
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
The synthesis of ezetimibe with high stereochemical purity
Krzysztof Bańkowski , Katarzyna Sidoryk , Katarzyna Filip , Joanna Zagrodzka
Pharmaceutical Research Institute (IF), Rydygiera 8, Warszawa 01-793, Poland
Ezetimibe, (3R,4S)-1-(4-fluorophenyl)-3-((3S)-3-(4-fluorophenyl)- 3-hydroxypropyl)-4-(4-hydroxyphenyl)-2-azetidinone, is an anti-hyperlipidemic drug which is used to lower cholesterol level. It acts by decreasing cholesterol absorption in the intestine.
The three chiral centers in the ezetimibe molecule give rise to eight stereoisomers and the synthesis of stereochemical pure ezetimibe is a significant challenge. The synthesis of ezetymibe is described in many patents and patent applications, however the problem of stereochemical purity of the final product and its intermediates is almost completely omitted.
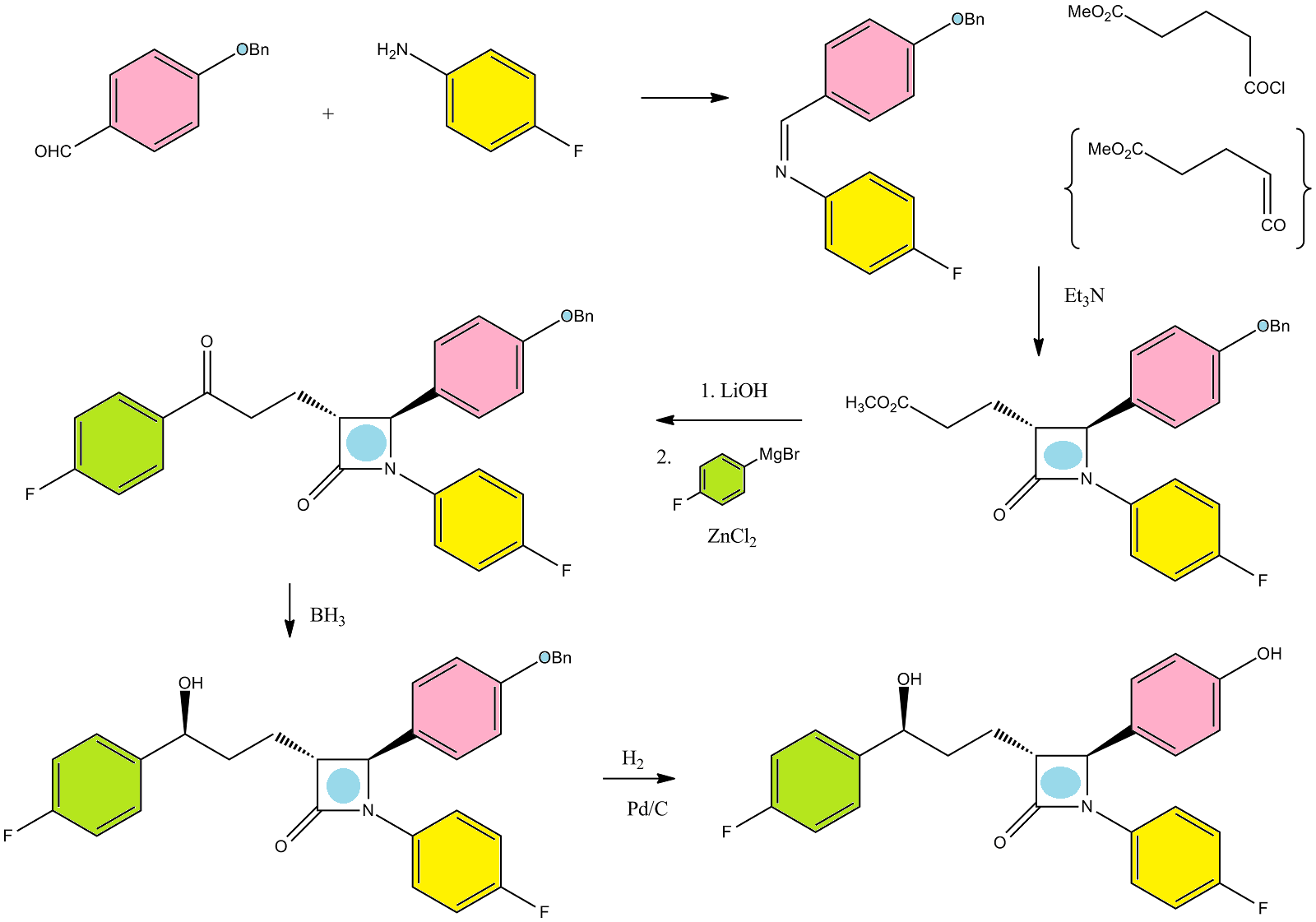
The synthesis of ezetimibe was realized by a procedure shown below, according to Schering Co. patents No US 6,207,822, EP 1137634:

We have investigated the sterochemical course of all steps of this process and found that for the preparation of optical pure ezetimibe the providing of pure (S,R,S,S) – EZ-6 is cru-cial. This diastereomer (product of anti-condensation of EZ-4 + EZ-5) is usually contaminated with (S,R,R,S) – EZ-6 isomer (syn-condensation), and also with (R,R,S,S) – EZ-6 isomer derived from small amount of (R,S)-alcohol EZ-4 which is usually occurring in required (S,S)-alcohol. The presence of (R,R,S,S) – EZ-6 diastereomer leads to (R,R,S) -“iso-ezetimibe” which is very difficult to remove from ezetimibe.
The synthesis of ezetimibe was optimized, all chemical and sterochemical impurities were isolated and/or synthesized and characterized by NMR, MS and HPLC techniques. The method for the purification of desired key intermediate (S,R,S,S)-6 was elaborated. These al-lowed us to develop the large scale efficient synthesis of pharmaceutical pure Ezetimibe (HPLC > 99,5 %, (R,R,S)-isomer < 0,1 %, single unknown impurity < 0,1 %, total impurities < 0,6 % ).
Ezetimibe has the chemical name 1-(4-fluorophenyl)-3(R)-[3-(4-fluorophenyl)-3(S)-hydroxypropyl]-4(S)-(4-hydroxyphenyl)-2-azetidinone (hereinafter referred to by its adopted name “ezetimibe”) and is structurally represented by Formula I.

Ezetimibe is in a class of lipid lowering compounds that selectively inhibit the intestinal absorption of cholesterol and related phytosterols. It is commercially available in products sold using the trademark ZETIA as a tablet for oral administration containing 10 mg of ezetimibe, and in combination products with simvastatin using the trademark VYTORIN.
U.S. Pat. No. 6,096,883 discloses generically and specifically ezetimibe and its related compounds along with their pharmaceutical compositions
The preparation of ezetimibe ezetimibe first disclosed in U.S. Patent US 5767115.

Hydrogen Debenzylation get ezetimibe, the method disclosed in this patent require the use of several key intermediates purified by column chromatography, increasing the difficulty and cost of industrial production.
US patent US5767115 to improve the synthesis process have also been reported. For example: W02006137080 US5767115 on the basis of synthesis of intermediate compound 3 were improved optimization, using pivaloyl chloride and the formation of a mixed anhydride intermediate compound 2, and then with the chiral auxiliary (S) -4- phenyl-2- oxazolidinone reaction intermediate compound 3; US Patent US6133001 discloses a microbial catalytic asymmetric reduction of carbonyl to give chiral hydroxy, instead US5767115 Synthesis of (R) -CBS catalyst;
W02008089984 reported the use of a rhodium catalyst [(S, S) -N- (piperidyl-N-sulfonyl) -I, 2-diphenyl ethylenediamine] (η 6-mesitylene) Ruthenium right of intermediate compound 9Said reduction.
W02008032338 reports by reacting the intermediate compound 8 with a salt of an aliphatic amine which was purified manner, although effectively improve the purity, but adds steps, and the yield was significantly reduced.
In addition to the synthetic route based on open Pu Xi US5767115, US Patent US6207822, US Patent US5856473, US patent US5886171, W02005066120, W02005113496, W02006050634, W02007017705 also disclose the ezetimibe different preparation methods.
Patent W02007072088 discloses another synthetic route for preparing ezetimibe ezetimibe, which is a small step synthesis reaction, the specific synthetic route is as follows:

Another US: 5739321; US: 1 5886171 reported the route: the (4S) – hydroxytetrahydrofuran _2_ one and N- (4- fluorophenyl) -4-benzyloxy-benzylidene methylamine as starting Preparation of raw ezetimibe, the reaction scheme is as follows:

…………

Since the first report since the synthesis method, there are already several ezetimibe ezetimibe synthetic route reports, such as document US 5856473, US 5739321, EP 1137634, EP 720599, WO 1995/08532 EP 0720599, provides ezetimibe ezetimibe synthetic route.
Example 9 Preparation of Compound 8 embodiment.
Hydrogenation bottle was added 7a (2.14 g, 4.3 mmol), methanol (30 mL), was added Pd / C (50 mg :), transferred into the autoclave, and replaced with hydrogen three times, filled with hydrogen 5 atm, room temperature and stirred for 6 hours, venting of hydrogen, filtered through Celite, with a small amount of methanol (10 mL), dried and concentrated, the residue was mixed solvent of methyl t-butyl ether, and recrystallized from n-hexane to give compound 8, 78% yield

8, 1H-NMR (300 MHz, DMSO 6 ) [delta] = 9.50 (s, 1H), 7.41-7.07 (m, 10H), 6.79 (d, J = 8.6 Hz, 2H), 5.27-5.25 (m, 1H) , 4.78-4.71 (m, 1H), 4.47-4.44 (m, 1H), 3.07-3.08 (m, 1H), 1.85-1.75 (m, 4H) ppm. 10 Compound la (P = Bn benzyl)
Example 3 (Preparation -2a of.
The reaction is as follows: Under an argon atmosphere, [Pd (C 3 H 5 ) Cl] 2 (54.8 mg, 0.15 mmol) and (&& 5 Lc (193 mg, 0.25 mmol) were added to a Schlenk tube, was added anhydrous CH 2 C1 2 C50 mL), stirred at room temperature for 10 minutes, the substrate was added successively lb (4.12 g, 10 mmol), K 2 C0 3(1.0 M solution, 30 mL, 30 mmol) and p-fluoroaniline (3.33 g, 30 mmol ). After stirring at room temperature for three hours, liquid separation, the aqueous phase was extracted with dichloromethane (3 x 50 mL), The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated

(I?) – 2a, 85% yield, 93% ee.
Example 4 Compound Example (Preparation -2b of.
The reaction is as follows: Under an argon atmosphere, [Pd (C 3 H 5 ) Cl] 2 (54.8 mg, 0.15 mmol) and (&& 5) -La (165 mg, 0.25 mmol) were added to a Schlenk tube, was added anhydrous CH 2 C1 2 C50 mL), stirred at room temperature for 10 minutes, the substrate was added successively lb (3.78 g, 10 mmol), K 2 C0 3(1.0 M solution, 30 mL, 30 mmol) and p-fluoroaniline (3.33 g, 30 mmol). After stirring at room temperature for three hours, liquid separation, the aqueous phase was extracted with dichloromethane (3 x 50 mL), The combined organic phase was dried over anhydrous sodium sulfate, filtered and concentrated, purified by column chromatography to give asymmetric amination the product of (i?) – 2b. The reaction formula is as follows:

(R) -2b, colorless liquid, yield 86%, [a] D 2Q = -89.1 (c 1.00, CHC1 3 ), EE 95% [determined by high-performance liquid chromatography, chiral AD-H column, n hexane / isopropanol = 95: 5, 1.0 mL / min, 254 nm; t R (minor) = 4.15 min; t R . (Major) = 4.60 min] 1H NMR (300 MHz, CDCl 3 ) [delta] = 7.20 (d, J = 8.4 Hz, 2H), 6.89-6.78 (m, 4H), 6.51-6.47 (m, 2H), 6.34 (s, 1H), 5.88 (s, 1H), 5.26 (s, 1H), 4.19-4.08 (m, 2H), 4.00 (s, br, 1H), 1.20 (t, J = 7.2 Hz, 3H), 0.97 (s, 9H), 0.18 (s, 6H) ppm; 13 C NMR (100 MHz, CDCl 3 ) [delta] = 166.1, 155.8 (d, J (F , C) = 234.3 Hz), 155.1 (s), 143.0 (d, J (F , C) = 1.9 Hz), 140.4 (s), 133.1 (s), 128.5 (s), 125.2 (s), 120.0 (s), 115.4 (d, J (F , C) = 22.3 Hz), 114.1 (d, J (F , C) = 7.4 Hz), 60.6 , 58.9, 25.5, 18.0, 13.9, -4.5 ppm; 19 F-NMR (376 MHz, CDCl 3 ) [delta] -127.5 ppm.
Preparation Example 5 Compound 4a embodiment.
(I?) – 2a (3.44 g, 8.48 mmol) and nucleophiles 3a (2.82 g, 12.7 mmol) was added in an eggplant-shaped flask, tetrahydrofuran (100 mL), DBU (4.25 g, 16.96 mmol was stirred at room temperature for 12 hours, thin layer chromatography until starting material disappeared by TLC the reaction mixture was concentrated and purified by column chromatography, to obtain compound 4a, 82% yield (Note: Allyl allyl).
ESI-MS m / z: 628.4 [M + H + ]; HRMS (ESI) m / z:. calcd for C 37 H 36 N0 6 F 2 +1 : 628.2505, Found:
+ ]. After the reaction system may also not treated directly in the next step. The reaction formula is as follows:

: Example 6 Preparation of Compound 5a.
To the reaction system of Example 5 is continued morpholine (4.43 g, 50.88 mmol) and Pd (PPh 3 ) 4 , and stirring was continued at room temperature for 6 hours, concentrated purified by chromatography (98 mg, 0.0848 mmol) after column .
The total yield from the compound 2a to 5a rate of 71%. Compound 5a is composed of a pair of non-enantiomer at a ratio of 2 or 3: 1. No need to separate the non-enantiomer, can be used directly in the next step.
ESI-MS m / z: 544.2 [M + H +]; HRMS (ESI) m / z:. Calcd for C33H31NO4 F 2 Na +1 : 566.2113, Found: 566.2113 [M + Na + ].
Preparation Example 7 Compound 6a embodiment.
Compound 5a (3.5 g, 6.4 mmol) and anhydrous tetrahydrofuran (50 mL) was added an eggplant-shaped flask, and cooled to -20 ° C under slowly added dropwise amino lithium hexamethyldisilazide (LiHMDS) (1.0 M THF, 14 ml, 14 mmol). The reaction system was stirred at this temperature continued for 40 minutes, 5 mL of water was added to quench the reaction, and extracted with dichloromethane (3 x 100 mL), the organic phase was dried over anhydrous sodium sulfate

6a, 77% yield. [A] D 2Q = +1.9 (c 1.00, MeOH), 95% EE [by the high performance liquid chromatography, chiral OD-H column n is isopropanol = 70:30, 1.0 mL / min, 254 nm; t R (Major) = 19.60 min; t R . (minor) = 25.83 min] 1H NMR (400 MHz, CDCl 3 ) [delta] = 7.98-7.94 (m, 2H), 7.41-7.30 ( m, 5H), 7.25-7.23 (m, 4H), 7.09 (t, J = 8.8 Hz, 2H), 6.96-6.88 (m, 4H), 5.02 (s, 1H), 4.67 (d, J = 2.4 Hz , 1H), 3.31-3.23 (m, 1H), 3.17-3.08 (m, 2H), 2.42-2.20 (m, 2H) ppm; 13 C NMR (100 MHz, CDCl 3 ) [delta] = 197.2, 167.1, 165.6 ( d, J (F , C) = 253.9 Hz), 158.9, 158.8 (d, J (F , C) = 242.2 Hz), 136.5, 133.7 (d, J (F , C) = 2.7 Hz), 132.9 (d , J (F , C) = 2.8 Hz), 130.5 (d, J (F , C) = 9.4 Hz), 129.3, 128.5, 127.9, 127.3, 127.1, 118.2 (d, J (F , C) = 7.9 Hz ), 115.7 (d, J (F , C) = 8.4 Hz), 115.5 (d, J (F , c) = 8.3 Hz), 115.3, 69.9, 60.9, 59.6, 35.4, 23.0 ppm; 19 F NMR (376 MHz, CDCl 3 ) [delta] -104.8, -117.9 ppm.
Compound 6a is the same as reported in the literature specific rotation direction, the same NMR data reported in the literature. References:
(A) Wu, G; Wong, Y;. Chen, X .; Ding, ZJ Org Chem 1999, 64, 3714. (b) Sasikala, CHVA;. Padi, PR; Sunkara, V; Ramayya, P .; Dubey , PK; Uppala, VBR;… Praveen, C. Org Process Res Dev 2009, 13, 907. (c) Sova, M .; Mravljak, J .; Kovac, A .; Pecar, S .; Casar, Z .; Gobec, S .; Synthesis, 2010, 20, 3433.
Preparation Example 8 Compound 7a embodiment.
In dichloromethane (40 mL) and tetrahydrofuran (5 mL) were added to an eggplant-shaped flask, and cooled to 0 ° C, was added borane dimethyl sulfide complex (0.46 mL, 7.23 mmol) and ![]() – (+) – 2-methyl–CBS- oxazaborolidine (133 mg, 0.482 mmol). Compound 6 (; 2.4 § , 4.82 11 ^ 101) was dissolved in dichloromethane (2011 ^) in the join. Stirred at the same temperature for 5 hours. After completion of the reaction with methanol (10 mL) quenched the reaction was concentrated, added to 1 mol per liter of dilute hydrochloric acid, methylene-wan (X) was extracted, the organic phase was washed with saturated sodium chloride wash paint, concentrated, ethyl acetate – n-hexane to give the compound 7a, 90% yield,> 99%. Reaction
– (+) – 2-methyl–CBS- oxazaborolidine (133 mg, 0.482 mmol). Compound 6 (; 2.4 § , 4.82 11 ^ 101) was dissolved in dichloromethane (2011 ^) in the join. Stirred at the same temperature for 5 hours. After completion of the reaction with methanol (10 mL) quenched the reaction was concentrated, added to 1 mol per liter of dilute hydrochloric acid, methylene-wan (X) was extracted, the organic phase was washed with saturated sodium chloride wash paint, concentrated, ethyl acetate – n-hexane to give the compound 7a, 90% yield,> 99%. Reaction

6a 7a
7a, 1H-NMR (300MHz, CDCI3) δ = 7.47-7.21 (m, 11H), 7.07-6.92 (m, 6H), 5.05 (s, 2H), 4.75-4.72 (m, 1H), 4.58 (m, 1H), 3.17-3.09 (m, 1H), 2.04-1.85 (m, 4H) ppm.


1H NMR spectrum of C24H21F2NO3 in CDCL3 at 400 MHz.
Patent
http://www.google.com/patents/CN103086938A?cl=en
Another report line 2: (5S) – acetyl-5- (4-fluorophenyl) valeric acid as reaction intermediates for the preparation of ezetimibe, the synthesis process is as follows:






Seventh Embodiment
The 7 (20g, 0.04mol) was dissolved in methanol (25OmL) was added ammonium formate (25g, 0.4mol), 10% palladium / carbon (Ig) and formic acid (2mL, 0.04mol), stirred at room temperature 20min, filtered palladium / carbon, the filtrate was concentrated to dryness. The residue was dissolved in ethyl acetate, washed with saturated brine, and dried. The organic phase was concentrated to approximately 40mL, was slowly added thereto at room temperature, methyl tert-butyl ether, stirring lh, floc filtered and the filtrate was concentrated to dryness. The residue was dissolved in ethyl acetate, petroleum ether was added, stirred at room temperature 2h, filtered, and dried to give a white solid Ezetimibe 6.4g, yield 38.9%, [a] 2 ° D = _23.7. IH-NMR (DMS0-d6) δ: 9.51 (s, 1Η), 7.32-7.08 (m, 10Η), 6.75 (d, J = 8.4, 2Η), 5.27 (d, J = 4.5, 1Η), 4.80 ( d, J = 2.1, 1Η), 4.49 (m, 1Η), 3.08 (m, 1Η), 1.68-1.82 (m, 4Η).
PATENT
http://www.google.com/patents/CN102675177A?cl=en

ezetimibe ezetimibe synthesis and purification methods:
A Method: IOOml reactor was added to 60ml of ethanol, was added glacial acetic acid and 6g 2. 4g compound 10, followed by stirring for 20 minutes, O. 6g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen , under hydrogen atmosphere, 10 ° C at atmospheric pressure for 18 hours the reaction inches, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor was concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml ko alcohol, filtered, the mother liquor 56ml of purified water was slowly added dropwise, after the large amount of solid precipitated, suction filtered, the filter cake rinsed with an aqueous solution of an ice drained, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml ko alcohol, purified water was slowly added dropwise 160ml a large number of solid precipitation, filtration, alcohol use ko – after pumping out water rinse, 60 ° C and dried under vacuum to obtain the product 4. Og, yield: 81 · 4%.
B method: to IOOml reaction flask 60ml of methanol, acetic acid 2. 4g and 6g compound 10,
Stirred for 20 minutes, added I. 2g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen under a hydrogen atmosphere, the reaction for 18 hours at ambient temperature and pressure inch, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml isopropanol, filtered, and the mother liquor was slowly added dropwise 56ml of purified water, a large number of solid precipitation, filtration, filter cake washed with isopropanol – water rinse after pumping dried, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml isopropanol was slowly added dropwise 160ml of purified water, large amount of solid precipitated, suction filtered, washed with isopropanol – water rinse after draining, 60 ° C under vacuum drying products 3. 7g, yield: 75.3%.
C Method: To a IOOml 60ml of methanol was added to the reaction vessel, was added glacial acetic acid and 6g 2. 4g compound 10, followed by stirring for 20 minutes, O. 8g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen , under hydrogen atmosphere, 30 ° C at atmospheric pressure for 18 hours the reaction inches, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor was concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml ko alcohol, filtered, the mother liquor 56ml of purified water was slowly added dropwise, after the large amount of solid precipitated, suction filtered, the filter cake washed with methanol – water rinsing after drained, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml of methanol, 160ml of purified water was slowly added dropwise a large number of solid precipitation, filtration, washed with methanol – after pumping out water rinse, 60 ° C under vacuum drying products 3. 5g, Yield: 71.3%.
PATENT
http://www.google.co.in/patents/CN102531985A?cl=en
Scheme 1

PATENT
http://www.google.com/patents/US20070049748
Processes for preparation of ezetimibe and its intermediates have also been described in U.S. Pat. Nos. 6,207,822, 5,856,473, 5,739,321, and 5,886,171, International Application Publication No. WO 2006/050634, and in Journal of Medicinal Chemistry 1998, 41, 973-980, Journal of Organic Chemistry 1999, 64, 3714-3718, and Tetrahedron Letters, 44(4), 801-804.

EXAMPLE 1 DETERMINATION OF IMPURITIES IN EZETIMIBE
Determining the level of impurities in ezetimibe using HPLC. The HPLC analysis conditions are as described in Table 1.
| TABLE 1 | |||
| HPLC method for detecting the level of the impurities. | |||
| Column: | Zorbax SB-C18 150 × 4.6 mm, 3.5 μm | ||
| Flow: | 1.0 ml/minute | ||
| Column oven | Ambient | ||
| temperature: | |||
| Wave length: | 230 nm | ||
| Injection volume: | 10 μl | ||
| Run time: | 65 minutes | ||
| Elution: | Gradient | ||
| Diluent: | Acetonitrile | ||
| Gradient Program: | Time | % B | % A |
| (in minutes) | concentration. | concentration. | |
| 0.01 | 35 | 65 | |
| 10.0 | 35 | 65 | |
| 35.0 | 80 | 20 | |
| 55.0 | 80 | 20 | |
| 60.0 | 35 | 65 | |
| 65.0 | 35 | 65 | |
| Mobile phase A = Buffer:Acetonitrile is 80:20 (v/v) | |||
| Mobile phase B = Buffer:Acetonitrile is 20:80 (v/v) | |||
| Buffer: 2.76 g of sodium dihydrogen phosphate monohydrate was | |||
| dissolved in 1000 ml of water and the pH was adjusted to 5.0 with | |||
| dilute NaOH solution. | |||
| IMPURITY NAME | RRT | ||
| Benzyl ezetimibe impurity | 2.6 | ||
| Benzyl ezetimibe diol impurity | 2.2 | ||
| Lactam cleaved alcohol impurity | 1.8 | ||
| Ezetimibe diol impurity | 0.66 | ||
| Lactam cleaved acid impurity | 1.5 | ||
PATENT
http://www.google.com/patents/CN104230978A?cl=en

Example 4. Synthesis of ezetimibe
[0042] To a 500ml bottle of single oral Compound I (PG1 = PG2 = trimethylsilyl) (10g, 13.3mmol), BSA (lOml), TBAF (0. 2g, 0. 66mmol) and methyl tert-butyl ether 100ml. Stirred for 15 minutes at room temperature, the disappearance of the test compound 8, the reaction was terminated. The pre-mixed solution of isopropanol and 2N sulfuric acid was added to the above solution and stirred at room temperature for 1 hour. Crystallized from aqueous isopropanol final product, the product was filtered and washed with aqueous isopropanol, washed with water until the eluate pH is less than 5. Drying at 60 ° to give the final product 5g, yield: 91%, optical yield was 100%. ΐ NMR (400MHz, d6-D MS0): δ 1. 68 (m, 2H), 1 · 82 (m, 2H), 3. 07 (m, 1H), 4. 47 (d, 1H), 4. 79 (d, 1H), 5. 25 (d, 1H), 6. 75 (d, 2H), 7. 10 (m, 4H), 7. 21 (m, 4H), 7. 29 (m, 2H ), 9. 43 (s, 1H).
Patent
http://www.google.com/patents/CN102531985B?cl=en
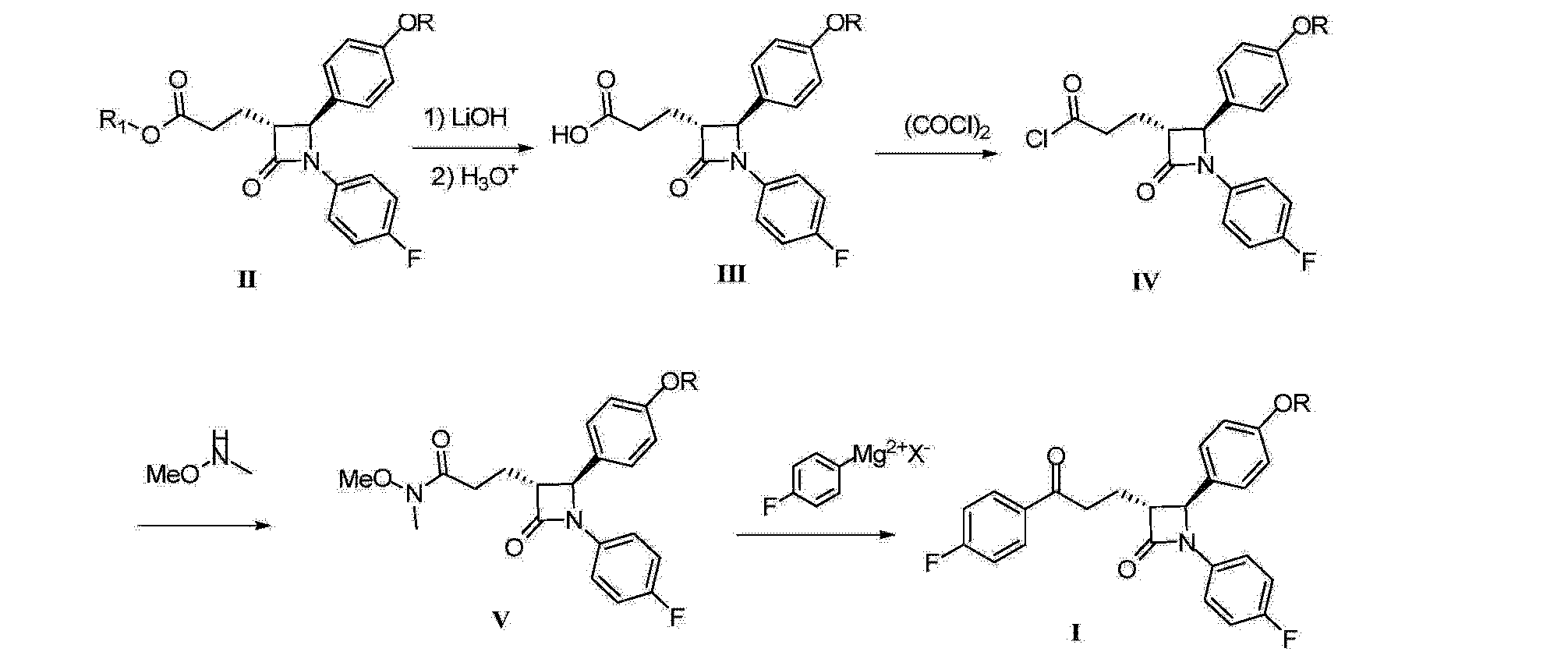
CN2006 / 10150638 discloses another improved synthetic process, the intermediate acid chloride (IV) first converted to the Weinreb amide, and then reacted with a Grignard reagent to give the key intermediate I.
………………….


……………
http://pubs.rsc.org/en/content/articlelanding/2014/ra/c3ra43861a#!divAbstract
The synthesis of four-membered azacycles is of importance because of the chemical and biological relevance of these compounds. Recent progress in copper-catalyzed reactions has been applicable to a variety of research fields, such as heterocyclic synthesis. The aim of the current review is to summarize the synthesis of strained four-membered ring taking advantage of copper catalyzed and mediated processes.


……………
http://www.google.com.tr/patents/WO2006137080A1?cl=en

Taken toluene (250 ml) into cleaned R.B.Flask under nitrogen atmosphere and cooled to 0-5°C. Borane DMS complex and (R)-tetrahydro-l-phenyl-3,3-diphenyl-l H,3H-pyrrol (l,2-c)(l,3,2) oxaza borolidine (R-phenyl CBS) is charged into the reaction mass at 0°C. 25 gm of Keto compound of formula-X is dissolved in toluene(50 ml) and added to the reaction mass at 0-5°C. Maintained the reaction mass for 3 hrs and quenched with methanol and followed by 1 N hydrochloric acid solution. Organic layer separated and washed with 5% hydrogen peroxide solution and 5% sodium sulfate solution and followed by with 10% sodium chloride solution. Distilled the solvent completely under reduced pressure at below 75°C. Product is isolated in diisopropyl ether and dried the product at 60-70°C for 6 hrs. (Yield: 15 gm). Example-2: Preparation of compound of hydroxy compound of formula-XL
Taken toluene (250 nil) into cleaned R.B.Flask under nitrogen atmosphere and cooled to 0-5°C. DIP Chloride (Mole ratio 1:1.5) into the reaction mass at O0C. 25 gm of keto compound of formula-X is dissolved in toluene(50 ml) and added to the reaction mass at 0-50C. Maintained the reaction mass for 3 hrs and quenched with ammonia solution. Organic layer separated and washed with 10% sodium chloride solution. Distilled the solvent completely under reduced pressure at below 750C. Residue is taken for next stage directly without any purification.
Step-h: Preparation of compound of formula-I (Ezetimibe).
Taken compound of formula-XII (10 gm) and isopropanol (100 ml) into a hydrogenation flask, added 5 % Pd/C ( 4gm) at 25°C and maintained at 45-500C for 3 hrs under hydrogen pressure, filtered through hyflow and washed the Pd/C with isopropanol(20 ml). Distilled the solvent completely under vacuum at below 7O0C, product is recrystallised in dichloromethane (Yield: 6 gm).
Purification of Ezetimibe (formula-1).
Ezetimibe (10 gm) is dissolved in 30 ml of methanol and filtered through hyflow and saturated with DM.Water(30 ml) and stirred for 1 hr at 20-250C. Product filtered and dried for 6-8 hrs at 80-850C (Yield:9 gm):
……………….
http://www.google.com.tr/patents/WO2005049592A1?cl=en
scheme A.

The compound of the formula 2

is an useful intermediate for the preparation of ezetimibe. The intermediates represented by the formula 2 can be prepared economically in good yields as represented by the scheme B.

wherein X is O or S; Y is O, S or N(lower alkyl); and R is alkyl, unsubstituted or substituted phenyl, unsubstituted or substituted naphthyl or lower alkoxy carbonyl, wherein substituents on phenyl and naphthyl are selected from the group consisting of lower alkyl and phenyl. The starting compounds of formula 3 are known or can be obtained from known methods. The reduction may be carried out in a neutral organic solvent or a combination of the neutral organic solvents. Neutral organic solvent means the solvent that is unreactive in the reduction reaction. The preferable organic solvents are chloroalkanes such as methylene dichloride, chloroform, carbon tetrachloride and ethylene dichloride; carbocyclic aromatics such as toluene and benzene; ethers such as methyl tert-butyl ether, diethylether and isopropyl ether; heterocyclic compound such as tetrahydrofuran; dimethylformamide; dimethylsulfoxide; alkanes such as pentane and hexane; and acetonitrile. More preferable solvents are toluene, diethyl ether, isopropyl ether, hexane, methylene dichloride and ethylene dichloride. The preferable reaction temperature is below the boiling temperature of the solvent used, more preferably between about -40°C and the boiling temperature of the solvent, still more preferably between about -20°C and 40°C and most preferably between about -10°C and 10°C. Quantity of (-)-DIP chloride used is preferably at least about 0.3 mole, more preferably about 0.5 to 10 mole, most preferably about 0.8 to 5 mole per mole of the keto compound of formula 3. Yield of the hydroxy compound of formula 2 is usually above 80%, typically between 90 % to 100%. The compounds of formula 2 wherein X is O; Y is O; and R is alkyl, unsubstituted or substituted phenyl are the preferred. Preferable conditions for obtaining a hydroxy compound of formula 2 from the corresponding keto compound of formula is that the keto compound of the formula 3 is mixed with a neutral solvent, reduced with (-)-DIP chloride at a temperature between -40°C and the boiling temperature of the solvent, more preferably between about -20°C and 40°C and most preferably between about -10°C and 10°C. The reaction mass may be subjected to usual work up. The reaction mass may be used directly in the next step to produce finally ezetimibe, or the hydroxy compound may be isolated and used in the next step. The invention will now be further described by the following examples, which are illustrative rather than limiting. Example 1 3-[5-(4-fluorophenyl)-1 ,5-dioxopentyl]-4-phenyl-2-oxazolidinone (100 gm) is dissolved in toluene (750 ml), the mixture of (-)-β- chlorodiisopinocampheylborane ((-)-DIP chloride) in heptane (545 ml, 1.5M) and toluene (750 ml) is added at 0°C to 5°C for 1 hour. The reaction mixture is stirred for 15 hours at 25°C to 30°C and 340 ml of 10% sodium chloride is then added at the same temperature. The layers are separated and the organic layer is washed with 5% sodium bicarbonate (300 ml), 1 N sulfuric acid (300 ml), and 10% sodium chloride (300 ml). Then the organic layer is dried on sodium sulfate to give 3-[(5S)-5-(4-fluorophenyl)-5-hydroxy-1-oxopentyl]-4-phenyI-2- oxazolidinone in 96% yield. Example 2 The organic layer of 3-[(5S)-5-(4-fluorophenyl)-5-hydroxy-1-oxopentyl]-4- phenyl-2-oxazolidinone from example 1 is mixed with 4-fluoro-N-(4- hydroxyphenyl)methylene-benzenamine (121 gm) and cooled to -10°C. Then diisopropylethylamine (260 ml) is added to the reaction mixture for 45 minutes at -10°C to -15°C, trimethylsilylchloride (135 ml) is added and stirred for 1 hour at -20°C to -25°C. The reaction mixture is cooled to -30°C, TiCI4 (35 ml) is slowly added to the reaction mixture at -30°C to -35°C and stirred for 3 hours at the same temperature. 5% Aq. tartaric acid solution (1700 ml) is added to the reaction mixture at 0°C, stirred for 1 hour and allowed the temperature to rise to 25°C. Then 20% Aq. NaHSO3 (350 ml) solution and stirred for 2 hours at 25°C to 30°C. The organic layer is separated and washed with 1000 ml water, concentrated to 250 ml volume and added 100 ml bistrimethylsilylacetamide. Then the reaction mixture is heated to reflux for 30 minutes. The organic layer is concentrated to remove methylene dichloride, crystallized from the mixture of ethyl acetate (250 ml) and n-heptane (250 ml), and filtered and dried to give 135 gm of compound 4 (prot = trimethylsilyl).
……………


| WO2005066120A2 * | 23 Ara 2004 | 21 Tem 2005 | Prosenjit Bose | Process for asymmetric synthesis of hydroxy-alkyl substituted azetidinone derivatives |
| WO2006137080A1 * | 17 Şub 2006 | 28 Ara 2006 | Kumar Muppa Kishore | Improved process for the preparation of ezetimibe |
| WO2008151324A1 * | 9 Haz 2008 | 11 Ara 2008 | Teva Pharma | Reduction processes for the preparation of ezetimibe |
| US7470678 | 1 Tem 2003 | 30 Ara 2008 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| US7842684 | 25 Nis 2007 | 30 Kas 2010 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7863265 | 19 Haz 2006 | 4 Oca 2011 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7871998 | 21 Ara 2004 | 18 Oca 2011 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7893048 | 21 Haz 2006 | 22 Şub 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502 | 21 Haz 2006 | 15 Mar 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US8013150 | 17 Şub 2006 | 6 Eyl 2011 | Msn Laboratories Ltd. | Process for the preparation of ezetimibe |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4044147 * | May 26, 1976 | Aug 23, 1977 | Pfizer Inc. | N-(acyl)-p-amino-N’-(monosubstituted)-benzamide anti-ulcer agents |
| US5248611 * | Jan 3, 1992 | Sep 28, 1993 | Scripps Clinic And Research Foundation | Stereoisomer separation method using antibody combing site-containing molecules |
| US5739321 * | May 31, 1996 | Apr 14, 1998 | Schering Corporation | 3-hydroxy γ-lactone based enantionselective synthesis of azetidinones |
| US5846991 * | Dec 29, 1997 | Dec 8, 1998 | Nissan Chemical Industries, Ltd. | Pyrazole derivatives |
| US5856473 * | Oct 31, 1996 | Jan 5, 1999 | Schering Corporation | Process for preparing 1-(4-fluorophenyl)-3(R)-(3(S)-hydroxy-3-( phenyl or 4-fluorophenyl!)-propyl)-4(S)-(4-hydroxyphenyl)-2-azetidinone |
| US5859035 * | Mar 27, 1997 | Jan 12, 1999 | Merck & Co., Inc. | Arylheteroaryl inhibitors of farnesyl-protein transferase |
| US5886171 * | May 28, 1997 | Mar 23, 1999 | Schering Corporation | 3-hydroxy gamma-lactone based enantioselective synthesis of azetidinones |
| US6194599 * | Apr 8, 1997 | Feb 27, 2001 | Catalytica, Inc. | Process for preparing biaryl compounds |
| US6207822 * | Dec 5, 1999 | Mar 27, 2001 | Schering Corporation | Process for the synthesis of azetidinones |
| US20030013699 * | May 22, 2002 | Jan 16, 2003 | Davis Harry R. | Methods for treating alzheimer’s disease and/or regulating levels of amyloid beta peptides in a subject |
| US20050124808 * | Sep 23, 2004 | Jun 9, 2005 | Dsm I.P. Assets B.V. | Process for preparing unsymmetrical biaryls and alkylated aromatic compounds from arylnitriles |
| US20060247273 * | Sep 24, 2004 | Nov 2, 2006 | Takayuki Kawaguchi | Carbamoyl-type benzofuran derivatives |
| US20090048441 * | Feb 17, 2006 | Feb 19, 2009 | Manne Satyanarayana Reddy | Process for the Preparation of Ezetimibe |
| USRE37721 * | Jun 15, 2000 | May 28, 2002 | Schering Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7470678 | Jul 1, 2003 | Dec 30, 2008 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| US7842684 | Apr 25, 2007 | Nov 30, 2010 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7863265 | Jun 19, 2006 | Jan 4, 2011 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7871998 | Dec 21, 2004 | Jan 18, 2011 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7893048 | Jun 21, 2006 | Feb 22, 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502 | Jun 21, 2006 | Mar 15, 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US8013150 * | Feb 17, 2006 | Sep 6, 2011 | Msn Laboratories Ltd. | Process for the preparation of ezetimibe |
| US8383810 | Dec 12, 2011 | Feb 26, 2013 | Merck Sharp & Dohme Corp. | Process for the synthesis of azetidinones |
| US20050239766 * | Jul 1, 2003 | Oct 27, 2005 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| US20060160785 * | Dec 5, 2005 | Jul 20, 2006 | Judith Aronhime | Ezetimibe polymorphs |
| US20060234996 * | Apr 13, 2006 | Oct 19, 2006 | Itai Adin | Novel crystalline form of ezetimibe and processes for the preparation thereof |
| US20110130378 * | May 26, 2009 | Jun 2, 2011 | Lek Pharmaceuticals D.D. | Ezetimibe process and composition |
| US20110183956 * | Jul 29, 2009 | Jul 28, 2011 | Janez Mravljak | Process for the synthesis of ezetimibe and intermediates useful therefor |
| CN102219803A * | Apr 20, 2011 | Oct 19, 2011 | 浙江大学 | Preparation method of ezetimibe intermediate |
| EP2128133A1 | May 26, 2008 | Dec 2, 2009 | Lek Pharmaceuticals D.D. | Ezetimibe process and composition |
| WO2008096372A2 * | Feb 6, 2008 | Aug 14, 2008 | Pranav Gupta | Process for preparing highly pure ezetimibe using novel intermediates |
| WO2009150038A1 | May 26, 2009 | Dec 17, 2009 | Lek Pharmaceuticals D.D. | Process for the preparation of ezetimibe and composition containing it |
| WO2009157019A2 * | Jun 23, 2009 | Dec 30, 2009 | Ind-Swift Laboratories Limited | Process for preparing ezetimibe using novel allyl intermediates |
| CN1131416A * | Sep 14, 1994 | Sep 18, 1996 | 先灵公司 | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| CN1330721A * | Oct 1, 1999 | Jan 9, 2002 | 先灵公司 | Resolution of trans-2-(alkoxycarbonylethyl)-laitams useful in synthesis of 1-(4-fluorophenyl)-3(R)-[3(s)-hydroxy-3-(flurophenyl)-propyl)]-4(s)-(4-hydroxyphenyl)-2-azetidinone |
| CN1931838A * | 20 Oct 2006 | 21 Mar 2007 | 屠勇军 | Azacyclo butanone derivative and its synthesis process |
| WO2007108007A1 * | 12 Sep 2006 | 27 Sep 2007 | Prabhavalkar Tirtha Suresh | A process for the preparation of ezetimibe via a novel intermediate |
| WO2008096372A2 * | 6 Feb 2008 | 14 Aug 2008 | Pranav Gupta | Process for preparing highly pure ezetimibe using novel intermediates |
| WO2009157019A2 * | 23 Jun 2009 | 30 Dec 2009 | Ind-Swift Laboratories Limited | Process for preparing ezetimibe using novel allyl intermediates |
| WO2008032338A2 * | 10 Eyl 2007 | 20 Mar 2008 | Reddy Manne Satyanarayana | Improved process for the preparation of ezetimibe and its intermediates |
| WO2008089984A2 * | 24 Oca 2008 | 31 Tem 2008 | Krka | Process for the preparation of ezetimibe and derivatives thereof |
| WO2008096372A2 * | 6 Şub 2008 | 14 Ağu 2008 | Pranav Gupta | Process for preparing highly pure ezetimibe using novel intermediates |
| WO2008106900A1 * | 3 Mar 2008 | 12 Eyl 2008 | Zentiva As | Method of manufacturing (3r, 4s) -1- (4-fluorophenyl) -3- [ (3s) -3- (4 -fluorophenyl) -3-hydroxypropyl) ] -4- (4-hyd roxyphenyl) -2-azetidinone |
| WO2008151324A1 * | 9 Haz 2008 | 11 Ara 2008 | Teva Pharma | Reduction processes for the preparation of ezetimibe |
| WO2009067960A2 * | 5 Kas 2008 | 4 Haz 2009 | Zentiva As | A method of manufacturing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone and its intermediates |
| WO2011158052A1 | 17 Haz 2011 | 22 Ara 2011 | Nanoform Cardiovascular Therapeutics Ltd. | Nanostructured ezetimibe compositions, process for the preparation thereof and pharmaceutical compositions containing them |
| WO2012155932A1 | 17 May 2011 | 22 Kas 2012 | Pharmathen S.A. | Improved process for the preparation of ezetimibe |
| CN102531986A * | 23 Şub 2012 | 4 Tem 2012 | 苏州朗科生物技术有限公司 | Preparation method for ezetimibe |
| CN103044305A * | 24 Oca 2013 | 17 Nis 2013 | 上海现代制药股份有限公司 | Preparation method of ezetimibe intermediate |
| EP1922304A2 † | 8 Eyl 2006 | 21 May 2008 | Teva Pharmaceutical Industries Ltd | Processes for the preparation of (3r,4s)-4-((4-benzyloxy)phenyl)-1-(4-fluorophenyl)-3-((s)-3-(4-fluorophenyl)-3-hydroxypropyl)-2-azetidinone, an intermediate for the synthesis of ezetimibe |
| EP1953140A1 * | 24 Oca 2007 | 6 Ağu 2008 | Krka | Process for the preparation of ezetimibe and derivatives thereof |
| US7842684 | 25 Nis 2007 | 30 Kas 2010 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7863265 | 19 Haz 2006 | 4 Oca 2011 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7871998 | 21 Ara 2004 | 18 Oca 2011 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7893048 | 21 Haz 2006 | 22 Şub 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502 | 21 Haz 2006 | 15 Mar 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US8383810 | 12 Ara 2011 | 26 Şub 2013 | Merck Sharp & Dohme Corp. | Process for the synthesis of azetidinones |
| WO2005049592A1 * | 24 Kas 2003 | 2 Haz 2005 | Hetero Drugs Ltd | A novel process for ezetimibe intermediate |
| US5856473 * | 31 Eki 1996 | 5 Oca 1999 | Schering Corporation | Process for preparing 1-(4-fluorophenyl)-3(R)-(3(S)-hydroxy-3-( phenyl or 4-fluorophenyl!)-propyl)-4(S)-(4-hydroxyphenyl)-2-azetidinone |
////

