WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
FDA approves first targeted treatment Tibsovo (ivosidenib) for patients with relapsed or refractory acute myeloid leukemia who have a certain genetic mutation
The U.S. Food and Drug Administration today approved Tibsovo (ivosidenib) tablets for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. This is the first drug in its class (IDH1 inhibitors) and is approved for use with an FDA-approved companion diagnostic used to detect specific mutations in the IDH1 gene in patients with AML.
“Tibsovo is a targeted therapy that fills an unmet need for patients with relapsed or refractory AML who have an IDH1 mutation,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The use of Tibsovo is associated with a complete remission in some patients and a reduction in the need for both red cell and platelet transfusions.”
The U.S. Food and Drug Administration today approved Tibsovo (ivosidenib) tablets for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. This is the first drug in its class (IDH1 inhibitors) and is approved for use with an FDA-approved companion diagnostic used to detect specific mutations in the IDH1 gene in patients with AML.
“Tibsovo is a targeted therapy that fills an unmet need for patients with relapsed or refractory AML who have an IDH1 mutation,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The use of Tibsovo is associated with a complete remission in some patients and a reduction in the need for both red cell and platelet transfusions.”
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of abnormal white blood cells in the bloodstream and bone marrow. The National Cancer Institute at the National Institutes of Health estimates that approximately 19,520 people will be diagnosed with AML this year; approximately 10,670 patients with AML will die of the disease in 2018.
Tibsovo is an isocitrate dehydrogenase-1 inhibitor that works by decreasing abnormal production of the oncometabolite 2-hydroxyglutarate (2-HG), leading to differentiation of malignant cells. If the IDH1 mutation is detected in blood or bone marrow samples using an FDA-approved test, the patient may be eligible for treatment with Tibsovo. Today the agency also approved the RealTime IDH1 Assay, a companion diagnostic that can be used to detect this mutation.
The efficacy of Tibsovo was studied in a single-arm trial of 174 adult patients with relapsed or refractory AML with an IDH1 mutation. The trial measured the percentage of patients with no evidence of disease and full recovery of blood counts after treatment (complete remission or CR), as well as patients with no evidence of disease and partial recovery of blood counts after treatment (complete remission with partial hematologic recovery or CRh). With a median follow-up of 8.3 months, 32.8 percent of patients experienced a CR orCRh that lasted a median 8.2 months. Of the 110 patients who required transfusions of blood or platelets due to AML at the start of the study, 37 percent went at least 56 days without requiring a transfusion after treatment with Tibsovo.
Common side effects of Tibsovo include fatigue, increase in white blood cells, joint pain, diarrhea, shortness of breath, swelling in the arms or legs, nausea, pain or sores in the mouth or throat, irregular heartbeat (QT prolongation), rash, fever, cough and constipation. Women who are breastfeeding should not take Tibsovo because it may cause harm to a newborn baby.
Tibsovo must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. The prescribing information for Tibsovo includes a boxed warning that an adverse reaction known as differentiation syndrome can occur and can be fatal if not treated. Signs and symptoms of differentiation syndrome may include fever, difficulty breathing (dyspnea), acute respiratory distress, inflammation in the lungs (radiographic pulmonary infiltrates), fluid around the lungs or heart (pleural or pericardial effusions), rapid weight gain, swelling (peripheral edema) or liver (hepatic), kidney (renal) or multi-organ dysfunction. At first suspicion of symptoms, doctors should treat patients with corticosteroids and monitor patients closely until symptoms go away.
Other serious warnings include a QT prolongation, which can be life-threatening. Electrical activity of the heart should be tested with an electrocardiogram during treatment. Guillain-Barré syndrome, a rare neurological disorder in which the body’s immune system mistakenly attacks part of its peripheral nervous system, has happened in people treated with Tibsovo, so patients should be monitored for nervous system problems.
The FDA granted this application Fast Track and Priority Review designations. Tibsovo also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Tibsovo to Agios Pharmaceuticals, Inc. The FDA granted the approval of the RealTime IDH1 Assay to Abbott Laboratories.
It is in a phase III clinical trial for acute myeloid leukemia (AML) with an IDH1 mutation and a phase III clinical trial for cholangiocarcinoma with an IDH1 mutation.[2]
OriginatorAgios Pharmaceuticals
DeveloperAbbVie; Agios Pharmaceuticals; University of Texas M. D. Anderson Cancer Center
ClassAntineoplastics; Cyclobutanes; Nitriles; Pyridines; Pyrrolidines; Small molecules
Mechanism of ActionIsocitrate dehydrogenase 1 inhibitors
Orphan Drug StatusYes – Acute myeloid leukaemia; Cholangiocarcinoma
28 Jun 2018Massachusetts General Hospital and Agios Pharmaceuticals plan a phase I trial for Acute myeloid leukaemia; Myelodysplastic syndromes and Chronic myelomonocytic leukaemia (Maintenance therapy) in USA (NCT03564821)
26 Jun 2018Ivosidenib licensed to CStone Pharmaceuticals in China, Hong Kong, Macau and Taiwan
14 Jun 2018Efficacy and adverse events data from a phase I trial in Acute myeloid leukaemia presented at the 23rd Congress of the European Haematology Association (EHA-2018)
Tecovirimat, sold under the brand name Tpoxx among others,[6] is an antiviral medication with activity against orthopoxviruses such as smallpox and monkeypox.[4][7][8] It is the first antipoxviral drug approved in the United States.[9][10] It is an inhibitor of the orthopoxvirus VP37 envelope wrapping protein.[4]
The drug works by blocking cellular transmission of the virus, thus preventing the disease.[11] Tecovirimat has been effective in laboratory testing; it has been shown to protect animals from monkeypox and rabbitpox and causes no serious side effects in humans.[6] Tecovirimat was first used for treatment in December 2018, after a laboratory-acquired vaccinia virus infection.[12]
The World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980. However, there have been longstanding concerns that smallpox may be used as a bioweapon.2,5 Tecovirimat is an antiviral drug that was identified via a high-throughput screen in 2002.2 It is effective against all orthopoxviruses, including vaccinia, cowpox, ectromelia, rabbitpox, monkeypox, and Variola (smallpox) virus.1,4
Tecovirimat was approved by the FDA in July 2018 as the first drug ever approved to treat smallpox.6,5 Tecovirimat was later approved by Health Canada in December 2021,7 followed by the approval from the European Commission in January 2022.9 Other than smallpox, tecovirimat is also indicated to treat complications due to replication of the vaccinia virus following vaccination against smallpox, and to treat monkeypox and cowpox in adults and children.8 Tecovirimat is available as both oral and intravenous formulations.10
Medical uses
In the United States, tecovirimat is indicated for the treatment of human smallpox disease.[4] In the European Union it is indicated for the treatment of smallpox, monkeypox, and cowpox.[5]
Mechanism of action
Tecovirimat inhibits the function of a major envelope protein required for the production of extracellular virus. The drug prevents the virus from leaving an infected cell, hindering the spread of the virus within the body.[16]
Chemistry
The first synthesis of tecovirimat was published in a patent filed by scientists at Siga Technologies in 2004. It is made in two steps from cycloheptatriene.[17]
The scheme has taken from SmartChem a knowledgebase by ROW2 Technologies, Inc. (www.row2technologies.com)
A perfect amalgamation of information on chemicals and global suppliers. A database where you can search for information on more than 150,000 chemicals and around 15,000 Global chemicals suppliers, including routes of synthesis, Applications, end uses, and validated contact details of global suppliers. For more information, please visit www.row2technologies.com or contact,
Reference: Dai, Dongcheng. Process for the preparation of tecovirimat. Assignee Siga Technologies, Inc., USA. WO 2014028545. (2014).
SYN 3
Synthetic Description
Reference: Medical composition containing ST-246, its preparation and anti-poxvirus application. Assignee Institute of Microbiology and Epidemiology, Academy of Military Medical Sciences, PLA, Peop. Rep. China. CN 101912389. (2010).
The present invention provides a process for making ST-246 outlined in Scheme 1
The present invention also provides a process for making ST-246 outlined in Scheme 2
The present invention further provides a process for making ST-246 outlined in Scheme 3
The present invention also provides a process for making ST-246 outlined in Scheme 4
The present invention further provides a process for making ST-246 outlined in Scheme 5
The present invention also provides the following compounds useful in the synthesis of ST-246:
EXAMPLE 1Synthetic Route I
Step A. Synthesis of Compound 6 (P=Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO04112718) in EtOH (80 mL, EMD, AX0441-3) was added tert-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc-hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCl3: δ 6.30 (br s, 1H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1.46 (s, 9H), 1.06-1.16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCl Salt)
Compound 6 (3.6 g, 11.83 mmol) was dissolved in i-PrOAc (65 mL, Aldrich, 99.6%). 4M HCl in dioxane (10.4 mL, 41.4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (15 mL) and dried under vacuum to yield HCl salt of compound 7 (1.9 g, 67% yield) as a white solid. The filtrate was concentrated to ⅓ its volume and stirred at 10-15° C. for 30 min. The solid was filtered, washed with minimal volume of i-PrOAc and dried to afford additional 0.6 g (21% yield) of compound 7. Total yield: 2.5 g (88% yield). 1H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1.07-1.17 (m, 2H), 0.18-0.29 (m, 1H), −0.01-0.07 (m, 1H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1.17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20° C. The resulting solution was stirred for 5 minutes at 15-20° C., to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15-20° C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH4Cl (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO04112718 and were consistent.
EXAMPLE 2Synthetic Route II
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (11.6%).
The reaction mixture was cooled to 45° C. and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1.5 g, 54% yield) as an off-white solid. 1H NMR in CDCl3: δ 8.44 (s, 1H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95° C. under nitrogen atmosphere. After 1.5 h at 95° C., LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo=94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95° C., LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 110° C. and the reaction was monitored. After heating at 110° C. for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo=94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO04112718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo=97:3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+
EXAMPLE 3Synthetic Route III
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and tert-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine by-product (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1H NMR in DMSO-d6: δ 9.61 (s, 1H), 7.16 (s, 2H), 1.42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
Step B. Synthesis of Compound 11 (HCl salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in i-PrOAc (57 mL, Aldrich, 99.6%). 4M HCl in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (10 mL) and dried at 45° C. under vacuum for 1 h to afford HCl salt of compound 11 (2.39 g, 89% yield) as a white solid. 1H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 113.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1.19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylamine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20° C. The resulting solution was stirred for 5 minute at 15-20° C. and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1.31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15-20° C. and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4Cl (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1.76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 110-115° C. under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo=94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo=93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25-35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo=99:1) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO04112718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo=91:9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
EXAMPLE 4Synthetic Route IV
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and tert-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 mL, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1.0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31.1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95° C. under nitrogen atmosphere. After 15 h at 95° C., LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105° C. overnight. After total 40 h at 95-105° C., LC-MS analysis at 254 nm showed ˜99% conversion to the desired product (endo:exo=93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25-50% EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo=91:9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCl salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in i-PrOAc (26 mL, Aldrich, 99.6%). 4M HCl in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (5 mL) and dried under vacuum to yield HCl salt of compound 7 (1.57 g, 97% yield) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1.34 mL, 7.7 mmol) keeping the temperature below 20° C. and the resulting solution was stirred for 5-10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20° C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4Cl (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO04112718.
EXAMPLE 5Synthetic Route V
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1.6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 mL,) was added triethylamine (2.04 mL, 14.63 mmol) keeping the temperature below 20° C. and the resulting solution was stirred for 5-10 minute. 4-Iodobenzoyl chloride 12 (1.95 g, 7.31 mmol, 1.1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20° C. After 24 h, additional 0.18 g (0.1 equiv, used total 1.6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and ˜5% of compound 7. The reaction was diluted with dichloromethane (100 mL). The organic phase was washed with saturated aqueous NH4Cl (100 mL), water (100 mL) and saturated aqueous NaHCO3 (100 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25-50% EtOAc in hexanes to afford compound 13 (1.63 g, 57% yield, HPLC area 93% pure) as a white solid. 1H NMR in DMSO-d6: δ 11.19 and 10.93 (two singlets with integration ratio of 1.73:1, total of 1H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1.18 (s, 2H), 0.27 (q, 1H), 0.06 (s, 1H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 mL) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (0.44 mL, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at −90° C. for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45° C. and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25-35% EtOAc in hexanes to afford ST-246 (55 mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO04112718.
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt) Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. Example 2: Synthetic Route II
Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co- injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III
ST-246 9
P = Boc Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+. duct
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+ Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:
P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III. Im ine by-product
Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV) To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:
Scheme 5 Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55 mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
As of 2009, the results of clinical trials support its use against smallpox and other related orthopoxviruses. It shows potential for a variety of uses including preventive healthcare, as a post-exposure therapeutic, as a therapeutic, and an adjunct to vaccination.[21][
Tecovirimat can be taken by mouth and as of 2008, was permitted for phase II trials by the U.S. Food and Drug Administration (FDA). In phase I trials, tecovirimat was generally well tolerated with no serious adverse events.[22] Due to its importance for biodefense, the FDA designated tecovirimat for fast-track status, creating a path for expedited FDA review and eventual regulatory approval. On 13 July 2018, the FDA announced approval of tecovirimat.[23]
Society and culture
Legal status
In November 2021, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization under exceptional circumstances for the medicinal product tecovirimat siga, intended for the treatment of orthopoxvirus disease (smallpox, monkeypox, cowpox, and vaccinia complications) in adults and in children who weigh at least 13 kilograms (29 lb)[24] The applicant for this medicinal product is Siga Technologies Netherlands B.V.[24] Tecovirimat was approved for medical use in the European Union in January 2022.[5][25]
In December 2021, Health Canada approved oral tecovirimat for the treatment of smallpox in people weighing at least 13 kilograms (29 lb).[26][27]
^ Jump up to:abc“Tecovirimat Siga EPAR”. European Medicines Agency. 10 November 2021. Archived from the original on 16 May 2022. Retrieved 23 April 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^ Jump up to:abAU patent 2004249250, Bailey, Thomas R.; Jordan, Robert & Rippin, Susan R., “Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases”, published 2004-12-29, assigned to Siga Pharmaceuticals Inc
^Hughes, David L. (2019). “Review of the Patent Literature: Synthesis and Final Forms of Antiviral Drugs Tecovirimat and Baloxavir Marboxil”. Organic Process Research & Development. 23 (7): 1298–1307. doi:10.1021/acs.oprd.9b00144. S2CID197172102.
^ Jump up to:ab“Tecovirimat Siga: Pending EC decision”. European Medicines Agency. 11 November 2021. Archived from the original on 13 November 2021. Retrieved 13 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
Prior to its eradication in 1980, variola virus, the virus that causes smallpox, was mainly spread by direct contact between people. Symptoms typically began 10 to 14 days after infection and included fever, exhaustion, headache and backache. A rash initially consisting of small, pink bumps progressed to pus-filled sores before finally crusting over and scarring. Complications of smallpox could include encephalitis (inflammation of the brain), corneal ulcerations (an open sore on the clear, front surface of the eye) and blindness.
TPOXX’s effectiveness against smallpox was established by studies conducted in animals infected with viruses that are closely related to the virus that causes smallpox, and was based on measuring survival at the end of the studies. More animals treated with TPOXX lived compared to the animals treated with placebo. TPOXX was approved under the FDA’s Animal Rule, which allows efficacy findings from adequate and well-controlled animal studies to support an FDA approval when it is not feasible or ethical to conduct efficacy trials in humans.
The safety of TPOXX was evaluated in 359 healthy human volunteers without a smallpox infection. The most frequently reported side effects were headache, nausea and abdominal pain.
The FDA granted this application Fast Track and Priority Review designations. TPOXX also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases and a Material Threat Medical Countermeasure Priority Review Voucher, which provides additional incentives for certain medical products intended to treat or prevent harm from specific chemical, biological, radiological and nuclear threats.
The FDA granted approval of TPOXX to SIGA Technologies Inc.
TPOXX was developed in conjunction with the U.S. Department of Health and Human Services’ Biomedical Advanced Research and Development Authority (BARDA).
Tecovirimat (Tpoxx) Tecovirimat is a drug used for the treatment or prophylaxis of viral infections, particularly those caused by the orthopoxvirus (Figure 12). In 2015, Dai described a procedure for the preparation of tecovirimat in a US patent (Scheme 33).[57 ] The developed method started with a cycloaddition reaction of cycloheptatriene with maleic anhydride in xylene to yield adduct 192, which after reaction with tert-butyl carbazate provided compound 193. Deprotection in acidic media gave rise to hydrazine derivative 194 and subsequent reaction with p-trifluoromethylbenzoyl chloride afforded tecovirimat (191).
57 [57] D. Dai, US Patent 0322010, 2015.
Synthesis
RAW MATERIAL
Key RM is, 4,6-Etheno-1H-cycloprop[f]isobenzofuran-1,3(3aH)-dione, 3a,4,4a,5,5a,6-hexahydro-, (3aR,4R,4aR,5aS,6S,6aS)-rel–
The present invention provides a process for making ST-246 outlined in Scheme 1
P = Boc
Scheme 1
The present invention also provides a process for making ST-246 outlined in, Scheme 2
Scheme 2
The present invention further provides a process for making ST-246 outlined in Scheme 3
ST-246
P = Boc
Scheme 3
P = Boc
Scheme 4
The present invention further provides a process for making ST-246 outlined in
Scheme 5
Scheme 5
Example 1 : Synthetic Route I:
P = Boc
Scheme 1
Step A. Synthesis of Compound 6 (P = Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt)
Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 -50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent.
Example 2: Synthetic Route II
Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III
ST-246 9
P = Boc
Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
duct
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:
P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Im ine by-product
Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:
Scheme 5
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55
mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
PAPER
N-(3,3a,4,4a,5,5a,6,6a-Octahydro-1,3-dioxo-4,6- ethenocycloprop[f]isoindol-2-(1H)-yl)carboxamides: Identification of Novel Orthopoxvirus Egress Inhibitors
ViroPharma Incorporated, 397 Eagleview Boulevard, Exton, Pennsylvania 19341, United States Army Medical Research Institute of Infectious Diseases, 1425 Porter Street, Frederick, Maryland 21702, University of Alabama, Birmingham, Alabama 35294, and SIGA Technologies, Inc., 4575 SW Research Way, Corvallis, Oregon 97333
J. Med. Chem., 2007, 50 (7), pp 1442–1444
DOI: 10.1021/jm061484y
A series of novel, potent orthopoxvirus egress inhibitors was identified during high-throughput screening of the ViroPharma small molecule collection. Using structure−activity relationship information inferred from early hits, several compounds were synthesized, and compound 14was identified as a potent, orally bioavailable first-in-class inhibitor of orthopoxvirus egress from infected cells. Compound 14 has shown comparable efficaciousness in three murine orthopoxvirus models and has entered Phase I clinical trials.
A mixture of 2.00 g (9.8 mmol) of 4-(trifluoromethyl) benzoic acid hydrazide, 1.86 g (9.8 mmol) of 4,4a,5,5a,6,6a-hexahydro-4,6-etheno-1Hcycloprop[f]isobenzofuran-1,3(3aH)-dione, and one drop of diisopropylethylamine in 40 mL of absolute ethanol was refluxed for 4.5 h. Upon cooling to rt, 4 mL of water was added, and the product began to crystallize. The suspension was cooled in an ice bath, and the precipitate collected by filtration. The crystalline solid was air-dried affording 3.20 g (87%) of the product as a white solid;
Preparation of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide
a. Preparation of Compounds 1(a) and 1(b).
A mixture of cycloheptatriene (5 g, 54.26 mmol) and maleic anhydride (6.13 g, 62.40 mmol) in xylenes (35 mL) was heated at reflux under argon overnight. The reaction was cooled to room temperature and a tan precipitate was collected by filtration and dried to give 2.94 grams (28%) of the desired product, which is a mixture of compounds 1(a) and 1(b). Compound 1(a) is normally predominant in this mixture and is at least 80% by weight. The purity of Compound 1(a) may be further enhanced by recrystallization if necessary. Compound 1(b), an isomer of compound 1(a) is normally less than 20% by weight and varies depending on the conditions of the reaction. Pure Compound 1(b) was obtained by concentrating the mother liquid to dryness and then subjecting the residue to column chromatography. Further purification can be carried out by recrystallization if necessary. 1H NMR (500 MHz) in CDCl3: δ 5.95 (m, 2H), 3.42 (m, 2H), 3.09 (m, 2H), 1.12 (m, 2H), 0.22 (m, 1H), 0.14 (m, 1H).
b. Preparation of N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. desired
A mixture of compound 1(a) (150 mg, 0.788 mmol) and 4-trifluoromethylbenzhydrazide (169 mg, 0.827 mmol) in ethanol (10 mL) was heated under argon overnight. The solvent was removed by rotary evaporation. Purification by column chromatography on silica gel using 1/1 hexane/ethyl acetate provided 152 mg (51%) of the product as a white solid.
c. Preparation of N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. UNWANTED
N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]4-(trifluoromethyl)-benzamide was prepared and purified in the same fashion as for N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide by replacing 1(a) with 1(b) and was obtained as a white solid. 1H NMR (300 MHz) in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+.
EXAMPLE 42 Characterization of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide (“ ”)
In the present application, ST-246 refers to: N-[(3aR,4R,4aR,5aS,65,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide.
Physico-Chemical Properties
Appearance: ST-246 is a white to off-white powder.
Melting Point: Approximately 196° C. by DSC.
Permeability: The calculated log P is 2.94. Based on the partition coefficient, ST-246 is expected to have good permeability.
Particle Size: The drug substance is micronized to improve its dissolution in the gastrointestinal fluids. The typical particle size of the micronized material is 50% less than 5 microns.
Solubility: The solubility of ST-246 is low in water (0.026 mg/mL) and buffers of the gastric pH range. Surfactant increases its solubility slightly. ST-246 is very soluble in organic solvents. The solubility data are given in Table 5.
Damon, Inger K.; Damaso, Clarissa R.; McFadden, Grant (2014). “Are We There Yet? The Smallpox Research Agenda Using Variola Virus”. PLoS Pathogens10 (5): e1004108.doi:10.1371/journal.ppat.1004108. PMID24789223.
Referenced by Citing Patent Filing date Publication date Applicant Title CN101912389A * Aug 9, 2010 Dec 15, 2010 中国人民解放军军事医学科学院微生物流行病研究所 Pharmaceutical composition containing ST-246 and preparation method and application thereof CN102406617A * Nov 30, 2011 Apr 11, 2012 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof CN102406617B Nov 30, 2011 Aug 28, 2013 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof CN103068232B * Mar 23, 2011 Aug 26, 2015 西佳科技股份有限公司 多晶型物形式st-246和制备方法 US8530509 Jul 29, 2011 Sep 10, 2013 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases US8802714 Aug 14, 2013 Aug 12, 2014 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases US9045418 Jul 3, 2014 Jun 2, 2015 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of Orthopoxvirus infections and associated diseases
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
CBD is a chemical component of the Cannabis sativa plant, more commonly known as marijuana. However, CBD does not cause intoxication or euphoria (the “high”) that comes from tetrahydrocannabinol (THC).
It is THC (and not CBD) that is the primary psychoactive component of marijuana.
“This approval serves as a reminder that advancing sound development programs that properly evaluate active ingredients contained in marijuana can lead to important medical therapies. And, the FDA is committed to this kind of careful scientific research and drug development,” said FDA Commissioner Scott Gottlieb, M.D. “Controlled clinical trials testing the safety and efficacy of a drug, along with careful review through the FDA’s drug approval process, is the most appropriate way to bring marijuana-derived treatments to patients. Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes. We’ll continue to support rigorous scientific research on the potential medical uses of marijuana-derived products and work with product developers who are interested in bringing patients safe and effective, high quality products. But, at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims. Marketing unapproved products, with uncertain dosages and formulations can keep patients from accessing appropriate, recognized therapies to treat serious and even fatal diseases.”
Dravet syndrome is a rare genetic condition that appears during the first year of life with frequent fever-related seizures (febrile seizures). Later, other types of seizures typically arise, including myoclonic seizures (involuntary muscle spasms). Additionally, status epilepticus, a potentially life-threatening state of continuous seizure activity requiring emergency medical care, may occur. Children with Dravet syndrome typically experience poor development of language and motor skills, hyperactivity and difficulty relating to others.
Lennox-Gastaut syndrome begins in childhood. It is characterized by multiple types of seizures. People with Lennox-Gastaut syndrome begin having frequent seizures in early childhood, usually between ages 3 and 5. More than three-quarters of affected individuals have tonic seizures, which cause the muscles to contract uncontrollably. Almost all children with Lennox-Gastaut syndrome develop learning problems and intellectual disability. Many also have delayed development of motor skills such as sitting and crawling. Most people with Lennox-Gastaut syndrome require help with usual activities of daily living.
“The difficult-to-control seizures that patients with Dravet syndrome and Lennox-Gastaut syndrome experience have a profound impact on these patients’ quality of life,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition.”
Epidiolex’s effectiveness was studied in three randomized, double-blind, placebo-controlled clinical trials involving 516 patients with either Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex, taken along with other medications, was shown to be effective in reducing the frequency of seizures when compared with placebo.
The most common side effects that occurred in Epidiolex-treated patients in the clinical trials were: sleepiness, sedation and lethargy; elevated liver enzymes; decreased appetite; diarrhea; rash; fatigue, malaise and weakness; insomnia, sleep disorder and poor quality sleep; and infections.
Epidiolex must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. As is true for all drugs that treat epilepsy, the most serious risks include thoughts about suicide, attempts to commit suicide, feelings of agitation, new or worsening depression, aggression and panic attacks. Epidiolex also caused liver injury, generally mild, but raising the possibility of rare, but more severe injury. More severe liver injury can cause nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice and/or dark urine.
Under the Controlled Substances Act (CSA), CBD is currently a Schedule I substance because it is a chemical component of the cannabis plant. In support of this application, the company conducted nonclinical and clinical studies to assess the abuse potential of CBD.
The FDA prepares and transmits, through the U.S. Department of Health and Human Services, a medical and scientific analysis of substances subject to scheduling, like CBD, and provides recommendations to the Drug Enforcement Administration (DEA) regarding controls under the CSA. DEA is required to make a scheduling determination.
The FDA granted Priority Review designation for this application. Fast-Track designation was granted for Dravet syndrome. Orphan Drug designation was granted for both the Dravet syndrome and Lennox-Gastaut syndrome indications.
The FDA granted approval of Epidiolex to GW Research Ltd.
Ipatasertib (RG7440) is an experimental cancer drug in development by Roche. It is a small molecule inhibitor of Akt. It was discovered by Array Biopharma and is currently in phase II trials for treatment of breast cancer.[1]
In vitro, ipatasertib showed activity against all three isoforms of Akt.[2]
Ipatasertib is an orally-available protein kinase B (PKB/Akt) inhibitor in phase III clinical development at Genentech for the treatment of metastatic castration-resistant prostate cancer in combination with abiraterone and prednisone.
In 2014, orphan drug designation was assigned in the U.S. for the treatment of gastric cancer including cancer of the gastro-esophageal junction.
Ipatasertib. An orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) with potential antineoplastic activity. Ipatasertib binds to and inhibits the activity of Akt in a non-ATP-competitive manner, which may result in the inhibition of the PI3K/Akt signaling pathway and tumor cell proliferation and the induction of tumor cell apoptosis. Activation of the PI3K/Akt signaling pathway is frequently associated with tumorigenesis and dysregulated PI3K/Akt signaling may contribute to tumor resistance to a variety of antineoplastic agents. Check for active clinical trials using this agent.
PROBLEM
It has been found that ipatasertib exhibits a very high solubility (>1 g/g water; >2 g/g water/ethanol 1:1) and a very high hygroscopicity (˜6% at 50% RH, >35% at 95% RH). Whereas poor solubility is often a limiting factor in the development of galenical formulations of other API’s (active pharmaceutical ingredient), a high solubility can equally be problematic for the process performance. Due to this very high intrinsic hygroscopicity of the API, ipatasertib drug substance tends to auto-dissolve to a honey-like viscous liquid at increased humidity. Such high solubility and hygroscopicity may pose serious problems for processing as well as for stability and shelf-life of the final product. Therefore, conventional pharmaceutical compositions comprising ipatasertib and processes for the manufacture of pharmaceutical compositions comprising wetting (e.g. wet granulation) are difficult due to the high solubility and high hygroscopicity of the API.
SYN
Bromination of (+)-(R)-pulegone (I) with Br2 in the presence of NaHCO3 in Et2O, followed by ring contraction via Favorskii rearrangement with NaOEt in EtOH, and treatment with semicarbazide hydrochloride and NaOAc in refluxing EtOH/H2O gives rise to cyclopentanecarboxylate (II) (1). Subsequent ozonolysis of olefin (II) by means of O3 in EtOAc at -78 °C, and reductive treatment with Zn in AcOH provides beta-ketoester (III). Reaction of ketoester (III) with ammonium acetate (IVa) in MeOH/CH2Cl2 yields enamine (V), which upon cyclization with ammonium formate (IVb) and formamide (VI) at 150 °C provides cyclopentapyrimidinol (VII). Chlorination of pyrimidinol (VII) using POCl3 in refluxing CH2Cl2 results in 4-chloro-5(R)-methyl-6,7-dihydro-5H-cyclopenta[d]pyrimidine (VIII), which is condensed with N-Boc-piperazine (IX) in the presence of DIEA in refluxing BuOH to produce piperazinyl cyclopentapyrimidine (X). Oxidation of compound (X) using mCPBA and NaHCO3 in CHCl3 furnishes N-oxide (XI). Subsequent rearrangement of N-oxide (XI) using Ac2O in CH2Cl2 at 100 °C yields acetate (XII). This compound (XII) is hydrolyzed with LiOH in H2O/THF to give alcohol (XIII), which upon Swern oxidation with (COCl)2, DMSO and Et3N in CH2Cl2 at -78 °C affords ketone (XIV) (1-6). Asymmetric transfer hydrogenation of ketone (XIV) in the presence of RuCl[(R,R)-TsDPEN(p-cymene)], HCOOH and Et3N in CH2Cl2, followed by protection with PNBCl in the presence of Et3N in CH2Cl2, and hydrolysis with LiOH in H2O/THF gives rise to alcohol (XV) (1-6). Also, intermediate (XV) can be produced by enzymatic reduction of ketone (XI) using KRED-101 in the presence of GDH, NADP, KOH and PEG-400, KRED-X1.1-P1F01 in the presence of glucose and NAD in DMSO/i-PrOH or KRED-X1.1-P1B06, KRED-X1.1-P1F01 or KRED-X1.1-P1H10 in the presence of NADP in DMSO/i-PrOH or i-PrOH (11,12). In an alternative method, asymmetric transfer hydrogenation of ketone (XIV) in the presence of RuCl[(R,R)-MsDPEN(p-cymene)], HCOOH and Et3N in CH2Cl2, followed by O-protection of the resultant cis/trans mixture of alcohols with PNBCl and Et3N or protection with pivaloyl chloride in the presence of DIEA in CH2Cl2, followed by separation of the resulting cis/trans mixture of esters by means of HPLC. Hydrolysis of trans ester with LiOH in THF yields alcohol (XV) (11). N-Deprotection of piperazine derivative (XV) by means of HCl in CH2Cl2, i-PrOH or toluene at 62 °C provides amine dihydrochloride (XVI) (1-7,11,12), which is then coupled with aminoacid derivative (XVIIa) (1-7,11) or its sodium salt (XVIIb) (12,13) in the presence of DIEA and HBTU in CH2Cl2 or NMM and T3P in i-PrOH or toluene to produce amide (XVIII) (1-7,11-13). Finally, Boc-deprotection of precursor (XVIII) by means of HCl in MeOH/Et2O, PrOH, i-PrOH or toluene at 57 °C furnishes the target GDC-0068
Synthesis of intermediate (XVII): Condensation of methyl (4-chlorophenyl)acetate (XIX) with formaldehyde (XX) in the presence of NaOMe in DMSO gives beta-hydroxyester (XXI). Subsequent dehydration of alcohol (XXI) using MsCl and Et3N in CH2Cl2 provides arylacrylate (XXII), which upon conjugate addition with isopropylamine (XXIII) in the presence of Boc2O in THF yields N-Boc beta-aminoester (XXIV). Basic hydrolysis of ester (XXIV) using KOSiMe3 in THF generates the potassium carboxylate (XXV), which upon condensation with 4(R)-benzyl-2-oxazolidinone (XXVI) via activation with pivaloyl chloride and BuLi in THF at -78 °C affords the N-acyl oxazolidinone (XXVII) (2-6). Finally, removal of the chiral auxiliary group of (XXVII) using LiOH and H2O2 in THF/H2O furnishes the key intermediate (XVII) (1-6,11). Alternative synthesis of intermediate (XXVII): Protection of isopropylamine (XXIII) with Boc2O in toluene affords tert-butyl isopropylcarbamate (XXVIII), which upon N-alkylation with bromomethyl methyl ether (XXIX) in the presence of NaHMDS in 2-MeTHF gives tert-butyl isopropyl(methoxymethyl)carbamate (XXX) (11). Condensation of 4(R)-benzyl-2-oxazolidinone (XXVI) with 2-(4-chlorophenyl)acetyl chloride (XXXIIa) using BuLi in THF at -50 °C (1) or with 2-(4-chlorophenyl)acetic acid (XXXIIb) via activation with pivaloyl chloride and Et3N in refluxing toluene (11) affords N-acyl oxazolidinone(XXXI). After conversion of intermediate (XXXI) to its titanium enolate with TiCl4 and DIEA in CH2Cl2 at -50 °C, diastereoselective Mannich reaction with formaldehyde hemiaminal (XXX) affords adduct (XXVII)
PAPER
Synthesis of Akt inhibitor ipatasertib. Part 2. Total synthesis and first kilogram scale-up
Org Process Res Dev 2014, 18(12): 1652
Herein, the first-generation process to manufacture Akt inhibitor Ipatasertib through a late-stage convergent coupling of two challenging chiral components on multikilogram scale is described. The first of the two key components is a trans-substituted cyclopentylpyrimidine compound that contains both a methyl stereocenter, which is ultimately derived from the enzymatic resolution of a simple triester starting material, and an adjacent hydroxyl group, which is installed through an asymmetric reduction of the corresponding cyclopentylpyrimidine ketone substrate. A carbonylative esterification and subsequent Dieckmann cyclization sequence was developed to forge the cyclopentane ring in the target. The second key chiral component, a β2-amino acid, is produced using an asymmetric aminomethylation (Mannich) reaction. The two chiral intermediates are then coupled in a three-stage endgame process to complete the assembly of Ipatasertib, which is isolated as a stable mono-HCl salt.
Herein, the route scouting and early process development of a key cyclopentylpyrimidine ketone intermediate toward the synthesis of Akt inhibitor Ipatasertib are described. Initial supplies of the intermediate were prepared through a method that commenced with the natural product (R)-(+)-pulegone and relied on the early construction of a methyl-substituted cyclopentyl ring system. The first process chemistry route, detailed herein, enabled the synthesis of the ketone on a hundred-gram scale, but it was not feasible for the requisite production of multikilogram quantities of this compound and necessitated the exploration of alternative strategies. Several new synthetic approaches were investigated towards the preparation of the cyclopentylpyrimidine ketone, in either racemic or chiral form, which resulted in the discovery of a more practical route that hinged on the initial preparation of a highly substituted dihydroxypyrimidine compound. The cyclopentane ring in the target was then constructed through a key carbonylative esterification and subsequent tandem Dieckmann cyclization–decarboxylation sequence that was demonstrated in a racemic synthesis. This proof-of-concept was later developed into an asymmetric synthesis of the cyclopentylpyrimidine ketone, which will be described in a subsequent paper, along with the synthesis of Ipatasertib.
PAPER
Discovery and preclinical pharmacology of a selective ATP-Competitive akt inhibitor (GDC-0068) for the treatment of human tumors
J Med Chem 2012, 55(18): 8110
PAPER
Asymmetric synthesis of akt kinase inhibitor ipatasertib
Org Lett 2017, 19(18): 4806
It has been found that ipatasertib exhibits a very high solubility (>1 g/g water; >2 g/g water/ethanol 1:1) and a very high hygroscopicity (˜6% at 50% RH, >35% at 95% RH). Whereas poor solubility is often a limiting factor in the development of galenical formulations of other API’s (active pharmaceutical ingredient), a high solubility can equally be problematic for the process performance. Due to this very high intrinsic hygroscopicity of the API, ipatasertib drug substance tends to auto-dissolve to a honey-like viscous liquid at increased humidity. Such high solubility and hygroscopicity may pose serious problems for processing as well as for stability and shelf-life of the final product. Therefore, conventional pharmaceutical compositions comprising ipatasertib and processes for the manufacture of pharmaceutical compositions comprising wetting (e.g. wet granulation) are difficult due to the high solubility and high hygroscopicity of the API.
Glasdegib (PF-04449913) is an experimental cancer drug developed by Pfizer. It is a small molecule inhibitor of the Sonic hedgehog pathway, which is overexpressed in many types of cancer. It inhibits smoothened receptor, as do most drug in its class.[1]
DeveloperGrupo Espanol de Trasplante Hematopoyetico y Terapia Celular; H. Lee Moffitt Cancer Center and Research Institute; Netherlands Cancer Institute; Pfizer
ClassAntineoplastics; Benzimidazoles; Phenylurea compounds; Piperidines; Small molecules
Mechanism of ActionHedgehog cell-signalling pathway inhibitors; SMO protein inhibitors
Orphan Drug StatusYes – Acute myeloid leukaemia; Myelodysplastic syndromes
20 Apr 2018Phase-III clinical trials in Acute myeloid leukaemia (Combination therapy, First-line therapy) in Japan (PO) (NCT03416179)
02 Apr 2018Pfizer terminates a phase II trial in Myelofibrosis (Second-line therapy or greater) in USA, Japan, Austria, France, Spain and United Kingdom (PO) (NCT02226172) (EudraCT2014-001048-40)
06 Feb 2018Phase-I/II clinical trials in Glioblastoma (Newly diagnosed) in Spain (PO) (EudraCT2017-002410-31)
Glasdegib is an orally bioavailable small-molecule inhibitor of the Hedgehog (Hh) signaling pathway with potential antineoplastic activity. Glasdegib appears to inhibit Hh pathway signaling. The Hh signaling pathway plays an important role in cellular growth, differentiation and repair. Constitutive activation of Hh pathway signaling has been observed in various types of malignancies.
Glasdegib is under investigation for the treatment of Acute Myeloid Leukemia.
SYNTHESIS
Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened
Inhibitors of the Hedgehog signaling pathway have generated a great deal of interest in the oncology area due to the mounting evidence of their potential to provide promising therapeutic options for patients. Herein, we describe the discovery strategy to overcome the issues inherent in lead structure 1 that resulted in the identification of Smoothened inhibitor 1-((2R,4R)-2-(1H-benzo[d]imidazol-2-yl)-1-methylpiperidin-4-yl)-3-(4-cyanophenyl)urea (PF-04449913, 26), which has been advanced to human clinical studies
Product was purified by Companion (ReadySep 40g, silica gel packed) with CH3OH/CH2Cl2 from 1-5% to give the title compound as an off-white solid 915mg (73%). LC-MS 375.3.
The dihydrochloride salt was prepared by adding 4M HCl in dioxane (1.22mL, 4.86 mmol) to a solution of 1-((2R,4R)-2-(1H-benzo[d]imidazol-2-yl)-1-methylpiperidin-4-yl)-3-(4- cyanophenyl)urea (910 mg’s, 2.43mmol) in methanol (10mL). The mixture was stirred at at 230C for 10 minutes. The solution was concentrated to give a white solid, 1082 mg’s as the 2 .HCl monohydrate salt. M.P. > 125 0C with dehydration above 130 0C. Analytical calculated for free base C21H22N6O: C 67.38%, H 5.88%, N 22.46%; Found: C 67.16%, H 5.54%, N 22.18%. Purity of the dihydrochloride monohydrate salt was determined to be > 99.9% by analytical HPLC using a Xbridge C18; 3.5µm column and eluting with 95:5 0.1% Perchloric Acid (HClO4) solution in water and acetonitrile, over a gradient of 25 minutes, with and ending solvent ratio of 5:95. Enantiomeric purity of the dihydrochloride monohydrate salt was > 99.9% by chiral HPLC using a Chiralcel OJ column and eluting with 96:4 Heptane:Ethanol(with 0.1% diethylamine).
Syn 2
Development of a Concise, Asymmetric Synthesis of a Smoothened Receptor (SMO) Inhibitor: Enzymatic Transamination of a 4-Piperidinone with Dynamic Kinetic Resolution
†Chemical Research & Development, ‡Analytical Research & Development, Pfizer Worldwide Research & Development, Eastern Point Road, Groton, Connecticut 06340, United States
A concise, asymmetric synthesis of a smoothened receptor inhibitor (1) is described. The synthesis features an enzymatic transamination with concurrent dynamic kinetic resolution (DKR) of a 4-piperidone (4) to establish the two stereogenic centers required in a single step. This efficient reaction affords the desired anti amine (3) in >10:1 dr and >99% ee. The title compound is prepared in only five steps with 40% overall yield.
To the crude solution of 3 in DMSO-H2O (UPLC assay ~55.0 mg/mL, 104 mL, ~5.74 g of 3, 24.9 mmol) from the enzymatic transamination reaction (vide supra) was added THF (57.0 mL) followed by 17 (mixture with imidazole, 9.31 gm, 74.0 wt%, 31.2 mmol). The mixture was then stirred at rt for three hours. Once the reaction was complete (<1 % of 3 remaining by UPLC), methanol (10.1 mL, 249 mmol) was added followed by 2-MeTHF (57.0 mL). The layers were separated and the aqueous was extracted with 2-MeTHF (57.0 mL). The combined organic layers were then washed with 2 × 50.0 mL water and 2 × 50.0 mL of 10% aqueous NaCl solution. The organic solution was then concentrated under vacuum and the solvent was switched to acetonitrile to give a slurry with a final volume of ~90.0 mL. The slurry was stirred at rt for three hours and filtered, and the solids were washed with 2 × 10.0 mL of acetonitrile and dried in oven at 60 °C for two hours. The solids (~7.90 gm) were then slurried in 70.0 mL of acetonitrile. The slurry was heated to 60 °C for two hours, cooled to rt, filtered, and the solids were dried in oven under vacuum at 60 °C for 12 hours to give 1 as white solids (7.64 g, 98.0 wt%, 80.0% corrected yield, > 98 UPLC area% purity). The analytical data were identical to that obtained with method A.
References
1. Lin TL, Matsui W. Hedgehog pathway as a drug target: smoothened inhibitors in development. Onco Targets Ther. 2012;5:47-58.
2. Munchhof MJ, Li Q, Shavnya A, et al. Discovery of PF-04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med Chem Lett. 2012;3(2):106-111.
3. Clement V, Sanchez P, de Tribolet N, et al. Hedgehog-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17(2):165-172.
4. Deschler, B. and Lübbert, M. (2006), Acute myeloid leukemia: Epidemiology and etiology. Cancer, 107: 2099–2107. doi: 10.1002/cncr.22233.
6. SEER Cancer Stat Facts: Acute Myeloid Leukemia. National Cancer Institute. Bethesda, MD, April 2017. Available at: http://seer.cancer.gov/statfacts/html/amyl.html. Accessed January 25, 2018.
7. Appelbaum FR, Gundacker H, Head DR, et al. Age and acute myeloid leukemia. Blood 2006; 107(9): 3481-5.
8. Estey E. Acute myeloid leukemia and myelodysplastic syndromes in older patients. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 2007; 25(14): 1908-15.
9. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 2012; 30(21): 2670-7.
10. Ornstein MC, Mukherjee S, Sekeres MA. More is better: combination therapies for myelodysplastic syndromes. Best Pract Res Clin Haematol. 2015;28(1):22-31.
CAS Registry Number: 192185-72-1; 192185-68-5 (unspecified stereo)
CAS Name: 6-[(R)-Amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-1-methyl-2(1H)-quinolinone
Manufacturers’ Codes: R-115777
Trademarks: Zarnestra (Janssen)
Molecular Formula: C27H22Cl2N4O
Molecular Weight: 489.40
Percent Composition: C 66.26%, H 4.53%, Cl 14.49%, N 11.45%, O 3.27%
Literature References: Farnesyl transferase inhibitor. Prepn: M. G. Venet et al., WO9721701; eidem, US6037350 (1997, 2000 both to Janssen). Review of syntheses: P. R. Angibaud et al.,Eur. J. Org. Chem.2004, 479-486. Inhibition of farnesyl protein transferase and antitumor effects in vivo: D. W. End et al., Cancer Res.61, 131 (2001). Clinical pharmacology and pharmacokinetics: J. Zujewski et al., J. Clin. Oncol.18, 927 (2000). Accelerator mass spec determn in biological samples: R. C. Garner et al., Drug Metab. Dispos.30, 823 (2002). Clinical evaluation in hematologic malignancies: J. Cortes et al., Blood101, 1692 (2003). Review of clinical experience: P. Norman, Curr. Opin. Invest. Drugs3, 313-319 (2002).
Properties: Crystals from 2-propanol, mp 234°. [a]D20 +22.86° (c = 0.98 in methanol).
Melting point: mp 234°
Optical Rotation: [a]D20 +22.86° (c = 0.98 in methanol)
Tipifarnib (R-115777) is a substance that is being studied in the treatment of acute myeloid leukemia (AML) and other types of cancer. It belongs to the family of drugs called farnesyltransferase inhibitors. It is also called Zarnestra. In June 2005, the FDA issued a Not Approvable Letter for Zarnestra.
Investigated for use/treatment in colorectal cancer, leukemia (myeloid), pancreatic cancer, and solid tumors.
Drug had been granted orphan drug designation by the FDA for the treatment of AML in 2004. In 2005, the Committee for Orphan Medicinal Products of the European Medicines Agency (EMEA) adopted a positive opinion on orphan medicinal product designation for the drug. In 2014, Eiger BioPharmaceuticals licensed the product for worldwide development for the treatment of viral diseases and Kura Oncology licensed development and commercialization rights for the treatment cancer indications.
Pharmacodynamics
R115777, a nonpeptidomimetic farnesyl transferase inhibitor, suppresses the growth of human pancreatic adenocarcinoma cell lines. This growth inhibition is associated with modulation in the phosphorylation levels of signal transducers and activators of transcription 3 (STAT3) and extracellular signal-regulated kinases (ERK)
Tipifarnib (INN,[1]:213 proposed trade name Zarnestra) is a farnesyltransferase inhibitor that is being investigated in patients 65 years of age and older with newly diagnosed acute myeloid leukemia (AML). It inhibits the Ras kinase in a post-translational modification step before the kinase pathway becomes hyperactive. It inhibits prenylation of the CaaX tail motif, which allows Ras to bind to the membrane where it is active. Without this step the protein cannot function.
It is also being tested in clinical trials in patients in certain stages of breast cancer.[2] It is also investigated as a treatment for multiple myeloma.[3]
Tipifarnib was submitted to the FDA by Johnson & Johnson for the treatment of AML in patients aged 65 and over with a new drug application (NDA) to the FDA on January 24, 2005.
In June 2005, the FDA issued a “not approvable” letter for tipifarnib.[6]Progeria
Confocal microscopy photographs of the descending aortas of two 15-month-old progeria mice, one untreated (left picture) and the other treated with the farnsyltransferase inhibitor drug tipifarnib (right picture). The microphotographs show prevention of the vascular smooth muscle cell loss that is otherwise rampant by this age. Staining was smooth muscle alpha-actin (green), lamins A/C (red) and DAPI (blue). (Original magnification, ×40)
It was shown on a mouse model of Hutchinson–Gilford progeria syndrome that dose-dependent administration of tipifarnib can significantly prevent both the onset of the cardiovascular phenotype as well as the late progression of existing cardiovascular disease.[7]
Crystalline form (I, II, III and IV) of tipifarnib . Useful for the treatment and/or prevention of abnormal cell growth diseases such as lung cancer, pancreatic cancer, colon cancer, melanoma, neuroblastoma or glioma. first filing from Solipharma claiming tipifarnib which was developing by Kura Oncology , under license from Johnson & Johnson subsidiary J&JPRD (now Janssen Research & Development).
Tipifarnib is a farnesyltransferase inhibitor that acts on H-RAS or N-RAS mutant cells and has antiproliferative effects. It can block the farnesylation modification of RAS protein, thereby disturbing its localization on the inner surface of the plasma membrane and subsequent activation of downstream signaling pathways, and has an effective anti-tumor disease activity.
Tipifarny’s chemical name is (R)-(+)-6-[amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chloro) Phenyl) 1-methyl-2(1H)-quinolinone, English name Tipifarnib; its chemical structure is shown below:
The patent document CN1101392C reports the preparation method of typrivadina, which is a racemate and does not disclose any characterization data; the patent document CN100567292C reports the preparation method of typ fenfanide, which is a mixture of certain enantiomeric excesses. Only the melting point of the mixture is mentioned; the patent document CN1246318C reports the preparation method of typifanidin and the method for the resolution and purification of tepifefene in its enantiomers. The present inventors have found that the form of typifene prepared according to the method provided by CN1246318C is in the crystalline state (herein referred to as “Form A”), but it has a defect of low crystallinity and poor stability of the crystal, and the patent The typifanibs reported in the documents CN1101392C and CN100567292C are both mixtures and lack the characteristic data accurately reflecting their physical form and cannot be fully disclosed.
Cyclization of 3-(3-chlorophenyl)-N-phenyl-2-propenamide by means of polyphosphoric acid (PPA) at 100 °C gives 4-(3-chlorophenyl)-1,2,3,4-tetrahydroquinolin-2-one ,
Which is condensed with 4-chlorobenzoic acid by means of PPA at 140 °C to yield 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-1,2,3,4-tetrahydroquinolin-2-one
The dehydrogenation of compound by means of Br2 in bromobenzene at 160 °C affords 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)quinolin-2-one,
Which is N-alkyalted with iodomethane in the presence of BnNMe3Cl and NaOH in THF to provide 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-1-methylquinolin-2-one.
Condensation of compound with 1-methylimidazole by means of BuLi in THF gives the triaryl carbinol (N-1),
Which is finally treated with NH3 in THF to afford the target Tipifarnib, R-115777 .
Farnesyltransf erase inhibitors block the main post-translational modification of the Ras protein, thus interfering with its localization to the inner surface of the plasma
10 membrane and subsequent activation of the downstream effectors. Although initially developed as a strategy to target Ras in cancer, farnesyltransferase inhibitors have
subsequently been acknowledged as acting by additional and more complex
mechanisms that may extend beyond Ras involving GTP-binding proteins, kinases,
centromere-binding proteins and probably other f arnesylated proteins.
15
A particular farnesyltransferase inhibitor is described in WO 97/21701, namely (R)-(+)- 6-[amino(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l- methyl-2(liϊ)-quinolinone. The absolute stereochemical configuration of the compound was not determined in the experiments described in the above-mentioned patent
20 specification, but the compound was identified by the prefix “(B)” to indicate that it was the second compound isolated from column chromatography. The compound thus obtained has been found to have the (R)-(+)-configuration. This compound will be
referred to below by its published code number Rl 15777 and has the following formula
Rl 15777 (Tipifamib) is a potent, orally active inhibitor of f arnesylprotein transferase.
It is one of the most advanced of the farnesylprotein transferase inhibitors currently
reported to be in clinical development, being one of the agents that have progressed to phase III studies.
30 Rl 15777 has been found to have very potent activity against neoplaslic diseases.
Antineoplastic activity in solid tumors, such as breast cancer, as well as in haematological malignancies, such as leukemia, have been observed. Also combination studies have been carried out demonstrating that R 115777 can be safely combined with several highly active anticancer drugs.
In WO 01/53289, the racemates (±) (4-(3-chloro-phenyl)-6-[(6-chloro-pyridin-3-yl)-(4-methoxy-benzylamino)-(3-methyl-3-f: -imidazol-4-yl)-methyl]-l-cyclopropylmethyl-liϊ-quinolin-2-one (racemate 1) and (±) 4-(3-chloro-phenyl)-6-[(6-chloro-pyridin-3-yl)-[(4-methoxy-benzylidene)-amino]-(3-methyl-3jr7-imidazol-4-yl)-methyl]-l-cyclopropylmethyl-liϊ-quinolin-2-one (racemate 2) are prepared.
racemate 1 racemate 2
After chiral molecule separation using column chromatography, either the benzylamino or the benzilidine moiety of the resulting (+) and /or (-) enantiomers are converted to an amino group under acidic conditions.
The synthesis of Rl 15777 as originally described in WO 97/21701, is presented in scheme 1.
Herein, in step 1, the intermediate 1-methyl imidazole in tetrahydrofuran, is mixed with a solution of ra-butyllithium in a hexane solvent to which is added chlorotriethylsilane (triethylsilyl chloride), followed by a further addition of ra-butyllithium in hexane, the resulting mixture being cooled to -78°C before the addition of a solution of a compound of formula (I), i.e. 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-l-methyl-2(12ϊ)-quinolinone in tetrahydrofuran. The reaction mixture is subsequently brought to room temperature, and then hydrolysed, extracted with ethyl acetate and the organic layer worked up to obtain a compound of formula (II), i.e. (±)-6-[hydroxy(4-chlorophenyl) (l-methyl-liϊ-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(lia- )-quinolinone.
In step 2, the hydroxy compound of formula (II) is chlorinated with thionylchloride to form a compound of formula (III), i.e. (±)-6-[chloro(4-chlorophenyl)(l -methyl- liJ-imidazol-5-yl)methyl]-4-(3-chloroρhenyl)-l-methyl-2(li3)-quinolinone.
In step 3, the chloro compound of formula (III) is treated, with NEaL OH in
tetrahydrofuran to form the amino compound of formula (IV), i.e. (±)-6-[amino(4-chlorophenyl)(l-methyl-l -imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl- 2(l/J)-quinolinone.
In step 4, the amino compound of formula (IV) is separated into its enantiomers by chiral column chromatography over Chiracel OD (25 cm; eluent: 100% ethanol; flow: 0.5 ml/rnin; wavelength: 220 nm). The pure (B)-fractions are collected and recrystallised from 2-propanol resulting in Rl 15777, the compound of formula (V).
Scheme 1
However, the procedure described in WO97/21701 has a number of disadvantages. For example, during the first step, the procedure results in the undesired formation of a corresponding compound of formula (XI), i.e. 6-[hydroxy(4-chlorophenyl) (1-methyl-lJrJ-imidazol-2-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(liϊ)-quinolinone)Jn which the imidazole ring is attached to the remainder of the molecule at the 2-position of the ring, instead of the desired 5-position. At the end of the procedure, this results in the formation of a compound of formula (XII), i.e.6-[amino(4-chlorophenyl)(l-methyl-lϊJ-imidazol-2-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(lβ -quinolinone.
(XI) CXH)
The use of n-butyllithium during the conversion of a compound of formula (I) in a compound of formula (II) is also undesirable in a commercial process in view of its pyrophoric nature and the formation of butane, a flammable gas, as the by-product. Also the carrying out of this process step, at a temperature as low as -78°C, is inconvenient and costly on a commercial scale.
Finally, the purification of compound (V) using chiral chromatography is expensive and disadvantageous in view of the large amounts of solvent needed and the specialised equipment required to perform a large scale chiral chromatography.
Another process for the synthesis of Rl 15777 as described in WO 02/072574, is presented in scheme 2.
Herein, in step 1, 1-methyl imidazole in tetrahydrofuran is mixed with a solution of n-hexyllithium in a hexane solvent to which is added tri-iso-butylsilyl chloride, followed by a further addition of n-hexyllithium in hexane. The compound of formula (I) in tetrahydrofuran is then added to the reaction mixture, keeping the temperature between -5°C and 0°C. The resulting product of formula (II) is isolated by salt formation.
In step 2, the chlorination reaction is effected by treatment of the compound of formula (II) with thionyl chloride in 1 ,3-dimethyl-2-imidazolidinone.
In step 3, the chloro compound of formula (III) is treated with a solution of ammonia in methanol. After the addition of water, the compound of formula (IV), precipitates and can be isolated.
In step 4, the compound of formula (IV) can be reacted with L-(-)-dibenzoyl tartaric acid (DBTA) to form the diastereomeric tartrate salt with formula (VI) i.e. R-(-)-6-[amino(4-chlorophenyl)(l-methyl-ljt–‘-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(l Z)-quinolinone [R-(R*,RH!)]-2,3-bis(benzoyloxy)butanedioate (2:3).
Finally, in step 5, the compound of formula (VI) is treated with aqueous ammonium hydroxide, to form the crude compound of formula (V) which is then purified by recrystallisation from ethanol to the pure compound (V).
(VI) (V)
Scheme 2
However, in view of the fact that water is present during the third and the fifth step of this procedure, there is significant formation of the hydroxy compound of formula (II).
This is important because the compounds of formula (II) and (V) are difficult to separate. In order to keep the quality of the final product (V) as high as possible, it is critical to limit the formation of compound (II).
The major drawback of the above described processes is the generation of large amounts of the other enantiomer that subsequently must be recycled.
Attempts were made to develop processes that solve this problem. One of the possibilities was to enter chirality in the first step of the procedure. A first study was carried out in order to determine if the conversion of an enantiomer of the hydroxy compound of formula (II) into a compound of formula (IV) could preserve chirality. Several experimental conditions have been tested starting with an enantiomer of a compound of formula (II), but racemisation always occurred.
Another possibility was to try entering chirality by adding N-methylimidazole under the reaction conditions described herein above under steps 1 of WO97/21701 and WO 02/072574, to an N-Ct-6alkyl-(S(R))-sulfinylketimine prepared from the compound of formula (I). It turned out that the resulting N-Cι-6alkyl-(S(R))-sulfinylamide of the compound of formula (I) was in the desired R-configuration and could be used for conversion into compound (V).
These results are completely unexpected, especially in view of Shaw et al.
(Tetrahedron Letters: 42, 7173-7176). Already in 2001, Shaw et al. disclosed an asymmetric synthesis process for the production of α-aryl-α-heteroaryl alkylamines using organometallic additions to N-tert-butanesulfinyl ketimines. However, the configuration and the yield of the final enantiomer formed with this process, was depending on the configuration of the N-tert-butanesulfinyl moiety of the ketimines, the composition of the aryl and/or the heteroaryl moieties of the ketimines, as well as on the organo- and the metallic moiety of the organometallic reagent. Furthermore, the use of heteroaryllithium reagents were described in this document, as being in particular disadvantageous, in view of their instability.
Thus the present invention solves the above described problems. It provides a new process for the preparation of the compound of formula (V) without the need to recycle one of the enantiomers while minimising the formation of undesired isomers and impurities and under conditions which offer economic advantages for operation on a commercial scale.
A. Preparation of intermediates
Example AJ
a) Preparation of /V-r(4-chlorophenyl‘)((,4- -chlorophenyl’)-l-methyl-l f-quinolin-2-one’)-6-yDmethylenel-2-methyl-2-propanesulfinamide TSfR-)! (com ound 15)
Ti(OEt) (0.0122 mol) was added to a mixture of compound (I) (0.0024 mol) and (R)-(+)-2-methyl-2-propane-sulfinamide (0.0024 mol) in DCM (15ml). The mixture was stirred and refluxed for 4 days, then cooled to room temperature. Ice water was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was extracted with saturated sodium chloride. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM/MeOH 98/2). The pure fractions were collected and the solvent was evaporated, yielding 0.95g of compound 15 _ (76%), melting point: 115°C.
b) Preparation of (R)-N-r(‘4-chlorophenyl1((4-(3-chlorophenyl)-l-methyl-lic/-quinoline- 2-one -6-ylVl-methyl-l/j‘-imidazole-5-yl’)methyll-2-methyl-2-propanesulfinamide rS(R)l (compound 161
(compound 16)
n-Butyllithium (1.34ml, 0.002 mol) was added dropwise at -70°C to a mixture of 1-methylimidazole (0.0021 mol) in THF (4.5ml). The mixture was stirred at -70°C for 15 minutes. Triethylsilyl chloride (0.0021 mol) was added. The mixture was stirred at -70°C for 15 minutes. n-Butyllithium (1.34ml, 0.0021 mol) was added dropwise. The mixture was stirred at -70°C for 15 minutes. A solution of compound 15 (0.0019 mol) in THF (5.5ml) was added. The mixture was stirred at -70°C for 45 minutes, poured out into ice water and extracted with EtOAc. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. The residue was purified by column chromatography over silica gel (15-40 m)(eluent: DCM/MeOH/ΝEUOH 95/5/0.5), yielding 0.59g (52%) of compound 16, diastereomeric excess 24%.
c) Preparation of the (B)-diastereomer (compound 18) of compound 16
(compound 18)
Compound 16 was purified by column chromatography over silica gel (15-40μm) (eluent: DCM/MeOH/NHtOH 95/5/0.5). Two fractions were collected and the solvent was evaporated, yielding 0.304g diastereomer (B) (compound 18) (27%), melting point 174°C.
Example A.2
a) Preparation of jV-r(4-chlorophenyl¥(4-(3-chlorophenyl‘)-l-methyl-l JJ-quinolin-2-one)-6-yl)methylene1-4-methylphenylsulfιnamidesulfιnamide fS(S‘)l (compound 17)
(compound 17)
Ti(OEt)4 (0.0122 mol) was added to a mixture of compound (I) (0.0123 mol) and (S)-(+)-j5-toluenesulfinamide (0.0123 mol) in DCM (80ml). The mixture was stirred and refluxed for 4 days, then cooled to room temperature. Satured sodium chloride was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. A fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM MeOH 98/2). The fractions were collected and the solvent was evaporated, yielding 0.65g of pure compound 17 .
The pure compound N-[(4-chlorophenyl)((4-(3-chlorophenyl)-l-methyl-l-tf-quinolin-2-one)-6-yl)methylene]-2-methyl-2-propanesulfinamide [S(R)] can be obtained in an analogues way.
B. Preparation of final compounds
Example BJ
a Preparation of compound (V)
Hydrochloric acid in isopropanol was added to a solution of compound 16 (0.00003 mol) in methanol (0J ml). The mixture was stirred at room temperature for 30 minutes. The mixture was added to potassium carbonate (10%) on ice. The organic layer was separated, washed with a solution of saturated sodium chloride, dried (MgS04), filtered, and evaporated giving 0,017 g (100%) of compound (V), enantiomeric excess 22%, content of compound (II) < 1%.
a) Preparation of N-r(4-chlorophenyl’)(l-methyl-lH-imidazol-5-yl)methylene‘)l-2- methyl-2-propanesulfinamide KSfl l (compound 25)
(compound 25) Ti(OEt)4 (0.0162 mol) was added to a mixture of (4-chlorophenyiχi-methyl-lH- imidazol-5-yl)methanone (0.0032 mol) and (R)-(+)-2-methyl-2-propane-sulfinamide (0.0032 mol) in DCE (7ml). The mixture was stirred and refluxed for 6 days, then cooled to room temperature. Ice water was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was extracted with saturated sodium chloride. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM/MeOH/NH OH 97/3/0.5), yielding 0.475g of compound 25 (46%).
The compound N-[(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methylene)]-2-methyl- 2-propanesulfinamide [(S(S)] can be obtained in an analogous way.
b) Preparation of N-r(4-chlorophenyl)((4-(3-chlorophenyl)-2-methoχy-quinoline-6- yl l-methyl-lH-imidazole-5-yl)methyn-2-methyl-2-propanesulfinamide TS(R)1 (compound 26)
(compound 26)
n-Butyllithium (0.00081 mol) in hexane, was added dropwise at -78°C to a mixture of 6-bromo-4-(3-chlorophenyl)-2-methoxy-quinoline (0.00081 mol) in THF (3 ml) under nitrogen flow. The mixture was stirred at -78°C for 30 minutes. A solution of compound 25 (0.00065 mol) in THF (0.6 ml) was added . The mixture was stirred at – 78°C for 1 hour and 30 minutes, poured out into ice water and extracted with EtOAc. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40μm)(eluent: DCM eOH/NB OH 97/3/0.1). The pure fractions were collected and the solvent was evaporated, yielding 0.138g (36 %) of compound 26, melting point 153°C.
The compound N-[(4-chlorophenyl)((4-(3-chlorophenyl)-2-methoxy-quinoline-6-yl)(l- methyl-lH-imidazole-5-yl)methyl]-2-methyl-2-propanesulfmamide [S(S)] can be obtained in an analogous way
c“) Preparation of (S)-l-,4-chlorophenylV l-r4-(3-chlorophenylV2-methoxy-quinoline-6- yll-l-(l-methyl-l/J-imidazole-5-yl)-methylamine (compound 27)
(compound 27) Hydrochloric acid in isopropanol was added to a solution of compound 26 (0.000018 mol) in methanol (4.2 ml). The mixture was stirred at room temperature for 30 minutes. The mixture was added to potassium carbonate (10%) on ice and extracted with ethyl acetate. The organic layer was separated, washed with a solution of saturated sodium chloride, dried (MgS0 ), filtered, and evaporated giving 0,086 g (100%) of compound 27, melting point 96°C, enantiomeric excess 88%. d) Preparation of (SV6-ramino(4-chlorophenyl¥l-methyl-l #-imidazol-5-yDmethyH-4- (3-chlorophenyD-lH)-quinorin-2-one (compound 28)
(compound 28) Compound 27 (0.00038 mol) in hydrochloric acid 3N (9.25 ml) and THF (9.25 ml), was stirred at 60°C for 24 hours and evaporated, giving 0,18 g (100%) of compound 28, melting point 210°C.
Example A.2
a) Preparation of N-r(4-chlorophenyl)(‘l-methyl-lH-imidazol-5-yl’)methylene‘)1-p-
(compound 29) Ti(OEt)4 (0.0419 mol) was added to a mixture of (4-chlorophenyl)(l-methyl-lH- imidazol-5-yl)methanone (0.0084 mol) and (S)-(+)-p-_toluenesulfinamide (0.0084 mol) in DCE (18ml). The mixture was stirred and refluxed for 7 days, then cooled to room temperature. Ice water was added. The mixture was filtered over celite. Celite was washed with DCM. The organic layer was extracted with saturated sodium chloride. The organic layer was separated, dried (MgS04), filtered, and the solvent was evaporated. This fraction was purified by column chromatography over silica gel (40 μm) (eluent: DCM/MeOH/ΝHiOH 97/3/0.5), yielding 1.15 g of compound 29 (38%).
The compound N-[(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methylene)]-p- toluenesulfinamide [(S(R)] can be obtained in an analogues way. B. Preparation of final compounds
Example B.l a) Preparation of (S)-6-ramino(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methyll-4-
Compound 28 (0.00038 mol) was added to a solution of THF (1.8 ml) and NaOH ION (1.8 ml). BTEAC (0.0019 mol) and methyliodide (0.00076 mol) were added and the mixture was stirred for 2 hours at room temperature. EtOAc was added. The organic layer was separated, dried (MgS04), filtered, and evaporated giving 0,149 g (83%) of compound 30, enantiomeric excess 86%.
110 ml of dry tetrahydrofuran was added to 7.6 ml of 1-methylimidazole (0.0946 mole) and the resulting solution cooled to -15°C.37.8 ml of n-hexyllithium 2.5 M in n-hexane (0.0946 mole) was added, while the temperature during addition was kept between – 5°C and 0°C. After addition, the reaction mixture was stirred for 15 minutes, while cooling to -12°C. 26.2 ml of tri-w o-butylsilyl chloride (0.0964 mole) was added, while the temperature during addition was kept between -5° and 0°C. After addition, the reaction mixture was stirred for 15 minutes, while cooling to -13°C. 37.2 ml of n- hexyllithium 2.5 M in n-hexane (0.0930 mole) was added, while the temperature during addition was kept between -5°C and 0°C (some precipitation occured). After addition, the reaction mixture was stirred for 15 minutes, while cooling to -14°C. 128 ml of dry tetrahydrofuran was added to 26.22 g of 6-(4-chlorobenzoyl)-4-(3-chlorophenyl)-l- methyl-2(lH)-quinolinone (compound (II)) (0.0642 mole) and stirred until dissolution. This solution was added to the reaction mixture, while the temperature during addition was kept between -5°C and 0°C. After addition, the reaction mixture was stirred for 15 minutes between -5°C and 0°C. 128 ml of water was added to the reaction mixture, followed by the addition of 10.6 ml of acetic acid. The mixture was then heated to 40°C and stirred for 2 hours. The layers were separated and the organic layer washed with 32 ml water. 64 ml water and 7.8 ml aqueous NaOΗ 50% were added to the organic layer which was stirred for 1 hour at ambient temperature. The layers were separated and the organic layer concentrated under reduced pressure, yielding 51.08 g of a brown oil (46.6 wt% 4-(3-chlorophenyl)-6-[(4-chlorophenyl)hydroxy(l-methyl-lH-imidazol-5- yl)methyl]-l-methyl-2(lH)-quinolinone (compound HI); 75.6 % yield).
The product can be isolated via the procedures mentioned above. The resulting product was analysed by hplc using the following conditions :-
Column: Ηypersil C18-BD 3μm, 100mm x 4 mm (i.d.)
Mobile phase:
Solvent A: 0.5% NΗLjOAc
Solvent B: CΗ3CN
Gradient: Time %A %B
0 100 0
15 0 100
18 0 100 19 100 0 23 100 0 Detector: UV 254nm Solvent: DMF The product was found to have a C5:C2 ratio of 99.8:0.2. In contrast using n-butyllithium in place of n-hexyllithium, triethylsilyl chloride in place of tri-i.ro- butylsilyl chloride and conducting the process at -70°C, i.e. generally in accordance with prior art procedures discussed above, the resulting product had a C5:C2 ratio of 95:5, a significant difference in commercial terms.
Preparation of compound (IV)
A 1 liter reaction vessel was charged with 105.4 g of 4-(3-chlorophenyl)-6-[(4- chlorophenyl)hydroxy ( 1 -methyl- 1 H-imidazol-5-yl)methyl] – 1 -methyl-2( 1 H)- quinolinone hydrochloric acid salt (compound (IΗ)and 400 ml of N,N- dimethylimidazolidinone added at 22°C. The mixture was stirred vigorously for 15 minutes at 22°C and became homogeneous. 32.1 ml of thionyl chloride was added over 10 minutes to the reaction mixture, the reaction temperature rising from 22°C to 40°C. After addition of the thionyl chloride, the reaction mixture was cooled from 40°C to 22°C and stirred for three hours at the latter temperature to provide a solution of 4-(3- chlorophenyl)-6-[chloro-(4-chlorophenyl)(l-methyl-lH-imidazol-5-yl)methyl]-l- methyl-2(lH)-quinolinone (compound (IN).
Preparation of unresolved compound (I)
429 ml of ammonia in methanol 7Ν was cooled to 5°C in a 3 liter reaction vessel and the solution of compound (IN), obtained in the previous stage, added, while stirring, over 10 minutes, with an exothermic reaction, the temperature rising from 5°C to 37°C. After the addition was complete, the reaction mixture was cooled to 22°C and stirred for 20 hours. 1000ml of water was then added over 20 minutes, the addition being slightly exothermic so the reaction mixture was cooled to keep the temperature below 30°C. The mixture was then stirred for 22 hours at 22°C, the resulting precipitate filtered off and the precipitate washed three times with 100ml of water to provide a yield of 70-75% of 6-[arnino(4-chlorophenyl)-l-methyl-lH-imidazol-5-ylmethyl]-4-(3- chlorophenyl)-l-methyl-2(lH)-quinolinone. Resolution of compound (I)
a) A 3 liter reaction vessel was charged with 146.8 g of 6-[amino(4-chlorophenyl)(l- methyl-lH-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(lH)-quinolinone and 301.1 g of L-(-)-dibenzoyl-tartaric acid monohydrate, 1200ml of acetone was added and the reaction mixture stirred vigorously for 10 minutes at 22°C to form a solution which was seeded with lOOmg of the final tartrate salt product (obtained from previous screening experiments) and then stirred for 22 hours at 22°C. The resulting precipitate was filtered off and the precipitate was washed twice with 75 ml of acetone and the product dried at 50°C in vacuo to yield 114.7g of R-(-)-6-[amino(4-chlorophenyl)(l- methyl-lΗ-imidazol-5-yl)methyl]-4-(3-chlorophenyl)-l-methyl-2(lΗ)-quinolinone [R- (R*,R*)]-2,3-bis(benzoyloxy)butanedioate (2:3).
b) 41.08 g of the product of stage a) and 80 ml ethanol were stirred for 15 minutes at 22°C. 12.0 ml concentrated aqueous ammonium hydroxide was added over 2 minutes, and the reaction mixture stirred for 1 hour at 25°C. 160 ml water was added over 10 minutes at 25 °C and the mixture heated to reflux and stirred at reflux for 1 hour. The reaction mixture was then cooled to 20°C and stirred for 16 hours at 20°C. The product was filtered, washed twice with 8 ml water and dried at 50°C in vacuo to yield 16.87 g of (R)-(+)-6-[amino(4-chloro-phenyl)(l-methyl-lH-imidazol-5-yl)methyl]-4-(3- chlorophenyl)-l-methyl-2(lH)-quinolinone (compound (I)).
Purification of compound (I)
265 ml of ethanol was added to 19.9g of compound (I), obtained as described in the previous stage, and the mixture warmed while stirring to reflux temperature (78 °C) and then stirred at reflux temperature for 15 minutes before cooling the solution to 75 °C. 1.0 g of activated carbon (Norit A Supra) was then added to the mixture which was stirred at reflux temperature for 1 hour, filtered while warm and the filter then washed with 20 ml warm ethanol. The filtrate and wash solvent were combined (the product spontaneously crystallizes at 48°C), and the mixture warmed to reflux temperature and concentrated by removing 203 ml of ethanol. The resulting suspension was cooled to 22°C, stirred for 18 hours at 22°C, cooled to 2°C and stirred for 5 more hours at 2°C. The precipitate was filtered and washed with 4 ml ethanol and the product dried at 50°C in vacuo to yield 17.25 g of purified compound (I) which complies with the infrared spectrum of reference material.
PAPER
Practical route to 2-quinolinones via a pd-catalyzed c-h bond Activation/C-C bond Formation/Cyclization cascade reaction
Org Lett 2015, 17(2): 222
Division of Chemistry and Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371
Quinolinone derivatives were constructed via a Pd-catalyzed C–H bond activation/C–C bond formation/cyclization cascade process with simple anilines as the substrates. This finding provides a practical procedure for the synthesis of quinolinone-containing alkaloids and drug molecules. The utility of this method was demonstrated by a formal synthesis of Tipifarnib.
0.5 mmol 4-Amino-4′-chlorodiphenylmethane 4, 1mmol acetic anhydride and 2 mL toluene were added into the Schlenk tuble. The mixture was stirred at r.t. for 5 minutes, then 0.5 mmol TsOH•H2O, 2.5 mmol (2E)-3-(3-chlorophenyl) propenoate, 1.5 mmol Na2S2O8 and 5 mmol % Pd(OAc)2 were added into the reaction system in one time. The mixture was heated at 100 oC for 36 h and cooled down to room temperature, quenched with 50 mL saturated sodium bicarbonate solution and extracted thrice with ethyl acetate (30 mL) and the combined organic phase was dried over Na2SO4. After evaporation of the solvents the residue was purified by silica gel chromatography to afford 5 as pale yellow solid (elute: hexane-EtOAc) (180 mg, 95%).
4-(3-chlorophenyl)-6-(4-chlorobenzyl)-2-quinolinone 5 (0.2 mmol), iodine (0.002 mmol), pyridine (0.002 mmol) and aqueous tert-butylhydroperoxide (70%, 0.5 ml) were sealed in a 5 mL tube, then stirred at 80 oC overnight. After cooling to room temperature, the mixture was purified by a short silica gel chromatography column to afford 6 as pale yellow solid (elute: DCM/acetone = 2/1) (77 mg, 98%).
Fedratinib had been in phase III clincial trials by Sanofi for the treatment of myelofibrosis.
However, Sanofi had discontinued this research because of the safety issues. Orphan drug designation was assigned in the U.S. and in Japan for this indication. In 2017, the clinical hold was lifted in the U.S. by Impact Biomedicines.
MOLECULAR FORMULA C27H36N6O3S
MOLECULAR WEIGHT 524.7
SPONSOR Sanofi
CODE DESIGNATIONS SAR302503; TG101348
CAS REGISTRY NUMBER……….936091-26-8
WHO 9707
TG-101348 , a dual-acting JAK2/FLT3 small molecule kinase inhibitor, has been evaluated in phase III clinical development at Sanofi (formerly known as sanofi-aventis) for the oral treatment of intermediate-2 or high risk primary myelofibrosis, post-polycythemia vera myelofibrosis or post-essential thrombocythemia myelofibrosis with splenomegaly. However, development of the compound has been discontinued due to safety issues.
In preclinical models of myeloproliferative diseases, TG-101348, administered orally, was shown to reduce V617F-expressing cell populations in a dose-dependent manner without adversely impacting normal hematopoiesis. The reduction of V617F- expressing cell populations correlated with improved survival and reduced morbidity. Orphan drug designation was assigned in the U.S. and in Japan for the treatment of secondary and primary myelofibrosis. In July 2010, TargeGen was acquired by Sanofi. In 2013, orphan drug designation was assigned by the FDA for the treatment of polycythemia vera.
Fedratinib is an orally bioavailable, small-molecule, ATP-competitive inhibitor of Janus-associated kinase 2 (JAK2) with potential antineoplastic activity. Fedratinib competes with JAK2 as well as the mutated form AK2V617F for ATP binding, which may result in inhibition of JAK2 activation, inhibition of the JAK-STAT signaling pathway, and the induction of tumor cell apoptosis. JAK2 is the most common mutated gene in bcr-abl-negative myeloproliferative disorders (MPDs); the mutated form JAK2V617F has a valine-to-phenylalanine modification at position 617 and plays a key role in tumor cell proliferation and survival.
Fedratinib has been used in trials studying the treatment and basic science of Solid Tumor, Myelofibrosis, Renal Impairment, Neoplasm Malignant, and Hepatic Impairment, among others.
Fedratinib (TG101348; SAR302503) is an orally available inhibitor of Janus kinase 2 (JAK-2) developed for the treatment of patients with myeloproliferative diseases including myelofibrosis. Fedratinib acts as a competitive inhibitor of protein kinase JAK-2 with IC50=6 nM; related kinases FLT3 and RET are also sensitive, with IC50=25 nM and IC50=17 nM, respectively. Significantly less activity was observed against other tyrosine kinases including JAK3 (IC50=169 nM).[1] In treated cells the inhibitor blocks downstream cellular signalling (JAK-STAT) leading to suppression of proliferation and induction of apoptosis.
Myelofibrosis is a myeloid malignancy associated with anemia, splenomegaly, and constitutional symptoms. Patients with myelofibrosis frequently harbor JAK-STAT activating mutations that are sensitive to TG101348. Phase I trial results focused on safety and efficacy of Fedratinib in patients with high- or intermediate-risk primary or post–polycythemia vera/essential thrombocythemia myelofibrosis have been published in 2011.[2]
Fedratinib was originally discovered at TargeGen. In 2010, Sanofi-Aventis acquired TargeGen and continued development of fedratinib until 2013. In 2016, Impact Biomedicines acquired the rights to fedratinib from Sanofi and continued its development for the treatment of myelofibrosis and polycythemia vera. In January 2018, Celgene acquired Impact Biomedicines.[3]
Condensation of 3-bromo-N-tertbutylbenzylsulfonamide with 2-chloro-5-methyl-pyrimidin-4-ylamine in the presence of Pd2(dba)3, Xantphos, Cs2CO3 in refluxing dioxane gives sulfonamide derivative , which is coupled with 4-[2-pyrrolidin-1-yl-ethoxy]phenylamine in AcOH at 150°C to provide the title compound
EXAMPLE 90. 7V-fe^-Butyl-3-{5-methyl-2-14-(2-pyrrolidm-l-yl-ethoxy)-phenylaminol- pyrimidin-4-ylaminol-benzenesuIfonamide (Compound LVII)
LVII
[0203] A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-l-yl- ethoxy)-phenylamine (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150 °C for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2 x 30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum atnount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
The compound and the pharmaceutical compositions described herein can be used for treating or delaying development of myelofibrosis in a subject. N-teft-Butyl-3-[(5-methyl-2-{ [4- (2-pyrrolidin-l-ylethoxy)phenyl]amino}pyrimidin-4-yl)amino]benzenesulfonamide has the following chemical structure:
Example 4. Synthesis of TG101348
Example 4.1 N-fer^-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)-benzenesulfonamide
(Intermediate)
Example 4.1(a)
1 2 Intermediate
[0162] A mixture of 2-chloro-5-methyl-pyrimidin-4-ylamine (1) (0.4 g, 2.8 mmol), 3-bromo-N- teft-butyl-benzenesulfonamide (2) (1.0 g, 3.4 mmol), Pd2(dba¾ (0.17 g, 0.19 mmol), Xantphos (0.2 g, 3.5 mmol) and cesium carbonate (2.0 g, 6.1 mmol) was suspended in dioxane (25 mL) and heated at reflux under the argon atmosphere for 3 h. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and the filtrate
concentrated in vacuo. The residue was dissolved in EtOAc and hexanes added until solid precipitated. After filtration, the title compound (1.2 g, 98%) was obtained as a light brown solid. It was used in the next step without purification. MS (ES+): m/z 355 (M+H)+.
Example 4.1(b)
SM2 Intermediate[0163] The Intermediate was synthesized from 2,4-dichloro-5-methylpyrimidine (SMI) and N-t- butyl-3-aminobenzenesulfonamide (SM2) in the following steps: (1) Mix MeOH (6.7UOa) and SMI (Combi Blocks) (UOa); (2) Add SM2 (1.15UOa, 082eq) and H20 (8.5UOa); (3) Heat 45°C, 20h, N2, IPC CPL SM2<2%; (4) Cool 20°C; (5) Centrifuge, N2; (6) Wash H20 (2.1UOa) + MeOH (1.7UOa); (7) Mix solid in H20 (4.3UOa) + MeOH (3.4UOa); (8) Centrifuge, N2; (9) Wash H20 (2.1UOa) + MeOH (1.7UOa); and (10) Dry 45°C, vacuum, 15h. Obtained
Intermediate, mass 49.6kg (UOb); Yield 79%; OP: 99.6%.
Example 4.2 N-½ri-Butyl-3-[(5-methyl-2-{ [4-(2-pyrrolidin-l- ylethoxy)phenyl]amino}pyrimidin-4-yl)amino]benzenesulfonamide
Intermediate TG101348
Example 4.2(a)
[0164] A mixture of N-ieri-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)- benzenesulfonamide (Intermediate) (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-l-yl-ethoxy)- phenylamine (3) (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150 °C for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCC^ solution (30 mL). The combined aqueous layers were extracted with EtOAc (2 x 30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2S04 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%). ]H NMR (500 MHz, DMSO-d6): δ 1.12 (s, 9H), 1.65-1.70 (m, 4H), 2.12 (s, 3H), 2.45-2.55 (m, 4H), 2.76 (t, /=5.8 Hz, 2H), 3.99 (t, 7=6.0 Hz, 2H), 6.79 (d, 7=9.0 Hz, 2H), 7.46-7.53 (m, 4H), 7.56 (s, 1H), 7.90 (s, 1H), 8.10-8.15 (m, 2H), 8.53 (s, 1H), 8.77 (s, 1H). MS (ES+): m/z 525 (M+H)+.
Example 4.2(b)
[0165] N-½ri-Butyl-3-[(5-methyl-2-{ [4-(2-pyrrolidin-l-ylethoxy)phenyl]amino}pyrimidin-4- yl)amino]benzenesulfonamide dihydrochloride monohydrate was prepared from 4-[2-(l- pyrrolidinyl)ethoxy] aniline dihydrochloride (SM3) and Intermediate following steps (A) and (B).
[0166] Step (A), preparation of free base of SM3 (3) from SM3, comprised steps (1) – (9): (1) Solubilize NaOH (0.42UOb) in H20 (9UOb); (2) Cool <20°C, N2; (3) Add TBME (6UOb) then SM3 (Malladi Drugs) (1.06UOb); (4) Mix >20mn then stop; (5) Drain Aq Ph then extract by TBME (3UOb); (6) Combine Or Ph; (7) Concentrate, vacuum, T<40°C, to an Oil; (8) Solubilize in IPA (2.5UOb); and (9) Calculate dry extract 23%.
[0167] Step (B) comprised the steps (1) – (6): (1) Mix IPA (10.5UOb) and Intermediate (UOb); (2) Add free base of SM3 (0.75UOb, 1.33eq/ interm); (3) add HC1 cone (0.413UOb); (4) Heat 70°C, 20h, N2, IPC CPL Interm<2%; (5) Cool <20°C; (2) Centrifuge, N2; (3) Wash IPA (3UOb); (4) Dry 50°C, vacuum, 26h; (5) De-lump in Fitzmill; and (6) polybag (x2) / poly drum. Obtained TG101348 dihydrochloride monohydrate, mass 83.8kg; Yield 98%; OP: 99.5%. Example 5 Capsule Form of TG101348 and Process of Making TG101348
Example 90 N-tert-Butyl-3-{5-methyl-2-[4-(2-pyrrolidin-1-yl-ethoxy)-phenylamino]-pyrimidin-4-ylamino}-benzenesulfonamide (Compound LVII)
A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-1-yl-ethoxy)-phenylamine (0.10 g, 0.49 mmol) in acetic acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150° C. for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaHCO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2×30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
Example 76 N-tert-Butyl-3-(2-chloro-5-methyl-pyrimidin-4-ylamino)-benzenesulfonamide (Intermediate 33)
A mixture of 2-chloro-5-methyl-pyrimidin-4-ylamine (0.4 g, 2.8 mmol), 3-bromo-N-tert-butyl-benzenesulfonamide (1.0 g, 3.4 mmol), Pd2(dba)3 (0.17 g, 0.19 mmol), Xantphos (0.2 g, 3.5 mmol) and cesium carbonate (2.0 g, 6.1 mmol) was suspended in dioxane (25 mL) and heated at reflux under the argon atmosphere for 3 h. The reaction mixture was cooled to room temperature and diluted with DCM (30 mL). The mixture was filtered and the filtrate concentrated in vacuo. The residue was dissolved in EtOAc and hexanes added until solid precipitated. After filtration, the title compound (1.2 g, 98%) was obtained as a light brown solid. It was used in the next step without purification. MS (ES+): m/z 355 (M+H)+.
Example 90N-tert-Butyl-3-{5-methyl-2-[4-(2-pyrrolidin-1-yl-ethoxy)-phenylamino]-pyrimidin-4-ylamino}-benenesulfonamide (Compound LVII)
[0308]
[0309]
A mixture of intermediate 33 (0.10 g, 0.28 mmol) and 4-(2-pyrrolidin-1-yl-ethoxy)-phenylamine (0.10 g, 0.49 mmol) in aeetie acid (3 mL) was sealed in a microwave reaction tube and irradiated with microwave at 150° C. for 20 min. After cooling to room temperature, the cap was removed and the mixture concentrated. The residue was purified by HPLC and the corrected fractions combined and poured into saturated NaIICO3 solution (30 mL). The combined aqueous layers were extracted with EtOAc (2×30 mL) and the combined organic layers washed with brine, dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated and the resulting solid dissolved in minimum amount of EtOAc and hexanes added until solid precipitated. After filtration, the title compound was obtained as a white solid (40 mg, 27%).
Methods for the treatment and management of myeloproliferative diseases using 4-(amino)-2-(2,6-dioxo(3-piperidyl)-isoindoline-1,3-dione in combination with other therapies
METHODS FOR TREATING TYROSINE-KINASE-INHIBITOR-RESISTANT MALIGNANCIES IN PATIENTS WITH GENETIC POLYMORPHISMS OR AHI1 DYSREGULATIONS OR MUTATIONS EMPLOYING DIANHYDROGALACTITOL, DIACETYLDIANHYDROGALACTITOL, DIBROMODULCITOL, OR ANALOGS OR DERIVATIVES THEREOF
In 2013, Array Biopharma licensed the product to Loxo Oncology for development and commercialization in the U.S. In 2016, breakthrough therapy designation was received in the U.S. for the treatment of unresectable or metastatic solid tumors with NTRK-fusion proteins in adult and pediatric patients who require systemic therapy and who have either progressed following prior treatment or who have no acceptable alternative treatments. In 2017, Bayer acquired global co-development and commercialization rights from Loxo Oncology.
Originator Array BioPharma
Developer Array BioPharma; Loxo Oncology; National Cancer Institute (USA)
Class Antineoplastics; Pyrazoles; Pyrimidines; Pyrrolidines; Small molecules
Mechanism of Action Tropomyosin-related kinase antagonists
Orphan Drug Status Yes – Solid tumours; Soft tissue sarcoma
LOXO-101 is a small molecule that was designed to block the ATP binding site of the TRK family of receptors, with 2 to 20 nM cellular potency against the TRKA, TRKB, and TRKC kinases. IC50 value: 2 – 20 nM Target: TRKA/B/C in vitro: LOXO-101 is an orally administered inhibitor of the TRK kinase and is highly selective only for the TRK family of receptors. LOXO-101 is evaluated for off-target kinase enzyme inhibition against a panel of 226 non-TRK kinases at a compound concentration of 1,000 nM and ATP concentrations near the Km for each enzyme. In the panel, LOXO-101 demonstrates greater than 50% inhibition for only one non-TRK kinase (TNK2 IC50, 576 nM). Measurement of proliferation following treatment with LOXO-101 demonstrates a dose-dependent inhibition of cell proliferation in all three cell lines. The IC50 is less than 100 nM for CUTO-3.29 and less than 10 nM for KM12 and MO-91, consistent with the known potency of this drug for the TRK kinase family. [1] LOXO-101 demonstrates potent and highly-selective inhibition of TRKA, TRKB, and TRKC over other kinase- and non-kinase targets. LOXO-101 is a potent, ATP-competitive TRK inhibitor with IC50s in low nanomolar range for inhibition of all TRK family members in binding and cellular assays, with 100x selectivity over other kinases. [2] in vivo: Athymic nude mice injected with KM12 cells are treated with LOXO-101 orally daily for 2 weeks. Dose-dependent tumor inhibition is observed, demonstrating the ability of this selective compound to inhibit tumor growth in vivo. [1]
N-Boc-pyrrolidine as starting material The method involves enantioselective deprotonation, transmetalation with ZnCl2, Negishi coupling with 2-bromo-1,4-difluorobenzene,
N-arylation with 5-chloropyrazolo[1,5-a]pyrimidine, nitration, nitro reduction and condensation with CDI and 3(S)-pyrrolidinol.
[00423] To a DCM (0.8 mL) solution of (R)-5-(2-(2,5-difiuorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-a]pyrimidin-3-amine (Preparation B; 30 mg, 0.095 mmol) was added CDI (31 mg, 0.19 mmol) at ambient temperature in one portion. After stirring two hours, (S)-pyrrolidin-3-ol (17 mg, 0.19 mmol) [purchased from Suven Life Sciences] was added in one portion. The reaction was stirred for 5 minutes before it was concentrated and directly purified by reverse-phase column chromatography, eluting with 0 to 50% acetonitrile/water to yield the final product as a yellowish foamy powder (30 mg, 74% yield). MS (apci) m/z = 429.2 (M+H).
[00424] To a solution of (S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo [ 1 ,5 -a]pyrimidin-3 -yl)-3 -hydroxypyrrolidine- 1 -carboxamide (4.5 mg, 0.011 mmol) in methanol (1 mL) at ambient temperature was added sulfuric acid in MeOH (105 μL, 0.011 mmol). The resulting solution was stirred for 30 minutes then concentrated to provide (S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-a]pyrimidin-3-yl)-3 -hydroxypyrrolidine- 1 -carboxamide sulfate (5.2 mg, 0.0099 mmol, 94 % yield) as a yellow solid.
(R,E)-N-(2,5-difluorobenzylidene)-2-methylpropane-2-sulfinamide (17): Compound 16 and (R)-2-methylpropane-2-sulfinamide (1.05 eq.) were charged to a reactor outfitted with a mechanical stirrer, reflux condensor, J-Kem temperature probe under N2. DCM (3 mL/g of 14) was added (endothermic from 22 °C to about 5 °C) followed by addition of cesium carbonate (0.70 eq.) (exothermic to -50 °C). Once the addition was complete, the reaction mixture was stirred at room temperature for 3 h (slowly cools from about 40 °C). When the reaction was called complete (HPLC) the mixture was filtered through Celite. The Celite pad (0.3 wt eq) was equilibrated with DCM (1 mL/g of 16), and the reaction mixture was poured through the pad. The Celite cake was washed with DCM (2 x 1 mL/g), and the filtrate concentrated partially to leave about 0.5 to 1 mL/g DCM remaining. The orange solution was stored at room temperature (generally overnight) and used directly in the next reaction. (100% yield was assumed).
2)
(R)-N-((R)-l-(2,5-difluorophenyl)-3-(l,3-dioxan-2-yl)propyl)-2-methylpropane-2-sulfinamide (19): To a reactor equipped with overhead stirring, reflux condensor, under
nitrogen, was added magnesium turnings (2.0 eq), and THF (8 mL/g of 17). The mixture was heated to 40 °C. Dibal-H (25% wt in toluene, 0.004 eq) was added to the solution, and the suspension heated at 40 °C for 25 minutes. A solution of 2-(2-bromoethyl)-l,3-dioxane (18) (2 eq) in THF (4.6 mL/g of 17) was added dropwise to the Mg solution via addition funnel. The solution temperature was maintained < 55 °C. The reaction progress was monitored by GC. When the Grignard formation was judged complete, the solution was cooled to -30 °C, and 17 (1.0 eq, in DCM) was added dropwise via addition funnel. The temperature was kept between -30 °C and -20 °C and the reaction was monitored for completion (FIPLC). Once the reaction was called complete, the suspension (IT = -27.7 °C) was vacuum transferred to a prepared and cooled (10 °C) 10% aqueous citric acid solution (11 mL/g of 17). The mixture temperature rose to 20 °C during transfer. The milky solution was allowed to stir at ambient temperature overnight. MTBE (5.8 mL/g) was added to the mixture, and it was transferred to a separatory funnel. The layers were allowed to separate, and the lower aqueous layer was removed. The organic layer was washed with sat. NaHC03 (11 mL/g) and then sat. NaCl (5.4 mL/g). The organic layer was removed and concentrated to minimum volume via vacuum distillation. MTBE (2 mL/g) was added, and the mixture again concentrated to minimum volume. Finally MTBE was added to give 2 mL/g total MTBE (GC ratio of MTBE:THF was about 9: 1), and the MTBE mixture was heated to 50 °C until full dissolution occurred. The MTBE solution was allowed to cool to about 35 °C, and heptane was added portion -wise. The first portion (2 mL/g) is added, and the mixture allowed to stir and form a solid for 1-2 h, and then the remainder of the heptane is added (8 mL/g). The suspension was allowed to stir for >lh. The solids were collected via filtration through polypropylene filter cloth (PPFC) and washed with 10% MTBE in heptane (4 mL/g. The wet solid was placed in trays and dried in a vacuum oven at 55 °C until constant weight (3101 g, 80.5%, dense white solid, 100a% and 100wt%).
3)
(R)-2-(2,5-difluorophenyl)pyrrolidine (R)-2-hydroxysuccinate (10): To a flask containing 4: 1 TFA:water (2.5 mL/g, pre-mixed and cooled to <35 °C before adding 19) was added (R)-N-((R)-l-(2,5-difluorophenyl)-3-(l,3-dioxan-2-yl)propyl)-2-methylpropane-2-sulfinamide (19) (1 eq). The mixture temperature rose from 34 °C to 48 °C and was stirred at ambient temperature for 1 h. Additional TFA (7.5 mL/g) was added, followed by triethylsilane (3 eq) over 5 minutes. The biphasic mixture was stirred vigorously under nitrogen for 21 h until judged complete (by GC, <5% of imine). The mixture was then concentrated under vacuum until -10 kg target mass (observed 10.8 kg after concentration). The resulting concentrate was transferred to a separatory funnel and diluted with MTBE (7.5 mL/g), followed by water (7.5 mL/g). The layers were separated. The MTBE layer was back-extracted with 1M HC1 (3 mL/g). The layers were separated, and the aqueous layers were combined in a round-bottomed flask with DCM (8 mL/g). The mixture was cooled in an ice bath and 40% NaOH was charged to adjust the pH to >12 (about 0.5 mL/g; the temperature went from 24 °C to 27 °C, actual pH was 13), and the layers separated in the separatory funnel. The aqueous layer was back-extracted twice with DCM (2 x 4 mL/g). The organic layers were concentrated to an oil (<0.5 mL/g) under vacuum (rotovap) and EtOH (1 mL/g based on product) was added. The yellow solution was again concentrated to an oil (81% corrected yield, with 3% EtOH, 0.2% imine and Chiral HPLC showed 99.7%ee).
Salt formation: To a solution of (R)-2-(2,5-difluorophenyl)pyrrolidine 10 (1 eq) in EtOH (15 mL/g) was added Z)-(+)-Malic Acid (1 eq). The suspension was heated to 70 °C for 30 minutes (full dissolution had occurred before 70 °C was reached), and then allowed to cool to room temperature slowly (mixture was seeded when the temperature was < 40 °C). The slurry was stirred at room temperature overnight, then cooled to <5 °C the next morning. The suspension was stirred at <5 °C for 2h, filtered (PPFC), washed with cold EtOH (2 x 2 mL/g), and dried (50-55 °C) under vacuum to give the product as a white solid (96% based on 91% potency, product is an EtOH solvate or hemi- solvate).
Compound 5 and 10 (1.05 eq) were charged to a reactor outfitted with a mechanical stirrer, J-Kem temperature probe, under N2. EtOH and THF (4: 1, 10 mL/g of 5) were added and the mixture was cooled to 15-25 °C. Triethylamine (3.5 eq) was added and the internal temp generally rose from 17.3 – 37.8 °C. The reaction was heated to 50 – 60 °C and held at that temperature for 7 h. Once the reaction is judged complete (HPLC), water (12 mL/g of 5) is added maintaining the temperature at 50 – 60 °C. The heat is removed and the suspension was slowly cooled to 21 °C over two h. After stirring at -21 °C for 2 h, the suspension was centrifuged and the cake was washed with water (3 x 3 mL/g of 5). The solid was transferred to drying trays and placed in a vacuum oven at 50 – 55 °C to give 11.
2)
(R)-5-(2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-a]pyrimidin-3-amine fumarate Pt/C hydrogenation (12 fumarate): To a Parr reactor was charged 11 (1.0 eq), 5% Pt/C ~ 50 wt% water (2 mol% Pt / Johnson Matthey B 103018-5 or Sigma Aldrich 33015-9), and MeOH (8 mL/g). The suspension was stirred under hydrogen at 25-30 psi and the temperature was maintained below 65 °C for ~8 h. When the reaction was called complete (HPLC), the reaction was cooled to 15 – 25 °C and the hydrogen atmosphere was replaced with a nitrogen atmosphere. The reaction mixture was filtered through a 2 micron bag filter and a 0.2 micron line filter in series. The filtrate from the Pt/C hydrogenation was transferred to a reactor under nitrogen with mechanical stirring and then MTBE (8 mL/g) and fumaric acid (1.01 eq) were charged. The mixture was stirred under nitrogen for 1 h and solids formed after -15 min. The mixture was cooled to -10 to -20 °C and stirred for 3 h. The suspension was filtered (PPFC), washed with MTBE (-2.5 mL/g), and the solids was dried under vacuum at 20-25 °C with a nitrogen bleed to yield an off-white solid (83% yield).
3)
Phenyl (5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)-3,3a-dihydropyrazolo[l,5-a]pyrimidin-3-yl)carbamate (13): To a 5 to 15°C solution of 12-fumarate (1.0 eq) in 2-MeTHF (15 mL/g) was added a solution of potassium carbonate (2.0 eq.) in water (5 mL/g) followed by phenyl chloroformate (1.22 eq.) (over 22 min, an exotherm from 7 °C to 11 °C occurred). The mixture was stirred for 2 h and then the reaction was called complete (HPLC). The stirring ceased and the aqueous layer was removed. The organic layer was washed with brine (5 mL/g) and concentrated to ca. 5 mL/g of 2-MeTHF under vacuum and with heating to 40 °C. To the 2-MeTHF solution was added heptanes (2.5 mL/g) followed by seeds (20 mg, 0.1 wt%). This mixture was allowed to stir at room temperature for 2 h (until a solid formed), and then the remainder of the heptanes (12.5 mL/g) was added. The mixture was stirred at ambient temperature for 2 h and then the solids were collected via filtration (PPFC), washed with 4: 1 heptanes :MeTHF (2 x 2 mL/g), and dried to give 13 (96%).
4)
(S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)pyrazolo[l,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-l-carboxamide hydrogen sulfate: To a flask containing 13 (1.0 eq) was added a solution of (S)-pyrrolidin-3-ol (1.1 eq.) in EtOH (10 mL/g). The mixture was heated at 50 – 60 °C for 5 h, called complete (HPLC), and then cooled to 20-35 °C. Once <35°C, the reaction was polish-filtered (0.2 micron) into a clean reaction vessel and the mixture was cooled to -5 to 5 °C. Sulfuric acid (1.0 eq.) was added over 40 minutes, the temperature rose to 2 °C and the mixture was seeded. A solid formed, and the mixture was allowed to stir at -5 to 5 °C for 6.5 h. Heptanes (10 mL/g) was added, and the mixture stirred for 6.5 h. The
suspension was filtered (PPFC), washed with 1 : 1 EtOH:heptanes (2 x 2 mL/g), and dried (under vacuum at ambient temperature) to give Formula I (92.3%).
Preparation of the hydrogen sulfate salt of the compound of Formula I:
Concentrated sulfuric acid (392 mL) was added to a solution of 3031 g of (S)-N-(5- ((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)-pyrazolo[l,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-l-carboxamide in 18322 mL EtOH to form the hydrogen sulfate salt. The solution was seeded with 2 g of (,S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-l-yl)-pyrazolo[l,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-l-carboxamide hydrogen sulfate and the solution was stirred at room temperature for at least 2 hours to form a slurry of the hydrogen sulfate salt. Heptane (20888 g) was added and the slurry was stirred at room temperature for at least 60 min. The slurry was filtered and the filter cake was washed with 1 : 1 heptane/EtOH. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius).
The dried hydrogen sulfate salt (6389 g from 4 combined lots) was added to a 5 :95 w/w solution of water/2-butanone (total weight 41652 g). The mixture was heated at about 68° Celsius with stirring until the weight percent of ethanol was about 0.5%, during which time a slurry formed. The slurry was filtered, and the filter cake was washed with a 5 :95 w/w solution of water/2-butanone. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius) to provide the crystalline form of (S)-N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-l-yl)-pyrazolo[l,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-l-carboxamide hydrogen sulfate.
Provided herein is a novel crystalline form of the compound of Formula I:
[0000]
also known as (S)—N-(5-((R)-2-(2, 5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide. In particular, the novel crystalline form comprises the hydrogen sulfate salt of the compound of Formula I in a stable polymorph form, hereinafter referred to as crystalline form (I-HS) and LOXO-101, which can be characterized, for example, by its X-ray diffraction pattern—the crystalline form (I-HS) having the formula:
[0000]
In some embodiments of the above step (c), the base is an alkali metal base, such as an alkali metal carbonate, such as potassium carbonate.
Preparation of 5-chloro-3-nitropyrazolo[1,5-a]pyrimidine Step A—Preparation of sodium pyrazolo[1,5-a]pyrimidin-5-olate
A solution of 1H-pyrazol-5-amine and 1,3-dimethylpyrimidine-2,4(1H,3H)-dione (1.05 equiv.) were charged to a round bottom flask outfitted with a mechanical stirrer, a steam pot, a reflux condenser, a J-Kem temperature probe and an N2 adaptor for positive N2 pressure control. Under mechanical stirring the solids were suspended with 4 vol. (4 mL/g) of absolute EtOH under a nitrogen atmosphere, then charged with 2.1 equivalents of NaOEt (21 wt % solution in EtOH), and followed by line-rinse with 1 vol. (1 mL/g) of absolute EtOH. The slurry was warmed to about 75° Celsius and stirred at gentle reflux until less than 1.5 area % of 1H-pyrazol-5-amine was observed by TRK1PM1 HPLC to follow the progression of the reaction using 20 μL of slurry diluted in 4 mL deionized water and 5 μL injection at 220 nm.
After 1 additional hour, the mixture was charged with 2.5 vol. (2.5 mL/g) of heptane and then refluxed at 70° Celsius for 1 hour. The slurry was then cooled to room temperature overnight. The solid was collected by filtration on a tabletop funnel and polypropylene filter cloth. The reactor was rinsed and charged atop the filter cake with 4 vol. (4 mL/g) of heptane with the cake pulled and the solids being transferred to tared drying trays and oven-dried at 45° Celsius under high vacuum until their weight was constant. Pale yellow solid sodium pyrazolo[1,5-a]-pyrimidin-5-olate was obtained in 93-96% yield (corrected) and larger than 99.5 area % observed by HPLC (1 mg/mL dilution in deionized water, TRK1PM1 at 220 nm).
Step B—Preparation of 3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one
A tared round bottom flask was charged with sodium pyrazolo[1,5-a]pyrimidin-5-olate that was dissolved at 40-45° Celsius in 3.0 vol. (3.0 mL/g) of deionized water, and then concentrated under high vacuum at 65° Celsius in a water-bath on a rotary evaporator until 2.4× weight of starting material was observed (1.4 vol/1.4 mL/g deionized water content). Gas chromatography (GC) for residual EtOH (30 μL of solution dissolved in ˜1 mL MeOH) was performed showing less than 100 ppm with traces of ethyl nitrate fumes being observed below upon later addition of HNO3. In some cases, the original solution was charged with an additional 1.5 vol. (1.5 mL/g) of DI water, then concentrated under high vacuum at 65° Celsius in a water-bath on a rotary evaporator until 2.4× weight of starting material was observed (1.4 vol/1.4 mL/g DI water content). Gas chromatograph for residual EtOH (30 μL of solution dissolved in about 1 mL MeOH) was performed showing <<100 ppm of residual EtOH without observing any ethyl nitrate fumes below upon later addition of HNO3.
A round bottom vessel outfitted with a mechanical stirrer, a steam pot, a reflux condenser, a J-Kem temperature probe and an N2 adaptor for positive N2 pressure control was charged with 3 vol. (3 mL/g, 10 equiv) of >90 wt % HNO3 and cooled to about 10° Celsius under a nitrogen atmosphere using external ice-water cooling bath under a nitrogen atmosphere. Using a pressure equalizing addition funnel, the HNO3solution was charged with the 1.75-1.95 volumes of a deionized water solution of sodium pyrazolo[1,5-a]pyrimidin-5-olate (1.16-1.4 mL DI water/g of sodium pyrazolo[1,5-a]pyrimidin-5-olate) at a rate to maintain 35-40° Celsius internal temperature under cooling. Two azeotropes were observed without any ethyl nitrate fumes. The azeotrope flask, the transfer line (if applicable) and the addition funnel were rinsed with 2×0.1 vol. (2×0.1 mL/g) deionized water added to the reaction mixture. Once the addition was complete, the temperature was gradually increased to about 45-50° Celsius for about 3 hours with HPLC showing >99.5 area % conversion of sodium pyrazolo[1,5-a]pyrimidin-5-olate to 3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one.
Step C—Preparation of 5-chloro-3-nitropyrazolo[1,5-a]pyrimidine
3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one was charged to a round bottom flask outfitted with a mechanical stirrer, a heating mantle, a reflux condenser, a J-Kem temperature probe and an N2 adaptor for positive N2pressure control. Under mechanical stirring the solids were suspended with 8 volumes (8 mL/g) of CH3CN, and then charged with 2,6-lutitine (1.05 equiv) followed by warming the slurry to about 50° Celsius. Using a pressure equalizing addition funnel, the mixture was dropwise charged with 0.33 equivalents of POCl3. This charge yielded a thick, beige slurry of a trimer that was homogenized while stirring until a semi-mobile mass was observed. An additional 1.67 equivalents of POCl3 was charged to the mixture while allowing the temperature to stabilize, followed by warming the reaction mixture to a gentle reflux (78° Celsius). Some puffing was observed upon warming the mixture that later subsided as the thick slurry got thinner.
The reaction mixture was allowed to reflux until complete dissolution to a dark solution and until HPLC (20 μL diluted in 5 mL of CH3CN, TRK1PM1 HPLC, 5 μL injection, 268 nm) confirmed that no more trimer (RRT 0.92) was present with less than 0.5 area % of 3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one (RRT 0.79) being observed by manually removing any interfering and early eluting peaks related to lutidine from the area integration. On a 1.9 kg scale, 0 area % of the trimer, 0.25 area % of 3-nitropyrazolo[1,5-a]pyrimidin-5(4H)-one, and 99.5 area % of 5-chloro-3-nitropyrazolo[1,5-a]pyrimidine was observed after 19 hours of gentle reflux using TRK1PM1 HPLC at 268 [0000]
Preparation of (R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxysuccinate Step A—Preparation of tert-butyl(4-(2,5-difluorophenyl)-4-oxobutyl)-carbamate
2-bromo-1,4-difluorobenzene (1.5 eq.) was dissolved in 4 volumes of THF (based on weight of tert-butyl 2-oxopyrrolidine-1-carboxylate) and cooled to about 5° Celsius. A solution of 2.0 M iPrMgCl in THF (1.4 eq.) was added over 2 hours to the mixture while maintaining a reaction temperature below 25° Celsius. The solution was allowed to cool to about 5° Celsius and stirred for 1 hour (GC analysis confirmed Grignard formation). A solution of tert-butyl 2-oxopyrrolidine-1-carboxylate (1.0 eq.) in 1 volume of THF was added over about 30 min while maintaining a reaction temperature below 25° Celsius. The reaction was stirred at about 5° Celsius for 90 min (tert-butyl 2-oxopyrrolidine-1-carboxylate was confirmed to be less than 0.5 area % by HPLC). The reaction was quenched with 5 volumes of 2 M aqueous HCl while maintaining a reaction temperature below 45° Celsius. The reaction was then transferred to a separatory funnel adding 10 volumes of heptane and removing the aqueous layer. The organic layer was washed with 4 volumes of saturated aqueous NaCl followed by addition of 2×1 volume of saturated aqueous NaCl. The organic layer was solvent-switched to heptane (<1% wt THF confirmed by GC) at a distillation temperature of 35-55° Celsius and distillation pressure of 100-200 mm Hg for 2×4 volumes of heptane being added with a minimum distillation volume of about 7 volumes. The mixture was then diluted to 10 volumes with heptane while heating to about 55° Celsius yielded a denser solid with the mixture being allowed to cool to room temperature overnight. The slurry was cooled to less than 5° Celsius and filtered through polypropylene filter cloth. The wet cake was washed with 2×2 volumes of heptane. The solids were dried under vacuum at 55° Celsius until the weight was constant, yielding tert-butyl(4-(2,5-difluorophenyl)-4-oxobutyl)-carbamate as a white solid at about 75% to 85% theoretical yield.
Step B—Preparation of 5-(2,5-difluorophenyl)-3,4-dihydro-2H-pyrrole
tert-butyl(4-(2,5-difluorophenyl)-4-oxobutyl)-carbamate was dissolved in 5 vol. of toluene with 2.2 eq. of 12M HCl being added observing a mild exotherm and gas evolution. The reaction was heated to 65° Celsius for 12-24 hours and monitored by HPLC. Upon completion the reaction was cooled to less than 15° Celsius with an ice/water bath. The pH was adjusted to about 14 with 3 equivalents of 2M aqueous NaOH (4.7 vol.). The reaction was stirred at room temperature for 1-2 hours. The mixture was transferred to a separatory funnel with toluene. The aqueous layer was removed and the organic layer was washed with 3 volumes of saturated aqueous NaCl. The organic layer was concentrated to an oil and redissolved in 1.5 volumes of heptane. The resulting suspension was filtered through a GF/F filter paper and concentrated to a light yellow oil of 5-(2,5-difluorophenyl)-3,4-dihydro-2H-pyrrole with a 90% to 100% theoretical yield.
Step C—Preparation of (R)-2-(2,5-difluorophenyl)-pyrrolidine
Chloro-1,5-cyclooctadiene iridium dimer (0.2 mol %) and (R)-2-(2-(diphenylphosphino)phenyl)-4-isopropyl-4,5-dihydrooxazole (0.4 mol %) were suspended in 5 volumes of MTBE (based on 5-(2,5-difluorophenyl)-3,4-dihydro-2H-pyrrole) at room temperature. The mixture was stirred for 1 hour and most of the solids dissolved with the solution turning dark red. The catalyst formation was monitored using an HPLC/PDA detector. The reaction was cooled to less than 5° Celsius and 5-(2,5-difluorophenyl)-3,4-dihydro-2H-pyrrole (1.0 eq.) was added using a 0.5 volumes of MTBE rinse. Diphenylsilane (1.5 eq.) was added over about 20 minutes while maintaining a reaction temperature below 10° Celsius. The reaction was stirred for 30 minutes below 10° Celsius and then allowed to warm to room temperature. The reaction was stirred overnight at room temperature. The completion of the reaction was confirmed by HPLC and then cooled to less than 5° Celsius. The reaction was quenched with 5 volumes of 2M aqueous HCl maintaining temperature below 20° Celsius. After 10 minutes the ice/water bath was removed and the reaction temperature was allowed to increase to room temperature while stirring for 2 hours. The mixture was transferred to a separatory funnel with 3 volumes of MTBE. The aqueous layer was washed with 3.5 volumes of MTBE followed by addition of 5 volumes of MTBE to the aqueous layer while adjusting the pH to about 14 by adding 0.75 volumes of aqueous 50% NaOH. The organic layer was washed with 5 volumes of aqueous saturated NaCl, then concentrated to an oil, and diluted with 3 volumes of MTBE. The solution was filtered through a polypropylene filter cloth and rinsed with 1 volume of MTBE. The filtrate was concentrated to an oil of (R)-2-(2,5-difluorophenyl)-pyrrolidine with a 95% to 100% theoretical yield and with 75-85% ee.
Step D—Preparation of (R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxy-succinate
(R)-2-(2,5-difluorophenyl)-pyrrolidine (1.0 eq.) was transferred to a round bottom flask charged with 15 volumes (corrected for potency) of EtOH (200 prf). D-malic acid (1.05 eq.) was added and the mixture was heated to 65° Celsius. The solids all dissolved at about 64° Celsius. The solution was allowed to cool to RT. At about 55° Celsius the solution was seeded with (R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxy-succinate (about 50 mg, >97% ee) and stirred at room temperature overnight. The suspension was then filtered through a polypropylene filter cloth and washed with 2×1 volumes of EtOH (200 prf). The solids were dried under vacuum at 55° Celsius, yielding (R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxy-succinate with a 75% to 90% theoretical yield and with >96% ee.
Referring to Scheme 1, suitable bases include tertiary amine bases, such as triethylamine, and K2CO3. Suitable solvents include ethanol, heptane and tetrahydrofuran (THF). The reaction is conveniently performed at temperatures between 5° Celsius and 50° Celsius. The reaction progress was generally monitored by HPLC TRK1PM1.
[0247]
Compounds II (5-chloro-3-nitropyrazolo[1,5-a]pyrimidine) and III ((R)-2-(2,5-difluorophenyl)-pyrrolidine (R)-2-hydroxysuccinate, 1.05 eq.) were charged to a round bottom flask outfitted with a mechanical stirrer, a J-Kem temperature probe and an N2 adaptor for positive N2 pressure control. A solution of 4:1 EtOH:THF (10 mL/g of compound II) was added and followed by addition of triethylamine (NEt3, 3.50 eq.) via addition funnel with the temperature reaching about 40° Celsius during addition. Once the addition was complete, the reaction mixture was heated to 50° Celsius and stirred for 0.5-3 hours to yield compound IV.
To a round bottom flask equipped with a mechanical stirrer, a J-Kem temperature probe, and an N2 inlet compound IV was added and followed by addition of tetrahydrofuran (10 mL/g of compound IV). The solution was cooled to less than 5° Celsius in an ice bath, and Zn (9-10 eq.) was added. 6M HCl (9-10 eq.) was then added dropwise at such a rate to keep the temperature below 30° Celsius (for 1 kg scale the addition took about 1.5 hours). Once the exotherm subsided, the reaction was allowed to warm to room temperature and was stirred for 30-60 min until compound IV was not detected by HPLC. At this time, a solution of potassium carbonate (K2CO3, 2.0 eq.) in water (5 mL/g of compound IV) was added all at once and followed by rapid dropwise addition of phenyl chloroformate (PhOCOCl, 1.2 eq.). Gas evolution (CO2) was observed during both of the above additions, and the temperature increased to about 30° Celsius after adding phenyl chloroformate. The carbamate formation was stirred at room temperature for 30-90 min. HPLC analysis immediately followed to run to ensure less than 1 area % for the amine being present and high yield of compound VI in the solution.
To the above solution amine VII ((S)-pyrrolidin-3-ol, 1.1 eq. based on theoretical yield for compound VI) and EtOH (10 mL/g of compound VI) was added. Compound VII was added before or at the same time as EtOH to avoid ethyl carbamate impurities from forming. The above EtOH solution was concentrated to a minimum volume (4-5 mL/g) using the batch concentrator under reduced pressure (THF levels should be <5% by GC), and EtOH (10 mL/g of compound VI) was back-added to give a total of 10 mL/g. The reaction was then heated at 50° Celsius for 9-19 hours or until HPLC shows that compound VI is less than 0.5 area %. The reaction was then cooled to room temperature, and sulfuric acid (H2SO4, 1.0 eq. to compound VI) was added via addition funnel to yield compound I-HS with the temperature usually exotherming at about 30° Celsius.
Example 1 Preparation of Crystalline Form (I-HS) (Method 1)
(S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide (0.500 g, 1.17 mmol) was dissolved in EtOH (2.5 mL) and cooled to about 5° Celsius. Concentrated sulfuric acid (0.0636 mL, 1.17 mmol) was added to the cooled solution and stirred for about 10 min, while warming to room temperature. Methyl tert-butyl ether (MTBE) (2 mL) was slowly added to the mixture, resulting in the product gumming out. EtOH (2.5 mL) was then added to the mixture and heated to about reflux until all solids were dissolved. Upon cooling to room temperature and stirring for about 1 hour, some solids formed. After cooling to about 5° Celsius, the solids were filtered and washed with MTBE. After filtration and drying at air for about 15 minutes, (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate was isolated as a solid.
Example 2 Preparation of Crystalline Form (I-HS) (Method 2)
Concentrated sulfuric acid (392 mL) was added to a solution of 3031 g of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide in 18322 mL EtOH to form the hydrogen sulfate salt. The solution was seeded with 2 g of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate and the solution was stirred at room temperature for at least 2 hours to form a slurry of the hydrogen sulfate salt. Heptane (20888 g) was added and the slurry was stirred at room temperature for at least 60 min. The slurry was filtered and the filter cake was washed with 1:1 heptane/EtOH. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius).
The dried hydrogen sulfate salt (6389 g from 4 combined lots) was added to a 5:95 w/w solution of water/2-butanone (total weight 41652 g). The mixture was heated at about 68° Celsius with stirring until the weight percent of ethanol was about 0.5%, during which time a slurry formed. The slurry was filtered, and the filter cake was washed with a 5:95 w/w solution of water/2-butanone. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius) to provide the crystalline form of (S)—N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate.
Example 3 Preparation of Amorphous Form AM(HS)
To a solution of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide (9.40 g, 21.94 mmol) in MeOH (220 mL) was slowly added sulfuric acid (0.1 M in MeOH, 219.4 mL, 21.94 mmol) at ambient temperature under rapid stirring. After 30 minutes, the reaction was first concentrated by rotary evaporator to near dryness, then on high vacuum for 48 h to provide amorphous form of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide sulfate (11.37 g, 21.59 mmol, 98.43% yield). LCMS (apci m/z 429.1, M+H).
WO 2010/048314 discloses in Example 14A a hydrogen sulfate salt of (S)—N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide. WO 2010/048314 does not disclose the particular form of the hydrogen sulfate salt described herein when prepared according to the method of Example 14A in that document. In particular, WO 2010/048314 does not disclose crystalline form (l-HS) as described below.
(S)—N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide having the formula (I):
Example 1 Preparation of Crystalline Form (I-HS) (Method 1)
(S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide (0.500 g, 1.17 mmol) was dissolved in EtOH (2.5 mL) and cooled to about 5° Celsius. Concentrated sulfuric acid (0.0636 mL, 1.17 mmol) was added to the cooled solution and stirred for about 10 min, while warming to room temperature. Methyl tert-butyl ether (MTBE) (2 mL) was slowly added to the mixture, resulting in the product gumming out. EtOH (2.5 mL) was then added to the mixture and heated to about reflux until all solids were dissolved. Upon cooling to room temperature and stirring for about 1 hour, some solids formed. After cooling to about 5° Celsius, the solids were filtered and washed with MTBE. After filtration and drying at air for about 15 minutes, (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidi n-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate was isolated as a solid.
Example 2 Preparation of Crystalline Form (I-HS) (Method 2)
Concentrated sulfuric acid (392 mL) was added to a solution of 3031 g of (S)—N-(5-((R)-2-(2, 5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1, 5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide in 18322 mL EtOH to form the hydrogen sulfate salt. The solution was seeded with 2 g of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate and the solution was stirred at room temperature for at least 2 hours to form a slurry of the hydrogen sulfate salt. Heptane (20888 g) was added and the slurry was stirred at room temperature for at least 60 min. The slurry was filtered and the filter cake was washed with 1:1 heptane/EtOH. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius).
The dried hydrogen sulfate salt (6389 g from 4 combined lots) was added to a 5:95 w/w solution of water/2-butanone (total weight 41652 g). The mixture was heated at about 68° Celsius with stirring until the weight percent of ethanol was about 0.5%, during which time a slurry formed. The slurry was filtered, and the filter cake was washed with a 5:95 w/w solution of water/2-butanone. The solids were then dried under vacuum at ambient temperature (oven temperature set at 15° Celsius) to provide the crystalline form of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)-pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide hydrogen sulfate.
Example 3 Preparation of Amorphous Form AM(HS)
To a solution of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide (9.40 g, 21.94 mmol) in MeOH (220 mL) was slowly added sulfuric acid (0.1 M in MeOH, 219.4 mL, 21.94 mmol) at ambient temperature under rapid stirring. After 30 minutes, the reaction was first concentrated by rotary evaporator to near dryness, then on high vacuum for 48 h to provide amorphous form of (S)—N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide sulfate (11.37 g, 21.59 mmol, 98.43% yield). LCMS (apci m/z 429.1, M+H).
CRYSTALLINE FORM OF (S)-N-(5-((R)-2-(2, 5-DIFLUOROPHENYL)-PYRROLIDIN-1-YL)-PYRAZOLO[1, 5-A]PYRIMIDIN-3-YL)-3-HYDROXYPYRROLIDINE-1-CARBOXAMIDE HYDROGEN SULFATE
CRYSTALLINE FORM OF (S)-N-(5-((R)-2-(2, 5-DIFLUOROPHENYL)-PYRROLIDIN-1-YL)-PYRAZOLO[1, 5-A]PYRIMIDIN-3-YL)-3-HYDROXYPYRROLIDINE-1-CARBOXAMIDE HYDROGEN SULFATE
CRYSTALLINE FORM OF (S)-N-(5-((R)-2-(2, 5-DIFLUOROPHENYL)-PYRROLIDIN-1-YL)-PYRAZOLO[1, 5-A]PYRIMIDIN-3-YL)-3-HYDROXYPYRROLIDINE-1-CARBOXAMIDE HYDROGEN SULFATE
CRYSTALLINE FORM OF (S)-N-(5-((R)-2-(2, 5-DIFLUOROPHENYL)-PYRROLIDIN-1-YL)-PYRAZOLO[1, 5-A]PYRIMIDIN-3-YL)-3-HYDROXYPYRROLIDINE-1-CARBOXAMIDE HYDROGEN SULFATE
STAMFORD, Conn., May 29, 2018 (GLOBE NEWSWIRE) — Loxo Oncology, Inc. (Nasdaq:LOXO), a biopharmaceutical company innovating the development of highly selective medicines for patients with genetically defined cancers, today announced that the U.S. Food and Drug Administration (FDA) has accepted the company’s New Drug Application (NDA) and granted Priority Review for larotrectinib for the treatment of adult and pediatric patients with locally advanced or metastatic solid tumors harboring an NTRK gene fusion. The FDA has set a target action date of November 26, 2018, under the Prescription Drug User Fee Act (PDUFA).
“We are excited the larotrectinib NDA has been accepted by FDA and granted Priority Review status,” said Josh Bilenker, M.D., chief executive officer of Loxo Oncology. “Larotrectinib marks an important shift towards treating cancer based on the tumor’s genetics rather than its site of origin in the body.”
The FDA grants Priority Review for the applications of medicines that, if approved, would provide significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions when compared to standard applications. Larotrectinib has also been granted Breakthrough Therapy Designation, Rare Pediatric Disease Designation and Orphan Drug Designation by the FDA.
Loxo Oncology and Bayer are engaged in a collaboration for the development and commercialization of larotrectinib. Bayer plans to submit a Marketing Authorization Application (MAA) in the European Union in 2018.
About Larotrectinib (LOXO-101)
Larotrectinib is an oral and highly selective investigational tropomyosin receptor kinase (TRK) inhibitor in clinical development for the treatment of patients with cancers that harbor a neurotrophic tyrosine receptor kinase (NTRK) gene fusion. Growing research suggests that the NTRK genes, which encode for TRKs, can become abnormally fused to other genes, resulting in growth signals that can lead to cancer in many sites of the body. In clinical trials, larotrectinib demonstrated anti-tumor activity in patients with tumors harboring NTRK gene fusions, regardless of patient age or tumor type. In an analysis of 55 RECIST-evaluable adult and pediatric patients with NTRK gene fusions, larotrectinib demonstrated a 75 percent centrally-assessed confirmed overall response rate (ORR) and an 80 percent investigator-assessed confirmed ORR, across many different types of solid tumors. The majority of all adverse events were grade 1 or 2.
Larotrectinib has been granted Priority Review, Breakthrough Therapy Designation, Rare Pediatric Disease Designation and Orphan Drug Designation by the U.S. FDA.
In November 2017, Loxo Oncology and Bayer entered into an exclusive global collaboration for the development and commercialization of larotrectinib and LOXO-195, a next-generation TRK inhibitor. Bayer and Loxo Oncologywill jointly develop the two products with Loxo Oncology leading the ongoing clinical studies as well as the filing in the U.S., and Bayer leading ex-U.S. regulatory activities and worldwide commercial activities. In the U.S., Loxo Oncology and Bayer will co-promote the products.
For additional information about the larotrectinib clinical trials, please refer to www.clinicaltrials.gov. Interested patients and physicians can contact the Loxo Oncology Physician and Patient Clinical Trial Hotline at 1-855-NTRK-123 or visit www.loxooncologytrials.com/trk-trials.
About TRK Fusion Cancer
TRK fusion cancer occurs when a neurotrophic tyrosine receptor kinase (NTRK) gene fuses with another unrelated gene, producing an altered tropomyosin receptor kinase (TRK) protein. The altered protein, or TRK fusion protein, is constantly active, triggering a permanent signal cascade. These proteins become the primary driver of the spread and growth of tumors in patients with TRK fusion cancer. TRK fusion cancer is not limited to certain types of cells or tissues and can occur in any part of the body. NTRK gene fusions occur in various adult and pediatric solid tumors with varying prevalence, including appendiceal cancer, breast cancer, cholangiocarcinoma, colorectal cancer, GIST, infantile fibrosarcoma, lung cancer, mammary analogue secretory carcinoma of the salivary gland, melanoma, pancreatic cancer, thyroid cancer, and various sarcomas. It may affect greater than 60 percent of both adult and pediatric patients with certain rare tumor types, such as secretory breast, secretory salivary gland and infantile fibrosarcoma. Only sensitive and specific tests can reliably detect TRK fusion cancer. Next-generation sequencing (NGS) can provide a comprehensive view of genomic alterations across a large number of genes. Fluorescence in situ hybridization (FISH) can also be used to test for TRK fusion cancer, and immunohistochemistry (IHC) can be used to detect the presence of TRK protein
About Loxo Oncology
Loxo Oncology is a biopharmaceutical company innovating the development of highly selective medicines for patients with genetically defined cancers. Our pipeline focuses on cancers that are uniquely dependent on single gene abnormalities, such that a single drug has the potential to treat the cancer with dramatic effect. We believe that the most selective, purpose-built medicines have the highest probability of maximally inhibiting the intended target, with the intention of delivering best-in-class disease control and safety. Our management team seeks out experienced industry partners, world-class scientific advisors and innovative clinical-regulatory approaches to deliver new cancer therapies to patients as quickly and efficiently as possible. For more information, please visit the company’s website at www.loxooncology.com.
larotrectinib
larotrectinib
Treatment for Solid Tumors
FDA Accepts Larotrectinib New Drug Application and Grants Priority Review
WHIPPANY, N.J., May 29, 2018 /PRNewswire/ — Bayer announced today that the U.S. Food and Drug Administration (FDA) has accepted the New Drug Application (NDA) submitted by its collaboration partner Loxo Oncology, Inc. (NASDAQ: LOXO), and granted Priority Review for larotrectinib for the treatment of adult and pediatric patients with locally advanced or metastatic solid tumors harboring a neurotrophic tyrosine receptor kinase (NTRK) gene fusion. The FDA has set a target action date of November 26, 2018, under the Prescription Drug User Fee Act (PDUFA).
NTRK gene fusions are genetic alterations that result in production of tropomyosin receptor kinase (TRK) fusion proteins, and lead to the development of tumor growth. Bayer and Loxo Oncology are jointly developing larotrectinib, which is being studied globally for the treatment of patients across a wide range of cancers that harbor an NTRK gene fusion. Bayer plans to submit a Marketing Authorization Application (MAA) in the European Union in 2018.
“TRK fusion cancer is not limited to any organ or site of the body and occurs in both adults and children,” said Scott Fields, MD, senior vice president and head of oncology development at Bayer’s Pharmaceutical Division. “The Priority Review designation for larotrectinib may help bring this treatment option to patients, facing a high unmet medical need, as soon as possible.”
The FDA grants Priority Review for the applications of medicines that, if approved, would provide significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions when compared to standard applications. Larotrectinib has also been granted Breakthrough Therapy Designation, which is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the medicine may demonstrate substantial improvement over available therapies on a clinically significant endpoint, Rare Pediatric Disease Designation and Orphan Drug Designation by the U.S. FDA.
About Larotrectinib (LOXO-101)
Larotrectinib is an investigational tropomyosin receptor kinase (TRK) inhibitor in clinical development for the treatment of patients with cancers that harbor a neurotrophic tyrosine receptor kinase (NTRK) gene fusion. Growing research suggests that the NTRK genes can become abnormally fused to other genes, producing a TRK fusion protein that can lead to the development of solid tumors across multiple sites of the body.
In November 2017, Bayer and Loxo Oncology entered into an exclusive global collaboration for the development and commercialization of larotrectinib and LOXO-195, a TRK inhibitor in clinical development. Bayer and Loxo Oncology will jointly develop the two products with Loxo Oncology leading the ongoing clinical studies as well as the filing in the U.S., and Bayer leading ex-U.S. regulatory activities and worldwide commercial activities. In the U.S., Bayer and Loxo Oncology will co-promote the products.
For additional information about the larotrectinib clinical trials, please refer to http://www.clinicaltrials.gov or visit http://www.loxooncologytrials.com. Larotrectinib has not been approved by the U.S. Food and Drug Administration, the European Medicines Agency or any other health authority.
About TRK Fusion Cancer
TRK fusion cancer occurs when a neurotrophic tyrosine receptor kinase (NTRK) gene fuses with another unrelated gene, producing an altered tropomyosin receptor kinase (TRK) protein. The altered protein, or TRK fusion protein, becomes active and triggers a signal cascade. These proteins become the primary oncogenic driver of the spread and growth of tumors. NTRK gene fusion has been identified in various adult and pediatric solid tumors with varying frequencies.
About Oncology at Bayer
Bayer is committed to delivering science for a better life by advancing a portfolio of innovative treatments. The oncology franchise at Bayer now includes four oncology products and several other compounds in various stages of clinical development. Together, these products reflect the company’s approach to research, which prioritizes targets and pathways with the potential to impact the way that cancer is treated.
About Bayer
Bayer is a global enterprise with core competencies in the Life Science fields of health care and agriculture. Its products and services are designed to benefit people and improve their quality of life. At the same time, the Group aims to create value through innovation, growth and high earning power. Bayer is committed to the principles of sustainable development and to its social and ethical responsibilities as a corporate citizen. In fiscal 2017, the Group employed around 99,800 people and had sales of EUR 35.0 billion. Capital expenditures amounted to EUR 2.4 billion, R&D expenses to EUR 4.5 billion. For more information, go to http://www.bayer.us.
///////////Larotrectinib, UNII:PF9462I9HX, ларотректиниб , 拉罗替尼 , ARRY-470, LOXO-101, PF9462I9HX, phase 3, Array BioPharma, Loxo Oncology, National Cancer Institute, BAYER, orphan drug designation, breakthrough therapy designation
(2-{[(1-{1-[(2R,3R)-3-[4-(4-Cyanophenyl)-1,3-thiazol-2-yl]-2-(2,5-difluorophenyl)-2-hydroxybutyl]-1H-1,2,4-triazol-4-ium-4-yl}ethoxy)carbonyl](methyl)amino}-3-pyridinyl)methyl N-methylglycinate hydrog en sulfate
FDA 2015, EU 2015, BAL8557-002, BCS CLASS I, RO-0098557 , AK-1820
fast track designation
QIDP
ORPHAN DRUG EU
1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47.
Isavuconazonium is a second-generation triazole antifungal approved on March 6, 2015 by the FDA for the treatment of invasive aspergillosis and invasive mucormycosis, marketed by Astellas under the brand Cresemba. It is the prodrug form of isavuconazole, the active moiety, and it is available in oral and parenteral formulations. Due to low solubility in waterof isavuconazole on its own, the isovuconazonium formulation is favorable as it has high solubility in water and allows for intravenous administration. This formulation also avoids the use of a cyclodextrin vehicle for solubilization required for intravenous administration of other antifungals such as voriconazole and posaconazole, eliminating concerns of nephrotoxicity associated with cyclodextrin. Isovuconazonium has excellent oral bioavailability, predictable pharmacokinetics, and a good safety profile, making it a reasonable alternative to its few other competitors on the market.
Originally developed at Roche, the drug candidate was subsequently acquired by Basilea. In 2010, the product was licensed to Astellas Pharma by Basilea Pharmaceutica for codevelopment and copromotion worldwide, including an option for Japan, for the treatment of fungal infection.
The U.S. Food and Drug Administration today approved Cresemba (isavuconazonium sulfate), a new antifungal drug product used to treat adults with invasive aspergillosis and invasive mucormycosis, rare but serious infections.
Isavuconazole, isavuconazonium, Voriconazole, and Ravuconazole are azole derivatives and known as antifungal drugs for treatment of systemic mycoses as reported in US 5,648,372, US 5,792,781, US 6,300,353 and US 6,812,238. The US patent No. 6,300,353 discloses Isavuconazole and its process. It has chemical name [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5- difluorophenyl)-butan-2-ol;
The Isavuconazonium iodide hydrochloride and Isavuconazonium sulfate can be prepared according to known methods, e.g. pending Indian Patent Applications IN 2424/MUM/2014 and IN 2588/MUM/2014.
Example-1: Preparation of Amorphous Isavuconazole
4-cyano Phenacyl bromide F F N N N OH N S CN Formula-I Formula-III In a round bottomed flask charged ethanol (250 ml), thioamide compound of formula-II (25.0 gm) and 4-cyano phenacyl bromide (18.4 gm) under stirring. The reaction mixture were heated to 70 0C. After completion of reaction the solvent was removed under vacuum distillation and water (250 ml) and Ethyl acetate (350 ml) were added to reaction mass. The reaction mixture was stirred and its pH was adjusted between 7 to 7.5 by 10 % solution of sodium bicarbonate. The layer aqueous layer was discarded and organic layer was washed with saturated sodium chloride solution (100 ml) and concentrated under vacuum to get residue. The residue was suspended in methyl tert-butyl ether (250 ml) and the reaction mixture was heated to at 40°C to make crystals uniform and finally reaction mass is cooled to room temperature filtered and washed with the methyl tert-butyl ether. The product was isolated dried to get pale yellowish solid product. Yield: 26.5 gm HPLC purity: 92.7%
CLIP
March 6, 2015
Release
The U.S. Food and Drug Administration today approved Cresemba (isavuconazonium sulfate), a new antifungal drug product used to treat adults with invasive aspergillosis and invasive mucormycosis, rare but serious infections.
Aspergillosis is a fungal infection caused by Aspergillus species, and mucormycosis is caused by the Mucorales fungi. These infections occur most often in people with weakened immune systems.
Cresemba belongs to a class of drugs called azole antifungal agents, which target the cell wall of a fungus. Cresemba is available in oral and intravenous formulations.
“Today’s approval provides a new treatment option for patients with serious fungal infections and underscores the importance of having available safe and effective antifungal drugs,” said Edward Cox, M.D., M.P.H, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Cresemba is the sixth approved antibacterial or antifungal drug product designated as a Qualified Infectious Disease Product (QIDP). This designation is given to antibacterial or antifungal drug products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act.
As part of its QIDP designation, Cresemba was given priority review, which provides an expedited review of the drug’s application. The QIDP designation also qualifies Cresemba for an additional five years of marketing exclusivity to be added to certain exclusivity periods already provided by the Food, Drug, and Cosmetic Act. As these types of fungal infections are rare, the FDA also granted Cresemba orphan drug designations for invasive aspergillosis and invasive mucormycosis.
The approval of Cresemba to treat invasive aspergillosis was based on a clinical trial involving 516 participants randomly assigned to receive either Cresemba or voriconazole, another drug approved to treat invasive aspergillosis. Cresemba’s approval to treat invasive mucormycosis was based on a single-arm clinical trial involving 37 participants treated with Cresemba and compared with the natural disease progression associated with untreated mucormycosis. Both studies showed Cresemba was safe and effective in treating these serious fungal infections.
The most common side effects associated with Cresemba include nausea, vomiting, diarrhea, headache, abnormal liver blood tests, low potassium levels in the blood (hypokalemia), constipation, shortness of breath (dyspnea), coughing and tissue swelling (peripheral edema). Cresemba may also cause serious side effects including liver problems, infusion reactions and severe allergic and skin reactions.
Cresemba is marketed by Astellas Pharma US, Inc., based in Northbrook, Illinois.
The active substance is isavuconazonium sulfate, a highly water soluble pro-drug of the active triazole isavuconazole. The chemical name of the active substance isavuconazonium sulfate is 1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47. The active substance has the following structure:
The structure of the active substance has been confirmed by elemental analysis, mass spectrometry, UV, IR, 1H-, 13C- and 19F-NMR spectrometry, and single crystal X-ray analysis, all of which support the chemical structure. It appears as a white, amorphous, hygroscopic powder. It is very soluble in water and over the pH range 1-7. It is also very soluble in methanol and sparingly soluble in ethanol. Two pKa values have been found and calculated to be 2.0 and 7.3. Its logPoct/wat calculated by software is 1.31.
Isavuconazonium sulfate has three chiral centres. The stereochemistry of the active substance is introduced by one of the starting materials which is controlled by appropriate specification. The two centres, C7 and C8 in the isavuconazole moiety and in an intermediate of the active substance, have R configuration. The third chiral centre, C29, is not located on isavuconazole moiety and has both the R and S configurations. The nondefined stereo centre at C29 has been found in all batches produced so far to be racemic. Erosion of stereochemical purity has not been observed in the current process. The active substance is a mixture of two epimers of C29.
An enantiomer of drug substance was identified as C7 (S), C8 (S) and C29 (R/S) structure. The control of the stereochemistry of isavuconazonium sulfate is performed by chiral HPLC on the active substance and its two precursors. Subsequent intermediates are also controlled by relevant specification in the corresponding steps. Two crystal forms have been observed by recrystallisation studies. However the manufacturing process as described yields amorphous form only.
Isavuconazonium (Cresemba ) is a water-soluble prodrug of the triazole antifungal isavuconazole (BAL4815), a 14-a-demethylase inhibitor, under development byBasilea Pharmaceutica International Ltd and Astellas Pharma Inc. Isavuconazonium, in both its intravenous and oral formulations, was approved for the treatment of invasive aspergillosis and invasive mucormycosis (formerly termed zygomycosis) in the US in March 2015. Isavuconazonium is under regulatory review in the EU for invasive aspergillosis and mucormycosis. It is also under phase III development worldwide for the treatment of invasive candidiasis and candidaemia. This article summarizes the milestones in the development of isavuconazonium leading to the first approval for invasive spergillosis and mucormycosis.
Introduction
The availability of both an intravenous (IV) and an oral formulation of isavuconazonium (Cresemba ), as a result of its water solubility, rapid hydrolysis to the active entity isavuconazole and very high oral bioavailability, provides maximum flexibility to clinicians for treating seriously ill patients with invasive fungal infections [1]. Both the IV and oral formulations have been approved by the US Food and Drug Administration (FDA) to treat adults with invasive aspergillosis and invasive mucormycosis [2]. The recommended dosages of each formulation are identical, consisting of loading doses of 372 mg (equivalent to 200 mg of isavuconazole) every eight hours for six doses, followed by maintenance therapy with 372 mg administered once daily [3]. The Qualified Infectious Disease Product (QIDP) designation of the drug with priority review status by the FDA isavuconazonium in the US provided and a five year extension of market exclusivity from launch. Owing to the rarity of the approved infections,
isavuconazonium was also granted orphan drug designation by the FDA for these indications [2]. It has also been granted orphan drug and QIDP designation in the US for the treatment of invasive candidiasis [4]. In July 2014, Basilea Pharmaceutica International Ltd submitted a Marketing Authorization Application to the European Medicines Agency (EMA) for isavuconazonium in the treatment of invasive aspergillosis and invasive mucormycosis, indications for which the EMA has granted isavuconazonium orphan designation [5, 6]. Isavuconazonium is under phase III development in many countries worldwide for the treatment of invasive candidiasis and candidaemia.
1.1 Company agreements
In 2010, Basilea Pharmaceutica International Ltd (a spinoff from Roche, founded in 2000) entered into a licence agreement with Astellas Pharma Inc in which the latter would co-develop and co-promote isavuconazonium worldwide, including an option for Japan. In return for milestone payments, Astellas Pharma was granted an exclusive right to commercialize isavuconazonium, while Basilea Pharmaceutica retained an option to co-promote the drug in the US, Canada, major European countries and China [7]. The companies amended their agreement in 2014, making Astellas Pharma responsible for all regulatory filings, commercialization and manufacturing of isavuconazonium in the US and Canada. Basilea Pharmaceutica waived its right to co-promote the product in the US and Canada, in order to assume all rights in the rest of the world [8]. However, Astellas Pharma remains as sponsor of the multinational, phase III ACTIVE trial in patients with invasive candidiasis.
2 Scientific Summary
Isavuconazonium (as the sulphate; BAL 8557) is a prodrug that is rapidly hydrolyzed by esterases (mainly butylcholinesterase) in plasma into the active moiety isavuconazole
(BAL 4815) and an inactive cleavage product (BAL 8728).
References
1. Falci DR, Pasqualotto AC. Profile of isavuconazole and its potential in the treatment of severe invasive fungal infections. Infect Drug Resist. 2013;6:163–74.
On 4 July 2014 orphan designation (EU/3/14/1284) was granted by the European Commission to Basilea Medical Ltd, United Kingdom, for isavuconazonium sulfate for the treatment of invasive aspergillosis.
Update: isavuconazonium sulfate (Cresemba) has been authorised in the EU since 15 October 2015. Cresemba is indicated in adults for the treatment of invasive aspergillosis.
Consideration should be given to official guidance on the appropriate use of antifungal agents.
The active substance is isavuconazonium sulfate, a highly water soluble pro-drug of the active triazole isavuconazole. The chemical name of the active substance isavuconazonium sulfate is 1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47. The active substance has the following structure
It appears as a white, amorphous, hygroscopic powder. It is very soluble in water and over the pH range 1-7. It is also very soluble in methanol and sparingly soluble in ethanol. Two pKa values have been found and calculated to be 2.0 and 7.3. Its logPoct/wat calculated by software is 1.31.
Isavuconazonium sulfate has three chiral centres. The stereochemistry of the active substance is introduced by one of the starting materials which is controlled by appropriate specification. The two centres, C7 and C8 in the isavuconazole moiety and in an intermediate of the active substance, have R configuration. The third chiral centre, C29, is not located on isavuconazole moiety and has both the R and S configurations. The nondefined stereo centre at C29 has been found in all batches produced so far to be racemic. Erosion of stereochemical purity has not been observed in the current process. The active substance is a mixture of two epimers of C29. An enantiomer of drug substance was identified as C7 (S), C8 (S) and C29 (R/S) structure. The control of the stereochemistry of isavuconazonium sulfate is performed by chiral HPLC on the active substance and its two precursors.
The present invention relates to a process for the preparation of stable Isavuconazonium or its salt thereof. In particular of the present invention relates to process for the preparing of isavuconazonium sulfate, Isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide has purity more than 90%. The process is directed to preparation of solid amorphous form of isavuconazonium sulfate, isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide. The present invention process of Isavuconazonium or its salt thereof is industrially feasible, simple and cost effective to manufacture of isavuconazonium sulfate with the higher purity and better yield.
Isavuconazonium sulfate is chemically known l-[[N-methyl-N-3-[(methylamino) acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl)thiazol-2-yl]butyl]-lH-[l,2,4]-triazo-4-ium Sulfate and is structurally represented by formula (I):
Formula I
Isavuconazonium sulfate (BAL8557) is indicated for the treatment of antifungal infection. Isavuconazonium sulfate is a prodrug of Isavuconazole (BAL4815), which is chemically known 4-{2-[(lR,2R)-(2,5-Difluorophenyl)-2-hydroxy-l-methyl-3-(lH-l ,2,4-triazol-l-yl)propyl]-l ,3-thiazol-4-yl}benzonitrile compound of Formula II
Formula II
US Ppatent No. 6,812,238 (referred to herein as ‘238); 7,189,858 (referred to herein as ‘858); 7,459,561 (referred to herein as ‘561) describe Isavuconazonium and its process for the preparation thereof.
The US Pat. ‘238 patent describes the process of preparation of Isavuconazonium chloride hydrochloride.
The US Pat. ‘238 described the process for the Isavuconazonium chloride hydrochloride, involves the condensation of Isavuconazole and [N-methyl-N-3((tert-butoxycarbonyl methylamino) acetoxymethyl) pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester. The prior art reported process require almost 15-16 hours, whereas the present invention process requires only 8-10 hours. Inter alia prior art reported process requires too many step to prepare isavuconazonium sulfate, whereas the present invention process requires fewer steps.
Moreover, the US Pat. ‘238 describes the process for the preparation Isavuconazonium hydrochloride, which may be used as the key intermediate for the synthesis of isavuconazonium sulfate, compound of formula I. There are several drawbacks in the said process, which includes the use of anionic resin to prepare Isavuconazonium chloride hydrochloride, consequently it requires multiple time lyophilization, which makes the said prior art process industrially, not feasible.
The inventors of the present invention surprisingly found that Isavuconazonium or a pharmaceutically acceptable salt thereof in yield and purity could be prepared by using substantially pure intermediates in suitable solvent.
Thus, an object of the present invention is to provide simple, cost effective and industrially feasible processes for manufacture of isavuconazonium sulfate. Inventors of the present invention surprisingly found that isavuconazonium sulfate prepared from isavuconazonium iodide hydrochloride, provides enhanced yield as well as purity.
The process of the present invention is depicted in the following scheme:
Formula I
Formula-IA
The present invention is further illustrated by the following example, which does not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present application.
Isavuconazole (20 g) and [N-methyl-N-3((tert-butoxycarbonylmethylamino)acetoxy methyl)pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester (24.7 g) were dissolved in acetonitrile (200ml). The reaction mixture was stirred to add potassium iodide (9.9 g). The reaction mixture was stirred at 47-50°C for 10-13 hour. The reaction mixture was cooled to room temperature. The reaction mass was filtered through celite bed and washed acetonitrile. Residue was concentrated under reduced pressure to give the crude solid product (47.7 g). The crude product was purified by column chromatography to get its pure iodide form (36.5 g).
Yield: 84.5 %
HPLC Purity: 87%
Mass: m/z 817.4 (M- 1)+
Example-2: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride
l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide (36.5 g) was dissolved in ethyl acetate (600 ml). The reaction mixture was cooled to -5 to 0 °C. The ethyl acetate hydrochloride (150 ml) solution was added to reaction mixture. The reaction mixture was stirred for 4-5 hours at room temperature. The reaction mixture was filtered and obtained solid residue washed with ethyl acetate. The solid dried under vacuum at room temperature for 20-24 hrs to give 32.0 gm solid.
Yield: 93 %
HPLC Purity: 86%
Mass: m/z 717.3 (M-HC1- 1)
Example-3: Preparation of Strong anion exchange resin (Sulfate).
Indion GS-300 was treated with aqueous sulfate anion solution and then washed with DM water. It is directly used for sulfate salt.
Example-4: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium Sulfate
Dissolved 10.0 g l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride in 200 ml deminerahzed water and 30 ml methanol. The solution was cooled to about 0 to 5°C. The strong anion exchange resin (sulfate) was added to the cooled solution. The reaction mixture was stirred to about 60-80 minutes. The reaction was filtered and washed with 50ml of demineralized water and methylene chloride. The aqueous layer was lyophilized to obtain
Crysvita (burosumab-twza) is a fibroblast growth factor 23 (FGF23) blocking antibody.
This drug is indicated for the treatment of X-linked hypophosphatemia with radiological evidence of bone disease in children of 1 year of age and older and adolescents with growing skeletons [4].
Burosumab (INN, trade name Crysvita) known as KRN23 is a human monoclonal antibody designed for the treatment of X-linked hypophosphatemia.[1][2][3] Burosumab was approved by the FDA for its intended purpose, in patients aged 1 year and older, on 17 April 2018.[4] The FDA approval fell under both the breakthrough therapy and orphan drug designations.[4]
This drug was developed by Ultragenyx and is in a collaborative license agreement with Kyowa Hakko Kirin.[5]
Burosumab (KRN23) is an entirely human monoclonal IgG1 antibody that binds excess fibroblast growth factor 23 (FGF23) and has been successfully tested in clinical trials in children with X-linked hypophosphatemic rickets [1].
The U.S. Food and Drug Administration approved Crysvita (burosumab) in April 2018. This is the first drug approved to treat adults and children ages 1 year and older with X-linked hypophosphatemia (XLH), which is a rare, inherited form of rickets. X-linked hypophosphatemia causes low circulating levels of phosphorus in the blood. It causes impaired bone growth and development in children and adolescents and issues with bone mineralization throughout a patient’s life [3].
XLH is a serious disease which affects about 3,000 children and 12,000 adults in the United States. Most children with XLH suffer from bowed or bent legs, short stature, bone pain and severe dental pain. Some adults with this condition suffer from persistent, unrelenting discomfort and complications, such as joint pain, impaired mobility, tooth abscesses and hearing loss [3]
Crysvita is specifically indicated for the treatment of X-linked hypophosphatemia (XLH) in adult and pediatric patients 1 year of age and older.
Crysvita is supplied as a subcutaneous injection. The recommended starting dose for pediatrics is 0.8 mg/kg of body weight, rounded to the nearest 10 mg, administered every two weeks. The minimum starting dose is 10 mg up to a maximum dose of 90 mg. After initiation of treatment with Crysvita, measure fasting serum phosphorus every 4 weeks for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is above the lower limit of the reference range for age and below 5 mg/dL, continue treatment with the same dose. Follow dose adjustment schedule per the drug label. The recommended dose regimen in adults is 1 mg/kg body weight, rounded to the nearest 10 mg up to a maximum dose of 90 mg, administered every four weeks. After initiation of treatment with Crysvita, assess fasting serum phosphorus on a monthly basis, measured 2 weeks post-dose, for the first 3 months of treatment, and thereafter as appropriate. If serum phosphorus is within the normal range, continue with the same dose. See drug label for specific dose adjustments.
Mechanism of Action
Crysvita (burosumab-twza) is a fibroblast growth factor 23 (FGF23) blocking antibody. X-linked hypophosphatemia is caused by excess fibroblast growth factor 23 (FGF23) which suppresses renal tubular phosphate reabsorption and the renal production of 1,25 dihydroxy vitamin D. Burosumab-twza binds to and inhibits the biological activity of FGF23 restoring renal phosphate reabsorption and increasing the serum concentration of 1,25 dihydroxy vitamin D.
Kutilek S: Burosumab: A new drug to treat hypophosphatemic rickets. Sudan J Paediatr. 2017;17(2):71-73. doi: 10.24911/SJP.2017.2.11. [PubMed:29545670]
Kinoshita Y, Fukumoto S: X-linked hypophosphatemia and FGF23-related hypophosphatemic diseases -Prospect for new treatment. Endocr Rev. 2018 Jan 26. pii: 4825438. doi: 10.1210/er.2017-00220. [PubMed:29381780]
FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia [Link]
//////////////Burosumab-twza, Crysvita FDA 2018, BLA 761068, Protein Based Therapies, Monoclonal antibody, mAb, KRN 23, breakthrough therapy, orphan drug designations, Peptide, ブロスマブ

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
















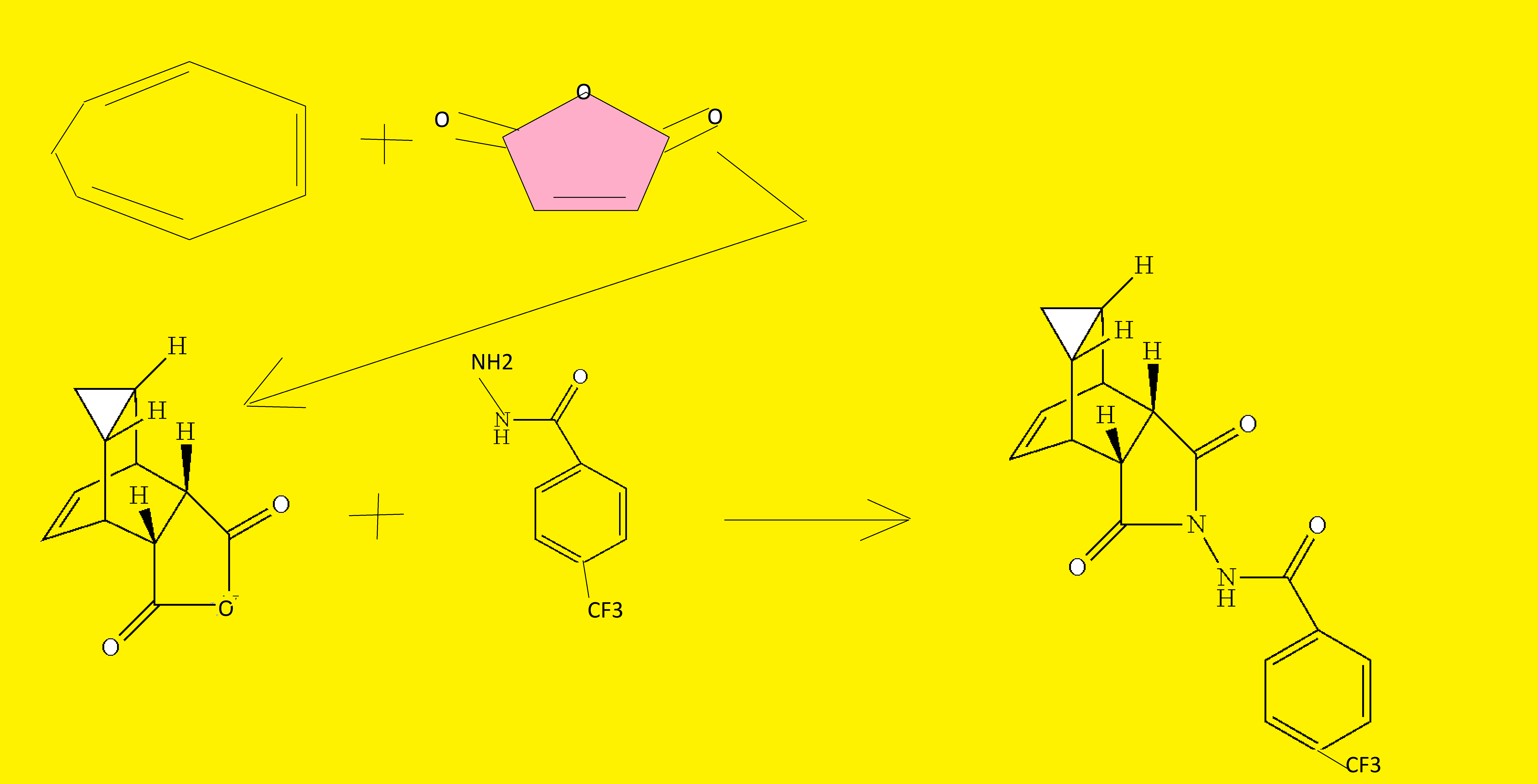




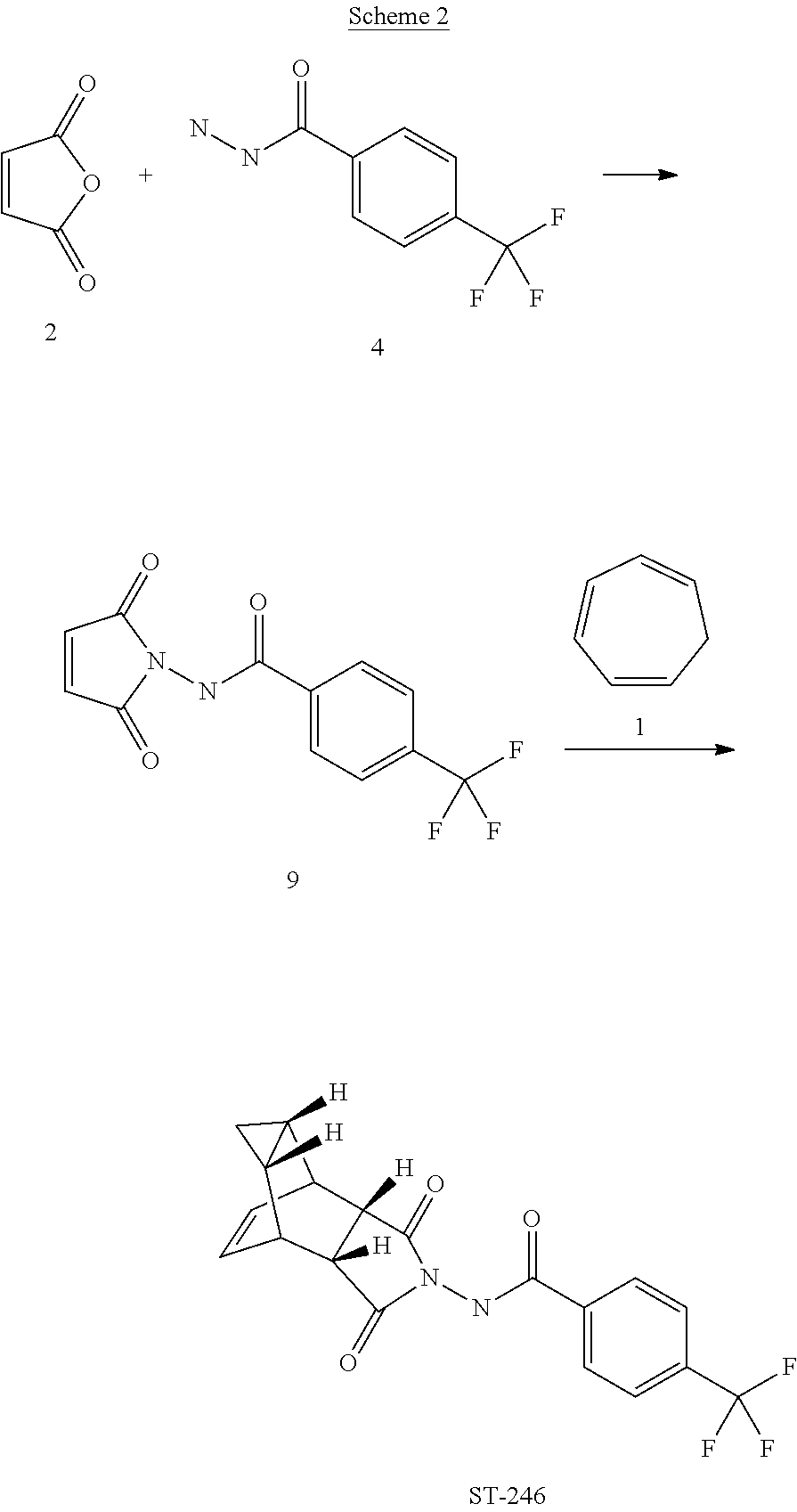
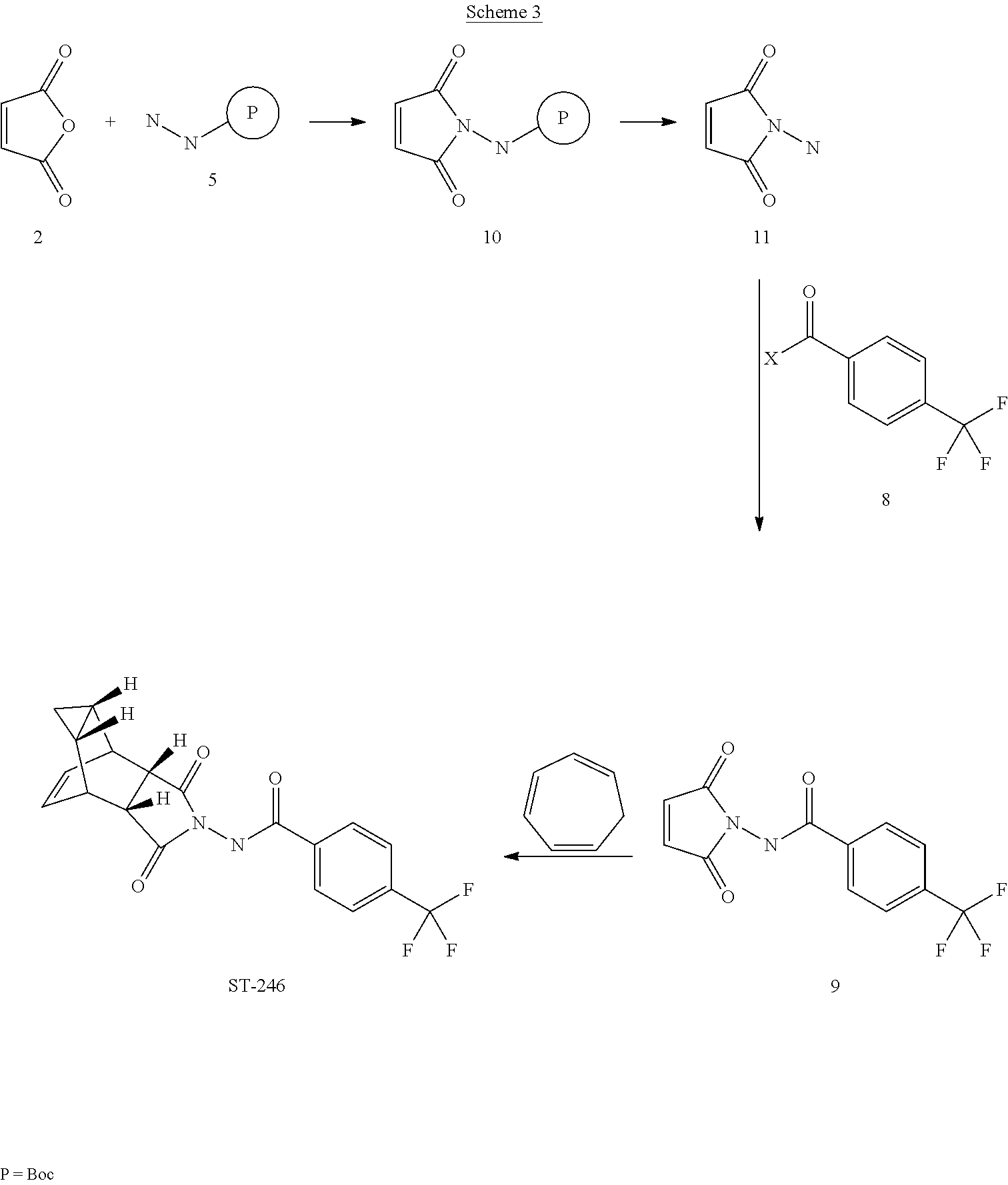
























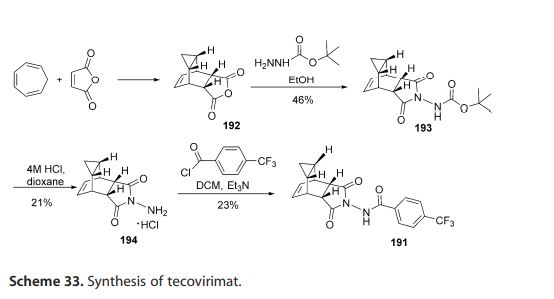
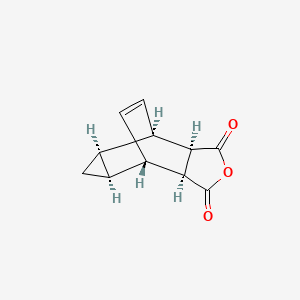























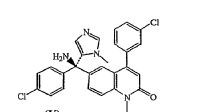

















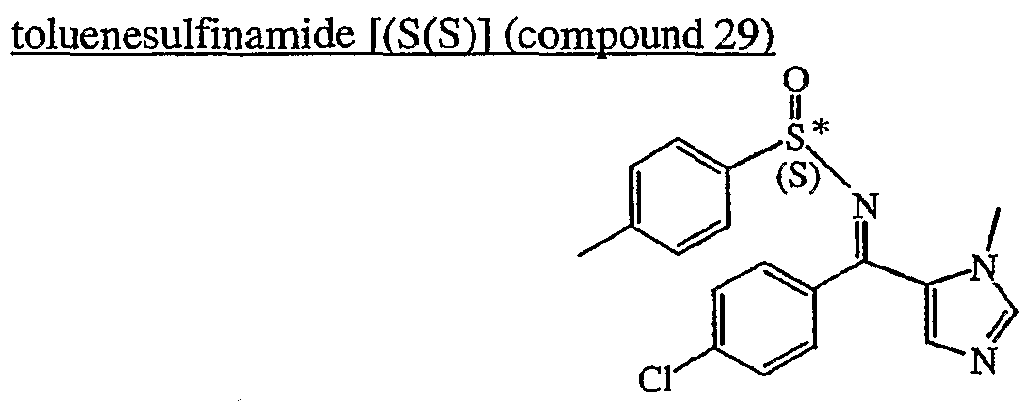



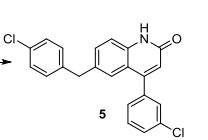
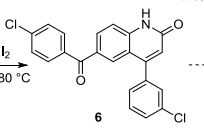



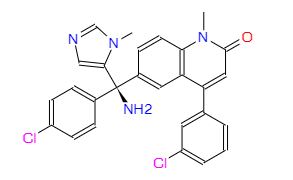

















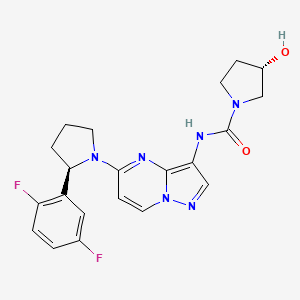

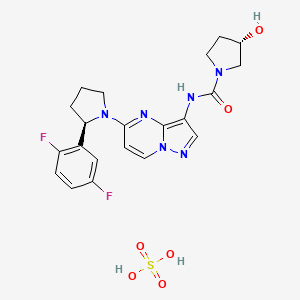





















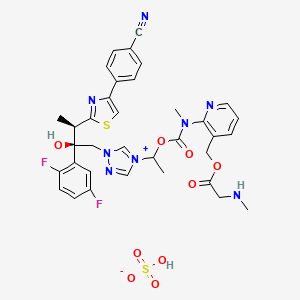





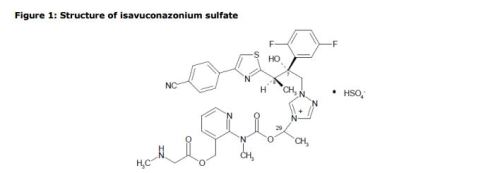

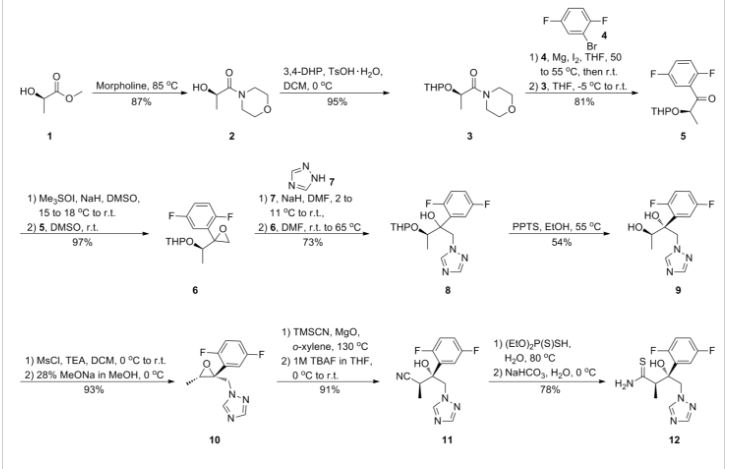
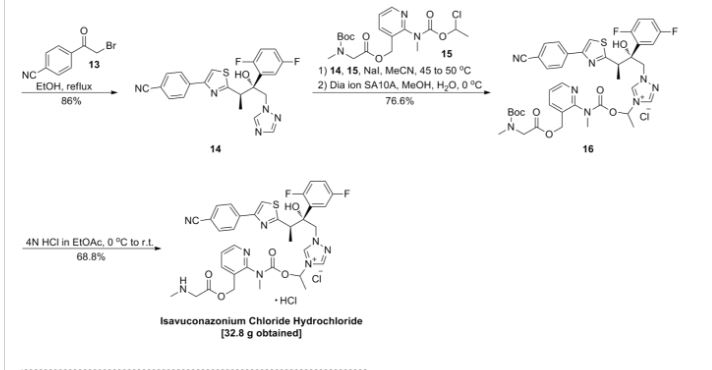
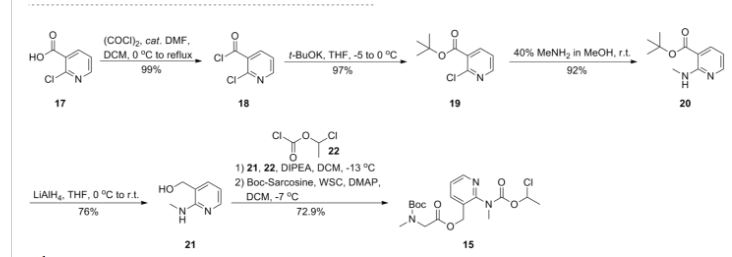




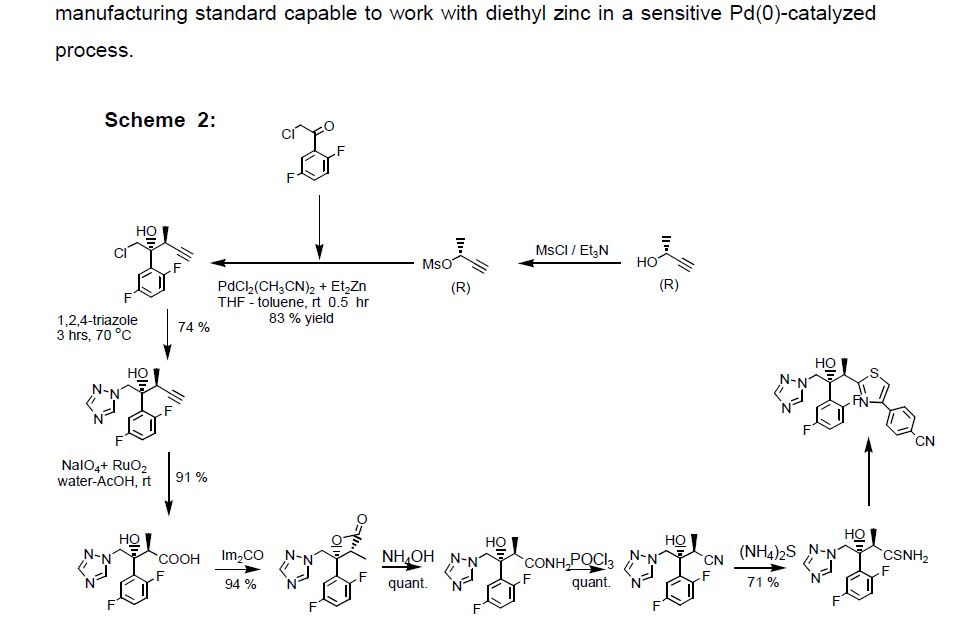


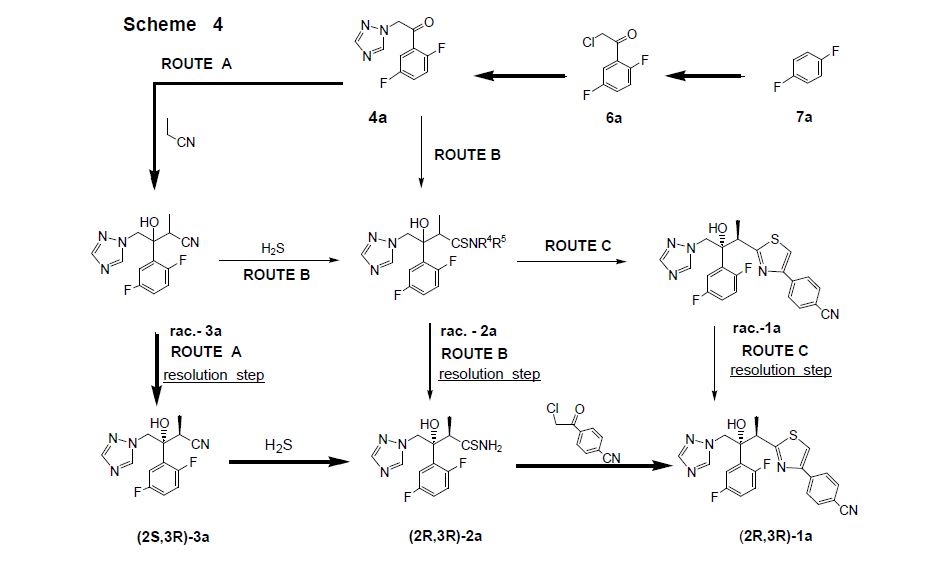
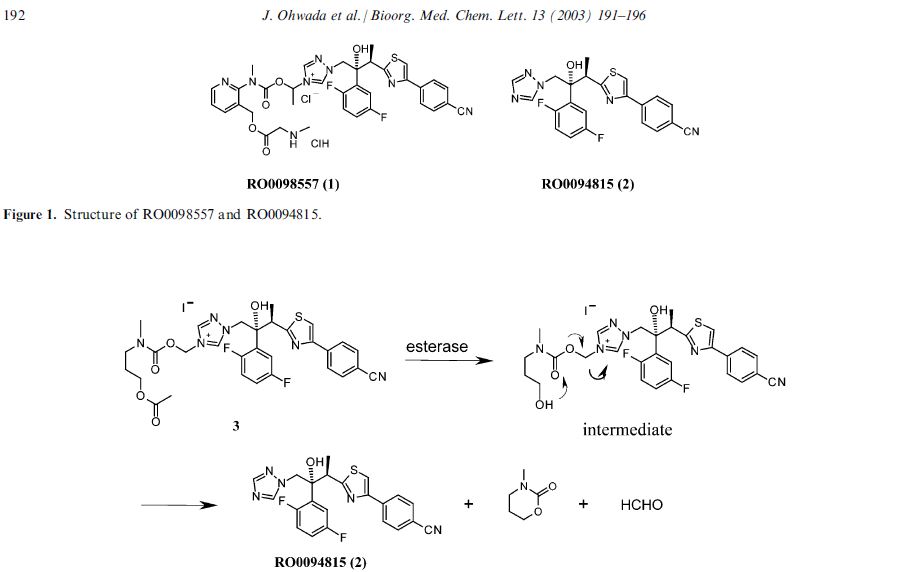
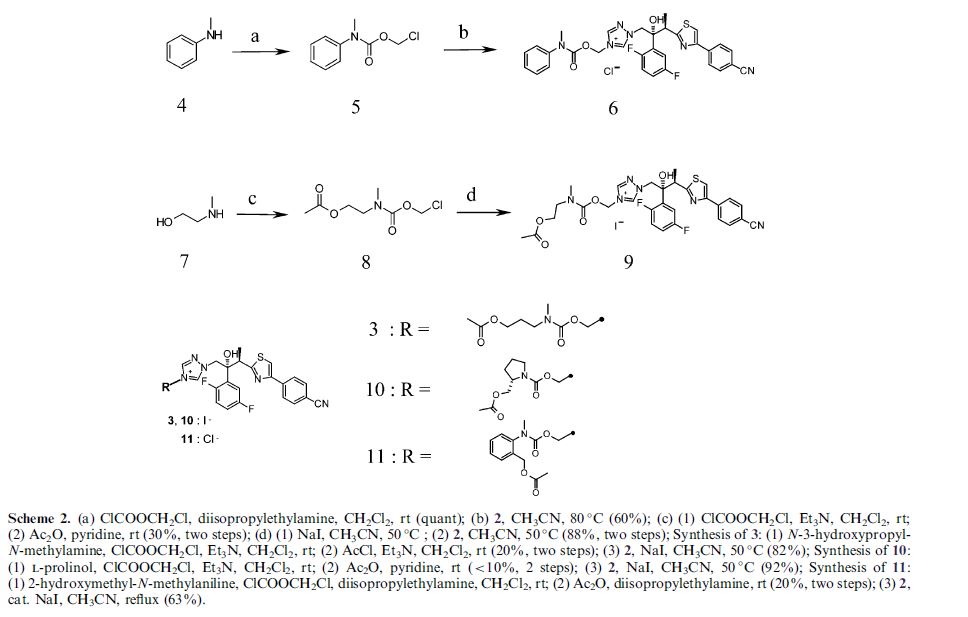

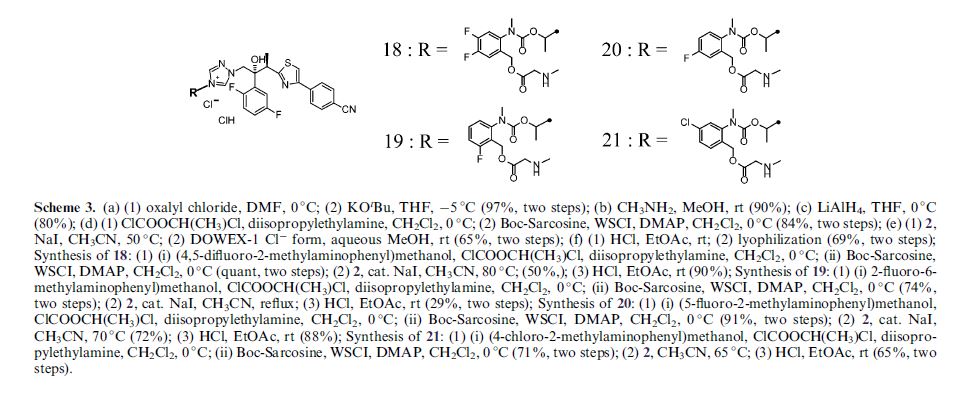



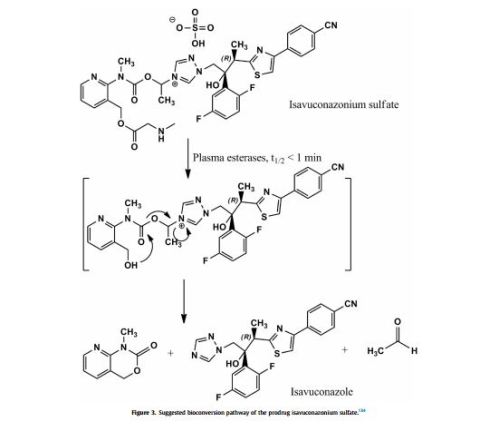








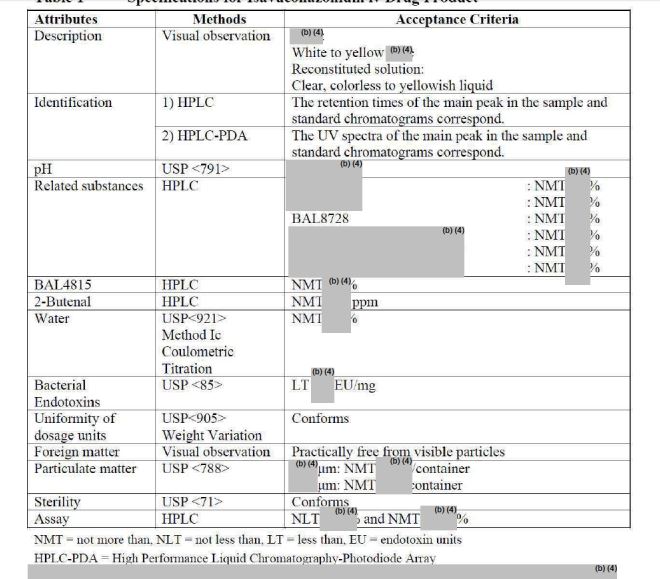
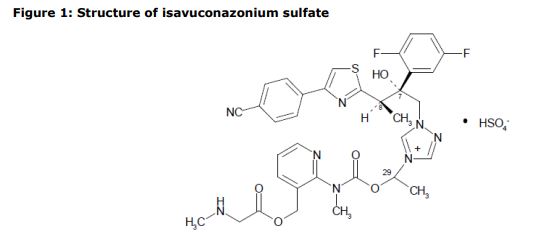
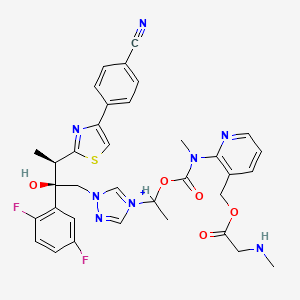









{kind=link}