









如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » 0rphan drug status (Page 22)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

Cannabidiol Seven Expanded Access INDs granted by FDA to U.S. physicians to treat with Epidiolex 125 children suffering from intractable epilepsy syndromes -
LONDON, Nov. 15, 2013
GW Pharmaceuticals plc (AIM: GWP, Nasdaq: GWPH, “GW”) announced today that the U.S. Food and Drug Administration (FDA) has granted orphan drug designation for Epidiolex(R), our product candidate that contains plant-derived Cannabidiol (CBD) as its active ingredient, for use in treating children with Dravet syndrome, a rare and severe form of infantile-onset, genetic, drug-resistant epilepsy syndrome. Epidiolex is an oral liquid formulation of a highly purified extract of CBD, a non-psychoactive molecule from the cannabis plant. Following receipt of this orphan designation, GW anticipates holding a pre-IND meeting with the FDA in the near future to discuss a development plan for Epidiolex in Dravet syndrome.
Dravet syndrome is a rare pediatric epilepsy syndrome with a distinctive but complex electroclinical presentation. Onset of Dravet syndrome occurs during the first year of life with clonic and tonic-clonic seizures in previously healthy and developmentally normal infants. Prognosis is poor and patients typically develop intellectual disability and life-long ongoing seizures. There are approximately 5,440 patients with Dravet in the United States and an estimated 6,710 Dravet patients in Europe. These figures may be an underestimate as this syndrome is reportedly underdiagnosed.
In addition to GW’s clinical development program for Epidiolex in Dravet syndrome, which is expected to commence in 2014, GW has also made arrangements to enable independent U.S. pediatric epilepsy specialists to treat high need pediatric epilepsy cases with Epidiolex immediately. To date in 2013, a total of seven “expanded access” INDs have been granted by the FDA to U.S. clinicians to allow treatment with Epidiolex of approximately 125 children with epilepsy. These children suffer from Dravet syndrome, Lennox-Gastaut syndrome, and other pediatric epilepsy syndromes. GW is aware of further interest from additional U.S. and ex-U.S. physicians to host similar INDs for Epidiolex. GW expects data generated under these INDs to provide useful observational data during 2014 on the effect of Epidiolex in the treatment of a range of pediatric epilepsy syndromes.
“I, together with many colleagues in the U.S. who specialize in the treatment of childhood epilepsy, very much welcome the opportunity to investigate Epidiolex in the treatment of Dravet syndrome. The FDA’s timely approval of the orphan drug designation for Epidiolex in Dravet syndrome is a key milestone that comes after many years of reported clinical cases that suggest encouraging evidence of efficacy for CBD in this intractable condition,” stated Dr. Orrin Devinsky, Professor of Neurology, Neurosurgery and Psychiatry in New York City. “With GW now making plans to advance Epidiolex through an FDA development program, we have the prospect for the first time of fully understanding the science of CBD in epilepsy with a view to making an appropriately tested and approved prescription medicine available in the future for children who suffer from this debilitating disease.”
“GW is proud to be at the forefront of this important new program to treat children with Dravet Syndrome and potentially other forms of intractable childhood epilepsy. For families in these circumstances, their lives are significantly impacted by constant and often times very severe seizures in children where all options to control these seizures have been exhausted,” stated Dr. Stephen Wright, GW’s R&D Director. “GW intends to advance a full clinical development program for Epidiolex in Dravet syndrome as quickly as possible, whilst at the same time helping families in the short term through supporting physician-led INDs to treat intractable cases. Through its efforts, GW aims to provide the necessary evidence to confirm the promise of CBD in epilepsy and ultimately enabling children to have access to an FDA-approved prescription CBD medicine.”
“This orphan program for Epidiolex in childhood epilepsy is an important corporate strategic priority for GW. Following receipt of today’s orphan designation, GW now intends to commence discussions with the FDA regarding the U.S. regulatory pathway for Epidiolex,” stated Justin Gover, GW’s Chief Executive Officer. “GW intends to pursue this development in-house and retains full commercial rights to Epidiolex.”
About Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition — generally a disease or condition that affects fewer than 200,000 individuals in the U.S. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication.
About GW Pharmaceuticals plc
Founded in 1998, GW is a biopharmaceutical company focused on discovering, developing and commercializing novel therapeutics from its proprietary cannabinoid product platform in a broad range of disease areas. GW commercialized the world’s first plant-derived cannabinoid prescription drug, Sativex(R), which is approved for the treatment of spasticity due to multiple sclerosis in 22 countries. Sativex is also in Phase 3 clinical development as a potential treatment of pain in people with advanced cancer. This Phase 3 program is intended to support the submission of a New Drug Application for Sativex in cancer pain with the U.S. Food and Drug Administration and in other markets around the world. GW has established a world leading position in the development of plant-derived cannabinoid therapeutics and has a deep pipeline of additional clinical-stage cannabinoid product candidates targeting epilepsy (including an orphan pediatric epilepsy program), Type 2 diabetes, ulcerative colitis, glioma and schizophrenia. For further information, please visit http://www.gwpharm.com.
Cannabidiol (CBD) is one of at least 85 cannabinoids found in cannabis.It is a major constituent of the plant, second to tetrahydrocannabinol (THC), and represents up to 40% in its extracts. Compared with THC, cannabidiol is not psychoactive in healthy individuals, and is considered to have a wider scope of medical applications than THC, including to epilepsy, multiple sclerosis spasms, anxiety disorders, bipolar disorder,schizophrenia,nausea, convulsion and inflammation, as well as inhibiting cancer cell growth. There is some preclinical evidence from studies in animals that suggests CBD may modestly reduce the clearance of THC from the body by interfering with its metabolism.Cannabidiol has displayed sedative effects in animal tests. Other research indicates that CBD increases alertness. CBD has been shown to reduce growth of aggressive human breast cancer cells in vitro, and to reduce their invasiveness.


Tasimelteon
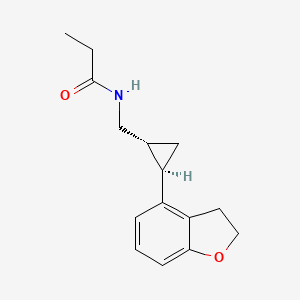
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide, 609799-22-6 cas
As expected, advisors to the US Food and Drug Administration have recommended approval of Vanda Pharmaceuticals’ tasimelteon, to be sold as Hetlioz, for the treatment of non-24-hour disorder in the totally blind.http://www.pharmatimes.com/Article/13-11-14/FDA_panel_backs_Vanda_body_clock_drug_for_blind.aspx
Tasimelteon (BMS-214,778) is a drug which is under development for the treatment of insomnia and other sleep disorders.[1] It is a selective agonistfor the melatonin receptors MT1 and MT2 in the suprachiasmatic nucleus of the brain, similar to older drugs such as ramelteon.[2] It has been through Phase III trials successfully and was shown to improve both onset and maintenance of sleep, with few side effects.[3]
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]

A drug being developed to treat transient insomnia in circadian rhythm sleep disorders (eg jet-lag. The drug appears to be effective in the dose range of 20 to 100mg with an advance in the melatonin rhythm of 2-3 hours with the higher dose
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the
suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of Cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands. This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder
(N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non- 24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping.
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary.
In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime. Tasimelteon
Tasimelteon is a circadian regulator which binds specifically to two high affinity melatonin receptors, Mella (MT1R) and Mellb (MT2R). These receptors are found in high density in the suprachiasmatic nucleus of the brain (SCN), which is responsible for synchronizing our sleep/wake cycle. Tasimelteon has been shown to improve sleep parameters in prior clinical studies, which simulated a desynchronization of the circadian clock. Tasimelteon has so far been studied in hundreds of individuals and has shown a good tolerability profile.
Tasimelteon has the chemical name: tr ns-N-[[2-(2,3-dihydrobenzofuran- 4-yl)cycloprop-lyl] methyl] propanamide, has the structure of Formula I:

Formula I
and is disclosed in US 5856529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78°C (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG- 400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MTIR. It’s affinity (¾) for MTIR is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
Metabolites of tasimelteon include, for example, those described in “Preclinical Pharmacokinetics and Metabolism of BMS-214778, a Novel
Melatonin Receptor Agonist” by Vachharajani et al., J. Pharmaceutical Sci., 92(4):760-772, which is hereby incorporated herein by reference. The active metabolites of tasimelteon can also be used in the method of this invention, as can pharmaceutically acceptable salts of tasimelteon or of its active metabolites. For example, in addition to metabolites of Formula II and III, above, metabolites of tasimelteon also include the monohydroxylated analogs M13 of Formula IV, M12 of Formula V, and M14 of Formula VI.
Formula IV

Formula V
MO

Formula VI
Thus, it is apparent that this invention contemplates entrainment of patients suffering free running circadian rhythm to a 24 hour circadian rhythm by administration of a circadian rhythm regulator (i.e., circadian rhythm modifier) capable of phase advancing and/or entraining circadian rhythms, such as a melatonin agonist like tasimelteon or an active metabolite oftasimelteon or a pharmaceutically acceptable salt thereof. Other MT1R and MT2R agonists, i.e., melatonin agonists, can have similar effects on the master body clock. So, for example, this invention further contemplates the use of melatonin agonists such as but not limited to melatonin, N-[l-(2,3-dihydrobenzofuran-4- yl)pyrrolidin-3-yl]-N-ethylurea and structurally related compounds as disclosed in US 6,211,225, LY-156735 ((R)-N-(2-(6-chloro-5-methoxy-lH-indol- 3yl) propyl) acetamide) (disclosed in U.S. Patent No. 4,997,845), agomelatine (N- [2-(7-methoxy-l-naphthyl)ethyl]acetamide) (disclosed in U.S. Patent No.
5,225,442), ramelteon ((S)-N-[2-(l,6,7,8-tetrahydro-2H-indeno- [5,4-b] furan-8- yl)ethyl]propionamide), 2-phenylmelatonin, 8-M-PDOT, 2-iodomelatonin, and 6- chloromelatonin.
Additional melatonin agonists include, without limitation, those listed in U.S. Patent Application Publication No. 20050164987, which is incorporated herein by reference, specifically: TAK-375 (see Kato, K. et al. Int. J.
Neuropsychopharmacol. 2000, 3 (Suppl. 1): Abst P.03.130; see also abstracts P.03.125 and P.03.127), CGP 52608 (l-(3-allyl-4-oxothiazolidine-2-ylidene)-4- met- hylthiosemicarbazone) (See Missbach et al., J. Biol. Chem. 1996, 271, 13515-22), GR196429 (N-[2-[2,3,7,8-tetrahydro-lH-fur-o(2,3-g)indol-l- yl] ethyl] acetamide) (see Beresford et al., J. Pharmacol. Exp. Ther. 1998, 285, 1239-1245), S20242 (N-[2-(7-methoxy napth-l-yl) ethyl] propionamide) (see Depres-Brummer et al., Eur. J. Pharmacol. 1998, 347, 57-66), S-23478 (see Neuropharmacology July 2000), S24268 (see Naunyn Schmiedebergs Arch. June 2003), S25150 (see Naunyn Schmiedebergs Arch. June 2003), GW-290569, luzindole (2-benzyl-N-acetyltryptamine) (see U.S. Patent No. 5,093,352), GR135531 (5-methoxycarbonylamino-N-acetyltrypt- amine) (see U.S. Patent Application Publication No. 20010047016), Melatonin Research Compound A, Melatonin Agonist A (see IMSWorld R&D Focus August 2002), Melatonin
Analogue B (see Pharmaprojects August 1998), Melatonin Agonist C (see Chem. Pharm. Bull. (Tokyo) January 2002), Melatonin Agonist D (see J. Pineal Research November 2000), Melatonin Agonist E (see Chem. Pharm. Bull. (Tokyo) Febrary 2002), Melatonin Agonist F (see Reprod. Nutr. Dev. May 1999), Melatonin Agonist G (see J. Med. Chem. October 1993), Melatonin Agonist H (see Famaco March 2000), Melatonin Agonist I (see J. Med. Chem. March 2000), Melatonin Analog J (see Bioorg. Med. Chem. Lett. March 2003), Melatonin Analog K (see MedAd News September 2001), Melatonin Analog L, AH-001 (2-acetamido-8- methoxytetralin) (see U.S. Patent No. 5,151,446), GG-012 (4-methoxy-2- (methylene propylamide)indan) (see Drijfhout et al., Eur. J. Pharmacol. 1999, 382, 157-66), Enol-3-IPA, ML-23 (N-2,4-dinitrophenyl-5-methoxy-tryptamine ) (see U.S. Patent No. 4,880,826), SL-18.1616, IP-100-9 (US 5580878), Sleep Inducing Peptide A, AH-017 (see U.S. Patent No. 5,151,446), AH-002 (8-methoxy- 2-propionamido-tetralin) (see U.S. Patent No. 5,151,446), and IP-101.
Metabolites, prodrugs, stereoisomers, polymorphs, hydrates, solvates, and salts of the above compounds that are directly or indirectly active can, of course, also be used in the practice of this invention.
Melatonin agonists with a MT1R and MT2R binding profile similar to that of tasimelteon, which has 2 to 4 time greater specificity for MT2R, are preferred.
Tasimelteon can be synthesized by procedures known in the art. The preparation of a 4-vinyl-2,3-dihydrobenzofuran cyclopropyl intermediate can be carried out as described in US7754902, which is incorporated herein by reference as though fully set forth.
Pro-drugs, e.g., esters, and pharmaceutically acceptable salts can be prepared by exercise of routine skill in the art.
In patients suffering a Non-24, the melatonin and Cortisol circadian rhythms and the natural day/night cycle become desynchronized. For example, in patients suffering from a free-running circadian rhythm, melatonin and Cortisol acrophases occur more than 24 hours, e.g., >24.1 hours, prior to each previous day’s melatonin and Cortisol acrophase, respectively, resulting in desynchronization for days, weeks, or even months, depending upon the length of a patient’s circadian rhythm, before the melatonin, Cortisol, and day /night cycles are again temporarily synchronized.
Chronic misalignment of Cortisol has been associated with metabolic, cardiac, cognitive, neurologic, neoplastic, and hormonal disorders. Such disorders include, e.g., obesity, depression, neurological impairments.


WASHINGTON, June 5, 2013 /PRNewswire/ — Vanda Pharmaceuticals Inc. (Vanda) presented additional entrainment and patient-level clinical data at SLEEP 2013, the 27th Annual Meeting of Associated Professional Sleep Societies in Baltimore, from its SET (Safety and Efficacy of Tasimelteon) and RESET (Randomized-withdrawal study of the Efficacy and Safety of Tasimelteon to treat Non-24-Hour Disorder) Phase III studies of tasimelteon, a circadian regulator for the treatment of Non-24-Hour Disorder (Non-24) in totally blind individuals. Non-24 is a serious, rare and chronic circadian rhythm disorder that affects a majority of totally blind individuals who lack light perception and cannot entrain (synchronize) their master body clock to the 24-hour day. Currently there is no approved FDA treatment for Non-24.
In the SET study, tasimelteon achieved the primary endpoints of entrainment (synchronizing) of the melatonin (aMT6s) rhythm as compared to placebo and clinical response as measured by entrainment plus a score of greater than or equal to 3 on the Non-24 Clinical Response Scale (N24CRS). Tasimelteon also demonstrated significant improvement versus placebo across a number of sleep and wake parameters including measures of total sleep time, nap duration, and timing of sleep, as well as in the Clinical Global Impression of Change (CGI-C), an overall global functioning scale. In treated patients, daytime naps decreased by 46 minutes per day in the worst 25% of days in a cycle and nighttime sleep increased by 57 minutes per day during the worst 25% of nights in a cycle.
The RESET study demonstrated that continued treatment with 20mg of tasimelteon was required to maintain entrainment of melatonin and cortisol circadian rhythms in individuals with Non-24. Patients treated with tasimelteon maintained their clinical benefits while patients who received placebo showed significant deterioration in measures of nighttime sleep, daytime naps and timing of sleep. Furthermore, discontinuation of tasimelteon resulted in a rapid relapse of circadian entrainment and a return to misaligned circadian rhythms, reinforcing the importance of chronic therapy.
Study investigator, Steven W. Lockley, Ph.D., Associate Professor of Medicine, Division of Sleep Medicine, Brigham and Women’s Hospital, Harvard Medical School, commented, “the results clearly demonstrate that tasimelteon can entrain the circadian clock, and that continued treatment is necessary to maintain entrainment.”
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
UPDATED ON JAN 2014
TASIMELTION, an orphan drug for non24
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide
(1R-trans)-N-[[2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]pro- pananamide VEC162
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
N-(((1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl)methyl)propanamide
Bristol-Myers Squibb Company
PRODUCT PATENT
U.S. Pat. No. 5,856,529
| CAS number | 609799-22-6 |
|---|
| Formula | C15H19NO2 |
|---|---|
| Mol. mass | 245.3 g/mol |
January 31, 2014 — The U.S. Food and Drug Administration today approved Hetlioz (tasimelteon), a melatonin receptor agonist, to treat non-24- hour sleep-wake disorder (“non-24”) in totally blind individuals. Non-24 is a chronic circadian rhythm (body clock) disorder in the blind that causes problems with the timing of sleep. This is the first FDA approval of a treatment for the disorder.
Non-24 occurs in persons who are completely blind. Light does not enter their eyes and they cannot synchronize their body clock to the 24-hour light-dark cycle.
VEC-162, BMS-214778, 609799-22-6, Hetlioz, Tasimelteon (USAN/INN), Tasimelteon [USAN:INN], UNII-SHS4PU80D9,
Tasimelteon
A year-long (2011-2012) study at Harvard is testing the use of tasimelteon in blind subjects with non-24-hour sleep–wake disorder.[4] In May 2013Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for Tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people.[5]
SEQUENCE
Discovered by Bristol-Myers Squibb (BMS) and co-developed with Vanda Pharmaceuticals, tasimelteon is a hypnotic family benzofuran. In Phase III development, it has an orphan drug status.
JAN2014.. APPROVED FDA
In mid-November 2013 the FDA announced their recommendation for the approval of Tasimelteon for the treatment of non-24-disorder.Tasimelteon effectively resets the circadian rhythm, helping to restore normal sleep patterns.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM374388.pdf
January 2010: FDA granted orphan drug tasimelteon to disturbed sleep / wake in blind without light perception.
February 2008: Vanda has completed enrollment in its Phase III trial in chronic primary insomnia.
June 2007: Results of a Phase III trial for transient insomnia tasimelteon presented by Vanda at the 21st annual meeting of the Associated Professional Sleep Societies. These results demonstrated improvements in objective and subjective measures of sleep and its maintenance.
2004 Vanda gets a license tasimelteon (or BMS-214778 and VEC-162) from Bristol-Myers Squibb.
About Tasimelteon: Tasimelteon is a circadian regulator in development for the treatment of Non-24. Tasimelteon is a dual melatonin receptor agonist (DMRA) with selective agonist activityat the MT1 and MT2 receptors.Tasimelteon’s ability to reset the master body clock in the suprachiasmatic nucleus (SCN) results in the entrainment of the body’s melatonin and cortisol rhythms with the 24-hour day-night cycle. The patent claiming tasimelteon as a new chemical entity extends through December 2022, assuming a 5-year extension to be granted under the Hatch-Waxman Act. Tasimelteon has been granted orphan drug designation for the treatment of Non-24 from both the U.S. and the European Union.
Previously, BMS-214778, identified as an agonist of melatonin receptors, has been the subject of pre-clinical studies for the treatment of sleep disorders resulting from a disturbance of circadian rhythms.The first Pharmacokinetic studies were performed in rats and monkeys.
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands.
This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder
Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non-24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping. TASIMELTION
TASIMELTION
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24 h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24 h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary. In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime.
INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
………………………..
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC

PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
…………………….
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58.
Curr Med Chem. 2012;19(21):3532-49. Review.
7 Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist.
Vachharajani NN, Yeleswaram K, Boulton DW.J Pharm Sci. 2003 Apr;92(4):760-72.
TASIMELTION
PATENTS
| US2010261786 | 10-15-2010 | PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE |
| US2009209638 | 8-21-2009 | TREATMENT FOR DEPRESSIVE DISORDERS |
| US6060506 | 5-10-2000 | Benzopyran derivatives as melatonergic agents |
| US5981571 | 11-10-1999 | Benzodioxa alkylene ethers as melatonergic agents |
| WO9825606 | 6-19-1998 | BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS |
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
|
 |

back to home for more updates


DR ANTHONY MELVIN CRASTO Ph.D

MST-188 is a purified form of a nonionic, triblock copolymer (poloxamer 188). It is an investigational agent that binds to hydrophobic surfaces on damaged cells and improves membrane hydration and lowers adhesion and viscosity, particularly under low shear conditions. MST-188 has the potential to reduce ischemic tissue injury and end-organ damage by restoring microvascular function, which is compromised in a wide range of serious and life-threatening diseases and conditions. We initially are developing MST-188 as a treatment for complications arising from sickle cell disease.

Background
Non-purified forms of poloxamer 188 (P188) have been used in foods, drugs and cosmetics since the 1950s. In the 1980s, extensive research on the mechanisms and potential clinical applications of P188 was conducted. Research has demonstrated that P188 binds to hydrophobic surfaces that develop when cells are damaged and restores normal hydrated surfaces, while having little or no activity in normal, healthy tissues. Research also has demonstrated that P188 prevents adhesion and aggregation of soluble fibrin and formed elements in the blood and maintains the deformability of red blood cells, the non-adhesiveness of unactivated platelets and granulocytes and the normal viscosity of blood. In addition, it is believed that P188 is not metabolized, but is excreted unchanged in the urine with a half-life of approximately four to six hours.
Formulations of P188 (non-purified and purified) have been studied in clinical trials involving nearly 4,000 individuals. It has been evaluated in the clinic to treat acute myocardial infarction, sickle cell disease and malaria, including a 2,950-patient, randomized, controlled study of P188 (non-purified) in acute myocardial infarction. The effectiveness of P188 also has been investigated in nonclinical studies of stroke, hemorrhagic shock, bypass surgery, adult respiratory distress syndrome, neurologic protection in deep hypothermic circulatory arrest, vasospasm, spinal cord injury, angioplasty, frostbite, amniotic fluid embolism, acute ischemic bowel disease and burns.
MST-188
Our(mast) purified form of P188, or purified P188, which is the active ingredient in MST-188, was designed to eliminate certain low molecular weight substances present in P188 (non-purified), which we believe were primarily responsible for the moderate to moderately severe elevations in serum creatinine levels (acute renal dysfunction) observed in prior clinical studies of P188 (non-purified). Purified P188 has been evaluated in multiple clinical studies by a prior sponsor, including a 255-patient, phase 3 study. In that study, purified P188 was generally well tolerated and there were no clinically significant elevations in serum creatinine among subjects who received purified P188 compared to placebo.
We believe that, as a rheologic, antithrombotic and cytoprotective agent, MST-188 has potential application in treating a wide range of diseases and conditions resulting from microvascular-flow abnormalities.
Sickle Cell Disease Market & Opportunity
More than $1.0 billion is spent annually in the U.S. to treat patients with sickle cell disease. Sickle cell disease is a genetic disorder characterized by the “sickling” of red blood cells, which normally are disc-shaped, deformable and move easily through the microvasculature carrying oxygen from the lungs to the rest of the body. Sickled, or crescent-shaped, red blood cells, on the other hand, are rigid and sticky and tend to adhere to each other and the vascular endothelium. Patients with sickle cell disease are known to experience severely painful episodes associated with the obstruction of small blood vessels by sickle-shaped red blood cells. These painful episodes are commonly known as acute crisis or vaso-occlusive crisis. Reduced blood flow to organs and bone marrow during vaso-occlusive crisis not only causes intense pain, but can result in tissue death, or necrosis. The frequency, severity and duration of these acute crises can vary considerably.
We (mast) estimate that, in the U.S., sickle cell disease results in over 95,000 hospitalizations and, in addition, approximately 69,000 emergency department treat-and-release encounters each year. When a patient with sickle cell disease makes an institutional visit, vaso-occlusive crisis is the primary diagnosis in approximately 77% of hospital admissions and 64% of emergency room treat-and-release encounters. In addition, although the number is difficult to measure, we estimate that the number of untreated sickle cell crisis events is substantial and in the hundreds of thousands in the U.S. each year. We believe that, if MST-188 is approved, as people with sickle cell disease are made aware of the new therapy, more people who suffer from acute crisis will seek treatment.
Development Status
We (mast) have initiated a Phase 3 clinical study of MST-188 for the treatment of sickle cell disease. The primary objective will be to demonstrate that MST-188 reduces the duration of vaso-occlusive crisis in patients with sickle cell disease. Please see our Clinical Trials page for more information regarding our phase 3 study of MST-188. In addition to the phase 3 study, we plan to conduct a number of smaller-scale clinical studies to further assess the efficacy, safety and tolerability of MST-188, and expect these studies to overlap with the phase 3 study.
US-based clinical-stage biotechnology firm Advaxis has received orphan drug designation from the US Food and Drug Administration (FDA) for its lead drug candidate ADXS-HPV to treat human papillomavirus (HPV) associated head and neck cancer patients.
Advaxis’s cancer vaccine gets FDA orphan status for treatment of HPV-associated head and neck cancer
PRINCETON, N.J., Nov 05, 2013 (BUSINESS WIRE) — Advaxis, Inc., /quotes/zigman/23528806/delayed/quotes/nls/adxs ADXS +2.61% , a leader in developing the next generation of cancer immunotherapies, announced that it has been granted Orphan Drug Designation from the U.S. Food and Drug Administration (FDA) Office of Orphan Products Development (OOPD) for ADXS-HPV, its lead drug candidate, for the treatment of human papillomavirus (HPV)-associated head and neck cancer.
Orphan Drug Designation is granted to drug therapies intended to treat diseases or conditions that affect fewer than 200,000 people in the United States. Orphan Drug Designation entitles the sponsor to clinical protocol assistance with the FDA, as well as federal grants, tax credits, and potentially a seven year market exclusivity period.
“We are very pleased to have been granted an orphan drug designation for ADXS-HPV in this unmet medical need,” commented Dr. Robert Petit, Chief Scientific Officer of Advaxis. “Patients with head and neck cancer have limited treatment options and we hope to improve their survival by developing ADXS-HPV for this indication. We plan to initiate an additional Phase 1/2 study in early stage head and neck cancer for ADXS-HPV with a nationally recognized center of excellence, and we will continue the ongoing Phase 1 study being sponsored by the University of Liverpool and Aintree University Hospitals NHS Foundation Trust that is evaluating the safety and efficacy of ADXS-HPV when combined with standard chemotherapy and radiation treatment in patients with head and neck cancer.”
“Receiving orphan drug designation for ADXS-HPV in head and neck cancer is excellent news for a technology that may offer the potential to treat an indication with few therapy options, and, importantly, it helps define a clear path forward to registration,” commented Daniel J. O’Connor, President and Chief Executive Officer of Advaxis.
About Orphan Drug Designation
Under the Orphan Drug Act (ODA), the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in the United States for this type of disease or condition will be recovered from sales of the product. The benefits of orphan drug designation can be substantial and include federal grants, tax credits, and potentially a seven year market exclusivity period once the product is approved, provided that the product is first to market.
In order for a sponsor to obtain orphan designation for a drug or biological product, an application must be submitted to OOPD, and the designation approved. The approval of an application for orphan designation is based upon the information submitted by the sponsor. A drug that has obtained orphan designation is said to have “orphan status.” Each designation request must stand on its own merit. Sponsors requesting designation of the same drug for the same indication as a previously designated product must submit their own data in support of their designation request. The approval of an orphan designation request does not alter the standard regulatory requirements and process for obtaining marketing approval. Safety and efficacy of a compound must be established through adequate and well-controlled studies.
About ADXS-HPV
ADXS-HPV is an immunotherapy that is designed to target cells expressing the HPV gene E7. Expression of the E7 gene from high-risk HPV variants is responsible for the transformation of infected cells into dysplastic and malignant tissues. Eliminating these cells can eliminate the dysplasia or malignancy. ADXS-HPV is designed to infect antigen-presenting cells and direct them to generate a powerful, cellular immune response to HPV E7. The resulting cytotoxic Tcells infiltrate and attack the tumors while specifically inhibiting tumor Tregs and MDSCs in the tumors that are protecting it.
About Head and Neck Cancer
Cancer of the head and neck includes cancers arising from mucosa lining the oral cavity, oropharynx, hypopharynx, larynx, sinonasal tract, and nasopharynx. The most common histologic type observed is squamous cell carcinoma; therefore, the term “head and neck squamous cell carcinoma” (HNSCC) is frequently used to imply squamous cell carcinomas involving these anatomical sites. Excessive tobacco and alcohol are important risk factors for HNSCCs overall, but human papillomavirus (HPV) is now recognized as the causative agent in a subset of HNSCCs.
While the incidence of head and neck cancers that are linked to alcohol and tobacco use as the primary risk factor has fallen in the past three decades, a trend attributed to decreasing tobacco use in the United States, the incidence of HPV-associated head and neck cancer has been increasing. The increase was observed particularly among young individuals (<60 years of age), men, and Caucasians. Studies have shown that oral HPV infection is likely to be sexually acquired, as the increase in the incidence of HPV-associated head and neck cancers may be attributed to changing sexual practices. According to the World Health Organization’s Human Papillomavirus and Related Cancers in the World Summary Report 2010, HPV is associated with 20-50% of oral squamous cell carcinomas. HPV-associated head and neck cancer is growing at an epidemic rate in western countries; and occurs more frequently (3:1) in men than women. In the United States, the number of HPV-positive head and neck cancer cases has already equaled the number of cervical cancer cases.
About Advaxis, Inc.
Advaxis is a clinical-stage biotechnology company developing the next generation of immunotherapies for cancer and infectious diseases. Advaxis immunotherapies are based on a novel platform technology using live, attenuated bacteria that are bio-engineered to secrete an antigen/adjuvant fusion protein(s) that is designed to redirect the powerful immune response all human beings have to the bacterium to the cancer itself.
ADXS-HPV is currently being evaluated in four clinical trials for human papillomavirus (HPV)-associated cancers: recurrent/refractory cervical cancer (India), locally advanced cervical cancer (GOG/NCI U.S. study, Clinical Trials.gov Identifier NCT01266460), head & neck cancer (CRUK study, Clinical Trials.gov Identifier NCT01598792), and anal cancer (BrUOG study, Clinical Trials.gov Identifier NCT01671488). Advaxis has over 15 distinct immunotherapies in various stages of development, developed directly by Advaxis and through strategic collaborations with recognized centers of excellence such as: the University of Pennsylvania, the Georgia Regents University Cancer Center, Brown University Oncology Group, and others.
ADXS-HPV is currently in Phase 1/2 clinical development for recurrent/refractory and advanced cervical cancer, HPV caused head and neck cancers, and anal cancer.
Links to ADXS-HPV trials:
ADXS-HPV is an immunotherapy that is designed to target cells expressing the HPV gene E7. Expression of the E7 gene from high-risk HPV variants is responsible for the transformation of infected cells into dysplastic and malignant tissues. Eliminating these cells can eliminate the dysplasia or malignancy. ADXS-HPV is designed to infect antigen-presenting cells and direct them to generate a powerful, cellular immune response to HPV E7. The resulting cytotoxic Tcells infiltrate and attack the tumors while specifically inhibiting tumor Tregs and MDSCs in the tumors that are protecting it.
The American Cancer Society estimates that there will be about 12,340 newly diagnosed cervical cancer cases and 7,060 newly diagnosed cases of anal cancer in the U.S. in 2013.
In 2009, the CDC reported that about 45% of women aged 20 to 24 had HPV. HPV causes a number of different types of cancer. The same types of genital HPV that cause cervical cancer (HPV-16, HPV-18) cause about 8 out of 10 squamous cell anal cancers. In addition, nearly half of cancers of the vulva and about 7 out of 10 vaginal cancers are HPV-related. Some other genital cancers (cancers of the penis and urethra) and some head and neck cancers (mostly the throat, tongue, and tonsils) are also related to high-risk types of HPV. For additional information about HPV, please visit: http://www.cancer.org/.
back to home for more updates
DR ANTHONY MELVIN CRASTO Ph.D

Macimorelin
CAS 381231-18-1
Chemical Formula: C26H30N6O3
Exact Mass: 474.23794
Molecular Weight: 474.55480
Elemental Analysis: C, 65.80; H, 6.37; N, 17.71; O, 10.11
945212-59-9 (Macimorelin acetate)
AEZS-130
ARD-07
D-87875
EP-01572
EP-1572
JMV-1843
USAN (ab-26)
MACIMORELIN ACETATE
THERAPEUTIC CLAIM
Diagnostic agent for adult growth hormone deficiency (AGHD)
CHEMICAL NAMES
1. D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-, acetate (1:1)
2. N2-(2-amino-2-methylpropanoyl-N1-[(1R)-1-formamido-2-(1H-indol-3-yl)ethyl]- D-tryptophanamide acetate
MOLECULAR FORMULA
C26H30N6O3.C2H4O2
MOLECULAR WEIGHT
534.6
SPONSOR
Aeterna Zentaris GmbH
CODE DESIGNATIONS
D-87575, EP 1572, ARD 07
CAS REGISTRY NUMBER
945212-59-9
Macimorelin (also known as AEZS-130, EP-1572) is a novel synthetic small molecule, acting as a ghrelin agonist, that is orally active and stimulates the secretion of growth hormone (GH). Based on results of Phase 1 studies, AEZS-130 has potential applications for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and AIDS. In addition to the therapeutic application, a Phase 3 trial with AEZS-130 as a diagnostic test for growth hormone deficiencies in adults has been completed.
http://www.ama-assn.org/resources/doc/usan/macimorelin-acetate.pdf
QUEBEC, Nov. 5, 2013 /PRNewswire/ – Aeterna Zentaris Inc. (the “Company”) today announced that it has submitted a New Drug Application (“NDA”) to the U.S. Food and Drug Administration (“FDA”) for its ghrelin agonist, macimorelin acetate (AEZS-130). Phase 3 data have demonstrated that the compound has the potential to become the first orally-approved product that induces growth hormone release to evaluate adult growth hormone deficiency (“AGHD”), with accuracy comparable to available intravenous and intramuscular testing procedures. read at
http://www.drugs.com/nda/macimorelin_acetate_131105.html
http://www.ama-assn.org/resources/doc/usan/macimorelin-acetate.pdf
macimorelin (JMV 1843), a ghrelin-mimetic growth hormone secretagogue in Phase III for adult growth hormone deficiency (AGHD)

Macimorelin, a growth hormone modulator, is currently awaiting registration in the U.S. by AEterna Zentaris as an oral diagnostic test of adult growth hormone deficit disorder. The company is also developing the compound in phase II clinical trials for the treatment of cancer related cachexia. The compound was being codeveloped by AEterna Zentaris and Ardana Bioscience; however, the trials underway at Ardana were suspended in 2008 based on a company strategic decision. AEterna Zentaris owns the worldwide rights of the compound. In 2007, orphan drug designation was assigned by the FDA for the treatment of growth hormone deficit in adults.
New active series of growth hormone secretagogues
J Med Chem 2003, 46(7): 1191
WO 2001096300
WO 2007093820
…………………………
J Med Chem 2003, 46(7): 1191
http://pubs.acs.org/doi/full/10.1021/jm020985q


Synthetic Pathway for JMV 1843 and Analoguesa
a Reagents and conditions: (a) IBCF, NMM, DME, 0 °C; (b) NH4OH; (c) H2, Pd/C, EtOH, HCl; (d) BOP, NMM, DMF, Boc-(d)-Trp-OH; (e) Boc2O, DMAP cat., anhydrous CH3CN; (f) BTIB, pyridine, DMF/H2O; (g) 2,4,5-trichlorophenylformate, DIEA, DMF; (h) TFA/anisole/thioanisole (8:1:1), 0 °C; (i) BOP, NMM, DMF, Boc-Aib-OH; (j) TFA/anisole/thioanisole (8:1:1), 0 °C; (k) RP preparative HPLC.
TFA, H-Aib-(d)-Trp-(d)-gTrp-CHO (7). 6 (1 g, 1.7 mmol) was dissolved in a mixture of trifluoroacetic acid (8 mL), anisole (1 mL), and thioanisole (1 mL) for 30 min at 0 °C. The solvents were removed in vacuo, the residue was stirred in ether, and the precipitated TFA, H-Aib-(d)-Trp-(d)-gTrp-CHO was filtered. 7 was purified by preparative HPLC and obtained in 52% yield. 1H NMR (400 MHz, DMSO-d6) + correlation 1H−1H: δ 1.21 (s, 3H, CH3 (Aib)), 1.43 (s, 3H, CH3 (Aib)), 2.97 (m, 2H, (CH2)β), 3.1 (m, 2H, (CH2)β‘), 4.62 (m, 1H, (CH)αA and (CH)αB), 5.32 (q, 0.4H, (CH)α‘B), 5.71 (q, 0.6H, (CH)α‘A), 7.3 (m, 4H, H5 and H6 (2 indoles)), 7.06−7.2 (4d, 2H, H2A and H2B (2 indoles)), 7.3 (m, 2H, H4 or H7 (2 indoles)), 7.6−7.8 (4d, 2H, H4A and H4B or H7A and H7B), 7.97 (s, 3H, NH2 (Aib) and CHO (formyl)), 8.2 (d, 0.4H, NH1B (diamino)), 8.3 (m,1H, NHA and NHB), 8.5 (d, 0.6H, NH1A (diamino)), 8.69 (d, 0.6H, NH2A (diamino)), 8.96 (d, 0.4H, NH2B (diamino)), 10.8 (s, 0.6H, N1H1A (indole)), 10.82 (s, 0.4H, N1H1B (indole)), 10.86 (s, 0.6H, N1H2A (indole)), 10.91 (s, 0,4H, N1H2B (indole)). MS (ES), m/z: 475 [M + H]+, 949 [2M + H]+. HPLC tR: 16.26 min (conditions A).
…………………………..
http://www.google.com/patents/US8192719
The inventors have now found that the oral administration of growth hormone secretagogues (GHSs) EP 1572 and EP 1573 can be used effectively and reliably to diagnose GHD.
EP 1572 (Formula I) or EP 1573 (Formula II) are GHSs (see WO 01/96300, Example 1 and Example 58 which are EP 1572 and EP 1573, respectively) that may be given orally.

EP 1572 and EP 1573 can also be defined as H-Aib-D-Trp-D-gTrp-CHO and H-Aib-D-Trp-D-gTrp-C(O)NHCH2CH3. Wherein, His hydrogen, Aib is aminoisobutyl, D is the dextro isomer, Trp is tryptophan and gTrp is a group of Formula III:

…………………………….
http://www.google.com/patents/US6861409
H-Aib-D-Trp-D-gTrp-CHO: 
Example 1 H-Aib-D-Trp-D-gTrp-CHO
Total synthesis (percentages represent yields obtained in the synthesis as described below):

Z-D-Tr-NH2
Z-D-Trp-OH (8.9 g; 26 mmol; 1 eq.) was dissolved in DME (25 ml) and placed in an ice water bath to 0° C. NMM (3.5 ml; 1.2 eq.), IBCF (4.1 ml; 1.2 eq.) and ammonia solution 28% (8.9 ml; 5 eq.) were added successively. The mixture was diluted with water (100 ml), and the product Z-D-Trp-NH2 precipitated. It was filtered and dried in vacuo to afford 8.58 g of a white solid.
Yield=98%.
C19H19N3O3, 337 g.mol−1.
Rf=0.46 {Chloroform/Methanol/Acetic Acid (180/10/5)}.
1H NMR (250 MHZ, DMSO-d6): δ 2.9 (dd, 1H, Hβ, Jββ′=14.5 Hz; Jβα=9.8 Hz); 3.1 (dd, 1H, Hβ′, Jβ′β=14.5 Hz; Jβ′α=4.3 Hz); 4.2 (sextuplet, 1H, Hα); 4.95 (s, 2H, CH2 (Z); 6.9-7.4 (m, 11H); 7.5 (s, 1H, H2); 7.65 (d, 1H, J=7.7 Hz); 10.8 (s, 1H, N1H).
Mass Spectrometry (Electrospray), m/z 338 [M+H]+, 360 [M+Na]+, 675 [2M+H]+, 697 [2M+Na]+.
Boc-D-Trp-D-Trp-NH2
Z-D-Trp-NH2 (3 g; 8.9 mmol; 1 eq.) was dissolved in DMF (100 ml). HCl 36% (845 μl; 1.1 eq.), water (2 ml) and palladium on activated charcoal (95 mg, 0.1 eq.) were added to the stirred mixture. The solution was bubbled under hydrogen for 24 hr. When the reaction went to completion, the palladium was filtered on celite. The solvent was removed in vacuo to afford HCl, H-D-Trp-NH2 as a colorless oil.
In 10 ml of DMF, HCl, H-D-Trp-NH2 (8.9 mmol; 1 eq.), Boc-D-Trp-OH (2.98 g; 9.8 mmol; 1.1 eq.), NMM (2.26 ml; 2.1 eq.) and BOP (4.33 g; 1.1 eq.) were added successively. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (100 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo to afford 4.35 g of Boc-D-Trp-D-Trp-NH2 as a white solid.
Yield=85%.
C27H31N5O4, 489 g.mol−1.
Rf=0.48 {Chloroform/Methanol/Acetic Acid (85/10/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.28 (s, 9H, Boc); 2.75-3.36 (m, 4H, 2 (CH2)β; 4.14 (m, 1H, CHα); 4.52 (m, 1H, CHα′); 6.83-7.84 (m, 14H, 2 indoles (10H), NH2, NH (urethane) and NH (amide)); 10.82 (d, 1H, J=2 Hz, N1H); 10.85 (d, 1H, J=2 Hz, N1H).
Mass Spectrometry (Electrospray), m/z 490 [M+H]+, 512 [M+Na]+, 979 [2M+H]+.
Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2
Boc-D-Trp-D-Trp-NH2 (3 g; 6.13 mmol; 1 eq.) was dissolved in acetonitrile (25 ml).
To this solution, di-tert-butyl-dicarbonate (3.4 g; 2.5 eq.) and 4-dimethylaminopyridine (150 mg; 0.2 eq.) were successively added. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/hexane {5/5} to afford 2.53 g of Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2 as a white solid.
Yield=60%.
C37H47N5O8, 689 g.mol−1.
Rf=0.23 {ethyl acetate/hexane (5/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.25 (s, 9H, Boc); 1.58 (s, 9H, Boc); 1.61 (s, 9H, Boc); 2.75-3.4 (m, 4H, 2 (CH2)β); 4.2 (m, 1H, CHα′); 4.6 (m, 1H, CHα); 7.06-8 (m, 14H, 2 indoles (10H), NH (urethane), NH and NH2 (amides)).
Mass Spectrometry (Electrospray), m/z 690 [M+H]+, 712 [M+Na]+, 1379 [2M+H]+, 1401 [2M+Na]+.
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-H
Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2 (3 g; 4.3 mmol; 1 eq.) was dissolved in the mixture DMF/water (18 ml/7 ml). Then, pyridine (772 μl; 2.2 eq.) and Bis(Trifluoroacetoxy)IodoBenzene (2.1 g; 1.1 eq.) were added. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and aqueous saturated sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. Boc-D-NiBoc)Trp-D-g(NiBoc)Trp-H was used immediately for the next reaction of formylation.
Rf=0.14 {ethyl acetate/hexane (7/3)}.
C36H47N5O7, 661 g.mol−1.
1H NMR (200 MHZ, DMSO-d6): δ 1.29 (s, 9H, Boc); 1.61 (s, 18H, 2 Boc); 2.13 (s, 2H, NH2 (amine)); 3.1-2.8 (m, 4H, 2 (CH2)β); 4.2 (m, 1H, CHα′); 4.85 (m, 1H, CHα); 6.9-8 (m, 12H, 2 indoles (10H), NH (urethane), NH (amide)).
Mass Spectrometry (Electrospray), m/z 662 [M+H]+, 684 [M+Na]+.
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-H (4.3 mmol; 1 eq.) was dissolved in DMF (20 ml). Then, N,N-diisopropylethylamine (815 μl; 1.1 eq.) and 2,4,5-trichlorophenylformate (1.08 g; 1.1 eq.) were added. After 30 minutes, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/hexane {5/5} to afford 2.07 g of Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO as a white solid.
Yield=70%.
C37H47N5O8, 689 g.mol−1.
Rf=0.27 {ethyl acetate/hexane (5/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.28 (s, 9H, Boc); 1.6 (s, 9H, Boc); 1.61 (s, 9H, Boc); 2.75-3.1 (m, 4H, 2 (CH2)β); 4.25 (m, 1H, (CH)αA&B); 5.39 (m, 0.4H, (CH)α′B); 5.72 (m, 0.6H, (CH)α′A); 6.95-8.55 (m, 14H, 2 indoles (10H), NH (urethane), 2 NH (amides), CHO (formyl)).
Mass Spectrometry (Electrospray), m/z 690 [M+H]+, 712 [M+Na]+, 1379 [2M+H]+.
Boc-Aib-D-Trp-D-gTrp-CHO
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO (1.98 g; 2.9 mmol; 1 eq.) was dissolved in a -mixture of trifluoroacetic acid (16 ml), anisole (2 ml) and thioanisole (2 ml) for 30 minutes at 0° C. The solvents were removed in vacuo, the residue was stirred with ether and the precipitated TFA, H-D-Trp-D-gTrp-CHO was filtered.
TFA, H-D-Trp-D-gTrp-CHO (2.9 mmol; 1 eq.), Boc-Aib-OH (700 mg; 1 eq.), NMM (2.4 ml; 4.2 eq.) and BOP (1.53 g; 1.2 eq.) were successively added in 10 ml of DMF. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate to afford 1.16 g of Boc-Aib-D-Trp-D-gTrp-CHO as a white solid.
Yield=70%.
C31H38N6O5, 574 g.mol−1.
Rf=0.26 {Chloroform/Methanol/Acetic Acid (180/10/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.21 (s, 6H, 2 CH3(Aib)); 1.31 (s, 9H, Boc); 2.98-3.12 (m, 4H, 2 (CH2)β); 4.47 (m, 1H, (CH)αA&B); 5.2 (m, 0.4H, (CH)α′B); 5.7 (m, 0.6H, (CH)α′A); 6.95-8.37 (m, 15H, 2 indoles (10H), 3 NH (amides), 1 NH (urethane) CHO (formyl)); 10.89 (m, 2H, 2 N1H (indoles)).
Mass Spectrometry (Electrospray), ml/z 575 [M+H]+, 597 [M+Na]+, 1149 [2M+H]+, 1171 [2M+Na]+.
H-Aib-D-Trp-D-gTrT-CHO
Boc-Aib-D-Trp-D-gTrp-CHO (1 g; 1.7 nmmol) was dissolved in a mixture of trifluoroacetic acid (8 ml), anisole (1 ml) and thioanisole (1 ml) for 30 minutes at 0° C. The solvents were removed in vacuo, the residue was stirred with ether and the precipitated TFA, H-Aib-D-Trp-D-gTrp-CHO was filtered.
The product TFA, H-Aib-D-Trp-D-gTrp-CHO was purified by preparative HPLC (Waters, delta pak, C18, 40×100 mm, 5 μm, 100 A).
Yield=52%.
C26H30N6O3, 474 g.mol−1.
1H NMR (400 MHZ, DMSO-d6)+1H/1H correlation: δ 1.21 (s, 3H, CH3 (Aib)); 1.43 (s, 3H, CH3 (Aib)); 2.97 (m, 2H, (CH2)β); 3.1 (m, 2H, (CH2)β′); 4.62 (m, 1H, (CH)αA&B); 5.32 (q, 0.4H, (CH)α′B); 5.71 (q, 0.6H, (CH)α′A); 7.3 (m, 4H5 and H6 (2 indoles)); 7.06-7.2 (4d, 2H, H2A et H2B (2 indoles)); 7.3 (m, 2H, H4 or H7 (2 indoles)); 7.6-7.8 (4d, 2H, H4A and H4B or H7A et H7B); 7.97 (s, 3H, NH2 (Aib) and CHO (Formyl));8.2 (d, 0.4H, NH1B (diamino)); 8.3 (m,1H, NHA&B); 8.5 (d, 0.6H, NH1A (diamino)); 8.69 (d, 0.6H, NH2A (diamino)); 8.96 (d, 0.4H, NH2B (diamino)); 10.8 (s, 0.6H, N1H1A (indole)); 10.82 (s, 0.4H, N1H1B (indole)); 10.86 (s, 0.6H, N1H2A (indole)); 10.91 (s, 0.4, N1H2B (indole)).
Mass Spectrometry (Electrospray), m/z 475 [M+H]+, 949 [2M+H]+.
………………………………
UPDATED INFO AS ON JAN 6 2014
Quebec City, Canada, January 6, 2014 – Aeterna Zentaris Inc. (NASDAQ: AEZS) (TSX: AEZS) (the “Company”) today announced that the U.S. Food and Drug Administration (“FDA”) has accepted for filing the Company’s New Drug Application (“NDA”) for its ghrelin agonist, macimorelin acetate, in Adult Growth Hormone Deficiency (“AGHD”). The acceptance for filing of the NDA indicates the FDA has determined that the application is sufficiently complete to permit a substantive review.
The Company’s NDA, submitted on November 5, 2013, seeks approval for the commercialization of macimorelin acetate as the first orally-administered product that induces growth hormone release to evaluate AGHD. Phase 3 data have demonstrated the compound to be well tolerated, with accuracy comparable to available intravenous and intramuscular testing procedures. The application will be subject to a standard review and will have a Prescription Drug User Fee Act (“PDUFA”) date of November 5, 2014. The PDUFA date is the goal date for the FDA to complete its review of the NDA.
David Dodd, President and CEO of Aeterna Zentaris, commented, “The FDA’s acceptance of this NDA submission is another significant milestone in our strategy to commercialize macimorelin acetate as the first approved oral product for AGHD evaluation. We are finalizing our commercial plan for this exciting new product. We are also looking to broaden the commercial application of macimorelin acetate in AGHD for use related to traumatic brain injury victims and other developmental areas, which would represent significant benefit to the evaluation of growth hormone deficiency, while presenting further potential revenue growth opportunities for the Company.”
Macimorelin acetate, a ghrelin agonist, is a novel orally-active small molecule that stimulates the secretion of growth hormone. The Company has completed a Phase 3 trial for use in evaluating AGHD, and has filed an NDA to the FDA in this indication. Macimorelin acetate has been granted orphan drug designation by the FDA for use in AGHD. Furthermore, macimorelin acetate is in a Phase 2 trial as a treatment for cancer-induced cachexia. Aeterna Zentaris owns the worldwide rights to this novel patented compound.
AGHD affects about 75,000 adults across the U.S., Canada and Europe. Growth hormone not only plays an important role in growth from childhood to adulthood, but also helps promote a hormonally-balanced health status. AGHD mostly results from damage to the pituitary gland. It is usually characterized by a reduction in bone mineral density, lean mass, exercise capacity, and overall quality of life.
Aeterna Zentaris is a specialty biopharmaceutical company engaged in developing novel treatments in oncology and endocrinology. The Company’s pipeline encompasses compounds from drug discovery to regulatory approval.
back to home for more updates
DR ANTHONY MELVIN CRASTO Ph.D

FUSIDIC ACID, 6990-06-3
2-[(1S,2S,5R,6S,7S,10S,11S,13S,14Z,15R,17R)-13-(acetyloxy)-5,17-dihydroxy-2,6,10,11-tetramethyltetracyclo[8.7.0.02,7.011,15]heptadecan-14-ylidene]-6-methylhept-5-enoic acid
Taksta (CEM-102)
Clinical-stage pharmaceutical firm Cempra has secured orphan drug status from the US Food and Drug Administration (FDA) for its drug candidate Taksta (CEM-102) to treat patients with prosthetic joint infections (PJI).
Cempra’s Taksta secures FDA orphan drug status for prosthetic joint infections treatment
Fusidic acid is a bacteriostatic antibiotic that is often used topically in creams and eyedrops, but may also be given systemically as tablets or injections. The global problem of advancing antimicrobial resistance has led to a renewed interest in its use recently.
Fusidic acid acts as a bacterial protein synthesis inhibitor by preventing the turnover ofelongation factor G (EF-G) from the ribosome. Fusidic acid is effective primarily ongram-positive bacteria such as Staphylococcus species, Streptococcus species, and Corynebacterium species. Fusidic acid inhibits bacterial replication and does not kill the bacteria, and is therefore termed bacteriostatic.
Fusidic acid is a true antibiotic, derived from the fungus Fusidium coccineum and was developed by Leo Laboratories in Ballerup, Denmark and released for clinical use in the 1960s. It has also been isolated from Mucor ramannianus and Isaria kogana. The drug is licensed for use as its sodium salt sodium fusidate, and it is approved for use under prescription in South Korea, Japan, UK, Canada, Europe, Australia, New Zealand, Thailand, India and Taiwan. A different oral dosing regimen, based on the compound’s Pharmacokinetic/pharmacodynamic (PK-PD) profile is in clinical development in the U.S. as Taksta.
Fusidic acid (TAKSTATM, CEM-102) is an antibiotic with a long history of safety and efficacy outside the United States. Cempra has exclusive rights to the supply of the compound for the U.S. market. Fusidic acid is orally active against gram-positive bacteria, including all S. aureus strains such as HA-MRSA and CA-MRSA. A novel dosing regimen has been successfully evaluated in a Phase II trial in patients with acute bacterial skin and skin structure infections (aBSSSI). Cempra is conducting a Phase II trial of TAKSTA for patients with prosthetic joint infections.
Profile of TAKSTA (CEM-102)
Prosthetic joint infections (PJI) occur in about 1% of hip replacements and 2% of knee replacements, translating to an incidence rate of about 10,000 per year in the U.S. at current hip and knee arthroplasty rates. There are few good options to treat these serious staphylococcal, often MRSA infections, which require long-term antibiotic treatment. Current therapy in the U.S. is with intravenous antibiotics such as vancomycin. An oral drug that can be safely administered for a long period of time could improve care and quality of life for these patients.
TAKSTA has shown potent activity against a large number of S. aureus strains, including CA-MRSA, HA-MRSA and linezolid-resistant strains, isolated in the U.S over a 10 year period. Its broad S. aureus coverage makes it useful for a broad range of clinical applications. Because of its safety and tolerability profile, TAKSTA could be ideal for patients suffering from staphylococcal infections that require long-term therapy such as patients with PJIs.
Cempra has developed a unique oral loading dose regimen to optimize key pathogen coverage and minimize drug resistance development. This regimen is incorporated in our Phase II trial to treat PJIs with TAKSTA in combination with rifampin, which is commonly used with injectible antibiotics such as vancomycin to treat PJIs.
Research on TAKSTA
Publications
The links for the articles go to subscription-based sites and may require a fee to view the article.
In Vitro Activity of CEM-102 (Fusidic Acid) Against Prevalent Clones and Resistant Phenotypes of Staphylococcus aureus
DF Sahm, J Deane, CM Pillar, P Fernandes
Antimicrobial Agents and Chemotherapy. June 2013 57: 4535-4346
http://aac.asm.org/content/57/9/4535
Efforts to Support the Development of Fusidic Acid in the United States
P Fernandes, D Pereira
Clinical Infectious Disease. June 2011 52:S542-6
http://www.ncbi.nlm.nih.gov/pubmed/21546632
Case report: Treatment of Chronic Osteomyelitis
CR Wolfe
Clinical Infectious Disease. June 2011 52:S538-41
http://cid.oxfordjournals.org/content/52/suppl_7/S538.long
The Safety Record of Fusidic Acid in Non-US markets: A Focus on Skin Infections
CN Kraus, BW Burnstead
Clinical Infectious Disease. June 2011 52:S527-37
http://cid.oxfordjournals.org/content/52/suppl_7/S527.long
A Randomized, Double-Blind Phase 2 Study Comparing the Efficacy and Safety of an Oral Fusidic Acid Loading-Dose Regimen to Oral Linezolid in the Treatment of Acute Bacterial Skin and Skin Structure Infections
JC Craft, SR Moriarty, K Clark, D Scott, TP Degenhardt, JG Still, GR Corey, A Das, P Fernandes
Clinical Infectious Disease. June 2011 52:S520-26
http://cid.oxfordjournals.org/content/52/suppl_7/S520.long
Application of Pharmacokinetic-Pharmacodynamic Modeling and the Justification of a Novel Fusidic Acid Dosing Regimen: Raising Lazarus from the Dead
BT Tsuji, OO Okusanya, JB Bulitta, A Forrest, SM Bhavnani, P Fernandes, PG Ambrose
Clinical Infectious Disease. June 2011 52:S513-19
http://cid.oxfordjournals.org/content/52/suppl_7/S513.long
Pharmacokinetics and Safety of Single, Multiple, and Loading Doses of Fusidic Acid in Healthy Subjects
JG Still, K Clark, TP Degenhardt, D. Scott, P. Fernandes, M. J. Gutierrez
Clinical Infectious Disease. June 2011 52:S504-12
http://cid.oxfordjournals.org/content/52/suppl_7/S504.long
Activity of Fusidic Acid Against Extracellular and Intracellular Staphylococcus aureus: Influence of pH and Comparison with Linezolid and Clindamycin
S Lemaire, F Van Bambeke, D Pierard, PC Appelbaum, PM Tulkens
Clinical Infectious Disease. June 2011 52:S493-503
http://cid.oxfordjournals.org/content/52/suppl_7/S493.long
Characterization of Global Patterns and the Genetics of Fusidic Acid Resistance
DJ Farrell, M Castanheira, I Chopra
Clinical Infectious Disease. June 2011 52:S487-92
http://cid.oxfordjournals.org/content/52/suppl_7/S493.long
In Vitro Antimicrobial Findings for Fusidic Acid Tested Against Contemporary (2008-2009) Gram-Positive Organisms Collected in the United States
RN Jones, RE Mendes, HS Sader, M Castanheira
Clinical Infectious Disease. June 2011 52:S477-86
http://cid.oxfordjournals.org/content/52/suppl_7/S477.long
New Rules for Clinical Trials in Patients with Acute Bacterial Skin and Skin Structure Iinfections: Do not Let the Perfect be the Enemy of the Good
GR Corey, ME Stryjewski
Clinical Infectious Disease. June 2011 52:S469-76
http://cid.oxfordjournals.org/content/52/suppl_7/S469.long
Introduction: Fusidic Acid Enters the United States
RC Moellering, GR Corey, ML Grayson
Clinical Infectious Disease. June 2011 52:S467-8
http://cid.oxfordjournals.org/content/52/suppl_7/S467.long
Evaluation of the Pharmacokinetics-Pharmacodynamics of Fusidic Acid Against Staphylococcus aureus and Streptococcus pyogenes Using In Vitro Infection Models: Implications for Dose Selection
OO Okusanya, BT Tsuji, JB Bulitta, A Forrest, CC Bulik, SM Bhavnani, P Fernandes, PG Ambrose
Diagnostic Microbiology & Infectious Disease. June 2011 70:101-11
http://www.ncbi.nlm.nih.gov/pubmed/21513848
In Vitro Activity of Fusidic Acid (CEM-102, Sodium Fusidate) Against Staphylococcus aureus Isolated from Cystic Fibrosis Patients and its Effect on the Activities of Tobramycin and Amikacin against Pseudomonas aeruginosa and Burkholderia cepacia
P McGhee, K Credito, L Beachel, PC Appelbaum, K Kosowaska-Shick
Antimicrobial Agents and Chemotherapy. June 2011 55:2417-19
http://www.ncbi.nlm.nih.gov/pubmed/21513848
Occurrence and Molecular Characterization of Fusidic Acid Resistance Mechanisms Among Staphylococcus spp. From European Countries (2008)
Castanheira, M., AA Watters, RE Mendes, DJ Farrell, RN Jones
Antimicrobial Agents and Chemotherapy. April 2010 65:1353-8
http://jac.oxfordjournals.org/content/65/7/1353.long
Update on Fusidic Acid (CEM-102) Tested Against Neisseria gonorrhoeae and Chlamydia trachomatis
R Jones, D Biedenbach, P Roblin, S Kohlhoff, M Hammerschlag
Antimicrobial Agents and Chemotherapy. October 2010 54: 4518-4519
http://aac.asm.org/cgi/content/citation/54/10/4518
Fusidic Acid Resistance Rates and Prevalence of Resistance Mechanisms Among Staphylococcus spp. Isolated in North America and Australia, 2007-2008
M Castanheira, AA Watters, JM Bell, JD Turnidge, RN Jones
Antimicrobial Agents and Chemotherapy. September 2010 54: 3614-3617
http://www.ncbi.nlm.nih.gov/pubmed/20566766
Spectrum of Activity, Mutation Rates, Synergistic Interactions, and the Effects of pH and Serum Proteins for Fusidic Acid (CEM-102)
D Biedenbach, P Rhomberg, R Mendes, R Jones
Diagnostic Microbiology & Infectious Disease. March 2010 66: 301-307
http://www.dmidjournal.com/article/S0732-8893(09)00424-6/abstract
Performance of Fusidic Acid (CEM-102) Susceptibility Testing Reagents: Broth Microdilution, Disk Diffusion, and Etest Methods as Applied to Staphylococcus aureus
R Jones, M Castanheira, P Rhomberg, L Woosley, M Pfaller
Journal of Clinical Microbiology. March 2010 48: 972-976
http://jcm.asm.org/cgi/content/abstract/48/3/972
Evaluation of the Activity of Fusidic Acid Tested Against Contemporary Gram-Positive Clinical Isolates From the USA and Canada
M Pfaller, M Castaneira, H Sader, R Jones
International Journal of Antimicrobial Agents. March 2010 35: 282-287
http://www.ijaaonline.com/article/S0924-8579(09)00510-X/abstract
Quantitative and qualitative assessment of antibiotic activity against Staphylococcus aureus biofilm.
Siala, W., M. P. Mingeot-Leclercq, P. M. Tulkens, and F. Van Bambeke.
Abstr. 6th Am. Soc. Microbiol. Conf. Biofilms, abstr A-179.
Download Poster 
Activity of Fusidic Acid Against Methicillin-resistant Staphylococcus Aureus (MRSA) Isolated from CF Patients
Prabhavathi Fernandes, Donald Anderson, K. Kosowska-Shick, P. McGhee, L. Beachel and P.C. Appelbaum
Download Abstract | Download Poster
Evaluation of L6 Ribosomal Protein Alterations in Fusidic Acid-Resistant Staphylococcus aureus: Fitness Cost and Time Kill Analysis
M Castanheira, RN Jones, LN Woosley, RE Mendes, GJ Moet, DJ Farrell
Download Abstract
Fusidic Acid Activity and Coverage of Gram-positive Pathogens Associated with Acute Bacterial Skin and Skin Structure Infections (ABSSSI) in the USA (2008-2010)
RN Jones, DJ Farrell, HS Sader, M Castanheira
Download Abstract | Download Poster
Activity of Fusidic Acid Tested Against Contemporary Staphylococcus aureus Collected from United States Hospitals
M. Castanheira, R.E. Mendes, P.R. Rhomberg, R.N. Jones
Download Abstract | Download Poster
Pharmacokinetics-Pharmacodynamics (PK-PD) of CEM- 102 (Sodium Fusidate) Against Streptococcus pyogenes Using In Vitro Pharmacodynamic Models (IVPM)
B. T. Tsuji, A. Forrest, P. A. Kelchlin, T. Brown, P. N. Holden, O. O. Okusanya, S. M. Bhavnani, P. Fernandes, P. G. Ambrose
Download Abstract | Download Poster
Activity of CEM-102 (sodium fusidate) against 40 MRSA from Cystic Fibrosis Patients
Cynthia Todd, Pamela Mcghee, and Peter Appelbaum
Download Abstract | Download Poster
Ability of CEM-102 (Fusidic Acid), Linezolid, Daptomycin to Select Resistant S.aureus Mutants at Steady-state Serum Levels
K. Kosowska-Shick, P. Mcghee, L. Beachel, P. C. Appelbaum;
Download Abstract | Download Poster
CEM-102 (Fusidic Acid) Maintains Potency against Resistant MRSA and Prevalent Hospital Acquired, Community Acquired,and Epidemic MRSA Clones
C.M. Pillar, M.K. Torres, D.F. Sahm and P. Fernandes
Download Abstract | Download Poster
In Vitro Activity Of Fusicic Acid (CEM-102) Against Resistant Strains Of Staphylococcus aureus
J. dubois, P. Fernandes
Download Abstract | Download Poster
MORE INFO

Fusidic acid (FA) is a tetracyclic triterpenoid or fusidane (steroidal) antibiotic derived from the fungus Fusidium coccineum that inhibits bacterial protein synthesis. FA is effective against gram-positive bacteria such as Staphylococcusspecies and Corynebacterium species (L. Verbist, J. Antimicro. Chemo. 25, Suppl. B, 1-5 (1990); A. Bryskier, Fusidic Acid, Chapter 23, in Antimicrobial Agents: Antibacterials and Antifungals (Andre Bryskier, Ed., ASM Press, Washington, USA, 2005)). FA also has moderate activity against Group A beta-hemolytic streptococci, or Streptococcus pyogenes (L. Verbist, J. Antimicro. Chemo. 25, Suppl. B, 1-5 (1990); A. Bryskier, Fusidic Acid, Chapter 23, inAntimicrobial Agents: Antibacterials and Antifungals (Andre Bryskier, Ed., ASM Press, Washington, USA, 2005); Skov et al., Diag. Micro. Infect. Dis. 40:111-116 (2001)).
FA was developed for clinical use in the 1960s and it is approved for human use outside of the United States, such as in the UK, Canada, Europe, Israel, Australia and New Zealand. It is typically prescribed at doses of 500 mg TID for treating skin and skin structure infections caused by Staphylococcus aureus (A. Bryskier,Fusidic Acid, Chapter 23, in Antimicrobial Agents: Antibacterials and Antifungals(Andre Bryskier, Ed., ASM Press, Washington, USA, 2005); Collignon et al., Int’l J. Antimicrobial Agents 12:S45-S58 (1999); D. Spelman, Int’l J. Antimicrobial Agents 12:S59-S66 (1999)), although some physicians have routinely prescribed the compound at 500 mg BID for treating skin and skin structure infections due to the long half-life of the compound (Fusidic Acid, in Principles and Practice of Infectious Diseases, 6th ed. (Mandell et al. eds., Elsevier, 2006)).
Treatment using FA has been well studied and it is generally regarded as safe when administered to humans, as evidenced by the fact that the drug has been in continuous use for more than 40 years. There are, however, several characteristics of FA that have prevented use of the drug against a wider spectrum of bacteria and in the treatment in additional types of infection. For example, approved dosing regimens have been shown to select for bacterial resistance, such as in S. aureus. Approved dosing regimens provide low multiples of the MIC and as a result, S. aureus resistant mutants can be selected after the first day of dosing. Once resistance has developed, FA is not effective against the resistant strains. Resistance is reported to occur if FA is used as a single drug as the resistance frequency at 4 and 8 times the MIC is in the range of 10−6 or 10−8 (Evans et al., J. Clin. Path. 19:555-560 (1966); Hansson et al., J. Mol. Biol.348:939-949 (2005), Jensen et al., Acta Pathol Microbiol Scand. 60:271-284 (1964); Besier et al., Antimicrob. Agents Chemo., 49(4):1426-1431 (2005); Gemmell et al., J. Antimicrobial Chemo. 57:589-608 (2006)).
The dosage of the drug cannot be simply increased as a means of avoiding development of resistance. It is difficult to achieve high concentrations of FA in the blood due to the substantial protein binding of the drug (approximately 95-97%) (K. Christiansen, International Journal of Antimicrobial Agents 12:S3-S9 (1999); Coutant et al., Diagn Microbiol Infect Dis 25:9-13 (1996); D. Reeves, J. Antimicrob. Chemo. 20:467-476 (1987); J. Turnidge, Int’l J. Antimicrobial Agents12:S23-S34 (1999); Rieutord et al., Int’l J. Pharmaceutics 119:57-64 (1995)). Moreover, high dosages of FA are not well-tolerated by patients receiving the drug. High doses of FA (e.g., 1 gram TID) are required if the drug is to be used in the treatment of bone and joint infections, less susceptible bacteria and other serious infections. However, treatment regimens using high doses of the drug induce nausea and vomiting and are rejected by patients (Fusidic Acid, inPrinciples and Practice of Infectious Diseases, 6th ed. (Mandell et al. eds., Elsevier, 2006); K. Christiansen, International Journal of Antimicrobial Agents 12:S3-S9 (1999); Nordin et al., Eur. J. Clin. Res. 5:97-106 (1994)).
In view of the tremendous costs associated with the de novo development of new anti-bacterials, expanding the indications for drugs that have already been demonstrated to be safe and effective is strongly needed. Overcoming the limitations on the uses of FA would broaden the population of bacterial infections against which it could be used and thus meet this need.
In a specific commercial pharmaceutical formulation, fusidic acid is presently marketed [see Monographs in the European Pharmacopeia 5.0] as a hemihydrate, which is the only hemihydrate form which has been described.
Patent GB 930,786 discloses salts of fusidic acid with organic and inorganic bases, solvates of fusidic acid, namely a benzene solvate and a methanol solvate. This patent further discloses an unspecified fusidic acid form with IR absorption bands (KBr) at 1265, 1385, 1695, 1730 and 3450 cm“1 and having a specific rotation [α]D 22 of minus 9 degrees (1% solution in CHCI3) obtainable by crystallisation of the methanol solvate of fusidic acid from ether. However, this form is distinct from the form of the present invention evident from the depicted IR spectrum in GB 930,786 which indicates that this form actually corresponds to the presently marketed hemihydrate form.
Solvates and salts of fusidic acid have also been disclosed in British patent GB 999,794. Patent ES 2208110 discloses two solvent free crystalline forms offusidic acid called Form I and Form II, and a crystalline hemihydrate called Form III which is identical to the presently marketed hemihydrate, respectively. The crystalline forms were identified and characterised by IR spectroscopy, differential scanning calorimetry, X-ray diffraction and melting points.
Patent WO 96/03128 discloses tablets containing a sodium salt form of fusidicacid and WO 86/03966 describes an ophthalmic gel composition comprising an undefined form of suspended fusidic acid.
GT Biologics, a developer of live biotherapeutics for the treatment of autoimmune diseases, has received orphan drug designation from the US Food and Drug Administration (FDA) for its lead product candidate, Thetanix.
read all at
read all on
http://microbewiki.kenyon.edu/index.php/Bacteroides_thetaiotaomicron

Isavuconazole (BAL4815; trade name Cresemba) is a triazole antifungal drug. Its prodrug, isavuconazonium sulfate (BAL8557), was granted approval by the U.S. Food and Drug Administration (FDA) on March 6, 2015[1]
During its Phase III drug trials, Astellas partnered with Basilea Pharmaceutica, the developer of the drug, for rights to co-development and marketing of isavuconazole. [2]
On May 28, 2013, Basilea Pharmaceutica announced it had been granted orphan drug status by the FDA for treatment of aspergillosis.[3] Since then, it has also been granted orphan drug status for the treatment of invasive candidiasis.[4]

Isavuconazonium sulfate (BAL8557)—a prodrug of isavuconazole.
CLINICAL TRIALS…LINK
PATENTS
|
6-27-2012
|
Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole
|
|
|
11-18-2011
|
Antifungal Composition
|
|
|
9-29-2010
|
PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS
|
|
|
12-3-2008
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
3-14-2007
|
N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
11-3-2004
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
10-10-2001
|
Azoles for treatment of fungal infections
|
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.

Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:



……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
http://www.sciencedirect.com/science/article/pii/S0960894X02008922
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.


We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.

Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%).
Figure options

Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).
read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
There have been three phase III clinical trials of isavuconazole, ACTIVE, VITAL and SECURE. As of June 2015, SECURE and VITAL have been presented in abstract form and results from ACTIVE have not been released.[9]
The SECURE trial compared voriconazole and isavuconazole in invasive fungal infections due to aspergillus. Isuvaconazole was found to be non-inferior to voriconazole, anothertriazole antifungal, with all cause mortality at 18.6%, compared to 20.2% in the voriconazole group. It additionally demonstrated a similar side effect profile.[10]
Data from the VITAL study showed that isavuconazole could be used in treatment of invasive mucormycosis, but did not evaluate its clinical efficacy for this indication.[11]
The ACTIVE trial is a comparison of isuvaconazole and caspofungin for invasive candida infections and results are anticipated in the second half of 2015.[12][13]
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{2-[(1R,2R)-(2,5-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
|
|
| Clinical data | |
| Trade names | Cresemba (prodrug form) |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, intravenous |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 6918485 |
| ChemSpider | 5293682 |
| UNII | 60UTO373KE |
| ChEBI | CHEBI:85979 |
| ChEMBL | CHEMBL409153 |
| NIAID ChemDB | 416566 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Molecular mass | 437.47 g/mol |


/////

The FDA today granted “breakthrough therapy” designation to ofatumumab for treatment of patients with chronic lymphocytic leukemia.
The designation applies to use of ofatumumab (Arzerra, GlaxoSmithKline) in combination with chlorambucil in patients with untreated CLL who unsuitable for fludarabine-based therapy.
Ofatumumab is a human monoclonal antibody that targets an epitope on the CD20 molecule encompassing parts of the small and large extracellular loops.
read all at
also read my post on newdrugapprovals
https://newdrugapprovals.wordpress.com/2013/07/08/gsk-tests-ofatumumab-in-rare-skin-disorder/
Ofatumumab (trade name Arzerra, also known as HuMax-CD20) is a human monoclonal antibody (for the CD20 protein) which appears to inhibit early-stage B lymphocyte activation. It is FDA approved for treating chronic lymphocytic leukemia that is refractory to fludarabine and alemtuzumab (Campath) and has also shown potential in treating Follicular non-Hodgkin’s lymphoma, Diffuse large B cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis. Ofatumumab has also received conditional approval in Europe for the treatment of refractory chronic lymphocytic leukemia. This makes ofatumumab the first marketing application for an antibody produced by Genmab, as well as the first human monoclonal antibody which targets the CD20 molecule that will be available for patients with refractory CLL.Designated an orphan drug by FDA for the treatment of B-CLL

MACITENTAN
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide,
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl] -N’-propylsulfamide
CAS NO 441798-33-0
ACT-064992, Opsumit,UNII-Z9K9Y9WMVL
Mechanism of Action: Endothelin receptor antagonist (ERA)
Date of Approval: October 18, 2013(US)
Indication: Pulmonary Hypertension (PAH)
Company: Actelion Pharmaceuticals Ltd
PCT patent application: WO2002053557
FDA N204410, MACITENTANTABLET; ORAL10MG, OPSUMIT, ACTELION PHARMS LTD
Macitentan is achiral
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
Macitentan (Opsumit® )is a novel dual endothelin receptor antagonist that resulted from a tailored drug discovery process. Macitentan has a number of potentially key beneficial characteristics – i.e., increased in vivo preclinical efficacy vs. existing ERAs resulting from sustained receptor binding and tissue penetration properties. A clinical pharmacology program indicated a low propensity of macitentan for drug-drug interactions.
Macitentan (ACT-064992) is a tissue-targeting dual ET(A)/ET(B) endothelin (ET) receptor antagonist designed for tissue targeting. Macitentan inhibited ET-1-induced contractions in isolated endothelium-denuded rat aorta (ET(A) receptors) and sarafotoxin S6c-induced contractions in isolated rat trachea (ET(B) receptors). In diabetic rats, chronic administration of macitentan decreased blood pressure and proteinuria and prevented end-organ damage. Treatment with macitentan enhanced the cytotoxicity mediated by paclitaxel as measured by the degree of apoptosis in tumor cells and tumor-associated endothelial cells. A Phase III clinical trial of macitentan was successfully completed in 2012.


Macitentan is an investigational drug being studied for the treatment of pulmonary arterial hypertension. It acts as a dualendothelin receptor antagonist and is being developed by Actelion.[1] A Phase III clinical trial was successfully completed in 2012.[2]
on 22 October 2012 – Actelion (SIX: ATLN) announced that it has submitted a New Drug Application (NDA) to the US Food and Drug Administration (FDA) seeking approval for macitentan (Opsumit®) for the treatment of patients with pulmonary arterial hypertension
Actelion’s experimental lung drug macitentan prolonged overall survival by more than a third according to detailed study data, which the company hopes will convince investors it has a viable follow-up product to secure its commercial future.
Europe’s largest standalone biotech company wants the drug, which treats pulmonary arterial hypertension — a disease that causes high blood pressure in the arteries of the lungs — to replace blockbuster Tracleer.
Tracleer currently makes up 87 percent of sales but loses patent protection in 2015 and has also seen its market share eroded by Gilead’s Letairis.

Macitentan has an active metabolite, ACT-132577, which is an oxidative depropylation product. Both macitentan and ACT-132577 are mainly excreted in form of hydrolysis products via urine (about 2/3 of all metabolites) and faeces (1/3).[3]
Co-administration of ciclosporin has only a slight effect on the concentrations of macitentan and its active metabolite, whilerifampicin decreases the area under the curve (AUC) of the drug’s blood plasma concentration by 79%, and ketoconazoleapproximately doubles it. This corresponds to the finding that macitentan is mainly metabolised via the liver enzyme CYP3A4.[4]

SYNTHESIS
The synthesis begins with the reaction of chlorosulfonyl isocyanate (1) (dissolved in dichloromethane at 0 ° C) with one equivalent of tert-butanol. This produces a by BOC protected Aminosulfonylchlorid (2). With one equivalent of n-propylamine (dissolved in 3 eq. Of triethylamine, dichloromethane, at 0 ° C, RT 16 h) is produced by a hydrochloric acid elimination BOC-protected sulfamide (3). This is dissolved in 5 M HCl and dioxane (4-8 h), the BOC protecting group is cleaved. The sulfamide formed (4) is potassium tert-butoxide-(dissolved in MeOH, 3h) is converted to the potassium salt (5). Tert-butoxide potassium acts as a very strong base for deprotonation. This sulfamide potassium salt reacts with the nucleophilic substituents on the heteroaromatic Dichlorpyrimidinderivat (6) (dissolved in dimethyl sulfoxide, at room temperature, RT 42-72 h) under KCl-cleavage to a Monochlorpyrimidin intermediate (7). By treatment with ethylene glycol (dissolved in dimethyl ether, potassium-tert-butoxide,), the ethylene glycol side chain is generated (8). With 2-chloro-5-bromo-pyrimidine (dissolved in tetrahydrofuran, close, at 60-75 ° C) is formed under elimination of HCl in an S N 1 reaction Macitentan (9)…………Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .

Synthesis of Macitentan
…………………………………………….
SYNTHESIS


YOU CAN READ AT YAOPHA.COM, lovely site to see for drugs


如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
………………………….
SYNTHESIS
(WO2006/051502A2, JMC2012, 7849). Chlorosulfonyl isocyanate ( 1 ) reaction with tert-butyl alcohol 2 , which is then reacted with n-propylamine 3 . 3 de-boc protected through the acid after reaction with potassium t-butoxide 4 . Another compound 5 with NaH after acidic protons off with dimethyl carbonate ( 6 ) to obtain 7 . 7 and formamidine hydrochloride ( 8 ) to ring chlorinated later POCl3 9 . 9 and 4 SNAr reaction occurs 10 . 10under basic conditions with ethylene glycol SNAr reaction occurs again in alkaline conditions with11 SNAr reaction occurs MACITENTAN.

………………………
http://www.google.com/patents/WO2014155304A1?cl=en
LC-MS (Agilent MS detector G1956B with Agilent 1200 Binary Pump and DAD).
Parameters of the LC-MS method:
Injection volume: 2 |jL
Column: Kinetex C18, 2.6 μιη, 2.1 x 50 mm
Column flow rate: 1 mL/min
Eluents: Eluent A: water + 0.08% TFA
Eluent B: MeCN + 0.012% TFA
Gradient: 2.0 min 95% B
2.8 min 95% B
3.0 min 5% B
Temperature: 40°C Detector wavelength 210 nm

Preparation B: N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]- 4-pyrimidinyl] -N’-propylsulfamide (macitentan):
N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)propane- 1-sulfamide (200 g; 0.46 mol; see Example 2 or 3) and 5-bromo-2-chloropyrimidine (117 g; 0.60 mol; 1.3 eq) were dissolved in toluene (3 L) and DMF (400 mL). The reaction mixture was warmed up to 50°C and toluene (approx. 400 mL) was distilled our under reduced pressure. The mixture was cooled to 0 °C and tBuOK (156 g, 3 eq, 1.38 mol) was added portionwise. It was stirred at 20 °C for 1 h. Water (1 L) was added and the pH of the solution was adjusted to 3-5 using 33% aq. HC1. The mixture was heated to 50°C and the layers were separated. The org. phase was treated with charcoal at 50°C and filtered over Celite. The filter cake was rinsed with toluene. At 50°C, water (1 L) was added to the org. layer. The layers were separated. The org. layer was concentrated under reduced pressure to a total volume of 1 L and cooled to 0°C. The solid obtained was filtered off. It was rinsed with toluene and MeOH. The crude material was suspended in EA (1 L) and heated to 50°C. 300 mL of EA were distilled out and MeOH (400 mL) was added. The suspension was cooled down to 0°C. The solid was filtered off, rinsed with MeOH and dried under reduced pressure to afford the title compound as a white solid (225 g; 83% yield).

……………………
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm3009103

Starting from the structure of bosentan (1), we embarked on a medicinal chemistry program aiming at the identification of novel potent dual endothelin receptor antagonists with high oral efficacy. This led to the discovery of a novel series of alkyl sulfamide substituted pyrimidines. Among these, compound 17 (macitentan, ACT-064992) emerged as particularly interesting as it is a potent inhibitor of ETA with significant affinity for the ETB receptor and shows excellent pharmacokinetic properties and high in vivo efficacy in hypertensive Dahl salt-sensitive rats. Compound 17 successfully completed a long-term phase III clinical trial for pulmonary arterial hypertension
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (17)
……………
WO 2015004265 click
Example 3 : N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)pr opane- 1- sulfamide (reaction in and work-up with MIBK):
EG (124 mL, 3.7 mol, 6.0 eq.) was added to a warm (40-50°C) suspension of the compound of Preparation A (150 g, 0.37 mol) in MIBK (600 mL). Solid KOtBu (114 g, 1.11 mol, 3.0 eq.) was added portionwise so that IT < 60°C. The mixture was stirred for
2- 3 h at 100-105°C. After completion of the reaction (LC-MS control), it was cooled to 50 °C. A 40%) aq. solution of citric acid monohydrate (300 mL) was added until pH 4 was reached. The layers were separated. The org. phase was washed with water (450 mL) and the layers were separated. Water (450 mL) was added and the mixture was warmed to 50°C. It was stirred at 50°C for 5 min. The layers were separated. The org. phase was concentrated under vacuum at 50°C until 200 mL of MIBK were removed. Hept (800 mL) was added dropwise at 70-75°C until turbidity was observed. The mixture was seeded with an analytically pure sample of N-(5-(4-bromophenyl)-6-(2 hydroxy ethoxy)pyrimidin-4-yl)propane-l-sulfamide and stirred at 60-65°C for 30 min. It was allowed to cool to 5°C within 5 h. It was filtered off, rinsed with a cold MIBK/Hept mixture (300 mL, 1 : 1) and dried under vacuum at 50°C to yield the title compound as a white solid (121 g; 76% yield).
The product had NMR data equivalent to those reported in Bolli et al, J. Med. Chem. (2012), 55, 7849-7861. [M+H]+ = 430 and 432. LC-MS: tR = 1.46 min; purity: 98.4% a/a. Residual ethylene glycol (GC-FID): 530 ppm.
…….
CN 104447572 click
(l) Martin H. Bolli et al. Reported the synthesis of Marcy cefotetan follows:

[0008] The method W 5- (4- desert phenyl) -4,6-dichloro-chewing clever as a starting material, N- propyl amine Lai ugly bell in DMS0 as a reaction solvent, an alcohol bell as t a base under substitution reaction conditions, the reaction temperature needs of 24-7 to give


The intermediate compound 15, compound 15 in hexylene glycol dimethyl off as the reaction solvent, a tertiary alcohol under conditions with a strong base clock as hexanediol substitution reaction, l〇 (TC Reaction of 18-2 to give compound 17, Compound 17 was then reacted with 5-chloro-chewing desert -2 clever substitution reaction at tetraammine Qiao Nan as a reaction solvent, ammoniated axis as the alkali conditions, the reaction to give the final product of Marcy cefotetan The route every step the higher the yield, the experimental use of N- propyl amine Lai ugly bell hygroscopic, unstable and a long time before the two-step reaction, the reaction at the second step requires l〇 (TC high temperature 18-2 technology is not suitable for industrial production.
[0009] International Patent W02002 / 053557 discloses some preparation methods and other Massey cefotetan column derivative method at each step of the preparation of the reaction times are longer, some reactions up to 4 days, and the resulting intermediate are purified by column chromatography method is not suitable for industrial production.



[00 pairs (3) N- [5- (4- desert) -6-mouth – [(5-desert -2- chew clever-yl) oxy] hexyl oxy] -4-chewing clever yl] -N ‘- Lai ugly propyl amine (Formula I) Synthesis
[0036] Weigh 20gN-5- (4- desert) -6- (2-2- light hexyl group -) 4- chew clever group -N ‘- Lai ugly propyl amine, 200ml dried DMS0 added to 1L H jar, add 20g of alcohol t-clock was added in portions, then add 17. 7g5- desert – dichloro chew clever, 30-4 (TC reduction reaction, the reaction and the reaction solution. a 10% sample skillfully acid to adjust PH value 3 to 4, the reaction mixture was added to 1000ml water, olive mix, suction. suction Massey cefotetan get wet crude product 42g, 450ml of methanol was added at room temperature and then beating 20min, filtration and dried 45C to give white solid was dried under vacuum to give 23.2 Marcy cefotetan yield;.. 85%
[0037] The compound (Formula I) relating to the physical and chemical properties, spectroscopic data are as follows:
[0038] branded point; 135-136 ° C; we NMR (300MHz, DMS0) 5 (egg m):… 9 8 (s, lH), 8 7 (s, 2H), 8 5 (s, l H,) 7. 5 (s, 2H), 7. 2 (s, IH), 7. 1 (s, 2H,) 4. 7 (s, 2H), 4. 6 (s, 2H,) 2. 8 (s, 2H,), 1. 5 (m, 2H,), 0. 81 (m, 3H), MS Qiaoqiao m / z 589 ([M + Tin +).
…………
see
WO 2002053557
http://www.google.com/patents/WO2002053557A1?cl=en
………..


Assignment of the signals mentioned in the text of the H-NMR spectrum of the drug Macitentan
Solvent: CDCl 3
δ 8.51 (s, 2H, CH) 11 , 8.49 (s, 1 H, CH) 10 , 7.58 to 7.63 (m, 2H, CH) 9 , 7.16 to 7.21 ( m, 2H, CH) 8 , 6.88 (s, 1H, NH) 7 , 5.61 (t, J = 6.2 Hz, 1H, NH) 6 , 4.72 to 4.76 (m, 2H , CH 2 ) 5 , 4.62 to 4.66 (m, 2H, CH 2 ) 4 , 2.99 (q, J = 6.8 Hz, 2H, CH 2 ) 3 , 1.61 (h, J = 7.3 Hz, 2H, CH2 ) 2 , 0.97 (t, J = 7.4 Hz, 3H, CH 3 ) 1 . [Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .]

Solvent: CDCl 3
δ 11.6, 22.7, 46.1, 65.3, 65.9, 104.8, 112.4, 123.7, 128.0, 131.7, 133.0, 155.7, 156 , 4, 159.7, 163.5, 166.3. [ Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 . ]
NMR PREDICT BY ME
1H NMR PREDICT

13C NMR PREDICT BY ME


COSY PREDICT BY ME, WORLDDRUGTRACKER ON A WHEELCHOPPER SCALING NEW HEIGHTS
REFERENCES
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion’s first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium – the single layer of cells separating every blood vessel from the blood stream. Actelion’s over 2,400 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SIX Swiss Exchange (ticker symbol: ATLN) as part of the Swiss blue-chip index SMI (Swiss Market Index SMI®).
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide
|
|
| Clinical data | |
| Trade names | Opsumit |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Metabolism | Hydrolysis, oxidation (CYP3A4) |
| Excretion | 2/3 urine, 1/3 faeces |
| Identifiers | |
| CAS Registry Number | 441798-33-0 |
| ATC code | C02KX04 |
| PubChem | CID: 16004692 |
| ChemSpider | 13134960 |
| ChEBI | CHEBI:76607 |
| Synonyms | ACT-064992 |
| Chemical data | |
| Formula | C19H20Br2N6O4S |
| Molecular mass | 588.273 g/mol |
| Patent | Submitted | Granted |
|---|---|---|
| Sulfamides and their use as endothelin receptor antagonists [US7094781] | 2004-04-22 | 2006-08-22 |
| Sulfamides and their use as endothelin receptor antagonists [US7285549] | 2006-08-10 | 2007-10-23 |
| Stable Pharmaceutical Compositions Comprising a Pyrimidine – Sulfamide [US2008233188] | 2008-09-25 | |
| Combination Comprising Paclitaxel for Treating Ovarian Cancer [US2010311774] | 2010-12-09 | |
| Stable pharmaceutical compositions comprising a pyrimidine-sulfamide [US2010004274] | 2010-01-07 | |
| SULFONYLUREA MODULATORS OF ENDOTHELIN RECEPTOR [US2011082151] | 2011-04-07 | |
| ENDOTHELIN RECEPTOR ANTAGONISTS FOR EARLY STAGE IDIOPATHIC PULMONARY FIBROSIS [US2010022568] | 2007-04-12 | 2010-01-28 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2011136818] | 2011-06-09 | |
| Therapeutic Compositions Comprising a Specific Endothelin Receptor Antagonist and a PDE5 Inhibitor [US2009318459] | 2009-12-24 |
Patent and Exclusivity
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
|
|---|---|---|---|---|---|---|---|
| N204410 | 001 | US7094781 | Oct 12, 2022 | Y | Y | ||
| N204410 | 001 | US8268847 | Apr 18, 2029 | U – 1446 | |||
| N204410 | 001 | US8367685 | Oct 4, 2028 | Y | U – 1445 |
| Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N204410 | 001 | ODE | Oct 18, 2020 |
| N204410 | 001 | NCE | Oct 18, 2018 |
U1446 METHOD OF TREATING PULMONARY HYPERTENSION COMPRISING ADMINISTERING MACITENTAN IN COMBINATION WITH A COMPOUND HAVING PHOSPHODIESTERASE-5 INHIBITORY PROPERTIES
U1445 METHOD OF TREATING PULMONARY ARTERIAL HYPERTENSION BY ADMINISTERING A PHARMACEUTICAL COMPOSITION COMPRISING MACITENTAN AND A POLYSORBATE, WHERIN THE POLYSORBATE REPRESENTS 0.1 TO 1% OF THE WEIGHT OF SAID PHARMACEUTICAL COMPOSITION
OPSUMIT (macitentan) is an endothelin receptor antagonist. The chemical name of macitentan is N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide. It has a molecular formula of C19H20Br2N6O4S and a molecular weight of 588.27. Macitentan is achiral and has the following structural formula:
 |
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
OPSUMIT is available as a 10 mg film-coated tablet for once daily oral administration. The tablets include the following inactive ingredients: lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 80, povidone, and sodium starch glycolate Type A. The tablets are film-coated with a coating material containing polyvinyl alcohol, soya lecithin, talc, titanium dioxide, and xanthan gum.
//////
