
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 438)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
OncoGenex Announces Plans for the Initiation of the Borealis-2 Clinical Trial Evaluating OGX-427 in Combination with Second-Line Therapy for Bladder Cancer
An antisense oligonucleotide of the form: GGGACGCGGCGCTCGTCAT

OGX-427 is a second generation antisense drug which, in preclinical experiments, inhibits production of Heat Shock Protein 27 (Hsp27), a cell survival protein found at elevated levels in many human cancers. The development program for OGX-427 aims to demonstrate inhibition of Hsp27 can lead to improved prognosis and treatment outcomes for cancer patients.
OncoGenex Announces Plans for the Initiation of the Borealis-2 Clinical Trial Evaluating OGX-427 in Combination with Second-Line Therapy for Bladder Cancer
Feb. 12, 2013
OncoGenex Pharmaceuticals, Inc. today announced plans for the initiation of the Borealis-2 clinical trial, an investigator-sponsored, randomized, controlled Phase 2 study evaluating OGX-427 in patients with advanced or metastatic bladder cancer who have disease progression following initial platinum-based chemotherapy treatment. The trial, which is the fourth Phase 2 study of OGX-427 in a genitourinary (GU) cancer, will investigate if combining OGX-427 with docetaxel, a standard option in salvage treatment for metastatic bladder cancer, improves survival compared to docetaxel alone.
“Bladder cancer is often sensitive to chemotherapy in the first-line setting, but, when patients relapse, resistance to chemotherapy is frequent,” stated Jonathan Rosenberg MD, Associate Physician, Memorial Sloan-Kettering Cancer Center and one of the primary investigators on the study. “This trial will evaluate the potential of OGX-427 to work synergistically with second- or third-line chemotherapy to overcome treatment resistance and prolong survival in patients with advanced bladder cancer.
“Limited options exist for both the first- and second-line treatment of advanced bladder cancer. Currently, first-line platinum-based chemotherapy regimens result in a median overall survival of approximately 12-15 months. Docetaxel is commonly used in second-line treatment, with a reported median overall survival of approximately six months. Given acquired treatment resistance and these short survival times, there continues to be a high unmet need for additional therapeutic options for this patient population.
OGX-427 is designed to inhibit Heat Shock Protein 27 (HSP27), a cell-survival protein found at elevated levels in many human cancers including prostate, bladder, breast and non-small cell lung cancer. Overexpression of Hsp27 is thought to be an important factor leading to the development of treatment resistance and is associated with negative clinical outcomes in patients with various tumor types.
“The launch of Borealis-2 marks OncoGenex’ continued commitment to expanding the OGX-427 clinical development program to better understand treatment resistance in GU cancers,” said Scott Cormack, President and Chief Executive Officer of OncoGenex. “Given the growing incidence of bladder cancer due to an aging population, we believe there is an urgent need to identify new strategies to address treatment resistance and potentially improve outcomes in this patient population.”
Borealis-2 will be the second randomized, controlled clinical trial of OGX-427 in advanced bladder cancer. The Borealis-1 clinical trial is the OncoGenex-sponsored, randomized, placebo-controlled Phase 2 study designed to evaluate a potential survival benefit, safety and tolerability of combining OGX-427 with gemcitabine and cisplatin in the first-line treatment of patients with advanced bladder cancer. If either Borealis trial shows a survival advantage, OncoGenex plans to initiate conversations with the Food and Drug Administration about the possibility of a Phase 3 study of OGX-427 in bladder cancer as part of the ORCA program.
About Borealis-2
The Borealis-2 clinical trial will randomize approximately 200 patients to receive either OGX-427 plus docetaxel treatment or docetaxel treatment alone. Patients may also continue weekly OGX-427 infusions as maintenance treatment until disease progression or unacceptable toxicity if they complete all 10 planned cycles of docetaxel or are discontinued from docetaxel due to docetaxel toxicity. The primary objective will be overall survival, with secondary objectives evaluating safety, tolerability, tumor response rates and the effect of therapy on Hsp27 levels and circulating tumor cells.Borealis-2 will be conducted at approximately 30 sites in the U.S. and is being sponsored by the Hoosier Oncology Group. Dr. Noah Hahn from the Indiana University Simon Cancer Center, Dr. Toni Choueiri from the Dana-Farber Cancer Institute and Dr. Jonathan Rosenberg from Memorial Sloan-Kettering Cancer Center will serve as the primary investigators on the study.
ABOUT ORCA
The “ORCA” (Overcoming Resistance in CAncer) program encompasses the on-going studies of OGX-427 aiming to demonstrate that inhibition of Hsp27 can lead to improved prognosis and treatment outcomes for cancer patients. Phase 2 clinical trials are underway in prostate and bladder cancers, with additional studies expected to initiate this year. For more information on OGX-427 and ORCA, please visit http://www.oncogenex.com.
Organic India launches single ingredient Moringa products in US

Moringa oleifera
The Drumstick Plant
08 February 2013, Organic India, a manufacturer of herb-based functional supplements, has launched organic single ingredient Moringa products in the US.
Available in both capsule and powder formulations, the product made from powdered leaves of Moringa oleifera tree contains vitamin A, B1, B3, B12, iron, magnesium, potassium, amino acids, and polyphenols and is used for restoring internal imbalances.
Organic India national sales manager Heather Henning said the ancient therapeutic Moringa oleifera plant has been used for years and has seen increasing popularity amongst mainstream consumers worldwide.
Moringa oleifera leaf powder
“Millions of people globally use Moringa for essential nutrition — now, the US distribution channel will have access to this extraordinary plant with USDA organic certification,” Henning added.
The company said Moringa supplement, which has more B12 than steak, more vitamin A than eggs, and more calcium than milk, will be unveiled to the public at Expo West 2013.
_leaves_with_flowers_at_Kolkata_W_IMG_2125.jpg)
Sonjna (Moringa oleifera) leaves with flowers
Moringa oleifera (synonym: Moringa pterygosperma) is the most widely cultivated species of the genus Moringa, which is the only genus in the family Moringaceae. English common names include moringa, and drumstick tree, from the appearance of the long, slender, triangular seed pods, horseradish tree, from the taste of the roots which resembles horseradish, or ben oil tree, from the oil derived from the seeds. The tree itself is rather slender, with drooping branches that grow to approximately 10m in height. In cultivation, it is often cut back annually to 1–2 meters and allowed to regrow so the pods and leaves remain within arm’s reach.[1][2]
In developing countries, moringa has potential to improve nutrition, boost food security, foster rural development, and support sustainable landcare.[3] It may be used as forage forlivestock, a micronutrient liquid, a natural anthelmintic and possible adjuvant.[2][4][5]

The moringa tree is grown mainly in semiarid, tropical, and subtropical areas, corresponding in the United States to USDA hardiness zones 9 and 10. While it grows best in dry, sandy soil, it tolerates poor soil, including coastal areas. It is a fast-growing, drought-resistant tree that is native to the southern foothills of the Himalayas in northwestern India.
Cultivation in Hawai’i, for commercial distribution in the United States, is in its early stages.[6]
“India is the largest producer of moringa, with an annual production of 1.1 to 1.3 million tonnes of tender fruits from an area of 380 km². Among the states, Andhra Pradesh leads in both area and production (156.65 km²) followed by Karnataka (102.8 km²) and Tamil Nadu(74.08 km²). In other states, it occupies an area of 46.13 km². Tamil Nadu is the pioneering state in·so·much as it has varied genotypes from diversified geographical areas and introductions from Sri Lanka.”[7]
Moringa is grown in home gardens and as living fences in Tamil Nadu Southern India and Thailand, where it is commonly sold in local markets.[8] In the Philippines, it is commonly grown for its leaves, which are used in soup.[9] Moringa is also actively cultivated by theWorld Vegetable Center in Taiwan, a center for vegetable research with a mission to reduce poverty and malnutrition in developing countries through improved production and consumption of vegetables. Tamil Nadu Southern India has Moringa in its folk stories and as well considered to be auspicious to grow in home. Interestingly the name in Tamil is Moorungai which sounds same as Moringa.
It is also widely cultivated in Africa, Cambodia, Nepal, Indonesia, Malaysia, Mexico, Central and South America, and Sri Lanka
.jpg)
An Indian drumstick (cut)
| Nutritional value per 100 g (3.5 oz) | |
|---|---|
| Energy | 64 kcal (270 kJ) |
| Carbohydrates | 8.28 g |
| – Dietary fiber | 2.0 g |
| Fat | 1.40 g |
| Protein | 9.40 g |
| Water | 78.66 g |
| Vitamin A equiv. | 378 μg (47%) |
| Thiamine (vit. B1) | 0.257 mg (22%) |
| Riboflavin (vit. B2) | 0.660 mg (55%) |
| Niacin (vit. B3) | 2.220 mg (15%) |
| Pantothenic acid (B5) | 0.125 mg (3%) |
| Vitamin B6 | 1.200 mg (92%) |
| Folate (vit. B9) | 40 μg (10%) |
| Vitamin C | 51.7 mg (62%) |
| Calcium | 185 mg (19%) |
| Iron | 4.00 mg (31%) |
| Magnesium | 147 mg (41%) |
| Manganese | 0.36 mg (17%) |
| Phosphorus | 112 mg (16%) |
| Potassium | 337 mg (7%) |
| Sodium | 9 mg (1%) |
| Zinc | 0.6 mg (6%) |
| Percentages are relative to US recommendations for adults. Source: USDA Nutrient Database |
|
| Nutritional value per 100 g (3.5 oz) | |
|---|---|
| Energy | 37 kcal (150 kJ) |
| Carbohydrates | 8.53 g |
| – Dietary fiber | 3.2 g |
| Fat | 0.20 g |
| Protein | 2.10 g |
| Water | 88.20 g |
| Vitamin A equiv. | 4 μg (1%) |
| Thiamine (vit. B1) | 0.0530 mg (5%) |
| Riboflavin (vit. B2) | 0.074 mg (6%) |
| Niacin (vit. B3) | 0.620 mg (4%) |
| Pantothenic acid (B5) | 0.794 mg (16%) |
| Vitamin B6 | 0.120 mg (9%) |
| Folate (vit. B9) | 44 μg (11%) |
| Vitamin C | 141.0 mg (170%) |
| Calcium | 30 mg (3%) |
| Iron | 0.36 mg (3%) |
| Magnesium | 45 mg (13%) |
| Manganese | 0.259 mg (12%) |
| Phosphorus | 50 mg (7%) |
| Potassium | 461 mg (10%) |
| Sodium | 42 mg (3%) |
| Zinc | 0.45 mg (5%) |
| Percentages are relative to US recommendations for adults. Source: USDA Nutrient Database
|
|

Lee Pharma buys China Rights for Kalbitor (ecallantide – for treatment of Hereditary Angioedema) from Dyax
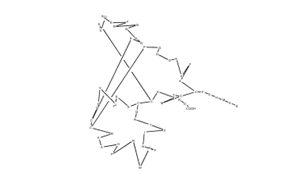

Ecallantide It is an inhibitor of the protein kallikrein and a 60-amino acid polypeptide.
-
Ecallantide
- CAS No.:460738-38-9
- Formula:C305H442N88O91S8
- Molecular Weight:7053.82798
- [Glu20,Ala21,Arg36,Ala38,His39,Pro40,Trp42]tissue factor pathway inhibitor (human)-(20-79)-peptide (modified on reactive bond region Kunitz inhibitor 1 domain containing fragment)
KALBITOR (ecallantide) is a human plasma kallikrein inhibitor for injection for subcutaneous use.
11 FEB 2013
Dyax Corp. a developer of novel biotherapeutics for unmet medical needs, and CVie Therapeutics (CVie), a subsidiary of Lee’s Pharmaceutical Holdings Ltd., announced today a strategic partnership for the development and commercialization of KALBITOR® (ecallantide) in the treatment of hereditary angioedema (HAE) and other angioedema indications in China, Hong Kong and Macau.
KALBITOR is currently marketed in United States for the treatment of acute attacks of HAE in
patients 16 years of age and older. Under the terms of the exclusive license agreement, Dyax will receive an upfront payment and is eligible to receive future development, regulatory and sales milestones. Dyax is also eligible to receive royalty on net product sales. CVie is solely responsible for all costs associated with development, regulatory activities, and the commercialization of KALBITOR in China, Hong Kong
and Macau. Additionally, CVie will purchase drug product from Dyax on a cost-plus basis for
commercial supply.
If approved in China, KALBITOR would become the first novel therapy available for HAE in China, where presently only steroids are used.
KALBITOR (ecallantide injection) is a clear and colorless, sterile, and nonpyrogenic solution. Each vial contains 10 mg ecallantide as the active ingredient, and the following inactive ingredients: 0.76 mg disodium hydrogen orthophosphate (dihydrate), 0.2 mg monopotassium phosphate, 0.2 mg potassium chloride, and 8 mg sodium chloride in water for injection, USP. KALBITOR (ecallantide injection) is preservative free, with a pH of approximately 7.0. A 30 mg dose is supplied as 3 vials each containing 1 mL of 10 mg/mL KALBITOR (ecallantide injection) . Each vial contains a slight overfill. Vials are intended for single use. Ecallantide is a 60-amino-acid protein produced in Pichia pastoris yeast cells by recombinant DNA technology.
The Ecallantide, with the IUPAC name of [Glu20,Ala21,Arg36,Ala38,His39,Pro40,Trp42]tissue factor pathway inhibitor (human)-(20-79)-peptide (modified on reactive bond region Kunitz inhibitor 1 domain containing fragment), is one kind of inhibitor. This chemical’s classification codes are Plasma Kallikrein Inhibitor; Reduction of Blood Loss During Cardiothoracic Surgery (Plasma Kallikrein Inhibitor); Treatment of Hereditary Angioedema. Ecallantide (trade name Kalbitor, investigational name DX-88) is an inhibitor of the protein kallikrein used for hereditary angioedema (HAE) and in the prevention of blood loss in cardiothoracic surgery. If approved for cardiothoracic surgery, it could become a replacement for aprotinin, which was withdrawn in 2007 after being shown to cause complications.
Ecallantide (trade name Kalbitor, investigational name DX-88) is a drug used for the treatment of hereditary angioedema (HAE) and in the prevention of blood loss incardiothoracic surgery.[1] It is an inhibitor of the protein kallikrein and a 60-amino acidpolypeptide which was developed from a Kunitz domain through phage display to mimic antibodies inhibiting kallikrein.[1] On November 27, 2009, ecallantide was approved by theU.S. Food and Drug Administration for the treatment of acute attacks of hereditary angioedema for persons over 16 years of age.[2]
If approved for cardiothoracic surgery, it could become a replacement foraprotinin, which was withdrawn in 2007 after being shown to cause complications.
- Lehmann A (August 2008). “Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery”. Expert Opin Biol Ther 8 (8): 1187–99. doi:10.1517/14712598.8.8.1187.PMID 18613770.
- Waknine, Yael (December 4, 2009). “FDA Approves Ecallantide for Hereditary Angioedema”. Medscape. Retrieved 2009-12-07.
- Dyax Corp. (2009). “Full prescibing information Kalbitor”. Retrieved 2010-05-02.
- Bhoola, K. D.; Figueroa, C. D.; Worthy, K. (1992). “Bioregulation of kinins: Kallikreins, kininogens, and kininases”. Pharmacological reviews 44 (1): 1–80. PMID 1313585. edit
- Stefan Offermanns; Walter Rosenthal (2008). Encyclopedia of Molecular Pharmacology. Springer. pp. 673–. ISBN 978-3-540-38916-3. Retrieved 11 December 2010.

Bayer Submits Riociguat for EU and US regulatory approval to treat treat patients with inoperable chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH)

Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
625115-55-1 CAS NO
Riociguat (BAY 63-2521) is a novel drug that is currently in clinical development by Bayer. It is a stimulator of soluble guanylate cyclase (sGC). At the moment Phase III clinical trialsinvestigate the use of riociguat as a new approach to treat two forms of pulmonary hypertension (PH): chronic thromboembolic pulmonary hypertension (CTEPH) andpulmonary arterial hypertension (PAH). Riociguat constitutes the first drug of a novel class of sGC stimulators.[1]
Sunday, February 10, 2013
In CHEST-1 patients treated with riociguat showed a statistically significant improvement from baseline in the six-minute walking test (6MWT) after 16 weeks, compared to those receiving placebo. The study included both patients with inoperable CTEPH and those with persistent or recurrent disease after a surgical procedure called pulmonary endarterectomy (PEA). The PATENT-1 study met its primary endpoint by demonstrating a statistically significant improvement from baseline in the 6MWT, after 12 weeks compared with placebo. PATENT-1 included both treatment naïve symptomatic PAH patients and those pre-treated with ERAs or non-iv prostanoid monotherapy.

Phase I clinical trials
One of the first studies was designed to test the safety profile, pharmacokinetics andpharmacodynamics of single oral doses of riociguat (0.25–5 mg). 58 healthy male subjects were given riociguat orally (oral solution or immediate-release tablet) in a randomised, placebo-controlled trial. Doses of riociguat were increased stepwise, and riociguat was well tolerated up to 2.5 mg.[7]
Phase II clinical trials
A proof-of-concept study, reported by the University of Gießen Lung Center, was the first small study (in 4 PAH patients) to investigate safety, tolerability, pharmacokinetics and efficacy parameters.[8] The drug was well-tolerated and superior to NO in efficacy and duration.
An open-label, non-controlled phase II trial of riociguat in 75 adult patients (42 with CTEPH and 33 with PAH, all in World Health Organization (WHO) functional class II or III) evaluated the safety and tolerability, and the effects on hemodynamics, exercise capacity and functional class. Riociguat was given three times daily for 12 weeks. Doses were titrated at 2-week intervals from 1.0 mg three times daily to a maximum of 2.5 mg three times daily. Riociguat had a favourable safety profile, and also significantly improved exercise capacity and hemodynamic parameters such as pulmonary vascular resistance, cardiac output and pulmonary arterial pressure compared to baseline values.[9]
In addition, a phase II study of riociguat is underway in patients suffering from other forms of PH such as associated with interstitial lung disease (PH-ILD). First results from this study are expected in 2011.[10]
Phase III clinical trials
The phase III trials on riociguat are multi-center studies. The study program includes large randomized, double-blind, placebo-controlled pivotal trial phase (CHEST-1 and PATENT-1), and open-label extensions of these studies (CHEST-2 and PATENT-2). Details of these studies are reported on ClinicalTrials.gov, a register of studies maintained by the National Institutes of Health (NIH).[6]
- “Background Riociguat”. Bayer HealthCare. Retrieved 15 December 2009.
- Yoshina S, Tanaka A, Kuo SC (March 1978). “Studies on heterocyclic compounds. XXXVI. Synthesis of furo[3,2-c]pyrazole derivatives. (4) Synthesis of 1,3-diphenylfuro[3,2-c]pyrazole-5-carboxaldehyde and its derivatives (author’s transl)” (in Japanese). Yakugaku Zasshi 98 (3): 272–9. PMID 650406.
- Stasch JP, Becker EM, Alonso-Alija C, et al. (March 2001). “NO-independent regulatory site on soluble guanylate cyclase”.Nature 410 (6825): 212–5. doi:10.1038/35065611.PMID 11242081.
- Evgenov OV, Pacher P, Schmidt PM, Haskó G, Schmidt HH, Stasch JP (September 2006). “NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential”. Nature Reviews. Drug Discovery 5 (9): 755–68. doi:10.1038/nrd2038. PMC 2225477.PMID 16955067.
- Mittendorf J, Weigand S, Alonso-Alija C, et al. (May 2009). “Discovery of riociguat (BAY 63-2521): a potent, oral stimulator of soluble guanylate cyclase for the treatment of pulmonary hypertension”. Chemmedchem 4 (5): 853–65.doi:10.1002/cmdc.200900014. PMID 19263460.
- ClinicalTrials.gov: Riociguat
- Frey R, Mück W, Unger S, Artmeier-Brandt U, Weimann G, Wensing G (December 2008). “Pharmacokinetics, pharmacodynamics, tolerability, and safety of the soluble guanylate cyclase activator cinaciguat (BAY 58-2667) in healthy male volunteers”. Journal of Clinical Pharmacology 48 (12): 1400–10. doi:10.1177/0091270008322906.PMID 18779378.
- Grimminger F, Weimann G, Frey R, et al. (April 2009). “First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension”. The European Respiratory Journal 33 (4): 785–92.doi:10.1183/09031936.00039808. PMID 19129292.
- “ATS International conference”. American Thoracic Society. 2009.
- ClinicalTrials.gov NCT00694850 Impact of Multiple Doses of BAY 63-2521 on Safety, Tolerability, Pharmacokinetics and Pharmacodynamics in Patients With Interstitial Lung Disease (ILD) Associated Pulmonary Hypertension
Codon AG submits application for approval for the articular cartilage product chondrosphere
Codon AG submits application for approval for the articular cartilage product chondrosphere
http://clinicaltrials.gov/ct2/show/NCT01222559
Sandoz launches first generic version of Cleocin Phosphate® in Dextrose 5%

Clindamycin rINN (pron.: /klɪndəˈmaɪsɨn/) is a lincosamide antibiotic. It is usually used to treat infections with anaerobic bacteria, but can also be used to treat some protozoal diseases, such as malaria. It is a common topical treatment for acne and can be useful against some methicillin-resistant Staphylococcus aureus (MRSA) infections.[1]
The most severe common adverse effect of clindamycin is Clostridium difficile-associated diarrhea (the most frequent cause of pseudomembranous colitis). Although this side effect occurs with almost all antibiotics, including beta-lactam antibiotics, it is classically linked to clindamycin use.[2]
Clindamycin is marketed under various trade names, including Dalacin, Lincocin (Bangladesh), and Daclin. Combination products include Duac, BenzaClin, Clindoxyl and Acanya (in combination with benzoyl peroxide), and Ziana (with tretinoin). Clindamycin is also available as a generic drug.
Clindamycin is a semisynthetic derivative of lincomycin, a natural antibiotic produced by the actinobacterium Streptomyces lincolnensis. It is obtained by 7(S)-chloro–substitution of the 7(R)-hydroxyl group of lincomycin.[34][35] The synthesis of clindamycin was first announced by BJ Magerlein, RD Birkenmeyer, and F Kagan on the fifth Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) in 1966.[36] It has been on the market since 1968.
- Daum RS (2007). “Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus”. N Engl J Med 357 (4): 380–90. doi:10.1056/NEJMcp070747. PMID 17652653.
- Thomas C, Stevenson M, Riley TV (2003). “Antibiotics and hospital-acquired Clostridium difficile-associated diarrhoea: a systematic review”. J Antimicrob Chemother 51 (6): 1339–50. doi:10.1093/jac/dkg254. PMID 12746372. http://jac.oxfordjournals.org/content/51/6/1339.full.pdf.
- Brook I, Lewis MA, Sándor GK, Jeffcoat M, Samaranayake LP, Vera Rojas J. Clindamycin in dentistry: more than just effective prophylaxis for endocarditis? Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005 ;100:550-8
- “Cleocin I.V. Indications & Dosage”. RxList.com. 2007. http://www.rxlist.com/cgi/generic/clindamyciniv_ids.htm. Retrieved 2007-12-01.
- Darley ES, MacGowan AP (2004). “Antibiotic treatment of gram-positive bone and joint infections”. J Antimicrob Chemother 53 (6): 928–35. doi:10.1093/jac/dkh191. PMID 15117932. http://jac.oxfordjournals.org/content/53/6/928.full.pdf.
- Feldman S, Careccia RE, Barham KL, Hancox J (May 2004). “Diagnosis and treatment of acne”. Am Fam Physician 69 (9): 2123–30. PMID 15152959. http://www.aafp.org/afp/2004/0501/p2123.pdf.
FDA Approves Pomalyst for Advanced Multiple Myeloma – February 8, 2013

Pomalyst (pomalidomide) Capsules
Company: Celgene Corporation
Date of Approval: February 8, 2013
Treatment for: Multiple Myeloma
Pomalyst (pomalidomide) is a thalidomide analogue indicated for the treatment of patients with multiple myeloma.
The U.S. Food and Drug Administration today approved Pomalyst (pomalidomide) to treat patients with multiple myeloma whose disease progressed after being treated with other cancer drugs.
Multiple myeloma is a form of blood cancer that primarily affects older adults and arises from plasma cells in the bone marrow. According to the National Cancer Institute, approximately 21,700 Americans are diagnosed with multiple myeloma and 10,710 die yearly from the disease.![]()
Pomalyst is a pill that modulates the body’s immune system to destroy cancerous cells and inhibit their growth. It is intended for patients who have received at least two prior therapies, including lenalidomide and bortezomib, and whose disease did not respond to treatment and progressed within 60 days of the last treatment (relapsed and refractory).
“Pomalyst is the third drug in a class of immunomodulatory agents that includes lenalidomide and thalidomide, and is the second drug approved in the past year to treat multiple myeloma,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in FDA’s Center for Drug Evaluation and Research. “Treatment for multiple myeloma is tailored to meet individual patient’s needs, and today’s approval provides an additional treatment option for patients who have not responded to other drugs.”
- FDA Approves Pomalyst for Advanced Multiple Myeloma – February 8, 2013
- Celgene Corporation Provides Update on FDA Advisory Committee for Pomalidomide – October 3, 2012
- The International Myeloma Foundation Says Pomalidomide, an Important New Drug for Patients, Has Been Submitted for FDA Approval – April 27, 2012
pomalidomide. 4-Amino-2-(2,6-dioxopiperidin-3-yl)isoindole-1,3-dione
Pomalidomide (INN, originally CC-4047 or 3-amino-thalidomide, marketed as Pomalyst by Celgene), is a derivative of thalidomide that is anti-angiogenic and also acts as an immunomodulator. Pomalidomide was approved on February 8, 2013 by the U.S. Food and Drug Administration (FDA) as a treatment for relapsed and refractory multiple myeloma.[1] An application for approval to treat multiple myeloma also has been submitted by Celgene to the European Medicines Agency, and a decision on that application is expected by the second half of 2013.[1]
Origin and development
The parent compound of pomalidomide, thalidomide, was originally discovered to inhibit angiogenesis in 1994.[2] Based upon this discovery, thalidomide was taken into clinical trials for cancer, leading to its ultimate FDA approval for multiple myeloma. Further structure activity studies done in Dr. Robert D’Amato’s lab at Boston Children’s Hospital led to the first report in 2001[3] that 3-amino-thalidomide was able to directly inhibit both the tumor cell and vascular compartments of myeloma cancers. This dual activity of pomalidomide makes it more efficacious than thalidomide in vitro and in vivo.[4]
Clinical trials
Phase I trial results showed tolerable side effects.[5]
Phase II clinical trials for multiple myeloma and myelofibrosis reported ‘promising results’.[6][7]
Phase III results were reported at ASH in 2012 and showed significant extension of progression-free survival (median 3.6 months vs. 1.8 months; P < 0.001), and overall survival in patients taking pomalidomide and dexamethasone.[8]
- “Pomalyst (Pomalidomide) Approved By FDA For Relapsed And Refractory Multiple Myeloma”. The Myeloma Beacon. Retrieved 2013-02-08.
- D’Amato, Robert J.; Loughnan, Michael S.; Flynn, Evelyn; Folkman, Judah (1994). “Thalidomide is an inhibitor of angiogenesis”. Proceedings of the National Academy of Sciences of the United States of America 91 (9): 4082–5. Bibcode 1994PNAS…91.4082D. doi:10.1073/pnas.91.9.4082. JSTOR 2364596. PMC 43727. PMID 7513432.
- D’Amato, R; Lentzsch, S; Anderson, KC; Rogers, MS (2001). “Mechanism of action of thalidomide and 3-aminothalidomide in multiple myeloma”. Seminars in Oncology 28 (6): 597–601. doi:10.1016/S0093-7754(01)90031-4. PMID 11740816.
- Lentzsch, S; Rogers, MS; Leblanc, R; Birsner, AE; Shah, JH; Treston, AM; Anderson, KC; D’Amato, RJ (2002). “S-3-Amino-phthalimido-glutarimide inhibits angiogenesis and growth of B-cell neoplasias in mice”. Cancer research 62 (8): 2300–5. PMID 11956087.
- Streetly, Matthew J.; Gyertson, Kylie; Daniel, Yvonne; Zeldis, Jerome B.; Kazmi, Majid; Schey, Stephen A. (2008). “Alternate day pomalidomide retains anti-myeloma effect with reduced adverse events and evidence of in vivo immunomodulation”. British Journal of Haematology 141 (1): 41–51. doi:10.1111/j.1365-2141.2008.07013.x. PMID 18324965.
- “Promising Results From 2 Trials Highlighting Pomalidomide Presented At ASH” (Press release). Celgene. December 11, 2008. Retrieved October 28, 2012.
- Tefferi, Ayalew (December 8, 2008). “Pomalidomide Therapy in Anemic Patients with Myelofibrosis: Results from a Phase-2 Randomized Multicenter Study”. 50th ASH Annual Meeting and Exposition. San Francisco. Retrieved October 28, 2012.
- “Phase III Study (MM-003) of Pomalidomide Plus Low-Dose Dexamethasone Demonstrates Significant Progression-Free and Overall Survival Improvement for Patients with Relapsed or Refractory Multiple Myeloma.”. 11 Dec 2012.
- This new drug is specifically indicated for patients who have received at least 2 prior therapies, including lenalidomide (Revlimid, Celgene) and bortezomib (Velcade, Millennium Pharmaceuticals), and whose disease did not respond to treatment and progressed within 60 days of the last treatment.

Warner Chilcott Announces FDA Approval of New Ulcerative Colitis Product Delzicol

Mesalazine (INN, BAN), also known as mesalamine (USAN) or 5-aminosalicylic acid (5-ASA), is an anti-inflammatory drug used to treat inflammatory bowel disease, such as ulcerative colitis[1] and mild-to-moderate Crohn’s disease.[2] Mesalazine is a bowel-specific aminosalicylate drug that acts locally in the gut and has its predominant actions there, thereby having few systemic side effects.[3]
As a derivative of salicylic acid, mesalazine is also thought to be an antioxidant that traps free radicals, which are potentially damaging byproducts of metabolism.[3]
Mesalazine is considered the active moiety of sulfasalazine, which is metabolized to sulfapyridine and mesalazine.[4]
About Delzicol
DUBLIN, Ireland, Feb. 5, 2013 — Warner Chilcott plc today announced that the United States Food and Drug Administration (FDA) has approved its new 400 mg mesalamine product indicated for the treatment of ulcerative colitis. The product will be marketed as Delzicol (mesalamine) 400 mg delayed-release capsules. The Company anticipates that it will commercially launch Delzicol in March 2013.Delzicol (mesalamine) delayed-release capsules are indicated for the treatment of mildly to moderately active ulcerative colitis and for the maintenance of remission of ulcerative colitis. For information on dosage and administration, contraindications, warnings and precautions, adverse reactions, and other important safety information, please see the prescribing information.
Warner Chilcott is a leading specialty pharmaceutical company currently focused on the women’s healthcare, gastroenterology, urology and dermatology segments of the branded pharmaceuticals market, primarily in North America. We are a fully integrated company with internal resources dedicated to the development, manufacture and promotion of our products.
- Kruis, W.; Schreiber, I.; Theuer; Brandes; Schütz; Howaldt; Krakamp; Hämling et al. (2001). “Low dose balsalazide (1.5 g twice daily) and mesalazine (0.5 g three times daily) maintained remission of ulcerative colitis but high dose balsalazide (3.0 g twice daily) was superior in preventing relapses”. Gut 49 (6): 783–789. doi:10.1136/gut.49.6.783. PMC 1728533. PMID 11709512. edit
- Sandborn WJ, Feagan BG, Lichtenstein GR (October 2007). “Medical management of mild to moderate Crohn’s disease: evidence-based treatment algorithms for induction and maintenance of remission”. Alimentary Pharmacology & Therapeutics 26 (7): 987–1003. doi:10.1111/j.1365-2036.2007.03455.x. PMID 17877506. Retrieved 2009-12-20.
- “mesalazine”. PharmGKB.
- Lippencott’s Illustrated Reviews: Pharmacology, 4th Ed. Finkel, Cubeddu and Clark
 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 amcrasto@gmail.com
amcrasto@gmail.com
TAKE A TOUR
KISUMU, KENYA

Kisumu – Wikipedia, the free encyclopedia
Kisumu is a port city in Kisumu County, Kenya 1,131 m (3,711 ft), with a population of 409,928 (2009 census). It is the third largest city in Kenya, the principal city …
Kisumu CountyKisumu County is one of the new devolved counties of Kenya. Its …
|
Kisumu International AirportKisumu International Airport is an airport in Kisumu, Kenya (IATA …
|

Boat riding in Kisumu

Local inhabitants near Kisumu, 1911


Clockwise: Lake Victoria Panorama, Kisumu Panorama, sunset at Oginga Odinga street, Downtown, Kiboko Point, Nighttime in Kisumu and Jomo Kenyatta Stadium.
|
|

Kisumu
|
|
Coordinates:  0°6′S 34°45′E 0°6′S 34°45′E |
|
| Country | |
|---|---|
| County | Kisumu County |

Kisumu panorama, viewed from Lake Victoria

Jomo Kenyatta Stadium

Kisumu Harbour. The green vegetation is water hyacinth

Nairobi University Kisumu Campus





Launch of Ipsen/Braintree’s Eziclen/Izinova in Europe expected by end 2013
Eziclen® / Izinova® a new bowel cleansing preparation, is a sulphate-based formulation (sodium, magnesium and potassium sulphates).
The dose for cleansing the colon will require two administrations of about 0.5 litre / 16 ounces of the product diluted in water, each followed by about 1 litre / 32 ounces, of clear liquids, each.
The drug will be registered under the trade name Eziclen® in the large majority of the concerned EU countries and under the name Izinova® in the other few countries including France & UK
7 feb 2013
French drugmaker Ipsen and US partner Braintree Laboratories yesterday announced today that Eziclen/Izinova (BLI-800) has successfully completed its European decentralized registration procedure involving 16 countries, paving the way for a launch of the product before the end of 2013.
The product will be indicated in adults for bowel cleansing prior to any procedure requiring a clean bowel (eg, bowel visualization including bowel endoscopy and radiology or surgical procedure). Each European Union member states (France [reference member state], Belgium, Czech Republic, Estonia, Germany, Greece, Italy, Latvia, Lithuania, Luxemburg, the Netherlands, Poland, Portugal, Romania, Spain and UK) should now adopt a national decision within 30 days. In practice, the grant of the national marketing authorization may vary from one to several months, Ipsen noted.
Jean Fabre, senior vice president Intercontinental Operations and Franchise “Primary Care” at Ipsen, stated: “The completion of the decentralized procedure for Eziclen/Izinova (BLI-800) is an important step forward to national marketing authorizations in Europe. The availability of Eziclen/Izinova (BLI-800) will provide physicians and patients with a valuable agent for pre-colonoscopy colonic cleansing, particularly in the screening of colorectal cancer. This decision gives also perspectives for our plant in Dreux where Eziclen/Izinova will be manufactured”.
In 2009, Ipsen acquired the exclusive manufacturing, marketing and distribution rights of Braintree’s proprietary formulation BLI-800. The agreement covers countries within the EU, Commonwealth of Independent States, selected Asian countries (including China) and some North African countries. This product was approved by US Food and Drug Administrtion in 2010 and is marketed as Suprep Bowel Prep Kit.
Chugai files Herceptin for post surgical Adjuvant treatment of HER2+ve breast cancer in Japan

Trastuzumab, monoclonal antibody
Thursday, 7 February 2013
Trastuzumab (INN; trade names Herclon, Herceptin) is a monoclonal antibody that interferes with the HER2/neu receptor. Its main use is to treat certain breast cancers.
The HER receptors are proteins that are embedded in the cell membrane and communicate molecular signals from outside the cell (molecules called EGFs) to inside the cell, and turn genes on and off. The HER proteins stimulate cell proliferation. In some cancers, notably certain types of breast cancer, HER2 is over-expressed, and causes cancer cells to reproduce uncontrollably.[1]
The original studies of trastuzumab showed that it improved overall survival in late-stage (metastatic) breast cancer from 20.3 to 25.1 months,[1] but there is controversy over whether trastuzumab is effective in earlier stage cancer.[2]Trastuzumab is also controversial because of its cost, as much as $100,000 per year,[3] and while certain private insurance companies in the U.S. and government health care systems in Canada, England and elsewhere have refused to pay for trastuzumab for certain patients, some companies have since accepted trastuzumab treatment as a covered preventative treatment.[4]
Trastuzumab is also being studied for the treatment of other cancers.[5] It has been used with some success in women with uterine papillary serous carcinomas that overexpress HER2/neu.[6]
References
- Hudis, CA (2007). “Trastuzumab–mechanism of action and use in clinical practice”. N Engl J Med. 357 (1): 39–51.doi:10.1056/NEJMra043186. PMID 17611206. Jul 5;357(1):39-51. Review /article
- 129 Herceptin and early breast cancer: a moment for caution [Editorial]. Lancet 2005;366:1673.
- “Herceptin or Trastuzumab: Efficacy, Side Effects”. Health and Life.
- “At last, Axa pays for Herceptin”. 2006.
- Vecchione L. Novel investigational drugs for gastric cancer.Expert Opin Investig Drugs. 2009 May 26. [Epub ahead of publication]. Review /article.
- Santin AD, Bellone S, Roman JJ, McKenney JK, Pecorelli S. (2008). “Trastuzumab treatment in patients with advanced or recurrent endometrial carcinoma overexpressing HER2/neu”.Int J Gynaecol Obstet 102 (2): 128–31.doi:10.1016/j.ijgo.2008.04.008. PMID 18555254.
