



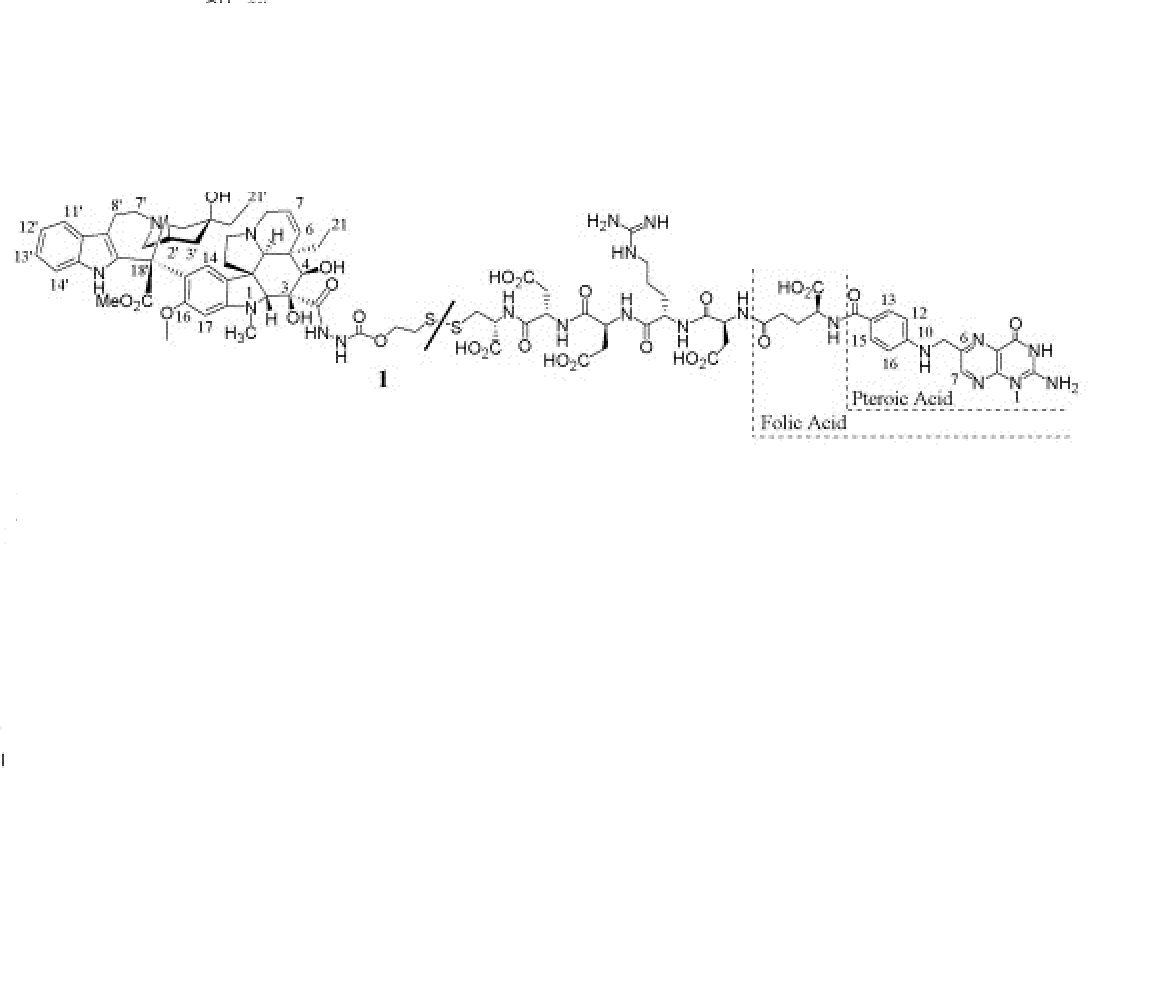
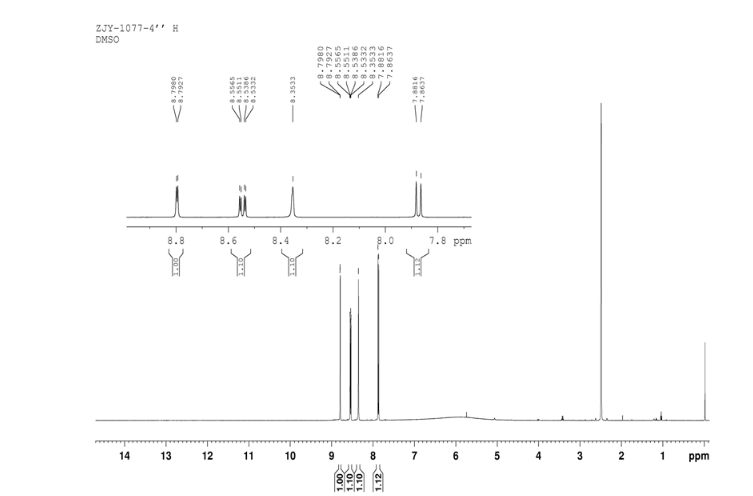
 4 nmr dmsod6
4 nmr dmsod6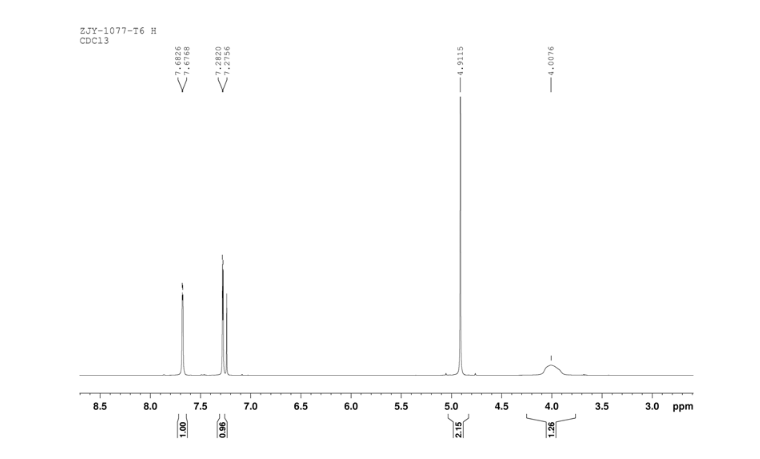 6 in dmsod6 1H NMR
6 in dmsod6 1H NMR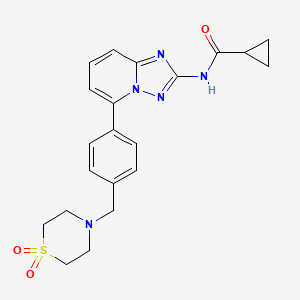
| Patent | Submitted | Granted |
|---|---|---|
| Compound useful for the treatment of degenerative and inflammatory diseases [US8088764] | 2010-12-30 | 2012-01-03 |
| NOVEL COMPOUNDS USEFUL FOR THE TREATMENT OF DEGENERATIVE AND INFLAMMATORY DISEASES [US2011190260] | 2011-08-04 |
Home » Phase2 drugs (Page 15)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sparsentan, PS433540, RE-021

Sparsentan (PS433540, RE-021)
- C32H40N4O5S
- Average mass592.749
FDA APPROVED 2023/2/17, Filspari
4′-((2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl)-N-(4,5-dimethylisoxazol-3-yl)-2′-(ethoxymethyl)-[1,1′-biphenyl]-2-sulfonamide
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide
Sparsentan
PS433540; RE-021, formerly known as DARA
CAS :254740-64-2
4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(4,5- dimethylisoxazol-3-yl)-2-(ethoxymethyl)biphenyl-2-sulfonamide
Mechanism of Action:acting as both an Endothelin Receptor Antagonist (ERA) and Angiotensin Receptor Blocker (ARB).
Indication: Focal Segmental Glomerulosclerosis (FSGS).Focal Segmental Glomerulosclerosis (FSGS) is a rare and severe nephropathy which affects approximately 50,000 patients in the United States. Most cases of FSGS are pediatric.
Development Stage: Phase II
Developer:Retrophin, Inc
- OriginatorBristol-Myers Squibb
- DeveloperRetrophin
- ClassAntihypertensives; Isoxazoles; Small molecules; Spiro compounds; Sulfonamides
- Mechanism of ActionAngiotensin type 1 receptor antagonists; Endothelin A receptor antagonists
- Orphan Drug Status Yes – Focal segmental glomerulosclerosis
-
- 09 Jan 2015 Sparsentan receives Orphan Drug status for Focal segmental glomerulosclerosis in USA
- 31 Dec 2013 Phase-II/III clinical trials in Focal segmental glomerulosclerosis in USA (PO)
- 07 May 2012I nvestigation in Focal segmental glomerulosclerosis in USA (PO)
Sparsentan is an investigational therapeutic agent which acts as both a selective endothelin receptor antagonist and an angiotensin receptor blocker. Retrophin is conducting the Phase 2 DUET trial of Sparsentan for the treatment of FSGS, a rare and severe nephropathy that is a leading cause of end-stage renal disease. There are currently no therapies approved for the treatment of FSGS in the United States. Ligand licensed worldwide rights of Sparsentan (RE-021) to Retrophin in 2012 .The Food and Drug Administration (FDA) has granted orphan drug designation for Retrophins sparsentan for the treatment of focal segmental glomerulosclerosis (FSGS) in January 2015.
In 2006, the drug candidate was licensed to Pharmacopeia by Bristol-Myers Squibb for worldwide development and commercialization. In 2012, a license was obtained by Retrophin from Ligand. In 2015, Orphan Drug Designation was assigned by the FDA for the treatment of focal segmental glomerulosclerosis.
Sparsentan, also known as RE-021, BMS346567, PS433540 and DARA-a, is a Dual angiotensin II and endothelin A receptor antagonist. Retrophin intends to develop RE-021 for orphan indications of severe kidney diseases including Focal Segmental Glomerulosclerosis (FSGS) as well as conduct proof-of-concept studies in resistant hypertension and diabetic nephropathy. RE-021, with its unique dual blockade of angiotensin and endothelin receptors, is expected to provide meaningful clinical benefits in mitigating proteinuria in indications where there are no approved therapies
Sparsentan, sold under the brand name Filspari, is a medication used for the treatment of primary immunoglobulin A nephropathy.[1] Sparsentan is an endothelin and angiotensin II receptor antagonist.[1][4] It is taken by mouth.[1]
The most common side effects include swelling of the extremities, low blood pressure, dizziness, high blood potassium, anemia, injury to the kidney, and increased liver enzymes in the blood.[5]
It was approved for medical use in the United States in February 2023.[5][6][7] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[8]
PATENT
WO 2000001389
https://www.google.co.in/patents/WO2000001389A1?cl=en


Example 41
4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.4! non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l, l’-biphenyl! -2-sulfonamide

A. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl)-N-[(2-trimethylsilylethoxy)methyl]-2′- hydroxym ethyl [1, l’-biphenyl] -2-sulfonamide P14 (243 mg, 0.41 mmol) was used to alkylate 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene hydrochloride according to General Method 4. 41A (100 mg, 35% yield) was isolated as a slightly yellow oil after silica gel chromatography using 1:1 hexanes/ethyl acetate as eluant. B. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methvn -N-0.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l,l’-biphenyn-2- sulfonamide
Deprotection of 41A (100 mg, 0.14 mmol) according to General Method 8 (ethanol) gave the title compound as white solid in 46% yield following silica gel chromatography (96:4 methanol/chloroform eluant):
MS m/e 565 (ESI+ mode); HPLC retention time 3.21 min (Method A);
HPLC purity >98%.
Example 42
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide

A. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3 ,4- dimethyl-5-isoxazolyl)-N-[(2-methoxyethoxy)methyll-2′- hvdroxym ethyl [1 , l’-biphenyl] -2-sulfonamide
Triethylsilane (6 ml) and TFA (6 ml) were added to a solution of 5F (960 mg, 1.5 mmol) in 15 ml dichloromethane at RT. The mixture was stirred at RT for 2 h and was then concentrated. The residue was taken up in ethyl acetate and was washed successively with aqueous sodium bicarbonate, water, and brine. The organic layer was dried over sodium sulfate and concentrated. The residue was chromatographed on silica gel using 100:2 dichloromethane/methanol to afford 42A (740 mg, 77%) as a colorless gum. Rf=0.13, silica gel, 100:5 dichloromethane/methanol. B. 4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-N-r(2-methoxyethoxy)methyll-2′- ethoxymethyl[l.l’-biphenyll-2-sulfonamide A mixture of 42A (100 mg, 0.15 mmol), iodoethane (960 mg, 6.1 mmol) and silver (I) oxide (180 mg, 0.77 mmol) in 0.7 ml DMF was heated at 40 ° C for 16 h.. Additional iodoethane (190 mg, 1.2 mmol) and silver (I) oxide (71 mg, 0.31 mmol) were added and the reaction mixture was heated at 40 ° C for an additional 4 h. The mixture was diluted with 1:4 hexanes/ethylacetate and was then washed with water and brine. The organic layer was dried over sodium sulfate and was then concentrated. The residue was chromatographed on silica gel using 200:3 dichloromethane/methanol as eluant to afford 42B (51mg, 49%) as a colorless gum. Rf=0.35, silica gel, 100:5 dichloromethane/methanol.
C. 4,-[(2-Butyl-4-oxo-1.3-diazaspirof4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl )-2′-ethoxym ethyl [ 1. l’-biphenyll -2-sulfonamide
42B (51 mg) was deprotected according to General Method 7 to afford the title compound in 80% yield following preparative reverse-phase HPLC purification: white solid; m.p. 74-80 ° C (amorphous); IH NMR (CDCL, )δ0.87(tr, J=7Hz, 3H), 0.99(tr, J=7Hz, 3H), 1.32(m, 2H), 1.59(m, 2H), 1.75-2.02(m, 11H), 2.16(s, 3H), 2.35(m, 2H), 3.38 (m, 2H), 4.23(m, 2H), 4.73(s, 2H), 7.11-7.85 (m, 7H); MS m/e 593 (ESI+ mode); HPLC retention time 18.22 min. (Method E); HPLC purity >97%.
PATENT
WO 2001044239
http://www.google.co.in/patents/WO2001044239A2?cl=en
……………………
Dual angiotensin II and endothelin A receptor antagonists: Synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides with improved potency and pharmacokinetics
J Med Chem 2005, 48(1): 171
J. Med. Chem., 2002, 45 (18), pp 3829–3835
DOI: 10.1021/jm020138n
 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPDThe ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a DIFFERENT COMPD
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
PATENT
WO 2010135350
http://www.google.com/patents/WO2010135350A2?cl=en
Compound 1 :

Scheme IV

Scheme V

Formula IV 1
Scheme VII

Formula Vl

A solution of 2-(2,4-dimethylphenyl)benzenesulfonic acid (Compound 12) (0.5 g, 1.9 mmol) in 50 mL of anhydrous acetonitrile was prepared and transferred to a round-bottom flask. After flushing with nitrogen gas, N-bromosuccinimide (0.75 g, 4.2 mmol) was added followed by 50 mg (0.2 mmol) of benzoyl peroxide. The solution was heated at reflux for 3 hours. The solvent was removed in-vacuo and the resulting syrup purified by silica gel chromatography (1 :1 hexanes/EtOAc) to yield Compound 13 as a white solid. 1H NMR (500 MHz, CD3CN) 8.12 (d, J = 7.5 Hz, IH), 7.92 (t, J = 7.5 Hz, IH), 7.78 (d, J= 7.5 Hz, IH), 7.74-7.71 (m, 2H), 7.68-7.65 (m, 2H), 5.12 (s, 2H), 4.70 (s, 2H). Example 4 2-(4-Bromomethyl-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 14)

A solution of 20 mg (0.058 mmol) of (l-bromomethylbenzo[3,4- d])benzo[l,2-f]-2-oxa-l,l-dioxo-l-thiocycloheptane (Compound 13) in ethanol was stirred at elevated temperature until the starting material was consumed to give crude product (compound 14) that was used directly in the next step without isolation or purification.
Example 5
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonic acid (Compound 15)

To the above ethanol solution of crude 2-(4-bromomethyl-2- ethoxymethylphenyl)benzenesulfonic acid (Compound 14) described in Example 4 was added approximately 25 mL of anhydrous DMF. The ethanol was removed from the system under reduced pressure. Approximately 15 mg (0.065 mmol) of 2-butyl-l,3- diazaspiro[4.4]non-l-en-4-one (compound 7 in Scheme IV) was added followed by 300 μL of a IM solution of lithium bis-trimethylsilylamide in THF. The solution was allowed to stir at room temperature for 3 hours. The solvents were removed under reduced pressure and the remaining residue purified by preparative RP-HPLC employing a Cl 8 column and gradient elution (H2O:MeCN) affording the title compound as a white solid; [M+H]+ calcd for C27H34N2O5S 499.21, found, 499.31 ; 1H NMR (500 MHz, CD3CN) 8.04 (t, J= 5.5 Hz, IH), 7.44-7.10 (m, 2H), 7.28 (s, IH), 7.22 (d, J= 8.0 Hz, 2H), 7.08- 7.04 (m, 2H), 4.74 (br s, 2H), 4.32 (d, J= 13.0 Hz IH), 4.13 (d, J= 13.0 Hz IH), 3.40- 3.31 (m, 2H), 2.66 (t, J= 8 Hz, 2H), 2.18-2.13 (m, 5H), 1.96-1.90 (m, 2H obscured by solvent), 1.48 (m, 2H), 1.27 (s, J= 7 Hz, 2H), 1.16 (t, J= 7 Hz, 3H), 0.78 (t, J= 7.5 Hz, 3H).
Example 6
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16)

To a solution of DMF (155 μL, 2 mmol, 2 equiv.) in dichloromethane (5 mL) at 0 0C was added dropwise oxalyl chloride (175 μL, 2 mmol, 2 equiv.) followed by a dichloromethane (5 mL) solution of 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l- en-3-yl)methyl)-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 15) (0.50 g, 1.0 mmol). The resulting mixture was stirred at 0 0C for ~2 hours, diluted with additional dichloromethane (25 mL), washed with saturated sodium bicarbonate solution (10 mL), water (10 mL), and brine (10 mL), dried over sodium sulfate, and then concentrated to give crude sulfonyl chloride (compound 16) that was used without purification.
Example 7
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

[0062] To a solution of 5-amino-3,4-dimethylisoxazole (60 mg, 0.54 mmol) in THF at -60 °C was added dropwise potassium tert-butoxide (1 mL of 1 M solution) followed by a solution of crude 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3- yl)methyl)-2-ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16) (0.28 g, 0.54 mmol) in THF (4 mL). The resulting mixture was stirred at about -60 °C for 1 hour, allowed to warm to room temperature overnight, and then quenched with IN HCl solution to about pH 4. Standard workup of extraction with ethyl acetate, washing with water, drying, and concentration provided the final compounds as a white solid. 1H NMR (400 MHz, CDCl3) 8.03 (dd, J = 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J = 7.5, 2H), 1.37 (sextet, J = 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
Example 8 l-Bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17)

To a solution of ethyl 4-bromo-3-ethoxymethylbenzoate (9.4 g, 33 mmol) in toluene (56 mL) at about -10 0C was added 51 g of a 20% diisobutylaluminum hydride solution in toluene (ca. 70 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy 5% NaOH solution to keep the temperature below about 10 °C. Organic phase of the resulting mixture was separated and the aqueous phase was extracted with toluene. The combined organic phase was concentrated in vacuo to a final volume of ~60 mL toluene solution of l-bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) that was used in next step without purification.
Example 9 l-Bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18)

To a solution of 1 -bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) (8.4 g, 33 mmol) in toluene (60 mL) prepared in Example 8 at about -10 °C was added methanesulfonyl chloride (7.9 g, 68 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy water to keep the temperature at about 0 °C. The organic layer was separated and washed again with icy water to provide a crude product solution of 1 – bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18) that was used without purification.
Example 10
1 -Bromo-4-((2-butyl-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 -yl)methy l)-2- ethoxymethylbenzene bisoxalic acid salt (Compound 19)

To the crude solution of 1 -bromo-2-ethoxymethyl-4- methanesulfonyloxymethylbenzene (Compound 18) (1 1 g, 33 mmol) in toluene (80 mL) prepared in Example 9 was added a 75% solution of methyltributylammonium chloride in water (0.47 mL). The resulting mixture was added to a solution of 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene (compound 7 in Scheme VI) (7.5 g, 32 mmol) in dichloromethane (33 mL) pretreated with a 10 M NaOH solution (23 mL). The reaction mixture was stirred at room temperature for 2 hours until compound 18 was not longer detectable by HPLC analysis and then was quenched with water (40 mL). After stirring about 10 minutes, the organic layer was separated and aqueous layer was extracted with toluene. The combined organic phase was washed with water and concentrated to a small volume. Filtration through a silica gel pad using ethyl acetate as solvent followed by concentration yielded 1 -bromo-4-((2-buty 1-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 – yl)methyl)-2-ethoxymethylbenzene as a crude oil product.
The crude oil was dissolved in ethyl acetate (22 mL) and warmed to around 50 °C. Anhydrous oxalic acid (4.6 g) was added to the warm solution at once and the resulting mixture was stirred until a solution was obtained. The mixture was cooled gradually and the bisoxalic acid salt (compound 19) was crystallized. Filtration and drying provided pure product (compound 19) in 50-60% yield from ethyl 4-bromo-3- ethoxymethylbenzoate in 3 steps. 1H NMR (400 MHz, CDCl3) 12.32 (bs, 4H), 7.58 (d, J = 7.8, IH), 7.36 (s, IH), 7.12 (d, J= 7.8, IH), 4.90 (s, 2H), 4.56 (s, 2H), 3.68 (q, J= 7.5, 2H), 2.87-2.77 (m, 2H), 2.40-1.95 (m, 8H), 1.62-1.53 (m, 2H), 1.38-1.28 (m, 4H), and 1.82 (t, J= 7.5, 3H).
Example 11
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

To a suspension of l-bromo-4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non- l-en-3-yl)methyl)-2-ethoxymethylbenzene bisoxalic acid salt (Compound 19) (5.0 g, 8.3 mmol) in toluene (20 niL) under nitrogen was added water (30 mL) and pH was adjusted to 8-9 by addition of a 2 M NaOH solution at room temperature. The organic phase was separated and mixed with 2-(N-(3,4-dimethyl-5-isoxazolyl)-N- methoxymethylamino)sulfonylphenylboronic acid pinacol ester (Scheme VII, Formula IX, where R8is methoxymethyl and M = boronic acid pinacol ester) (3.6 g, 8.5 mmol), bis(dibenzylideneacetone)palladium(0) (Pd(dba)2) (0.12 g), and a standard phosphine ligand. After a 2 M sodium carbonate solution was added, the reaction mixture was warmed to 70 0C and stirred until the reaction was complete by HPLC analysis. The reaction was cooled to room temperature and quenched with water, and then separated in phases. The organic phase was treated with activated carbon, filtered through a pad of silica gel, and was concentrated to afford a crude mixture.
The crude reaction mixture was dissolved in ethanol (40 mL) after palladium catalyst was removed and was treated with 6 M HCl solution (ca. 40 mL). The mixture was warmed to 75-80 °C and stirred for about 2 hours until the reaction was completed by HPLC analysis. After the mixture was cooled to room temperature, the pH of the mixture was adjusted to 8 by addition of 10 M NaOH solution. The mixture was stirred for 2 more hours and the pH was adjusted to 6 by adding 2 M HCl and the crystal seeds. Filtration of the crystalline solid followed by drying provided N-(3,4-dimethyl-5- isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en-3yl)methyl-2- ethoxymethylphenyl)phenylsulfonamide (Compound 1) as a white solid.1H NMR (400 MHz, CDCIa) 8.03 (dd, J= 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J= 7.5, 2H), 1.37 (sextet, J= 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
| US20040002493 * | Aug 20, 2001 | Jan 1, 2004 | Kousuke Tani | Benzoic acid derivatives and pharmaceutical agents comprising the same as active ingredient |
| US20070054806 * | Sep 6, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Novel sulfonamide-comprising solid formulations |
| US20070054807 * | Sep 8, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Storage-stable formulations of sulfonamides |
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Filspari |
| Other names | RE-021, PS433540 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623018 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.275.317 |
| Chemical and physical data | |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
References
- ^ Jump up to:a b c d e f “Filspari- sparsentan tablet, film coated”. DailyMed. 17 February 2023. Retrieved 6 March 2023.
- ^ Jump up to:a b c d “Filspari EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Filspari Product information”. Union Register of medicinal products. 23 April 2024. Retrieved 7 September 2024.
- ^ Chiu AW, Bredenkamp N (September 2023). “Sparsentan: A First-in-Class Dual Endothelin and Angiotensin II Receptor Antagonist”. The Annals of Pharmacotherapy. 58 (6): 645–656. doi:10.1177/10600280231198925. PMID 37706310. S2CID 261743204.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Drug Trials Snapshots: Filspari”. U.S. Food and Drug Administration (FDA). 17 February 2023. Retrieved 7 September 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Travere Therapeutics Announces FDA Accelerated Approval of Filspari (sparsentan), the First and Only Non-immunosuppressive Therapy for the Reduction of Proteinuria in IgA Nephropathy” (Press release). Travere Therapeutics. 17 February 2023. Retrieved 17 February 2023 – via GlobeNewswire.
- ^ Syed YY (April 2023). “Sparsentan: First Approval”. Drugs. 83 (6): 563–568. doi:10.1007/s40265-023-01864-x. PMC 10232600. PMID 37022667.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ “PHARMACOPEIA LAUNCHES STUDY OF DARA COMPOUND | FDAnews”. http://www.fdanews.com.
- ^ “Ligand Licenses DARA Program to Retrophin”. investor.ligand.com. 21 February 2012.
- ^ https://www.fiercebiotech.com/biotech/retrophin-sheds-shkreli-connection-new-name-travere-therapeutics.
{{cite news}}: Missing or empty|title=(help) - ^ “Ongoing Non-malignant Hematological, Neurological, and Other Disorder Indications Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 21 August 2024. Retrieved 7 September 2024.
- ^ “Travere Therapeutics Announces Full FDA Approval of Filspari (sparsentan), the Only Non-Immunosuppressive Treatment that Significantly Slows Kidney Function Decline in IgA Nephropathy” (Press release). Travere Therapeutics. 5 September 2024. Retrieved 7 September 2024 – via GlobeNewswire.
- ^ “Despite trial scare, Travere’s Filspari gains full FDA nod in kidney disease showdown with Novartis”. fiercepharma.com.
External links
- Clinical trial number NCT03762850 for “A Study of the Effect and Safety of Sparsentan in the Treatment of Patients With IgA Nephropathy (PROTECT)” at ClinicalTrials.gov
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Sparsentan (Filspari). Sparsentan (27), marketed by Travere Therapeutics, is an oral, dual endothelin angiotensin receptor antagonist that received accelerated USFDA approval in February 2023 for reducing proteinuria in adults with primary immunoglobulin A (IgA) nephropathy who are at risk of rapid
disease progression.205206,207 Also known as Berger’s disease, IgAnephropathy is an immune-complex mediated disease characterized by deposits of IgA in the kidneys, resulting in inflammation and damage which can eventually lead to kidney failure. Typical treatment of IgA nephropathy has focused
on supportive care to slow kidney decline, for example, lowering blood pressure, reducing proteinuria, and minimizing lifestyle risk factors; immunosuppressive therapy has also been utilized, though it is controversial and carries risks.208 Sparsentan is the first nonimmunosuppressive treatment for IgA nephropathy and has received first-in-class and orphan drug designations. Accelerated approval was based on reduction of proteinuria (which is a risk factor for disease progression) during interim
analysis in phase III clinical trials. 209 endothelin type A (ETASparsentan blocks ) and angiotensin II type 1 receptors(AT1), interrupting the signaling pathway that contributes to disease progression. 210
The structure of the drug combines 211,212 elements that target both of these receptor types.
213 Thesynthesis of sparsentan (27), as shown in Scheme 50 and Scheme 51, was disclosed by Retrophin Pharmaceuticals (now Travere Therapeutics). Its telescoped sequences and isolation of intermediates as salts suggest that this route may be suitable for large-scale manufacturing.
The synthesis of the spirocyclic imidazolinone intermediate 27.7 is shown in Scheme 50.
Displacement of the benzylic bromide in 27.1 with sodium ethoxide produced ether 27.2. Reduction of the ester with sodium borohydride and zinc chloride yielded alcohol 27.3 which was then converted to mesylate 27.4. Reaction with spirocyclic imidazolinone 27.5 under phase transfer conditions
yielded 27.6 whichwasisolatedasthebisoxalatesalt (27.7).The sequence from 27.1 to 27.7 is telescoped, and no yields were given in the patent.
The construction of the biphenyl framework is shown in Scheme 51. Treatment of aryl bromide 27.8 with n-BuLi and triisopropyl borate followed by reaction with pinacol yielded boronic ester 27.9. Intermediates 27.7 and 27.9 were coupled via a Suzuki reaction to form the biphenyl which was isolated as
the camphorsulfonate salt (27.10). The synthesis was finished with deprotection of the methoxymethyl group under acidic conditions followed by recrystallization from isopropanol and heptane to yield sparsentan (27).
(206) Donadio, J. V.; Grande, J. P. IgA nephropathy. N. Engl. J. Med.2002, 347, 738−748.
(207) Fabiano, R. C. G.; Pinheiro, S. V. B.; Simões e Silva, A. C.Immunoglobulin A nephropathy: a pathophysiology view. Inflammation Res. 2016, 65, 757−770.
(208) Floege, J.; Rauen, T.; Tang, S. C. W. Current treatment of IgAnephropathy. Springer Semin. Immunopathol. 2021, 43, 717−728.
(209) Rovin, B.H.; Barratt, J.; Heerspink, H. J. L.; Alpers, C. E.; Bieler,S.; Chae, D.-W.; Diva, U. A.; Floege, J.; Gesualdo, L.; Inrig, J. K.; et al.Efficacy and safety of sparsentan versus irbesartan in patients with IgA
nephropathy (PROTECT): 2-year results from a randomised, active controlled, phase 3 trial. Lancet 2023, 402, 2077−2090.
(210) Komers, R.; Plotkin, H. Dual inhibition of renin-angiotensin aldosterone system and endothelin-1 in treatment of chronic kidney disease. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 2016, 310, R877−
R884.
(211) Murugesan, N.; Tellew, J. E.; Gu, Z.; Kunst, B. L.; Fadnis, L.;Cornelius, L. A.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; et al. Discovery of N-isoxazolyl biphenylsulfonamides as potent dual
angiotensin II and endothelin A receptor antagonists. J. Med. Chem.2002, 45, 3829−3835.
(212) Murugesan, N.; Gu, Z.; Fadnis, L.; Tellew, J. E.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; Dickinson, K. E.; Valentine,M.T.; et al. Dual angiotensin II and endothelin A receptor antagonists:
synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides withimproved potencyandpharmacokinetics. J. Med. Chem. 2005, 48, 171−179.
(213) Komers, R.; Shih, A. Biphenyl sulfonamide compounds for the treatment of kidney diseases or disorders. WO 2018071784, 2018.

//////////////Sparsentan, PS433540, RE-021, Bristol-Myers Squibb, ORPHAN DRUG, Retrophin, FDA 2023, APPROVALS 2023
O=S(C1=CC=CC=C1C2=CC=C(CN3C(CCCC)=NC4(CCCC4)C3=O)C=C2COCC)(NC5=NOC(C)=C5C)=O,
Ciraparantag, Aripazine
Ciraparantag
PER977, Aripazine
CAS Number:1438492-26-2
Chemical Name:N1,N1-[piperazine-1,4-diylbis(propane-1,3-diyl)]bis-L-argininamide
(2S,2’S)-N,N’-(Piperazine-1,4-diyldipropane-3,1-diyl)bis(2-amino-5-carbamimidamidopentanamide)
2S,2’S)-N,N’-(piperazine-1,4-diylbis(propane-3,1-diyl))bis(2-amino-5-guanidinopentanamide)
C22H48N12O2
Mw: 512.40232
Mechanism of Action: an intravenously administered anticoagulant Reversal Agent
Blood coagulation factor modulators; Factor Xa inhibitors
Indication: Anticoagulant Reversal
Development Stage: Phase II
Developer:Perosphere, Inc..Perosphere Inc.
Highest Development Phases
- Phase IIHaemorrhage
Most Recent Events
- 02 Apr 2015Ciraparantag receives Fast Track designation for Haemorrhage [IV] (In volunteers) in USA
- 05 Nov 2014Efficacy and adverse events data from a phase I/II trial in Haemorrhage released by Perosphere
- 06 Oct 2014Aripazine is available for licensing as of 06 Oct 2014. http://www.perosphere.com/
Aripazine(PER977, ciraparantag)

Ciraparantag, also known as PER977, is a A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. PER977 is water-soluble, cationic molecule that is designed to bind specifically to unfractionated heparin and low-molecular-weight heparin through noncovalent hydrogen bonding and charge–charge interactions.
PER-977 is an intravenous heparin neutralizer in phase II clinical trials at Perosphere to reverse edoxaban’s induced anticoagulation.
In April 2015, fast track designation was assigned in the U.S. as an investigational anticoagulant reversal agent.
WO 2013082210
http://www.google.com/patents/WO2013082210A1?cl=en
In one scheme, the compound of Formula V (DAP)

is synthesized by reacting excess equivalents (e.g., at least about two equivalents) of compound 1

with one equivalent of compound 2

in the presence of a peptide coupling reagent, to obtain a compound 3

wherein PI is a protecting group and P2 is a protecting group or is a hydrogen.
the coupling involved reacting compound 1, wherein PI was Boc and P2 was a hydrogen (depicted as Boc-Arg-OH HCl below), with compound 2 as depicted below:

The resultant crude product was more than 95% pure by thin layer
chromatography (TLC).
Subsequently, the deprotection step was carried out as depicted below:

The deprotected product was purified by preparative HPLC using 1% acetic acid buffer. Product purity of >98% was observed. Residual TFA was removed by low quantity of DOWEX resin. The molecular weight of DAP (the compound of Formula V) is 512.4, and the compound synthesized according to the above scheme exhibited the following primary peak by mass spectroscopy: [M+H]+=513.4.
|
References |
1: Dzik WH. Reversal of oral factor Xa inhibitors by prothrombin complex concentrates: a re-appraisal. J Thromb Haemost. 2015 Jun;13 Suppl 1:S187-94. doi: 10.1111/jth.12949. PubMed PMID: 26149022.
2: Crowther M, Crowther MA. Antidotes for Novel Oral Anticoagulants: Current Status and Future Potential. Arterioscler Thromb Vasc Biol. 2015 Aug;35(8):1736-45. doi: 10.1161/ATVBAHA.114.303402. Epub 2015 Jun 18. PubMed PMID: 26088576.
3: Sullivan DW Jr, Gad SC, Laulicht B, Bakhru S, Steiner S. Nonclinical Safety Assessment of PER977: A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. Int J Toxicol. 2015 Jun 15. pii: 1091581815590667. [Epub ahead of print] PubMed PMID: 26079256.
4: Mo Y, Yam FK. Recent advances in the development of specific antidotes for target-specific oral anticoagulants. Pharmacotherapy. 2015 Feb;35(2):198-207. doi: 10.1002/phar.1532. Epub 2015 Feb 3. PubMed PMID: 25644580.
5: Yates SW. Interrupting anticoagulation in patients with nonvalvular atrial fibrillation. P T. 2014 Dec;39(12):858-80. PubMed PMID: 25516695; PubMed Central PMCID: PMC4264672.
6: Vanden Daelen S, Peetermans M, Vanassche T, Verhamme P, Vandermeulen E. Monitoring and reversal strategies for new oral anticoagulants. Expert Rev Cardiovasc Ther. 2015 Jan;13(1):95-103. doi: 10.1586/14779072.2015.987126. Epub 2014 Nov 28. PubMed PMID: 25431993.
7: Costin J, Ansell J, Laulicht B, Bakhru S, Steiner S. Reversal agents in development for the new oral anticoagulants. Postgrad Med. 2014 Nov;126(7):19-24. doi: 10.3810/pgm.2014.11.2829. Review. PubMed PMID: 25387210.
8: Ansell JE, Bakhru SH, Laulicht BE, Steiner SS, Grosso M, Brown K, Dishy V, Noveck RJ, Costin JC. Use of PER977 to reverse the anticoagulant effect of edoxaban. N Engl J Med. 2014 Nov 27;371(22):2141-2. doi: 10.1056/NEJMc1411800. Epub 2014 Nov 5. PubMed PMID: 25371966.
9: Hankey GJ. Intracranial hemorrhage and novel anticoagulants for atrial fibrillation: what have we learned? Curr Cardiol Rep. 2014 May;16(5):480. doi: 10.1007/s11886-014-0480-9. Review. PubMed PMID: 24643903.
///////
BEXAGLIFLOZIN


Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
FDA APPROVED
| 1/20/2023 |
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise
Drug Trials Snapshot
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)

Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.

Example 33
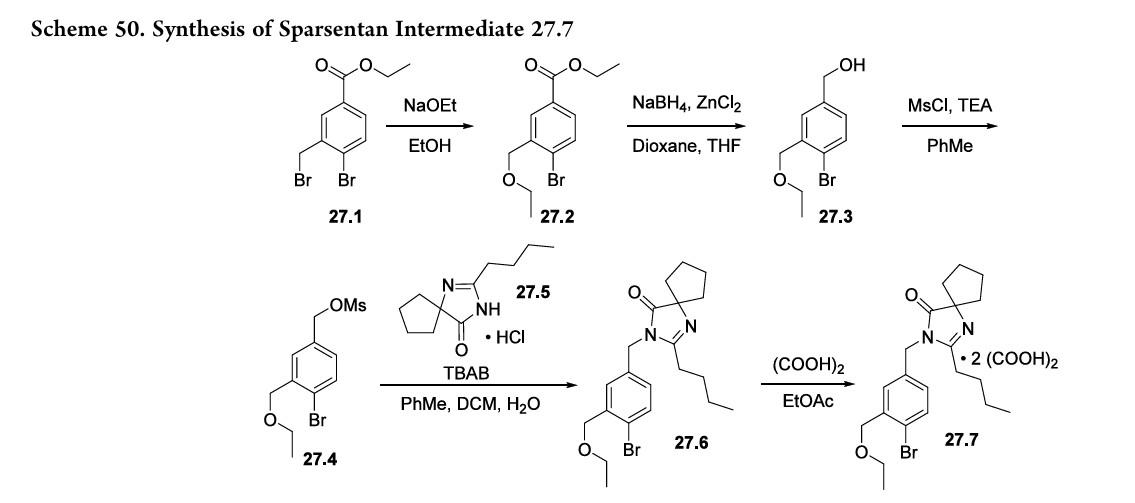
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
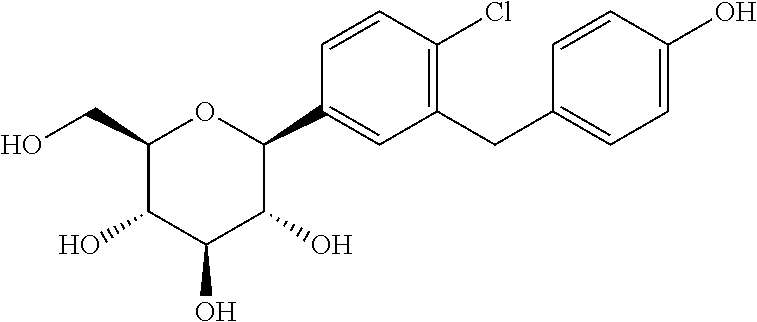
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694

Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.

Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.

(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Example 1A
Preparation of 2-cyclopropoxyethanol (1)

To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
////////BEXAGLIFLOZIN, APPROVALS 2023, FDA 2023
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079J.Med.Chem.2025,68,2147−2182
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof
type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting
SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe
reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates,
whichmaybeacommonchallengetootherSGLT2inhibitors.31
Anotherpatent disclosedbyPiramal Enterprises suggesteda
similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34
Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2
whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith
activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal
functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane,
whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was
reported in 92% yield. Details on stereoselectivity of this
approachwerenotdisclosed.
Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol
3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether
3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.

(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447−
453.
(29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium
glucose co-transporter-2 inhibitors for the treatment of diabetes
mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79.
(30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive
review of small-molecule drugs for the treatment of type 2 diabetes
mellitus: Synthetic approaches and clinical applications. Eur. J. Med.
Chem. 2024, 267, No. 116185.
(31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F.
Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A.
Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev.
2018, 22, 467−488.
(32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.;
Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′
position as potent and selective renal sodium-dependent glucose co
transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes.
Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470.
(33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the
preparation of benzyl-benzene C-glycosides via coupling reaction as
potential SGLT2 inhibitors. US 20130267694, 2013.
(34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal,
D. Aprocess for the preparation of SGLT2 inhibitor and intermediates
thereof. WO 2018207113, 2018.
(35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu,
B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside
derivatives as antidiabetic agents. WO 2009026537, 2009.
.
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
Bexagliflozin (Brenzavvy)
On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin
can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which
lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108].
The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring.
BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol.
Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453.
[105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng,
B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and
prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293.
[106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M.
P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin.
Pharmacother. 22 (2021) 2095–2103.
[107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am.
Soc. Nephrol. 18 (2023) 279–289.
[108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr.
Drug Saf. 13 (2018) 84–91.
[109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd,
J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives
as Antidiabetic Agents, 2009. WO2009026537A1.
.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Lemborexant

Lemborexant
E2006
CAS Number: 1369764-02-2
MF C22 H20 F2 N4 O2
MW 410.42
Chemical Name: (1R, 2S) -2 – {[(2,4-dimethylpyrimidin-5-yl) oxy] methyl} -2- (3-fluorophenyl ) N (5-fluoropyridin-2-yl) cyclopropanecarboxamide
Cyclopropanecarboxamide, 2-[[(2,4-dimethyl-5-pyrimidinyl)oxy]methyl]-2-(3-fluorophenyl)-N-(5-fluoro-2-pyridinyl)-, (1R,2S)-
(1R,2S)-2-{[(2,4-dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropanecarboxamide
Indication: Insomnia
Company: Eisai
Lemborexant (INN) (code name E-2006) is a dual antagonist of the orexinOX1 and OX2receptors which is under development byEisai for the treatment of insomnia.[1][2][3] As of December 2014, it is in phase IIclinical trials.[4]
Orexin receptors are G-protein coupled receptors found predominately in the brain. Their endogenous ligands, orexin-A and orexin-B, are expressed by neurons localized in the hypothalamus. Orexin-A is a 33 amino acid peptide; orexin-B consists of 28 amino acids. (Sakurai T. et al., Cell, 1998, 92, 573-585). There are two subtypes of orexin receptors, OXi and OX2; OX) binds orexin-A preferentially, while OX2 binds both orexin-A and -B. Orexins stimulate food consumption in rats, and it has been suggested that orexin signaling could play a role in a central feedback mechanism for regulating feeding behavior (Sakurai et al., supra). It has also been observed that orexins control wake-sleep conditions (Chemelli R.M. et al., Cell, 1999, 98, 437-451). Orexins may also play roles in brain changes associated with opioid and nicotine dependence (S.L. Borgland et al, Neuron, 2006, 49, 598-601; C.J. Winrow et al., Neuropharmacology, 2010, 58, 185-194), and ethanol dependence (J.R. Shoblock et al, Psychopharmacology, 2011, 215, 191-203). Orexins have additionally been suggested to play a role in some stress reactions (T. Ida et al, Biochem. Biophys. Res. Commun., 2000, 270, 318- 323).
Compounds such as (lR,2S)-2-(((2,4-dimethylpyrimidin-5-yl)oxy)methyl)-2-(3- fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropanecarboxamide (Compound A, below) have been found to be potent orexin receptor antagonists, and may be useful in the treatment of sleep disorders such as insomnia, as well as for other therapeutic uses.

……………….
paper
Journal of Medicinal Chemistry (2015), 58(11), 4648-4664.

The orexin/hypocretin receptors are a family of G protein-coupled receptors and consist of orexin-1 (OX1) and orexin-2 (OX2) receptor subtypes. Orexin receptors are expressed throughout the central nervous system and are involved in the regulation of the sleep/wake cycle. Because modulation of these receptors constitutes a promising target for novel treatments of disorders associated with the control of sleep and wakefulness, such as insomnia, the development of orexin receptor antagonists has emerged as an important focus in drug discovery research. Here, we report the design, synthesis, characterization, and structure–activity relationships (SARs) of novel orexin receptor antagonists. Various modifications made to the core structure of a previously developed compound (–)-5, the lead molecule, resulted in compounds with improved chemical and pharmacological profiles. The investigation afforded a potential therapeutic agent, (1R,2S)-2-{[(2,4-dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropanecarboxamide (E2006), an orally active, potent orexin antagonist. The efficacy was demonstrated in mice in an in vivo study by using sleep parameter measurements.
(1R,2S)-2-{[(2,4-dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropanecarboxamide
(1R,2S)-2-{[(2,4-Dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropanecarboxamide (34)
The title compound was synthesized as a white solid (3.66 g, 56.4% yield) from (1R,2S)-2-{[(2,4-dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)cyclopropanecarboxylic acid 18c by adapting the procedure described for compound 23.
1H NMR (400 MHz, DMSO-d) δ (ppm): 1.46–1.50 (m, 1H), 1.68 (t, J = 6.0 Hz, 1H), 2.01 (s, 3H), 2.36 (s, 3H), 2.59–2.63 (m, 1H), 4.27 (d, J = 10.4 Hz, 1H), 4.66 (d, J = 10.4 Hz, 1H), 7.06–7.11 (m, 1H), 7.37–7.44 (m, 3H), 7.60–7.65 (m, 1H), 7.85–7.89 (m, 1H), 8.11 (s, 1H), 8.30 (d, J = 3.2 Hz, 1H), 11.20 (br s, 1H).
13C NMR (150 MHz, CDCl3) δ (ppm): 18.7, 18.7, 25.0, 29.0, 34.9, 70.7, 114.5, 114.7, 115.9, 124.2, 125.4, 130.2, 135.5, 138.9, 144.1, 147.3, 149.1, 156.4, 157.0, 159.8, 162.8, 167.9.
HRMS (ESI(+)) calcd for C22H21F2N4O2 [M + H]+, 411.1627; found, 411.1622. Purity: >95%.
………………………….
WO 2013123240
E. Preparation of Compounds of Formula V


((lR,2S)-2-(((2,4-dimethylpyrimidin-5-yI)oxy)methyl)-2-(3-fluorophenyl)-cyclopropyl) methanol (11). ((lR,2S)-2-(3-fluorophenyl)-2-((tosyloxy)methyl)cyclopropyl)metliyl acetate (8, 11.05 g, 0.028 mol, 1.0 equiv.), 2,4-dimethylpyrimidin-5-ol (3.74 g, 0.030 mol, 1.07 equiv.), and cesium carbonate (22.94 g, 1.8 equiv.) were dissolved in ACN (110.5 mL), under nitrogen. The solution was stirred vigorously and heated to 65-70 °C for 2-3 hours. The reaction was monitored by HPLC and TLC (EtO Ac/Heptane = 1/1). Once complete, aqueous 1 N NaOH solution (71.82 mL) was added to the reaction mixture. The reaction mixture was stirred at 20-25 °C for 10-16 h, and was monitored by HPLC and TLC (EtO Ac/Heptane = 1/1). Once the hydrolysis reaction was complete, the reaction mixture was diluted with MTBE (110.50 mL) and stirred for at least 15 min. The aqueous layer was back extracted once with MTBE (55.25 mL). The organic layers were combined and washed once with saturated aqueous NaCl solution (33.15 mL). The solvent was removed under reduced pressure to afford the title compound; ((lR,2S)-2-(((2,4- dimethylpyrimidin-5-yl)oxy)methyl)-2-(3-fluorophenyl)cyclopi pyl)methanol: (11, 8.51 g).
((lR,2S)-2-(((2,4-dimethylpyrimidin-5-yl)oxy)methyl)-2-(3-fluorophenyl)- cyclopropyl)methanol: 1H NMR (500 MHz, DMSO-d6) δ 8.21 (s, 1H), 7.33 (td, J = 8.0, 6.5 Hz, 1H), 7.20 (d, J= 7.9 Hz, 1H), 7.19 – 7.14 (m, 1H), 7.01 (ddd, J= 8.3, 2.6, 1.2 Hz, 1H), 4.63 (t, J = 5.4 Hz, 1H), 4.36 (dd, J= 22.5, 10.5 Hz, 2H), 3.72 – 3.61 (m, 2H), 2.45 (s, 3H), 2.22 (s, 3H), 1.51 – 1.43 (m, 1H), 1.23 (dd, J= 8.9, 5.0 Hz, 1H), 1.01 (dd, J= 6.0, 5.3 Hz, 1H). 13C NMR (126 MHz, DMSO-dfi) δ 162.48 (d, JCF = 243.0 Hz), 158.91, 156.26, 149.51, 147.47 (d, JCF = 7.5 Hz), 139.85, 130.35 (d, JCF = 8.5 Hz), 124.72 (d, JCF = 2.5 Hz), 115.54 (d, JCF = 21.3 Hz), 113.43 (d, JCF = 20.9 Hz), 72.73, 60.70, 29.23, 28.64, 24.94, 18.77, 17.06.
HRMS Calculated for C17H20FN2O2 [M+H]+ 303.1590; found 303.1517.
F. Preparation of Compounds of Formula VII

(lR,2S)-2-(((2,4-dimethylpyrimidin-5-yl)oxy)methyl)-2-(3-fluorophenyl)cyclopropane- carboxylic acid (13). ((lR,2S)-2-(((2,4-dimethylpyrimidin-5-yl)oxy)methyl)-2-(3- fluorophenyl)cyclopropyl)methanol (11, 87.5 g, 290 mmol, 1.0 equiv.) was dissolved in toluene (390 mL). To the mixture was added pH 7 buffer (107 g, prepared from 4.46 g of sodium phosphate dibasic and 7.79 g of sodium phosphate monobasic in 94.4 mL of water) and 2,2,6,6- tetramethylpiperidine 1-oxyl (TEMPO) (0.93 g, 5.9 mmol, 0.02 equiv.). The mixture was cooled to 0 °C and sodium hypochlorite solution (5% active chlorine, 383 mL, 304 mmol, 1.05 equiv.) was added dropwise, maintaining the internal temperature below 9 °C. The mixture was allowed to warm to room temperature and stirred for 2 h. To the mixture was added aqueous hydrochloric acid (2.0 M, 8.73 mL, 0.05 equiv.) followed by a solution of sodium chlorite (36.0 g, 318 mmol, 1.1 equiv.) in water (87 mL), maintaining the internal temperature below 26 °C. The mixture was stirred at room temperature for 4 h, and then cooled to 10 °C. A solution of sodium thiosulfate (92 g, 579 mmol, 2.0 equiv.) in water (177 mL) was added, maintaining the internal temperature below 20 °C. The mixture was stirred for 20 min, and then aqueous sodium hydroxide solution (4 N, 87 mL, 348 mmol, 1.2 equiv.) was added to achieve ca. pH = 13. The mixture was heated to 80 °C for 4 hours, then cooled to room temperature. Stirring was halted and the phases allowed to split. The lower aqueous phase was collected and the upper organic phase was washed once with 4 N sodium hydroxide solution (17 mL). The combined aqueous phases were acidified with aqueous hydrochloric acid solution (4 N, 17 mL) to pH = 4 and extracted with ethyl acetate (2 x 470 mL). The combined organic phases were washed with ca. 20% aqueous NaCl solution (175 mL). The organic phases were concentrated by rotary evaporation to yield 96.84 g of crude oil. A portion (74 g) of this crude oil was dissolved in acetonitrile (400 mL) and concentrated to dryness by rotary evaporation. Another portion of acetonitrile (400 mL) was added and the mixture was again concentrated to dryness. To the residue was added acetonitrile (370 mL). The mixture was heated to 65 °C resulting in a clear solution. The mixture was cooled to room temperature, then to 0 °C and held at this temperature for 6 h. The mixture was filtered and the wet cake was washed with acetonitrile (2 x 74 mL). The cake was dried under vacuum with a nitrogen sweep, then in a vacuum oven at 20 torr and 40 °C to afford (lR,2S)-2-(((2,4- dimethylpyrimidin-5-yl)oxy)methyl)-2-(3-fluorophenyl)cyclopropanecarboxylic acid (13, 56.9 g, 80% yield) as an off-white crystalline solid.
(lR,2S)-2-(((2,4-dimethylpyrimidin-5-yl)oxy)methyl)-2-(3-fluoi phenyl)- cyclopropanecarboxylic acid: 1H NMR (500 MHz, DMSO-d6) δ 12.47 (s, 1H), 8.17 (s, 1H), 7.39 (td, J= 8.0, 6.4 Hz, 1H), 7.29 (d, J= 7.9 Hz, 1H), 7.27 – 7.22 (m, 1H), 7.10 (td, J – 8.3, 2.1 Hz, 1H), 4.63 (d, J= 10.2 Hz, 1H), 4.30 (d, J= 10.2 Hz, 1H), 2.46 (s, 3H), 2.26 (s, 3H), 2.13 (dd, J= 7.7, 6.6 Hz, 1H), 1.63 – 1.54 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 172.65, 162.48 (d, JCF = 243.6 Hz), 159.08, 156.24, 149.45, 145.15 (d, JCF = 7.5 Hz), 139.60, 130.71 (d, JCF = 8.5 Hz), 124.79 (d, JCF = 2.6 Hz), 115.60 (d, JCF = 21.8 Hz), 114.32 (d, JCF = 20.8 Hz), 71.15, 33.92 (d, JCF = 2.0 Hz), 26.46, 24.96, 19.72, 18.70.
HRMS Calculated for Ci7Hi8FN203 [M+H]+ 317.1301; found 317.1298.
G. Preparation of Compounds of Formula IX

(lR,2S)-2-(((2,4-dimethylpyrimidin-5-yl)oxy)methyl)-2-(3-fluorophenyl)-N-(S- fluoropyridin-2-yl)cyclopropanecarboxamide (14). (lR,2S)-2-(((2,4-dimethylpyrimidin- 5-yl)oxy)methyl)-2-(3-fluorophenyl)-cyclopropanecarboxylic acid (13, 12.80 g, 0.040 mol, 1.0 equiv.), and 2-amino-5-fluoiOpyridine (4.76 g, 0.0425 mol, 1.05 equiv.) were dissolved in ethyl acetate (102.4 mL), under nitrogen. The solution was cooled to 0-5 °C, and N,N- diisopropylethylamine (14.10 mL, 0.081 mol, 2.0 equiv.) was added to the reaction mixture while maintaining the internal temperature at 0-15 °C. The reaction mixture was stirred at 0-10 °C for 20-30 minutes. n-Propylphosphonic anhydride (T3P; 50% w/w solution in ethyl acetate, 36.1 g, 1.4 equiv.) was added to the reaction mixture while maintaining the internal temperature at 0-15 °C. The reaction was stirred at 20-25 °C for at least 20-24 hour and monitored by HPLC and TLC (EtO Ac/Heptane = 1/1). Upon completion of the reaction, the reaction mixture was cooled to 0-5 °C and then was quenched with water (64.0 mL) while maintaining the internal temperature below 10-15 °C. The aqueous layer was back extracted once with MTBE (76.8 mL). The organic layers were combined and washed once with saturated aqueous NaHC03 solution (38.4 mL) and once with water (38.4 mL). The organic layer was polish filtered and the filter rinsed with MTBE (12,8 mL). The organic layer was then concentrated under reduced pressure to a minimum stirrable volume. Ethyl acetate (60.8 mL) was added to the reaction mixture and the mixture was heated to no more than 50 °C to achieve a clear solution. n-Heptane (86.3 mL) was added slowly with agitation. The reaction mixture was cooled to 20-25 °C, and the suspension was stirred for at least 1 h at 20-25 °C and then stirred at least for 1 h at 0-5 °C. The suspension was filtered and the cake was washed two times with 5 : 1 heptane/ethyl acetate (2 x
12.8 mL). The cake was dried under nitrogen and/or vacuum to provide the title compound, (lR,2S)-2-(((2,4-dimethylpyrimidin-5-yl)oxy)methyl)-2-(3-fluoiOphenyl)-N-(5-fiuoropyridin^ yl)cyclopropanecarboxamide, (14, 12.54 g, >99% ee) as a white to off white solid.
(lR,2S)-2-(((2,4-dimethylpyrimidin-5-yl)oxy)methyl)-2-(3-fluoiOphenyl)-N-(5- fluoropyridin-2-yl)cyclopropanecarboxamide:
1H NMR (500 MHz, DMSO-d6) δ 11.19 (s, 1H), 8.31 (d, J = 3.0 Hz, 1H), 8.12 (s, 1H), 7.94 – 7.85 (m, 1H), 7.62 (tt, J = 8.7, 3.1 Hz, 1H), 7.44 (dd, J = 10.6, 1.5 Hz, 1H), 7.41 – 7.40 (m, 1H), 7.39 (s, 1H), 7.14 – 7.06 (m, 1H), 4.67 (d, J = 10.2 Hz, 1H), 4.29 (t, J= 9.9 Hz, 1H), 2.63 (t, J= 7.0 Hz, 1H), 2.38 (s, 3H), 2.03 (s, 3H), 1.76 – 1.64 (m, 1H), 1.49 (dd, J = 8.0, 4.8 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 168.68, 161.98 (d, JcF = 242.3 Hz), 158.46, 155.15, 155.38 (d, JCF = 247.9 Hz), 148.90, 148.51, 145.00 (d, JCF = 7.7 Hz), 139.37, 135.15 (d, JCF = 24.9 Hz), 130.06 (d, JCF = 8.4 Hz), 125.05 (d, JCF = 19.5 Hz), 124.70 (d, JCF = 2.6 Hz), 115.71 (d, JCF = 21.7 Hz), 114.20 (d, JCF = 4.1 Hz), 113.70 (d, JCF =
20.9 Hz), 70.80, 34.09 (d, JCF = 1.9 Hz), 26.90, 24.38, 18.37, 17.78.
HRMS Calculated for C22H21F2N402 [M+H]+ 411.1627; found 411.1632.
……………….
WO 2012039371
Production Example 14
(1R, 2S) -2 – Synthesis of {[(2,4-dimethyl-pyrimidin-5-yl) oxy] methyl} -2- (3-fluorophenyl) cyclopropanecarboxylic acid (Prep14-6)

(1) (1S, 5R) -1- (3- fluorophenyl) -3-hexane-2-one to oxabicyclo [3.1.0] (Prep14-1)
3-fluorophenyl acetonitrile (70g) was dissolved in THF (500ml), ice – salt bath under cooling, was added dropwise NaHMDS (1000ml, 1.06M). After allowed to stir 1 hour, R – (-) – it was added dropwise epichlorohydrin (40.6ml) (approximately 10 minutes, the internal temperature <10 ℃). After it was allowed to stirred for 2 hours (maintained before and after the internal temperature 0 ℃), and stirred at room temperature for 14 hours. The reaction was I was dropping a small amount of water cooled with ice. The reaction solution was concentrated under reduced pressure, the residue in ethanol (700ml), 1N potassium hydroxide aqueous solution (1000ml) was added and heated to reflux for 5 hours. After returning to room temperature, it was added 5N hydrochloric acid (400ml), and stirred for 1 hour at 60 ℃. The reaction mixture was concentrated under reduced pressure, it was added thereto to carry out a liquid separation with ethyl acetate and water. The organic layer saturated aqueous sodium hydrogen carbonate solution, it was washed successively with saturated sodium chloride aqueous solution. Dried over magnesium sulfate, and the solvent was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to obtain a purified by (n- heptane-ethyl acetate) The title compound (84.9g).
1 H-NMR (400MHz, CDCl 3) δ (ppm): 1.41 (t, J = 5.2Hz, 1H), 1.64 (dd, J = 8.0,5.2Hz, 1H), 2 .56-2.63 (m, 1H), 4.30 (d, J = 9.2Hz, 1H), 4.47 (dd, J = 9.2,4.8Hz, 1H), 6.96- 7.02 (m, 1H), 7.16-7.21 (m, 2H), 7.28-7.35 (m, 1H).
(2) (1S, 2R) -1- (3- fluorophenyl) cyclopropane-1,2-dimethanol (Prep14-2)
THF- methanol compound Prep14-1 (72.7g) (440ml-220ml) sodium borohydride solution (25g) was added at 0 ℃, and the mixture was stirred for 65 hours at room temperature. Under ice-cooling, water and 5N hydrochloric acid were added to the reaction solution, followed by extraction with ethyl acetate. The organic layer was washed with a saturated sodium chloride aqueous solution, and then dried with magnesium sulfate. The solvent was concentrated under reduced pressure, the residue was purified by silica gel column chromatography to obtain a purified by (n- heptane-ethyl acetate) The title compound (72.7g).
1 H-NMR (400MHz, CDCl 3) δ (ppm): 0.80 (t, J = 5.0Hz, 1H), 1.10 (dd, J = 8.6,5.0Hz, 1H), 1 .62-1.71 (m, 1H), 3.41 (t, J = 11.4Hz, 1H), 3.58 (d, J = 12.0Hz, 1H), 4.12-4.25 ( m, 2H), 6.90-6.96 (m, 1H), 7.08-7.14 (m, 1H), 7.16-7.21 (m, 1H) 7.24-7.32 (m, 1H).
(3) {(1S, 2R) – [2- (tert- butyldiphenylsilyloxy) -1- (3-fluorophenyl) cyclopropyl]} methanol (Prep14-3)
Compound Prep14-2 a (42.4g) was dissolved triethylamine (33.0ml) in dichloromethane (216ml), was cooled to -20 ℃, was added dropwise tert- butyldiphenylsilyl chloride (56.3ml) (about 30 minute, almost at the same time insoluble matter is deposited with the completion of the dropping). After stirring for 1 hour, further stirred at room temperature for 20 hours.Water was added to the reaction mixture, and the mixture was extracted with dichloromethane. Washed with water and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel column chromatography to obtain a purified by (n- heptane ethyl acetate) The title compound (67.8g).
1 H-NMR (400MHz, CDCl 3) δ (ppm): 0.73 (t, J = 5.2Hz, 1H), 1.04 (dd, J = 8.4,5.2Hz, 1H), 1 .09 (s, 9H), 1.48-1.53 (m, 1H), 3.52 (t, J = 12.0Hz, 1H), 3.56 (dd, J = 9.6,1. 6Hz, 1H), 3.70 (dd, J = 9.6,1.6Hz, 1H), 4.18 (t, J = 12.0Hz, 1H), 4.20 (dd, J = 12.0 , 5.2Hz, 1H), 6.93 (tdd, J = 8.0,2.4,1.2Hz, 1H), 7.11 (dt, J = 9.6,2.4Hz, 1H), 7.20 (dt, J = 8.0,1.2Hz, 1H), 7.28 (td, J = 8.0,6.0Hz, 1H), 7.37-7.49 (m, 6H) , 7.69-7.74 (m, 4H).
(4) {(1R, 2S) -2 – {[(-5- 2,4- dimethyl-pyrimidin-yl) oxy] methyl} -2- (3-fluorophenyl) cyclopropyl} methanol (Prep14-4)
Compound Prep14-3 (581mg), triphenylphosphine (1.3g) and Preparation Example 4 to give 2,4-dimethyl – THF (10ml) solution of diisopropyl azodicarboxylate pyrimidin-5-ol (183mg) ( The 0.316ml) was added dropwise at 0 ℃, and the mixture was stirred at room temperature for 2 days. The reaction mixture was concentrated under reduced pressure, silica gel column chromatography (n- heptane: ethyl acetate = 19: 1 → 7: 3) was purified by. The resulting (1S, 2R) -2- (tert- butyldiphenylsilyloxy-methyl) -1 – {[(2,4-dimethyl-pyrimidin-5-yl) oxy] methyl} -1- (3-fluorophenyl) cyclopropane was dissolved in THF (15ml), tetrabutylammonium fluoride (1M-THF solution: 1.61ml) was added dropwise at room temperature and stirred at room temperature for 14 hours. The reaction mixture was concentrated under reduced pressure, silica gel column chromatography (n- heptane: ethyl acetate = 10: 1 → 0: 1) to obtain purified by the title compound (238mg).
1 H-NMR (400MHz, CDCl 3) δ (ppm): 1.00 (t, J = 5.6Hz, 1H), 1.25-1.33 (m, 1H), 1.78-1.88 (m, 1H), 2.39 (s, 3H), 2.61 (s, 3H), 3.58 (dd, J = 12.0,9.6Hz, 1H), 4.02-4.11 (m, 1H), 4.12 (d, J = 10.4Hz, 1H), 4.43 (d, J = 9.6Hz, 1H), 6.92-6.98 (m, 1H), 7 .10-7.16 (m, 1H), 7.18-7.23 (m, 1H), 7.29 (td, J = 8.0,6.0Hz, 1H), 8.00 (s, 1H).
(4 alternative method)
((1R, 2S) -2 – {[(2,4- dimethyl-pyrimidin-5-yl) oxy]} methyl] -2- (3-fluorophenyl) cyclopropyl} methanol (Prep14-4) (alternative method)
Triethylamine (14.5ml) was added in dichloromethane (200ml) solution of compound Prep14-3 (41.3g), cooled to 0 ℃. It was added dropwise methanesulfonyl chloride (7.34ml), and stirred for 1 hour. Water was added to the reaction mixture, and the mixture was extracted with dichloromethane. Dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue in acetonitrile (200ml) solution obtained in Production Example 4- (2) 2,4-dimethyl – pyrimidin-5-ol (14.1g) and cesium carbonate (61.8g) was added, 70 ℃ It was heated to. After 4 hours of stirring at 70 ℃, the reaction solution was cooled to 0 ℃, tetrabutylammonium fluoride (1M-THF solution: 190ml) was added dropwise, and the mixture was stirred for 1 hour at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. Dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by NH- silica gel column chromatography (n- heptane: ethyl acetate = 9: 1 to 1: 1) to give the title compound (20.7g) was purified by.
(5) (1R, 2S) -2 – {[(2,4- dimethyl-pyrimidin-5-yl) oxy] methyl} -2- of (3-fluorophenyl) cyclopropane carbaldehyde (Prep14-5)
Oxalyl dichloromethane solution of chloride (137μl) a (7ml) was cooled to -78 ℃, there was added dropwise dimethyl sulfoxide (226μl) (internal temperature below -60 ℃). After stirring for 10 minutes at the same temperature, dichloromethane (3ml) solution of the compound to the reaction mixture Prep14-4 (238mg) was dropped at -78 ℃, and the mixture was stirred at the same temperature for 30 minutes. After stirring for 15 minutes triethylamine (671μl) was added to the reaction mixture, and the temperature was raised to room temperature. Saturated sodium chloride aqueous solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was dried anhydrous magnesium sulfate and concentrated under reduced pressure to give the crude title compound (236mg).
1 H-NMR (400MHz, CDCl 3) δ (ppm): 1.67 (dd, J = 8.0,4.8Hz, 1H), 1.96-2.00 (m, 1H), 2.36 (s, 3H), 2.49-2.55 (m, 1H), 2.59 (s, 3H), 4.19 (d, J = 9.6Hz, 1H), 4.44 (d, J = 10.0Hz, 1H), 6.97-7.04 (m, 1H), 7.14-7.20 (m, 1H), 7.21-7.25 (m, 1H), 7.30 -7.37 (m, 1H), 7.95 (s, 1H), 9.87 (d, J = 3.2Hz, 1H).
(6) (1R, 2S) -2 – {[(2,4- dimethyl-pyrimidin-5-yl) oxy] methyl} -2- (3-fluorophenyl) cyclopropanecarboxylic acid (Prep14-6)Compound Prep14- 5 (18.9g) and 2-methyl-2-butene (26.1ml), sodium dihydrogen phosphate the (9.07g) was dissolved in acetone-water mixed solvent (200ml · 40ml), sodium chlorite ( 6.26g) and I were added little by little. After stirring for 2 hours at room temperature, the reaction solution was concentrated under reduced pressure. The precipitated solid was filtered off, washed with dichloromethane, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (n- heptane: After 1, ethyl acetate: ethyl acetate = 1: 1-0 methanol = 10: 1) to give the title compound (16.2g) was purified by.
1 H-NMR (400MHz, CDCl 3) δ (ppm): 1.55 (dd, J = 8.4,5.6Hz, 1H), 1.76 (t, J = 5.6Hz, 1H), 2 .25 (dd, J = 8.4,6.4Hz, 1H), 2.33 (s, 3H), 2.55 (s, 3H), 4.47 (t, J = 9.6Hz, 1H) , 4.50 (d, J = 9.6Hz, 1H), 6.99 (tdd, J = 8.0,2.4,1.2Hz, 1H), 7.21 (dt, J = 9.6 , 2.4Hz, 1H), 7.26 (td, J = 8.0,1.2Hz, 1H), 7.32 (td, J = 8.0,6.0Hz, 1H), 8.21 ( s, 1H).
Compound Prep14-6 can be prepared directly by the following method from the compound Prep14-4.
Compound Prep14-4 (300mg) and TEMPO (5mol%, 7.74mg) was dissolved in phosphate buffer solution of acetonitrile · pH6.4 (5ml · 5ml), 2N- hydrochloric acid (150μl), sodium chlorite (180mg ) and it was added. After heating to 40 °, 5w% of the hypochlorite solution (2mol%, 26.5μl) were added and stirred for 2 hours. Cooled to room temperature, the reaction mixture was stirred for 5 minutes was added an excess of 2-methyl-2-butene in. The reaction solution was extracted with dichloromethane, the solvent was distilled off under reduced pressure, the residue was purified by silica gel column chromatography (n- heptane: ethyl acetate = 1: 1 to 0: After 1, ethyl acetate: methanol = 9: 1) in was purified to give the title compound (215mg).
Example 95
(1R, 2S) -2 – {[(2,4- dimethyl-pyrimidin-5-yl) oxy] methyl} -2- (3-fluorophenyl) -N- (5- fluoro-2-yl) cyclopropane The synthesis of carboxamide (95)

Acid Prep14-6 a (226mg) was dissolved in dichloromethane (10ml), oxalyl chloride (122μl), and stirred for 1 hour at room temperature was added DMF (a few drops). The reaction mixture was concentrated under reduced pressure to give the crude acid chloride. N in THF (10ml) solution of 2-amino-5-fluoro pyridine (96.1mg), N- diisopropylethylamine (283μl) was added mixture was heated to 60 ℃, the temperature of intact dropwise a THF solution of the crude acid chloride in it was allowed to stir for 1 hour. The reaction mixture was allowed to cool to room temperature and allowed to stir for 1 hour, after which the reaction mixture was concentrated under reduced pressure, partitioned between ethyl acetate and water, the organic layer was separated. The organic layer was dried over anhydrous magnesium sulfate, and the filtrate was concentrated under reduced pressure. The residue was purified by NH- silica gel column chromatography (n- heptane: ethyl acetate = 2: 1) to give diethyl ether to the obtained target compound was added. The precipitated solid was filtered dried to give the title compound (130mg).
1 H-NMR (400MHz, d-DMSO) δ (ppm): 1.46-1.50 (m, 1H), 1.68 (t, J = 6.0Hz, 1H), 2.01 (s, 3H), 2.36 (s, 3H), 2.59-2.63 (m, 1H), 4.27 (d, J = 10.4Hz, 1H), 4.66 (d, J = 10. 4Hz, 1H), 7.06-7.11 (m, 1H), 7.37-7.44 (m, 3H), 7.60-7.65 (m, 1H), 7.85-7. 89 (m, 1H), 8.11 (s, 1H), 8.30 (d, J = 3.2Hz, 1H), 11.20 (brs, 1H)
MS [M + H] + = 411
Synthesis coming…….watch out
References
- Christopher, John A (2014). “Small-molecule antagonists of the orexin receptors”. Pharmaceutical Patent Analyst 3 (6): 625–638.doi:10.4155/ppa.14.46. ISSN 2046-8954.
- Cristoph Boss, Catherine Ross (2015). “Recent Trends in Orexin Research – 2010 to 2015”. ScienceDirect.doi:10.1016/j.bmcl.2015.05.012.
- Boss, Christoph (2014). “Orexin receptor antagonists – a patent review (2010 to August 2014)”. Expert Opinion on Therapeutic Patents 24 (12): 1367–1381.doi:10.1517/13543776.2014.978859. ISSN 1354-3776.
- AdisInsight. “Lemborexant”. Springer. Retrieved 2015-05-23.

External links
| Systematic (IUPAC) name | |
|---|---|
|
(1R,2S)-2-[(2,4-dimethylpyrimidin-5-yl)oxymethyl]-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropane-1-carboxamide
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 1369764-02-2 |
| ATC code | None |
| PubChem | CID: 56944144 |
| ChemSpider | 34500836 |
| Chemical data | |
| Formula | C22H20F2N4O2 |
| Molecular mass | 410.417 g/mol |
////////
Vintafolide

Vintafolide, EC-145 , MK-8109
mw 1917.041, cas 742092-03-1, mf C86 H109 N21 O26 S2
(2S)-2-[(4-{[(2-amino-4-oxo-3H-pteridin-6-yl)methyl]amino}phenyl)formamido]-4-{[(1S)-1-{[(1S)-4-carbamimidamido-1-{[(1S)-2-carboxy-1-{[(1S)-2-carboxy-1-{[(1R)-1-carboxy-2-({2-[({[(1R,9R,10S,11R,12R,19R)-12-ethyl-4-[(13S,15R,17S)-17-ethyl-17-hydroxy-13-(methoxycarbonyl)-1,11-diazatetracyclo[13.3.1.04,12.05,10]nonadeca-4(12),5,7,9-tetraen-13-yl]-10,11-dihydroxy-5-methoxy-8-methyl-8,16-diazapentacyclo[10.6.1.01,9.02,7.016,19]nonadeca-2,4,6,13-tetraen-10-yl]formohydrazido}carbonyl)oxy]ethyl}disulfanyl)ethyl]carbamoyl}ethyl]carbamoyl}ethyl]carbamoyl}butyl]carbamoyl}-2-carboxyethyl]carbamoyl}butanoic acid
Vincaleukoblastin-23-oic acid, O4-deacetyl-, 2-[(2-mercaptoethoxy)carbonyl]hydrazide, disulfide with N-[4-[[(2-amino-1,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine
Endocyte innovator
Vintafolide is an investigational targeted cancer therapeutic currently under development by Endocyte and Merck & Co.[1] It is a small molecule drug conjugate consisting of a small molecule targeting the folate receptor, which is overexpressed on certain cancers, such as ovarian cancer, and a potent chemotherapy drug, vinblastine.[2] It is being developed with a companion imaging agent, etarfolatide, that identifies patients that express the folate receptor and thus would likely respond to the treatment with vintafolide.[3] A Phase 3 study evaluating vintafolide for the treatment of platinum-resistant ovarian cancer (PROCEED trial) and a Phase 2b study(TARGET trial) in non-small-cell lung carcinoma (NSCLC) are ongoing.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
A Marketing Authorization Application (MAA) filing for vintafolide and etarfolatide for the treatment of patients withfolate receptor-positive platinum-resistant ovarian cancer in combination with doxorubicin, pegylated liposomal doxorubicin (PLD), has been accepted by the European Medicines Agency.[6] The drug received an orphan drug status in Europe in March 2012.[1] Merck & Co. acquired the development and marketing rights to this experimental cancer drug from Endocyte in April 2012.[1] The drug received orphan drug status in Europe in March 2012.[3]Endocyte remains responsible for the development and commercialization of etarfolatide, a non-invasive companion imaging agent used to identify patients expressing the folate receptor that will likely respond to treatment with vintafolide.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
In 2014 Merck and Endocyte stopped a late-stage study of vintafolide in treating ovarian cancer on the recommendation of a data safety monitoring board, saying that the drug failed to improve progression-free survival.[7]
Vintafolide is folate-conjugated with DAVBLH, which is a derivative of the vinca alkaloid vinblastine.Vinblastine is a microtubule-destabilizing agent that binds tubulin and causes M phase-specific cell cycle arrest and apoptosis of mitotically active cells. Vinblastine is an extremely potent chemotherapeutic agent but has significant toxicities including bone marrow suppression, neurotoxicity, gastrointestinal toxicity and vesicant injury.
Endocyte’s desacetylvinblastinehydrazide/folate conjugate (EC-145) is a folate-targeted cytotoxic anticancer drug in early development for the treatment of non-small cell lung cancer (NSCLC) and breast cancer. The compound had been pre-registered in the E.U. by Merck for the treatment of ovarian cancer, but the application was withdrawn due to lack of efficacy.
In 2012, the product was licensed to Merck & Co. by Endocyte for worldwide exclusive development and commercialization. In 2014, however, this license agreement was terminated and Endocyte regained all rights.
Folates can serve as one-carbon donors in reactions that are critical in the de novo biosynthesis of purines and thymidylate, amino acid metabolism and methylation reactions. Folate can enter a cell by two routes: RFC or by membrane-bound FRs. RFC is a bidirectional anion transporter that is the normal entry method for reduced folates in most cells. By contrast, FRs are expressed in a limited distribution in normal tissues but are overexpressed in multiple cancers including ovarian, lung, breast and colorectal cancer. FRs bind folate derivatives with high affinity and mediate their internalization by endocytosis. Given that FRs are not typically expressed on the luminal surface of epithelial cells, making them inaccessible to normal circulation, they are attractive therapeutic targets with limited toxicity. In addition to the therapeutic agent vintafolide, a radiodiagnostic agent (99mTc-etarfolatide [EC20]) has been developed to allow single-photon emission computed tomography (SPECT) imaging to identify FR-expressing tissues (tumors).
In 2012, orphan drug designations were assigned in the E.U. for the treatment of ovarian cancer and to be used with folic acid for the diagnosis of positive folate-receptor status in ovarian cancer. In 2013, orphan drug designation was assigned in the U.S. for the treatment of ovarian cancer.
Vintafolide is a water-soluble derivative of folic acid and the vinca alkaloid DAVLBH. The molecules are connected through a hydrophilic L-peptide spacer and a disulfide linker (Figure 1). The disulfide linker serves as a cleavable bond that is necessary for drug release following receptor mediated endocytosis. The disulfide bond is reduced in the acidic environment of the endosome, leading to efficient release of vinblastine.
Vintafolide.
DAVBLH: Desacetylvinblastine hydrazide

Structure of vintafolide and mechanism of release of the payload in the endosome.

Mechanism of action
Folate is required for cell division, and rapidly dividing cancer cells often express folate receptors in order to capture enough folate to support rapid cell growth. Elevated expression of the folate receptor occurs in many diseases, including other aggressively growing cancers and inflammatory disorders.[8] Vintafolide binds to the folate receptor and is subsequently taken up by the cell through a natural internalization process called endocytosis. Once inside the cell, vintafolide’s linker releases the chemotherapy drug which kills the cell.[3]
……………
Bioorganic & Medicinal Chemistry Letters (2006), 16(19), 5093-5096
http://www.sciencedirect.com/science/article/pii/S0960894X06008079
An efficient synthesis of the folate receptor (FR) targeting conjugate EC145 is described. EC145 is a water soluble derivative of the vitamin folic acid and the potent cytotoxic agent, desacetylvinblastine monohydrazide. Both molecules are connected in regioselective manner via a hydrophilic peptide spacer and a reductively labile disulfide linker.

………approach for the design and regioselective synthesis of a FA-vinca alkaloid conjugate 1 (EC145,BELOW). As indicated in the retrosynthetic scheme, 1 can be assembled by tethering a FA-Spacer unit 2 to the highly potent cytotoxic molecule, desacetylvinblastine monohydrazide 3, via a linker containing a reducible disulfide bond. The latter is important for drug delivery applications since real-time imaging using a fluorescence resonance energy transfer technique has recently demonstrated that reduction-mediated release of the drug cargo from a disulfide linked FA-conjugate efficiently occurs within the endosomes of cancer cells.

-
Scheme 1.
Reagents and conditions: (i) a—Fmoc-Asp(OtBu)-OH, PyBOP, DIPEA, RT, 1 h; b—20% piperidine/DMF, rt, 10 min; (ii) a—Fmoc-Arg(Pbf)-OH, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iii) a—Fmoc-Glu-OtBu, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iv) N10-TFA-pteroic acid, PyBOP, DIPEA, rt, 1.5 h; (v) TFA/H2O/TIPS/EDT (92.5:2.5:2.5:2.5), rt, 1 h; (vi) aq NH4OH, pH 9.3, rt, 1 h.
- Selected 1H NMR data for 2 (D2O, 300 MHz): δ 8.68 (s, 1H, FA H-7), 7.57 (d, 2H,J = 8.4 Hz, FA H-12 & 16), 6.67 (d, 2H, J = 9 Hz, FA H-13 & 15), 4.40–4.75 (series of m, 5H), 4.35 (m, 2H), 4.16 (m, 1H), 3.02 (m, 2H), 2.55–2.95 (series of m, 8H), 2.42 (m, 2H), 2.00–2.30 (m, 2H), 1.55–1.90 (m, 2H), 1.48 (m, 2H).
- 1H NMR for compound 6 (DMSO-d6, 300 MHz): δ 8.38 (m, 1H), 8.16 (dt, 1H, J = 8 Hz, 1 Hz), 8.02 (dt, 1H, J = 8 Hz, 1 Hz), 7.88 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz), 7.7 (m, 2H), 7.63 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz,), 7.4–7.2 (br, 1H), 7.2 (m, 1H), 4.72 (t, 2H,J = 6 Hz), 3.36 (t, 2H, J = 6 Hz).
- Selected 1H NMR data for
EC145 (D2O, 300 MHz): δ 8.67 (s, 1H, FA H-7), 7.50 (br s, 1H, VLB H-11′), 7.30–7.40 (br s, 1H, VLB H-14′), 7.35 (d, 2H, J = 7.8 Hz, FA H-12 & 16), 7.25 (m, 1H, VLB H-13′), 7.05 (br s, 1H, VLB H-12′), 6.51 (d, 2H, J = 8.7 Hz, FA H-13 & 15), 6.4 (s, 2H, VLB H-14 & 17), 5.65 (m, 1H, VLB H-7), 5.5 (m, 1H, VLB H-6), 4.15 (m,1H, VLB H-8′), 3.82 (s, 3H, VLB C18′ –CO2CH3), 3.69 (s, 3H, VLB C16 –OCH3), 2.8 (s, 3H, VLB N–CH3), 1.35 (br s, 1H, VLB H-3′), 1.15 (m, 1H, VLB H-2′), 0.9 (t, 3H, J = 7 Hz, VLB H-21′), 0.55 (t, 3H, J = 6.9 Hz, VLB H-21).
 CLICK ON IMAGE FOR CLEAR VIEW
CLICK ON IMAGE FOR CLEAR VIEW
…………
WO 2004069159
http://www.google.com/patents/WO2004069159A2?cl=en
EXAMPLE 16b

The compounds of Examples 16a and 16b were prepared from the peptidyl fragment Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH , prepared according to the general procedure described in Scheme 12. The Michael addition of this peptidyl fragment to the maleimido derivative of seco-CBI-bis-indole resulted in the folate conjugates Example 16a. The peptidyl fragment also reacted with either the thiosulfonate or pyridyldithio-activated vinblastine to form Example 16b. The maleimido derivative of seco-CBI-bis-indole, and the thiosulfonate and pyridyldithio- activated vinblastine intermediates were prepared using the procedures described herein for other examples.
……………..
https://www.google.com/patents/WO2012142281A1?cl=en
Folate-targeted drugs have been developed and are being tested in clinical trials as cancer therapeutics. EC145, also known as vintafolide, comprises a highly potent vinca alkaloid cytotoxic compound, desacetylvinblastine hydrazide (DAVLBH), conjugated to folate. The EC 145 molecule targets the folate receptor found at high levels on the surface of epithelial tumors, including non-small cell lung carcinomas (NSCLC), ovarian, endometrial and renal cancers, and others, including fallopian tube and primary peritoneal carcinomas. It is believed that EC 145 binds to tumors that express the folate receptor delivering the vinca moiety directly to cancer cells while avoiding normal tissue. Thus, upon binding, EC 145 enters the cancer cell via endocytosis, releases DAVLBH and causes cell death or inhibits cell function. EC 145 has the following formula

EC145
and has been accorded the Chemical Abstracts Registry Number 742092-03-1. As used herein, according to the context, the term EC 145 means the compound, or a pharmaceutically acceptable salt thereof; and the compound may be present in a solid, solution or suspension in an ionized form, including a protonated form. EC145 is disclosed in U.S. Patent No. 7,601,332; and particular uses and an aqueous liquid pH 7.4, phosphate-buffered formulation for intravenous administration are disclosed in WO 2011/014821. As described in WO 2011/014821, it is necessary to store the aqueous liquid formulation in the frozen state to ensure its stability. To avoid this necessity, a formulation is needed which has adequate stability at ambient temperature.
As one aspect of the invention described herein, there is provided a pharmaceutical composition of EC145 which is a lyophilized solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
In another aspect of the invention, there is provided a pharmaceutical composition of EC 145 which is an X-ray amorphous solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
| Systematic (IUPAC) name | |
|---|---|
|
N-(4-{[(2-Amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoyl)-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine disulfide with methyl (5S,7R,9S)-5-ethyl-9-[(3aR,4R,5S,5aR,10bR,13aR)-3a-ethyl-4,5-dihydroxy-8-methoxy-6-methyl-5-({2-[(2-sulfanylethoxy)carbonyl]hydrazinyl}carbonyl)-3a,4,5,5a,6,11,12,13a-octahydro-1H-indolizino[8,1-cd]carbazol-9-yl]-5-hydroxy-1,4,5,6,7,8,9,10-octahydro-2H-3,7-methanoazacycloundecino[5,4-b]indol-9-carboxylate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 742092-03-1 |
| ATC code | L01CA06 |
| ChemSpider | 27444385 |
| Synonyms | EC-145 |
| Chemical data | |
| Formula | C86H109N21O26S2 |
| Molecular mass | 1917 g/mol |
References
- Sridharan, Balaji (Apr 16, 2012). “Endocyte soars on cancer drug deal with Merck”. Reuters.
- Statement on a nonproprietary name adopted by the USAN Council, United States Adopted Names (USAN) Council, 6 April 2012
- Kuo, Phillip H. (February 2013). “Companion Imaging Diagnostics for Targeted Therapies”. Radiology Today 14 (2): 32.
- “Merck, Endocyte in Development Deal”. Drug Development & Discovery magazine. 2012-04-25.
- Dosio, F.; Milla, P.; Cattel, L. (2010). “EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers”. Current opinion in investigational drugs (London, England : 2000) 11 (12): 1424–1433. PMID 21154124.
- “EMA Accepts For Review MAA Filings For Vintafolide And Etarfolatide”. rttnews.com. 2012-11-27.
- Garde, Damian (2014-05-02). “Merck halts study of the billion-dollar cancer drug vintafolide”. Fierce Biotech. Retrieved 21 April 2015.
- “Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay” 338 (2). March 2005. pp. 284–93. doi:10.1016/j.ab.2004.12.026.PMID 15745749.
-
WO2008098970A1 * Feb 13, 2008 Aug 21, 2008 Pf Medicament Anhydrous crystalline vinflunine salts, method of preparation and use thereof as a drug and means of vinflunine purification WO2010150100A1 * Jun 23, 2010 Dec 29, 2010 Entarco Sa The use of spinosyns and spinosyn compositions against diseases caused by protozoans, viral infections and cancer WO2011014821A1 * Jul 30, 2010 Feb 3, 2011 Endocyte, Inc. Folate-targeted diagnostics and treatment US20100247669 * Sep 30, 2010 Cerulean Pharma Inc. Polymer-agent conjugates, particles, compositions, and related methods of use
////////Vintafolide, BMS-753493, DAVBLH, Desacetylvinblastine hydrazide, EC-145 , MK-8109 , phase 2
Bempedoic Acid
Bempedoic Acid
ETC-1002, ESP-55016
CAS 738606-46-7
- C19H36O5
- MW 344.486 Da
8-Hydroxy-2,2,14,14-tetramethylpentadecanedioic acid
8-Hydroxy-2.2.14,14-tetramethylpentadecanedioic acid
ATP Citrate Lyase Inhibitor and AMP-activated Protein kinase (AMPK) activator
Indication: Hypercholesterolemia
Development Stage: Phase II
Developer: Esperion Therapeutics
Esperion Therapeutics was founded in April 2008 by former executives of, and investors in, the original Esperion Therapeutics which was founded in July 1998 and was bought by Pfizer for $ 1.3 billion in 2004 and then spun out in 2008. ETC-1002 was first discovered at the original Esperion, and Esperion subsequently acquired the rights to it from Pfizer in 2008. Esperion own the exclusive worldwide rights to ETC-1002.

Bempedoic Acid ( ETC-1002) has a UNIQUE Dual mechanism of Action That has the Potential to Regulate Both lipid and Carbohydrate Metabolism. ETC-1002 Appears to Work by inhibitin ATP citrate lyase (ACL), a Key Enzyme in the Cholesterol biosynthetic pathway, and activating a Complementary Enzyme, 5′-adenosine monophosphate-activated Protein kinase (AMPK). Both Enzymes are Known to Play Significant roles in the synthesis of Cholesterol and glucose in the liver. By inhibitin Cholesterol synthesis in the liver, Causes ETC-1002 the liver to take up LDL particles from the blood, which reduces LDL-C levels.

WO 2004067489


6.13
7-Bromo-2,2-dimethylheptanoic acid ethyl ester
 7-Bromo-2,2-dimethylheptanoic acid ethyl ester
7-Bromo-2,2-dimethylheptanoic acid ethyl ester
Under argon atmosphere and cooling with an ice-bath, a solution of lithium diisopropylamide in THF (1.7 L, 2.0 M, 3.4 mol) was slowly dropped into a solution of 1 ,5- dibromopentane (950 g, 4.0 mol) and ethyl isobutyrate (396 g, 3.4 mol) in THF (5 L) while keeping the temperature below +5 DC. The reaction mixture was stiπed at room temperature for 20 h and quenched by slow addition of saturated ammonium chloride solution (3 L). The resulting solution was divided into three 4-L portions. Each portion was diluted with saturated ammonium chloride solution (5 L) and extracted with ethyl acetate (2 ‘ 2 L). Each 4-L portion of ethyl acetate was washed with saturated sodium chloride solution (2 L), 1 N hydrochloric acid (2 L), saturated sodium chloride solution (2 L), saturated sodium bicarbonate solution (2 L), and saturated sodium chloride solution (2 L). The three separate ethyl acetate layers were combined into a single 12-L portion, dried over magnesium sulfate, and concenfrated in vacuo to give the crude material (1.7 L) which was purified by vacuum distillation. Two fractions were obtained: the first boiling at 88 – 104 °C / 0.6 ton (184.2 g), the second at 105 – 120 °C / 1.4 ton (409.6 g) for atotal yield of 60 %. 1H NMR (300 MHz, CDC13/TMS): δ (ppm): 4.11 (q, 2 H, J = 7.2 Hz), 3.39 (t, 2 H, J = 6.8 Hz), 1.85 (m, 2 H), 1.56 – 1.35 (m, 4 H), 1.24 (t, 3 H, J = 7.2 Hz), 1.31 – 1.19 (m, 2 H), 1.16 (s, 6 H). 13C NMR (75 MHz, CDCI3/TMS): δ (ppm): 177.9, 60.2, 42.1, 40.5, 33.8, 32.6, 28.6, 25.2, 24.2, 14.3. HRMS (El, pos): Calcd. for CπH22Brθ2 (MH+): 265.0803, found: 265.0810.
6.18
2,2,14.14-Tetramethyl-8-oxo-pentadecanedioic acid diethyl ester
 p- toluenesulfonyl methyl isocyanide
p- toluenesulfonyl methyl isocyanide
 8-isocyano-2,2,14,14-teframethyl-8-(toluene-4-sulfonyl)-pentadecanedioic acid diethyl ester
8-isocyano-2,2,14,14-teframethyl-8-(toluene-4-sulfonyl)-pentadecanedioic acid diethyl ester
 2,2,14,14-tetramethyl-8-oxo-pentadecanedioic acid diethyl ester
2,2,14,14-tetramethyl-8-oxo-pentadecanedioic acid diethyl ester
Under Ar atmosphere, to a solution of 7-bromo-2,2-dimethylheptanoic acid ethyl ester (26.50 g, 100 mmol), tetra-n-butylammonium iodide (3.69 g, 10 mmol) and p- toluenesulfonyl methyl isocyanide (9.80 g, 50 mmol) in anhydrous DMSO (300 mL) was added sodium hydride (4.80 g, 20.5 mmol, 60 % dispersion in mineral oil) at 5 – 10 oC. The reaction mixture was stiπed at room temperature for 20 h and quenched with ice-water (300 mL). The product was extracted with dichloromethane (3 D 100 mL). The combined organic layers were washed with water (200 mL), half-saturated NaCl solution (2 ‘ 200 ■– ■ ■• •• ■• .. <i„ ‘ir ι., – ib,
mL), and saturated NaCl solution (200 mL), dried over MgS04, and concentrated in vacuo to get the crude 8-isocyano-2,2,14,14-teframethyl-8-(toluene-4-sulfonyl)-pentadecanedioic acid diethyl ester (36.8 g) as an orange oil, which was used in the next step without purification. To a solution of this crude product (36.8 g) in dichloromethane (450 mL) was added concentrated hydrochloric acid (110 mL) and the mixture was stiπed at room temperature for 1 h. The solution was diluted with water (400 mL) and the aqueous layer was extracted with dichloromethane (200 mL). The combined organic layers were washed with saturated NaHC0 solution (2 x 150 mL) and saturated NaCl solution (150 mL). The organic solution was dried over Na2S04 and concenfrated in vacuo. The residue was subjected to column chromatography (silica gel, hexanes : ethyl acetate = 11 : 1) to give 2,2,14,14-tetramethyl-8-oxo-pentadecanedioic acid diethyl ester (12.20 g, 66 % over two steps) as a colorless oil. lH NMR (300 MHz, CDC13/TMS): δ (ppm): 4.11 (q, 4 H, J – 6.9 Hz), 2.37 (t, 4 H, J – 7.5 Hz), 1.58 – 1.47 (m, 8 H), 1.35 – 1.10 (m, 8 H), 1.24 (t, 6 H, J = 7.2 Hz), 1.15 (s, 12 H). 13C NMR (75 MHz, CDC13/TMS): δ (ppm): 211.6, 178.3, 60.5, 43.1, 42.5, 40.9, 30.1, 25.5, 25.1, 24.1, 14.7. HRMS (LSIMS, nba): Calcd. for C23IL3O5 (MH+): 399.3110, found: 399.3129.
6.19
8-Oxo-2,2,14,14-tetramethylpentadecanedioic acid
A solution of KOH (25 g) in water (50 mL) was added to a solution of 2,2,14,14-tetramethyl-8-oxo-pentadecanedioic acid diethyl ester (10.69 g, 155 mmol) in ethanol (400 mL), then heated at reflux for 4 h. After cooling, the solution was evaporated to a volume of ca. 50 mL and diluted with water (800 mL). The organic impurities were removed by extracting with dichloromethane (2 x 200 mL). The aqueous layer was acidified to pH 2 with concentrated hydrochloric acid (50 mL) and extracted with methyl tert.-butyl ether (MTBE, 3 x 200 mL). The combined organic layers were dried over magnesium sulfate and concenfrated in vacuo to give the crude product (9.51 g) as an oil. Crystallization from hexanes / MTBE (50 mL : 25 mL) afforded 8-oxo-2,2,14,14- teframethylpentadecanedioic acid (6.92 g, 79 %) as waxy, white crystals. M.p.: 83 – 84 °C. 1H NMR (300 MHz, CDCI3/TMS): δ (ppm): 12.03 (s, 2 H), 2.37 (t, 4 H, J = 7.3 Hz), 1.52 – 1.34 (m, 8 H), 1.28 – 1.10 (m, 8 H), 1.06 (s, 12 H). 13C NMR (75 MHz, CDCI3/TMS): δ (ppm): 210.5, 178.8, 41.7, 41.2, 29.1, 25.0, 24.4, 23.1. HRMS (LSIMS, gly): Calcd. for C19H3505 (MH+): 343.2484, found: 343.2485.
6.20
8-Hydroxy-2.2.14,14-tetramethylpentadecanedioic acid
Under nitrogen atmosphere, sodium borohydride (0.06 g, 1.6 mmol) was added to a stiπed solution of 8-oxo-2,2,14,14-tetramethylpentadecanedioic acid (1.18 g, 3.4 mmol) in methanol (50 mL) at 0 °C. The reaction progress was momtored by thin layer chromatography (silica; hexanes : ethyl acetate = 50 : 50). Additional sodium borohydride was added after 1 h (0.48 g, 13 mmol). After 8 h, the reaction mixture was hydrolyzed with water (50 mL) and acidified with concenfrated hydrochloric acid (3 mL) to pH 1. The solution was diluted with water (50 mL) and exfracted with dichloromethane (4 x 25 mL). The combined organic layers were washed with saturated sodium chloride solution (2 x 30 mL), dried over magnesium sulfate, concentrated in vacuo, and dried in high vacuo to give 8-hydroxy-2,2,14,14-tetramethylpentadecanedioic acid (0.7 g, 60 %) as a very viscous oil.
!H NMR (300 MHz, CDC13/TMS): δ (ppm): 7.42 (br. s, 3 H), 3.59 (br. s, 1 H), 1.65 – 1.00 (m, 20 H), 1.18 (s, 12 H). 13C NMR (75 MHz, CDC13/TMS): δ (ppm): 184.5, 71.8, 42.1, 40.5, 37.0, 29.8, 25.2, 25.1, 24.9, 24.8.
HRMS (FAB): Calcd. for Cι9H3705 (MH+): 345.2635, found: 345.2646. HPLC: 83.8 % purity.
………………..
PAPER
Journal of Medicinal Chemistry, 47 (24), 6082-6099;. 2004
http://pubs.acs.org/doi/abs/10.1021/jm040006p

Keto-substituted hydrocarbons with 11−19 methylene and bis-terminal hydroxyl and carboxyl groups have been synthesized and evaluated in both in vivo and in vitro assays for their potential to favorably alter lipid disorders including metabolic syndrome. Compounds were assessed for their effects on the de novo incorporation of radiolabeled acetate into lipids in primary cultures of rat hepatocytes as well as for their effects on lipid and glycemic variables in obese female Zucker fatty rats [Crl:(ZUC)-faBR] following 1 and 2 weeks of oral administration. The most active compounds were found to be symmetrical with four to five methylene groups separating the central ketone functionality and the gem dimethyl or methyl/aryl substituents. Furthermore, biological activity was found to be greatest in both in vivo and in vitro assays for the tetramethyl-substituted keto diacids and diols (e.g., 10c, 10g,14c), and the least active were shown to be the bis(arylmethyl) derivatives (e.g., 10e, 10f,14f). Compound 14c dose-dependently elevated HDL-cholesterol, reduced triglycerides, and reduced NEFA, with a minimum effective dose of 30 mg/kg/day. Compound 10g dose-dependently modified non-HDL-cholesterol, triglycerides, and nonesterified fatty acids, with a minimum effective dose of 10 mg/kg/day. At this dose, compound 10g elevated HDL-cholesterol levels 2−3 times higher than pretreatment levels, and a dose-dependent reduction of fasting insulin and glucose levels was observed.
ONLY KETO COMPD DESCRIBED
2,2,14,14-Tetramethyl-8-oxopentadecanedioic Acid (10g). According to the procedure given for 10f, 9g (8.54 g, 21.4 mmol) was saponified with KOH (85%, 4.53 g, 68.6 mmol) in EtOH (13 mL) and water (5 mL) at reflux for 4 h. The solid product obtained after usual workup was recrystallized from Et2O/hexanes (50 mL/50 mL), affording 10g (4.16 g, 57%) as colorless needles.
Mp: 82−83 °C.
1H NMR (CDCl3): δ 11.53 (br, 2H), 2.39 (t, 4H, J = 7.3), 1.60−1.50 (m, 8 H), 1.30−1.20 (m, 8 H), 1.18 (s, 12 H).
13C NMR (CDCl3): δ 211.7, 185.0, 42.8, 42.3, 40.4, 29.7, 25.1, 24.8, 23.8.
HRMS (LSIMS, gly): calcd for C19H35O5 (MH+) 343.2484, found 343.2444.
HPLC: Alltima C-8 column, 250 × 4.6 mm, 5 μm; 60% acetonitrile/40% 0.05 M KH2PO4, flow rate 1.0 mL/min; RI, tR 6.50 min, 92.6% pure.
Anal. (C19H34O5): C, H.
A PRECURSOR OF Bempedoic Acid
PATENTs/PAPERS
WO2005068412,
WO2004067489,
Journal of Medicinal Chemistry, 47 (24), 6082-6099;. 2004
US20040198814
Esperion Therapeutics


Ringing the bell were Roger Newton, Esperion’s founder and chief science officer, and Tim Mayleben, the CEO and president.
Esperion raised about $73 million in its offering on June 26. It hopes to use the funds to conduct two Phase 3 U.S. Food and Drug Administration trials next year on a cholesterol-lowering drug with the working name of ETC-1002.
ETC-1002 has shown good results in preliminary human trials in lowering LDL, the so-called bad cholesterol, in patients who are either intolerant or resistant to such statin drugs as Lipitor and Torvast. More results are expected this summer.
This was the second IPO for a drug company called Esperion. The first Esperion was founded in 1998 to create a drug to raise HDL, the so-called good cholesterol. It went public in 2000 and was sold to Pfizer Inc. for $1.3 billion in 2004.
In 2008, as part of closing its Michigan operations, Pfizer sold the name and rights to some small molecules back to Newton.
 Esperion Therapeutics founder and chief scientific officer Roger Newton, left, and CEO and President Tim Mayleben celebrate the company’s initial public …
Esperion Therapeutics founder and chief scientific officer Roger Newton, left, and CEO and President Tim Mayleben celebrate the company’s initial public …


 Esperion cofounder Roger Newton was one of the Key players in the Development of LDL-Cholesterol Lowering Pfizer’s statin atorvastatin (Lipitor), the Biggest Selling Drug of All time with Annual Sales of Almost $ 13 Billion Dollars in 2006 at ITS Peak.
Esperion cofounder Roger Newton was one of the Key players in the Development of LDL-Cholesterol Lowering Pfizer’s statin atorvastatin (Lipitor), the Biggest Selling Drug of All time with Annual Sales of Almost $ 13 Billion Dollars in 2006 at ITS Peak.

Esperion President and CEO Tim Mayleben (left) and Chief Science Officer Roger Newton in the company’s labs at the Michigan Life Science and Innovation Center.Mayleben previously told AnnArbor.com that the drug being developed by the company, which is housed at the Michigan Life Science and Innovation Center in Plymouth Township, is undergoing the second round of “phase two” clinical tests. Most drugs go through three phases of testing before the results are submitted to the Food and Drug Administration. Mayleben said he does not expect the company to submit ETC-1002 to the FDA for approval for at least another three years.,Newton, a co-inventor of Lipitor and founder of the first Esperion, raised more than $22 million to buy the intellectual property from the original company back from Pfizer when the company closed its Ann Arbor offices in 2007…….http://www.annarbor.com/business-review/ann-arbor-pharmaceutical-company-esperion-therapeutics-to-ring-nasdaq-opening-bell-wednesday/
……………………
Michigan Life Science and Innovation Center in Plymouth Township
 SRI International’s Helen Parish (from left), David Sahner and Elizabeth Wood in November 2013 at the site of the nonprofit’s new clinical laboratory at the Michigan Life Science and Innovation Center in Plymouth Township.
SRI International’s Helen Parish (from left), David Sahner and Elizabeth Wood in November 2013 at the site of the nonprofit’s new clinical laboratory at the Michigan Life Science and Innovation Center in Plymouth Township.
 Michigan Life Science and Innovation Center
Michigan Life Science and Innovation Center
 Esperion Therapeutics CEO Roger Newton in his laboratory at the Michigan Life Science Innovation Center in Plymouth Township.
Esperion Therapeutics CEO Roger Newton in his laboratory at the Michigan Life Science Innovation Center in Plymouth Township.
 Pfizer Inc. announced Jan. 22, 2007 that it would close its Ann Arbor research campus on Plymouth Road and Huron Parkway. In the photo at left, then-Ann Arbor SPARK CEO Michael Finney, then Gov. Jennifer Granholm and Ann Arbor Mayor John Hieftje speak at a press conference addressing Pfizer’s announcement.
Pfizer Inc. announced Jan. 22, 2007 that it would close its Ann Arbor research campus on Plymouth Road and Huron Parkway. In the photo at left, then-Ann Arbor SPARK CEO Michael Finney, then Gov. Jennifer Granholm and Ann Arbor Mayor John Hieftje speak at a press conference addressing Pfizer’s announcement.
///////Bempedoic Acid, PHASE 2, Esperion Therapeutics, Roger Newton, Tim Mayleben, ETC 1002, ESP 55016
ASLAN Pharmaceuticals Gains Orphan Designation for Rare Cancer Drug ASLAN001 (varlitinib)

(R)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine

ASLAN001 , Varlitinib
C22H19ClN6O2S
Molecular Weight: 466.94
Elemental Analysis: C, 56.59; H, 4.10; Cl, 7.59; N, 18.00; O, 6.85; S, 6.87
CAS: 845272-21-1 (Varlitinib); 1146629-86-8 (Varlitinib tosylate).
ASLAN001; ASLAN-001; ASLAN 001; AR 00334543; ARRY-334543; ARRY334543; ARRY-543; ARRY543; ARRY 543.
(R)-N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)-N6-(4-methyl-4,5-dihydrooxazol-2-yl)quinazoline-4,6-diamine.
(R)-4-[[3-Chloro-4-[(thiazol-2-yl)methoxy]phenyl]amino]-6-[(4-methyl-4,5-dihydrooxazol-2-yl)amino]quinazoline
4,6-Quinazolinediamine, N4-[3-chloro-4-(2-thiazolylmethoxy)phenyl]-N6-[(4R)-4,5-dihydro-4-methyl-2-oxazolyl]-
ASLAN Pharmaceuticals, a Singapore-based drugmaker, announced The Food and Drug Administration (FDA) gave an orphan drug designation on August 13 to its pan-HER inhibitor ASLAN001 (varlitinib), a drug candidate created to treat a destructive form of bile duct cancer called cholangiocarcinoma that has no known cure. ………http://www.dddmag.com/news/2015/08/aslan-pharmaceuticals-gains-orphan-designation-rare-cancer-drug
Current developer: Array Biopharma Inc,
![]()
Varlitinib, also known as ARRY-543 and ASLAN001, is an orally bioavailable inhibitor of the epidermal growth factor receptor family with potential antineoplastic activity.
Varlitinib (ASLAN-001) is an oncolytic drug in phase II clinical trials at ASLAN Pharmaceuticals for the treatment of gastric cancer and for the treatment of metastatic breast cancer in combination with capecitabine. Clinical development is also ongoing for the treatment of solid tumors in combination with cisplatin/FU and cisplatin/capecitabine. The product had been in phase I/II clinical trials at Array BioPharma for the treatment of patients with advanced pancreatic cancer. Phase II clinical trials had also been ongoing for the treatment of solid tumors. No recent development has been reported for this research
Varlitinib selectively and reversibly binds to both EGFR (ErbB-1) and Her-2/neu (ErbB-2) and prevents their phosphorylation and activation, which may result in inhibition of the associated signal transduction pathways, inhibition of cellular proliferation and cell death. EGFR and Her-2 play important roles in cell proliferation and differentiation and are upregulated in various human tumor cell types. Due to the dual inhibition of both EGFR and Her-2, this agent may be therapeutically more effective than agents that inhibit EGFR or Her-2 alone.
The drug is a dual inhibitor of the ErB-2 and EGFR receptor kinases, both of which have been shown to stimulate aberrant growth, prolong survival and promote differentiation of many tumor types. The compound behaves as a reversible ATP-competitive inhibitor with nanomolar potency both in vitro and in cell-based proliferation assays.
In 2011, the compound was licensed to Aslan Pharmaceuticals by Array BioPharma worldwide for the treatment of solid tumors, initially targeting patients with gastric cancer through a development program conducted in Asia.
In 2015, orphan drug designation was assigned to the compound in the U.S. for the treatment of cholangiocarcinoma.
SEE NMR ………….http://www.medkoo.com/Product-Data/Varlitinib/Varlitinib-QC-KB20121128web.pdf
……………..
https://www.google.co.in/patents/US20050043334
Example 52
(R)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine
Prepared using (R)-2-aminopropan-1-o1. MS APCI (+) m/z 467, 469 (M+1, Cl pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, 1H), 8.47 (s, 1H), 8.09 (s, 1H), 7.86 (d, 1H), 7.81 (d, 1H), 7.77 (d, 1H), 7.69 (m, 3H), 7.32 (d, 1H), 7.02 (s, 1H), 5.54 (s, 2H), 4.47 (m, 1H), 3.99 (m, 1H), 3.90 (m, 1H), 1.18 (d, 3H).
Example 53

(S)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine
Prepared using (S)-2-amino-propan-1-o1. MS APCI (+) m/z 467, 469 (M+1, Cl pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, 1H), 8.47 (s, 1H), 8.09 (s, 1H), 7.86 (d, 1H), 7.81 (d, 1H), 7.77 (d, 1H), 7.69 (m, 3H), 7.32 (d, 1H), 7.02 (s, 1H), 5.54 (s, 2H), 4.47 (m, 1H), 3.99 (m, 1H), 3.90 (m, 1H), 1.18 (d, 3H).
………………
PATENT
http://www.google.co.in/patents/WO2005016346A1?cl=en
Example 52

R VN4-r3-Chloro-4-(‘thiazol-2-v-metho-xy)-phenyll-N6-(4-methyl-4,5-dihvdro-oxazol- 2-yl)-quinazoUne-4,6-diamine
[00194] Prepared using (R)-2-aminopropan- 1 -ol. MS APCI (+) m/z 467, 469
(M+l, CI pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, IH), 8.47 (s, IH), 8.09 (s, IH), 7.86 (d, IH), 7.81 (d, IH), 7.77 (d, IH), 7.69 (m, 3H), 7.32 (d, IH), 7.02 (s, IH), 5.54 (s, 2H), 4.47 (m, IH), 3.99 (m, IH), 3.90 (m, IH), 1.18 (d, 3H). Example 53

(S)-N4-|“3-Chloro-4- thiazol-2-ylmethoxy)-phenyll-N6-(‘4-methyl-4,5-dihvdro-oxazol- 2-yl)-quinazoline-4,6-diamine [00195] Prepared using (S)-2-amino-propan- 1 -ol. MS APCI (+) m z 467, 469
(M+l, CI pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, IH), 8.47 (s, IH), 8.09 (s, IH), 7.86 (d, IH), 7.81 (d, IH), 7.77 (d, IH), 7.69 (m, 3H), 7.32 (d, IH), 7.02 (s, IH), 5.54 (s, 2H), 4.47 (m, IH), 3.99 (m, IH), 3.90 (m, IH), 1.18 (d, 3H).
………
CAUTION a very similar molecule but not same
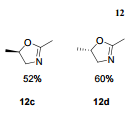 NOTE……..METHYL NEXT TO OXYGEN ATOM
NOTE……..METHYL NEXT TO OXYGEN ATOM
Design, Synthesis and Bioactivities Evaluation of Novel Quinazoline Analogs Containing Oxazole Units
DOI: 10.1002/cjoc.201400271,…………http://onlinelibrary.wiley.com/doi/10.1002/cjoc.201400271/abstract;jsessionid=04211D7526D97240F44E4355C6C57F50.f01t01
CAUTION …THIS IS NOT SAME
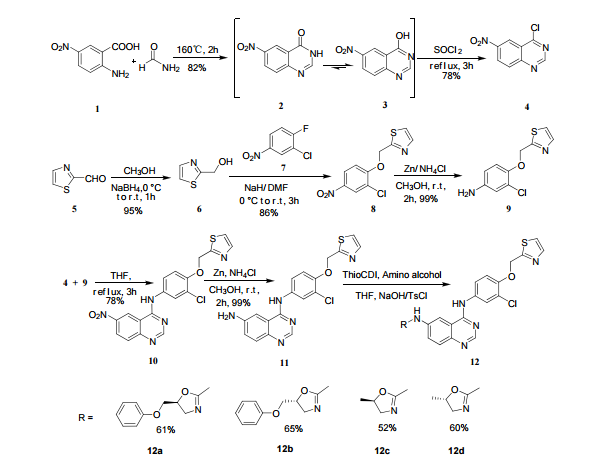
A novel type of quinazoline derivatives, which were designed by the combination of quinazoline as the backbone and oxazole scaffold as the substituent, have been synthesized and their biological activities were evaluated for anti-proliferative activities and EGFR inhibitory potency. Compound 12b demonstrated the most potent inhibitory activity (IC50=0.95 µmol/L for EGFR), which could be optimized as a potential EGFR inhibitor in the further study. The structures of the synthesized quinazoline analogs and all intermediates were comfirmed by 1H and 13C NMR, 2D NMR spectra, IR spectra and MS spectra.
12c: Employing the same method as above, compound 12c was prepared and the amino alcohol was (S)-2-amino-propan-1-ol. Yellow solid, yield 52 %. m.p. 243-244 °C; [α] 20D =﹢22.5 ° (c 1.0, CH3CN); 1 H NMR (DMSO-D6): δ 9.54 (s, 1 H), 8.46 (s, 1 H), 8.06 (s, 2 H), 7.85 (d, 2 H, J=3.3 Hz), 7.79 (d, 2 H, J=3.3 Hz), 7.75 (d, 1 H, J=8.9 Hz), 7.64 (d, 1 H, J=8.3 Hz), 7.30 (d, 1 H, J=9.0 Hz), 5.54 (s, 2 H), 4.76 (m, 1 H), 3.72 (s, 1 H), 3.19 (s, 1 H), 1.34 (d, 3 H, J=6.15 Hz). 13C NMR (DMSO-D6) δ: 165.8, 156.9, 152.0, 148.8, 145.3, 142.6, 134.3, 128.7, 128.0, 123.5, 121.7, 121.3, 121.0, 115.6, 114.6, 72.5, 67.7, 63.0, 29.8, 29.0, 20.0, 13.9. IR (KBr) ν: 3439, 3278, 3101, 2925, 1660, 1631, 1601, 1557, 1500, 1428, 1404, 1384, 1329, 1291, 1257, 1225, 1052 cm-1. Anal. calcd for C22H19N6O2SCl: C 55.59, H 4.10, N 18.00, O 6.85; found C 55.55, H 4.13, N 18.02, O 6.78; MS (ESI) m/z: 467.2 (M+H).
12d: Employing the same method as above, compound 12d was prepared and the amino alcohol was (R)-2-amino-propan-1-ol. Yellow solid, yield 60%. m.p. 242-243 °C; [α] 20D = ﹣22.3 ° (c 1.0, CH3CN); 1 H NMR (DMSO-D6): δ 9.52 (s, 1 H), 8.80 (s, 1 H), 8.52 (dd, 1 H, J=2.7 Hz, J=8.9 Hz), 8.45 (s, 1 H), 8.30 (s, 1 H), 8.07 (s, 1 H), 7.85 (d, 1 H, J=3.2 Hz), 7.79 (d, 1 H, J=3.2 Hz), 7.75 (s, 1 H), 7.63 (d, 1 H, J=8.2 Hz), 7.31 (d, 1 H, J=9.0 Hz), 5.53 (s, 2 H), 4.76 (m, 1 H), 3.81 (s, 1 H), 3.19 (s, 1 H), 1.34 (d, 3 H, J=6.2 Hz). 13C NMR (DMSO-D6) δ: 165.8, 156.9, 152.0, 148.8, 145.3, 142.6, 134.3, 128.7, 128.0, 123.5, 121.7, 121.3, 121.0, 115.6, 114.6, 72.5, 67.7, 63.0, 29.8, 29.0, 20.0, 13.9. IR (KBr) ν: 3439, 3278, 3101, 2925, 1660, 1631, 1601, 1557, 1500, 1428, 1404, 1384, 1329, 1291, 1257, 1225, 1052 cm-1. Anal. calcd for C22H19N6O2SCl: C 55.59, H 4.10, N 18.00, O 6.85; found C 55.55, H 4.13, N 18.02, O 6.78; MS (ESI) m/z: 467.20 (M+H).
The above paper allows you to synthesize the key amino int 11 ………N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)quinazoline-4,6-diamine (11)
this can be applied to varlitinib till int 11
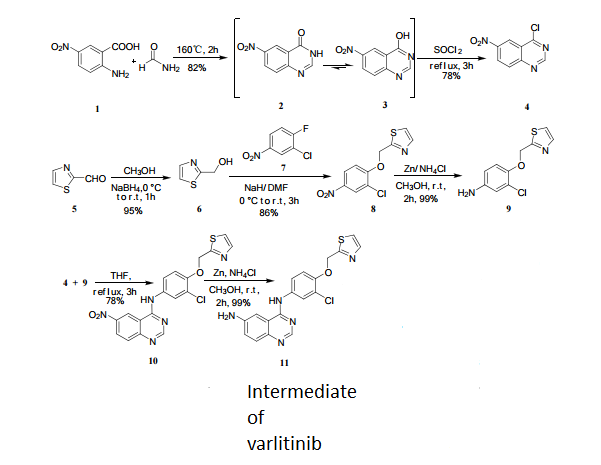
6-Nitro-4-hydroxyquinazoline (3)
2-amino-5-nitrobenzoic acid (5.46 g, 30 mmol) was added to a 250 mL flask equipped with a reflux condenser. Then 50 mL formamide was added. The mixture was heated with vigorous stirring at 160 °C for 3 h. After cooling the solution was poured in ice-water to give 3 in almost pure form (Yellow solid 4.70 g, yield 82.0%). m.p. 317-318 °C; 1 H NMR (DMSO-d6): δ 12.74 (1 H, s, OH, exchangeable), 8.78 (1 H, d, J=2.4 Hz), 8.53 (1 H, dd, J=2.6 Hz, 9.0 Hz), 8.30 (s, 1 H), 7.84 (1 H, d, J=9.0 Hz); 13C NMR (DMSO-d6) δ: 160.1, 152.9, 148.9, 145.0, 129.1, 128.3, 122.7, 121.9. IR (KBr) ν: 3172, 3046, 2879, 1674, 1615, 1577, 1514, 1491, 1469, 1343, 1289, 1242, 1167, 1112, 928, 920, 901, 803, 753, 630, 574, 531 cm-1. Anal. calcd for C8H5N3O3: C 50.27, H 2.64, N 21.98; found C 50.30, H 2.65, N 21.96; MS (ESI) m/z: 189.97 (M-H).
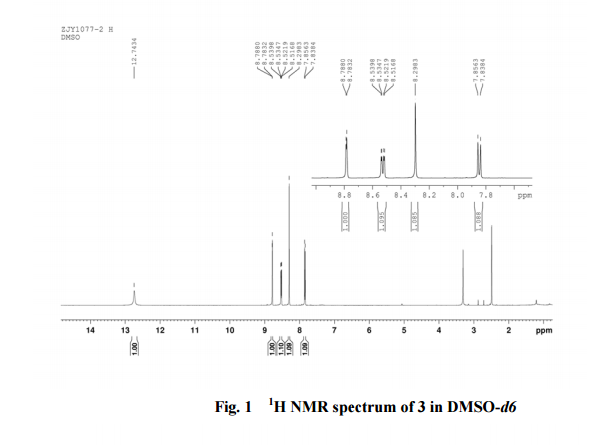
 13C NMR OF 3 IN DMSOD6
13C NMR OF 3 IN DMSOD6
IR
4-chloro-6-Nitroquinazoline (4)
In a 100 mL flask equipped with a reflux condenser, 6-nitroquinazolin-4-one (2.86 g, 15 mmol) and thionyl chloride (SOCl2) 25 mL were added. The mixture was heated under reflux with vigorous stirring for 2 h. After the solution was clear, the reaction mixture was heated for another 2 h. Then, 150 mL of ice MeOH was dropped into it carefully, the mixture was extracted with CH2Cl2. The organic layer was S3 dried under MgSO4, filtered and the solvent removed to give 4-chloro-6-nitroquinazoline (4). Yellow solid 2.45 g, yield 78%. m.p. 134-135 °C; 1 H NMR (DMSO-d6): δ 8.80 (1 H, d, J=3.0 Hz), 8.54(1 H, dd, J=2.7 Hz, 9.0 Hz), 8.35(s, 1 H), 7.87 (1 H, d, J= 9.0 Hz); 13C NMR (DMSO-d6) δ: 160.0, 152.5, 149.1, 145.1, 128.7, 128.4, 122.7, 122.0. IR (KBr) ν: 3431, 3082, 3038, 2664, 2613, 2567, 1724, 1685, 1676, 1646, 1617, 1578, 1526, 1468, 1359, 1346, 1269 cm-1. Anal. calcd for C8H4N3O2Cl: C 45.84, H 1.92, N 20.05, O 15.27; found C 45.81, H 1.97, N 20.02, O 15.21; MS (ESI) m/z: 207.96 (M-H).
13C NMR OF4 IN DMSOD6
IR
Thiazol-2-yl-methano1 (6)
Sodium borohydride (16.0 g, 140 mmol) was added to a stirred solution of thiazole-2-carbaldehyde (24.2 g, 214 mmol) in MeOH (400 mL) at 0 °C . The reaction mixture was warmed to room temperature. After 1 hour, the reaction mixture was quenched by the addition of water and the organics were removed by concentration. The resulting aqueous mixture was extracted with EtOAc. The combined organic extracts were dried under Na2SO4 and concentrated to give thiazol-2-yl-methano1 (23.39 g, 95%). bp:75-76 °C (0.2 mmHg) [lit.[19] bp:70-80 °C (0.2 mmHg)]; m. p. 63-64 °C. 1 H NMR (CDCl3) δ 4.91 (s, 2 H), 5.1(br, l H), 7.28(d, 1 H, J=3.2 Hz), 7.68 (d, 1 H, J=2.9 Hz). IR (KBr) ν: 3135, 3099, 3082, 2814, 1509, 1446, 1351, 1189, 1149, 1073, 1050, 977, 775, 745, 613, 603 cm-1. Anal. calcd for C4H5NOS: C 41.72, H 4.38, N 12.16; found C 41.74, H 4.33, N 12.18; MS (ESI) m/z: 116.11 (M+H).
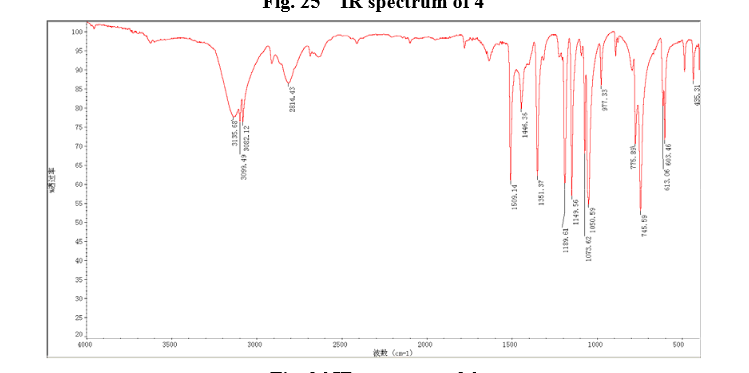
2-((2-Chloro-4-nitrophenoxy)methyl)thiazole (8)
2-(2-chloro-4-nitro-phenoxymethy1)-thiazole was prepared by adding thiazol-2-yl-methanol (5.48 g, 47.65 mmol) to a slurry of sodium hydride (2.42 g of a 60% dispersion in oil, 60.5 mmol) in THF (50 ml) at 0 °C After several minutes, 2-chloro-1-fluoro- 4-nitro-benzene (7.58 g, 43.60 mmol) was added and the reaction mixture warmed to room temperature. The reaction mixture was stirred at room temperature for 3 h, and 60 °C for 16 h. After cooling to room temperature, the reaction mixture was poured into 300 mL water. The resulting precipitate was collected by filtration, washed with water, and dried in vacuo to give 2-(2- chloro-4-nitrophenoxymethy1)-thiazole (11.06 g, 86%) which was used in next step without further purification. m.p. 170-171 °C; 1 H NMR (DMSO-d6): δ 8.35 (1 H, d, J=2.8 Hz), 8.25 (1 H, dd, J=2.8 Hz, 9.15 Hz), 7.87 (1 H, d, J=3.3 Hz), 7.83(1 H, d, J=3.3 Hz), 7.54 (1 H, d, J=9.2 Hz), 5.73(s, 1 H); 13C NMR (DMSO-d6) δ: 164.2, 158.5, 143.2, 141.7, 125.9, 124.9, 122.4, 122.2, 114.6, 68.4; IR (KBr) ν: 3112, 3009, 1587, 1509, 1500, 1354, 1319, 1284, 1255, 1154, 1125, 1054, 1006, 894, 780, 746, 728 cm-1. Anal. calcd for C10H7N2O3SCl: C 44.37, H 2.61, N 10.35, O 17.73; found C 44.31, H 2.67, N 10.29; MS (ESI) m/z: 268.89 (M-H).
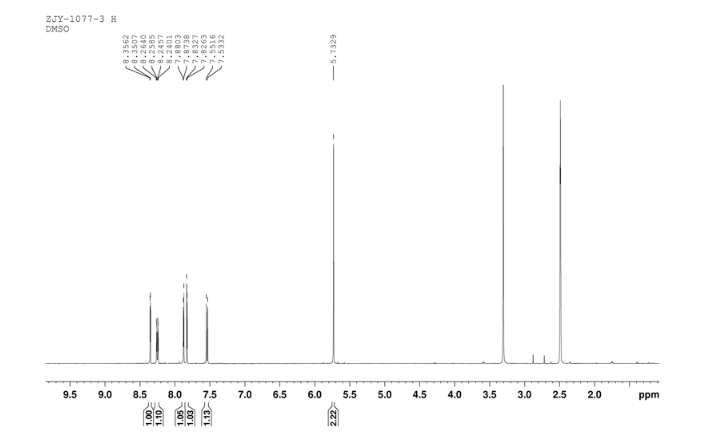 1H NMR 8 DMSOD6
1H NMR 8 DMSOD6
13C NMR OF 8 IN DMSOD6
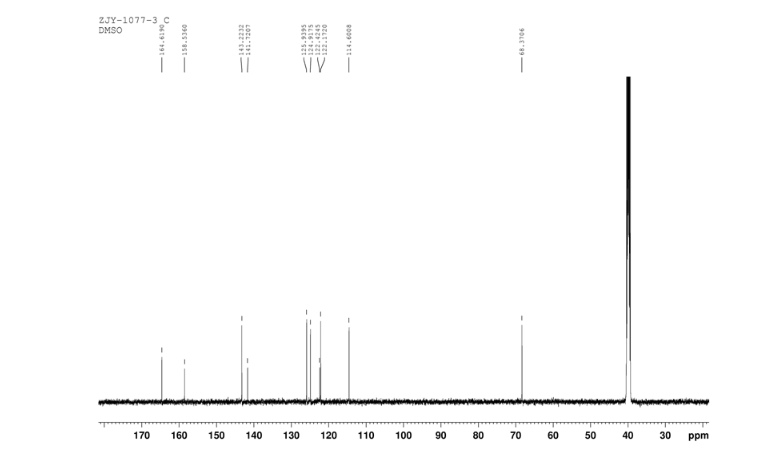
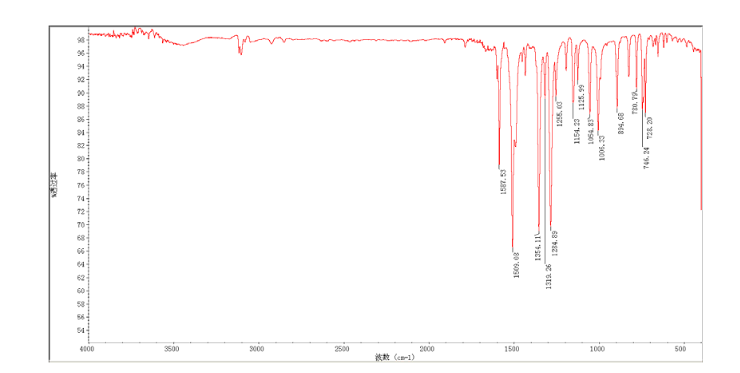
3-Chloro-4-(thiazol-2-ylmethoxy)aniline (9)
In a flask equipped with a reflux condenser, the compound 8 15.00 g (55.6 mmol), reduced zinc powder 14.44 g (222.0 mmo1, 4 eq), saturated ammonia chloride (5 mL) and methanol (100 mL) were mixed. The mixture was stirred at a temperature of 40 °C for 1.5 h. Then the zinc powder was filtered off, the filtrate was concentrated to obtain yellow solid 13.21 g, yield 99%. m.p. 60-61 °C; 1 H NMR (DMSO-d6): δ 7.80 (1 H, d, J=3.3 Hz), 7.75 (1 H, d, J=3.3 Hz), 6.96 (1 H, d, J=8.8 Hz), 6.64(1 H, d, J=2.7 Hz), 6.46 (1 H, dd, J=2.7 Hz, J=8.7 Hz), 5.30 (s, 2 H), 5.04 (s, 2 H, NH2, exchangeable); 13C NMR (DMSO-d6) δ: 166.8, 145.1, 144.1, 142.80, 123.1, 121.5, 117.7, 115.2, 113.6, 69.1. IR (KBr) ν: 3322, 3192, 3112, 1607, 1499, 1457, 1436, 1291, 1274, 1221, 1191, 1144, 1057, 1027, 857, 797, 767, 733, 584 cm-1. Anal. calcd for C10H9N2OSCl: C 49.90, H 3.77, N 11.64, O 6.65; found C 49.95, H 3.76, N 11.66, O 6.60; MS (ESI) m/z: 239.01 (M-H).
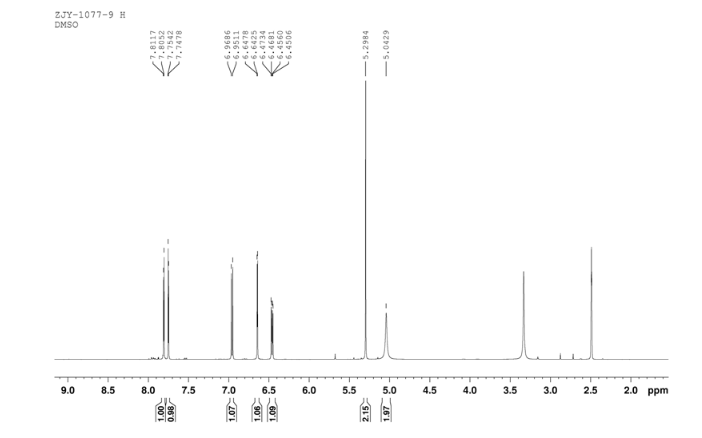 1H NMR DMSOD6 OF 9
1H NMR DMSOD6 OF 9
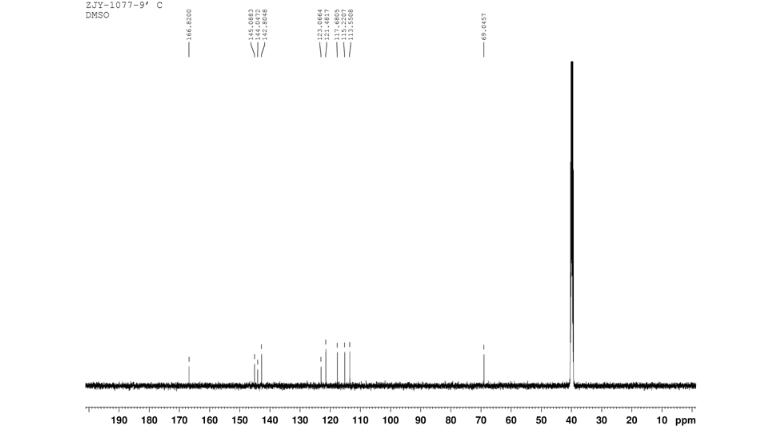 13C NMR OF 9 IN DMSOD6
13C NMR OF 9 IN DMSOD6
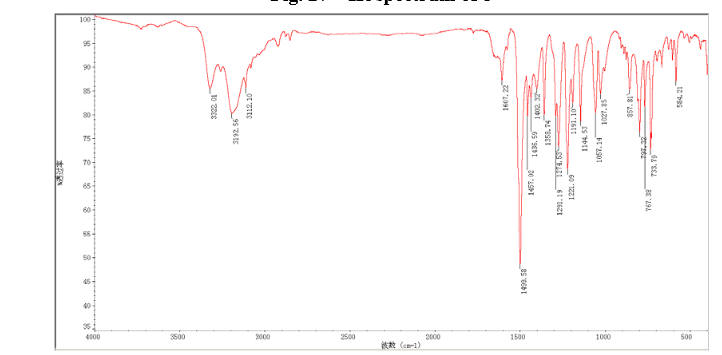
N-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)-6-nitro- quinazolin-4-amine(10)
In a flask equipped with a reflux condenser, 6-nitro-4-chloro- quinazoline 8.0 g (38.3 mmol) and 3-Chloro-4-(thiazol-2-ylmethoxy)aniline 8.9 g (37.0 mmol) were dissolved into 150 mL of THF, and the solution was refluxed for 3 h.Then a lot of yellow solid was deposited. Then it was filtered affording to yellow solid 12.8 g, yield 81%. m.p. 183-184 °C (decompose); 1 H NMR (DMSO-d6): δ 11.97(s, 1 H, exchangeable), 9.84 (s, 1 H), 9.00 (s, 1 H), 8.76 (1 H, d, J=9.1 Hz), 8.12-8.14 (m, 1 H), 7.94 (1 H, d, J=2.3 Hz), 7.87 (1 H, d, J=3.2 Hz), 7.81 (1 H, d, J=3.2 Hz), 7.44 (1 H, d, J=9.0 Hz), 7.69 (1 H, dd, J=2.5 Hz, J=8.9 Hz), 5.61 (s, 2 H); 13C NMR (DMSO-d6) δ: 166.8, 145.1, 144.1, 142.8, 123.1, 121.5, 117.7, 115.2, 113.7, 69.1. IR (KBr) ν: 3442, 3100, 1636, 1618, 1570, 1552, 1523, 1492, 1442, 1400, 1377, 1344, 1301, 1267, 1069, 805 cm-1. Anal. calcd for C18H12N5O3SCl: C 52.24, H 2.92, N 16.92, O 11.60; found C 52.26, H 2.93, N 16.96, O 11.58; MS (ESI) m/z: 412.84 (M-H).
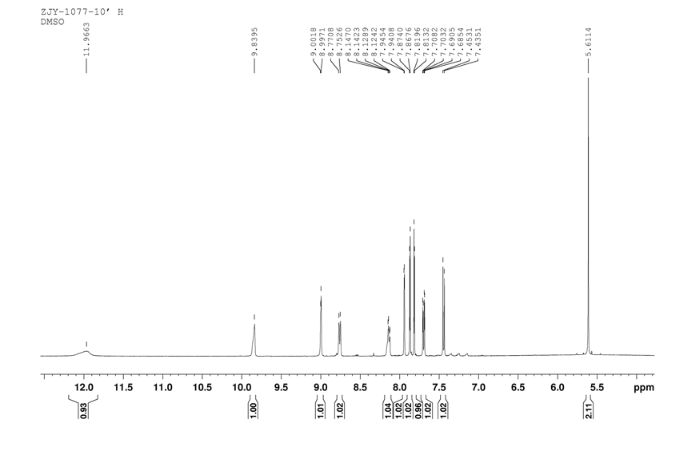 1H NMR DMSOD6 OF 10
1H NMR DMSOD6 OF 10
 13C NMR OF 10 IN DMSOD6
13C NMR OF 10 IN DMSOD6
N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)quinazoline-4,6-diamine (11)
In a flask equipped with a reflux condenser, the compound 10 5.00 g (12.1 mmol), reduced zinc powder 3.2 g (48.5 mmo1, 4 eq), saturated ammonia chloride (3 mL) and methanol (60 mL) were mixed. The mixture was stirred at room temperature for 30 min. Then the zinc powder was filtered off, the filtrate was concentrated to obtain yellow solid 4.58 g, yield 98%. m.p. 197-198 °C (decompose); 1 H S4 NMR (DMSO-d6): δ 9.33(s, 1 H, exchangeable), 8.31 (s, 1 H), 8.05 (d, 1 H, J=2.6 Hz), 7.85 (d, 1 H, J=3.3 Hz), 7.79 (1 H, d, J=3.3 Hz), 7.73 (1 H, dd, J=2.5 Hz, J=9.0 Hz), 7.51 (1 H, d, J=8.9 Hz), 7.30 (1 H, d, J=2.4 Hz), 7.29 (1 H, d, J=4.7 Hz), 7.23 (1 H, dd, J=2.3 Hz, J=8.9 Hz), 5.57 (s, 2 H, exchangeable), 5.52 (s, 2 H); 13C NMR (DMSO-d6) δ: 165.9, 155.8, 149.7, 148.5, 147.3, 142.6, 142.5, 134.6, 128.7, 123.6, 123.2, 121.4, 121.3, 121.1, 116.5, 114. 7, 100.9, 67.8. IR (KBr) ν: 3443, 3358, 3211, 3100, 1631, 1596, 1577, 1560, 1530, 1494, 1431, 1383, 1217, 910 cm-1. Anal. calcd for C18H14N5OSCl: C 56.32, H 3.68, N 18.24, O 4.17; found C 56.34, H 3.70, N 18.22, O 4.14; MS (ESI) m/z: 382.66 (M-H).
 11 1HNMR DMSOD6
11 1HNMR DMSOD6
 13C NMR OF 11 IN DMSOD6
13C NMR OF 11 IN DMSOD6
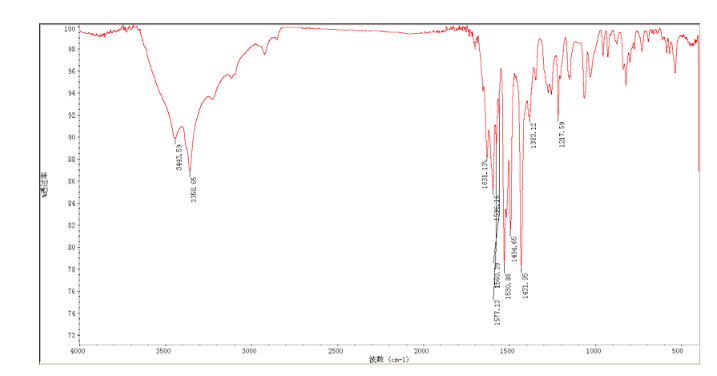
Construction finally as per patent ……….US20050043334
Treatment of N4-[3-chloro-4-(thiazol-2-ylmethoxy)phenyl]quinazoline-4,6-diamine (11) with 1,1′-thiocarbonyldiimidazole , followed by condensation with 2(R)-amino-1-propanol in THF/CH2Cl2 affords thiourea derivative , which finally undergoes cyclization in the presence of TsCl and NaOH in THF/H2O to furnish varlitinib .
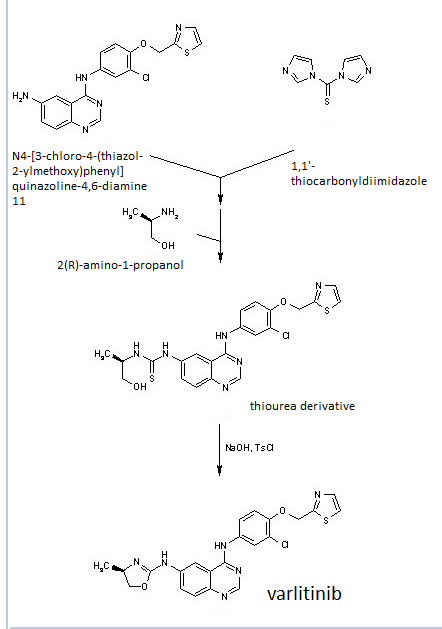
-
ASLAN Pharmaceuticals
-
Address: 10 Bukit Pasoh Rd, Singapore 089824Phone:+65 6222 4235


 carl fith
carl fith
Mr Carl Firth, CEO, Aslan Pharmaceuticals, Singapore (left) and Mr Dan Devine, CEO, Patrys, Australia (right)
///////ASLAN001, varlitinib, ASLAN Pharmaceuticals, Orphan Designation, ARRY-534, ARRY-334543 , PHASE 2, ORPHAN DRUG DESIGNATION, array
Filgotinib

Filgotinib
EU APPROVED 2020/9/24, JYSELECA
JAPAN APPROVED2020/9/25
- C21H23N5O3S
- MW425.504
- Elemental Analysis: C, 59.28; H, 5.45; N, 16.46; O, 11.28; S, 7.54
1206161-97-8
Cyclopropanecarboxamide, N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]-
G146034
GLPG0634
N-(5-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide
Galapagos Nv INNOVATOR
PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulcerative colitis
Filgotinib is an orally available inhibitor of JAK1/JAK2 and TYK2 in phase III clinical development at Galapagos and Gilead for the treatment of rheumatoid arthritis, moderate or severe Crohn’s disease and ulcerative colitis
IL-6 antagonist; Jak1 tyrosine kinase inhibitor; Tyk2 tyrosine kinase inhibitor; Jak3 tyrosine kinase inhibitor; Jak2 tyrosine kinase inhibitor
Autoimmune disease; Cancer; Colitis; Crohns disease; Inflammatory disease; Neoplasm; Rheumatoid arthritis; Transplant rejection
In 2017, orphan drug designation was assigned to the compound in the U.S. for the treatment of pediatric Crohn’s disease and pediatric ulcerative colitis.
GlaxoSmithKline had been developing filgotinib preclinically for the treatment of rheumatoid arthritis pursuant to a license; however, in 2010, the compound was re-acquired by Galapagos. In 2012, the product was licensed to Abbott for development and marketing. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. The license agreement between Galapagos and Abbott was terminated in September 2015, Galapagos regaining all rights to the product. The same year, Galapagos and Gilead entered into a global partnership and Gilead obtained the global rights of codevelopment and commercialization for the treatment of inflammatory diseases
Filgotinib (GLPG0634), by the Belgian biotech company Galápagos NV, is a drug which is currently under investigation for the treatment of rheumatoid arthritis and Crohn’s disease.
Filgotinib (GLPG0634) is an orally-available, selective inhibitor of JAK1 (Janus kinase 1) for the treatment of rheumatoid arthritis and potentially other inflammatory diseases. Filgotinib (GLPG0634) dose-dependently inhibited Th1 and Th2 differentiation and to a lesser extent the differentiation of Th17 cells in vitro. GLPG0634 was well exposed in rodents upon oral dosing, and exposure levels correlated with repression of Mx2 expression in leukocytes. The JAK1 selective inhibitor GLPG0634 (Filgotinib) is a promising novel therapeutic with potential for oral treatment of rheumatoid arthritis and possibly other immune-inflammatory diseases. Filgotinib (GLPG0634) is currently in a Phase 2 study in Crohn’s disease.

Mechanism of action
Filgotinib is a Janus kinase inhibitor with selectivity for subtype JAK1 of this enzyme. It is considered a promising agent as it inhibits JAK1 selectively. Less selective JAK inhibitors (e.g. tofacitinib) are already being marketed. They show long-term efficacy in the treatment of various inflammatory diseases. However, their lack of selectivity leads to dose-limiting side effects.[1] It is thought that inhibition of all JAK isoenzymes is beneficial in rheumatoid arthritis. However, pan-JAK inhibition might also lead to unwanted side effects that might not outweigh its benefits. This is the rationale for the development of newer and more selective inhibitors like filgotinib.
The signal transmission of large numbers of proinflammatory cytokines is dependent on JAK1. Inhibition of JAK2 may also contribute to the efficacy against RA. Nonetheless it is thought that JAK2 inhibition might lead to anemia and thrombopenia by interference witherythropoietin and thrombopoietin and granulocyte-macrophage colony-stimulating factor. Therefore one might prefer to choose a more selective JAK1 inhibitor as a primary therapeutic option. Filgotinib exerts a 30-fold selectivity for JAK1 compared to JAK2.[2] It is however still to be seen to what extent JAK2 inhibition should be avoided.
Novel crystalline forms of filgotinib salts, particularly hydrochloride salt, useful for treating JAK-mediated diseases eg inflammatory diseases, autoimmune diseases, proliferative diseases, allergy and transplant rejection. Galapagos and licensee AbbVie are developing filgotinib, a selective JAK-1 inhibitor, for treating rheumatoid arthritis (RA) and Crohn’s disease (CD). In August 2015, the drug was reported to be in phase 2 clinical development for treating RA and CD. The drug is also being investigated for the treatment of colitis and was discovered as part of the company’s arthritis alliance with GSK; however in August 2010 Galapagos reacquired the full rights. See WO2013189771, claiming use of filgotinib analog for treating inflammatory diseases. Also see WO2010010190 (co-assigned with GSK and Abbott) and WO2010149769 (assigned to Galapagos) claiming filgotinib, generically and specifically, respectively.
Clinical trials and approval
The efficacy of filgotinib is currently studied in a phase2b program (DARWIN trial 1, 2) with involvement of 886 rheumatoid arthritis patients and 180 Crohn’s disease patients.
Phase 1 study
It was shown in phase 1 studies that the pharmacokinetics of filgotinib metabolism is independent of hepatic CYP450 enzymatic degradation. The drug metabolism is however mediated by carboxylesterases. There is no interference reported with the metabolism of methotrexate nor with any of the investigated transport proteins.[3]
Phase 2 study: Proof of concept (2011)
In november 2011 Galápagos released the results of their phase 2 study (identification: NCT01384422, Eudract: 2010-022953-40) in which 36 patients were treated who showed a suboptimal clinical response to methotrexate treatment. Three groups of twelve patients were treated either with 200 mg filgotinib in a single dose, 200 mg divided in two doses or placebo. The primary end-point was the ACR20 score, which monitors improvements in the symptomatology of the patient. After the scheduled 4 weeks of treatment, 83% of the respondents showed an improved ACR20-score. Half of the treated patients showed a complete (or near complete) remission of the disease. There were no reports ofanemia nor changes in lipidemia. The company stated in their press release that filgotinib is the first selective JAK1 inhibitor that shows clinical efficacy. As a result of this study, the company stated that “GLPG0634 shows one of the highest initial response rates ever reported for rheumatoid arthritis treatments”.[4]
DARWIN 1 trial
The DARWIN 1 trial is a 24 week double blind placebo-controlled trial with 599 rheumatoid arthritis patients enrolled. All participants have moderate to severe RA and showed an insufficient response to standard methotrexate treatment. The trial compares three dosages of filgotinib as a once or twice per day regimen. During the trial all participants remain on their methotrexate treatment. According to the company, the results of this trial are expected in July 2015.[5]
DARWIN 2 trial
The DARWIN 2 trial is a double blind placebo-controlled trial with 280 rheumatoid arthritis patients enrolled who show an insufficient response to standard methotrexate treatment. This trial, in contrast to the previous DARWIN 1 trial, methotrexate is discontinued. Therefore, this trial investigates filgotinib as a monotherapy.[6] The recruitment of DARWIN trial 2b ended in november 2014.[7] Preliminary results are expected in the second quarter of 2015 and a full completion of the study is expected in the third quarter of 2015.
DARWIN 3 trial
Patients who complete DARWIN 1 and 2 will be eligible for DARWIN 3.
COSY PREDICT
Time line
- june 2011: results of first phase 2 trial
- november 2014: initiation of DARWIN 1 and 2 trials
- april 2015: expected date of DARWIN 1 trial results
- june 2015: expected date of DARWIN 2 trial results
NMR FROM NET….ABMOLE, DMSOD6
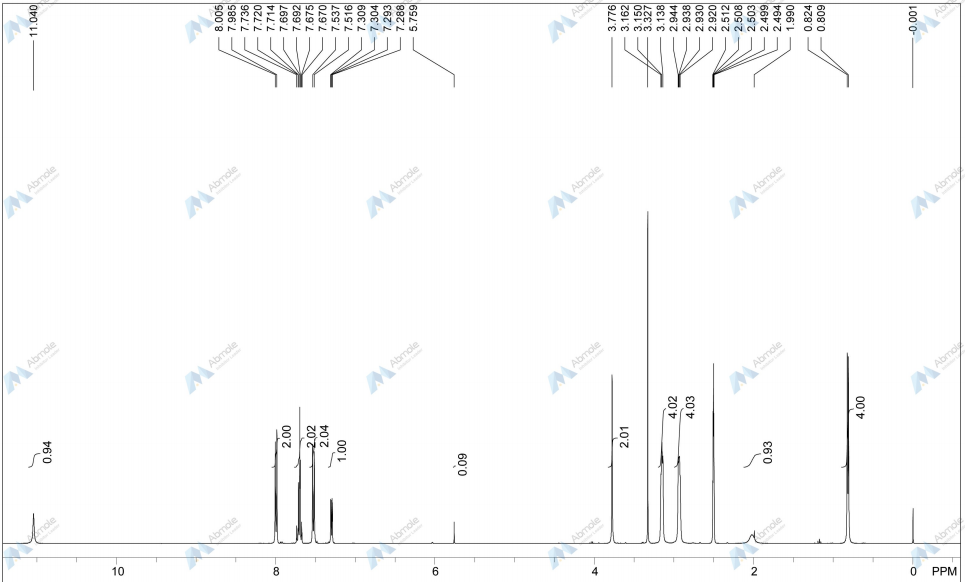
NMR MEDKOO DMSOD6

CHEMIETEK
1H NMR PREDICT


13C NMR PREDICT
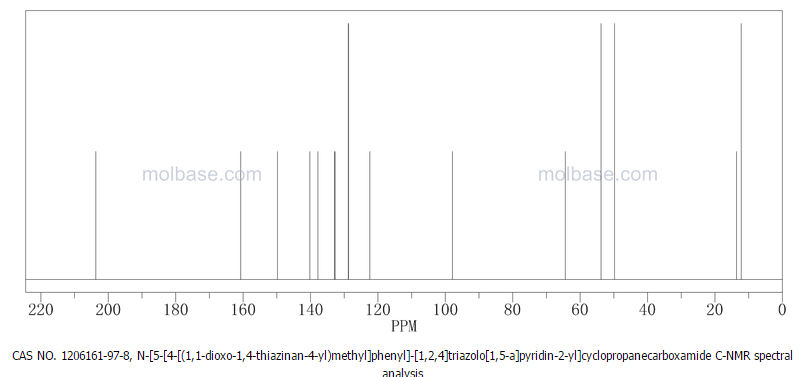

……………………
MORE PREDICTS
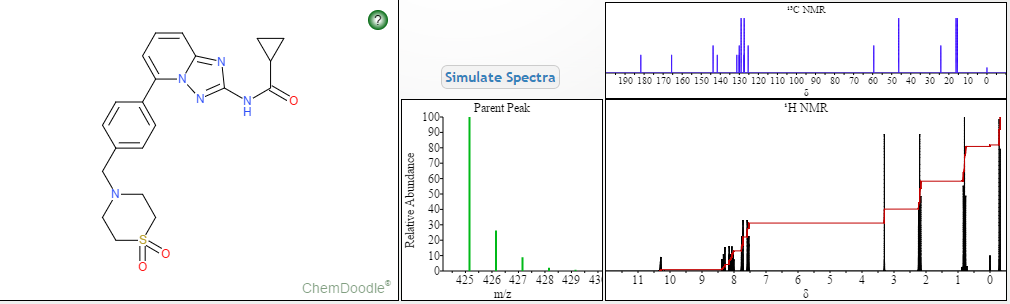
1H NMR PREDICT
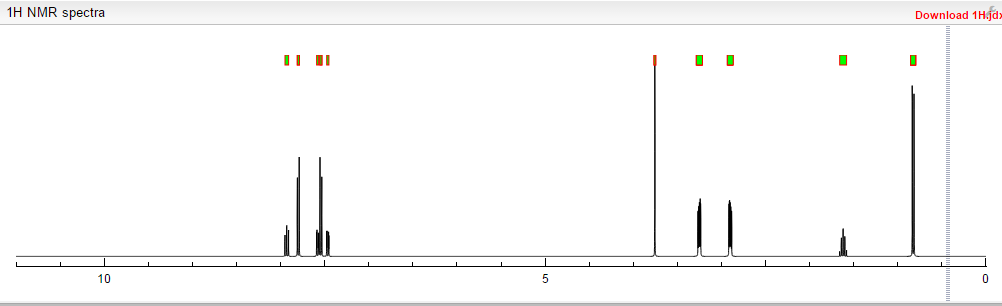
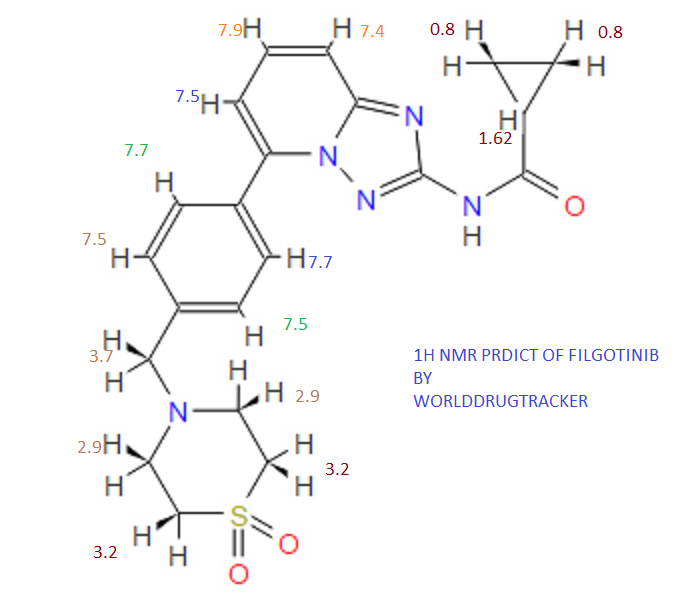
13C NMR PREDICT
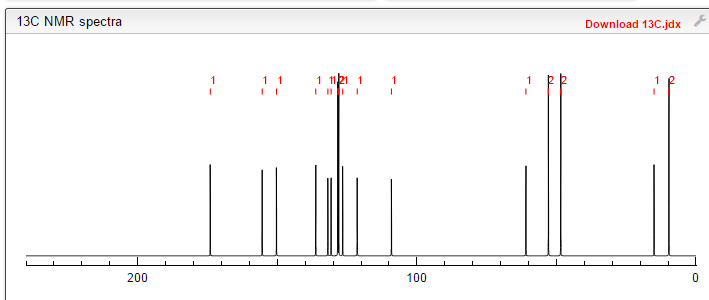
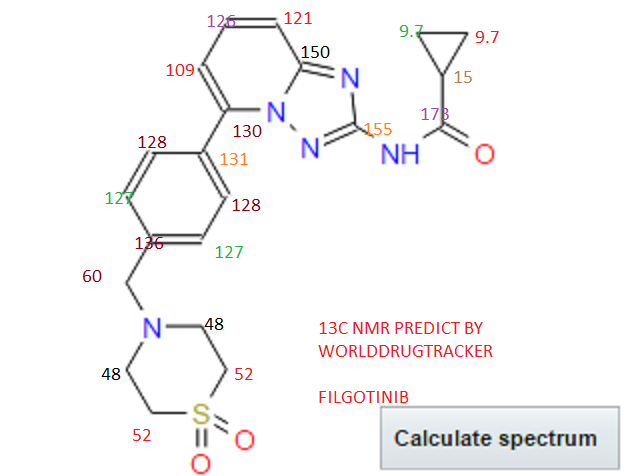
PRODUCT PATENT
http://www.google.com/patents/WO2010149769A1?cl=en
| Applicants: | GALAPAGOS NV [BE/BE]; Generaal De Wittelaan L11/A3 B-2800 Mechelen (BE) (For All Designated States Except US). MENET, Christel Jeanne Marie [FR/BE]; (BE) (For US Only). SMITS, Koen Kurt [BE/BE]; (BE) (For US Only) |
| Inventors: | MENET, Christel Jeanne Marie; (BE). SMITS, Koen Kurt; (BE) |
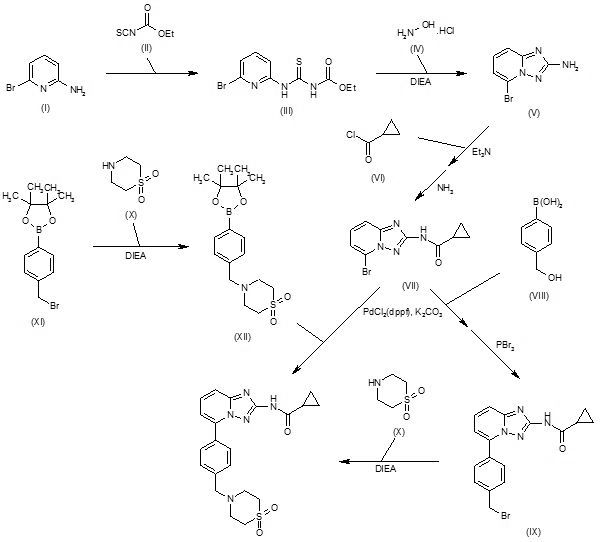
| International Filing Date: | 25.06.2010 |
ESTIMATED EXP 2030
Condensation of 2-amino-6-bromopyridine (I) with ethoxycarbonyl isothiocyanate (II) in CH2Cl2 gives 1-(6-bromopyridin-2-yl)-3-carboethoxythiourea (III), which upon cyclization with hydroxylamine hydrochloride (IV) in the presence of DIEA in EtOH/MeOH yields 2-amino-5-bromo[1,2,4]triazolo[1,5-a]pyridine (V). N-Acylation of amine (V) with cyclopropanecarbonyl chloride (VI) using Et3N in acetonitrile, and subsequent treatment with methanolic ammonia furnishes the carboxamide (VII) (1-3), which upon Suzuki coupling with 4-(hydroxymethyl)phenylboronic acid (VIII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C, followed by bromination with PBr3 in CHCl3 affords intermediate (IX). Condensation of benzyl bromide derivative (IX) with thiomorpholine-1,1-dioxide (X) using DIEA in CH2Cl2/MeOH yields filgotinib (1,2). Alternatively, condensation of (4-bromomethylphenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (XI) with thiomorpholine 1,1-dioxide (X) in the presence of DIEA in CH2Cl2/MeOH gives intermediate (XII), which undergoes Suzuki coupling with aryl bromide (VII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C to afford the target filgotinib
The present invention is based on the discovery that the compound of the invention is able to act as an inhibitor of JAK and that it is useful for the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6. In a specific aspect the compound is an inhibitor of JAKl and JAK2. The present invention also provides methods for the production of this compound, a pharmaceutical composition comprising this compound and methods for treating inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 by administering the compound of the invention.
Accordingly, in a first aspect of the invention, a compound of the invention is provided having a formula (I):

[0017] The compound of the invention is a novel inhibitor of JAK that appears to exhibit a dramatically improved in vivo potency as compared to structurally similar compounds. In a particular embodiment the compound of the invention is an inhibitor of JAKl and JAK2. In particular it appears to exhibit this increase in potency at lower in vivo exposure levels compared to structurally similar compounds. The use of a compound with these improvements is expected to result in a lower dosage requirement (and therefore an improved dosing schedule).
General Synthetic Method Scheme 1

1. RCOCI, Et3N 2. NH3 / MeOH CH3CN, 20 0C 2O 0C

wherein Ar represents phenyl-Ll-heterocycloalkyl, where Ll is a bond, -CH2– or -CO- and the heterocycloalkyl group is optionally substituted.
General
1.1.1 l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

(2)
[00117] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5 0C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20 0C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
7.7.2 5-Bromo-[l, 2, 4]triazolo[l, 5-a]pyridin-2-ylamine (3)

[00118] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH
(1 :1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 0C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition Of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-t/β) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (IH, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 :1, M+H+, 100%).
7.7.3 General procedure for mono-acylation to afford intermediate (4):

[00119] To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN
(150 mL) at 5 0C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2x50mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A
Preparation of compounds of the invention via Suzuki coupling (5):
[00120] An appropriate boronic acid (2eq.) is added to a solution of bromo intermediate (4) in
1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters
XBridge Prep Cl 8 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using
MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

Bl. 4 4-[2-(Cyclopropanecarbonyl-amino)-[ 1 , 2, 4]triazolo[l, 5-a] pyridin-5-yl] -benzoyl chloride

[00121] 2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)- [l,2,4]triazolo[l,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide formation (General Method)

[00122] An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 00C. The acid chloride (Bl, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method C

Wherein R3a or R3b together with the nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reductive alkylation (general method)
[00123] An appropriate amine (2 eq.), cyclopropanecarboxylic acid (for example cyclopropanecarboxylic acid [5-(4-formyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridine-2-yl]-amide) prepared by method A (1 eq.) and Ti(OPr)4 are mixed and stirred at room temperature for 3 hrs. The mixture is diluted in ethanol and Na(CN)BH3 (leq.) is added. The resulting solution is stirred at room temperature for 16 hrs. The mixture is diluted in water and filtered. The filtrate is washed with ethanol. The combined solvent phases are evaporated under vacuum. The final compound is isolated by preparative HPLC.
Method D 
wherein R1 and R2 together with the Nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reaction ofalkylation

[00124] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and Et3N (2 eq) (or AgCO3) are dissolved in DCM/MeOH (4:1 v:v) under N2 and an amine (2 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSθ4. Organic layers are filtered and evaporated. The final compound is isolated by flash chromatography.
Suzuki coupling

[00125] The obtained boronic acid (2eq.) is added to a solution of cyclopropanecarboxylic acid
(5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide (4) in 1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (This reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Synthesis of the compound of the invention and comparative examples
Compound l(the compound of the invention)
Step 1:

[00126] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and DIPEA
(2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgS O4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
Step 2: Suzuki coupling

[00127] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide
(l.leq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00128] Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 500C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative route to Compound l(the compound of the invention):
Step 1:

[00129] 4-(Hydroxymethyl)phenylboronic acid (l.leq.) was added to a s o luti o n o f cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

[00130] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)- [l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSθ4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

[00131] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin- 2-yl]-amide (leq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO^ Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
PATENT
WO 2010010190
WO 2013173506
WO 2013189771
WO 2015117980
WO 2015117981
POLYMORPH
CN 105061420
https://encrypted.google.com/patents/CN105061420A?cl=en
JAK inhibitor N-(5-(4-(1,1-dioxothiomorpholinyl)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide, and methods for preparing the four crystal forms, wherein the four crystal forms respectively are a crystal form H1, a crystal form H2, a crystal form H3 and a crystal form H4,
POLYMORPH
E CRYSTAL
CN 105111206
D CRYSTAL
CN 105111207
H CRYSYAL
CN 105198876
G
CN 105198877
F CN 105198878
C CN 105198880
POLYMORPH
WO 2016105453
POLYMORPH
Preparation of solid forms of filgotinib free base
WO 2017012773
The present invention relates to cryst. filgotinib free base (I), a method of its prepn. and a pharmaceutical compn. comprising the same. Thus, reacting the compd. II with thiomorpholine dioxide afforded 92% I (purity of 98.6%) which was then converted into its hydrochloride salt. Pharmaceutical compn. comprising compd. I was disclosed.
POLYMORPH
CN 105669669
The present invention provides a crystal form A, B, D, G and M of N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide hydrochloride.
PAPER
Future Medicinal Chemistry (2015), 7(2), 203-235. | Language: English, Database: CAPLUSA review. The discovery of the JAK-STAT pathway was a landmark in cell biol. The identification of these pathways has changed the landscape of treatment of rheumatoid arthritis and other autoimmune diseases. The two first (unselective) JAK inhibitors have recently been approved by the US FDA for the treatment of myelofibrosis and rheumatoid arthritis and many other JAK inhibitors are currently in clin. development or at the discovery stage. Research groups have demonstrated the different roles of JAK member and the therapeutic potential of targeting them selectively. ………..
https://www.future-science.com/doi/10.4155/fmc.14.149
PAPER
Journal of Pharmaceutical Sciences (Philadelphia, PA, United States) (2018), 107(6), 1624-1632.
PATENT
US2010/331319 A1, ; Page/Page column 13-14
http://www.google.com/patents/US20100331319
Synthetic Preparation of the Compound of the Invention and Comparative Examples
The compound of the invention and the comparative examples can be produced according to the following scheme.

wherein Ar represents phenyl-L1-heterocycloalkyl, where L1 is a bond, —CH2— or —CO— and the heterocycloalkyl group is optionally substituted.
General 1.1.1 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5° C. is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20° C.) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (1H, br s, NH), 8.81 (1H, d, J=7.8 Hz, H-3), 8.15 (1H, br s, NH), 7.60 (1H, t, J=8.0 Hz, H-4), 7.32 (1H, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.2 5-Bromo-[1,2,4]triazolo[1,5-a]pyridin-2-ylamine (3)

To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1:1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20° C.) for 1 h. 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1:1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-d6) δ 7.43-7.34 (2H, m, 2×aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1:1, M+H+, 100%).
1.1.3 General Procedure for Mono-Acylation to Afford Intermediate (4)

To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN (150 mL) at 5° C. is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp. (for 1-16 h) to hydrolyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2×50 mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A Preparation of Compounds of the Invention Via Suzuki Coupling (5):
An appropriate boronic acid (2 eq.) is added to a solution of bromo intermediate (4) in 1,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140° C. for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 90° C. for 16 h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSO4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

B1. 4 4-[2-(Cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoyl chloride

2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide Formation (General Method)

An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 0° C. The acid chloride (B1, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
…
Synthesis of the Compound of the Invention and Comparative Examples Compound 1 (the Compound of the Invention) Step 1:

2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
STEP 2: Suzuki coupling

4-[4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-1,1-dioxide (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 50° C., the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative Route to Compound 1 (the Compound of the Invention): Step 1:

4-(Hydroxymethyl)phenylboronic acid (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSO4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
…………………….
PATENT
Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders
Also claims a method for preparing filgotinib hydrochloride trihydrate. The present filing forms a pair with this week’s filing, WO2015117980, claiming a tablet composition comprising filgotinib hydrochloride.
The compound cyclopropanecarboxylic acid {5-[4-(l,l-dioxo-thiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5-a]pyridin-2-yl -amide (Compound 1), which has the chemical structure:

is disclosed in our earlier application WO 2010/149769 (Menet C. J., 2010) as being an inhibitor of JAK and as being useful in the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, allergy, transplant rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 or interferons. Hereafter this compound is named Compound 1. The data presented in WO 2010/149769 demonstrate that despite similar in vitro activities, Compound 1 has unexpectedly high in vivo potency compared with structurally similar compounds.
Example 1. Preparation of Compound 1
1.1. Route 1
1.1.1. 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide

[00205] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) is added portionwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated without further purification.
1.1.2. Cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide

1.1.2.1. Step i): l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea
[00206] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5°C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction
mixture is then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3 x 600 niL) and air-dried to afford the desired product. The thiourea may be used as such for the next step without any purification. lH (400 MHz, CDC13) δ 12.03 (1H, br s), 8.81 (1H, d), 8.15 (1H, br s), 7.60 (1H, t), 7.32 (1H, dd), 4.31 (2H, q), 1.35 (3H, t).
1.1.2.2. Step ii): 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine
[00207] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) is added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids are washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2.3. Step Hi): Cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide
[00208] To a solution of the 2-amino-triazolopyridine obtained in the previous step (7.10 g, 33.3 mmol) in dry MeCN (150 mL) at 5°C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et20 (50 mL). The solids are collected by filtration, washed with H20 (2x50mL), acetone (50 mL) and Et20 (50 mL), then dried in vacuo to give the desired compound.
1.1.3. Compound 1

[00209] 4-[4-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)-benzyl] hiomoφholine , l -dioxide (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo.
[00210] The final compound is obtained after purification by flash chromatography.
[00211] Alternatively, after completion of the reaction, a palladium scavenger such as 1 ,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cool down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HC1 is added, and after stirring at room temperature, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at room temperature, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H20, treated with a palladium scavenger (e.g. SMOPEX 234) at 50°C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the desired compound as a free base.
1.2. Route 2
1.2.1. Step 1: cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2, 4]triazolo[l, 5- a] pyridin-2-yl] -amide

[00212] 4-(Hydroxymethyl)phenylboronic acid (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water
(4:1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo. The resulting mixture is used without further purification.
1.2.2. Step 2: Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5- a Jpyridin-2-ylJ -amide

[00213] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl] -amide (1.0 eq) in chloroform is slowly added phosphorus tribromide (1.0 eq.). The reaction mixture is stirred at room temperature for 20 h, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer is dried over anhydrous MgSO i, filtered and concentrated to dryness. The resulting white residue is triturated in dichloromethane/diethyl ether 2:1 to afford the desired product.
1.2.3. Step 3:

[00214] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (l eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5: 1 v:v) under N2 and thiomorpho line 1,1-dioxide (1.1 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is dissolved in DCM, washed with water and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated by column chromatography using EtOAc to afford the desired product.
…………………
PATENT
http://www.google.co.in/patents/WO2013189771A1?cl=en
Example 1. Synthesis of the compounds
1.1. Route 1
1.1.1. Synthesis of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (Intermediate 3)

led to 5 °C was added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture was then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gave a solid which was collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea was used as such in the next step without any purification.
[00157] lH (400 MHz, CDC13) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.1.2. 5-Bromo-f 1,2, 4]triazolo[ 1 ,5-a] pyridin-2-ylamine (3)
[00158] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) was added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture was stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) was then added and the mixture slowly heated to reflux (Note: bleach scrubber was required to quench H2S evolved). After 3 h at reflux, the mixture was allowed to cool and filtered to collect the precipitated solid. Further product was collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids were washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound was used as such in the next step without any purification.
[00159] lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2. Synthesis of 4-[ 4-(4, 4, 5, 5-Tetramethyl-f 1, 3,2] ‘ dioxaborolan-2-yl) -benzyl] ‘- thiomor holine- 1, 1 -dioxide (Intermediate 4)

[00160] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portion wise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
1.1.3. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- a ridin-2-ylamine (Formula I)

[00161] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide (l .leq.) was added to a solution of 5-bromo-[l,2,4]triazolo[l,5-a]pyrid in-2-ylamine (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90°C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhydrous MgSC>4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00162] lH (400 MHz, CDC13) δ 7.94-7.92 (d, 2H), 7.52-7.48 (m, 3H), 7.37-7.34 (m, 1H), 7.02-7.00 (m, 1H), 6.00 (d, 2H), 3.76 (d, 2H), 3.15-3.13 (m, 4H), 2.93-2.91 (m, 4H).
[00163] m/z 358.2 (M+H+, 100%). 1.2. Route 2
1.2.1. Cyclopropanecarboxylic acid {5-[4-(l, l-dioxo-thiomorpholin-4-ylmethyl)-phenylJ- [l,2,4]triazolo[l,5-a]pyridin-2-yl}-amide (Formula II)
[00164] The compound according to Formula II may be synthesized according to the procedure described in WO 2010/149769.
1.2.2. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- aJpyridin-2-ylamine (Formula I)
[00165] The compound according to Formula I can also be produced by hydrolysis of the compound accor ing to Formula II:

[00166] Hydrochloric acid 30% aq (12.06 kg; 3.9 rel. volumes) was added to a slurry of the compound according to Formula II (3.45 kg; 1.0 equiv.) in demineralized water (10.0 kg; 3.0 rel. volumes). Subsequently, a line rinse was performed with demineralized water (3.4 kg; 1.0 rel. volumes). The reaction mixture was heated to 80±5°C for 14.5 h. After completion of the reaction (conversion > 99%>), the reaction mixture was cooled to 20±5°C. The reaction mixture was diluted with demineralized water (6.8 kg; 2.0 rel. volumes) and sodium hydroxide 33%> aq (9.52 kg; 3.7 rel volumes) was dosed at such a rate that the temperature of the reactor contents remained below 35°C. An additional amount of sodium hydroxide 33%> aq (2.55 kg; 1.0 rel. volumes) was needed to get the pH > 10. The product was filtered off, washed twice with demineralized water (1.5 rel. volumes) and dried under vacuum for 1 h, thus yielding the crude compound according to Formula I.
[00167] The crude compound according to Formula I (5.70 kg) was re-slurried in demineralized water (23.0 kg; 8.5 rel. volumes). Hydrochloric acid 30%> aq (1.65 kg; 0.7 rel. volumes) and demineralized water (4.3 kg; 1.6 rel. volumes) were added and the reaction mixture was stirred at 20±5°C for 45 min. As the compound according to Formula I was not dissolved completely, the reaction mixture was stirred at 45±5°C for 1 h. The reaction mixture was filtered and the residue was washed with demineralized water (2.0 kg 0.75 rel. volumes). Sodium hydroxide 33%> aq (1.12 kg; 0.6 rel volumes) was added to the filtrate. An additional amount of sodium hydroxide 33%> aq (1.01 kg) was needed to get the pH > 10. The resulting reaction mixture was stirred at 20±5°C for about 3 h. The product was filtered off, washed twice with demineralized water (4.1 kg; 1.5 rel. volumes), and twice with methyl tert-butyl ether (MTBE; 3.0 kg; 1.5 rel. volumes) and dried under vacuum for 15.5 h on the filter. The product was further dried in a vacuum oven at 40±5°C for 202 h, thus affording the desired compound according to Formula I.
Update
Scheme 1: General S nthesis of Compounds of Formula I or A

Formula A
Scheme 7.

(16) (17) (18)
(18a): R3a=R3b=R2a=R (18b): R3a=R3b=D; R2a 18c): R3a=R3b=H; R2a

References
- Namour, Florence; Diderichsen, Paul Matthias; Cox, Eugène; Vayssière, Béatrice; Van der Aa, Annegret; Tasset, Chantal; Van’t Klooster, Gerben (2015-02-14). “Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection”. Clin Pharmacokinet. Epub ahead of print.doi:10.1007/s40262-015-0240-z.
- Van Rompaey, L; Galien, R; Van der Aar, E; Clement-Lacroix, P; Van der Aar, E; Nelles, L; Smets, B; Lepescheux, L; Cristophe, T; Conrath, K; Vandeghinste, N; Vayssiere, B; De Vos, S; Fletcher, S; Brys, R; Van’t Klooster, G; Feyen, J; Menet, C (2013-10-01). “Preclinical characterization of GLPG0634, a selective inhibitor of JAK1 for the treatment of inflammatory diseases”. J Immunol. 191(7). doi:10.4049/jimmunol.1201348.
- http://acrabstracts.org/abstracts/phase-1-and-phase-2-data-confirm-that-glpg0634-a-selective-jak1-inhibitor-has-a-low-potential-for-drug-drug-interactions/
- “Galapagos’ GLPG0634 shows excellent efficacy and safety in rheumatoid arthritis Phase II study” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos reports that the last patient in DARWIN 1 has completed 12 weeks of treatment” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos completes recruitment for Darwin 1 study with GLPG0634 (filgotinib) in RA”. EuroInvestor. Retrieved 2015-02-26.
- NASDAQ OMX Corporate Solutions. “Galapagos completes recruitment for Darwin 2 monotherapy study with GLPG0634 (filgotinib) in RA”. Yahoo Finance. Retrieved 2015-02-26.
| US8551980 | Nov 17, 2010 | Oct 8, 2013 | Bayer Intellectual Property Gmbh | Substituted triazolopyridines |
| US8796457 | Jun 25, 2010 | Aug 5, 2014 | Galapagos Nv | Compound useful for the treatment of degenerative and inflammatory diseases |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Biological half-life | 6 hours[1] |
| Identifiers | |
| CAS Registry Number | 1206161-97-8 |
| ATC code | L01XE18 |
| IUPHAR/BPS | 7913 |
| ChemSpider | 28189566 |
| UNII | 3XVL385Q0M |
| ChEMBL | CHEMBL3301607 |
| Chemical data | |
| Formula | C21H23N5O3S |
| Molecular mass | 425.50402 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
/////////Galapagos, GLPG0634, Filgotinib, PHASE 2, orphan drug designation, PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulceraticolitis
ve SMILES code: O=C(C1CC1)NC2=NN3C(C4=CC=C(CN5CCS(CC5)(=O)=O)C=C4)=CC=CC3=N2
Orilotimod
Orilotimod
(2R)-2-amino-5-{[(1R)-1-carboxy-2-(1H-indol-3-yl)ethyl]amino}-5-oxopentanoic acid
Apo805,UNII-Q66Z43C5XM; Thymodepressin; Orilotimod [USAN]; AC1OIBUF;
- C16H19N3O5
- MW 333.339
Apotex Technologies Inc. INNOVATOR
Orilotimod potassium,
-
APO805K1
D-Tryptophan, D-gamma-glutamyl-, potassium salt (1:1), CAS 960155-19-5
The drug, orilotimod, was originally developed and launched by Immunotech Developments; however, ApoPharma (a subsidiary of Apotex) is developing orilotimod, presumably a topical formulation, for the treatment of psoriasis. In August 2015, the ApoPharma’s drug was reported to be in phase 2 clinical development.
Thymodepressin is the free diacid having Chemical Abstracts Service (CAS) Registry Number@ of 186087-26-3. U.S. Pat. No. 5,736,519 discloses H-D-iGlu-D-Trp-OH and a process for its preparation wherein it is purified by ion exchange chromatography. It is an immunosuppressant and selectively inhibits proliferation of hemopoietic precursor cells and stimulates granulocyte and lymphocyte apoptosis (Sapuntsova, S. G., et al. (May 2002), Bulletin of Experimental Biology and Medicine, 133(5), 488-490).
Thymodepressin is currently being sold in Russia as the disodium salt of D-isoglutamyl-D-tryptophan in liquid formulation for injection and intranasal administration for the treatment of psoriasis and atopic dermatitis. The solid form of the disodium salt of D-isoglutamyl-D-tryptophan is an amorphous powder which is hygroscopic and very difficult to handle. The disodium salt of D-isoglutamyl-D-tryptophan has the molecular formula C16H17N3Na2O5 and is reported in Kashirin, D. M., et al. (2000), Pharmaceutical Chemistry Journal, 34(11), 619-622.
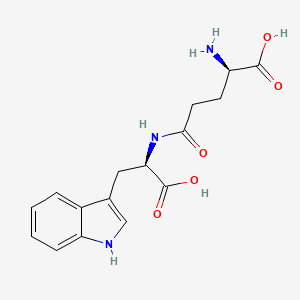
PAPENT
BEAWARE EXAMPLE WITH AN ESTER GP
http://www.google.im/patents/WO2012129671A1?cl=en
Preparation of H-D-Glu( -Trp-OH)-0-Et hydrochloride salt (Apo836.HCI)

A. Preparation of Boc-D-Glu(D-Trp-0-Bzl)-0-Et
Proceeding in a similar manner as described under Example 3A, Boc-D- Glu(D-Trp-0-Bzl)-0-Et was prepared in 87% yield.1H NMR ( DMSO-D6l 400 MHz) δ ppm: 10.87, (s, 1 H), 8.35 (d, J = 7.2 Hz, 1 H), 7.48 (d, J = 7.8 Hz, 1 H), 7.35 (d, J = 7.9 Hz, 1 H), 7.29-7.33 (m, 3H), 7.23 (d, J = 7.7 Hz, 1H), 7.09-7.22 (m, 3H), 7.08 (t, J = 7.6 Hz, 1H), 6.98 (t, J = 7,7 Hz, 1 H), 4.98 – 5.06 (m, 2H), 4.55 (apparent q, J = 7.3 Hz, 1 H), 4.04 – 4.11 (m, 2H), 3.90 – 3.95 (m, 1 H), 3.04 – 3.19 (m, 2H), 2.18 – 2.23 (m, 2H), 1.84 – 1.89 (m, 1 H), 1.70 – 1.77 (m, 1 H), 1.38 (s, 9H), 1.16 (t, J = 7.1 Hz, 3H); MS-ESI (m/z): 552 [ +1]+.
B. Preparation of Boc-D-Glu(D-Trp-OH)-0-Et
Proceeding in a similar manner as described under Example 3B, Boc-D-
Glu(D-Trp-OH)-0-Et was prepared in quantitative yield. 1H NMR ( DMSO-D6, 400 MHz) δ ppm: 12.62 (br. 1H), 10.82, (s, 1 H), 8.10 (d, J = 7.7 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1 H), 7.33 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 7.5 Hz, 1 H), 7.12 (s, 1 H), 7.06 (t, J = 7.3 Hz, 1 H), 6.98 (t, J = 7.5 Hz, 1 H)„ 4.45 (apparent q, J = 7.7 Hz, 1 H), 4.03 – 4.11 (m, 2H), 3.87 – 3.92 (m, 1 H), 3.13 – 3.18 (m, 1H), 2.96 – 3.03 (m,
1 H), 2.13 – 2.20 (m, 2H), 1.82 – 1.88 (m, 1H), 1.69-1.75 (m, 1 H), 1.38 (s, 9H>, 1.17 (t, J = 7.1 Hz, 3H); MS-ESI (m/z): 462 [M+1]+.
C. Preparation of H-D-Glu(D-Trp-OH)-0-Et.HCI (Apo836 HCI)
To an ice-cooled solution of Boc-D-Glu(D-Trp-OH)-0-Et (4.55 g, 9.8 mmol) obtained in Section B above in dichloromethane (100 mL) was bubbled HCI gas for 15 min. The reaction mixture was concentrated under vacuum by rotary evaporation to give H-D-Glu(D-Trp-OH)-0-Et hydrochloride (Apo836.HCI, 4.0 g) as a foamy solid. 1 H NMR ( DMSO-D6, 400 MHz) δ ppm: 12.68 (br. s, 1 H), 10.90, (s, 1H), 8.66 (br, s, 3H), 8.33 (d, J = 7.8 Hz, 1 H), 7.52 (d, J = 7.8 Hz, 1 H), 7.33 (d, J = 8.0 Hz, 1 H), 7.12 (d, J = 1.5 Hz, 1H), 7.06 (t, J = 7.2 Hz, 1 H), 6.98 (t, J = 7.2 Hz, 1 H), 4.47 (apparent q, J = 4.8 Hz, 1 H), 4.13 – 4.19 (m, 2H), 3.90 (br, 1 H), 3.16 – 3.20 (m, 1H), 2.98 – 3.04 (m, 1 H), 2.29 – 2.33 (m, 2H), 1.94 – 1.98
(m, 2H), 1.20 (t, J = 7.1 Hz, 3H); MS-ESI (m/z): 362 [M+1]+ (free base).
……………………..
file:///H:/ORILOTIMODUS20150225341A1.pdf
Novel crystalline and amorphous salts of thymodepressin (orilotimod), particularly potassium salt, useful for treating psoriasis and atopic dermatitis. Also claims salt exchange method for preparing thymodepressin salts.
hymodepressin is the free diacid having Chemical Abstracts Service (CAS) Registry Number@ of 186087-26-3. U.S. Pat. No. 5,736,519 discloses H-D-iGlu-D-Trp-OH and a process for its preparation wherein it is purified by ion exchange chromatography. It is an immunosuppressant and selectively inhibits proliferation of hemopoietic precursor cells and stimulates granulocyte and lymphocyte apoptosis (Sapuntsova, S. G., et al. (May 2002), Bulletin of Experimental Biology and Medicine, 133(5), 488-490).
Thymodepressin is currently being sold in Russia as the disodium salt of D-isoglutamyl-D-tryptophan in liquid formulation for injection and intranasal administration for the treatment of psoriasis and atopic dermatitis. The solid form of the disodium salt of D-isoglutamyl-D-tryptophan is an amorphous powder which is hygroscopic and very difficult to handle. The disodium salt of D-isoglutamyl-D-tryptophan has the molecular formula C16H17N3Na2O5 and which is reported in Kashirin, D. M., et al. (2000), Pharmaceutical Chemistry Journal, 34(11), 619-622.
Through investigations in our laboratory, we have determined that the freeze-dried disodium salt of D-isoglutamyl-D-tryptophan is extremely hygroscopic turning into a gel in a matter of minutes in air and cannot easily be handled.
A powdery or amorphous form of a compound, intended for pharmaceutical use may give rise to manufacturing problems due to bulk density issues, hygroscopicity and variable water content that cannot be corrected by vacuum drying. D-isoglutamyl-D-tryptophan is a dipeptide and the drying of an amorphous form at elevated temperature, for example, 80-100° C. under vacuum is not recommended. Thus, there are serious difficulties experienced during the purification of the disodium salt of D-isoglutamyl-D-tryptophan and obtaining the pure disodium salt on a manufacturing scale. Further, there is no published procedure for its preparation.
The monosodium salt of D-isoglutamyl-D-tryptophan is identified by the CAS Registry System and is listed in the CAS REGISTRYSM File with a CAS Registry Number@ of 863988-88-9. However, there are no references citing the substance and thus no publication of its identity, its physical and/or chemical properties, its characterization or a procedure for its preparation. Freeze-dried powders of mono sodium and disodium salts of peptide drugs may not have controllable powder bulk density ranges for formulation. They may require significant investment in freeze-dried dispersion technology.
EXAMPLES
Example 1
Preparation of potassium salt of D-isoglutamyl-D-tryptophan (1:1) from D-isoglutamyl-D-tryptophan and potassium hydroxide
In a 100-mL round bottom flask equipped with a magnetic stir bar was placed 5 mL of potassium hydroxide solution (0.5 N). The solution was cooled to 0° C. in an ice-water bath, and solid H-D-iGlu-D-Trp-OH (1.00 g, 3 mmol) was added. The mixture was stirred while the pH of the solution was adjusted to ca. 6.0 by adding a few drops of potassium hydroxide solution (0.5 N). The solution was filtered to remove any solid particulates. The filtrate was evaporated to dryness at a bath temperature of about 30° C. to afford a solid. After drying under vacuum at room temperature for overnight, the salt was obtained in quantitative yield, with a HPLC purity (peak area percent) of 98.3%. HPLC method; Column: XTerra MS C18; 5 μm, 4.6×250 mm; Mobile phase: A=the aqueous phase: 4 mM Tris, 2 mM EDTA, pH 7.4; B=the organic phase: CH3CN; gradient: B %: 0 min. 5%, 15 min. 55%, 30 min. 55%, 32 min. 5%, 35 min. 5%; Flow rate: 1 mL/min; injection volume: 5 μL; λ: 222, 254, 282, 450 nm; retention time of the product: 6.41 min. The XRPD pattern of this crystalline material is shown in FIG. 1A; the water content by Karl-Fischer test is 0.7%; UV (water, c=23.8 ρM, λmax nm): 221 (ε 33270), 280 (ε 5417); MS (m/z): 372.0 [M]+, 334.2 [C16H20N3O5]+, 187.9 (100%). The FT-IR (KBr) spectrum is shown in FIG. 1B.
Example 2
A. Preparation of mono potassium salt of D-isoglutamyl-D-tryptophan (1:1) from the mono ammonium salt of D-isoglutamyl-D-tryptophan (1:1)
A solution of H-D-iGlu-D-Trp-OH, mono ammonium salt (1:1), (1.66 g, 4.05 mmol) and potassium hydroxide (253 mg, 4.50 mmol) in water (20 mL) was stirred at room temperature for 15 min. The pH of the solution was about 9. The reaction mixture was evaporated under reduced pressure to a volume of about 1 mL. After cooling to room temperature, isopropanol was added until a solid precipitated out. The resulting suspension was stirred at room temperature for 15 min, then filtered. The solid was washed with isopropanol (2×20 mL) and ethyl acetate (20 mL), then dried under vacuum in an oven at 42° C. overnight. An off white solid was obtained (1.49 g, 99% yield). The water content by Karl-Fischer test is 2.5%. Analytical data (XRPD pattern, FT-IR and MS spectra) are similar to those described in Example 1.
B. Preparation of amorphous form of potassium salt of D-isoglutamyl-D-tryptophan (1:1) from the mono ammonium salt of D-isoglutamyl-D-tryptophan (1:1)
A solution of H-D-iGlu-D-Trp-OH, mono ammonium salt (1:1), (517 mg, 1.40 mmol) and potassium hydroxide (82 mg, 1.46 mmol) in water (10 mL) was stirred at room temperature for 30 minutes. The resulting mixture was freeze-dried overnight. An off white solid was obtained in quantitative yield. The XRPD pattern spectrum confirmed that this material is amorphous.
1H NMR (D2O) δ: 7.69 (d, J=7.9 Hz, 1H), 7.48 (d, J=8.2 Hz, 1H), 7.23 (t, J=7.6 Hz, 1H), 7.22 (s, 1H), 7.16 (t, J=7.4 Hz, 1H), 4.59 (dd, J=8.7, 4.8 Hz, 1H), 3.51 (dd, J=6.8, 5.8 Hz, 1H), 3.38 (dd, J=14.8, 4.8 Hz, 1H), 3.11 (dd, J=14.8, 8.8 Hz, 1H), 2.20-2.49 (m, 2H) and 1.85-1.94 (m, 2H);
13C NMR (D2O) δ: 181.4, 177.0, 176.6, 138.8, 129.9, 126.9, 124.5, 121.9, 121.4, 114.5, 113.2, 58.6, 57.0, 34.6 (CH2), 30.2 (CH2) and 29.3 (CH2);
the water content by Karl-Fischer test is 5.4%;
the FT-IR (KBr) spectrum is shown in FIG. 1C;
MS (m/z): 371.7 [M]+, 334.2 [C16H20N3O5]+, 187.9 (100%);
HPLC purity (peak area percent): 99.8%, Retention time: 5.04 min; HPLC conditions: Column Waters Symmetry C18, 3.9×150 mm, 5 μm; Mobile phase: 0.035% HClO4, pH 2/CH3CN, 85/15, isocratic, Flow rate: 1 mL/min; λ: 220, 254, 280 nm.
| Patent | Submitted | Granted |
|---|---|---|
| GAMMA-GLUTAMYL AND BETA-ASPARTYL CONTAINING IMMUNOMODULATOR COMPOUNDS AND METHODS THEREWITH [EP1042286] | 2000-10-11 | 2010-08-25 |
| CRYSTALLINE D-ISOGLUTAMYL-D-TRYPTOPHAN AND THE MONO AMMONIUM SALT OF D-ISOGLUTAMYL-D-TRYPTOPHAN [US8119606] | 2010-01-21 | 2012-02-21 |
| Pharmaceutically Acceptable Salts of Thymodepressin and Processes for their Manufacture [US8138221] | 2010-03-04 | 2012-03-20 |
| CRYSTALLINE FORMS OF THE MONO-SODIUM SALT OF D-ISOGLUTAMYL-D-TRYPTOPHAN [US8207217] | 2010-02-04 | 2012-06-26 |
////////Orilotimod, PHASE 2, thymodepressin, APO 805K1
C1=CC=C2C(=C1)C(=CN2)CC(C(=O)O)NC(=O)CCC(C(=O)O)N
Odalasvir
ACH-3102 , Odalasvir
Odalasvir
ACH-0143102; ACH-3102
CAS : 1415119-52-6
Dimethyl N, N ‘- (tricyclo [8.2.2.24,7] hexadeca-1 (12), 4,6, 10,13,15-hexaene-5,11-diylbis {1H-benzimidazole-5,2-diyl [(2S, 3aS, 7aS) -octahydro-1H-indole-2,1-diyl] [(1S) -1 – (1-methylethyl) -2-oxoethylene]}) biscarbamate
Carbamic acid, N,N’-(tricyclo(8.2.2.24,7)hexadeca-4,6,10,12,13,15-hexaene-5,11-diylbis(1H-benzimidazole-6,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl)))bis-, C,C’-dimethyl ester
Dimethyl N,N’-(1,4(1,4)-dibenzenacyclohexaphane-12,42-diylbis(1hbenzimidazole-5,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((2S)-3-methyl-1-oxobutan-1,2-diyl)))biscarbamate
Mechanism of Action: HCV NS5A Protein inhibitor
Indication: Hepatitis C
Developer: Achillion Pharmaceuticals, Inc.

-
C60-H72-N8-O6
- 1001.2788
Odalasvir[1] is an investigational new drug in development for the treatment hepatitis C.
Achillion Pharmaceuticals Inc’s Odalasvir (ACH-3102) is an investigational new drug in development for the treatment hepatitis C. Achillion’s ongoing study tests its NS5A inhibitor, ACH-3102, with Sovaldi in previously untreated genotype 1 hepatitis C patients over six and eight weeks of therapy. The main goal is to achieve a cure, or sustained virological response, 12 weeks after the completion of therapy.
Odalasvir is a hepatitis C virus (HCV NS5A) inhibitor in phase II clinical studies at Achillion for the treatment of hepatitis C.
In 2012, fast track designation was assigned to the compound in the U.S. for the treatment of chronic hepatitis C.
WILL BE UPDATED………….
WO 2012166716
http://www.google.com/patents/US20120302538

General Considerations
All nonaqueous reactions were performed under an atmosphere of dry argon gas using oven-dried glassware and anhydrous solvents. The progress of reactions and the purity of target compounds were determined using one of the following two HPLC methods: (1) Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with an isocratic elution of 0.24 min at 90:10 water:acetonitrile containing 0.05% formic acid followed by a 4.26-min linear gradient elution from 90:10 to 10:90 at a flow rate of 1.0 mL/min with UV (PDA), ELS, and MS (SQ in APCI mode) detection (method 1); and (2) Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with an isocratic elution of 0.31 min at 95:5 water:acetonitrile containing 0.05% formic acid followed by a 17.47-min linear gradient elution from 95:5 to 5:95 at a flow rate of 0.4 mL/min with UV (PDA), ELS, and MS (SQ in APCI mode) detection (method 2).
Target compounds were purified via preparative reverse-phase HPLC using a YMC Pack Pro C18 5 μm 150×20 mm column with an isocratic elution of 0.35 min at 95:5 water:acetonitrile containing 0.1% trifluoroacetic acid followed by a 23.3-min linear gradient elution from 95:5 to 5:95 at a flow rate of 18.9 mL/min with UV and mass-based fraction collection.

Example 1
Synthesis of Compound 10
Compound 10 was prepared via bromination of [2.2]paracyclophane as outlined previously (Reich, H. J.; Cram, D. J. J. Am. Chem. Soc. 1969, 91, 3527-3533; Reich, H. J.; Cram, D. J. J. Am. Chem. Soc. 1969, 91, 3534-3543). Compounds 1, 2, 6, 8, and 10 can be obtained from commercial sources. Compounds 3-7 and 9 were prepared using general synthetic methods known in the art.
Example 2Synthesis of Compound 11
A deoxygenated (argon) mixture of 9 (284.2 mg), 10 (52.3 mg), K3PO4 (248.1 mg), and PdCl2dppf.CH2Cl2 (7.4 mg) in dioxane/water (5.5 mL/0.55 mL) was irradiated in a microwave for 2 h at 80° C. The resulting mixture was evaporated under reduced pressure and the remaining solid was extracted with DCM. This crude material was purified by PTLC (20 cm×20 cm×2000 μm glass plates; eluted with 45:50:5 v/v/v DCM:EtOAc:MeOH, Rf 0.28) to give 75.3 mg of 11. The purity of 11 was determined via analytical reverse-phase HPLC using a 3.5-min gradient elution of increasing concentrations of ACN in water (10-90%) containing 0.05% formic acid with a flow rate of 1.0 mL/min on a Waters AQUITY HPLC BEH C18 1.7 μm 2.1×50 mm column with UV (PDA), ELS, and MS (SQ in APCI mode) detection. HPLC: tR 1.57 min (98% purity). MS m/z calculated for C56H64N8O6 ([M]+), 945. found, 946 ([M+1]+).
SEE ALSO
US 2012302538
http://www.google.com/patents/US20120302538
……………
see
US 20150023913
http://www.google.com/patents/US20150023913
…………..
see
WO 2015005901
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Dimethyl N,N′-(1,4(1,4)-Dibenzenacyclohexaphane-12,42-diylbis(1hbenzimidazole-5,2-diyl((2S,3aS,7aS)-octahydro-1H-indole-2,1-diyl)((2S)-3-methyl-1-oxobutan-1,2-diyl)))biscarbamate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 1415119-52-6 |
| ATC code | None |
| Chemical data | |
| Formula | C60H72N8O6 |
| Molecular mass | 1001.28 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook 
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////////