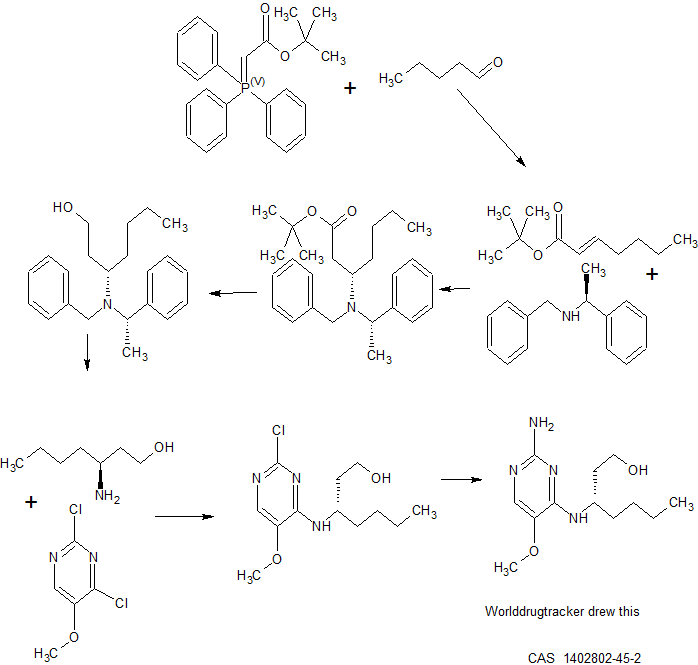
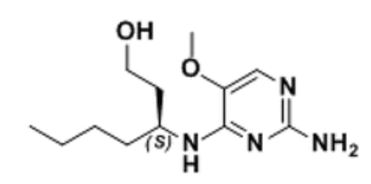
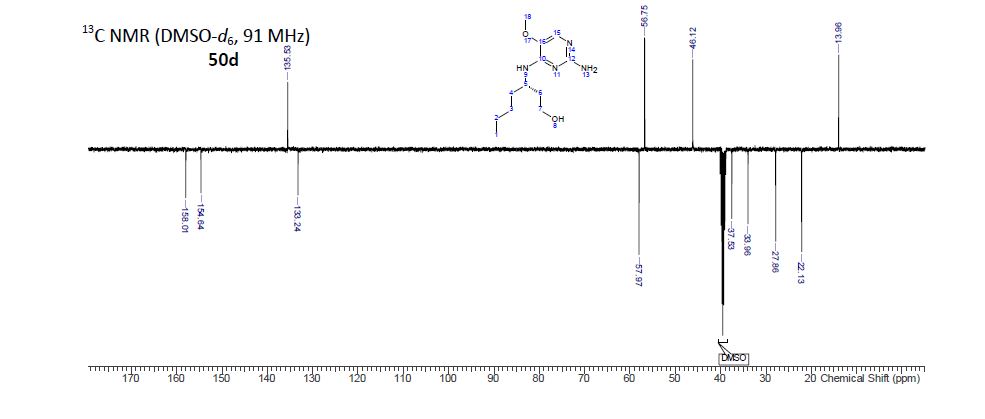
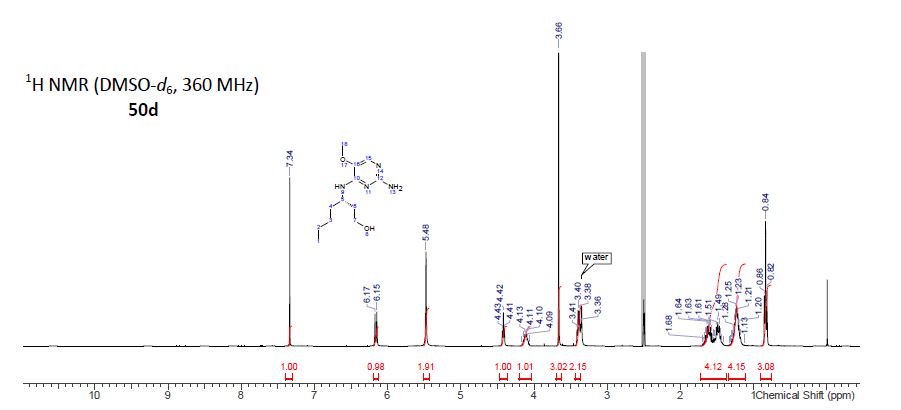
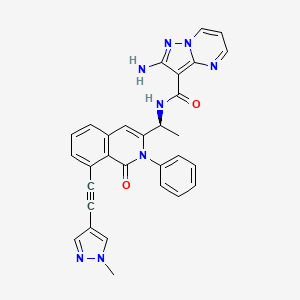
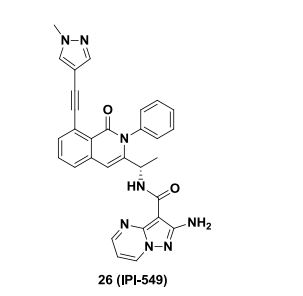
BMS-960
PRECLINICAL
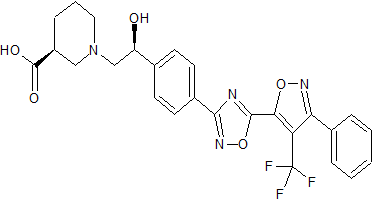
(S)-1-((S)-2-Hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic Acid
3-Piperidinecarboxylic acid, 1-[(2S)-2-hydroxy-2-[4-[5-[3-phenyl-4-(trifluoromethyl)-5-isoxazolyl]-1,2,4-oxadiazol-3-yl]phenyl]ethyl]-, (3S)-
(S)-1-((S)-2-Hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic Acid
CAS 1265321-86-5 FREE FORM
FREE FORM 528.48, C26 H23 F3 N4 O5
CAS 1265323-40-7 HCL SALT
BASIC PATENT WO201117578, 2011, (US Patent 8399451)

Sphingosine-1-phosphate (S1P) is the endogenous ligand for the sphingosine-1-phophate receptors (S1P1–5) and triggers a number of cellular responses through their stimulation. S1P and its interaction with the S1P receptors play a significant role in a variety of biological processes including vascular stabilization, heart development, lymphocyte homing, and cancer angiogenesis. Agonism of S1P1, especially, has been shown to play an important role in lymphocyte trafficking from the thymus and secondary lymphoid organs, inducing immunosuppression, which has been established as a novel mechanism of treatment for immune diseases and vascular diseases
Sphingosine-1 -phosphate (SlP) has been demonstrated to induce many cellular effects, including those that result in platelet aggregation, cell proliferation, cell morphology, tumor cell invasion, endothelial cell and leukocyte chemotaxis, endothelial cell in vitro angiogenesis, and lymphocyte trafficking. SlP receptors are therefore good targets for a wide variety of therapeutic applications such as tumor growth inhibition, vascular disease, and autoimmune diseases. SlP signals cells in part via a set of G protein-coupled receptors named SlPi or SlPl, SlP2 or S1P2, SlP3 or S1P3, SlP4 Or S1P4, and SlP5 or S1P5 (formerly called EDG-I, EDG-5, EDG-3, EDG-6, and EDG-8, respectively).
SlP is important in the entire human body as it is also a major regulator of the vascular and immune systems. In the vascular system, SlP regulates angiogenesis, vascular stability, and permeability. In the immune system, SlP is recognized as a major regulator of trafficking of T- and B-cells. SlP interaction with its receptor SlPi is needed for the egress of immune cells from the lymphoid organs (such as thymus and lymph nodes) into the lymphatic vessels. Therefore, modulation of SlP receptors was shown to be critical for immunomodulation, and SlP receptor modulators are novel immunosuppressive agents.
The SlPi receptor is expressed in a number of tissues. It is the predominant family member expressed on lymphocytes and plays an important role in lymphocyte trafficking. Downregulation of the SlPi receptor disrupts lymphocyte migration and homing to various tissues. This results in sequestration of the lymphocytes in lymph organs thereby decreasing the number of circulating lymphocytes that are capable of migration to the affected tissues. Thus, development of an SlPi receptor agent that suppresses lymphocyte migration to the target sites associated with autoimmune and aberrant inflammatory processes could be efficacious in a number of autoimmune
Among the five SlP receptors, SlPi has a widespread distribution and is highly abundant on endothelial cells where it works in concert with SIP3 to regulate cell migration, differentiation, and barrier function. Inhibition of lymphocyte recirculation by non-selective SlP receptor modulation produces clinical immunosuppression preventing transplant rejection, but such modulation also results in transient bradycardia. Studies have shown that SlPi activity is significantly correlated with depletion of circulating lymphocytes. In contrast, Sl P3 receptor agonism is not required for efficacy. Instead, SIP3 activity plays a significant role in the observed acute toxicity of nonselective SlP receptor agonists, resulting in the undesirable cardiovascular effects, such as bradycardia and hypertension. (See, e.g., Hale et al, Bioorg. Med. Chem. Lett., 14:3501 (2004); Sanna et al., J. Biol. Chem., 279: 13839 (2004); Anliker et al., J. Biol. Chem., 279:20555 (2004); Mandala et al., J. Pharmacol. Exp. Ther., 309:758 (2004).)
An example of an SlPi agonist is FTY720. This immunosuppressive compound FTY720 (JPI 1080026-A) has been shown to reduce circulating lymphocytes in animals and humans, and to have disease modulating activity in animal models of organ rejection and immune disorders. The use of FTY720 in humans has been effective in reducing the rate of organ rejection in human renal transplantation and increasing the remission rates in relapsing remitting multiple sclerosis (see Brinkman et al., J. Biol. Chem., 277:21453 (2002); Mandala et al., Science, 296:346 (2002); Fujino et al., J.
Pharmacol. Exp. Ther., 305:45658 (2003); Brinkman et al, Am. J. Transplant., 4: 1019 (2004); Webb et al., J. Neuroimmunol, 153: 108 (2004); Morris et al., Eur. J. Immunol, 35:3570 (2005); Chiba, Pharmacology & Therapeutics, 108:308 (2005); Kahan et al., Transplantation, 76: 1079 (2003); and Kappos et al., N. Engl. J. Med., 335: 1124 (2006)). Subsequent to its discovery, it has been established that FTY720 is a prodrug, which is phosphorylated in vivo by sphingosine kinases to a more biologically active agent that has agonist activity at the SlPi, SIP3, SlP4, and SIP5 receptors. It is this activity on the SlP family of receptors that is largely responsible for the pharmacological effects of FTY720 in animals and humans. [0007] Clinical studies have demonstrated that treatment with FTY720 results in bradycardia in the first 24 hours of treatment (Kappos et al, N. Engl. J. Med., 335: 1124 (2006)). The observed bradycardia is commonly thought to be due to agonism at the SIP3 receptor. This conclusion is based on a number of cell based and animal experiments. These include the use of SIP3 knockout animals which, unlike wild type mice, do not demonstrate bradycardia following FTY720 administration and the use of SlPi selective compounds. (Hale et al., Bioorg. Med. Chem. Lett., 14:3501 (2004); Sanna et al., J. Biol. Chem., 279: 13839 (2004); and Koyrakh et al., Am. J. Transplant, 5:529 (2005)).
The following applications have described compounds as SlPi agonists: WO 03/061567 (U.S. Patent Publication No. 2005/0070506), WO 03/062248 (U.S. Patent No. 7,351,725), WO 03/062252 (U.S. Patent No. 7,479,504), WO 03/073986 (U.S. Patent No. 7,309,721), WO 03/105771, WO 05/058848, WO 05/000833, WO 05/082089 (U.S. Patent Publication No. 2007/0203100), WO 06/047195, WO 06/100633, WO 06/115188, WO 06/131336, WO 2007/024922, WO 07/109330, WO 07/116866, WO 08/023783 (U.S. Patent Publication No. 2008/0200535), WO 08/029370, WO 08/114157, WO 08/074820, WO 09/043889, WO 09/057079, and U.S. Patent No. 6,069,143. Also see Hale et al., J. Med. Chem., 47:6662 (2004).
There still remains a need for compounds useful as SlPi agonists and yet having selectivity over Sl P3.
Applicants have found potent compounds that have activity as SlPi agonists. Further, applicants have found compounds that have activity as SlPi agonists and are selective over SIP3. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
SYNTHESIS


(S)-1-((S)-2-Hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic acid, HCl (BMS-960). CAS 1265323-40-7
(S)-1-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic acid, HCl (BMS-960)
1H NMR (400 MHz, DMSO-d6) δ 12.88 (br. s, 1H), 10.5 (br. s, 1H), 8.14 (d, J = 8.6 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.69–7.57 (m, 5H), 6.43 (br. s., 1H), 5.37 (d, J = 10.8 Hz, 1H), 3.89–3.60 (m, 2H), 3.50–2.82 (m, 6H), 2.14–1.99 (m, 1H), 1.97–1.75 (m, 1H), 1.63–1.35 (m, 1H);
13C NMR (101 MHz, CDCl3) δ 172.8, 168.5, 164.0, 161.6, 155.4, 156.2, 131.2, 129.0, 128.9, 127.4, 127.2, 125.5, 124.3, 122.2, 111.6, 66.6. 63.0, 52.9, 52.2, 38.8, 25.0, 21.7;
19F NMR (376 MHz, DMSO-d6) δ −54.16;
Anal. calcd for C26H23F3N4O5·HCl: C, 54.71; H, 4.36; N, 9.80. Found: C, 54.76; H, 3.94; N, 9.76;
HRMS (ESI) m/e 529.17040 [(M + H)+, calcd for C26 H24 N4 O5 F3 529.16933].
PATENT
WO 2011017578
Example 14
(S)-l-((S)-2-Hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-l,2,4- oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic acid
Preparation 14A: (3S)-Ethyl l-(2-(4-cyanophenyl)-2-hydroxyethyl)piperidine-3- carboxylate

(14A)-isomer A (14A)-isomer B [00210] To a mixture of (S)-ethyl piperidine-3-carboxylate (1.3 g, 8.27 mmol) in toluene (50 mL) was added 4-(2-bromoacetyl)benzonitrile (2.4 g, 10.71 mmol). The reaction mixture was stirred overnight. LCMS indicated completion of reaction. MeOH (10 mL) was added to the mixture, followed by the portionwise addition of sodium borohydride (0.313 g, 8.27 mmol). After 1 hour, LCMS show complete reduction to the desired alcohol. The reaction was quenched with water. The reaction mixture was diluted with ethyl acetate and washed with saturated NaCl. The organic layer was dried with MgSO4, filtered, concentrated, and purified on a silica gel cartridge using an EtOAc/hexanes gradient to yield 2.0 g of solid product. The product was separated by chiral HPLC (Berger SFC MGIII instrument equipped with a CHIRALCEL® OJ (25 x 3 cm, 5 μM). Temp: 30 0C; Flow rate: 130 mL/min; Mobile phase: C(V(MeOH +
0.1%DEA) in 9: 1 ratio isocratic:
[00211] Peak 1 (Isomer A): RT = 2.9 min. for (S)-ethyl l-((S)-2-(4-cyanophenyl)-2- hydroxyethyl)piperidine-3-carboxylate (>99% d.e.). The absolute and relative stereochemistry of compound 14A-isomer A was assigned (S,S) by X-ray crystal structure (see Alternative Route data). 1H NMR (400 MHz, CDCl3) δ ppm 7.63 (2 H, m, J=8.35 Hz), 7.49 (2 H, m, J=8.35 Hz), 4.77 (1 H, dd, J=10.55, 3.52 Hz), 4.17 (2 H, q, J=7.03 Hz), 3.13 (1 H, d, J=9.23 Hz), 2.53-2.67 (3 H, m), 2.44 (2 H, dd, J=18.68, 9.89 Hz), 2.35 (1 H, dd, J=12.74, 10.55 Hz), 1.87-2.01 (1 H, m), 1.71-1.82 (1 H, m), 1.52-1.70 (2 H, m), 1.28 (3 H, t, J=7.03 Hz).
[00212] Peak 2 (Isomer B): RT = 3.8 min for (S)-ethyl l-((R)-2-(4-cyanophenyl)-2- hydroxyethyl)piperidine-3-carboxylate (>99% d.e.). The absolute and relative stereochemistry of 14A-isomer B was assigned (S,R) based on the crystal structure of 14A-isomer A. 1H NMR (400 MHz, CDCl3) δ ppm 7.63 (2 H, m, J=8.35 Hz), 7.49 (2 H, m, J=8.35 Hz), 4.79 (1 H, dd, J=10.55, 3.52 Hz), 4.16 (2 H, q, J=7.03 Hz), 2.69-2.91 (3 H, m), 2.60-2.68 (1 H, m), 2.56 (1 H, dd, J=12.30, 3.52 Hz), 2.36 (1 H, dd, J=12.52, 10.77 Hz), 2.25 (1 H, t, J=8.79 Hz), 1.65-1.90 (3 H, m), 1.52-1.64 (1 H, m, J=12.69, 8.49, 8.49, 4.17 Hz), 1.27 (3 H, t, J=7.25 Hz).
[00213] (S)-Ethyl l-((S)-2-(4-cyanophenyl)-2-hydroxyethyl)piperidine-3-carboxylate (14A-isomer A) was carried forward to make Example 14 and (S)-ethyl l-((R)-2-(4- cyanophenyl)-2-hydroxyethyl)piperidine-3-carboxylate (14A-isomer B) was carried forward to make Example 15.
Preparation 14B: (S)-Ethyl l-((S)-2-hydroxy-2-(4-((Z)-N’-hydroxycarbamimidoyl) phenyl)ethyl)piperidine-3 -carboxylate
[00214] To a mixture of ((S)-ethyl l-((S)-2-hydroxy-2-(4-((Z)-N’- hydroxycarbamimidoyl) phenyl)ethyl)piperidine-3 -carboxylate (14A-Isomer A) (58 mg, 0.192 mmol) and hydroxylamine hydrochloride (26.7 mg, 0.384 mmol) in 2-propanol (10 mL) was added sodium bicarbonate (64.5 mg, 0.767 mmol). The reaction mixture was heated at 85 0C. The reaction mixture was diluted with ethyl acetate and washed with sat NaCl. The organic layer was dried with MgSO4, filtered, and concentrated to yield 56 mg. MS (M+l) = 464. HPLC Peak RT = 1.50 minutes.
Preparation 14C: (S)-Ethyl l-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl) isoxazol-5-yl)-l,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylate

[00215] 3-Phenyl-4-(trifluoromethyl)isoxazole-5-carbonyl fluoride, InM-G (214 mg, 0.78 mmol) was dissolved in acetonitrile (5.00 mL). DIEA (0.272 mL, 1.555 mmol) and (S)-ethyl- 1 -((S)-2-hydroxy-2-(4-((Z)-N’-hydroxycarbamimidoyl) phenyl)ethyl)- piperidine-3-carboxylate (261 mg, 0.778 mmol) were added. The reaction mixture was stirred for 2 hours, then IM TBAF in THF (0.778 mL, 0.778 mmol) was added. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and purified by HPLC in three batches. HPLC conditions: PHENOMENEX® Luna C18 5 micron column (250 x 30mm); 25-100% CH3CN/water (0.1% TFA); 25 minute gradient; 30 mL/min. Isolated fractions with correct mass were partitioned between EtOAc and saturated NaHCO3 with back extracting aqueous layer once. The organic layer was dried with MgSO4, filtered, and concentrated to give 155mg of (S)- ethyl l-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-l,2,4- oxadiazol-3-yl)phenyl)ethyl) piperidine-3-carboxylate. 1H NMR (400 MHz, MeOH-d3) δ ppm 8.04 (2 H, d, J=8.13 Hz), 7.55-7.60 (2 H, m), 7.41-7.54 (5 H, m), 4.81 (1 H, ddd, J=8.35, 4.06, 3.84 Hz), 3.96-4.10 (2 H, m), 2.82-3.08 (1 H, m), 2.67-2.82 (1 H, m), 2.36- 2.61 (3 H, m), 2.08-2.33 (2 H, m), 1.73-1.87 (1 H, m, J=8.54, 8.54, 4.45, 4.17 Hz), 1.32- 1.70 (3 H, m), 1.09-1.19 (3 H, m). MS (m+l) = 557. HPLC Peak RT = 3.36 minutes. Purity = 99%.
Example 14: [00216] (S)-Ethyl l-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5- yl)-l,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylate (89 mg, 0.16 mmol) was heated at 50 0C in 6N HCl (5 mL) in acetonitrile (5 mL). The reaction mixture was stirred overnight and then filtered and purified by HPLC. HPLC conditions:
PHENOMENEX® Luna C 18 5 micron column (250 x 30mm); 25-100% CH3CN/water (0.1% TFA); 25 minute gradient; 30 mL/min. Isolated fractions with correct mass were freeze-dried overnight to yield 36 mg of (S)-l-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4- (trifluoromethyl)isoxazol-5-yl)-l,2,4-oxadiazol-3-yl)phenyl)ethyl) piperidine-3- carboxylic acid as a TFA salt. 1H NMR (400 MHz, MeOH-d3) δ ppm 8.23 (2 H, d, J=8.35 Hz), 7.65-7.74 (4 H, m), 7.54-7.65 (3 H, m), 5.29 (1 H, t, J=7.03 Hz), 4.00 (1 H, br. s.), 3.43-3.75 (1 H, m), 3.34-3.41 (2 H, m), 2.82-3.24 (2 H, m), 2.26 (1 H, d, J=I 1.86 Hz), 1.84-2.14 (2 H, m), 1.52-1.75 (1 H, m). MS (m+1) = 529. HPLC Peak RT = 3.24 minutes. Purity = 98%. Example 14-Alternate Synthesis Route 1
Preparation 14D (Alternate Synthesis Route 1): (S)-4-(Oxiran-2-yl)benzonitrile
[00217] To 800 mL of 0.2M, pH 6.0 sodium phosphate buffer in a 2 L flask equipped with an overhead stirrer was added D-glucose (38.6 g, 1.2 eq), β-nicotinamide adenine dinucleotide, free acid (1.6 g, mmol), glucose dehydrogenase (36 mg, 3.2 kU,
CODEXIS® GDH- 102, 90 U/mg), and enzyme KRED-NADH-110 (200 mg,
CODEXIS®, 25 U/mg). The vessels containing the reagents above were rinsed with 200 mL of fresh sodium phosphate buffer and added to the reaction which was stirred to dissolution and then heated to 40 0C. To this mixture was added a solution of 2-bromo- 4′-cyanoacetophenone (40 g, 178.5 mmol) in 100 mL DMSO through an addition funnel in about 30 min. The container was rinsed with 20 mL DMSO and the rinse was added to the reactor. A pH of 5.5-6.0 was maintained by adding 1 M NaOH through a fresh addition funnel (total volume of 200 mL over 6h) after which HPLC showed complete consumption of the starting material. The reaction mixture was extracted with 800 mL MTBE x 2 and the combined extracts were washed with 300 mL of 25% brine. The crude alcohol was transferred to a 3L 3-neck flask and treated with solid NaOtBu (34.3 g, 357 mmol) stirring for 1 h and then additional NaOtBu (6.9 g, 357 mmol) and stirring for 30 min. The reaction mixture was filtered and the solution was washed with 300 mL 0.2 M pH 6.0 sodium phosphate buffer, brine, and then the solvent was removed in vacuo and the resulting white solid was dried in a vacuum oven to give (S)-4-(oxiran-2- yl)benzonitrile (23 g, 90% yield, 100% e.e.). 1H NMR (400 MHz, CDCl3) δ ppm 7.62 (2 H, d), 7.35 (2 H, d), 3.88 (1 H, dd), 3.18 (1 H, app t), 2.73 (1 H, dd) Purity = 99%.
[00218] Chiral HPLC was done on a CHIRALP AK® AD-RH 4.6x150mm (Daicel Chemical Industries Ltd.) column using gradient of solvent A (10 mM NH4OAc in water/acetonitrile, 90: 10) and solvent B (10 mM NH4OAc in water/acetonitrile, 10:90) with 70% to 90% in 40 min at a flow rate of 0.5 ml/min at ambient temperature. The detection employed UV at 235 nm. The retention times are as follows:
[00219] Peak 1 (Isomer A): RT = 16.7 min. for (S)-4-(oxiran-2-yl)benzonitrile
[00220] Peak 2 (Isomer B): RT = 14.0 min. for (R)-4-(oxiran-2-yl)benzonitrile Preparation of 14A-isomer A (Alternate Synthesis Route 1): (S)-Ethyl l-((S)-2-(4- cyanophenyl)-2 -hydroxy ethyl)piperidine-3-carboxylate
(14A)-isomer A
[00221] (S)-4-(Oxiran-2-yl)benzonitrile (10.00 g, 68.9 mmol), (S)-ethyl piperidine-3- carboxylate (10.83 g, 68.9 mmol) and iPrOH (100 mL) was charged into a round bottom flask under N2. After heating at 55 0C for 4 hours, 4-dimethylaminopyridine (1.683 g, 13.78 mmol) was then added. The reaction mixture was then heated to 50 0C for an additional 12 hours. At this time HPLC indicated the starting material was completely converted to the desired product. The reaction mixture was then cooled to room temperature. EtOAc (120 ml) was added, followed by 100 ml of water. The organic layer was separated, extracted with EtOAc (2x 100 mL) and concentrated under vacuo to give a crude product. The crude product was recrystallized from EtOH/EtOAc/H2O (3/2/2) (8ml/lg) to give a crystalline off-white solid 14A-alt (15 g, 72% yield, 99.6% e.e.). The absolute and relative stereochemistry was determined by single X-ray crystallography employing a wavelength of 1.54184 A. The crystalline material had an orthorhombic crystal system and unit cell parameters approximately equal to the following:
a = 5.57 A α = 90.0°
b = 9.7l A β = 90.0°
c = 30.04 A γ = 90.0°
Space group: P212121
Molecules/asymmetric unit: 2
Volume/Number of molecules in the unit cell = 1625 A3
Density (calculated) = 1.236 g/cm3
Temperature 298 K.
Preparation 14E (Alternate Route 1): (S)-Ethyl l-((S)-2-(tert-butyldimethylsilyloxy)-2- (4-cyanophenyl)ethyl)piperidine-3-carboxylate

[00222] To a mixture of (S)-ethyl 1 -((S)-2-(4-cyanophenyl)-2-hydroxy ethyl) piperidine-3-carboxylate (17.0 g, 56.2 mmol) and DIPEA (17.68 ml, 101 mmol) in CH2Cl2 (187 mL) was added tert-butyldimethylsilyl trifluoromethanesulfonate (16 ml, 69.6 mmol) slowly. The reaction was monitored with HPLC. The reaction completed in 2 hours. The reaction mixture (a light brown solution) was quenched with water, the aqueous layer was extracted with DCM. The organic phase was combined and dried with Na2SO4. After concentration, the crude material was further purified on a silica gel cartridge (33Og silica, 10-30% EtOAc/hexanes gradient) to afford a purified product (S)- ethyl 1 -((S)-2-(tert-butyldimethylsilyloxy)-2-(4-cyanophenyl)ethyl) piperidine-3 – carboxylate (22.25 g, 53.4 mmol, 95 % yield). 1H NMR (400 MHz, CDCl3) δ ppm 7.61 (2 H, d), 7.45 (2 H, d), 4.79 (1 H, m), 4.15 (2 H, m), 2.88 (1 H, m), 2.75 (1 H, m), 2.60 (1 H, dd), 2.48 (1 H, m), 2.40 (1 H, dd), 2.33 (1 H, tt), 2.12 (1 H, tt), 1.90 (1 H, m), 1.68 (1 H, dt), 1.52 (1 H, m), 1.48 (1 H, m), 1.27 (3 H, t), 0.89 (9 H, s), 0.08 (3 H, s), -0.07 (3 H, s).
Preparation 14F (Alternate Route 1): (S)-Ethyl l-((S)-2-(tert-butyldimethylsilyloxy)-2- (4-((Z)-N’-hydroxycarbamimidoyl)phenyl)ethyl)piperidine-3-carboxylate
[00223] (S)-Ethyl- 1 -((S)-2-(tert-butyldimethylsilyloxy)-2-(4-cyanophenyl)ethyl) piperidine-3-carboxylate (31.0 g, 74.4 mmol) was dissolved in EtOH (248 mL).
Hydroxylamine (50% aq) (6.84 ml, 112 mmol) was added and stirred at room temperature overnight. Then all volatiles were removed with ROTA VAPOR®. The residue was purified with on a silica gel cartridge (33Og silica, 0-50% EtOAc/hexanes gradient) to give (S)-ethyl l-((S)-2-(tert-butyldimethylsilyloxy)-2-(4-((Z)-N’- hydroxycarbamimidoyl)phenyl)ethyl)piperidine-3-carboxylate (31 g, 68.9 mmol, 93 % yield) as a white foam. 1H NMR (400 MHz, CDCl3) δ ppm 8.38 (1 H, br s), 7.58 (2 H, d), 7.37 (2 H, d), 4.88 (2 H, br s), 4.81 (1 H, m), 4.13 (2 H, m), 2.96 (1 H, m), 2.82 (1 H, m), 2.61 (1 H, dd), 2.51 (1 H, m), 2.42 (1 H, dd), 2.32 (1 H, tt), 2.13 (1 H, dt), 1.91 (1 H, m), 1.66 (1 H, dt), 1.58 (1 H, m), 1.48 (1 H, m), 1.27 (3 H, t), 0.89 (9 H, s), 0.08 (3 H, s), -0.09 (3 H, s). Preparation 14G (Alternate Route 1): (S)-Ethyl l-((S)-2-(tert-butyldimethylsilyloxy)-2- (4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-l,2,4-oxadiazol-3- yl)phenyl)ethyl)piperidine-3-carboxylate

[00224] (S)-Ethyl- 1 -((S)-2-(tert-butyldimethylsilyloxy)-2-(4-((Z)-N’- hydroxycarbamimidoyl)phenyl)ethyl)piperidine-3-carboxylate (32.6g, 72.5 mmol) was dissolved in acetonitrile (145 ml) (anhydrous) and cooled to ~3 0C with ice-bath. 3- phenyl-4-(trifluoromethyl)isoxazole-5-carbonyl chloride (19.98 g, 72.5 mmol) was dissolved in 5OmL anhydrous acetonitrile and added dropwise. The internal temperature was kept below 10 0C during addition. After addition, the reaction mixture was allowed to warm to room temperature. At 30 minutes, HPLC showed completion of the first reaction step. The reaction mixture was re-cooled to below 10 0C. DIEA (18.99 ml, 109 mmol) was added slowly. After the addition, the reaction mixture was heated up to 55 0C for 17 hr s. HPLC/LCMS showed completion of the reaction. The solvents were removed by ROTA VAPOR®. The residue was stirred in 25OmL 20% EtOAc/hexanes and the DIPEA HCl salt precipitated from solution and was removed via filtration. The filtrate was concentrated and purified using a silica gel cartridge (3X33Og silica, 0-50%
EtOAc/hexanes gradient). (S)-ethyl l-((S)-2-(tert-butyldimethylsilyloxy)-2-(4-(5-(3- phenyl-4-(trifluoromethyl)isoxazol-5-yl)-l,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3- carboxylate (43g, 64.1 mmol, 88 % yield) was obtained a light yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm 8.16 (2 H, d), 7.68 (2 H, d), 7.57 (5 H, m), 4.85 (1 H, m), 4.14 (2 H, m), 2.95 (1 H, m), 2.82 (1 H, m), 2.64 (1 H, dd), 2.51 (1 H, m), 2.49 (1 H, dd), 2.35 (1 H, tt), 2.14 (1 H, dt), 1.91 (1 H, m), 1.66 (1 H, dt), 1.57 (1 H, m), 1.48 (1 H, m), 1.27 (3 H, t), 0.92 (9 H, s), 0.11 (3 H, s), -0.05 (3 H, s).
Example 14 (Alternate Route 1): (S)-l-((S)-2-Hydroxy-2-(4-(5-(3-phenyl-4- (trifluoromethyl)isoxazol-5-yl)-l,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3- carboxylic acid

[00225] (S)-Ethyl l-((S)-2-(tert-butyldimethylsilyloxy)-2-(4-(5-(3-phenyl-4- (trifluoromethyl)isoxazol-5-yl)-l,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3- carboxylate (42g, 62.6 mmol) was dissolved in dioxane (150 ml) and treated with 6M HCl (150 ml). The reaction mixture was heated to 65 0C for 6 hours (the reaction was monitored with HPLC, EtOH was distilled out to push the equilibrium forward). Dioxane was removed and the residue was redissolved in ACN/water and lyophilized separately to give crude (S)-l-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl) isoxazol-5-yl)- l,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic acid, HCl, (37g crude foamy solid). The crude solid (36 g, 63.7 mmol) was suspended in acetonitrile (720 mL) and heated to 60 0C and water (14.4 mL) was added dropwise. A clear solution was obtained, which was cooled to room temperature and concentrated to a viscous oil, treated with ethyl acetate (1.44 L) with vigorously stirring, heated to 60 0C, and cooled to room temperature. (S)-l-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)- l,2,4-oxadiazol-3-yl)phenyl)ethyl) piperidine-3-carboxylic acid, HCl (28g, 49.3 mmol, 77 % yield) was collected and vacuum dried. Characterization of product by 1H NMR and chiral HPLC matched Example 14 prepared in previous synthesis.
Preparation of Intermediate (14A)-isomer A-Alternate Route 2; 2-Steps: (S)-Ethyl 1- ((S)-2-(4-cyanophenyl)-2-hydroxyethyl)piperidine-3-carboxylate
(14A)-isomer A
Step 1 : Preparation (14D) (Alternate Route 2): (S)-Ethyl l-(2-(4-cyanophenyl)-2- oxoethyl)piperidine-3-carboxylate hydrobromide
(14D)-isomer A
[00226] To a solution of commercially available (S)-ethyl piperidine-3-carboxylate (10 g, 63.6 mmol) in 200 mL toluene was added 4-(2-bromoacetyl)benzonitrile (17g, 76 mmol). The reaction mixture was stirred overnight. The next day, the precipitated solid was collected by filtration and washed with ethyl acetate (x3) and dried under vacuum to give 15.2g of (S)-ethyl l-(2-(4-cyanophenyl)-2-oxoethyl)piperidine-3-carboxylate hydrobromide. MS (M+ 1) = 301. HPLC Peak RT = 1.51 minutes.
Step 2: Preparation of 14 A-isomer A (Alternate Route 2): (S)-Ethyl l-((S)-2-(4- cyanophenyl)-2-hydroxyethyl)piperidine-3 -carboxylate
[00227] Phosphate buffer (1100 mL, BF045, pH 7.0, 0. IM) was added into two liter jacketed glass reactor. The temperature of the reactor was adjusted to 20 0C with the help of a circulator and the reaction mixture was stirred with a magnetic stirrer. Dithiothretol (185.2 mg, 1 mM), magnesium sulfate (288.9 mg, 2 mM), and D-glucose (11.343 g, 62.95 m moles) were added into the reactor. (5*)-Ethyl l-(2-(4-cyanophenyl)-2-oxoethyl) piperidine-3 -carboxylate HBr salt (12 g, 31.47 m moles dissolved in 60 mL DMSO) was added into the reactor slowly with continuous stirring, β-nicotinamide adenine dinucleotide phosphate sodium salt (NADP), 918.47 mg, glucose dehydrogenase, 240 mg (total 18360 U, 76.5 U/mg, ~ 15U/mL, Amano Lot. GDHY1050601) and KRED-114, 1.2 g (CODEXIS® assay 7.8 U/mg of solid), were dissolved in 2.0 mL, 2.0 mL and 10 ml of the same buffer, respectively. Next, NADP, GDH and KRED-114 were added to the reactor in that order. The remaining 26 mL of same buffer was used to wash the NADP, GDH and KRED-114 containers and buffer was added into the same reactor. The starting pH of the reaction was 7.0 which decreased with the progress of the reaction and was maintained at pH 6.5 during the course of the reaction (used pH stat, maintained with IM NaOH). The reaction was run for 4.5 hours and immediately stopped and extracted with ethyl acetate. The ethyl acetate solution was evaporated under reduced pressure and weight of the dark brown residue was 12.14 g. The product was precipitated with dichloromethane and heptane to give 9 g of crude product which was further purified by dissolving it in minimum amount of dichloromethane and re-precipitating by the addition of excess amount of heptane to give 5.22 g. The process was repeated to give an additional 2.82 g of highly pure product for a total of 8.02 g of de > 99.5%.
[00228] Chiral HPLC was done on a CHIRALP AK® AD-RH 4.6x150mm (Daicel Chemical Industries Ltd.) column using gradient of solvent A (10 mM NH4OAc in water/acetonitrile, 90: 10) and solvent B (IO mM NH4OAc in water/acetonitrile, 10:90) with 70% to 90% in 40 min at a flow rate of 0.5 ml/min at ambient temperature. The detection was done by UV at 235 nm. The retention times are as follows: [00229] Peak 1 (14A-isomer A): RT = 20.7 min. for (S)-ethyl l-((S)-2-(4- cyanophenyl)-2-hydroxyethyl)piperidine-3-carboxylate.
[00230] Peak 2 (14B-isomer B): RT = 30.4 min. for (S)-ethyl l-((R)-2-(4- cyanophenyl)-2-hydroxyethyl)piperidine-3-carboxylate.
[00231] Compound 14A-isomer A prepared using this asymmetric method was unambiguously assigned since it was identical to the 14A-isomer A (by 1H NMR and chiral HPLC retention time) that was prepared above and determined by X-ray crystallography. Synthesis of Example 14 from this material followed the same route as described above.
paper
Regioselective Epoxide Ring Opening for the Stereospecific Scale-Up Synthesis of BMS-960, A Potent and Selective Isoxazole-Containing S1P1Receptor Agonist
Xiaoping Hou*†  , Huiping Zhang†, Bang-Chi Chen†, Zhiwei Guo‡, Amarjit Singh‡, Animesh Goswami‡, John L. Gilmore†, James E. Sheppeck†, Alaric J. Dyckman† , Percy H. Carter†, and Arvind Mathur†
, Huiping Zhang†, Bang-Chi Chen†, Zhiwei Guo‡, Amarjit Singh‡, Animesh Goswami‡, John L. Gilmore†, James E. Sheppeck†, Alaric J. Dyckman† , Percy H. Carter†, and Arvind Mathur†
† Discovery Chemistry, Bristol-Myers Squibb, Princeton, New Jersey 08540, United States
‡ Chemical & Synthetic Development, Bristol-Myers Squibb, New Brunswick, New Jersey 08903, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.6b00366

This article presents a stereospecific scale-up synthesis of (S)-1-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic acid (BMS-960), a potent and selective isoxazole-containing S1P1 receptor agonist. The process highlights an enzymatic reduction of α-bromoketone toward the preparation of (S)-bromo alcohol, a key precursor of (S)-4-(oxiran-2-yl)benzonitrile. A regioselective and stereospecific epoxide ring-opening reaction was also optimized along with improvements to 1,2,4-oxadiazole formation, hydrolysis, and crystallization. The improved process was utilized to synthesize batches of BMS-960 for Ames testing and other toxicological studies.
PAPER
Journal of Medicinal Chemistry (2016), 59(13), 6248-6264.
Discovery and Structure–Activity Relationship (SAR) of a Series of Ethanolamine-Based Direct-Acting Agonists of Sphingosine-1-phosphate (S1P1)
John L. Gilmore*, James E. SheppeckII, Scott H. Watterson, Lauren Haque, Parag Mukhopadhyay, Andrew J. Tebben, Michael A. Galella, Ding Ren Shen, Melissa Yarde, Mary Ellen Cvijic, Virna Borowski, Kathleen Gillooly, Tracy Taylor, Kim W. McIntyre, Bethanne Warrack, Paul C. Levesque, Julia P. Li, Georgia Cornelius, Celia D’Arienzo, Anthony Marino, Praveen Balimane, Luisa Salter-Cid, Joel C. Barrish, William J. Pitts, Percy H. Carter, Jenny Xie, and Alaric J. Dyckman
Research and Development, Bristol-Myers Squibb, P.O. Box 4000, Princeton, New Jersey 08543, United States
J. Med. Chem., 2016, 59 (13), pp 6248–6264
DOI: 10.1021/acs.jmedchem.6b00373
Abstract

Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid metabolite that regulates a multitude of physiological processes such as lymphocyte trafficking, cardiac function, vascular development, and inflammation. Because of the ability of S1P1 receptor agonists to suppress lymphocyte egress, they have great potential as therapeutic agents in a variety of autoimmune diseases. In this article, the discovery of selective, direct acting S1P1 agonists utilizing an ethanolamine scaffold containing a terminal carboxylic acid is described. Potent S1P1 agonists such as compounds 18a and 19a which have greater than 1000-fold selectivity over S1P3 are described. These compounds efficiently reduce blood lymphocyte counts in rats through 24 h after single doses of 1 and 0.3 mpk, respectively. Pharmacodynamic properties of both compounds are discussed. Compound 19a was further studied in two preclinical models of disease, exhibiting good efficacy in both the rat adjuvant arthritis model (AA) and the mouse experimental autoimmune encephalomyelitis model (EAE).
BASE
(S)-1-((S)-2-Hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl) isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic Acid (18a)
(S)-ethyl 1-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylate (36%).
1H NMR (400 MHz, MeOH-d3) δ ppm 8.04 (2 H, d, J = 8.13 Hz), 7.55–7.60 (2 H, m), 7.41–7.54 (5 H, m), 4.81 (1 H, ddd, J = 8.35, 4.06, 3.84 Hz), 3.96–4.10 (2 H, m), 2.82–3.08 (1 H, m), 2.67–2.82 (1 H, m), 2.36–2.61 (3 H, m), 2.08–2.33 (2 H, m), 1.73–1.87 (1 H, m, J = 8.54, 8.54, 4.45, 4.17 Hz), 1.32–1.70 (3 H, m), 1.09–1.19 (3 H, m).
MS (M + H)+ at m/z 557. HPLC purity: 99%, tr = 3.36 min (method B).
TFA salt
(S)-1-((S)-2-hydroxy-2-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)phenyl)ethyl)piperidine-3-carboxylic acid, TFA salt (18a, 61%) as a white solid.
1H NMR (400 MHz, MeOH-d3) δ ppm 8.23 (2 H, d, J = 8.35 Hz), 7.65–7.74 (4 H, m), 7.54–7.65 (3 H, m), 5.29 (1 H, t, J = 7.03 Hz), 4.00 (1 H, br s), 3.43–3.75 (1 H, m), 3.34–3.41 (2 H, m), 2.82–3.24 (2 H, m), 2.26 (1 H, d, J = 11.86 Hz), 1.84–2.14 (2 H, m), 1.52–1.75 (1 H, m).
MS (M + H)+ at m/z 529.
HPLC tr = 3.27 min (method B). HPLC purity: 99.4%, tr = 8.78 min (method E); 99.0%, tr = 7.29 min (method F).
HCL SALT
This material was converted to the HCl salt for the following analyses: mp: 219.2 °C. Anal. Calcd for C26H23N4O5F3·HCl: 0.14% water: C, 55.2; H, 4.31; N, 9.87; Cl, 6.25. Found: C, 55.39; H, 4.10; N, 9.88; Cl, 6.34. [α]D20 + 30.47 (c 0.336, MeOH). HPLC with chiral stationary phase (A linear gradient using CO2 (solvent A) and IPA with 0.1% DEA (solvent B); t = 0 min, 30% B, t = 10 min, 55% B was employed on a Chiralcel AD-H 250 mm × 4.6 mm ID, 5 μm column; flow rate was 2.0 mL/min): tr = 5.38 min with >99% ee.
References
Gilmore, J. L.; Sheppeck, J. E.; Watterson, S. H.; Haque, L.; Mukhopadhyay, P.; Tebben, A. J.; Galella, M. A.; Shen, D. R.; Yarde, M.; Cvijic, M. E.; Borowski, V.; Gillooly, K.; Taylor, T.; McIntyre, K. W.; Warrack, B.; Levesque, P. C.; Li, J. P.; Cornelius, G.; D’Arienzo, C.; Marino, A.; Balimane, P.; Salter-Cid, L.; Barrish, J. C.; Pitts, W. J.; Carter, P. H.; Xie, J.; Dyckman, A. J.Discovery and Structure Activity Relationship (SAR) of a Series of Ethanolamine-Based Direct-Acting Agonists of Sphingosine-1-Phosphate (S1P1) J. Med. Chem. 2016, 59, 6248– 6264, DOI: 10.1021/acs.jmedchem.6b00373
Gilmore, J. L.; Sheppeck, J. E. Preparation of 3-(4-(1-hydroxyethyl)phenyl)-1,2,4-oxadiazole derivatives as sphingosine-1-phosphate receptor agonists for the treatment of autoimmune disease and inflammation. PCT Int. Appl. 2011, WO 2011017578.
//////BMS-960, PRECLINICAL, BMS 960
Cl.O=C(O)[C@H]1CCCN(C1)C[C@@H](O)c2ccc(cc2)c3nc(on3)c5onc(c4ccccc4)c5C(F)(F)F

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....





























































































































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..