
Home » 0rphan drug status (Page 5)
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Idecabtagene vicleucel
Idecabtagene vicleucel
CAS 2306267-75-2
STN: BLA 125736
An autologous T lymphocyte-enriched cell transduced ex vivo with an anti-BCMA CAR lentiviral vector encoding a chimeric antigen receptor CAR, comprising a CD8 hinge and TM domain, 4-1BB costimulatory domain and CD3ζ signaling domain, targeting human B cell maturation antigen for cancer immunotherapy (Celgene Corp., NJ)
- Bb2121
| Name | Idecabtagene vicleucel (USAN); Abecma (TN) |
|---|---|
| Product | ABECMA (Celgene Corporation) |
| CAS | 2306267-75-2 |
| Efficacy | Antineoplastic, Anti-BCMA CAR-T cell |
| Disease | Multiple myeloma [DS:H00010] |
| Comment | Cellular therapy product |
USFDA 2021/4/21 APPROVED
Dendritic cells (DCs) are antigen-presenting cells (APCs) that process antigens and display them to other cells of the immune system. Specifically, dendritic cells are capable of capturing and presenting antigens on their surfaces to activate T cells such as cytotoxic T cells (CTLs). Further, activated dendritic cells are capable of recruiting additional immune cells such as macrophages, eosinophils, natural killer cells, and T cells such as natural killer T cells.
Despite major advances in cancer treatment, cancer remains one of the leading causes of death globally. Hurdles in designing effective therapies include cancer immune evasion, in which cancer cells escape destructive immunity, as well as the toxicity of many conventional cancer treatments such as radiation therapy and chemotherapy, which significantly impacts a patient’s ability to tolerate the therapy and/or impacts the efficacy of the treatment.
Given the important role of dendritic cells in immunity, derailed dendritic cell functions have been implicated in diseases such as cancer and autoimmune diseases. For example, cancer cells may evade immune detection and destruction by crippling dendritic cell functionality through prevention of dendritic cell recruitment and activation. In addition, dendritic cells have been found in the brain during central nervous system inflammation and may be involved in the pathogenesis of autoimmune diseases in the brain.
One mechanism by which cancers evade immune detection and destruction is by crippling dendritic cell functionality through prevention of dendritic cell (DC) recruitment and activation. Accordingly, there remains a need for cancer therapies that can effectively derail tumor evasion and enhance anti-tumor immunity as mediated, for example, by dendritic cells.
NEW DRUG APPROVALS
ONE TIME
$10.00
DESCRIPTION
ABECMA is a BCMA-directed genetically modified autologous T cell immunotherapy product consisting of a patient’s own T cells that are harvested and genetically modified ex vivo through transduction with an anti-BCMA02 chimeric antigen receptor (CAR) lentiviral vector (LVV). Autologous T cells transduced with the anti-BCMA02 CAR LVV express the anti-BCMA CAR on the T cell surface. The CAR is comprised of a murine extracellular single-chain variable fragment (scFv) specific for recognizing B cell maturation antigen (BCMA) followed by a human CD8α hinge and transmembrane domain fused to the T cell cytoplasmic signaling domains of CD137 (4-1BB) and CD3ζ chain, in tandem. Binding of ABECMA to BCMA-expressing target cells leads to signaling initiated by CD3ζ and 4-1BB domains, and subsequent CAR-positive T cell activation. Antigen-specific activation of ABECMA results in CAR-positive T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA-expressing cells.
ABECMA is prepared from the patient’s peripheral blood mononuclear cells (PBMCs), which are obtained via a standard leukapheresis procedure. The mononuclear cells are enriched for T cells, through activation with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, which are then transduced with the replication-incompetent lentiviral vector containing the anti-BCMA CAR transgene. The transduced T cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved. The product must pass a sterility test before release for shipping as a frozen suspension in one or more patient-specific infusion bag(s). The product is thawed prior to infusion back into the patient [see DOSAGE AND ADMINISTRATION and HOW SUPPLIED/Storage And Handling].
The ABECMA formulation contains 50% Plasma-Lyte A and 50% CryoStor® CS10, resulting in a final DMSO concentration of 5%.
FDA approves idecabtagene vicleucel for multiple myeloma
On March 26, 2021, the Food and Drug Administration approved idecabtagene vicleucel (Abecma, Bristol Myers Squibb) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies; 88% had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel in the dose range of 300 to 460 x 106 CAR-positive T cells. Efficacy was established based on overall response rate (ORR), complete response (CR) rate, and duration of response (DOR), as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The ORR was 72% (95% CI: 62%, 81%) and CR rate was 28% (95% CI 19%, 38%). An estimated 65% of patients who achieved CR remained in CR for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome (CRS), neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. The most common side effects of idecabtagene vicleucel include CRS, infections, fatigue, musculoskeletal pain, and hypogammaglobulinemia.
Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities that dispense the therapy must be specially certified to recognize and manage CRS and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
The recommended dose range for idecabtagene vicleucel is 300 to 460 × 106 CAR-positive T cells. View full prescribing information for Abecma.
This application was granted breakthrough therapy designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
FDA D.I.S.C.O. Burst Edition: FDA approval of ABECMA (idecabtagene vicleucel) the first FDA approved cell-based gene therapy for the treatment of adult patients with relapsed or refractory multiple myeloma
Welcome back to the D.I.S.C.O., FDA’s Drug Information Soundcast in Clinical Oncology, Burst Edition, brought to you by FDA’s Division of Drug Information in partnership with FDA’s Oncology Center of Excellence. Today we have another quick update on a recent FDA cancer therapeutic approval.
On March 26, 2021, the FDA approved idecabtagene vicleucel (brand name Abecma) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen-directed genetically modified autologous chimeric antigen receptor T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies, 88% of whom had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel and was established based on overall response rate, complete response rate, and duration of response, as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The overall response rate was 72% and complete response rate was 28%. An estimated 65% of patients who achieved complete response remained in complete response for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome, neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities dispensing the therapy must be specially certified to recognize and manage cytokine release syndrome and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
Full prescribing information for this approval can be found on the web at www.fda.gov, with key word search “Approved Cellular and Gene Therapy Products”.
Health care professionals should report serious adverse events to FDA’s MedWatch Reporting System at www.fda.gov/medwatch.
Follow the Division of Drug Information on Twitter @FDA_Drug_InfoExternal Link Disclaimer and the Oncology Center of Excellence @FDAOncologyExternal Link Disclaimer. Send your feedback via email to FDAOncology@fda.hhs.gov. Thanks for tuning in today to the DISCO Burst Edition.
PAT
WO 2019148089
In various aspects, the present invention relates to XCR1 binding agents having at least one targeting moiety that specifically binds to XCR1. In various embodiments, these XCR1 binding agents bind to, but do not functionally modulate ( e.g . partially or fully neutralize) XCR1. Therefore, in various embodiments, the present XCR1 binding agents have use in, for instance, directly or indirectly recruiting a XCR1-expressing cell to a site of interest while still allowing the XCR1-expressing cell to signal via XCR1 (i.e. the binding of the XCR1 binding agent does not reduce or eliminate XCR1 signaling at the site of interest). In various embodiments, the XCR-1 binding agent functionally modulates XCR1. In an embodiment, the targeting moiety is a single domain antibody (e.g. VHH, HUMABODY, scFv, on antibody). In various embodiments, the XCR1 binding agent further comprises a signaling agent, e.g., without limitation, an interferon, an interleukin, and a tumor necrosis factor, that may be modified to attenuate activity. In various embodiments, the XCR1 binding agent comprises additional targeting moieties that bind to other targets (e.g. antigens, receptor) of interest. In an embodiment, the other targets (e.g. antigens, receptor) of interest are present on tumor cells. In another embodiment, the other targets (e.g. antigens, receptor) of interest are present on immune cells. In some embodiments, the present XCR1 binding agent may directly or indirectly recruit an immune cell (e.g. a dendritic cell) to a site of action (such as, by way of non-limiting example, the tumor microenvironment). In some embodiments, the present XCR1 binding agent facilitates the presentation of antigens (e.g., tumor antigens) by dendritic cells.
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises the heavy chain of SEQ ID NO: 223 and/or the light chain of SEQ ID NO: 224, or a variant thereof (e.g. an amino acid sequence having at least about 90%, or at least about 93%, at least about 95%, at least about 97%, at least about 98%, at least about 99%, identity with SEQ ID NO: 223 and/or SEQ ID NO: 224).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227) and/or a light chain CDR 1 of RSSLGLVHRNGNTYLH (SEQ ID NO: 228), light chain CDR 2 of KVSHRFS (SEQ ID NO: 229), and light chain CDR 3 of SQSTFIVPWT (SEQ ID NO: 230), or a variant thereof (e.g. with four or fewer amino acid substitutions, or with three or fewer amino acid substitutions, or with two or fewer amino acid substitutions, or with one amino acid substitution).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227).
Illustrative Disease Modifying Therapies
EXAMPLES
Example 1. Identification and Characterization of Human XCR1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 5G7 antibody and human IFNa2 with an R149A mutation.
AFNs were made based on the 5G7 anti-hXcr1 Ab using the intact (full) Ab or a scFv format.
The 5G7 heavy chain is:
QAYLQQSGAELVRPGASVKMSCKASGYTFTSHNLHWVKQTPRQGLQWIGAIYPGNGNTAYNQKFKGKATLTVD
KSSSTAYMQLSSLTSDDSAVYFCARWGSVVGDWYFDVWGTGTTVTVSSASTKGPSVFPLAPCSRSTSESTAAL
GCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSWTVPSSNFGTQTYTCNVDHKPSNTKVDKTVE
RKCCVECPPCPAPPAAAPSVFLFPPKPKDTLMISRTPEVTCVWDVSHEDPEVQFNWYVDGVEVHNAKTKPREE
QFNSTFRVVSVLTWHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLV
KGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLS
LSPGK (SEQ ID NO: 223)
The 5G7 light chain is:
DWMTQTPLSLPVTLGNQASIFCRSSLGLVHRNGNTYLHWYLQKPGQSPKLLIYKVSHRFSGVPDRFSGSGSGT DFTLKISRVEAEDLGVYFCSQSTHVPWTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASWCLLNNFYPREAK VQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 224)
5G7 Heavy chain CDR 1 is SHNLH (SEQ ID NO: 225), Heavy chain CDR 2 is AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), Heavy chain CDR 3 is WGSVVGDWYFDV (SEQ ID NO: 227). 5G7 Light chain CDR 1 is RSSLGLVHRNGNTYLH (SEQ ID NO: 228), Light chain CDR 2 is KVSHRFS (SEQ ID NO: 229), and Light chain CDR 3 is SQSTHVPWT (SEQ ID NO: 230).
The sequence of hulFNa2(R149A) is:
CDLPQTHSLGSRRTLMLLAQMRKISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMIQQIFNLFSTKDSSAA WDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVRKYFQRITLYLKEKKYSPCAWEVVRAEIMASF SLSTNLQESLRSKE (SEQ ID NO: 231).
In case of the intact Ab AFN, the 5G7 Ab heavy chain was fused to h I FN a2_R149A (human IFNal with a R149A mutation) via a flexible (GGS)2oG-linker and co-expressed with the 5G7 Ab light chain (sequences shown below). 5G7 scFv-AFN was constructed by linking the Ab VL and VH domains via a (GGGS)4 linker and followed by a (GGS)2o-linker and the sequence encoding hlFNa2_R149A. Recombinant proteins, cloned in the pcDNA3.4 expression-vector, were produced in ExpiCHO cells (Thermo Fisher Scientific) and purified on HisPUR spin plates (Thermo Fisher Scientific) according to the manufacturer’s instructions.
To test binding of the AFNs, parental HL1 16 and HL1 16 cells stably expressing hXcrl (HL116-hXcr1) were incubated with a serial dilution AFN for two hours at 4°C. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figures 1A and 1 B clearly show that both 5G7 Ab-AFN and 5G7 scFv bind specifically to hXcrl expressing cells.
Biological activity was measured on parental HL1 16 cells (an IFN responsive cell-line stably transfected with a p6-16 luciferase reporter) and the derived HL116-hXcr1 cells. Cells were seeded overnight and stimulated for 6 hours with a serial dilution 5G7 AFNs. Luciferase activity was measured on an EnSight Multimode Plate Reader (Perkin Elmer). Data in Figures 2A and 2B clearly illustrate that 5G7 AFNs, in the intact Ab format or as scFv, are clearly more active on cells expressing hXcrl compared to parental cells, illustrating that it is possible to restore signaling of an IFNa2 mutant by specific targeting to hXcrl .
Example 2. Identification and Characterization of Mouse Xcr1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 MAARX10 antibody and human IFNa2 with Q124R mutation.
Similar to the anti-human Xcr1 Ab, AFNs based on the MARX10 anti-mouse Xcr1 Ab were made, as intact Ab or as scFv. In case of the intact Ab AFN, the MARX10 Ab heavy chain was fused to hlFNa2_Q124R (human IFNa2 with Q124R mutation) via a flexible (GGS)2oG-linker and co-expressed with the MARX10 Ab light chain. scFv-AFN was constructed by linking the Ab VL and VH domains, in VH-VL (scFv(1 )) or VL-VH (scFv(2)) orientation, via a (GGGS)4 linker and followed by a (GGS)2o-linker and h I FN a2_Q 124R.
Selectivity of AFNs (produced and purified as described above for the human Xcr1 Ab AFNs) was tested by comparing binding at 2.5 pg/ml to MOCK or mouse Xcr1 transfected Hek293T cells. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figure 3 clearly show that all three specifically bind to mXcrl expressing cells.
REF
New England Journal of Medicine (2021), 384(8), 705-716
https://www.rxlist.com/abecma-drug.htm#indications
///////////Idecabtagene vicleucel, breakthrough therapy designation, orphan drug designation, FDA 2021, APPROVALS 2021, Bb2121, Bb , ABECMA
Manufacturer: Celgene Corporation, a Bristol-Myers Squibb Company
Indications:
- Treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.
Product Information
- Package Insert – ABECMA
- Demographic Subgroup Information – idecabtagene vicleucel [ABECMA]
Refer to Section 1.1 of the Clinical Review Memo for information about participation in the clinical trials and any analysis of demographic subgroup outcomes that is notable.
Supporting Documents
Dasiglucagon




Dasiglucagon
Treatment of Hypoglycemia in Type 1 and Type 2 Diabetes Patients
| Formula | C152H222N38O50 |
|---|---|
| CAS | 1544300-84-6 |
| Mol weight | 3381.6137 |
FDA APPROVED, 2021/3/22, Zegalogue
Zealand Pharma A/S
UNIIAD4J2O47FQ
HypoPal rescue pen
(4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S,3R)-2-[[2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]acetyl]amino]-3-hydroxybutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]amino]-2-methylpropanoyl]amino]propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-carboxy-1-[[(2S)-1-[[(1S,2R)-1-carboxy-2-hydroxypropyl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-5-oxopentanoic acid
. [16-(2-methylalanine)(S>X),17-L-alanine(R>A),20-L-α-glutamyl(Q>E),21-L-αglutamyl(D>E),24-L-lysyl(Q>K),27-L-α-glutamyl(M>E),28-L-serine(N>S)]human glucagon
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L- phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L- lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L- arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L- valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-alpha-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-alpha-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-alpha-glutamyl-L-alphaC152 H222 N38 O50L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-Molecular Weight3381.61
Other Names
- L-Histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-L-threonine
- Developer Beta Bionics; Zealand Pharma
- ClassAntihyperglycaemics; Antihypoglycaemics; Peptides
- Mechanism of ActionGlucagon receptor agonists
- Orphan Drug StatusYes – Hypoglycaemia; Congenital hyperinsulinism
- RegisteredHypoglycaemia
- Phase IIICongenital hyperinsulinism
- Phase II/IIIType 1 diabetes mellitus
- 22 Mar 2021Registered for Hypoglycaemia (In children, In adolescents, In adults, In the elderly) in USA (SC) – First global approval
- 22 Mar 2021Zealand Pharma anticipates the launch of dasiglucagon in USA (SC, Injection) in June 2021
- 22 Mar 2021Pooled efficacy and safety data from three phase III trials in Hypoglycaemia released by Zealand Pharma
NEW DRUG APPROVALS
one time
$10.00
PATENTS
WO 2014016300
US 20150210744
PAPER
Pharmaceutical Research (2018), 35(12), 1-13
Dasiglucagon, sold under the brand name Zegalogue, is a medication used to treat severe hypoglycemia in people with diabetes.[1]
The most common side effects include nausea, vomiting, headache, diarrhea, and injection site pain.[1]
Dasiglucagon was approved for medical use in the United States in March 2021.[1][2][3] It was designated an orphan drug in August 2017.[4]
Dasiglucagon is under investigation in clinical trial NCT03735225 (Evaluation of the Safety, Tolerability and Bioavailability of Dasiglucagon Following Subcutaneous (SC) Compared to IV Administration).
Medical uses
Dasiglucagon is indicated for the treatment of severe hypoglycemia in people aged six years of age and older with diabetes.[1][2]
Contraindications
Dasiglucagon is contraindicated in people with pheochromocytoma or insulinoma.[1]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214231s000lbl.pdf
- ^ Jump up to:a b “Dasiglucagon: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 March 2021.
- ^ “Zealand Pharma Announces FDA Approval of Zegalogue (dasiglucagon) injection, for the Treatment of Severe Hypoglycemia in People with Diabetes” (Press release). Zealand Pharma. 22 March 2021. Retrieved 22 March 2021 – via GlobeNewswire.
- ^ “Dasiglucagon Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 10 August 2017. Retrieved 22 March 2021.
External links
- “Dasiglucagon”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03378635 for “A Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Type 1 Diabetes Subjects” at ClinicalTrials.gov
- Clinical trial number NCT03688711 for “Trial to Confirm the Clinical Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Subjects With T1DM” at ClinicalTrials.gov
- Clinical trial number NCT03667053 for “Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in T1DM Children” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zegalogue |
| AHFS/Drugs.com | Zegalogue |
| License data | US DailyMed: Dasiglucagon |
| Routes of administration | Subcutaneous |
| Drug class | Glucagon receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1544300-84-6 |
| PubChem CID | 126961379 |
| DrugBank | DB15226 |
| UNII | AD4J2O47FQ |
| KEGG | D11359 |
| Chemical and physical data | |
| Formula | C152H222N38O50 |
| Molar mass | 3381.664 g·mol−1 |
| 3D model (JSmol) | Interactive image |
///////////Dasiglucagon, FDA 2021, APPROVALS 2021, Zegalogue, ダシグルカゴン, ZP 4207, ZP-GA-1, Hypoglycemia, Type 1, Type 2 , Diabetes Patients, Zealand Pharma A/S, Orphan Drug Status, Hypoglycaemia, Congenital hyperinsulinism, HypoPal rescue pen, DIABETES
#Dasiglucagon, #FDA 2021, #APPROVALS 2021, #Zegalogue, #ダシグルカゴン, #ZP 4207, ZP-GA-1, #Hypoglycemia, #Type 1, #Type 2 , #Diabetes Patients, #Zealand Pharma A/S, #Orphan Drug Status, #Hypoglycaemia, #Congenital hyperinsulinism, #HypoPal rescue pen, #DIABETESSMILES
- C[C@H]([C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CC2=CC=C(C=C2)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC3=CC=C(C=C3)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(=O)O)C(=O)NC(C)(C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC4=CC=CC=C4)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC5=CNC6=CC=CC=C65)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)NC(=O)CNC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CO)NC(=O)[C@H](CC7=CNC=N7)N)O
Fosdenopterin hydrobromide

Fosdenopterin hydrobromide
FDA APPR 2021/2/26, NULIBRY
BBP-870/ORGN001
a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.
| ホスデノプテリン臭化水素酸塩水和物; |
| Formula | C10H14N5O8P. 2H2O. HBr |
|---|---|
| CAS | 2301083-34-9DIHYDRATE |
| Mol weight | 480.1631 |
2301083-34-9
(1R,10R,12S,17R)-5-amino-11,11,14-trihydroxy-14-oxo-13,15,18-trioxa-2,4,6,9-tetraza-14λ5-phosphatetracyclo[8.8.0.03,8.012,17]octadeca-3(8),4-dien-7-one;dihydrate;hydrobromide
1,3,2-DIOXAPHOSPHORINO(4′,5′:5,6)PYRANO(3,2-G)PTERIDIN-10(4H)-ONE, 8-AMINO-4A,5A,6,9,11,11A,12,12A-OCTAHYDRO-2,12,12-TRIHYDROXY-, 2-OXIDE, HYDROBROMIDE, HYDRATE (1:1:2), (4AR,5AR,11AR,12AS)-
| CYCLIC PYRANOPTERIN MONOPHOSPHATE MONOHYDROBROMIDE DIHYDRATE |
(4aR,5aR,11aR,12aS)-8-Amino-2,12,12-trihydroxy-4a,5a,6,7,11,11a,12,12aoctahydro-2H-2lambda5-(1,3,2)dioxaphosphinino(4′,5′:5,6)pyrano(3,2-g)pteridine-2,10(4H)-dione, hydrobromide (1:1:2)
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide, hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide,hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780



C10H14N5O8P, Average: 363.223
150829-29-1
- ALXN-1101
- WHO 11150
- Synthesis ReferenceClinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, Tiwari A, Schwarz G, Saotome Y, Storek M, Belaidi AA, Santamaria-Araujo JA: Synthesis of cyclic pyranopterin monophosphate, a biosynthetic intermediate in the molybdenum cofactor pathway. J Med Chem. 2013 Feb 28;56(4):1730-8. doi: 10.1021/jm301855r. Epub 2013 Feb 19.
Fosdenopterin (or cyclic pyranopterin monophosphate, cPMP), sold under the brand name Nulibry, is a medication used to reduce the risk of death due to a rare genetic disease known as molybdenum cofactor deficiency type A (MoCD-A).[1]
Adverse effects
The most common side effects include complications related to the intravenous line, fever, respiratory infections, vomiting, gastroenteritis, and diarrhea.[1]
Mechanism of action
People with MoCD-A cannot produce cyclic pyranopterin monophosphate (cPMP) in their body.[1] Fosdenopterin is an intravenous medication that replaces the missing cPMP.[1][2] cPMP is a precursor to molybdopterin, which is required for the enzyme activity of sulfite oxidase, xanthine dehydrogenase/oxidase and aldehyde oxidase.[3]
History
Fosdenopterin was developed by José Santamaría-Araujo and Guenter Schwarz at the German universities TU Braunschweig and the University of Cologne.[4][5]
The effectiveness of fosdenopterin for the treatment of MoCD-A was demonstrated in thirteen treated participants compared to eighteen matched, untreated participants.[1][6] The participants treated with fosdenopterin had a survival rate of 84% at three years, compared to 55% for the untreated participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for fosdenopterin priority review, breakthrough therapy, and orphan drug designations along with a rare pediatric disease priority review voucher.[1] The FDA granted the approval of Nulibry to Origin Biosciences, Inc., in February 2021.[1] It is the first medication approved for the treatment of MoCD-A.[1]
References
- ^ Jump up to:a b c d e f g h i j “FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A”. U.S. Food and Drug Administration (FDA) (Press release). 26 February 2021. Retrieved 26 February 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ DrugBank DB16628 . Accessed 2021-03-05.
- ^ Santamaria-Araujo JA, Fischer B, Otte T, Nimtz M, Mendel RR, Wray V, Schwarz G (April 2004). “The tetrahydropyranopterin structure of the sulfur-free and metal-free molybdenum cofactor precursor”. The Journal of Biological Chemistry. 279 (16): 15994–9. doi:10.1074/jbc.M311815200. PMID 14761975.
- ^ Schwarz G, Santamaria-Araujo JA, Wolf S, Lee HJ, Adham IM, Gröne HJ, et al. (June 2004). “Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli”. Human Molecular Genetics. 13 (12): 1249–55. doi:10.1093/hmg/ddh136. PMID 15115759.
- ^ Tedmanson S (5 November 2009). “Doctors risk untried drug to stop baby’s brain dissolving”. TimesOnline.
- ^ Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, et al. (November 2015). “Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study”. Lancet. 386 (10007): 1955–63. doi:10.1016/S0140-6736(15)00124-5. PMID 26343839. S2CID 21954888.
External links
- “Fosdenopterin”. Drug Information Portal. U.S. National Library of Medicine.
Molybdenum cofactor deficiency (MoCD) is an exceptionally rare autosomal recessive disorder resulting in a deficiency of three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde oxidase.1 Signs and symptoms begin shortly after birth and are caused by a build-up of toxic sulfites resulting from a lack of SOX activity.1,5 Patients with MoCD may present with metabolic acidosis, intracranial hemorrhage, feeding difficulties, and significant neurological symptoms such as muscle hyper- and hypotonia, intractable seizures, spastic paraplegia, myoclonus, and opisthotonus. In addition, patients with MoCD are often born with morphologic evidence of the disorder such as microcephaly, cerebral atrophy/hypodensity, dilated ventricles, and ocular abnormalities.1 MoCD is incurable and median survival in untreated patients is approximately 36 months1 – treatment, then, is focused on improving survival and maintaining neurological function.
The most common subtype of MoCD, type A, involves mutations in MOCS1 wherein the first step of molybdenum cofactor synthesis – the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP) – is interrupted.1,3 In the past, management strategies for this disorder involved symptomatic and supportive treatment,5 though efforts were made to develop a suitable exogenous replacement for the missing cPMP. In 2009 a recombinant, E. coli-produced cPMP was granted orphan drug designation by the FDA, becoming the first therapeutic option for patients with MoCD type A.1
Fosdenopterin was approved by the FDA on Februrary 26, 2021, for the reduction of mortality in patients with MoCD type A,5 becoming the first and only therapy approved for the treatment of MoCD. By improving the three-year survival rate from 55% to 84%,7 and considering the lack of alternative therapies available, fosdenopterin appears poised to become a standard of therapy in the management of this debilitating disorder.
Fosdenopterin replaces an intermediate substrate in the synthesis of molybdenum cofactor, a compound necessary for the activation of several molybdenum-dependent enzymes including sulfite oxidase (SOX).1 Given that SOX is responsible for detoxifying sulfur-containing acids and sulfites such as S-sulfocysteine (SSC), urinary levels of SSC can be used as a surrogate marker of efficacy for fosdenopterin.7 Long-term therapy with fosdenopterin has been shown to result in a sustained reduction in urinary SSC normalized to creatinine.7
Animal studies have identified a potential risk of phototoxicity in patients receiving fosdenopterin – these patients should avoid or minimize exposure to sunlight and/or artificial UV light.7 If sun exposure is necessary, use protective clothing, hats, and sunglasses,7 in addition to seeking shade whenever practical. Consider the use of a broad-spectrum sunscreen in patients 6 months of age or older.8
Molybdenum cofactor deficiency (MoCD) is a rare autosomal-recessive disorder in which patients are deficient in three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde dehydrogenase.1 The loss of SOX activity appears to be the main driver of MoCD morbidity and mortality, as the build-up of neurotoxic sulfites typically processed by SOX results in rapid and progressive neurological damage. In MoCD type A, the disorder results from a mutation in the MOCS1 gene leading to deficient production of MOCS1A/B,7 a protein that is responsible for the first step in the synthesis of molybdenum cofactor: the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP).1,4
Fosdenopterin is an exogenous form of cPMP, replacing endogenous production and allowing for the synthesis of molybdenum cofactor to proceed.7
- Mechler K, Mountford WK, Hoffmann GF, Ries M: Ultra-orphan diseases: a quantitative analysis of the natural history of molybdenum cofactor deficiency. Genet Med. 2015 Dec;17(12):965-70. doi: 10.1038/gim.2015.12. Epub 2015 Mar 12. [PubMed:25764214]
- Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, Hennermann JB, Jameson E, Konig K, McGregor TL, Font-Montgomery E, Santamaria-Araujo JA, Santra S, Vaidya M, Vierzig A, Wassmer E, Weis I, Wong FY, Veldman A, Schwarz G: Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet. 2015 Nov 14;386(10007):1955-63. doi: 10.1016/S0140-6736(15)00124-5. Epub 2015 Sep 3. [PubMed:26343839]
- Iobbi-Nivol C, Leimkuhler S: Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim Biophys Acta. 2013 Aug-Sep;1827(8-9):1086-101. doi: 10.1016/j.bbabio.2012.11.007. Epub 2012 Nov 29. [PubMed:23201473]
- Mendel RR: The molybdenum cofactor. J Biol Chem. 2013 May 10;288(19):13165-72. doi: 10.1074/jbc.R113.455311. Epub 2013 Mar 28. [PubMed:23539623]
- FDA News Release: FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A [Link]
- OMIM: MOLYBDENUM COFACTOR DEFICIENCY, COMPLEMENTATION GROUP A (# 252150) [Link]
- FDA Approved Drug Products: Nulibry (fosdenopterin) for intravenous injection [Link]
- Health Canada: Sun safety tips for parents [Link]
SYN
Journal of Biological Chemistry (1995), 270(3), 1082-7.
https://linkinghub.elsevier.com/retrieve/pii/S0021925818829696
PATENT
WO 2005073387
PATENT
WO 2012112922
PAPER

Journal of Medicinal Chemistry (2013), 56(4), 1730-1738
https://pubs.acs.org/doi/10.1021/jm301855r

Cyclic pyranopterin monophosphate (1), isolated from bacterial culture, has previously been shown to be effective in restoring normal function of molybdenum enzymes in molybdenum cofactor (MoCo)-deficient mice and human patients. Described here is a synthesis of 1 hydrobromide (1·HBr) employing in the key step a Viscontini reaction between 2,5,6-triamino-3,4-dihydropyrimidin-4-one dihydrochloride and d-galactose phenylhydrazone to give the pyranopterin (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one (10) and establishing all four stereocenters found in 1. Compound 10, characterized spectroscopically and by X-ray crystallography, was transformed through a selectively protected tri-tert-butoxycarbonylamino intermediate into a highly crystalline tetracyclic phosphate ester (15). The latter underwent a Swern oxidation and then deprotection to give 1·HBr. Synthesized 1·HBr had in vitro efficacy comparable to that of 1 of bacterial origin as demonstrated by its enzymatic conversion into mature MoCo and subsequent reconstitution of MoCo-free human sulfite oxidase–molybdenum domain yielding a fully active enzyme. The described synthesis has the potential for scale up.
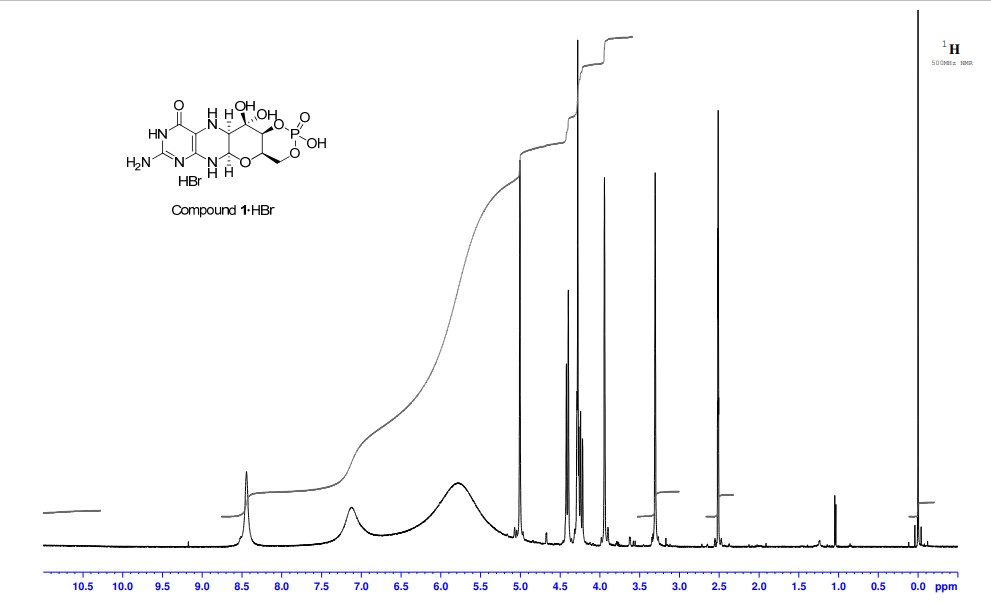
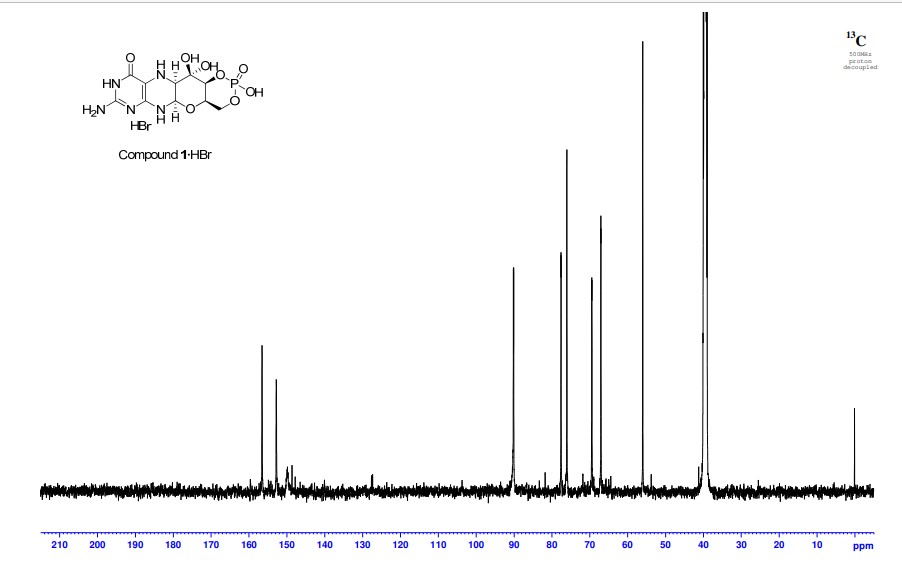





PAPER
European Journal of Organic Chemistry (2014), 2014(11), 2231-2241.
https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/ejoc.201301784
Abstract
The first synthesis of an oxygen‐stable analogue of the natural product cyclic pyranopterin monophosphate (cPMP) is reported. In this approach, the hydropyranone ring is annelated to pyrazine by a sequence comprising ortho‐lithiation/acylation of a 2‐halopyrazine, followed by nucleophilic aromatic substitution. The tetrose substructure is introduced from the chiral pool, from D‐galactose or D‐arabitol.

Abstract
Molybdenum cofactor (Moco) deficiency is a lethal hereditary metabolic disease. A recently developed therapy requires continuous intravenous supplementation of the biosynthetic Moco precursor cyclic pyranopterin monophosphate (cPMP). The limited stability of the latter natural product, mostly due to oxidative degradation, is problematic for oral administration. Therefore, the synthesis of more stable cPMP analogues is of great interest. In this context and for the first time, the synthesis of a cPMP analogue, in which the oxidation‐labile reduced pterin unit is replaced by a pyrazine moiety, was achieved starting from the chiral pool materials D‐galactose or D‐arabitol. Our synthesis, 13 steps in total, includes the following key transformations: i) pyrazine lithiation, followed by acylation; ii) closure of the pyrane ring by nucleophilic aromatic substitution; and iii) introduction of phosphate.
Patent
https://patents.google.com/patent/US9260462B2/en
Molybdenum cofactor (Moco) deficiency is a pleiotropic genetic disorder. Moco consists of molybdenum covalently bound to one or two dithiolates attached to a unique tricyclic pterin moiety commonly referred to as molybdopterin (MPT). Moco is synthesized by a biosynthetic pathway that can be divided into four steps, according to the biosynthetic intermediates precursor Z (cyclic pyranopterin monophosphate; cPMP), MPT, and adenylated MPT. Mutations in the Moco biosynthetase genes result in the loss of production of the molybdenum dependent enzymes sulfite-oxidase, xanthine oxidoreductase, and aldehyde oxidase. Whereas the activities of all three of these cofactor-containing enzymes are impaired by cofactor deficiency, the devastating consequences of the disease can be traced to the loss of sulfite oxidase activity. Human Moco deficiency is a rare but severe disorder accompanied by serious neurological symptoms including attenuated growth of the brain, untreatable seizures, dislocated ocular lenses, and mental retardation. Until recently, no effective therapy was available and afflicted patients suffering from Moco deficiency died in early infancy.
It has been found that administration of the molybdopterin derivative precursor Z, a relatively stable intermediate in the Moco biosynthetic pathway, is an effective means of therapy for human Moco deficiency and associated diseases related to altered Moco synthesis (see U.S. Pat. No. 7,504,095). As with most replacement therapies for illnesses, however, the treatment is limited by the availability of the therapeutic active agent.
Scheme 3.

Scheme 4.

(I).
Scheme 6.

(I).

Scheme 8.

(I).

Scheme 10.

EXAMPLESExample 1Preparation of Precursor Z (cPMP)


Experimental
Air sensitive reactions were performed under argon. Organic solutions were dried over anhydrous MgSO4 and the solvents were evaporated under reduced pressure. Anhydrous and chromatography solvents were obtained commercially (anhydrous grade solvent from Sigma-Aldrich Fine Chemicals) and used without any further purification. Thin layer chromatography (t.l.c.) was performed on glass or aluminum sheets coated with 60 F254 silica gel. Organic compounds were visualized under UV light or with use of a dip of ammonium molybdate (5 wt %) and cerium(IV) sulfate 4H2O (0.2 wt %) in aq. H2SO4 (2M), one of I2 (0.2%) and KI (7%) in H2SO4 (1M), or 0.1% ninhydrin in EtOH. Chromatography (flash column) was performed on silica gel (40-63 μm) or on an automated system with continuous gradient facility. Optical rotations were recorded at a path length of 1 dm and are in units of 10−1 deg cm2 g−1; concentrations are in g/100 mL. 1H NMR spectra were measured in CDCl3, CD3OD (internal Me4Si, δ 0 ppm) or D2O(HOD, δ 4.79 ppm), and 13C NMR spectra in CDCl3 (center line, δ 77.0 ppm), CD3OD (center line, δ 49.0 ppm) or DMSO d6 (center line δ 39.7 ppm), D2O (no internal reference or internal CH3CN, δ 1.47 ppm where stated). Assignments of 1H and 13C resonances were based on 2D (1H—1H DQF-COSY, 1H—13C HSQC, HMBC) and DEPT experiments. 31P NMR were run at 202.3 MHz and are reported without reference. High resolution electrospray mass spectra (ESI-HRMS) were recorded on a Q-TOF Tandem Mass
Spectrometer. Microanalyses were performed by the Campbell Microanalytical Department, University of Otago, Dunedin, New Zealand.
A. Preparation of (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one mono hydrate (1)
2,5,6-Triamino-3,4-dihydropyrimidin-4-one dihydrochloride (Pfleiderer, W.; Chem. Ber. 1957, 90, 2272; Org. Synth. 1952, 32, 45; Org. Synth. 1963, Coll. Vol. 4, 245, 10.0 g, 46.7 mmol), D-galactose phenylhydrazone (Goswami, S.; Adak, A. K. Tetrahedron Lett. 2005, 46, 221-224, 15.78 g, 58.4 mmol) and 2-mercaptoethanol (1 mL) were stirred and heated to reflux (bath temp 110° C.) in a 1:1 mixture of MeOH—H2O (400 mL) for 2 h. After cooling to ambient temperature, diethyl ether (500 mL) was added, the flask was shaken and the diethyl ether layer decanted off and discarded. The process was repeated with two further portions of diethyl ether (500 mL) and then the remaining volatiles were evaporated. Methanol (40 mL), H2O (40 mL) and triethylamine (39.4 mL, 280 mmol) were successively added and the mixture seeded with a few milligrams of 1. After 5 min a yellow solid was filtered off, washed with a little MeOH and dried to give 1 as a monohydrate (5.05 g, 36%) of suitable purity for further use. An analytical portion was recrystallized from DMSO-EtOH or boiling H2O. MPt 226 dec. [α]D 20 +135.6 (c1.13, DMSO). 1H NMR (DMSO d6): δ 10.19 (bs, exchanged D2O, 1H), 7.29 (d, J=5.0 Hz, slowly exchanged D2O, 1H), 5.90 (s, exchanged D2O, 2H), 5.33 (d, J=5.4 Hz, exchanged D2O, 1H), 4.66 (ddd, J˜5.0, ˜1.3, ˜1.3 Hz, 1H), 4.59 (t, J=5.6 Hz, exchanged D2O, 1H), 4.39 (d, J=10.3 Hz, exchanged D2O, 1H), 3.80 (bt, J˜1.8 Hz, exchanged D2O, 1H), 3.70 (m, 1H), 3.58 (dd, J=10.3, 3.0 Hz, 1H), 3.53 (dt, J=10.7, 6.4 Hz, 1H), 3.43 (ddd, J=11.2, 5.9, 5.9 Hz, 1H), 3.35 (t, J=6.4 Hz, 1H), 3.04 (br m, 1H). 13C NMR (DMSO d6 center line 6 39.7): δ 156.3 (C), 150.4 (C), 148.4 (C), 99.0 (C), 79.4 (CH), 76.5 (CH), 68.9 (CH), 68.6 (CH), 60.6 (CH2), 53.9 (CH). Anal. calcd. for C10H15N5O5H2O 39.60; C, 5.65; H, 23.09; N. found 39.64; C, 5.71; H, 22.83; N.
B. Preparation of Compounds 2 (a or b) and 3 (a, b or c)
Di-tert-butyl dicarbonate (10.33 g, 47.3 mmol) and DMAP (0.321 g, 2.63 mmol) were added to a stirred suspension of 1 (1.5 g, 5.26 mmol) in anhydrous THF (90 mL) at 50° C. under Ar. After 20 h a clear solution resulted. The solvent was evaporated and the residue chromatographed on silica gel (gradient of 0 to 40% EtOAc in hexanes) to give two product fractions. The first product to elute was a yellow foam (1.46 g). The product was observed to be a mixture of two compounds by 1H NMR containing mainly a product with seven Boc groups (2a or 2b). A sample was crystallized from EtOAc-hexanes to give 2a or 2b as a fine crystalline solid. MPt 189-191° C. [α]D 20 −43.6 (c 0.99, MeOH). 1H NMR (500 MHz, CDCl3): δ 5.71 (t, J=1.7 Hz, 1H), 5.15 (dt, J=3.5, ˜1.0, 1H), 4.97 (t, J=3.8, 1H), 4.35 (br t, J=˜1.7, 1H), 4.09-3.97 (m, 3H), 3.91 (m, 1H), 1.55, 1.52, 1.51, 1.50, 1.45 (5s, 45H), 1.40 (s, 18H). 13C NMR (125.7 MHz, CDCl3): δ 152.84 (C), 152.78 (C), 151.5 (C), 150.9 (C), 150.7 (2×C), 150.3 (C), 149.1 (C), 144.8 (C), 144.7 (C), 118.0 (C), 84.6 (C), 83.6 (C), 83.5 (C), 82.7 (3×C), 82.6 (C), 76.3 (CH), 73.0 (CH), 71.4 (CH), 67.2 (CH), 64.0 (CH2), 51.4 (CH), 28.1 (CH3), 27.8 (2×CH3), 27.7 (CH3), 27.6 (3×CH3). MS-ESI+ for C45H72N5O19 +, (M+H)+, Calcd. 986.4817. found 986.4818. Anal. calcd. for C45H71N5O19H2O 54.39; C, 7.39; H, 6.34; N. found 54.66; C, 7.17; H, 7.05; N. A second fraction was obtained as a yellow foam (2.68 g) which by 1H NMR was a product with six Boc groups present (3a, 3b or 3c). A small amount was crystallized from EtOAc-hexanes to give colorless crystals. [α]D 2O −47.6 (c, 1.17, CHCl3). 1H NMR (500 MHz, CDCl3): δ 11.10 (br s, exchanged D2O, 1H), 5.58 (t, J=1.8 Hz, 1H), 5.17 (d, J=3.4 Hz, 1H), 4.97 (t, J=3.9 Hz, 1H), 4.62 (s, exchanged D2O, 1H), 4.16 (dd, J=11.3, 5.9 Hz, 1H), 4.12 (dd, J=11.3, 6.4 Hz, 1H), 3.95 (dt, J=6.1, 1.1 Hz, 1H), 3.76 (m, 1H), 1.51, 1.50, 1.49, 1.48, 1.46 (5s, 54H). 13C NMR (125.7 MHz, CDCl3): δ 156.6 (C), 153.0 (C), 152.9 (C), 151.9 (C), 150.6 (C), 149.4 (2×C), 136.2 (C), 131.8 (C), 116.9 (C), 85.0 (2×C), 83.3 (C), 82.8 (C), 82.49 (C), 82.46 (C), 73.3 (CH), 71.5 (CH), 67.2 (CH), 64.5 (CH2), 51.3 (CH), 28.0, 27.72, 27.68, 27.6 (4×CH3). MS-ESI+ for C40H64N5O17 +, (M+H)+calcd. 886.4287. found 886.4289.
C. Preparation of Compound 4a, 4b or 4c
Step 1—The first fraction from B above containing mainly compounds 2a or 2b (1.46 g, 1.481 mmol) was dissolved in MeOH (29 mL) and sodium methoxide in MeOH (1M, 8.14 mL, 8.14 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
Step 2—The second fraction from B above containing mainly 3a, 3b or 3c (2.68 g, 3.02 mmol) was dissolved in MeOH (54 mL) and sodium methoxide in MeOH (1M, 12.10 mL, 12.10 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
The products from step 1 and step 2 above were combined and chromatographed on silica gel (gradient of 0 to 15% MeOH in CHCl3) to give 4a, 4b or 4c as a cream colored solid (1.97 g). 1H NMR (500 MHz, DMSO d6): δ 12.67 (br s, exchanged D2O, 1H), 5.48 (d, J=5.2 Hz, exchanged D2O, 1H), 5.43 (t, J=˜1.9 Hz, after D2O exchange became a d, J=1.9 Hz, 1H), 5.00 (br s, exchanged D2O, 1H), 4.62 (d, J=5.7 Hz, exchanged D2O, 1H), 4.27 (d, J=6.0 Hz, exchanged D2O, 1H), 3.89 (dt, J=5.2, 3.8 Hz, after D2O became a t, J=3.9 Hz, 1H), 3.62 (dd, J=6.0, 3.7 Hz, after D2O exchange became a d, J=3.7 Hz, 1H), 3.52-3.39 (m, 4H), 1.42 (s, 9H), 1.41 (s, 18H). 13C NMR (125.7 MHz, DMSO d6): δ 157.9 (C), 151.1, (C), 149.8 (2×C), 134.6 (C), 131.4 (C), 118.8 (C), 83.5 (2×C), 81.3 (C), 78.2 (CH), 76.5 (CH), 68.1 (CH), 66.8 (CH), 60.6 (CH2), 54.4 (CH), 27.9 (CH3), 27.6 (2×CH3). MS-ESI+ for C25H40N5O11 +, (M+H)+ calcd. 586.2719. found 586.2717.
D. Preparation of Compound 5a, 5b or 5c
Compound 4a, 4b or 4c (992 mg, 1.69 mmol) was dissolved in anhydrous pyridine and concentrated. The residue was dissolved in anhydrous CH2Cl2 (10 mL) and pyridine (5 mL) under a nitrogen atmosphere and the solution was cooled to −42° C. in an acetonitrile/dry ice bath. Methyl dichlorophosphate (187 μL, 1.86 mmol) was added dropwise and the mixture was stirred for 2 h 20 min. Water (10 mL) was added to the cold solution which was then removed from the cold bath and diluted with ethyl acetate (50 mL) and saturated NaCl solution (30 mL). The organic portion was separated and washed with saturated NaCl solution. The combined aqueous portions were extracted twice further with ethyl acetate and the combined organic portions were dried over MgSO4 and concentrated. Purification by silica gel flash column chromatography (eluting with 2-20% methanol in ethyl acetate) gave the cyclic methyl phosphate 5a, 5b or 5c (731 mg, 65%). 1H NMR (500 MHz, CDCl3,): δ 11.72 (bs, exchanged D2O, 1H), 5.63 (t, J=1.8 Hz, 1H), 5.41 (s, exchanged D2O, 1H), 4.95 (d, J=3.2 Hz, 1H), 4.70 (dt, J=12.4, 1.8 Hz, 1H), 4.42 (dd, J=22.1, 12.1 Hz, 1H). 4.15 (q, J=3.7 Hz, 1H), 3.82 (s, 1H), 3.75 (s, 1H), 3.58 (d, J=11.7 Hz, 3H), 2.10 (bs, exchanged D20, 1H+H2O), 1.50 (s, 9H), 1.46 (s, 18H). 13C NMR (125.7 MHz, CDCl3, centre line δ 77.0): δ 157.5 (C), 151.2 (C), 149.6 (2×C), 134.5 (C), 132.3 (C), 117.6 (C), 84.7 (2×C), 82.8 (C), 77.3 (CH), 74.8 (d, J=4.1 Hz, CH), 69.7 (CH2), 68.8 (d, J=4.1 Hz, CH), 68.6 (d, J=5.9 Hz, CH), 56.0 (d, J=7.4 Hz, CH3), 51.8 (CH), 28.1 (CH3), 27.8 (CH3). MS-ESI+ for C26H40N5NaO13P+ (M+Na)+, calcd. 684.2252. found 684.2251.
E. Preparation of Compound 6a, 6b or 6c
Compound 5a, 5b or 5c (223 mg, 0.34 mmol) was dissolved in anhydrous CH2Cl2 (7 mL) under a nitrogen atmosphere. Anhydrous DMSO (104 μL, 1.46 mmol) was added and the solution was cooled to −78° C. Trifluoroacetic anhydride (104 μL, 0.74 mmol) was added dropwise and the mixture was stirred for 40 min. N,N-diisopropylethylamine (513 μL, 2.94 mmol) was added and the stirring was continued for 50 min at −78° C. Saturated NaCl solution (20 mL) was added and the mixture removed from the cold bath and diluted with CH2Cl2 (30 mL). Glacial acetic acid (170 μL, 8.75 mmol) was added and the mixture was stirred for 10 min. The layers were separated and the aqueous phase was washed with CH2Cl2 (10 mL). The combined organic phases were washed with 5% aqueous HCl, 3:1 saturated NaCl solution:10% NaHCO3 solution and saturated NaCl solution successively, dried over MgSO4, and concentrated to give compound 6a, 6b or 6c (228 mg, quant.) of suitable purity for further use. 1H NMR (500 MHz, CDCl3): δ 5.86 (m, 1 H), 5.07 (m, 1 H), 4.70-4.64 (m, 2 H), 4.49-4.40 (m, 1 H), 4.27 (m, 1 H), 3.56, m, 4 H), 1.49 (s, 9 H), 1.46 (s, 18 H) ppm. 13C NMR (500 MHz, CDCl3): δ 157.5 (C), 151.1 (C), 150.6 (2 C), 134.6 (C), 132.7 (C), 116.6 (C), 92.0 (C), 84.6 (2 C), 83.6 (C), 78.0 (CH), 76.0 (CH), 70.4 (CH2), 67.9 (CH), 56.2 (CH3) δ6.0 (CH), 28.2 (3CH3), 26.8 (6 CH3) ppm. 31P NMR (500 MHz, CDCl3): δ−6.3 ppm.
F. Preparation of compound 7: (4aR,5aR,11aR,12aS)-1,3,2-Dioxaphosphorino[4′,5′:5,6]pyrano[3,2-g]pteridin-10(4H)-one,8-amino-4-a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-2-oxide
Compound 6a, 6b or 6c (10 mg, 14.8 μmol was dissolved in dry acetonitrile (0.2 mL) and cooled to 0° C. Bromotrimethylsilane (19.2 μL, 148 μmol) was added dropwise and the mixture was allowed to warm to ambient temperature and stirred for 5 h during which time a precipitate formed. HCl(aq) (10 μl, 37%) was added and the mixture was stirred for a further 15 min. The mixture was centrifuged for 15 min (3000 g) and the resulting precipitate collected. Acetonitrile (0.5 mL) was added and the mixture was centrifuged for a further 15 min. The acetonitrile wash and centrifugation was repeated a further two times and the resulting solid was dried under high vacuum to give compound 7 (4 mg, 75%). 1H NMR (500 MHz, D2O): δ 5.22 (d, J=1.6 Hz, 1H), 4.34 (dt, J=13, 1.6 Hz, 1H), 4.29-4.27 (m, 1H), 4.24-4.18 (m, 1H), 3.94 (br m, 1H), 3.44 (t, J=1.4 Hz, 1H). 31P NMR (500 MHz, D2O): δ −4.8 MS-ESI+ for C10H15N5O8P+, (M+H)+calcd. 364.0653. found 364.0652.
Example 2Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. Coli in the In vitro Synthesis of Moco
In vitro synthesis of Moco was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. Moco synthesis also involved the use of the purified components E. coli MPT synthase, gephyrin, molybdate, ATP, and apo-sulfite oxidase. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. The assay is based on the conversion of cPMP into MPT, the subsequent molybdate insertion using recombinant gephyrin and ATP, and finally the reconstitution of human apo-sulfite oxidase.
As shown in FIG. 1, Moco synthesis from synthetic cPMP was confirmed, and no differences in Moco conversion were found in comparison to E. coli purified cPMP.
Example 3Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. coli in the In vitro Synthesis of MPT
In vitro synthesis of MPT was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. MPT synthesis also involved the use of in vitro assembled MPT synthase from E. coli. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. Three repetitions of each experiment were performed and are shown in FIGS. 2 and 3.
As shown in FIGS. 2 and 3, MPT synthesis from synthetic cPMP confirmed, and no apparent differences in MPT conversion were found when compared to E. coli purified cPMP. A linear conversion of cPMP into MPT is seen in all samples confirming the identity of synthetic cPMP (see FIG. 2). Slight differences between the repetitions are believed to be due to an inaccurate concentration determination of synthetic cPMP given the presence of interfering chromophores.
Example 4Preparation of Precursor Z (cPMP)
A. Preparation of Starting Materials

B. Introduction of the protected Phosphate

The formation of the cyclic phosphate using intermediate [10] (630 mg) gave the desired product [11] as a 1:1 mixture of diastereoisomers (494 mg, 69%).

C. Oxidation and Overall Deprotection of the Molecule
Oxidation of the secondary alcohol to the gem-diol did prove successful on intermediate [12], but the oxidized product [13] did show significant instability and could not be purified. For this reason, deprotection of the phosphate was attempted before the oxidation. However, the reaction of intermediate [11] with TMSBr led to complete deprotection of the molecule giving intermediate [14]. An attempt to oxidize the alcohol to the gem-diol using Dess-Martin periodinane gave the aromatized pteridine [15].
Oxidation of intermediate [11] with Dess-Martin periodinane gave a mixture of starting material, oxidized product and several by-products. Finally, intermediate [11] was oxidized using the method described Example 1. Upon treatment, only partial oxidation was observed, leaving a 2:1 mixture of [11]/[16]. The crude mixture was submitted to the final deprotection. An off white solid was obtained and analyzed by 1H-NMR and HPLC-MS. These analyses suggest that cPMP has been produced along with the deprotected precursor [11].
Because the analytical HPLC conditions gave a good separation of cPMP from the major impurities, this method will be repeated on a prep-HPLC in order to isolate the final material.
CLIP
BridgeBio Pharma And Affiliate Origin Biosciences Announces FDA Acceptance Of Its New Drug Application For Fosdenopterin For The Treatment Of MoCD Type A
Application accepted under Priority Review designation with Breakthrough Therapy Designation and Rare Pediatric Disease Designation previously grantedThere are currently no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury for infants and children.This is BridgeBio’s first NDA acceptanceSAN FRANCISCO, September 29, 2020 – BridgeBio Pharma, Inc. (Nasdaq: BBIO) and affiliate Origin Biosciences today announced the US Food and Drug Administration (FDA) has accepted its New Drug Application (NDA) for fosdenopterin (previously BBP-870/ORGN001), a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.The NDA has been granted Priority Review designation. Fosdenopterin has previously been granted Breakthrough Therapy Designation and Rare Pediatric Disease Designation in the US and may be eligible for a priority review voucher if approved. It received Orphan Drug Designation in the US and Europe. This is BridgeBio’s first NDA acceptance.“We want to thank the patients, families, scientists, physicians and all others involved who helped us reach this critical milestone,” said BridgeBio CEO and founder Neil Kumar, Ph.D. “MoCD Type A is a devastating disease with a median survival of less than four years and we are eager for our investigational therapy to be available to patients, who currently have no approved treatment options. BridgeBio exists to help as many patients as possible afflicted with genetic diseases, no matter how rare. We are grateful that the FDA has accepted our first NDA for priority review and we look forward to submitting our second NDA later this year for infigratinib for second line treatment of cholangiocarcinoma.”About Fosdenopterin
Fosdenopterin is being developed for the treatment of patients with MoCD Type A. Currently, there are no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury with a median survival between 3 to 4 years. Fosdenopterin is a first-in-class cPMP hydrobromide dihydrate and is designed to treat MoCD Type A by replacing cPMP and permitting the two remaining MoCo synthesis steps to proceed, with activation of MoCo-dependent enzymes and elimination of sulfites.About Molybdenum Cofactor Deficiency (MoCD) Type A
MoCD Type A is an ultra-rare, autosomal recessive, inborn error of metabolism caused by disruption in molybdenum cofactor (MoCo) synthesis which is vital to prevent buildup of s-sulfocysteine, a neurotoxic metabolite of sulfite. Patients are often infants with severe encephalopathy and intractable seizures. Disease progression is rapid with a high infant mortality rate.Those who survive beyond the first few month’s experience profuse developmental delays and suffer the effects of irreversible neurological damage, including brain atrophy with white matter necrosis, dysmorphic facial features, and spastic paraplegia. Clinical presentation that can be similar to hypoxic-ischemic encephalopathy (HIE) or other neonatal seizure disorders may lead to misdiagnosis and underdiagnosis. Immediate testing for elevated sulfite levels and S-sulfocysteine in the urine and very low serum uric acid may help with suspicion of MoCD.About Origin Biosciences
Origin Biosciences, an affiliate of BridgeBio Pharma, is a biotechnology company focused on developing and commercializing a treatment for Molybdenum Cofactor Deficiency (MoCD) Type A. Origin is led by a team of veteran biotechnology executives. Together with patients and physicians, the company aims to bring a safe, effective treatment for MoCD Type A to market as quickly as possible. For more information on Origin Biosciences, please visit the company’s website at www.origintx.com.
About BridgeBio Pharma
BridgeBio is a team of experienced drug discoverers, developers and innovators working to create life-altering medicines that target well-characterized genetic diseases at their source. BridgeBio was founded in 2015 to identify and advance transformative medicines to treat patients who suffer from Mendelian diseases, which are diseases that arise from defects in a single gene, and cancers with clear genetic drivers. BridgeBio’s pipeline of over 20 development programs includes product candidates ranging from early discovery to late-stage development. For more information visit bridgebio.com.
| Clinical data | |
|---|---|
| Trade names | Nulibry |
| Other names | Precursor Z, ALXN1101 |
| License data | US DailyMed: Fosdenopterin |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 150829-29-1 |
| PubChem CID | 135894389 |
| DrugBank | DB16628 |
| ChemSpider | 17221217 |
| UNII | 4X7K2681Y7 |
| KEGG | D11779 |
| ChEMBL | ChEMBL2338675 |
| CompTox Dashboard (EPA) | DTXSID90934067 |
| Chemical and physical data | |
| Formula | C10H14N5O8P |
| Molar mass | 363.223 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESNC1=NC(=O)C2=C(N[C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]4C(O)(O)[C@@H]3N2)N1 | |
| hideInChIInChI=1S/C10H14N5O8P/c11-9-14-6-3(7(16)15-9)12-4-8(13-6)22-2-1-21-24(19,20)23-5(2)10(4,17)18/h2,4-5,8,12,17-18H,1H2,(H,19,20)(H4,11,13,14,15,16)/t2-,4-,5+,8-/m1/s1Key:CZAKJJUNKNPTTO-AJFJRRQVSA-N |
//////////Fosdenopterin hydrobromide, ホスデノプテリン臭化水素酸塩水和物 , ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780, BBP-870/ORGN001, Priority Review designation, Breakthrough Therapy Designation, Rare Pediatric Disease Designation, Orphan Drug Designation, molybdenum cofactor deficiency, ALXN-1101, WHO 11150, FDA 2021, APPROVALS 2021
#Fosdenopterin hydrobromide, #ホスデノプテリン臭化水素酸塩水和物 , #ALXN1101 HBr, #UNII-X41B5W735T, X41B5W735T, #D11780, #BBP-870/ORGN001, #Priority Review designation, #Breakthrough Therapy Designation, #Rare Pediatric Disease Designation, #Orphan Drug Designation, #molybdenum cofactor deficiency, #ALXN-1101, #WHO 11150, #FDA 2021, #APPROVALS 2021
C1C2C(C(C3C(O2)NC4=C(N3)C(=O)NC(=N4)N)(O)O)OP(=O)(O1)O.O.O.Br
Devimistat
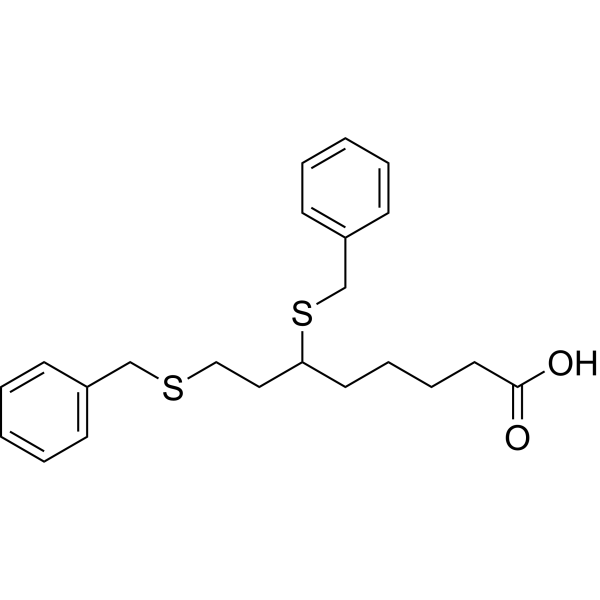

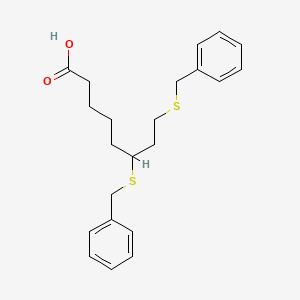
Devimistat
CPI-613
| Molecular Weight | 388.59 |
|---|---|
| Formula | C₂₂H₂₈O₂S₂ |
| CAS No. | 95809-78-2 |
| SMILES | O=C(O)CCCCC(SCC1=CC=CC=C1)CCSCC2=CC=CC=C2 |
phase III, hematological cancer
6,8-Bis(benzylsulfanyl)octanoic acid
Octanoic acid, 6,8-bis[(phenylMethyl)thio]-
Octanoic acid, 6,8-bis((phenylmethyl)thio)-
Rafael Pharmaceuticals (formerly Cornerstone Pharmaceuticals), a subsidiary of Rafael Holdings, is developing devimistat, the lead candidate from a program of thioctans and their derivatives that act as pyruvate dehydrogenase and alpha-ketoglutarate inhibitors and stimulators of pyruvate dehydrogenase kinase (PDK), using the company’s proprietary Altered Energy Metabolism Directed (AEMD) platform, for the iv treatment of hematological cancer [phase III, January 2021].
Devimistat (INN; development code CPI-613) is an experimental anti-mitochondrial drug being developed by Rafael Pharmaceuticals.[1] It is being studied for the treatment of patients with metastatic pancreatic cancer and relapsed or refractory acute myeloid leukemia (AML).
Devimistat’s mechanism of action differs from other drugs, operating on the tricarboxylic acid cycle and inhibiting enzymes involved with cancer cell energy metabolism. A lipoic acid derivative different from standard cytotoxic chemotherapy, devimistat is currently being studied in combination with modified FOLFIRINOX to treat various solid tumors and heme malignancies.
Regulation
The U.S. Food and Drug Administration (FDA) has designated devimistat as an orphan drug for the treatment of pancreatic cancer, AML, myelodysplastic syndromes (MDS), peripheral T-cell lymphoma, and Burkitt’s lymphoma, and given approval to initiate clinical trials in pancreatic cancer and AML.
Clinical trials
Clinical trials of the drug are underway including a Phase III open-label clinical trial[2] to evaluate efficacy and safety of devimistat plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas.
Developed as part of Rafael’s proprietary Altered Metabolism Directed (AMD) drug platform, CPI-613® was discovered at Stony Brook University. CPI-613® is designed to target the mitochondrial tricarboxylic acid (TCA) cycle, an indispensable process essential to tumor cell multiplication and survival, selectively in cancer cells.
The attacks of CPI-613® on the TCA cycle also substantially increases the sensitivity of cancer cells to a diverse range of chemotherapeutic agents. This synergy allows for combinations of CPI-613® with lower doses of these generally toxic drugs to be highly effective with lower patient side effects. Combinations with CPI-613® represent a diverse range of potential opportunities to substantially improve patient benefit in many different cancers.
The U.S. Food and Drug Administration (FDA) has given Rafael approval to initiate pivotal clinical trials in pancreatic cancer and acute myeloid leukemia (AML), and has designated CPI-613® as an orphan drug for the treatment of pancreatic cancer, AML, Myelodysplastic syndromes (MDS), peripheral T-cell lymphoma and Burkitt’s lymphoma. The EMA has granted orphan drug designation to CPI-613® for pancreatic cancer and AML.
Learn more about recent developments involving CPI-613®: CPI-613® (devimistat) Fact Sheet
he FDA granted a Fast Track designation to devimistat for the treatment of patients with acute myeloid leukemia.
The FDA has granted a Fast Track designation to devimistat (CPI-613) for the treatment of patients with acute myeloid leukemia (AML), Rafael Pharmaceuticals, announced in a press release.1
“This designation underscores the pressing need to find new ways to combat this aggressive disease,” said Jorge Cortes, MD, director of the Georgia Cancer Center at Augusta University, and principal investigator on the phase 3 clinical trial, in a statement. “It brings hope not only to clinicians, but to patients who hear that they have been diagnosed.”
The first-in-class agent devimistat targets enzymes that are involved in cancer cell energy metabolism. This therapy substantially increases the sensitivity of cancer cells to a diverse range of chemotherapies, and this synergy allows for potential combinations that could be more effective with devimistat and lower doses of drugs that are generally toxic.
“Receiving Fast Track designation, especially during a pandemic that has created significant challenges for many trials across the globe, is a testament to the dedicated work of the Rafael team,” stated Sanjeev Luther, president and CEO of Rafael Pharmaceuticals, Inc.
Devimistat combinations appear promising with a diverse range of potential opportunities to improve benefit in patients with various cancer types. Two pivotal phase 3 clinical trials, including the AVENGER 500 study in pancreatic cancer (NCT03504423) and ARMADA 2000 for AML (NCT03504410), have been approved for initiation by the FDA.
The primary end point of the multicenter, open-label, randomized ARMADA 2000 study is complete response (CR), and secondary end points include overall survival and CR plus CR with partial hematologic recovery rate. To be eligible to enroll to the study, patients must be aged ≥50 years with a documented AML diagnosis that has relapsed from or became refractory to previous standard therapy. Patients must have an ECOG performance status of 0 to 2 and an expected survival longer than 3 months.
Five hundred patients are expected to be enrolled and randomized in the study. To enroll, patients could not have received prior radiotherapy or cytotoxic chemotherapy for their current AML. Those with active central nervous system involvement, active uncontrolled bleeding, history of other malignancy, or known hypersensitivity to study drugs are ineligible to enroll to the trial as well.
This study aims to determine the safety and efficacy of devimistat in combination with high-dose cytarabine and mitoxantrone in older patients with relapsed/refractory AML compared with high-dose cytarabine and mitoxantrone therapy alone. Other control groups include patients treated with mitoxantrone, etoposide, and cytarabine and the combination of fludarabine, cytarabine, and filgrastim. The addition of devimistat is expected to improve the CR rate in patients who are aged 50 years or older with relapsed/refractory AML.
In a prior phase 1 study of devimistat plus high-dose cytarabine and mitoxantrone in patients with relapsed/refractory AML, the addition of devimistat sensitized AML cells to chemotherapy treatment.2
The objective response rate was 50% including CRs in 26 of 62 evaluable patients. Median overall survival was 6.7 months. In patients above age 60, the CR or CR with incomplete hematologic recovery rate was 47% and the median survival was 6.9 months.
This designation for this experimental anti-mitochondrial agent follows news of another Fast Track designation granted to devimistat for the treatment of patients with metastatic pancreatic cancer in November 2020, as well as an Orphan Drug designation granted in October 2020 for the treatment of patients with soft tissue sarcoma.
References
1. Rafael Pharmaceuticals Receives FDA Fast Track Designation for CPI-613® (devimistat) for the treatment of acute myeloid leukemia (AML). News Release. Rafael Pharmaceuticals, Inc. December 15, 2020. Accessed December 15, 2020. https://bit.ly/34g6YsR
2. Pardee TS, Anderson RG, Pladna KM, et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin Cancer Res. 2018;24(9):2060-2073. doi:10.1158/1078-0432.CCR-17-2282 P[APERJournal of the American Chemical Society (1954), 76, 4109-12.https://pubs.acs.org/doi/abs/10.1021/ja01645a016
PAPERJournal of the American Chemical Society (1955), 77, 416-19.https://pubs.acs.org/doi/abs/10.1021/ja01607a057PAPERJustus Liebigs Annalen der Chemie (1958), 614, 66-83.https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/jlac.19586140108PATENTWO 2009123597WO 2009110859WO 2010110771PATENTCN 111362848
PATENT
WO-2021011334
Deuterated derivatives of 6,8-bis(benzylsulfanyl)octanoic acid (CPI-613 or devimistat ) or its salts for treating cancer.
CPI-613 (6,8-bis(benzylsulfanyl)octanoic acid) is a first-in-class investigational small-molecule (lipoate analog), which targets the altered energy metabolism unique to many cancer cells. CPI-613 is currently being evaluated in two phase III clinical trials, and has been granted orphan drug designation for the treatment of pancreatic cancer, acute myeloid leukemia (AML), peripheral T-cell lymphoma (PTCL), Burkitt lymphoma and myelodysplastic syndromes (MDS).
[0004] One limitation to the clinical utility of CPI-613 is its very rapid metabolism. After IV dosing the half-life of 6,8-bis(benzylsulfanyl)octanoic acid is only about 1-2 hours (Pardee,
T.S. et al, Clin Cancer Res. 2014, 20, 5255-64). The short half-life limits the patient’s overall exposure to the drug and necessitates administration of relatively high doses. For safety reasons, CPI-613 is administered via a central venous catheter as an IV infusion over 30-120 minutes, with higher doses requiring longer infusion times.
The terms“6,8-bis(benzylsulfanyl)octanoic acid” and“ 6,8-bis-benzylthio-octanoic acid” refer to the compound known as CPI-613 or devimistat, having the chemical structure
PATENT
WO2020132397
claiming the use of CPI-613 in combination with an autophagy inhibitor eg chloroquine for treating eg cancers.
CPI-613 (6,8-bis-benzylthio-octanoic acid) is a first-in-class investigational small-molecule (lipoate analog), which targets the altered energy metabolism that is common to many cancer cells. CPI-613 has been evaluated in multiple phase I, I/II, and II clinical studies, and has been granted orphan drug designation for the treatment of pancreatic cancer, acute myeloid leukemia (AML), peripheral T-cell lymphoma (PTCL), Burkitt lymphoma and myelodysplastic syndromes (MDS).
PAPER
https://pubs.acs.org/doi/10.1021/op200091t
An Efficient, Economical Synthesis of the Novel Anti-tumor Agent CPI-613
Cite this: Org. Process Res. Dev. 2011, 15, 4, 855–857
Publication Date:May 2, 2011
https://doi.org/10.1021/op200091t

An efficient and practical synthesis of the novel anti-tumor compound 6,8-dithiobenzyl octanoic acid, CPI-613 (2), was developed and executed on a practical scale. CPI-613 can be made in a single vessel from (±)-lipoic acid (1) via reductive opening of the disulfide ring followed by benzylation of the sulfhydryls with benzyl bromide. CPI-613 was isolated by simple crystallization in high yield and purity. The process is scaleable and has been demonstrated at up to 100 kg.CPI-613 (2) was isolated [4.7 kg (90%)] with an HPLC purity of 99.8 area %. Mp 66–67 °C. IR: 3050, 1710, 1400, 668 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.40–7.20 (m, 10 H), 3.80–3.60 (m, 4 H), 2.60–2.50 (m, 2 H), 2.44 (t, J = 8.7, 2 H), 2.23 (t, J = 8.1, 2 H) 2.03–1.30 (m, 8 H). Anal. Calc for C22H28O2S2: C, 68.00; H, 7.26; S, 16.50. Found: C, 67.99; H, 7.31; S, 16.37.

References
- ^ “CPI-613”. Rafael Pharmaceuticals.
- ^ Philip PA, Buyse ME, Alistar AT, Rocha Lima CM, Luther S, Pardee TS, Van Cutsem E (October 2019). “A Phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas”. Future Oncology. 15 (28): 3189–3196. doi:10.2217/fon-2019-0209. PMC 6854438. PMID 31512497.
| Clinical data | |
|---|---|
| Other names | CPI-613 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 95809-78-2 |
| PubChem CID | 24770514 |
| DrugBank | 12109 |
| ChemSpider | 28189062 |
| UNII | E76113IR49 |
| ChEMBL | ChEMBL3186849 |
| CompTox Dashboard(EPA) | DTXSID70914807 |
| ECHA InfoCard | 100.231.125 |
| Chemical and physical data | |
| Formula | C22H28O2S2 |
| Molar mass | 388.58 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C1=CC=C(C=C1)CSCCC(CCCCC(=O)O)SCC2=CC=CC=C2 |
//////////devimistat, CPI-613, CPI 613, phase 3, hematological cancer , Fast Track designation, ORPHAN DRUG,
Lurbinectedin

Lurbinectedin
(1’R,6R,6aR,7R,13S,14S,16R)-5-(Acetyloxy)-2′,3′,4′,6,6a,7,9′-decahydro-8,14-dihydroxy-6′,9-dimethoxy-4,10,23-trimethyl-spiro(6,16-(epithiopropaneoxymethano)-7.13-imino-12H-1,3-dioxolo[7,8]soquino[3,2-b][3]benzazocine-20,1′-[1H]pyrido[3,4-b]indol]-19-one
| Molecular Weight | 784.87 |
| Formula | C41H44N4O10S |
| CAS No. | 497871-47-3 (Lurbinectedin); |
| Chemical Name | Spiro[6,16-(epithiopropanoxymethano)-7,13-imino-12H-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1′-[1H]pyrido[3,4-b]indol]-19-one, 5-(acetyloxy)-2′,3′,4′,6,6a,7,9′,13,14,16-decahydro-8,14-dihydroxy-6′,9-dimethoxy-4,10,23-trimethyl-, (1’R,6R,6aR,7R,13S,14S,16R)- (9CI) |
fda approved , 6/15/2020 , ZEPZELCA, Pharma Mar S.A.
To treat metastatic small cell lung cancer
Drug Trials Snapshot
Research Code:PM-01183; PM-1183
MOA:RNA polymerase inhibitor
Indication:Ovarian cancer; Breast cancer; Non small cell lung cancer (NSCLC)лурбинектединلوربينيكتيدين芦比替定(1R,1’R,2’R,3’R,11’S,12’S,14’R)-5′,12′-Dihydroxy-6,6′-dimethoxy-7′,21′,30′-trimethyl-27′-oxo-2,3,4,9-tetrahydrospiro[β-carboline-1,26′-[17,19,28]trioxa[24]thia[13,30]diazaheptacyclo[12.9.6.13,11. 02,13.04,9.015,23.016,20]triaconta[4,6,8,15,20,22]hexaen]-22′-yl acetate [ACD/IUPAC Name]2CN60TN6ZS497871-47-3[RN]9397
Lurbinectedin is in phase III clinical development for the treatment of platinum refractory/resistant ovarian cancer.
Phase II clinical trials are also ongoing for several oncology indications: non-small cell lung cancer, breast cancer, small cell lung cancer, head and neck carcinoma, neuroendocrine tumors, biliary tract carcinoma, endometrial carcinoma, germ cell tumors and Ewing’s family of tumors.
Lurbinectedin, sold under the brand name Zepzelca, is a medication for the treatment of adults with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.[1][2][3]
The most common side effects include leukopenia, lymphopenia, fatigue, anemia, neutropenia, increased creatinine, increased alanine aminotransferase, increased glucose, thrombocytopenia, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.[1][2][3]
Lurbinectedin is a synthetic tetrahydropyrrolo [4, 3, 2-de]quinolin-8(1H)-one alkaloid analogue with potential antineoplastic activity.[4] Lurbinectedin covalently binds to residues lying in the minor groove of DNA, which may result in delayed progression through S phase, cell cycle arrest in the G2/M phase and cell death.[4]
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Structure
Lurbinectedin is structurally similar to trabectedin, although the tetrahydroisoquinoline present in trabectedin is replaced with a tetrahydro β-carboline which enables lurbinectedin to exhibit increased antitumor activity compared with trabectedin.[7]
Biosynthesis
Lurbinectedin a marine agent isolated from the sea squirt species Ecteinascidia turbinata. Synthetic production is necessary because very small amounts can be obtained from sea organisms. For example, one ton (1000 kg) of sea squirts are required to produce one gram of trabectedin, which is analogue of lurbinectedin. Complex synthesis of lurbinectedin starts from small, common starting materials that require twenty-six individual steps to produce the drug with overall yield of 1.6%.[8][9]
Mechanism of action
According to PharmaMar,[10] lurbinectedin inhibits the active transcription of the encoding genes. This has two consequences. On one hand, it promotes tumor cell death, and on the other it normalizes tumor microenvironment. Active transcription is the process by which there are specific signal where information contained in the DNA sequence is transferred to an RNA molecule. This activity depends on the activity of an enzyme called RNA polymerase II. Lurbinectedin inhibits transcription through a very precise mechanism. Firstly, lurbinectedin binds to specific DNA sequences. It is at these precise spots that slides down the DNA to produce RNA polymerase II that is blocked and degraded by lurbinectedin. Lurbinectedin also has important role in tumor microenvironment. The tumor cells act upon macrophages to avoid them from behaving like an activator of the immune system. Literally, macrophages work in any tumor’s favor. Macrophages can contribute to tumor growth and progression by promoting tumor cell proliferation and invasion, fostering tumor angiogenesis and suppressing antitumor immune cells.[11][12] Attracted to oxygen-starved (hypoxic) and necrotic tumor cells they promote chronic inflammation. So, not only that macrophages inhibit immune system avoiding the destruction of tumor cells, but they also create tumor tissue that allows tumor growth. However, macrophages associated with tumors are cells that are addicted to the transcription process. Lurbinectedin acts specifically on the macrophages associated with tumors in two ways: firstly, by inhibiting the transcription of macrophages that leads to cell death and secondly, inhibiting the production of tumor growth factors. In this way, lurbinectedin normalizes the tumor microenvironment.
History
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 participants with metastatic SCLC who had disease progression on or after platinum-based chemotherapy.[3][6] Participants received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.[3] The trial was conducted at 26 sites in the United States, Great Britain, Belgium, France, Italy, Spain and Czech Republic.[6]
The U.S. Food and Drug Administration (FDA) granted the application for lurbinectedin priority review and orphan drug designations and granted the approval of Zepzelca to Pharma Mar S.A.[3][13]
Research
Clinical Trials
Lurbinectedin can be used as monotherapy in the treatment of SCLC. Lurbinectedin monotherapy demonstrated the following clinical results in relapsed extensive stage SCLC:
- For sensitive disease (chemotherapy-free interval of ≥ 90 days) overall response rate (ORR) was 46.6% with 79.3% disease control rate and median overall survival (OS) being increased to 15.2 months.[14]
- For resistant disease (chemotherapy-free interval of < 90 days) overall response rate (ORR) was 21.3% with 46.8% disease control rate and 5.1 months median overall survival (OS).[14]
Lurbinectedin is also being investigated in combination with doxorubicin as second-line therapy in a randomized Phase III trial.[medical citation needed] While overall survival in this trial is not yet known, response rates at second line were
- 91.7% in sensitive disease with median progression-free survival of 5.8 months, and
- 33.3% in resistant disease with median progression-free of 3.5 months.[15]
Lurbinectedin is available in the U.S. under Expanded Access Program (EAP).[15][16]
SYN
SYN
WO2011/147828
Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01 /771 15; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 1 1456- 1 1460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001 , Curr. Med. Chem. Anti-Cancer Agents, 1 , pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731 , ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641 , and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources.
At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to
specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum- sensitive ovarian cancer in combination with pegylated liposomal doxorubicin.
ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula
It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought.
The first synthetic process for producing ecteinascidin compounds was described in US Patent 5,721 ,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence.
An improvement in the preparation of one intermediate used in such process was disclosed in US Patent 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale.
A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens.
Cyanosafracin B
An improvement in such hemisynthetic process was disclosed in
EP 1.287.004.
To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600.
WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula:
Total synthesis strategies for the synthesis of the pentacyclic core -743 are overviewed in Figure I.
X = OH or CI
R = Protecting Group
WO2007087220 JOC 2008, 73, 9594-9600
EXAMPLE 3: SYNTHESIS OF COMPOUND 17.
Scheme X above provides an example of the synthesis of compound 17 from intermediate 10.
Compounds 16 and 17 are obtainable from intermediate 15 using the same procedures than those previously described in WO03/014127.
SYN

Reference:
1. WO2003014127A1.
https://patents.google.com/patent/WO2003014127A1/en
The ecteinascidins are exceedingly potent antitumour agents isolated from the marine tunicate Ecteinascidia turbinata. Several ecteinascidins have been reported previously in the patent and scientific literature. See, for example:
U.S. Patent No 5.256.663, which describes pharmaceutical compositions comprising matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins, and the use of such compositions as antibacterial, antiviral, and/ or antitumour agents in mammals.
U.S. Patent No 5.089.273, which describes novel compositions of matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins 729, 743, 745, 759A, 759B and 770. These compounds are useful as antibacterial and/or antitumour agents in mammals.
U.S. Patent No 5.149.804 which describes Ecteinascidins 722 and 736 (Et’s 722 and 736) isolated from the Caribbean tunicate Ecteinascidia turbinata and their structures. Et’s 722 and 736 protect mice in vivo at very low concentrations against P388 lymphoma, B 16 melanoma, and Lewis lung carcinoma.
U.S. Patent No 5.478.932, which describes ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX- 1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.654.426, which describes several ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX-1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.721.362 which describes a synthetic process for the formation of ecteinascidin compounds and related structures.
U.S. Patent No 6.124.292 which describes a series of new ecteinascidin- like compounds.
WO 0177115, WO 0187894 and WO 0187895, which describe new synthetic compounds of the ecteinascidin series, their synthesis and biological properties.
See also: Corey, E.J., J. Am. Chem. Soc, 1996, 118 pp. 9202-9203; Rinehart, et al., Journal of Natural Products, 1990, “Bioactive Compounds from Aquatic and Terrestrial Sources”, vol. 53, pp. 771- 792; Rinehart et al., Pure and Appl. Chem., 1990, “Biologically active natural products”, vol 62, pp. 1277- 1280; Rinehart, et al., J. Org. Chem., 1990, “Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent Antitumour Agents from the Caribbean Tunicate Ecteinascidia tuminata”, vol. 55, pp. 4512-4515; Wright et al., J. Org. Chem., 1990, “Antitumour Tetrahydroisoquinoline Alkaloids from the Colonial ascidian Ecteinascidia turbinata”, vol. 55, pp. 4508-4512; Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional anitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 1 1456- 1 1460; Science 1994, “Chemical Prospectors Scour the Seas for Promising Drugs”, vol. 266, pp.1324; Koenig, K.E., “Asymmetric Synthesis”, ed. Morrison, Academic Press, Inc., Orlando, FL, vol. 5, 1985, p. 71; Barton, et al., J. Chem Soc. Perkin Trans., 1 , 1982, “Synthesis and Properties of a Series of Sterically Hindered Guanidine bases”, pp. 2085; Fukuyama et al., J. Am. Chem. Soc, 1982, “Stereocontrolled Total Synthesis of (+)-Saframycin B”, vol. 104, pp. 4957; Fukuyama et al., J. Am. Chem. Soc, 1990, “Total Synthesis of (+) – Saframycin A”, vol. 112, p. 3712; Saito, et al., J. Org. Chem., 1989, “Synthesis of Saframycins. Preparation of a Key tricyclic Lactam Intermediate to Saframycin A”, vol. 54, 5391; Still, et al., J Org. Chem., 1978, “Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution”, vol. 43, p. 2923; Kofron, W.G.; Baclawski, L.M., J. Org. Chem., 1976, vol. 41, 1879; Guan et al., J. Biomolec Struc & Dynam., vol. 10, pp. 793-817 (1993); Shamma et al., “Carbon- 13 NMR Shift Assignments of Amines and Alkaloids”, p. 206 (1979); Lown et al., Biochemistry, 21, 419-428 (1982); Zmijewski et al., Chem. Biol. Interactions, 52, 361-375 (1985); Ito, CRC Crit. Rev. Anal. Chem., 17, 65- 143 (1986); Rinehart et al., “Topics in Pharmaceutical Sciences 1989”, pp. 613-626, D. D. Breimer, D. J. A. Cromwelin, K. K. Midha, Eds., Amsterdam Medical Press B. V., Noordwijk, The Netherlands (1989); Rinehart et al., “Biological Mass Spectrometry”, 233-258 eds. Burlingame et al., Elsevier Amsterdam (1990); Guan et al., Jour. Biomolec. Struct. & Dynam., vol. 10 pp. 793-817 (1993); Nakagawa et al., J. Amer. Chem. Soc, 11 1 : 2721-2722 (1989);; Lichter et al., “Food and Drugs from the Sea Proceedings” (1972), Marine Technology Society, Washington, D.C. 1973, 117- 127; Sakai et al., J. Amer. Chem. Soc, 1996, 1 18, 9017; Garcϊa-Rocha et al., Brit. J. Cancer, 1996, 73: 875-883; and pommier et al., Biochemistry, 1996, 35: 13303- 13309;
In 2000, a hemisynthetic process for the formation of ecteinascidin compounds and related structures such as phthalascidin starting from natural bis(tetrahydroisoquinoline) alkaloids such as the saframycin and safracin antibiotics available from different culture broths was reported; See Manzanares et al., Org. Lett., 2000, “Synthesis of Ecteinascidin ET-743 and Phthalascidin Pt-650 from Cyanosafracin B”, Vol. 2, No 16, pp. 2545-2548; and International Patent Application WO 00 69862.
Ecteinascidin 736 was first discovered by Rinehart and features a tetrahydro-β-carboline unit in place of the tetrahydroisoquinoline unit more usually found in the ecteinascidin compounds isolated from natural sources; See for example Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional antitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 11456-11460.
Et-736
WO 9209607 claims ecteinascidin 736, as well as ecteinascidin 722 with hydrogen in place of methyl on the nitrogen common to rings C and D of ecteinascidin 736 and O-methylecteinascidin 736 with methoxy in place of hydroxy on ring C of ecteinascidin 736.
Despite the positive results obtained in clinical applications in chemotherapy, the search in the field of ecteinascidin compounds is still open to the identification of new compounds with optimal features of cytotoxicity and selectivity toward the tumour and with a reduced systemic toxicity and improved pharmacokinetic properties.
PATENT
WO2001087894A1.
PATENT
US 20130066067
https://patents.google.com/patent/US20130066067A1/en
- Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01/77115; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 11456-11460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001, Curr. Med. Chem. Anti–Cancer Agents, 1, pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731, ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641, and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
- [0003]
The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources. - [0004]
At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum-sensitive ovarian cancer in combination with pegylated liposomal doxorubicin. - [0005]
ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula - [0006]
It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought. - [0007]
The first synthetic process for producing ecteinascidin compounds was described in U.S. Pat. No. 5,721,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence. - [0008]
An improvement in the preparation of one intermediate used in such process was disclosed in U.S. Pat. No. 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale. - [0009]
A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens. - [0010]
An improvement in such hemisynthetic process was disclosed in EP 1.287.004. - [0011]
To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600. - [0012]
WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula: - [0013]
Total synthesis strategies for the synthesis of the pentacyclic core of ET-743 are overviewed in FIG. 1.
PAPER
Angewandte Chemie, International Edition (2019), 58(12), 3972-3975.
https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201900035
An efficient and scalable approach is described for the total synthesis of the marine natural product Et‐743 and its derivative lubinectedin, which are valuable antitumor compounds. The method delivers 1.6 % overall yield in 26 total steps from Cbz‐protected (S)‐tyrosine. It features the use of a common advanced intermediate to create the right and left parts of these compounds, and a light‐mediated remote C−H bond activation to assemble a benzo[1,3]dioxole‐containing intermediate.

Synthesis of lactone SI-5. A mixture of 19 (98.0 mg, 0.16 mmol, 1.0 equiv), 2-(5-methoxy-1H-indol-3-yl) ethanamine hydrochloride salt (357.8 mg, 1.58 mmol, 10.0 equiv) and NaOAc (144 mg, 1.74 mmol, 11.0 equiv) in anhydrous EtOH (5.0 mL) was stirred at 60 oC for 5 h. The cooled mixture was extracted with ethyl acetate, and the organic layer was dried over sodium sulfate and concentrated. The residue was purified by flash column chromatography (eluting with DCM/MeOH = 20:1) to afford compound SI-5 (109 mg, 87%). [α]𝐷 20 = -27.7 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 2.2 Hz, 1H), 6.75 (dd, J = 8.8, 2.4 Hz, 1H), 6.66 (s, 1H), 6.22 (d, J = 1.0 Hz, 1H), 6.02 (d, J = 1.0 Hz, 1H), 5.78 (s, 1H), 5.08 (d, J = 11.7 Hz, 1H), 4.55 (s, 1H), 4.32 (s, 1H), 4.27 (d, J = 3.8 Hz, 1H), 4.23–4.15 (m, 2H), 3.81 (s, 3H), 3.79 (s, 3H), 3.47–3.39 (m, 2H), 3.20–3.10 (m, 1H), 3.06 (d, J = 18.1 Hz, 1H), 2.93 (dd, J = 18.2, 9.1 Hz, 1H), 2.86–2.76 (m, 1H), 2.62 (dt, J = 14.9, 4.8 Hz, 1H), 2.56–2.47 (m, 2H), 2.37 (s, 3H), 2.30–2.27 (m, 1H), 2.26 (s, 3H), 2.22 (s, 3H), 2.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.6, 168.8, 154.0, 148.2, 145.8, 143.1, 141.3, 140.5, 131.4, 130.8, 130.7, 129.4, 127.3, 120.9, 120.8, 118.4, 118.4, 113.9, 113.8, 112.2, 111.8, 110.2, 102.2, 100.5, 62.6, 61.4, 60.7, 60.5, 59.6, 59.6, 55.9, 54.9, 54.8, 42.1, 41.6, 39.9, 39.5, 29.5, 24.0, 20.8, 16.0, 9.9; HRMS (ESI) m/z calcd. for C42H43N5O9S [M + H]+ 794.2860, found 794.2858

Lurbinectedin: To a solution of SI-5 (80 mg, 0.1 mmol, 1.0 equiv) in acetonitrile and water (3:2, v/v, 10 mL) was added silver nitrate (514 mg, 3 mmol, 30.0 equiv). The suspension was stirred at 25 oC for 24 h before a mixture of saturated brine (5.0 mL) and saturated sodium hydrogen carbonate (5 mL) were added. The resultant mixture was stirred at 25 oC for 15 min before it was filtered through celite and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried over sodium sulfate and concentrated, and the residue was purified by flash column chromatography (eluting with DCM/MeOH = 20:1) to afford Lurbinectedin (71 mg, 89%). [α]𝐷 20 = -45.0 (c = 1.0, CHCl3) 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 1H), 7.13 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 2.2 Hz, 1H), 6.74 (dd, J = 8.8, 2.4 Hz, 1H), 6.67 (s, 1H), 6.19 (d, J = 1.1 Hz, 1H), 5.99 (d, J = 1.1 Hz, 1H), 5.77 (br s, 1H), 5.20 (d, J = 11.3 Hz, 1H), 4.82 (s, 1H), 4.53–4.40 (m, 2H), 4.18–4.08 (m, 2H), 3.81 (s, 3H), 3.79 (s, 3H), 3.49 (d, J = 4.2 Hz, 1H), 3.24–3.13 (m, 2H), 3.01 (d, J = 17.9 Hz, 1H), 2.88–2.79 (m, 2H), 2.63 (dt, J = 15.0, 4.9 Hz, 1H), 2.56–2.47 (m, 2H), 2.37 (s, 3H), 2.32–2.27 (m, 1H), 2.26 (s, 3H), 2.19 (s, 3H), 2.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.4, 168.8, 153.8, 147.9, 145.5, 142.9, 141.1, 140.7, 131.8, 131.3, 130.7, 129.1, 127.3, 121.4, 121.0, 118.2, 115.6, 112.9, 111.9, 111.7, 110.0, 101.8, 100.4, 82.0, 62.4, 61.9, 60.4, 57.8, 57.5, 56.0, 55.8, 55.0, 42.2, 41.3, 39.8, 39.3, 29.3, 23.6, 20.6, 15.9, 9.7; HRMS (ESI) m/z calcd. for C41H44N4O10S [M – OH]+ 767.2745, found 767.2742.
References
- ^ Jump up to:a b c d e “Zepzelca- lurbinectedin injection, powder, lyophilized, for solution”. DailyMed. 15 June 2020. Retrieved 24 September 2020.
- ^ Jump up to:a b c d “Jazz Pharmaceuticals Announces U.S. FDA Accelerated Approval of Zepzelca (lurbinectedin) for the Treatment of Metastatic Small Cell Lung Cancer” (Press release). Jazz Pharmaceuticals. 15 June 2020. Retrieved 15 June 2020 – via PR Newswire.
- ^ Jump up to:a b c d e f g “FDA grants accelerated approval to lurbinectedin for metastatic small”. U.S. Food and Drug Administration (FDA). 15 June 2020. Retrieved 16 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Lurbinectedin”. National Cancer Institute. Retrieved 15 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Zepzelca: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 June 2020.
- ^ Jump up to:a b c d “Drug Trials Snapshots: Zepzelca”. U.S. Food and Drug Administration (FDA). 15 June 2020. Retrieved 28 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ Takahashi, Ryoko; Mabuchi, Seiji; Kawano, Mahiru; Sasano, Tomoyuki; Matsumoto, Yuri; Kuroda, Hiromasa; Kozasa, Katsumi; Hashimoto, Kae; Sawada, Kenjiro; Kimura, Tadashi (17 March 2016). “Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary”. PLOS ONE. 11 (3): e0151050. Bibcode:2016PLoSO..1151050T. doi:10.1371/journal.pone.0151050. PMC 4795692. PMID 26986199.
- ^ Total synthesis of marine antitumor agents trabectedin and lurbinectedin | https://www.sciencedaily.com/releases/2019/02/190219111659.htm
- ^ A Scalable Total Synthesis of the Antitumor Agents Et‐743 and Lurbinectedin | https://onlinelibrary.wiley.com/doi/full/10.1002/anie.201900035
- ^ PharmaMar presentation of Lurbinectedin’s Mechanism of Action Lurbinectedin Mechanisim of Action | https://www.youtube.com/watch?v=8daELhxAXcQ
- ^ Qian BZ, Pollard JW (April 2010). “Macrophage diversity enhances tumor progression and metastasis”. Cell. 141 (1): 39–51. doi:10.1016/j.cell.2010.03.014. PMC 4994190. PMID 20371344.
- ^ Engblom C, Pfirschke C, Pittet MJ (July 2016). “The role of myeloid cells in cancer therapies”. Nature Reviews. Cancer. 16 (7): 447–62. doi:10.1038/nrc.2016.54. PMID 27339708. S2CID 21924175.
- ^ “Lurbinectedin Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 1 August 2018. Retrieved 16 June 2020.
- ^ Jump up to:a b Paz-Ares, Luis G.; Trigo Perez, Jose Manuel; Besse, Benjamin; Moreno, Victor; Lopez, Rafael; Sala, Maria Angeles; Ponce Aix, Santiago; Fernandez, Cristian Marcelo; Siguero, Mariano; Kahatt, Carmen Maria; Zeaiter, Ali Hassan; Zaman, Khalil; Boni, Valentina; Arrondeau, Jennifer; Martinez Aguillo, Maite; Delord, Jean-Pierre; Awada, Ahmad; Kristeleit, Rebecca Sophie; Olmedo Garcia, Maria Eugenia; Subbiah, Vivek (20 May 2019). “Efficacy and safety profile of lurbinectedin in second-line SCLC patients: Results from a phase II single-agent trial”. Journal of Clinical Oncology. 37 (15_suppl): 8506. doi:10.1200/JCO.2019.37.15_suppl.8506.
- ^ Jump up to:a b Calvo, E.; Moreno, V.; Flynn, M.; Holgado, E.; Olmedo, M.E.; Lopez Criado, M.P.; Kahatt, C.; Lopez-Vilariño, J.A.; Siguero, M.; Fernandez-Teruel, C.; Cullell-Young, M.; Soto Matos-Pita, A.; Forster, M. (October 2017). “Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study”. Annals of Oncology. 28 (10): 2559–2566. doi:10.1093/annonc/mdx357. PMC 5834091. PMID 28961837. Lay summary.
- ^ Farago, Anna F; Drapkin, Benjamin J; Lopez-Vilarino de Ramos, Jose Antonio; Galmarini, Carlos M; Núñez, Rafael; Kahatt, Carmen; Paz-Ares, Luis (January 2019). “ATLANTIS: a Phase III study of lurbinectedin/doxorubicin versus topotecan or cyclophosphamide/doxorubicin/vincristine in patients with small-cell lung cancer who have failed one prior platinum-containing line”. Future Oncology. 15 (3): 231–239. doi:10.2217/fon-2018-0597. PMC 6331752. PMID 30362375.
External links
- “Lurbinectedin”. Drug Information Portal. U.S. National Library of Medicine.
- “Lurbinectedin”. NCI Dictionary of Cancer Terms. National Cancer Institute.
- Clinical trial number NCT02454972 for “Clinical Trial of Lurbinectedin (PM01183) in Selected Advanced Solid Tumors” at ClinicalTrials.gov
FDA grants accelerated approval to lurbinectedin for metastatic small cell lung cancer
On June 15, 2020, the Food and Drug Administration granted accelerated approval to lurbinectedin(ZEPZELCA, Pharma Mar S.A.) for adult patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 patients with metastatic SCLC who had disease progression on or after platinum-based chemotherapy. Patients received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.
The main efficacy outcome measures were confirmed overall response rate (ORR) determined by investigator assessment using RECIST 1.1 and response duration. Among the 105 patients, the ORR was 35% (95% CI: 26%, 45%), with a median response duration of 5.3 months (95% CI: 4.1, 6.4). The ORR as per independent review committee was 30% (95% CI: 22%, 40%) with a median response duration of 5.1 months (95% CI: 4.9, 6.4).
The most common adverse reactions (≥20%), including laboratory abnormalities, were myelosuppression, fatigue, increased creatinine, increased alanine aminotransferase, increased glucose, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.
The recommended lurbinectedin dose is 3.2 mg/m2 every 21 days.
View full prescribing information for ZEPZELCA.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this application, a modified Project Orbis was undertaken because of the timing of submission to other regulatory agencies. FDA is collaborating with the Australian Therapeutic Goods Administration (TGA). FDA approved this application 2 months ahead of the goal date. The review is ongoing for the Australian TGA.
FDA granted lurbinectedin orphan drug designation for the treatment of SCLC and priority review to this application. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
REFERENCES
1: Calvo E, Moreno V, Flynn M, Holgado E, Olmedo ME, Lopez Criado MP, Kahatt C, Lopez-Vilariño JA, Siguero M, Fernandez-Teruel C, Cullell-Young M, Soto Matos-Pita A, Forster M. Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study. Ann Oncol. 2017 Oct 1;28(10):2559-2566. doi: 10.1093/annonc/mdx357. PubMed PMID: 28961837.
2: Erba E, Romano M, Gobbi M, Zucchetti M, Ferrari M, Matteo C, Panini N, Colmegna B, Caratti G, Porcu L, Fruscio R, Perlangeli MV, Mezzanzanica D, Lorusso D, Raspagliesi F, D’Incalci M. Ascites interferes with the activity of lurbinectedin and trabectedin: Potential role of their binding to alpha 1-acid glycoprotein. Biochem Pharmacol. 2017 Nov 15;144:52-62. doi: 10.1016/j.bcp.2017.08.001. Epub 2017 Aug 4. PubMed PMID: 28782526.
3: Belgiovine C, Bello E, Liguori M, Craparotta I, Mannarino L, Paracchini L, Beltrame L, Marchini S, Galmarini CM, Mantovani A, Frapolli R, Allavena P, D’Incalci M. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br J Cancer. 2017 Aug 22;117(5):628-638. doi: 10.1038/bjc.2017.205. Epub 2017 Jul 6. PubMed PMID: 28683469; PubMed Central PMCID: PMC5572168.
4: Jimeno A, Sharma MR, Szyldergemajn S, Gore L, Geary D, Diamond JR, Fernandez Teruel C, Soto Matos-Pita A, Iglesias JL, Cullell-Young M, Ratain MJ. Phase I study of lurbinectedin, a synthetic tetrahydroisoquinoline that inhibits activated transcription, induces DNA single- and double-strand breaks, on a weekly × 2 every-3-week schedule. Invest New Drugs. 2017 Aug;35(4):471-477. doi: 10.1007/s10637-017-0427-2. Epub 2017 Jan 20. PubMed PMID: 28105566.
5: Paz-Ares L, Forster M, Boni V, Szyldergemajn S, Corral J, Turnbull S, Cubillo A, Teruel CF, Calderero IL, Siguero M, Bohan P, Calvo E. Phase I clinical and pharmacokinetic study of PM01183 (a tetrahydroisoquinoline, Lurbinectedin) in combination with gemcitabine in patients with advanced solid tumors. Invest New Drugs. 2017 Apr;35(2):198-206. doi: 10.1007/s10637-016-0410-3. Epub 2016 Nov 21. PubMed PMID: 27873130.
6: Harlow ML, Maloney N, Roland J, Guillen Navarro MJ, Easton MK, Kitchen-Goosen SM, Boguslawski EA, Madaj ZB, Johnson BK, Bowman MJ, D’Incalci M, Winn ME, Turner L, Hostetter G, Galmarini CM, Aviles PM, Grohar PJ. Lurbinectedin Inactivates the Ewing Sarcoma Oncoprotein EWS-FLI1 by Redistributing It within the Nucleus. Cancer Res. 2016 Nov 15;76(22):6657-6668. doi: 10.1158/0008-5472.CAN-16-0568. Epub 2016 Oct 3. PubMed PMID: 27697767; PubMed Central PMCID: PMC5567825.
7: Céspedes MV, Guillén MJ, López-Casas PP, Sarno F, Gallardo A, Álamo P, Cuevas C, Hidalgo M, Galmarini CM, Allavena P, Avilés P, Mangues R. Lurbinectedin induces depletion of tumor-associated macrophages, an essential component of its in vivo synergism with gemcitabine, in pancreatic adenocarcinoma mouse models. Dis Model Mech. 2016 Dec 1;9(12):1461-1471. Epub 2016 Oct 20. PubMed PMID: 27780828; PubMed Central PMCID: PMC5200894.
8: Santamaría Nuñez G, Robles CM, Giraudon C, Martínez-Leal JF, Compe E, Coin F, Aviles P, Galmarini CM, Egly JM. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol Cancer Ther. 2016 Oct;15(10):2399-2412. Epub 2016 Sep 14. PubMed PMID: 27630271.
9: Metaxas Y, Cathomas R, Mark M, von Moos R. Combination of cisplatin and lurbinectedin as palliative chemotherapy in progressive malignant pleural mesothelioma: Report of two cases. Lung Cancer. 2016 Dec;102:136-138. doi: 10.1016/j.lungcan.2016.07.012. Epub 2016 Jul 14. PubMed PMID: 27440191.
10: Lima M, Bouzid H, Soares DG, Selle F, Morel C, Galmarini CM, Henriques JA, Larsen AK, Escargueil AE. Dual inhibition of ATR and ATM potentiates the activity of trabectedin and lurbinectedin by perturbing the DNA damage response and homologous recombination repair. Oncotarget. 2016 May 3;7(18):25885-901. doi: 10.18632/oncotarget.8292. PubMed PMID: 27029031; PubMed Central PMCID: PMC5041952.
11: Takahashi R, Mabuchi S, Kawano M, Sasano T, Matsumoto Y, Kuroda H, Kozasa K, Hashimoto K, Sawada K, Kimura T. Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary. PLoS One. 2016 Mar 17;11(3):e0151050. doi: 10.1371/journal.pone.0151050. eCollection 2016. PubMed PMID: 26986199; PubMed Central PMCID: PMC4795692.
12: Pernice T, Bishop AG, Guillen MJ, Cuevas C, Aviles P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J Pharm Biomed Anal. 2016 May 10;123:37-41. doi: 10.1016/j.jpba.2016.01.043. Epub 2016 Jan 21. PubMed PMID: 26871278.
13: Elez ME, Tabernero J, Geary D, Macarulla T, Kang SP, Kahatt C, Pita AS, Teruel CF, Siguero M, Cullell-Young M, Szyldergemajn S, Ratain MJ. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin Cancer Res. 2014 Apr 15;20(8):2205-14. doi: 10.1158/1078-0432.CCR-13-1880. Epub 2014 Feb 21. PubMed PMID: 24563480.
14: Romano M, Frapolli R, Zangarini M, Bello E, Porcu L, Galmarini CM, García-Fernández LF, Cuevas C, Allavena P, Erba E, D’Incalci M. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Int J Cancer. 2013 Nov;133(9):2024-33. doi: 10.1002/ijc.28213. Epub 2013 May 25. PubMed PMID: 23588839.
15: Vidal A, Muñoz C, Guillén MJ, Moretó J, Puertas S, Martínez-Iniesta M, Figueras A, Padullés L, García-Rodriguez FJ, Berdiel-Acer M, Pujana MA, Salazar R, Gil-Martin M, Martí L, Ponce J, Molleví DG, Capella G, Condom E, Viñals F, Huertas D, Cuevas C, Esteller M, Avilés P, Villanueva A. Lurbinectedin (PM01183), a new DNA minor groove binder, inhibits growth of orthotopic primary graft of cisplatin-resistant epithelial ovarian cancer. Clin Cancer Res. 2012 Oct 1;18(19):5399-411. doi: 10.1158/1078-0432.CCR-12-1513. Epub 2012 Aug 15. PubMed PMID: 22896654.
| Clinical data | |
|---|---|
| Pronunciation | LOOR-bih-NEK-teh-din |
| Trade names | Zepzelca |
| Other names | PM-01183 |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a620049 |
| License data | US DailyMed: Lurbinectedin |
| Pregnancy category | US: N (Not classified yet) |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic agent |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 497871-47-3 |
| PubChem CID | 57327016 |
| DrugBank | 12674 |
| ChemSpider | 32701856 |
| UNII | 2CN60TN6ZS |
| KEGG | D11644 |
| ChEMBL | ChEMBL4297516 |
| CompTox Dashboard (EPA) | DTXSID30198065 |
| Chemical and physical data | |
| Formula | C41H44N4O10S |
| Molar mass | 784.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]CC1=CC2=C([C@@H]3[C@@H]4[C@H]5C6=C(C(=C7C(=C6[C@@H](N4[C@H]([C@H](C2)N3C)O)COC(=O)[C@@]8(CS5)C9=C(CCN8)C2=C(N9)C=CC(=C2)OC)OCO7)C)OC(=O)C)C(=C1OC)O | |
| InChI[hide]InChI=1S/C41H44N4O10S/c1-17-11-20-12-25-39(48)45-26-14-52-40(49)41(38-22(9-10-42-41)23-13-21(50-5)7-8-24(23)43-38)15-56-37(31(45)30(44(25)4)27(20)32(47)33(17)51-6)29-28(26)36-35(53-16-54-36)18(2)34(29)55-19(3)46/h7-8,11,13,25-26,30-31,37,39,42-43,47-48H,9-10,12,14-16H2,1-6H3/t25-,26-,30+,31+,37+,39-,41+/m0/s1Key:YDDMIZRDDREKEP-HWTBNCOESA-N |
//////////lurbinectedin, FDA 2020, 2020 APPROVALS, ORPHAN, priority review , ZEPZELCA, Pharma Mar, PM-1183, PM 1183, PM 01183, лурбинектедин , لوربينيكتيدين , 芦比替定
Cc1cc2c(c(c1OC)O)[C@@H]3[C@@H]4[C@H]5c6c(c7c(c(c6OC(=O)C)C)OCO7)[C@@H](N4[C@H]([C@H](C2)N3C)O)COC(=O)[C@@]8(CS5)c9c(c1cc(ccc1[nH]9)OC)CCN8
Naxitamab

(Heavy chain)
QVQLVESGPG VVQPGRSLRI SCAVSGFSVT NYGVHWVRQP PGKGLEWLGV IWAGGITNYN
SAFMSRLTIS KDNSKNTVYL QMNSLRAEDT AMYYCASRGG HYGYALDYWG QGTLVTVSSA
STKGPSVFPL APSSKSTSGG TAALGCLVKD YFPEPVTVSW NSGALTSGVH TFPAVLQSSG
LYSLSSVVTV PSSSLGTQTY ICNVNHKPSN TKVDKRVEPK SCDKTHTCPP CPAPELLGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSRDEL
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ
QGNVFSCSVM HEALHNHYTQ KSLSLSPGK
(Light chain)
EIVMTQTPAT LSVSAGERVT ITCKASQSVS NDVTWYQQKP GQAPRLLIYS ASNRYSGVPA
RFSGSGYGTE FTFTISSVQS EDFAVYFCQQ DYSSFGQGTK LEIKRTVAAP SVFIFPPSDE
QLKSGTASVV CLLNNFYPRE AKVQWKVDNA LQSGNSQESV TEQDSKDSTY SLSSTLTLSK
ADYEKHKVYA CEVTHQGLSS PVTKSFNRGE C
(Disulfide bridge: H22-H95, H146-H202, H222-L211, H228-H’228, H231-H’231, H263-H323, H369-H427, H’22-H’95, H’146-H’202, H’222-L’211, H’263-H’323, H’369-H’427, L23-L88, L131-L191, L’23-L’88, L’131-L’191)
Naxitamab
ナキシタマブ;
Antineoplastic, Anti-GD2 antibody
| Formula | C6414H9910N1718O1996S44 |
|---|---|
| CAS | 1879925-92-4 |
| Mol weight | 144434.4882 |
FDA APPROVED 2020/11/25, Danyelza
FDA grants accelerated approval to naxitamab for high-risk neuroblastoma in bone or bone marrow
On November 25, 2020, the Food and Drug Administration granted accelerated approval to naxitamab (DANYELZA, Y-mAbs Therapeutics, Inc.) in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) for pediatric patients one year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow demonstrating a partial response, minor response, or stable disease to prior therapy.
Efficacy was evaluated in patients with relapsed or refractory neuroblastoma in the bone or bone marrow enrolled in two single-arm, open-label trials: Study 201 (NCT 03363373) and Study 12-230 (NCT 01757626). Patients with progressive disease following their most recent therapy were excluded. Patients received 3 mg/kg naxitamab administered as an intravenous infusion on days 1, 3, and 5 of each 4-week cycle in combination with GM-CSF subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. At the investigator’s discretion, patients were permitted to receive pre-planned radiation to the primary disease site in Study 201 and radiation therapy to non-target bony lesions or soft tissue disease in Study 12-230.
The main efficacy outcome measures were confirmed overall response rate (ORR) per the revised International Neuroblastoma Response Criteria (INRC) and duration of response (DOR). Among 22 patients treated in the multicenter Study 201, the ORR was 45% (95% CI: 24%, 68%) and 30% of responders had a DOR greater or equal to 6 months. Among 38 patients treated in the single-center Study 12-230, the ORR was 34% (95% CI: 20%, 51%) with 23% of patients having a DOR greater or equal to 6 months. For both trials, responses were observed in either the bone, bone marrow or both.
The prescribing information contains a Boxed Warning stating that naxitamab can cause serious infusion-related reactions and neurotoxicity, including severe neuropathic pain, transverse myelitis and reversible posterior leukoencephalopathy syndrome (RPLS). To mitigate these risks, patients should receive premedication prior to each naxitamab infusion and be closely monitored during and for at least two hours following completion of each infusion.
The most common adverse reactions (incidence ≥25% in either trial) in patients receiving naxitamab were infusion-related reactions, pain, tachycardia, vomiting, cough, nausea, diarrhea, decreased appetite, hypertension, fatigue, erythema multiforme, peripheral neuropathy, urticaria, pyrexia, headache, injection site reaction, edema, anxiety, localized edema, and irritability. The most common Grade 3 or 4 laboratory abnormalities (≥5% in either trial) were decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased platelet count, decreased potassium, increased alanine aminotransferase, decreased glucose, decreased calcium, decreased albumin, decreased sodium and decreased phosphate.
The recommended naxitamab dose is 3 mg/kg/day (up to 150 mg/day) on days 1, 3, and 5 of each treatment cycle, administered after dilution as an intravenous infusion in combination with GM-CSF, subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. Treatment cycles are repeated every 4 to 8 weeks.
View full prescribing information for DANYELZA. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761171lbl.pdf
This review used the Real-Time Oncology Review (RTOR) pilot program and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted accelerated approval based on overall response rate and duration of response. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials.
This application was granted priority review, breakthrough therapy, and orphan drug designation. A priority review voucher was issued for this rare pediatric disease product application. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
////////////Naxitamab, priority review, breakthrough therapy, orphan drug, FDA 2020, 2020 APPROVALS, Danyelza, MONOCLONAL ANTIBODY, PEPTIDE, ナキシタマブ,
Ansuvimab-zykl

Ansuvimab-zykl
FDA APPROVED, 12/21/2020, EBANGA
To treat ebola
https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-treatment-ebola-virus
The U.S. Food and Drug Administration approved Ebanga (Ansuvimab-zykl), a human monoclonal antibody, for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga blocks binding of the virus to the cell receptor, preventing its entry into the cell.
Zaire ebolavirus is one of four Ebolavirus species that can cause a potentially fatal human disease. It is transmitted through blood, body fluids, and tissues of infected people or wild animals, and through surfaces and materials, such as bedding and clothing, contaminated with these fluids. Individuals who care for people with the disease, including health care workers who do not use correct infection control precautions, are at the highest risk for infection.
During an Ebola outbreak in the Democratic Republic of the Congo (DRC) in 2018-2019, Ebanga was evaluated in a clinical trial (the PALM trial). The PALM trial was led by the U.S. National Institutes of Health and the DRC’s Institut National de Recherche Biomédicale with contributions from several other international organizations and agencies.
In the PALM trial, the safety and efficacy of Ebanga was evaluated in a multi-center, open-label, randomized controlled trial. 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. The primary analysis population was all patients who were randomized and concurrently eligible to receive either Ebanga or the investigational control during the same time period of the trial. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
The most common symptoms experienced while receiving Ebanga include: fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection. Hypersensitivity, including infusion-related events, can occur in patients taking Ebanga, and treatment should be discontinued in the event of a hypersensitivity reaction.
Patients who receive Ebanga should avoid the concurrent administration of a live virus vaccine against Ebolavirus. There is the potential for Ebanga to inhibit replication of a live vaccine virus and possibly reduce the efficacy of this vaccine.
Ebanga was granted an Orphan Drug designation, which provides incentives to assist and encourage drug development for rare diseases. Additionally, the agency granted Ebanga a Breakthrough Therapy designation.
FDA granted the approval to Ridgeback Biotherapeutics, LP.
Ansuvimab, sold under the brand name Ebanga, is a monoclonal antibody medication for the treatment of Zaire ebolavirus (Ebolavirus) infection.[1][2]
The most common symptoms include fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection.[1]
Ansuvimab was approved for medical use in the United States in December 2020.[1][2]
Chemistry
The drug is composed of a single monoclonal antibody (mAb) and was initially isolated from immortalized B-cells that were obtained from a survivor of the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo.[3] In work supported by the United States National Institutes of Health and the Defense Advanced Projects Agency, the heavy and light chain sequences of ansuvimab mAb was cloned into CHO cell lines and initial production runs were produced by Cook Phamica d.b.a. Catalent under contract of Medimmune.[4][5]
Mechanism of action
Neutralization
Ansuvimab is a monoclonal antibody therapy that is infused intravenously into patients with Ebola virus disease. Ansuvimab is a neutralizing antibody,[3] meaning it binds to a protein on the surface of Ebola virus that is required to infect cells. Specifically, ansuvimab neutralizes infection by binding to a region of the Ebola virus envelope glycoprotein that, in the absence of ansuvimab, would interact with virus’s cell receptor protein, Niemann-Pick C1 (NPC1).[6][7][8] This “competition” by ansuvimab prevents Ebola virus from binding to NPC1 and “neutralizes” the virus’s ability to infect the targeted cell.[6]
Effector function
Antibodies have antigen-binding fragment (Fab) regions and constant fragment (Fc) regions. The Neutralization of virus infection occurs when the Fab regions of antibodies binds to virus antigen(s) in a manner that blocks infection. Antibodies are also able to “kill” virus particles directly and/or kill infected cells using antibody-mediated “effector functions” such as opsonization, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis. These effector functions are contained in the Fc region of antibodies, but is also dependent on binding of the Fab region to antigen. Effector functions also require the use of complement proteins in serum or Fc-receptor on cell membranes. Ansuvimab has been found to be capable of killing cells by antibody-dependent cell-mediated cytotoxicity.[3] Other functional killing tests have not been performed.
History
Ansuvimab is a monoclonal antibody that is being evaluated as a treatment for Ebola virus disease.[9] Its discovery was led by the laboratory of Nancy Sullivan at the United States National Institute of Health Vaccine Research Center and J. J. Muyembe-Tamfum from the Institut National pour la Recherche Biomedicale (INRB) in the Democratic Republic of Congo, working in collaboration with the Institute of Biomedical Research and the United States Army Medical Research Institute of Infectious Diseases.[3][10] Ansuvimab was isolated from the blood of a survivor of the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo roughly ten years later.[3]
In 2018, a Phase 1 clinical trial of ansuvimab was conducted by Martin Gaudinski within the Vaccine Research Center Clinical Trials Program that is led by Julie E. Ledgerwood.[5][4][11] Ansuvimab is also being evaluated during the 2018 North Kivu Ebola outbreak.[12]
Ansuvimab has also shown success with lowering the mortality rate from ~70% to about 34%. In August 2019, Congolese health authorities, the World Health Organization, and the U.S. National Institutes of Health promoted the use of ansuvimab, alongside REGN-EB3, a similar Regeneron-produced monoclonal antibody treatment, over other treatments yielding higher mortality rates, after ending clinical trials during the outbreak.[13][14]
Discovery
A 2016 paper describes the efforts of how ansuvimab was originally developed as part of research efforts lead by Dr. Nancy Sullivan at the United States National Institute of Health Vaccine Research Center and Dr. J. J. Muyembe-Tamfum from the Institut National de Recherche Biomedicale (INRB) in the Democratic Republic of Congo.[3][10] This collaborative effort also involved researchers from Institute of Biomedical Research and the United States Army Medical Research Institute of Infectious Diseases.[3][10] A survivor from the 1995 outbreak of Ebola virus disease in Kikwit, Democratic Republic of Congo donated blood to the project that began roughly ten years after they had recovered.[3] Memory B cells isolated from the survivor’s blood were immortalized, cultured and screened for their ability to produce monoclonal antibodies that reacted with the glycoprotein of Ebola virus. Ansuvimab was identified from one of these cultures and the antibody heavy and light chain gene sequences were sequenced from the cells.[3] These sequences were then cloned into recombinant DNA plasmids and purified antibody protein for initial studies was produced in cells derived from HEK 293 cells.[3]
Ansuvimab and mAb100 combination
In an experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and treated with a combination of ansuvimab and another antibody isolated from the same subject, mAb100. Three doses of the combination were given once a day starting 1 day after the animals were infected. The control animal died and the treated animals all survived.[3]
Ansuvimab monotherapy
In a second experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and only treated with ansuvimab. Three doses of ansuvimab were given once a day starting 1 day or 5 days after the animals were infected. The control animals died and the treated animals all survived.[3] Unpublished data referred to in a publication of the 2018 Phase I clinical trial results of ansuvimab, reported that a single infusion of ansuvimab provided full protection of rhesus macaques and was the basis of the dosing used for human studies.[5][4]
Development
Ansuvimab was developed by the Vaccine Research Center with support of the United States National Institutes of Health and the Defense Advanced Projects Agency. The heavy and light chain sequences of ansuvimab mAb were cloned into CHO cell lines to enable large-scale production of antibody product for use in humans.[4][5]
Human safety testing
In early 2018,[9] a Phase 1 clinical trial of ansuvimab’s safety, tolerability and pharmacokinetics was conducted by Dr. Martin Gaudinski within the Vaccine Research Center Clinical Trials Program that is led by Dr. Julie E. Ledgerwood.[5][4][11] The study was performed in the United States at the NIH Clinical Center and tested single dose infusions of ansuvimab infused over 30 minutes. The study showed that ansuvimab was safe, had minimal side effects and had a half-life of 24 days.[5][4]
Ridgeback Biotherapeutics
A license for ansuvimab was obtained by Ridgeback Biotherapeutics in 2018, from the National Institutes of Health–National Institute of Allergy and Infectious Diseases.[15] Ansuvimab was given orphan drug status in May 2019 and March 2020.[16][17][18]
Experimental use in the Democratic Republic of Congo
During the 2018 Équateur province Ebola outbreak, ansuvimab was requested by the Democratic Republic of Congo (DRC) Ministry of Public Health. Ansuvimab was approved for compassionate use by the World Health Organization MEURI ethical protocol and at DRC ethics board. Ansuvimab was sent along with other therapeutic agents to the outbreak sites.[19][20][11] However, the outbreak came to a conclusion before any therapeutic agents were given to patients.[11]
Approximately one month following the conclusion of the Équateur province outbreak, a distinct outbreak was noted in Kivu in the DRC (2018–20 Kivu Ebola outbreak). Once again, ansuvimab received approval for compassionate use by WHO MEURI and DRC ethic boards and has been given to many patients under these protocols.[11] In November 2018, the Pamoja Tulinde Maisha (PALM [together save lives]) open-label randomized clinical control trial was begun at multiple treatment units testing ansuvimab, REGN-EB3 and remdesivir to ZMapp. Despite the difficulty of running a clinical trial in a conflict zone, investigators have enrolled 681 patients towards their goal of 725. An interim analysis by the Data Safety and Monitoring Board (DSMB) of the first 499 patient found that ansuvimab and REGN-EB3 were superior to the comparator ZMapp. Overall mortality of patients in the ZMapp and remdesivir groups were 49% and 53% compared to 34% and 29% for ansuvimab and REGN-EB3. When looking at patients who arrived early after disease symptoms appeared, survival was 89% for ansuvimab and 94% for REGN-EB3. While the study was not powered to determine whether there is any difference between REGN-EB3 and ansuvimab, the survival difference between those two therapies and ZMapp was significant. This led to the DSMB halting the study and PALM investigators dropping the remdesivir and ZMapp arms from the clinical trial. All patients in the outbreak who elect to participate in the trial will now be given either ansuvimab or REGN-EB3.[21][22][13][12]
In October 2020, the U.S. Food and Drug Administration (FDA) approved atoltivimab/maftivimab/odesivimab (Inmazeb, formerly REGN-EB3) with an indication for the treatment of infection caused by Zaire ebolavirus.[23]
FDA approves ansuvimab-zykl for Ebola virus infection
DECEMBER 21, 2020 BY JANICE REICHERThttps://www.antibodysociety.org/antibody-therapeutic/fda-approves-ansuvimab-zykl-for-ebola-virus-infection/embed/#?secret=zWW0Sr0BdW
On December 21, 2020, the US Food and Drug Administration approved Ebanga (ansuvimab-zykl) for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga had been granted US Orphan Drug designation and Breakthrough Therapy designations. Ansuvimab is a human IgG1 monoclonal antibody that binds and neutralizes the virus.
The safety and efficacy of Ebanga were evaluated in the multi-center, open-label, randomized controlled PALM trial. In this study, 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
Ebanga is the 12th antibody therapeutic to be granted a first approval in the US or EU during 2020.
The Antibody Society maintains a comprehensive table of approved monoclonal antibody therapeutics and those in regulatory review in the EU or US. The table, which is located in the Web Resources section of the Society’s website, can be downloaded in Excel format.
References
- ^ Jump up to:a b c d “FDA Approves Treatment for Ebola Virus”. U.S. Food and Drug Administration. 21 December 2020. Retrieved 23 December 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “Ridgeback Biotherapeutics LP Announces the Approval of Ebanga for Ebola” (Press release). Ridgeback Biotherapeutics LP. 22 December 2020. Retrieved 23 December 2020– via Business Wire.
- ^ Jump up to:a b c d e f g h i j k l Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, Wollen S, et al. (March 2016). “Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody”. Science. 351 (6279): 1339–42. Bibcode:2016Sci…351.1339C. doi:10.1126/science.aad5224. PMID 26917593.
- ^ Jump up to:a b c d e f Clinical trial number NCT03478891 for “Safety and Pharmacokinetics of a Human Monoclonal Antibody, VRC-EBOMAB092-00-AB (MAb114), Administered Intravenously to Healthy Adults” at ClinicalTrials.gov
- ^ Jump up to:a b c d e f Gaudinski MR, Coates EE, Novik L, Widge A, Houser KV, Burch E, et al. (March 2019). “Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody ansuvimab targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study”. Lancet. 393 (10174): 889–898. doi:10.1016/S0140-6736(19)30036-4. PMC 6436835. PMID 30686586.
- ^ Jump up to:a b Misasi J, Gilman MS, Kanekiyo M, Gui M, Cagigi A, Mulangu S, et al. (March 2016). “Structural and molecular basis for Ebola virus neutralization by protective human antibodies”. Science. 351 (6279): 1343–6. Bibcode:2016Sci…351.1343M. doi:10.1126/science.aad6117. PMC 5241105. PMID 26917592.
- ^ Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, et al. (August 2011). “Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection”. Nature. 477 (7364): 344–8. Bibcode:2011Natur.477..344C. doi:10.1038/nature10380. PMC 3230319. PMID 21866101.
- ^ Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, et al. (August 2011). “Ebola virus entry requires the cholesterol transporter Niemann-Pick C1”. Nature. 477 (7364): 340–3. Bibcode:2011Natur.477..340C. doi:10.1038/nature10348. PMC 3175325. PMID 21866103.
- ^ Jump up to:a b “NIH begins testing Ebola treatment in early-stage trial”. National Institutes of Health (NIH). 2018-05-23. Retrieved 2018-10-15.
- ^ Jump up to:a b c Hayden EC (2016-02-26). “Ebola survivor’s blood holds promise of new treatment”. Nature. doi:10.1038/nature.2016.19440. ISSN 1476-4687.
- ^ Jump up to:a b c d e “NIH VideoCast – CC Grand Rounds: Response to an Outbreak: Ebola Virus Monoclonal Antibody (mAb114) Rapid Clinical Development”. videocast.nih.gov. Retrieved 2019-08-09.
- ^ Jump up to:a b Kingsley-Hall A. “Congo’s experimental mAb114 Ebola treatment appears successful: authorities | Central Africa”. http://www.theafricareport.com. Retrieved 2018-10-15.
- ^ Jump up to:a b McNeil DG (12 August 2019). “A Cure for Ebola? Two New Treatments Prove Highly Effective in Congo”. The New York Times. Retrieved 13 August 2019.
- ^ Molteni M (12 August 2019). “Ebola is Now Curable. Here’s How The New Treatments Work”. Wired. Retrieved 13 August 2019.
- ^ “Ridgeback Biotherapeutics LP announces licensing of mAb114, an experimental Ebola treatment, from the National Institute of Allergy and Infectious Diseases” (Press release). Ridgeback Biotherapeutics LP. Retrieved 2019-08-17 – via PR Newswire.
- ^ “Ansuvimab Orphan Drug Designations and Approvals”. accessdata.fda.gov. 8 May 2019. Retrieved 24 December 2020.
- ^ “Ansuvimab Orphan Drug Designations and Approvals”. accessdata.fda.gov. 30 March 2020. Retrieved 24 December 2020.
- ^ “Ridgeback Biotherapeutics LP Announces Orphan Drug Designation for mAb114”(Press release). Ridgeback Biotherapeutics LP. Retrieved 2019-08-17 – via PR Newswire.
- ^ Check Hayden, Erika (May 2018). “Experimental drugs poised for use in Ebola outbreak”. Nature. 557 (7706): 475–476. Bibcode:2018Natur.557..475C. doi:10.1038/d41586-018-05205-x. ISSN 0028-0836. PMID 29789732.
- ^ WHO: Consultation on Monitored Emergency Use of Unregistered and Investigational Interventions for Ebola virus Disease. https://www.who.int/emergencies/ebola/MEURI-Ebola.pdf
- ^ Mole B (2019-08-13). “Two Ebola drugs boost survival rates, according to early trial data”. Ars Technica. Retrieved 2019-08-17.
- ^ “Independent monitoring board recommends early termination of Ebola therapeutics trial in DRC because of favorable results with two of four candidates”. National Institutes of Health (NIH). 2019-08-12. Retrieved 2019-08-17.
- ^ “FDA Approves First Treatment for Ebola Virus”. U.S. Food and Drug Administration(FDA) (Press release). 14 October 2020. Retrieved 14 October 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Ansuvimab”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Zaire ebolavirus |
| Clinical data | |
| Trade names | Ebanga |
| Other names | Ansuvimab-zykl, mAb114 |
| License data | US DailyMed: Ansuvimab |
| Routes of administration | Intravenous |
| Drug class | Monoclonal antibody |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 2375952-29-5 |
| DrugBank | DB16385 |
| UNII | TG8IQ19NG2 |
| KEGG | D11875 |
| Chemical and physical data | |
| Formula | C6368H9924N1724O1994S44 |
| Molar mass | 143950.15 g·mol−1 |
//////////Ansuvimab-zykl , EBANGA, FDA 2020, 2020 APPROVALS, MONOCLONAL ANTIBODY, Orphan Drug designation, , Breakthrough Therapy designation , Ridgeback Biotherapeutics,
Lumasiran
 |
The molecular formula of lumasiran sodium is C530H669F10N173O320P43S6Na43 and the molecular weight is 17,286 Da.
lumasiran
CAS 1834610-13-7
FDA APPROVED, 11/23/2020, Oxlumo
To treat hyperoxaluria type 1
Press Release
Drug Trials Snapshot
RNA, (Gm-sp-Am-sp-Cm-Um-Um-Um-(2′-deoxy-2′-fluoro)C-Am-(2′-deoxy-2′-fluoro)U-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)C-Um-Gm-Gm-Am-Am-Am-Um-Am-Um-Am), 3′-[[(2S,4R)-1-[29-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[3-[[5-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-1-oxopentyl]amino]propyl]amino]-3-oxopropoxy]methyl]-1,12,19,25-tetraoxo-16-oxa-13,20,24-triazanonacos-1-yl]-4-hydroxy-2-pyrrolidinyl]methyl hydrogen phosphate], complex with RNA (Um-sp-(2′-deoxy-2′-fluoro)A-sp-Um-Am-Um-(2′-deoxy-2′-fluoro)U-Um-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)C-Am-Gm-Gm-Am-(2′-deoxy-2′-fluoro)U-Gm-(2′-deoxy-2′-fluoro)A-Am-Am-Gm-Um-Cm-sp-Cm-sp-Am) (1:1)
Nucleic Acid Sequence
Sequence Length: 44, 23, 2115 a 8 c 7 g 14 umultistranded (2); modified
OXLUMO is supplied as a sterile, preservative-free, clear, colorless-to-yellow solution for subcutaneous administration containing the equivalent of 94.5 mg of lumasiran (provided as lumasiran sodium) in 0.5 Ml of water for injection and sodium hydroxide and/or phosphoric acid to adjust the pH to ~7.0.
Lumasiran An investigational RNAi Therapeutic for Primary Hyperoxaluria Type 1 (PH1)
Overview • Lumasiran (ALN-GO1) is an investigational, subcutaneously administered (under the skin) RNA interference (RNAi) therapeutic targeting glycolate oxidase (GO) in development for the treatment of primary hyperoxaluria type 1 (PH1).
• PH1 is a rare, life-threatening disease that can cause serious damage to kidneys and progressively to other organs.1
• PH1 is characterized by the pathologic overproduction of oxalate by the liver. Oxalate is an end product of metabolism that, when in excess, is toxic and accumulates in the kidneys forming calcium oxalate crystals.1,2
• Symptoms of PH1 are often associated with recurrent kidney stones and include flank pain, urinary tract infections, painful urination, and blood in the urine.2,3
• Currently, the only curative treatment is a liver transplant, to correct the metabolic defect, combined with a kidney transplant, to replace the terminally damaged kidneys.1,3 Clinical Development
• The safety and efficacy of lumasiran are being evaluated in a randomized, double-blind, placebo-controlled, global, multicenter Phase 3 study of approximately 30 PH1 patients, called ILLUMINATE-A (NCT03681184).
• The primary endpoint is percent change in 24-hour urinary oxalate excretion from baseline to Month 6.
• Key secondary and exploratory endpoints in ILLUMINATE-A will evaluate additional measures of urinary oxalate, estimated glomerular filtration rate (eGFR), safety, and tolerability.
Regulatory Designations • Breakthrough Therapy Designation by the U.S. Food and Drug Administration (FDA) • Priority Medicines (PRIME) Designation from the European Medicines Agency (EMA) • Orphan Drug Designations in both the U.S. and the European Union
/////////lumasiran, fda 2020, 2020 approvals, Oxlumo, Breakthrough Therapy Designation, Orphan Drug, Priority Medicines (PRIME) Designation
Setmelanotide

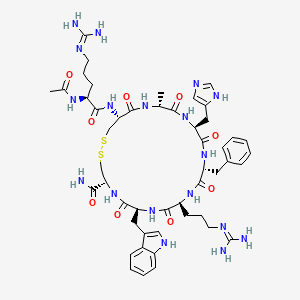

Setmelanotide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
- Molecular FormulaC49H68N18O9S2
- Average mass1117.309 Da
- N-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide (2->8)-disulfide
1,2-Dithia-5,8,11,14,17,20-hexaazacyclotricosane-4-carboxamide, 22-[[(2S)-2-(acetylamino)-5-[(diaminomethylene)amino]-1-oxopentyl]amino]-10-[3-[(diaminomethylene)amino]propyl]-16-(1H-imidazol-5-ylmeth yl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-13-(phenylmethyl)-, (4R,7S,10S,13R,16S,19R,22R)- [ACD/Index Name]10011920014-72-8[RN]Imcivree [Trade name]N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-Ltryptophyl- L-cysteinamide, cyclic (2-8)-disulfideN7T15V1FUYRM-493, BIM-22493UNII-N7T15V1FUYсетмеланотид [Russian] [INN]سيتميلانوتيد [Arabic] [INN]司美诺肽 [Chinese] [INN](4R,7S,10S,13R,16S,19R,22R)-22-[[(2S)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-13-benzyl-10-[3-(diaminomethylideneamino)propyl]-16-(1H-imidazol-5-ylmethyl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-1,2-dithia-5,8,11,14,17,20-hexazacyclotricosane-4-carboxamide
FDA 11/25/2020, Imcivree, To treat obesity and the control of hunger associated with pro-opiomelanocortin deficiency, a rare disorder that causes severe obesity that begins at an early age
Drug Trials Snapshot, 10MG/ML, SOLUTION;SUBCUTANEOUS, Orphan


update Imcivree EMA APPROVED 2021/7/16
DESCRIPTION
IMCIVREE contains setmelanotide acetate, a melanocortin 4 (MC4) receptor agonist. Setmelanotide is an 8 amino acid cyclic peptide analog of endogenous melanocortin peptide α-MSH (alpha-melanocyte stimulating hormone).
The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-Lhistidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C49H68N18O9S2 (anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide is:
 |
IMCIVREE injection is a sterile clear to slightly opalescent, colorless to slightly yellow solution. Each 1 mL of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with 2 to 4 molar equivalents of acetate, and the following inactive ingredients: 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-glycero-3phosphoethanolamine sodium salt, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 11 mg mannitol, 5 mg phenol, 10 mg benzyl alcohol, 1 mg edetate disodium dihydrate, and Water for Injection. The pH of IMCIVREE is 5 to 6.
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor. Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects. Setmelanotide increases resting energy expenditure in both obese animals and humans. Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).
Setmelanotide, sold under the brand name Imcivree, is a medication for the treatment of obesity.[1]
The most common side effects include injection site reactions, skin hyperpigmentation (skin patches that are darker than surrounding skin), headache and gastrointestinal side effects (such as nausea, diarrhea, and abdominal pain), among others.[1] Spontaneous penile erections in males and adverse sexual reactions in females have occurred with treatment.[1] Depression and suicidal ideation have also occurred with setmelanotide.[1]
SYN
WO 2011060355



Medical uses
Setmelanotide is indicated for chronic weight management (weight loss and weight maintenance for at least one year) in people six years and older with obesity due to three rare genetic conditions: pro-opiomelanocortin (POMC) deficiency, proprotein subtilisin/kexin type 1 (PCSK1) deficiency, and leptin receptor (LEPR) deficiency confirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes considered pathogenic (causing disease), likely pathogenic, or of uncertain significance.[1] Setmelanotide is the first FDA-approved treatment for these genetic conditions.[1]
Setmelanotide is not approved for obesity due to suspected POMC, PCSK1, or LEPR deficiency with variants classified as benign (not causing disease) or likely benign or other types of obesity, including obesity associated with other genetic syndromes and general (polygenic) obesity.[1]
Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects.[2] In addition to reducing appetite, setmelanotide increases resting energy expenditure in both obese animals and humans.[3] Importantly, unlike certain other MC4 receptor agonists, such as LY-2112688, setmelanotide has not been found to produce increases in heart rate or blood pressure.[4]
Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).[5] (19.6-fold selectivity for MC4 over MC3, the second target of highest activity.)
History
Setmelanotide was evaluated in two one-year studies.[1] The first study enrolled participants with obesity and confirmed or suspected POMC or PCSK1 deficiency while the second study enrolled participants with obesity and confirmed or suspected LEPR deficiency; all participants were six years or older.[1] The effectiveness of setmelanotide was determined by the number of participants who lost more than ten percent of their body weight after a year of treatment.[1]
The effectiveness of setmelanotide was assessed in 21 participants, ten in the first study and eleven in the second.[1] In the first study, 80 percent of participants with POMC or PCSK1 deficiency lost ten percent or more of their body weight.[1] In the second study, 46 percent of participants with LEPR deficiency lost ten percent or more of their body weight.[1]
The study also assessed the maximal (greatest) hunger in sixteen participants over the previous 24 hours using an eleven-point scale in participants twelve years and older.[1] In both studies, some, but not all, of participants’ weekly average maximal hunger scores decreased substantially from their scores at the beginning of the study.[1] The degree of change was highly variable among participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for setmelanotide orphan disease designation, breakthrough therapy designation, and priority review.[1] The FDA granted the approval of Imcivree to Rhythm Pharmaceutical, Inc.[1]
Research
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor.[6][4] Its peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It was first discovered at Ipsen and is being developed by Rhythm Pharmaceuticals for the treatment of obesity and diabetes.[6] In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity.[7] As of December 2014, the drug is in phase II clinical trials for obesity and PWS.[6][8][9][needs update] So far, preliminary data has shown no benefit of Setmelanotide in Prader-Willi syndrome.[10]
PATENT
WO 2007008704
WO 2011060355
WO 2011060352
US 20120225816
PAPER
Journal of Medicinal Chemistry, 61(8), 3674-3684; 2018
PATENT
https://patents.google.com/patent/US9314509
Synthesis of Example 1i.e., Ac-Arg-cyclo(Cys-D-Ala-His-D-Phe-Arg-Trp-Cys)-NH2

The title peptide having the above structure was assembled using Fmoc chemistry on an Apex peptide synthesizer (Aapptec; Louisville, Ky., USA). 220 mg of 0.91 mmol/g (0.20 mmoles) Rink Amide MBHA resin (Polymer Laboratories; Amherst, Mass., USA) was placed in a reaction well and pre-swollen in 3.0 mL of DMF prior to synthesis. For cycle 1, the resin was treated with two 3-mL portions of 25% piperidine in DMF for 5 and 10 minutes respectively, followed by 4 washes of 3-mL DMF—each wash consisting of adding 3 mL of solvent, mixing for 1 minute, and emptying for 1 minute. Amino acids stocks were prepared in NMP as 0.45M solutions containing 0.45M HOBT. HBTU was prepared as a 0.45M solution in NMP and DIPEA was prepared as a 2.73M solution in NMP. To the resin, 2 mL of the first amino acid (0 9 mmoles, Fmoc-Cys(Trt)-OH) (Novabiochem; San Diego, Calif., USA) was added along with 2 mL (0.9 mmoles) of HBTU and 1.5 mL (4.1 mmoles) of DIPEA. After one hour of constant mixing, the coupling reagents were drained from the resin and the coupling step was repeated. Following amino acid acylation, the resin was washed with two 3-mL aliquots of DMF for 1 minute. The process of assembling the peptide (deblock/wash/acylate/wash) was repeated for cycles 2-9 identical to that as described for cycle 1. The following amino acids were used: cycle 2) Fmoc-Trp(Boc)-OH (Genzyme; Cambridge, Mass., USA); cycle 3) Fmoc-Arg(Pbf)-OH (Novabiochem); cycle 4) Fmoc-DPhe-OH (Genzyme); cycle 5) Fmoc-His(Trt)-OH (Novabiochem); cycle 6) Fmoc-D-Ala-OH (Genzyme); cycle 7) Fmoc-Cys(Trt)-OH, (Novabiochem); and cycle 8) Fmoc-Arg(Pbf)-OH (Genzyme). The N-terminal Fmoc was removed with 25% piperidine in DMF as described above, followed by four 3-mL DMF washes for 1 minute. Acetylation of the N-terminus was performed by adding 0.5 mL of 3M DIPEA in NMP to the resin along with 1.45 mL of 0.45M acetic anhydride in NMP. The resin was mixed for 30 minutes and acetylation was repeated. The resin was washed with 3 mL of DMF for a total of 5 times followed with 5 washes with 5 mL of DCM each.
To cleave and deprotect the peptide, 5mL of the following reagent was added to the resin: 2% TIS/5% water/5% (w/v) DTT/88% TFA. The solution was allowed to mix for 3.5 hours. The filtrate was collected into 40 mL of cold anhydrous ethyl ether. The precipitate was pelleted for 10 minutes at 3500 rpm in a refrigerated centrifuge. The ether was decanted and the peptide was re-suspended in fresh ether. The ether workup was performed three times. Following the last ether wash, the peptide was allowed to air dry to remove residual ether.
The peptide was dissolved in 10% acetonitrile and analyzed by mass spectrometry and reverse-phase HPLC employing a 30×4.6 cm C18 column (Vydac; Hesperia, Calif., USA) with a gradient of 2-60% acetonitrile (0.1% TFA) over 30 minutes. This analysis identified a product with ˜53% purity. Mass analysis employing electrospray ionization identified a main product containing a mass of 1118.4 corresponding to the desired linear product. The crude product (˜100 mg) was diluted to a concentration of 2 mg/mL in 5% acetic acid. To this solution, 0.5M iodine/methanol was added dropwise with vigorous stirring until a pale yellow color was achieved. The solution was vigorously stirred for another 10 minutes. Excess iodine was then quenched by adding 1.0M sodium thiosulfate under continuous mixing until the mixture was rendered colorless. The peptide was re-examined by mass spectrometry analysis and HPLC. Mass spectrometry analysis identified a main species with a mass of 1116.4 which indicated successful oxidation to form the cyclic peptide. The peptide solution was purified on a preparative HPLC equipped with a C18 column using a similar elution gradient. The purified product was re-analyzed by HPLC for purity (>95%) and mass spectrometry (1116.9 which is in agreement with the expected mass of 1117.3) and subsequently lyophilized. Following lyophilization, 28 mg of purified product was obtained representing a 24% yield.
The other exemplified peptides were synthesized substantially according to the procedure described for the above-described synthetic process. Physical data for select exemplified peptides are given in Table 1.
TABLE 1 Example Mol. Wt. Mol. Wt. Purity Number (calculated) (ES-MS) (HPLC) 1 1117.3 1116.9 95.1% 2 1117.3 1116.8 99.2% 3 1280.5 1280.6 98.0% 5 1216.37 1216.20 99.9%
Preparation of Pamoate Salt of Example 1
The acetate salt of Example 1 (200 mg, 0.18 mmole) was dissolved in 10 mL of water. Sodium pamoate (155 mg, 0.36 mmole) was dissolved in 10 mL of water. The two solutions were combined and mixed well. The precipitates were collected by centrifugation at 3000 rpm for 20 minutes, washed for three times with water, and dried by lyophilization.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment for weight management for people with certain rare genetic conditions”. U.S. Food and Drug Administration (FDA) (Press release). 27 November 2020. Retrieved 27 November 2020. This article incorporates text from this source, which is in the public domain.
- ^ Kim GW, Lin JE, Blomain ES, Waldman SA (January 2014). “Antiobesity pharmacotherapy: new drugs and emerging targets”. Clinical Pharmacology and Therapeutics. 95 (1): 53–66. doi:10.1038/clpt.2013.204. PMC 4054704. PMID 24105257.
- ^ Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, et al. (April 2015). “RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals”. The Journal of Clinical Endocrinology and Metabolism. 100 (4): 1639–45. doi:10.1210/jc.2014-4024. PMC 4399297. PMID 25675384.
- ^ Jump up to:a b Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, et al. (February 2013). “Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques”. Diabetes. 62 (2): 490–7. doi:10.2337/db12-0598. PMC 3554387. PMID 23048186.
- ^ Muniyappa R, Chen K, Brychta R, Abel B, Mullins K, Staker P, et al. (June 2014). “A Randomized, Double-Blind, Placebo-Controlled, Crossover Study to Evaluate the Effect of a Melanocortin Receptor 4 (MC4R) Agonist, RM-493, on Resting Energy Expenditure (REE) in Obese Subjects” (PDF). Endocrine Reviews. Rhythm Pharmaceuticals. 35 (3). Retrieved 2015-05-21.
- ^ Jump up to:a b c Lee EC, Carpino PA (2015). “Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014)”. Pharmaceutical Patent Analyst. 4 (2): 95–107. doi:10.4155/ppa.15.1. PMID 25853469.
- ^ “Obesity and Diabetes Caused by Genetic Deficiencies in the MC4 Pathway”. Rhythm Pharmaceuticals. Retrieved 2015-05-21.
- ^ Jackson VM, Price DA, Carpino PA (August 2014). “Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies”. Expert Opinion on Investigational Drugs. 23 (8): 1055–66. doi:10.1517/13543784.2014.918952. PMID 25000213. S2CID 23198484.
- ^ “RM-493: A First-in-Class, Phase 2-Ready MC4 Agonist: A New Drug Class for the Treatment of Obesity and Diabetes”. Rhythm Pharmaceuticals. Archived from the original on 2015-06-14. Retrieved 2015-05-21.
- ^ Duis J, van Wattum PJ, Scheimann A, Salehi P, Brokamp E, Fairbrother L, et al. (March 2019). “A multidisciplinary approach to the clinical management of Prader-Willi syndrome”. Molecular Genetics & Genomic Medicine. 7 (3): e514. doi:10.1002/mgg3.514. PMC 6418440. PMID 30697974.
ADDITIONAL INFORMATION
The peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It is being researched by Rhythm Pharmaceuticals for the treatment of obesity and diabetes. In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity. As of December 2014, the drug is in phase II clinical trials for obesity and PWS.
L-Cysteinamide, N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-, cyclic (2->8)-disulfide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
REFERENCES
1: Lee EC, Carpino PA. Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014). Pharm Pat Anal. 2015;4(2):95-107. doi: 10.4155/ppa.15.1. PubMed PMID: 25853469.
2: Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LH, Reitman ML, Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015 Apr;100(4):1639-45. doi: 10.1210/jc.2014-4024. Epub 2015 Feb 12. PubMed PMID: 25675384; PubMed Central PMCID: PMC4399297.
3: Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, Hofmann SM, Tschöp MH, Van der Ploeg LH, Müller TD. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015 Feb 4;7(3):288-98. doi: 10.15252/emmm.201404508. PubMed PMID: 25652173; PubMed Central PMCID: PMC4364946.
4: Jackson VM, Price DA, Carpino PA. Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies. Expert Opin Investig Drugs. 2014 Aug;23(8):1055-66. doi: 10.1517/13543784.2014.918952. Epub 2014 Jul 7. Review. PubMed PMID: 25000213.
5: Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013 Feb;62(2):490-7. doi: 10.2337/db12-0598. Epub 2012 Oct 9. PubMed PMID: 23048186; PubMed Central PMCID: PMC3554387.
6: Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA, Culler MD, Yang H, Dixit VD, Butler AA. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides. 2009 Oct;30(10):1892-900. doi: 10.1016/j.peptides.2009.07.012. Epub 2009 Jul 29. PubMed PMID: 19646498; PubMed Central PMCID: PMC2755620.
External links
- “Setmelanotide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Imcivree |
| Other names | RM-493; BIM-22493; IRC-022493; N2-Acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide, cyclic (2-8)-disulfide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 920014-72-8 |
| PubChem CID | 11993702 |
| ChemSpider | 10166169 |
| UNII | N7T15V1FUY |
| KEGG | D11927 |
| Chemical and physical data | |
| Formula | C49H68N18O9S2 |
| Molar mass | 1117.32 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@@H]1C(=O)N[C@H](C(=O)N[C@@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)C)C(=O)N)Cc2c[nH]c3c2cccc3)CCCN=C(N)N)Cc4ccccc4)Cc5cnc[nH]5 | |
| InChI[hide]InChI=1S/C49H68N18O9S2/c1-26-41(70)63-37(20-30-22-55-25-59-30)46(75)64-35(18-28-10-4-3-5-11-28)44(73)62-34(15-9-17-57-49(53)54)43(72)65-36(19-29-21-58-32-13-7-6-12-31(29)32)45(74)66-38(40(50)69)23-77-78-24-39(47(76)60-26)67-42(71)33(61-27(2)68)14-8-16-56-48(51)52/h3-7,10-13,21-22,25-26,33-39,58H,8-9,14-20,23-24H2,1-2H3,(H2,50,69)(H,55,59)(H,60,76)(H,61,68)(H,62,73)(H,63,70)(H,64,75)(H,65,72)(H,66,74)(H,67,71)(H4,51,52,56)(H4,53,54,57)/t26-,33+,34+,35-,36+,37+,38+,39+/m1/s1Key:HDHDTKMUACZDAA-PHNIDTBTSA-N |
///////////Setmelanotide, FDA 2020, 2020 APPROVALS, Imcivree, Orphan, PEPTIDE, ANTIOBESITY, UNII-N7T15V1FUY, сетмеланотид , سيتميلانوتيد , 司美诺肽 , BIM 22493, RM 493
CC1C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(CSSCC(C(=O)N1)NC(=O)C(CCCN=C(N)N)NC(=O)C)C(=O)N)CC2=CNC3=CC=CC=C32)CCCN=C(N)N)CC4=CC=CC=C4)CC5=CN=CN5
Berotralstat

Berotralstat
CAS 1809010-50-1
DIHCl 1809010-52-3
Molecular Formula, C30-H26-F4-N6-O, Molecular Weight, 562.5684
1-(3-(Aminomethyl)phenyl)-N-(5-((R)-(3-cyanophenyl)((cyclopropylmethyl)amino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide
1H-Pyrazole-5-carboxamide, 1-(3-(aminomethyl)phenyl)-N-(5-((R)-(3-cyanophenyl)((cyclopropylmethyl)amino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-
To treat patients with hereditary angioedema
FDA APPROVED 12/4/2020, Orladeyo, 110MG CAPSULE 0RAL
New Drug Application (NDA): 214094
Company: BIOCRYST PHARMACEUTICALS INC
New Drug Application (NDA): 214094
Company: BIOCRYST PHARMACEUTICALS INC
Berotralstat Hydrochloride
1-[3-(Aminomethyl)phenyl]-N-(5-{(1R)-(3-cyanophenyl)[(cyclopropylmethyl)amino]methyl}-2-fluorophenyl)-3-(trifluoromethyl)-1H-pyrazole-5-carboxamide dihydrochloride
C30H26F4N6O▪2HCl : 635.48
[1809010-52-3]
Berotralstat, also known as BCX-7353, is a kallikrein inhibitor. BCX7353 is a synthetic, once-daily, small molecule drug that can be taken as an oral capsule to treat HAE attacks and for prophylaxis.
Hereditary angioedema (HAE) is rare disorder caused by a SERPING1 gene mutation that triggers severe swelling of the skin and upper airway. Treatment options for HAE with deficient and dysfunctional C1-inhibitor are expanding to include small-molecule drugs that inhibit protein interactions in the kallikrein-kinin system
Serine proteases make up the largest and most extensively studied group of proteolytic enzymes. Their critical roles in physiological processes extend over such diverse areas as blood coagulation, fibrinolysis, complement activation, reproduction, digestion, and the release of physiologically active peptides. Many of these vital processes begin with cleavage of a single peptide bond or a few peptide bonds in precursor protein or peptides. Sequential limited proteolytic reactions or cascades are involved in blood clotting, fibrinolysis, and complement activation. The biological signals to start these cascades can be controlled and amplified as well. Similarly, controlled proteolysis can shut down or inactivate proteins or peptides through single bond cleavages.
Kallikreins are a subgroup of serine proteases. In humans, plasma kallikrein (KLKB1) has no known homologue, while tissue kallikrein-related peptidases (KLKs) encode a family of fifteen closely related serine proteases. Plasma kallikrein participates in a number of pathways relating to the intrinsic pathway of coagulation, inflammation, and the complement system.
Coagulation is the process by which blood forms clots, for example to stop bleeding. The physiology of coagulation is somewhat complex insofar as it includes two separate initial pathways, which converge into a final common pathway leading to clot formation. In the final common pathway, prothrombin is converted into thrombin, which in turn converts fibrinogen into fibrin, the latter being the principal building block of cross- linked fibrin polymers which form a hemostatic plug. Of the two initial pathways upstream of the final common pathway, one is known as the contact activation or intrinsic pathway, and the other is known as the tissue factor or extrinsic pathway.
The intrinsic pathway begins with formation of a primary complex on collagen by high-molecular- weight kininogen (HMWK), prekallikrein, and FXII (Factor XII; Hageman factor). Prekallikrein is converted to kallikrein, and FXII is activated to become FXIIa. FXIIa then converts Factor XI (FXI) into FXIa, and FXIa in turn activates Factor IX (FIX), which with its co-factor F Villa form the“tenase” complex, which activates Factor X (FX) to FXa. It is FXa which is responsible for the conversion of prothrombin into thrombin within the final common pathway.
Prekallikrein, the inactive precursor of plasma kallikrein, is synthesized in the liver and circulates in the plasma bound to FDVTWK or as a free zymogen. Prekallikrein is cleaved by activated factor XII(FXIIa) to release activated plasma kallikrein (PK). Activated plasma kallikrein displays endopeptidase activity towards peptide bonds after arginine (preferred) and lysine. PK then generates additional FXIIa in a feedback loop which in turn activates factor XI (FXI) to FXIa to connect to the common pathway. Although the initial activation of the intrinsic pathway is through a small amount of FXIIa activating a small amount of PK, it is the subsequent feedback activation of FXII by PK that controls the extent of activation of the intrinsic pathway and hence downstream coagulation. Hathaway, W. E., et al. (1965) Blood 26:521-32.
Activated plasma kallikrein also cleaves HMWK to release the potent vasodilator peptide bradykinin. It is also able to cleave a number of inactive precursor proteins to generate active products, such as plasmin (from plasminogen) and urokinase (from prourokinase). Plasmin, a regulator of coagulation, proteolytically cleaves fibrin into fibrin degradation products that inhibit excessive fibrin formation.
Patients who have suffered acute myocardial infarction (MI) show clinical evidence of being in a hypercoagulable (clot-promoting) state. This hypercoagulability is
paradoxically additionally aggravated in those receiving fibrinolytic therapy. Increased generation of thrombin, as measured by thrombin-antithrombin III (TAT) levels, is observed in patients undergoing such treatment compared to the already high levels observed in those receiving heparin alone. Hoffmeister, H. M. et al. (1998) Circulation 98:2527-33. The increase in thrombin has been proposed to result from plasmin-mediated activation of the intrinsic pathway by direct activation of FXII by plasmin.
Not only does the fibrinolysis-induced hypercoagulability lead to increased rates of reocclusion, but it is also probably responsible, at least in part, for failure to achieve complete fibrinolysis of the clot (thrombus), a major shortcoming of fibrinolytic therapy (Keeley, E. C. et al. (2003) Lancet 361 : 13-20). Another problem in fibrinolytic therapy is the accompanying elevated risk of intracranial hemorrhage. Menon, V. et al. (2004) (Chest l26:549S-575S; Fibrinolytic Therapy Trialists’ Collaborative Group (1994) Lancet 343 :311-22. Hence, an adjunctive anti -coagulant therapy that does not increase the risk of bleeding, but inhibits the formation of new thrombin, would be greatly beneficial. Plasma kallikrein inhibitors also have therapeutic potential for treating hereditary angioedema (HAE). HAE is is a serious and potentially life-threatening rare genetic illness, caused by mutations in the Cl -esterase inhibitor (C1INH) gene, located on chromosome 1 lq. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by CllNH gene mutations which produce low levels of Cl -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective Cl protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; l00(Suppl2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herein by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that CllNH is the most important inhibitor of kallikrein in human plasma and that CllNH deficiency leads to unopposed activation of the kallikrein- bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack. All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
Therefore, a need exists to develop inhibitors of PK that can tip the balance of fibrinolysis/thrombosis at the occluding thrombus toward dissolution, thereby promoting reperfusion and also attenuating the hypercoagulable state, thus preventing thrombus from reforming and reoccluding the vessel. In particular, the creation of plasma kallikrein inhibitors that are specific and capable of being formulated for in vivo use could lead to a new class of therapeutics. Thus, what is needed are improved compositions and methods for preparing and formulating plasma kallikrein inhibitors.
For example, in patients with angioedema conditions, small polypeptide PK inhibitor DX-88 (ecallantide) alleviates edema in patients with hereditary angioedema (HAE). Williams, A. et al. (2003) Transfus. Apher. Sci. 29:255-8; Schneider, L. et al.
(2007) J Allergy Clin Immunol. 120:416-22; and Levy, J. H. et al. (2006) Expert Opin. Invest. Drugs 15: 1077-90. A bradykinin B2 receptor antagonist, Icatibant, is also effective in treating HAE. Bork, K. et al. (2007) J. Allergy Clin. Immunol. 119:1497-1503. Because plasma kallikrein generates bradykinin, inhibition of plasma kallikrein is expected to inhibit bradykinin production.
For example, in coagulation resulting from fibrinolytic treatment (e.g., treatment with tissue plasminogen activator or streptokinase), higher levels of plasma kallikrein are found in patients undergoing fibrinolysis. Hoffmeister, H. M. et al. (1998) J. Cardiovasc. Pharmacol. 31 :764-72. Plasmin-mediated activation of the intrinsic pathway has been shown to occur in plasma and blood and was markedly attenuated in plasma from individuals deficient in any of the intrinsic pathway components. Ewald, G. A. et al. (1995) Circulation 91 :28-36. Individuals who have had an acute MI were found to have elevated levels of activated plasma kallikrein and thrombin. Hoffmeister, H. M., et al. (1998) Circulation 98:2527-33.
DX-88 reduced brain edema, infarct volume, and neurological deficits in an animal model of ischemic stroke. Storini, C. et al. (2006) J Pharm. Exp. Ther. 318:849-854. Cl- inhibitor reduced infarct size in a mouse model of middle cerebral artery occlusion
(MCAO). De Simoni, M. G. et al. (2004) Am. J. Pathol. 164: 1857-1863; and Akita, N. et al. (2003) Neurosurgery 52:395-400). B2 receptor antagonists were found to reduce the infarct volume, brain swelling, and neutrophil accumulation and were neuroprotective in an MCAO animal model. Zausinger, S. et al. (2003 ) Acta Neurochir. Suppl. 86:205-7;
Lumenta, D. B. et al. (2006) Brain Res. 1069:227-34; Ding-Zhou, L. et al. (2003) Br. J Pharmacol. 139: 1539-47.
Regarding blood loss during cardiopulmonary bypass (CPB), it has been found that the kallikrein-kinin (i.e., contact) system is activated during CABG. Wachtfogel, Y. T. (1989) Blood 73:468. Activation of the contact system during CPB results in up to a 20- fold increase in plasma bradykinin. Cugno, M. et al. (2006) Chest 120:1776-82; and Campbell, D. J. et al. (2001 ) Am. J. Physiol. Reg. Integr. Comp. Physiol. 281 : 1059-70.
Plasma kallikrein inhibitors P8720 and PKSI-527 have also been found to reduce joint swelling in rat models of arthritis. De La Cadena, R. A. et al. (1995) FASEB J. 9:446- 52; Fujimori, Y. (1993) Agents Action 39:42-8. It has also been found that inflammation in animal models of arthritis was accompanied by activation of the contact system. Blais, C. Jr. et al. (1997) Arthritis Rheum. 40: 1327-33.
Additionally, plasma kallikrein inhibitor P8720 has been found to reduce inflammation in an acute and chronic rat model of inflammatory bowel disease (IBD). Stadnicki, A. et al. (1998) FASEB J. 12:325-33; Stadnicki, A. et al. (1996) Dig. Dis. Sci.
41 :9l2-20; and De La Cadena, R. A., et al. (1995) FASEB J. 9:446-52. The contact system is activated during acute and chronic intestinal inflammation. Sartor, R. B. et al. (1996) Gastroenterology 110: 1467-81. It has been found that B2 receptor antagonist, an antibody to high molecular weight kininogen, or reduction in levels of kininogen reduced clinicopathology in animal models of IBD. Ibid !; Arai, Y. et al. (1999) Dig. Dis. Sci.
44:845-51; and Keith, J. C. et al. (2005) Arthritis Res. Therapy 7 :R769-76.
H-D-Pro-Phe-Arg-chloromethylketone (CMK), an inhibitor of PK and FXII and a physiological inhibitor (Cl -inhibitor), has been found to reduce vascular permeability in multiple organs and reduce lesions in lipopolysaccharide (LPS)- or bacterial-induced sepsis in animals. Liu, D. et al. (2005) Blood 105:2350-5; Persson, K. et al. (2000) J. Exp. Med. 192: 1415-24. Clinical improvement was observed in sepsis patients treated with Cl- inhibitor. Zeerleder, S. et al. (2003) Clin. Diagnost. Lab. Immunol. 10:529-35; Caliezi, C., et al. (2002) Crit. Care Med. 30:1722-8; and Marx, G. et al. (1999) Intensive Care Med.
25: 1017-20. Fatal cases of septicemia are found to have a higher degree of contact activation. Martinez-Brotons, F. et al. (1987) Thromb. Haemost. 58:709-713; and Kalter, E. S. et al. (1985) J. Infect. Dis. 151 : 1019-27.
It has also been found that prePK levels are higher in diabetics, especially those with proliferative retinopathy, and correlate with fructosamine levels. Gao, B.-B., et al. (2007) Nature Med. 13: 181-8; and Kedzierska, K. et al. (2005) Archives Med. Res. 36:539- 43. PrePK is also found to be highest in those with a sensorimotor neuropathy. Christie,
M. et al. (1984) Thromb. Haemostas. (Stuttgart) 52:221-3. PrePK levels are elevated in diabetics and are associated with increased blood pressure. PrePK levels independently correlate with the albumin excretion rate and are elevated in diabetics with
macroalbuminuria, suggesting prePK may be a marker for progressive nephropathy. Jaffa, A. A. et al. (2003) Diabetes 52: 1215-21. Bl receptor antagonists have been found to decrease plasma leakage in rats treated with streptozotocin. Lawson, S. R. et al. (2005)
Eur. J. Pharmacol. 514:69-78. Bl receptor antagonists can also prevent streptozotocin- treated mice from developing hyperglycemia and renal dysfunction. Zuccollo, A. et al. (1996) Can. J. Physiol. Pharmacol. 74:586-9.
PATENT
WO 2015134998
https://patents.google.com/patent/WO2015134998A1/en
PATENT
WO 2020092898
https://patents.google.com/patent/WO2020092898A1/en
Example 1 : Synthetic protocol for racemic compound 54e
Reproduced from WO 2015/134998 and U.S. Patent Application Publication No. 2017/0073314 A1 (both incorporated by reference)

Preparation of 1 -(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide
(54e)
Step-l : Preparation of 3-((3-amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b)
To a solution of 3-formylbenzonitrile (54a) (29 g, 217 mmol) in tetrahydrofuran (200 mL) cooled to 0 °C was added freshly prepared Grignard reagent (52c) (245 mL, 221 mmol, ~ 0.9 M in THF) stirred at 0 °C for 1 h, and room temperature for 18 h. The reaction mixture was quenched with 1 N HC1 (aq. 440 mL), stirred for 3 h, neutralized with NaOH (2 N, aq.) to pH = ~ 8. The reaction mixture was extracted with ethyl acetate (600, 300 mL). The combined extracts were washed with brine (120 mL), dried over MgS04, filtered and concentrated in vacuum. The crude product was purified by flash column
chromatography [silica gel, eluting with hexanes/ethyl acetate (1 :0 to 1 : 1) to give 3-((3- amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b) (36.28 g) as a brown gum which was used as such for next step; MS (ES+) 265.3 (M+23).
Step-2: Preparation of tert-butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2- fluorophenylcarbamoyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c)
To a solution of 3-((3-amino-4-fluorophenyl)(hydroxy)methyl)benzonitrile (54b) (24.682 g, 102 mmol) in DMF (480 mL) was added l-(3-((tert- butoxycarbonylamino)methyl)phenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxylic acid (lOd) (35.0 g, 91 mmol), N-ethyl-N-isopropylpropan-2-amine (132 mL, 758 mmol), bromotripyrrolidin-l-ylphosphonium hexafluorophosphate(V) (PyBrOP, 42.8 g, 91 mmol) and stirred at room temperature for 19 h. The reaction mixture was diluted with ethyl acetate (1000 mL), washed with water (500, 400 mL), brine (400 mL), dried over MgS04, filtered and concentrated in vacuum. The crude product was purified by flash column chromatography [silica gel, eluting with hexanes/ethyl acetate (1 :0 to 1 : 1)] to afford tert- butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c) (4.583 g, 5% for two steps) as a yellow solid; ¾ NMR (300 MHz, DMSO-i¾) d 10.57 (s, 1H), 7.81 (t, J= 1.7 Hz, 1H), 7.73 – 7.66 (m, 2H), 7.64 – 7.19 (m, 10H), 6.25 (d, J= 4.0 Hz, 1H), 5.78 (d, J= 4.0 Hz, 1H), 4.19 (d, J= 6.1 Hz, 2H), 1.37 (s, 9H); 19F NMR (282 MHz, DMSO-i¾) d -60.81 , -123.09; MS (ES+) 632.3 (M+23).
Step-3: Preparation of tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d)
To a solution of tert-butyl 3-(5-(5-((3-cyanophenyl)(hydroxy)methyl)-2- fluorophenylcarbamoyl)-3-(trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54c) (1.333 g, 2.187 mmol) in dichloromethane (40 mL) at 0°C was added thionyl chloride (0.340 mL, 4.59 mmol) and warmed to room temperature over 2 h. The reaction mixture was quenched with triethyl amine (2.0 mL, 14.35 mmol) stirred at room temperature for 1 h. It was then treated with cyclopropylmethanamine (4.30 mL, 48.0 mmol), concentrated to remove most of dichloromethane followed by addition of acetonitrile (30 mL), stirring at 70 °C for 14 h, and concentration in vacuum to dryness. The residue was treated with chlorofrom (200 mL), washed with water (100 mL), dried over MgS04 followed by filtration and
concentration. The crude product was purified by flash column chromatography [silica gel eluting with hexanes/ethyl acetate (1 :0 to 2: 1)] to afford tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d) (184 mg, 13%) as colorless gum; ¾ NMR (300 MHz, DMSO-ά) d 10.56 (s, 1H), 7.89 (t, J= 1.7 Hz, 1H), 7.77 – 7.71 (m, 1H), 7.70 – 7.30 (m, 10H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 4.19 (d, J= 6.2 Hz, 2H), 2.26 (d, J= 6.6 Hz, 2H), 1.37 (s, 9H), 1.00 – 0.80 (m, 1H), 0.45 – 0.28 (m, 2H), 0.12 – -0.01 (m, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.80 , -123.20; MS (ES+) 663.4 (M+l). Step-4: Preparation of l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e)
To a solution of tert-butyl 3-(5-(5-((3- cyanophenyl)(cyclopropylmethylamino)methyl)-2-fluorophenylcarbamoyl)-3- (trifluoromethyl)-lH-pyrazol-l-yl)benzylcarbamate (54d) (161 mg, 0.243 mmol) in 1,4- Dioxane (18 mL) was added hydrogen chloride (2.60 mL, 10.40 mmol, 4 M in l,4-dioxane) and stirred at room temperature for 16 h. the reaction mixture was treated with hexanes, decanted, washed with hexanes, and decanted again. The insoluble crude product was purified by flash column chromatography [silica gel, eluting with chloroform/CMA80 (1 :0 to 2:1)] to afford l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e). The pure product was dissolved in methanol (10 mL) and added 4 N HC1 (aq. 0.14 mL) followed by concentration in vacuum to dryness to give HC1 salt of l-(3- (aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl-methylamino)methyl)-2- fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (54e) (74 mg, 48%) white solid; ¾ NMR (300 MHz, DMSO- d, D20 ex NMR) d 8.13 (t, J = 1.7 Hz, 1H), 7.98 – 7.84 (m, 3H), 7.73 – 7.64 (m, 3H), 7.63 – 7.48 (m, 4H), 7.44 (dd, J = 10.2, 8.6 Hz, 1H),
5.75 (s, 1H), 4.12 (s, 2H), 2.76 (d, J = 7.2 Hz, 2H), 1.17 – 0.94 (m, 1H), 0.68 – 0.47 (m, 2H), 0.34-0.24 (m, 2H); 19F NMR (282 MHz, DMSO- d) d -60.82, -120.02; MS (ES+): 563.3 (M+l); Analysis calculated for C30H26F4N6O2.O HCT3.0 H2O: C, 52.26; H, 4.97; N, 12.19; Found: C, 52.26; H, 5.00; N, 11.72.
Example 2: Separation of enantiomers of racemic compound 54e
Reproduced from WO 2015/134998 and U.S. Patent Application Publication No. 2017/0073314 A1 (both incorporated by reference)

Compound I (free base) Separation of (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lFl-pyrazole-5-carboxamide (Compound I), and (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lFl-pyrazole-5-carboxamide ((-
)-enantiomer)
Isomers of Racemic l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lF[- pyrazole-5-carboxamide (54e) (0.4 g) were separated by using preparative SFC method using the following conditions to furnish:
Preparative SFC Method used:
Column 20mm x 25.0 cm ChromegaChiral CCS from
Regis Technologies (Morton Grove, IL)
CO2 Co-solvent (Solvent B) Methanol: Isopropanol (1 : 1) with 1%
Isopropylamine
Isocratic Method 20 % Co-solvent at 80 mL/min
System Pressure 200 bar
Column Temperature 25 °C
Sample Diluent Methanol: Isopropanol
Chiral Purity of peaks was determined by following Analytical SFC Method:
Column 4.6 x 100 mm ChiralPak AS from Chiral
Technologies (West Chester, PA)
CO2 Co-solvent (Solvent B) Methanol: Isopropanol (1 : 1) with 0.1%
Isopropylamine
Isocratic Method 5-65 % Co-solvent Gradient at 4 mL/min System Pressure 100 bar
Column Temperature 25 °C
Sample Diluent Methanol
Peak-l (Compound I) 2.1 min 144 mg >95% ee (UV 254)
98.6 % purity (UV 254)
Peak-2 ((-)-enantiomer) 2.4 min 172 mg 95.5 % ee (UV 254)
96.5 % purity (UV 254) 1. Peak-l assigned as (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3- (trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (144 mg, >95%ee) free base as white solid; Optical rotation: [O]D = (+) 6.83 [CH3OH, 1.2]; ‘H NMR (300 MHz, DMSO-£¾) d 10.53 (s, 1H, D2O exchangeable), 7.88 (t, J= 1.7 Hz, 1H), 7.77 – 7.71 (m, 1H), 7.67 (dt, 7= 7.7, 1.4 Hz, 1H), 7.63 (dd, J= 7.5, 2.1 Hz, 1H), 7.56 (s, 1H), 7.54 – 7.47 (m, 2H), 7.47 – 7.38 (m, 2H), 7.34 (ddt, J= 8.6, 5.9, 2.8 Hz, 2H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 3.77 (s, 2H), 2.31 – 2.21 (m, 2H), 0.97 – 0.80 (m, 1H), 0.42 – 0.33 (m, 2H), 0.10 – -0.02 (m, 2H); 19F NMR (282 MHz, DMSO-Ts) d -60.73 , -123.20; MS (ES+) 563.3 (M+l), 561.3 (M-l). To a solution of free base mixture of (+)-l-(3-(aminomethyl)phenyl)-N-(5-((3- cyanophenyl)(cyclopropyl-methylamino)methyl)-2-fluorophenyl)-3- (trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (120 mg) in methanol (15 mL) was added hydrogen chloride (0.969 mL, 1.938 mmol), stirred at room temperature for 10 min, evaporated to dryness to afford (+)-l-(3- (aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl-methylamino)methyl)-2- fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide (Compound I) (100 mg) hydrochloride salt as white solid; ¾ NMR (300 MHz, DMSO-Ts) d 10.84 (s, 1H, D2O exchangeable), 10.44 (s, 2H, D2O exchangeable), 8.44 (s, 3H, D2O exchangeable), 8.30 (s, 1H, D2O exchangeable), 8.09 (d, J= 7.9 Hz, 1H), 7.99 (d, J = 6.8 Hz, 1H), 7.91 – 7.83 (m, 1H), 7.80 – 7.50 (m, 7H), 7.42 (dd, J= 10.3, 8.6 Hz, 1H), 5.78 (d, J= 6.9 Hz, 1H), 4.13 (d, J= 5.7 Hz, 2H), 2.88 – 2.62 (m, 2H), 1.42 – 0.99 (m, 1H), 0.73 – 0.46 (m, 2H), 0.32 (d, J= 4.4 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.81 , -119.99; MS (ES+): MS (ES+) 563.3 (M+l), MS (ES-) 561.3 (M-l), 597.3 (M+Cl); Analysis calculated for C30H26F4N6O 2HC1 l.75H20: C, 54.02; H, 4.76; Cl, 10.63; N, 12.60; Found: C, 54.12; H, 4.83; Cl, 10.10; N, 11.97. Peak-2 assigned as (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (172 mg, 95.5 % ee) as free base was repurified by flash column chromatography (silica gel 12 g, eluting 0-30% MeOH in chloroform for 15 min) to afford (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) free base as an off-white solid; Optical rotation: [O]D = (-) 5.44
[CH3OH, 1.25]; ¾ NMR (300 MHz, DMSO-i¾) d 7.88 (t, J= 1.6 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.70 – 7.61 (m, 2H), 7.57 (s, 1H), 7.54 – 7.47 (m, 2H), 7.45 – 7.41 (m,
2H), 7.34 (ddq, J= 8.7, 6.1, 3.5, 2.8 Hz, 2H), 7.22 (dd, J= 10.3, 8.5 Hz, 1H), 4.93 (s, 1H), 3.78 (s, 2H), 2.25 (d, J= 6.9 Hz, 2H), 0.90 (ddd, J= 9.8, 8.0, 5.2 Hz, 1H), 0.47 – 0.29 (m, 2H), 0.04 (dd, J= 5.0, 1.5 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.73 , -123.19; MS (ES+) 563.3 (M+l), MS (ES-), 561.3 (M-l). To a solution of free base of (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (0.124 g, 0.220 mmol) in methanol (15 mL) was added hydrogen chloride (1.102 mL, 2.204 mmol), stirred at room temperature for 10 min, evaporated to dryness to afford (-)-l-(3-(aminomethyl)phenyl)-N-(5-((3-cyanophenyl)(cyclopropyl- methylamino)methyl)-2-fluorophenyl)-3-(trifluoromethyl)-lH-pyrazole-5-carboxamide ((-)-enantiomer) (0.121 g) hydrochloride salt as an off-white solid; Ή NMEE ¾ NMR (300 MHz, DMSO-i¾) d 10.82 (s, 1H, D20 exchangeable), 10.36 (s, 2H, D2O exchangeable), 8.38 (s, 3H, D2O exchangeable), 8.27 (s, 1H), 8.06 (d, J= 7.9 Hz, 1H), 7.98 (d, J= 6.7 Hz, 1H), 7.87 (d, J= 7.7 Hz, 1H), 7.78 – 7.49 (m, 7H), 7.48 – 7.37 (m, 1H), 5.78 (s, 1H), 4.13 (d, j= 5.7 Hz, 2H), 2.72 (s, 2H), 1.14 (s, 1H), 0.56 (d, j= 7.7 Hz, 2H), 0.31 (d, J= 5.0 Hz, 2H); 19F NMR (282 MHz, DMSO-i¾) d -60.82 , -120.03; MS (ES+): MS (ES+) 563.3 (M+l), MS (ES-), 561.3 (M-l), 597.2 (M+Cl); Analysis calculated for C30H26F4N6O.2HCI. I .75H2O: C, 54.02; H, 4.76; Cl, 10.63; N, 12.60; Found: C, 54.12; H, 4.83; Cl, 10.10; N, 11.97.
Example 3 : Preparation of a Seed Crystal of Compound I*2

A solution of Compound I ( see Example 2) in methyl tert-butyl ether (MTBE) (1 equiv) is added to a solution of HC1 (aq) (2 equiv) in methanol (cold), followed by heating to about 30°C, and keeping it at about 30°C for not longer than 5 hours while stirring at about 115 rpm. Compound I bis(HCl) is collected by filtration and dried. The crystalline material obtained can be used as a seed for the crystallization protocol described in
Example 4. Example 4: Large-Scale Synthetic & Crystallization Protocol for Compound I*2(HC1 )

Compound I (free base) Compound I bis(HCI)
37% Aqueous hydrochloric acid (38.1 kg, 32.3 L, 2.14 equiv.) was charged to a clean and empty crystallization vessel, methanol (228.9 kg, 39.5 equiv.) was added, and the contents were cooled to -7 ± 3°C. A solution of Compound I free base (approx. 101.8 kg; 180.9 moles) in MTBE (approx. 1,300 L) was filtered through a polish filter into the crystallization vessel at temperature -5 ± 5°C. After rinse with MTBE, pre-weighed Compound I»2(HCl) seed crystals (1.39 kg, 0.012 equiv.; Example 3) were charged to the crystallization vessel via the manhole. The vessel content was heated to 30-33°C, and the agitation speed was set to 25-50 rpm. After confirmed crystallization, the slurry was agitated for another three to four hours. The product slurry was transferred to centrifuge and isolated by centrifugation. The product was washed with MTBE (585 L). After dry spinning the wet product, Compound I*2(HC1), it was discharged from the centrifuge, and the product was dried at < 40°C under vacuum in a cone drier. Product Compound I»2(HCl) yield: 100 kg; 157.4 mol; approx. 85%.
‘H NMR (300 MHz, DMSO-c/i,) data is shown in the following table:

19F NMR (282 MHz, DMSO- is) data is shown in the following table:

Compound I has two basic sites. The conjugate acid of the primary amine was calculated to have a pKa value of 8.89, and the conjugate acid of the secondary amine was calculated to have a pKa value of 7.86.
The XRPD pattern of Compound I»2(HCl) is shown in Fig. 1. Compound I»2(HCl) has characteristic peaks in its XRPD pattern at values of two theta (°2Q) of 5.28, 8.96, 14.27, 16.18, 19.79, 21.16, 22.01, 23.31, 24.64, and 30.31. TG-IR analysis indicated two, distinct weight loss regions: the first was completed by 125 °C while the second began at approximately 208 °C. IR analysis of the off gasses from this experiment detected only trace amounts of water at the initial weight loss while HC1 gas was detected at the 208°C event. No other solvents were detected in the sample. Thus, it was determined that Compound I*2(HC1) initially loses water when heated and, when heated to above 200°C, the salt begins to break apart and HC1 gas is evolved. The IR signal for all these events is very weak indicating that they are occurring over a range and not at a specified temperature. An exemplary TG-IR spectrum is shown in Fig. 2.
REFERENCES
1: Sohtome Y, Sodeoka M. Development of Chaetocin and S-Adenosylmethionine Analogues as Tools for Studying Protein Methylation. Chem Rec. 2018 Dec;18(12):1660-1671. doi: 10.1002/tcr.201800118. Epub 2018 Oct 16. Review. PubMed PMID: 30324709.
2: Bensussen A, Torres-Sosa C, Gonzalez RA, Díaz J. Dynamics of the Gene Regulatory Network of HIV-1 and the Role of Viral Non-coding RNAs on Latency Reversion. Front Physiol. 2018 Sep 28;9:1364. doi: 10.3389/fphys.2018.01364. eCollection 2018. PubMed PMID: 30323768; PubMed Central PMCID: PMC6172855.
////////berotralstat, Orladeyo, BIOCRYST, APPROVALS 2020, FDA 2020, ORPHAN DRUG, CX-7353, CX 7353,
NCc1cccc(c1)n2nc(cc2C(=O)Nc3cc(ccc3F)[C@H](NCC4CC4)c5cccc(c5)C#N)C(F)(F)F

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}