
Home » Preclinical drugs (Page 8)
Category Archives: Preclinical drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PNQ 103 from Advinus for the potential treatment of COPD,; sickle cell disease (SCD)

Formula I and Formula II
OR

PNQ 103
STRUCTURE COMING…………
for the potential treatment of COPD & sickle cell disease (SCD)
Adenosine A2b receptor antagonist
Advinus Therapeutics Ltd
KEEP WATCHING THIS POST……….
PNQ-103 is a proprietary A2B Adenosine receptor (A2BAdoR antagonist), currently in the pre-clinical development stage for the potential treatment of COPD & sickle cell disease (SCD). Advinus is looking for partnering/co-development opportunities.
A2BAdenosine Receptor (A2BAdoR) Antagonist PNQ-103 for COPD and SCD
COPD
Chronic Obstructive Pulmonary Disease (COPD) is a disease that damages lung tissue or restricts airflow through the bronchioles and bronchi, and commonly leads to chronic bronchitis and emphysema. COPD, along with asthma, forms the third leading cause of death in both developed and developing countries and an annual direct and indirect cost of healthcare of more than $50 billion in the US alone. Current therapies suffer from lack of long term efficacy, patient compliance and a narrow therapeutic index.
Adenosine is a powerful bronchoconstrictor and pro-inflammatory agent in COPD and asthma. Adenosine regulates tissue function by activating its receptors: A1AdoR and A2AAdoR are high affinity receptors and A2BAdoR and A3AdoR are low affinity receptors. During pathological conditions in lung, local adenosine concentrations rise to high levels and activate A2BAdoR. A2BAdoR agonized by adenosine induces both bronchoconstriction and pro-inflammatory effects in lung by acting on multiple cell types that lead to airway hyperreactivity and chronic inflammation. Therefore, A2BAdoR antagonists are expected to be beneficial in COPD and asthma.
PNQ-103 is a proprietary A2BAdoR antagonist, currently in the pre-clinical development stage for the potential treatment of COPD. It is a potent, selective, orally bio-available agent with low clearance and small volume of distribution. PNQ-103 is efficacious in standard rodent asthma and lung fibrosis models. PNQ-103 was found to be safe in exploratory safety studies including a Drug Matrix Screen, mini-AMES test, and a test for cardiovascular liability in dog telemetry as well as a 30- day repeat dose study in rats.
SCD
Sickle Cell Disease (SCD) affects millions of people worldwide. It is caused by an autosomal mutation in the hemoglobin gene (substitution of amino-acid valine [Hb A] for glutamic acid [Hb S]. Hb S in low O2 condition polymerizes, leading to distortion of the cell membrane of red blood cells (RBC) into an elongated sickle shape. Sickled RBCs accumulate in capillaries causing occlusions, impair circulation and cause tissue damage and severe disabilities. Unfortunately, there is no targeted therapy for SCD.
Adenosine levels are elevated in SCD patients. Activation of the A2BAdoR by adenosine increases 2,3-DPG levels in RBCs, which reduces Hb S affinity to O2 and promotes its polymerization leading to RBC sickling. A recent study published in Nature Medicine (2011; 17:79-86) demonstrated potential utility of an A2BAdoR antagonist for the treatment of SCD, through selective inhibition of 2,3-DPG production in RBCs. Therefore, PNQ-103, a selective A2BAdoR antagonist, is expected to be useful for the treatment of SCD. In support, ex vivo PoC (selective inhibition of 2,3-DPG production) has been established for PNQ-103 in RBCs from normal and SCD patients.
EXAMPLES………
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012035548
Example 1: Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyraz -4-yl]-l,6-dihydr»-purin-7-ylmethyl} ester

Step I: Synthesis of l-(3-Trifiuoroirethyl-ben:ijl)-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,354-tetrahydro-pyrimidin-5-yl)-amide
A mixture of 5,6-diamino-3-propyl-l H-pyrimidine-2,4-dione (4.25 g, 0.023 mol), l-(3-Trifluoromethyl-benzyl)-lH-pyrazole-4-carboxylic acid (6.23 g, 0.023 mol), prepared by conventional methods starting from pyrazole-4-carboxylic ester, in methanol (50 ml) were cooled to 0 °C and added EDCI.HC1 (8.82 g, 0.046 mol). The reaction mixture was stirred at 25 °C for 6 h and the organic volatiles were evaporated. To this residue water (50 ml) was added and the precipitate was filtered off, and washed with cold water (50 ml) to obtain l-(3-Trifluoromethyl-benzyl)- 1 H-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (7.2 g, 72 %) as a pale yellow solid.
‘HNMR(400MHz, DMSO d6): δ 0.82 (t, J=7.6Hz, 3H); 1.46-1.51 (m, 2H); 3.64 (t, J=7.2Hz, 2H); 5.49 (s, 2H); 6.01 (s, 2H); 7.55-7.63 (m, 2H); 7.68-7.72 (m, 2H); 7.99 (s, 1H); 8.37 (s, 1H); 8.55 (s, 1H); 10.42 (s, 1H).
Step II: Preparation of l-Propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazoI-4-yl]-3,7-dihydro-purine-2,6-dione
A mixture of l-(3-Trifluoromethyl-benzyl)-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (30 g, 0.068 mol), P205(34.0g, 0.240.8 mol) and DMF (300ml) were heated at 100 °C for 30 minutes. The reaction mixture was cooled to 20-25 °C. The reaction mixture was slowly poured into water (1.5 L) with vigorous stirring. Solid material separated was filtered off, and washed with water (200ml) to obtain 1 -Propyl-8-[l -(3-trifluoromethyl-benzyl)-l H-pyrazol-4-yl]-3,7-dihydro-purine-2,6-dione (25 g, 88 %) as a pale yellow solid.
‘HNMR(400MHz, DMSO d6): δ 0.87 (t, J=7.2Hz, 3H); 1.53-1.60 (m, 2H); 3.98 (t, J=7.2Hz, 2H); 5.53 (s, 2H); 7.57-7.64 (m, 2H); 7.69-7.71 (m, 2H); 8.08 (s, 1H); 8.47 (s, 1H); 1 1.83 (s, 1H); 13.39 (s, 1H)
Step III: Preparation of 2-ChIoro-l-propyI-8-[l-(3-trifluoromethyI-benzyl)-lH-pyrazol-4-yl]-l,7-dihydro-purin-6-one
A mixture of l-Propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-3,7-dihydro-purine-2,6-dione (7.2 g, 0.017 mol), NH4C1 (4.54 g, 0.085 mol) and POCl3 (220 ml) were heated at 120-125 °C for 72 h. Reaction mixture was cooled to 20-25 °C. It was then concentrated under vacuum and quenched with cold water slowly and solid material was separated. It was filtered off and washed with water. The solid material was dried under vacuum. The crude product was purified by column chromatography using silica gel (230-400 mesh) and 0.5 to 4 % methanol in chloroform as an eluent to obtain 2-Chloro-l-propyl-8-[l-(3-trifluoromethyl-benzyl)- lH-pyrazol-4-yl]-l,7-dihydro-purin-6-one (4.2 g, 58 %) as a pale yellow solid.
‘HNMR(400MHz, CD3OD): 6 1.02 (t, J=7.2Hz, 3H); 1.78-1.84 (m, 2H); 4.29 (t, J=7.6Hz,
2H); 5.52 (s, 2H); 7.56-7.57 (m, 2H); 7.63 (m, 2H); 8.12 (s, 1H); 8.35 (s, 1 H)
Step IV: Preparation of 6-Oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-purine-2-carbonitrile
A mixture of 2-Chloro-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-l H-pyrazol-4-yl]-l ,7-dihydro-purin-6-one (O. lg, 0.23 mmol), NaCN (0.016 g, 0.35 mmol), Nal (0.069g, 0.46 mmol) and DMF (2 ml) were stirred for 48 h at 65-70 °C. Reaction mixture was cooled to 20-25 °C and water was added. Solid material was separated. It was filtered off and washed with water. The product was dried under vacuum to obtain 6-Oxo-l-propyl-8-[l-(3-
trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-puriiAe-2-carbonitrile (0.075 g, 77 %) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 0.97 (t, J=7.6Hz, 3H); 1.71-1.77 (m, 2H); 4.12 (t, J=7.6Hz, 2H); 5.51 (s, 2H); 7.57-7.67 (m, 4H); 8.14 (s, 1H); 8.55 (s, 1H); 14.01 (bs, 1H)
Preparation of hosphoric acid di-tert-butyl ester chloromethyl ester:

Step I: Phosphoric acid di-tert-butyl ester
A mixture of di-tert-butylphosphite (5 g, 0.026 mol), NaHC03 (3.71 g, 0.044 mol) and water (50 ml) were taken and cooled to 0-(-5 , °C. KMn04 (6.18 g, 0.039 mol) was added to the reaction mixture in portion wise over ¾ period of 30 minutes at that temperature. The reaction mixture was allowed to warm to 20-25 °C ana stirred for 1.5 hours at that temperature. To this reaction mixture activated charcoal (25 g) was added and stirred at 55-60 °C for 1 hour. The reaction mixture was cooled to room temperature and filtered off and washed with water (200 ml). The filtrate was concentrated to half of its volume and cooled to 0 °C. It was then acidified with con. HC1 (pH~l-2) to obtain solid. The solid material was filtered off, washed with ice cold water and dried under vacuum to obtain Phosphoric acid di-tert-butyl ester as white solid (3.44 g, 63 %).
Step II. Phosphoric acid di-tert-butyl ester chloromethyl ester
A mixture of Phosphoric acid di-tert-butyl ester (1 g, 0.0048 mol), NaHC03 (0.806 g, 0.0096 mol), tetra butyl ammonium hydrogen sulphate (0.163 g, 0.00048 mol), water (40 ml) and DCM (25 ml) were taken. The mixture was cooled to 0 °C and stirred at that temperature for 20 minutes. Chloromethyl chlorosulphatc (0.943g, 0.0057 mol) in DCM (15 ml) was added to it at 0 °C. The reaction mixture allc ed to warm to room temperature and stirred for 18 hours. The organic layer was separated and aqueous layer was extracted with DCM (30 ml). The organic layer was washed with brine (60 ml) solution and dried over Na2SC>4. The organic layer was evaporated to obtain Phosphoric acid di-tert-butyl ester chloromethyl ester as colorless oil (0.79 g, 64%).

Step I: Phosphoric acid di-tert-butyl ester 2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl ester
A mixture of 6-Oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-purine-2-carbonitrile (0.5 g, 0.0012mol), K2C03 (0.485 g, 0.0036 mol ) and acetone ( 10 ml) were taken and stirred for 20 minutes at room temperature. Nal (0.702 g, 0.0047 mol) was added and then Phosphoric acid di-ten-butyl ester chloromethyl ester (0.619 g, 0.0024 mol in 2 ml acetone) was added to the reaction mixture drop wise. The reaction mixture was heated at 45 °C for 16 h. The reaction mixture was filtered through celite and washed with acetone. The organic layer was concentrated and the residue was taken in ethyl acetate (30 ml) and saturated NaHC03 solution (20 ml). The organic layer was separated and washed with saturated sodium thiosulphate solution (20 ml). The organic layer was washed with 0.5 N HC1 solution (20 ml) and brine solution (20 ml). The organic layer was dried over sodium sulphate and evaporated to obtain brown colored mass. The crude product, which is a mixture of N7 and N9 isomers was purified by column chromatography (230-400 mesh silica gel and it was first treated with 5% triethyl amine in hexane) using 5-20 % acetone in hexane (with 0.5 to 1% triethyl amine) as an eluent to obtain N7 isomer (0.34g, 45 % ) and N9 isomer ( 0.1 lg, 14 % )
Phosphoric acid di-tert-butyl ester 2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl ester (N7-isomer).
Ή NMR (400MHz, DMSO d6):6 0.95 (t J=8Hz, 3H); 125 (s, 18 H); 1.75-1.80 (m, 2H); 4.18 (t, J=7.2Hz, 2H); 5.58 (s, 2H); 6.34 (d, ![]()
2H); 7.61-7.63 (m, 2H); 7.70-7.73 (m, 2H); 8.19 (s, 1H); 8.75 (s, 1H)
Phosphoric acid di-tert-butyl ester 2-cyano-8-[l-(3-trifluoromethyI-benzyl)-lH-pyrazol-4-yl]-6-oxo-l-propyl-l,6-dihydro-purin-9-ylmethyl ester (N9-isomer)
Ή NMR (400MHz, DMSO d6): δ 0.94 (t, J=8Hz, 3H); 125 (s, 18 H); 1.74-1.78 (m, 2H); 4.21 (t, J=7.2Hz, 2H); 5.59 (s, 2H); 6.05 (d, J=10.8Hz, 2H); 7.62-7.63 (m, 2H); 7.69-7.71 (m, 2H); 8.16 (s, 1H); 8.71 (s, 1H)
Step II: Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl} ester (N7-isomer).
The above product, N7 isomer (0.34 g, 0.52 mmol) was dissolved in DCM (20 ml) and TFA (0.29 ml, 4.2 mmol) was added to it. The reaction mixture was stirred at room temperature for 7 hours. The organic volatiles were evaporated and the residue was stirred with pentane: diethyl ether (3:1, 10 ml) and the solid material obtained was filtered off and washed with 10 % diethyl ether in pentane (10 ml) to obtain Phosphoric acid mono- {2-cyano-6-oxo-l -propyls’ [ 1 -(3 -trifluoromethyl-benzyl)- 1 H-pyrazol-4-yl]- 1 ,6-dihydro-purin-7-ylmethyl } ester (0.239g, 85 %) as an off white solid.
(400MHz, DMSO d6): δ 0.96 (t, J=7.6Hz, 3H); 1.75-1.81 (m, 2H); 4.16 (t, J=7.2Hz, 2H); 5.58 (s, 2H); 6.23 (d, J=6Hz, 2H); 7.61-7.63 (m, 2H); 7.69-7.75 (m, 2H); 8.22 (s, 1 H); 8.80 (s, 1H); (M+1): 538.2
Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-9-ylmethyl} ester (N9-isomer, 28%)
(400MHz, DMSO d6): δ 0.93 (t, J=7.6Hz, 3H); 1.72-1.80 (m, 2H); 4.16 (t, J=7.2Hz, 2H); 5.54 (s, 2H); 5.95 (d, J=6Hz, 2H); 7.59-7.60 (m, 2H); 7.67-7.73 (m, 2H); 8.17 (s, 1H); 8.72 (s, 1H).
Step III: Phosphoric acid mon -{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyI)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-yimethyl} ester di sodium salt
The above product (0.239g, 0.44 mmol) and water (25 ml) were taken. To the suspension formed, NaHC03 solution (0.1 12g, 1.3 mmol in 20 ml water) was added. The reaction mixture was stirred at room temperature for 1.5 h and the solid material obtained was filtered off. The clear solution was passed through reverse phase column chromatography (LCMS). The fraction obtained was evaporated. It was lyophilized to obtain pure Phosphoric acid mono-{2-cyano-6-oxo- 1 -propyl-8-[ 1 -(3 -trifluoromethyl-benzyl)- 1 H-pyrazol-4-yl]- 1 ,6-dihydro-purin-7-ylmethyl} ester di sodium salt (0.208g; 80%) as an off white solid.
Ή NMR: (400MHz, D20): δ 0.97 (t, J=7.6Hz, 3H); 1.80-1.86 (m, 2H); 4.28 (t, J=7.6Hz, 2H); 5.53 (s, 2H); 6.04 (d, J=3.2Hz, 2H); 7.52-7.53 (m, 2H); 7.62-7.64 (m, 2H); 8.22 (s, 1H); 8.74 (s, 1H)
31P NMR: (400MHz, D20): δ 0.447
EXAMPLES…………..
Patent
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009118759
Example Al: 1, 3-Dipropyl-8-[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yI]-3, 7-dihydro-purine-2, 6-dione

Step I: l-(3-p-ToIyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid ethyl ester
A mixture of l-prop-2-ynyl-lH-pyrazole-4-carboxylic acid ethyl ester obtained as given in example Bl (0.20Og, l.lmmol), 4-iodo toluene (0.254g, 1.1 mol), copper iodide (0.021g, O.l lmmol), dichlorobis (triphenylphosphine)-palladium (II) (39mg, O.Oόmmol), triethylamine (2ml), DMF (2ml) was degassed for lOmin. and stirred for 20hrs at 25-25 0C. Reaction mixture was diluted with water (10ml) and extracted with
• ethyl acetate. Organic layer was washed with brine solution and dried over Na2SO4.
The solvent was evaporated and crude product was purified by column chromatography
(Ethyl acetate: hexane-12:78) to obtain pure l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4- carboxylic acid ethyl ester compound (0.226g, 75%). 1HNMR^OOMHZ, CDCl3): δ 1.35 (t, J=6.8Hz, 3H); 2.37 (s, 3H); 4.31 (q, J=6.8Hz, 2H); 5.18 (s, 2H); 7.16 (d, J=7.6Hz, 2H); 7.38 (d, J=8Hz, 2H); 7.95 (s, IH); 8.21 (s, IH)
Step II: l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxy!ic acid l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid ethyl ester (0.226g, 0.84 mmol) was dissolved in a mixture of solvents THF: methanol: water (3:1:1, 10ml) and LiOH (0.07 Ig, 1.7mol) was added to the reaction mixture with stirring. The reaction mixture was then stirred at 20-25 0C for 2 hours. Solvents were evaporated and the residue was diluted with water (0.5 ml) and acidified with dil. HCl, filtered and dried to obtain off white precipitate, l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (0.182g, 90%).
1HNMR^OOMHZ, CDCl3): δ 2.37 (s, 3H); 5.2 (s, 2H); 7.16 (d, J=7.6Hz, 2H); 7.38 (d, J=8Hz, 2H); 8.01 (s, IH); 8.29 (s, IH) Step III: 1, 3-Dipropyl-8-[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yI]-3, 7-dihydro-‘ purine-2, 6-dione
A mixture of 5,6-diamino-l,3-dipropyl-lH-pyrimidine-2,4-dione (0.075g, 0.33 mmol), l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (0.080gm, 0.33mmol), methanol (5ml), EDCI (0.089g, 0.46mmol) were taken and stirred for 12 hours at 20-25 0C. The reaction mixture was concentrated to obtain intermediate l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (6-amino-2, 4-dioxo-l, 3-dipropyl)-l, 2, 3, 4-tetrahydro-pyrimidine-5yl) amide (50mg, 34%) which was dissolved in hexamethyldisilazane (HMDS). To this reaction mixture ammonium sulphate (0.01 Og) was added. The reaction mixture was refluxed at 140 0C for 18hrs. The organic volatiles were evaporated and the residue was treated with crushed ice, the precipitate formed was filtered off. The product was then purified by column chromatography (l%MeOH in CHCl3) to obtain 1, 3-dipropyl-8~[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yl]-3, 7-dihydro-purine-2, 6-dione (0.035g, 92%). ‘HNMR(400MHz, DMSO d6): δ 0.76-0.87 (m, 6H); 1.51-1.57 (m, 2H); 1.68-1.74 (m, 2H); 2.29 (s, 3H); 3.82 (t, J=7.2Hz, 2H); 3.95 (t, J=7.2Hz, 2H); 5.36 (s, 2H); 7.18 (d, J=8Hz, 2H); 7.35 (d. J=8Hz, 2H); 8.08 (s, IH); 8.49 (s, IH); 13.9 (bs,lH)
Happy new year wishes 2016


/////////
PNQ 201 from Advinus for for potential treatment of IBD.
formula I

PNQ 201
STRUCTURE COMING……
Adenosine A2b receptor antagonist
Advinus Therapeutics Ltd
KEEP WATCHING THIS POST……………
PNQ-201 is a proprietary orally active A2B Adenosine receptor (A2BAdoR) antagonist, currently in pre-clinical development for potential treatment of IBD. Advinus is looking for partnering/co-development opportunities.
A2BAdenosine Receptor (A2BAdoR) Antagonist PNQ-201 for IBD
Inflammatory Bowel Disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), is a multifactorial disease of an etiology not fully understood. It includes chronic inflammation of the gut, characterized by dysfunction of mucosal immunity. Current oral therapies are ineffective, non-specific, and have significant adverse effects. As such, there is a large unmet medical need for the development of new and specific therapies for IBD.
Adenosine is a stimulator of pro-inflammatory effects in the gastro-intestinal tract. Adenosine regulates tissue function by activating its receptors: A1AdoR and A2AAdoR are high affinity receptors and A2BAdoR and A3AdoR are low affinity receptors. A2BAdoR is highly expressed in cecum and colon, with expression increased even further in epithelial cells in human and murine colitis. A2BAdoR, agonized by adenosine induces cytokine secretion at the mucosal surface, inflammatory cell infiltration into intestinal wall, focal crypt damage and ulceration. Therefore, A2BAdoR antagonists are expected to be beneficial in IBD patients.
PNQ-201 is a proprietary orally active A2BAdoR antagonist, currently in pre-clinical development for the potential treatment of IBD. PNQ-201 is a potent and selective A2B antagonist. It is selected for development on the basis of poor systemic bioavailability and high exposure in colon/cecum. Negligible systemic bioavailability and maximum exposure at the sites of action in the lower gastrointestinal tract is expected to offer maximum therapeutic benefits while minimizing potential side effects. PNQ-201 has shown a robust efficacy profile in standard models of IBD, namely, the mouse DSS-induced colitis model and the rat TNBS-induced colitis model. PNQ-201 was found to be safe in exploratory safety studies including a Drug Matrix Screen, mini-AMES test, and a 14- day repeat dose toxicology study in rats.
PATENT
Example 1 : 8-(l-Benzyl~lH-pyrazol-4-yl)-l-propyl-l,4,5,7-tetrahydro-purin-6-one

Step 1: l-Benzyl-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide
A mixture of 5,6-diamino-3-propyl-lH-pyrimidine-2,4-dione (1.6g, 8.55mmol), 1-benzyl-lH-pyrazole-4-carboxylic acid (1.75g, 8.65mmol) in methanol (10ml) were cooled to 0 0C and added EDCLHCl (2.32g, 12.11mmol). The reaction mixture was stirred at 25 0C for 20 hours and the solvents were removed under reduced pressure. To this residue water (10ml) was added and the precipitate was filtered off, and was washed sequentially with cold water (20ml) and DCM (25ml) to obtain l-Benzyl-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3 -propyl- 1 ,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (1.5 g, 47 %) as a pale yellow solid.
1HNMR^OOMHZ5 DMSO d6): δ 0.82 (t, J=7.6Hzs 3H); 1.46-1.51 (m, 2H); 3.64 (t, J=7.2Hz, 2H);^5.36 (s, 2H); 6.01 (s, 2H); 7.26-7.38 (m, 5H); 7.96 (s, IH); 8.31 (s, IH); 8.54 (s, IH); 10.43 (s, IH).
Step 2 : 8-(l-Benzyl-lH-pyrazol-4-yl)-2-chloro-l-propyH,7-dihydro-purin-6-one A mixture of l-benzyl-lH-pyrazole-4-carboxylicacid(6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-ρyrimidin-5-yl)-amide (0.5g, 13.5mmol)s POCl3 (10ml) and DMF (0.1ml) were heated at 125-130 0C for 20 hours. Reaction mixture was cooled to 20-25 0C. It was then concentrated under vacuum. The residue was triturated with diethyl ether, dried. The crude product was purified by column chromatography using silica gel (100-200 mesh) and 2 to 4 % methanol in DCM as an eluent to obtain 8-( 1 -Benzyl- 1 H-pyrazol-4-yl)-2-chloro-l -propyl- l,7-dihydro-purin-6-one (0.04g, 8%) as a pale brown solid.
1HNMR^OOMHZ5 DMSO d6): δ 0.93 (t, J=7.6Hz, 3H); 1.67-1.73 (m, 2H); 4.15 (t, J=7.6Hz, 2H); 5.42 (s, 2H); 7.29-7.39 (m, 5H); 8.14 (s, IH); 8.49 (s, IH); 13.68 (bs, IH). Step 3: 8-(l-Benzyl-lH-pyrazol-4-yl)-l-propyl-l,7-dihydro-purin-6-one
A mixture of 8-(l -benzyl- lH-pyrazol-4-yl)-2-chloro-l -propyl- l,7-dihydro-purin-6-one (0.035 g, 0.094 mmol), Pd\C (10%) (0.025g), in ethanol (20ml) were stirred under hydrogen atmosphere for 20 hours. Reaction mixture was filtered through celite bed washed with methanol (20ml), and the solvents were removed under vacuum. The crude product was purified by column chromatography using silica gel (100-200 mesh) and 2 to 4 % methanol in DCM as an eluent to obtain 8-(l-Benzyl-lH-pyrazol-4-yl)-l-propyl-l,7-dihydro-purin-6-one (0.012g, 39%) as off white solid.
1HNMR^OOMHZ, DMSO d6): δ 0.89 (t, J=7.2Hz, 3H); 1.66-1.72 (m, 2H); 3.94 (t, J=7.6Hz, 2H); 5.41 (s, 2H); 7.302-7.38 (m, 5H); 8.03 (s, IH); 8.16 (s, IH); 8.34 (s, IH).
PATENT
WO 2011055391
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011055391
Preparation 1: 2-chloro-8-cyclopentyl-l-propyI-l, 7-dihydro-purin-6-one:

Step 1: Cyclopentane carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetra hydro-pyriniidin-S-y -amide
To a solution of 5, 6-diamino-3-propyI-lH-pyrimidine-2, 4-dione (0.6 g, 2.72 mmol) in methanol (50 ml) was added cyclopentane carboxylic acid (0.310 g, 2.72 mmol). The reaction mixture was cooled to 0°C and then l-ethyl-3(3′-dimethylaminopropyl) carbodiimide hydrochloride (EDCI.HC1) (0.78 g, 4.1 mmol) was added. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in water. The solid was filtered and washed thoroughly with water followed by diethyl ether. The product obtained was dried under high vacuum. The crude product (0.40 g) was used for the next step without further purification.
Step 2: Preparation of 8-cyclopentyI-2-chloro-l-propyl-l, 7-dihydro-purin-6-one
To a suspension of cyclopentanecarboxylic acid (6-amino-2,4-dioxo-3-propyl- 1,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (0.40 g, crude) obtained from step 1 in phosphorus oxychloride (25 ml) was added phosphorus pentachloride (0.10 g) and the resulting reaction mixture was refluxed overnight. Phosphorus oxychloride was evaporated under reduced pressure. The residue was slowly quenched with water. Ethyl acetate was added and the organic layer was separated and washed thoroughly with water followed by brine. The ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under vacuum. The crude product was purified by preparative TLC using dichloromethane, methanol (9:1) as the solvent system to give 0.075 g (19% over two steps) of the product as a white solid.
•H MR (400 MHz, DMSO d6): δ 0.9 (t, J = 8 Hz, 3H), 1.59-1.82 (m, 8H), 1.99 (m, 2H), 3.15 (t, J = 8 Hz, 1H), 4.12 (t, J = 8 Hz, 2H).
Preparations 2 to 7 were prepared following the experimental procedure as given for Preparation 1.
Preparation 2: 2-Chloro-8-cyclohexyl- 1 -propyl- 1 ,7-dihydro-purin-6-one,
Preparation 3: 2-Chloro-8-cyclopropyl-l -propyl- 1 ,7-dihydro-purin-6-one,
Preparation^ 2-Chloro-8-(hexahydro-2,5-methano-pentalen-3a-yl)-l -propyl- 1,7- dihydro-purin-6-one,
Preparation 5: 8-Bicyclo-[2.2.1]-hept-2-yl-2-chloro-l -propyl- 1, 7-dihydro-purin-6- one,
Preparation 6: 8-Adamantan-2-yl-2-chloro-l -propyl- 1, 7-dihydro-purin-6-one, Preparation7:3-[4-(2-Chloro-6-ox0-l-propyl-6,7-dihydro-lH-purin-8-yl)- bicyclo[2.2.2]oct-l-yl]-propionic acid.
Example 1: 8-Cyclopentyl-2-(3, 4-difluoro-phenoxy)-l-propyl-l, 7-dihydro-purin- 6-one:

To a solution of 8-cyclopentyl-2-chloro- 1 -propyl- l,7-dihydro-purin-6-one (0.06 g, 0.21 mmol) in N-methyl-2-pyrrolidone (0.2 ml) was added K2CO3 (0.044g, 0.32 mmol) followed by 3, 4-difluoro phenol and the reaction mixture was heated at 130 °C overnight. The reaction mixture was diluted with ethyl acetate and water. The layers were separated and ethyl acetate layer was washed with water. The ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under vacuum. The crude product was purified by preparative TLC using 3% methanol in DCM to give the product (0.015 g, 19 %) as a white solid.
‘HNMR (400 MHz, DMSO d6): δ 0.94 (t, J = 8 Hz, 3H), 1.59-1.74 (m, 6H), 1.94 (br.s, 2H), 3.12 (m, 2H), 4.09 (br. s, 2H), 7.21 (d, J = 8 Hz, 1H), 7.53-7.65 (m, 2H), 12.74 (br.s, 1H).
PNQ 370 useful in treating Parkinson’s disease from ADVINUS
2016

PNQ 370
Advinus Therapeutics Ltd
Adenosine A2a receptor antagonist
for treating disease or disorder susceptible to improvement by antagonism of A2A receptor.
Advinus Therapeutics is investigating PNQ-370, presumed to be lead from a series of small molecule therapeutics including PD-2 and PD-3, as adenosine A2a receptor antagonist, for the potential treatment of Parkinson’s disease . In November 2012, this drug was in preclinical development .
![]()
KEEP WATCHING THIS POST AS I ARRIVE AT THE STRUCTURE…………..








ONE OF THE ABOVE OR SIMILAR
INTRODUCTION
The effects of adenosine are mediated through at least four specific cell membrane receptors so far identified and classified as Ai, A2A, A2B and A3 belonging to G protein-coupled receptor family. The Ai and A3 receptors down-regulate cellular cAMP levels through their coupling to G protein, which inhibit adenylate cyclase. In contrast, A2A and A2B receptors couple to G protein that activate adenylate cyclase and increase intracellular levels of cAMP. Through these receptors, adenosine regulates the wide range of physiological functions.
Advances in understanding the role of adenosine and its receptors in physiology and pathophysiology, as well as new developments in medicinal chemistry of these receptors have identified potential therapeutic areas for drug development. With the combination of pharmacological data, using selective ligands and genetically modified mice, important progress has been made toward an understanding of the role of ARs in a variety of diseases, such as inflammatory conditions, sepsis, heart attack, ischemia-reperfusion injury, vascular injury, spinal cord injury, chronic obstructive pulmonary disease (COPD), asthma, diabetes, obesity, inflammatory bowel disease, retinopathy, and Parkinson’s Disease (PD).
Happy new year wishes 2016
Movement disorder constitutes a serious health problem, especially among the elderly. These movement disorders can often be the result of brain lesions. Disorders involving the basal ganglia which result in movement disorders include Parkinson’s disease, Huntington’s chorea and Wilson’s disease. Tremor, rigidity, akinesia and postural changes are four classic symptoms of Parkinson’s disease, it is also associated with depression, dementia and overall cognitive decline. Parkinson’s disease has a prevalence of 1 per 1000 of the total population and increases to 1 per 100 for those aged over 60 years. Degeneration of dopaminergic neurons in the substantia nigra and the subsequent reductions in the interstitial concentrations of dopamine in the striatum are critical to the development of Parkinson’s disease. About 80% of cells from the substantia nigra can be destroyed before the clinical symptoms of Parkinson’s disease become apparent
PD is a progressive, incurable disorder with no definite preventive treatment, although drugs are available to alleviate the symptoms and/or slow down the progress of the disease. Current therapy is based on dopamine replacement therapy, the most common drug treatments being dopaminomimetic agents, including L-DOPA, a dopamine precursor, as well as direct or indirect dopamine receptor agonists. L-DOPA is the mainstay in the treatment of PD, but because of tolerance problems and a wide range of adverse reactions, including involuntary movements and vomiting, a strong demand for new therapies exists. Among the various strategies, A2A AR blockers are considered a potential approach to treatment of the disease. Within the brain A2A ARs are richly expressed in the striatum, nucleus accumbens, and olfactory tubercle. A coexpression of A2A with D2 dopamine receptors has been reported in the GABAergic striatopallidal neurons where adenosine and dopamine agonists exert antagonistic effects in the regulation of locomotor activity. Activation of A2A ARs in striatopallidal neurons decreases the affinity of D2 receptors for dopamine, antagonizing the effects of D2 receptors.
The negative interaction between A2A and D2 receptors is at the basis of the use of A2A antagonists as a novel therapeutic approach in the treatment of PD. (Pharmacol. Ther. 2005, 105, 267). The recent discovery that the A2A can form functional heteromeric receptor complexes with other Gprote in-coupled receptors such as D2 and the mGlu5 receptors has also suggested new opportunities for the potential of A2A antagonists in PD. (J. Mol. Neurosci. 2005, 26, 209).
A2A knockout (KO) mice transient focal ischemia caused less neuronal damage in comparison to their wild-type (WT) littermates (J. Neurosci. 1999, 19, 9192.). Therefore, it seems that tonic activation of A2A ARs may be responsible for dangerous signal during injury, in contrast to the neuroprotective effects induced by endogenous Al activation. Recently, selective inactivation or reconstitution of A2A ARs in bone-marrow cells revealed their contribution to the development of ischemic brain injury (J.F. Nat. Med. 2004, 10, 1081) Blockade of A2A ARs has recently been implicated in the treatment of movement disorders such as Parkinson’s disease (Trends Pharmacol. Sci. 1997, 18, 338-344) and in the treatment of cerebral ischaemia (Life Sci. 1994, 55, 61-65).
The potential utility of A2A AR antagonists in the treatment of Parkinson’s disease has been reviewed (CNS drugs, 1998, 10, 31 1-320). One advantage of A2A AR antagonist therapy is that the underlying neurodegenerative disorder may also be treated ((Ann. N. Y. Acad. Sci. 1997, 825 (Neuroprotective Agents), 3048). In particular, blockade of A2A AR function confers neuroprotection against MPTP-induced neurotoxicity in mice (Neurosci. 2001, 21, RC143).
Alzheimer’s disease (AD) is a neurodegenerative disorder of the central nervous system manifested by cognitive and memory deterioration, a variety of neuropsychiatric symptoms, behavioral disturbances, and progressive impairment of daily life activities. Recent research suggests that adenosine receptors play important roles in the modulation of cognitive function. Epidemiological studies have found an association between coffee (a nonselective adenosine receptor antagonist) consumption and improved cognitive function in AD patients and in the elderly. Long-term administration of caffeine in transgenic animal models showed a reduced amyloid burden in brain with better cognitive performance.

Antagonists of adenosine A2A receptors mimic these beneficial effects of caffeine on cognitive function. Neuronal cell cultures with amyloid beta in the presence of an A2A receptor antagonist completely prevented amyloid beta-induced neurotoxicity. These findings suggest that the adenosinergic system constitutes a new therapeutic target for AD, and caffeine and A2A receptor antagonists may have promise to manage cognitive dysfunction in AD (Curr Neuropharmacol. 2009 September; 7(3): 207-216).
High expression of A2A ARs has been found in platelets, leukocytes, vascular smooth muscle, and endothelial cells with important implications in the regulation of inflammatory responses. It is now well established that stimulation of the A2A AR in immune cells induces anti-inflammatory effects, mostly due to its ability to increase cAMP levels, which has strong immunosuppressive effects (Trends Immunol. 2005, 26, 299). Stimulation of A2A ARs inhibits neutrophil adherence to the endothelium, degranulation of activated neutrophils and monocytes, plus superoxide anion generation. A2A ARs have been recently defined as sensors and terminators of proinflammatory activities. The strongest evidence for the key role of A2A in inflammation is derived by the elegant study using mice deficient in A2A ARs (Nature 2001, 414, 916).

In this model the lack of A2A subtype leads to increased tissue inflammation and damage, thus suggesting a negative and nonredundant regulatory role for the A2A AR. This model permits one to appreciate that adenosinergic regulation of immune cells is fundamental in normal physiological control of inflammation in vivo in spite of the fact that other Gs-protein-coupled receptors and cAMP elevating ligands are present, such as cathecolamines, prostaglandins, dopamine, and histamine (Trends Immunol. 2005, 26, 299). Interestingly, the A2A AR has been demonstrated to be involved in promotion of wound healing and angiogenesis in healing wounds (Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R283).
Moreover, it plays an active role in the pathogenesis of dermal fibrosis, suggesting a role for antagonists as novel therapeutic approach in the treatment and prevention of dermal fibrosis in diseases such as scleroderma (Arthritis Rheum. 2006, 54, 2632) as well as hepatic fibrosis (Br. J. Pharmacol. 2006 Aug; 148(8): 1 144-55). Studies also suggest that A2A receptor antagonists may be beneficial for social memory impairment and hypertension (Behav Brain Res. 2005 Apr 30;159(2):197-205), sepsis (J Immunol. 2006 May 1 ; 176(9): 5616-26), spinal cord injury and neuroprotection (J Neuroinflammation. 201 1 Apr 12;8:31), retinopathy (IVOS, Jan. 2000, vol. 41 (1), 230-243, depression (Neurology. 2003 Dec 9;61(1 1 Suppl 6):S82-7), narcolepsy and other sleep related disorders (Prog Neurobiol. 2007 Dec;83(5):332-47), attention-deficit hyperactivity disorder (ADHD) (Behav Pharmacol. 2009 Mar;20(2): 134-45; Clinical Genetics (2000), 58(1), 31-40 and references therein),

Dr Rashmi Barbhaiya, CEO & Managing Director

… Dr Rashmi Barbhaiya, CEO & Managing Director and Dr Kasim Mookthiar, Chief Scientific Officer and SVP, Drug Discovery, Advinus Therapeutics …
Antagonists of the A2A receptor are potentially useful therapies for the treatment of addiction. Major drugs of abuse (opiates, cocaine, ethanol, and the like) either directly or indirectly modulate dopamine signaling in neurons particularly those found in the nucleus accumbens, which contain high levels OfA2A adenosine receptors. Dependence has been shown to be augmented by the adenosine signaling pathway, and it has been shown that administration of an A2A receptor antagonist redues the craving for addictive substances (“The Critical Role of Adenosine A2A Receptors and Gi βγ Subunits in Alcoholism and Addiction: From Cell Biology to Behavior”, by Ivan Diamond and Lina Yao, (The Cell Biology of Addiction, 2006, pp 291-316) and “Adaptations in Adenosine Signaling in Drug Dependence: Therapeutic Implications”, by Stephen P. Hack and Macdonald J. Christie, Critical Review in Neurobiology, Vol. 15, 235-274 (2003)). See also Alcoholism: Clinical and Experimental Research (2007), 31(8), 1302-1307.
A2A receptors may be beneficial for the treatment or prevention of disorders such as a movement disorder, for example, Parkinson’s disease or progressive supernuclear palsy, Restless leg syndrome, nocturnal myoclonus, cerebral ischaemia, Huntington’s disease, multiple system atrophy, corticobasal degeneration, Wilson’s disease or other disorders of basal ganglia which results in dyskinesias, post traumatic stress disorder. See for example WO200013682, WO200012409, WO2009156737, WO20091 1442, WO2008121748, WO2001092264, WO2007038284, WO2008002596, WO20091 1 1449, WO20091 1 1442, WO2008121748, WO2009156737, WO2003022283, WO2005044245, WO2008077557, WO20091 1 1449, WO2009705138, WO20091 1 1442, WO2007035542, WO20080870661, WO2008070529, WO20051 16026, WO2009055548, WO2007133983, WO2010045006, WO2010045015, WO2010045008 WO2009015236.
![]()

centre: Mr Ratan Tata, Chairman, Tata Sons, flanked by Dr Rashmi Barbhaiya (left), Managing Director and CEO, Advinus, and Mr R. Gopalakrishnan, …
ONE EXAMPLE………..
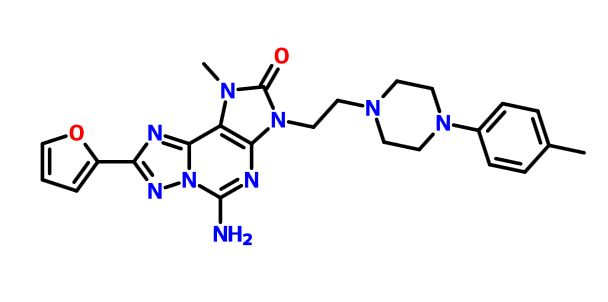
| Molecular Formula: | C26H31N9O4 |
|---|---|
| Molecular Weight: | 533.58224 g/mol |
 A1
A15-amino-8-(furan-2-yl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-1-yl]ethyl]-1-methyl-[1,2,4]triazolo[5,1-f]purin-2-one
Example Al :
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
Step-1 : 2-[(2,5-Diamino-6-chloro-pyrimidin-4-yI)amino]ethanol
A mixture of 4,6-dichloropyrimidine-2,5-diamine (28g, 156mmol), ethanolamine (18ml, 312mmol) and ethanol (250ml) were heated at 100-1 10 °C for 16 hours. The mixture was cooled and solvent was removed. To the residue methanol (100ml) was added and stirred for 20 minutes. The solid was filtered off to obtain 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol (22.0g, 70%).
‘H MR(400MHz, DMSO d6): δ 3.36-3.40 (m, 2H); 3.50-3.54 (m, 2H); 3.88 (bs, 2H); 4.74 (t, J=5.6Hz, 1H); 5.63 (bs, 2H); 6.51 (t, J=5.6Hz, 1H)
Step-2: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one
A mixture of 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol obtained in step 1 (l O.Og, 49.26mmol) in acetonitrile (400ml) were cooled to 0 °C. To this reaction mixture K2C03 (20.39gm, 147.7mmol) and 4-nitrophenyl chloroformate (19.8g, 98.52mmol)was added and stirred at 25-27 °C for 24 hours. This reaction mixture was filtered and washed with acetonitrile (300ml) and diethyl ether (300ml) respectively. Solid obtained was dried to obtain crude 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one as a yellow solid. Small amount of crude material was purified by column chromatography to obtain pure product. ‘HNMR(400MHz, DMSO d6): δ 3.61-3.66 (m, 2H); 3.72-3.75 (m, 2H); 4.85 (t, J=6Hz, 1H); 6.60 (s, 2H); 1 1.21 (s, 1 H)
Step-3: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one obtained in step 2 (13g, 56.7mmol) , K2C03 (1 1.5g, 84mmol), methyl iodide (12g, 85.15mmol) and DMF (130ml) were stirred at 25-30 °C for 16 hours. The reaction mixture was concentrated and purified by column chromatography using 60-120 silica gel and 4% methanol in DCM as an eluent to obtain 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one (8g, 58%) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.42 (s, 3H); 3.65 (t, J=5.6Hz, 2H); 3.78 (t, J=5.6Hz, 2H); 4.85 (t, J=5.6Hz, 1H); 6.69 (bs, 2H).
Step-4: 2-Amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyI-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one obtained in step 3 (8g, 32.9mmol) , Hydrazine hydrate (16ml ,32.9mmol) and ethanol (300ml) were heated at 100-1 10 °C for 16 hours. The reaction mixture was concentrated and solid obtained was filtered off and dried to obtain 2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (7g, 89 %) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.37 (s, 3H); 3.58-3.61 (m, 2H); 3.71 (t, J=6Hz, 2H); 4.29 (bs, 2H); 4.87 (t, J=5.6Hz, 1H), 6.00 (bs, 2H); 7.63 (s, 1H).
Step-5: N’-[2-Amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide
2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (4.5g, 18.18mmol) obtained in step 4, 2-furoic acid (2.53g, 22.5mmol), HOBT (2.53g, 18.8 mmol) and N-methylmorpholine were taken in dimethylformamide (40ml). l-Ethyl-3(3′-dimethylaminopropryl)carbodiimide hydrochloride (EDCI.HCl) (5.4g, 28.2mmol) was added to the reaction mixture and stirred at 25-27 °C for 14 hours. The reaction mixture was evaporated and residue was purified by column chromatography to obtain N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide (5.3g, 84%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.43 (s, 3H); 3.59-3.63 (m, 2H); 3.74 (t, J=6Hz, 2H); 4.88 (t, J=5.6Hz, 1H); 5.98 (bs, 2H); 6.67 (bs, 1H); 7.25 (d, J=3.2Hz, 1H); 7.90 (s, 1H); 8.35 (s, 1H); 10.28 (s, lH).
Step-6: 5-Amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l^,4]triazolo[5,l-flpurin-2-one
A mixture of N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide obtained in step 5 (5.3g, 15.9mmol), Ν,Ο-bistrimethylsilylacetamide (27ml, 1 1 1.4mmol) and hexamethyldisilazane (83ml, 397mmol) were heated at 1 10-120 °C for 16 hours. The reaction mixture was quenched with methanol (100ml) and water (100ml) and organic volatiles were evaporated. The solid obtained was filtered off and washed with water (30ml) followed by diethyl ether (100ml) to obtain 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l,2,4]triazolo[5,l-f]purin-2-one (3.50g, 71%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.56 (s, 3H); 3.67-3.70 (m, 2H); 3.84-3.87 (m, 2H); 4.88 (t, J=5.6Hz, 1H); 6.73 (bs, 1H); 7.20 (bs, 1H); 7.79 (bs, 2H); 7.94 (bs, 1H).
Step-7: 2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5,l-fJpurin-3-yl]ethyl 4-methylbenzenesulfonate
A mixture of 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l -methyl-[l,2,4]triazolo[5, l-fJpurin-2-one obtained in step 6 (3.5g, l lmmol), p-toluene sulphonylchloride (5.2 g, 27mmol) were taken in pyridine (30ml)and stirred at 25-27 °C for 16 hours. To the reaction mixture hexane (100ml) was added and solid obtained was filtered off and washed with water (100ml) followed by hexane (100ml) to obtain 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate (4.1g, 78%) as a brown solid. ‘HNMR (400MHz, DMSO d6): δ 2.02 (s, 3H); 3.49 (s, 3H); 3.99 (t, J=4.8Hz, 2H); 4.71 (t, J=4.8Hz, 2H); 6.73-6.75 (m, 1H); 7.01 (d, J=8Hz, 2H); 7.23 (d, J=3.2Hz, 1H); 7.41 (d, J=8.4Hz, 2H); 7.78 (bs, 2H); 7.96 (d, J=1.2Hz, 1H).
Step-8: : 5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-l-methyl-[l,2,4]triazolo[5,l-f)purin-2-one
A mixture of 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l ,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate obtained in step 7 (0.25g, 0.533mmol), l-[4-(2-Methoxy-ethoxy)-phenyl]-piperazine (0.188g, 0.799mmol) and DIPEA (0.27ml, 1.599mmol) were taken in DMF (5ml) and stirred at 80 °C for 16 hours. To the reaction mixture water (100ml) was added and solid obtained was filtered off. The crude product was purified by column chromatography to obtain 5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one (0.135g, 47%) as an off white solid
‘HNMR (400MHz, DMSO d6): δ 2.60 (bs, 4H); 2.68 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.29 (s, 3H); 3.56 (s, 3H); 3.59-3.62 (m, 2H); 3.94-4.00 (m, 4H); 6.71 -6.73 (m, 1H); 6.79-6.86 (m, 4H); 7.19 (dd, J=3.2Hz, 1.2Hz, 1H); 7.80 (bs, 2H); 7.94 (bs, 1H).
ANOTHER……..
Example Gl: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-[l,2,4]triazolo[5,l-i]purin-2-one

Step-1 : 2-Amino-6-chloro-7-ethyl-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-3 in example Al)
‘HNMR (400MHz, DMSO d6): δ 1.21 (t, J=7.2Hz, 3H); 3.64 (s, 2H); 3.78 (t, J=6Hz, 2H);
3.92 (q, J=7.2Hz, 2H); 4.92 (bs, I H); 6.7 (bs, 2H).
Step-2 : 2-Amino-7-ethyl-6-hydrazino-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-4 in example Al)
‘ HNMR (400MHz, DMSO d6): δ 1.07 (t, J=6.8Hz, 3H); 3.59 (q, J=6Hz, 2H); 3.72 (t, J=6Hz,
2H); 3.91 (q, J=6.8Hz, 2H); 4.32 (bs, 2H); 4.86 (t, J=5.6Hz, IH); 5.99 (bs, 2H), 7.55 (bs, IH).
Step-3: N’-[2-Amino-7-ethyl-9-(2-hydroxyethyl)-8-oxo-purin-6-yl]furan- 2carbohydrazide (Procedure is same as step-5 in example Al)
Crude product was used in next step
Step-4: 5-Amino-l-ethyI-8-(2-furyl)-3-(2-hydroxyethyl)-[l,2,4]triazolo[5,l-flpurin-2-one
(Procedure is same as step-6 in example Al)
‘H MR (400MHZ, DMSO d6): δ 1.34 (t, J=7.2Hz, 3H); 3.67 (q, J=5.6Hz, 2H); 3.84 (t, J=5.6Hz, 2H); 4.01 (q, J=7.2Hz, 2H); 4.87 (t, J=6Hz, IH); 6.70 (bs, IH); 7.17 (d, J=2.8Hz, I H); 7.18 (bs, 2H); 7.92 (bs, IH).
Step-5: 2-[5-Amino-l-ethyl-8-(2-furyl)-2-oxo-[l,2,4]triazoIo[5,l-f|purin-3-yl]ethyl 4- methylbenzenesulfonate (procedure is same as step-7 in example Al)
lHNMR (400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.00 (s, 3H); 3.95-4.00 (m, 4H); 4.47 (bs, 2H); 6.74 (s, IH); 7.00 (d, J=7.6Hz, 2H); 7.22 (s, IH); 7.42 (d, J=7.6Hz, 2H); 7.78 (bs, 2H); 7.97 (bs, IH).
Step-6: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyi]piperazin-l- yl]ethyl]-[l,2,4]triazolo[5,l-f]purin-2-one (procedure is same as step-8 in example Al)
HNMR(400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.60 (bs, 4H); 2.68 (t, J=6.8Hz, 2H); 2.95 (bs, 4H); 3.28(s, 3H);3.61 (t, J=4.4Hz, 2H); 3.94-4.04 (m, 6H); 6.72 (dd, J=2Hz, 3.6Hz, I H); 6.78-6.85 (m, 4H); 7.19 (d, J=3.2Hz, IH); 7.81(bs, 2H); 7.94 (s, IH).
Representative compounds of the present disclosure were tested and had micromolar to nanomolar activity.

 A1 ABOVE
A1 ABOVE
A7 ABOVE
A9 ABOVE
A13 ABOVE
A31 ‘HNMR (400MHz, DMSO d6): δ 2.62 (bs,4H); 2.68 (t, J=6.8Hz, 2H); 2.85 (bs, 4H); 3.28 (s, 3H); 3.57 (s, 3H); 3.59-3.62 (m, 2H); o 3.95 (t, J=6.8Hz, 2H); 4.01-4.04 (m, 2H);
5-Amino-3-[2-[4-[2-fluoro-4-(2- 6.66-6.68 (m, 1H); 6.72 (dd, J=2 Hz,3.6Hz, methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-8- 1H); 6.79 (dd, J=2.8Hz, 14Hz, 1H); 6.92 (t, (2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f|purin-2- J=9.6Hz, 1H); 7.19 (d, J=3.2Hz, 1 H); 7.93 one (bs, 2H); 7.93-7.94 (m, 1H).
A31 ABOVE
A32 HNM (400MHz, DMSO d6): δ 2.59 (bs,
4H); 2.68(t, J=6.4Hz, 2H); 3.27(t, J=4.8Hz, 4H); 3.56 (s, 3H); 3.96 (t, J=6.4Hz, 2H);
0 6.72(dd, J=2Hz, 3.6Hz, 1H); 6.99 (d, J=8.8Hz,
4-[4-[2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo- 2H); 7.19 (d, J=3.6Hz, 1H);7.56 (d, J=8.8Hz, [ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]piperazin- 2H); 7.80 (bs, 2H); 7.93 (bs, lH).
l-yl]benzonitrile
A32 ABOVE
A36 ‘HNMR(400MHz, CDCI3): δ θ.09 (d,
J=4.4Hz, 2H); 0.50 (d, J=6.8Hz, 2H); 0.82- 0.89 (m, 1H); 2.24 (d, J=6.0Hz, 2H): 2.52- 2.72 (m, 8H); 2.80 (t, J=6.4Hz, 2H); 3.76 (s,
5-Amino-3-[2-[4-(cyclopropylmethyl)piperazin- 3H); 4.07 (t, J=6.8Hz, 2H); 5.89 (bs, 2H); l -yl]ethyl]-8-(2-furyl)-l-methyl- 6.61 (bs, 1H); 7.22 (d, J=2.4Hz, 1H); 7.64 (s, [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 1H).
A36 ABOVE
A38 ‘HNMR(400MHz, CDCI3): δ 2.62 . (t,
J=4.4Hz, 4H); 2.79 (t, J=6.4Hz, 2H); 2.81 (s, 6H); 3.22 (t, J=4.4Hz, 4H): 3.77 (s, 3H); 4.06 (t, J=6.8Hz, 2H); 5.74 (bs, 2H); 6.60 (dd,
4-[2-[5-Amino-8-(2-fiiryl)- 1 -methyl-2-oxo- J=2.0Hz, 3.2Hz, 1H); 7.24 (d, J=3.6Hz, 1H);
[ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]-N,N- 7.65 (s, 1H).
dimethy l-piperazine- 1 -sulfonamide
A38 ABOVE
A39 ‘HNMR(400MHZ, DMSO d6): δ 1.89-1.94
im, 1H); 2.09-2.18 .(m, 1 H); 2.60 (bs, 4H); 2.67 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.56 (s, 3H); 3.69-3.85 (m, 4H); 3.95 (t, J=6.4Hz,
2H); 4.89 (bs, 1H); 6.72 (dd, J=2.0, 3.2Hz,
5-Amino-8-(2-furyl)-l -methyl-3-[2-[4-(4- 1H); 6.78 (d, J=9.2Hz, 2H); 6.85 (d, J=9.2Hz, tetrahydrofuran-3-yloxyphenyl)piperazin- 1 – 2H): 7.20 (d, J=3.2Hz, 1 H); 7.80 (bs, 2H); yl]ethyl]-[l ,2,4]triazolo[5,l-f]purin-2-one
7.93 (s, 1H).
A39 ABOVE
A42 ‘HNMR(400MHz, CDCI3): δ
2.26 (s,3H); 2.94-2.97 (m, 6H); 3.72 (s, 2H); 3.75 (s, 3H); 4.17 (t, J=6.4Hz, 2H); 5.74 (bs, 2H); 6.59 (dd, J=1.6Hz, 3.6Hz, 1H);7.13 (s, J=3.6Hz, IH); 7.21-7.24 (m, IH); 7.63 (s,
5-Amino-8-(2-furyl)-l-methyl-3-[2-(3-methyl- IH); 8.20 (bs, IH),
7,8-dihydro-5H- 1 ,6-naphthyridin-6-yl)ethyl]- [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
A42 ABOVE
A57 HNMR(400MHz, DMSO d6): δ 2.95 (t,
J=8Hz, 2H); 3.52 (s, 3H); 3.69 (s, 3H ), 3.97 (t, J=8Hz, 2H); 6.71 (dd, J=2Hz, 3.6Hz, I H );
5-Amino-8-(2-furyl)-3-[2-(4- 6.80 (dd, J=2Hz, 6.8Hz, 2H); 7.10 (d, methoxyphenyl)ethyl]- 1 -methyl- J=8.8Hz, 2H); 7.18 (dd, J=0.8Hz, 3.2Hz, I H );
[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 7.80 (bs, 2H), 7.94 (dd, J=lHz, 2Hz, I H ).
A57 ABOVE
A58 HNMR(400MHz, DMSO d6): δ 2.61 (bs,
4H); 2.68 (bs, 2H); 3.05(bs, 4H); 3.57 (s, 3H ), 3.96 (bs, 2H); 6.72 (bs, IH); 6.92 (d, J=8Hz, 2H); 7.01 (d, J=10Hz, 2H );7.03(d, J=148Hz, IH); 7.19 (bs , 1 H); 7.80 (bs, 2H); 7.94 (s,
5-amino-3-[2-[4-[4- IH).
(difluoromethoxy)phenyl]piperazin-l-yl]ethyl]- 8-(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin- 2-one
A58 ABOVE
A62 O ‘HNMR (400MHz, DMSO d6): δ 0.66-0.70
(m, 4H); 1.90-1.94 (m, lH); 2.41 (bs, 4H); 2.65 (t, J=6Hz, 2H); 3.38 (bs, 2H); 3.56 (bs, 5H); 3.93 (t, J=6.4 Hz, 2H); 6.71 (bs, 1H );
5-Amino-3-[2-[4- 7.19 (d, J=2.4Hz, 1H); 7.79 (bs, 2H); 7.93 (bs,
(cyclopropanecarbonyl)piperazin- 1 -yl]ethyl]-8- 1H).
(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin-2- one
A62 ABOVE
A63 ‘HNMR (400MHz, DMSO d6): δ 0.07-0.10
(m, 2H); 0.40-0.44 (m, 2H); 0.88-0.94 (m,lH); 2.21 (d, J=6.4Hz, 2H); 2.41-2.45 (m, 4H); 2.64 (t, J=6.4Hz, 2H); 3.38 (bs,4H); 3.56
5-Amino-3-[2-[4-(2- (s, 3H); 3.93 (t, J=6.4Hz, 2H); 6.72 (dd, cyclopropylacetyl)piperazin-l -yl]ethyl]-8-(2- J=2Hz,3.6 Hz, 1H); 7.19-7.20 (m, 1H); 7.80 fury 1)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fJpurin-2- (bs, 2H); 7.93 (d, J=0.8 Hz, 1H).
one
A63 ABOVE
C1 ABOVE

E1 ABOVE

D3 ABOVE
G1 ABOVE
G2
H2
M1
M2
M3
M6
ETC AS IN TABLE……………..


/////////
n21c(nc4c(c1nc(n2)c3occc3)N(C(N4CCN5CCN(CC5)c6ccc(cc6)OCCOC)=O)C)N
CN1C2=C(N=C(N3C2=NC(=N3)C4=CC=CO4)N)N(C1=O)CCN5CCN(CC5)C6=CC=C(C=C6)OCCOC
Lefucoxib (乐福昔布)

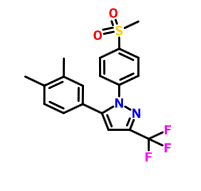
Lefucoxib (乐福昔布)
5-(3,4-dimethyl-phenyl)-1-methanesulfonyl-3-trifluoromethol-pyrazole
1 [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole
CAS 849048-84-6
![]()
| Molecular Formula: | C19H17F3N2O2S |
|---|---|
| Molecular Weight: | 394.41069 g/mol |
IND FILED
Prostaglandin G/H Synthase 2 (PTGS2; COX-2) Inhibitors
A COX-2 inhibitor potentially for the treatment of rheumatoid arthritis.
cyclooxygenase-2 (COX-2) inhibitor
National Center of Biomedical Analysis
![]()
 CHINA FLAG
CHINA FLAG

PATENT
CN 1468854
http://www.google.com/patents/CN1468854A?cl=en
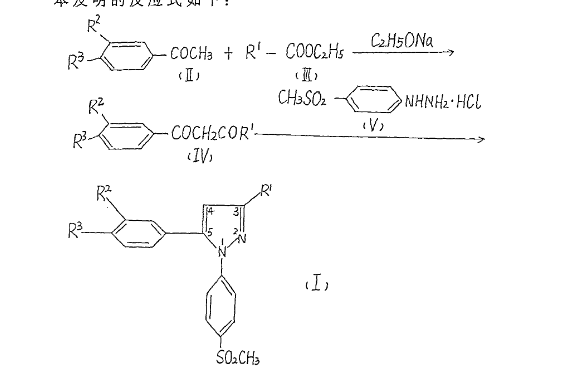
Example 1
1 [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole (I1)
1- (3,4- two toluene-yl) -4,4,4-trifluoro-methyl – D-1,3-dione (IV1) of sodium metal was weighed 2.3g (0.1mol) was added 50ml of anhydrous toluene to prepare a sodium sand. After cooling, ethanol was added dropwise 12ml, and then heated at 60 ℃, complete reaction of sodium metal. After cooling to room temperature, was added 3,4-dimethylphenyl ethanone 23.8g (0.1mol) and trifluoroacetic ethyl acetate 20ml (0.2mol), reacted at 100 ℃ 5 hours. Toluene was distilled off under reduced pressure, a 10% aqueous hydrochloric acid was added, the pH was adjusted to 2-3, extracted with ethyl acetate, washed with water, dried over anhydrous MgSO4, ethyl acetate was distilled off under reduced pressure. Then under reduced pressure, distillation, collecting fractions 105-107 ℃ / 0.7mmHg, was 14.6g, 60% yield.
1- [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole (I1) take the above-prepared substituted (IV1) 2.38g (0.01mol ), 15ml of ethanol, then added p-methanesulfonyl phenyl hydrazine salt alkoxide 2.3g (0.01ml). Was refluxed for 15 hours. Place the refrigerator overnight, the crystals were collected by filtration, recrystallized from ethanol, mp 129-31 ℃, to give 3.1 g.
Elemental analysis: C19H17F3N2O2S Calculated: C, 57.86; H, 4.34; N, 7.10 Found: C, 57.97; H, 4.29; N, 7.20MS (m / z): 395 (M + 1)

References
Cheng, Feixiong, Edited by Lee, Philip W, From Handbook of Metabolic Pathways of Xenobiotics (2014), 4, 1655-1656
Bi, X.; Meng, Z.; Chen, H.; Zhu, X.; Dou, G.
In vivo and in vitro metabolism of lefucoxib in rats, J Pharm Biomed Anal. 2008 Sep 10;48(1):134-9. doi: 10.1016/j.jpba.2008.04.024. Epub 2008 Apr 30.
Bi, X.; Meng, Z.; Dou, G. Determination of lefucoxib in rat plasma, urine, and feces by high-performance liquid chromatography with fluorescence detection: Application in pharmacokinetic studies
J Chromatogr B Anal Technol Biomed Life Sci 2007, 850(1-2): 199
Talanta (2011), 85(1), 8-27
Jiefangjun Yaoxue Xuebao (2009), 25(6), 496-498.
Yaowu Fenxi Zazhi (2006), 26(9), 1222-1224.
Zhongguo Yaolixue Yu Dulixue Zazhi (2007), 21(2), 147-151.
| CN101497585B | Jan 31, 2008 | Jan 12, 2011 | 中国科学院理化技术研究所 | Method for photocatalytic synthesis of 1,3,5-trisubstituted-2-pyrazole derivative |

 .
.
//////////c1c(ccc(c1C)C)c2n(nc(c2)C(F)(F)F)c3ccc(cc3)S(=O)(=O)C
CC1=C(C=C(C=C1)C2=CC(=NN2C3=CC=C(C=C3)S(=O)(=O)C)C(F)(F)F)C
Pfizer’s PF 04937319 glucokinase activators for the treatment of Type 2 diabetes

PF 04937319
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.

Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes
New Antibacterial oxazolidinones in pipeline by Wockhardt

WCK ?
(5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
(5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
MF C19 H25 F2 N3 O5, MW 413.42
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-hydroxy-4-(methoxymethyl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
CAS 957796-51-9
Antibacterial oxazolidinones
Wockhardt Ltd, Innovator
THIS MAY BE WCK 4086?????….WATCHOUT THIS POST FOR UPDATION
PATENTS
WO 2015173664, US8217058, WO 2012059823, IN 2011MU03726

Oxazolidinone represent a novel chemical class of synthetic antimicrobial agents. Linezolid represents the first member of this class to be used clinically. Oxazolidinones display activity against important Gram-positive human and veterinary pathogens including Methicillin-Resistant Staphylococcus aureus (MRSA), Vancomycin Resistant Enterococci (VRE) and β-lactam Resistant Streptococcus pneumoniae (PRSP). The oxazolidinones also show activity against Gram-negative aerobic bacteria, Gram-positive and Gram-negative anaerobes. (Diekema D J et al., Lancet 2001 ; 358: 1975-82).
Various oxazolidinones and their methods of preparation are disclosed in the literature. International Publication No. WO 1995/25106 discloses substituted piperidino phenyloxazolidinones and International Publication No. WO 1996/13502 discloses phenyloxazolidinones having a multisubstituted azetidinyl or pyrrolidinyl moiety. US Patent Publication No. 2004/0063954, International Publication Nos. WO 2004/007489 and WO 2004/007488 disclose piperidinyl phenyl oxazolidinones for antimicrobial use.
Pyrrolidinyl/piperidinyl phenyl oxazohdinone antibacterial agents are also described in Kim H Y et al., Bioorg. & Med. Chem. Lett., (2003), 13:2227-2230. International Publication No. WO 1996/35691 discloses spirocyclic and bicyclic diazinyl and carbazinyl oxazolidinone derivatives. Diazepeno phenyloxazolidinone derivatives are disclosed in the International Publication No. WO 1999/24428. International Publication No. WO 2002/06278 discloses substituted aminopiperidino phenyloxazolidinone derivatives.
Various other methods of preparation of oxazolidinones are reported in US Patent No. 7087784, US Patent No. 6740754, US Patent No. 4948801 , US Patent No. 3654298, US Patent No. 5837870, Canadian Patent No. 681830, J. Med. Chem., 32, 1673 (1989), Tetrahedron, 45, 1323 (1989), J. Med. Chem., 33, 2569 (1990), Tetrahedron Letters, 37, 7937-40 (1996) and Organic Process Research and Development, 11 , 739-741(2007).
Indian Patent Application No. 2534/MUM/2007 discloses a process for the preparation of substituted piperidino phenyloxazolidinones. International Publication No. WO2012/059823 further discloses the process for the preparation of phosphoric acid mono-(L-{4-[(5)-5-(acetylaminomethyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}4-methoxymethyl piperidine-4-yl)ester.
US Patent No. 8217058 discloses (5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide as an antibacterial agent and its process for preparation.
PATENT
WO2015173664

In some embodiments, there is provided a process for preparation of a compound of Formula (I) as shown in Scheme 1

(I I) (I N)
Scheme 1

Example 1
Preparation of (55)-iV-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide (I)
To a stirred solution of lithium teri-butoxide (59.1 g, 0.74 mol) in tetrahydrofuran (500 ml) was added a solution of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-carbamic acid benzyl ester (II) (100 g, 0.25 mol) in 500 ml of tetrahydrofuran slowly at room temperature. The resulting mixture was stirred for 3 hours at room temperature (formation of lumps observed). The reaction mixture was cooled to temperature of 10°C to 15°C and acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) (95.2 g, 0.49 mol) was added in one lot, after 5 minutes methanol (2.36 g, 0.075 mol) was added in one portion. The resulting mixture was stirred further at temperature of 10°C to 15°C. After 5 hours the reaction mixture was allowed to warm to room temperature and stirring continued further for 16 hours. An aqueous solution of saturated ammonium chloride (100 ml) was added to the reaction mixture, the resulting mixture was stirred well and the solvent evaporated under reduced pressure (35°C, 150 mm Hg). The residual mixture was diluted with water (1 L stirred well and filtered under suction, the residual solid was washed with additional fresh water (100 ml). The residual mass was suspended in acetone (500 ml), stirred well and the mixture diluted with hexane (1 L), slowly. The mixture was stirred further for 1 hour and filtered under suction. The residual solid was washed with a 2:1 mixture of acetone and water (100 ml). The residual solid was dried at 45°C, for 3.5 hour at 4 mm Hg, to obtain the 78 g of (55)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l -yl)-phenyl]-2-oxo-oxazolidin-5-ylmethylj -acetamide (I) as white solid, in 77% yield.
Analysis:
Mass: 414 (M+l ); for Molecular Weight: 413 and Molecular Formula: ![]()
Melting Point: 178-179°C;
1H NMR (400 MHz, DMSO): δ 8.18-8.21 (m, 1H), 7.19-7.25 (d, 2H), 4.07-4.71 (m, 1H), 4.32 (s, 1H), 4.02-4.07 (t, 1H), 3.64-3.68 (t, 1H), 3.14 (s, 2H), 2.81-2.83 (d, 2H), 1.81 (s, 3H), 1.63-1.69 (t, 2H), 1.42-1.45 (d, 2H);
Purity as determined by HPLC: 97.65%.
Example 2
Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III)
Step-I: Preparation of l-amino-3-chloro-propan-2-ol hydrochloride (VI)
Benzaldehyde (118.67 g, 1.03 mol) was dissolved in ethanol (297 ml) under stirring and the solution was cooled to 18-19°C. To this solution aqueous ammonia solution (25%) (101.58 ml) was added slowly, followed by slow addition of S-epichlorohydrin (100 g, 1 mol). The resulting mixture was warmed to 40°C and stirred for 7 hours. The mixture was allowed to cool to room temperature and stirred further. After 16 hours, the reaction mixture was concentrated to 50% volume under reduced pressure. Toluene (228 ml) was added to the reaction mixture followed by addition of aqueous hydrochloric acid (162 ml of concentrated hydrochloric acid diluted with 152 ml of water). The mixture thus obtained for 3 hours at 45°C, the resulting mixture was allowed to cool to room temperature and the toluene layer separated. The toluene layer was further extracted with water (56 ml). The combined aqueous layer was diluted with ethanol (56 ml) and the mixture evaporated under reduced pressure. This process was repeated again. To the final concentrate was added ethanol (180 ml), stirred for 10 minutes and the mixture cooled to -28°C to -30°C and maintained at this temperature for 2 hours. The separated solid was filtered under suction and the residue washed with cold (-30°C) ethanol (50 ml). The residue was dried at 45°C, under reduced pressure (4 mm Hg) for 3 hours, to obtain 96 g of l-amino-3-chloro-propan-2-ol hydrochloride (VI) as white solid in 61% yield.
Analysis:
Mass: 110 (M+l) as free base; for Molecular Weight: 145.5 and Molecular Formula: ![]()
1H NMR (400 MHz, D20): δ 4.02-4.08 (m, 1H), 3.51-3.61 (m, 2H), 3.12-3.16 (dd, 1H), 2.93 -2.99 (dd, 1H).
Step-II: Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III).
A stirred solution of dichloromethane (220.8 ml) containing the step-I salt (96 g, 0.66 mol) was cooled to 18-20°C. Acetic anhydride (154.78 g, 1.5175 mol) was added slowly (slight exothermic). Pyridine (67.76 g, 0.8577 mol) was added slowly (exothermic) while maintaining the temperature at 18-20°C. The resulting mixture was heated to 40°C for 5 hours. The reaction mixture was allowed to cool to room temperature and stirring continued for further 16 hours. The reaction mass was cooled to 3-6°C and diluted with 170 ml of fresh water. To this was added an aqueous solution of potassium carbonate (191.2 g of K2CO3 in 382 ml water). The reaction mixture was further diluted with additional dichloromethane (170 ml) and water (425 ml). The reaction mass was stirred well and the dichloromethane layer separated. The aqueous layer was further extracted with 2×170 ml dichloromethane. The combined dichloromethane layer was washed with aqueous sodium chloride solution (13.6 g of sodium chloride in 493 ml water). The solvent was evaporated till a volume of 170 ml and the residual layer was diluted with toluene (340 ml), stirred well and the solvent was evaporated completely at 40°C under reduced pressure (4 mm Hg). To the residue ethyl acetate (170 ml) and hexane (187 ml) were added and the mixture stirred for 30 minute. The separated solid was filtered under suction and the residue washed with 50 ml of a 1 :1 mixture of ethyl acetate and hexane. The solid obtained was dried under reduced pressure (4 mm Hg) at 45°C for 3.5 hours, to obtain 96 g of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) as a white solid, in 75% yield.
Analysis:
Mass: 194 (M+l); for Molecular Weight: 193 and Molecular Formula: C7Hi2ClN03; 1H NMR (400 MHz, CDC13): 5 5.69 (s, 1H), 5.0-5.1 (m, 1H), 3.4-3.7 (m, 4H), 2.1 (s, 3H), 1.9 (s, 3H).
PATENT
http://www.google.st/patents/WO2007132314A2?cl=en



(3) (4)
Scheme -1

(6) Formula π Scheme-2

Formula II Formula in

Formula I(a) Scheme-4
Example -11 : (5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
The example- 10 (54.86 g, 0.144 mol) was suspended in methanol (1100 ml) under stirring at RT. Sodium metal (4 g, 0.174 mol) was added in small lots in 2 min to the above suspension under stirring. The reaction mixture was warmed to 40-420C and was stirred at this temperature for about 40 hrs. After completion of the reaction (TLC), the solvent was evaporated under reduced pressure to obtain a thick slurry. The thick slurry thus obtained was gradually added to water (1100 ml) under stirring. After the complete addition, the pH of the aqueous suspension was adjusted to 7 by adding sufficient quantity of glacial acetic acid. The separated solid was filtered and the residue was washed with water. The obtained solid was further purified by column chromatography over silica gel to obtain the product as a white solid, 32.7 g, 55 % yield.
M.P.: 173-1740C;
MS : M+l= 414(MH+, 100%); for M.F.: Ci9H25F2N3O5
1H-NMR (400 MHz, CDCl3): δ 7.0-7.1 (m, 2H5Ar-H), 6.0 (t, IH, NH), 4.70-4.80 (m, IH), 4.00 (t,lH), 3.70-3.75 (m, 2H), 3.5-3.7 (m, IH), 3.43 (s, 3H, OCH3), 3.37-3.42 (m, 2H), 3.30 (s, 2H, -OCH2), 3.0-3.05 (m, 2H), 2.22(bs,lH ,-OH),2.04 (s, 3H, COCH3), 1.70-1.75 (m, 4H).
Patent
INDIAN 3049/MUM/2010
Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxy methyl-piperidin-4-yl) ester

Specific intermediate compounds of the invention include:
6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane;
1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol;
[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one;
(5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one; and
(5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide.
Examples
Preparation of Intermediate-1: 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one
Chloroform (9.3 L) was charged in a 20 L reaction assembly and 4-piperidone hydrochloride (1.17 Kg, 7.62 mol) was added under stirring followed by triethylamine (2.14 Kg, 2.95 L, 21.1 mol). After 30 minutes of stirring, 3,4,5-trifluoronitrobenzene (1.5 Kg, 8.47 mol) was added to the mixture in one lot and the contents were heated to 65-70ºC for 8 h. After completion of the reaction, chloroform was removed under vacuum to obtain a syrupy mass. At this stage, water (10 L) was added to the mass and the chloroform recovery was continued under vacuum below 65oC till the chloroform was removed completely. The slurry was cooled to RT and filtered. The solid product was washed with water (3 L) followed by hexanes (2 L). The product was dried in a vacuum oven below 70oC to obtain the product as a yellow solid, 1.88 Kg ; Yield 97%.
M.P.: 130-132oC; MS: 257(M+1); M.F.: C11H10F2N2O3.
Preparation of Intermediate 3: 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
Method A:
Preparation of Intermediate–2: (Stage-I): 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane
A solution of trimethylsulfoxonium iodide (1.504kg, 6.836mol) in acetonitrile (7L) was cooled to 0 to 5oC. , under argon atmosphere. Potassium tert-butoxide (0.736kg, 6.552 mol) was added in small lots over 0.5h. The resulting solution was stirred for 2h at the same temperature. To this solution was added 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one ( 1.4kg, 5.46mol) in small lots over a period of 1h, while maintaining the temp. between 5-10oC. The resulting mixture was stirred for 1h. The solvent was evaporated to a minimum amount possible, under reduced pressure while maintaining the temperature below 10oC. The residue was poured in water( 18L) and the pH adjusted to neutral with dilute acetic acid. The resulting slurry was stirred well and the separated solid filtered under suction. The solid was washed with fresh water till the filtrate was free of acetic acid. The solid was dried at 80oC, for 6h, under reduced pressure to obtain the product as pale yellow solid, 1.264kgs, yield 85%.
M.P.: 96-97oC; MS: M+1: 271; M.F.: C12H12F2N2O3,.
Preparation of Intermediate-3: (Stage-II): 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
To a solution of sodium methoxide (236g, 4.35mol) in methanol (3L), at RT, was added 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro [2.5]octane (964g, 3.57mol) in small portions and the reaction mixture was stirred for 26h at RT. Acetic acid (265g, 4.44mol) was added slowly to neutralize the pH of the solution. The resulting mixture was poured into chilled water(18L) and stirred for 1h. The separated solid was filtered under suction. The solid was washed with additional water till the filtrate was free of acetic acid. The solid was dried for 10hat RT under reduced pressure, to obtain the product as a pale yellow solid, 973g, yield, 90%
M.P.: 84-86oC; MS: 303 (M+1); M.F.: C13H16F2N2O4
Method B:
Dimethylsulfoxide (DMSO, 100 ml) and methanol (500 ml) were charged in a 1 L glass reaction assembly. Potassium hydroxide (59.2g, 0.898 mol) was charged in the assembly followed by trimethylsulfoxonium iodide (94.5 g, 0.43 mol) and the contents were stirred for 30 minutes and then cooled to 10oC-15oC. To the cooled contents was added 1-(2,6-difluoro-4-nitrophenyl)-piperidin-4-one (100 g, 0.39 mol) in small lots. After the addition, the temperature was allowed to raise to RT and the contents were further stirred for 24 h (ring opening of the epoxide intermediate viz. 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane takes place).
[Physical data of the intermediate: M.P.: 96-970C, MS: 271(M+1); M.F.: C12H12F2N2O3, .
After completion of the reaction the contents were poured slowly in ice-water (600g crushed ice in 600 ml water). The precipitated solid product was filtered and was washed with water:methanol, 2:1 (100 ml X 2). The wet product was used in the next step.
M.P.: 84-86oC; MS: 303 (M+1);.M.F.: C13H16F2N2O4,:
Preparation of Intermediate -5: [3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester
Method A: Preparation of Intermediate 4: ( Stage-I)
Water (1.19 L) and methanol (595 ml) were charged in a 3 L glass reaction assembly, followed by 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (85 g, 0.281 mol) and the contents were stirred. Sodium dithionite (288 g, 1.407 mol) was added in one lot and the reaction mixture was heated to 80oC for 8 h. After completion of the reaction (TLC), methanol was recovered under vacuum below 65oC. After the recovery, the aqueous residue was extracted with chloroform (400 ml X 3). The combined chloroform extract (containing the intermediate 1-(4-amino-2,6-difluoro-phenyl)-4-methoxymethyl-piperidin-4-ol) was dried over anhydrous Sodium sulfate and used in the next step (carbamate formation).
Preparation of Intermediate -5: (Stage-II):
The above chloroform extract was charged in a 3 L glass reaction assembly. Sodium bicarbonate (70 g, 0.843 mol) was added to the extract and the contents were cooled to 15oC-20oC. Benzylchloroformate solution (50% in toluene, 48 g, 96 ml, 0.281 mol) was added slowly to the above mixture under stirring. After completion of the addition, the reaction mixture was stirred at RT for 2 h. After completion of the reaction (TLC), the contents were filtered on a Buchner assembly and the solid cake was washed with chloroform (85 ml X 2). The combined filtrate was evaporated under vacuum below 50oC to obtain yellowish oily mass, which was poured slowly in hexanes (850 ml) under stirring to obtain a precipitate. The precipitated product was filtered and washed with hexanes (100 ml X 2). The product was dried in a vacuum oven below 65oC to obtain 60.2 g brownish product (Yield = 38% on the basis of step-I input).
M.P.: 138-140oC; MS: 407(M+1); M.F.: C21H24F2N2O4.:.
Method B: : Preparation of Intermediate 4: ( Stage-I): To a solution of 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (973g, 3.22 mol) in ethyl acetae (10L) was added 10% Pd-C, (250g, 50% wet) and the resulting miture was hydrogenated in a pressure at 30 PSI, 45-55oC, for 3h. The catakyst was filtered and the residue was washed with additional ethyl acetate( 200ml). The combined filtrates were used as such for the next reaction (carbamate formation)
Preparation of Intermediate -5: (Stage-II):
To the above filtrate was added sodium bicarbonate(406g, 4.83 mol) and the mixture warmed to 40-45oC. To this mixture was added a 50% solution of Benzyl chloroformate in toluene(1.373L, 4.025 mol), drop-wise, over a period of 1h. Stir the resulting mixture for 1h and filter the insoluble material. The residue was washed with 300ml of ethyl acetate. The filtrates were combined and the solvent evaporated under reduced pressure, below 55oC.. Cool the residue and dilute it with hexane(10L). The resulting slurry was stirred well and the separated solid was filtered under suction. The residue was washed with additional hexane ( 2L). The solid was dried for 10h at RT, to obtain the product as dark brown solid, 1200g, yield, 96%.
M.P.: 138-140oC; MS: 407( M+1); M.F.: C21H24F2N2O.
Preparation of Intermediate -6:
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one
To a mixture of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester (100g, 0.237 mol) in dry tetrahydrofuran (THF) (2 L) at 40ºC was added drop-wise n-BuLi in hexane (1.6M, 45.5 g, 455 ml, 0.711 mol) under nitrogen atmosphere. The contents were stirred for 1 h at 40ºC and R-(-)-glycidyl butyrate (68.25 g, 0.474 mol) was added gradually. After the addition of R-(-)-glycidyl butyrate, the reaction mixture was stirred for 5-6 h at 40oC till completion of the reaction (TLC). After completion of the reaction, a solution of sodium methoxide (2 g) in methanol (66 ml) was added to the contents followed by water (8 ml) and the contents were stirred for an additional 0.5 h. Water (1 L) was added to the solution and the contents were extracted with ethyl acetate (1 L). The aqueous layer was further extracted with ethyl acetate (3 X 500 ml). The combined organic layer was evaporated under vacuum to obtain a thick residue. tert-Butyl methyl ether (1 L) was added to the residue and the contents were stirred for about 1 h to obtain a solid product, which was filtered and washed with tert-butyl methyl ether (2 X 100 ml). The product was dried under vacuum below 60ºC to obtain the product as a 46.5 g dark brown compound, 46.5g ,yield 51%.
M.P.: 117-119oC; MS: 373(M+1); M.F.: C17H22F2N2O5..
Preparation of Intermediate -7: (5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester
To a mixture of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one (45 g, 0.121 mol) in dichloromethane (0.3 L), was added triethylamine (24.5 g, 34 ml, 0.242 mol) while stirring. Methanesulfonyl chloride (18 g, 12.2 ml, 0.157 mol) was added to the above solution over a period of 1 h at 10oC -20oC and the reaction mixture was stirred for additional 2 h at RT. After completion of the reaction (TLC), the contents were evaporated under vacuum at 40oC to obtain an oily residue. Water (450 ml) was added to the residue and the traces of dichloromethane were removed under vacuum. The solid product thus obtained was filtered, washed with water (2 X 50 ml) and dried under vacuum at 70oC to obtain 50.6 g brownish compound. Yield = 93%; M.P.:106-108oC; MS: 451(M+1); M.F.: C18H24F2N2O7S.
Preparation of Intermediate 8a: (5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one
Method A:
To a solution of (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl)oxazolidin-2-one (2g, 5.3 mmol),in tetrahydrofuran (20 mL), under argon , was added diphenylphosphoryl azide (1.63mL, 5.9 mmol). The solution was cooled to 0oC in an ice-bath. 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) (0.76mL, 4.9mmol) was added drop-wise over 15min..The reaction was stirred at same temperature for 1 hr, and then warmed to room temperature and stirred under for 16 hr. The reaction mixture was diluted with ethyl acetate (20 mL), and water (20mL). After separation of water layer, the organic layer was washed with water and 0.5M citric acid monohydrate (10 mL). The organic layer was dried over sodium sulfate and the solvent evaporated under reduced pressure.The residue was triturated with ether to obtain the product as a buff colored solid, 1.32g (62%).
M.P.: 106-108oC; M.S.- 398(M+1); M.F.- C17H21F2N5O4,
Method B:
To a solution of (5R)-methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester (20 g, 0.044 mol, wet) in N,N-dimethylformamide (30 ml), was added sodium azide (8.6 g, 0.133 mol) in a single lot. The reaction mixture was gradually heated and the temperature was maintained at 70ºC for 8 h. After completion of the reaction (TLC), the contents were cooled to 20-25ºC and poured slowly into chilled water (300 ml). The solid product thus obtained was filtered and washed with water (2 x 50 ml). The wet product was air dried to obtain 16.5g dark brown compound (being an azide, it was NOT exposed to heat during drying) Yield ~ 93%.
M.P.: 106-108oC; MS : 398(M+1); M.F.: C17H21F2N5O4;:
Preparation of Intermediate 8b: (5S)-N-2-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-phthalimide
Method A:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate(10g, 0.022 mol), Potassium phthalimide (12.2g, 0.066 mol) and DMF (50ml) was heated, with stirring, at 90oC for 4h. The resulting mixture was cooled to RT and poured over ice-water mixture. The separated solid was filtered, washed with water and dried under suction to obtain the product as a white solid, 9.46g, in 85% yield.
M.P.: 154-156 oC; MS: 502 (M+1); M.F. C25H25F2N3O6.
Method B:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 minute phthalimide (1.18g, 8 mmol)) was added and after a further stirring for 10 minute, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 8 hrs ice-cold water (4 ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 1.56g, yield 58%.
M.P.: 154-156 oC; MS : 502 (M+1); M.F. C25H25F2N3O6.
Preparation of Intermediate 10: (5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40oC to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HCl solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70oC to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50oC.. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume.
To the above ethyl acetate solution was added Triethyl amine (19.1g, 0.189 mol), and acetic anhydride (16.1g, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5oC, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate(100ml) and dried at 70oC under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.011mol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(1.11g, 0.011mol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate (1gm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 ºC. for 15hrs. Then a mixture of conc. HCl (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75ºC for 5hrs. The mixture was concentrated under reduced pressure at 60-75 ºC. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 ºC for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50ºC to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Preparation of Intermediate -11: (S)-N-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryloxy-piperi din-1yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
To a solution of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)-phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.2 mmol) and tetrazole (0.6 mmol) in dichloromethane (5 ml) was added dibenzyl N,N,diisopropylphosphoramidite (0.4 mmol) and the resulting mixture was stirred for 4h. The resulting solution was cooled to 0 oC and 0.6 ml of 0.5M m-chloroperbenzoic acid solution in dichloromethane was added. After 4h, the solvent was evaporated under residue pressure and the residue chromatographed on a column of silica gel to obtain the product as a off-white solid in 75% yield,
MS: 674 (M+1); M.F. C33H38F2N3O8P;
Example A: Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxymethyl-piperidin-4-yl) ester
To a suspension of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryl- oxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.15 mmol) and 20 % palladium hydroxide (20 mg) in 20 ml of a mixture of dichloromethane /aqueous methanol was stirred at room temperature for 6h. The catalyst was filtered and the residue evaporated under reduced pressure. The residue obtained was triturated with acetone to obtain a white solid as product in 70% yield.
MP. >140 °C; MS : 494(M+1) M.F.: C19H26F2N3O8P.
PATENT
http://www.google.co.in/patents/WO2012059823A1?cl=en


c) Converting intermediate of Formula (5) into intermediate of structure (6)



f) Converting intermediate of Formula (11) into compound of Formula (A) or Pharmaceutically acceptable salts thereof



ormu a-
Scheme-1
Preparation of Intermediate 10: (5S)- N-{ 3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl- piperidin- 1 -yl)-phenyl] -2-oxo-oxazolidin-5-ylmethyl } -acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l- yl)-phenyl] -oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40°C to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HC1 solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4- hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70°C to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-5- azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50°C. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume. To the above ethyl acetate solution was added Triethyl amine (19. lg, 0.189 mol), and acetic anhydride (16. lg, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5°C, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate( 100ml) and dried at 70°C under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine- lyl)phenyl]-2-oxo-oxazolidin-5-yl methyl }-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.01 lmol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(l.l lg, 0.01 lmol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5. Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)phenyl]- 2-oxo-oxazolidin-5-yl methyl }-methanesulfonate (lgm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 °C. for 15hrs. Then a mixture of cone. HC1 (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75°C for 5hrs. The mixture was concentrated under reduced pressure at 60-75 °C. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 °C for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50°C to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.1 lg, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3- (3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-l-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5.
PATENT
http://www.google.co.in/patents/WO2008038092A2?cl=en

IV

V
‘ Scheme-1 ‘
/////////
SEE FULL ZOLID SERIES…………http://drugsynthesisint.blogspot.in/p/zolid.html
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorobiphenyl-2-carboxamide
Cas 1820758-44-8
C24 H18 F N3 O4 S
4′-((5-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl-thio)-methyl)-4-fluorobiphenyl-2-carboxamide


Glycogen synthase kinase-3 (GSK-3) is a constitutively active, ubiquitous serine/threonine kinase that takes part in a number of physiological processes ranging from glycogen metabolism to apoptosis. GSK-3 is a key mediator of various signaling pathways, such as the Wnt and the insulin/AKT signaling pathways.
Therefore, dysregulation of GSK-3 has been linked to various human diseases, such as cancer, diabetes, and neurodegenerative diseases.Two related isoforms of GSK-3 exist in mammals, GSK-3α and -β, which share a sequence identity within their catalytic domains of 98%.
Beyond the catalytic domains they show significant differences. Although these isoforms are structurally related, they are not functionally equivalent, and one cannot compensate for loss of the other.
The debate on the respective contributions of the isoforms GSK-3α and GSK-3β on the pathogenesis of different diseases is ongoing.
Various studies indicate that the therapies of certain diseases benefit from specific targeting of GSK-3α and GSK-3β. GSK-3α was recently identified as a differentiation target in acute myeloid leukemia (AML). AML is a hematopoietic malignancy defined by uncontrolled proliferation and disrupted myeloid differentiation. AML is the second most common form of leukemia in adults.
The current treatment of AML with conventional chemotherapy is very aggressive yet ineffective for the majority of patients with the disease.Thus, alternative targeted treatment approaches for AML are highly desirable. GSK-3α recently emerged as a potential target in this disease.
PAPER

The challenge for glycogen synthase kinase-3 (GSK-3) inhibitor design lies in achieving high selectivity for one isoform over the other. The therapy of certain diseases, such as acute myeloid leukemia (AML), may require α-isoform specific targeting. The scorpion shaped GSK-3 inhibitors developed by our group achieved the highest GSK-3α selectivity reported so far but suffered from insufficient aqueous solubility. This work presents the solubility-driven optimization of our isoform-selective inhibitors using a scorpion shaped lead. Among 15 novel compounds, compound 27 showed high activity against GSK-3α/β with the highest GSK-3α selectivity reported to date. Compound 27 was profiled for bioavailability and toxicity in a zebrafish embryo phenotype assay. Selective GSK-3α targeting in AML cell lines was achieved with compound 27, resulting in a strong differentiation phenotype and colony formation impairment, confirming the potential of GSK-3α inhibition in AML therapy
Evaluation of Improved Glycogen Synthase Kinase-3α Inhibitors in Models of Acute Myeloid Leukemia
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01200
http://pubs.acs.org/doi/suppl/10.1021/acs.jmedchem.5b01200/suppl_file/jm5b01200_si_001.pdf
compound 27 as a colorless solid. HPLC: 96%, tR = 6.93 min.
1H NMR (DMSO-d6, 500 MHz, 300 K): δ (ppm) = 4.32 (td, J = 5.2 Hz, J = 3.7 Hz, 4H), 4.60 (s, 2H), 7.05 (d, J = 8.4 Hz, 1H), 7.25 (dd, J = 9.1 Hz, J = 2.7 Hz, 1H), 7.31 (td, J = 8.6 Hz, J = 2.8 Hz, 1H), 7.38 (m, 3H), 7.41 (d, J = 2.0 Hz, 1H), 7.45 (dd, J = 8.4 Hz, J = 2.1 Hz, 1H), 7.49 (d, J = 8.2 Hz, 2H), 7.73 (s, 1H).
13C NMR (DMSO, 125 MHz, 300 K): δ (ppm) = 35.6, 64.1, 64.4, 114.3 (d, JC–F = 21 Hz), 115.0, 115.9 (d, JC–F = 21 Hz), 115.9, 118.1, 120.0, 128.6 (2C), 128.8 (2C), 132.0 (d, JC–F = 8 Hz), 134.8, 135.5, 138.9, 139.0 (d, JC–F = 7 Hz), 143.8, 146.7, 160.9 (d, JC–F = 247 Hz), 162.7, 164.9, 169.5.
EI-MS: m/z = 463 (100, [M+]), 464 (26, [M+ + H]), 465 (7, [M+ + 2H].
ABOUT Boris Schmidt
Boris Schmidt
-
Clemens Schöpf-Institute of Chemistry and BiochemistryDarmstadt, Hessen, Germany*Phone: +49 6151 163075. Fax: +49 6151 163278.E-mail: Schmidt_boris@t-online.de.
………………………………………….
ABOUT Theresa Neumann
-
Clemens Schöpf-Institute of Chemistry and BiochemistryDarmstadt, Hessen, Germany
////////FC(C=C1C(N)=O)=CC=C1C(C=C2)=CC=C2CSC3=NN=C(O3)C4=CC5=C(OCCO5)C=C4
New 5-Substituted-N-(piperidin-4-ylmethyl)-1H-indazole-3-carboxamides: Potent Glycogen Synthase Kinase-3 (GSK-3) Inhibitors in Model of Mood Disorders
CAS 1452582-16-9, 428.47, C23 H26 F2 N4 O2
1H-Indazole-3-carboxamide, 5-(2,3-difluorophenyl)-N-[[1-(2-methoxyethyl)-4-piperidinyl]methyl]-
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
![]()
1 H-indazole-3-carboxamide compounds acting as glycogen synthase kinase 3 beta (GSK-33) inhibitors and to their use in the treatment of GSK-33-related disorders such as (i) insulin-resistance disorders; (ii) neurodegenerative diseases; (iii) mood disorders; (iv) schizophrenic disorders; (v) cancerous disorders; (vi) inflammation, (vii) substance abuse disorders; (viii) epilepsies; and (ix) neuropathic pain.
Protein kinases constitute a large family of structurally related enzymes, which transfer phosphate groups from high-energy donor molecules (such as adenosine triphosphate, ATP) to specific substrates, usually proteins. After phosphorylation, the substrate undergoes to a functional change, by which kinases can modulate various biological functions.
In general, protein kinases can be divided in several groups, according to the substrate that is phosphorylated. For example, serine/threonine kinase phosphorylates the hydroxyl group on the side chain of serine or threonine aminoacid.
Glycogen synthase kinases 3 (GSK-3) are constitutively active multifunctional enzymes, quite recently discovered, belonging to the serine/threonine kinases group.
Human GSK-3 are encoded by two different and independent genes, which leads to GSK-3a and GSK-33 proteins, with molecular weights of about 51 and 47 kDa, respectively. The two isoforms share nearly identical sequences in their kinase domains, while outside of the kinase domain, their sequences differ substantially (Benedetti et al., Neuroscience Letters, 2004, 368, 123-126). GSK-3a is a multifunctional protein serine kinase and GSK-33 is a serine-threonine kinase.
It has been found that GSK-33 is widely expressed in all tissues, with widespread expression in the adult brain, suggesting a fundamental role in neuronal signaling pathways (Grimes and Jope, Progress in Neurobiology, 2001, 65, 391-426). Interest in glycogen synthase kinases 3 arises from its role in various physiological pathways, such as, for example, metabolism, cell cycle, gene expression, embryonic development oncogenesis and neuroprotection (Geetha et al., British Journal Pharmacology, 2009, 156, 885-898).
GSK-33 was originally identified for its role in the regulation of glycogen synthase for the conversion of glucose to glycogen (Embi et al., Eur J Biochem, 1980, 107, 519-527). GSK-33 showed a high degree of specificity for glycogen synthase.
Type 2 diabetes was the first disease condition implicated with GSK- 3β, due to its negative regulation of several aspects of insulin signaling pathway. In this pathway 3-phosphoinositide-dependent protein kinase 1 (PDK-1 ) activates PKB, which in turn inactivates GSK-33. This inactivation of GSK-33 leads to the dephosphorylation and activation of glycogen synthase, which helps glycogen synthesis (Cohen et al., FEBS Lett, 1997, 410, 3-10). Moreover, selective inhibitors of GSK-33 are expected to enhances insulin signaling in prediabetic insulin- resistant rat skeletal muscle, thus making GSK-33 an attractive target for the treatment of skeletal muscle insulin resistance in the pre-diabetic state (Dokken et al., Am J. Physiol. Endocrinol. Metab., 2005, 288, E1 188-E1 194).
GSK-33 was also found to be a potential drug target in others pathological conditions due to insulin-resistance disorders, such as syndrome X, obesity and polycystic ovary syndrome (Ring DB et al., Diabetes, 2003, 52: 588-595).
It has been found that GSK-33 is involved in the abnormal phosphorylation of pathological tau in Alzheimer’s disease (Hanger et al., Neurosci. Lett, 1992, 147, 58-62; Mazanetz and Fischer, Nat Rev Drug Discov., 2007, 6, 464-479; Hong and Lee, J. Biol. Chem., 1997, 272, 19547- 19553). Moreover, it was proved that early activation of GSK-33, induced by apolipoprotein ApoE4 and β-amyloid, could lead to apoptosis and tau hyperphosphorylation (Cedazo-Minguez et al., Journal of Neurochemistry, 2003, 87, 1 152- 1 164). Among other aspect of Alzheimer’s disease, it was also reported the relevance of activation of GSK-33 at molecular level (Hernandez and Avila, FEBS Letters, 2008, 582, 3848-3854).
Moreover, it was demonstrated that GSK-33 is involved in the genesis and maintenance of neurodegenerative changes associated with Parkinson’s disease (Duka T. et al., The FASEB Journal, 2009; 23, 2820- 2830).
Accordingly to these experimental observations, inhibitors of GSK-33 may find applications in the treatment of the neuropathological consequences and the cognitive and attention deficits associated with tauopathies; Alzheimer’s disease; Parkinson’s disease; Huntington’s disease (the involvement of GSK-33 in such deficits and diseases is disclosed in Meijer L. et al., TRENDS Pharm Sci, 2004; 25, 471 -480); dementia, such as, but not limited to, vascular dementia, post-traumatic dementia, dementia caused by meningitis and the like; acute stroke; traumatic injuries; cerebrovascular accidents; brain and spinal cord trauma; peripheral neuropathies; retinopathies and glaucoma (the involvement of GSK-33 in such conditions is disclosed in WO 2010/109005).
The treatment of spinal neurodegenerative disorders, like amyotrophic lateral sclerosis, multiple sclerosis, spinal muscular atrophy and neurodegeneration due to spinal cord injury has been also suggested in several studies related to GSK-33 inhibition, such as, for example in Caldero J. et al., “Lithium prevents excitotoxic cell death of motoneurons in organotypic slice cultures of spinal cord”, Neuroscience. 2010 Feb 17;165(4):1353-69, Leger B. et al., “Atrogin-1 , MuRF1 , and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients”, Muscle Nerve, 2009 Jul; 40(1 ):69-78, and Galimberti D. et al., “GSK33 genetic variability in patients with Multiple Sclerosis”, Neurosci Lett. 201 1 Jun 1 5;497(1 ):46- 8. Furthermore, GSK-33 has been linked to the mood disorders, such as bipolar disorders, depression, and schizophrenia.
Inhibition of GSK-33 may be an important therapeutic target of mood stabilizers, and regulation of GSK-33 may be involved in the therapeutic effects of other drugs used in psychiatry. Dysregulated GSK-33 in mood disorder, bipolar disorder, depression and schizophrenia could have multiple effects that could impair neural plasticity, such as modulation of neuronal architecture, neurogenesis, gene expression and the ability of neurons to respond to stressful, potentially lethal conditions (Jope and Ron, Curr. Drug Targets, 2006, 7, 1421- 1434).
The role of GSK-33 in mood disorder was highlighted by the study of lithium and valproate (Chen et al., J. Neurochem., 1999, 72, 1327- 1330; Klein and Melton, Proc. Natl. Acad. Sci. USA, 1996, 93, 8455-8459), both of which are GSK-33 inhibitors and are used to treat mood disorders. There are also existing reports from the genetic perspective supporting the role of GSK-33 in the disease physiology of bipolar disorder (Gould, Expert. Opin. Ther. Targets, 2006, 10, 377-392).
It was reported a decrease in AKT1 protein levels and its phosphorylation of GSK-33 at Serine-9 in the peripheral lymphocytes and brains of individuals with schizophrenia. Accordingly, this finding supports the proposal that alterations in AKT1 -GSK-33 signaling contribute to schizophrenia pathogenesis (Emamian et al., Nat Genet, 2004, 36, 131- 137).
Additionally, the role of GSK-33 in cancer is a well-accepted phenomenon.
The potential of small molecules that inhibit GSK-33 has been evidenced for some specific cancer treatments (Jia Luo, Cancer Letters, 2009, 273, 194-200). GSK-33 expression and activation are associated with prostate cancer progression (Rinnab et al., Neoplasia, 2008, 10, 624-633) and the inhibition of GSK3b was also proposed as specific target for pancreatic cancer (Garcea et al., Current Cancer Drug Targets, 2007, 7, 209-215) and ovarian cancer (Qi Cao et al., Cell Research, 2006, 16 671 -677). Acute inhibition of GSK-33 in colon-rectal cancer cells activates p53-dependent apoptosis and antagonizes tumor growth (Ghosh et al., Clin Cancer Res 2005, 1 1 , 4580-4588).
The identification of a functional role for GSK-33 in MLL-associated leukaemia suggests that GSK-33 inhibition may be a promising therapy that is selective for transformed cells that are dependent on HOX overexpression (Birch et al., Cancer Cell, 2010, 1 7, 529-531 ).
GSK-33 is involved in numerous inflammatory signalling pathways, for example, among others GSK-33 inhibition has been shown to induce secretion of the anti-inflammatory cytokine IL-1 0. According to this finding, GSK-33 inhibitors could be useful to regulate suppression of inflammation (G. Klamer et al., Current Medicinal Chemistry, 2010, 17(26), 2873-2281, Wang et al., Cytokine, 2010, 53, 130-140).
GSK-33 inhibition has been also shown to attenuate cocaine-induced behaviors in mice. The administration of cocaine in mice pretreated with a GSK-33 inhibitor demonstrated that pharmacological inhibition of GSK3 reduced both the acute behavioral responses to cocaine and the long- term neuroadaptations produced by repeated cocaine (Cocaine-induced hyperactivity and sensitization are dependent on GSK3, Miller JS et al. Neuropharmacology. 2009 Jun; 56(8):1 1 16-23, Epub 2009 Mar 27).
The role of GSK-33 in the development of several forms of epilepsies has been demonstrated in several studies, which suggest that inhibition of GSK-33 could be a pathway for the treatment of epilepsy (Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy, Lohi H et al., Hum Mol Genet. 2005 Sep 15;14(18):2727-36 and Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease, Purl R et al., J Biol Chem. 2009 Aug 21 ;284(34) 22657-63). The relationship between GSK-33 inhibition and treatment of neuropathic pain has been demonstrated in Mazzardo-Martins L. et al., “Glycogen synthase kinase 3-specific inhibitor AR-A014418 decreases neuropathic pain in mice: evidence for the mechanisms of action”, Neuroscience. 2012 Dec 13;226, and Xiaoping Gu et al., “The Role of Akt/GSK33 Signaling Pathway in Neuropathic Pain in Mice”, Poster A525, Anesthesiology 2012 October 13-17, 2012 Washington.
A review on GSK-33, its function, its therapeutic potential and its possible inhibitors is given in “GSK-33: role in therapeutic landscape and development of modulators” (S. Phukan et al., British Journal of Pharmacology (2010), 160, 1- 19).
WO 2004/014864 discloses 1 H-indazole-3-carboxamide compounds as selective cyclin-dependant kinases (CDK) inhibitors. Such compounds are assumed to be useful in the treatment of cancer, through a mechanism mediated by CDK2, and neurodegenerative diseases, in particular Alzheimer’s disease, through a mechanism mediated by CDK5, and as anti-viral and anti-fungine, through a mechanism mediated by CDK7, CDK8 and CDK9.
Cyclin-dependant kinases (CDKs) are serine/threonine kinases, first discovered for their role in regulating the cell cycle. CDKs are also involved in regulating transcription, mRNA processing, and the differentiation of nerve cells. Such kinases activate only after their interaction and binding with regulatory subunits, namely cyclins.
Moreover, 1 H-indazole-3-carboxamide compounds were also described as analgesics in the treatment of chronic and neuropathic pain (see, for example, WO 2004/074275 and WO 2004/101 548) and as 5-HT4 receptor antagonists, useful in the treatment of gastrointestinal disorders, central nervous system disorders and cardiovascular disorders (see, for example, WO 1994/101 74).
Patent
WO 2013124158
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
SEE ENTRY 8

DMSO-de; δ 13.09 (s, 1 H), 8.23-8.42 (m, 2H), 7.72 (dd, J=0.82, 8.69 Hz, 1 H), 7.55 (td, J=1.76, 8.74 Hz, 1 H), 7.24-7.49 (m, 3H), 3.40 (t, J=6.04 Hz, 2H), 3.22 (s, 3H), 3.18 (d, J=6.40 Hz, 2H), 2.84 (d, J=11.53 Hz, 2H), 2.42 (t, J=5.95 Hz, 2H), 1.82- 2.02 (m, 2H), 1.41 -1.71 (m, 3H), 1.06-1.31 (m, 2H)
PAPER

Hit Optimization of 5-Substituted-N-(piperidin-4-ylmethyl)-1H-indazole-3-carboxamides: Potent Glycogen Synthase Kinase-3 (GSK-3) Inhibitors with in Vivo Activity in Model of Mood Disorders
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01208

http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b01208
Aziende Chimiche Riunite Angelini Francesco A.C.R.A.F. S.P.A.
Angelini S.p.A., Angelini Research Center,
![]()
/////
COCCN1CCC(CNC(=O)c2n[nH]c3ccc(cc23)c4cccc(F)c4F)CC1
BMS-248360, A NEW SARTAN ON HORIZON

2-[4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-[(3,3-dimethyl-2-oxopyrrolidin-1-yl)methyl]phenyl]-N-(3,4-dimethyl-1,2-oxazol-5-yl)benzenesulfonamide
4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide,
4′- . (2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non-l-en-3-yl)methyll -N-C3.4- dimethyl-5-isoxazolyl)-2,-[(3.3-dimethyl-2-oxo-l- pyrrolidinvDmethyll [1.1 ‘-biphenyl] -2-sulfonamide
4‘-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N–(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide
BMS-248360
PRECLINICAL …..treating hypertension
Bristol Myers Squibb Co, INNOVATOR
Hypertension remains one of the largest unmet medical needs in the 21st century, especially when one considers that hypertension is the portent of future debilitating cardiovascular disease. While many drugs are available for treating the disease, approximately one-third of the hypertensive population is still not adequately treated. Of the more recent avenues explored for treating hypertension, disruption of the effects of either angiotensin II (AII) or endothelin-1 (ET-1) has shown promise. These endogenous vasoactive peptides are among the most potent vasoconstrictors and cell proliferative factors identified to date. AII is the effector molecule of the renin−angiotensin system (RAS), and a large number of AII receptor (AT1) antagonists, including irbesartan , have been developed for treating hypertension


SYNTHESIS
picked from…….http://www.drugfuture.com/synth/syndata.aspx?ID=324487

EP 1094816; JP 2002519380; US 2002143024; WO 0001389
The intermediate biphenyl aldehyde (XI) is prepared by two related methods. 4-Bromo-3-methylbenzonitrile (I) is oxidized to aldehyde (II) via radical bromination with N-bromosuccinimide/benzoyl peroxide, followed by treatment with trimethylamine N-oxide. Suzuki coupling of aryl bromide (II) with the pinacol boronate (III) affords biphenyl (IV). After protection of the aldehyde moiety of (IV) as the corresponding ethylene ketal (V), its cyano group is reduced to aldehyde (VI) employing DIBAL in THF. Subsequent reduction of (VI) with NaBH4 leads to alcohol (VII), which is further converted into the benzyl bromide (VIII) by means of CBr4/PPh3. Bromide (VIII) is condensed with the spiro imidazolone (IX) in the presence of NaH, to produce (X). Then acidic hydrolysis of the ethylene ketal and SEM groups of (X) gives rise to the intermediate aldehyde (XI)
NEXT

Alternatively, reduction of 4-bromo-3-formylbenzonitrile ethylene ketal (XII) by means of DIBAL leads to aldehyde (XIII), which is further reduced to alcohol (XIV) with NaBH4. After bromination of (XIV) with CBr4/PPh3, the resultant benzyl bromide (XV) is condensed with the spiro imidazolone (IX), yielding (XVI). Then, acidic ketal hydrolysis in (XVI) furnishes aldehyde (XVII). Suzuki coupling between aryl bromide (XVII) and boronic acid (XVIII) gives biphenyl (XIX). The SEM group of (XIX) is then removed under acidic conditions to provide (XI)

Reductive amination of the biphenyl aldehyde (XI) with 4-amino-2,2-dimethylbutanoic acid (XX) in the presence of NaBH(OAc)3 produces aminoacid (XXI). This is finally cyclized to the corresponding lactam by treatment with DIC

Coupling of 2-bromobenzenesulfonyl chloride (I) with 5-amino-3,4-dimethylisoxazole (II) affords sulfonamide (III), which is further protected as the N-methoxyethoxymethyl derivative (IV) employing MEM-chloride in DMF. Lithiation of bromosulfonamide (IV), followed by treatment with trimethyl borate and acidic work up leads to the boronic acid intermediate (V). This is then subjected to Suzuki coupling with 4-bromo-3-methylbenzaldehyde (VI) to yield the biphenyl adduct (VII). After reduction of aldehyde (VII) to the benzylic alcohol (VIII) with NaBH4, reaction with methanesulfonyl chloride and diisopropylethylamine gives rise to the mesylate (IX) (1-3).

Mesylate (IX) is condensed with ethyl 2-propyl-4-ethylimidazole-5-carboxylate (X) yielding (XI). Simultaneous ester group hydrolysis and MEM group deprotection under acidic conditions gives rise to the imidazolecarboxylic acid (XII). This is finally coupled with methylamine via activation with CDI to produce the desired N-methyl carboxamide (1-3).
Reductive amination of the biphenyl aldehyde (XI) with 4-amino-2,2-dimethylbutanoic acid (XX) in the presence of NaBH(OAc)3 produces aminoacid (XXI). This is finally cyclized to the corresponding lactam by treatment with DIC
PAPER
 BMS 248360
BMS 248360The ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
15 as a white solid (40 mg, 31%):
mp 104−110 °C;
1H NMR (CDCl3) δ 0.90 (t, J = 7.0 Hz, 3H), 1.08 (s, 3H), 1.14 (s, 3H), 1.36 (m, 2H), 1.61 (m, 2H), 1.75−2.06 (m, 13H), 2.17 (s, 3H), 2.39 (m, 2H), 4.18 (m, 2H), 4.71 (m, 2H), 7.02−7.93 (m, 7H);
13CNMR (CDCl3 ) δ 7.82, 11.91, 14.79, 23.36, 25.50, 25.61, 27.11, 28.81, 29.88, 35.33, 38.42, 41.48, 44.59, 46.24, 46.47, 109.29, 125.15, 125.76, 129.68, 130.58, 131.76, 133.20, 134.07, 137.15, 138.27, 139.11, 139.57, 155.81, 162.68, 162.91, 181.25, 187.83.
Anal. (C36H45N5O5S) C, H, N, S.
……………………………
PATENT
US 2002143024
http://www.google.com/patents/US20020143024
 Zhang, H.-Y. et al., Tetrahedron, 1994, 50, 11339-11362.
Zhang, H.-Y. et al., Tetrahedron, 1994, 50, 11339-11362.

N-(3,4-Dimethyl-5-iso-xazolyl)-2′-formyl-4′-(hydroxy-methyl)-N-[[2-(tri-methylsilyl)ethoxy]- methyl][1,1′- biphenyl]-2- sulfonamide
Example 3 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
[0414]

Example 3 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
A. 4′-Cyano-2′-(1,3-dioxolan-2-yl)-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
A mixture of 2B (1.28 g, 2.73 mmol), ethylene glycol (1.69 g, 27.3 mmol) and p-toluenesulfonic acid (38 mg) in toluene (30 mL) was heated at 130° C. for 5 h, while a Dean-Stark water separator was used. After cooling, the mixture was diluted with EtOAc. The organic liquid was separated and washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 5:4 hexane/EtOAc to afford 3A (1.1 g, 79%) as a colorless gum: Rf=0.57, silica gel, 1:2 hexane/EtOAc.
B. 2′-(1,3-Dioxolan-2-yl)-4′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
To 3A (1.1 g, 2.14 mmol) in THF (21 mL) at 0° C. was added DIBAL-H (1M in CH2Cl2, 4.28 mL 4.28 mmol) dropwise. The reaction was stirred at RT overnight. MeOH (20 mL) was added and the reaction was stirred for 5 min. The mixture was poured into cold 0.1 N HCl solution (150 mL), shaken for 5 min, and then extracted with 3:1 EtOAc/hexane. The combined organic extracts were washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 3:4 hexane/EtOAc to afford 3B (710 mg, 64%) as a colorless gum: Rf=0.45, silica gel, 2:3 hexane/EtOAc.
C. 2′-(1,3-Dioxolan-2-yl)-4′-hydroxymethyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) [1,1′-biphenyl]-2-sulfonamide
3B (710 mg, 1.4 mmol) was subjected to sodium borohydride reduction according to General Method 11 to afford 3C, which was used for the next reaction step without further purification.
D. 4′-Bromomethyl-2′-(1,3-dioxolan-2-yl)-N-(3,4′-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) [1,1′-biphenyl]-2-sulfonamide
3C was treated with carbon tetrabromide and triphenylphosphine according to General Method 2. The crude residue was chromatographed on silica gel using 3:2 hexane/EtOAc to afford 3D (750 mg, 94%) as a colorless gum: Rf=0.74, silica gel, 1:2 hexane/EtOAc.
E. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-(1,3-dioxolan-2-yl)-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
3D (750 mg, 1.3 mmol) was treated with 2-n-butyl-1,3-diazaspiro[4.4]non-1-en-4-one hydrochloride (387 mg, 1.68 mmol) according to General Method 4. The crude residue was chromatographed on silica gel using 100:1.7 CH2Cl2/MeOH to afford 3E as a gum (830 mg, 93%): Rf=0.40, silica gel, 100:5 CH2Cl2/MeOH.
F. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
3E (830 mg, 1.20 mmol) was subjected to deprotection according to General Method 7. The crude residue was chromatographed on silica gel using 100:1.5 and then 100:4 CH2Cl2 /MeOH to afford the title compound as a gum (480 mg, 72%): Rf=0.16, silica gel, 100:5 CH2Cl2/MeOH.
Example 4 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2′-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl][1,1′-biphenyl]-2-sulfonamide

To 3F (110 mg, 0.20 mmol) in CH2Cl2 (4 mL) was added 4-amino-2,2-dimethylbutanoic acid hydrochloride (98 mg, 0.59 mmol) [Scheinmann, et al., J. Chem. Research (S), 414-415 (1993)] and 3 Å molecular sieves, followed by glacial acetic acid (35 mg, 0.59 mmol) and then sodium acetate (48 mg, 0.59 mmol). The mixture was stirred for 8 minutes, and NaB(AcO)3H (124 mg, 0.59 mmol) was then added. The reaction mixture was stirred at RT for 2 h, diluted with EtOAc and filtered through celite. The filtrate was washed with H2O and brine, dried and concentrated. This material was dissolved in CH2Cl2 (6 mL) and 1,3-diisopropylcarbodiimide (32 mg, 0.25 mmol) was added. The reaction mixture was stirred at RT for 2 h and diluted with CH2Cl2, washed with H2O and brine, dried and concentrated. The residue was purified by preparative HPLC to provide the title compound as a white solid (40 mg, 31%, for two steps): mp 104-110° C. Analysis calculated for C36H45N5O5S.0.8 H2O: Calc’d: C, 64.13; H, 6.97; N, 10.39; S, 4,75. Found: C, 64.18; H, 6.60; N, 10.23; S, 4.50.
Example 5 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide (Alternative Preparation for 3F)
A. 2-[(2′-Bromo-5′-formyl)phenyl)]-1,3-dioxolane
DIBAL-H (1.0 M solution in toluene, 445 mL, 445 mmol, 1.1 eq) was added over 30 minutes to a solution of 2-[(2′-bromo-5′-cyano)phenyl)]-1,3-dioxolane (103 g, 404 mmol, 1.0 eq) [Zhang, H.-Y. et al., Tetrahedron, 50, 11339-11362 (1994)] in toluene (2.0 L) at −78° C. The solution was allowed to warm to 0° C. After 1 hour, a solution of Rochelle’s salt (125 g) in water (200 mL) was added, and the mixture was allowed to warm to room temperature and was stirred vigorously for 16 h. The organic layer was concentrated and the residue partitioned between ethyl acetate (1 L) and 1 N hydrochloric acid (800 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (800 mL), dried over sodium sulfate, and then concentrated to give 70.5 g of crude 5A as a yellow solid, which was used without further purification.
B. 2-[(2′-Bromo-5′-hydroxymethyl)phenyl)]-1,3-dioxolane
Sodium borohydride (3.66 g, 96.7 mmol, 0.5 eq) was added to a solution of crude 5A (49.7 g, approximately 193 mmol, 1.0 eq) in absolute ethanol (1300 mL) at 0° C. After 2 hours, a solution of 10% aqueous sodium dihydrogen phosphate (50 mL) was added and the mixture was stirred and allowed to warm to room temperature. The mixture was concentrated, then partitioned between ethyl acetate (800 mL) and saturated aqueous sodium bicarbonate (500 mL). The organic layer was dried over sodium sulfate and concentrated to give 49.0 g of crude 5B as a yellow oil, which was used without further purification.
C. 2-[(2′-Bromo-5′-bromomethyl)phenyl)]-1,3-dioxolane
Triphenylphosphine (52.7 g, 199 mmol, 1.05 eq) was added in portions over 15 minutes to a solution of crude 5B (49.0 g, approximately 189 mmol, 1.0 eq) and carbon tetrabromide (69.0 g, 208 mmol, 1.1 eq) in THF at 0° C. After 2 hours, saturated aqueous sodium bicarbonate solution (20 mL) was added, and the mixture was allowed to warm to room temperature and was then concentrated. Ether (500 mL) was added, and the resulting mixture was filtered. The filtrate was dried over magnesium sulfate and concentrated. The residue was chromatographed on silica gel (8:1 hexanes/ethyl acetate as eluant) to give 5C as a white solid (31.1 g, 51% yield from 2-[(2′-bromo-5′-cyano)phenyl)]-1,3-dioxolane).
D. 2-(1,3-Dioxolan-2-yl)-4-[(2-n-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]bromobenzene
[0436] Sodium hydride (60% dispersion in mineral oil, 9.65 g, 241 mmol, 2.5 eq) was added in portions over 15 minutes to a mixture of 2-n-butyl-1,3-diazaspiro[4.4]non-1-en-4-one hydrochloride (18.7 g, 96.5 mmol, 1.0 eq) in DMF (400 mL) at 0° C. The mixture was stirred and allowed to warm to room temperature over 15 minutes. To this mixture was added via canula a solution of 5C (31.1 g, 96.5 mmol, 1.0 eq) in DMF (100 mL). After 14 hours, the mixture was concentrated in vacuo and partitioned between ethyl acetate (500 mL) and 10% aqueous sodium dihydrogen phosphate (300 mL). The organic layer was dried over sodium sulfate and concentrated to give crude 5D as an orange oil (42.7 g), which was used without further purification.
E. 4-[(2-n-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2-formyl-bromobenzene
A solution of crude 5D (6.0 g, approximately 13.6 mmol, 1.0 eq) in THF (180 mL) and 1N hydrochloric acid (30 mL) was heated at 65° C. for 1.5 hours. The mixture was cooled and then treated with saturated aqueous sodium carbonate solution (75 mL) and ethyl acetate (200 mL). The organic layer was removed and dried over sodium sulfate, concentrated, and then further dried azeotropically with toluene to give 5E as a crude yellow oil (8.2 g) which contained a small amount of toluene. This material was used without further purification.
F. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl)[1,1′-biphenyl]-2-sulfonamide
Palladium catalyzed Suzuki coupling of 5E and [2-[[(3,4-dimethyl-5-isoxazolyl)[(2-methoxyethoxy)methyl]amino]sulfonyl]phenyl]boronic acid was performed according to General Method 1 to yield 5F in 60% yield.
G. 4′-[(2-Butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-2′-formyl-N-(3,4-dimethyl-5-isoxazolyl)-[1,1′-biphenyl]-2-sulfonamide
Deprotection of 5F according to General Method 7 provided the title compound (5G=3F) in 73% yield: Rf=0.2 (silica gel using CH2Cl2/MeOH [100:5]).
PATENT
EP 1237888; WO 0144239
Example 3 4′-r(2-Butyl-4-oxo-1.3-diazaspiror4.41non-l-en-3-yl)methvn-2′-formyl-N-
(3, 4-dimethyl-5-isoxazolyl)-[ 1,1 ‘-biphenyl] -2-sulfonamide

A. 4′-Cvano-2>-(1.3-dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [1.1 ‘-biphenyl] -2-sulfonamide
A mixture of 2B (1.28 g, 2.73 mmol), ethylene glycol (1.69 g, 27.3 mmol) and p-toluenesulfonic acid (38 mg) in toluene (30 mL) was heated at 130°C for 5 h, while a Dean-Stark water separator was used. After cooling, the mixture was diluted with EtOAc. The organic liquid was separated and washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 5:4 hexane/EtOAc to afford 3A (1.1 g, 79%) as a colorless gum: R^0.57, silica gel, 1:2 hexane EtOAc.
B. 2,-(1.3-Dioxolan-2-yl)-4′-formyl-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [1 , l’-biphenyl] -2-sulfonamide To 3A (1.1 g, 2.14 mmol) in THF (21 mL) at 0°C was added DIBAL- H (IM in CH2C12, 4.28 mL 4.28 mmol) dropwise. The reaction was stirred at RT overnight. MeOH (20 mL) was added and the reaction was stirred for 5 min. The mixture was poured into cold 0.1 N HCI solution (150 mL), shaken for 5 min, and then extracted with 3:1 EtOAc/hexane. The combined organic extracts were washed with H2O and brine, dried and concentrated. The residue was chromatographed on silica gel using 3:4 hexane/EtOAc to afford 3B (710 mg, 64%) as a colorless gum: R^O.45, silica gel, 2:3 hexane/EtOAc. C. 2′-(1.3-Dioxolan-2-yl)-4′-hvdroxymethyl-N-(3.4-dimethyl-5- isoxazolyl)-N-(2-methoxyethoxymethyl) [1.1 ‘-biphenyl] -2- sulfonamide
3B (710 mg, 1.4 mmol) was subjected to sodium borohydride reduction according to General Method 11 to afford 3C, which was used for the next reaction step without further purification.
D. 4l-Bromomethyl-2,-(1.3-dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)- N-(2-methoxyethoxymethyl) [1 , l’-biphenyl] -2-sulfonamide 3C was treated with carbon tetrabromide and triphenylphosphine according to General Method 2. The crude residue was chromatographed on silica gel using 3:2 hexane/EtOAc to afford 3D (750 mg, 94%) as a colorless gum: R^0.74, silica gel, 1:2 hexane/EtOAc.
E. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn- 2,-(1.3- dioxolan-2-yl)-N-(3.4-dimethyl-5-isoxazolyl)-N-(2- methoxyethoxymethyl) [ 1. l’-biphenyll -2-sulfonamide 3D (750 mg, 1.3 mmol) was treated with 2-re-butyl-l,3- diazaspiro[4.4]non-l-en-4-one hydrochloride (387 mg, 1.68 mmol) according to General Method 4. The crude residue was chromatographed on silica gel using 100:1.7 CH2CL/MeOH to afford 3E as a gum (830 mg, 93%): R^O.40, silica gel, 100:5 CH2Cl2/MeOH.
F. 4′-r(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2,– formyl-N-(3.4-dimethyl-5-isoxazolyl)-[l.l’-biphenyl1-2-sulfonamide
3E (830 mg, 1.20 mmol) was subjected to deprotection according to General Method 7. The crude residue was chromatographed on silica gel using 100:1.5 and then 100:4 CH2C12 /MeOH to afford the title compound as a gum (480 mg, 72%): R^O.16, silica gel, 100:5 CH.Cl MeOH.
Example 4
4′- . (2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non-l-en-3-yl)methyll -N-C3.4- dimethyl-5-isoxazolyl)-2,-[(3.3-dimethyl-2-oxo-l- pyrrolidinvDmethyll [1.1 ‘-biphenyl] -2-sulfonamide
To 3F (110 mg, 0.20 mmol) in CH2C12 (4 mL) was added 4-amino- 2,2-dimethylbutanoic acid hydrochloride (98 mg, 0.59 mmol) [Scheinmann, et al., J. Chem. Research (S), 414-415 (1993)] and 3A molecular sieves, followed by glacial acetic acid (35 mg, 0.59 mmol) and then sodium acetate (48 mg, 0.59 mmol). The mixture was stirred for 8 minutes, and NaB(AcO)3H (124 mg, 0.59 mmol) was then added. The reaction mixture was stirred at RT for 2 h, diluted with EtOAc and filtered through celite. The filtrate was washed with H2O and brine, dried and concentrated. This material was dissolved in CH2C12 (6 mL) and 1,3-diisopropylcarbodiimide (32 mg, 0.25 mmol) was added. The reaction mixture was stirred at RT for 2 h and diluted with CH2C12, washed with H2O and brine, dried and concentrated. The residue was purified by preparative HPLC to provide the title compound as a white solid (40 mg, 31%, for two steps): mp 104- 110°C. Analysis calculated for C36H45N5O5S • 0.8 H2O: Calc’d: C, 64.13; H, 6.97; N, 10.39; S, 4,75. Found: C, 64.18; H, 6.60; N, 10.23; S, 4.50.
Example 5
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2,-formyl-N-
(3,4-dimethyl-5-isoxazolyl)-[l,l’-biphenyl]-2-sulfonamide (Alternative
Preparation for 3F)
A. 2-[(2′-Bromo-5′-formyl)phenyl)1-1.3-dioxolane
DIBAL-H (1.0 M solution in toluene, 445 mL, 445 mmol, 1.1 eq) was added over 30 minutes to a solution of 2-[(2′-bromo-5′-cyano)phenyl)]-l,3- dioxolane (103 g, 404 mmol, 1.0 eq) [Zhang, H.-Y. et al., Tetrahedron, 50, 11339-11362 (1994)] in toluene (2.0 L) at -78 °C. The solution was allowed to warm to 0 °C. After 1 hour, a solution of Rochelle’s salt (125 g) in water (200 mL) was added, and the mixture was allowed to warm to room temperature and was stirred vigorously for 16 h. The organic layer was concentrated and the residue partitioned between ethyl acetate (1 L) and 1 N hydrochloric acid (800 mL). The organic layer was washed with saturated aqueous sodium bicarbonate (800 mL), dried over sodium sulfate, and then concentrated to give 70.5 g of crude 5A as a yellow solid, which was used without further purification.
B. 2-[(2′-Bromo-5′-hvdroxymethyl)phenyl)l-1.3-dioxolane
Sodium borohydride (3.66 g, 96.7 mmol, 0.5 eq) was added to a solution of crude 5A (49.7 g, approximately 193 mmol, 1.0 eq) in absolute ethanol (1300 mL) at 0 °C. After 2 hours, a solution of 10% aqueous sodium dihydrogen phosphate (50 mL) was added and the mixture was stirred and allowed to warm to room temperature. The mixture was concentrated, then partitioned between ethyl acetate (800 mL) and saturated aqueous sodium bicarbonate (500 mL). The organic layer was dried over sodium sulfate and concentrated to give 49.0 g of crude 5B as a yellow oil, which was used without further purification. C. 2-[(2′-Bromo-5′-bromomethyl)phenyl)]-l,3-dioxolane Triphenylphosphine (52.7 g, 199 mmol, 1.05 eq) was added in portions over 15 minutes to a solution of crude 5B (49.0 g, approximately 189 mmol, 1.0 eq) and carbon tetrabromide (69.0 g, 208 mmol, 1.1 eq) in THF at 0 °C. After 2 hours, saturated aqueous sodium bicarbonate solution (20 mL) was added, and the mixture was allowed to warm to room temperature and was then concentrated. Ether (500 mL) was added, and the resulting mixture was filtered. The filtrate was dried over magnesium sulfate and concentrated. The residue was chromatographed on silica gel (8:1 hexanes/ethyl acetate as eluant) to give 5C as a white solid (31.1 g, 51% yield from 2-[(2′-bromo-5′-cyano)phenyl)]-l,3-dioxolane).
D. 2-( 1 ,3-Dioxolan-2-yl)-4- [ (2-re-butyl-4-oxo- 1 ,3-diazaspiro [4.4] non- 1- en-3-yl)methyl] bromobenzene Sodium hydride (60% dispersion in mineral oil, 9.65 g, 241 mmol,
2.5 eq) was added in portions over 15 minutes to a mixture of 2-rc-butyl- l,3-diazaspiro[4.4]non-l-en-4-one hydrochloride (18.7 g, 96.5 mmol, 1.0 eq) in DMF (400 mL) at 0°C. The mixture was stirred and allowed to warm to room temperature over 15 minutes. To this mixture was added via canula a solution of 5C (31.1 g, 96.5 mmol, 1.0 eq) in DMF (100 mL). After 14 hours, the mixture was concentrated in vacuo and partitioned between ethyl acetate (500 mL) and 10% aqueous sodium dihydrogen phosphate (300 mL). The organic layer was dried over sodium sulfate and concentrated to give crude 5D as an orange oil (42.7 g), which was used without further purification.
E. 4-[(2-n-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyl1-2- formyl-bromobenzene
A solution of crude 5D (6.0 g, approximately 13.6 mmol, 1.0 eq) in THF (180 mL) and IN hydrochloric acid (30 mL) was heated at 65°C for 1.5 hours. The mixture was cooled and then treated with saturated aqueous sodium carbonate solution (75 mL) and ethyl acetate (200 mL). The organic layer was removed and dried over sodium sulfate, concentrated, and then further dried azeotropically with toluene to give 5E as a crude yellow oil (8.2 g) which contained a small amount of toluene. This material was used without further purification.
F. 4′-.(2-Butyl-4-oxo-1.3-diazaspiro■4.41non-l-en-3-yl)methyl1-2,– formyl-N-(3,4-dimethyl-5-isoxazolyl)-N-(2-methoxyethoxymethyl) f 1.1 ‘-biphenyl] -2-sulfonamide Palladium catalyzed Suzuki coupling of 5E and [2-[[(3,4-dimethyl-5- isoxazolyl) [(2-methoxyethoxy)methyl] amino] sulfonyl] phenyl]boronic acid was performed according to General Method 1 to yield 5F in 60% yield.
G. 4’-[ 2-Butyl-4-oxo-1.3-diazaspiro[4■41non-l-en-3-yl)methvn-2,– formyl-N-(3 ,4-dimethyl-5-isoxazolyl)- fi .1 ‘-biphenyl] -2-sulfonamide
Deprotection of 5F according to General Method 7 provided the title compound (5G = 3F) in 73% yield: R^0.2 (silica gel using CH2ClJ eOH [100:5]).
| Patent | Submitted | Granted |
|---|---|---|
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6638937] | 2002-10-03 | 2003-10-28 |
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6835741] | 2004-06-03 | 2004-12-28 |
| Biphenyl sulfonamides as dual angiotensin endothelin receptor antagonists [US6852745] | 2004-07-01 | 2005-02-08 |
///////////BMS-248360, Preclinical, SARTAN, BMS, HYPERTENTION
CCCCC1=NC2(CCCC2)C(=O)N1CC3=CC(=C(C=C3)C4=CC=CC=C4S(=O)(=O)NC5=C(C(=NO5)C)C)CN6CCC(C6=O)(C)C
GSK2334470

GSK2334470
GSK2334470; 1227911-45-6; GSK-2334470; GSK 2334470;
(3S,6R)-1-[6-(3-Amino-1H-indazol-6-yl)-2-(methylamino)-4-pyrimidinyl]-N-cyclohexyl-6-methyl-3-piperidinecarboxamide
(3S.6/?V1-r6-(3-Amino-1 H-indazol-6-ylV2-(methylaminoV4-pyrimidinyll-Λ/-cvclohexyl-6- methyl-3-piperidinecarboxamide
| Molecular Weight | 462.59 |
| Formula | C25H34N8O |
| CAS Number | 1227911-45-6 |
![]()
Phosphoinositide Dependent Kinase (PDK) 1 Inhibitors
[α]20D = – 32.6 o (c 1.17, MeOH)
[α] D = -27.6 (Concentration = 1.16, Solvent = Methanol)
SOL………DMSO to 100 mM
ethanol to 100 mM
nmr……http://www.chemietek.com/Files/Line2/Chemietek,%20GSK2334470%20(1),%20NMR-DMSO.pdf
http://file.selleckchem.com/downloads/nmr/S708702-GSK2334470-HNMR-Selleck.pdf


GSK2334470 is a potent and selective PDK1 (3-Phosphoinositide dependent protein kinase-1) inhibitor. GSK2334470 blocks the phosphorylation of known PDK1 substrates, but surprisingly find that the potency and kinetics of inhibition vary for different PDK1 targets. GSK2334470 subsequent activation of PDK1 substrates S6K1, SGK and RSK in HEK293, U87 and mouse embryonic fibroblast cell lines.
GSK2334470 inhibited activation of an Akt1 mutant lacking the PH domain (pleckstrin homology domain) more potently than full-length Akt1, suggesting that GSK2334470 is more effective at inhibiting PDK1 substrates that are activated in the cytosol rather than at the plasma membrane. GSK2334470 also suppressed T-loop phosphorylation and activation of RSK2 (p90 ribosomal S6 kinase 2), another PDK1 target activated by the ERK (extracellular-signal-regulated kinase) pathway.
GSK2334470 is a highly specific and potent inhibitor of PDK1 (3-Phosphoinositide dependent protein kinase-1) with IC50 of 10 nM. It does not suppress activity on other 96 kinases, including Aurora, ROCK, p38 MAPK and PI3K. GSK2334470 has been used in cells to ablate T-loop phosphorylation and activate SGK, S6K1 and RSK as well as suppress the activation of Akt.
PATENT
WO 2010059658
http://www.google.com/patents/WO2010059658A1?cl=en
Example 78
(3S.6/?V1-r6-(3-Amino-1 H-indazol-6-ylV2-(methylaminoV4-pyrimidinyll-Λ/-cvclohexyl-6- methyl-3-piperidinecarboxamide
To (3S,6R)-1-[6-(4-cyano-3-fluorophenyl)-2-(methylamino)-4-pyrimidinyl]-Λ/-cyclohexyl-6- methyl-3-piperidinecarboxamide (260 mg, 0.58 mmol) in EtOH (10 ml.) as a suspension at room temperature in a microwave vial was added hydrazine monohydrate (807 uL, 16.7 mmol, 30 equiv) in one portion. The mixture was capped and heated at 100 0C for 48 hours. A duplicate run was performed. The crude reactions from both runs were combined, and concentrated in vacuo. The residue was taken up in 10 ml. of water. The resulting suspension was sonicated briefly, and filtered. The solids collected were dried under vacuum at room temperature over P2O5 for 18 hours, and then at 65 0C under vacuum for another 18 hours to afford the title compound (410 mg) as a cream-colored solid. LC-MS (ES) m/z = 463 [M+H]+. 1H NMR (400 MHz, CD3OD): δ 1.16 – 1.32 (m, 3H),1.29 (d, J = 6.8 Hz, 3H), 1.34 – 1.45 (m, 2H), 1.65 – 1.68 (m, 1 H), 1.76 – 1.81 (m, 5H), 1.85 – 1.92 (m, 2H), 1.97 – 2.05 (m, 1 H), 2.35 – 2.42 (m, 1 H), 2.97 (s, 3H), 3.1 1 – 3.15 (m, 1 H),3.64 – 3.70 (m, 1 H), 4.45 – 4.65 (bs, 1 H), 4.72 – 4.92 (bs, 1 H), 6.45 (s, 1 H), 7.52 (dd, J =8.5, 1.14 Hz, 1 H), 7.75 (d, J = 8.3 Hz, 1 H), 7.85 (s, 1 H).
ntermediate 112
Cis- methyl-6-methyl-3-piperidinecarboxylate

A solution of cis-3-methyl 1-(phenylmethyl)-6-methyl-1 ,3-piperidinedicarboxylate (69 g, 237 mol) in EtOH (50 mL) and EtOAc (300 mL) was added to a slurry of 10% Pd/C (3.7 g) in EtOAc (30 mL) and EtOH (10 mL) EtOH under nitrogen in a Parr Shaker bottle. The mixture was hydrogenated under 65 psi at room temperature for 4 hours. The mixture was filtered through celite, and washed with EtOAc. The filtrate was concentrated in vacuo to give 37 g of the title compound as a liquid. LC-MS (ES) m/z = 158 [M+H]+.
Intermediate 113
Methyl (3S,6f?)-6-methyl-3-piperidinecarboxylate L-(+)-tartaric acid salt 
L-(+)-Tartaric acid salt A suspension of L-(+)-tartaric acid (39 g, 260 mmol, 1.05 equiv) in IPA (200 ml.) and water (13 mL) water was heated in a water bath at 600C until all dissolved. To this hot stirred solution was added neat racemic methyl (3S,6R)-6-methyl-3-piperidinecarboxylate (39 g, 248 mmol), followed by addition of 25 mL of IPA rinse. The resulting mixture was heated to 60 0C, resulting in a clear solution, and then cooled to room temperature, while the hot water bath was removed. This hot solution was seeded with a sample of methyl (3S,6R)-6-methyl-3-piperidinecarboxylate L-(+)-tartaric acid salt that had a chiral purity of 98% ee, and aged at ambient temperature (with the water bath removed) for 20 minutes. The mixture turned into an oily texture with seeds still present. To the mixture was added 5 mL of water, and heated in the warm water bath at 43 0C. The mixture became clear with the seeds still present. The heating was stopped, and the mixture was stirred in the warm water bath. After 20 minutes, the mixture gradually turned into a paste. After another 10 min, the water bath was removed, and the mixture was stirred at ambient temperature for another 1 hour. The resulting paste was filtered. The cake was washed with 50 mL of IPA, giving 62 g of wet solids. This cake was taken up in 150 mL of IPA and 8 mL of water, and stirred as a slurry while being heated in a water bath to 60 0C (internal temp 55 0C) for 5 minutes. The heating was turned off while the mixture was still stirred in the warm water bath. After 30 min, the mixture was filtered. The cake was washed with 100 mL of IPA. Drying under house vacuum at room temperature for 48 hours gave 46.7 g of solids. An analytical sample was derivatised to the corresponding N-Cbz derivative (as in the preparation of intermediate 1 11 ), which was determined by chiral HPLC (methods used to analyze the resolution of intermediate 11 1 above) to have 85% ee. This material was taken up in IPA (420 mL) and water (38 mL) as a suspension. The mixture was heated in a water bath to 65 0C, at which time the mixture became a clear solution. The heating bath was removed. The mixture was seeded and aged at ambient temp for 20 hours. The solids formed were filtered, and washed with 100 mL of IPA. The solids collected were dried under house vacuum at room temperature for 24 h, and then under vacuum at room temperature for another 24 hours to give 28.5 g of the title compound. An analytical sample was converted to the N-Cbz derivative. The ee was determined to be 97.7%. LC-MS (ES) m/z = 158 [M+H]+.
Intermediate 114 4,6-Dichloro-Λ/-methyl-2-pyrimidinamine

Methylamine (2M solution, 113 ml_, 217 mmol, 2.05 equiv) was charged to a 1 L 3-neck flask fitted with a magnetic stirrer and a thermometer. The mixture was chilled in an ice bath. To this stirred solution was added via addition funnel a solution of 4,6-dichloro-2-(methylsulfonyl)pyrimidine (25 g, 1 10 mmol) in EtOAc (250 ml.) portionwise over a 25 minutes period. The temp was between 5-10 0C. After completion of addition, the ice bath was removed, and the mixture was stirred for 1 hour at ambient temperature. LCMS showed conversion complete. The suspension was filtered, and washed with EtOAc. The filtrate was concentrated in vacuo. The residue was partitioned between water (100 ml.) and EtOAc (450 ml_). The organic was washed with brine, dried over MgSO4, filtered and concentrated in vacuo to give white solids, which were triturated in 150 ml. of CH2CI2. These solids were collected by filtration and washing with cold CH2CI2 (50 ml_). Drying under house vacuum at room temperature for 20 hours, and then high vacuum at room temperature for 3 hours gave 9.31 g of the title compound as a solid. LC-MS (ES) m/z = 179 [M+H]+.
Intermediate 121 (3S,6/?)-1-r6-Chloro-2-(methylamino)-4-pyrimidinyll-Λ/-cvclohexyl-6-methyl-3-piperidinecarboxamide

To a suspension of (3S,6/?)-1-[6-chloro-2-(methylamino)-4-pyrimidinyl]-6-methyl-3-piperidinecarboxylic acid (3.05 g, 10.71 mmol) in CH2CI2 (50 ml.) at room temperature was added Hunig’s base (2.70 ml_, 15.43 mmol, 1.3 equiv) and cyclohexylamine (1.60 ml_, 14.2 mmol, 1.2 equiv), and the resulting mixture was chilled in an ice bath. To this stirred solution was added HATU (4.96 g, 13.1 mmol, 1.1 equiv) in one portion, and the resulting suspension was stirred in the ice bath for 30 minutes. LCMS showed conversion complete. The mixture was diluted with CH2CI2 (50 ml.) and filtered through celite. The filtrate was washed water (2 X 25 ml.) and then brine. The organic was dried over Na2SO4, filtered, and concentrated in vacuo. Silica gel column chromatography using gradient elution of 1 % EtOAc in CHCI3 to 50% EtOAc in CHCI3 afforded the title compound (4.26 g) as a foam. LC-MS (ES) m/z = 366 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2011), 54(6), 1871-1895.
http://pubs.acs.org/doi/full/10.1021/jm101527u

Phosphoinositide-dependent protein kinase-1(PDK1) is a master regulator of the AGC family of kinases and an integral component of the PI3K/AKT/mTOR pathway. As this pathway is among the most commonly deregulated across all cancers, a selective inhibitor of PDK1 might have utility as an anticancer agent. Herein we describe our lead optimization of compound 1toward highly potent and selective PDK1 inhibitors via a structure-based design strategy. The most potent and selective inhibitors demonstrated submicromolar activity as measured by inhibition of phosphorylation of PDK1 substrates as well as antiproliferative activity against a subset of AML cell lines. In addition, reduction of phosphorylation of PDK1 substrates was demonstrated in vivo in mice bearing OCl-AML2 xenografts. These observations demonstrate the utility of these molecules as tools to further delineate the biology of PDK1 and the potential pharmacological uses of a PDK1 inhibitor.
REFERENCES
Najafov, et al., Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem.J. (2011), 433 (2) 357.
For a PDK1 inhibitor, the substrate matters.
Knight ZA. Biochem J. 2011 Jan 15;433(2):e1-2. PMID: 21175429.
Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1.
Najafov A, et al. Biochem J. 2011 Jan 15;433(2):357-69. PMID: 21087210.

Jeffrey Axten
Director, Medicinal Chemistry, Virtual Proof of Concept DPU at GlaxoSmithKline
/////
