
Home » 0rphan drug status (Page 6)
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Teprotumumab-trbw

Tepezza (teprotumumab-trbw)
Company: Horizon Therapeutics plc
Date of Approval: January 21, 2020
Treatment for: Thyroid Eye Disease
UNIIY64GQ0KC0A
CAS number1036734-93-6
R-1507 / R1507 / RG-1507 / RG1507 / RO-4858696 / RO-4858696-000 / RO-4858696000 / RO4858696 / RO4858696-000 / RV-001 / RV001
Tepezza (teprotumumab-trbw) is a fully human monoclonal antibody (mAb) and a targeted inhibitor of the insulin-like growth factor 1 receptor (IGF-1R) for the treatment of active thyroid eye disease (TED).
FDA Approves Tepezza (teprotumumab-trbw) for the Treatment of Thyroid Eye Disease (TED) – January 21, 2020
Today, the U.S. Food and Drug Administration (FDA) approved Tepezza (teprotumumab-trbw) for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis). Today’s approval represents the first drug approved for the treatment of thyroid eye disease.
“Today’s approval marks an important milestone for the treatment of thyroid eye disease. Currently, there are very limited treatment options for this potentially debilitating disease. This treatment has the potential to alter the course of the disease, potentially sparing patients from needing multiple invasive surgeries by providing an alternative, non surgical treatment option,” said Wiley Chambers, M.D., deputy director of the Division of Transplant and Ophthalmology Products in the FDA’s Center for Drug Evaluation and Research. “Additionally, thyroid eye disease is a rare disease that impacts a small percentage of the population, and for a variety of reasons, treatments for rare diseases are often unavailable. This approval represents important progress in the approval of effective treatments for rare diseases, such as thyroid eye disease.”
Thyroid eye disease is associated with the outward bulging of the eye that can cause a variety of symptoms such as eye pain, double vision, light sensitivity or difficulty closing the eye. This disease impacts a relatively small number of Americans, with more women than men affected. Although this condition impacts relatively few individuals, thyroid eye disease can be incapacitating. For example, the troubling ocular symptoms can lead to the progressive inability of people with thyroid eye disease to perform important daily activities, such as driving or working.
Tepezza was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive Tepezza or a placebo. Of the patients who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than 2 millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.
The most common adverse reactions observed in patients treated with Tepezza are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache. Tepezza should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for 6 months following the last dose of Tepezza.
The FDA granted this application Priority Review, in addition to Fast Track and Breakthrough Therapy Designation. Additionally, Tepezza received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases or conditions. Development of this product was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and efficacy of products for use in rare diseases or conditions.
The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.
Teprotumumab (RG-1507), sold under the brand name Tepezza, is a medication used for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis).[1]
The most common adverse reactions observed in people treated with teprotumumab-trbw are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache.[1] Teprotumumab-trbw should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for six months following the last dose of teprotumumab-trbw.[1]
It is a human monoclonal antibody developed by Genmab and Roche. It binds to IGF-1R.
Teprotumumab was first investigated for the treatment of solid and hematologic tumors, including breast cancer, Hodgkin’s and non-Hodgkin’s lymphoma, non-small cell lung cancer and sarcoma.[2][3] Although results of phase I and early phase II trials showed promise, research for these indications were discontinued in 2009 by Roche. Phase II trials still in progress were allowed to complete, as the development was halted due to business prioritization rather than safety concerns.
Teprotumumab was subsequently licensed to River Vision Development Corporation in 2012 for research in the treatment of ophthalmic conditions. Horizon Pharma (now Horizon Therapeutics, from hereon Horizon) acquired RVDC in 2017, and will continue clinical trials.[4] It is in phase III trials for Graves’ ophthalmopathy (also known as thyroid eye disease (TED)) and phase I for diabetic macular edema.[5] It was granted Breakthrough Therapy, Orphan Drug Status and Fast Track designations by the FDA for Graves’ ophthalmopathy.[6]
In a multicenter randomized trial in patients with active Graves’ ophthalmopathy Teprotumumab was more effective than placebo in reducing the clinical activity score and proptosis.[7] In February 2019 Horizon announced results from a phase 3 confirmatory trial evaluating teprotumumab for the treatment of active thyroid eye disease (TED). The study met its primary endpoint, showing more patients treated with teprotumumab compared with placebo had a meaningful improvement in proptosis, or bulging of the eye: 82.9 percent of teprotumumab patients compared to 9.5 percent of placebo patients achieved the primary endpoint of a 2 mm or more reduction in proptosis (p<0.001). Proptosis is the main cause of morbidity in TED. All secondary endpoints were also met and the safety profile was consistent with the phase 2 study of teprotumumab in TED.[8] On 10th of July 2019 Horizon submitted a Biologics License Application (BLA) to the FDA for teprotumumab for the Treatment of Active Thyroid Eye Disease (TED). Horizon requested priority review for the application – if so granted (FDA has a 60-day review period to decide) it would result in a max. 6 month review process.[9]
History[edit]
Teprotumumab-trbw was approved for use in the United States in January 2020, for the treatment of adults with thyroid eye disease.[1]
Teprotumumab-trbw was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive teprotumumab-trbw or a placebo.[1] Of the subjects who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than two millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.[1]
The U.S. Food and Drug Administration (FDA) granted the application for teprotumumab-trbw fast track designation, breakthrough therapy designation, priority review designation, and orphan drug designation.[1] The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.[1]
References
- ^ Jump up to:a b c d e f g h “FDA approves first treatment for thyroid eye disease”. U.S. Food and Drug Administration (FDA) (Press release). 21 January 2020. Retrieved 21 January 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ https://clinicaltrials.gov/ct2/show/NCT01868997
- ^ http://adisinsight.springer.com/drugs/800015801
- ^ http://www.genmab.com/product-pipeline/products-in-development/teprotumumab
- ^ http://adisinsight.springer.com/drugs/800015801
- ^ http://www.genmab.com/product-pipeline/products-in-development/teprotumumab
- ^ Smith, TJ; Kahaly, GJ; Ezra, DG; Fleming, JC; Dailey, RA; Tang, RA; Harris, GJ; Antonelli, A; Salvi, M; Goldberg, RA; Gigantelli, JW; Couch, SM; Shriver, EM; Hayek, BR; Hink, EM; Woodward, RM; Gabriel, K; Magni, G; Douglas, RS (4 May 2017). “Teprotumumab for Thyroid-Associated Ophthalmopathy”. The New England Journal of Medicine. 376 (18): 1748–1761. doi:10.1056/NEJMoa1614949. PMC 5718164. PMID 28467880.
- ^ “Horizon Pharma plc Announces Phase 3 Confirmatory Trial Evaluating Teprotumumab (OPTIC) for the Treatment of Active Thyroid Eye Disease (TED) Met Primary and All Secondary Endpoints”. Horizon Pharma plc. Retrieved 22 March 2019.
- ^ “Horizon Therapeutics plc Submits Teprotumumab Biologics License Application (BLA) for the Treatment of Active Thyroid Eye Disease (TED)”. Horizon Therapeutics plc. Retrieved 27 August 2019.
External links
- “Teprotumumab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IGF-1R |
| Clinical data | |
| Other names | teprotumumab-trbw, RG-1507 |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.081.384 |
| Chemical and physical data | |
| Formula | C6476H10012N1748O2000S40 |
| Molar mass | 145.6 kg/mol g·mol−1 |
/////////Teprotumumab-trbw, APPROVALS 2020, FDA 2020, ORPHAN, BLA, fast track designation, breakthrough therapy designation, priority review designation, and orphan drug designation, Tepezza, Horizon Therapeutics, MONOCLONAL ANTIBODY, 2020 APPROVALS, active thyroid eye disease, Teprotumumab
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-thyroid-eye-disease
Brilliant blue G , ブリリアントブルーG ,
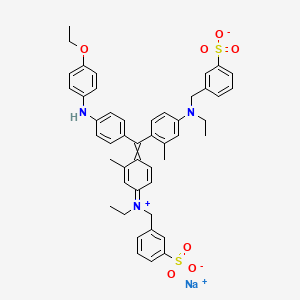
Brilliant blue G
FDA 2019, 12/20/2019, TISSUEBLUE, New Drug Application (NDA): 209569
Company: DUTCH OPHTHALMIC, PRIORITY; Orphan
OPQ recommends APPROVAL of NDA 209569 for commercialization of TissueBlue (Brilliant Blue G Ophthalmic Solution), 0.025%
Neuroprotectant
sodium;3-[[4-[[4-(4-ethoxyanilino)phenyl]-[4-[ethyl-[(3-sulfonatophenyl)methyl]azaniumylidene]-2-methylcyclohexa-2,5-dien-1-ylidene]methyl]-N-ethyl-3-methylanilino]methyl]benzenesulfonate
| Formula |
C47H48N3O7S2. Na
|
|---|---|
| CAS |
6104-58-1
|
| Mol weight |
854.0197
|
ブリリアントブルーG, C.I. Acid Blue 90
UNII-M1ZRX790SI
M1ZRX790SI
6104-58-1
Brilliant Blue G
Derma Cyanine G
SYN


////////////Brilliant blue G , ブリリアントブルーG , C.I. Acid Blue 90, FDA 2019, PRIORITY, Orphan
CCN(CC1=CC(=CC=C1)S(=O)(=O)[O-])C2=CC(=C(C=C2)C(=C3C=CC(=[N+](CC)CC4=CC(=CC=C4)S(=O)(=O)[O-])C=C3C)C5=CC=C(C=C5)NC6=CC=C(C=C6)OCC)C.[Na+]
- Benzenemethanaminium, N-[4-[[4-[(4-ethoxyphenyl)amino]phenyl][4-[ethyl[(3-sulfophenyl)methyl]amino]-2-methylphenyl]methylene]-3-methyl-2,5-cyclohexadien-1-ylidene]-N-ethyl-3-sulfo-, hydroxide, inner salt, monosodium salt
- Benzenemethanaminium, N-[4-[[4-[(4-ethoxyphenyl)amino]phenyl][4-[ethyl[(3-sulfophenyl)methyl]amino]-2-methylphenyl]methylene]-3-methyl-2,5-cyclohexadien-1-ylidene]-N-ethyl-3-sulfo-, inner salt, monosodium salt (9CI)
- Brilliant Indocyanine G (6CI)
- C.I. Acid Blue 90 (7CI)
- C.I. Acid Blue 90, monosodium salt (8CI)
- Acid Blue 90
- Acid Blue G 4061
- Acid Blue PG
- Acid Bright Blue G
- Acid Brilliant Blue G
- Acid Brilliant Cyanine G
- Acidine Sky Blue G
- Amacid Brilliant Cyanine G
- Anadurm Cyanine A-G
- BBG
- Benzyl Cyanine G
- Biosafe Coomassie Stain
- Boomassie blue silver
- Brilliant Acid Blue G
- Brilliant Acid Blue GI
- Brilliant Acid Blue J
- Brilliant Acid Cyanine G
- Brilliant Blue G
- Brilliant Blue G 250
- Brilliant Blue J
- Brilliant Indocyanine GA-CF
- Bucacid Brilliant Indocyanine G
- C.I. 42655
- CBB-G 250
- Colocid Brilliant Blue EG
- Coomassie Blue G
- Coomassie Blue G 250
- Coomassie Brilliant Blue G
- Coomassie Brilliant Blue G 250
- Coomassie G 250
- Cyanine G
- Daiwa Acid Blue 300
- Derma Cyanine G
- Derma Cyanine GN 360
- Dycosweak Acid Brilliant Blue G
- Eriosin Brilliant Cyanine G
- Fenazo Blue XXFG
- Impero Azure G
- Kayanol Cyanine G
- Lerui Acid Brilliant Blue G
- Milling Brilliant Blue 2J
- NSC 328382
- Optanol Cyanine G
- Orient Water Blue 105
- Orient Water Blue 105S
- Polar Blue G
- Polar Blue G 01
- Polycor Blue G
- Sandolan Cyanine N-G
- Sellaset Blue B
- Serva Blue G
- Serva Blue G 250
- Silk Fast Cyanine G
- Simacid Blue G 350
- Sumitomo Brilliant Indocyanine G
- Supranol Cyanin G
- Supranol Cyanine G
- TissueBlue
- Triacid Fast Cyanine G
- Water Blue 105
- Water Blue 105S
- Water Blue 150
- Xylene Brilliant Cyanine G
Avapritinib, アバプリチニブ , авапритиниб , أفابريتينيب ,

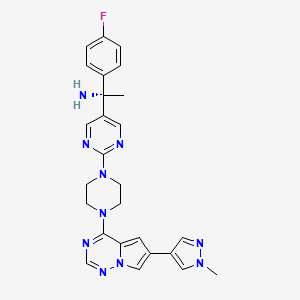
Avapritinib
BLU-285, BLU285
Antineoplastic, Tyrosine kinase inhibitor
アバプリチニブ
(1S)-1-(4-fluorophenyl)-1-[2-[4-[6-(1-methylpyrazol-4-yl)pyrrolo[2,1-f][1,2,4]triazin-4-yl]piperazin-1-yl]pyrimidin-5-yl]ethanamine
| Formula |
C26H27FN10
|
|---|---|
| CAS |
1703793-34-3
|
| Mol weight |
498.558
|
| No. | Drug Name | Active Ingredient | Approval Date | FDA-approved use on approval date* |
|---|---|---|---|---|
| 1. | Ayvakit | avapritinib | 1/9/2020 | To treat adults with unresectable or metastatic gastrointestinal stromal tumor (GIST) |
PRIORITY; Orphan, NDA 212608
Avapritinib, sold under the brand name Ayvakit, is a medication used for the treatment of tumors due to one specific rare mutation: It is specifically intended for adults with unresectable or metastatic ( y) gastrointestinal stromal tumor (GIST) that harbor a platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutation.[1]
Common side effects are edema (swelling), nausea, fatigue/asthenia (abnormal physical weakness or lack of energy), cognitive impairment, vomiting, decreased appetite, diarrhea, hair color changes, increased lacrimation (secretion of tears), abdominal pain, constipation, rash. and dizziness.[1]
Ayvakit is a kinase inhibitor.[1]
History
The U.S. Food and Drug Administration (FDA) approved avapritinib in January 2020.[1] The application for avapritinib was granted fast track designation, breakthrough therapy designation, and orphan drug designation.[1] The FDA granted approval of Ayvakit to Blueprint Medicines Corporation.[1]
Avapritinib was approved based on the results from the Phase I NAVIGATOR[2][3] clinical trial involving 43 patients with GIST harboring a PDGFRA exon 18 mutation, including 38 subjects with PDGFRA D842V mutation.[1] Subjects received avapritinib 300 mg or 400 mg orally once daily until disease progression or they experienced unacceptable toxicity.[1] The recommended dose was determined to be 300 mg once daily.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] For subjects harboring a PDGFRA exon 18 mutation, the overall response rate was 84%, with 7% having a complete response and 77% having a partial response.[1] For the subgroup of subjects with PDGFRA D842V mutations, the overall response rate was 89%, with 8% having a complete response and 82% having a partial response.[1] While the median duration of response was not reached, 61% of the responding subjects with exon 18 mutations had a response lasting six months or longer (31% of subjects with an ongoing response were followed for less than six months).[1]
PATENT
WO 2015057873
https://patents.google.com/patent/WO2015057873A1/en
Example 7: Synthesis of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, 1 -f\ [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine and (S)- 1 – (4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (Compounds 43 and 44)


Step 1 : Synthesis of (4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f] [ 1 ,2,4] triazin-4-yl)piperazin- 1 -yl)pyrimidin-5-yl)methanone:

4-Chloro-6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/] [l,2,4]triazine (180 mg, 0.770 mmol), (4-fluorophenyl)(2-(piperazin-l-yl)pyrimidin-5-yl)methanone, HC1 (265 mg, 0.821 mmol) and DIPEA (0.40 mL, 2.290 mmol) were stirred in 1,4-dioxane (4 mL) at room temperature for 18 hours. Saturated ammonium chloride was added and the products extracted into DCM (x2). The combined organic extracts were dried over Na2S04, filtered through Celite eluting with DCM, and the filtrate concentrated in vacuo. Purification of the residue by MPLC (25- 100% EtOAc-DCM) gave (4-fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methanone (160 mg, 0.331 mmol, 43 % yield) as an off-white solid. MS (ES+) C25H22FN90 requires: 483, found: 484 [M + H]+.
Step 2: Synthesis of (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-p razol-4-yl)p rrolo[2, l- ] [l,2,4]triazin-4- l)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide:

(S)-2-Methylpropane-2-sulfinamide (110 mg, 0.908 mmol), (4-fluorophenyl)(2-(4-(6-(l- methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5- yl)methanone (158 mg, 0.327 mmol) and ethyl orthotitanate (0.15 mL, 0.715 mmol) were stirred in THF (3.2 mL) at 70 °C for 18 hours. Room temperature was attained, water was added, and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0- 10% MeOH-EtOAc) gave (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)methylene)-2- methylpropane-2-sulfinamide (192 mg, 0.327 mmol, 100 % yield) as an orange solid. MS (ES+) C29H3iFN10OS requires: 586, found: 587 [M + H]+.
Step 3: Synthesis of (lS’)-N-(l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- l)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-

(lS’,Z)-N-((4-Fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide (190 mg, 0.324 mmol) was taken up in THF (3 mL) and cooled to 0 °C. Methylmagnesium bromide (3 M solution in diethyl ether, 0.50 mL, 1.500 mmol) was added and the resulting mixture stirred at 0 °C for 45 minutes. Additional methylmagnesium bromide (3 M solution in diethyl ether, 0.10 mL, 0.300 mmol) was added and stirring at 0 °C continued for 20 minutes. Saturated ammonium chloride was added and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0-10% MeOH-EtOAc) gave (lS’)-N-(l-(4-fluorophenyl)-l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol, 61.5 % yield) as a yellow solid (mixture of diastereoisomers). MS (ES+) C3oH35FN10OS requires: 602, found: 603 [M + H]+. Step 4: Synthesis of l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f\ [ 1 ,2,4] triazin-4- l)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine:

(S)-N- ( 1 – (4-Fluorophenyl)- 1 -(2- (4- (6-( 1 -methyl- 1 H-pyrazol-4-yl)pyrrolo [2,1- /] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol) was stirred in 4 M HCl in 1,4-dioxane (1.5 mL)/MeOH (1.5 mL) at room temperature for 1 hour. The solvent was removed in vacuo and the residue triturated in EtOAc to give l-(4-fluorophenyl)- l-(2-(4-(6-(l -methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4- yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine, HCl (110 mg, 0.206 mmol, 103 % yield) as a pale yellow solid. MS (ES+) C26H27FN10requires: 498, found: 482 [M- 17 + H]+, 499 [M + H]+.
Step 5: Chiral separation of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine and (5)-1-(4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- 1 -yl)pyrimidin- -yl)ethanamine:

The enantiomers of racemic l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (94 mg, 0.189 mmol) were separated by chiral SFC to give (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH- pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (34.4 mg, 0.069 mmol, 73.2 % yield) and (lS,)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (32.1 mg, 0.064 mmol, 68.3 % yield). The absolute stereochemistry was assigned randomly. MS (ES+)
C26H27FN10 requires: 498, found: 499 [M + H]+.
References
- ^ Jump up to:a b c d e f g h i j k l m “FDA approves the first targeted therapy to treat a rare mutation in patients with gastrointestinal stromal tumors”. U.S. Food and Drug Administration (FDA) (Press release). 9 January 2020. Archived from the original on 11 January 2020. Retrieved 9 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Blueprint Medicines Announces FDA Approval of AYVAKIT (avapritinib) for the Treatment of Adults with Unresectable or Metastatic PDGFRA Exon 18 Mutant Gastrointestinal Stromal Tumor”. Blueprint Medicines Corporation (Press release). 9 January 2020. Archived from the original on 11 January 2020. Retrieved 9 January 2020.
- ^ “Blueprint Medicines Announces Updated NAVIGATOR Trial Results in Patients with Advanced Gastrointestinal Stromal Tumors Supporting Development of Avapritinib Across All Lines of Therapy”. Blueprint Medicines Corporation (Press release). 15 November 2018. Archived from the original on 10 January 2020. Retrieved 9 January 2020.
Further reading
- Wu CP, Lusvarghi S, Wang JC, et al. (July 2019). “Avapritinib: A Selective Inhibitor of KIT and PDGFRα that Reverses ABCB1 and ABCG2-Mediated Multidrug Resistance in Cancer Cell Lines”. Mol. Pharm. 16 (7): 3040–3052. doi:10.1021/acs.molpharmaceut.9b00274. PMID 31117741.
- Gebreyohannes YK, Wozniak A, Zhai ME, et al. (January 2019). “Robust Activity of Avapritinib, Potent and Highly Selective Inhibitor of Mutated KIT, in Patient-derived Xenograft Models of Gastrointestinal Stromal Tumors”. Clin. Cancer Res. 25 (2): 609–618. doi:10.1158/1078-0432.CCR-18-1858. PMID 30274985.
External links
- “Avapritinib”. Drug Information Portal. U.S. National Library of Medicine (NLM).
| Clinical data | |
|---|---|
| Trade names | Ayvakit |
| Other names | BLU-285, BLU285 |
| License data | |
| Routes of administration |
By mouth |
| Drug class | Antineoplastic agents |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C26H27FN10 |
| Molar mass | 498.570 g·mol−1 |
| 3D model (JSmol) | |
///////Avapritinib, 2020 APPROVALS, PRIORITY, Orphan, BLU-285, BLU285, FDA 2020, Ayvakit, アバプリチニブ , авапритиниб , أفابريتينيب ,
FDA approves novel treatment Oxbryta (voxelotor) to target abnormality in sickle cell disease
Sickle cell disease is a lifelong, inherited blood disorder in which red blood cells are abnormally shaped (in a crescent, or “sickle” shape), which restricts the flow in blood vessels and limits oxygen delivery to the body’s tissues, leading to severe pain and organ damage. It is also characterized by severe and chronic inflammation that worsens vaso-occlusive crises during which patients experience episodes of extreme pain and organ damage. Nonclinical studies have demonstrated that Oxbryta inhibits red blood cell sickling, improves red blood cell deformability (ability of a red blood cell to change shape) and improves the blood’s ability to flow.
“Oxbryta is an inhibitor of deoxygenated sickle hemoglobin polymerization, which is the central abnormality in sickle cell disease,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research. “With Oxbryta, sickle cells are less likely to bind together and form the sickle shape, which can cause low hemoglobin levels due to red blood cell destruction. This therapy provides a new treatment option for patients with this serious and life-threatening condition.”
Oxbryta’s approval was based on the results of a clinical trial with 274 patients with sickle cell disease. In the study, 90 patients received 1500 mg of Oxbryta, 92 patients received 900 mg of Oxbryta and 92 patients received a placebo. Effectiveness was based on an increase in hemoglobin response rate in patients who received 1500 mg of Oxbryta, which was 51.1% for these patients compared to 6.5% in the placebo group.
/////////fda 2019, Fast Track designation, Oxbryta, Orphan Drug designation, voxelotor, Global Blood Therapeutics, sickle cell disease
FDA approves first treatment Givlaari (givosiran) for inherited rare disease
///////////Givlaari, givosiran, fda 2019, Breakthrough Therapy designation, Priority Review, Orphan Drug
Blarcamesine, ブラルカメシン ,

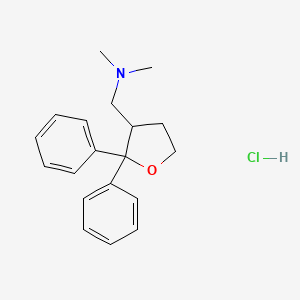
Blarcamesine
ブラルカメシン;
[(2,2-diphenyloxolan-3-yl)methyl]dimethylamine
- Anavex 2-73
- Tetrahydro-N,N-dimethyl-2,2-diphenyl-3-furanemethanamine
- THD-DP-FM
- AE-37 / AE37 / ANAVEX 2-73 FREE BASE
- UNII 9T210MMZ3F
| Formula |
C19H23NO
|
|---|---|
| Cas |
195615-83-9
195615-84-0 HCL
|
| Mol weight |
281.392
|
Treatment of Rett syndrome, Investigated for use/treatment in breast cancer.
Anti-amnesic, Muscarinic/sigma receptor agonist
- Originator Anavex Life Sciences
- Developer ABX-CRO; Anavex Life Sciences; The Michael J. Fox Foundation for Parkinsons Research
- Class Antidementias; Antidepressants; Antiepileptic drugs; Antiparkinsonians; Anxiolytics; Behavioural disorder therapies; Dimethylamines; Furans; Neuroprotectants; Neuropsychotherapeutics; Nootropics; Small molecules
- Mechanism of Action Muscarinic receptor modulators; Sigma-1 receptor agonists
- Orphan Drug Status Yes – Epilepsy; Rett syndrome
- Phase II/III Alzheimer’s disease
- Phase II Parkinson’s disease; Rett syndrome
- Preclinical Amyotrophic lateral sclerosis; Angelman syndrome; Anxiety disorders; Autistic disorder; Fragile X syndrome; Multiple sclerosis
- No development reported Cognition disorders; Epilepsy; Stroke
- 28 Oct 2019 No recent reports of development identified for phase-I development in Cognition-disorders in USA
- 09 Oct 2019 Anavex Life Sciences initiates enrolment in the long term extension ATTENTION-AD trial for Alzheimer’s disease in (country/ies)
- 02 Oct 2019 Anavex Life Sciences has patent protection covering compositions of matter and methods of treating Alzheimer’s disease for blarcamesine in USA
- Anavex Life Sciences is developing ANAVEX-2-73 and its active metabolite ANAVEX-19-144, for treating Alzheimer’s disease, epilepsy, stroke and Rett syndrome.
ANAVEX2-73 is an experimental drug is in Phase II trials for Alzheimer’s disease, phase I trials for epilepsy, and in preclinical trials for amyotrophic lateral sclerosis, Parkinson’s disease, Rett syndrome, stroke.[1][2] ANAVEX2-73 acts as a muscarinic receptor and a moderate sigma1 receptor agonist.[1] ANAVEX2-73 may function as a pro-drug for ANAVEX19-144 as well as a drug itself. ANAVEX19-144 is the active metabolite of ANAVEX 1-41, which is similar to ANAVEX2-73 but it is not as selective for sigma receptor.[2]
Properties and uses
ANAVEX2-73 has an inhibitory constant (ki) lower than 500 nM for all M1–M4 muscarinic acetylcholine receptor subtypes, demonstrating that it acts as a powerful antimuscarinic compound.[2] ANAVEX2-73 was originally tested in mice against the effect of the muscarinic receptor antagonist scopolamine, which induces learning impairment.[1] M1 receptor agonists are known to reverse the amnesia caused by scopolamine.[3] Scopolamine is used in the treatment of Parkinson’s disease and motion sickness by reducing the secretions of the stomach and intestines and can also decreases nerve signals to the stomach.[3] This is via competitive inhibition of muscarinic receptors.[3] Muscarinic receptors are involved in the formation of both short term and long term memories.[1] Experiments in mice have found that M1 and M3 receptor agonists inhibit the formation of amyloid-beta and target GSK-3B.[clarification needed]Furthermore, stimulation of the M1 receptor activates AF267B, which in turn blocks β-secretase, which cleaves the amyloid precursor protein to produce the amyloid-beta peptide. These amyloid-beta peptides aggregate together to form plaques. This enzyme[clarification needed] is involved in the formation of Tau plaques, which are common in Alzheimer’s disease.[clarification needed][4]Therefore. M1 receptor activation appears to decreases tau hyperphosphorylation and amyloid-beta accumulation.[4]
Sigma1 activation appears to be only involved in long-term memory processes. This partly explains why ANAVEX2-73 seems to be more effective in reversing scopolamine-induced long-term memory problems compared to short-term memory deficits.[1] The sigma-1 receptor is located on mitochondria-associated endoplasmic reticulum membranes and modulates the ER stress response and local calcium exchanges with the mitochondria. ANAVEX2-73 prevented Aβ25-35-induced increases in lipid peroxidation levels, Bax/Bcl-2ratio and cytochrome c release into the cytosol, which are indicative of elevated toxicity.[clarification needed] ANAVEX2-73 inhibits mitochondrial respiratory dysfunction and therefore prevents against oxidative stress and apoptosis. This drug prevented the appearance of oxidative stress. ANAVEX2-73 also exhibits anti-apoptotic and anti-oxidant activity. This is due in part because sigma-1 agonists stimulate the anti-apoptoic factor Bcl-2 due to reactive oxygen species dependent transcriptional activation of nuclear factor kB.[5] Results from Marice (2016) demonstrate that sigma1 compounds offer a protective potential, both alone and possibly with other agents like donepezil, an acetylcholinesterase inhibitor, or the memantine, a NMDA receptor antagonist.[6]
PATENT
WO9730983
PATENT
Novel crystalline forms of A2-73 (blarcamesine hydrochloride, ANAVEX2-73, AV2-73), a mixed muscarinic receptor ligand and Sig-1 R agonist useful for treating Alzheimer’s disease.
PATENT
WO2017013498
SYN
By Foscolos, George B. et alFrom Farmaco, 51(1), 19-26; 1996

References
- ^ Jump up to:a b c “ANAVEX 2-73 – AdisInsight”. Adisinsight.springer.com. Retrieved 2016-05-25.
- ^ Jump up to:a b c Malviya, M; Kumar, YC; Asha, D; Chandra, JN; Subhash, MN; Rangappa, KS (2008). “Muscarinic receptor 1 agonist activity of novel N-arylthioureas substituted 3-morpholino arecoline derivatives in Alzheimer’s presenile dementia models”. Bioorg Med Chem. 16: 7095–7101. doi:10.1016/j.bmc.2008.06.053.
- ^ Jump up to:a b Leal, NS; Schreiner, B; Pinho, CM; Filadi, R; Wiehager, B; Karlström, H; Pizzo, P; Ankarcrona, M (2016). “Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production”. J Cell Mol Med. 20: 1686–1695. doi:10.1111/jcmm.12863. PMC 4988279. PMID 27203684.
- ^ Lahmy, V; Long, R; Morin, D; Villard, V; Maurice, T (2015-09-28). “Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model”. Front Cell Neurosci. 8: 463. doi:10.3389/fncel.2014.00463. PMC 4299448. PMID 25653589.
- ^ Maurice, T (2015-09-28). “Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments”. Behav. Brain Res. 296: 270–8. doi:10.1016/j.bbr.2015.09.020. PMID 26386305.
//////////Blarcamesine, ブラルカメシン , Orphan Drug Status, PHASE 2
CN(C)CC1CCOC1(C1=CC=CC=C1)C1=CC=CC=C1
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
Today, the U.S. Food and Drug Administration approved Inrebic (fedratinib) capsules to treat adult patients with certain types of myelofibrosis.
“Prior to today, there was one FDA-approved drug to treat patients with myelofibrosis, a rare bone marrow disorder. Our approval today provides another option for patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The FDA is committed to encouraging the development of treatments for patients with rare diseases and providing alternative options, as not all patients respond in the same way.”
Myelofibrosis is a chronic disorder where scar tissue forms in the bone marrow and the production of the blood cells moves from the bone marrow to the spleen and liver, causing organ enlargement. It can cause extreme fatigue, shortness of breath, pain below the ribs, fever, night sweats, itching and bone pain. When myelofibrosis occurs on its own, it is called primary myelofibrosis. Secondary myelofibrosis occurs when there is excessive red blood cell production (polycythemia vera) or excessive platelet production (essential thrombocythemia) that evolves into myelofibrosis.
Jakafi (ruxolitinib) was approved by the FDA in 2011. The approval of Inrebic for intermediate-2 or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis was based on the results of a clinical trial where 289 patients with myelofibrosis were randomized to receive two different doses (400 mg or 500 mg daily by mouth) of fedratinib or placebo. The clinical trial showed that 35 of 96 patients treated with the fedratinib 400 mg daily dose (the dose recommended in the approved label) experienced a significant therapeutic effect (measured by greater than or equal to a 35% reduction from baseline in spleen volume at the end of cycle 6 (week 24) as measured by an MRI or CT scan with a follow-up scan four weeks later). As a result of treatment with Inrebic, 36 patients experienced greater than or equal to a 50% reduction in myelofibrosis-related symptoms, such as night sweats, itching, abdominal discomfort, feeling full sooner than normal, pain under ribs on left side, and bone or muscle pain.
The prescribing information for Inrebic includes a Boxed Warning to advise health care professionals and patients about the risk of serious and fatal encephalopathy (brain damage or malfunction), including Wernicke’s, which is a neurologic emergency related to a deficiency in thiamine. Health care professionals are advised to assess thiamine levels in all patients prior to starting Inrebic, during treatment and as clinically indicated. If encephalopathy is suspected, Inrebic should be immediately discontinued.
Common side effects for patients taking Inrebic are diarrhea, nausea, vomiting, fatigue and muscle spasms. Health care professionals are cautioned that patients may experience severe anemia (low iron levels) and thrombocytopenia (low level of platelets in the blood). Patients should be monitored for gastrointestinal toxicity and for hepatic toxicity (liver damage). The dose should be reduced or stopped if a patient develops severe diarrhea, nausea or vomiting. Treatment with anti-diarrhea medications may be recommended. Patients may develop high levels of amylase and lipase in their blood and should be managed by dose reduction or stopping the mediation. Inrebic must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
The FDA granted this application Priority Review designation. Inrebic also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Inrebic to Impact Biomedicines, Inc., a wholly-owned subsidiary of Celgene Corporation.
LINK
///////Inrebic , fedratinib, FDA 2019, Priority Review , Orphan Drug, Biomedicines, Celgene , bone marrow disorder
FDA approves third oncology drug Rozlytrek (entrectinib) that targets a key genetic driver of cancer, rather than a specific type of tumor
FDA also approves drug for second indication in a type of lung cancer
The U.S. Food and Drug Administration today granted accelerated approval to Rozlytrek (entrectinib), a treatment for adult and adolescent patients whose cancers have the specific genetic defect, NTRK (neurotrophic tyrosine receptor kinase) gene fusion and for whom there are no effective treatments.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” said FDA Acting Commissioner Ned Sharpless, M.D. “Using the FDA’s expedited review pathways, including breakthrough therapy designation and accelerated approval process, we’re supporting this innovation in precision oncology drug development and the evolution of more targeted and effective treatments for cancer patients. We remain committed to encouraging the advancement of more targeted innovations in oncology treatment and across disease types based on our growing understanding of the underlying biology of diseases.”
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than the location in the body where the tumor originated. The approval marks a new paradigm in the development of cancer drugs that are “tissue agnostic.” It follows the policies that the FDA developed in a guidance document released in 2018. The previous tissue agnostic indications approved by the FDA were pembrolizumab for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib for NTRK gene fusion tumors in 2018.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK-fusion-positive tumors by relying on efficacy information obtained primarily in adults. The FDA continues to encourage the inclusion of adolescents in clinical trials. Traditionally, clinical development of new cancer drugs in pediatric populations is not started until development is well underway in adults, and often not until after approval of an adult indication,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
The ability of Rozlytrek to shrink tumors was evaluated in four clinical trials studying 54 adults with NTRK fusion-positive tumors. The proportion of patients with substantial tumor shrinkage (overall response rate) was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for nine months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid and colon/rectum.
Rozlytrek was also approved today for the treatment of adults with non-small cell lung cancer whose tumors are ROS1-positive (mutation of the ROS1 gene) and has spread to other parts of the body (metastatic). Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
Rozlytrek’s common side effects are fatigue, constipation, dysgeusia (distorted sense of taste), edema (swelling), dizziness, diarrhea, nausea, dysesthesia (distorted sense of touch), dyspnea (shortness of breath), myalgia (painful or aching muscles), cognitive impairment (confusion, problems with memory or attention, difficulty speaking, or hallucinations), weight gain, cough, vomiting, fever, arthralgia and vision disorders (blurred vision, sensitivity to light, double vision, worsening of vision, cataracts, or floaters). The most serious side effects of Rozlytrek are congestive heart failure (weakening or damage to the heart muscle), central nervous system effects (cognitive impairment, anxiety, depression including suicidal thinking, dizziness or loss of balance, and change in sleep pattern, including insomnia and excessive sleepiness), skeletal fractures, hepatotoxicity (damage to the liver), hyperuricemia (elevated uric acid), QT prolongation (abnormal heart rhythm) and vision disorders. Health care professionals should inform females of reproductive age and males with a female partner of reproductive potential to use effective contraception during treatment with Rozlytrek. Women who are pregnant or breastfeeding should not take Rozlytrek because it may cause harm to a developing fetus or newborn baby.
Rozlytrek was granted accelerated approval. This approval commits the sponsor to provide additional data to the FDA. Rozlytrek also received Priority Review, Breakthrough Therapy and Orphan Drug designation. The approval of Rozlytrek was granted to Genentech, Inc.
///////////////Rozlytrek, entrectinib, accelerated approval, priority Review, Breakthrough Therapy, Orphan Drug designation, fda 2019, Genentech, cancer
Tanzisertib
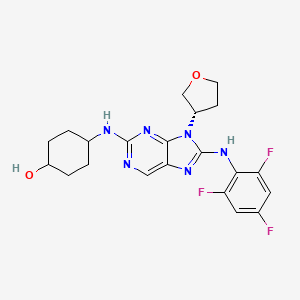
Tanzisertib
CAS 899805-25-5
trans-4-((9-((3S)-Tetrahydrofuran-3-yl)-8-((2,4,6-trifluorophenyl)amino)-9H-purin-2-yl)amino)cyclohexanol
4-[[9-[(3S)-oxolan-3-yl]-8-(2,4,6-trifluoroanilino)purin-2-yl]amino]cyclohexan-1-ol
C21-H23-F3-N6-O2, 448.4467
- CC 930
- CC-930
- Tanzisertib
- UNII-M5O06306UO
- A c-Jun amino-terminal kinase inhibitor.UNII, M5O06306UO
Treatment of Idiopathic Pulmonary Fibrosis (IPF)
- Originator Celgene Corporation
- Class Antifibrotics; Small molecules
- Mechanism of ActionJ NK mitogen-activated protein kinase inhibitors
- Orphan Drug Status Yes – Idiopathic pulmonary fibrosis
- Discontinued Discoid lupus erythematosus; Idiopathic pulmonary fibrosis
- 16 Jul 2012 Celgene Corporation terminates a phase II trial in Discoid lupus erythematosus in USA (NCT01466725)
- 23 Feb 2012 Celgene initiates enrolment in a phase II trial for Discoid lupus erythematosus in the USA (NCT01466725)
- 08 Nov 2011The Committee for Orphan Medicinal Products (COMP) recommends orphan drug designation for tanzisertib in European Union for Idiopathic pulmonary fibrosis
Tanzisertib has been granted orphan drug status by the FDA for the treatment of idiopathic pulmonary fibrosis. A positive opinion has been received from the EU Committee for Orphan Medicinal Products (COMP
Tanzisertib has been used in trials studying the treatment of Fibrosis, Discoid Lupus, Pulmonary Fibrosis, Interstitial Lung Disease, and Lung Diseases, Interstitial, among others.
PATENT
https://patents.google.com/patent/US20090048275A1/de

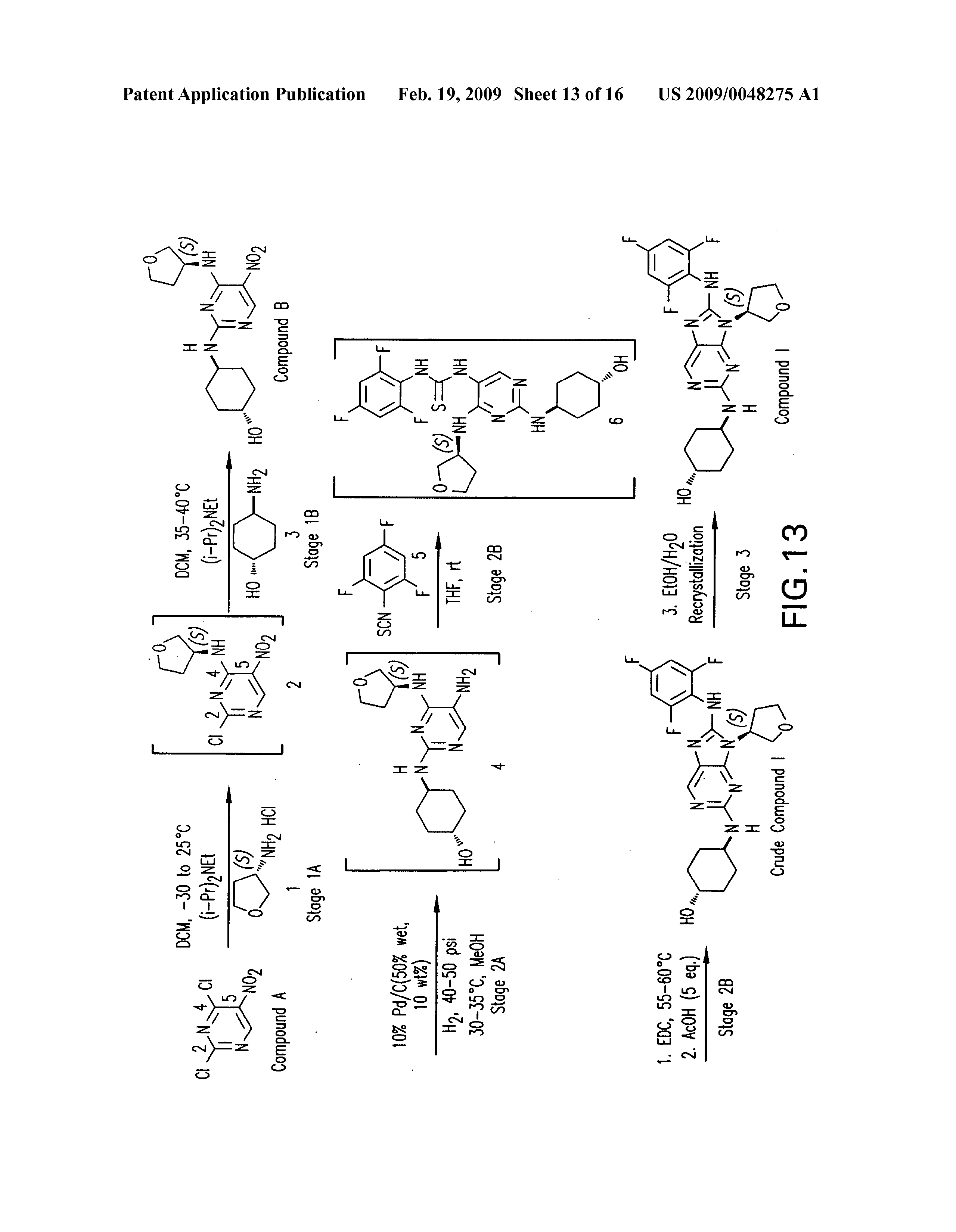

PATENT
WO 2006076595
US 20070060598
WO 2008057252
US 20080021048
US 20140094456
WO 2014055548
PATENT
WO 2015153683
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015153683
/////////Tanzisertib, CC 930, Idiopathic Pulmonary Fibrosis, Orphan Drug, phase II, CELGENE
c1c(c(c(cc1F)F)Nc2n(c3nc(ncc3n2)N[C@H]4CC[C@@H](CC4)O)[C@@H]5COCC5)F
Reldesemtiv
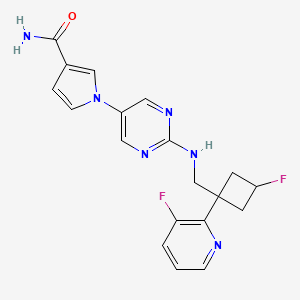
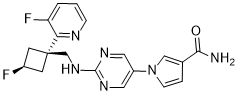
Reldesemtiv
CK-2127107
CAS 1345410-31-2
UNII-4S0HBYW6QE, 4S0HBYW6QE
MW 384.4 g/mol, MF C19H18F2N6O
1-[2-({[trans-3-fluoro-1-(3-fluoropyridin-2-yl)cyclobutyl]methyl}amino)pyrimidin-5-yl]-1H-pyrrole-3- carboxamide
1-[2-[[3-fluoro-1-(3-fluoropyridin-2-yl)cyclobutyl]methylamino]pyrimidin-5-yl]pyrrole-3-carboxamide
Reldesemtiv, also known as CK-2127107, is a skeletal muscle troponin activator (FSTA) and is a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue such as SMA, COPD, and ALS.
Cytokinetics , in collaboration with Astellas , is developing reldesemtiv, the lead from a program of selective fast skeletal muscle troponin activators, in an oral suspension formulation, for the treatment of indications associated with neuromuscular dysfunction, including spinal muscular atrophy and amyotrophic lateral sclerosis.
- Originator Cytokinetics
- Developer Astellas Pharma; Cytokinetics
- Class Pyridines; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Troponin stimulants
- Orphan Drug Status Yes – Spinal muscular atrophy
- Phase II Amyotrophic lateral sclerosis; Chronic obstructive pulmonary disease; Spinal muscular atrophy
- Suspended Muscle fatigue
- No development reported Muscular atrophy
- 05 May 2019 Safety and efficacy data from the phase II FORTITUDE-ALS trial in Amyotrophic lateral sclerosis presented at the American Academy of Neurology Annual Meeting (AAN-2019)
- 07 Mar 2019 Cytokinetics completes the phase III FORTITUDE-ALS trial for Amyotrophic lateral sclerosis in USA, Australia, Canada, Spain, Ireland and Netherlands (PO) (NCT03160898)
- 22 Jan 2019 Cytokinetics plans a phase I trial in Healthy volunteers in the first quarter of 2019
Reldesemtiv, a next-generation, orally-available, highly specific small-molecule is being developed by Cytokinetics, in collaboration with Astellas Pharma, for the improvement of skeletal muscle function associated with neuromuscular dysfunction, muscle weakness and/or muscle fatigue in spinal muscular atrophy (SMA), chronic obstructive pulmonary disease (COPD) and amyotrophic lateral sclerosis (ALS). The drug candidate is a fast skeletal muscle troponin activator (FSTA) or troponin stimulant intended to slow the rate of calcium release from the regulatory troponin complex of fast skeletal muscle fibers. Clinical development for ALS, COPD and SMA is underway in the US, Australia, Canada, Ireland, Netherlands and Spain. No recent reports of development had been identified for phase I development for muscular atrophy in the US. Due to lack of of efficacy determined at interim analysis Cytokinetics suspended phase I trial in muscle fatigue in the elderly.
The cytoskeleton of skeletal and cardiac muscle cells is unique compared to that of all other cells. It consists of a nearly crystalline array of closely packed cytoskeletal proteins called the sarcomere. The sarcomere is elegantly organized as an interdigitating array of thin and thick filaments. The thick filaments are composed of myosin, the motor protein responsible for transducing the chemical energy of ATP hydrolysis into force and directed movement. The thin filaments are composed of actin monomers arranged in a helical array. There are four regulatory proteins bound to the actin filaments, which allows the contraction to be modulated by calcium ions. An influx of intracellular calcium initiates muscle contraction; thick and thin filaments slide past each other driven by repetitive interactions of the myosin motor domains with the thin actin filaments.
[0003] Of the thirteen distinct classes of myosin in human cells, the myosin-II class is responsible for contraction of skeletal, cardiac, and smooth muscle. This class of myosin is significantly different in amino acid composition and in overall structure from myosin in the other twelve distinct classes. Myosin-II forms homo-dimers resulting in two globular head domains linked together by a long alpha-helical coiled-coiled tail to form the core of the sarcomere’s thick filament. The globular heads have a catalytic domain where the actin binding and ATPase functions of myosin take place. Once bound to an actin filament, the release of phosphate (cf. ADP-Pi to ADP) signals a change in structural conformation of the catalytic domain that in turn alters the orientation of the light-chain binding lever arm domain that extends from the globular head; this movement is termed the powerstroke. This change in orientation of the myosin head in relationship to actin causes the thick filament of which it is a part to move with respect to the thin actin filament to which it is bound. Un-binding of the globular head from the actin filament (Ca2+ regulated) coupled with return of the catalytic domain and light chain to their starting conformation/orientation completes the catalytic cycle, responsible for intracellular movement and muscle contraction.
Tropomyosin and troponin mediate the calcium effect on the interaction on actin and myosin. The troponin complex is comprised of three polypeptide chains: troponin C, which binds calcium ions; troponin I, which binds to actin; and troponin T, which binds to tropomyosin. The skeletal troponin-tropomyosin complex regulates the myosin binding sites extending over several actin units at once.
Troponin, a complex of the three polypeptides described above, is an accessory protein that is closely associated with actin filaments in vertebrate muscle. The troponin complex acts in conjunction with the muscle form of tropomyosin to mediate the
Ca2+ dependency of myosin ATPase activity and thereby regulate muscle contraction. The troponin polypeptides T, I, and C, are named for their tropomyosin binding, inhibitory, and calcium binding activities, respectively. Troponin T binds to tropomyosin and is believed to be responsible for positioning the troponin complex on the muscle thin filament. Troponin I binds to actin, and the complex formed by troponins I and T, and tropomyosin inhibits the interaction of actin and myosin. Skeletal troponin C is capable of binding up to four calcium molecules. Studies suggest that when the level of calcium in the muscle is raised, troponin C exposes a binding site for troponin I, recruiting it away from actin. This causes the tropomyosin molecule to shift its position as well, thereby exposing the myosin binding sites on actin and stimulating myosin ATPase activity.
U.S. Patent No. 8962632 discloses l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide, a next-generation fast skeletal muscle troponin activator (FSTA) as a potential treatment for people living with debilitating diseases and conditions associated with neuromuscular or non-neuromuscular dysfunction, muscular weakness, and/or muscle fatigue.
PATENT
WO 2011133888

PATENT
WO2016039367 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016039367&tab=FULLTEXT
claiming the use of a similar compound for treating stress urinary incontinence.
PATENT
WO-2019133605
Process for preparing reldesemtiv , a myosin, actin, tropomyosin, troponin C, troponin I, troponin T modulator, useful for treating neuromuscular disorders, muscle wasting, claudication and metabolic syndrome.
Scheme 1
[0091] Scheme 1 illustrates a scheme of synthesizing the compound of Formula (1C).
Scheme 2
[0092] Scheme 2 illustrates an alternative scheme of synthesizing the compound of Formula (1C).
M
TFAA DS, toluene
Et
to
2
HCI, H20
50°C
Scheme 3
[0093] Scheme 3 illustrates a scheme of converting the compound of Formula (1C) to the compound of Formula (II).
H2
Ni Raney
NH3
Scheme 4
[0094] Scheme 4 illustrates a scheme of converting the compound of Formula (II) to the compound of Formula (1).
Examples
[0095] To a flask was added N-methylpyrrolidone (30 mL), tert-butyl cyanoacetate (8.08 g) at room temperature. To a resulting solution was added potassium tert-butoxide (7.71 g), l,3-dibromo-2,2-dimethoxy propane (5.00 g) at 0 °C. To another flask, potassium iodide (158 mg), 2,6-di-tert-butyl-p-cresol (42 mg), N-methylpyrrolidone (25 mL) were added at room temperature and then resulting solution was heated to 165 °C. To this solution, previously prepared mixture was added dropwise at 140-165 °C, then stirred for 2 hours at 165 °C. To the reaction mixture, water (65 mL) was added. A resulting solution was extracted with toluene (40 mL, three times) and then combined organic layer was washed with water (20 mL, three times) and 1N NaOH aq. (20 mL). A resulting organic layer was concentrated below 50 °C under reduced pressure to give 3, 3 -dimethoxy cyclobutane- l-carbonitrile (66% yield,
GC assay) as toluene solution. 1H MR (CDCl3, 400 MHz) d 3.17 (s, 3H), 3.15 (s, 3H), 2.93-2.84 (m, 1H), 2.63-2.57 (m, 2H), 2.52-2.45 (m, 2H).
Example 2 Synthesis of methyl 3,3-dimethoxycyclobutane-l-carboxylate
[0096] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. MeOH (339.00 kg), 3-oxocyclobutanecarboxylic acid (85.19 kg, 746.6 mol, 1.0 eq.), Amberlyst-l5 ion exchange resin (8.90 kg, 10% w/w), and
trimethoxymethane (196.00 kg, 1847.3 mol, 2.5 eq.) were charged into the reactor and the resulting mixture was heated to 55±5°C and reacted for 6 hours to give methyl 3,3-dimethoxycyclobutane-l-carboxylate solution in MeOH. 1H NMR (CDCl3, 400 MHz) d 3.70 (s, 3H), 3.17 (s, 3H), 3.15 (s, 3H), 2.94-2.85 (m, 1H), 2.47-2.36 (m, 4H).
Example 3 Synthesis of 3, 3-dimethoxycyclobutane-l -carboxamide
[0097] The methyl 3, 3 -dimethoxy cyclobutane- l-carboxylate solution in MeOH prepared as described in Example 2 was cooled to below 25°C and centrifuged. The filter cake was washed with MeOH(7.00 kg) and the filtrate was pumped to the reactor. The solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH
(139.40 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. MeOH (130.00 kg) was charged to the reactor and the solution was concentrated under vacuum below 55°C until the system had no more than 2 volumes. Half of the resulting solution was diluted with MeOH (435.00 kg) and cooled to below 30°C. NH3 gas (133.80 kg) was injected into the reactor below 35°C for
24 hours. The mixture was stirred at 40±5°C for 72 hours. The resulting solution was
concentrated under vacuum below 50°C until the system had no more than 2 volumes.
MTBE(l8l.OO kg) was charged into the reactor. The resulting solution was concentrated under vacuum below 50°C until the system had no more than 2 volumes. PE (318.00 kg) was charged into the reactor. The resulting mixture was cooled to 5±5°C, stirred for 4 hours at 5±5°C, and centrifuged. The filter cake was washed with PE (42.00 kg) and the wet filter cake was put into a vacuum oven. The filter cake was dried at 30±5°C for at least 8 hours to give 3,3-dimethoxycyclobutane-l-carboxamide as off-white solid (112.63 kg, 94.7% yield). 1H NMR (CDCf, 400 MHz) d 5.76 (bs, 1H), 5.64 (bs, 1H), 3.18 (s, 3H), 3.17 (s, 3H), 2.84-2.76 (m, 1H), 2.45-2.38 (m, 4H).
[0098] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Toluene (500.00 kg), 3,3-dimethoxycyclobutane-l-carboxamide (112.54kg, 706.9 mol, 1.0 eq.), and TEA (158.00 kg, 1561.3 mol, 2.20 eq) were charged into the reactor and the resulting mixture was cooled to 0+ 5°C. TFAA (164.00 kg, 781 mol, 1.10 eq.) was added dropwise at 0±5°C. The resulting mixture was stirred for 10 hours at 20±5°C and cooled below 5±5°C. H20 (110.00 kg) was charged into the reactor at below 15 °C. The resulting mixture was stirred for 30 minutes and the water phase was separated. The aqueous phase was extracted with toluene (190.00 kg) twice. The organic phases were combined and washed with H20 (111.00 kg). H20 was removed by azeotrope until the water content was no more than 0.03%. The resulting solution was cooled to below 20°C to give 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene (492.00 kg with 17.83% assay content, 87.9% yield).
Example 5 Synthesis of l-(3-fluoropyridin-2-yl)-3,3-dimethoxycyclobutane-l-carbonitrile
[0099] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The 3,3-dimethoxycyclobutane-l-carbonitrile solution in toluene prepared as described in Example 4 (246.00 kg of a 17.8% solution of 3,3-dimethoxycyclobutane-l-carbonitrile in toluene, 1.05 eq.) and 2-chloro-3-fluoropyridine (39.17 kg, 297.9 mol, 1.00 eq.) were charged into the reactor. The reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. The mixture was slowly cooled to -20±5°C. NaHDMS (2M in THF) (165.71 kg, 1.20 eq) was added
dropwise at -20±5°C. The resulting mixture was stirred at -l5±5°C for 1 hour. The mixture was stirred until the content of 2-chloro-3-fluoropyridine is no more than 2% as measured by HPLC. Soft water (16.00 kg) was added dropwise at below 0°C while maintaining the reactor temperature. The resulting solution was transferred to another reactor. Aq. NH4Cl (10% w/w, 88.60 Kg) was added dropwise at below 0°C while maintaining the reactor temperature. Soft water (112.00 kg) was charged into the reactor and the aqueous phase was separated and collected. The aqueous phase was extracted with ethyl acetate (70.00 kg) and an organic phase was collected. The organic phase was washed with sat. NaCl (106.00 kg) and collected. The above steps were repeated to obtain another batch of organic phase. The two batches of organic phase were concentrated under vacuum below 70°C until the system had no more than 2 volumes. The resulting solution was cooled to below 30°C to give a l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution. 1H NMR (CDC13, 400 MHz) d 8.42-8.38 (m, 1H), 7.50-7.45 (m, 1H), 7.38-7.33 (m, 1H), 3.28 (s, 3 H), 3.13 (s, 3H), 3.09-3.05 (m, 4H).
Example 6 Synthesis of I-(3-fluoropyridin-2-yl)-3-oxocyclohutanecarhonitrile
[0100] A reactor was vacuumed to 0.02 MPa and less and then inerted with nitrogen to atmosphere for three times. Water (603.00 kg) was added to the reactor and was stirred.
Concentrated HC1 (157.30 kg) was charged into the reactor at below 35°C. The l-(3-fluoropyridin-2-yl)-3, 3 -dimethoxy cyclobutane- l-carbonitrile solution prepared as described in Example 5 (206.00 kg) was charged into the reactor and the resulting mixture was heated to 50±5°C and reacted for 3 hours at 50±5°C. The mixture was reacted until the content of 1-(3 -fluoropyridin-2-yl)-3, 3 -dimethoxycyclobutane- l-carbonitrile was no more than 2.0% as measured by HPLC. The reaction mixture was cooled to below 30°C and extracted with ethyl acetate (771.00 kg). An aqueous phase was collected and extracted with ethyl acetate (770.00 kg). The organic phases were combined and the combined organic phase was washed with soft water (290.00 kg) and brine (385.30 kg). The organic phase was concentrated under vacuum at below 60°C until the system had no more than 2 volumes. Propan-2-ol (218.00 kg) was charged into the reactor. The organic phase was concentrated under vacuum at below
60°C until the system had no more than 1 volume. PE (191.00 kg) was charged into the reactor at 40±5 °C and the resulting mixture was heated to 60±5 °C and stirred for 1 hour at 60±5 °C. The mixture was then slowly cooled to 5±5 °C and stirred for 5 hours at 5±5 °C. The mixture was centrifuged and the filter cake was washed with PE (48.00 kg) and the wet filter cake was collected. Water (80.00 kg), concentrated HC1 (2.20 kg), propan-2-ol (65.00 kg), and the wet filter cake were charged in this order into a drum. The resulting mixture was stirred for 10 minutes at 20±5 °C. The mixture was centrifuged and the filter cake was washed with a mixture solution containing 18.00 kg of propan-2-ol, 22.50 kg of soft water, and 0.60 kg of concentrated HC1. The filter cake was put into a vacuum oven and dried at 30±5°C for at least 10 hours. The filter cake was dried until the weight did not change to give l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile as off-white solid (77.15 kg, 68.0% yield). 1H NMR (CDCl3, 400 MHz) d 8.45-8.42 (m, 1H), 7.60-7.54 (m, 1H), 7.47-7.41 (m, 1H), 4.18-4.09 (m, 2H), 4.02-3.94 (m, 2H).
Example 7 Synthesis of I-(3-fhtoropyridin-2-yl)-3-hydroxycyclobulanecarbonilrile
[0101] To a solution of l-(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile (231 g,
1.22 mol) in a mixture ofDCM (2 L) and MeOH (200 mL) was added NaBH4 portionwise at -78° C. The reaction mixture was stirred at -78°C. for 1 hour and quenched with a mixture of methanol and water (1 : 1). The organic layer was washed with water (500 mL><3), dried over Na2S04, and concentrated. The residue was purified on silica gel (50% EtO Ac/hexanes) to provide the title compound as an amber oil (185.8 g, 77.5%). Low Resolution Mass
Spectrometry (LRMS) (M+H) m/z 193.2.
Example 8 Synthesis of (ls,3s)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutane-l-carbonitrile
[0102] To a solution of 1 -(3 -fluoropyridin-2-yl)-3 -hydroxy cyclobutanecarbonitrile (185 g, 0.96 mol) in DCM (1 L) was added DAST portionwise at 0-10 °C. Upon the completion of addition, the reaction was refluxed for 6 hours. The reaction was cooled to rt and poured onto sat. NaHCCf solution. The mixture was separated and the organic layer was washed with water, dried over Na2S04, and concentrated. The residue was purified on silica gel (100% DCM) to provide the title compound as a brown oil (116g) in a 8: 1 transxis mixture. The above brown oil (107 g) was dissolved in toluene (110 mL) and hexanes (330mL) at 70 °C. The solution was cooled to 0 °C and stirred at 0 °C overnight. The precipitate was filtered and washed with hexanes to provide the trans isomer as a white solid (87.3 g). LRMS (M+H) m/z 195.1.
Example 9 Synthesis of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine
[0103] A mixture of ( 1.v,3.v)-3-fluoro- 1 -(3-fluoropyridin-2-yl)cyclobutane- 1 -carbonitrile (71 g, 0.37 mol) and Raney nickel (~7 g) in 7N ammonia in methanol (700 mL) was charged with hydrogen (60 psi) for 2 days. The reaction was filtered through a celite pad and washed with methanol. The filtrate was concentrated under high vacuum to provide the title compound as a light green oil (70 g, 97.6%). LRMS (M+H) m/z 199.2.
Example 10 Synthesis of t-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate
[0104] A mixture of ((lr,3r)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methanamine (37.6 g, 190 mmol), 5-bromo-2-fluoropyrimidine (32.0 g, 181 mmol), DIPEA (71 mL, 407 mmol), and NMP (200 mL) was stirred at rt overnight. The reaction mixture was then diluted with EtOAc (1500 mL) and washed with saturated sodium bicarbonate (500 mL). The
organic layer was separated, dried over Na2S04, and concentrated. The resultant solid was dissolved in THF (600 mL), followed by the slow addition of DMAP (14 g, 90 mmol) and Boc20 (117.3 g, 542 mmol). The reaction was heated to 60° C. and stirred for 3 h. The reaction mixture was then concentrated and purified by silica gel chromatography
(EtO Ac/hex) to give 59.7 g oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a white solid.
Example 11 Synthesis of t-butyl 5-(3-cyano- 1 H -pyrrol- 1 -yl)pyrimidin-2-yl(((lrans)-3-fhtoro-l-(3-fluoropyridin-2-yl)cyclohutyl)methyl)carhamate
[0105] To a solution oft-butyl 5-bromopyrimidin-2-yl((trans-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl) carbamate (1.0 g, 2.8 mmol) in 15 mL of toluene (degassed with nitrogen) was added copper iodide (100 mg, 0.6 mmol), potassium phosphate (1.31 g, 6.2 mmol), trans-N,N’-dimethylcyclohexane-l, 2-diamine (320 mg, 2.2 mmol), and 3-cyanopyrrole (310 mg, 3.6 mmol). The reaction was heated to 100 °C and stirred for 2 h. The reaction was then concentrated and purified by silica gel chromatography (EtOAc/hexanes) to afford 1.1 g of t-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate as a clear oil.
Example 12 Synthesis of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3-carboxamide
[0106] To a solution oft-butyl 5-(3-cyano-lH-pyrrol-l-yl)pyrimidin-2-yl(((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)carbamate (1.1 g, 3.1 mmol) in DMSO (10 mL) was added potassium carbonate (1.3 g, 9.3 mmol). The mixture was cooled to 0 °C and hydrogen peroxide (3 mL) was slowly added. The reaction was warmed to rt and stirred for 90 min. The reaction was diluted with EtO Ac (75 mL) and washed three times with brine (50 mL). The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10% MeOH/CH2Cl2) to afford 1.07 g of a white solid compound. This compound was dissolved in 25% TFA/CH2CI2 and stirred for 1 hour. The reaction was then concentrated, dissolved in ethyl acetate (75 mL), and washed three times with saturated potassium carbonate solution. The organic layer was then dried over Na2S04, filtered, and concentrated to give a crude solid that was triturated with 75% ethyl acetate/hexanes. The resultant slurry was sonicated and filtered to give 500 mg of l-(2-((((trans)-3-fluoro-l-(3-fluoropyridin-2-yl)cyclobutyl)methyl)amino)pyrimidin-5-yl)-lH-pyrrole-3 -carboxamide as a white solid. LRMS (M+H=385).
REFERENCES
1: Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, Wolff AA, Malik FI. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018 May;57(5):729-734. doi: 10.1002/mus.26017. Epub 2017 Dec 11. PubMed PMID: 29150952.
2: Gross N. The COPD Pipeline XXXII. Chronic Obstr Pulm Dis. 2016 Jul 14;3(3):688-692. doi: 10.15326/jcopdf.3.3.2016.0150. PubMed PMID: 28848893; PubMed Central PMCID: PMC5556764.
//////////////CK-2127107, CK 2127107, CK2127107, Reldesemtiv, Cytokinetics, Astellas, neuromuscular disorders, muscle wasting, claudication, metabolic syndrome, spinal muscular atrophy, amyotrophic lateral sclerosis, Orphan Drug Status, Spinal muscular atrophy, Phase II
C1C(CC1(CNC2=NC=C(C=N2)N3C=CC(=C3)C(=O)N)C4=C(C=CC=N4)F)F

{kind=link}