
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
LERIGLITAZONE
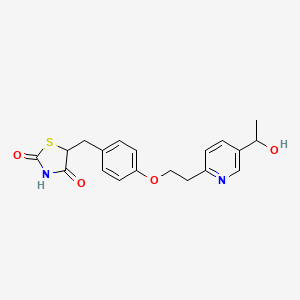
LERIGLITAZONE
C19H20N2O4S,
| MW 372.4 |
Hydroxypioglitazone, CAS 146062-44-4
MIN 102, Hydroxy Pioglitazone (M-IV)лериглитазон [Russian] [INN]ليريغليتازون [Arabic] [INN]乐立格列酮 [Chinese] [INN]
5-[[4-[2-[5-(1-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione
Hydroxypioglitazone is a member of the class of thiazolidenediones that is the hydroxy derivative of pioglitazone. It has a role as a human xenobiotic metabolite. It is a member of thiazolidinediones, a member of pyridines and an aromatic ether. It derives from a pioglitazone.
- OriginatorIDIBELL
- DeveloperMinoryx Therapeutics
- ClassNeuroprotectants; Phenyl ethers; Pyridines; Small molecules; Thiazolidinediones
- Mechanism of ActionPeroxisome proliferator-activated receptor gamma agonists
- Orphan Drug StatusYes – Adrenoleucodystrophy; Friedreich’s ataxia
- Phase II/IIIAdrenoleucodystrophy
- Phase IIFriedreich’s ataxia
- PreclinicalCNS disorders
- 23 Sep 2020Leriglitazone receives Rare Pediatric Disease designation from the US FDA for X-linked adrenoleukodystrophy before September 2020
- 23 Sep 2020Minoryx Therapeutics licenses leriglitazone to Sperogenix Therapeutics in China, Hong Kong and Macau for X-linked adrenoleukodystrophy (X-ALD)
- 14 Sep 2020Minoryx Therapeutics completes the phase II FRAMES trial in Friedreich’s ataxia (In adolescents, In adults) in Spain, Germany, France and Belgium (PO) (NCT03917225)

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Leriglitazone (Hydroxypioglitazone), a metabolite of pioglitazone. Leriglitazone (Hydroxypioglitazone) PioOH is a PPARγ agonist, stabilizes the PPARγ activation function-2 (AF-2) co-activator binding surface and enhances co-activator binding, affording slightly better transcriptional efficacy. Leriglitazone (Hydroxypioglitazone) binds to the PPARγ C-terminal ligand-binding domain (LBD) with Ki of 1.2 μM,induces transcriptional efficacy of the PPARγ (LBD) with EC50 of 680 nM.
Leriglitazone is under investigation in clinical trial NCT03917225 (A Clinical Study to Evaluate the Effect of MIN-102 on the Progression of Friedreich’s Ataxia in Male and Female Patients).
Treatment of X-Linked Adrenoleukodystrophy
PATENT
WO 9218501
WO 9322445
PAPER
Chemical & Pharmaceutical Bulletin (1995), 43(12), 2168-72
https://www.jstage.jst.go.jp/article/cpb1958/43/12/43_12_2168/_article
The metabolites of (±)-5-[p-[2-(5-ethyl-2-pyridyl)ethoxy]benzyl]-2, 4-thiazolidinedione (1, pioglitazone), which is a representative insulin-sensitizing agent, were synthesized to confirm their structures and for studies of their pharmacological properties. Of the metabolites identified, a compound hydroxylated at the 2-position of the ethoxy chain (3) and compounds oxygenated at the ethyl side chain attached to the pyridine ring (4, 5) were found to be active, although the potency was slightly lower than that of the parent compound.




PAPER
Journal of Medicinal Chemistry (1996), 39(26), 5053-5063.
https://pubs.acs.org/doi/10.1021/jm9605694
Pioglitazone (5-(4-(2-(5-ethyl-2-pyridyl)ethoxy)benzyl)-2,4-thiazolidinedione, 2) is a prototypical antidiabetic thiazolidinedione that had been evaluated for possible clinical development. Metabolites 6−9 have been identified after dosing of rats and dogs. Ketone 10 has not yet been identified as a metabolite but has been added to the list as a putative metabolite by analogy to alcohol 6 and ketone 7. We have developed improved syntheses of pioglitazone (2) metabolites 6−9 and the putative metabolite ketone 10. These entities have been compared in the KKAy mouse model of human type-II diabetes to pioglitazone (2). Ketone 10 has proven to be the most potent of these thiazolidinediones in this in vivo assay. When 6−10 were compared in vitro in the 3T3-L1 cell line to 2, for their ability to augment insulin-stimulated lipogenesis, 10 was again the most potent compound with 6, 7, and 9 roughly equivalent to 2. These data suggest that metabolites 6, 7, and 9 are likely to contribute to the pharmacological activity of pioglitazone (2), as had been previously reported for ciglitazone (1).
PATENT
WO 2015150476
Compound 5-[4-[2-(5-(1 -hydroxyethyl)-2-pyridinyl)ethoxy]benzyl]-2,4-thiazolidinedione of formula (1 ) can be prepared according to Scheme 1 (see e.g. J.Med.Chem. 1996, 39(26),5053).
Scheme 1
Scheme 2
Yet another method to prepare mixtures (c) – comprising compound (2) and (4) – and (d) – comprising compounds (3) and (5) – (scheme 3), includes the resolution of the racemic mixture VIII using the already described methods (chiral HPLC separation, enzymatic resolution, chiral resolution, etc) followed by double bond reduction in each of the enantiomers Villa and Vlllb.
Scheme 4
Compounds of formula (2), (3), (4) and (5) may be obtained from mixtures (c) and (d) (Scheme 45) by chiral HPLC separation. Alternatively, the desired enantiomerically pure compounds can be prepared by chiral synthetic procedures known to those skilled in the art (for example: asymmetric hydrogenolysis of the corresponding single isomer of compound VI).
HPLC Method
Column: Symmetry Shield RP-18, 5 μηη (4.6 x 250 mm); wavelength: 210 nm; flow: 1 mL/min; run time: 28 min; mobile phase-gradient: (t/%B): 0/10, 8/10, 12/60, 16/80, 20/80, 24/10, 28/10 [A: Water (potassium dihydrogen o-phosphate (pH~3)), B: Acetonitrile]
A mixture of compounds (2) and (4) (mixture (c)) and a mixture of compounds (3) and (5) (mixture (d)) were prepared according to Scheme 7.
Example 6: Preparation of diastereomeric mixtures D-1 and D-2 of M-IV:
Scheme 1 :
Ent-1 (VIII) Ent-2 (VIII)
Step 3 Step 3
MIV D-1 MIV D-2
Step 1 : Synthesis of compound VIII: HCI (48 ml, 2N) was added to a solution of compound VI (10 g, 0.024 mol) in methanol (200 ml) and the mixture was heated to reflux. After 4 h of reflux, the reaction mixture was cooled to r.t. and concentrated under reduced pressure to afford a yellow solid. The solid was suspended in water (70 ml) and neutralized using a saturated NaHC03 solution. The resulting pale yellow precipitate was collected by filtration and vacuum dried to afford compound VIII (7.5 g; 84% yield).
ES-MS [M+1]+: 371.0.
Step 2: Chiral prep. HPLC
Compound VIII (1 .0 g) was dissolved in a mixture containing equal volumes of acetonitrile, methanol and dichloromethane; injected (150 μΙ injections) in chiral prep-HPLC column (Chiralpak-IA 250 x 20 mm, 5 micron) and separated [Mobile phase- n-Hexane/0.05% Et3N in EtOH (50:50); flow Rate: 18ml/min; run time: 60 min]. The fractions containing the enantiomers Villa and Vlllb were separately concentrated under reduced pressure to minimum volume and the respective residues were diluted with EtOAc (100 ml), followed by water (50 ml). The resultant organic phases were
dried over anhydrous Na2S04 and concentrated to afford compounds Villa and Vlllb as off-white solids. Enantiomers Villa and Vlllb were isolated but the absolute configuration of each enantiomer has not been determined.
Compound Ent-1 (VIII): 250 mg (Yield: 50%); tR (Chiral HPLC) = 14.8 min; ES-MS [M+1]+: 371 .0; 1H NMR (400 MHz, DMSO-d6): δ 12.5 (br S, 1 H), 8.47 (s, 1 H), 7.71 (s, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.53 (d , J = 9.2 Hz, 2H), 7.31 (d, J = 7.6 Hz, 1 H), 7.08 (d, J = 8.8 Hz, 2H), 5.25 (d, J = 4.0 Hz, 1 H), 4.74-4.76 (m, 1 H), 4.43 (dd, J = 6.8, 6.4 Hz, 2H), 3.18 (t, J = 6.4 Hz, 2H), 1.34 (d, J = 6.4 Hz, 3H).
Compound Ent-2 (VIII): 237 mg (Yield: 47%); tR (Chiral HPLC) = 16.7 min; ES-MS [M+1]+: 371 .0; 1H NMR (400 MHz, DMSO-d6): δ 12.5 (br S, 1 H), 8.47 (s, 1 H), 7.71 (s, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.53 (d , J = 8.8 Hz, 2H), 7.31 (d, J = 8.0 Hz, 1 H), 7.08 (d, J = 9.2 Hz, 2H), 5.23 (d, J = 3.6Hz, 1 H), 4.75 (m, 1 H), 4.43 (dd, J = 6.8, 6.4 Hz, 2H), 3.18 (dd, J = 6.8, 6.4 Hz, 2H), 1 .34 (d, J = 6.4 Hz, 3H).
Synthesis of diastereomeric mixtures of M-IV
Synthesis of D-1 MIV
Step 3: A solution of NaBH4 (77 mg, 2.02 mmol) in 0.1 N NaOH (2 ml) was added slowly to a stirred solution of compound Ent-1 (VIII) (250 mg, 0.675 mmol), dimethylglyoxime (32 mg, 0.27 mmol) and CoCI2.6H20 (16 mg, 0.067 mmol) in a mixture of water (10 ml), THF (10 ml) and 1 M NaOH (0.5ml) solution at 10 °C, and the reaction mixture was stirred at r.t. for 1 h. After color of the reaction medium faded, additional quantity of NaBH4 (26 mg, 0.675 mmol) and CoCI2.6H20 (16 mg, 0.067 mmol) were added and stirring was continued at r.t. [additional quantities of CoC|2 and NaBH4 were added at 12 h intervals till the starting material was consumed, as monitored by LCMS]. After 90-96 h, the reaction mixture was neutralized with AcOH (pH~7); diluted with water (10 ml) and extracted in EtOAc (3 χ 50 ml). The combined organic extract was dried over anhydrous Na2S04 and concentrated to afford crude compound which was purified by flash column chromatography (Si02; 4% methanol in CH2CI2) to afford diastereomeric mixture of MIV D-1 (125 mg) as off-white solid.
Synthesis of D-2 MIV
Step 3: A solution of NaBH4 (72 mg, 1 .921 mmol) in 0.1 N NaOH (2 ml) was added slowly to a stirred solution of compound Ent-2 (VIII) (237 mg, 0.64 mmol), dimethylglyoxime (30 mg, 0.256 mmol) and CoCI2.6H20 (15 mg, 0.064 mmol) in a mixture of water (10 ml), THF (10 ml), and 1 M NaOH (0.5ml) solution at 10 °C, and the
reaction mixture was stirred at r.t. for 1 h. After color of the reaction medium faded, additional quantity of NaBH4 (24 mg, 0.64 mmol) and CoCI2.6H20 (15 mg, 0.064 mmol) were added and stirring was continued at r.t. [additional quantities of CoCI2.6H20 and NaBH4 were added at 12 h intervals till the starting material was consumed, as monitored by LCMS]. After 96 h, the reaction mixture was neutralized with AcOH (pH~7); diluted with water (10 ml) and extracted in EtOAc (3 χ 50 ml). The combined organic extract was dried over anhydrous Na2S04 and concentrated to afford crude compound, which was purified by flash column chromatography (Si02; 4% methanol in CH2CI2) to afford diastereomeric mixture of MIV D-2 (100 mg) as off-white solid.
MIV D-1 : yield: 125 mg (50%); tR (Chiral HPLC) = 17.8, 14.7 min; ES-MS [M+1]+: 373.0, 1H NMR (400 MHz, DMSO-d6): δ 12.00 (br s, NH), 8.46 (d, J = 2.0 Hz, 1 H), 7.67 (dd, J = 8.0, 2.4 Hz, 1 H), 7.30 (d, J = 8.0 Hz, 1 H), 7.13 (d, J = 8.8Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.27 (d, J = 4.0 Hz, 1 H), 4.88-4.85 (m, 1 H), 4.76-4.74 (m, 1 H), 4.30 (t, J = 6.8 Hz, 2H), 3.30 (m, 1 H), 3.14 (dd, J = 6.8, 6.4 Hz, 2H), 3.08-3.02 (m, 1 H), 1 .34 (d, J = 6.4 Hz, 3H).
MIV D-2: yield: 100 mg (42%); tR (Chiral HPLC) = 19.4, 16.5 min; ES-MS [M+1]+: 373.0; 1H NMR (400 MHz, DMSO-d6): δ 12.01 (br s, -NH), (d, J = 2.0 Hz, 1 H), 7.67 (dd, J = 8.0, 2.0 Hz, 1 H), 7.31 (d, J = 8.0 Hz, 1 H), 7.13 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.27 (d, J = 4.0 Hz, 1 H), 4.88-4.85 (m, 1 H), 4.76-4.74 (m, 1 H), 4.30 (dd, J = 6.8, 6.4 Hz, 2H), 3.30 (m, 1 H), 3.14 (dd, J = 6.8, 6.4 Hz, 2H), 3.08-3.02 (m, 1 H), 1.34 (d, J = 6.8 Hz, 3H).
Diastereomeric mixtures D-1 and D-2 of MIV correspond to mixtures (c) and (d) described above, but the specific diastereomers present in each diastereomeric mixture have not been assigned.
Example 7: in vitro ADME and toxicological characterization
Protocol: The assays performed include cytochrome P450 inhibition with the different isoforms, microsomal and hepatocyte stability, neurotoxicity in neural cells and hERG safety assays using a patch clamp electrophysiology measurement (FDA Draft Guidance for Industry. Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labelling Recommendations 2012, The European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions Adopted in 2012, Schroeder K et al. 2003 J Biomol Screen 8 (1 ); 50-64, Barter ZE et al. 2007
Curr Drug Metab 8 (1 ); 33-45, LeCluyse EL and Alexandre E 2010 Methods Mol Biol 640; 57-82). The results indicate a safe and favourable ADME profile for the compounds of the invention.
Example 8: The brain plasma ratios of Pioglitazone, MIV, Mill and Mil following oral dosing of a single administration of Pioglitazone at 4.5 mg/kg in male C57BL/6 mice.
The brain-plasma ratio was calculated based on levels of Pioglitazone, MIV, Mill and Mllin plasma and brain quantified at C max (maximal concentration) following oral dosing of a single administration of Pioglitazone at 4.5 mg/kg in male C57BL/6 mice. The percentage brain plasma ratio was 9, 13, 7 and 1 %, respectively, for Pioglitazone, Mil and Mill as shown in the Figure 4. Thus, active metabolites Mill and Mil crossed the BBB at much lower extent than Pioglitazone as it was predicted based on the physicochemical properties of the compounds (see Tablel ). In contrast, unexpectedly metabolite MIV crossed the BBB in a higher percentage than the parent compound Piolgitazone
The calculations of the both indexes (ClogP and QPIogBB) for Pioglitazone and its metabolites Mil and Mill are shown in Table 1 . For both indexes the 2 metabolites are lower than for pioglitazone, suggesting for Mil, and Mill a less favored penetration and distribution within CNS.
TABLE 1
PATENT
WO 2018116281
https://patents.google.com/patent/WO2018116281A1/enPioglitazone is a “dirty” drug which is converted to many metabolites in vivo. The metabolic pathway of pioglitazone after oral administration has been studied in several animal species and in humans and the metabolites have been described in the literature (see e.g. Sohda et al, Chem. Pharm. Bull., 1995, 43(12), 2168-2172) and Maeshiba et al, Arzneim.-Forsch/Drug Res, 1997, 47 (I), 29-35). At least six metabolites have been identified, named M-I to M-VI. Amongst these metabolites, M-II, M-III and M-IV show some pharmacological activity but are less active than Pioglitazone in diabetic preclinical models.
[0005] 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione has the following structure:

[0006] Tanis et al. (J. Med. Chem. 39(26 ):5053-5063 (1996)) describe the synthesis of 5-[[4-[2-[5-( 1 -hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione as follows:Scheme 1

[0007] Tanis et al. describe that the intermediate 14 was obtained in a 27% yield by reacting compound 13 in an aqueous 37% formaldehyde at 170°C for 6 hours. In this process, 5-[[4- [2-[5-( 1 -hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione (compound 6 in Scheme 1) was obtained in a 2.47% overall yield.[0008] WO 2015/150476 Al describes the use of 5-[[4-[2-[5-(l-hydroxyethyl)-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione, and its pharmaceutically acceptable salts, in the treatment of central nervous system (CNS) disorders. WO 2015/150476 Al describes that 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione was prepared according to the process of Tanis et al. (supra) where the intermediate corresponding to compound 14 of Tanis et al. was prepared similarly at 160°C for 5 hours providing a 17% yield. The overall yield of 5-[[4-[2-[5-(l-hydroxyethyl)-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione was about 1.5%.[0009] Due to the low yield of the intermediate 2-[5-(l-methoxymethoxy-ethyl)pyridine-2- yl]ethanol, the process step for preparing this intermediate is critical for the overall yield of the product, 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione. In addition, the prior art process to obtain compound 14 is difficult to scale because the reaction is carried out in a pressure vessel at a very high temperature and it is a very dirty reaction.[0010] Accordingly, the processes described in the art afford the product 5-[[4-[2-[5-(l- hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione only in a very low overall yield and, therefore, they are not suitable for large scale synthesis. In addition, the prior art process employs CH3OCH2CI, a known carcinogen, for protecting the hydroxyl group in the key intermediate. There is a need for an improved process for synthesizing 5- [[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione, and its pharmaceutically acceptable salts.Formula I illustrated by Scheme 2:Scheme 2 r
B


deprotectionoptional saltformation

I (HCI salt)[0255] In another embodiment, the disclosure provides a process for preparing the compound of Formula I illustrated by Scheme 3 : Scheme 3C
Br. e

step ‘< step b step c

step step g

[0256] In another embodiment in Scheme 3, step c, the order of mixing of the reagents can be as follows: 1. n-BuLi, 2. ethylene oxide, and 3. Cul. This order of mixing is described in Example 2.[0257] In the step a, 2,5-dibromopyridine (1) is reacted with i-PrMgCl in THF and then further with acetaldehyde to obtain compound 2. The reaction mixture is preferably filtered over Celite® after the reaction to remove most of the salts. In one embodiment, the addition of acetaldehyde is conducted at a temperature between -15°C and -10°C to control the exothermic reaction. [0258] In the step b, compound 2 is reacted with TBDMS-C1 in the presence of imidazole having DMF as a solvent. The crude product 3 is advantageously purified by a short plug filtration.[0259] In the step c, the hydroxyl protected compound 3 is reacted with ethylene oxide in the presence of n-BuLi and Cu(I)iodide while maintaining the reaction temperature, i.e., the reaction mixture temperature, below -20°C. In one embodiment, the reaction temperature is maintained below -55°C while adding n-BuLi and Cu(I)iodide into the reaction mixture. In another embodiment, the temperature of the reaction mixture is maintained below -55°C while adding n-BuLi, followed by ethylene oxide and then Cu(I)iodide into the reaction mixture. In another embodiment, the temperature of the reaction mixture is maintained below -55°C while adding n-BuLi into the reaction mixture, followed by ethylene oxide. In this embodiment, Cu(I)iodide is added then into the reaction mixture while the reaction mixture temperature is maintained below -20°C, and preferably below -55 °C. The reaction mixture is then allowed to slowly warm to room temperature after the addition of the reagents and stirred at room temperature, e.g., 20-25°C, overnight. This process is described in detail in Example 2. After the reaction, the complexed copper is advantageously removed by washing with 10% ammonia. The crude compound 4 can be purified by column chromatography to give >99% pure product with a yield of about 52%.[0260] The following examples are illustrative, but not limiting, of the methods of the present invention. Suitable modifications and adaptations of the variety of conditions and parameters normally encountered in clinical therapy and which are obvious to those skilled in the art in view of this disclosure are within the spirit and scope of the invention.ExamplesCOMPARATIVE EXAMPLE 1Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]- 2,4-thiazolidinedione (9a) according to the process described in WO 2015/150476 Al Scheme 4

8a 9a[0261] (a) Synthesis of l-(6-methyl-pyridin-3-yl)-ethanol (3a)[0262] LiHMDS (1.0 M in tetrahydrofuran, 463 ml, 0.463 mol) was added drop wise to a cooled solution of methyl 6-methylnicotinate (la) (20 g, 0.132 mol) and ethyl acetate (82 g, 0.927 mol) in dimethylformamide at -50°C; gradually raised the temperature to room temperature and stirred at the same temperature. After 1 h, the reaction mixture was cooled to 0°C; slowly diluted with 20% sulphuric acid and heated to reflux. After 4 h, the reaction mixture was cooled to room temperature, and further to 0°C and basified with potassium carbonate. The reaction medium was diluted with water and extracted in ethyl acetate (3×50 mL). Combined organic extract was dried over sodium sulphate and concentrated to afford crude l-(6-methylpyridin-3-yl)ethan-l-one (2a) (20.0 g) which was taken to the next step without any purification. ES-MS [M+l]+: 136.1.Sodium borohydride (2.3 g, 0.06 mol) was added in small portions over 30 min, to a solution of compound 2a (16.4 g, 0.121 mol) in ethanol (160 mL) at 0°C and the reaction mixture was stirred at same temperature. After 1 h, the reaction mixture was diluted with sodium bicarbonate solution (sat) (2×200 mL) and extracted with dichloromethane (2×500 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford a pale yellow oil, which was purified by flash column chromatography (5% methanol/dichloromethane) to afford compound 3a (17.0 g; 93% yield over 2 steps) as a pale yellow oil. ES-MS [M+l]+: 138.1. 1H NMR (400 MHz, CDC13): δ 8.35 (d, J = 2.0 Hz, 1H), 7.63 (dd, J = 8.0, 2.4 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 4.89 (q, J = 6.5 Hz, 1H), 3.30 (br s, 1H), 2.50 (s, 3H), 1.48 (d, J = 6.5 Hz, 3H).[0263] (b) Synthesis of 5-(l-methoxymethoxy-ethyl)-2-methyl-pyridine (4a):Compound 3a (15 g, 0.109 mol) was added, drop wise, to a cooled suspension of sodium hydride (6.56 g, 0.164 mol) in tetrahydrofurane (150 mL) and stirred at 0°C. After 30 min, chloromethyl methyl ether (13.2 g, 0.164 mol) was added drop wise while stirring and keeping the internal temperature around 0°C. After addition is over, the reaction mixture was stirred at the same temperature for 1 h. The reaction was quenched with ice cold water (80 mL) and extracted with ethyl acetate (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford an orange color oil, which was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 4a (10.0 g; 51% yield) as a pale yellow oil. ES-MS [M+l]+: 182.2. 1H NMR (400 MHz, CDC13): δ 8.45 (d, J = 2.0 Hz, 1H), 7.56 (dd, J = 8.0, 2.0 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 4.75 (q, J = 6.4 Hz, 1H), 4.57 (ABq, 2H), 3.36 (s, 3H), 2.53 (s, 3H), 1.48 (d, J = 6.6 Hz, 3H).[0264] (c) Synthesis of 2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethanol (5a):A mixture of compound 4a (7.0 g, 0.0386 mol) and 37% formaldehyde solution (5.8 g, 0.077 mol) was heated to 160°C in a sealed glass tube for 5 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford a crude compound which was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 5 (1.2 g; 17% yield) as pale yellow oil. ES-MS [M+l]+: 212.1. 1H NMR (400 MHz, CDC13): δ 8.42 (d, J = 2.0 Hz, 1H), 7.65 (dd, J = 8.0, 2.4 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 4.72 (q, J = 6.6 Hz, 1H), 4.65 (t, J = 5.6 Hz, 1H), 4.52 (ABq, 2H), 3.73 (m, 2H), 3.24 (s, 3H), 2.86 (t, J = 7.2 Hz, 2H), 1.49 (d, J = 6.4 Hz, 3H).[0265] The total yield for compound 5a from compound la was 8% molar.[0266] (d) Synthesis of 4-{2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethoxy}- benzaldehyde (6a): Methanesulphonylchloride (1.19 g, 0.01 mol) was added, drop wise, to a cooled suspension of compound 5a (1.7 g, 0.008 mol) and triethylamine (1.79 ml, 0.013 mol) in dichloromefhane (20 mL) at 0°C and stirred at same temperature for 1 h. The reaction mixture was diluted with water (50 mL) and extracted with dichloromethane (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford 2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethyl methanesulfonate (2.04 g; 88% yield) as a yellow oil, which was taken to next step without purification. ES-MS [M+l]+: 290.[0267] 2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethyl methanesulfonate was added (2.3 g, 0.008 mol) to a stirred suspension of 4-hydroxybenzaldehyde (1.65 g, 0.0137 mol) and potassium carbonate (1.86 g, 0.0137 mol) in mixture of toluene (25 mL) and ethanol (25 mL); stirred at 85°C for 5 h. After consumption of the starting materials, the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate (2×100 mL). The combined organic extract was washed with water; dried over anhydrous sodium sulphate and concentrated to afford a crude dark yellow liquid. The crude was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 6a (1.5 g; 60% yield) as pale yellow liquid. ES-MS [M+l]+: 316.1.[0268] (e) Synthesis of 5-(4-{2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethoxy}- benzylidene)-thiazolidine-2,4-dione (7a):Piperidine (80 mg, 0.95 mmol) was added to a solution of compound 6a (0.6 g, 1.9 mmol) and thiazolidine-2,4-dione (0.22 g, 1.9 mmol) in ethanol (15 mL) and the mixture was heated to reflux overnight. After 15 h, the reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford crude mixture, which was purified by flash column chromatography (2% methanol/dichloromethane) to afford compound 7 (500 mg; 64% yield) as a yellow solid. ES-MS [M+l]+: 415.1. 1H NMR (400 MHz, DMSO-d6): δ 12.25 (br s, 1H), 8.47 (d, J = 2.0 Hz, 1H), 7.70 (dd, J = 8.0, 2.0 Hz, 1H), 7.54 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 8.8 Hz, 2H), 4.73 (m, 1H), 4.60-4.40 (m, 4H), 4.22 (t, J = 6.2 Hz, 1H), 3.24 (s, 3H), 3.20 (t, J = 6.8 Hz, 2H), 1.41 (d, J = 6.0 Hz, 3H).[0269] (f) Synthesis of 5-(4-{2-[5-(l-hydroxy-ethyl)-pyridin-2-yl]-ethoxy}-benzyl)- thiazolidine-2,4-dione (9a): [0270] A solution of sodium borohydride (115 mg, 3.017 mmol) in 0.2N sodium hydroxide(1.2 mL) was added slowly to a stirred solution of compound 7 (0.5 g, 1.207 mmol), dimethylglyoxime (42 mg, 0.36 mmol) and C0CI2.6H2O (23 mg, 0.096 mmol) in a mixture of water (6 mL): tetrahydrofurane (6 mL) and 1M sodium hydroxide (1 mL) solution at 10°C and after addition, the reaction mixture was stirred at room temperature. After 1 h, the reaction color lightened and additional quantities of sodium borohydride (46 mg, 1.207 mmol) and C0CI2.6H2O (22 mg, 0.096 mmol) were added and stirring was continued at room temperature. After 12 h, the reaction was neutralized with acetic acid (pH~7); diluted with water (10 mL) and extracted in ethyl acetate (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford crude compound 8a, 5-(4- (2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethoxy)benzyl)thiazolidine-2,4-dione, (0.4 g) as pale yellow semi solid, which was taken to next step without purification. ES-MS [M+l]+: 417.5.[0271] 2N HC1 (2 mL) was added to a solution of compound 8a (0.4 g, 0.96 mmol) in methanol (20 ml) and the mixture was heated to reflux. After 4 h, the reaction mixture was cooled to room temperature and then concentrated under reduced pressure to afford a residue which was dissolved in water and the solution was neutralized using sodium bicarbonate solution (sat). The resulting white precipitate was collected by filtration to afford compound 9a (250 mg; 56% yield over 2 steps) as an off-white solid. ES-MS [M+l]+: 373.4. 1H NMR (400 MHz, DMSO-de): δ 12.00 (br s, -NH), 8.46 (d, J = 2.0 Hz, 1H), 7.66 (dd, J = 8.0, 2.4 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.25 (d, J = 4.4 Hz, 1H), 4.86 (m, 1H), 4.75 (m, 1H), 4.30 (t, J = 6.8 Hz, 2H), 3.30 (m, 1H), 3.14 (t, J = 6.4 Hz, 2H), 3.04 (m, 1H), 1.34 (d, J = 6.4 Hz, 3H).[0272] The overall yield of compound 9a was 1.5% molar.EXAMPLE 2Synthesis of 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol[0273] The synthesis of 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol was conducted according to the Scheme 5 using the reagents and solvents listed in Table 1 below: Scheme 5TBDMS-CI OTBDMS 1 . n-BuLi, <-55°C OTBDMSImidazole
DMF

[0274] The 1H-NMR spectra were recorded with Agilent MercuryPlus 300 NMR spectrometer.[0275] LC-MS data were obtained on an Agilent 1290 series with UV detector and HP 6130MSD mass detector using as column Waters XB ridge BEH XP (2.1 x 50 mm; 2.5 μιτι) and as eluent Ammonium acetate (10 mM); Water/ Methanol/ Acetonitrile.[0276] (a) l-(6-bromopyridin-3-yl)ethan-l-ol (2)[0277] A 20 L vessel was placed under nitrogen atmosphere and charged with tetrahydrofuran (5.5 L) and 2,5-dibromopyridine (1) (2000 g, 8.44 mol, 1.0 eq) (OxChem Corporation). The mixture was cooled to -10°C and isopropyl magnesium chloride (20% in THF, 6.02 L, 11.82 mol, 1.4 eq) (Rockwood Lithium) was added slowly over 1 h, keeping the reaction temperature below 5°C. After addition, the cooling bath was removed and the temperature was kept below 30°C (some additional cooling was needed to achieve this) and the reaction mixture was stirred overnight. After 16 h, a sample was taken; quenched with saturated aqueous ammonium chloride and extracted with methyl tert-buty\ ether (TBME). The TBME was evaporated under vacuum. 1H-NMR in deuterated chloroform showed complete conversion.[0278] The reaction mixture was cooled to -15°C and a solution of acetaldehyde (472 g,10.72 mol, 1.27 eq) (Acros) in tetrahydrofuran (200 mL) was added dropwise, while keeping temperature below -10°C. After the addition was complete, the cooling bath was removed and the temperature was allowed to rise to maximum of 5-8°C. After 1.5 h, a sample was taken and the reaction was quenched with aqueous ammonium chloride as described above. 1H-NMR showed the reaction was complete.[0279] Two batches were combined for work up.[0280] The reaction mixture was quenched by pouring the mixture into a solution of aqueous ammonium chloride (1 kg in 5 L water) and stirred for 15 min, filtered over Celite and rinsed thoroughly with toluene. The filtrate was transferred to a separation funnel and the obtained two layers system was separated. The aqueous layer was extracted with toluene (2 L). The combined organic layers were dried over sodium sulfate and filtered. Evaporation of the filtrate to dryness under vacuum yielded 3.49 kg (99%) of the desired crude material. XH NMR (300 MHz, CDC13): δ 8.30 (d, J = 2.5 Hz, 1H), 7.59 (dd, J = 8.0, 2.5 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 4,91 (q, J = 6.5 Hz, 1H), 1.49 (d, J = 6.5 Hz, 3H).[0281] (b) 2-bromo-5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridine (3)[0282] A 50 L reactor under nitrogen atmosphere was charged with compound 2 (10.0 kg, around 49.5 mol) and DMF (16 L). The mixture was cooled to 10°C and imidazole (6.74 kg, 99 mol, 2.0 eq) (Apollo Scientific Ltd.) was added portion wise within 30 min. The mixture was cooled to 0°C and TBDMS-Cl (7.46 kg, 49.5 mol, 1.0 eq) (Fluorochem) was added portion wise within 5 h, keeping the temperature below 3°C. The mixture reaction temperature was allowed to reach room temperature and stirred overnig ht. H NMR of a sample showed complete conversion.[0283] The reaction mixture was transferred to a 100 L extraction-vessel and the product was extracted with heptane (2×7.5 L, 10 L). The combined heptane-layers were washed with water (2×6 L, 3 L) to remove small amounts of DMF, dried over sodium sulfate and evaporated under vacuum to give crude compound 3 (15.5 kg, 49.0 mol) in a 99.0% yield. This crude product was purified by a short plug filtration, using 10 kg silica/heptane and eluted with heptane (approx. 50 L). The product-fractions were combined and evaporated under vacuum to give 12.0 kg of purified compound 3 (38 mol) as a brown oil in a 76.8% molar yield. (Average yield for 3 experiments was 78%). HPLC-MS: Rt= 2.6 min, M+l=316.1 and 318.1; 1H NMR (300 MHz, CDC13): δ 8.55 (d, J = 2.2 Hz, 1H), 7.54 (dd, J = 8.2, 2.2 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 4,86 (q, J = 6.5 Hz, 1H), 1.40 (d, J = 6.5 Hz, 3H), 0.88 (s, 9H), 0.02 (d, J = 26 Hz, 2x3H).[0284] (c) 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol (4)[0285] The ethylene oxide solution in diethylether was prepared in advance. Diethylether(1.2 L) in a 3 L three-necked flask was cooled at -65 °C and ethylene oxide (462.3 g, 10.5 mol, 1.06 eq) (Linde) was added and stirred at -70°C. Alternatively, the ethylene oxide solution can be made at about -20°C and then added gradually to the reaction mixture having a temperature at about -60°C. [0286] To a solution of 2-bromo-5-(l-((ieri-butyldimethylsilyl)oxy)ethyl)pyridine (3) (3.13 kg, 9.90 mol, 1.0 eq) in diethylether (7.5 L) cooled at -59°C, n-butyllithium (4 L, 10.0 mol, 2.5M in hexanes, 1.01 eq) (Aldrich Chemistry) was added while keeping temperature between -58°C and -62°C. After addition, the mixture was stirred for 1 h while keeping temperature between -60°C and -68°C. The upfront prepared ethylene oxide solution was added at once to the reaction mixture, while temperature was around -62°C. Subsequently, copper(I) iodide (962.3 g, 5.05 mol, 0.51 eq) (Acros Organics) was added in portions of 120 g, every 10 min, keeping the temperature between -61°C and -63°C. Stirring was continued for 1 h after addition keeping temperature between -61°C and -63°C. The cooling bath was removed and allowing the temperature to rise to about 15°C and further to 25 °C with a water bath overnight.[0287] Workup: The reaction-mixture was poured into a solution of 1 kg ammonium- chloride in 5 L water and stirred for 30 min, then the layers were separated. The organic layer was washed with aqueous ammonium hydroxide (10%, 2.5 L, 4x) to remove Cu-complex (blue color disappeared). The combined organic layers were dried over sodium sulfate and evaporated to give 3.12 kg (max. 9.90 mol) crude compound 4 as a brown oil. The crude compound was purified over 20 kg silica (heptane/EtOAc) by eluting with 80 L heptane/EtOAc, 20 L EtOAc, 25 L EtOAc/MeOH 95/5, 25 L EtOAc/MeOH 9/1 and 10 L EtOAc/MeOH 8/2, to give 1.47 kg of purified compound 4 (5.22 mol) as a brown oil (with tendency to solidify) in a 52.7% average molar yield (HPLC-purity of 99.5%). (Average yield over 12 experiments 52%). HPLC-MS: Rt= 2.3 min, M+l=282.1; 1H NMR (300 MHz, CDC13): δ 8.42 (d, J = 2.1 Hz, 1H), 7.61 (dd, J = 8.3, 2.1 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 4,88 (q, J = 7.0 Hz, 1H), 4.01 (t, J=6.0 Hz, 2 H), 3.00 (t, J=6.0 Hz, 2 H), 1.41 (d, J =7.0 Hz, 3H), 0.90 (s, 9H), 0.02 (d, J = 26 Hz, 2x3H).[0288] Another 2.5% of the product was isolated by re -purifying impure product fraction.The total yield of compound 4 from compound 1 was 39.6% molar.EXAMPLE 3Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]- 2,4-thiazolidinedione hydrochloride (9) 2. Sodium bisulfiteethanol/water mixture
3. Addition 10% aqueous sodium hydroxide solution

until pH 12

from step e dimethylglyoxime7step g step f

step h[0289] The 1H-NMR spectra were recorded with a 400 MHz Avance Bruker NMR spectrometer. LC-MS data were obtained on a Agilent Technologies 6130 Quadrapole LC/MS using as column Agilent XDB-C18 and as eluent 0.1% formic acid (aq) and 0.05% formic acid in acetonitrile.[0290] Steps d and e: Synthesis of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl] ethoxy] -benzaldehyde (6)[0291] To a well stirred solution of 5-[[[(l,l-dimethylethyl)dimethylsilyl]-oxy]ethyl]-2- pyridineethanol (4) (obtained as described in Example 2) (1.91 kg) in toluene (8.6 L) at 5°C were added sodium hydroxide (30% aqueous, 2.79 L) and tetrabutylammonium bromide (7.2 g). p-Toluenesulfonyl chloride (1.62 kg) was next added in portions during 5 min. After the addition, the reaction mixture was allowed to reach room temperature in 0.5 h and stirred at this temperature for 18 h. Water (7.3 L) was then added and the mixture was mixed well. Once the solids were dissolved, the layers were allowed to settle and the organic layer was separated. This organic phase was washed with water (5.7 L, 2x), followed by washing with a solution of sodium chloride (57 g) in water (5.7 L). The solvents were concentrated at reduced pressure to an amount of 2.5 kg of a brown oil (compound 5).[0292] To this well stirred brown oil were added subsequently ethanol (7.8 L), water (0.86L), 4-hydroxybenzaldehyde (0.88 kg) and potassium carbonate (1.17 kg) and then the mixture was heated at 75 °C for 18 h. Then, the solvent was evaporated while adding toluene (7.7 L) during 6 h and then the reaction mixture was allowed to cool. At 30°C, water (7.6 L) was added, stirred until all solids were dissolved and the mixture was cooled to room temperature. The layers were allowed to settle and separated. The organic layer was washed with water (7.6 L). The first aqueous extract was extracted with toluene (2.8 L) and this organic extract was used to also extract the aqueous washing. The organic extracts were combined and concentrated under vacuum to give 3.49 kg of a black oil (crude title compound 6).[0293] 1.73 kg of this black oil was dissolved in ethanol (0.74 L) and added to a well stirred solution of sodium bisulfite (1.36 kg) in a mixture of water (3.27 L) and ethanol (0.74 L). The container of the black oil was rinsed with ethanol (0.37 L) twice and these two rinses were also added to the bisulfite reaction mixture. After 75 min, heptane (5.3 L) was added, well mixed for 5 min, and the layers were allowed to settle and separated. To the organic layer was added a solution of sodium bisulfite (0.55 kg) in water (2.65 L), and ethanol (1.06 L). After stirring for 30 min, the layers were allowed to settle and separated. The two bisulfide aqueous extracts were combined and flasks rinsed with water (2.12 L). Next, toluene (4.5 L) and heptane (4.5 L) were added, the mixture was well stirred and the pH was adjusted to 12 using sodium hydroxide (10% aq) (temperature became 32°C). After stirring for an additional 5 min, the layers were allowed to settle and separated at 30°C. The aqueous layer was extracted with a mixture of toluene (1.5 L) and heptane (3.0 L). The layers were separated and the organic layers were combined. The combined organic layers were washed with water (5 L, 2x) and concentrated under vacuum to give the purified title compound 6. This procedure was repeated with another 1.73 kg of the black oil (crude title compound 6) to give in total 2.77 kg of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl]ethoxy]-benzaldehyde (6) as brown oil which contained 24% m/m of toluene according to 1H NMR (yield = 80%, calculated from compound 4 and corrected for residual toluene). [0294] 1H NMR (CDC13) δ: 0.00 (s, 3H), 0.09 (s, 3H), 0.91 (s, 9H), 1.44 (d, = 6 Hz, 3H),3.30 (t, = 7 Hz, 2H), 4.47 (t, = 7 Hz, 2H), 4.92 (q, = 6 Hz, 1H), 6.99 – 7.30 (m, 3H), 7.62- 7.67 (m, 1H), 7.80 – 7.85 (m, 2H), 8.5- 8.54 (m, 1H) and 9.88 (s, 1H).[0295] LC-MS; rt 7.5 min: ES: M+ 387, 386.[0296] Step f: Synthesis of (5Z)-5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methylene]-2,4-thiazolidinedione (7)[0297] A solution of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]- ethoxy]-benzaldehyde (6) (2.75 kg, containing 24% m/m of toluene) and piperidine (6.0 g) in methanol (3.16 L) was concentrated at 40°C under reduced pressure. The residue was dissolved in methanol (10.4 L) and 2,4-thiazolidinedione (759 g) and piperidine (230 g) were added. The mixture was heated at 47°C. After 25 h, the reaction mixture was allowed to cool to room temperature. The mixture was kept at pH 5-6 by adjusting it with acetic acid, if necessary. After a night at room temperature, water (1.56 L) was added and the suspension was stirred at room temperature for additional 2 h. The solids were isolated by filtration, washed with methanol (1 L, 2x) and dried under vacuum to give crude compound 7 (1.65 kg). The crude compound was mixed with methanol (10 L) and dichloromethane (8.6 L) and heated at 32°C until all solids dissolved. Then, the solvents were removed by distillation until the temperature of the mixture reached 34°C at a pressure of 333 mbar. Then, it was allowed to cool to room temperature overnight and stirred at 2°C for additional 2 h. The solids were isolated by filtration, washed with methanol (0.5 L, 2x) and dried under vacuum to give title compound 7 (1.50 kg) (yield = 61%).[0298] 1H NMR (CDCI3) δ 0.00 (s, 3H), 0.08 (s, 3H), 0.90 (s, 9H), 1.43 (d, = 6 Hz, 3H),3.32 (t, = 7 Hz, 2H), 4.48 (t, = 7 Hz, 2H), 4.92 (q, = 6 Hz, 1H), 6.95 – 7.00 (m, 2H), 7.24 – 7,28 (m, 1H), 7.38 – 7.42 (m, 2H), 7.67 (s, 1H), 7.69 – 7.73 (m, 1H) and 8.48 (d, = 3 Hz, 1H).[0299] LC-MS; rt 7.5 min: ES: M+ 487, 486, 485.[0300] Step g: Synthesis of 5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione (8)[0301] To a stirred suspension of (5Z)-5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]- ethyl]-2-pyridinyl]ethoxy]phenyl]methylene]-2,4-thiazolidinedione (7) (10 g) in THF (10 mL) and sodium hydroxide (IN aq, 21 mL) was added of a solution of cobalt chloride (26 mg) and of dimethylglyoxime (930 mg) in THF (2.3 mL) and water (1.0 mL). Then the suspension was put under a nitrogen atmosphere by applying the sequence of vacuum and flushing with nitrogen (4x). Thereafter, the suspension was heated to 30°C. Then, a stock solution of sodium borohydride was prepared by dissolving sodium borohydride (2.7 g) in a mixture of water (15.8 mL) and a solution of sodium hydroxide (1 N aq, 3.5 mL), which was put under a nitrogen atmosphere by applying a sequence of vacuum and flushing with nitrogen (3x). This was added to the suspension of compound 7 at a rate of 4.5 mL/h. Simultaneously, nitrogen gas-saturated acetic acid was added to the suspension at a rate of 0.7 mL/h to maintain a pH of 10.0-10.5. After 1 h 30 min the rate of addition of the sodium borohydride solution and acetic acid were both reduced by half. Next, 3 h 45 min after start of addition, the addition of sodium borohydride and acetic acid were stopped. The mixture was allowed to cool down to room temperature and acetone (2.5 mL) was added over a period of 1 minute. After stirring the reaction mixture for 15 min acetic acid was added until the pH was 5.5-6.0 (about 3 mL required). Next, a mixture of ethyl acetate/toluene (1/3 v/v, 30 mL) was added, well mixed and layers were allowed to settle. The aqueous layer was separated and washed with ethyl acetate/toluene (1/3 v/v, 10 mL). Both organic extracts were pooled and water (40 mL) was added, well mixed and layers were allowed to settle. The pH of the aqueous layer was adjusted to 5.5-6 using saturated sodium hydrogen carbonate solution (aq) and again mixed with the organic layer. Layers were allowed to settle and the organic layer was separated and concentrated under vacuum to give 11.09 g of yellow oil (crude mixture containing title compound 8 and its borane complex). Several batches were combined for work up.33.1 g of the crude mixture containing title compound 8 and its borane complex (not corrected for residual solvents) was dissolved in toluene (30 mL) and filtered. The filtrate was submitted to column chromatography (silica gel, gradient of toluene to toluene/ethyl acetate 1/1) to give 30.0 g of mixture of 5-[[4-[2-[5-[[[(l,l- dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione (8) and its borane complex as a slightly yellow oil (yield = 100% from compound 4, not corrected for residual solvents). [0303] 1H NMR (CDC13) δ: -0.03 – 0.10 (m, 6H), 0.87 – 0.93 (m, 9H), 1.42 (d, / = 6 Hz, 3H),3.05-3.71 (m, 4H), 4.30 – 4.51 (m, 3H), 4.87 – 4.94 (m, 1H), 6.82 – 6.88 (m, 2H), 7.10-7.92 (m, 5H), 8.49 (d, / = 3 Hz, 0.6H) and 8.72 (brs, 0.4H).[0304] LC-MS; rt 6.8 min: ES: M+ 489, 488, 487, M“ 487, 486, 485; rt 8.1 min: ES M“ 501,500, 499, 498, 485.[0305] Step h: Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]- methyl]-2,4-thiazolidinedione hydrochloride (9)[0306] To a stirred solution of the mixture of (5-[[4-[2-[5-[[[(l,l-dimethylethyl)- dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione and its borane complex (8) (5.17 g) in methanol (25.2 mL) at 22°C was added hydrochloric acid (30%, 2.75 mL) in about 5 min to give a temperature rise to 28°C. This solution was heated to 40 °C. Three hours after addition, the 11 g of volatiles were removed under reduced pressure. Then, acetonitrile (40.3 mL) was added and the mixture was heated at reflux for 0.5 h. Next, the suspension was allowed to cool down to room temperature and stirred for 1 h at room temperature. Solids were isolated by filtration, washed with a mixture of acetonitrile/water (20/1 v/v, 10 mL) and with acetonitrile (10 mL) and dried under vacuum at 40 °C to give 4.00 g of white solids (crude 9) (yield = 77%, not corrected for residual solvents).[0307] Purification of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione hydrochloride (9):[0308] The crude mixture of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]- methyl]-2,4-thiazolidinedione hydrochloride (3.95 g, crude 9) was dissolved in methanol/water (7/2 v/v, 80 mL) by heating it to 49°C. To this solution was added washed norit (obtained by heating a suspension of norit (6 g) in methanol/water (7/2 v/v, 90 mL) at 45°C for 1 h, then isolating the norit by filtration and washing it twice with methanol/water (7/2 v/v, 30 mL) and drying it under vacuum at 40°C). Equipment was rinsed with methanol/water (7/2 v/v, 18 mL). After 0.5 h of stirring at 46°C, the warm suspension was filtered to remove the norit and filter was washed twice with methanol/water (7/2 v/v, 18 mL). The filtrate was concentrated under vacuum at a bath temperature of 60°C to a mass of 11.8 g (1 v of compound and 2 v of water). To the suspension was added butanone (19.7 mL, 5 v) and the mixture was heated at a bath temperature of 95°C. Under distillation at a constant volume, butanone (95 mL) was added. Next, heating was stopped and the suspension was allowed to reach room temperature in about 0.5 h. Subsequently it was stirred for 0.75 h at room temperature. The solids were isolated by filtration, washed with a mixture of butanone/water (95/5 v/v, 18 mL) and butanone (18 mL) and dried under vacuum at 40°C to give 3.57 g of compound 9 as white solids (yield = 91%).[0309] 1H NMR (DMSO-de): δ 12.00 (br s, -NH), 8.71 (d, = 2.0 Hz, 1H), 8.45 (dd, = 8.3,1.7 Hz, 1H), 7.98 (d, = 8.3 Hz, 1H), 7.15 (d, = 8.7 Hz, 2H), 6.88 (d, = 8.7 Hz, 2H), 5.57 (s, OH), 4.95 (q, = 6.5 Hz, 1H), 4.86 (dd, = 8.9, 4.4 Hz, 1H), 4.40 (t, = 6.3 Hz, 2H), 3.49 (t, = 6.2 Hz, 2H), 3.29 (dd, = 14.2, 4.4 Hz, 1H), 3.06 (dd, = 14.2, 9.0 Hz, 1H), 1.41 (d, = 6.5 Hz, 3H).[0310] LC-MS; rt 3.5 min: ES: M+ 374, 373, M“ 372, 371.EXAMPLE 4Conditions tested in the preparation of compound 5 in the Step d[0311] The conditions described in Table 2 below were tested in the step d in the preparation of compound 5 from compound 4 providing a good yield of compound 5:Table 2Entry Reaction Conditions Amount of p-Ts-Cl / Eq1 Toluene/water/Bu4NBr/NaOH 1.052 1.083 1.074 1.07+0.035 1.076 Et3N / DCM 1.187 1.408 Pyridine / DCM 1.40 EXAMPLE 5Conditions tested in the preparation of compound 6 in the Step e[0312] The conditions described in Table 3 below were tested in the step e in the preparation of compound 6 from compound 5 providing a good yield of compound 6:Table 3

PATENT
Compound 1 is administered to the subject. The structure of 5-[[4-[2-[5-(l -hydroxy ethyljpyri din-2 – yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione is:
[0047] The present disclosure encompasses the use of stereoisomers of 5-[[4-[2-[5-(l- hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione. 5-[[4-[2-[5- (l-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione has two asymmetric centers and thus four stereoisomers are possible as follows:
//////////LERIGLITAZONE, MIN 102 , лериглитазон , ليريغليتازون , 乐立格列酮 , Hydroxy Pioglitazone, M-IV, PHASE 2
CC(C1=CN=C(C=C1)CCOC2=CC=C(C=C2)CC3C(=O)NC(=O)S3)O

NEW DRUG APPROVALS
ONE TIME
$10.00
DOMPERIDONE
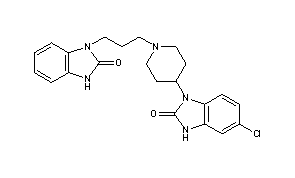
DOMPERIDONE
- Molecular FormulaC22H24ClN5O2
- Average mass425.911 Da
1H-Benzimidazol-2-ol, 5-chloro-1-[1-[3-(2-hydroxy-1H-benzimidazol-1-yl)propyl]-4-piperidinyl]-
260-968-7[EINECS]
2H-Benzimidazol-2-one, 5-chloro-1-[1-[3-(2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)propyl]-4-piperidinyl]-1,3-dihydro-
4-(5-Chloro-2-oxo-1-benzimidazolinyl)-1-[3-(2-oxobenzimidazolinyl)propyl]piperidine
57808-66-9[RN]домперидон
دومبيريدون
多潘立酮
CAS Registry Number: 57808-66-9
CAS Name: 5-Chloro-1-[1-[3-(2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)propyl]-4-piperidinyl]-1,3-dihydro-2H-benzimidazol-2-one
Additional Names: 5-chloro-1-[1-[3-(2-oxo-1-benzimidazolinyl)propyl]-4-piperidyl]-2-benzimidazolinone
Manufacturers’ Codes: R-33812
Trademarks: Euciton (Roux-Ocefa); Evoxin (Sterling Winthrop); Gastronorm (Janssen); Mod (Irbi); Motilium (Janssen); Nauzelin (Janssen); Peridon (Italchimici); Peridys (Robapharm)
Molecular Formula: C22H24ClN5O2
Molecular Weight: 425.91
Percent Composition: C 62.04%, H 5.68%, Cl 8.32%, N 16.44%, O 7.51%
Literature References: A novel in vitro dopamine antagonist with antinauseant properties.Prepn: J. Vandenberk et al.,DE2632870; eidem,US4066772 (1977, 1978 both to Janssen). Pharmacology: C. Ennis et al.,J. Pharm. Pharmacol.31, Suppl., 14P (1979). Gastrokinetic properties: J. M. Van Neuten et al.,Life Sci.23, 453 (1978). 3H-domperidone studies: M. P. Martres et al.,ibid. 1781; M. Baudry et al.,Arch. Pharmacol.308, 231 (1979). Clinical studies: A. J. Reyntjens et al.,Arzneim.-Forsch.28, 1194 (1978); D. B. Wilson, J. W. Dundee, Anaesthesia34, 765 (1979). Review of pharmacology, pharmacokinetics and therapeutic efficacy: R. N. Brogden et al.,Drugs24, 360-400 (1982).
Properties: Crystals from DMF/water, mp 242.5°.
Melting point: mp 242.5°
Therap-Cat: Antiemetic.
Keywords: Antiemetic; Dopamine Receptor Antagonist.
Domperidone, sold under the brand name Motilium among others, is a medication used as an antiemetic, gastric prokinetic agent, and galactagogue.[1][6][7] It may be taken by mouth or rectally, and is available as a tablet, orally disintegrating tablets,[8] suspension, and suppositories.[9] The drug is used to relieve nausea and vomiting; to increase the transit of food through the stomach (by increasing gastrointestinal peristalsis); and to promote lactation (breast milk production) by release of prolactin.[1][7]
It is a peripherally selective dopamine D2 receptor antagonist and was developed by Janssen Pharmaceutica.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
syn
Prepn: J. Vandenberk et al., DE 2632870; eidem, US 4066772 (1977, 1978 both to Janssen).

syn
Ruben Vardanyan, in Piperidine-Based Drug Discovery, 2017
Domperidone (5565)
Domperidone (7.1.6) (Motilium), a peripherally selective D2-like receptor antagonist, regulates the motility of the gastric and small intestinal smooth muscles and has been shown to have some effects on the motor function of the esophagus. It effectively prevents bile reflux but does not affect gastric secretion. As a result of the blockade of dopamine receptors in the chemoreceptor trigger zone it also has an antiemetic activity. Domperiodone provided relief of such symptoms as anorexia, nausea, vomiting, abdominal pain, early satiety, bloating, and distension in patients with symptoms of diabetic gastropathy. It also provided short-term relief of symptoms in patients with dyspepsia or gastroesophageal reflux, prevented nausea and vomiting associated with emetogenic chemotherapy, and prevented the gastrointestinal and emetic adverse effects of antiparkinsonian drugs. Because domperidone does not readily cross the blood brain barrier and does not inhibit dopamine receptors in the brain, reports of adverse effects on the CNS, such as dystonic reactions, are rare [52–61]. Domperidone is widely used in many countries and can now be officially prescribed to patients in the United States. There are very few treatment options currently available for patients with gastrointestinal motility disorders, especially for patients with gastroparesis. Domperidone has been successfully used in the United States and in many countries as a second-line treatment option for the treatment of gastroparesis.
Synthesis of domperidone (7.1.6) started with arylation of ethyl 4-aminopiperidine-1-carboxylate (7.1.28) with 1,4-dichloro-2-nitrobenzene (7.1.29) on heating at 150°C in cyclohexanol in the presence of sodium carbonate and potassium iodide (in a later disclosure in toluene in presence of sodium carbonate [62]) to give compound (7.1.30), which on reflux in 48% hydrobromic acid solution yielded N-(4-chloro-2-nitrophenyl)piperidin-4-amine (7.1.31). The obtained product was alkylated with 1-(3-chloropropyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (7.1.32) on reflux in MBIK in the presence of sodium carbonate and potassium iodide to give compound (7.1.33). The ring closure could be effected by heating o-phenylene diamine (7.1.33) with an appropriate cyclizing agent, such as phosgene, urea, potassium isocyanate [63], and the like. In this patent potassium isocyanate dissolved in water was carefully added to a solution of compound (7.1.34) in 10 N hydrochloric acid solution (exothermic reaction) to give desired domperidone (7.1.6) [64,65] (Scheme 7.4).

Medical uses
Nausea and vomiting
There is some evidence that domperidone has antiemetic activity.[10] It is recommended by the Canadian Headache Society for treatment of nausea associated with acute migraine.[11]
Gastroparesis
Gastroparesis is a medical condition characterised by delayed emptying of the stomach when there is no mechanical gastric outlet obstruction. Its cause is most commonly idiopathic, a diabetic complication or a result of abdominal surgery. The condition causes nausea, vomiting, fullness after eating, early satiety (feeling full before the meal is finished), abdominal pain and bloating.
Domperidone may be useful in diabetic and idiopathic gastroparesis.[12][13]
However, increased rate of gastric emptying induced by drugs like domperidone does not always correlate (equate) well with relief of symptoms.[14]
Parkinson’s disease
Parkinson’s disease is a chronic neurological condition where a decrease in dopamine in the brain leads to rigidity (stiffness of movement), tremor and other symptoms and signs. Poor gastrointestinal function, nausea and vomiting is a major problem for people with Parkinson’s disease because most medications used to treat Parkinson’s disease are given by mouth. These medications, such as levodopa, can cause nausea as a side effect. Furthermore, anti-nausea drugs, such as metoclopramide, which do cross the blood–brain barrier may worsen the extra-pyramidal symptoms of Parkinson’s disease.
Domperidone can be used to relieve gastrointestinal symptoms in Parkinson’s disease; it blocks peripheral D2 receptors but does not cross the blood–brain barrier in normal doses (the barrier between the blood circulation of the brain and the rest of the body) so has no effect on the extrapyramidal symptoms of the disease.[15]
Functional dyspepsia
Domperidone may be used in functional dyspepsia in both adults and children.[16][17]
Lactation
The hormone prolactin stimulates lactation (production of breast milk). Dopamine, released by the hypothalamus stops the release of prolactin from the pituitary gland. Domperidone, by acting as an anti-dopaminergic agent, results in increased prolactin secretion, and thus promotes lactation (that is, it is a galactogogue). Domperidone moderately increases the volume of expressed breast milk in mothers of preterm babies where breast milk expression was inadequate, and appears to be safe for short-term use for this purpose.[18][19][20] In the United States, domperidone is not approved for this or any other use.[21][22]
A study called the EMPOWER trial was designed to assess the effectiveness and safety of domperidone in assisting mothers of preterm babies to supply breast milk for their infants.[23] The study randomized 90 mothers of preterm babies to receive either domperidone 10 mg orally three times daily for 28 days (Group A) or placebo 10 mg orally three times daily for 14 days followed by domperidone 10 mg orally three times daily for 14 days (Group B). Mean milk volumes at the beginning of the intervention were similar between the 2 groups. After the first 14 days, 78% of mothers receiving domperidone (Group A) achieved a 50% increase in milk volume, while 58% of mothers receiving placebo (Group B) achieved a 50% increase in milk volume.[24]
To induce lactation, domperidone is used at a dosage of 10 to 20 mg 3 or 4 times per day by mouth.[25] Effects may be seen within 24 hours or may not be seen for 3 or 4 days.[25] The maximum effect occurs after 2 or 3 weeks of treatment, and the treatment period generally lasts for 3 to 8 weeks.[25] A 2012 review shows that no studies support prophylactic use of a galactagogue medication at any stage of pregnancy, including domperidone.[26]
Reflux in children
Domperidone has been found effective in the treatment of reflux in children.[27] However some specialists consider its risks prohibitory of the treatment of infantile reflux.[28]
Contraindications
- QT-prolonging drugs like amiodarone[29]
Side effects
Side effects associated with domperidone include dry mouth, abdominal cramps, diarrhea, nausea, rash, itching, hives, and hyperprolactinemia (the symptoms of which may include breast enlargement, galactorrhea, breast pain/tenderness, gynecomastia, hypogonadism, and menstrual irregularities).[25] Due to blockade of D2 receptors in the central nervous system, D2 receptor antagonists like metoclopramide can also produce a variety of additional side effects including drowsiness, akathisia, restlessness, insomnia, lassitude, fatigue, extrapyramidal symptoms, dystonia, Parkinsonian symptoms, tardive dyskinesia, and depression.[1][7] However, this is not the case with domperidone, because, unlike other D2 receptor antagonists, it minimally crosses the blood-brain-barrier, and for this reason, is rarely associated with such side effects.[1][7]
Excess prolactin levels
Due to D2 receptor blockade, domperidone causes hyperprolactinemia.[30] Hyperprolactinemia can suppress the secretion of gonadotropin-releasing hormone (GnRH) from the hypothalamus, in turn suppressing the secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) and resulting in hypogonadism (low sex hormone (e.g., testosterone, estradiol) levels).[31] As such, male patients may experience low libido, erectile dysfunction, and impaired spermatogenesis.[31] Also in accordance with hyperprolactinemia, 10–15% of female patients have been reported to experience mammoplasia (breast enlargement), mastodynia (breast pain/tenderness), galactorrhea (inappropriate or excessive milk production/secretion), and amenorrhea (cessation of menstrual cycles) with domperidone treatment.[30] Gynecomastia has been reported in males treated with domperidone,[32] and galactorrhea could occur in males as well.[31]
Rare reactions
Cardiac reactions
Domperidone use is associated with an increased risk of sudden cardiac death (by 70%)[33] most likely through its prolonging effect of the cardiac QT interval and ventricular arrhythmias.[34][35] The cause is thought to be blockade of hERG voltage-gated potassium channels.[36][37] The risks are dose-dependent, and appear to be greatest with high/very high doses via intravenous administration and in the elderly, as well as with drugs that interact with domperidone and increase its circulating concentrations (namely CYP3A4 inhibitors).[38][39] Conflicting reports exist, however.[40] In neonates and infants, QT prolongation is controversial and uncertain.[41][42]
UK drug regulatory authorities (MHRA) have issued the following restriction on domperidone in 2014 due to increased risk of adverse cardiac effects:
Domperidone (Motilium) is associated with a small increased risk of serious cardiac side effects. Its use is now restricted to the relief of nausea and vomiting and the dosage and duration of use have been reduced. It should no longer be used for the treatment of bloating and heartburn. Domperidone is now contraindicated in those with underlying cardiac conditions and other risk factors. Patients with these conditions and patients receiving long-term treatment with domperidone should be reassessed at a routine appointment, in light of the new advice.
However, a 2015 Australian review concluded the following:[39]
Based on the results of the two TQT (the regulatory agency gold standard for assessment of QT prolongation) domperidone does not appear to be strongly associated with QT prolongation at oral doses of 20 mg QID in healthy volunteers. Further, there are limited case reports supporting an association with cardiac dysfunction, and the frequently cited case-control studies have significant flaws. While there remains an ill-defined risk at higher systemic concentrations, especially in patients with a higher baseline risk of QT prolongation, our review does not support the view that domperidone presents intolerable risk.
Possible central toxicity in infants
In Britain a legal case involved the death of two children of a mother whose three children had all had hypernatraemia. She was charged with poisoning the children with salt. One of the children, who was born at 28 weeks gestation with respiratory complications and had a fundoplication for gastroesophageal reflux and failure to thrive was prescribed domperidone. An advocate for the mother suggested the child may have suffered neuroleptic malignant syndrome as a side effect of domperidone due to the drug crossing the child’s immature blood-brain-barrier.[43]
Interactions
In healthy volunteers, ketoconazole increased the Cmax and AUC concentrations of domperidone by 3- to 10-fold.[44] This was accompanied by a QT interval prolongation of about 10–20 milliseconds when domperidone 10 mg four times daily and ketoconazole 200 mg twice daily were administered, whereas domperidone by itself at the dosage assessed produced no such effect.[44] As such, domperidone with ketoconazole or other CYP3A4 inhibitors is a potentially dangerous combination.[44]
Pharmacology
Pharmacodynamics
Domperidone is a peripherally selective dopamine D2 and D3 receptor antagonist.[7] It has no clinically significant interaction with the D1 receptor, unlike metoclopramide.[7] The medication provides relief from nausea by blocking D receptors.[10] It blocks dopamine receptors in the anterior pituitary gland increasing release of prolactin which in turn increases lactation.[45][46] Domperidone may be more useful in some patients and cause harm in others by way of the genetics of the person, such as polymorphisms in the drug transporter gene ABCB1 (which encodes P-glycoprotein), the voltage-gated potassium channel KCNH2 gene (hERG/Kv11.1), and the α1D—adrenoceptor ADRA1D gene.[47]
Effects on prolactin levels
A single 20 mg oral dose of domperidone has been found to increase mean serum prolactin levels (measured 90 minutes post-administration) in non-lactating women from 8.1 ng/mL to 110.9 ng/mL (a 13.7-fold increase).[7][48][49][50] This was similar to the increase in prolactin levels produced by a single 20 mg oral dose of metoclopramide (7.4 ng/mL to 124.1 ng/mL; 16.7-fold increase).[49][50] After two weeks of chronic administration (30 mg/day in both cases), the increase in prolactin levels produced by domperidone was reduced (53.2 ng/mL; 6.6-fold above baseline), but the increase in prolactin levels produced by metoclopramide, conversely, was heightened (179.6 ng/mL; 24.3-fold above baseline).[7][50] This indicates that acute and chronic administration of both domperidone and metoclopramide is effective in increasing prolactin levels, but that there are differential effects on the secretion of prolactin with chronic treatment.[49][50] The mechanism of the difference is unknown.[50] The increase in prolactin levels observed with the two drugs was, as expected, much greater in women than in men.[49][50] This appears to be due to the higher estrogen levels in women, as estrogen stimulates prolactin secretion.[51]
For comparison, normal prolactin levels in women are less than 20 ng/mL, prolactin levels peak at 100 to 300 ng/mL at parturition in pregnant women, and in lactating women, prolactin levels have been found to be 90 ng/mL at 10 days postpartum and 44 ng/mL at 180 days postpartum.[52][53]
Pharmacokinetics
With oral administration, domperidone is extensively metabolized in the liver (almost exclusively by CYP3A4/5, though minor contributions by CYP1A2, CYP2D6, and CYP2C8 have also been reported)[54] and in the intestines.[5] Due to the marked first-pass effect via this route, the oral bioavailability of domperidone is low (13–17%);[1] conversely, its bioavailability is high via intramuscular injection (90%).[1] The terminal half-life of domperidone is 7.5 hours in healthy individuals, but can be prolonged to 20 hours in people with severe renal dysfunction.[1] All of the metabolites of domperidone are inactive as D2 receptor ligands.[1][5] The drug is a substrate for the P-glycoprotein (ABCB1) transporter, and animal studies suggest that this is the reason for the low central nervous system penetration of domperidone.[55]
Chemistry
Domperidone is a benzimidazole derivative and is structurally related to butyrophenone neuroleptics like haloperidol.[56][57]
History
- 1974 – Domperidone synthesized at Janssen Pharmaceutica[58] following the research on antipsychotic drugs.[59] Janssen pharmacologists discovered that some of antipsychotic drugs had a significant effect on dopamine receptors in the central chemoreceptor trigger zone that regulated vomiting and started searching for a dopamine antagonist that would not pass the blood–brain barrier, thereby being free of the extrapyramidal side effects that were associated with drugs of this type.[59] This led to the discovery of domperidone as a strong anti-emetic with minimal central effects.[59][60]
- 1978 – On 3 January 1978 Domperidone was patented in the United States under patent US4066772 A. The application has been filed on 17 May 1976. Jan Vandenberk, Ludo E. J. Kennis, Marcel J. M. C. Van der Aa and others has been cited as the inventors.
- 1979 – Domperidone marketed under trade name “Motilium” in Switzerland and (Western) Germany.[61]
- 1999 – Domperidone was introduced in the forms of orally disintegrating tablets (based on Zydis technology).[62]
- Janssen Pharmaceutical has brought domperidone before the United States Federal Drug Administration (FDA) several times, including in the 1990s.
- 2014 – In April 2014 Co-ordination Group for Mutual Recognition and Decentralised Procedures – Human (CMDh) published official press-release suggesting to restrict the use of domperidone-containing medicines. It also approved earlier published suggestions by Pharmacovigilance Risk Assessment Committee (PRAC) to use domperidone only for curing nausea and vomiting and reduce maximum daily dosage to 10 mg.[9]
Society and culture
Generic names
Domperidone is the generic name of the drug and its INN, USAN, BAN, and JAN.[63][6][64]
Regulatory approval
It was reported in 2007 that domperidone is available in 58 countries, including Canada,[65] but the uses or indications of domperidone vary between nations. In Italy it is used in the treatment of gastroesophageal reflux disease and in Canada, the drug is indicated in upper gastrointestinal motility disorders and to prevent gastrointestinal symptoms associated with the use of dopamine agonist antiparkinsonian agents.[66] In the United Kingdom, domperidone is only indicated for the treatment of nausea and vomiting and the treatment duration is usually limited to 1 week.
In the United States, domperidone is not currently a legally marketed human drug and it is not approved for sale in the U.S. On 7 June 2004, FDA issued a public warning that distributing any domperidone-containing products is illegal.[67]
It is available over-the-counter to treat gastroesophageal reflux and functional dyspepsia in many countries, such as Ireland, the Netherlands, Italy, South Africa, Mexico, Chile, and China.[68]
Domperidone is not generally approved for use in the United States. There is an exception for use in people with treatment-refractory gastrointestinal symptoms under an FDA Investigational New Drug application.[1]
Formulations
| showFormulations |
|---|
Research
Domperidone has been studied as a potential hormonal contraceptive to prevent pregnancy in women.[72]
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s Reddymasu, Savio C.; Soykan, Irfan; McCallum, Richard W. (2007). “Domperidone: Review of Pharmacology and Clinical Applications in Gastroenterology”. The American Journal of Gastroenterology. 102 (9): 2036–2045. ISSN 0002-9270. PMID 17488253.
- ^ “БРЮЛІУМ ЛІНГВАТАБС” [BRULIUM LINGUATABS]. Нормативно-директивні документи МОЗ України (in Ukrainian). 18 March 2014. Retrieved 29 May 2015.
- ^ “Domperidone”. Archived from the original on 22 May 2013. Retrieved 30 June 2013.
- ^ Jump up to:a b Suzanne Rose (October 2004). Gastrointestinal and Hepatobiliary Pathophysiology. Hayes Barton Press. pp. 523–. ISBN 978-1-59377-181-2.
- ^ Jump up to:a b c d Simard, C.; Michaud, V.; Gibbs, B.; Massé, R.; Lessard, É; Turgeon, J. (2008). “Identification of the cytochrome P450 enzymes involved in the metabolism of domperidone”. Xenobiotica. 34 (11–12): 1013–1023. doi:10.1080/00498250400015301. ISSN 0049-8254. PMID 15801545. S2CID 27426219.
- ^ Jump up to:a b Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. pp. 366–. ISBN 978-3-88763-075-1.
- ^ Jump up to:a b c d e f g h Barone JA (1999). “Domperidone: a peripherally acting dopamine2-receptor antagonist”. The Annals of Pharmacotherapy. 33 (4): 429–40. doi:10.1345/aph.18003. PMID 10332535. S2CID 39279569.
- ^ “MOTILIUM INSTANTS PL 13249/0028” (PDF). Medicines and Healthcare products Regulatory Agency. 23 February 2010. Archived from the original (PDF) on 31 October 2014. Retrieved 2014-10-31.
- ^ Jump up to:a b “CMDh confirms recommendations on restricting use of domperidone-containing medicines: European Commission to take final legal decision”. European Medicines Agency. 25 April 2014. Archived from the original on 30 January 2016. Retrieved 2014-10-31.
- ^ Jump up to:a b Reddymasu SC, Soykan I, McCallum RW. (2007). “Domperidone: review of pharmacology and clinical applications in gastroenterology”. Am J Gastroenterol. 102 (9): 2036–45. PMID 17488253.
- ^ Worthington I, Pringsheim T, Gawel MJ, Gladstone J, Cooper P, Dilli E, Aube M, Leroux E, Becker WJ (September 2013). “Canadian Headache Society Guideline: acute drug therapy for migraine headache”. The Canadian Journal of Neurological Sciences. 40(5 Suppl 3): S1–S80. doi:10.1017/S0317167100118943. PMID 23968886.
- ^ Stevens JE, Jones KL, Rayner CK, Horowitz M (June 2013). “Pathophysiology and pharmacotherapy of gastroparesis: current and future perspectives”. Expert Opinion on Pharmacotherapy. 14(9): 1171–86. doi:10.1517/14656566.2013.795948. PMID 23663133. S2CID 23526883.
- ^ Silvers D, Kipnes M, Broadstone V, Patterson D, Quigley EM, McCallum R, Leidy NK, Farup C, Liu Y, Joslyn A (1998). “Domperidone in the management of symptoms of diabetic gastroparesis: efficacy, tolerability, and quality-of-life outcomes in a multicenter controlled trial. DOM-USA-5 Study Group”. Clinical Therapeutics. 20 (3): 438–53. doi:10.1016/S0149-2918(98)80054-4. PMID 9663360.
- ^ Janssen P, Harris MS, Jones M, Masaoka T, Farré R, Törnblom H, Van Oudenhove L, Simrén M, Tack J (September 2013). “The relation between symptom improvement and gastric emptying in the treatment of diabetic and idiopathic gastroparesis”. The American Journal of Gastroenterology. 108 (9): 1382–91. doi:10.1038/ajg.2013.118. PMID 24005344. S2CID 32835351.
- ^ Ferrier J (2014). “Domperidone as an unintended antipsychotic”. Can Pharm J. 147 (2): 76–7. doi:10.1177/1715163514521969. PMC 3962062. PMID 24660005.
- ^ Xiao M, Qiu X, Yue D, Cai Y, Mo Q (2013). “Influence of hippophae rhamnoides on two appetite factors, gastric emptying and metabolic parameters, in children with functional dyspepsia”(PDF). Hellenic Journal of Nuclear Medicine. 16 (1): 38–43. PMID 23529392.
- ^ Huang X, Lv B, Zhang S, Fan YH, Meng LN (December 2012). “Itopride therapy for functional dyspepsia: a meta-analysis”. World Journal of Gastroenterology. 18 (48): 7371–7. doi:10.3748/wjg.v18.i48.7371. PMC 3544044. PMID 23326147.
- ^ Grzeskowiak LE, Smithers LG, Amir LH, Grivell RM (October 2018). “Domperidone for increasing breast milk volume in mothers expressing breast milk for their preterm infants: a systematic review and meta-analysis”. BJOG. 125 (11): 1371–1378. doi:10.1111/1471-0528.15177. hdl:2440/114203. PMID 29469929.
- ^ Grzeskowiak LE, Lim SW, Thomas AE, Ritchie U, Gordon AL (February 2013). “Audit of domperidone use as a galactogogue at an Australian tertiary teaching hospital”. Journal of Human Lactation. 29 (1): 32–7. doi:10.1177/0890334412459804. hdl:2440/94368. PMID 23015150. S2CID 26535783.
- ^ Donovan TJ, Buchanan K (2012). “Medications for increasing milk supply in mothers expressing breastmilk for their preterm hospitalised infants”. The Cochrane Database of Systematic Reviews. 3 (3): CD005544. doi:10.1002/14651858.CD005544.pub2. PMID 22419310.
- ^ da Silva OP, Knoppert DC (September 2004). “Domperidone for lactating women”. CMAJ. 171 (7): 725–6. doi:10.1503/cmaj.1041054. PMC 517853. PMID 15451832.
- ^ “FDA warns against women using unapproved drug, domperidone to increase milk production.” U.S. Food and Drug Administration 7 June 2004.
- ^ Asztalos EV, Campbell-Yeo M, daSilva OP, Kiss A, Knoppert DC, Ito S (2012). “Enhancing breast milk production with Domperidone in mothers of preterm neonates (EMPOWER trial)”. BMC Pregnancy and Childbirth. 12: 87. doi:10.1186/1471-2393-12-87. PMC 3532128. PMID 22935052.
- ^ Asztalos EV, Campbell-Yeo M, da Silva OP, Ito S, Kiss A, Knoppert D, et al. (EMPOWER Study Collaborative Group) (2017). “Enhancing human milk production with Domperidone in mothers of preterm infants”. Journal of Human Lactation. 33 (1): 181–187. doi:10.1177/0890334416680176. PMID 28107101. S2CID 39041713.
- ^ Jump up to:a b c d Henderson, Amanda (2003). “Domperidone: Discovering New Choices for Lactating Mothers”. AWHONN Lifelines. 7 (1): 54–60. doi:10.1177/1091592303251726. ISSN 1091-5923. PMID 12674062.
- ^ Donovan, Timothy J; Buchanan, Kerry (14 March 2012). “Medications for increasing milk supply in mothers expressing breastmilk for their preterm hospitalised infants”. Cochrane Database of Systematic Reviews (3): CD005544. doi:10.1002/14651858.cd005544.pub2. PMID 22419310.
- ^ Kapoor, A.K.; Raju, S.M. (2013). “7.2 Gastrointestinal Drugs”. Illustrated Medical Pharmacology. JP Medical Ltd. p. 677. ISBN 978-9350906552. Retrieved 31 October 2014. (Google Books)
- ^ Rebecca Smith (1 August 2014). “Fear that reflux treatment for babies will be denied under new Nice guidance”. The Daily Telegraph. Archived from the original on 31 October 2014. Retrieved 2014-10-31.
- ^ Jump up to:a b Swannick G. (ed.) “MIMS Australia.” December 2013
- ^ Jump up to:a b Elliott Proctor Joslin; C. Ronald Kahn (2005). Joslin’s Diabetes Mellitus: Edited by C. Ronald Kahn … [et Al.]. Lippincott Williams & Wilkins. pp. 1084–. ISBN 978-0-7817-2796-9.
- ^ Jump up to:a b c Edmund S. Sabanegh, Jr. (20 October 2010). Male Infertility: Problems and Solutions. Springer Science & Business Media. pp. 83–. ISBN 978-1-60761-193-6.
- ^ Gerald G. Briggs; Roger K. Freeman; Sumner J. Yaffe (28 March 2012). Drugs in Pregnancy and Lactation: A Reference Guide to Fetal and Neonatal Risk. Lippincott Williams & Wilkins. pp. 442–. ISBN 978-1-4511-5359-0.
- ^ Leelakanok N, Holcombe A, Schweizer ML (2015). “Domperidone and Risk of Ventricular Arrhythmia and Cardiac Death: A Systematic Review and Meta-analysis”. Clin Drug Investig. 36 (2): 97–107. doi:10.1007/s40261-015-0360-0. PMID 26649742. S2CID 25601738.
- ^ van Noord C, Dieleman JP, van Herpen G, Verhamme K, Sturkenboom MC (November 2010). “Domperidone and ventricular arrhythmia or sudden cardiac death: a population-based case-control study in the Netherlands”. Drug Safety. 33 (11): 1003–14. doi:10.2165/11536840-000000000-00000. PMID 20925438. S2CID 21177240.
- ^ Johannes CB, Varas-Lorenzo C, McQuay LJ, Midkiff KD, Fife D (September 2010). “Risk of serious ventricular arrhythmia and sudden cardiac death in a cohort of users of domperidone: a nested case-control study”. Pharmacoepidemiology and Drug Safety. 19(9): 881–8. doi:10.1002/pds.2016. PMID 20652862. S2CID 20323199.
- ^ Rossi M, Giorgi G (2010). “Domperidone and long QT syndrome”. Curr Drug Saf. 5 (3): 257–62. doi:10.2174/157488610791698334. PMID 20394569.
- ^ Doggrell SA, Hancox JC (2014). “Cardiac safety concerns for domperidone, an antiemetic and prokinetic, and galactogogue medicine” (PDF). Expert Opin Drug Saf. 13 (1): 131–8. doi:10.1517/14740338.2014.851193. PMID 24147629. S2CID 30668496.
- ^ Marzi, Marta; Weitz, Darío; Avila, Aylén; Molina, Gabriel; Caraballo, Lucía; Piskulic, Laura (2015). “Efectos adversos cardíacos de la domperidona en pacientes adultos: revisión sistemática”. Revista Médica de Chile. 143 (1): 14–21. doi:10.4067/S0034-98872015000100002. ISSN 0034-9887. PMID 25860264.
- ^ Jump up to:a b Buffery PJ, Strother RM (2015). “Domperidone safety: a mini-review of the science of QT prolongation and clinical implications of recent global regulatory recommendations”. N. Z. Med. J. 128(1416): 66–74. PMID 26117678.
- ^ Ortiz, Arleen; Cooper, Chad J.; Alvarez, Alicia; Gomez, Yvette; Sarosiek, Irene; McCallum, Richard W. (2015). “Cardiovascular Safety Profile and Clinical Experience With High-Dose Domperidone Therapy for Nausea and Vomiting”. The American Journal of the Medical Sciences. 349 (5): 421–424. doi:10.1097/MAJ.0000000000000439. ISSN 0002-9629. PMC 4418779. PMID 25828198.
- ^ Djeddi D, Kongolo G, Lefaix C, Mounard J, Léké A (November 2008). “Effect of domperidone on QT interval in neonates”. The Journal of Pediatrics. 153 (5): 663–6. doi:10.1016/j.jpeds.2008.05.013. PMID 18589449.
- ^ Günlemez A, Babaoğlu A, Arisoy AE, Türker G, Gökalp AS (January 2010). “Effect of domperidone on the QTc interval in premature infants”. Journal of Perinatology. 30 (1): 50–3. doi:10.1038/jp.2009.96. PMC 2834362. PMID 19626027.
- ^ Coulthard MG, Haycock GB (January 2003). “Distinguishing between salt poisoning and hypernatraemic dehydration in children”. BMJ (Clinical Research Ed.). 326 (7381): 157–60. doi:10.1136/bmj.326.7381.157. PMC 1128889. PMID 12531853.
- ^ Jump up to:a b c Jeffrey K. Aronson (27 November 2009). Meyler’s Side Effects of Antimicrobial Drugs. Elsevier. pp. 2244–. ISBN 978-0-08-093293-4.
- ^ Saeb-Parsy K. “Instant pharmacology.” John Wiley & Sons, 1999 ISBN 0471976393, 9780471976394 p216.
- ^ Sakamoto Y, Kato S, Sekino Y, Sakai E, Uchiyama T, Iida H, Hosono K, Endo H, Fujita K, Koide T, Takahashi H, Yoneda M, Tokoro C, Goto A, Abe Y, Kobayashi N, Kubota K, Maeda S, Nakajima A, Inamori M (2011). “Effects of domperidone on gastric emptying: a crossover study using a continuous real-time 13C breath test (BreathID system)”. Hepato-gastroenterology. 58 (106): 637–41. PMID 21661445.
- ^ Parkman HP, Jacobs MR, Mishra A, Hurdle JA, Sachdeva P, Gaughan JP, Krynetskiy E (January 2011). “Domperidone treatment for gastroparesis: demographic and pharmacogenetic characterization of clinical efficacy and side-effects”. Digestive Diseases and Sciences. 56 (1): 115–24. doi:10.1007/s10620-010-1472-2. PMID 21063774. S2CID 39632855.
- ^ Gabay MP (2002). “Galactogogues: medications that induce lactation”. J Hum Lact. 18 (3): 274–9. doi:10.1177/089033440201800311. PMID 12192964. S2CID 29261467.
- ^ Jump up to:a b c d Hofmeyr GJ, Van Iddekinge B, Blott JA (1985). “Domperidone: secretion in breast milk and effect on puerperal prolactin levels”. Br J Obstet Gynaecol. 92 (2): 141–4. doi:10.1111/j.1471-0528.1985.tb01065.x. PMID 3882143. S2CID 25489895.
- ^ Jump up to:a b c d e f Brouwers JR, Assies J, Wiersinga WM, Huizing G, Tytgat GN (1980). “Plasma prolactin levels after acute and subchronic oral administration of domperidone and of metoclopramide: a cross-over study in healthy volunteers”. Clin. Endocrinol. 12 (5): 435–40. doi:10.1111/j.1365-2265.1980.tb02733.x. PMID 7428183. S2CID 27266775.
- ^ Fujino T, Kato H, Yamashita S, Aramaki S, Morioka H, Koresawa M, Miyauchi F, Toyoshima H, Torigoe T (1980). “Effects of domperidone on serum prolactin levels in human beings”. Endocrinol. Jpn. 27 (4): 521–5. doi:10.1507/endocrj1954.27.521. PMID 7460861.
- ^ Jan Riordan (January 2005). Breastfeeding and Human Lactation. Jones & Bartlett Learning. pp. 76–. ISBN 978-0-7637-4585-1.
- ^ Kenneth L. Becker (2001). Principles and Practice of Endocrinology and Metabolism. Lippincott Williams & Wilkins. pp. 147–. ISBN 978-0-7817-1750-2.
- ^ Youssef AS, Parkman HP, Nagar S (2015). “Drug-drug interactions in pharmacologic management of gastroparesis”. Neurogastroenterol. Motil. 27 (11): 1528–41. doi:10.1111/nmo.12614. PMID 26059917. S2CID 34728070.
- ^ Stan K. Bardal; Jason E. Waechter; Douglas S. Martin (2011). Applied Pharmacology. Elsevier Health Sciences. pp. 184–. ISBN 978-1-4377-0310-8.
- ^ Hospital Formulary. HFM Publishing Corporation. 1991. p. 171.
Domperidone, a benzimidazole derivative, is structurally related to the butyrophenone tranquilizers (eg, haloperidol (Haldol, Halperon]).
- ^ Giovanni Biggio; Erminio Costa; P. F. Spano (22 October 2013). Receptors as Supramolecular Entities: Proceedings of the Biannual Capo Boi Conference, Cagliari, Italy, 7-10 June 1981. Elsevier Science. pp. 3–. ISBN 978-1-4831-5550-0.
- ^ Wan EW, Davey K, Page-Sharp M, Hartmann PE, Simmer K, Ilett KF (27 May 2008). “Dose-effect study of domperidone as a galactagogue in preterm mothers with insufficient milk supply, and its transfer into milk”. British Journal of Clinical Pharmacology. 66(2): 283–289. doi:10.1111/j.1365-2125.2008.03207.x. PMC 2492930. PMID 18507654.
- ^ Jump up to:a b c Sneader, Walter (2005). “Plant Product Analogues and Compounds Derived from Them”. Drug discovery : a history. Chichester: John Wiley & Sons Ltd. p. 125. ISBN 978-0-471-89979-2.
- ^ Corsini, Giovanni Umberto (2010). “Apomorphine: from experimental tool to therpeutic aid” (PDF). In Ban, Thomas A; Healy, David & Shorter, Edward (eds.). The Triumph of Psychopharacology and the Story of CINP. CINP. p. 54. ISBN 978-9634081814. Archived from the original (PDF) on 1 November 2014.
- ^ “Domperidone”. Pharmaceutical Manufacturing Encyclopedia, 3rd Edition (Vol. 1-4). William Andrew Publishing. 2013. p. 138. ISBN 9780815518563. Retrieved 12 December 2014.
- ^ Rathbone, Michael J.; Hadgraft, Jonathan; Roberts, Michael S. (2002). “The Zydis Oral Fast-Dissolving Dosage Form”. Modified-Release Drug Delivery Technology. CRC Press. p. 200. ISBN 9780824708696. Retrieved 31 October 2014.
- ^ Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 466–. ISBN 978-1-4757-2085-3.
- ^ “Domperidone”.
- ^ Reddymasu SC, Soykan I, McCallum RW (2007). “Domperidone: review of pharmacology and clinical applications in gastroenterology”. Am. J. Gastroenterol. 102 (9): 2036–45. PMID 17488253.
- ^ “Domperidone – heart rate and rhythm disorders.” Canadian adverse reactions newsletter. Government of Canada. January 2007 17(1)
- ^ “How to Obtain”. Food and Drug Administration. 10 February 2015. Retrieved 24 February 2016.
- ^ Fais, Paolo; Vermiglio, Elisa; Laposata, Chiara; Lockwood, Robert; Gottardo, Rossella; De Leo, Domenico (2015). “A case of sudden cardiac death following Domperidone self-medication”. Forensic Science International. 254: e1–e3. doi:10.1016/j.forsciint.2015.06.004. ISSN 0379-0738. PMID 26119456.
- ^ “De Standaard: “Motilium from now on only with prescription””. standaard.be. 7 May 2013. Retrieved 3 October 2013.
- ^ “ipcalabs.com”. ipcalabs.com. Retrieved 30 June 2013.
- ^ “torrentpharma.com”. torrentpharma.com. Retrieved 30 June2013.
- ^ Hofmeyr, G. J.; Van Iddekinge, B.; Van Der Walt, L. A. (2009). “Effect of domperidone-induced hyperprolactinaemia on the menstrual cycle; a placebo-controlled study”. Journal of Obstetrics and Gynaecology. 5 (4): 263–264. doi:10.3109/01443618509067772. ISSN 0144-3615.
External links
//////////////DOMPERIDONE, Antiemetic, Dopamine Receptor Antagonist, домперидон , دومبيريدون , 多潘立酮

NEW DRUG APPROVALS
ONE TIME
$10.00
MEBEVERINE
MEBEVERINE
- Molecular FormulaC25H35NO5
- Average mass429.549 Da
3,4-Dimethoxybenzoic Acid 4-[Ethyl[2-(4-methoxyphenyl)-1-methylethyl]amino]butyl Ester
3625-06-7[RN]
222-830-4[EINECS]
3,4-Diméthoxybenzoate de 4-{éthyl[1-(4-méthoxyphényl)-2-propanyl]amino}butyle
мебеверин
ميبيفيرين
美贝维林
- EINECS:222-830-4
- LD50:24 mg/kg (M, i.v.); 995 mg/kg (M, p.o.)
Derivatives
hydrochloride
- Formula:C25H35NO5 • HCl
- MW:466.02 g/mol
- CAS-RN:2753-45-9
- EINECS:220-400-0
- LD50:17.7 mg/kg (R, i.v.); 1540 mg/kg (R, p.o.)
Mebeverine
CAS Registry Number: 3625-06-7
CAS Name: 3,4-Dimethoxybenzoic acid 4-[ethyl[2-(4-methoxyphenyl)-1-methylethyl]amino]butyl ester
Additional Names: veratric acid 4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl ester;3,4-dimethoxybenzoic acid 4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl ester;4-[ethyl(p-methoxy-a-methylphenethyl)amino]butyl 3,4-dimethoxybenzoate;4-[N-[2-(p-methoxyphenyl)-1-methylethyl]-N-ethylamino]butyl 3,4-dimethoxybenzoate
Molecular Formula: C25H35NO5
Molecular Weight: 429.55
Percent Composition: C 69.90%, H 8.21%, N 3.26%, O 18.62%
Literature References: Smooth muscle relaxant. Prepn: BE609490C.A.59, 517b (1963) and T. Kralt et al.,DE1126889; eidem,US3265577 (1962, 1962, 1966 to N. V. Philips). Pharmacology: G. Bertaccini et al.,Farmaco Ed. Sci.30, 823 (1975).
Derivative Type: Hydrochloride
CAS Registry Number: 2753-45-9
Trademarks: Colofac (Duphar); Duspatalin (Duphar); Duspatal (Duphar)
Molecular Formula: C25H35NO5.HCl
Molecular Weight: 466.01
Percent Composition: C 64.43%, H 7.79%, N 3.01%, O 17.17%, Cl 7.61%
Properties: Crystals from ethyl methyl ketone, mp 105-107° (Ger. patent); also reported as mp 129-131° (Belg. patent).
Melting point: mp 105-107° (Ger. patent); mp 129-131° (Belg. patent)
Therap-Cat: Antispasmodic.
Keywords: Antispasmodic.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PAT
Indian Pat. Appl., 201841023171

PAPER
Pharma Chemica, 2(2), 366-378; 2010
In a quest of novel antispasmodic agents with antimicrobial properties, the present study describes design and synthesis of novel analogs for veratric acid ester 4-[ethyl-{2-(4- methoxyphenyl)-1-methylethyl} amino] butan-1-ol, an antispasmodic drug which is expected to be a potent antimicrobial agent may be due to the presence of two benzene rings and a secondary or tertiary nitrogen in the basic structural framework of the molecule. The reaction between substituted 2-ethylamino-1-(4’-methoxyphenyl) propane and various haloaryl benzoates derivatives obtained from reaction between different homologs of benzoic acid and dibromoalkanes in a two step process to give corresponding structurally diverse analogs of lead compound has been achieved. The structures of these novel analogs were confirmed by different structure elucidation techniques. All the compounds have been screened for their anti-spasmodic activity and the study extended further to evaluate their sedative, antibacterial and antifungal potency. The novel analogs of lead compound exhibited pronounced antispasmodic activities and also gave encouraging results of antimicrobial and sedative activity as anticipated.
General method of preparation of veratric acid ester 4-[ethyl-{2-(4-methoxyphenyl)-1- methylethyl} amino] butan-1-ol hydrochloride (5) and its analogs (5a-5p) A mixture of compound (3) (149 g, 0.47 mol) and Compound (4) (183 g, 0.95 mol) in ethyl methyl ketone (MEK) was refluxed for a period of 30 h at 75-80oC. The progress of the reaction was monitored by TLC to ensure formation of product and complete conversion of starting. On reaction completion solvent was distilled off and water (750 ml) was added to the reaction mass followed by toluene (300 ml). The resulting solution was cooled to 30oC and stirred for 30 minutes before layer separation. The organic layer was washed further with water (2×100 ml) and dried over sodium sulphate. To the organic layer IPA-HCl (72 g, 20 %) was added till pH is acidic (2-2.5).The product precipitated as solid hydrochloride salt was isolated by filtration and recrystallized from methanol. Yield: 181 g, 82% m.p., 105-107°C.
-C-H stretching (2959-2840), -C=O stretching (1717), -C=C stretching (1605, 1514, 1459), asymmetrical -C-O-C and –C-O stretching (1265-1130), symmetrical -C-O-C stretching (1023)
Chemical Shift ð, 1.25-1.22 ppm (t, 3H, -CH3), 1.57-1.51 (m, 3H, -CH3), 1.90-1.81 (d, 2H, -CH2), 2.16-2.11(m, 2H, – CH2), 2.54-2.25 (m, 1H, -CH), 3.11-3.05 (m, 4H, -CH2), 3.58-3.54 (t, 2H, -CH2), 3.77 (s, 3H, -OCH3), 3.91 (s, 6H, – OCH3), 4.37-4.33 (d, 2H, -CH2), 6.87-6.79 (m, 3H, Ar-H), 7.14-7.12 (d, 2H, Ar-H), 7.52-7.50 (d, 1H, Ar-H), 7.68-7.65 (m, 1H, Ar-H)
Antispasmodic drugs relieve cramps or spasms of the stomach, intestines, and bladder. Antispasmodics are classes (group) of drugs that can help to control some symptoms that arise from the gut, in particular, gut spasm. There are two main types namely “Antimuscarinics” and “Smooth muscle relaxants”. Antispasmodics are commonly used in “Irritable bowel syndrome” (IBS) to help relieve some of the symptoms of IBS such as spasm (colic), bloating and abdominal (stomach) pain and to reduce the motility (movement) of the intestines (gut) [1].
After understanding further the medicinal importance of antispasmodics and their ever increasing demand worldwide, we pursue to undertake the detailed synthetic and pharmacological study of antispasmodics to identify novel candidates as potential drug substances. Our parallel interest also lies on identifying novel antimicrobials since over the years; antibiotics are known to be the major protective agents against bacterial infections. However, the usage of antibiotics and antibacterial chemotherapeutics is becoming more and more restricted in the present age, despite the fact that there exist a large number of antibiotics. This is largely attributed to the emergence of drug-resistant bacteria, which render even some of the most broad spectrum antibiotics ineffective. In addition, most antibiotics have side effects. Thus, it becomes essential to investigate newer drugs with less resistance. Different studies on search of newer antimicrobials and antibacterial have revealed that moderate to remarkable antimicrobial or antibacterial action is present in several compounds, belonging to various pharmacological categories, such as antihistamines [2-4], tranquilizers [5], antihypertensive [6], anti-psychotics [7-11] anti-spasmodic [12] and anti-inflammatory agents [13]. Such compounds, having antibacterial properties in addition to their predesignated pharmacological actions, are termed as non-antibiotics [12]. Many of these compounds possess two or three benzene rings and nitrogen in the secondary or tertiary state in their molecular structure which is expected to be one of the bases for exhibiting antimicrobial potency [14]. Based on this rationale and to pursue our interest to identify newer antispasmodic agents with sedative and antimicrobial properties
[1] M. H. Pittler, E. Ernst, Am. J. Gastroenterol., 1998, 93 (7), 1131–5. [2] S. G. Dastidar, P. K. Saha, B. Sanyamat, A. N. Chakrabarty, J. Appl. Bacteriol., 1976, 41, 209- 214. [3] D. Chattopadyay, S. G. Dastidar, A. Chakrabarty, Arzneimittelforschang, 1988, 38, 869-872. [4] A. Chakrabarty, D. P. Acharya, D. K. Neogi, S. G. Dastidar, Indian J. Med. Res., 1989, 89, 233-237. [5] S. K. Dash, S. G. Dastidar, A. Chakrabarty, Indian J. Exp. Biol., 1977, 15, 324-326. [6] S. G. Dastidar, U. Mondal, S. Niyogi, A. Chakrabarty, Indian J. Med. Res., 1986, 84, 142- 147. [7] J. Molnar, Y. Mandi, J. Kiral, Acta Microbiol Acad Sci Hung., 1976, 23, 45-54. [8] J. E. Kristiansen, Acta Pathol. Microbial Immunol. Scand., 1992, 100 (Suppl. 30), 7-14 [9] S. G. Dastidar, A. Chaudhury, S. Annadurai, M. Mookerjee, A. Chakrabarty, J. Chemother., 1995, 7, 201-206. [10] V. Radhakrishnan, K. Ganguly, M. Ganguly, S. G. Dastidar, A. Chakrabarty, Indian J. Exp. Biol., 1999, 37, 671-675. [11] P. Bourlioux, J. M. Moreaux, W. J. Su, H. Boureau, Acta Pathol. Microbial. Immunol Scand., 1992, 100 (Suppl. 30), 40-43. [12] S. G. Dastidar, A. Chakrabarty, J. Molnar, N. Motohashi, National Institute of Science Communication (NISCOM), New Delhi, 1998, pp. 15. [13] S. Annadurai, S. Basu, S. Ray, S. G. Dastidar, A. C
Mebeverine is a drug used to alleviate some of the symptoms of irritable bowel syndrome. It works by relaxing the muscles in and around the gut.[1]
Medical use
Mebeverine is used to alleviate some of the symptoms of irritable bowel syndrome (IBS) and related conditions; specifically stomach pain and cramps, persistent diarrhoea, and flatulence.[2]
Data from controlled clinical trials have not found a difference from placebo or statistically significant results in the global improvement of IBS.[3][4]
It has not been tested in pregnant women nor in pregnant animals so pregnant women should not take it; it is expressed at low levels in breast milk, while no adverse effects have been reported in infants, breastfeeding women should not take this drug.[1]
Adverse effects
Adverse effects include hypersensitivity reactions and allergic reactions, immune system disorders, skin disorders including hives, oedema and widespread rashes.[2]
Additionally, the following adverse effects have been reported: heartburn, indigestion, tiredness, diarrhoea, constipation, loss of appetite, general malaise, dizziness, insomnia, headache, and decreased pulse rate.[1]
It does not have systemic anticholinergic side effects.[2]
Mebeverine can, on highly rare occasions, cause drug-induced acute angle closure glaucoma.[5]
Mechanism of action
Mebeverine is an anticholinergic but its mechanism of action is not known; it appears to work directly on smooth muscle within the gastrointestinal tract and may have an anaesthetic effect, may affect calcium channels, and may affect muscarinic receptors.[2]
It is metabolized mostly by esterases, and almost completely. The metabolites are excreted in urine.[2]
Mebeverine exists in two enantiomeric forms. The commercially available product is a racemic mixture of them. A study in rats indicates that the two have different pharmacokinetic profiles.[6]
History
It is a second generation papaverine analog, and was first synthesized around the same time as verapamil.[7]
It was first registered in 1965.[8]
Availability
Mebeverine is a generic drug and is available internationally under many brand names.[9]
SYN
Anon., Belgian Patent 609,490 (1962); T. Kralt,
H. O. Moes, A. Lindner and W. J. Asma, German Patent 1,126,889 (1962); Chem. Abstr., 59: 517b
(1963).
SYN

SYN
https://www.sciencedirect.com/science/article/abs/pii/S0731708502000237
References
- ^ Jump up to:a b c “Colofac data sheet” (PDF). New Zealand Medicines and Medical Devices Safety Authority. 14 June 2017. Retrieved 21 July2017.
- ^ Jump up to:a b c d e “Colofac Tablets 135mg – Summary of Product Characteristics (SPC)”. UK Electronic Medicines Compendium. 26 August 2016. Retrieved 21 July 2017.
- ^ Annaházi A, Róka R, Rosztóczy A, Wittmann T (May 2014). “Role of antispasmodics in the treatment of irritable bowel syndrome”. World Journal of Gastroenterology. 20 (20): 6031–43. doi:10.3748/wjg.v20.i20.6031. PMC 4033443. PMID 24876726.
- ^ Darvish-Damavandi M, Nikfar S, Abdollahi M (February 2010). “A systematic review of efficacy and tolerability of mebeverine in irritable bowel syndrome”. World Journal of Gastroenterology. 16(5): 547–53. doi:10.3748/wjg.v16.i5.547. PMC 2816265. PMID 20128021.
- ^ Lachkar Y, Bouassida W (March 2007). “Drug-induced acute angle closure glaucoma”. Current Opinion in Ophthalmology. 18 (2): 129–33. doi:10.1097/ICU.0b013e32808738d5. PMID 17301614. S2CID 30903966.
- ^ Hatami M, Farhadi K, Tukmechi A (August 2012). “Fiber-based liquid-phase micro-extraction of mebeverine enantiomers followed by chiral high-performance liquid chromatography analysis and its application to pharmacokinetics study in rat plasma”. Chirality. 24(8): 634–9. doi:10.1002/chir.22057. PMID 22700279.
- ^ Sneader W (2005). Drug Discovery: A History. John Wiley & Sons. p. 132. ISBN 9780471899792.
- ^ “Mebeverine”. druginfosys. Retrieved 1 February 2015.
- ^ “Mebeverine”. International. drugs.com. Retrieved 1 February2015.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | By mouth |
| ATC code | A03AA04 (WHO) |
| Legal status | |
| Legal status | UK: P (Pharmacy medicines)US: Not approvedIn general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 3625-06-7 HCl: 2753-45-9 |
| PubChem CID | 4031 |
| ChemSpider | 3891 |
| UNII | 7F80CC3NNVHCl: 15VZ5AL4JN |
| KEGG | D04868 |
| ChEMBL | ChEMBL282121 |
| CompTox Dashboard (EPA) | DTXSID6023238 |
| ECHA InfoCard | 100.020.756 |
| Chemical and physical data | |
| Formula | C25H35NO5 |
| Molar mass | 429.557 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI | |
| (verify) |
//////////mebeverine, мебеверин ,ميبيفيرين ,美贝维林 , Antispasmodic

NEW DRUG APPROVALS
ONE TIME
$10.00
Sotradecol, Sodium tetradecyl sulfate

Sotradecol
Sodium tetradecyl sulfate
cas 139-88-8, Na FORM
free form 300-52-7
- Molecular FormulaC14H29NaO4S
- Average mass316.432 Da
139-88-8 [RN], cas 1191-50-0,
- CAS No.68043-79-8
4-Undecanol, 7-ethyl-2-methyl-, hydrogen sulfate, sodium salt (1:1)
7-Ethyl-2-methyl-4-undecyl sulfate sodium salt
UNII:Q1SUG5KBD6
натрия тетрадецилсульфат [Russian] [INN]
تتراديسيل سولفات صوديوم [Arabic] [INN]
十四烷硫酸钠 [Chinese] [INN]
CAS Registry Number: 139-88-8
CAS Name: 7-Ethyl-2-methyl-4-undecanol hydrogen sulfate sodium salt
Additional Names: 7-ethyl-2-methyl-4-hendecanol sulfate sodium salt; sodium 2-methyl-7-ethyl-4-undecyl sulfate; sodium 7-ethyl-2-methylundecyl-4-sulfate
Trademarks: Sotradecol (Elkins-Sinn); Tergitol 4; Trombavar; Trombovar
Molecular Formula: C14H29NaSO4, Molecular Weight: 316.43
Percent Composition: C 53.14%, H 9.24%, Na 7.27%, S 10.13%, O 20.22%
Properties: White, waxy solid. Sol in water, alcohol, ether. The pH of a 5% soln is from 6.5 to 9.0. Surface tension (dynes/cm) of aq soln at 25°: 56.5 dynes/cm (0.05% w/w); 52 (0.10%); 47 (0.20%); 40 (0.50%); 35 (1.0%). LD50 orally in rats: 4.95 g/kg, H. F. Smyth, C. P. Carpenter, J. Ind. Hyg. Toxicol.30, 63 (1948).
Toxicity data: LD50 orally in rats: 4.95 g/kg, H. F. Smyth, C. P. Carpenter, J. Ind. Hyg. Toxicol.30, 63 (1948)
Use: Wetting agent.
Therap-Cat: Sclerosing agent., Keywords: Sclerosing Agent.
Synonyms of Sodium Tetradecyl Sulfate [INN]
- 4-Ethyl-1-isobutyloktylsiran sodny
- EINECS 205-380-3
- Natrii tetracylis sulfas
- Natrii tetradecylis sulfas
- Natrii tetradecylis sulfas [Latin]
- Natrii tetradecylsulfas
- NSC 755887
- Obliterol
- Sodium sotradecol
- Sodium tetradecyl sulfate
- Sotradecol
- Tergitol
- Tergitol 4
- Tergitol anionic 4
- Tergitol penetrant 4
- Tetradecilsulfato sodico
- Tetradecilsulfato sodico [Spanish]
- Tetradecyl sulfate de sodium
- Trombovar
- UNII-Q1SUG5KBD6
- Varicol
An anionic surface-active agent used for its wetting properties in industry and used in medicine as an irritant and sclerosing agent for hemorrhoids and varicose veins.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Sodium tetradecyl sulfate is an anionic surfactant which occurs as a white, waxy solid. The structural formula is as follows:
 |
C14H28NaS04 7-Ethyl -2-methyl -4-hendecanol sulfate sodium salt MW 316.44Sotradecol® (sodium tetradecyl sulfate injection) is a sterile nonpyrogenic solution for intravenous use as a sclerosing agent.
1% (10 mg/mL): Each mL contains sodium tetradecyl sulfate 10 mg, benzyl alcohol 0.02 mL and dibasic sodium phosphate, anhydrous 4.0 mg in Water for Injection. pH 7.9; monobasic sodium phosphate and/or sodium hydroxide added, if needed, for pH adjustment.
3% (30 mg/mL): Each mL contains sodium tetradecyl sulfate 30 mg, benzyl alcohol 0.02 mL and dibasic sodium phosphate, anhydrous 9.0 mg in Water for Injection. pH 7.9; monobasic sodium phosphate and/or sodium hydroxide added, if needed, for pH adjustment.
Sodium tetradecyl sulfate (STS) is a commonly used synonym for 7-ethyl-2-methyl-4-undecanyl sulfate sodium salt[1] which is anionicsurfactant that is the active component of the sclerosant drug Sotradecol. It is commonly used in the treatment of varicose and spider veins of the leg, during the procedure of sclerotherapy.[2] Being a detergent, its action is on the lipid molecules in the cells of the vein wall, causing inflammatory destruction of the internal lining of the vein and thrombus formation eventually leading to sclerosis of the vein. It is used in concentrations ranging from 0.1% to 3% for this purpose. It is occasionally used for the treatment of stabilisation of joints that regularly dislocate, particularly in patients with Ehlers-Danlos syndrome.[3] In the UK, Ireland, Italy, Australia, New Zealand and South Africa, it is sold under the trade-name Fibro-Vein in concentrations of 0.2%, 0.5%, 1.0%, and 3%.[4]
Synthesis
It may be prepared by the aldol condensation of methyl isobutyl ketone and 2-ethylhexanal (which is itself formed by the aldol self-concensation of butyraldehyde), followed by sulfonation of the resulting alcohol.
SYN

RSC Advances, 10(22), 12788-12799; 2020
https://pubs.rsc.org/en/content/articlelanding/2020/RA/D0RA00386G

PAT
CN 106278961
CN 106278958
U.S.S.R., 1051067,
NMR
| Compound name: | Sodium Tetradecyl Sulfate |
|---|---|
| Spectrum type: | 1H NMR Spectrum (1D, 400 MHz, DMSO-d6, experimental) |

References
- ^ “SOTRADECOL® (Sodium tetradecyl sulfate)” (PDF). Retrieved 29 August 2014.
- ^ Jenkinson HA, Wilmas KM, Silapunt S (November 2017). “Sodium Tetradecyl Sulfate: A Review of Clinical Uses”. Dermatologic Surgery. 43 (11): 1313–1320. doi:10.1097/DSS.0000000000001143. PMID 28430735.
- ^ Burling F (2019). “Comparison of tetradecyl sulfate versus polidocanol injections for stabilisation of joints that regularly dislocate in an Ehlers-Danlos population”. BMJ Open Sport & Exercise Medicine. 5 (1): e000481. doi:10.1136/bmjsem-2018-000481. PMC 6350757. PMID 30792884.
- ^ Fibro-Vein history and details
| Clinical data | |
|---|---|
| Other names | 7-Ethyl-2-methyl-4-hendecanol sulfate sodium salt |
| AHFS/Drugs.com | Consumer Drug Information |
| Routes of administration | Intravenous injection |
| ATC code | C05BB04 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 139-88-8 |
| PubChemCID | 14492 |
| ChemSpider | 8440 |
| UNII | Q1SUG5KBD6 |
| ChEMBL | ChEMBL1200354 |
| CompTox Dashboard (EPA) | DTXSID3041530 |
| ECHA InfoCard | 100.004.892 |
| Chemical and physical data | |
| Formula | C14H29NaO4S |
| Molar mass | 316.43 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////Sodium tetradecyl sulfate, sotradecol, Sclerosing Agent, varicose veins
CCCCCCCCCCCCCCOS(=O)(=O)[O-].[Na+]

NEW DRUG APPROVALS
ONE TIME
$10.00
POLIDOCANOL


POLIDOCANOL
Synonym: Polidocanol; C12E9, Dodecyl nonaethylene glycol ether, Dodecylnonaglycol, Polidocanol, Polyoxyethylene (9) lauryl ether; trade names: Asclera, Aethoxysklerol and Varithena; Laureth-9; Dodecylnonaoxyethylene glycol monoether
IUPAC/Chemical Name: 3,6,9,12,15,18,21,24,27-nonaoxanonatriacontan-1-ol
3055-99-0
Chemical Formula: C30H62O10
Exact Mass: 582.4343Polidocanol
CAS Registry Number: 9002-92-0
CAS Name: a-Dodecyl-w-hydroxypoly(oxy-1,2-ethanediyl)
Additional Names: polyethylene glycol (9) monododecyl ether; dodecyl alcohol polyoxyethylene ether; hydroxypolyethoxydodecane; laureth 9; polyoxyethylene lauryl ether
Trademarks: Aethoxysklerol (Kreussler); Aetoxisclerol (Dexo); Atlas G-4829 (ICI); Hetoxol L-9 (Heterene Chem.)Line Formula: C12H25(OCH2CH2)nOH
Literature References: Contains an average of nine ethylene oxide units and has an average mol wt ~600. Prepd by reaction of ethylene oxide and dodecyl alcohol: Pertsemlides, Soehring, Arzneim.-Forsch.10, 990 (1960). Toxicology: H. S. Zipf et al.,ibid.7, 162 (1957). Review of clinical experience: P. M. Goldman, J. Dermatol. Surg. Oncol.15, 204-209 (1989).
Properties: Sol in water, ethanol, toluene. Miscible with hot mineral, natural and synthetic oils; with fats and fatty alcohols. LD50 in mice (mg/kg): 1170 orally, 125 i.v. (Zipf).
Toxicity data: LD50 in mice (mg/kg): 1170 orally, 125 i.v. (Zipf)
Use: Solvent; nonionic emulsifier; pharmaceutic aid (surfactant); spermaticide.
Therap-Cat: Anesthetic (topical); antipruritic; sclerosing agent.
Keywords: Anesthetic (Local); Antipruritic; Sclerosing Agent.
| EINECS | 221-284-4 | ||
| CAS No. | 3055-99-0 | Density | 1.007 g/cm3 |
| PSA | 103.30000 | LogP | 4.04900 |
| Solubility | Melting Point | 33-36 °C | |
| Formula | C30H62O10 | Boiling Point | 615.857 °C at 760 mmHg |
| Molecular Weight | 582.43 | Flash Point | 326.259 °C |
Polidocanol is a local anaesthetic and antipruritic component of ointments and bath additives. It relieves itching caused by eczema and dry skin.[1] It has also been used to treat varicose veins,[2] hemangiomas, and vascular malformations.[3] It is formed by the ethoxylation of dodecanol.
Polidocanol is a local anaesthetic and antipruritic component of ointments and bath additives. It relieves itching caused by eczema and dry skin. It is formed by the ethoxylation of dodecanol. The substance is also used as a sclerosant, an irritant injected to treat varicose veins, under the trade names Asclera, Aethoxysklerol and Varithena. Polidocanol causes fibrosis inside varicose veins, occluding the lumen of the vessel, and reducing the appearance of the varicosity. The FDA has approved polidocanol injections for the treatment of small varicose (less than 1 mm in diameter) and reticular veins (1 to 3 mm in diameter). Polidocanol works by damaging the cell lining of blood vessels, causing them to close and eventually be replaced by other types of tissue.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

Yu, Zeqiong; Bo, Shaowei; Wang, Huiyuan; Li, Yu; Yang, Zhigang; Huang, Yongzhuo; Jiang, Zhong-Xing. Application of Monodisperse PEGs in Pharmaceutics: Monodisperse Polidocanols. Molecular Pharmaceutics. Volume 14. Issue 10. Pages 3473-3479. 2017.
SYN 2

Jiang, Zhongxing; Yu, Zeqiong. Process for preparation of monodisperse nona-polyethylene glycol dodecyl alcohol monoether and sulfate. Assignee Wuhan University, Peop. Rep. China. CN 106316802. (2017).
Sclerotherapy
Polidocanol is also used as a sclerosant, an irritant injected to treat varicose veins, under the trade names Asclera, Aethoxysklerol[4] and Varithena.[5] Polidocanol causes fibrosis inside varicose veins, occluding the lumen of the vessel, and reducing the appearance of the varicosity.
The FDA has approved polidocanol injections for the treatment of small varicose (less than 1 mm in diameter) and reticular veins (1 to 3 mm in diameter). Polidocanol works by damaging the cell lining of blood vessels, causing them to close and eventually be replaced by other types of tissue.[6][7] Polidocanol in the form of Varithena injected in the greater saphenous vein can cause the eruption of varicose and spider veins throughout the lower leg. This procedure should be done with caution and with the knowledge that the appearance of the leg may be forever compromised.

Pure polidocanol for pharmaceutical use
On March 30th,2010 the FDA approved Polidocanol under the trade name Asclera. Polidocanol is a sclerosing agent indicated to treat uncomplicated spider veins (varicose veins ≤1 mm in diameter) and uncomplicated reticular veins (varicose veins 1 to 3 mm in diameter) in the lower extremities. Varicose veins develop when the small valves inside the veins no longer work properly, allowing the blood to flow backwards and then pool in the vein.
When injected intravenously, Polidocanol works by locally damaging the endothelium of the blood vessel, causing platelets to aggregate at the site of damage and attach to the venous wall. Eventually, a dense network of platelets, cellular debris and fibrin occludes the vessel, which is then replaced with connective fibrous tissue. As one would expect for this type of molecule and also the mechanism of action, there is believed to be no specific molecular target for Polidocanol.
Polidocanol is a large ‘small molecule’ drug (Molecular Weight of 583 g.mol-1), with a mean half-life of 1.5 hr. Polidocanol is administrated intravenously and the strength of the solution and the volume injected depend on the size and extent of the varicose veins. Thus, the recommended dosage is 0.1 to 0.3 mL for each injection (Asclera 0.5% for spider veins and Asclera 1% for reticular veins) into each varicose vein, and a maximum recommended volume per treatment session of 10 mL.
Polidocanol’s chemical structure is 2-[2-[2-[2-[2-[2-[2-[2-[2-(dodecyloxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol. It is a non-ionic detergent, similar to polyethylene glycol (PEG) in structure, consisting of two components, a polar hydrophilic (dodecyl alcohol) and an apolar hydrophobic (polyethylene oxide – the part in brackets in the chemical structure) chain.
References
- ^ “E45 itch relief cream”. netdoctor.co.uk. Retrieved 2007-07-12.
- ^ Star P, Connor DE, Parsi K (April 2018). “Novel developments in foam sclerotherapy: Focus on Varithena® (polidocanol endovenous microfoam) in the management of varicose veins”. Phlebology. 33 (3): 150–162. doi:10.1177/0268355516687864. PMID 28166694.
- ^ Gao Z, Zhang Y, Li W, Shi C (January 2018). “Effectiveness and safety of polidocanol for the treatment of hemangiomas and vascular malformations: A meta-analysis”. Dermatologic Therapy. 31 (1). doi:10.1111/dth.12568. PMID 29082587.
- ^ Sclerotherapy, Laurence Z Rosenberg, MD, eMedicine.com
- ^ “Varithena™ (polidocanol injectable foam) For Intravenous Use. Full Prescribing Information” (PDF). Biocompatibles, Inc. Archived from the original (PDF) on 4 August 2016. Retrieved 1 October 2015.
- ^ Facts and Companies: Varicose Vein Treatment Approved
- ^ “Asclera Full Prescribing Information in Drug Reference Encyclopedia”. Retrieved 2010-04-11.
| Clinical data | |
|---|---|
| Other names | PolydocanolLaureth 9Macrogol lauryl etherLauromacrogolPEG-9 lauryl alcoholPOE-9 lauryl alcoholDodecylpolyethyleneglycoletherHydroxyl polyethoxy dodecaneOxypolyethoxydodecane |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category | Topical: allowed Injection: contraindication in months 1–3 and after week 36 |
| Routes of administration | topical, subcutaneous injection |
| ATC code | C05BB02 (WHO) |
| Legal status | |
| Legal status | OTC (topical), ℞ (injection) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 9002-92-0 3055-99-0 |
| PubChem CID | 656641 |
| ChemSpider | 570993 |
| UNII | 0AWH8BFG9A |
| KEGG | D01993 |
| ChEMBL | ChEMBL1201751 |
| ECHA InfoCard | 100.019.351 |
| Chemical and physical data | |
| Formula | C30H62O10 |
| Molar mass | 582.816 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
////////POLIDOCANOL, Anesthetic , Antipruritic, Sclerosing Agent,
CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO

NEW DRUG APPROVALS
ONE TIME
$10.00
DOCUSATE
DOCUSATE
1,4-Bis(2-ethylhexyl) sulfosuccinate
- Molecular FormulaC20H38O7S
- Average mass422.577 Da
1,4-Bis[(2-ethylhexyl)oxy]-1,4-dioxobutane-2-sulfonic acid
10041-19-7[RN]
233-124-0[EINECS]
Docusate Sodium
Dioctyl sodium sulfosuccinate
sodium;1,4-bis(2-ethylhexoxy)-1,4-dioxobutane-2-sulfonate
CAS Registry Number: 577-11-7
CAS Name: Sulfobutanedioic acid 1,4-bis(2-ethylhexyl) ester sodium salt
Additional Names: sulfosuccinic acid 1,4-bis(2-ethylhexyl) ester S-sodium salt; bis(2-ethylhexyl)sodium sulfosuccinate; dioctyl sodium sulfosuccinate; sodium dioctyl sulfosuccinate; DSS
Trademarks: Aerosol OT (Cyanamid); Colace (Roberts); Comfolax (Searle); Coprola (Dunster); Dioctylal (Continental Pharma); Dioctyl (Medo); Diotilan (Chinoin); Disonate (Lannett); Doxinate (Hoechst); Doxol (Blair); Dulcivac (Harvey); Jamylène (Thaplix); Molatoc; Molcer (Wallace); Nevax; Regutol (Schering-Plough); Soliwax (Concept Pharm.); Velmol (Berlex); Waxsol (Norgine); Yal (Ritter)
Molecular Formula: C20H37NaO7S
Molecular Weight: 444.56
Percent Composition: C 54.03%, H 8.39%, Na 5.17%, O 25.19%, S 7.21%
Literature References: Prepn: Jaeger, US2028091; US2176423 (1936, 1939, both to Am. Cyanamid). Structure and wetting power: Caryl, Ind. Eng. Chem.33, 731 (1941). Comprehensive description: S. Ahuja, J. Cohen, Anal. Profiles Drug Subs.2, 199-219 (1973); 12, 713-720 (1983). For structure see Docusate calcium.
Properties: Available as wax-like solid, usually in rolls of tissue-thin material; also as 50-75% solns in various solvents. Soly in water (g/l): 15 (25°), 23 (40°), 30 (50°), 55 (70°). Sol in CCl4, petr ether, naphtha, xylene, dibutyl phthalate, liq petrolatum, acetone, alcohol, vegetable oils. Very sol in water + alcohol, water + water-miscible organic solvents. Stable in acid and neutral solns; hydrolyzes in alkaline solns.
Derivative Type: Docusate potassium
CAS Registry Number: 7491-09-0
Trademarks: Rectalad (Carter-Wallace)
Molecular Formula: C20H37KO7S
Molecular Weight: 460.67
Percent Composition: C 52.14%, H 8.10%, K 8.49%, O 24.31%, S 6.96%
NOTE: Ingredient of the laxative Peri-Colace (Roberts) which also contains casanthranol.Use: Sodium salt as pharmaceutic aid (surfactant); as wetting agent in industrial, pharmaceutical, cosmetic and food applications; dispersing and solubilizing agent in foods; adjuvant in tablet formation.
Therap-Cat: Stool softener.
Therap-Cat-Vet: Stool softener.
Keywords: Laxative/Cathartic.
Docusate Calcium
CAS Registry Number: 128-49-4
CAS Name: Sulfobutanedioic acid 1,4-bis(2-ethylhexyl)ester calcium salt
Additional Names: bis[2-ethylhexyl]calcium sulfosuccinate; calcium dioctyl sulfosuccinate; dioctyl calcium sulfosuccinate
Trademarks: Surfak (HMR)
Molecular Formula: C40H74CaO14S2
Molecular Weight: 883.22
Percent Composition: C 54.40%, H 8.44%, Ca 4.54%, O 25.36%, S 7.26%
Literature References: Prepd from dioctyl sodium sulfosuccinate dissolved in isopropanol and from calcium chloride dissolved in methanol: Klotz, US3035973 (1962 to Lloyd Brothers).
Properties: White precipitate. Sol in mineral and vegetable oils, liq polyethylene glycol. Practically insol in glycerol. Claimed to have greater surface-active wetting properties than the sodium salt.
NOTE: Ingredient of Doxidan (HMR) which also contains phenolphthalein.
Therap-Cat: Stool softener.
Keywords: Laxative/Cathartic.
Derivatives
free acid
- Formula:C20H38O7S
- MW:422.58 g/mol
- CAS-RN:10041-19-7
- EINECS:233-124-0
calcium salt
- Formula:C40H74CaO14S2
- MW:883.23 g/mol
- CAS-RN:128-49-4
- EINECS:204-889-8
potassium salt
- Formula:C20H37KO7S
- MW:460.67 g/mol
- CAS-RN:7491-09-0
- EINECS:231-308-5
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 141-02-6 | C20H36O4 | bis(2-ethylhexyl) fumarate | 2-Butenedioic acid (E)-, bis(2-ethylhexyl) ester |
| C4H4O4 | (E)-2-butenedioic acid | ||
| 104-76-7 | C8H18O | 2-ethyl-1-hexanol | 1-Hexanol, 2-ethyl- |
SYN
https://scialert.net/fulltext/?doi=jas.2011.1396.1400
 | |
| Fig. 1: | Synthesis of Trihexyltetradecylphosphonium octylsulfosuccinate [P6, 6, 6, 14][docusate] |
SYN

Docusate is the common chemical and pharmaceutical name of the anionbis(2-ethylhexyl) sulfosuccinate, also commonly called dioctyl sulfosuccinate (DOSS).[2][3][4]
Salts of this anion, especially docusate sodium, are widely used in medicine as laxatives and as stool softeners, by mouth or rectally.[1] It is on the World Health Organization’s List of Essential Medicines.[5][6] Some studies claim that docusate is not more effective than a placebo for improving constipation.[7][8][9][10] Other docusate salts with medical use include those of calcium and potassium.[11][1][2]
Docusate salts are also used as food additives, emulsifiers, dispersants, and wetting agents, among other uses.[12]
History
Sodium docusate was patented in 1937 by Coleman R. Caryl and Alphons O. Jaeger for American Cyanamid,[3] which commercialized it for many years as a detergent under the brand name Aerosol OT.
Its use for the treatment of constipation was first proposed in 1955 by James L. Wilson and David G. Dickinson,[4] and quicky popularized under the name Doxinate.[13]
Medical use
Constipation
The main medical use of docusate sodium is to treat constipation, acting as a laxative and stool softener. In painful anorectal conditions such as hemorrhoid and anal fissures, it can help avoid pain caused by straining during bowel movements.
When administered by mouth, a bowel movement often occurs in 1 to 3 days,[1] while rectal use may be effective within 20 minutes.[14]
Sodium docusate is recommended as a stool softener for children.[1]
However, its effectiveness for constipation is poorly supported by evidence.[7][8] Multiple studies have found docusate to be no more effective than a placebo for improving constipation.[7][8][9][10] Others have found it to be less useful for the treatment of chronic constipation than psyllium.[10][15][16]
The medication may be given to people who are receiving opioid medication, although prolonged use may cause irritation of the gastrointestinal tract.[10][16]
Other medical uses
Docusate sodium, when used with ear syringing, may help with earwax removal, particularly in the case of impaction.[17]
Sodium docusate is also used as a lubricant in the production of tablets and as an emulsifier in topical preparations and other suspensions.[18]
Precautions and contraindications
Docusate sodium is approved and recommended as safe during pregnancy and breastfeeding.[19][20]
Docusate is not recommended in people with appendicitis, acute abdomen, or ileus.[16]
When taken by mouth it should be ingested with plenty of water.
Side effects
Side effects are uncommon and typically mild,[1] and may include stomach pain, abdominal cramps or diarrhea,[1] Efficacy decreases with long-term use, and may cause poor bowel function.[11]
Serious allergic reactions may occur with the drug. The most severe side effect of docusate, although very rare, is rectal bleeding.[21]
Interactions
Docusate might increase resorption of other drugs, for example, dantron (1,8-dihydroxyanthraquinone).[16]
Mechanism of action
Docusate sodium works by allowing more water to be absorbed by the stool.[11][22]
Docusate does not stay in the gastrointestinal tract, but is absorbed into the bloodstream and excreted via the gallbladder[16] after undergoing extensive metabolism.
The effect of docusate may not necessarily be all due to its surfactant properties. Perfusion studies suggest that docusate inhibits fluid absorption or stimulates secretion in the portion of the small intestine known as the jejunum.
Pharmaceutical brand names
In the U.S., docusate sodium for pharmaceutical use is available under multiple brand names: Aqualax, Calube, Colace, Colace Micro-Enema, Correctol Softgel Extra Gentle, DC-240, Dialose, Diocto, Dioctocal, Dioctosoftez, Dioctyn, Dionex, Doc-Q-Lace, Docu Soft, Docucal, Doculax, Docusoft S, DOK, DOS, Doss-Relief, DSS, Dulcolax – Stool Softener (not to be confused with another drug marketed under the Dulcolax brand, bisacodyl, which is a stimulant laxative), Ex-Lax Stool Softener, Fleet Sof-Lax, Genasoft, Kasof, Laxa-basic, Modane Soft, Octycine-100, Pedia-Lax, Preferred Plus Pharmacy Stool Softener, Regulax SS, Sulfalax Calcium, Sur-Q-Lax, Surfak Stool Softener, and Therevac-SB. Generic preparations are also available.
In the UK, dioctyl sodium sulfosuccinate is sold under the brand name Docusol (Typharm Ltd) and DulcoEase (Boehringer Ingelheim).
In Australia, dioctyl sodium sulfosuccinate is sold as Coloxyl and Coloxyl with senna.
In India, preparations include Laxatin by Alembic, Doslax by Raptakos Laboratories, Cellubril by AstraZeneca, and Laxicon by Stadmed.
Other uses
Dioctyl sodium sulfosuccinate is used as a surfactant in a wide range of applications, often under the name Aerosol-OT.[4][23] It is unusual in that it is able to form microemulsions without the use of co-surfactants, and it has a rich variety of aqueous-phase behavior including multiple liquid crystalline phases.[24]
Food additive
Dioctyl sodium sulfosuccinate has been approved by the US FDA as a “generally recognized as safe” (GRAS) additive.[25] It is used in a variety of food products, as a surface active agent, stabilizer, thickener, wetting agent, processing aid, solubilizing agent, emulsifier, and dispersant. The highest amount found in food products is 0.5% by weight, which include pasteurized cheese spreads, cream cheeses and salad dressings.[26] The FDA also approved its use as a wetting agent or solubilizer for flavoring agents in carbonated and non-carbonated drinks at levels up to 10 parts per million.[25]
Microencapsulation
Sodium docusate is the most widely used surfactant in reverse micelleencapsulation studies.[27]
Non-medical brand names
As a surfactant, docusate sodium is or has been commercialized under many brand names, including DSSj Aerosol OT, Alphasol OT, Colace, Complemix, Coprol, Dioctylal, Dioctyl-Medo Forte, Diotilan, Diovac, Disonate, Doxinate, Doxol, Dulsivac, Molatoc, Molofac, Nevax, Norval, Regutol, Softili, Solusol, Sulfimel DOS, Vatsol OT, Velmol, and Waxsol[28]
Chemistry
Structure and properties
The structural formula of the docusate anion is R−O−C(=O)−CH(SO−
3)−CH
2−C(=O)−O−R, where R is the 2-ethylhexyl groupH
3C−(CH
2)
3−C(−CH
2−CH
3)H−CH
2−. The conjugate acid can be described as the twofold carboxylate ester of sulfosuccinic acid with 2-ethylhexanol.
The compound is a white, wax-like, plastic solid, with an odor suggestive of octyl alcohol. It starts to decompose at about 220 °C.[28]
Solubility of dioctyl sodium sulfosuccinate in water is 14 g/L at 25 °C, increasing to 55 g/L at 70 °C.[28] Solubility is better in less polar solvents: 1:30 in ethanol, 1:1 in chloroform and diethylether, and practically unlimited in petroleum ether (25 °C). It also is highly soluble in glycerol, although this is a rather polar solvent. It is also highly soluble in xylene, oleic acid, acetone, diacetone alcohol, methanol, isopropanol, 2-butanol, methyl acetate, ethyl acetate, furfurol, and vegetable oils.[28]
The ester groups are easily cleaved under basic conditions, but are stable against acids.[16]
Synthesis
Sodium dioctyl sulfosuccinate can be obtained by treating dioctyl maleate with sodium bisulfite. The bisulfite anion adds to the double bond:−CH=CH− + HSO−
3 → −CH(−SO−
3)−CH
2−
Toxicity
Ingestion may cause the side effects described above, such as diarrhea, intestinal bloating, and occasionally cramping pains. Dioctyl sodium sulfosuccinate is not known to be carcinogenic, mutagenic, or teratogenic.[29]
Marine species
Dioctyl sodium sulfosuccinate is of low toxicity for crustaceans such as the hermit crabClibanarius erythropus and the shrimp Crangon crangon. Toxicity for molluscs varies widely, with 48-hour LD50 found between 5 mg/l for the common limpet and 100 mg/l for the common periwinkle. Various species of phytoplankton have an LD50 around 8 mg/l.
In a 2010 study, dioctyl sodium sulfosuccinate exhibited higher toxicity against bacteria (Vibrio fischeri, Anabaena sp.) and algae (Pseudokirchneriella subcapitata) than did a number of fluorinated surfactants (PFOS, PFOA, or PFBS). Measuring bioluminescence inhibition of the bacteria and growth inhibition of the algae, the LD50 were in the range of 43–75 mg/l. Combinations of the fluorinated compounds with dioctyl sodium sulfosuccinate showed mid to highly synergistic effects in most settings, meaning that such combinations are significantly more toxic than the individual substances.[30]
Freshwater species
The substance is highly toxic for rainbow trout with a median lethal concentration (LC50) of 0.56 mg/l after 48 hours for the pure substance. It is only slightly to moderately toxic for rainbow trout fingerlings, and slightly toxic for harlequin rasboras (LC50 27 mg/l of a 60% formulation after 48 hours).
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d e f g h “Docusate Salts”. The American Society of Health-System Pharmacists. Archived from the original on 23 September 2015. Retrieved 11 August 2015.
- ^ Jump up to:a b American Society of Health-System Pharmacists (15 August 2011). “Stool Softeners”. Archived from the original on 5 September 2015.
- ^ Jump up to:a b US 2181087, Caryl CR, Jaeger AO, “Detergent composition”, issued 21 November 1939, assigned to American Cyanamid
- ^ Jump up to:a b c Wilson JL, Dickinson DG (May 1955). “Use of dioctyl sodium sulfosuccinate (aerosol O.T.) for severe constipation”. Journal of the American Medical Association. 158 (4): 261–3. doi:10.1001/jama.1955.02960040019006a. PMID 14367076.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
- ^ “Docusate – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b c Fakheri RJ, Volpicelli FM (February 2019). “Things We Do for No Reason: Prescribing Docusate for Constipation in Hospitalized Adults”. Journal of Hospital Medicine. 14 (2): 110–113. doi:10.12788/jhm.3124. PMID 30785419.
- ^ Jump up to:a b c “Dioctyl Sulfosuccinate or Docusate (Calcium or Sodium) for the Prevention or Management of Constipation: A Review of the Clinical Effectiveness”. CADTH Rapid Response Reports. 26 June 2014. PMID 25520993.
- ^ Jump up to:a b Candy B, Jones L, Larkin PJ, Vickerstaff V, Tookman A, Stone P (May 2015). “Laxatives for the management of constipation in people receiving palliative care” (PDF). The Cochrane Database of Systematic Reviews. 13 (5): CD003448. doi:10.1002/14651858.CD003448.pub4. PMC 6956627. PMID 25967924.
- ^ Jump up to:a b c d Ramkumar D, Rao SS (April 2005). “Efficacy and safety of traditional medical therapies for chronic constipation: systematic review”. The American Journal of Gastroenterology. 100 (4): 936–71. PMID 15784043.
- ^ Jump up to:a b c 2013 Nurse’s Drug Handbook. Burlington, MA: Jones & Bartlett Learning. 2013. p. 366. ISBN 9781449642846.
- ^ Ash M, Ash I (2004). Handbook of preservatives. Endicott, N.Y.: Synapse information resources. p. 375. ISBN 9781890595661.
- ^ Friedman M (October 1956). “Dioctyl sodium sulfosuccinate (doxinate) in chronic functional constipation”. American Practitioner and Digest of Treatment. 7 (10): 1588–91. PMID 13362832.
- ^ “Docusate sodium”. 18 December 2004. Archived from the original on 21 July 2011. Retrieved 6 March 2019.
- ^ Portalatin M, Winstead N (March 2012). “Medical management of constipation”. Clinics in Colon and Rectal Surgery. 25 (1): 12–9. doi:10.1055/s-0032-1301754. PMC 3348737. PMID 23449608.
- ^ Jump up to:a b c d e f Dinnendahl V, Fricke U, eds. (2010). Arzneistoff-Profile(in German). 2 (23 ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. ISBN 978-3-7741-9846-3.
- ^ “How effective is docusate as a cerumenolytic agent?”. GlobalRPH.com. Archived from the original on 23 November 2010.
- ^ Jasek W, ed. (2008). Austria-Codex Stoffliste (in German) (41 ed.). Vienna: Österreichischer Apothekerverlag. p. 316. ISBN 978-3-85200-190-6.
- ^ Yaffe SJ (2011). Drugs in pregnancy and lactation : a reference guide to fetal and neonatal risk (9 ed.). Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. p. 1651. ISBN 9781608317080.
- ^ Mahadevan U, Kane S (July 2006). “American gastroenterological association institute medical position statement on the use of gastrointestinal medications in pregnancy”. Gastroenterology. 131(1): 278–82. doi:10.1053/j.gastro.2006.04.048. PMID 16831610.
- ^ drugs.com: Docusate Archived 16 July 2010 at the Wayback Machine
- ^ Hamilton RJ (2013). Tarascon pocket pharmacopoeia : 2013 classic shirt-pocket edition (27 ed.). Burlington, Ma.: Jones & Bartlett Learning. p. 112. ISBN 9781449665869.
- ^ Whiffen AJ (1946). “Aerosol OT in the preparation of microscopic mounts of fungi”. Mycologia. 38: 346. doi:10.1080/00275514.1946.12024063. PMID 20983186.
- ^ Nave S, Eastoe J, Penfold J (November 2000). “What Is So Special about Aerosol-OT? 1. Aqueous Systems”. Langmuir. 16(23): 8733–8740. doi:10.1021/la000341q.
- ^ Jump up to:a b “GRAS Notice Inventory Agency Response Letter GRAS Notice No. GRN 000006”. Center for Food Safety and Applied Nutrition. 20 July 1998. Archived from the original on 31 October 2017. Retrieved 24 January 2020.
- ^ “CFR – Code of Federal Regulations Title 21”. http://www.accessdata.fda.gov. Retrieved 29 January 2020.
- ^ Flynn PF (2004). “Multidimensional multinuclear solution NMR studies of encapsulated macromolecules”. Prog. Nucl. Magn. Reson. Spectrosc. 45 (1–2): 31–51. doi:10.1016/j.pnmrs.2004.04.003.
- ^ Jump up to:a b c d Ahuja S, Cohen J (January 1973). “Dioctyl Sodium Sulfosuccinate”. InAnalytical Profiles of Drug Substances. Analytical Profiles of Drug Substances. 2. Academic Press. pp. 199–219. doi:10.1016/S0099-5428(08)60040-4. ISBN 9780122608025.
- ^ ScienceLab.com: Docusate sodium Material Safety Data SheetArchived 2006-10-17 at the Wayback Machine
- ^ Rosal R, Rodea-Palomares I, Boltes K, Fernández-Piñas F, Leganés F, Petre A (September 2010). “Ecotoxicological assessment of surfactants in the aquatic environment: combined toxicity of docusate sodium with chlorinated pollutants”. Chemosphere. 81 (2): 288–93. Bibcode:2010Chmsp..81..288R. doi:10.1016/j.chemosphere.2010.05.050. PMID 20579683.
External links
- “Docusate”. Drug Information Portal. U.S. National Library of Medicine.
- “Docusate sodium”. Drug Information Portal. U.S. National Library of Medicine.
- Stool Softeners at the N.I.H.PubMed Health resource.
//////////DOCUSATE, Stool softener, Laxative, Cathartic,
CCCC(CC)COC(=O)CC(C(=O)OCC(CC)CCCC)S(=O)(=O)[O-].[Na+]

NEW DRUG APPROVALS
one time
$10.00
LANSOPRAZOLE
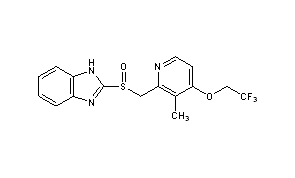

Lansoprazole
- Molecular FormulaC16H14F3N3O2S
- Average mass369.362 Da
Lansoprazole, AG-1749, ABT-006, CG-4801, A-65006, Ogast, Lanzor, Lanzo, Agopton, Opiren, Bamalite, Takepron, Lansox, Lansox, Ogastro, Monolitum, Prevacid, Zoton103577-45-3[RN]
1H-Benzimidazole, 2-[[[3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridinyl]methyl]sulfinyl]-
лансопразол [Russian] [INN], لانسوبرازول [Arabic] [INN], 兰索拉唑 [Chinese] [INN]
CAS Registry Number: 103577-45-3
CAS Name: 2-[[[3-Methyl-4-(2,2,2-trifluoro-ethoxy)-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazole
Additional Names: 2-(2-benzimidazolylsulfinylmethyl)-3-methyl-4-(2,2,2-trifluoroethoxy)pyridine
Manufacturers’ Codes: A-65006; AG-1749
Trademarks: Agopton (Takeda); Lansox (Takeda); Lanzor (Aventis); Limpidex (Sigma-Tau); Ogast (Takeda); Prevacid (TAP); Takepron (Takeda); Zoton (Wyeth)
Molecular Formula: C16H14F3N3O2S, Molecular Weight: 369.36
Percent Composition: C 52.03%, H 3.82%, F 15.43%, N 11.38%, O 8.66%, S 8.68%
Literature References: Gastric proton-pump inhibitor. Prepn: A. Nohara, Y. Maki, EP174726; eidem,US4628098 (both 1986 to Takeda).HPLC determn in plasma: T. Uno et al., J. Chromatogr. B816, 309 (2005). Pharmacology: H. Satoh et al.,J. Pharmacol. Exp. Ther.248, 806 (1989). Mechanism of action study: H. Nagaya et al.,ibid.252, 1289 (1990). Clinical pharmacology and effect on human gastric acid secretion: P. Müller et al.,Aliment. Pharmacol. Ther.3, 193 (1989). Review of pharmacology and clinical experience: H. D. Langtry, M. I. Wilde, Drugs54, 473-500 (1997). Comparative clinical trial with esomeprazole in erosive esophagitis: C. W. Howden et al., Clin. Drug Invest.22, 99 (2002).
Properties: mp 178-182° (dec).
Melting point: mp 178-182° (dec)
Therap-Cat: Antiulcerative., Keywords: Antiulcerative; Gastric Proton Pump Inhibitor.
Lansoprazole, sold under the brand name Selanz SR among others, is a medication which reduces stomach acid.[2] It is used to treat peptic ulcer disease, gastroesophageal reflux disease, and Zollinger–Ellison syndrome.[3] Effectiveness is similar to other proton pump inhibitors (PPIs).[4] It is taken by mouth.[2] Onset is over a few hours and effects last up to a couple of days.[2]
Common side effects include constipation, abdominal pain, and nausea.[2][5] Serious side effects may include osteoporosis, low blood magnesium, Clostridium difficile infection, and pneumonia.[2][5] Use in pregnancy and breastfeeding is of unclear safety.[1] It works by blocking H+/K+-ATPase in the parietal cells of the stomach.[2]
Lansoprazole was patented in 1984 and came into medical use in 1992.[6] It is available as a generic medication.[3] In 2017, it was the 188th most commonly prescribed medication in the United States, with more than three million prescriptions.[7][8]
Medical uses
Lansoprazole is used for treatment of:[5]
- Ulcers of the stomach and duodenum, and NSAID-induced ulcers
- Helicobacter pylori infection, alongside antibiotics (adjunctive treatment), treatment to kill H. pylori causing ulcers or other problems involves using two other drugs besides lansoprazole known as “triple therapy“, and involves taking twice daily for 10 or 14 days lansoprazole, amoxicillin, and clarithromycin
- Gastroesophageal reflux disease
- Zollinger-Ellison syndrome[9]
There is no good evidence that it works better than other PPIs.[4]
Side effects
Side effects of PPIs in general[10] and lansoprazole in particular[11] may include:[5]
- Common: diarrhea, abdominal pain[12]
- Infrequent: dry mouth, insomnia, drowsiness, blurred vision, rash, pruritus
- Rarely and very rarely: taste disturbance, liver dysfunction, peripheral oedema, hypersensitivity reactions (including bronchospasm, urinary, angioedema, anaphylaxis), photosensitivity, fever, sweating, depression, interstitial nephritis, blood disorders (including leukopenia, leukocytosis, pancytopenia, thrombocytopenia), arthralgia, myalgia, skin reactions[13] including (erythroderma[14] Stevens–Johnson syndrome, toxic epidermal necrolysis, bullous eruption)
PPIs may be associated with a greater risk of hip fractures and Clostridium difficile-associated diarrhea.[5]: 22
Interactions
Lansoprazole interacts with several other drugs, either due to its own nature or as a PPI.[15]
- PPIs reduce absorption of antifungals (itraconazole and ketoconazole) [16] and possibly increase digoxin in plasma
- Increases plasma concentrations of cilostazol (risk of toxicity)
Lansoprazole possibly interacts with, among other drugs:
- sucralfate
- ampicillin
- bisacodyl
- clopidogrel
- delavirdine
- fluvoxamine
- iron salts
- voriconazole
- aminophylline and theophylline
- astemizole
Chemistry
It is a racemic 1:1 mixture of the enantiomers dexlansoprazole and levolansoprazole.[17] Dexlansoprazole is an enantiomerically pure active ingredient of a commercial drug as a result of the enantiomeric shift. Lansoprazole’s plasma elimination half-life (1.5 h) is not proportional to the duration of the drug’s effects to the person (i.e. gastric acid suppression).[18]
History
Main article: Discovery and development of proton pump inhibitors
Lansoprazole , available in the name of Selanz SR, was originally synthesized at Takeda and was given the development name AG 1749.[19] Takeda patented it in 1984 and the drug launched in 1991.[20] In the United States, it was approved for medical use in 1995.[21]
Society and culture

Prevacid 30 mg
Patents
The lansoprazole molecule is off-patent and so generic drugs are available under many brand names in many countries;[22] there are patents covering some formulations in effect as of 2015.[23] Patent protection expired on 10 November 2009.[24][25]
Availability
Since 2009, lansoprazole has been available over the counter (OTC) in the U.S. as Prevacid 24HR[26][27] and as Lansoprazole 24HR.[28] In Australia, it is marketed by Pfizer as Zoton.[citation needed]
Research
In vitro experiments have shown that lansoprazole binds to the pathogenic form of tau protein.[29] As of 2015 laboratory studies were underway on analogs of lansoprazole to explore their use as potential PET imaging agents for diagnosing tauopathies including Alzheimer’s disease.[29]
SYN
English: doi: 10.1248/cpb.38.2853
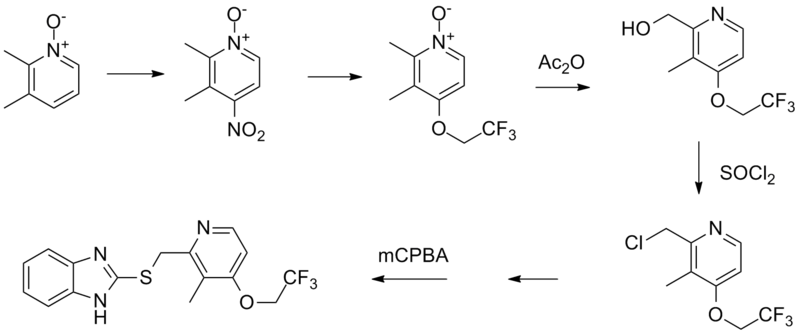
SYN
Method of synthesis
i. 2,3-dimethyl-4-nitropyridine-1-oxide is reacted with 2,2,2-trifluoroethanol in presence of potassium carbonate to give 2,3-dimethyl-4-(2,2,2-trifluoro-ethoxy)pyridine-1-oxide.
ii. The compound so formed is treated with acetic anhydride in acidic conditions followed by nutrilizing with sodium hydroxide solution to get 2-hydroxymethyl-3-methyl-4-(2,2,2-trifluoroethoxy)-pyridine
iii. Last is treated with thionyl chloride followed by reaction with 2-mercaptobenzimidazole to get 2-[3-methyl-4-(2,2,2-trifluoroethoxy)pyrid-2-ylmethylthio]benzimidazole.
iv. Above formed compound is reacted with m-chloro-perbenzoic acid to get lansoprazole.[2]
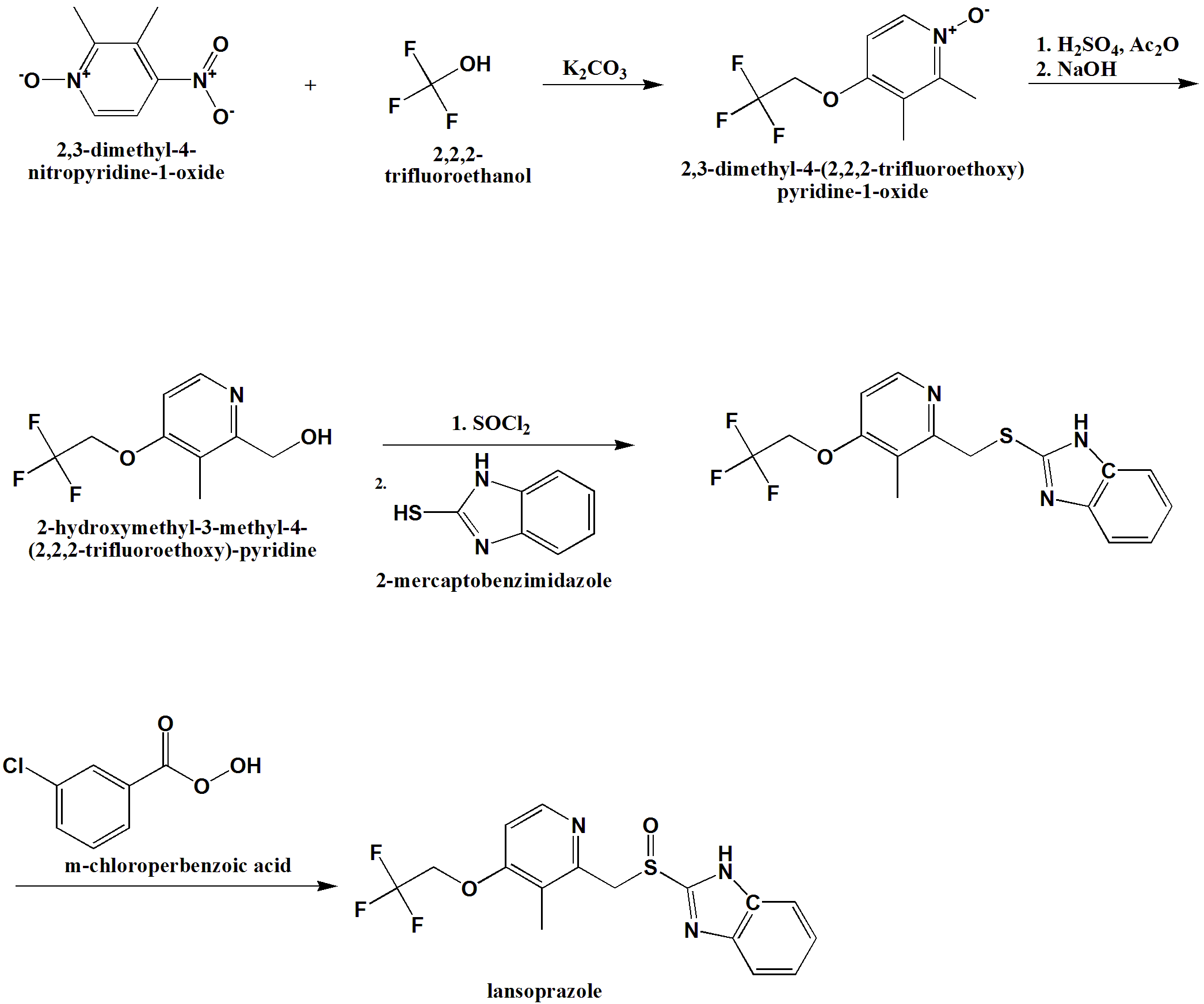
SYN
Proton Pump Inhibitors
Ruben Vardanyan, Victor Hruby, in Synthesis of Best-Seller Drugs, 2016
Lansoprazole–Prevacid
Lansoprazole (37.3) is the second approved gastric acid pump inhibitor. The common approach for the synthesis of lansoprazole involves coupling of mercapto-benzimidazole (37.24) with a new 2-chloromethylpyridine derivative (37.32) followed by oxidation of the prochiral sulfide group with m-chloroperbenzoic acid or hydrogen peroxide was first disclosed by Nohara and Maki [73], with followed improvements in patents [74-78] and briefly summed up in papers [79-80].
Lansoprazole synthesis is represented on the Scheme 37.4.

In principle it repeats the synthesis Scheme of omeprazole, differing in details and characteristics, for example, in place of 2,3,5-collidine (37.15) as a starting material, 2,3-lutidine (37.27) was selected, and the methoxy group in the fourth position of pyridine ring was replaced by the 2,2,2-trifluoroethoxy group.
Another interesting approach has been demonstrated [81]. In this case, 2-chloromethyl-3-methyl-4-(2,2,2-trifluoroethoxy)pyridine (37.32) was prepared starting with 3-picoline (37.34), which was oxidized using peracids (i.e., m-chloroperoxybenzoic acid) to produce 3-methylpyridineN-oxide (37.35). The obtained product was nitrated with fuming nitric acid to produce 3-methyl-4-nitropyridine N-oxide (37.36). The prepared N-oxide was treated with dimethylsulfate at 65 to 70°C to form N-methoxypyridinium salt (37.37), the aqueous solution of which on cooling was treated with sodium cyanide to produce an after formation of intermediate (37.38) and elimination of methanol 2-cyano-3-methyl-4-nitropyridine (37.39). This method for the synthesis of 2-cyanopyridines via addition of cyanide ion to N-alkoxy-quaternary salts of pyridines, supplements the plethora of Reissert-Kaufmann reactions in the quinoline and isoquinoline series previously described [82]. The nitro group in (37.39) was replaced by the 2,2,2-trifluoroethoxy group by a direct reaction with sodium trifluoroethoxide in trifluoroethanol that produced ether (37.40). The next step—transformation of nitrile group in prepared 2-cyanopyridine (37.40) to 2-carboxypyridine (37.41)—was carried out in a one-pot procedure by heating the 2-cyano compound in the presence of concentrated sulfuric acid followed by reaction of the intermediate amide with sodium nitrite under aqueous acidic conditions [83,84]. The obtained acid was esterified in methanol with a catalytic amount of sulfuric acid to produce ester (37.42). The ester (37.42) was reduced by NaBH4, producing the above-described 2-hydroxymethyl- pyridine derivative (37.31) followed by a reaction with thionyl chloride in dioxane that produced the required 2-chloromethylpyridine compound (37.32). Direct reaction of the last with 2-mercaptobenzimidazole (37.34) in methanol, even without use of any base, produced a sulfide (37.33) in high yield. The oxidation of the last to lansoprazole (37.3) has been carried out by various oxidants and catalysts, which, together with the desired sulfoxide, produced a certain amount of overoxidized product. Oxidizing sulfide (37.33) with a new oxidation method made up of the use of the composite metal oxide catalyst, LiNbMoO6, in methanol and 35% H2O2 as an oxidant sulfide (37.33) was successfully oxidized to desired lansoprazole (37.3) (Scheme 37.5.).

Lansoprazole is the second inhibitor of the gastric H+/K+-ATPase to be marketed for the treatment of peptic ulcer disease and reflux esophagitis, erosive esophagitis, and Zollinger-Ellison syndrome. It is an inhibitor of gastric acid secretion and also exhibits antibacterial activity against H. pylori in vitro. More common side effects of lansoprazole are diarrhea and skin rash or itching. Less-common side effects are abdominal pain, joint pain, nausea, vomiting, and increased or decreased appetite [85-91].
SYN
| AU 8545895; EP 0174726; ES 8607288; JP 1986050978; US 4628098; US 4689333 |
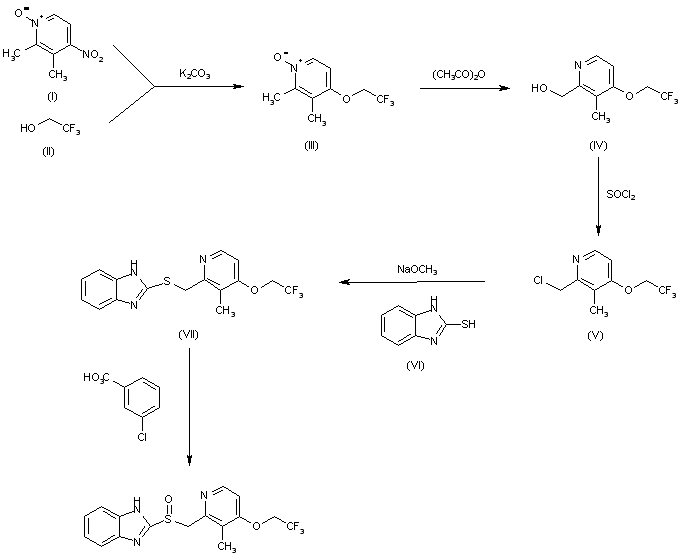
The condensation of 2,3-dimethyl-4-nitropyridine N-oxide (I) with 2,2,2-trifluoroethanol (II) by means of K2CO3 in hot HMPT gives 2,3-dimethyl-4-(2,2,2-trifluoroethoxy)pyridine N-oxide (III), which by isomerization in acetic anhydride at 100 C is converted to 2-(hydroxymethyl)-3-methyl-4-(2,2,2-trifluoroethoxy)pyridine (IV). The reaction of (IV) with SOCl2 in refluxing CHCl3 affords the corresponding chloromethyl derivative (V), which is condensed with 2-mercaptobenzimidazole (VI) by means of sodium methoxide in refluxing methanol to yield 2-(2-benzimidazolylthiomethyl)-3-methyl-4-(2,2,2-trifluoroethoxy)pyridin (VII). Finally, this compound is oxidized with m-chloroperbenzoic acid in CHCl3.
SYN
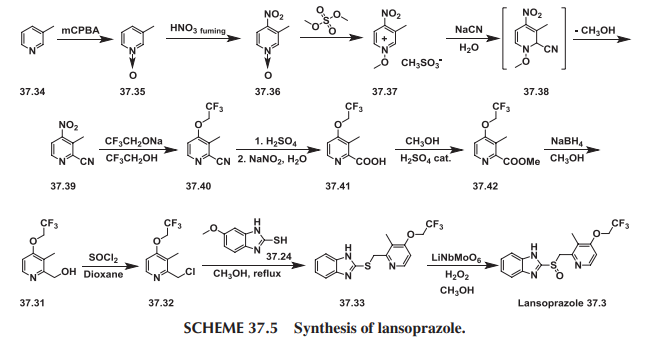
SYN
Chemical Synthesis
Similar to the synthesis of the chiral sulfoxide of armodafinil vide supra, the preparation of the chiral sulfoxide of lansoprazole utilized the catalytic oxidation method developed by Kagan and co-workers (the Scheme). Two routes have been reported that describe the preparation of dexlansoprazole on large scale. The first route developed by Takeda reacts commercially available thioether 29, also used to make lansoprazole, under the Kagan asymmetric oxidation conditions and the alternative route utilizes the cheaper commercial intermediate nitrosulfide 30 in the analogous asymmetric oxidation by Kagan). Thus, the catalyst complex consisting of (+)-DET, Ti(OiPr)4 and water was formed in the presence of thioether 29 in toluene at 30–40°C. The reaction mixture was then cooled to 5 °C and DIPEA and cumene hydroperoxide (CMHP) were added to give, after aqueous work-up and in situ crystallization from the organic layer, dexlansoprazole (VI) in 98% ee. No yield was given in the patent. An alternate, but similar, sequence was also described wherein the nitrosulfide intermediate 30 was subjected to similar oxidative conditions that gave intermediate nitro compound 31 in 80% yield and 98% ee. Compound 31 was treated with KOH and trifluoroethanol to provide dexlansoprazole (VI).
R-(+)-Lansoprazole Preparation Products And Raw materials
PATENT
https://patents.google.com/patent/WO2008087665A2/enA number of substituted 2-(2-pyridylmethyl) sulfinyl-lH-benzimidazole derivatives are reported as gastric proton pump inhibitors. These benzimidazole derivatives include lansoprazole, omeprazole, pantoprazole, and rabeprazole. The Lansoprazole is generally represented by the following chemical formula I

US 4,628,098 & 4,689,333 describes lansoprazole having its chemical name (2-[[[3-methyl-4-(2, 2, 2-trifluoro-ethoxy)-2-pyridinyl] methyl] sulfinyl]-lH-benzimidazole. As a characteristic shared with other benzimidazole derivatives (e.g., omeprazole and pantoprazole), lansoprazole can inhibit gastric acid secretion, and thus commonly used as an antiulcer agent. Several methods for preparing Lansoprazole are known. The majority of these methods involve the use of a lansoprazole precursor that contains a thioether group. The thioether group is oxidized in the last step of preparation to form the lansoprazole. These patents (‘098 and ‘333) further describes the oxidation of the thioether group using m-chloroperbenzoic acid, per acid, sodium bromite, sodium hypochlorite, or hydrogen peroxide as the oxidizing agent and the reaction solvent is halogenated hydrocarbon, ether, amide, alcohol, or water.US 6,002,011 describe the crystallization of Lansoprazole from the same ethanol: water system, containing traces of ammonia. This patent discloses a reslurry method in water, which permits to obtain more stable “solvent free” Lansoprazole. This patent fails to disclose the level of purity for Lansoprazole. In addition, the ethanol and water are difficult to eliminate. Even after intensive drying, Lansoprazole still contains solvent and is unstable under storage. US 6,180,652 describe the presence of sulfone derivative. Formation of sulfone derivative brings about the drawback of low yield of the desired sulfoxide. Although attempts have been made to separate the sulfone derivative from Lansoprazole, it is not a simple task, given their very similar structures and physicochemical properties. This patent also describes a method for separation of Lansprazole from its sulfone derivative, by converting to an acetone complex of the Lansoprazole salt & hence is purified in this method. Lansoprazole and other 2-(2- pyridylmethyl) sulfinylbenzimidazole derivatives tend to lose stability and undergo decomposition when contaminated with traces of a solvent, particularly water, in their crystal structure. It is desirable that the benzimidazole crystals be solvent free (i.e., residual solvent should be reduced to a minimum).US 6,909,004 describes the method of purifying Lansoprazole, comprising the steps of: a) providing a solution of lansoprazole in a solvent selected from an organic solvent or a mixture of organic solvent and water in the presence of an amine compound; b) combining the provided solution with an acid, and c)isolating the purified Lansoprazole. The amine compound is present in 1:1, mole: mole, ratio relative to the lansoprazole. Solution is in an organic solvent selected from the group consisting of alcohols, acetone, 2-butanone, dimethylformamide and tetrahydrofuran. The alcohol consisting of ethanol, methanol, n-propanol, & iso-propanol.US 7022859 & US 7060837 provides a method for preparing a substantially pure Lansoprazole containing less than about 0.2% (wt/wt) impurities including sulfone/sulfide derivatives. The present invention also provides a process for recrystallizing Lansoprazole to obtain a Lansoprazole containing less than about 0.1% (wt/wt) water.US 2004/010151 disclose a method of preparing crystalline Lansoprazole form A, comprising the steps of: a) preparing a solution of Lansoprazole in a solvent selected from the group consisting of methanol, n-butanol, acetone, methylethylketone, ethyl acetate, dimethyl sulfoxide, dimethylforniamide and their mixtures optionally with water; and b) isolating crystalline Lansoprazole form A.US 2005/020638 describe the process of preparing a stable Lansoprazole, comprising the steps of: a) crystallizing a Lansoprazole from an organic solvent or a mixture of organic solvent and water in the presence of a weak base; and b) isolating a stable Lansoprazole. An amorphous form of Lansoprazole prepared by spray drying method has been described (Farm. Vest. vol. 50, p. 347 (1999)). Curin et al. describe an ethanole solvate form and an ethanole-hydrate form of Lansoprazole (Farm. Vest. vol. 48, pp. 290-291 (1997). Kotar et al. describe two lansoprazole polymorphs, designated as crystalline Lansoprazole forms A and B, (Eur. J. Pharm. Sci. vol. 4, p. 182 (1996 Supp). According to Kotar, each of the crystalline Lansoprazole forms A and B exhibits a different DSC curve. In fact, crystalline Lansoprazole form B is unstable and can undergo a solid-solid transition to form crystalline Lansoprazole form A. No XRD data for crystalline Lansoprazole forms A and B, and fails to disclose processes for preparing these crystalline forms. No indication was found in the literature regarding the existence of other crystalline Lansoprazole forms other than the known forms A, B, ethanolate and ethanolate- hydrate.WO 00/78729 is discloses a phenomenon of polymorphism in Lansoprazole. The crystalline forms , I and II. The form I find application as an active ingredient of pharmaceutical compositions.WO 03/082857 disclose a method of preparing crystalline Lansoprazole form A, comprising the steps of: a) preparing a solution of Lansoprazole in a solvent selected from the group consisting of methanol, n-butanol, acetone, methylethylketone, ethyl acetate, dimethyl sulfoxide, dimethylformamide and their mixtures optionally with water; and b) isolating crystalline Lansoprazole form A.WO 2004/046135 describe the process for preparing a stable Lansoprazole compound, comprising the steps of: a) crystallizing a Lansoprazole from an organic solvent or a mixture of organic solvent and water in the presence of an amine; and b) isolating a stable Lansoprazole compound, wherein the stable Lansoprazole compound comprises greater than 500 ppm and not more than about 3,000 ppm water.Since proton pump inhibitors of the benzimidazole-type are very susceptible to degradation under acidic or neutral conditions, the reaction mixture is usually worked-up under basic conditions. These basic conditions may decompose any unwanted oxidizing agent still present in the reaction mixture and may also neutralize any acid formed when the oxidizing agent is consumed in the oxidation reaction. The main problem with the oxidation reaction to convert the sulfide intermediates of formula (II) into the sulfoxide compounds of formula (I) is over- oxidation, i.e. oxidation from sulfoxides of formula (I) to sulfones of formula (III) ; N-oxide of formula (IV) & chlorinating impurities ( V).The formation of sulfones of formula (III) due to over-oxidation is almost impossible to avoid and can be kept to a minimum by performing the oxidation reaction at a low temperature and restricting the amount of oxidizing agent. Typically the amount of oxidizing agent is less than 1 molar equivalent of the starting material, i.e. sulfide intermediates of formula (II), which inevitably results in a less than 100% conversion of starting material. Usually the amount of oxidizing agent is a compromise between maximum conversion of starting material, maximum formation of sulfoxides of formula (I) and minimum formation of unwanted sulfones of formula (III). Chlorinating impurities (V) are observed when chlorinating oxidizing agent such as sodium hypochlorite is used for oxidation reaction. Furthermore removal of the sulfones of formula (III) & chlorinating impurities (V) has often proved to be difficult, time-consuming and costly, in particular when high performance chromatography on an industrial scale is needed. Another problem with the benzimidazole-type is very susceptible to degradation when exposed to high temperatures for removal of solvents during distillation.Thus, there is continuing need to obtain 2-(2-pyridylmethyl) sulfϊnyl-lH-benzirnidazoles (e.g., Lansoprazole) that are free of contaminants including sulfone and sulfide derivatives. There has also -been a long-felt need for a method to prepare Lansoprazole having reduced water content (<0.1% wt/wt water).SCHEME : ]

LANSOPRAZOLE (I) SULPHONE (III)

N-OXIDE (IV)

SULPHIDE (II)+ Chlorinated Impurities(V)General Example10 g of 2- [3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridyl] methylthio-lH-benzimidazole was suspended in 100 ml chloroform and cooled to -100C. To the above suspension 3.4 g m- chloroperbenzoic acid solution in chloroform was added over a period of 2 hrs at -10 C. After completion of reaction, reaction mass was added to sodium bicarbonate solution (500 ml) and both layers were separated. Organic layer was washed with 2 x 50 ml of hypo solution followed by washing with 3 x 200 ml sodium bicarbonate solution. Both the layers were separated. Chloroform layer was washed with sodium bicarbonate solution (0.5%; 500 ml) at room temperature. Various co-solvents mentioned in Table- 1 were added to organic layer cool slowly to -10 to 100C. Filtered and washed with chilled chloroform (10 ml) followed by sodium bicarbonate solution (0.5%, 100 ml) & dried to get pure Lansoprazole.SYNhttp://www.ijmca.com/File_Folder/116-120.pdf


join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Literatures:
Chemical and Pharmaceutical Bulletin, , vol. 38, # 10 p. 2853 – 2858
Literatures:
RECORDATI INDUSTRIA CHIMICA E FARMACEUTICA SPA Patent: WO2008/77866 A1, 2008 ; Location in patent: Page/Page column 16-17; 19 ;
Yield: ~92%
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4628098A *1984-08-161986-12-09Takeda Chemical Industries, Ltd.2-[2-pyridylmethylthio-(sulfinyl)]benzimidazolesWO2004018454A1 *2002-08-212004-03-04Teva Pharmaceutical Industries Ltd.A method for the purification of lansoprazoleUS20040049045A1 *2000-12-012004-03-11Hideo HashimotoProcess for the crystallization of (r)-or (s)-lansoprazole
Publication numberPriority datePublication dateAssigneeTitleWO2012004802A12009-07-072012-01-12Council Of Scientific & Industrial ResearchContinuous flow process for the preparation of sulphoxide compoundsCN107964005A *2017-11-102018-04-27扬子江药业集团江苏海慈生物药业有限公司A kind of preparation method of Lansoprazole
References
- ^ Jump up to:a b c “Lansoprazole Use During Pregnancy”. Drugs.com. Retrieved 3 March 2019.
- ^ Jump up to:a b c d e f “Lansoprazole Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ Jump up to:a b British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 79–80. ISBN 9780857113382.
- ^ Jump up to:a b “[99] Comparative effectiveness of proton pump inhibitors | Therapeutics Initiative”. 28 June 2016. Retrieved 14 July 2016.
- ^ Jump up to:a b c d e “Lansoprazole capsule, delayed release pellets”. DailyMed. 11 October 2016. Retrieved 31 December 2019.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 445. ISBN 9783527607495.
- ^ “The Top 300 of 2020”. ClinCalc. Retrieved 11 April 2020.
- ^ “Lansoprazole – Drug Usage Statistics”. ClinCalc. Retrieved 11 April 2020.
- ^ Hirschowitz BI, Mohnen J, Shaw S (August 1996). “Long-term treatment with lansoprazole for patients with Zollinger-Ellison syndrome”. Aliment. Pharmacol. Ther. 10 (4): 507–22. doi:10.1046/j.1365-2036.1996.10152000.x. PMID 8853754. S2CID 10668517.
- ^ British National Formulary (Free registration required) 1.3.5 Proton pump inhibitors
- ^ British National Formulary (Free registration required) Lansoprazole
- ^ “Prevacid (Lansoprazole) Drug Information: Side Effects and Drug Interactions – Prescribing Information at RxList”. RxList. Retrieved 9 February 2016.
- ^ K C Singhal & S Z Rahman, Lansoprazole Induced Adverse Effects on the Skin, Indian Medical Gazette, July 2001, Vol. CXXXV. N0. 7: 223-225
- ^ Sterry W, Assaf C (2007). “Erythroderma”. In Bolognia JL (ed.). Dermatology. St. Louis: Mosby. p. 154. ISBN 978-1-4160-2999-1..
- ^ British National Formulary (Free registration required) Lansoprazole interactions
- ^ Piscitelli, S. C.; Goss, T. F.; Wilton, J. H.; d’Andrea, D. T.; Goldstein, H; Schentag, J. J. (1991). “Effects of ranitidine and sucralfate on ketoconazole bioavailability”. Antimicrobial Agents and Chemotherapy. 35 (9): 1765–1771. doi:10.1128/aac.35.9.1765. PMC 245265. PMID 1952845.
- ^ “Pharmacy Benefit Update”. Retrieved 2 July 2014.
- ^ “Prevacid Pharmacology, Pharmacokinetics, Studies, Metabolism”. RxList.com. 2007. Archived from the original on 16 August 2000. Retrieved 14 April 2007.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 102. ISBN 9783527607495.
- ^ Chorghade, Mukund S. (2006). Drug Discovery and Development, Volume 1: Drug Discovery. John Wiley & Sons. p. 201. ISBN 9780471780090.
- ^ Mosby’s Drug Consult: Lansoprazole
- ^ drugs.com International availability of lansoprazole Page accessed 3 February 2015
- ^ drugs.com Generic lansoprazole Page accessed 3 February 2015
- ^ “Prevacid Drug Profile”. Drugpatentwatch.com. Retrieved 30 April 2020.
- ^ Teva to release Prevacid version when patent expires
- ^ “Prevacid 24 HR- lansoprazole capsule, delayed release”. DailyMed. 7 August 2019. Retrieved 31 December 2019.
- ^ “Prevacid 24 HR- lansoprazole capsule, delayed release”. DailyMed. 11 December 2019. Retrieved 31 December 2019.
- ^ “Lansoprazole 24 HR- lansoprazole capsule, delayed release”. DailyMed. 21 December 2017. Retrieved 31 December 2019.
- ^ Jump up to:a b Villemagne, VL; Fodero-Tavoletti, MT; Masters, CL; Rowe, CC (January 2015). “Tau imaging: early progress and future directions”. The Lancet. Neurology. 14 (1): 114–24. doi:10.1016/s1474-4422(14)70252-2. PMID 25496902. S2CID 10502833.
External links
- “Lansoprazole”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | /lænˈsoʊprəzoʊl/ lan-SOH-prə-zohl |
| Trade names | Prevacid, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a695020 |
| License data | EU EMA: by INNUS DailyMed: LansoprazoleUS FDA: Lansoprazole |
| Pregnancy category | AU: B3[1] |
| Routes of administration | By mouth, intravenous (IV) |
| Drug class | Proton pump inhibitor |
| ATC code | A02BC03 (WHO) |
| Legal status | |
| Legal status | AU: S2, S3, & S4UK: POM (Prescription only)US: OTC / Rx-only |
| Pharmacokinetic data | |
| Bioavailability | 80% or more |
| Protein binding | 97% |
| Metabolism | Liver (CYP3A4– and CYP2C19-mediated) |
| Elimination half-life | 1.0–1.5 hours |
| Excretion | Kidney and fecal |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 103577-45-3 |
| PubChem CID | 3883 |
| IUPHAR/BPS | 7208 |
| DrugBank | DB00448 |
| ChemSpider | 3746 |
| UNII | 0K5C5T2QPG |
| KEGG | D00355 |
| ChEBI | CHEBI:6375 |
| ChEMBL | ChEMBL480 |
| CompTox Dashboard (EPA) | DTXSID4023200 |
| ECHA InfoCard | 100.173.220 |
| Chemical and physical data | |
| Formula | C16H14F3N3O2S |
| Molar mass | 369.36 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI | |
| (verify) |
////LANSOPRAZOLE, A-65006, AG-1749, A 65006, AG 1749, лансопразол , لانسوبرازول , 兰索拉唑 , Antiulcerative, Gastric Proton Pump Inhibitor,

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
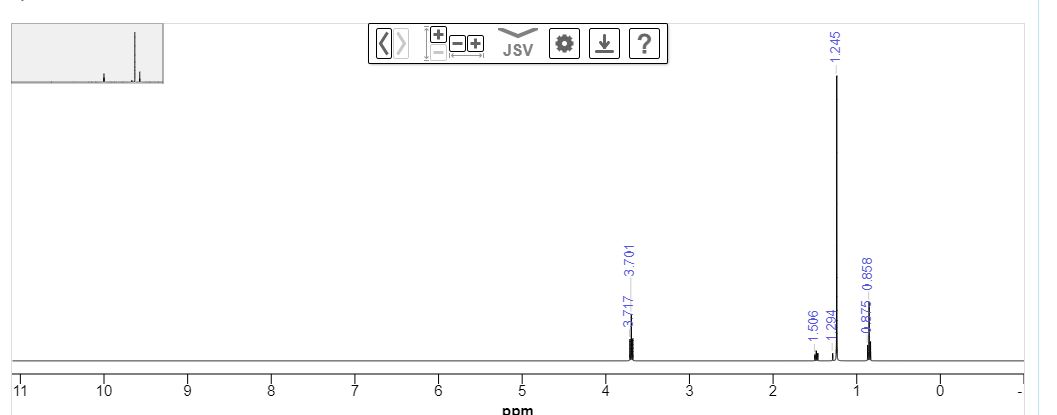{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
-lansoprazole-from-xtal-3D-bs-17.png){kind=link}