposed mechanism of action is as the follow steps: (1) after administration, INNO-206 rapidly binds endogenous circulating albumin through the EMCH linker. (2) circulating albumin preferentially accumulates in tumors, bypassing uptake by other non-specific sites including heart, bone marrow and gastrointestinal tract; (3) once albumin-bound INNO-206 reaches the tumor, the acidic environment of the tumor causes cleavage of the acid sensitive linker; (4) free doxorubicin is released at the site of the tumor.
INNO-206 – status of clinical trials:
CytRx has announced that, in December 2011, CytRx initiated its international Phase 2b clinical trial to evaluate the preliminary efficacy and safety of INNO-206 as a first-line therapy in patients with soft tissue sarcoma who are ineligible for surgery. The Phase 2b clinical trial will provide the first direct clinical trial comparison of INNO-206 with native doxorubicin, which is dose-limited due to toxicity, as a first-line therapy. (source:http://cytrx.com/inno_206, accessed date: 02/01/2012).
Results of Phase I study:
In a phase I study a starting dose of 20 mg/m2 doxorubicin equivalents was chosen and 41 patients with advanced cancer disease were treated at dose levels of 20–340 mg/m2 doxorubicin equivalents . Treatment with INNO-206 was well tolerated up to 200 mg/m2 without manifestation of drug-related side effects which is a ~3-fold increase over the standard dose for doxorubicin (60 mg/kg). Myelosuppression and mucositis were the predominant adverse effects at dose levels of 260 mg/m2 and became dose-limiting at 340 mg/m2. 30 of 41 patients were assessable for analysis of response. Partial responses were observed in 3 patients (10%, small cell lung cancer, liposacoma and breast carcinoma). 15 patients (50%) showed a stable disease at different dose levels and 12 patients (40%) had evidence of tumor progression. (source: Invest New Drugs (2010) 28:14–19)
phase 2
CytRx Corporation (CYTR), a biopharmaceutical research and development company specializing in oncology, today announced that its oral presentation given by Sant P. Chawla, M.D., F.R.A.C.P., Director of the Sarcoma Oncology Center, titled “Randomized phase 2b trial comparing first-line treatment with aldoxorubicin versus doxorubicin in patients with advanced soft tissue sarcomas,” was featured in The Lancet Oncology in its July 2014 issue (Volume 15, Issue 8) in a review of the major presentations from the 2014 American Society of Clinical Oncology (ASCO) Annual Meeting.
“We are honored to have been included in The Lancet Oncology’s review of major presentations from ASCO and pleased that these important clinical findings are being recognized by one of the world’s premier oncology journals,” said Steven A. Kriegsman, CytRx President and CEO. “In clinical trials, aldoxorubicin has been shown to be a well-tolerated and efficacious single agent for the treatment of soft tissue sarcoma (STS) that lacks the cardiotoxicity associated with doxorubicin therapy, the current standard of care. We remain on track to report the full overall survival results from this trial prior to year-end 2014.”
The data presented at ASCO 2014 were updated results from CytRx’s ongoing multicenter, randomized, open-label global Phase 2b clinical trial investigating the efficacy and safety of aldoxorubicin compared with doxorubicin as first-line therapy in subjects with metastatic, locally advanced or unresectable STS. The updated trial results demonstrated that aldoxorubicin significantly increases progression-free survival (PFS), PFS at 6 months, overall response rate (ORR) and tumor shrinkage, compared to doxorubicin, the current standard-of-care, as a first-line treatment in patients with STS. The data trended in favor of aldoxorubicin for all of the major subtypes of STS
phase 3
Aldoxorubicin is currently being studied in a pivotal global Phase 3 clinical trial evaluating the efficacy and safety of aldoxorubicin as a second-line treatment for patients with STS under a Special Protocol Assessment with the FDA. CytRx is also conducting two Phase 2 clinical trials evaluating aldoxorubicin in patients with late-stage glioblastoma (GBM) and HIV-related Kaposi’s sarcoma and expects to start a phase 2b study in patients with relapsed small cell lung cancer
PATENTS WO 2000076551, WO 2008138646, WO 2011131314,
…………………….
WO 2014093815
http://www.google.com/patents/WO2014093815A1?cl=en
Anthracyclines are a class of antibiotics derived from certain types of Streptomyces bacteria. Anthracyclines are often used as cancer therapeutics and function in part as nucleic acid intercalating agents and inhibitors of the DNA repair enzyme topoisomerase II, thereby damaging nucleic acids in cancer cells, preventing the cells from replicating. One example of an anthracycline cancer therapeutic is doxorubicin, which is used to treat a variety of cancers including breast cancer, lung cancer, ovarian cancer, lymphoma, and leukemia. The 6-maleimidocaproyl hydrazone of doxorubicin (DOXO-EMCH) was originally synthesized to provide an acid-sensitive linker that could be used to prepare immunoconjugates of doxorubicin and monoclonal antibodies directed against tumor antigens (Willner et al., Bioconjugate Chem 4:521-527 (1993)). In this context, antibody disulfide bonds are reduced with dithiothreitol to form free thiol groups, which in turn react with the maleimide group of DOXO-EMCH to form a stable thioether bond. When administered, the doxorubicin-antibody conjugate is targeted to tumors containing the antigen recognized by the antibody. Following antigen-antibody binding, the conjugate is internalized within the tumor cell and transported to lysosomes. In the acidic lysosomal environment, doxorubicin is released from the conjugate intracellularly by hydrolysis of the acid-sensitive hydrazone linker. Upon release, the doxorubicin reaches the cell nucleus and is able to kill the tumor cell. For additional description of doxorubicin and
DOXO-EMCH see, for example, U.S. Patents 7,387,771 and 7,902,144 and U.S. Patent Application No. 12/619,161, each of which are incorporated in their entirety herein by reference.
[0003] A subsequent use of DOXO-EMCH was developed by reacting the molecule in vitro with the free thiol group (Cys-34) on human serum albumin (HSA) to form a stable thioether conjugate with this circulating protein (Kratz et al, J Med Chem 45:5523-5533 (2002)). Based on these results, it was
hypothesized that intravenously-administered DOXO-EMCH would rapidly conjugate to HSA in vivo and that this macromolecular conjugate would preferentially accumulate in tumors due to an “enhanced permeability and retention” (EPR) intratumor effect (Maeda et al., J Control Release 65:271-284 (2000)).
[0004] Acute and repeat-dose toxicology studies with DOXO-EMCH in mice, rats, and dogs identified no toxicity beyond that associated with doxorubicin, and showed that all three species had significantly higher tolerance for DOXO-EMCH compared to doxorubicin (Kratz et al, Hum Exp Toxicol 26: 19-35 (2007)). Based on the favorable toxicology profile and positive results from animal tumor models, a Phase 1 clinical trial of DOXO-EMCH was conducted in 41 advanced cancer patients (Unger et al, Clin Cancer Res 13:4858-4866 (2007)). This trial found DOXO-EMCH to be safe for clinical use. In some cases, DOXO-EMCH induced tumor regression.
[0005] Due to the sensitivity of the acid-cleavable linker in DOXO-EMCH, it is desirable to have formulations that are stable in long-term storage and during reconstitution (of, e.g., previously lyophilized compositions) and administration. DOXO-EMCH, when present in compositions, diluents and administration fluids used in current formulations, is stable only when kept at low temperatures. The need to maintain DOXO-EMCH at such temperatures presents a major problem in that it forces physicians to administer cold (4°C) DOXO-EMCH compositions to patients. Maintaining DOXO-EMCH at low temperatures complicates its administration in that it requires DOXO-EMCH to be kept at 4°C and diluted at 4°C to prevent degradation that would render it unsuitable for patient use. Further, administration at 4°C can be harmful to patients whose body temperature is significantly higher (37°C).
[0006] Lyophilization has been used to provide a stable formulation for many drugs. However, reconstitution of lyophilized DOXO-EMCH in a liquid that does not maintain stability at room temperature can result in rapid decomposition of DOXO-EMCH. Use of an inappropriate diluent to produce an injectable composition of DOXO-EMCH can lead to decreased stability and/or solubility. This decreased stability manifests itself in the cleavage of the linker between the doxorubicin and EMCH moieties, resulting in degradation of the DOXO-EMCH into two components: doxorubicin and linker-maleimide. Thus, stable,
reconstituted lyophilized solutions of anthracycline-EMCH (e.g., DOXO-EMCH), and injectable compositions containing the same, are required to solve these problems and to provide a suitable administration vehicle that can be used reasonably in treating patients both for clinical trials and commercially.
DOXO-EMCH. The term “DOXO-EMCH,” alone or in combination with any other term, refers to a compound as depicted by the following structure:

OH
DOXO-EMCH is also referred to as (E)-N’-(l-((2S,4S)-4-(4-amino-5-hydroxy-6- methyl-tetrahydro-2H-pyran-2-yloxy-2,5 , 12-trihydroxy-7-methoxy-6, 11- dioxol,2,3,4,6,l l-hexahydrotetracen-2-yl)-2-hydroxyethylidene)-6-(2,5-dioxo-2H- pyrrol- 1 (5H)yl)hexanehydrazide»HCl.
………………………………
CN 102675385
http://www.google.com/patents/CN102675385A?cl=en
According to literature reports, (eg see David Willner et al, “(6_Maleimidocaproyl) hydrazoneof Doxorubicm-A New Derivative for the Preparation ofImmunoconjugates oiDoxorubicin,” Bioconjugate Chem. 1993,4, 521-527; JK Tota Hill, etc. man, “The method of preparation of thioether compounds noir,” CN1109886A, etc.), adriamycin 13 – bit hydrazone derivative synthesis and the main process are as follows:
[0004]

[0005] First, maleic anhydride and 6 – aminocaproic acid was refluxed in a large number of acid reaction ko ni acid I; agent under the action of the ring after the cyclization maleimidocaproic acid 2 (yield 30-40% ), cyclic acid anhydride mixture is generally ko, trimethyl silyl chloride and tri-amines such ko; maleimido aminocaproic acid tert-butyl ester with hydrazine to condensation to give 2 – (6 – aminocaproic maleimido ) hydrazine carboxylic acid tert-butyl ester 3 (yield 70-85%), the condensing agent is N-methylmorpholine and isobutyl chloroformate; 3 in a large number of trifluoroacetic acid deprotection ko maleimido ko has trifluoroacetic acid hydrazide 4 (yield 70%); the doxorubicin hydrochloride salt with a ko in trifluoroacetic acid catalyzed condensation in methanol solvent to doxorubicin hydrazone product was obtained (yield 80%) .
[0006] The synthetic method the yield is low (in particular, by maleic acid imido step 2), the total yield of not more than 20%, and the solvent consumption is large, adriamycin hydrazone product per Malek consumes about ko acid reaction solvent, 70mL, tetrahydrofuran 300mL, ko trifluoroacetic acid 40mL, and because the 2 – (6 – maleimido hexanoyl)-hydrazine carboxylic acid tert-butyl ester was purified by column chromatography required, but also to consume a large amount of Solvent. This has resulted in synthesis post-processing complex process, complicated operation. And because the end product of the synthesis of doxorubicin hydrazone ko using trifluoroacetic acid, inevitably there will be in the product ko trifluoroacetic acid impurities, not divisible. Based on the high cost of such a route exists, yield and production efficiency is low, Eri Arts route operational complexity and other shortcomings, is obviously not suitable for mass production, it is necessary to carry out improvements or exploring other Eri Arts synthesis methods.
doxorubicin hydrazone derivative,

Wherein n is an integer of 1-15, characterized in that said method comprises the steps of: (1) the maleic acid chloride of the formula H2N-(CH2) n-COOH amino acid I b in the presence of a base prepared by condensation of maleimido group steps I c acid,

(2) maleic acid imido group I c and then with an acylating reagent of tert-butyl carbazate in the presence of a base in the reaction of step I d,

(3) I d deprotection with trifluoroacetic acid, the alkali and removing trifluoroacetic acid to obtain the maleimido group I e hydrazide steps

(4) an imido group of maleic hydrazide I e and doxorubicin hydrochloride catalyzed condensation of hydrogen chloride to obtain a final product hydrazone derivative of doxorubicin,

[0028]


[0049] The butene-ni chloride 15. 2g (0. Imol) was dissolved in 25mL of chloroform was dried by adding anhydrous potassium carbonate 27. 6g (0. 2mol), the gas and gas protection and conditions of 0 ° C was added dropwise 6 – aminocaproic acid 13. 2g (0. ImoI) in chloroform (50mL) solution, add after reaction at room temperature for 3 hours. Washed with saturated brine, dried over anhydrous magnesium sulfate, suction filtered, concentrated under reduced pressure. The residue was recrystallized from alcohol ko maleimido acid (Compound c) 18. 8g, 90% yield, m.p. :85-87 ° C.
[0050] Compound c 10. 5g (50mmol) and thionyl chloride crab 5. 3mL (75mmol) was heated under reflux the mixture I. 5 hours and concentrated under reduced pressure in an argon atmosphere under the conditions of 0 ° C and added dropwise to the hydrazine carboxylic acid tert-butyl ester 6.6g (50mmol) amine with a three ko
10. 8mL (75mmol) in anhydrous ko ether (50mL) solution added after the reaction was continued at room temperature for I. 5 hours. Washed with 5% hydrochloric acid, 5% sodium bicarbonate, and saturated brine, dried over anhydrous magnesium sulfate overnight, filtered with suction to give the compound of d ko ether solution. The solution was cooled to 0 ° C, was added dropwise trifluoroacetic acid ko 7. 4mL (100mmOl), After the addition the reaction was continued for 10 minutes, suction filtered, the filter cake was washed twice with ether, ko and dried in vacuo to give 6 – maleic acid sub-aminocaproic acid hydrazide trifluoro-ko 12. 2g, 72% yield, m.p. 99-102 ° C. IOmL this salt is added to sodium hydroxide (10%) solution, stirred for a while, with ko extracted with ether, the organic layer was washed with water, dried over anhydrous magnesium sulfate, and concentrated to give 6 – aminocaproic maleimido hydrazide (compound e) 7. Sg, 70% yield.
[0051] The doxorubicin hydrochloride 0. 58g (Immol) with compound e 0. 45g (2mmol) was dissolved in 150mL of anhydrous methanol, passing about 2mmol of dry hydrogen chloride, under argon, at room temperature protected from light and reaction conditions 24 inches. Concentrated under reduced pressure at room temperature, the solid was washed with about IOOmL ko nitrile, and dried in vacuo doxorubicin 6 – aminocaproic maleimido hydrazone O. 63g, 80% yield. 1H NMR (CD3OD) δ: 7. 94 (bd, 1H), 7. 82 (t, 1H), 7. 55 (d, 1H), 6. 78 (s, 2H), 5. 48 (s, 1H ), 5. 07 (t, 1H), 4 · 59 (d, 1H), 4 · 21 (m, 1Η), 4 · 02 (s, 3H), 3 · 63-3. 30 (m, 5H) , 2 · 55-2. 26 (m, 4H), 2. 19-1. 88 (m, 3Η), I. 69-1. 18 (m, 12Η, I. 26). [0052] Although specific reference to the above embodiments of the present invention will be described, it will be understood that in the appended claims without departing from the invention as defined by the spirit and scope of the skilled person can be variously truncated, substitutions and changes. Accordingly, the present invention encompasses these deletions, substitutions and changes.
………………………………….
US 5622929
http://www.google.co.in/patents/US5622929
http://www.google.co.in/patents/EP0554708A1
Method A:

As noted below, Method A is the preferred method when the Michael Addition Receptor is a maleimido moiety.
Alternatively, the Formula (IIa) compound may be prepared by reaction of the drug with a hydrazide to form an intermediate hydrazone drug derivative followed by reaction of this compound with a Michael Addition Receptor containing moiety according to the general process described in Method B:

…………………………………….
http://www.google.co.in/patents/WO2012167255A1?cl=en
Synthesis of DOXO-EMCH
The synthesis of DOXO-EMCH was done initially in accordance with that previously published by Willner and co-workers (Bioconjugate Chem., 4:521-527, 1993). Problems arose in the initial addition of the 6-maleimidocaproylhydrazine to the C-13 ketone of doxorubicin. HPLC results did not give a good yield of product, only 50-60%. Upon further analysis, we determined TFA was not needed to catalyze the reaction, and instead used pyridine. With pyridine, chromatograms from the HPLC showed 90% DOXO-EMCH relative to 10% DOX. The pyridine may have improved the yield by serving as a base to facilitate formation of the hydrazone. Another problem we encountered in the synthesis was purification of the final product. According to Willner’ s method, 5 volumes of acetonitrile (ACN) were to be added to a concentrated methanolic solution of crude DOXO-EMCH to achieve crystallization after 48 h at 4 °C. By this method, only 10-20%) of the desired product precipitated. To obtain a better yield, the crystallization step was done 4 times with 6 volumes of ACN used in each step. A lesser amount of methanol was needed each time, as less product remained in solution. Even with the multiple crystallizations, a final yield of only 59% was obtained. Various other methods for crystallization were explored, including using different solvents and increasing the initial solubility in methanol by heat, but none gave better results. 1.2 Rate of Hydrolysis of DOXO-EMCH at Varying pH
Subsequent pH studies to determine the rate of hydrolysis of the hydrazone were carried out as a benchmark for later hydrolysis experiments with PPD-EMCH. The results of the hydrolysis experiments showed that at lower pH, the hydrolysis reaction proceeded very quickly in the formation of DOX. Moreover, at higher pH the hydrazone proved to be very robust in that its degradation is very slow.
General HPLC instruments and methods
Analytical HPLC methods were performed using a Hewlett-Packard/ Aligent 1050/1100 chromatograph with an auto injector, diode array UV-vis absorption detector. Method 1.1 : Analytical HPLC injections were onto an Aligent Zorbax Eclipse XDB-C18 reversed phase column, 4.6 mm x 150 mm, eluting at 1.0 mL/min. A gradient of acetonitrile/20 mM sodium phosphate buffer (pH 6.9), 80% buffer to 55% at 10 min, 55% to 40% at 12 min, 40% to 80% at 13 min. Retention times: at 480 nm, DOX (9.4 min), DOXO-EMCH (1 1.2 min).
Synthesis of DOXO-EMCH
The synthesis of DOXO-EMCH was accomplished using the procedure reported by Willner et al, with several changes to improve the yield (Willner, D., et al.,
Bioconjugate Chem., 4:521-27, 1993). DOX’HCl (20 mg, 34 μιηοΐ) was dissolved in 6 mL of methanol. Pyridine (12.53 μί) was added to the solution, followed by 35.4 mg
EMCH’TFA. The reaction was stirred at room temperature overnight. By HPLC, the reaction was 90% complete. The solvent was evaporated to dryness by rotary evaporation. A minimal amount of methanol was used to dissolve the solid, and six volumes of acetonitrile at 4 °C were added to the solution. The resulting solution was allowed to sit undisturbed at 4 °C for 48 h for crystallization. The precipitate was collected, and the crystallization method was repeated 4 times. The resulting solids were combined and washed three times with 1 : 10 methanokacetonitrile. The final yield of DOXO-EMCH was 11.59 mg, 58%. HPLC Method 1.1 was used. NMR spectra corresponded to those previously given by Willner (Bioconjugate Chem. 4:521-27. 1993).
…………………………….
http://www.google.co.in/patents/US20070219351
DOXO-EMCH, the structural formula of which is shown below,

…………………………………
SEE
(6-Maleimidocaproyl)hydrazone of doxorubicin – A new derivative for the preparation of immunoconjugates of doxorubicin
Bioconjugate Chem 1993, 4(6): 521
| References |
1: Kratz F, Azab S, Zeisig R, Fichtner I, Warnecke A. Evaluation of combination therapy schedules of doxorubicin and an acid-sensitive albumin-binding prodrug of doxorubicin in the MIA PaCa-2 pancreatic xenograft model. Int J Pharm. 2013 Jan 30;441(1-2):499-506. doi: 10.1016/j.ijpharm.2012.11.003. Epub 2012 Nov 10. PubMed PMID: 23149257.
2: Walker L, Perkins E, Kratz F, Raucher D. Cell penetrating peptides fused to a thermally targeted biopolymer drug carrier improve the delivery and antitumor efficacy of an acid-sensitive doxorubicin derivative. Int J Pharm. 2012 Oct 15;436(1-2):825-32. doi: 10.1016/j.ijpharm.2012.07.043. Epub 2012 Jul 28. PubMed PMID: 22850291; PubMed Central PMCID: PMC3465682.
3: Kratz F, Warnecke A. Finding the optimal balance: challenges of improving conventional cancer chemotherapy using suitable combinations with nano-sized drug delivery systems. J Control Release. 2012 Dec 10;164(2):221-35. doi: 10.1016/j.jconrel.2012.05.045. Epub 2012 Jun 13. PubMed PMID: 22705248.
4: Sanchez E, Li M, Wang C, Nichols CM, Li J, Chen H, Berenson JR. Anti-myeloma effects of the novel anthracycline derivative INNO-206. Clin Cancer Res. 2012 Jul 15;18(14):3856-67. doi: 10.1158/1078-0432.CCR-11-3130. Epub 2012 May 22. PubMed PMID: 22619306.
5: Kratz F, Elsadek B. Clinical impact of serum proteins on drug delivery. J Control Release. 2012 Jul 20;161(2):429-45. doi: 10.1016/j.jconrel.2011.11.028. Epub 2011 Dec 1. Review. PubMed PMID: 22155554.
6: Elsadek B, Kratz F. Impact of albumin on drug delivery–new applications on the horizon. J Control Release. 2012 Jan 10;157(1):4-28. doi: 10.1016/j.jconrel.2011.09.069. Epub 2011 Sep 16. Review. PubMed PMID: 21959118.
7: Kratz F, Fichtner I, Graeser R. Combination therapy with the albumin-binding prodrug of doxorubicin (INNO-206) and doxorubicin achieves complete remissions and improves tolerability in an ovarian A2780 xenograft model. Invest New Drugs. 2012 Aug;30(4):1743-9. doi: 10.1007/s10637-011-9686-5. Epub 2011 May 18. PubMed PMID: 21590366.
8: Boga C, Fiume L, Baglioni M, Bertucci C, Farina C, Kratz F, Manerba M, Naldi M, Di Stefano G. Characterisation of the conjugate of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin with lactosaminated human albumin by 13C NMR spectroscopy. Eur J Pharm Sci. 2009 Oct 8;38(3):262-9. doi: 10.1016/j.ejps.2009.08.001. Epub 2009 Aug 18. PubMed PMID: 19695327.
9: Graeser R, Esser N, Unger H, Fichtner I, Zhu A, Unger C, Kratz F. INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Invest New Drugs. 2010 Feb;28(1):14-9. doi: 10.1007/s10637-008-9208-2. Epub 2009 Jan 8. PubMed PMID: 19148580.
10: Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008 Dec 18;132(3):171-83. doi: 10.1016/j.jconrel.2008.05.010. Epub 2008 May 17. Review. PubMed PMID: 18582981.
11: Unger C, Häring B, Medinger M, Drevs J, Steinbild S, Kratz F, Mross K. Phase I and pharmacokinetic study of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin. Clin Cancer Res. 2007 Aug 15;13(16):4858-66. PubMed PMID: 17699865.
12: Lebrecht D, Walker UA. Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7(2):108-13. Review. PubMed PMID: 17652814.
13: Kratz F. DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials. Expert Opin Investig Drugs. 2007 Jun;16(6):855-66. Review. PubMed PMID: 17501697.
14: Kratz F, Ehling G, Kauffmann HM, Unger C. Acute and repeat-dose toxicity studies of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH), an albumin-binding prodrug of the anticancer agent doxorubicin. Hum Exp Toxicol. 2007 Jan;26(1):19-35. PubMed PMID: 17334177.
15: Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Kratz F, Walker UA. The 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH) is superior to free doxorubicin with respect to cardiotoxicity and mitochondrial damage. Int J Cancer. 2007 Feb 15;120(4):927-34. PubMed PMID: 17131338.
16: Di Stefano G, Lanza M, Kratz F, Merina L, Fiume L. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization and preliminary biological properties of the conjugate. Eur J Pharm Sci. 2004 Dec;23(4-5):393-7. PubMed PMID: 15567293.
| EP0169111A1 * | Jun 18, 1985 | Jan 22, 1986 | Sanofi | Cytotoxic conjugates useful in therapy, and process for obtaining them |
| EP0269188A2 * | Jun 18, 1985 | Jun 1, 1988 | Elf Sanofi | Cytotoxic conjugates useful in therapy, and process for obtaining them |
| EP0306943A2 * | Sep 8, 1988 | Mar 15, 1989 | Neorx Corporation | Immunconjugates joined by thioether bonds having reduced toxicity and improved selectivity |
| EP0328147A2 * | Feb 10, 1989 | Aug 16, 1989 | Bristol-Myers Squibb Company | Anthracycline immunoconjugates having a novel linker and methods for their production |
| EP0398305A2 * | May 16, 1990 | Nov 22, 1990 | Bristol-Myers Squibb Company | Anthracycline conjugates having a novel linker and methods for their production |
| EP0457250A2 * | May 13, 1991 | Nov 21, 1991 | Bristol-Myers Squibb Company | Novel bifunctional linking compounds, conjugates and methods for their production |
KEY words
Aldoxorubicin, CytRx, CANCER, INNO-206, PHASE 3, oncology, Soft Tissue Sarcoma
ORGANIC SPECTROSCOPY
Idoxuridine

Idoxuridine, NSC-39661, IdUrd, IDUR, IDU, IUDR, Herpid
Idoxuridine is an anti-herpesvirus antiviral drug.
It is a nucleoside analogue, a modified form of deoxyuridine, similar enough to be incorporated into viral DNA replication, but theiodine atom added to the uracil component blocks base pairing. It is used only topically due to cardiotoxicity. It was synthesized byWilliam Prusoff in the late 1950s.[1] Initially developed as an anticancer drug, idoxuridine became the first antiviral agent in 1962.[2]
Clinical use
Idoxuridine is mainly used topically to treat herpes simplex keratitis.[3] Epithelial lesions, especially initial attacks presenting with a dendritic ulcer, are most responsive to therapy, while infection with stromal involvement are less responsive.[4] Idoxuridine is ineffective against herpes simplex virus type 2 and varicella-zoster.[3]
Side effects
Common side effects of the eye drops include irritation, blurred vision and photophobia.[5] Corneal clouding and damage of the corneal epithelium may also occur.[citation needed]
Formulations and dosage
Idoxuridine is available as either a 0.5% ophthalmic ointment or as a 0.1% ophthalmic solution.[3] The dosage of the ointment is every 4 hours during day and once before bedtime.[3] The dosage of the solution is 1 drop in the conjunctival sac hourly during the day and every 2 hours during the night until definitive improvement, then 1 drop every 2 hours during the day and every 4 hours during the night.[3] Therapy is continued for 3-4 days after healing is complete, as demonstrated by fluorescein staining.[3]
UV – spectrum

|
||||
| Conditions : Concentration – 2 mg / 100 ml | ||||
| Solvent designation schedule | Methanol  |
Water  |
0.1 M HCl  |
0.1M NaOH  |
|---|---|---|---|---|
| The absorption maximum | 284 nm | 288 nm | 288 nm | 280 nm |
 |
227 | 217 | 217 | 164 |
| ε | 8040 | 7680 | 7680 | 5810 |
IR – spectrum
| Wavelength (μm) |
|---|
 |
| Wavenumber (cm -1 ) |
ass Spectrum
| Spectrum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 |
||||||||||
| The 10 largest peaks: | ||||||||||
| Peak | 28 | 39 | 40 | 43 | 69 | 112 | 127 | 195 | 238 | 254 |
| Meaning | 436 | 291 | 393 | 300 | 295 | 319 | 608 | 570 | 999 | 34 |
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | D06BB01 J05AB02 S01AD01 |
C 9 H 11 IN 2 O 5 | 354.10 g / mol | 54-42-2 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-iodo-1,2,3,4-tetrahydropyrimidine-2,4-dione | |
| Clinical data | |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a601062 |
| Pregnancy cat. | C (US) B1 (topical), B3 (ophthalmologic) [AU] |
| Legal status | Prescription Only (S4) (AU) |
| Routes | topically |
| Identifiers | |
| CAS number | 54-42-2 |
| ATC code | D06BB01 J05AB02,S01AD01 |
| PubChem | CID 5905 |
| DrugBank | DB00249 |
| ChemSpider | 10481938 |
| UNII | LGP81V5245 |
| KEGG | D00342 |
| ChEMBL | CHEMBL788 |
| NIAID ChemDB | 001857 |
| Synonyms | Iododeoxyuridine; IUdR |
| Chemical data | |
| Formula | C9H11IN2O5 |
| Mol. mass | 354.099 g/mol |
Application
-
antiviral ( Herpes simplex )
Classes of substances
-
Iodine compounds
-
Uridine and deoxyuridine
-
Synthesis pathway
| Synthesis a) |
|---|
   |
| Synthesis of b) |
 |


Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| Germany | Virunguent | Almirall Hermal |
| France | Iduviran | Chauvin |
| United Kingdom | Gerpid | Astellas |
| Italy | Iduher | Farmigea |
| Idustatin | Sanofi-Aventis | |
| Japan | IMU | Kaken |
| I.D.U. | Senju; Takeda | |
| USA | Dendrid | Alcon |
| Gerpleks | Allergan | |
| Stokes | SmithKline & French | |
| Ukraine | No | No |
Formulations
-
eye drops 0.1%;
-
eye ointment 0.25%;
-
Ointment 0.2%, 0.5%;
-
solution of 5%, 10%, 40%
Links
-
Chang, PK; Welch, AD: J. Med. Chem. (JMCMAR) 6, 428 (1963).
-
FR 1,336,866 (Roussel-Uclaf; appl. 27.7.1962).
-
GB 1024156 (Roussel-Uclaf; appl. 24.7.1963; F-prior. 27.7.1962).
References
- Jump up^ Prusoff, W.H. (1959) Synthesis and biological activities of iododeoxyuridine, an analog of thymidine. Biochim Biophys Acta. March; 32(1): 295–296.
- Jump up^ Wilhelmus KR (2010). “Antiviral treatment and other therapeutic interventions for herpes simplex virus epithelial keratitis”. Cochrane Database Syst Rev 12: CD002898. doi:10.1002/14651858.CD002898.pub4. PMID 21154352.
- ^ Jump up to:a b c d e f Goodman and Gilman’s The Pharmacological Basis of Therapeutics. Edited by Gilman AG, Rall TW, Nies AS, Taylor P. McGraw-Hill. 8th ed. 1990.
- Jump up^ Maxwell E. Treatment of herpes keratitis with 5-iodo-2-deoxyuridine (IDU): a clinical evaluation of 1500 cases. Am. J. Ophthalmol., 1963, 56, 571-573.
- Jump up^ Drugs.com: Idoxuridine ophthalmic
Further Reading
- Seth A, Misra A, Umrigar D (2004). “Topical liposomal gel of idoxuridine for the treatment of herpes simplex: pharmaceutical and clinical implications”. Pharm Dev Technol 9 (3): 277–289. doi:10.1081/PDT-200031432. PMID 15458233.
- Otto S (1998). “Radiopharmaceuticals (Strontium 89) and radiosensitizers (idoxuridine)”. J Intraven Nurs 21 (6): 335–7. PMID 10392098.
- Fauth E, Zankl H (1999). “Comparison of spontaneous and idoxuridine-induced micronuclei by chromosome painting”. Mutat Res 440 (2): 147–56. doi:10.1016/s1383-5718(99)00021-2. PMID 10209337.
Idarubicin hydrochloride

Idarubicin hydrochloride
NSC-256439, IMI-30, DMDR, Idamycin, Zavedos

Idarubicin /ˌaɪdəˈruːbɨsɪn/ or 4-demethoxydaunorubicin is an anthracycline antileukemic drug. It inserts itself into DNA and prevents DNA from unwinding by interfering with the enzyme topoisomerase II. It is an analog of daunorubicin, but the absence of a methoxy group increases its fat solubility and cellular uptake.[1] Similar to other anthracyclines, it also induces histone eviction fromchromatin.[2]
It belongs to the family of drugs called antitumor antibiotics.
It is currently combined with cytosine arabinoside as a first line treatment of acute myeloid leukemia.
It is distributed under the trade names Zavedos (UK) and Idamycin (USA).

UV – spectrum
|
|
||||
| Conditions : Concentration – 1 mg / 100 ml | ||||
| Solvent designation schedule | Methanol  |
Water  |
0.1 M HCl  |
0.1M NaOH  |
|---|---|---|---|---|
| The absorption maximum | 481 nm, 287 nm, 251 nm |
484 nm 289 nm 257 nm |
484 nm 289 nm 257 nm |
Observed decay |
 |
207 179 816 |
194 180 743 |
194 180 738 |
– |
| ε | 11100 9540 43600 |
10400 9620 39700 |
10400 9620 39400 |
– |

IR – spectrum
| Wavelength (μm) |
|---|
 |
| Wavenumber (cm -1 ) |
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | L01DB06 | C 26 H 27 NO 9 | 497.50 g / mol | 58957-92-9 |
Idarubicin is the 4-demethoxy derivative of daunorubicin. Idarubicin is an antineoplastic agent that has been used to treat various cancers, including those of the breast, lung, stomach, ovaries, and lymph system. Idarubicin is marketed as an intravenous injection of Idarubicin hydrochloride of the formula,

under the brand name IDAMYCIN®. Idarubicin hydrochloride is a red-orange crystalline powder, soluble in water, methanol, and other polar solvents like dimethylformamide. It is practically insoluble in acetone, chloroform, and methylene chloride. Idarubicin hydrochloride has a melting point of 175-180°C, and apH of 5.0-6.5 in a 0.5% w/v solution in water.
Application
-
antitumor agent
-
anthracycline antibiotic
Classes of substances
-
Naftatsenovye antibiotics
Synthesis pathway
| Preparation |
|---|
    |
Synthesis a)
|
 |
Synthesis of b)
|
 |


condensation of chiral tetraline (I) with phthalic anhydride (II) by means of AlCl3 at 180 C gives the naphthacenedione (III), acetyl group which is ketalized with ethylene glycol and p-toluenesulfonic acid yielding the dioxolane (IV). The hydroxylation of (IV) with Br2 and AIBN in CCl4/CHCl3 affords the 4-demethoxy-7-epidaunomycinone (V), which is isomerized with TFA yielding 4-demethoxydaunomycinone (VI) . The condensation of (VI) with the acylated hexopyranosyl chloride (VII) by means of CF3SO3Ag of Br2Hg affords the trifluoroacetylated 4-demethoxydaunomycin (VIII), which is finally deprotected by treated with NaOH to eliminate the trifluoroacetyl groups
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| Germany | Zavedos | Pharmacia |
| France | – “- | Pfizer |
| United Kingdom | – “- | Pharmacia |
| Italy | – “- | Pharmacia & Upjohn |
| Japan | Idamitsin | Pfizer |
| Ukraine | Zavedos | Actavis Italy SpA, Italy |
| Idalek | CJSC “Biolik”, Ukraine | |
| Zavedos | Pfizer Іtaliya Srl, Іtaliya | |
| Rubidium | NGO “Lance Farm”, Russia | |
| other generic drugs | ||
Formulations
-
Capsules of 5 mg, 10 mg, 25 mg;
-
vial of 5 mg, 10 mg (hydrochloride)
IDAMYCIN PFS Injection contains idarubicin hydrochloride and is a sterile, semi-synthetic, preservative-free solution (PFS) antineoplastic anthracycline for intravenous use. Chemically, idarubicin hydrochloride is 5, 12-Naphthacenedione, 9-acetyl-7-[(3-amino-2,3,6-trideoxy-α-L-lyxo-hexopyranosyl)oxy]-7,8,9,10-tetrahydro-6,9,11-trihydroxyhydrochloride, (7S-cis). The structural formula is as follows:
 |
C26H27NO9•Hcl M.W 533.96
IDAMYCIN PFS (idarubicin hydrochloride injection) is a sterile, red-orange, isotonic parenteral preservative-free solution, available in 5 mL (5 mg), 10 mL (10 mg) and 20 mL (20 mg) single-use-only vials.
Each mL contains Idarubicin HCL, USP 1 mg and the following inactive ingredients: Glycerin, USP 25 mg and Water for Injection, USP q.s. Hydrochloric Acid, NF is used to adjust the pH to a target of 3.5.
| Product Name |
Idarubicin Hydrochloride
|
| Chemical Name |
(7S,9S)-9-Acetyl-7-[(3-amino-2,3,6-trideoxy-a-L- lyxo-hexopyranosyl)oxy]-7,8,9,10-tetrahydro-6,9,11- trihydroxy-5,12-naphthacenedione hydrochloride
|
| Synonym |
Idamycin; Zavedos
|
| Formula Wt. |
533.96
|
| Melting Point |
183oC-185oC
|
| Purity |
≥98%
|
| Solubility |
Soluble in water and methanol.
|
| Store Temp |
-20oC
|
| References |
Ganzina, F., Pacciarini, MA., Di Pietro, N. Invest New Drugs. 4:85-105 (1986). Tsuruo, T., Oh-Hara, T., Sudo, Y., Naito, M. Anticancer Res. 13:357-61 (1993). Belaud-Rotureau, MA., Durrieu, F., Labroille, G. et al Leukemia 14:1266-75 (2000).
|
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (1S,3S)-3-acetyl-3,5,12-trihydroxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl 3-amino-2,3,6-trideoxo-α-L–lyxo-hexopyranoside | |
| Clinical data | |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a691004 |
| Pregnancy cat. | D (US) |
| Legal status | ℞-only (US) |
| Pharmacokinetic data | |
| Protein binding | 97% |
| Half-life | 22 hours |
| Identifiers | |
| CAS number | 58957-92-9 |
| ATC code | L01DB06 |
| PubChem | CID 42890 |
| DrugBank | DB01177 |
| ChemSpider | 39117 |
| UNII | ZRP63D75JW |
| KEGG | D08062 |
| ChEBI | CHEBI:42068 |
| ChEMBL | CHEMBL1117 |
| Synonyms | 9-acetyl-7-(4-amino-5-hydroxy-6-methyl-tetrahydropyran-2-yl)oxy-6,9,11-trihydroxy-7,8,9,10-tetrahydrotetracene-5,12-dione |
| Chemical data | |
| Formula | C26H27NO9 |
| Mol. mass | 497.494 g/mol |
Links
-
Synthesis a)
-
US 4,471,052 (Adria; 9.11.1984; appl. 18.1.1982).
-
-
Synthesis of b)
-
DOS 2,525,633 (Soc. Farmaceutici; appl. 06.09.1975; GB -prior. 16.12.1974).
-
US 4,046,878 (Soc. Farmaceutici; 09/06/1977; appl. 05/22/1975; GB -prior. 12.6.1974).
-
-
-
UV and IR Spectra. H.-W. Dibbern, RM Muller, E. Wirbitzki, 2002 ECV
-
NIST / EPA / NIH Mass Spectral Library 2008
-
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman.Academic Press, 2000.
-
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
-
References
- Package insert
- Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, Nieuwland M, Ovaa H, Rottenberg S, van Tellingen O, Janssen J, Huijgens P, Zwart W, Neefjes J (2013). “Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin”. Nature Communications 4: 1908. doi:10.1038/ncomms2921. PMID 23715267.
External links
- Idarubicin bound to proteins in the PDB
 |
|
Title: Idarubicin
CAS Registry Number: 58957-92-9
CAS Name: (7S,9S)-9-Acetyl-7-[(3-amino-2,3,6-trideoxy-a-L-lyxo-hexopyranosyl)oxy]-7,8,9,10-tetrahydro-6,9,11-trihydroxy-5,12-naphthacenedione
Additional Names: (1S,3S)-3-acetyl-1,2,3,4,6,11-hexahydro-3,5,12-trihydroxy-6,11-dioxo-1-naphthacenyl-3-amino-2,3,6-trideoxy-a-L-lyxo-hexopyranoside; 4-demethoxydaunomycin; 4-demethoxydaunorubicin; DMDR
Manufacturers’ Codes: IMI-30; NSC-256439
Molecular Formula: C26H27NO9
Molecular Weight: 497.49
Percent Composition: C 62.77%, H 5.47%, N 2.82%, O 28.94%
Literature References: Orally active anthracycline; analog of daunorubicin, q.v. Prepn: B. Patelli et al. DE 2525633; eidem, US4046878 (1976, 1977 both to Soc. Farmac. Ital.); and antitumor activity: F. Arcamone et al., Cancer Treat. Rep. 60, 829 (1976). Total synthesis for larger scale preparation: M. J. Broadhurst et al., Chem. Commun. 1982, 158. Synthesis of optically pure isomers: Y. Kimura et al., Bull. Chem. Soc. Jpn. 59, 423 (1986). Metabolism and biodistribution in rats: G. Zini et al., Cancer Chemother. Pharmacol. 16, 107 (1986). HPLC determn in plasma: S. S. N. De Graaf et al., J. Chromatogr. 491, 501 (1989). Clinical pharmacokinetics: H. C. Gillies et al., Br. J. Clin. Pharmacol. 23, 303 (1987). Clinical evaluation of cardiac toxicity: F. Villani et al., Eur. J. Cancer Clin. Oncol. 25, 13 (1989). Reviews of pharmacology and antitumor efficacy: A. M. Casazza, Cancer Treat. Rep. 63, 835-844 (1979); F. Ganzina et al., Invest. New Drugs 4, 85-105 (1986). Symposium on clinical experience in acute leukemias: Semin. Oncol. 17, Suppl. 2, 1-36 (1989).
Derivative Type: Hydrochloride
CAS Registry Number: 57852-57-0
Trademarks: Idamycin (Pharmacia & Upjohn); Zavedos (Pharmacia & Upjohn)
Molecular Formula: C26H27NO9.HCl
Molecular Weight: 533.95
Percent Composition: C 58.48%, H 5.29%, N 2.62%, O 26.97%, Cl 6.64%
Properties: Orange crystalline powder, mp 183-185° (Arcamone); also reported as mp 172-174° (Broadhurst). [a]D20 +205° (c = 0.1 in methanol) (Arcamone); also reported as [a]D20 +188° (c = 0.10 in methanol) (Kimura).
Melting point: mp 183-185° (Arcamone); mp 172-174° (Broadhurst)
Optical Rotation: [a]D20 +205° (c = 0.1 in methanol) (Arcamone); [a]D20 +188° (c = 0.10 in methanol) (Kimura)
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Antibiotics and Analogs; Anthracyclines; Topoisomerase II Inhibitor.
|
Fighting prostate cancer with a tomato-rich diet
PUBLIC RELEASE DATE:
27-Aug-2014
Men who eat over 10 portions a week of tomatoes have an 18 per cent lower risk of developing prostate cancer, new research suggests.
With 35,000 new cases every year in the UK, and around 10,000 deaths, prostate cancer is the second most common cancer in men worldwide.
Rates are higher in developed countries, which some experts believe is linked to a Westernised diet and lifestyle.
To assess if following dietary and lifestyle recommendations reduces risk of prostate cancer, researchers at the Universities of Bristol, Cambridge and Oxford looked at the diets and lifestyle of 1,806 men aged between 50 and 69 with prostate cancer and compared with 12,005 cancer-free men.
The NIHR-funded study, published in the medical journal Cancer Epidemiology, Biomarkers and Prevention, is the first study of its kind to develop a prostate cancer ‘dietary index’ which consists of dietary components – selenium, calcium…
View original post 373 more words
Best practice paper on visual inspection to be published in September 2014
DRUG REGULATORY AFFAIRS INTERNATIONAL

The ECA working group on visual inspection, which was founded this year, is going to publish its first document during the ECA event Particles in Parenterals and beyond. Read more.
http://www.gmp-compliance.org/eca_mitt_4410_8398,Z-PEM_n.html
Press Announcement: Best practice paper on visual inspection to be published in September 2014
|
View original post 93 more words
Handling of OOS Results in Europe
DRUG REGULATORY AFFAIRS INTERNATIONAL

FDA’s Guidance on Out-of-Specification Results has been seen as the state of the art regarding the handling of OOS results. In the meantime, Europe – through the British MHRA and the German ZLG – has also developed requirements on that topic. Read more here about the most important regulations of the respective guidance documents.
GMP News: Handling of OOS Results in Europe
http://www.gmp-compliance.org/enews_4461_Handling-of-OOS-Results-in-Europe_8360,8430,Z-QCM_n.html
|
View original post 317 more words
Iroko Pharmaceuticals Gains FDA Approval of Zorvolex for Management of Osteoarthritis Pain
Iroko Pharmaceuticals Gains FDA Approval of Zorvolex for Management of Osteoarthritis Pain

August 25, 2014 — Iroko Pharmaceuticals, LLC, a global specialty pharmaceutical company dedicated to advancing the science of analgesia, announced today the United States Food and Drug Administration (FDA) has approved Zorvolex (diclofenac) capsules, a nonsteroidal anti-inflammatory drug (NSAID), for the management of osteoarthritis pain. This marks the second indication for Zorvolex, approved by FDA in October 2013 for the treatment of mild to moderate acute pain in adults1.
“Given the dose-related adverse events associated with NSAIDs as a class and the widespread use of NSAIDs for osteoarthritis, we are delighted to gain approval for our first SoluMatrix® NSAID for the management of osteoarthritis pain,” said Dr. Clarence Young, Chief Medical Officer of Iroko Pharmaceuticals. “Iroko has already made great strides to help fill the need for low dose NSAID options in patients with acute pain and we are continuing to expand our portfolio to also address chronic pain indications.”
Zorvolex was developed to align with recommendations from FDA and several professional medical organizations that NSAIDs be used at the lowest effective dose for the shortest possible duration consistent with individual patient treatment goals2. Zorvolex is the first FDA-approved low dose NSAID developed using proprietary SoluMatrix Fine Particle Technology™ and is now available by prescription. Zorvolex contains diclofenac as submicron particles that are approximately 20 times smaller than their original size. The reduction in particle size provides an increased surface area, leading to faster dissolution.
“Expanding the use of Zorvolex beyond acute pain to osteoarthritis pain, a chronic condition, is a testament to Iroko’s continued commitment to developing a low dose NSAID portfolio to address a broad range of unmet patient needs,” said John Vavricka, President and CEO of Iroko Pharmaceuticals. “This second approval for Zorvolex continues to lay the groundwork for our future portfolio, which utilizes a new approach to pain management.”

The approval of Zorvolex for the management of osteoarthritis pain was supported by data from a 12-week, multi-center, randomized, double-blind, parallel-group, placebo-controlled trial that enrolled 305 patients, aged 41-90 years, with osteoarthritis of the hip or knee. Half of the patients were between the ages of 61-90. Participants were randomized to Zorvolex 35mg three times daily or 35mg twice daily, or placebo3. The Supplemental New Drug Application (sNDA) also included data from a 12-month open-label safety study that enrolled 602 patients1.
“NSAIDs continue to be an integral part of the management for osteoarthritis, the most common type of arthritis4, and their use is likely to increase as the U.S. population continues to age and the incidence of osteoarthritis rises5,” said Dr. Roy Altman, Professor of Medicine in Rheumatology at UCLA. “The approval of Zorvolex is a welcome and meaningful advance and is the first SoluMatrix® NSAID option approved by the FDA for osteoarthritis pain.”

About Iroko Pharmaceuticals, LLC
Iroko is a global specialty pharmaceutical company, based in Philadelphia, dedicated to advancing the science of analgesia. The company develops and globally commercializes pharmaceutical products.
Iroko is at the forefront of the development of SoluMatrix® NSAIDs – new low dose drug products based on existing NSAIDs – using iCeutica Inc.’s proprietary SoluMatrix Fine Particle Technology™ exclusively licensed to Iroko for NSAIDs. Zorvolex is the first SoluMatrix® NSAID and is available in pharmacies; a second was approved by FDA in February 2014. For more information, visit http://www.iroko.com.

A CASE OF H.I.V. POSITIVE WITH TUBERCULER INFECTION ; एच० आई०वी० के साथ टी०बी० से इन्फेक्टेड मरीज का केस
कुछ दिन पहले एच०आई०वी० से infected एक मरीज का ई०टी०जी० आयुर्वेदास्कैन और इसके तत्सम्बन्धित परीक्षण किये गये /
मरीज की उम्र देखकर मुझे बहुत आश्चर्य हुआ क्योन्कि उसकी उमर के वल २१ साल की है / रोगी के रोग इतिहास को जानने की इछ्छा हुयी और मैने उत्सुकतावश उससे सारी तकलीफ बताने के लिये कहा / रोगी के साथ उसके गार्जियन भी थे /
मुझे यह मालूम करना था कि इतनी छोटी अवस्था मे इस नवयुवक को क्यो H.I.V. जैसा सन्क्रमण हुआ है /
जैसा मुझे बताया गया कि इस रोगी का ROAD accident हुआ था / इस दुर्घटना के बाद उसे अस्पताल मे भरती कराया गया था जहां इस रोगी को कुछ लोगो का रक्त चढाया गया था / इन्ही रक्त दाताओ मे कोई भी व्यक्ति एच०आई०वी० से इन्फेक्टेड होगा इसीलिये जब इस रोगी को रक्त चढाया गया तो इसके भी HIV Infection पैदा हो गया /
कुछ दिनो…
View original post 156 more words
