
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
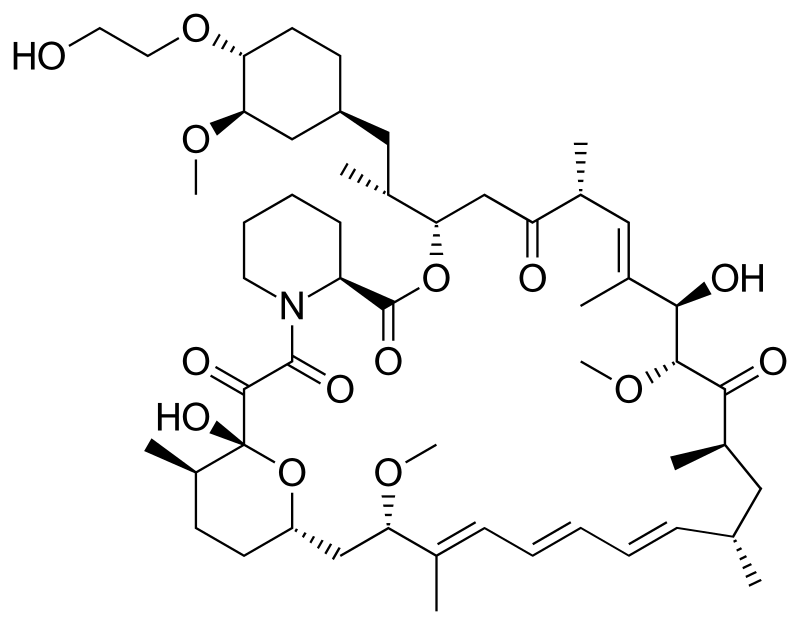
Cipla, New Patent, WO 2016020664, Everolimus



CIPLA LIMITED [IN/IN]; Peninsula Business Park Ganpatrao Kadam Marg Lower Parel Mumbai 400 013 (IN).
KING, Lawrence [GB/GB]; (GB) (MW only)
RAO, Dharmaraj Ramachandra; (IN).
MALHOTRA, Geena; (IN).
PULLELA, Venkata Srinivas; (IN).
ACHARYA, Vinod Parameshwaran; (IN)
WO2016020664, PROCESS FOR THE SYNTHESIS OF EVEROLIMUS AND INTERMEDIATES THEREOF

Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),

It is a derivative of sirolimus of formula III),

and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress™ (USA) and Certican™ (Europe and other countries) in transplantation medicine, and Afinitor™ in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV), 
wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),

was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Scheme

Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2-Hydroxy)ethyl rapamycin] of formula (I).
However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),

as the alkylation agent. The overall yield reported was 25%.
International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),

wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus.
European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step.
The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity.


Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl
t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl
t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and
solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC.
Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS-ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane
solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to -25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol-triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.

////Cipla, New Patent, WO 2016020664, Everolimus, INDIA

