
Home » 0rphan drug status (Page 4)
Category Archives: 0rphan drug status
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lonapegsomatropin
FPTIPLSRLF DNAMLRAHRL HQLAFDTYQE FEEAYIPKEQ KYSFLQNPQT SLCFSESIPT
PSNREETQQK SNLELLRISL LLIQSWLEPV QFLRSVFANS LVYGASDSNV YDLLKDLEEG
IQTLMGRLED GSPRTGQIFK QTYSKFDTNS HNDDALLKNY GLLYCFRKDM DKVETFLRIV
QCRSVEGSCG F
(Disulfide bridge: 53-165, 182-189)

Lonapegsomatropin, ロナペグソマトロピン
FDA APPROVED, 25/8/21, Skytrofa, Treatment of growth hormone deficiency
To treat short stature due to inadequate secretion of endogenous growth hormone
1934255-39-6 CAS, UNII: OP35X9610Y
Molecular Formula, C1051-H1627-N269-O317-S9[-C2-H4-O]4n
ACP 001; ACP 011; lonapegsomatropin-tcgd; SKYTROFA; TransCon; TransCon growth hormone; TransCon hGH; TransCon PEG growth hormone; TransCon PEG hGH; TransCon PEG somatropin,
WHO 10598
PEPTIDE
Biologic License Application (BLA): 761177
Company: ACENDIS PHARMA ENDOCRINOLOGY DIV A/S
SKYTROFA is a human growth hormone indicated for the treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH) (1).
- OriginatorAscendis Pharma
- DeveloperAscendis Pharma; VISEN Pharmaceuticals
- ClassGrowth hormones; Hormonal replacements; Polyethylene glycols
- Mechanism of ActionSomatotropin receptor agonists
- Orphan Drug StatusYes – Somatotropin deficiency
- RegisteredSomatotropin deficiency
- 25 Aug 2021Registered for Somatotropin deficiency (In children, In infants) in USA (SC)
- 27 May 2021Ascendis Pharma expects European Commission decision on the Marketing Authorisation Application (MAA) for Somatotropin deficiency (In children, In infants, In neonates) in fourth quarter of 2021
- 27 May 2021Phase-III clinical trials in Somatotropin deficiency (In children, Treatment-naive) in Japan (SC)
Ascendis Pharma A/S Announces U.S. Food and Drug Administration Approval of SKYTROFA® (lonapegsomatropin-tcgd), the First Once-weekly Treatment for Pediatric Growth Hormone Deficiency
SKYTROFA, the first FDA approved treatment utilizing TransCon™ technology, is a long-acting prodrug of somatropin that releases the same somatropin used in daily therapies –
– Once weekly SKYTROFA demonstrated higher annualized height velocity (AHV) at week 52 compared to a daily growth hormone with similar safety and tolerability –
– Availability in the U.S. expected shortly supported by a full suite of patient support programs –
– Ascendis Pharma to host investor conference call today, Wednesday, August 25 at 4:30 p.m. E.T. –
COPENHAGEN, Denmark, Aug. 25, 2021 (GLOBE NEWSWIRE) — Ascendis Pharma A/S (Nasdaq: ASND), a biopharmaceutical company that utilizes its innovative TransCon technologies to potentially create new treatments that make a meaningful difference in patients’ lives, today announced that the U.S. Food and Drug Administration (FDA) has approved SKYTROFA (lonapegsomatropin-tcgd) for the treatment of pediatric patients one year and older who weigh at least 11.5 kg (25.4 lb) and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
As a once-weekly injection, SKYTROFA is the first FDA approved product that delivers somatropin (growth hormone) by sustained release over one week.
“Today’s approval represents an important new choice for children with GHD and their families, who will now have a once-weekly treatment option. In the pivotal head-to-head clinical trial, once-weekly SKYTROFA demonstrated higher annualized height velocity at week 52 compared to somatropini,” said Paul Thornton, M.B. B.Ch., MRCPI, a clinical investigator and pediatric endocrinologist in Fort Worth, Texas. “This once-weekly treatment could reduce treatment burden and potentially replace the daily somatropin therapies, which have been the standard of care for over 30 years.”
Growth hormone deficiency is a serious orphan disease characterized by short stature and metabolic complications. In GHD, the pituitary gland does not produce sufficient growth hormone, which is important not only for height but also for a child’s overall endocrine health and development.
The approval includes the new SKYTROFA® Auto-Injector and cartridges which, after first removed from a refrigerator, allow families to store the medicine at room temperature for up to six months. With a weekly injection, patients switching from injections every day can experience up to 86 percent fewer injection days per year.
“SKYTROFA is the first product using our innovative TransCon technology platform that we have developed from design phase through non-clinical and clinical development, manufacturing and device optimization, and out to the patients. It reflects our commitment and dedication to addressing unmet medical needs by developing a pipeline of highly differentiated proprietary products across multiple therapeutic areas,” said Jan Mikkelsen, Ascendis Pharma’s President and Chief Executive Officer. “We are grateful to the patients, caregivers, clinicians, clinical investigators, and our employees, who have all contributed to bringing this new treatment option to children in the U.S. with GHD.”
In connection with the commercialization of SKYTROFA, the company is committed to offering a full suite of patient support programs, including educating families on proper injection procedures for SKYTROFA as the first once-weekly treatment for children with GHD.
“It is wonderful that patients and their families now have the option of a once-weekly growth hormone therapy,” said Mary Andrews, Chief Executive Officer and co-founder of the MAGIC Foundation, a global leader in endocrine health, advocacy, education, and support. “GHD is often overlooked and undertreated in our children and managing it can be challenging for families. We are excited about this news as treating GHD is important, and children have a short time to grow.”
The FDA approval of SKYTROFA was based on results from the phase 3 heiGHt Trial, a 52-week, global, randomized, open-label, active-controlled, parallel-group trial that compared once-weekly SKYTROFA to daily somatropin (Genotropin®) in 161 treatment-naïve children with GHDii. The primary endpoint was, AHV at 52 weeks for weekly SKYTROFA and daily hGH treatment groups. Other endpoints included adverse events, injection-site reactions, incidence of anti-hGH antibodies, annualized height velocity, change in height SDS, proportion of subjects with IGF-1 SDS (0.0 to +2.0), PK/PD in subjects < 3 years, and preference for and satisfaction with SKYTROFA.
At week 52, the treatment difference in AHV was 0.9 cm/year (11.2 cm/year for SKYTROFA compared with 10.3 cm/year for daily somatropin) with a 95 percent confidence interval [0.2, 1.5] cm/year. The primary objective of non-inferiority in AHV was met for SKYTROFA in this trial and further demonstrated a higher AHV at week 52 for lonapegsomatropin compared to daily somatropin, with similar safety, in treatment-naïve children with GHD.
No serious adverse events or discontinuations related to SKYTROFA were reported. Most common adverse reactions (≥ 5%) in pediatric patients include: infection, viral (15%), pyrexia (15%), cough (11%), nausea and vomiting (11%), hemorrhage (7%), diarrhea (6%), abdominal pain (6%), and arthralgia and arthritis (6%)ii. In addition, both arms of the study reported low incidences of transient, non-neutralizing anti-hGH binding antibodies and no cases of persistent antibodies.
Conference Call and Webcast Information
| Date | Wednesday, August 25, 2021 |
| Time | 4:30 p.m. ET/1:30 p.m. Pacific Time |
| Dial In (U.S.) | 844-290-3904 |
| Dial In (International) | 574-990-1036 |
| Access Code | 8553236 |
A live webcast of the conference call will be available on the Investors and News section of the Ascendis Pharma website at www.ascendispharma.com. A webcast replay will be available on this website shortly after conclusion of the event for 30 days.
The Following Information is Intended for the U.S. Audience Only
INDICATION
SKYTROFA® is a human growth hormone indicated for the treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
IMPORTANT SAFETY INFORMATION
- SKYTROFA is contraindicated in patients with:
- Acute critical illness after open heart surgery, abdominal surgery or multiple accidental trauma, or if you have acute respiratory failure due to the risk of increased mortality with use of pharmacologic doses of somatropin.
- Hypersensitivity to somatropin or any of the excipients in SKYTROFA. Systemic hypersensitivity reactions have been reported with post-marketing use of somatropin products.
- Closed epiphyses for growth promotion.
- Active malignancy.
- Active proliferative or severe non-proliferative diabetic retinopathy.
- Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea or have severe respiratory impairment due to the risk of sudden death.
- Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic doses of somatropin. Safety of continuing SKYTROFA treatment in patients receiving replacement doses for the approved indication who concurrently develop these illnesses has not been established.
- Serious systemic hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with post-marketing use of somatropin products. Do not use SKYTROFA in patients with known hypersensitivity to somatropin or any of the excipients in SKYTROFA.
- There is an increased risk of malignancy progression with somatropin treatment in patients with active malignancy. Preexisting malignancy should be inactive with treatment completed prior to starting SKYTROFA. Discontinue SKYTROFA if there is evidence of recurrent activity.
- In childhood cancer survivors who were treated with radiation to the brain/head for their first neoplasm and who developed subsequent growth hormone deficiency (GHD) and were treated with somatropin, an increased risk of a second neoplasm has been reported. Intracranial tumors, in particular meningiomas, were the most common of these second neoplasms. Monitor all patients with a history of GHD secondary to an intracranial neoplasm routinely while on somatropin therapy for progression or recurrence of the tumor.
- Because children with certain rare genetic causes of short stature have an increased risk of developing malignancies, practitioners should thoroughly consider the risks and benefits of starting somatropin in these patients. If treatment with somatropin is initiated, carefully monitor these patients for development of neoplasms. Monitor patients on somatropin therapy carefully for increased growth, or potential malignant changes of preexisting nevi. Advise patients/caregivers to report marked changes in behavior, onset of headaches, vision disturbances and/or changes in skin pigmentation or changes in the appearance of preexisting nevi.
- Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses. New onset type 2 diabetes mellitus has been reported in patients taking somatropin. Undiagnosed impaired glucose tolerance and overt diabetes mellitus may be unmasked. Monitor glucose levels periodically in all patients receiving SKYTROFA. Adjust the doses of antihyperglycemic drugs as needed when SKYTROFA is initiated in patients.
- Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in a small number of patients treated with somatropin. Symptoms usually occurred within the first 8 weeks after the initiation of somatropin and resolved rapidly after cessation or reduction in dose in all reported cases. Fundoscopic exam should be performed before initiation of therapy and periodically thereafter. If somatropin-induced IH is diagnosed, restart treatment with SKYTROFA at a lower dose after IH-associated signs and symptoms have resolved.
- Fluid retention during somatropin therapy may occur and is usually transient and dose dependent.
- Patients receiving somatropin therapy who have or are at risk for pituitary hormone deficiency(s) may be at risk for reduced serum cortisol levels and/or unmasking of central (secondary) hypoadrenalism. Patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA therapy. Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism.
- Undiagnosed or untreated hypothyroidism may prevent response to SKYTROFA. In patients with GHD, central (secondary) hypothyroidism may first become evident or worsen during SKYTROFA treatment. Perform thyroid function tests periodically and consider thyroid hormone replacement.
- Slipped capital femoral epiphysis may occur more frequently in patients undergoing rapid growth. Evaluate pediatric patients with the onset of a limp or complaints of persistent hip or knee pain.
- Somatropin increases the growth rate and progression of existing scoliosis can occur in patients who experience rapid growth. Somatropin has not been shown to increase the occurrence of scoliosis. Monitor patients with a history of scoliosis for disease progression.
- Cases of pancreatitis have been reported in pediatric patients receiving somatropin. The risk may be greater in pediatric patients compared with adults. Consider pancreatitis in patients who develop persistent severe abdominal pain.
- When SKYTROFA is administered subcutaneously at the same site over a long period of time, lipoatrophy may result. Rotate injection sites when administering SKYTROFA to reduce this risk.
- There have been reports of fatalities after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. SKYTROFA is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome.
- Serum levels of inorganic phosphorus, alkaline phosphatase, and parathyroid hormone may increase after somatropin treatment.
- The most common adverse reactions (≥5%) in patients treated with SKYTROFA were: viral infection (15%), pyrexia (15%), cough (11%), nausea and vomiting (11%), hemorrhage (7%), diarrhea (6%), abdominal pain (6%), and arthralgia and arthritis (6%).
- SKYTROFA can interact with the following drugs:
- Glucocorticoids: SKYTROFA may reduce serum cortisol concentrations which may require an increase in the dose of glucocorticoids.
- Oral Estrogen: Oral estrogens may reduce the response to SKYTROFA. Higher doses of SKYTROFA may be required.
- Insulin and/or Other Hypoglycemic Agents: SKYTROFA may decrease insulin sensitivity. Patients with diabetes mellitus may require adjustment of insulin or hypoglycemic agents.
- Cytochrome P450-Metabolized Drugs: Somatropin may increase cytochrome P450 (CYP450)-mediated antipyrine clearance. Carefully monitor patients using drugs metabolized by CYP450 liver enzymes in combination with SKYTROFA.
You are encouraged to report side effects to FDA at (800) FDA-1088 or www.fda.gov/medwatch. You may also report side effects to Ascendis Pharma at 1-844-442-7236.
Please click here for full Prescribing Information for SKYTROFA.
About SKYTROFA® (lonapegsomatropin-tcgd)
SKYTROFA® is a once-weekly prodrug designed to deliver somatropin over a one-week period. The released somatropin has the same 191 amino acid sequence as daily somatropin.
SKYTROFA single-use, prefilled cartridges are available in nine dosage strengths, allowing for convenient dosing flexibility. They are designed for use only with the SKYTROFA® Auto-Injector and may be stored at room temperature for up to six months. The recommended dose of SKYTROFA for treatment-naïve patients and patients switching from daily somatropin is 0.24 mg/kg body weight, administered once weekly. The dose may be adjusted based on the child’s weight and insulin-like growth factor-1 (IGF-1) SDS.
SKYTROFA has been studied in over 300 children with GHD across the Phase 3 program which consists of the heiGHt Trial (for treatment-naïve patients), the fliGHt Trial (for treatment-experienced patients), and the enliGHten Trial (an ongoing long-term extension trial). Patients who completed the heiGHt Trial or the fliGHt Trial were able to continue into the enliGHten Trial and some have been on SKYTROFA for over four years.
SKYTROFA is being evaluated for pediatric GHD in Phase 3 trials in Japan and Greater China, including the People’s Republic of China, Hong Kong, Macau and Taiwan. Ascendis Pharma is also conducting the global Phase 3 foresiGHt Trial in adults with GHD. SKYTROFA has been granted orphan designation for GHD in both the U.S. and Europe.
About TransCon™ Technologies
TransCon refers to “transient conjugation.” The proprietary TransCon platform is an innovative technology to create new therapies that are designed to potentially optimize therapeutic effect, including efficacy, safety and dosing frequency. TransCon molecules have three components: an unmodified parent drug, an inert carrier that protects it, and a linker that temporarily binds the two. When bound, the carrier inactivates and shields the parent drug from clearance. When injected into the body, physiologic conditions (e.g., pH and temperature) initiate the release of the active, unmodified parent drug in a predictable manner. Because the parent drug is unmodified, its original mode of action is expected to be maintained. TransCon technology can be applied broadly to a protein, peptide or small molecule in multiple therapeutic areas, and can be used systemically or locally.
About Ascendis Pharma A/S
Ascendis Pharma is applying its innovative platform technology to build a leading, fully integrated biopharma company focused on making a meaningful difference in patients’ lives. Guided by its core values of patients, science and passion, the company utilizes its TransCon technologies to create new and potentially best-in-class therapies.
Ascendis Pharma currently has a pipeline of multiple independent endocrinology rare disease and oncology product candidates in development. The company continues to expand into additional therapeutic areas to address unmet patient needs.
Ascendis is headquartered in Copenhagen, Denmark, with additional facilities in Heidelberg and Berlin, Germany, in Palo Alto and Redwood City, California, and in Princeton, New Jersey.
Please visit www.ascendispharma.com (for global information) or www.ascendispharma.us (for U.S. information).

NEW DRUG APPROVALS
ONE TIME
$10.00
///////////Lonapegsomatropin, Skytrofa, APPROVALS 2021, FDA 2021, PEPTIDE, ロナペグソマトロピン , ACP 00, ACP 011, lonapegsomatropin-tcgd, TransCon, TransCon growth hormone, TransCon hGH, TransCon PEG growth hormone, TransCon PEG hGH, TransCon PEG somatropin, ORPHAN DRUG
Avalglucosidase alfa
QQGASRPGPR DAQAHPGRPR AVPTQCDVPP NSRFDCAPDK AITQEQCEAR GCCYIPAKQG
LQGAQMGQPW CFFPPSYPSY KLENLSSSEM GYTATLTRTT PTFFPKDILT LRLDVMMETE
NRLHFTIKDP ANRRYEVPLE TPRVHSRAPS PLYSVEFSEE PFGVIVHRQL DGRVLLNTTV
APLFFADQFL QLSTSLPSQY ITGLAEHLSP LMLSTSWTRI TLWNRDLAPT PGANLYGSHP
FYLALEDGGS AHGVFLLNSN AMDVVLQPSP ALSWRSTGGI LDVYIFLGPE PKSVVQQYLD
VVGYPFMPPY WGLGFHLCRW GYSSTAITRQ VVENMTRAHF PLDVQWNDLD YMDSRRDFTF
NKDGFRDFPA MVQELHQGGR RYMMIVDPAI SSSGPAGSYR PYDEGLRRGV FITNETGQPL
IGKVWPGSTA FPDFTNPTAL AWWEDMVAEF HDQVPFDGMW IDMNEPSNFI RGSEDGCPNN
ELENPPYVPG VVGGTLQAAT ICASSHQFLS THYNLHNLYG LTEAIASHRA LVKARGTRPF
VISRSTFAGH GRYAGHWTGD VWSSWEQLAS SVPEILQFNL LGVPLVGADV CGFLGNTSEE
LCVRWTQLGA FYPFMRNHNS LLSLPQEPYS FSEPAQQAMR KALTLRYALL PHLYTLFHQA
HVAGETVARP LFLEFPKDSS TWTVDHQLLW GEALLITPVL QAGKAEVTGY FPLGTWYDLQ
TVPIEALGSL PPPPAAPREP AIHSEGQWVT LPAPLDTINV HLRAGYIIPL QGPGLTTTES
RQQPMALAVA LTKGGEARGE LFWDDGESLE VLERGAYTQV IFLARNNTIV NELVRVTSEG
AGLQLQKVTV LGVATAPQQV LSNGVPVSNF TYSPDTKVLD ICVSLLMGEQ FLVSWC
(Disulfide bridge:26-53, 36-52, 47-71, 477-502, 591-602, 882-896)
Avalglucosidase alfa
アバルグルコシダーゼアルファ (遺伝子組換え)
Avalglucosidase alfa (USAN/INN);
Avalglucosidase alfa (genetical recombination) (JAN);
Avalglucosidase alfa-ngpt
To treat late-onset Pompe disease
| Formula | C4490H6818N1197O1299S32 |
|---|---|
| CAS | 1802558-87-7 |
| Mol weight | 99375.4984 |
FDA APPROVED Nexviazyme, 2021/8/6, Enzyme replacement therapy product
Treatment of Pompe disease
Biologic License Application (BLA): 761194
Company: GENZYME CORP
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-pompe-diseaseFor Immediate Release:August 06, 2021
Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
Patients with Pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure. Normally, glycogen—the stored form of glucose—breaks down to release glucose into the bloodstream to be used as fuel for the cells.
“Pompe disease is a rare genetic disease that causes premature death and has a debilitating effect on people’s lives,” said Janet Maynard, M.D., deputy director of the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine in the FDA’s Center for Drug Evaluation and Research. “Today’s approval brings patients with Pompe disease another enzyme replacement therapy option for this rare disease. The FDA will continue to work with stakeholders to advance the development of additional new, effective and safe therapies for rare diseases, including Pompe disease.”
Nexviazyme, an enzyme replacement therapy, is an intravenous medication that helps reduce glycogen accumulation. The effectiveness of Nexviazyme for the treatment of Pompe disease was demonstrated in a study of 100 patients who were randomized to take Nexviazyme or another FDA-approved enzyme replacement therapy for Pompe disease. Treatment with Nexviazyme improved lung function similar to the improvement seen with the other therapy.
The most common side effects included headache, fatigue, diarrhea, nausea, joint pain (arthralgia), dizziness, muscle pain (myalgia), itching (pruritus), vomiting, difficulty breathing (dyspnea), skin redness (erythema), feeling of “pins and needles” (paresthesia) and skin welts (urticaria). Serious reactions included hypersensitivity reactions like anaphylaxis and infusion-associated reactions, including respiratory distress, chills and raised body temperature (pyrexia). Patients susceptible to fluid volume overload or with compromised cardiac or respiratory function may be at risk for serious acute cardiorespiratory failure.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Nexviazyme also received an orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Nexviazyme to Genzyme Corporation.
###
NEW DRUG APPROVALS
one time
$10.00
FDA grants priority review for avalglucosidase alfa, a potential new therapy for Pompe disease
- The FDA decision date for avalglucosidase alfa, an investigational enzyme replacement therapy, is set for May 18, 2021
- Regulatory submission based on positive data from two trials in patients with late-onset and infantile-onset Pompe disease, respectively
- Avalglucosidase alfa received FDA Breakthrough Therapy and Fast Track designations for the treatment of people with Pompe Disease
- Pompe disease, a rare degenerative muscle disorder, affects approximately 3,500 people in the U.S.
- Milestone reinforces 20+year commitment to Pompe disease community
PARIS – November 18, 2020 – The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application (BLA) for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease (acid α-glucosidase deficiency). The target action date for the FDA decision is May 18, 2021.
Avalglucosidase alfa is an investigational enzyme replacement therapy designed to improve the delivery of acid alpha-glucosidase (GAA) enzyme to muscle cells, and if approved, would offer a potential new standard of care for patients with Pompe disease.
In October, the European Medicines Agency accepted for review the Marketing Authorization Application for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease. The Medicines and Healthcare Products Regulatory Agency in the UK has granted Promising Innovative Medicine designation for avalglucosidase alfa.
“The hallmarks of Pompe disease are the relentless and debilitating deterioration of the muscles, which causes decreased respiratory function and mobility,” said Karin Knobe, Head of Development for Rare Diseases and Rare Blood Disorders at Sanofi. “Avalglucosidase alfa is specifically designed to deliver more GAA enzyme into the lysosomes of the muscle cells. We have been greatly encouraged by positive clinical trial results in patients with late-onset and infantile-onset Pompe disease.”
Pompe disease is a rare, degenerative muscle disorder that can impact an individual’s ability to move and breathe. It affects an estimated 3,500 people in the U.S. and can manifest at any age from infancy to late adulthood.i
The BLA is based on positive data from two trials:
- Pivotal Phase 3, double-blind, global comparator-controlled trial (COMET), which evaluated the safety and efficacy of avalglucosidase alfa compared to alglucosidase alfa (standard of care) in patients with late-onset Pompe disease. Results from this trial were presented during a Sanofi-hosted virtual scientific session in June 2020 and in October 2020 at World Muscle Society and the American Association of Neuromuscular and Electrodiagnostic Medicine.
- The Phase 2 (mini-COMET) trial evaluated the safety and exploratory efficacy of avalglucosidase alfa in patients with infantile-onset Pompe disease previously treated with alglucosidase alfa. Results from this trial were presented at the WORLDSymposium, in February 2020.
Delivery of GAA to Clear Glycogen
Pompe disease is caused by a genetic deficiency or dysfunction of the lysosomal enzyme GAA, which results in build-up of complex sugars (glycogen) in muscle cells throughout the body. The accumulation of glycogen leads to irreversible damage to the muscles, including respiratory muscles and the diaphragm muscle supporting lung function, and other skeletal muscles that affect mobility.
To reduce the glycogen accumulation caused by Pompe disease, the GAA enzyme must be delivered into the lysosomes within muscle cells. Research led by Sanofi has focused on ways to enhance the delivery of GAA into the lysosomes of muscle cells by targeting the mannose-6-phosphate (M6P) receptor that plays a key role in the transport of GAA.
Avalglucosidase alfa is designed with approximately 15-fold increase in M6P content, compared to standard of care alglucosidase alfa, and aims to help improve cellular enzyme uptake and enhance glycogen clearance in target tissues.ii The clinical relevance of this difference has not been confirmed.
Avalglucosidase alfa is currently under clinical investigation and its safety and efficacy have not been evaluated by any regulatory authority worldwide.
| About Sanofi Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, Empowering Life |
/////////Avalglucosidase alfa, FDA 2021, Nexviazyme, APPROVALS 2021, PEPTIDE, Enzyme replacement therapy , Pompe disease, アバルグルコシダーゼアルファ (遺伝子組換え), Fast Track, Priority Review, Breakthrough Therapy, orphan drug designation, genzyme, sanofi
MIRDAMETINIB
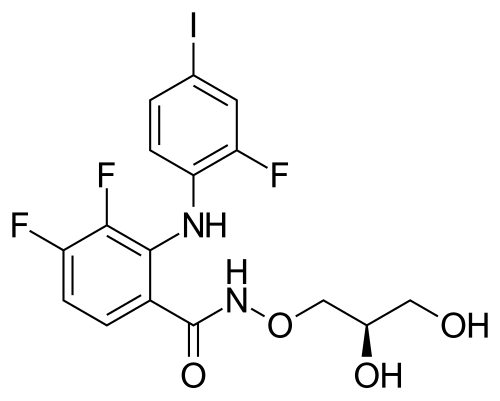

MIRDAMETINIB
391210-10-9
Chemical Formula: C16H14F3IN2O4
Molecular Weight: 482.19
PD0325901; PD 0325901; PD-325901; mirdametinib
FDA APPROVED 2/11/2025, Gomekli, To treat neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection
IUPAC/Chemical Name: (R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-((2-fluoro-4-iodophenyl)amino)benzamide
SpringWorks Therapeutics (a spin out of Pfizer ) is developing mirdametinib, a second-generation, non-ATP competitive, allosteric MEK1 and MEK2 inhibitor derived from CI-1040, for treating type 1 neurofibromatosis (NF1) and advanced solid tumors. In June 2021, a phase I/II trial was initiated in patients with low grade glioma.
- OriginatorPfizer
- DeveloperAstraZeneca; BeiGene; BIOENSIS; Pfizer; SpringWorks Therapeutics; St. Jude Childrens Research Hospital; University of Oxford
- ClassAniline compounds; Anti-inflammatories; Antineoplastics; Benzamides; Immunotherapies; Small molecules
- Mechanism of ActionMAP kinase kinase 1 inhibitors; MAP kinase kinase 2 inhibitors
- Orphan Drug StatusYes – Neurofibromatosis 1
- Phase IINeurofibromatosis 1
- Phase I/IIGlioma
- Phase ISolid tumours
- PreclinicalChronic obstructive pulmonary disease
- No development reportedCervical cancer
- DiscontinuedBreast cancer; Cancer; Colorectal cancer; Malignant melanoma; Non-small cell lung cancer
- 22 Jul 2021SpringWorks Therapeutics receives patent allowance for mirdametinib from the US Patent and Trademark Office for the treatment of Neurofibromatosis type 1-associated plexiform neurofibromas
- 16 Jun 2021SpringWorks Therapeutics and St. Jude Children’s Research Hospital agree to develop mirdametinib in USA for glioma
- 15 Jun 2021Efficacy and safety data from the phase IIb RENEU trial for Neurofibromatosis type 1-associated plexiform neurofibromas released by SpringWorks Therapeutics
Mirdametinib, sold under the brand name Gomekli, is a medication used for the treatment of people with neurofibromatosis type 1.[1] Mirdametinib is a kinase inhibitor.[1][2] It is taken by mouth.[1]
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib was approved for medical use in the United States in February 2025.[1][3]
SCHEME
SIDE CHAIN

MAIN
Medical uses
Mirdametinib is indicated for the treatment of people with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection.[1]
Adverse effects
The most common adverse reactions in adults include rash, diarrhea, nausea, musculoskeletal pain, vomiting, and fatigue.[3] The most common grade 3 or 4 laboratory abnormalities include increased creatine phosphokinase.[3] The most common adverse reactions in children include rash, diarrhea, musculoskeletal pain, abdominal pain, vomiting, headache, paronychia, left ventricular dysfunction, and nausea.[3] The most common grade 3 or 4 laboratory abnormalities include decreased neutrophil count and increased creatine phosphokinase.[3]
Mirdametinib can cause left ventricular dysfunction and ocular toxicity including retinal vein occlusion, retinal pigment epithelial detachment, and blurred vision.[3]
History
The efficacy of mirdametinib was evaluated in ReNeu (NCT03962543), a multicenter, single-arm trial in 114 participants aged two years of age and older (58 adults, 56 pediatric participants) with symptomatic, inoperable NF1-associated plexiform neurofibromas causing significant morbidity.[3] An inoperable plexiform neurofibromas was defined as a plexiform neurofibromas that could not be completely surgically removed without risk for substantial morbidity due to encasement or close proximity to vital structures, invasiveness, or high vascularity.[3]
The US Food and Drug Administration (FDA) granted the application for mirdametinib priority review, fast track, and orphan drug designations along with a priority review voucher.[3]
Society and culture
Legal status
Mirdametinib was approved for medical use in the United States in February 2025.[3][4][5]
PATENT
US-11066358
On July 20, 2021, SpringWorks Therapeutics announced that the United States Patent and Trademark Office (USPTO) has issued US11066358 , directed to mirdametinib , the Company’s product candidate in development for several oncology indications, including as a monotherapy for patients with neurofibromatosis type 1-associated plexiform neurofibromas (NF1-PN) and was assigned to Warner-Lambert Company (a subsidiary of Pfizer ).This patent was granted on July 20, 2021, and expires on Feb 17, 2041. Novel crystalline forms of mirdametinib and compositions comprising them are claimed.
| N—((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (“mirdametinib”, or “PD-0325901”) is a small molecule drug which has been designed to inhibit mitogen-activated protein kinase kinase 1 (“MEK1”) and mitogen-activated protein kinase kinase 2 (“MEK2”). MEK1 and MEK2 are proteins that play key roles in the mitogen-activated protein kinase (“MAPK”) signaling pathway. The MAPK pathway is critical for cell survival and proliferation, and overactivation of this pathway has been shown to lead to tumor development and growth. Mirdametinib is a highly potent and specific allosteric non-ATP-competitive inhibitor of MEK1 and MEK2. By virtue of its mechanism of action, mirdametinib leads to significantly inhibited phosphorylation of the extracellular regulated MAP kinases ERK1 and ERK2, thereby leading to impaired growth of tumor cells both in vitro and in vivo. In addition, evidence indicates that inflammatory cytokine-induced increases in MEK/ERK activity contribute to the inflammation, pain, and tissue destruction associated with rheumatoid arthritis and other inflammatory diseases. |
Example 1: Production of Essentially Pure Form IV
Lab Scale Production of Essentially Pure Form IV
| All reactions were performed in toluene other than otherwise stated. Triflic anhydride gave the best yield. |
[TABLE-US-00002]TABLE 1 Coupling Agents for Step 1Entry No.Coupling AgentYieldNotes 1Mesyl Chloridedid not react 2Benzyl chloride27Had to heat 70° C. for 166 hr34-fluorobenzensulfonylchloride27Ran 93 hrs. at 70° C.44-chlorobenzensulfonylchloride35Complete after 68 hrs. 50° C.5Tosyl Chloride36Had to heat to 70° C. for 164 hrs6Benzyl chloride52study solvent effects: DMF, DMSO, NMP – all similar DMSO fastest all complete after 110 hrs., heated to 70° C. after 66 hrs.7Triflic anhydride91Cooled to −74° C. |
| [TABLE-US-00004]TABLE 3 Yields for base deprotection ReagentYield* Methyl hydrazine85-95% Anhydrous NH3 (sparged)78-90% Anhydrous NH3 (50 psi)80-92% Aqueous NH390-97% *from PD-0333760 |
Step 2: Fluoride Displacement
Pilot Plant Preparation of Essentially Pure Form IV
Step 1: Preparation of “Side Chain”, PD-0337792
Step 2: Preparation of PD-0315209
Step 3: Preparation of PD-0325901
Polymorph Transformation
| 21.4 kg PD-0315209, 9.7 kg CDI (1.05 equiv.), 91 kg solution of 9.7% PD-0337792 in Toluene (1.1 equiv.) were used and resulted in 12.74 kg of PD-0325901 (assay 99.4%, 100% Form IV, Yield 48%). |
PATENT
WO2006134469 , claiming methods of preparing MEK inhibitor, assigned to Warner-Lambert Co .
https://patents.google.com/patent/WO2006134469A1/enThe compound Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide represented by formula 1

i is a highly specific non-ATP-competitive inhibitor of MEK1 and MEK2. The compound of formula ± (Compound I) is also known as the compound PD 0325901. Compound I is disclosed in WO 02/06213; WO 04/045617; WO 2005/040098; EP 1262176; U.S. Patent Application Pub. No. 2003/0055095 A1 ; U.S. Patent Application Pub. No. 2004/0054172 A1; U.S. Patent Application Pub. No. 2004/0147478 A1 ; and U.S. Patent Application No. 10/969,681, the disclosures of which are incorporated herein by reference in their entireties.Numerous mitogen-activated protein kinase (MAPK) signaling cascades are involved in controlling cellular processes including proliferation, differentiation, apoptosis, and stress responses. Each MAPK module consists of 3 cytoplasmic kinases: a mitogen-activated protein kinase (MAPK), a mitogen-activated protein kinase kinase (MAPKK), and a mitogen-activated protein kinase kinase kinase (MAPKKK). MEK occupies a strategic downstream position in this intracellular signaling cascade catalyzing the phosphorylation of its MAP kinase substrates, ERK1 and ERK2. Anderson et al. “Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase.” Nature 1990, v.343, pp. 651-653. In the ERK pathway, MAPKK corresponds with MEK (MAP kinase ERK Kinase) and the MAPK corresponds with ERK (Extracellular Regulated Kinase). No substrates for MEK have been identified other than ERK1 and ERK2. Seger et al. “Purification and characterization of mitogen-activated protein kinase activator(s) from epidermal growth factor-stimulated A431 cells.” J. Biol. Chem., 1992, v. 267, pp. 14373-14381. This tight selectivity in addition to the unique ability to act as a dual-specificity kinase is consistent with MEK’s central role in integration of signals into the MAPK pathway. The RAF-MEK-ERK pathway mediates proliferative and anti-apoptotic signaling from growth factors and oncogenic factors such as Ras and Raf mutant phenotypes that promote tumor growth, progression, and metastasis. By virtue of its central role in mediating the transmission of growth- promoting signals from multiple growth factor receptors, the Ras-MAP kinase cascade provides molecular targets with potentially broad therapeutic applications.One method of synthesizing Compound I is disclosed in the above-referenced WO 02/06213 andU.S. Patent Application Pub. No. 2004/0054172 A1. This method begins with the reaction of 2-fluoro-4- iodo-phenylamine and 2,3,4-trifluoro-benzoic acid in the presence of an organic base, such as lithium diisopropylamide, to form 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzoic acid, which is then reacted with (R)-0-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of a peptide coupling agent (e.g., diphenylphosphinic chloride) and a tertiary amine base (e.g., diisopropylethylamine). The resulting product is hydrolyzed under standard acidic hydrolysis conditions (e.g., p-TsOH in MeOH) to provide Compound 1. (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine is prepared by reaction of [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol with N-hydroxyphthalimide in the presence of Ph3P and diethyl azodicarboxylate.Another method of synthesizing Compound I, which is disclosed in the above-referenced U.S.Patent Application No. 10/969,681, comprises reaction of 3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzoic acid with (R)-O-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-hydroxylamine in the presence of N1N1– carbonyldiimidazole. The resulting product is hydrolyzed with aqueous acid and crystallized to provide polymorphic form IV of Compound I.Although the described methods are effective synthetic routes for small-scale synthesis of Compound I, there remains a need in the art for new synthetic routes that are safe, efficient and cost effective when carried out on a commercial scale.The present invention provides a new synthetic route including Steps I through Step III to the MEK inhibitor Λ/-[(R)-2,3-dihydroxy-propoxy]-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I).Step I: Preparation of 0-{r(4RV2.2-dimethyl-1.3-dioxolan-4-ynmethyl}hydroxylanπine (6) The method of the present invention comprises a novel Step I of preparing of 0-{[(4R)-2,2- dimethyl-1 ,3-dioxolan-4-yl]methyl}hydroxylamine (6) from [(4S)-2,2-dimethyl-1 ,3-dioxoIan-4-yl]methanol (1) through the formation of [(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methyl trifluoromethanesulfonate (3) and its coupling with N-hydroxyphthalimide (4) to afford 2-{[(4R)-2,2-dimethyl-1 ,3-dioxolan-4-yl]methoxy}-1 H- isoindole-1 ,3(2H)-dione (5), which is subsequently de-protected to give 6 as shown in Scheme 1.Scheme 1



The reaction of compound (1) with trifluoromethanesulfonic anhydride (2) is carried out in the presence of a non-nucleophilic base, such as, for example, a tertiary organic amine, in an aprotic solvent at a temperature of from -5O0C to 50C, preferably, at a temperature less than -150C, to form triflate (3). A preferred tertiary organic amine is triethylamine, and a preferred solvent is toluene. Treatment of triflate (3) with N-hydroxyphthalimide (4) furnishes phthalimide (5), which can be isolated if desired. However, in order to minimize processing time and increase overall yield, 0-{[(4R)- 2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) can be prepared in a one-pot process with no phthalimide (S) isolation. Cleavage of the phthalimide function could be achieved by methods known in the art, for example, by hydrazinolysis. However, the use of less hazardous aqueous or anhydrous ammonia instead of methyl hydrazine (CH3NHNH2) is preferred.Step II: Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) As shown in Scheme 2, Step Il of the method of the present invention provides 3,4-difluoro-2-(2- fluoro-4-iodophenylamino)-benzoic acid (9).Scheme 2

Preparation of compound (9) can be carried out by reacting compound (7), wherein X is halogen, or O-SC^R^ or 0-P(3O)(OR^, wherein R^ is alkyl or aryl, with compound (8) optionally in a solvent, and in the presence of from about 1 mol equivalent to about 10 mol equivalents of at least one base, wherein the base is selected from: a Group I metal cation hydride or a Group 2 metal cation hydride, including lithium hydride, sodium hydride, potassium hydride, and calcium hydride, a Group I metal cation dialkylamide or a Group 2 metal cation dialkylamide, including lithium diisopropylamide, a Group I metal cation amide or a Group 2 metal cation amide, including lithium amide, sodium amide, potassium amide, a Group I metal cation alkoxide or a Group 2 metal cation alkoxide, including sodium ethoxide, potassium terf-butoxide, and magnesium ethoxide, and a Group I metal cation hexamethyldisilazide, including lithium hexamethyldisilazide; for a time, and at a temperature, sufficient to yield compound (9).Preferably, preparation of compound (9) is carried out by reacting compound (7), wherein X is halogen, more preferably, X is fluorine, in an aprotic solvent with compound (8) in the presence of from about 3 mol equivalents to about 5 mol equivalents of a Group I metal cation amide at a temperature of from 2O C to 55°C, more preferably, at a temperature from 45°C to 55°C. A catalytic amount of Group I metal cation dialkylamide can be added if necessary. A preferred Group I metal cation amide is lithium amide, a preferred Group I metal cation dialkylamide is lithium diisopropylamide, and a preferred solvent is tetrahydrofuran. Preferably, the reaction is performed by adding a small amount of compound (7) and compound (8) to lithium amide in tetrahydrofuran followed by slow continuous addition of the remaining portion. This procedure minimizes the risk of reactor over-pressurization due to gas side product (ammonia) generation.Step III: Preparation of N-((RV2.3-dihydroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I)Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using a carboxylic acid activating reagent such as, for example, COCI2, S(O)C^, S(O)2Cl2, P(O)Cl3, triphenylphosphine/diethylazodicarboxylate, diphenylphosphinic chloride, N, N’-dicyclohexylcarbodiimide, (benzotriazol-1 -yloxy)tripyrolidinophosphonium hexafluorophosphate, (benzotriazol-1 – yloxy)tris(dimethylamino)phosphonium hexafluorophosphate, N-ethyl-N’-(3- dimethylaminopropyl)carbodiimide hydrochloride, or 1,1′-carbonyldiimidazole (CDI).A preferred carboxylic acid activating reagent is 1,1′-carbonyldimidazole (CDI) shown in Scheme 3. Preparation of the desirable polymorphic Form IV of Compound I using CDI is described in the above- referenced U.S. Patent Application No. 10/969,681.Scheme 3

10

10 11 Compound IIn according to the present invention, the method was modified to include the advantageous procedure for product purification and isolation, which procedure is performed in single-phase systems such as, for example, toluene/acetonitrile for the first isolation/crystallization and ethanol/toluene for the second recrystallization. Water addition, implemented in the previous procedure, was omitted to avoid the two-phase crystallization from the immiscible water-toluene system that caused inconsistent product purity. The one-phase procedure of the present invention provides consistent control and removal of un- reacted starting material and side products. Alternatively, Compound I can be obtained by coupling 0-{[(4R)-2,2-dimethyl-1,3-dioxolan-4- yl]methyl}hydroxylamine (6) with 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) using thionyl chloride (SOCI2) as shown in Scheme 4.Scheme 4


Compound IExamplesThe reagents and conditions of the reactions described herein are merely illustrative of the wide variety of starting materials, their amounts and conditions which may be suitably employed in the present invention as would be appreciated by those skilled in the art, and are not intended to be limiting in any way.HPLC (Conditions A): 10 μL injection volume onto Agilent Zorbax RX-C18 150 mm x 4.6 mm x 3.5 μm column at 30°C column temperature, 1.0 mL/min flow rate and detection at 246 nm. Mobile phase A (v/v): 25 mM Acetate Buffer, pH 6.0; Mobile phase B (v/v): Acetonitrile, and Linear Gradient Table:

Sample Preparation: Dilute 100 μL reaction mixture to 10 mL with acetonitrile. Mix in a vial 200 μL of this sample solution with 300 μL carbonate buffer pH 10.0 and 300 μL solution of 2-mercaptopyridine in acetonitrile (18 mM), heat the vial for 10 minutes at 500C and dilute to 1:1 ratio in mobile phase A.GC (Conditions B): 1 μL injection onto an RTX-5 column (30 m x 0.25 mm x 0.25 μm) with initial oven temperature of 120°C for 2 min. to final temperature of 250°C in 15°C/minute ramping and a final time of 2.33 min; Flow rate: 1 mL/min.HPLC (Conditions C): 5 μL injection onto Phenomenex Luna C18(2) 150 mm x 4.6 mm x 3μm column ; flow rate : 1.0 mL/min; detection at 225 nm; mobile phase A: 95/5 v/v Water/Acetonitrile with 0.1% Trifluoroacetic acid (TFA), mobile phase B: 5/95 v/v Water/Acetonitriie with 0.1% TFA; Linear Gradient Table:

Sample preparation: Dilute 1 ml_ reaction mixture to 100 mL with acetonitrile and dilute 1 mL of this solution to 10 mL with 50:50 Water/Acetonitrile.HPLC (Conditions D): 5 μL injection onto Waters SymmetryShield RP 18, 150 mm x 4.6 mm x 3.5 μm column; flow rate: 1.0 mL/min; detection at 235 nm; mobile phase A: 25 mM Acetate Buffer adjusted to pH 5.5, mobile phase B: Acetonitrile; Linear Gradient Table:

Sample preparation: Dilute 40 μL of reaction mixture in 20 mL acetonitrile.HPLC (Conditions E): 10 μL sample injection onto YMC ODS-AQ 5 μm, 250 mm x 4.6 mm column; flow rate: 1.0 ml_/min; detection at 280 nm; temperature 30°C; mobile phase : 75/25 v/v Acetonitrile/Water with 0.1% Formic acid.Sample preparation: Quench reaction mixture sample with dipropylamine and stir for about 5 minutes before further dilution with mobile phase.DSC measurement was performed using a Mettler-Toledo DSC 822, temperature range 25° to 150°C with 5°C/min heating rate in a 40 μL aluminum pan. Experimental Conditions for Powder X-Rav Diffraction (XRD):A Rigaku Miniflex+ X-ray diffractometer was used for the acquisition of the powder XRD patterns. The instrument operates using the Cu Ka1 emission with a nickel filter at 1.50451 units. The major instrumental parameters are set or fixed at:X-ray: Cu / 30 kV (fixed) / 15 mA (fixed)Divergence Slit: Variable Scattering Slit: 4.2° (fixed) Receiving Slit: 0.3 mm (fixed) Scan Mode: FT Preset Time: 2.0 s Scan Width: 0.050° Scan Axis: 2Theta/Theta Scan Range: 3.000° to 40.000°Jade Software Version: 5.0.36(SP1) 01/05/01 (Materials Data, Inc.) Rigaku Software: Rigaku Standard Measurement for Windows 3.1 Version 3.6(1994-1995) Example 1. Preparation of 0-ffl4R)-2.2-dimethyl-1.3-dioxolan-4-vπmethyl}hvdroxylamine (6)A solution containing [(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol (1) (13.54 ml_, 0.109 mol) (DAISO Co., Ltd., CAS# 22323-82-6) and triethylamine (18.2 ml_, 0.131 mol) in 115 mL toluene was cooled to -15 C, then trifluoromethanesulfonic anhydride (2) (18.34 mL, 30.75 g, 0.109 mol) (Aldrich, Catalog # 17,617-6 ) was added drop wise while maintaining the temperature at less than -15°C. The mixture was then stirred for 2 hours, and transferred to a separate flask containing a mixture (slurry) of N- hydroxyphthalimide (4) (18.99 g, 0.116 mol) (Aldrich, Catalog # H5.370-4) and 18.2 mL (0.13 mol) triethylamine in 95 mL toluene. The resulting mixture was warmed to 20-25°C and stirred for at least 5 hours or until reaction completion (determined by HPLC (Conditions A)). Water (93 mL) was then added to quench the reaction mixture, the phases were separated, and the bottom aqueous layer was discarded. The water quench was repeated two more times resulting in a pale yellow organic layer. The organic layer was heated to 35 C and treated with 36.7 mL ammonium hydroxide solution (contains about 28-29% wt/wt ammonia). The mixture was stirred for at least 12 hours or until the reaction was deemed complete as determined by GC (Conditions B). The water was then removed under reduced pressure by co- distilling it with toluene to about half of the original volume at temperatures around 35-45 C. Toluene (170 mL) was added to the concentrated solution and the distillation was repeated. A sample was drawn for water content determination by Karl Fisher method (using EM Science Aquastar AQV-2000 Titrator with a sample injected to a pot containing methanol and salicylic acid). The distillation was repeated ifl water content was more than 0.1%. The concentrated solution was filtered to remove the white solid side product, and the filtrate was stored as 112mL (98 g) product solution containing 9.7% w/w compound 6 in toluene. This solution was ready for use in the final coupling step (Example 3). Overall chemical yield was 59%. A small sample was evaporated to yield a sample for NMR identification.1H NMR (400 MHz, CDCI3): δ 5.5 (bs, 2H), 4.35 (m, 1H), 4.07 (dd, 1H), 3.77 (m, 2H), 3.69 (dd, 1H), 1.44 (s, 3H), 1.37 (s, 3H).Example 2. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9)A solution of 2-fluoro-4-iodoaniline (8) (16.4 g, 0.069 mol) (Aldrich, Catalog # 30,660-6) and 2,3,4- trifluorobenzoic acid (7) (11.98 g, 0.068 mol) (Aldrich, Cat # 33,382-4) in 38 mL tetrahydrofuran (THF) was prepared and a portion (about 5%) of this solution was added to a stirring slurry of lithium amide (5 g, 0.22 mol) in 40 mL THF at 50-55 C. After about 15-30 min. an exotherm followed by gas release and color change are observed. The remaining portion of the (8) and (7) solution was added slowly over 1-2 hr while maintaining temperatures within 45-55°C. The mixture was stirred until the reaction was deemed complete (by HPLC (Conditions C). The final mixture was then cooled to 20-25°C and transferred to another reactor containing 6 N hydrochloric acid (47 mL) followed by 25 mL acetonitrile, stirred, and the bottom aqueous phase was discarded after treatment with 40 mL 50% sodium hydroxide solution. The organic phase was concentrated under reduced pressure and 57 mL acetone was added. The mixture was heated to 50°C, stirred, and added with 25 mL warm (40-50°C) water and cooled to 25-30°C to allow crystallization to occur (within 1-4 hours). Once the crystallization occurred, the mixture was further cooled to 0 to -5°C and stirred for about 2 hours. The solid product was filtered and the wet cake was dried in vacuum oven at about 55°C. Overall chemical yield was 21.4 g, 80%. 1H NMR (400 MHz, (CD3)2SO): δ 13.74 (bs, 1H), 9.15 (m, 1 H), 7.80 (dd, 1H), 7.62 (d, 1H), 7.41 (d, 1H), 7.10 (q, 1H), 6.81 (m, 1H).Example 2B. Preparation of 3.4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) by the solid addition of lithium amide methodTo a stirring solution of 2,3,4-trifluorobenzoic acid (13) (5.0 g, 28.4 mmol) and 2-fluoro-4- iodoaniline (14) (6.73 g, 28.4 mmol) in MeCN (100 mL), under N2 atmosphere was added lithium amide (2.61 g, 113.6 mmol) in small portions. The reaction mixture was heated to reflux for 45 minutes, cooled to ambient temperature and quenched with 1 N HCI and then water. The yellowish white precipitate was filtered, washed with water. The solid was triturated in CH2CI2 (30 mL) for 1h, filtered and dried in a vacuum oven at 45°C for 14 hours to give 8.Og (72%) of compound (9) as an off-white solid, mp 201.5-203 °C.Example 3. Preparation of N-((R)-2.3-dihvdroxypropoxy)-3.4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound \)3,4-Difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (20 g, 0.051 mol) in 100 mL acetonitrile was treated with 1,1′-carbonyldiimidazole (CDI) (8.66 g, 0.053 mol) (Aldrich, Cat # 11,553-3) and stirred for about 2 hours at 20-25°C until the reaction was deemed complete by HPLC (Conditions D). 94 mL (84.9 g) of 9.7% w/w solution of O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) in toluene was then added and stirred for about 4 hours or until the reaction was deemed complete by HPLC (Conditions D). To this mixture was added 66 mL of 5.6 % hydrochloric acid solution, and after stirring, the bottom aqueous phase was discarded. Again 66 mL of 5.6 % hydrochloric acid solution was added to the organic phase and stirred at 20-25°C for 12-18 hours or until the reaction was deemed complete by HPLC (Conditions D). The bottom layer was then discarded and the remaining organic layer was concentrated under reduced pressure to remove about 10-20% solvent, and the volume was adjusted to about 9-11 mL/g with toluene (80 mL). Crude product was then crystallized at 10-15°C. The slurry was allowed to stir for about 2 hours and the crude solid product was filtered, and dried. The dried crude product was recharged to the reactor and dissolved into 150 mL of 5% v/v ethanol/toluene mixture at 55- 67°C. The solution was then clarified at this temperature through filter (line filter) to remove any remaining particulate matter. The solution was then cooled slowly to 5°C to crystallize and stirred for at least 2 h, filtered and dried. The dried solid product was redissolved in EtOH (60 mL) at 35°C, and product was precipitated out by adding water (300 mL) at 35°C followed by cooling to 200C. The slurry was stirred for at least 2 hours to transform the crystals to the desired polymorphic Form IV as determined by DSC and Powder X-ray Diffraction pattern (PXRD). The slurry was filtered and dried under vacuum oven at 70- 90°C to yield the final N-((R)-2,3-dihydroxypropoxy)-3,4-difluoro-2-(2-fluoro-4-iodo-phenylamino)- benzamide (Compound I) product. Overall chemical yield was 13 g, 53%. Melting point (DSC): 112+1° C. Appearance: White to off-white crystals.Shown in Figure 1, PXRD conforms to polymorphic crystal Form IV disclosed in the above mentioned U.S. Patent Application No. 10/969,681 1H NMR (400 MHz, (CD3)2SO): δ 11.89 (bs, 1H), 8.71 (bs, 1H), 7.57 (d, 1H), 7.37 (m, 2H), 7.20 (q, 1H), 6.67 (m, 1H), 4.84 (bs, 1H), 4.60 (m, 1H), 3.87 (m, 1 H), 3.7 (m, 2H), 3.34 (m, 2H).Example 4. Preparation of N-((R)-2.3-dihydroxypropoxyV3.4-difluoro-2-(2-fluoro-4-iodo-phenylanrιinoV benzamide (Compound \)To a stirring solution of 3,4-difluoro-2-(2-fluoro-4-iodophenylamino)-benzoic acid (9) (120 g, 0.30 mol) in a mixture of 1 mL N,N-dimethylformamide and 1000 mL toluene was added thionyl chloride (55 g, 0.462 mol). The mixture was heated to 50-65 C and stirred for 2 hours or until reaction completion as determined by HPLC (Conditions E). The final reaction mixture was then cooled and concentrated under reduced pressure to a slurry keeping the temperature below 35°C. Toluene (600 mL) was added to dissolve the slurry and vacuum distillation was repeated. Additional toluene (600 mL) was added to the slurry dissolving all solids and the solution was then cooled to 5° -10°C. The solution was then treated with O-{[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl}hydroxylamine (6) (63 g, 0.43 mol) solution in 207 mL toluene followed by potassium carbonate (65 g) and water (200 mL), stirred for at least 2 hours at 20- 25°C. The stirring was stopped to allow phase separation and the bottom phase was discarded. The remaining organic layer was treated with hydrochloric acid solution (7.4%, 240 mL) until pH was less than 1 and stirred for 2 hours. The final reaction mixture was slightly concentrated under vacuum collecting about 100 mL distillate and the resulting organic solution was cooled to 5°C to crystallize the product and filtered. The filter cake was washed with toluene (1000 mL) followed by water (100 mL) and the wet cake (crude product Compound I) was charged back to the flask. Toluene (100 mL), ethanol (100 mL) and water (100 mL) are then added, stirred at 30-35°C for about 15 min, and the bottom aqueous phase was discarded. Water (200 mL) was then added to the organic solution and the mixture was stirred at about 3O C to allow for crystallization. The stirring was continued for 2 hours after product crystallized, then it was further cooled to about 0°C and stirred for at least 2 hours. The slurry was filtered and wet cake was dried under reduced pressure at 55-85°C to yield the final product N-((R)-2,3-dihydroxypropoxy)-3,4- difluoro-2-(2-fluoro-4-iodo-phenylamino)-benzamide (Compound I) product. Overall chemical yield was 86 g, 58%.
PATENT
WO2002/006213 describes crystalline Forms I and II. U.S. Pat. No. 7,060,856 (“the ‘856 patent”)
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2002006213
| Clinical data | |
|---|---|
| Trade names | Gomekli |
| Other names | PD-0325901 |
| AHFS/Drugs.com | Gomekli |
| License data | US DailyMed: Mirdametinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | L01EE05 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 391210-10-9 |
| PubChem CID | 9826528 |
| IUPHAR/BPS | 7935 |
| DrugBank | DB07101 |
| ChemSpider | 10814340 |
| UNII | 86K0J5AK6M |
| KEGG | D11675 |
| ChEBI | CHEBI:9826528 |
| ChEMBL | ChEMBL507361 |
| PDB ligand | 4BM (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C16H14F3IN2O4 |
| Molar mass | 482.198 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f “Gomekli- mirdametinib capsule; Gomekli- mirdametinib tablet, for suspension”. DailyMed. 27 February 2025. Retrieved 2 April 2025.
- ^ Armstrong AE, Belzberg AJ, Crawford JR, Hirbe AC, Wang ZJ (June 2023). “Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas”. BMC Cancer. 23 (1): 553. doi:10.1186/s12885-023-10996-y. PMC 10273716. PMID 37328781.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection”. U.S. Food and Drug Administration (FDA). 11 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “UPDATE: SpringWorks Therapeutics Announces FDA Approval of Gomekli (mirdametinib) for the Treatment of Adult and Pediatric Patients with NF1-PN” (Press release). SpringWorks Therapeutics. 12 February 2025. Archived from the original on 13 February 2025. Retrieved 16 February 2025 – via GlobeNewswire News Room.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 14 February 2025. Retrieved 16 February 2025.
External links
- “Mirdametinib (Code C52195)”. NCI Thesaurus.
- Clinical trial number NCT03962543 for “MEK Inhibitor Mirdametinib (PD-0325901) in Patients With Neurofibromatosis Type 1 Associated Plexiform Neurofibromas (ReNeu)” at ClinicalTrials.gov
- Moertel CL, Hirbe AC, Shuhaiber HH, Bielamowicz K, Sidhu A, Viskochil D, Weber MD, Lokku A, Smith LM, Foreman NK, Hajjar FM, McNall-Knapp RY, Weintraub L, Antony R, Franson AT, Meade J, Schiff D, Walbert T, Ambady P, Bota DA, Campen CJ, Kaur G, Klesse LJ, Maraka S, Moots PL, Nevel K, Bornhorst M, Aguilar-Bonilla A, Chagnon S, Dalvi N, Gupta P, Khatib Z, Metrock LK, Nghiemphu PL, Roberts RD, Robison NJ, Sadighi Z, Stapleton S, Babovic-Vuksanovic D, Gershon TR: ReNeu: A Pivotal, Phase IIb Trial of Mirdametinib in Adults and Children With Symptomatic Neurofibromatosis Type 1-Associated Plexiform Neurofibroma. J Clin Oncol. 2025 Feb 20;43(6):716-729. doi: 10.1200/JCO.24.01034. Epub 2024 Nov 8. [Article]
- Weiss BD, Wolters PL, Plotkin SR, Widemann BC, Tonsgard JH, Blakeley J, Allen JC, Schorry E, Korf B, Robison NJ, Goldman S, Vinks AA, Emoto C, Fukuda T, Robinson CT, Cutter G, Edwards L, Dombi E, Ratner N, Packer R, Fisher MJ: NF106: A Neurofibromatosis Clinical Trials Consortium Phase II Trial of the MEK Inhibitor Mirdametinib (PD-0325901) in Adolescents and Adults With NF1-Related Plexiform Neurofibromas. J Clin Oncol. 2021 Mar 1;39(7):797-806. doi: 10.1200/JCO.20.02220. Epub 2021 Jan 28. [Article]
- Ioannou M, Lalwani K, Ayanlaja AA, Chinnasamy V, Pratilas CA, Schreck KC: MEK Inhibition Enhances the Antitumor Effect of Radiotherapy in NF1-Deficient Glioblastoma. Mol Cancer Ther. 2024 Sep 4;23(9):1261-1272. doi: 10.1158/1535-7163.MCT-23-0510. [Article]
- FDA Approved Drug Products: GOMEKLI (mirdametinib) capsules and tablets for oral and oral suspension use (Feb 2024) [Link]
- FDA News: FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection [Link]
////////MIRDAMETINIB, Orphan Drug Status, Neurofibromatosis 1, PHASE 2, PD0325901, PD 0325901, PD-325901, FDA 2025, GOMEKLI, APPROVALS 2025
O=C(NOC[C@H](O)CO)C1=CC=C(F)C(F)=C1NC2=CC=C(I)C=C2F
BELUMOSUDIL

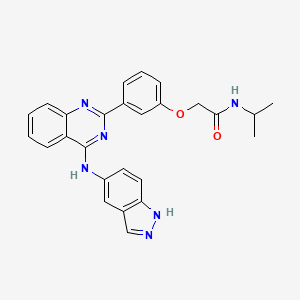
BELUMOSUDIL
MW 452.5
911417-87-3, SLx-2119, KD-025, KD 025, WHO 11343
2-[3-[4-(1H-indazol-5-ylamino)quinazolin-2-yl]phenoxy]-N-propan-2-ylacetamide
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
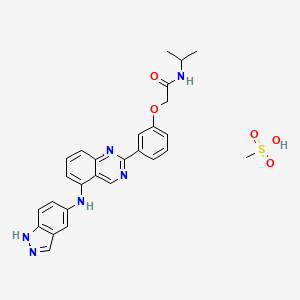
Belumosudil mesylate
KD025 mesylate
2109704-99-4
UPDATE FDA APPROVED 7/16/2021 To treat chronic graft-versus-host disease after failure of at least two prior lines of systemic therapy, Rezurock
New Drug Application (NDA): 214783
Company: KADMON PHARMA LLC
200 MG TABLET
FDA approves belumosudil for chronic graft-versus-host disease
On July 16, 2021, the Food and Drug Administration approved belumosudil (Rezurock, Kadmon Pharmaceuticals, LLC), a kinase inhibitor, for adult and pediatric patients 12 years and older with chronic graft-versus-host disease (chronic GVHD) after failure of at least two prior lines of systemic therapy.
Efficacy was evaluated in KD025-213 (NCT03640481), a randomized, open-label, multicenter dose-ranging trial that included 65 patients with chronic GVHD who were treated with belumosudil 200 mg taken orally once daily.
The main efficacy outcome measure was overall response rate (ORR) through Cycle 7 Day 1 where overall response included complete response (CR) or partial response (PR) according to the 2014 criteria of the NIH Consensus Development Project on Clinical Trials in Chronic Graft-versus-Host Disease. The ORR was 75% (95% CI: 63, 85); 6% of patients achieved a CR, and 69% achieved a PR. The median time to first response was 1.8 months (95% CI: 1.0, 1.9). The median duration of response, calculated from first response to progression, death, or new systemic therapies for chronic GVHD, was 1.9 months (95% CI: 1.2, 2.9). In patients who achieved response, no death or new systemic therapy initiation occurred in 62% (95% CI: 46, 74) of patients for at least 12 months since response.
The most common adverse reactions (≥ 20%), including laboratory abnormalities, were infections, asthenia, nausea, diarrhea, dyspnea, cough, edema, hemorrhage, abdominal pain, musculoskeletal pain, headache, phosphate decreased, gamma glutamyl transferase increased, lymphocytes decreased, and hypertension.
The recommended dosage of belumosudil is 200 mg taken orally once daily with food.
View full prescribing information for Rezurock.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Australia’s Therapeutic Goods Administration, Health Canada, Switzerland’s Swissmedic, and the United Kingdom’s Medicines and Healthcare products Regulatory Agency.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application 6 weeks ahead of the FDA goal date.
This application was granted priority review and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Belumosudil mesylate is an orally available rho kinase 2 (ROCK 2) inhibitor being developed at Kadmon. In 2020, the drug candidate was submitted for a new drug application (NDA) in the U.S., under a real-time oncology review pilot program, for the treatment of chronic graft-versus-host disease (cGVHD). The compound is also in phase II clinical development for the treatment of idiopathic pulmonary fibrosis and diffuse cutaneous systemic sclerosis. Formerly, the company had also been conducting clinical research for the treatment of psoriasis and non-alcoholic steatohepatitis (NASH); however, no further development has been reported for these indications. Originally developed by Nano Terra, the product was licensed to Kadmon on an exclusive global basis in 2011. In 2019, Kadmon entered into a strategic partnership with BioNova Pharmaceuticals and established a joint venture, BK Pharmaceuticals, to exclusively develop and commercialize KD-025 for the treatment of graft-versus-host disease in China. The compound has been granted breakthrough therapy designation in the U.S. for the treatment of cGVHD and orphan drug designations for cGVHD and systemic sclerosis. In the E.U. belumosudil was also granted orphan drug status in the E.U. for the treatment of cGVHD.
Kadmon , under license from NT Life Sciences , is developing belumosudil as mesylate salt, a ROCK-2 inhibitor, for treating IPF, chronic graft-versus-host disease, hepatic impairment and scleroderma. In July 2021, belumosudil was reported to be in pre-registration phase.
Belumosudil (formerly KD025 and SLx-2119) is an experimental drug being explored for the treatment of chronic graft versus host disease (cGvHD), idiopathic pulmonary fibrosis (IPF), and moderate to severe psoriasis. It is an inhibitor of Rho-associated coiled-coil kinase 2 (ROCK2; ROCK-II).[1] Belumosudil binds to and inhibits the serine/threonine kinase activity of ROCK2. This inhibits ROCK2-mediated signaling pathways which play major roles in pro- and anti-inflammatory immune cell responses. A genomic study in human primary cells demonstrated that the drug also has effects on oxidative phosphorylation, WNT signaling, angiogenesis, and KRAS signaling.[2] Originally developed by Surface Logix, Inc,[1] Belumosudil was later acquired by Kadmon Corporation. As of July 2020 the drug was in completed or ongoing Phase II clinical studies for cGvHD, IPF and psoriasis.[3]
cGvHD is a complication that can follow stem cell or hematopoietic stem cell transplantation where the transplanted cells (graft) attack healthy cells (host). This causes inflammation and fibrosis in multiple tissues. Two cytokines controlled by the ROCK2 signaling pathway, IL-17 and IL-21, have a major role in the cGvHD response. In a 2016 report using both mouse models and a limited human clinical trial ROCK2 inhibition with belumosudil targeted both the immunologic and fibrotic components of cGvHD and reversed the symptoms of the disease.[4] In October 2017 KD025 was granted orphan drug status in the United States for treatment of patients with cGvHD.[5]
IPF is a progressive fibrotic disease where the lining of the lungs become thickened and scarred.[6] Increased ROCK activity has been found in the lungs of humans and animals with IPF. Treatment with belumosudil reduced lung fibrosis in a bleomycin mouse model study.[7] Belumosudil may have a therapeutic benefit in IPF by targeting the fibrotic processes mediated by the ROCK signaling pathway.
Psoriasis is an inflammatory skin condition where patients experiences eruptions and remissions of thickened, erythematous, and scaly patches of skin. Down-regulation of pro-inflammatory responses was observed with KD025 treatment in Phase 2 clinical studies in patients with moderate to severe psoriasis.[8]
“Substance Name:Substance Name: Belumosudil [USAN]”.
PATENT
| WO2012040499 |
https://patents.google.com/patent/WO2012040499A2/en
PATENT
| CN106916145 |
https://patents.google.com/patent/CN106916145A/en

WO 2014055996, WO 2015157556



Patent
WO-2021129589
Novel crystalline polymorphic forms (N1, N2 and N15) of KD-025 (also known as belumosudil ), useful as a Rho A kinase 2 (ROCK-2) inhibitor for treating multiple sclerosis, psoriasis, rheumatoid arthritis, idiopathic pulmonary fibrosis (IPF), atherosclerosis, non-alcoholic fatty liver and systemic sclerosis. Represents the first filing from Sunshine Lake Pharma or its parent HEC Pharm that focuses on belumosudil.KD-025 is a selective ROCK2 (Rho-associated protein kinase 2, Rho-related protein kinase 2) inhibitor. It has multiple clinical indications such as the treatment of multiple sclerosis, psoriasis, rheumatoid arthritis, and Primary pulmonary fibrosis, atherosclerosis, non-alcoholic fatty liver, etc., among which many indications are in clinical phase I, and psoriasis and systemic sclerosis are in clinical phase II.
The structure of KD-025 is shown in the following formula (1).
Example 1 Preparation method of crystal form N1 of KD-025[0222]300mg of KD-025 solid was suspended and stirred in 10mL methanol at room temperature. After 22h, it was filtered, suction filtered and placed in a drying oven at 50°C under vacuum overnight to obtain 262mg of powder. The obtained crystal was detected by XPRD and confirmed to be KD-025 crystal form N1; its X-ray powder diffraction pattern was basically the same as that of Fig. 1, its DSC pattern was basically the same as that of Fig. 2, and the TGA pattern was basically the same as that of Fig. 3.
PATENT
WO2006105081 ,
Belumosudil product pat,
protection in the EU states until March 2026, expires in the US in May 2029 with US154 extension.
Example 82
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
[0257] A suspension of 2-(3-(4-(lH-indazol-5-ylamino)qumazolin-2-yl)ρhenoxy)acetic acid (70 mg, 0.14 mmol), PyBOP® (40 mg, 0.077 mmol), DlEA (24 μL, 0.14 mmol) in dry CH2Cl2 : DMF (2 : 0.1 mL) was stirred at RT for 15 minutes. To this solution of activated acid was added propan-2-amine (5.4 mg, 0.091 mmol). After 30 minutes, 1.0 equivalent of DIEA and 0.55 equivalents of PyBOP® were added. After stirring the solution for 15 minutes, 0.65 equivalents of propan-2-aminewere added and the mixture was stirred for an additional 30 minutes. The solvent was removed in vacuo and the crude product was purified using prep HPLC (25-50 90 rnins) to afford 2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide. (40 mg, 0.086 mmol, 61 %).
References
- ^ Jump up to:a b Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, et al. (October 2008). “Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin”. Blood Coagulation & Fibrinolysis. 19 (7): 709–18. doi:10.1097/MBC.0b013e32830b2891. PMC 2713681. PMID 18832915.
- ^ Park J, Chun KH (5 May 2020). “Identification of novel functions of the ROCK2-specific inhibitor KD025 by bioinformatics analysis”. Gene. 737: 144474. doi:10.1016/j.gene.2020.144474. PMID 32057928.
- ^ “KD025 – Clinical Trials”. ClinicalTrials.gov. Retrieved 25 July 2020.
- ^ Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. (April 2016). “Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism”. Blood. 127 (17): 2144–54. doi:10.1182/blood-2015-10-678706. PMC 4850869. PMID 26983850.
- ^ Shanley M (October 6, 2017). “Therapy to Treat Transplant Complications Gets Orphan Drug Designation”. RareDiseaseReport. Retrieved 25 July 2018.
- ^ “Pulmonary Fibrosis”. The Mayo Clinic. Retrieved July 25, 2018.
- ^ Semedo D (June 5, 2016). “Phase 2 Study of Molecule Inhibitor for Idiopathic Pulmonary Fibrosis Begins”. Lung Disease News. BioNews Services, LLC. Retrieved 25 July 2018.
- ^ Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, et al. (May 2017). “Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10”. Journal of Immunology. 198 (10): 3809–3814. doi:10.4049/jimmunol.1602142. PMC 5421306. PMID 28389592.
| Clinical data | |
|---|---|
| Routes of administration |
Oral administration (tablets or capsules) |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 911417-87-3 |
| PubChem CID | 11950170 |
| UNII | 834YJF89WO |
| CompTox Dashboard (EPA) | DTXSID80238425 |
| Chemical and physical data | |
| Formula | C26H24N6O2 |
| Molar mass | 452.518 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////BELUMOSUDIL, SLx-2119, KD-025, KD 025, WHO 11343, PHASE 2, cGvHD, IPF, psoriasis, Breakthrough Therapy, Orphan Drug Designation
CC(C)NC(=O)COC1=CC=CC(=C1)C2=NC3=CC=CC=C3C(=N2)NC4=CC5=C(C=C4)NN=C5
NEW DRUG APPROVALS
ONE TIME
$10.00
Asparaginase erwinia chrysanthemi (recombinant)-rywn
Sequence:
1ADKLPNIVIL ATGGTIAGSA ATGTQTTGYK AGALGVDTLI NAVPEVKKLA51NVKGEQFSNM ASENMTGDVV LKLSQRVNEL LARDDVDGVV ITHGTDTVEE101SAYFLHLTVK SDKPVVFVAA MRPATAISAD GPMNLLEAVR VAGDKQSRGR151GVMVVLNDRI GSARYITKTN ASTLDTFKAN EEGYLGVIIG NRIYYQNRID201KLHTTRSVFD VRGLTSLPKV DILYGYQDDP EYLYDAAIQH GVKGIVYAGM251GAGSVSVRGI AGMRKAMEKG VVVIRSTRTG NGIVPPDEEL PGLVSDSLNP301AHARILLMLA LTRTSDPKVI QEYFHTY
>Protein sequence for asparaginase (Erwinia chrysanthemi) monomer ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGEQFSNM ASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTVKSDKPVVFVAA MRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSARYITKTNASTLDTFKAN EEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKVDILYGYQDDPEYLYDAAIQH GVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNP AHARILLMLALTRTSDPKVIQEYFHTY
References:
- Therapeutic Targets Database: TTD Biologic drug sequences in fasta format [Link]
Asparaginase erwinia chrysanthemi (recombinant)-rywn
JZP458-201
JZP458
CAS Registry Number 1349719-22-7
Protein Chemical FormulaC1546H2510N432O476S9
Protein Average Weight 140000.0 Da
Rylaze, FDA APPROVED 6/30/2021, BLA 761179
L-Asparaginase (ec 3.5.1.1, L-asparagine amidohydrolase) erwinia chrysanthemi tetramer alpha4Asparaginase (Dickeya chrysanthemi subunit)
Other Names
- Asparaginase Erwinia chrysanthemi
- Crisantaspase
- Cristantaspase
- Erwinase
- Erwinaze
- L-Asparagine amidohydrolase (Erwinia chrysanthemi subunit)
Asparaginase erwinia chrysanthemi [USAN]
L-Asparaginase, erwinia chrysanthemi
Asparaginase (erwinia chrysanthemi)
Asparaginase erwinia chrysanthemi
L-Asparaginase (ec 3.5.1.1, L-asparagine amidohydrolase) erwinia chrysanthemi tetramer alpha4
Asparaginase erwinia chrysanthemi (recombinant) [USAN]
Asparaginase erwinia chrysanthemi (recombinant)
A hydrolase enzyme that converts L-asparagine and water to L-aspartate and NH3.
NCI: Asparaginase Erwinia chrysanthemi. An enzyme isolated from the bacterium Erwinia chrysanthemi (E. carotovora). Asparagine is critical to protein synthesis in leukemic cells, which cannot synthesize this amino acid due to the absence of the enzyme asparagine synthase. Asparaginase hydrolyzes L-asparagine to L-aspartic acid and ammonia, thereby depleting leukemic cells of asparagine and blocking protein synthesis and tumor cell proliferation, especially in the G1 phase of the cell cycle. This agent also induces apoptosis in tumor cells. The Erwinia-derived product is often used for those patients who have experienced a hypersensitivity reaction to the E. Coli formulation. (NCI Thesaurus)
- Treatment of Acute Lymphoblastic Leukemia (ALL)
- Antineoplastic Agents
| 10MG/0.5ML | INJECTABLE;INTRAMUSCULAR |
PATENT
WO 2011003633
https://patents.google.com/patent/WO2011003633A1/en
The present invention concerns a conjugate of a protein having substantial L-asparagine aminohydrolase activity and polyethylene glycol, particularly wherein the polyethylene glycol has a molecular weight less than or equal to about 5000 Da, particularly a conjugate wherein the protein is a L-asparaginase from Erwinia, and its use in therapy.Proteins with L-asparagine aminohydrolase activity, commonly known as L- asparaginases, have successfully been used for the treatment of Acute Lymphoblastic Leukemia(ALL) in children for many years. ALL is the most common childhood malignancy (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393).[0003] L-asparaginase has also been used to treat Hodgkin’s disease, acute myelocytic leukemia, acute myelomonocytic leukemia, chronic lymphocytic leukemia, lymphosarcoma, reticulosarcoma, and melanosarcoma (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).The anti-tumor activity of L-asparaginase is believed to be due to the inability or reduced ability of certain malignant cells to synthesize L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669). These malignant cells rely on an extracellular supply of L-asparagine. However, the L-asparaginase enzyme catalyzes the hydrolysis of L-asparagine to aspartic acid and ammonia, thereby depleting circulating pools of L-asparagine and killing tumor cells which cannot perform protein synthesis without L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0004] L-asparaginase from E. coli was the first enzyme drug used in ALL therapy and has been marketed as Elspar® in the USA or as Kidrolase® and L-asparaginase Medac® in Europe. L- asparaginases have also been isolated from other microorganisms, e.g., an L-asparaginase protein from Erwinia chrysanthemi, named crisantaspase, that has been marketed as Erwinase® (Wriston Jr., J.C. (1985) “L-asparaginase” Meth. Enzymol. 113, 608-618; Goward, CR. et al. (1992) “Rapid large scale preparation of recombinant Erwinia chrysanthemi L-asparaginase”, Bioseparation 2, 335-341). L-asparaginases from other species of Erwinia have also been identified, including, for example, Erwinia chrysanthemi 3937 (Genbank Accession#AAS67028), Erwinia chrysanthemi NCPPB 1125 (Genbank Accession #CAA31239), Erwinia carotovora (Genbank Accession #AAP92666), and Erwinia carotovora subsp. Astroseptica (Genbank Accession #AAS67027). These Erwinia chrysanthemi L-asparaginases have about 91-98% amino acid sequence identity with each other, while the Erwinia carotovora L- asparaginases have approximately 75-77% amino acid sequence identity with the Erwinia chrysanthemi L-asparaginases (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0005] L-asparaginases of bacterial origin have a high immunogenic and antigenic potential and frequently provoke adverse reactions ranging from mild allergic reaction to anaphylactic shock in sensitized patients (Wang, B. et al. (2003) “Evaluation of immunologic cross reaction of anti- asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),Leukemia 17, 1583-1588). E. coli L-asparaginase is particularly immunogenic, with reports of the presence of anti-asparaginase antibodies to E. coli L-asparaginase following i.v. or i.m. administration reaching as high as 78% in adults and 70% in children (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0006] L-asparaginases from Escherichia coli and Erwinia chrysanthemi differ in their pharmacokinetic properties and have distinct immunogenic profiles, respectively (Klug Albertsen, B. et al. (2001) “Comparison of intramuscular therapy with Erwinia asparaginase and asparaginase Medac: pharmacokinetics, pharmacodynamics, formation of antibodies and influence on the coagulation system” Brit. J. Haematol. 115, 983-990). Furthermore, it has been shown that antibodies that developed after a treatment with L-asparaginase from E. coli do not cross react with L-Asparaginase from Erwinia (Wang, B. et al., Leukemia 17 (2003) 1583-1588). Thus, L-asparaginase from Erwinia (crisantaspase) has been used as a second line treatment of ALL in patients that react to E. coli L-asparaginase (Duval, M. et al. (2002) “Comparison of Escherichia co/z-asparaginase with £Vwzm‘α-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment ofCancer, Children’s Leukemia Group phase 3 trial” Blood 15, 2734-2739; Avramis and Panosyan,Clin. Pharmacokinet. (2005) 44:367-393).[0007] In another attempt to reduce immunogenicity associated with administration of microbial L-asparaginases, an E. coli L-asparaginase has been developed that is modified with methoxy- polyethyleneglycol (mPEG). This method is commonly known as “PEGylation” and has been shown to alter the immunological properties of proteins (Abuchowski, A. et al. (1977) “Alteration of Immunological Properties of Bovine Serum Albumin by Covalent Attachment of Polyethylene Glycol,” J.Biol.Chem. 252 (11), 3578-3581). This so-called mPEG-L- asparaginase, or pegaspargase, marketed as Oncaspar® (Enzon Inc., USA), was first approved in the U.S. for second line treatment of ALL in 1994, and has been approved for first- line therapy of ALL in children and adults since 2006. Oncaspar® has a prolonged in vivo half-life and a reduced immunogenicity/antigenicity.[0008] Oncaspar® is E. coli L-asparaginase that has been modified at multiple lysine residues using 5 kDa mPEG-succinimidyl succinate (SS-PEG) (U.S. Patent No. 4,179,337). SS-PEG is aPEG reagent of the first generation that contains an instable ester linkage that is sensitive to hydro lysis by enzymes or at slightly alkaline pH values (U.S. Patent No. 4,670,417; Makromol. Chem. 1986, 187, 1131-1144). These properties decrease both in vitro and in vivo stability and can impair drug safety.[0009] Furthermore, it has been demonstrated that antibodies developed against L-asparaginase from E. coli will cross react with Oncaspar® (Wang, B. et al. (2003) “Evaluation of immunologic cross-reaction of anti-asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),” Leukemia 17, 1583-1588). Even though these antibodies were not neutralizing, this finding clearly demonstrated the high potential for cross-hypersensitivity or cross-inactivation in vivo. Indeed, in one report 30-41% of children who received pegaspargase had an allergic reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0010] In addition to outward allergic reactions, the problem of “silent hypersensitivity” was recently reported, whereby patients develop anti-asparaginase antibodies without showing any clinical evidence of a hypersensitivity reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588). This reaction can result in the formation of neutralizing antibodies to E. coli L-asparaginase and pegaspargase; however, these patients are not switched to Erwinia L-asparaginase because there are not outward signs of hypersensitivity, and therefore they receive a shorter duration of effective treatment (Holcenberg, J., J. Pediatr. Hematol. Oncol. 26 (2004) 273-274).[0011] Erwinia chrysanthemi L-asparaginase treatment is often used in the event of hypersensitivity to E. co/z-derived L-asparaginases. However, it has been observed that as many as 30-50% of patients receiving Erwinia L-asparaginase are antibody-positive (Avramis andPanosyan, Clin. Pharmacokinet. (2005) 44:367-393). Moreover, because Erwinia chrysanthemi L-asparaginase has a significantly shorter elimination half-life than the E. coli L-asparaginases, it must be administered more frequently (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393). In a study by Avramis et al., Erwinia asparaginase was associated with inferior pharmacokinetic profiles (Avramis et al., J. Pediatr. Hematol. Oncol. 29 (2007) 239-247). E. coli L-asparaginase and pegaspargase therefore have been the preferred first-line therapies for ALL over Erwinia L-asparaginase.[0012] Numerous biopharmaceuticals have successfully been PEGylated and marketed for many years. In order to couple PEG to a protein, the PEG has to be activated at its OH terminus. The activation group is chosen based on the available reactive group on the protein that will bePEGylated. In the case of proteins, the most important amino acids are lysine, cysteine, glutamic acid, aspartic acid, C-terminal carboxylic acid and the N-terminal amino group. In view of the wide range of reactive groups in a protein nearly the entire peptide chemistry has been applied to activate the PEG moiety. Examples for this activated PEG-reagents are activated carbonates, e.g., p-nitrophenyl carbonate, succinimidyl carbonate; active esters, e.g., succinimidyl ester; and for site specific coupling aldehydes and maleimides have been developed (Harris, M., Adv. Drug – A -DeI. Rev. 54 (2002), 459-476). The availability of various chemical methods for PEG modification shows that each new development of a PEGylated protein will be a case by case study. In addition to the chemistry the molecular weight of the PEG that is attached to the protein has a strong impact on the pharmaceutical properties of the PEGylated protein. In most cases it is expected that, the higher the molecular weight of the PEG, the better the improvement of the pharmaceutical properties (Sherman, M. R., Adv. Drug Del. Rev. 60 (2008), 59-68; Holtsberg, F. W., Journal of Controlled Release 80 (2002), 259-271). For example, Holtsberg et al. found that, when PEG was conjugated to arginine deaminase, another amino acid degrading enzyme isolated from a microbial source, pharmacokinetic and pharmacodynamic function of the enzyme increased as the size of the PEG attachment increased from a molecular weight of 5000Da to 20,000 Da (Holtsberg, F.W., Journal of Controlled Release 80 (2002), 259-271).[0013] However, in many cases, PEGylated biopharmaceuticals show significantly reduced activity compared to the unmodified biopharmaceutical (Fishburn, CS. (2008) Review “The Pharmacology of PEGylation: Balancing PD with PK to Generate Novel Therapeutics” J. Pharm. Sd., 1-17). In the case of L-asparaginase from Erwinia carotovora, it has been observed that PEGylation reduced its in vitro activity to approximately 57% (Kuchumova, A.V. et al. (2007) “Modification of Recombinant asparaginase from Erwinia carotovora with Polyethylene Glycol 5000” Biochemistry (Moscow) Supplement Series B: Biomedical Chemistry, 1, 230-232). The L-asparaginase from Erwinia carotovora has only about 75% homology to the Erwinia chrysanthemi L-asparaginase (crisantaspase). For Oncaspar® it is also known that its in vitro activity is approximately 50% compared to the unmodified E. coli L-asparaginase.[0014] The currently available L-asparaginase preparations do not provide alternative or complementary therapies— particularly therapies to treat ALL— that are characterized by high catalytic activity and significantly improved pharmacological and pharmacokinetic properties, as well as reduced immunogenicity. L-asparaginase protein has at least about 80% homology or identity with the protein comprising the sequence of SEQ ID NO:1, more specifically at least about 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% homology or identity with the protein comprising the sequence of SEQ ID NO:1. SEQ ID NO:1 is as follows:ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGE QFSNMASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTV KSDKPVVFVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSA RYITKTNASTLDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKV DILYGYQDDPEYLYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY (SEQ ID NO:1) [0048] The term “comprising the sequence of SEQ ID NO:1” means that the amino-acid sequence of the protein may not be strictly limited to SEQ ID NO:1 but may contain additional amino-acids.ExamplesExample 1 : Preparation of Recombinant Crisantaspase [0100] The recombinant bacterial strain used to manufacture the naked recombinant Erwinia chrysanthemi L-asparaginase protein (also referred to herein as “r-crisantaspase”) was an E. coli BL21 strain with a deleted ansB gene (the gene encoding the endogenous E. coli type II L- asparaginase) to avoid potential contamination of the recombinant Erwinia chrysanthemi L- asparaginase with this enzyme. The deletion of the ansB gene relies on homologous recombination methods and phage transduction performed according to the three following steps:1) a bacterial strain (NMI lOO) expressing a defective lambda phage which supplies functions that protect and recombine electroporated linear DNA substrate in the bacterial cell was transformed with a linear plasmid (kanamycin cassette) containing the kanamycin gene flanked by an FLP recognition target sequence (FRT). Recombination occurs to replace the ansB gene by the kanamycin cassette in the bacterial genome, resulting in a ΛansB strain; 2) phage transduction was used to integrate the integrated kanamycin cassette region from the ΛansB NMI lOO strain to the ansB locus in BL21 strain. This results in an E. coli BL21 strain with a deleted ansB gene and resistant to kanamycin; 3) this strain was transformed with a FLP -helper plasmid to remove the kanamycin gene by homologous recombination at the FRT sequence. The genome of the final strain (BL21 ΛansB strain) was sequenced, confirming full deletion of the endogenous ansB gene.[0101] The E. co/z‘-optimized DNA sequence encoding for the mature Erwinia chrysanthemi L- asparaginase fused with the ENX signal peptide from Bacillus subtilis was inserted into an expression vector. This vector allows expression of recombinant Erwinia chrysanthemi L- asparaginase under the control of hybrid T5/lac promoter induced by the addition of Isopropyl β- D-1-thiogalactopyranoside (IPTG) and confers resistance to kanamycin.[0102] BL21 ΛansB strain was transformed with this expression vector. The transformed cells were used for production of the r-crisantaspase by feed batch glucose fermentation in Reisenberg medium. The induction of the cell was done 16h at 23°C with IPTG as inducer. After cell harvest and lysis by homogenization in 1OmM sodium phosphate buffer pH6 5mM EDTA (Buffer A), the protein solution was clarified by centrifugation twice at 1500Og, followed by 0.45μm and 0.22μm filtration steps. The recombinant Erwinia chrysanthemi L-asparaginase was next purified using a sequence of chromatography and concentration steps. Briefly, the theoretical isoelectric point of the Erwinia chrysanthemi L-asparaginase (7.23) permits the recombinant enzyme to adsorb to cation exchange resins at pH6. Thus, the recombinant enzyme was captured on a Capto S column (cation exchange chromatography) and eluted with salt gradient in Buffer A. Fractions containing the recombinant enzyme were pooled. The pooled solution was next purified on Capto MMC column (cation exchange chromatography) in Buffer A with salt gradient. . The eluted fractions containing Erwinia chrysanthemi L-asparaginase were pooled and concentrated before protein separation on Superdex 200pg size exclusion chromatography as polishing step. Fractions containing recombinant enzymes were pooled, concentrated, and diafiltered against 10OmM sodium phosphate buffer pH8. The purity of the final Erwinia chrysanthemi L-asparaginase preparation was evaluated by SDS-PAGE (Figure 1) and RP-HPLC and was at least 90%. The integrity of the recombinant enzyme was verified byN-terminal sequencing and LC-MS. Enzyme activity was measured at 37°C using Nessler’s reagent. The specific activity of the purified recombinant Erwinia chrysanthemi L-asparaginase was around 600 U/mg. One unit of enzyme activity is defined as the amount of enzyme that liberates lμmol of ammonia from L-asparagine per minute at 37°C. Example 2: Preparation of 10 kDa mPEG-L- Asparaginase Conjugates[0103] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration between 2.5 and 4 mg/mL, in the presence of 150 mg/mL or 36 mg/mL 10 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 10 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 10 kDa mPEG-L-asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) residues being conjugated corresponding to PEGylation of 78% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (39% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 50% of accessible amino groups (e.g., lysine residues and/or the N-terminus)) . SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 3: Preparation of 5 kDa mPEG-L-Asparaginase Conjugates[0104] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration of 4 mg/mL, in the presence of 150 mg/mL or 22.5 mg/mL 5 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 5 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 5 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) being conjugated corresponding to PEGylation of 84% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (36% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 43% of accessible amino groups (e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 4: Preparation of 2 kDa mPEG-L-Asparaginase Conjugates[0105] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer pH 8.0 at a protein concentration of 4 mg/mL in the presence of150 mg/mL or 22.5 mg/mL 2 kDa mPEG-NHS for 2 hours at 22°C. The resulting crude 2 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 2 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as reference, one corresponding to maximum PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N- terminus) being conjugated corresponding to PEGylation of 86% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (47% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 55% of accessible amino groups {e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 5: Activity of mPEG-r-Crisantaspase Conjugates[0106] L-asparaginase aminohydrolase activity of each conjugate described in the proceeding examples was determined by Nesslerization of ammonia that is liberated from L-asparagine by enzymatic activity. Briefly, 50μL of enzyme solution were mixed with 2OmM of L-asparagine in a 50 mM Sodium borate buffer pH 8.6 and incubated for 10 min at 37°C. The reaction was stopped by addition of 200μL of Nessler reagent. Absorbance of this solution was measured at 450 nm. The activity was calculated from a calibration curve that was obtained from Ammonia sulfate as reference. The results are summarized in Table 2, below:Table 2: Activity of mPEG-r-crisantaspase conjugates

* the numbers “40%” and “100%” indicate an approximate degree of PEGylation of respectively 40-55% and 100% of accessible amino groups (see Examples 2-4, supra).** the ratio mol PEG / mol monomer was extrapolated from data using TNBS assay, that makes the assumption that all amino groups from the protein (e.g., lysine residues and the N-terminus) are accessible.[0107] Residual activity of mPEG-r-crisantaspase conjugates ranged between 483 and 543 Units/mg. This corresponds to 78-87% of L-asparagine aminohydrolase activity of the unmodified enzyme. Example 6: L-Asparagine-Depleting Effect of Unmodified Crisantaspase
PAPER
Biotechnology and Applied Biochemistry (2019), 66(3), 281-289. |
https://iubmb.onlinelibrary.wiley.com/doi/10.1002/bab.1723
Crisantaspase is an asparaginase enzyme produced by Erwinia chrysanthemi and used to treat acute lymphoblastic leukemia (ALL) in case of hypersensitivity to Escherichia coli l-asparaginase (ASNase). The main disadvantages of crisantaspase are the short half-life (10 H) and immunogenicity. In this sense, its PEGylated form (PEG-crisantaspase) could not only reduce immunogenicity but also improve plasma half-life. In this work, we developed a process to obtain a site-specific N-terminal PEGylated crisantaspase (PEG-crisantaspase). Crisantaspase was recombinantly expressed in E. coli BL21(DE3) strain cultivated in a shaker and in a 2-L bioreactor. Volumetric productivity in bioreactor increased 37% compared to shaker conditions (460 and 335 U L−1 H−1, respectively). Crisantaspase was extracted by osmotic shock and purified by cation exchange chromatography, presenting specific activity of 694 U mg−1, 21.7 purification fold, and yield of 69%. Purified crisantaspase was PEGylated with 10 kDa methoxy polyethylene glycol-N-hydroxysuccinimidyl (mPEG-NHS) at different pH values (6.5–9.0). The highest N-terminal pegylation yield (50%) was at pH 7.5 with the lowest poly-PEGylation ratio (7%). PEG-crisantaspase was purified by size exclusion chromatography and presented a KM value three times higher than crisantaspase (150 and 48.5 µM, respectively). Nonetheless, PEG-crisantaspase was found to be more stable at high temperatures and over longer periods of time. In 2 weeks, crisantaspase lost 93% of its specific activity, whereas PEG-crisantaspase was stable for 20 days. Therefore, the novel PEG-crisantaspase enzyme represents a promising biobetter alternative for the treatment of ALL.
ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGEQFSN
MASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTVKSDKPVV
FVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSARYITKTNAST
LDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKVDILYGYQDDPEY
LYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEE
LPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY
Figure S1 – Amino acid sequence of the enzyme crisantaspase without the signal peptide and with the lysines highlighted in red (Swiss-Prot/TrEMBL accession number: P06608|22-348 AA).
……………………………………………………………………………………………………………………………..
As a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in patients who are allergic to E. coli-derived asparaginase products
Press ReleaseFor Immediate Release:June 30, 2021
FDA Approves Component of Treatment Regimen for Most Common Childhood Cancer
Alternative Has Been in Global Shortage Since 2016
Today, the U.S. Food and Drug Administration approved Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) as a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in adult and pediatric patients who are allergic to the E. coli-derived asparaginase products used most commonly for treatment. The only other FDA-approved drug for such patients with allergic reactions has been in global shortage for years.
“It is extremely disconcerting to patients, families and providers when there is a lack of access to critical drugs for treatment of a life-threatening, but often curable cancer, due to supply issues,” said Gregory Reaman, M.D., associate director for pediatric oncology in the FDA’s Oncology Center of Excellence. “Today’s approval may provide a consistently sourced alternative to a pivotal component of potentially curative therapy for children and adults with this type of leukemia.”
Acute lymphoblastic leukemia occurs in approximately 5,700 patients annually, about half of whom are children. It is the most common type of childhood cancer. One component of the chemotherapy regimen is an enzyme called asparaginase that kills cancer cells by depriving them of substances needed to survive. An estimated 20% of patients are allergic to the standard E. coli-derived asparaginase and need an alternative their bodies can tolerate.
Rylaze’s efficacy was evaluated in a study of 102 patients who either had a hypersensitivity to E. coli-derived asparaginases or experienced silent inactivation. The main measurement was whether patients achieved and maintained a certain level of asparaginase activity. The study found that the recommended dosage would provide the target level of asparaginase activity in 94% of patients.
The most common side effects of Rylaze include hypersensitivity reactions, pancreatic toxicity, blood clots, hemorrhage and liver toxicity.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Health Canada, where the application review is pending.
Rylaze received Fast Track and Orphan Drug designations for this indication. Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fulfill an unmet medical need. Orphan Drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted approval of Rylaze to Jazz Pharmaceuticals.
REF
DUBLIN, June 30, 2021 /PRNewswire/ — Jazz Pharmaceuticals plc (Nasdaq: JAZZ) today announced the U.S. Food and Drug Administration (FDA) approval of Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn) for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase.1 Rylaze is the only recombinant erwinia asparaginase manufactured product that maintains a clinically meaningful level of asparaginase activity throughout the entire duration of treatment, and it was developed by Jazz to address the needs of patients and healthcare providers with an innovative, high-quality erwinia-derived asparaginase with reliable supply.
“We are excited to bring this important new treatment to patients who are in critical need, and we are grateful to FDA for the approval of Rylaze based on its established safety and efficacy profile. We are pleased Rylaze was approved before the trial is complete and are diligently working to advance additional clinical trial data. We are committed to quickly engaging with FDA to evolve the Rylaze product profile with additional dosing options and an IV route of administration,” said Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals. “Thank you to our collaborators within the Children’s Oncology Group, the clinical trial investigators, patients and their families, and all of the other stakeholders who helped us achieve this significant milestone.”
Rylaze was granted orphan drug designation for the treatment of ALL/LBL by FDA in June 2021. The Biologics Licensing Application (BLA) approval followed review under the Real-Time Oncology Review (RTOR) program, an initiative of FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The company expects Rylaze will be commercially available in mid-July.
“The accelerated development and approval of Rylaze marks an important step in bringing a meaningful new treatment option for many ALL patients – most of whom are children – who cannot tolerate E. coli-derived asparaginase medicine,” said Dr. Luke Maese, assistant professor at the University of Utah, Primary Children’s Hospital and Huntsman Cancer Institute. “Before the approval of Rylaze, there was a significant need for an effective asparaginase medicine that would allow patients to start and complete their prescribed treatment program with confidence in supply.”
Recent data from a Children’s Oncology Group retrospective analysis of over 8,000 patients found that patients who did not receive a full course of asparaginase treatment due to associated toxicity had significantly lower survival outcomes – regardless of whether those patients were high risk or standard risk, slow early responders.2
About Study JZP458-201
The FDA approval of Rylaze, also known as JZP458, is based on clinical data from an ongoing pivotal Phase 2/3 single-arm, open-label, multicenter, dose confirmation study evaluating pediatric and adult patients with ALL or LBL who have had an allergic reaction to E. coli-derived asparaginases and have not previously received asparaginase erwinia chrysanthemi. The study was designed to assess the safety, tolerability and efficacy of JZP458. The determination of efficacy was measured by serum asparaginase activity (SAA) levels. The Phase 2/3 study is being conducted in two parts. The first part is investigating the intramuscular (IM) route of administration, including a Monday-Wednesday-Friday dosing schedule. The second part remains active to further confirm the dose and schedule for the intravenous (IV) route of administration.
The FDA approval of Rylaze was based on data from the first of three IM cohorts, which demonstrated the achievement and maintenance of nadir serum asparaginase activity (NSAA) greater than or equal to the level of 0.1 U/mL at 48 hours using IM doses of Rylaze 25 mg/m2. The results of modeling and simulations showed that for a dosage of 25 mg/m2 administered intramuscularly every 48 hours, the proportion of patients maintaining NSAA ≥ 0.1 U/mL at 48 hours after a dose of Rylaze was 93.6% (95% CI: 92.6%, 94.6%).1
The most common adverse reactions (incidence >15%) were abnormal liver test, nausea, musculoskeletal pain, fatigue, infection, headache, pyrexia, drug hypersensitivity, febrile neutropenia, decreased appetite, stomatitis, bleeding and hyperglycemia. In patients treated with the Rylaze, a fatal adverse reaction (infection) occurred in one patient and serious adverse reactions occurred in 55% of patients. The most frequent serious adverse reactions (in ≥5% of patients) were febrile neutropenia, dehydration, pyrexia, stomatitis, diarrhea, drug hypersensitivity, infection, nausea and viral infection. Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received Rylaze. Adverse reactions resulting in permanent discontinuation included hypersensitivity (6%) and infection (3%).1
The company will continue to work with FDA and plans to submit additional data from a completed cohort of patients evaluating 25mg/m2 IM given on Monday and Wednesday, and 50 mg/m2 given on Friday in support of a M/W/F dosing schedule. Part 2 of the study is evaluating IV administration and is ongoing. The company also plans to submit these data for presentation at a future medical meeting.
Investor Webcast
The company will host an investor webcast on the Rylaze approval in July. Details will be announced separately.
About Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn)
Rylaze, also known as JZP458, is approved in the U.S. for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase. Rylaze has orphan drug designation for the treatment of ALL/LBL in the United States. Rylaze is a recombinant erwinia asparaginase that uses a novel Pseudomonas fluorescens expression platform. JZP458 was granted Fast Track designation by the U.S. Food and Drug Administration (FDA) in October 2019 for the treatment of this patient population. Rylaze was approved as part of the Real-Time Oncology Review program, an initiative of the FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The full U.S. Prescribing Information for Rylaze is available at: <http://pp.jazzpharma.com/pi/rylaze.en.USPI.pdf>
Important Safety Information
RYLAZE should not be given to people who have had:
- Serious allergic reactions to RYLAZE
- Serious swelling of the pancreas (stomach pain), serious blood clots, or serious bleeding during previous asparaginase treatment
RYLAZE may cause serious side effects, including:
- Allergic reactions (a feeling of tightness in your throat, unusual swelling/redness in your throat and/or tongue, or trouble breathing), some of which may be life-threatening
- Swelling of the pancreas (stomach pain)
- Blood clots (may have a headache or pain in leg, arm, or chest)
- Bleeding
- Liver problems
Contact your doctor immediately if any of these side effects occur.
Some of the most common side effects with RYLAZE include: liver problems, nausea, bone and muscle pain, tiredness, infection, headache, fever, allergic reactions, fever with low white blood cell count, decreased appetite, mouth swelling (sometimes with sores), bleeding, and too much sugar in the blood.
RYLAZE can harm your unborn baby. Inform your doctor if you are pregnant, planning to become pregnant, or nursing. Females of reproductive potential should use effective contraception (other than oral contraceptives) during treatment and for 3 months following the final dose. Do not breastfeed while receiving RYLAZE and for 1 week after the final dose.
Tell your healthcare provider if there are any side effects that are bothersome or that do not go away.
These are not all the possible side effects of RYLAZE. For more information, ask your healthcare provider.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088 (1-800-332-1088).
About ALL
ALL is a cancer of the blood and bone marrow that can progress quickly if not treated.3 Leukemia is the most common cancer in children, and about three out of four of these cases are ALL.4 Although it is one of the most common cancers in children, ALL is among the most curable of the pediatric malignancies due to recent advancements in treatment.5,6 Adults can also develop ALL, and about four of every 10 cases of ALL diagnosed are in adults.7 The American Cancer Society estimates that almost 6,000 new cases of ALL will be diagnosed in the United States in 2021.7 Asparaginase is a core component of multi-agent chemotherapeutic regimens in ALL.8 However, asparaginase treatments derived from E. coli are associated with the potential for development of hypersensitivity reactions.9
About Lymphoblastic Lymphoma
LBL is a rare, fast-growing, aggressive subtype of Non-Hodgkin’s lymphoma, most often seen in teenagers and young adults.8 LBL is a very aggressive lymphoma – also called high-grade lymphoma – which means the lymphoma grows quickly with early spread to different parts of the body.10,11
About Jazz Pharmaceuticals plc
Jazz Pharmaceuticals plc (NASDAQ: JAZZ) is a global biopharmaceutical company whose purpose is to innovate to transform the lives of patients and their families. We are dedicated to developing life-changing medicines for people with serious diseases – often with limited or no therapeutic options. We have a diverse portfolio of marketed medicines and novel product candidates, from early- to late-stage development, in neuroscience and oncology. We actively explore new options for patients including novel compounds, small molecules and biologics, and through cannabinoid science and innovative delivery technologies. Jazz is headquartered in Dublin, Ireland and has employees around the globe, serving patients in nearly 75 countries. For more information, please visit www.jazzpharmaceuticals.com and follow @JazzPharma on Twitter.
About The Children’s Oncology Group (COG)
COG (childrensoncologygroup.org), a member of the NCI National Clinical Trials Network (NCTN), is the world’s largest organization devoted exclusively to childhood and adolescent cancer research. COG unites over 10,000 experts in childhood cancer at more than 200 leading children’s hospitals, universities, and cancer centers across North America, Australia, and New Zealand in the fight against childhood cancer. Today, more than 90% of the 14,000 children and adolescents diagnosed with cancer each year in the United States are cared for at COG member institutions. Research performed by COG institutions over the past 50 years has transformed childhood cancer from a virtually incurable disease to one with a combined 5-year survival rate of 80%. COG’s mission is to improve the cure rate and outcomes for all children with cancer.
Caution Concerning Forward-Looking Statements
This press release contains forward-looking statements, including, but not limited to, statements related to Jazz Pharmaceuticals’ belief in the potential of Rylaze to provide a reliable therapeutic option for adult and pediatric patients to maximize their chance for a cure, plans for a mid-July 2021 launch of Rylaze, the availability of a reliable supply of Rylaze and other statements that are not historical facts. These forward-looking statements are based on Jazz Pharmaceuticals’ current plans, objectives, estimates, expectations and intentions and inherently involve significant risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in such forward-looking statements as a result of these risks and uncertainties, which include, without limitation, effectively launching and commercializing new products; obtaining and maintaining adequate coverage and reimbursement for the company’s products; delays or problems in the supply or manufacture of the company’s products and other risks and uncertainties affecting the company, including those described from time to time under the caption “Risk Factors” and elsewhere in Jazz Pharmaceuticals’ Securities and Exchange Commission filings and reports (Commission File No. 001-33500), including Jazz Pharmaceuticals’ Annual Report on Form 10-K for the year ended December 31, 2020 and future filings and reports by Jazz Pharmaceuticals. Other risks and uncertainties of which Jazz Pharmaceuticals is not currently aware may also affect Jazz Pharmaceuticals’ forward-looking statements and may cause actual results and the timing of events to differ materially from those anticipated. The forward-looking statements herein are made only as of the date hereof or as of the dates indicated in the forward-looking statements, even if they are subsequently made available by Jazz Pharmaceuticals on its website or otherwise. Jazz Pharmaceuticals undertakes no obligation to update or supplement any forward-looking statements to reflect actual results, new information, future events, changes in its expectations or other circumstances that exist after the date as of which the forward-looking statements were made.
Jazz Media Contact:
Jacqueline Kirby
Vice President, Corporate Affairs
Jazz Pharmaceuticals plc
CorporateAffairsMediaInfo@jazzpharma.com
Ireland, +353 1 697 2141
U.S. +1 215 867 4910
Jazz Investor Contact:
Andrea N. Flynn, Ph.D.
Vice President, Head, Investor Relations
Jazz Pharmaceuticals plc
investorinfo@jazzpharma.com
Ireland, +353 1 634 3211
References
- Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) injection, for intramuscular use Prescribing Information. Palo Alto, CA: Jazz Pharmaceuticals, Inc.
- Gupta S, Wang C, Raetz EA et al. Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children’s Oncology Group. J Clin Oncol. 2020 Jun 10;38(17):1897-1905. doi: 10.1200/JCO.19.03024
- National Cancer Institute. Adult Acute Lymphoblastic Leukemia Treatment (PDQ®)–Patient Version. Available at www.cancer.gov/types/leukemia/patient/adult-all-treatment-pdq. Accessed June 29, 2021
- American Cancer Society. Key Statistics for Childhood Leukemia. Available at https://www.cancer.org/cancer/leukemia-in-children/about/key-statistics.html. Accessed June 29, 2021.
- American Cancer Society. Cancer Facts & Figures 2019. www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html. Accessed June 29, 2021.
- Pui C, Evans W. A 50-Year Journey to Cure Childhood Acute Lymphoblastic Leukemia. Seminars in Hematology. 2013;50(3), 185-196.
- American Cancer Society. Key Statistics for Acute Lymphocytic Leukemia (ALL). Available at https://cancerstatisticscenter.cancer.org/?_ga=2.8163506.1018157754.1621008457-1989786785.1621008457#!/data-analysis/NewCaseEstimates. Accessed June 29, 2021.
- Salzer W, Bostrom B, Messinger Y et al. 2018. Asparaginase activity levels and monitoring in patients with acute lymphoblastic leukemia. Leukemia & Lymphoma. 59:8, 1797-1806, DOI: 10.1080/10428194.2017.1386305.
- Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57(4):748–757. DOI: 10.3109/10428194.2015.1101098.
- Leukemia Foundation. Lymphoblastic Lymphoma. Available at https://www.leukaemia.org.au/disease-information/lymphomas/non-hodgkin-lymphoma/other-non-hodgkin-lymphomas/lymphoblastic-lymphoma/. Accessed June 29, 2021.
- Mayo Clinic. Acute Lymphocytic Leukemia Diagnosis. Available at https://www.mayoclinic.org/diseases-conditions/acute-lymphocytic-leukemia/diagnosis-treatment/drc-20369083. Accessed June 29, 2021.
SOURCE Jazz Pharmaceuticals plc
Related Links
CLIP
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4776285/

/////////////asparaginase erwinia chrysanthemi (recombinant)-rywn, Rylaze, Jazz Pharmaceuticals, JZP458-201, JZP458, FDA 2021, APPROVALS 2021, ORPHAN, Fast Track, Acute Lymphoblastic Leukemia, ALL, Antineoplastic Agents
https://chem.nlm.nih.gov/chemidplus/id/1349719227
https://go.drugbank.com/drugs/DB08886

NEW DRUG APPROVALS
ONE TIME
$10.00
Piflufolastat F 18 injection, Dcfpyl F-18

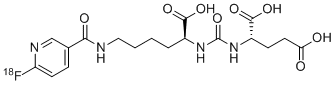
Piflufolastat F 18 injection
Dcfpyl F-18
CAS 207181-29-0
PLAIN F 1423758-00-2 WITHOUT RADIO LABELC18 H23 F N4 O8, 441.4L-Glutamic acid, N-[[[(1S)-1-carboxy-5-[[[6-(fluoro-18F)-3-pyridinyl]carbonyl]amino]pentyl]amino]carbonyl]-2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl) amino]-pentyl}ureido)-pentanedioic acid
Other Names
- N-[[[(1S)-1-Carboxy-5-[[[6-(fluoro-18F)-3-pyridinyl]carbonyl]amino]pentyl]amino]carbonyl]-L-glutamic acid
- [18F]DCFPyl
Dcfpyl F-18
(18F)Dcfpyl
UNII-3934EF02T7
18F-DCFPyL
3934EF02T7
Progenics Pharmaceuticals, Inc.
APPROVED 5/26/2021 fda, Pylarify
For positron emission tomography imaging of prostate-specific membrane antigen-positive lesions in men with prostate cancer
For positron emission tomography (PET) of prostatespecific membrane antigen (PSMA) positive lesions in men with prostate cancer: • with suspected metastasis who are candidates for initial definitive therapy. • with suspected recurrence based on elevated serum prostate-specific antigen (PSA) level.
- Originator Johns Hopkins University School of Medicine
- Developer Curium Pharma; Progenics Pharmaceuticals
- Class Amides; Carboxylic acids; Fluorinated hydrocarbons; Imaging agents; Pyridines; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules; Urea compounds
- Mechanism of ActionPositron-emission tomography enhancers
- Orphan Drug StatusNo
- MarketedProstate cancer
- 28 May 2021Registered for Prostate cancer (Diagnosis) in USA (IV) – First global approval
- 28 May 2021Adverse events data from phase III CONDOR and phase II/III OSPREY trials in prostate cancer released by Lantheus Holdings
- 27 May 2021Lantheus Holdings intends to launch Fluorine-18 DCFPyL in USA at end of 2021
PYLARIFY contains fluorine 18 (F 18), radiolabeled prostate-specific membrane antigen inhibitor imaging agent. Chemically piflufolastat F 18 is 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl) amino]-pentyl}ureido)-pentanedioic acid. The molecular weight is 441.4 and the structural formula is:

The chiral purity of the unlabeled piflufolastat F 18 precursor is greater than 99% (S,S). PYLARIFY is a sterile, non-pyrogenic, clear, colorless solution for intravenous injection. Each milliliter contains 37 to 2,960 MBq (1 to 80 mCi) piflufolastat F 18 with ≤0.01 µg/mCi of piflufolastat at calibration time and date, and ≤ 78.9 mg ethanol in 0.9% sodium chloride injection USP. The pH of the solution is 4.5 to 7.0. PYLARIFY has a radiochemical purity of at least 95% up to 10 hours following end of synthesis, and specific activity of at least 1000 mCi/µmol at the time of administration.
PYLARIFY contains fluorine 18 (F 18), radiolabeled prostate-specific membrane antigen inhibitor imaging agent. Chemically piflufolastat F 18 is 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)amino]-pentyl}ureido)-pentanedioic acid. The molecular weight is 441.4 and the structural formula is:
 |
The chiral purity of the unlabeled piflufolastat F 18 precursor is greater than 99% (S,S).
PYLARIFY is a sterile, non-pyrogenic, clear, colorless solution for intravenous injection. Each milliliter contains 37 to 2,960 MBq (1 to 80 mCi) piflufolastat F 18 with ≤0.01 μg/mCi of piflufolastat at calibration time and date, and ≤ 78.9 mg ethanol in 0.9% sodium chloride injection USP. The pH of the solution is 4.5 to 7.0.
PYLARIFY has a radiochemical purity of at least 95% up to 10 hours following end of synthesis, and specific activity of at least 1000 mCi/μmol at the time of administration.
Physical Characteristics
PYLARIFY is radiolabeled with fluorine 18 (F 18), a cyclotron produced radionuclide that decays by positron emission to stable oxygen 18 with a half-life of 109.8 minutes. The principal photons useful for diagnostic imaging are the coincident pair of 511 keV gamma photons, resulting from the interaction of the emitted positron with an electron (Table 3).
Table 3: Principal Radiation Produced from Decay of Fluorine 18
| Radiation Energy (keV) | Abundance (%) | |
| Positron | 249.8 | 96.9 |
| Gamma | 511 | 193.5 |
FDA
- Approval Letter(s) (PDF)
- Printed Labeling (PDF)
- Product Quality Review(s) (PDF)
- Multi-Discipline Review (PDF)
- Proprietary Name Review(s) (PDF)
- Officer/Employee List (PDF)
- Other Review(s) (PDF)
- Risk Assessment and Risk Mitigation Review(s) (PDF)
- Administrative and Correspondence Documents (PDF)
PATENT
WO 2016030329
WO 2017072200
PAPER
Journal of Labelled Compounds and Radiopharmaceuticals (2016), 59(11), 439-450
CLIP
https://ejnmmires.springeropen.com/articles/10.1186/s13550-016-0195-6
![Radiosynthesis of [ 18 F]DCFPyL](https://www.researchgate.net/publication/301830909/figure/fig4/AS:362696278069251@1463484939571/Radiosynthesis-of-18-FDCFPyL.png)
Radiosynthesis of [ 18 F]DCFPyL

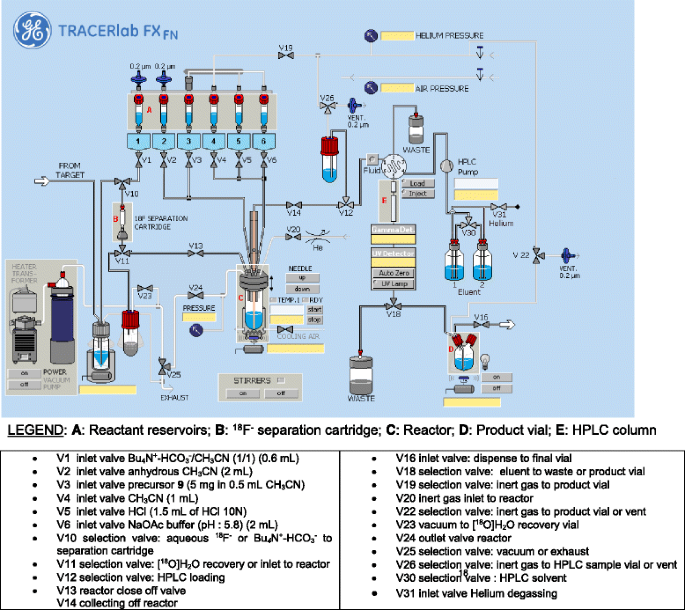
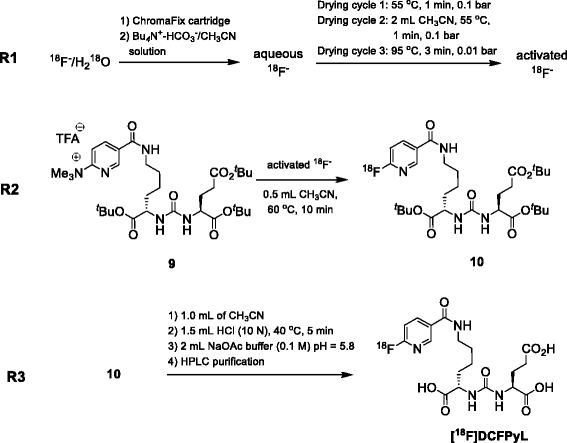
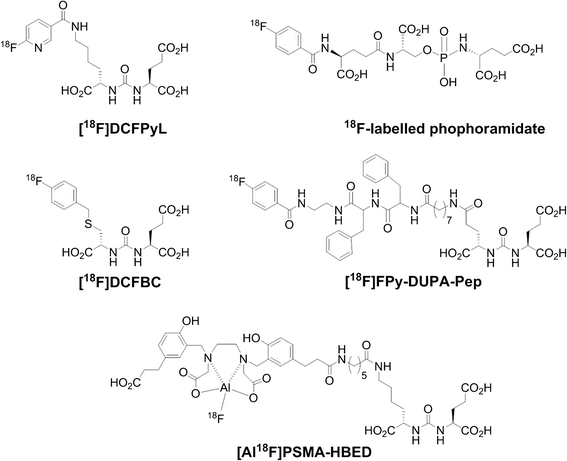
Structure of 18F-labeled small-molecule PSMA inhibitors
/////////piflufolastat F 18, injection, Orphan Drug , Prostate cancer, [18F]DCFPyL, 18F-DCFPYL, DCFPYL F-18, fda 2021, approvals 2021
- Product Quality Review(s) (PDF)
- Multi-Discipline Review (PDF)
- Proprietary Name Review(s) (PDF)
- Officer/Employee List (PDF)
- Other Review(s) (PDF)
- Risk Assessment and Risk Mitigation Review(s) (PDF)
- Administrative and Correspondence Documents (PDF)
- Label (PDF)
C1=CC(=NC=C1C(=O)NCCCCC(C(=O)O)NC(=O)NC(CCC(=O)O)C(=O)O)F

NEW DRUG APPROVALS
one time
$10.00
Pegcetacoplan
Sequence:
1ICVWQDWGAH RCTXK
Sequence:
1ICVWQDWGAH RCTXK
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| terminal mod. | Lys-15 | C-terminal amide |
| terminal mod. | Lys-15′ | C-terminal amide |
| bridge | Cys-2 – Cys-12 | disulfide bridge, dimer |
| bridge | Lys-15 – Lys-15′ | covalent bridge, dimer |
| bridge | Cys-2′ – Cys-12′ | disulfide bridge, dimer |
| uncommon | Oaa-14 | – |
| uncommon | Oaa-14′ | – |
Pegcetacoplan
ペグセタコプラン;
FDA APPROVED Empaveli, 2021/5/14
Protein Sequence
Sequence Length: 30, 15, 15multichain; modifiedPoly(oxy-1,2-ethanediyl), α-hydro-ω-hydroxy-, 15,15′-diester with N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-α-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-[2-(2-aminoethoxy)ethoxy]acetyl-N6-carboxy-L-lysinamide cyclic (2→12)-(disulfide)Polymer
Poly(oxy-1,2-ethanediyl), alpha-hydro-omega-hydroxy-, 15,15′-diester with N-acetyl-Lisoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-N6-carboxy-L-lysinamide cyclic (2�->12)-(disulfide)
O,O’-bis((S2,S12-cyclo(N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-Ltryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-L-lysinamide))-N6.15-carbonyl)polyethylene glycol(n = 800-1100)
- APL-2
- WHO 10743
| Formula | C170H248N50O47S4. (C2H4O)n3872.40 g·mol−1 |
|---|---|
| EfficacyDisease | Complement inhibitorParoxysmal nocturnal hemoglobinuria |
| CAS | 2019171-69-6 |
| Comment | Treatment of paroxysmal nocturnal hemoglobinuria (PNH), complement-mediated nephropathies, and age-related macular degeneration (AMD) |
- OriginatorApellis Pharmaceuticals
- ClassAnti-inflammatories; Anti-ischaemics; Antianaemics; Cyclic peptides; Eye disorder therapies; Polyethylene glycols; Urologics
- Mechanism of ActionComplement C3 inhibitors
- Orphan Drug StatusYes – Paroxysmal nocturnal haemoglobinuria; Autoimmune haemolytic anaemia; Glomerulonephritis
- RegisteredParoxysmal nocturnal haemoglobinuria
- Phase IIIAge-related macular degeneration
- Phase IIAmyotrophic lateral sclerosis; Autoimmune haemolytic anaemia; Glomerulonephritis; IgA nephropathy; Lupus nephritis; Membranous glomerulonephritis
- Phase I/IIWet age-related macular degeneration
- DiscontinuedIschaemia
- 02 Jun 2021Apellis Pharmaceuticals plans a phase III trial for Glomerulonephritis in the second half of 2021
- 25 May 2021Top-line efficacy and safety results from the phase III PRINCE trial for Paroxysmal nocturnal haemoglobinuria released by Apellis Pharmaceuticals
- 18 May 2021Registered for Paroxysmal nocturnal haemoglobinuria in USA (SC) – First global approval
Pegcetacoplan, sold under the brand name Empaveli, is a medication used to treat paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
The most common side effects include injection-site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.[2]
Paroxysmal nocturnal hemoglobinuria is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells).[1]
Pegcetacoplan is the first treatment for paroxysmal nocturnal hemoglobinuria that binds to complement protein C3.[1] Pegcetacoplan was approved for medical use in the United States in May 2021.[1][3]
Pegcetacoplan is a complement inhibitor indicated in the treatment of paroxysmal nocturnal hemoglobinuria (PNH).5,7 Prior to its FDA approval, patients with PNH were typically treated with the C5 inhibiting monoclonal antibody eculizumab.5 Patients given eculizumab experienced less hemolysis caused by the membrane attack complex, but were still somewhat susceptible to hemolysis caused by C3b opsonization.5,6 Pegcetacoplan was developed out of a need for an inhibitor of complement mediated hemolysis further upstream of C5.5,6 Pegcetacoplan is a pegylated C3 inhibitor that can disrupt the processes leading to both forms of hemolysis that threaten patients with PNH.5
Pegcetacoplan was granted FDA approval on 14 May 2021.7
Medical uses
Pegcetacoplan is indicated to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
EMPAVELI contains pegcetacoplan, a complement inhibitor. Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kiloDalton (kDa) PEG molecule. The peptide portions of pegcetacoplan contain 1-methyl-L-tryptophan (Trp(Me)) in position 4 and amino(ethoxyethoxy)acetic acid (AEEA) in position 14.
The molecular weight of pegcetacoplan is approximately 43.5 kDa. The molecular formula is C1970H3848N50O947S4. The structure of pegcetacoplan is shown below.
 |
EMPAVELI injection is a sterile, clear, colorless to slightly yellowish aqueous solution for subcutaneous use and is supplied in a 20-mL single-dose vial. Each 1 mL of solution contains 54 mg of pegcetacoplan, 41 mg of sorbitol, 0.384 mg of glacial acetic acid, 0.490 mg of sodium acetate trihydrate, and Water for Injection USP. EMPAVELI may also contain sodium hydroxide and/or additional glacial acetic acid for adjustment to a target pH of 5.0.
FDA approves new treatment for adults with serious rare blood disease..
FDA has approved Empaveli (pegcetacoplan) injection to treat adults with paroxysmal nocturnal hemoglobinuria (PNH), a rare, life-threatening blood disease. Empaveli is the first PNH treatment that binds to compliment protein C3.
PNH is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells). The disease affects 1-1.5 people per million. Individuals are typically diagnosed around ages 35 to 40. PNH can be serious, with median survival of 10 years after diagnosis. However, some patients live for decades with only minor symptoms.
PNH is caused by gene mutations that affect red blood cells. Red blood cells in people with these mutations are defective and can be destroyed by the immune system, which causes anemia.
The effectiveness of Empaveli was evaluated in a study enrolling 80 patients with PNH and anemia who had been taking eculizumab, a treatment previously approved for PNH. Patients first completed a four-week period during which they received Empaveli 1,080 mg twice weekly in addition to eculizumab at their previous dose. After the first four weeks, patients were randomly assigned to receive either Empaveli or their current dose of eculizumab for 16 weeks.
After 16 weeks, the severity of anemia was compared in the two treatment groups on the basis of hemoglobin concentration (a laboratory measure of anemia). In both treatment groups, the average hemoglobin was 8.7 g/dL at baseline, indicating severe anemia. (Normal hemoglobin values in adult men are 14 g/dL or above; normal values in adult women are 12 g/dL or above.) During the 16 weeks of treatment, patients in the Empaveli group had an average increase in their hemoglobin of 2.4 g/dL. Meanwhile, patients in the eculizumab group had an average decrease in their hemoglobin of 1.5 g/dL.
Empaveli is available only through a restricted program under a risk evaluation and mitigation strategy. Meningococcal (a type of bacteria) infections can occur in patients taking Empaveli and can become life-threatening or fatal if not treated early. Empaveli may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria. Patients should be monitored for infusion-related reactions. Empaveli can interfere with certain laboratory tests. The most common side effects are injection site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.
Empaveli received priority review, fast track and orphan drug designations for this indication.
FDA granted the approval of Empaveli to Apellis Pharmaceuticals.
Adverse effects
Meningococcal (a type of bacteria) infections can occur in people taking pegcetacoplan and can become life-threatening or fatal if not treated early.[1] Pegcetacoplan may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria.[1]
History
The effectiveness of pegcetacoplan was evaluated in a study enrolling 80 participants with paroxysmal nocturnal hemoglobinuria and anemia who had been taking eculizumab, a treatment previously approved for paroxysmal nocturnal hemoglobinuria.[1]
References
- ^ Jump up to:a b c d e f g h i “FDA approves new treatment for adults with serious rare blood disease”. U.S. Food and Drug Administration (FDA). 14 May 2021. Retrieved 14 May 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d https://pi.apellis.com/files/PI_Empaveli.pdf
- ^ “Apellis Announces U.S. Food and Drug Administration (FDA) Approval of Empaveli (pegcetacoplan) for Adults with Paroxysmal Nocturnal Hemoglobinuria (PNH)” (Press release). Apellis Pharmaceuticals. 14 May 2021. Retrieved 14 May 2021 – via GlobeNewswire.
External links
- “Pegcetacoplan”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03500549 for “Study to Evaluate the Efficacy and Safety of APL-2 in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Empaveli |
| Other names | APL-2 |
| License data | US DailyMed: Pegcetacoplan |
| Routes of administration | Subcutaneous infusion |
| Drug class | Complement inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2019171-69-6 |
| UNII | TO3JYR3BOU |
| KEGG | D11613 |
| ChEMBL | ChEMBL4298211 |
| Chemical and physical data | |
| Formula | C170H248N50O47S4 |
| Molar mass | 3872.40 g·mol−1 |
/////////Pegcetacoplan, ペグセタコプラン , FDA 2021, APPROVALS 2021, APL-2, WHO 10743, Apellis Pharmaceuticals, Empaveli, priority review, fast track, orphan drug
https://www.sec.gov/Archives/edgar/data/1492422/000156459020007350/apls-10k_20191231.htm
NEW DRUG APPROVALS
ONE TIME
$10.00
Sotorasib

![4-((S)-4-Acryloyl-2-methylpiperazin-1-yl)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=137278711&t=l)
Sotorasib
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-propan-2-ylpyridin-3-yl)-4-[(2S)-2-methyl-4-prop-2-enoylpiperazin-1-yl]pyrido[2,3-d]pyrimidin-2-one
AMG 510
AMG-510
AMG510
| Formula | C30H30F2N6O3 |
|---|---|
| CAS | 2296729-00-3 |
| Mol weight | 560.5944 |
FDA APPROVED, 2021/5/28 Lumakras
Antineoplastic, Non-small cell lung cancer (KRAS G12C-mutated)
ソトラシブ (JAN);
Sotorasib
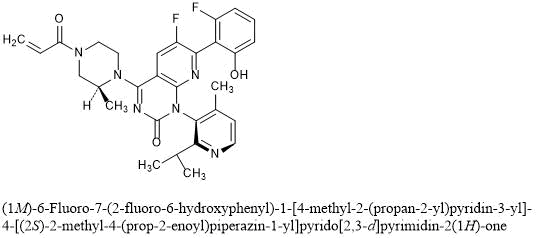
(1M)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one
C30H30F2N6O3 : 560.59
[2296729-00-3]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
 |
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras™, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
View full prescribing information for Lumakras.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
This application was granted priority review, fast-track, breakthrough therapy and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Sotorasib, sold under the brand name Lumakras is an anti-cancer medication used to treat non-small-cell lung cancer (NSCLC).[1][2] It targets a specific mutation, G12C, in the protein KRAS which is responsible for various forms of cancer.[3][4]
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
The U.S. Food and Drug Administration (FDA) granted the application for sotorasib orphan drug, fast track, priority review, and breakthrough therapy designations.[2] The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA).[2] The application reviews are ongoing at the other regulatory agencies.[2]
The FDA granted approval of Lumakras to Amgen Inc.[2]
Society and culture
Economics
Sotorasib costs US$17,900 per month.[5]
Names
Sotorasib is the recommended international nonproprietary name (INN).[14]
PAPER
Nature (London, United Kingdom) (2019), 575(7781), 217-223
https://www.nature.com/articles/s41586-019-1694-1
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310540

KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180

KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
AMG 510 [(R)-38]. (1R)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one
………… concentrated in vacuo. Chromatographic purification of the residue (silica gel; 0–100% 3:1 EtOAc–EtOH/heptane) followed by chiral supercritical fluid chromatography (Chiralpak IC, 30 mm × 250 mm, 5 μm, 55% MeOH/CO2, 120 mL/min, 102 bar) provided (1R)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510; (R)-38; 2.25 g, 43% yield) as the first-eluting peak. 1H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ −115.6 (d, J = 5.2 Hz, 1 F), −128.6 (br s, 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M + H]+ calcd for C30H30F2N6O3 561.24202. Found 561.24150.

d (1R)-6-Fluoro7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1- piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one ((R)-38; AMG 510; 2.25 g, 43% yield) as the first-eluting peak.1 H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ –115.6 (d, J = 5.2 Hz, 1 F), –128.6 (br. s., 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M+H]+ Calcd for C30H30F2N6O3 561.24202; Found 561.24150. Atropisomer configuration (R vs. S) assigned crystallographically.The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180.
PATENT
WO 2021097212
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
PATENT
WO 2020102730
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020102730
PATENT
US 20180334454
References
- ^ Jump up to:a b c d e “Lumakras- sotorasib tablet, coated”. DailyMed. Retrieved 6 June 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy”. U.S. Food and Drug Administration (FDA). 28 May 2021. Retrieved 28 May 2021. This article incorporates text from this source, which is in the public domain.
- ^ “KRAS mutant-targeting AMG 510”. NCI Drug Dictionary. National Cancer Institute. 2 February 2011. Retrieved 16 November2019.
- ^ Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (November 2019). “The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity”. Nature. 575 (7781): 217–23. Bibcode:2019Natur.575..217C. doi:10.1038/s41586-019-1694-1. PMID 31666701.
- ^ Jump up to:a b “FDA approves Amgen drug for lung cancer with specific mutation”. CNBC. 28 May 2021. Retrieved 28 May 2021.
- ^ Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). “KRASG12C inhibition with sotorasib in advanced solid tumors”. N Engl J Med. doi:10.1056/NEJMoa1917239. PMC 7571518.
- ^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
- ^ “The Discovery Of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published In Nature” (Press release). Amgen. 30 October 2019. Retrieved 16 November 2019.
- ^ Irving M (24 December 2019). “Drug targeting common cancer cause enters phase 2 clinical trials”. New Atlas. Retrieved 24 December 2019.
- ^ Jump up to:a b c d Halford B (3 April 2019). “Amgen unveils its KRas inhibitor in human clinical trials: AMG 510 shuts down a mutant version of the cancer target via covalent interaction”. Chemical & Engineering News. 97 (4). Retrieved 16 November 2019.
- ^ Al Idrus A (9 September 2019). “Amgen’s KRAS drug continues to deliver but faces ‘curse’ of high expectations”. fiercebiotech.com. Retrieved 16 November 2019.
- ^ Kaiser J (30 October 2019). “Two new drugs finally hit ‘undruggable’ cancer target, providing hope for treatments”. Science Magazine. AAAS. Retrieved 16 November 2019.
- ^ Astor L (9 September 2019). “FDA Grants AMG 510 Fast Track Designation for KRAS G12C+ NSCLC”. targetedonc.com. Retrieved 16 November 2019.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85” (PDF). WHO Drug Information. 35 (1).
Further reading
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (September 2020). “KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors”. N Engl J Med. 383 (13): 1207–17. doi:10.1056/NEJMoa1917239. PMC 7571518. PMID 32955176.
- Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. (January 2020). “Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors”. J Med Chem. 63 (1): 52–65. doi:10.1021/acs.jmedchem.9b01180. PMID 31820981.
External links
- “Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lumakras |
| Other names | AMG 510 |
| License data | US DailyMed: Sotorasib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2252403-56-6 |
| PubChem CID | 137278711 |
| DrugBank | DB15569 |
| ChemSpider | 72380148 |
| UNII | 2B2VM6UC8G |
| KEGG | D12055 |
| Chemical and physical data | |
| Formula | C30H30F2N6O3 |
| Molar mass | 560.606 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////Sotorasib, ソトラシブ , FDA 2021, APPROVALS 2021, Lumakras, CANCER, ANTINEOPLASTIC, AMG 510, AMG-510, AMG510, AMGEN, priority review, fast-track, breakthrough therapy, orphan drug
CC1CN(CCN1C2=NC(=O)N(C3=NC(=C(C=C32)F)C4=C(C=CC=C4F)O)C5=C(C=CN=C5C(C)C)C)C(=O)C=C
Sotorasib
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-propan-2-ylpyridin-3-yl)-4-[(2S)-2-methyl-4-prop-2-enoylpiperazin-1-yl]pyrido[2,3-d]pyrimidin-2-one
AMG 510
AMG-510
AMG510
| Formula | C30H30F2N6O3 |
|---|---|
| CAS | 2296729-00-3 |
| Mol weight | 560.5944 |
FDA APPROVED, 2021/5/28 Lumakras
Antineoplastic, Non-small cell lung cancer (KRAS G12C-mutated)
ソトラシブ (JAN);
Sotorasib
(1M)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one
C30H30F2N6O3 : 560.59
[2296729-00-3]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
|
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras™, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
View full prescribing information for Lumakras.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
This application was granted priority review, fast-track, breakthrough therapy and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Sotorasib, sold under the brand name Lumakras is an anti-cancer medication used to treat non-small-cell lung cancer (NSCLC).[1][2] It targets a specific mutation, G12C, in the protein KRAS which is responsible for various forms of cancer.[3][4]
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
The U.S. Food and Drug Administration (FDA) granted the application for sotorasib orphan drug, fast track, priority review, and breakthrough therapy designations.[2] The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA).[2] The application reviews are ongoing at the other regulatory agencies.[2]
The FDA granted approval of Lumakras to Amgen Inc.[2]
Society and culture
Economics
Sotorasib costs US$17,900 per month.[5]
Names
Sotorasib is the recommended international nonproprietary name (INN).[14]
PAPER
Nature (London, United Kingdom) (2019), 575(7781), 217-223
https://www.nature.com/articles/s41586-019-1694-1
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310540
KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180
KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
AMG 510 [(R)-38]. (1R)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one
………… concentrated in vacuo. Chromatographic purification of the residue (silica gel; 0–100% 3:1 EtOAc–EtOH/heptane) followed by chiral supercritical fluid chromatography (Chiralpak IC, 30 mm × 250 mm, 5 μm, 55% MeOH/CO2, 120 mL/min, 102 bar) provided (1R)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510; (R)-38; 2.25 g, 43% yield) as the first-eluting peak. 1H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ −115.6 (d, J = 5.2 Hz, 1 F), −128.6 (br s, 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M + H]+ calcd for C30H30F2N6O3 561.24202. Found 561.24150.
d (1R)-6-Fluoro7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1- piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one ((R)-38; AMG 510; 2.25 g, 43% yield) as the first-eluting peak.1 H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ –115.6 (d, J = 5.2 Hz, 1 F), –128.6 (br. s., 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M+H]+ Calcd for C30H30F2N6O3 561.24202; Found 561.24150. Atropisomer configuration (R vs. S) assigned crystallographically.The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180.
PATENT
WO 2021097212
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
PATENT
WO 2020102730
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020102730
PATENT
US 20180334454
References
- ^ Jump up to:a b c d e “Lumakras- sotorasib tablet, coated”. DailyMed. Retrieved 6 June 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy”. U.S. Food and Drug Administration (FDA). 28 May 2021. Retrieved 28 May 2021. This article incorporates text from this source, which is in the public domain.
- ^ “KRAS mutant-targeting AMG 510”. NCI Drug Dictionary. National Cancer Institute. 2 February 2011. Retrieved 16 November2019.
- ^ Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (November 2019). “The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity”. Nature. 575 (7781): 217–23. Bibcode:2019Natur.575..217C. doi:10.1038/s41586-019-1694-1. PMID 31666701.
- ^ Jump up to:a b “FDA approves Amgen drug for lung cancer with specific mutation”. CNBC. 28 May 2021. Retrieved 28 May 2021.
- ^ Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). “KRASG12C inhibition with sotorasib in advanced solid tumors”. N Engl J Med. doi:10.1056/NEJMoa1917239. PMC 7571518.
- ^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
- ^ “The Discovery Of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published In Nature” (Press release). Amgen. 30 October 2019. Retrieved 16 November 2019.
- ^ Irving M (24 December 2019). “Drug targeting common cancer cause enters phase 2 clinical trials”. New Atlas. Retrieved 24 December 2019.
- ^ Jump up to:a b c d Halford B (3 April 2019). “Amgen unveils its KRas inhibitor in human clinical trials: AMG 510 shuts down a mutant version of the cancer target via covalent interaction”. Chemical & Engineering News. 97 (4). Retrieved 16 November 2019.
- ^ Al Idrus A (9 September 2019). “Amgen’s KRAS drug continues to deliver but faces ‘curse’ of high expectations”. fiercebiotech.com. Retrieved 16 November 2019.
- ^ Kaiser J (30 October 2019). “Two new drugs finally hit ‘undruggable’ cancer target, providing hope for treatments”. Science Magazine. AAAS. Retrieved 16 November 2019.
- ^ Astor L (9 September 2019). “FDA Grants AMG 510 Fast Track Designation for KRAS G12C+ NSCLC”. targetedonc.com. Retrieved 16 November 2019.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85” (PDF). WHO Drug Information. 35 (1).
Further reading
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (September 2020). “KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors”. N Engl J Med. 383 (13): 1207–17. doi:10.1056/NEJMoa1917239. PMC 7571518. PMID 32955176.
- Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. (January 2020). “Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors”. J Med Chem. 63 (1): 52–65. doi:10.1021/acs.jmedchem.9b01180. PMID 31820981.
External links
- “Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lumakras |
| Other names | AMG 510 |
| License data | US DailyMed: Sotorasib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2252403-56-6 |
| PubChem CID | 137278711 |
| DrugBank | DB15569 |
| ChemSpider | 72380148 |
| UNII | 2B2VM6UC8G |
| KEGG | D12055 |
| Chemical and physical data | |
| Formula | C30H30F2N6O3 |
| Molar mass | 560.606 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////Sotorasib, ソトラシブ , FDA 2021, APPROVALS 2021, Lumakras, CANCER, ANTINEOPLASTIC, AMG 510, AMG-510, AMG510, AMGEN, priority review, fast-track, breakthrough therapy, orphan drug
CC1CN(CCN1C2=NC(=O)N(C3=NC(=C(C=C32)F)C4=C(C=CC=C4F)O)C5=C(C=CN=C5C(C)C)C)C(=O)C=C
NEW DRUG APPROVALS
ONE TIME
$10.00
Infigratinib phosphate


Infigratinib phosphate
FDA APPR Truseltiq 2021/5/28
インフィグラチニブリン酸塩;
3-(2,6-dichloro-3,5-dimethoxyphenyl)-1-[6-[4-(4-ethylpiperazin-1-yl)anilino]pyrimidin-4-yl]-1-methylurea;phosphoric acid
- BGJ 398
- BGJ-398
- BGJ398
- NVP-BGJ398
- WHO 10032
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Infigratinib acetate | 03D0789NYP | 1310746-17-8 | XHCQHOGMMJKLRU-UHFFFAOYSA-N |
| Infigratinib hydrochloride | WY8VD4RV77 | 1310746-15-6 | VBAIJSJSFCXDJB-UHFFFAOYSA-N |
| Infigratinib mesylate | E223Z0KWCC | 1310746-12-3 | BXJJFNXYWJLBOS-UHFFFAOYSA-N |
| Infigratinib phosphate | 58BH47BV6S | 1310746-10-1 | GUQNHCGYHLSITB-UHFFFAOYSA-N |
International/Other BrandsTruseltiq (BridgeBio Pharma, Inc.)
| Formula | C26H31Cl2N7O3. H3PO4 |
|---|---|
| CAS | 1310746-10-1FREE form 872511-34-7 |
| Mol weight | 658.4706 |
- Originator Novartis
- Developer Array BioPharma; Novartis; Novartis Oncology; QED Therapeutics
- Class Aniline compounds; Antineoplastics; Chlorobenzenes; Methylurea compounds; Phenyl ethers; Piperazines; Pyrimidines; Small molecules
- Mechanism of Action Type 1 fibroblast growth factor receptor antagonists; Type 3 fibroblast growth factor receptor antagonists; Type 4 fibroblast growth factor receptor antagonists; Type-2 fibroblast growth factor receptor antagonists
- Orphan Drug Status Yes – Cholangiocarcinoma
- RegisteredCholangiocarcinoma
- Phase IIIBladder cancer; Urogenital cancer
- Phase IIAchondroplasia; Head and neck cancer
- Phase IBreast cancer
- Phase 0Glioblastoma
- DiscontinuedHaematological malignancies; Malignant melanoma; Solid tumours
- 31 May 2021Clinical development is ongoing in Bladder cancer (QED Therapeutics pipeline, May 2020)
- 28 May 2021Registered for Cholangiocarcinoma (Second-line therapy or greater, Metastatic disease, Inoperable/Unresectable, Late-stage disease) in USA (PO) – First global approval (under Project Orbis using RTOR program)
- 28 May 2021Efficacy and safety data from a phase II trial in Cholangiocarcinoma released by QED Therapeutics
Infigratinib, sold under the brand name Truseltiq, is an anti-cancer medication used to treat cholangiocarcinoma (bile duct cancer).[1][2]
Infigratinib is a receptor tyrosine kinase inhibitor (and more specifically an inhibitor of the fibroblast growth factor receptors FGFR1, FGFR2, FGFR3).[3][1][2] It was designated an orphan drug by the U.S. Food and Drug Administration (FDA) in 2019,[4] and it was approved for medical use in the United States in May 2021.[2]
Infigratinib is a pan-fibroblast growth factor receptor (FGFR) kinase inhibitor. By inhibiting the FGFR pathway, which is often aberrated in cancers such as cholangiocarcinoma, infigratinib suppresses tumour growth.1 Cholangiocarcinoma is the most common primary malignancy affecting the biliary tract and the second most common primary hepatic malignancy.2 Infitratinib is a pan-FGFR inhibitor, as it is an ATP-competitive inhibitor of all four FGFR receptor subtypes.1
On May 28, 2021, the FDA granted accelerated approval to infigratinib – under the market name Truseltiq – for the treatment of previously treated, unresectable locally advanced or metastatic cholangiocarcinoma in adults with a fibroblast growth factor receptor 2 (FGFR2) fusion or another rearrangement as detected by an FDA-approved test.5 This approval follows pemigatinib, another FGFR inhibitor approved by the FDA for the same therapeutic indication.
Infigratinib is indicated for the treatment of previously treated, unresectable locally advanced or metastatic cholangiocarcinoma in adults with a fibroblast growth factor receptor 2 (FGFR2) fusion or another rearrangement as detected by an FDA-approved test.4
Medical uses
Infigratinib is indicated for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma (bile duct cancer) with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test.[1]
PAPER
Journal of Medicinal Chemistry (2011), 54(20), 7066-7083.
https://pubs.acs.org/doi/10.1021/jm2006222

A novel series of N-aryl-N′-pyrimidin-4-yl ureas has been optimized to afford potent and selective inhibitors of the fibroblast growth factor receptor tyrosine kinases 1, 2, and 3 by rationally designing the substitution pattern of the aryl ring. On the basis of its in vitro profile, compound 1h (NVP-BGJ398) was selected for in vivo evaluation and showed significant antitumor activity in RT112 bladder cancer xenografts models overexpressing wild-type FGFR3. These results support the potential therapeutic use of 1h as a new anticancer agent.

PATENT
US 9067896
PATENT
WO 2020243442
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020243442
In 2018, it was estimated that 150,350 new patients would be diagnosed with urinary system cancer: 81,190 urinary bladder; 65,340 kidney and renal pelvis; and, 3,820 ureter and other urinary organs. Excluding non-urothelial kidney cancers, approximately 5 to 10% of all urothelial carcinomas are upper tract urothelial carcinomas (UTUC). The incidence of UTUC is 2 to 3 times greater in men than women (Siegel et al, 2018; Roupret et al, 2015).
[0003] In contrast to invasive urinary bladder cancer (UCB), UTUC has a more aggressive clinical course. At the time of diagnosis, 60% of patients with UTUC have invasive cancer compared to 15% to 25% of patients with UCB (Roupret et al, 2015; Margulis et al., 2009). Thirty-six percent have regional disease and 9% distant disease (Raman et al., 2010). A large retrospective review of 1363 patients with UTUC who underwent radical nephroureterectomy (RNU) at 12 centers demonstrated that 28% of the total population had recurrence after RNU (Margulis et al, 2009).
[0004] To reduce the morbidity and mortality in patients with UTUC, neoadjuvant or adjuvant treatment is needed. The POUT study, a large randomized trial in UTUC supports the use of standard-of-care adjuvant cisplatin-based chemotherapy (Birtle et al., 2020). Because many patients with UTUC will have one remaining kidney following RNU and frequently have other significant co-morbid conditions, cisplatin-based therapy is not well tolerated (NCCN Guidelines Version 3, 2018). Renal function before and after RNU greatly limits the number of patients with UTUC who are eligible for platinum-based neoadjuvant or adjuvant therapy. Therefore, targeted therapies are needed for treating UTUC (Lane et al., 2010).
[0005] Despite demonstrated survival benefit for neoadjuvant treatment of invasive UCB, many patients with invasive UCB are unlikely to receive (neo)adjuvant cisplatin-based chemotherapy, due in part to cisplatin ineligibility (Porter et al., 2011). In addition, residual disease following neoadjuvant therapy is associated with a poor prognosis (Grossman et al, 2003). Therefore,
there remains an unmet need for a substantial proportion of patients with invasive UCB who are ineligible or refuse to receive cisplatin-based adjuvant chemotherapy or who have residual disease following neoadjuvant therapy.
Infigratinib, as depicted in formula (I), is a selective and ATP-competitive pan-fibroblast growth factor receptor (FGFR) kinase inhibitor, also known as 3-(2,6-dichloro-3,5-dimethoxyphenyl)- 1 – { 6- [4-(4-ethyl- 1 -piperazin- 1 -yljphenylamino] -pyrimidinyl-4-yl } – 1 -methylurea. Infigratinib selectively inhibits the kinase activity of FGFR1, FGFR2, FGFR3, and
FGFR4.
PATENT
WO 2011071821
https://patents.google.com/patent/WO2011071821A1/en3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-l-{6-[4-(4-ethyl-piperazin-l-yl)- phenylamino]-pyrimidin-4-yl}-l -methyl-urea (described in USSN 11/570983, filed June 23, 2005, and incorporated by reference in its entirety herein) has the structure of Formula I:

The compound of Formula I is a protein kinase inhibitor and is useful in the treatment of proliferative diseases mediated by protein kinases. In particular, the compound of Formula I inhibits FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2, and Ret kinases. It is therefore useful in the treatment of cancers including AML, melanocyte neoplasia, breast cancer, colon cancer, lung cancer (especially small-cell lung cancer), cancer of the prostate or Kaposi’s sarcoma.[0003] It is well known that the crystalline form of the active pharmaceutical ingredient (API) of a particular drug is often an important determinant of the drug’s ease of preparation, hygroscopicity, stability, solubility, storage stability, ease of formulation, rate of dissolution in gastrointestinal fluids and in vivo bioavailability. Crystalline forms occur where the same composition of matter crystallizes in a different lattice arrangement resulting in different thermodynamic properties and stabilities specific to the particular crystalline form.Crystalline forms may also include different hydrates or solvates of the same compound. In deciding which form is preferable, the numerous properties of the forms are compared and the preferred form chosen based on the many physical property variables. It is entirely possible that one form can be preferable in some circumstances where certain aspects such as ease of preparation, stability, etc. are deemed to be critical. In other situations, a different form may be preferred for greater dissolution rate and/or superior bioavailability. It is not yet possible to predict whether a particular compound or salt of a compound will form polymorphs, whether any such polymorphs will be suitable for commercial use in a therapeutic composition, or which polymorphs will display such desirable properties.Example 2: Manufacture of the Free Base of the Compound of Formula I

IA. N- [4-(4-ethyl-piperazin- 1 -yl)-phenyl] -N’ -methyl-pyrimidine-4,6-diamine[0077] A mixture of 4-(4-ethylpiperazin-l-yl)-aniline (1 g, 4.88 mmol), (6-chloro- pyrimidin-4-yl)-methyl-amine (1.81 g, 12.68 mmol, 1.3 eq.), and 4N HC1 in dioxane (15 ml) is heated in a sealed tube to 150°C for 5h. The reaction mixture is concentrated, diluted with DCM and a saturated aqueous solution of sodium bicarbonate. The aqueous layer is separated and extracted with DCM. The organic phase is washed with brine, dried (sodium sulfate), filtered and concentrated. Purification of the residue by silica gel column chromatography (DCM/MeOH, 93:7) followed by trituration in diethyl ether affords the title compound as a white solid: ESI-MS: 313.2 [MH]+; tR= 1.10 min (gradient J); TLC: Rf = 0.21 (DCM/MeOH, 93:7).B. 4-(4-Ethylpiperazin- 1 -yl)-aniline[0078] A suspension of l-ethyl-4-(4-nitro-phenyl)-piperazine (6.2 g, 26.35 mmol) and Raney Nickel (2 g) in MeOH (120 mL) is stirred for 7 h at RT, under a hydrogen atmosphere. The reaction mixture is filtered through a pad of celite and concentrated to afford 5.3 g of the title compound as a violet solid: ESI-MS: 206.1 [MH]+; TLC: Rf = 0.15 (DCM/MeOH + 1 % NH3aq, 9:l).C. 1 -Ethyl-4-(4-nitro-phenyl)-piperazine[0079] A mixture of l-bromo-4-nitrobenzene (6 g, 29.7 mmol) and 1-ethylpiperazine (7.6 ml, 59.4 mmol, 2 eq.) is heated to 80°C for 15h. After cooling to RT, the reaction mixture is diluted with water and DCM/MeOH, 9:1. The aqueous layer is separated and extracted with DCM/MeOH, 9:1. The organic phase is washed with brine, dried (sodium sulfate), filtered and concentrated. Purification of the residue by silica gel column chromatography(DCM MeOH + 1 % NH3aq, 9:1) affords 6.2 g of the title compound as a yellow solid: ESI- MS: 236.0 [MH]+; tR= 2.35 min (purity: 100%, gradient J); TLC: Rf = 0.50 (DCM/MeOH + 1 % NH3aq, 9:1).D. (6-chloro-pyrimidin-4-yl)-methyl-amine[0080] This material was prepared by a modified procedure published in the literature (J. Appl. Chem. 1955, 5, 358): To a suspension of commercially available 4,6- dichloropyrimidine (20 g, 131.6 mmol, 1.0 eq.) in isopropanol (60 ml) is added 33% methylamine in ethanol (40.1 ml, 328.9 mmol, 2.5 eq.) at such a rate that the internal temperature does not rise above 50°C. After completion of the addition the reaction mixture was stirred for lh at room temperature. Then, water (50 ml) is added and the suspension formed is chilled in an ice bath to 5°C. The precipitated product is filtered off, washed with cold isopropanol/water 2:1 (45 ml) and water. The collected material is vacuum dried over night at 45°C to afford the title compound as colorless powder: tR = 3.57 min (purity: >99%, gradient A), ESI-MS: 144.3 / 146.2 [MH]+.E. (3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-l-{6-[4-(4-ethyl-piperazin-l-yl)-phenylamino]- pyrimidin-4-yl} – 1 -methyl-urea)[0081] The title compound was prepared by adding 2,6-dichloro-3,5-dimethoxyphenyl- isocyanate (1.25 eq.) to a solution of N-[4-(4-ethyl-piperazin-l-yl)-phenyl]-N’-methyl- pyrimidine-4,6-diamine (2.39 g, 7.7 mmol, 1 eq.) in toluene and stirring the reaction mixture for 1.5h at reflux. Purification of the crude product by silica gel column chromatography (DCM MeOH + 1 % NH3aq, 95:5) affords the title compound as a white solid: ESI-MS: 560.0 / 561.9 [MHf; tR= 3.54 min (purity: 100%, gradient J); TLC: Rf = 0.28 (DCM/MeOH + 1 % NH3aq, 95:5). Analysis: C26H3iN703Cl2, calc. C 55.72% H 5.57% N 17.49% O 8.56% CI 12.65%; found C 55.96% H 5.84% N 17.17% O 8.46% CI 12.57%. The title compound was characterized by XRPD, thermal and other methods as described below. Example 3: Manufacture of the Monophosphoric Acid Salt Form A of the Compound of Formula I.[0082] To a round bottom flask was added 3-(2,6-dichloro-3,5-dimethoxyphenyl)-l-{6-[4- (4-ethylpiperazin-l-yl)phenylamino]-pyrimidine-4-yl}-l -methyl-urea (134 g, 240 mmol) and IPA (2000 ml). The suspension was stirred and heated to 50°C and a solution of phosphoric acid (73.5 g, 750 mmol) in water (2000 ml) added to it portions. The mixture was stirred at 60°C for 30 min. and filtered through a polypropylene pad. The pad was washed with warm IP A/water (1:1, 200 ml) and the filtrates were combined. To this clear solution, IPA (6000 ml) was added and the mixture was stirred under reflux for 20 min, cooled slowly to room temperature (25° C), and stirred for 24 hours. The white salt product was collected by filtration, washed with IPA (2 χ 500 ml) and dried in the oven at 60° C under reduced pressure for two days to provide the phosphate salt (form A) 110 g. Yield 70%. Purity >98% by HPLC. Analysis: C26H34 707C12P, calc. C 47.42% H 5.20% N 14.89% O 17.01% CI 10.77% P 4.70%; found C 47.40% H 5.11% N 14.71% O 17.18% CI 10.73% P 4.87%. The title compound was characterized by XRPD, thermal and other methods as described below.
References
- ^ Jump up to:a b c d “Infigratinib prescribing information” (PDF). U.S. Food and Drug Administration. May 2021.
- ^ Jump up to:a b c “BridgeBio Pharma’s Affiliate QED Therapeutics and Partner Helsinn Group Announce FDA Approval of Truseltiq (infigratinib) for Patients with Cholangiocarcinoma” (Press release). BridgeBio Pharma. 28 May 2021. Retrieved 28 May 2021 – via GlobeNewswire.
- ^ Botrus G, Raman P, Oliver T, Bekaii-Saab T (April 2021). “Infigratinib (BGJ398): an investigational agent for the treatment of FGFR-altered intrahepatic cholangiocarcinoma”. Expert Opinion on Investigational Drugs. 30 (4): 309–316. doi:10.1080/13543784.2021.1864320. PMID 33307867.
- ^ “Infigratinib Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 11 September 2019. Retrieved 30 May 2021.
External links
- “Infigratinib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02150967 for “A Phase II, Single Arm Study of BGJ398 in Patients With Advanced Cholangiocarcinoma” at ClinicalTrials.gov
| Efficacy | Antineoplastic, Angiogenesis inhibitor |
|---|---|
| Disease | Cholangiocarcinoma (FGFR2 fusion or other rearrangement) |
| Clinical data | |
|---|---|
| Trade names | Truseltiq |
| Other names | BGJ-398 |
| License data | US DailyMed: Infigratinib |
| Routes of administration | By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 872511-34-7 |
| PubChem CID | 53235510 |
| DrugBank | DB11886 |
| ChemSpider | 26333103 |
| UNII | A4055ME1VK |
| KEGG | D11589 |
| CompTox Dashboard (EPA) | DTXSID70236238 |
| Chemical and physical data | |
| Formula | C26H31Cl2N7O3 |
| Molar mass | 560.48 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCCN1CCN(CC1)C2=CC=C(C=C2)NC3=CC(=NC=N3)N(C)C(=O)NC4=C(C(=CC(=C4Cl)OC)OC)Cl | |
| hideInChIInChI=1S/C26H31Cl2N7O3/c1-5-34-10-12-35(13-11-34)18-8-6-17(7-9-18)31-21-15-22(30-16-29-21)33(2)26(36)32-25-23(27)19(37-3)14-20(38-4)24(25)28/h6-9,14-16H,5,10-13H2,1-4H3,(H,32,36)(H,29,30,31)Key:QADPYRIHXKWUSV-UHFFFAOYSA-N |
////////Infigratinib phosphate, FDA 2021 APPROVALS 2021, Truseltiq, インフィグラチニブリン酸塩 , Orphan Drug, Cholangiocarcinoma, BGJ 398, BGJ-398, BGJ398, NVP-BGJ398, WHO 10032

NEW DRUG APPROVALS
one time
$10.00
Ibrexafungerp citrate, Brexafemme
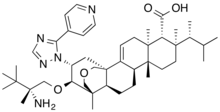

Ibrexafungerp citrate
| アイブレキサフンジェルプクエン酸塩; |
| Formula | C44H67N5O4. C6H8O7 |
|---|---|
| cas | Citrate1965291-08-0 free 1207753-03-4 |
| Mol weight | 922.1574 |
Brexafemme, fda approved 2021, 2021/6/1
Antifungal, Cell wall biosynthesis inhibitor, Treatment of invasive fungal infections due to Candida spp. or Aspergillus spp., vulvovaginal candidiasis
SCY-078 citrate, MK-3118; SCY-078,
- WHO 10597
(1R,5S,6R,7R,10R,11R,14R,15S,20R,21R)-21-[(2R)-2-amino-2,3,3-trimethylbutoxy]-5,7,10,15-tetramethyl-7-[(2R)-3-methylbutan-2-yl]-20-(5-pyridin-4-yl-1,2,4-triazol-1-yl)-17-oxapentacyclo[13.3.3.01,14.02,11.05,10]henicos-2-ene-6-carboxylic acid;2-hydroxypropane-1,2,3-tricarboxylic acid
- Originator Merck & Co; SCYNEXIS
- Class Antifungals; Glycosides; Triterpenes
- Mechanism of ActionBeta-1,3-D glucan synthetase inhibitors
- Orphan Drug StatusYes – Invasive bronchopulmonary aspergillosis; Candidiasis
- RegisteredVulvovaginal candidiasis
- Phase IIICandidiasis
- Phase IIInvasive bronchopulmonary aspergillosis
- Phase IUnspecified
- PreclinicalPneumocystis pneumonia
- 01 Jun 2021Registered for Vulvovaginal candidiasis (In adolescents, In children, In the elderly, In adults) in USA (PO)
- 01 May 2021Ibrexafungerp – SCYNEXIS receives Qualified Infectious Disease Product status for Vulvovaginal candidiasis (Recurrent, Prevention) in USA
- 30 Apr 2021Efficacy data from phase III VANISH-303 and VANISH-306 trials in Vulvovaginal Candidiasis presented at the 2021 American College of Obstetricians and Gynecologists Annual Meeting (ACOG-2021)
Ibrexafungerp, sold under the brand name Brexafemme, is an antifungal medication used to treat vulvovaginal candidiasis (VVC) (vaginal yeast infection).[1] It is taken by mouth.[1]
Ibrexafungerp is a triterpenoid antifungal.[1]
Ibrexafungerp was approved for medical use in the United States in June 2021.[1][2] It is the first approved drug in a novel antifungal class.[2]
Medical uses
Ibrexafungerp is indicated for the treatment of adult and postmenarchal pediatric females with vulvovaginal candidiasis (VVC).[1][2]
Syn
https://www.sciencedirect.com/science/article/abs/pii/S0960894X20307721

Abstract
We previously reported medicinal chemistry efforts that identified MK-5204, an orally efficacious β-1,3-glucan synthesis inhibitor derived from the natural product enfumafungin. Further extensive optimization of the C2 triazole substituent identified 4-pyridyl as the preferred replacement for the carboxamide of MK-5204, leading to improvements in antifungal activity in the presence of serum, and increased oral exposure. Reoptimizing the aminoether at C3 in the presence of this newly discovered C2 substituent, confirmed that the (R) t-butyl, methyl aminoether of MK-5204 provided the best balance of these two key parameters, culminating in the discovery of ibrexafungerp, which is currently in phase III clinical trials. Ibrexafungerp displayed significantly improved oral efficacy in murine infection models, making it a superior candidate for clinical development as an oral treatment for Candida and Aspergillus infections.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
SYN
Bioorg. Med. Chem. Lett. 2021, 32, 127661.
PATENT
WO 2010019204
https://patents.google.com/patent/WO2010019204A1/en
SYN
https://doi.org/10.1021/acs.jmedchem.3c00501
Ibrexafungerp (Brexafemme). Ibrexafungerp (1), formerly SCY-078 or MK-3118 and developed by Scynexis Inc., is a first-in-class triterpenoid antifungal that inhibits the biosynthesis of β-(1,3)
D-glucan in the fungal cell wall. This mechanism of action provides an opportunity for the treatment
of fungal infections that are azole- or echinocandrin-resistant strains. In June 2021, ibrexafungerp received its first approval by the United States Food and Drug Administration (USFDA) for
the treatment of vulvovaginal candidiasis in adult and postmenarchal pediatric females.
24,25 Ibrexafungerp is a semisynthetic derivative of the natural product enfumafungin that
incorporates a pyridine triazole moiety on the core phenanthropyran ring system as well as a pendant 2-amino-2,3,3trimethyl-butyl ether. The drug demonstrates potent, broad spectrum activity against Candida sp. and is orally bioavailable.
As shown in Scheme 1, the synthesis of ibrexafungerp started with the natural product enfumafungin (1.1). The lactol of enfumafungin was reduced using triethylsilane and trifluoroacetic acid to give pyran 1.2. Treatmentwith H2SO4 in methanol resulted in cleavage of the glucose moiety to generate 1.3 in 87%
yield over 2 steps. Carboxylic acid 1.3 was converted to the corresponding benzyl ester upon treatment with benzyl bromide to give compound 1.4in an89%yield. Reaction of 1.4 with (R)
N-sulfonyl aziridine 1.5 (prepared as shown in Scheme 2) in the presence of potassium t-pentylate and the cation complexing agent 18-crown-6 provided ether 1.6 in 78% yield. Metal reduction with sodiumin liquid ammoniaconcurrently removed the N-sulfonyl benzyl groups to generate compound 1.7, which
was converted to hydrazine intermediate 1.8 with anhydrous hydrazine and BF32·OEt 28-30 in 1,2-dichloroethane (DCE). Cyclocondensation of 1.8 with acyl amidine derivative 1.9 upon heating in acetic acid then provided ibrexafungerp (1) in 66% yield.
Thepreparationof(R)-N-sulfonylaziridine1.5 isdescribedin Scheme 2. Condensation of 3,3-dimethylbutan-2-one (1.10)with (R)-p-toluenesulfinamide (1.11) gave an 84% yield of compound 1.12, which cyclized upon treatment with trimethylsulfoxonium chloride and n-butyllithium to give chiral toluenesulfinyl aziridine 1.13 in 64% yield. Oxidation of 1.13 with meta-chloroperoxybenzoic acid afforded the tosyl-pro
tected (R)-alpha-disubstituted aziridine 1.5..
(24) Lee, A. Ibrexafungerp: First approval. Drugs 2021, 81, 1445−
1450.
(25) Jallow, S.; Govender, N. P. Ibrexafungerp: A first-in-class oral
triterpenoid glucan synthase inhibitor. J. Fungi 2021, 7, 163.
(26) Lamoth, F.; Alexander, B. D. Antifungal activities of SCY-078
(MK-3118) and standard antifungal agents against clinical non
aspergillus mold isolates. Antimicrob. Agents Chemother. 2015, 59,
4308−4311
(27) Scorneaux, B.; Angulo, D.; Borroto-Esoda, K.; Ghannoum, M.;
Peel, M.; Wring, S. SCY-078 is fungicidal against Candida species in
time-kill studies. Antimicrob. Agents Chemother. 2017, 61, e01961-16.
(28) Apgar, J. M.; Wilkening, R. R.; Parker, D. L.; Meng, D.;
Wildonger, K. J.; Sperbeck, D.; Greenlee, M. L.; Balkovec, J. M.;
Flattery, A. M.; Abruzzo, G. K.; Galgoci, A. M.; Giacobbe, R. A.; Gill, C.
J.; Hsu, M.-J.; Liberator, P.; Misura, A. S.; Motyl, M.; Nielsen Kahn, J.;
Powles, M.; Racine, F.; Dragovic, J.; Fan, W.; Kirwan, R.; Lee, S.; Liu,
H.; Mamai, A.; Nelson, K.; Peel, M. Ibrexafungerp: an orally active β
1,3-glucan synthesis inhibitor. Bioorg. Med. Chem. Lett. 2021, 32,
127661.
(29) Greenlee, M. L.; Wilkening, R.; Apgar, J.; Sperbeck, D.;
Wildonger, K. J.; Meng, D.; Parker, D. L.; Pacofsky, G. J.; Heasley, B.
H.; Mamai, A.; Nelson, K. Antifungal Agents. WO 2010019204, 2010.
(30) Greenlee, M. L.; Wilkening, R.; Apgar, J.; Wildonger, K. J.; Meng,
D.; Parker, D. L. Antifungal Agents. WO 2010019203A1, 2010.
(31) Imran, M.; Khan, S. A.; Alshammari, M. K.; Alqahtani, A. M.;
Alanazi, T. A.; Kamal, M.; Jawaid, T.; Ghoneim, M. M.; Alshehri, S.;
Shakeel, F. Discovery, development, inventions and patent review of
fexinidazole: The first all-oral therapy for human African trypanoso
miasis. Pharmaceuticals 2022, 15, 128.


SYN
European Journal of Medicinal Chemistry 245 (2023) 114898
The gram-scale synthesis of this drug is demonstrated in Scheme 3 [50]. Starting with triterpene glycoside enfumafungin 14, a reduction of the bridging hemiacetal with triethylsilane provided the intermediate 15, followed by hydrolysis, etherification and benzyl protection, gave compound 16 in 76% yield over 2 steps. Subsequent ring-opening alkylation reaction of 16 with tosyl protected aziridine 17 gave com pound 18, which then underwent Borch reduction to provide the in termediate 19. Treatment of 19 with biaryl 20 in the presence of boron trifluoride diethyl etherate gave rise to the substitution product ibrexafungerp. In this synthetic method, the pyridine-triazolium biaryl and chiral benzene sulfonamide were elegantly introduced into the triterpene enfumafungin through ring-opening and substitution reactions to give the triterpene derivative. These elegant and practical synthetic
methods could be employed as the versatile tools for the synthesis of other drug molecules.
[50] J.M. Apgar, R.R. Wilkening, D.L. Parker, J.D. Meng, K.J. Wildonger, D. Sperbeck,
M.L. Greenlee, J.M. Balkovec, A.M. Flattery, G.K. Abruzzo, A.M. Galgoci, R.
A. Giacobbe, C.J. Gill, M.J. Hsu, P. Liberator, A.S. Misura, M. Motyl, J.N. Kahn,
M. Powles, F. Racine, J. Dragovic, W. Fan, R. Kirwan, S. Lee, H. Liu, A. Mamai,
K. Nelson, M. Peel, Ibrexafungerp: an orally active β-1, 3-glucan synthesis
inhibitor, Bioorg, Med. Chem. Lett. 32 (2021), 127661.

.
References
- ^ Jump up to:a b c d e f g https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214900s000lbl.pdf
- ^ Jump up to:a b c “Scynexis Announces FDA Approval of Brexafemme (ibrexafungerp tablets) as the First and Only Oral Non-Azole Treatment for Vaginal Yeast Infections”. Scynexis, Inc. (Press release). 2 June 2021. Retrieved 2 June 2021.
Further reading
- Azie N, Angulo D, Dehn B, Sobel JD (September 2020). “Oral Ibrexafungerp: an investigational agent for the treatment of vulvovaginal candidiasis”. Expert Opin Investig Drugs. 29 (9): 893–900. doi:10.1080/13543784.2020.1791820. PMID 32746636.
- Davis MR, Donnelley MA, Thompson GR (July 2020). “Ibrexafungerp: A novel oral glucan synthase inhibitor”. Med Mycol. 58 (5): 579–592. doi:10.1093/mmy/myz083. PMID 31342066.
- Petraitis V, Petraitiene R, Katragkou A, Maung BB, Naing E, Kavaliauskas P, et al. (May 2020). “Combination Therapy with Ibrexafungerp (Formerly SCY-078), a First-in-Class Triterpenoid Inhibitor of (1→3)-β-d-Glucan Synthesis, and Isavuconazole for Treatment of Experimental Invasive Pulmonary Aspergillosis”. Antimicrob Agents Chemother. 64 (6). doi:10.1128/AAC.02429-19. PMC 7269506. PMID 32179521.
External links
- “Ibrexafungerp”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03734991 for “Efficacy and Safety of Oral Ibrexafungerp (SCY-078) vs. Placebo in Subjects With Acute Vulvovaginal Candidiasis (VANISH 303)” at ClinicalTrials.gov
- Clinical trial number NCT03987620 for “Efficacy and Safety of Oral Ibrexafungerp (SCY-078) vs. Placebo in Subjects With Acute Vulvovaginal Candidiasis (Vanish 306)” at ClinicalTrials.gov
Ibrexafungerp, also known as SCY-078 or MK-3118, is a novel enfumafungin derivative oral triterpene antifungal approved for the treatment of vulvovaginal candidiasis (VVC), also known as a vaginal yeast infection.1,9 It was developed out of a need to treat fungal infections that may have become resistant to echinocandins or azole antifungals.1 Ibrexafungerp is orally bioavailable compared to the echinocandins caspofungin, micafungin, and anidulafungin; which can only be administered parenterally.1,2 Similar to echinocandins, ibrexafungerp targets the fungal β-1,3-glucan synthase, which is not present in humans, limiting the chance of renal or hepatic toxicity.6,9
Ibrexafungerp was granted FDA approval on 1 June 2021.9
β-1,3-glucan synthase is composed of a catalytic subunit, FKS1 or FKS2, and a GTP-binding regulatory subunit, Rho1.5,6 This synthase is involved in the synthesis of β-1,3-glucan, a fungal cell wall component.6
Ibrexafungerp acts similarly to the echinocandin antifungals, by inhibiting the synthesis of β-1,3-glucan synthase.1,9 While echinocandins bind to the FKS1 domain of β-1,3-glucan synthase, enfumafungin and its derivatives bind at an alternate site which allows them to maintain their activity against fungal infections that are resistant to echinocandins.3,4
Ibrexafungerp has been shown in animal studies to distribute well to vaginal tissue, making it a favourable treatment for vulvovaginal candidiasis.4
- Wring SA, Randolph R, Park S, Abruzzo G, Chen Q, Flattery A, Garrett G, Peel M, Outcalt R, Powell K, Trucksis M, Angulo D, Borroto-Esoda K: Preclinical Pharmacokinetics and Pharmacodynamic Target of SCY-078, a First-in-Class Orally Active Antifungal Glucan Synthesis Inhibitor, in Murine Models of Disseminated Candidiasis. Antimicrob Agents Chemother. 2017 Mar 24;61(4). pii: AAC.02068-16. doi: 10.1128/AAC.02068-16. Print 2017 Apr. [Article]
- Hector RF, Bierer DE: New beta-glucan inhibitors as antifungal drugs. Expert Opin Ther Pat. 2011 Oct;21(10):1597-610. doi: 10.1517/13543776.2011.603899. Epub 2011 Jul 25. [Article]
- Kuhnert E, Li Y, Lan N, Yue Q, Chen L, Cox RJ, An Z, Yokoyama K, Bills GF: Enfumafungin synthase represents a novel lineage of fungal triterpene cyclases. Environ Microbiol. 2018 Sep;20(9):3325-3342. doi: 10.1111/1462-2920.14333. Epub 2018 Sep 13. [Article]
- Larkin EL, Long L, Isham N, Borroto-Esoda K, Barat S, Angulo D, Wring S, Ghannoum M: A Novel 1,3-Beta-d-Glucan Inhibitor, Ibrexafungerp (Formerly SCY-078), Shows Potent Activity in the Lower pH Environment of Vulvovaginitis. Antimicrob Agents Chemother. 2019 Apr 25;63(5). pii: AAC.02611-18. doi: 10.1128/AAC.02611-18. Print 2019 May. [Article]
- Ha YS, Covert SF, Momany M: FsFKS1, the 1,3-beta-glucan synthase from the caspofungin-resistant fungus Fusarium solani. Eukaryot Cell. 2006 Jul;5(7):1036-42. doi: 10.1128/EC.00030-06. [Article]
- Perlin DS: Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci. 2015 Sep;1354:1-11. doi: 10.1111/nyas.12831. Epub 2015 Jul 17. [Article]
- Wring S, Murphy G, Atiee G, Corr C, Hyman M, Willett M, Angulo D: Clinical Pharmacokinetics and Drug-Drug Interaction Potential for Coadministered SCY-078, an Oral Fungicidal Glucan Synthase Inhibitor, and Tacrolimus. Clin Pharmacol Drug Dev. 2019 Jan;8(1):60-69. doi: 10.1002/cpdd.588. Epub 2018 Jun 27. [Article]
- Ghannoum M, Arendrup MC, Chaturvedi VP, Lockhart SR, McCormick TS, Chaturvedi S, Berkow EL, Juneja D, Tarai B, Azie N, Angulo D, Walsh TJ: Ibrexafungerp: A Novel Oral Triterpenoid Antifungal in Development for the Treatment of Candida auris Infections. Antibiotics (Basel). 2020 Aug 25;9(9). pii: antibiotics9090539. doi: 10.3390/antibiotics9090539. [Article]
- FDA Approved Drug Products: Brexafemme (Ibrexafungerp) Oral Tablet [Link]
| Clinical data | |
|---|---|
| Pronunciation | /aɪˌbrɛksəˈfʌndʒɜːrp/ eye-BREKS-ə-FUN-jurp |
| Trade names | Brexafemme |
| Other names | SCY-078 |
| License data | US DailyMed: Ibrexafungerp |
| Pregnancy category | Contraindicated[1] |
| Routes of administration | oral, intravenous |
| Drug class | Antifungal |
| ATC code | J02AX07 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Pharmacokinetic data | |
| Protein binding | >99%[1] |
| Metabolism | Hydroxylation (CYP3A4) then conjugation (glucuronidation, sulfation)[1] |
| Elimination half-life | 20 hours[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1207753-03-4as citrate: 1965291-08-0 |
| PubChem CID | 46871657as citrate: 137552087 |
| DrugBank | DB12471as citrate: DBSALT003185 |
| UNII | A92JFM5XNUas citrate: M4NU2SDX3E |
| KEGG | D11544as citrate: D11545 |
| ChEMBL | ChEMBL4297513as citrate: ChEMBL4298168 |
| CompTox Dashboard (EPA) | DTXSID901336871 |
| Chemical and physical data | |
| Formula | C44H67N5O4 |
| Molar mass | 730.051 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Ibrexafungerp citrate, Brexafemme, アイブレキサフンジェルプクエン酸塩 , SCY-078 citrate, UNII-M4NU2SDX3E, M4NU2SDX3E, MK-3118; SCY-078, Orphan Drug, Merck, SCYNEXIS, WHO 10597, ANTI FUNGAL
CC(C)C(C)C1(CCC2(C3CCC4C5(COCC4(C3=CCC2(C1C(=O)O)C)CC(C5OCC(C)(C(C)(C)C)N)N6C(=NC=N6)C7=CC=NC=C7)C)C)C.C(C(=O)O)C(CC(=O)O)(C(=O)O)O
NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}