
Brexpiprazole
ブレクスピプラゾール
OPC-34712, UNII-2J3YBM1K8C, OPC34712,
CAS 913611-97-9,
Molecular Weight:433.56578 g/mol
7-[4-[4-(1-benzothiophen-4-yl)piperazin-1-yl]butoxy]-1H-quinolin-2-one
7-[4-[4-(1-Benzothiophen-4-yl)piperazin-1-yl]butoxy]quinolin-2(1H)-one
2(1H)-Quinolinone, 7-[4-(4-benzo[b]thien-4-yl-1-piperazinyl)butoxy]-
7- [ 4- ( 4-benzo[b]thiophen-4- yl-piperazin-l-yl)butoxy] -lH-quinolin-2-one
7-[4-(4-benzo[b]thiophen-4-yl-piperazin-1-yl)butoxy]-1H-quinolin-2-one
NDA is considered filed as of September 9, 2014 (60 days after submission). The PDUFA date is July 11, 2015.
UPDATE JULY 2015 ON STATUS OF APPROVAL
depression and schizophrenia

Brexpiprazole (// breks-pip-rə-zohl; also called OPC-34712) is a novel D2 dopamine partial agonist investigational product currently in clinical trials for the treatment of depression, schizophrenia, and attention deficit hyperactivity disorder(ADHD).[1]Although it failed Stage 2 trials for ADHD, it has been designed to provide improved efficacy and tolerability (e.g., lessakathisia, restlessness and/or insomnia) over established adjunctive treatments for major depressive disorder (MDD).[2]
OPC-34712 is an antidepressant and antipsychotic drug candidate awaiting approval in the U.S. for the treatment of schizophrenia and also as adjunctive treatment of major depressive disorder (MDD). The product is in phase III clinical trials for the treatment of agitation associated with Alzheimer’s disease. Phase III clinical trials are also underway for the treatment of post-traumatic stress disorder (PTSD).
brexpiprazole (pre-registration, as of April 2015), which is being developed by Otsuka and Lundbeck, useful for treating schizophrenia, agitation associated with Alzheimer’s disease, major depressive disorder and attention deficit hyperactivity disorder. Family members of the product case, WO2006112464, hold protection in EU states until 2026 and its US equivalent, US7888362, has US154 extension, expiring in 2027. Suzhou Vigonvita Life Sciences appears to be new to patenting and is the first collaborative filing from the three assignees.

Phase II clinical trials are also ongoing for use as adjunctive therapy in adults with attention deficit hyperactivity disorder (ADHD). The compound is being developed by Otsuka Pharmaceutical. In 2011, a codevelopment and commercialization agreement was signed by Lundbeck and Otsuka Pharmaceutical in Latin and North America, Australia and Europe for the treatment of psychiatric disorders.
The drug is being developed by Otsuka, and is considered to be a successor[3] of its top-selling antipsychotic agent aripiprazole(brand names: Abilify, Aripiprex). Otsuka’s US patent on aripiprazole expired on October 20, 2014;[4] however, due to a pediatric extension, a generic will not become available until at least April 20, 2015.[5]

Brexpiprazole (1) , a serotonin–dopamine activity modulator, is an investigational new drug currently in phase-III clinical trials for the treatment of depression, schizophrenia, and attention deficit hyperactivity disorder.(1A) Brexpiprazole is also considered to be a possible successor to the top-selling antipsychotic agent aripiprazole.(2A)
-
-
Brexpiprazole
In the clinical program, brexpiprazole demonstrated improvement in symptoms in both schizophrenia and as adjunctive therapy in major depressive disorder (MDD)
July 2015 is the anticipated completion timing of the FDA’s review (based on PDUFA timeline)Otsuka Pharmaceutical Co., Ltd. (Otsuka) and H. Lundbeck A/S (Lundbeck) today announced that the U.S. Food and Drug Administration (FDA) has determined that the New Drug Application (NDA) for brexpiprazole for monotherapy in adult patients with schizophrenia and for adjunctive treatment of major depressive disorder (MDD) in adult patients is sufficiently complete to allow for a substantive review, and the NDA is considered filed as of September 9, 2014 (60 days after submission). The PDUFA date is July 11, 2015.The NDA is supported by seven completed placebo-controlled clinical phase II or III studies in the proposed indications – three studies in schizophrenia and four studies with brexpiprazole as adjunctive therapy in MDD. The dossier included data from more than 6,000 participants of whom more than 5,000 received brexpiprazole.
Brexpiprazole in adult patients with schizophreniaOne clinical phase II and two clinical phase III placebo-controlled studies have been completed using brexpiprazole in adult patients suffering from schizophrenia. Across the three studies more than 1,700 patients have been randomized.In the first pivotal phase III study randomizing approximately 625 patients, brexpiprazole 2mg/day and 4 mg/day both demonstrated greater improvement of symptoms relative to placebo as measured by change from baseline in the Positive and Negative Syndrome Scale (PANSS) Total Score at week 6 (p<0.05). Results of the key secondary endpoint supported primary results.In the second pivotal phase III study randomizing approximately 650 patients, brexpiprazole 4 mg/day again demonstrated greater improvement of symptoms relative to placebo (p<0.05) in change from baseline in the PANSS Total Score at Week 6. Brexpiprazole 2 mg/day showed numerical improvement (p>0.05) over placebo at Week 6.The results from the clinical phase II studyi were presented at the 24th Annual US Psychiatric and Mental Health Congress in November 2011. The study showed a clinically meaningful improvement from baseline measured by PANSS total score at week 6, although it did not achieve statistical separation from placeboii.In the placebo-controlled phase II and III studies, the rates of discontinuation due to adverse events were 8.1% for patients receiving brexpiprazole compared to 12.7% of patients receiving placebo; the only adverse event that occurred in more than 5% of brexpiprazole patients and more frequently than placebo was akathisia (5.8% vs. 4.5%).
Brexpiprazole as adjunctive therapy in major depressive disorder (MDD) Four studies have been included in the dossier using brexpiprazole as adjunctive therapy for adult patients suffering from MDD who had demonstrated a consistent, inadequate response to at least two regimens of prior antidepressant treatment. Patients with MDD and an inadequate response to one to three antidepressants were enrolled and received antidepressants for 8 weeks, single blinded, in the two phase III studies. Patients with an inadequate response during this prospective phase were provided antidepressant therapy and randomized adjunctive treatment with either brexpiprazole or placebo for 6 weeks. The primary efficacy endpoint was the change in MADRS (Montgomery–Åsberg Depression Rating Scale) Total Score from baseline at week 6. MADRS is a commonly used scale to assess the range of symptoms in patients with MDD. Across the four studies, more than 3,900 patients entered the prospective phase and more than 1,800 patients were included in the randomized phase of the studies.The first pivotal phase III results were presented in a poster session at the 22nd European Psychiatry Association Congress (EPA) in March 2014. This two-arm phase III study randomized approximately 380 patients and demonstrated an improvement of symptoms with an antidepressant plus 2 mg brexpiprazole that was greater than an antidepressant plus placebo (p<0.001)The second pivotal phase III study was a three-arm study in which approximately 675 patients were randomized to treatment with an antidepressant plus either placebo, 1 mg brexpiprazole or 3 mg brexpiprazole.v Patients in both brexpiprazole treatment groups showed greater improvement in symptoms as measured by the MADRS compared to placebo (1 mg p>0.05, 3 mg p<0.05). Results of the second pivotal phase III study in MDD have not yet been published.
The first clinical phase IIvi study randomized approximately 425 patients in four arms and was presented at the 164th Annual Meeting of the American Psychiatric Association in May 2011. Patients exhibited greater improvements than adjunctive placebo in MADRS Total score with the 1.5 (±0.5) mg/day dose of brexpiprazole after six weeks of treatment (p
About brexpiprazole (OPC-34712)Brexpiprazole is a novel investigational psychotropic compound discovered by Otsuka and under co-development with Lundbeck. Brexpiprazole is a serotonin-dopamine activity modulator (SDAM) that acts as a partial agonist at 5-HT1A and dopamine D2 receptors at similar potency, and an antagonist at 5-HT2A and noradrenaline alpha1B/2C receptors.

Partnership with Lundbeck
In November 2011, Otsuka and Lundbeck have announced a global alliance.[6] Lundbeck has given Otsuka an upfront payment of $200 million, and the deal includes development, regulatory and sales payments, for a potential total of $1.8 billion. Specifically for OPC-34712, Lundbeck will obtain 50% of net sales in Europe and Canada and 45% of net sales in the US from Otsuka.
The partnership has been presented by Otsuka to its investors as a good fit for several reasons:[7]
- Geographic strategy: Otsuka in Japan, Asia, US; Lundbeck in Europe, South America and emerging markets
- Research strategy: Otsuka has knowledge in antipsychotics, Lundbeck in anti-depressant and anxiolytic.
- CNS strategy: Otsuka has a robust portfolio in next-generation CNS drugs, while Lundbeck covers a wide range of CNS conditions from Alzheimer’s to schizophrenia.
- Similar corporate culture

Clinical trials
OPC-34712 is currently in clinical trials for adjunctive treatment of MDD, adjunctive treatment of adult ADHD and schizophrenia.[8]
Major depression
Phase II
The Phase 2 multicenter, double-blind, placebo-controlled study randomized 429 adult MDD patients who exhibited an inadequate response to one to three ADTs in the current episode. The study was designed to assess the efficacy and safety of OPC-34712 as an adjunctive treatment to standard ADT. The ADTs included in the study were desvenlafaxine, escitalopram, fluoxetine, paroxetine, sertraline, and venlafaxine.[9]
Phase III
A new Phase III study is currently in the recruiting stage: “Study of the Safety and Efficacy of Two Fixed Doses of OPC-34712 as Adjunctive Therapy in the Treatment of Adults With Major Depressive Disorder (the Polaris Trial)”.[10] Its goal is “to compare the effect of OPC-34712 to the effect of placebo (an inactive substance) as add on treatment to an assigned FDA approved antidepressant treatment (ADT) in patients with Major Depressive Disorder who demonstrate an incomplete response to a prospective trial of the same assigned FDA approved ADT”. Estimated enrollment is 1250 volunteers.
Brexpiprazole, code: OPC-34712) is Otsuka Pharmaceutical Co., Ltd. developed a new generation of anti-psychotic drug candidates, and its role in multiple receptors, dopamine D2 receptor partial agonist (improving positive and negative symptoms, cognitive impairment and depressive symptoms), 5-HT2A receptor antagonist (improving negative symptoms, cognitive dysfunction, symptoms of depression, insomnia), α1 adrenoceptor antagonists (improving positive symptoms of schizophrenia), 5 – serotonin uptake / reuptake inhibitors (improve depressive symptoms); at the same time, but also a 5-HT1A partial agonist (anxiolytic and antidepressant activity) and 5-HT7 antagonist (temperature, circadian rhythms, learning and memory, sleep) . Currently, in the United States and Europe as adjuvant treatment of severe depression (MDD) Phase III clinical trial; III clinical trial for the treatment of schizophrenia in the United States, Europe and Japan, meanwhile, is still the United States Phase II adult ADHD Clinical Trials.
Adult ADHD
Phase II
- Study of the Safety and Efficacy of OPC-34712 as a Complementary Therapy in the Treatment of Adult Attention Deficit/Hyperactivity Disorder (STEP-A)[11] The company did not move the product to Phase III, and it is presumed this drug failed Phase II trials for the disorder.
Schizophrenia
Phase I
- Trial to Evaluate the Effects of OPC-34712 on QT/QTc in Subjects With Schizophrenia or Schizoaffective Disorder[12]
Phase II
- A Dose-finding Trial of OPC-34712 in Patients With Schizophrenia[13]
Phase III
- Efficacy Study of OPC-34712 in Adults With Acute Schizophrenia (BEACON)[14]
- Safety and Tolerability Study of Oral OPC-34712 as Maintenance Treatment in Adults With Schizophrenia (ZENITH)[15]
- Study of the Effectiveness of Three Different Doses of OPC-34712 in the Treatment of Adults With Acute Schizophrenia (VECTOR)[16]
- A Long-term Trial of OPC-34712 in Patients With Schizophrenia[17]
Conferences
- Phase II results were presented at the American Psychiatric Association’s 2011 annual meeting in May 2011.[18]
- The drug has been presented at the 2nd Congress of Asian College of Neuropsychopharmacology[19] in September 2011.
- At the US Psychiatric and Mental Health Congress in November 2011 in Vegas, Robert McQuade presented the Phase II Trial results for Schizophrenia[20]
Pharmacology
Brexpiprazole acts as a partial agonist of the 5-HT1A, D2, and D3 receptors, and as an antagonist of the 5-HT2A, 5-HT2B, 5-HT7, α1A–, α1B–, α1D–, and α2C-adrenergic, and H1receptors.[22] It has negligible affinity for the mACh receptors.[22]
Patents
| Patent |
Submitted |
Granted |
| PIPERAZINE-SUBSTITUTED BENZOTHIOPHENES FOR TREATMENT OF MENTAL DISORDERS [US2011152286] |
2011-06-23 |
|
| Piperazine-substituted benzothiophenes for treatment of mental disorders [US7888362] |
2010-07-15 |
2011-02-15 |
Otsuka Pharmaceutical Co., Ltd. are disclosed in PCT Application WO2006112464A1 in the preparation route see Scheme 1, the difficulty of the route is the first reaction generates byproducts easily separated by column chromatography is not easy to obtain high-purity intermediates, thus affecting the final product Bray prazosin purity and yield.Scheme 1:
Subsequently, Otsuka Pharmaceutical Co., Ltd. are disclosed in PCT Application WO2013015456A1 in the alternative method of preparing the reaction of this step, see Scheme 2, the route the reagents are more expensive, high-cost, environmentally unfriendly and not suitable for industrial production.
Reaction Scheme 2:
Due to the above production process there is a high cost, and difficult to separate impurities and other shortcomings, it is necessary to find an economical, practical, environmental protection, new routes to improve process stability, reduce costs, improve product quality.
Synthesis
WO 2013015456
IN THIS BELOW PIC WE SEE
click on pics below to view
Synthesis of A
1 BROMO 4 CHLORO BUTANE WAS REACTED WITH 7 HYDROXY 1H QUINOLINE -2-ONE TO GIVE A
7 ( 4 CHLORO BUTOXY)-1H -QUINOLINE-2-ONE, WHICH WILL BE USED FOR COUPLING AT LAST STAGE
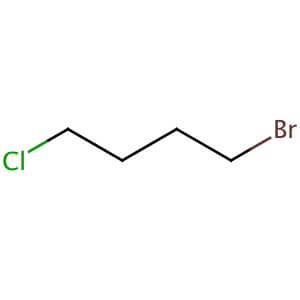 1 BROMO 4 CHLORO BUTANE
1 BROMO 4 CHLORO BUTANE

IN THE BELOW PIC 2,6-Dichlorobenzaldehyde AND RHODANINE WERE REACTED TO GIVE 2,6-dichlorobenzylidenerhodanine.
 2,6-Dichlorobenzaldehyde
2,6-Dichlorobenzaldehyde
 RHODANINE
RHODANINE
NEXT WAS
2,6-dichlorobenzylidenerhodanine, GAVE (Z)-3-(2,6-dichlorophenyl)-2-mercapto-2-propenoic acid.
1H-NMR (DMSO-d6) d
ppm; 7.23-7.67 (4H, m), 3.5-5.7 (1H, br.), 11.7-14.5 (1H, br.).
Next was prepration of K salt
(Z)-3-(2,6-dichlorophenyl-2-mercapto-2-propenoic acid and potassium hydroxide gave ((Z)-3-(2,6-dichlorophenyl-2-mercapto-2-propenoic acid potassium salt).
Next stage
((Z)-3-(2,6-dichlorophenyl-2-mercapto-2-propenoic acid potassium
salt) GAVE 2-carboxy-4-chlorobenzo[b]thiophene.
Yield: 48.8 g. 1H-NMR (DMSO-d6) d ppm; 7.53 (1H, t, J = 7.7 Hz), 7.58 (1H, dd, J = 7.7, 1.3
Hz), 8.03 (1H, d, J = 0.5 Hz), 8.07 (1H, d, J = 7.6 Hz).
NEXT IS DECARBOXYLATION
A mixture of 2-carboxy-4-chlorobenzo[b]thiophene, 1,3-dimethyl-2-imidazolidinone, and 1,8-
diazabicyclo[5.4.0]-undec-7-ene GAVE compound. 4-chlorobenzo[b]thiophene. 1H-NMR (DMSO-d6) d ppm; 7.38 (1H, t, J = 8.4
Hz), 7.51 (1H, dd, J = 5.5, 0.8 Hz), 7.48 (1H, dd, J = 7.7, 0.9 Hz), 7.94 (1H, dd, J = 5.5, 0.4
Hz), 8.02 (1H, dt, J = 8.0, 0.9 Hz).

BETTER REPRESENTATION OF ABOVE PIC
CLIPS FROM PATENT
Synthesis of 2,6-dichlorobenzylidenerhodanine
2,6-Dichlorobenzaldehyde (77.0 g) , rhodanine (58.6 g) , and acetic acid (539 ml) were suspended with stirring at room temperature. Anhydrous sodium acetate (116 g) was added to the suspension, and the resulting mixture was heated under reflux for 3 hours. The reaction mixture was cooled to 45°C, and ice water (700 ml) was added. After the mixture was stirred for 0.2 hours, the precipitated crystals were collected by filtration, washed with water, and then dried to obtain 2,6- dichlorobenzylidenerhodanine. Even in non-dried form, this product could be subjected to the subsequent step.
Yield: 125.4 g^- MR (CDC13) 6ppm;7.30-7.44 (3H, m) , 7.70 (1H. s), 9.6 (1H, br.).
Reference Example 3
• Synthesis of (Z)-3-(2,6-dichlorophenyl)-2-mercapto-2-propenoic acid
A suspension of 2,6- dichlorobenzylidenerhodanine (160.4 g) and water (800 ml) was stirred at room temperature, and sodium hydroxide (83.0 g) was added over a period of 1 hour. The resulting mixture was heated with stirring for another 0.5 hours. The reaction mixture was cooled with ice (10°C), and concentrated hydrochloric acid (192 ml) was added. After the mixture was stirred while cooling with ice for 0.5 hours, the precipitated crystals were collected by filtration. The crystals obtained by filtration were washed with water and then dried to obtain an equivalent amount of (Z)-3-(2,6-dichlorophenyl)-2-mercapto-2- propenoic acid.
Yield: 138.9 g l-NMR (DMSO-de) δρρπΐ;7.23-7.67 (4H, m) , 3.5-5.7 (1H, br.), 11.7-14.5 (1H, br.).
Reference Example 4
• Synthesis of 2-carboxy-4-chlorobenzo[b] thiophene
A suspension of (Z)-3-(2,6-dichlorophenyl-2-mercapto-2- propenoic acid (72.4 g) and water (362 ml) was stirred at room temperature. Further, potassium hydroxide (40.8 g) was added, and the mixture was heated under reflux for 4 hours . After the mixture was allowed to cool, the mixture was stirred for 1 hour while cooling with ice. The precipitated crystals ((Z)-3-(2,6- dichlorophenyl-2-mercapto-2-propenoic acid potassium salt) were collected by filtration and washed with cold water. After the crystals were suspended in water, 35% concentrated hydrochloric acid (32 ml) was added (pH = 1), and the mixture was stirred at room temperature for 1 hour. The precipitated crystals were collected by filtration and dried to obtain 2-carboxy-4- chlorobenzo[b] thiophene.
Yield of 48.8 g ^- MRiDMSO-de) 6ppm; 7.53 (1H, t, J = 7.7 Hz), 7.58 (1H, dd, J = 7.7, 1.3 Hz), 8.03 (1H, d, J = 0.5 Hz), 8.07 (1H, d, J = 7.6 Hz).
Reference Example 5
• Synthesis of K salt 4-chlorobenzo[b] thiophen-2-carboxylate
Reference Example 6
· Synthesis of 2-carboxy-4-chlorobenzo[b]thiophene
Sodium 4-chlorobenzo[b] thiophen-2-carboxylate (2.40 g) was dissolved in water (33 ml) at 60°C. Concentrated hydrochloric acid (1.3 ml) was added to the solution at the same temperature, and the resulting mixture was stirred. The precipitated crystals were collected by filtration, washed with water, and then dried to obtain 2-carboxy-4-chlorobenzo[b] thiophene.
Yield: 1.61 g ^- MR (DMS0-d6);7.53 (1H, t, J = 7.7 Hz), 7.58 (1H, dd, J = 7.7, 1.3 Hz), 8.03 (1H, d, J = 0.5 Hz), 8.07 (1H, d, J = 7.6 Hz).


Elaborate description
IN THIS BELOW PIC WE SEE
Synthesis of 4-(1-piperazinyl)benzo[b]thiophene
4-Chlorobenzo[b]thiophene and xylene , Subsequently, piperazine, sodium tert-butoxide, palladium acetate (II), and 2-dicyclohexylphosphino-2′,6′-di-iso-propoxy-1,1′-biphenyl (RuPhos) …… producing 4-(1-piperazinyl)benzo[b]thiophene.
NEXT IS PREPARATION OF HYDROCHLORIDE
4-(1-piperazinyl)benzo[b]thiophene hydrochloride. 1H-NMR (DMSO-d6) d ppm;
3.30 (4H, br.s), 3.61 (4H, br.s), 6.97 (1H, d, J = 7.8 Hz), 7.32 (1H, br. dd, J = 8.4, 7.8 Hz),
7.53 (1H, d, J = 5.6 Hz), 7.70 (1H, d, J = 8.4 Hz), 7.76 (1H, d, J = 5.6 Hz), 9.37 (1H, br.s).
NEXT IS REACTION WITH A TO GIVE BREXPIPRAZOLE
1-benzo[b]thiophen-4-yl-piperazine hydrochloride, potassium carbonate
and DMF and 7-(4-chlorobutoxy)-1H-quinolin-2-one A FROM PIC 1 and potassium iodide GAVE BREXPIPRAZOLE, ie 7-[4-(4-benzo[b]thiophen-4-yl-piperazin-1-yl)butoxy]-1H-quinolin-2-one.
1H-NMR (DMSO-d6) d ppm; 1.6-1.75 (2H, m), 1.75-1.9 (2H, m), 2.44
(2H, t, J = 7.0 Hz), 2.55-2.70 (4H, m), 3.00-3.15 (4H, m), 4.06 (2H, t, J = 6.3 Hz), 6.30 (1H,
d, J = 9.5 Hz), 6.75-6.85 (2H, m), 6.88 (1H, d, J = 7.5 Hz), 7.27 (1H, dd, J = 8 Hz, 8 Hz),
7.40 (1H, d, J = 5.5 Hz), 7.55 (1H, d, J = 9.5 Hz), 7.61 (1H, d, J = 8 Hz), 7.69 (1H, d, J = 5.5
Hz), 7.80 (1H, d, J = 9.5 Hz), 11.58 (1H, bs).

BETTER REPRESENTATION OF PIC
Example 2
• Synthesis of 4- (l-piperazinyl)benzo[b]thiophene hydrochloride
4-Chlorobenzo[b] thiophene (5.00 g), piperazine (5.11 g) , palladium acetate (II) (2.7 mg), tri-tert-butylphosphonium
tetraphenylborate (6.2 mg), sodium tert-butoxide (8.548 g), and xylene (70 ml) were stirred at 120 to 130°C for 5 hours. After the reaction mixture was cooled to room temperature, water was added thereto, and the layers were separated. The xylene layer was washed with water, and then with saline. After addition of activated carbon, the mixture was stirred at room temperature for 30 minutes. After filtration of the mixture, concentrated
hydrochloric acid was added to the filtrate, and the resulting mixture was stirred at room temperature for 30 minutes. The precipitated crystals were collected by filtration and dried to obtain 4- ( l-piperazinyl)benzo[b] thiophene hydrochloride.
Yield: 6.94 g !H-NMRiDMSO-de) 6ppm; 3.30 (4H, br.s), 3.61 (4H, br.s), 6.97 (1H, d, J= 7.8 Hz), 7.32 (1H, br.dd, J= 8.4. 7.8 Hz), 7.53 (1H, d, J= 5.6 Hz), 7.70 (1H, d, J= 8.4 Hz), 7.76 (1H, d, J= 5.6 Hz), 9.37 (1H, br.s).
Example 3
• Synthesis of 4- ( 1-piperazinyl)benzo[b] thiophene hydrochloride
4-Chlorobenzo[b] thiophene (10.0 g) and xylene (100 ml) were placed in a reaction vessel. The reaction vessel was
evacuated and then purged with argon. Subsequently, piperazine (15.3 g) , sodium tert-butoxide (17.1 g) , palladium acetate (II) (13.0 mg) , and 2-dicyclohexylphosphino-2′,6′-di-iso-propoxy-1,1′- biphenyl (RuPhos) (69.0 mg) were added. After evacuation and purging with argon, the mixture was refluxed for 2 hours. After the reaction mixture was cooled to about 80°C, water (50 ml) and silica #600H (0.65 g) were added. The mixture was stirred at approximately 60°C for about 10 minutes, and then filtered. After the filtrate was separated into layers, the xylene layer was washed with water. Subsequently, the xylene layer was placed into the reaction vessel again. After addition of water (200 ml) and concentrated hydrochloric acid (8.0 ml) , the mixture was heated with stirring for dissolution. The layers were separated at 75°C or more. After the aqueous layer was collected, toluene (150 ml) and 25% aqueous sodium hydroxide solution (16 ml) were added, and the mixture was stirred. The layers were separated, and the organic layer was collected. The organic layer was washed with water and concentrated with an evaporator. Methanol (150 ml) was added to the concentrated oil to dissolve the oil, thus producing a methanol solution. 2-Propanol (150 ml) and concentrated
hydrochloric acid (7 ml) were placed into another reaction vessel, and the methanol solution was added dropwise over a period of 15 minutes or more. After completion of the dropwise addition, the mixture was cooled and stirred at 10°C or less for about 30 minutes, and then filtered (washed with a mixture of 5 ml of methanol and 5 ml of 2-propanol) . The crystals were collected, and then dried to obtain 4-(l-piperazinyl)benzo[b]thiophene hydrochloride.
Yield: 11.61 g
^-NMRfDMSO-de) oppm;
3.25-3.40 (8H, br.s), 6.96 (1H, d, J = 7.5 Hz), 7.32 (1H, dd, J = 8.0, 7.5 Hz), 7.52 (1H, d, J = 5.5 Hz ) . 7.70 (1H, d, J = 8.0 Hz), 7.75 (1H, d, J = 5.5 Hz), 9.35 (1H, br.s).
Reference Example 9
· Synthesis of 7- ( 4-chlorobutoxy) -lH-quinolin-2-one
After 7-hydroxy-lH-quinolin-2-one (10 g) and DMF (50 ml) were heated to approximately 30°C, an aqueous potassium carbonate solution (potassium carbonate: 8.6 g, water: 10 ml) was added. After the mixture was stirred at 30 to 40°C for about 15 minutes, l-bromo-4-chlorobutane (14.3 ml) was added and stirred at approximately 40°C for 5 hours. Water (100 ml) was added dropwise over a period of 30 minutes or more while the
temperature was maintained at 30°C or more. After the mixture was stirred at approximately 30°C for 30 minutes, stirring was continued at 10°C or less for 1 hour, after which the precipitated crystals were collected by filtration. After methanol (100 ml) was added to the precipitated crystals, the mixture was stirred under reflux to ensure dissolution. This solution was cooled and stirred at 30 to 40°C for 30 minutes and then at 5°C or less for about 1 hour, after which the precipitated crystals were
collected by filtration. The crystals were dried at 60°C to obtain 7- (4-chlorobutoxy) -lH-quinolin-2-one as white powder.
Yield: 12.3 g
^I-NMR (300 MHz; CDC13) oppm; 1.95-2.05 (4H, m) , 3.64 (2H, t, J = 6.0Hz), 4.10 (2H, t. J = 5.5 Hz), 6.56 (1H, d, J = 9.5 Hz), 6.80 (1H. dd, J = 9.0 Hz, 2.5 Hz), 6.84 (1H, d, J = 2.5 Hz), 7.45 (1H, d, J = 9.0 Hz), 7.73 (1H, d, J = 9.5 Hz), 12.45 (1H, brs).
Example 4
· Synthesis of 7- [4- (4-benzo[b]thiophen-4-yl-piperazin-l- yl)butoxy] -lH-quinolin-2-one
After 1-benzo[b] thiophen-4-yl-piperazine hydrochloride (10.6 g), potassium carbonate (5.8 g) , and DMF (50 ml) were stirred at 30 to 40°C for about 30 minutes, 7-(4-chlorobutoxy) -1H- quinolin-2-one (10.0 g) and potassium iodide (6.9 g) were added. The mixture was stirred at 90 to 100°C for 2 hours. While the temperature of the mixture was maintained at 60°C or more, water (150 ml) was added dropwise over a period of 10 minutes or more.
After the mixture was cooled to 10°C or less, the precipitated crystals were collected by filtration, and washed with water and then with ethanol.
After ethanol (325 ml) and acetic acid (25 ml) were added to the precipitated crystals, the mixture was stirred under reflux for dissolution. Concentrated hydrochloric acid (3.6 ml) was added at around 70°C, and the mixture was cooled. After confirming the precipitation of crystals, the mixture was heated again and stirred under reflux for 1 hour. After the mixture was cooled to 10°C or less, the precipitated crystals were collected by filtration and washed with ethanol.
After ethanol (191 ml) and water (127 ml) were added to the precipitated crystals, the mixture was stirred under reflux for dissolution. After activated carbon (0.89 g) was added, the mixture was stirred under reflux for 30 minutes and then hot filtered. After activated carbon was removed, the mixture was heated again for dissolution. After 25% aqueous sodium hydroxide solution (5.8 ml) was added at approximately 70°C, the mixture was stirred under reflux for 30 minutes, after which water (64 ml) was added at approximately 70°C. After the mixture was stirred at 40°C for 30 minutes, the precipitated crystals were collected by filtration at 40°C or less, then washed with water, and dried to obtain 7- [4-(4-benzo[b]thiophen-4-yl-piperazin-l-yl)butoxy] -1H- quinolin-2-one as white crystals.
Yield: 14.30 g ^-NMRfDMSO-de) 6ppm; 1.6-1.75 (2H, m) . 1.75-1.9 (2H, m) , 2.44 (2H, t, J = 7.0 Hz),2.55-2.70 (4H, m) , 3.00-3.15 (4H, m) , 4.06 (2H, t, J = 6.3 Hz), 6.30 (1H, d, J = 9.5 Hz), 6.75-6.85 (2H, m) , 6.88 (1H, d, J = 7.5 Hz), 7.27 (1H, dd, J = 8 Hz, 8 Hz), 7.40 (1H, d, J = 5.5 Hz), 7.55 (1H, d, J = 9.5 Hz), 7.61 (1H, d, J = 8 Hz), 7.69 (1H, d, J = 5.5 Hz), 7.80 (1H, d, J = 9.5 Hz), 11.58 (1H, bs) .


IH NMR PREDICT
![7-[4-[4-(1-benzothiophen-4-yl)piperazin-1-yl]butoxy]-1H-quinolin-2-one NMR spectra analysis, Chemical CAS NO. 913611-97-9 NMR spectral analysis, 7-[4-[4-(1-benzothiophen-4-yl)piperazin-1-yl]butoxy]-1H-quinolin-2-one H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-29/002/477/2477980_1h.png)
13 C NMR PREDICT
![7-[4-[4-(1-benzothiophen-4-yl)piperazin-1-yl]butoxy]-1H-quinolin-2-one NMR spectra analysis, Chemical CAS NO. 913611-97-9 NMR spectral analysis, 7-[4-[4-(1-benzothiophen-4-yl)piperazin-1-yl]butoxy]-1H-quinolin-2-one C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-29/002/477/2477980_13c.png)
Patent
https://patentscope.wipo.int/search/en/detail.jsf;jsessionid=D842B4D68D66F641E505E9690CF876D0.wapp2nB?docId=WO2015054976&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=FullText
Wherein, X is halogen, such as fluorine, chlorine, bromine, iodine; R and R 1 as defined above in the definition of the compounds of formula I the same;
Wherein, X is fluorine, chlorine, bromine or iodine; R 1 as defined above, with a compound of formula I as defined for the same;
Wherein, X is fluorine, chlorine, bromine or iodine; R 1 is the same as defined in the compounds shown above, and R are as defined for formula I. The present invention also provides processes for preparing key intermediates Bray prazosin/ Brexpiprazole or a salt thereof, the method as shown in Scheme 6:
Example 26
7- [4- (benzothiazol-4-yl-1-piperazinyl) butoxy] -3,4-dihydro -2 (1H) – quinolinone
Preparation of
The product (400mg, 0.83mmol) of Example 25 will be implemented, silver carbonate (46mg, 0.16mmol) was dissolved in DMSO (5mL) and the acetic acid was heated to 120 ℃ overnight. Cooling, water was added, extracted with ethyl acetate, ethyl acetate layer was washed with saturated sodium bicarbonate and brine each wash again, dried over anhydrous sodium sulfate, and silica gel column chromatography, to give a solid (80mg, yield 22%).
1 HNMR (400 MHz, DMSO-d 6 ): δ10.00 (s, 1H), 7.69 (d, 1H), 7.61 (d, 1H), 7.40 (d, 1H), 7.27 (t, 1H), 7.04 ( d, 1H), 6.89 (d, 1H), 6.50 (dd, 1H), 6.45 (d, 1H), 3.93 (t, 2H), 3.06 (br, 4H), 2.78 (t, 2H), 2.60 (br , 4H), 2.41 (t, 4H), 1.74 (t, 2H), 1.60 (t, 2H) ESI: [M + 1] + = 436.3.
Example 27
7- [4- (2-carboxy-benzothiophen-4-yl-1-piperazinyl) butoxy] -2 (1H) – quinolinone
Preparation of
A mixture of 2-chloro-6- (4- (4 – ((2-oxo-1,2-dihydro-quinolin-7-yl) oxy) butyl) piperazin-1-yl) benzaldehyde (80mg , 0.18mmol) was dissolved in DMF (5mL) was added sodium hydroxide (29mg, 0.73mmol) and thioglycolic acid (0.025mL, 0.36mmol), 120 ℃ stirred for 16 hours. Cooling, water was added, adjusted with 1N HCl aqueous solution is about pH = 5, extracted with ethyl acetate, the ethyl acetate layer was washed with saturated brine, dried over anhydrous sodium sulfate, and silica gel column chromatography, to give a solid (40mg, yield 46 %).
Piperazine hydrochloride – (2-carboxy-benzothiophen-4-yl)
Example 28 1-
The product of Example 17 (100mg, 0.25mmol) was dissolved in acetic acid (3mL) and concentrated hydrochloric acid (0.5 mL) in, 100 ℃ stirred for 10 hours. The reaction solution was poured into ice water, stirred for 10min after filtration, to obtain the target substance (38mg, 50% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 8.04 (s, 1H), 7.69 (d, 1H), 7.43 (t, 1H), 7.00 (d, 1H), 3.30 ( bs, 8H) ESI: [M + 1] + = 262.9.
Preparation of tert-butyl piperazine-1 – Example 224- (2-carboxy-benzothiophen-4-yl)
Under nitrogen, to N, at room temperature was added N- dimethylformamide (5mL) within the reference product (200g, 0.62mmol) of Example 1, thioglycolic acid (114mg, 1.23mmol), sodium methoxide (133mg, 2.45mmol ), and the mixture was stirred at 105 ℃ 18 hours. Cooling, water was added, extracted with ethyl acetate, separated and the aqueous phase was adjusted pH = 5 or so, the precipitated solid was filtered and dried to obtain the target substance (130mg, 58% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 7.98 (s, 1H), 7.64 (d, 1H), 7.42 (t, 1H), 6.95 (d, 1H), 3.53 (bs, 4H), 3.035 ( bs, 4H) ESI: [M-1] – = 361.1.
Preparation of piperazine-1-carboxylic acid tert-butyl ester – (2-carboxy-benzothiophen-4-yl) Example 234-
Under nitrogen, to N, at room temperature was added N- dimethylformamide (5mL) within the reference product (200g, 0.62mmol) of Example 1, thioglycolic acid (114mg, 1.23mmol), sodium hydroxide (99mg, 2.45 mmol), the mixture was stirred at 105 ℃ 18 hours. Cooling, water was added, extracted with ethyl acetate, separated and the aqueous phase was adjusted pH = 5 or so, the precipitated solid was filtered and dried to obtain the target substance (180mg, yield 81%).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 7.98 (s, 1H), 7.64 (d, 1H), 7.42 (t, 1H), 6.95 (d, 1H), 3.53 (bs, 4H), 3.035 ( bs, 4H) ESI: [M-1] – = 361.1.
Example 24 7- [4- (2-ethoxycarbonyl-4-phenyl and thienyl-1-piperazinyl) butoxy] -2 (1H) – quinolinone Preparation of
A mixture of 2-chloro-6- (4- (4 – ((2-oxo-1,2-dihydro-quinolin-7-yl) oxy) butyl) piperazin-1-yl) benzaldehyde (80mg , 0.18mmol) was dissolved in DMF (5mL) was added DIPEA (94mg, 0.73mmol) and ethyl mercaptoacetate (0.024mL, 0.22mmol), 110 ℃ stirred for 16 hours. Cooling, water was added, extracted with ethyl acetate, the ethyl acetate layer was washed with saturated brine, dried over anhydrous sodium sulfate, and silica gel column chromatography, to give a solid (40mg, 46% yield).
Preparation of piperazine-1-carboxylic acid tert-butyl ester – Example 184- (2-ethoxycarbonyl phenyl and thien-4-yl)
Under nitrogen, was added at room temperature to DMF (5mL) within the reference product (200mg, 0.62mmol) of Example 1, ethyl mercaptoacetate (0.081ml, 0.74mmol), DIPEA (342mg, 2.48mmol), the mixture was at 105 ℃ stirred for 18 hours, 1N HCl aqueous solution was added adjust pH = 7, and extracted with methyl tert-butyl ether, the ether layer was washed three times with saturated brine, dried over anhydrous sodium sulfate, the drying agent was filtered off, and concentrated by column chromatography to obtain the target (170mg, yield 71%).
1 HNMR (400 MHz, CDCl 3 ): [delta] 8.40 (s, 1H), 7.58 (d, 1H), 7.37 (t, 1H), 6.95 (d, 1H), 4.44 (q, 2H), 3.64 (m, 4H), 3.15 (m, 4H) ESI: [M + 1] + = 391.1.
Preparation of piperazine-1-carboxylic acid tert-butyl ester – Example 194- (2-ethoxycarbonyl phenyl and thien-4-yl)
Under nitrogen at room temperature was added the product of Reference Example 1 to ethanol (5mL) inside (200mg, 0.62mmol), ethyl mercaptoacetate (0.081ml, 0.74mmol), sodium hydroxide (100mg, 2.48mmol), the mixture 85 ℃ stirred for 6 hours, and concentrated by column chromatography to obtain the target substance (70mg, 30% yield).
1 HNMR (400 MHz, CDCl 3 ): [delta] 8.40 (s, 1H), 7.58 (d, 1H), 7.37 (t, 1H), 6.95 (d, 1H), 4.44 (q, 2H), 3.64 (m, 4H), 3.15 (m, 4H) ESI: [M + 1] + = 391.1.
Piperazine hydrochloride – (2-carboxy-benzothiophen-4-yl) Example 201-
The product of Example 2 (200mg, 0.55mmol), was dissolved in THF (5mL) was added concentrated hydrochloric acid (0.5mL), 50 ℃ heated 6h.Cooling, methyl tert-butyl ether (5mL), filtered to give the target (130mg, yield 79%).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 8.04 (s, 1H), 7.69 (d, 1H), 7.43 (t, 1H), 7.00 (d, 1H), 3.30 ( bs, 8H) ESI: [M + 1] + = 262.9.
Piperazine hydrochloride – (benzothiophen-4-yl) Example 211-
The product of Example 20 (130mg, 0.43mmol) was added to diphenyl ether (3mL) in, 260 ℃ heating 0.5h. Cooling, filtration object (60mg, 55% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 7.75 (d, 1H), 7.69 (d, 1H), 7.53 (t, 1H), 7.31 (t, 1H), 6.97 ( t, 1H), 3.30 (bs, 8H) ESI: [M + 1] + = 219.2.
PAPER
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00027

Figure 1. Brexpiprazole (1) and intermediate 18.
2-Chloro-6-fluorobenzaldehyde was converted to 4-(1-piperazinyl)benzo[b]thiophene dihydrochloride (18), an intermediate in the synthesis of brexpiprazole, via a five-step sequence in 54% overall yield. This procedure requires no expensive catalyst and avoids the side products produced in the coupling step in the reported process. Several kilograms of compound 18 were prepared using this economical and scalable process.
1-(Benzo[b]thiophen-4-yl)piperazine Dihydrochloride (18)
Compound 10 (1.5 kg, 4.71 mol) was dissolved in ………………..DELETED…………………, and then dried to give compound 18 (1.17 kg, 85% yield). HPLC for compound 18 (tR = 6.3 min, identical to authentic sample) 99.8% purity; HPLC method B.
18:
1H NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 9.65 (s, 2H), 7.75 (d, J = 5.5 Hz, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.53 (d, J = 5.5 Hz, 1H), 7.30 (t, J = 7.9 Hz, 1H), 6.96 (d, J = 7.6 Hz, 1H), 3.30 (s, 8H).
13C NMR (100 MHz, DMSO-d6): δ 146.92, 140.62, 133.40, 126.50, 125.06, 121.91, 117.73, 112.56, 48.52, 43.00.
MS (ESI, eV): m/z = 219.1 [M + H]+.
………..
PATENT
http://www.google.com/patents/US20140187782
A 4-(1-piperazinyl)benzo[b]thiophene compound represented by Formula (1):
is useful for various medicines such as antipsychotic drugs. Moreover, a 4-(1-piperazinyl)benzo[b]thiophene compound represented by Formula (4):
wherein R1 is a hydrogen atom or a protecting group, is useful as an intermediate for synthesizing the compound represented by Formula (1).
Reference Example 30 and Example 1 of PTL 1 specifically disclose a method for producing a benzo[b]thiophene compound (the reaction scheme shown below). In Reference Example 30, 4-(1-piperazinyl)benzo[b]thiophene is produced by heating under reflux a mixture comprising 14.4 g of 4-bromobenzo[b]thiophene, 29.8 g of anhydrous piperazine, 9.3 g of sodium tert-butoxide, 0.65 g of (R)-(+)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), 0.63 g of tris(dibenzylideneacetone)dipalladium (0), and 250 ml of toluene (step X).

However, the reaction of the step X produces a relatively large amount of by-products that can hardly be separated, and the purity of the compound (4a) is thus inevitably reduced. Moreover, although column purification is performed to increase the purity of the compound (4a), it is very difficult to completely remove by-products, even by column chromatography purification. For this reason, there is a demand for the development of a novel method for producing the compound (4a) with high yield and high purity.
Furthermore, by-products contained in the compound (4a) inevitably reduce the purity of the compound (1) in the subsequent step Y. Since the method described in PTL 1 requires purification by column chromatography to obtain the target compound (1) with high purity, the method is not suitable for the industrial process of mass production. In addition, according to the method, incorporation of by-products that can hardly be separated is inevitable, and high-purity products usable as active pharmaceutical ingredients cannot be produced without purification by column chromatography.
CITATION LISTPatent Literature
- PTL 1: Japanese Unexamined Patent Publication No. 2006-316052 Non Patent Literature
- NPL 1: Tetrahedron Lett., 2004, 45, 9645



Example 4
Synthesis of 7-[4-(4-benzo[b]thiophen-4-yl-piperazin-1-yl)butoxy]-1H-quinolin-2-one
After 1-benzo[b]thiophen-4-yl-piperazine hydrochloride (10.6 g), potassium carbonate (5.8 g), and DMF (50 ml) were stirred at 30 to 40° C. for about 30 minutes, 7-(4-chlorobutoxy)-1H-quinolin-2-one (10.0 g) and potassium iodide (6.9 g) were added. The mixture was stirred at 90 to 100° C. for 2 hours. While the temperature of the mixture was maintained at 60° C. or more, water (150 ml) was added dropwise over a period of 10 minutes or more. After the mixture was cooled to 10° C. or less, the precipitated crystals were collected by filtration, and washed with water and then with ethanol.
After ethanol (325 ml) and acetic acid (25 ml) were added to the precipitated crystals, the mixture was stirred under reflux for dissolution. Concentrated hydrochloric acid (3.6 ml) was added at around 70° C., and the mixture was cooled. After confirming the precipitation of crystals, the mixture was heated again and stirred under reflux for 1 hour. After the mixture was cooled to 10° C. or less, the precipitated crystals were collected by filtration and washed with ethanol.
After ethanol (191 ml) and water (127 ml) were added to the precipitated crystals, the mixture was stirred under reflux for dissolution. After activated carbon (0.89 g) was added, the mixture was stirred under reflux for 30 minutes and then hot filtered. After activated carbon was removed, the mixture was heated again for dissolution. After 25% aqueous sodium hydroxide solution (5.8 ml) was added at approximately 70° C., the mixture was stirred under reflux for 30 minutes, after which water (64 ml) was added at approximately 70° C. After the mixture was stirred at 40° C. for 30 minutes, the precipitated crystals were collected by filtration at 40° C. or less, then washed with water, and dried to obtain 7-[4-(4-benzo[b]thiophen-4-yl-piperazin-1-yl)butoxy]-1H-quinolin-2-one as white crystals.
Yield: 14.30 g
1H-NMR (DMSO-d6) δ ppm;
1.6-1.75 (2H, m), 1.75-1.9 (2H, m), 2.44 (2H, t, J=7.0 Hz), 2.55-2.70 (4H, m), 3.00-3.15 (4H, m), 4.06 (2H, t, J=6.3 Hz), 6.30 (1H, d, J=9.5 Hz), 6.75-6.85 (2H, m), 6.88 (1H, d, J=7.5 Hz), 7.27 (1H, dd, J=8 Hz, 8 Hz), 7.40 (1H, d, J=5.5 Hz), 7.55 (1H, d, J=9.5 Hz), 7.61 (1H, d, J=8 Hz), 7.69 (1H, d, J=5.5 Hz), 7.80 (1H, d, J=9.5 Hz), 11.58 (1H, bs).
………………………
PATENT
http://www.google.com/patents/WO2006112464A1?cl=en
Example 1
Preparation of 7- [4- (4-benzo [b] thiophen-4-yl- piperazin-1-yl) butoxy] -lH-quinolin-2-one
A mixture of 9.0 g of 7- ( 4-chlorobutoxy) -IH- quinolin-2-one, 10 g of 1-benzo [b] thiophene-4-yl- piperazine hydrochloride, 14 g of potassium carbonate, 6 g of sodium iodide and 90 ml of dimethylformamide was stirred for 2 hours at 😯0C. Water was added to the reaction solution and precipitated crystals were separated by filtration. The crystals were dissolved in a mixed solvent of dichloromethane and methanol, dried over magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (dichloromethane .-methanol = 100:3). Recrystallized from ethanol, 13.6 g of 7- [4- (4-benzo [b] thiophen-4-yl- piperazin-1-yl) butoxy] -lH-quinolin-2-one in the form of a white powder was obtained.
Melting point 183.5-184.50C
1H-NMR ( DMSO-dg) δppm:
1.6-1.75 (2H, m) , 1.75-1.9(2H, m) , 2.44(2H, t, J=7Hz) , 2.5-2.8(4H, m) , 2.9-3.2(4H, m) , 4.06(2H, t, J=6.5Hz), 6.3O(1H, d, J=9.5Hz), 6.75-6.85 (2H, m) , 6.88(1H, d, J=7.5Hz), 7.27 (IH, dd, J=8Hz, 8Hz), 7.40 (IH, d, J=5.5Hz), 7.55 (IH, d, J=9.5Hz), 7.61(1H, d, J=8Hz) , 7.69(1H, d, J=5.5Hz), 7.8O(1H, d, J=9.5Hz), 11.59(1H, bs) .
……………….
PATENT
 7- [ 4- ( 4-benzo[b]thiophen-4- yl-piperazin-l-yl)butoxy] -lH-quinolin-2-one
7- [ 4- ( 4-benzo[b]thiophen-4- yl-piperazin-l-yl)butoxy] -lH-quinolin-2-one
The dihydrate of the benzothiophene compound represented by Formula (I) or of a salt thereof according to the present invention can be produced from an anhydride of the benzothiophene compound or of a salt thereof.
The benzothiophene compound (in the form of an
anhydride) of Formula (I), from which the dihydrate of the present invention is produced, is a known compound, and can be obtained by the production method disclosed in Example 1 of
JP2006-316052A or according to Reference Examples 1 and 2
Fig. 1 shows the ^-NMR spectrum of the dihydrate of the benzothiophene compound represented by Formula (I) prepared in Example 1.
Fig. 2 shows the X-ray powder diffraction pattern of the dihydrate of the benzothiophene compound represented by
Formula (I) prepared in Example 1.
Fig. 3 shows the infrared absorption spectrum of the dihydrate of the benzothiophene compound represented by Formula (I) prepared in Example 1.
Fig. 4 shows the Raman spectrum of the dihydrate of the benzothiophene compound represented by Formula (I) prepared in Example 1.
Fig. 5 shows the XH- MR spectrum of the benzothiophene compound represented by Formula (I) prepared in Example 2.
Reference Example 1: Synthesis of 7-(4-chlorobutoxy)-lH-quinolin- 2-one Methanol (149 L) , 7-hydroxy-lH-quinolin-2-one (14.87 kg), and potassium hydroxide (6.21 kg) were mixed and stirred. After dissolution, l-bromo-4-chlorobutane (47.46 kg) was further added thereto and the resulting mixture was stirred under reflux for seven hours. Thereafter, the mixture was stirred at 10° C for one hour. The precipitated crystal was centrifuged and washed with methanol (15 L). The wet crystal was collected and placed in a tank. Water (149 L) was added thereto, followed by stirring at room temperature. After centrifugation, the resulting solid was washed with water (30 L). The wet crystal was collected and placed in a tank. After adding methanol (74 L), the mixture was stirred under reflux for one hour, cooled to 10° C, and then stirred. The precipitated crystal was centrifuged and washed with methanol (15 L). The separated crystal was dried at 60° C to obtain 7- (4-chlorobutoxy) -lH-quinolin-2-one (15.07 kg).
Reference Example 2: Synthesis of 7- [ 4- ( 4-benzo[b] thiophen-4-yl- piperazin-l-yl)butoxy] -lH-quinolin-2-one
Water (20 L), potassium carbonate (1.84 kg), 1- benzo[b] thiophen-4-yl-piperazine hydrochloride (3.12 kg), and ethanol (8 L) were mixed and stirred at 50° C. 7- ( 4-Chlorobutoxy) – lH-quinolin-2-one (2.80 kg) obtained in Reference Example 1 was added to the mixture and stirred under reflux for nine hours.
After concentrating the solvent (8 L) under ordinary pressure, the mixture was stirred at 90° C for one hour and then cooled to 9° C . The precipitated crystal was centrifuged and then
sequentially washed with water (8 L) and ethanol (6 L). The separated crystal was dried at 60° C to obtain a crude product. The crude product (4.82 kg) and ethanol (96 L) were mixed in a reaction vessel, and acetic acid (4.8 L) was introduced into the reaction vessel. The mixture was stirred under reflux for one hour to dissolve the crude product. After introducing
hydrochloric acid (1.29 kg), the mixture was cooled to 10° C. The mixture was heated again, refluxed for one hour, and cooled to 7° C. The precipitated crystal was centrifuged and washed with ethanol (4.8 L). The separated crystal was dried at 60° C to obtain 7- [4- (4-benzo[b] thiophen-4-yl-piperazin-l-yl)butoxy] -1H- quinolin-2-one hydrochloride (5.09 kg). The resulting 7- [4- (4- benzo[b]thiophen-4-yl-piperazin-1-yl)butoxy] -lH-quinolin-2-one hydrochloride (5.00 kg), ethanol (45 L), and water (30 L) were mixed in a reaction vessel. The mixture was stirred under reflux to dissolve the 7-[4-(4-benzo[b]thiophen-4-yl-piperazin-l- yl)butoxy] -lH-quinolin-2-one hydrochloride. Activated carbon (500 g) and water (5 L) were added thereto, and an activated carbon treatment was conducted under reflux for 30 minutes. After performing hot filtration, a solution containing sodium hydroxide (511 g) dissolved in water (1.5 L) was flowed into the reaction vessel while stirring the filtrate under reflux. After stirring under reflux for 30 minutes, water (10 L) was introduced thereto and the mixture was cooled to approximately 40° C. The
precipitated crystal was centrifuged and washed with water (125 L). The separated crystal was dried at 80° C to obtain 7- [4- (4- benzo[b]thiophen-4-yl-piperazin-1-yl)butoxy] – lH-quinolin-2-one (3.76 kg).
Example 1: Preparation of 7- [ 4- ( 4-benzo[b]thiophen-4-yl- piperazin-l-yl)butoxy] -lH-quinolin-2-one dihydrate
The 7- [4- (4-benzo[b] thiophen-4-yl-piperazin-1- yl)butoxy] -lH-quinolin-2-one (3.2 kg) obtained in Reference
Example 2, ethanol (64 L) , water (74 L) , and acetic acid (1.77 kg) were mixed in a reaction vessel to prepare an acidic liquid mixture. The mixture was stirred under reflux to dissolve the 7- [ 4- ( 4-benzo[b] thiophen-4-yl-piperazin-1-yl)butoxy] -1H-quinolin-2- one (reflux temperature: 84° C). After cooling to -5°C, the solution obtained above was introduced, over a period of 30 minutes, into a solution containing 25% sodium hydroxide (5.9 kg) and water (54 L) that was cooled to 0°C, to prepare a liquid mixture with pHlO. After being stirred at 5° C or below for one hour, the mixture was heated to 20 to 30° C and further stirred for-seven hours . The precipitated crystal was filtered and washing with water (320 L) was performed, until alkali in the solid component disappeared (i.e.. until the pH value of the filtrate became 7 ) . The solid component was then air-dried until its weight became constant to obtain a white solid 7-[4-(4- benzofb] thiophen-4-yl-piperazin-l-yl)butoxy] -lH-quinolin-2-one dihydrate (unground, 3.21 kg).
Fig. 1 shows the XH-NMR spectrum (D SO-d6, TMS) of the dihydrate prepared by the aforesaid method. As shown in Fig. 1, in the ^- MR spectrum (DMSO-d6, TMS) , peaks were observed at 1.64 ppm (tt, J = 7.4 Hz, J = 7.4 Hz, 2H) , 1.80 ppm (tt, J = 7.0 Hz, J = 7.0 Hz, 2H), 2.44 ppm (t, J = 7.5 Hz, 2H) , 2.62 ppm (br, 4H) , 3.06 ppm (br, 4H) , 3.32 ppm (s, 4H + H20) , 4.06 ppm (t, J = 6.5 Hz, 2H), 6.29 ppm (d, J = 9.5 Hz, 1H), 6.80 ppm (d, J = 2.5 Hz, 1H) , 6.80 ppm (dd, J = 2.5 Hz, J = 9.0 Hz, 1H) , 6.88 ppm (d, J = 7.5 Hz, 1H), 7.27 ppm (dd, J = 7.8 Hz, J = 7.8 Hz, 1H) , 7.40 ppm (dd, J = 0.5 Hz, J = 5.5 Hz, 1H), 7.55 ppm (d, J = 9.0 Hz, 1H) , 7.61 ppm (d, J = 8.0 Hz, 1H) , 7.69 ppm (d, J = 5.5 Hz, 1H) , 7.80 ppm (d, J = 9.5 Hz, 1H), and 11.57 ppm (s, 1H) .
The X-ray powder diffraction spectrum of the dihydrate prepared by the aforesaid method was measured using an X-ray diffractometer (D8 ADVANCE, available from Bruker AXS). Fig. 2 shows the X-ray powder diffraction spectrum. As shown in Fig. 2, in the X-ray powder diffraction spectrum, diffraction peaks were observed at 2Θ = 8.1° , 8.9° , 15.1° , 15.6° , and 24.4° . Other than those mentioned above, the diffraction peaks were also observed at 2Θ = 11.6°.. 12.2°, 14.0°, 16.3°, 18.1°, 18.4°, 18.9°, 19.5°, 20.5°, 21.5°, 22.6°, 23.3°, 25.0°, 26.1°, 26.4°, 27.1°. 28.1°, 28.5°, 28.9°, 29.8°, 30.4°, 30.7°, 31.6°, 32.9°, 33.9°, 34.4°, 35.2°, 36.0°, 36.7°, 37.4° , and 38.3°.
The IR (KBr) spectrum of the dihydrate prepared by the aforesaid method was measured. Fig. 3 shows the IR (KBr) spectrum. As shown in Fig. 3, in the IR (KBr) spectrum, absorption bands were observed in the vicinity of wavenumbers 3509 cm“1, 2934 cm“1, 2812 cm“1, 1651 cm“1, 1626 cm“1, 1447 cm“1, 1223 cm“1 and 839 cm“1.
The Raman spectrum of the dihydrate prepared by the aforesaid method was measured. Fig. 4 shows the Raman spectrum. As shown in Fig. 4, in the Raman spectrum, absorption bands were observed in the vicinity of wavenumbers 1497 cm“1, 1376 cm“1, 1323 cm“1, 1311 cm“1, 1287 cm“1, 1223 cm“1, and 781 cm“1.
Other than those mentioned above, absorption was also observed in the vicinity of wavenumbers 1656 cm“1, 1613 cm“1, 1563 cm“1, 1512 cm“1, 1468 cm“1, 1446 cm“1, 1241 cm“1, 1203 cm“1, 1145 cm“1, 1096 cm“1, 1070 cm“1, 971 cm“1, and 822 cm“1.
The water content of the dihydrate prepared by the aforesaid method was measured using a moisture meter (CA-100, available from Mitsubishi Chemical Analytech Co., Ltd.) by the Karl Fischer method. As a result, the dihydrate had a water content of 7.79% by weight.
Example 2; Preparation of finely ground dihydrate
Dihydrate crystal (2.73 kg) obtained in Example 1 was ground using a jet mill. Here, the air pressure was set at 5 kgf/cm2, and the rotational speed of the feeder was set at 20 rpm. As a result, finely ground 7-[4-(4-benzo[b]thiophen-4-yl- piperazin-1-yl)butoxy] -1H-quinoli -2-one dihydrate (2.61 kg,
95.6%) was obtained.
The dihydrate (finely ground product) thus obtained had a mean particle diameter of 5.5 um. The mean particle diameter was measured using a Microtrack HRA, manufactured by Nikkiso Co., Ltd.
Fig. 5 shows the ^-NMR spectrum (DMSO-d6, TMS) of the dihydrate prepared by the above method. As shown in Fig. 5, in the ^- MR spectrum (DMSO-d6, TMS), peaks were observed at 1.64 ppm (tt, J = 7.3 Hz, J = 7.3 Hz, 2H), 1.80 ppm (tt, J = 6.9 Hz, J = 6.9 Hz, 2H), 2.44 ppm (t, J = 7.3 Hz, 2H) , 2.62 ppm (br, 4H) , 3.06 ppm (br, 4H) , 3.32 ppm (s, 4H + H20) , 4.06 ppm (t, J = 6.5 Hz, 2H), 6.29 ppm (d, J = 9.5 Hz, 1H) , 6.80 ppm (d, J = 2.5 Hz , 1H) , 6.80 ppm (dd, J = 2.3 Hz, J = 9.3 Hz, 1H) , 6.88 ppm (d, J = 7.5 Hz, 1H), 7.27 ppm (dd, J = 8.0 Hz, J = 8.0 Hz, 1H) , 7.40 ppm (d, J = 5.5 Hz, 1H), 7.55 ppm (d, J = 9.5 Hz , 1H) , 7.61 ppm (d, J = 8.0 Hz, 1H), 7.69 ppm (d, J = 5.5 Hz, 1H) , 7.80 ppm (d, J = 9.5
Hz, 1H), and 11.57 ppm (s, 1H) .
The X-ray powder diffraction spectrum of the dihydrate prepared by the aforesaid method was measured in the same manner as in Example 1. Fig. 6 shows the X-ray powder diffraction spectrum. As shown in Fig. 6, in the X-ray powder diffraction spectrum, diffraction peaks were observed at 2Θ = 8.2° , 8.9° ,
15.2° , 15.7° and 24.4° .
Other than those mentioned above, the diffraction peaks were also observed at 2Θ = 6.8°, 12.2°, 14.0°, 14.5″, 17.4°,
18.1°, 18.5°, 19.0°, 19.2°, 19.6°, 20.3°, 20.6°, 21.5°, 22.7°,
23.4°, 25.0°, 26.1°, 27.1°, 28.6°, 29.0°, 30.4°, 34.0°, 34.5°,
35.3° , and 36.7° .
The IR (KBr) spectrum of the dihydrate prepared by the aforesaid method was measured in the same manner as in Example 1.
Fig. 7 shows the IR (KBr) spectrum. As shown in Fig. 7, in the IR
(KBr) spectrum, absorption bands were observed in the vicinity of wavenumbers 3507 cm“1, 2936 cm“1, 2812 cm“1, 1651 cm“1, 1626 cm“1,
1447 cm“1, 1223 cm“1 and 839 cm“1.
The Raman spectrum of the dihydrate prepared by the aforesaid method was measured. Fig. 8 shows the Raman spectrum.
As shown in Fig. 8, in the Raman spectrum, absorption bands were observed in the vicinity of wavenumbers 1496 cm‘1, 1376 cm“1, 1323 cm‘1, 1311 cm“1, 1286 cm“1, 1223 cm“1, and 781cm“1.
Other than those mentioned above, absorption was also observed in the vicinity of wavenumbers 1656 cm“1, 1614 cm“1, 1563 cm“1, 1512 cm“1, 1467 cm“1, 1446 cm“1, 1241 cm“1, 1203 cm“1, 1145 cm“1,
1095 cm“1, 1069 cm“1, 971 cm“1, and 822 cm“1.
The water content of the dihydrate prepared by the aforesaid method was measured using a moisture meter (CA-100, available from Mitsubishi Chemical Analytech Co., Ltd.) by the
Karl Fischer method. As a result, the dihydrate had a water content of 6.74% by weight . Example 3 : Preparation of 7- [ 4- ( 4-benzo[b] thiophen-4-yl- piperazin-l-yl)butoxy] -lH-quinolin-2-one dihydrate
7- [ 4- ( 4-Benzo[ ] thiophen-4-yl-piperazin-1-yl)butoxy] – lH-quinolin-2-one (5.0 kg), ethanol (100 L), water (115 L), and DL-lactic acid (2.29 kg) were mixed to prepare an acidic liquid mixture. The liquid mixture was stirred under reflux to dissolve the 7- [4- (4-benzo[b] thiophen-4-yl-piperazin-l-yl)butoxy] -1H- quinolin-2-one (reflux temperature: 82° C). After cooling to -5°C, the solution obtained above was introduced, over a period of about 15 minutes, into a solution containing sodium hydroxide (1.48 kg) and water (135 L) that was cooled to 1°C, to prepare a liquid mixture with pHll. After being stirred at approximately 2 to 5° C for three hours, the mixture was heated to 45° C and
further stirred at 45 to 50° C for two hours. The precipitated crystal was filtered and washing with water (200 L) was performed until alkali in the solid component disappeared (i.e., until the pH value of the filtrate became 7). The solid component was further washed with a liquid mixture of ethanol (15 L) and water (20 L). The solid component was then dried at room temperature until its weight became constant to obtain a white solid 7- [4- (4- benzo[b] thiophen-4-yl-piperazin-1-yl)butoxy] -1H-quinolin-2-one dihydrate (unground, 5.11 kg).
The dihydrate thus obtained was the same as that obtained in Example 1.
The Raman spectrum of the dihydrate prepared by the aforesaid method was measured. Fig. 9 shows the Raman spectrum. As shown in Fig. 9, in the Raman spectrum, absorption bands were observed in the vicinity of wavenumbers 1497 cm“1, 1376 cm“1, 1323 cm“1, 1311 cm“1, 1287 cm“1, 1223 cm“1, and 782 cm“1.
Other than those mentioned above, absorption was also observed in the vicinity of wavenumbers 1656 cm“1, 1614 cm“1, 1563 cm“1, 1512 cm“1, 1468 cm“1, 1446 cm“1, 1241 cm“1, 1203 cm“1, 1145 cm“1, 1126 cm“1, 1096 cm“1, 1070 cm“1, 972 cm“1, and 822 cm“1.
…………………….
PATENT
http://www.google.com/patents/WO2006112464A1?cl=en
…………………..
PATENT
http://www.google.com/patents/US20110152286
References
- “Phase II and Phase III Drugs in U.S. Development for Depression, Anxiety, Sleep Disorders, Psychosis, & ADHD”. Retrieved 9 February 2012.
- “Otsuka Pharmaceutical Development & Commercialization, Inc.”. Bloomberg Businessweek. Retrieved 10 February 2012.
- “Otsuka HD places top priority on development of OPC-34712.”. Chemical Business Newsbase. January 3, 2011. Retrieved 10 February 2012.
- Patent 5006528, Oshiro, Yasuo; Seiji Sato & Nobuyuki Kurahashi, “Carbostyril derivatives”, published October 20, 1989
- “Patent and Exclusivity Search Results”. Electronic Orange Book. US Food and Drug Administration. Retrieved 8 December 2008.
- “Lundbeck and Otsuka Pharmaceutical sign historic agreement to deliver innovative medicines targeting psychiatric disorders worldwide”. Lundbeck. Retrieved 10 February2012.
- “Otsuka Holdings Financial Results Presentation Q2 FY2011”. Retrieved 10 February2012.
- “OPC-34712 search results”. Retrieved 10 February 2012.
- “Study of the Safety and Efficacy of OPC-34712 as Adjunctive Therapy in the Treatment of Patients With Major”. Retrieved 15 February 2012.
- “Study of the Safety and Efficacy of Two Fixed Doses of OPC-34712 as Adjunctive Therapy in the Treatment of Adults With Major Depressive Disorder (the Polaris Trial)”. Retrieved 10 February 2012.
- ^ Jump up to:a b “Study of the Safety and Efficacy of OPC-34712 as a Complementary Therapy in the Treatment of Adult Attention Deficit/Hyperactivity Disorder (STEP-A)”. Retrieved10 February 2012.
- “Trial to Evaluate the Effects of OPC-34712 on QT/QTc in Subjects With Schizophrenia or Schizoaffective Disorder”. Retrieved 10 February 2012.
- “A Dose-finding Trial of OPC-34712 in Patients With Schizophrenia”. Retrieved10 February 2012.
- “Efficacy Study of OPC-34712 in Adults With Acute Schizophrenia (BEACON)”. Retrieved 10 February 2012.
- Jump up^ “Safety and Tolerability Study of Oral OPC-34712 as Maintenance Treatment in Adults With Schizophrenia (ZENITH)”. Retrieved 10 February 2012.
- “Study of the Effectiveness of Three Different Doses of OPC-34712 in the Treatment of Adults With Acute Schizophrenia (VECTOR)”. Retrieved 10 February 2012.
- “A Long-term Trial of OPC-34712 in Patients With Schizophrenia”. Retrieved10 February 2012.
- “Otsuka Pharmaceutical Co., Ltd. Announces Results from a Phase 2 Study of Investigational Product OPC-34712 as Adjunctive Therapy in Adults with Major Depressive Disorder”. Retrieved 16 February 2012.
- “Preclinical Pharmacology of Brexpiprazole (Opc-34712): A Novel Compound with Dopamine D2 Receptor Partial Agonist Activity”. Retrieved 16 February 2012.
- “2011 U.S. Psych Congress Poster Session Abstracts”. Retrieved 16 February 2012.
- “Otsuka Pharmaceutical reports OPC-34712 Phase 2 trial results in major depressive disorder”. News-Medical.Net. Retrieved 10 February 2012.
- Maeda K, Sugino H, Akazawa H et al. (September 2014). “Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator”. J. Pharmacol. Exp. Ther. 350 (3): 589–604. doi:10.1124/jpet.114.213793.PMID 24947465.
- “Trial to Evaluate the Effects of OPC-34712 on QT/QTc in Subjects With Schizophrenia or Schizoaffective Disorder”. Retrieved 10 February 2012.
- “Canadian Patents Database 2620688”. Retrieved 16 February 2012.
- http://zliming2004.blog.163.com/blog/static/6456372120149963644472/
………
| JP2006316052A |
|
|
|
Title not available |
| US20110152286 * |
Dec 16, 2010 |
Jun 23, 2011 |
Otsuka Pharmaceutical Co., Ltd. |
Piperazine-substituted benzothiophenes for treatment of mental disorders |
UPDATED
SUZHOU VIGONVITA LIFE SCIENCES CO., LTD. [CN/CN]; 398, Ruoshui Road, Suzhou Industrial Park Suzhou, Jiangsu 215123 (CN).
TOPHARMAN SHANGHAI CO., LTD. [CN/CN]; 1088, Chuansha Road, Pudong Shanghai 201209 (CN).
SHANGHAI INSTITUTE OF MATERIA MEDICA, CHINESE ACADEMY OF SCIENCES [CN/CN]; 555, Zuchongzhi Road, Zhangjiang, Pudong Shanghai 201203 (CN)
(EN)The present invention relates to methods of preparing brexpiprazole, analogs thereof, key intermediates and salts thereof. Specifically, the present invention relates to new methods of preparing brexpiprazole, analogs thereof, key intermediates and salts thereof, and to the key intermediates and salts thereof used in the methods. The methods involve mild reaction conditions, stable intermediates, easy operations, and widely available reagents, thereby allowing for reduced synthesis costs, a shortened production cycle, high yield, and high product quality. The methods are suited for use in large-scale production.
Bray prazosin (Brexpiprazole, code: OPC-34712) is Otsuka Pharmaceutical Co., Ltd. developed a new generation of anti-psychotic drug candidates that act on more than one receptor, dopamine D2 receptor partial agonist (improving positive and negative symptoms, cognitive impairment and depressive symptoms), 5-HT2A receptor antagonist (improving negative symptoms, cognitive dysfunction, symptoms of depression, insomnia), α1 adrenoceptor antagonists (improving positive symptoms of schizophrenia), 5 – serotonin uptake / reuptake inhibitors (symptoms of depression); at the same time, but also a 5-HT1A partial agonist (anxiolytic and antidepressant activity) and 5-HT7 antagonist (temperature, circadian rhythms, learning and memory, sleep) . Currently, in the United States and Europe as an adjunctive treatment of severe depression (MDD) Phase III clinical trials; III clinical trials the treatment of schizophrenia in the United States, Europe and Japan, meanwhile, is still the United States II Adult ADHD clinical trials.
Otsuka Pharmaceutical Co., Ltd. are disclosed in PCT Application WO2006112464A1 in the preparation route Bray prazosin, see Scheme 1, the difficulty of the route is the first step in the reaction by-products easily separated by column chromatography is not easy to obtain high-purity intermediates, thus affecting the final product Bray prazosin purity and yield.
Subsequently, Otsuka Pharmaceutical Co., Ltd. are disclosed in PCT Application WO2013015456A1 in the alternative method of preparing the reaction of this step, see Scheme 2, along the route of the reagents are more expensive, high-cost, environmentally unfriendly and not suitable for industrial production.
Due to the above production process there is a high cost, and difficult to separate impurities and other shortcomings, it is necessary to find an economical, practical, environmental protection, new routes to improve process stability, reduce costs, improve product quality.
In response to these deficiencies, an object of the present invention is to provide a new, simple operation, high yield, low cost, environmentally friendly and suitable for industrial mass production Bray prazosin and the like, key intermediates and preparing a salt thereof.
Another object of the present invention is to provide novel compounds and salts thereof of the manufacturing process.
To achieve the above objects, the present invention provides compounds of formula I, the structure is as follows:
Wherein, R is C1 ~ C6 straight or branched chain alkyl, benzyl; preferably, R is C1 ~ C4 straight or branched chain alkyl group; most preferably, R is methyl, ethyl, t-butyl group;
R
1 is acyl amino-protecting groups (e.g. formyl ( ), an acetyl group, a propionyl group, a benzoyl group, haloacetyl group, phthaloyl) or class alkoxycarbonyl amino-protecting group (e.g. t-butoxycarbonyl , benzyloxycarbonyl, 9-fluorenyl methoxy carbonyl); said haloacetyl group is a fluorinated acetyl, bromoacetyl, chloroacetyl or iodoacetyl group; preferably, R
1 is formyl, acetyl and tert-butoxycarbonyl groups;



The present invention also provides a method for preparing a compound of formula I, the compound of formula II with a thioglycolate as shown in the reaction, the compound, the method shown in Formula I as shown in Scheme 3,
Wherein, X is halogen, such as fluorine, chlorine, bromine, iodine; R and R 1 are as defined above, the compound of formula I as defined the same;
The above reaction in the presence of a base, in particular, is an inorganic base (e.g. sodium hydroxide, potassium hydroxide, strontium hydroxide, lithium hydroxide, barium hydroxide, calcium hydroxide, cesium hydroxide, sodium hydrogen carbonate, potassium bicarbonate, potassium carbonate, sodium carbonate, strontium carbonate, cesium carbonate, sodium sulfide, sodium hydroxide, etc.) or an organic base (e.g. sodium alkoxide, potassium alkoxide, butyl lithium, 1,8-diazabicyclo [5,4 0] undecene -7 (DBU), pyridine, quinoline, 4-dimethylaminopyridine (DMAP) or an organic amine, etc.) performed in the presence of, wherein the sodium alkoxide may be sodium methoxide, sodium ethoxide, propoxy sodium alkoxide, sodium isopropoxide, n-butoxide, sodium tert-butoxide and the like; may be the potassium alkoxide, potassium methoxide, potassium ethoxide, potassium-propanol, potassium isopropoxide, n-butoxide, potassium tert-butoxide , the organic amine may be triethylamine, diethylamine, n-butylamine, tripropylamine, diisopropylamine, diisopropylethylamine, etc., preferably, the base may be an inorganic alkali sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate, strontium carbonate, sodium sulfide, sodium hydroxide, or organic bases as sodium methoxide, sodium ethoxide, tert-butoxide potassium, triethylamine, diethylamine, diisopropylamine or diisopropylethylamine;
The above reaction is carried out in a suitable solvent, the solvent is water, C 1 ~ C 5 lower alcohol (such as methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, tert-butanol, n-amyl alcohol, amyl alcohol, ethylene glycol, propylene glycol, glycerol), N, N- dimethylformamide (DMF), dimethyl sulfoxide (DMSO), tetrahydrofuran (THF), acetonitrile, dioxane, N- methylpyrrolidone, methylene chloride, chloroform, ethylene glycol dimethyl ether, diethylene glycol dimethyl ether or ethylene glycol monomethyl ether, and the like, one or more, preferably, the solvent is water , methanol, ethanol, N, N- dimethylformamide (DMF), dimethylsulfoxide (DMSO), tetrahydrofuran (THF), acetonitrile, dioxane or ethylene glycol dimethyl ether or a species; the reaction time from 1 hour to 24 hours, preferably 2 hours to 12 hours. The reaction temperature is 0 ℃ ~ 150 ℃, preferably room temperature ~ 100 ℃.
To achieve the above object, the present invention also provides a compound of formula III, is structured as follows:
Wherein, R
1 is acyl amino protecting groups (e.g. formyl, acetyl, propionyl, benzoyl, halo acetyl, phthaloyl) or class alkoxycarbonyl amino-protecting group (e.g. t-butoxycarbonyl , benzyloxycarbonyl, 9-fluorenyl methoxycarbonyl), said haloacetyl group is a fluorinated acetyl, bromoacetyl, chloroacetyl or iodoacetyl;

Preferably, R
1 is formyl, acetyl or t-butoxycarbonyl;

The present invention also provides a method of preparing compounds of Formula III are shown, thioglycolic acid compound and reacting compound of formula III as shown in the method shown by the formula II as shown in Scheme 4,
Wherein, X is fluorine, chlorine, bromine or iodine; R 1 as defined above, with a compound of formula I as defined the same;
Bray prazosin (Brexpiprazole, code: OPC-34712) is Otsuka Pharmaceutical Co., Ltd. developed a new generation of anti-psychotic drug candidates that act on more than one receptor, dopamine D2 receptor partial agonist (improving positive and negative symptoms, cognitive impairment and depressive symptoms), 5-HT2A receptor antagonist (improving negative symptoms, cognitive dysfunction, symptoms of depression, insomnia), α1 adrenoceptor antagonists (improving positive symptoms of schizophrenia), 5 – serotonin uptake / reuptake inhibitors (symptoms of depression); at the same time, but also a 5-HT1A partial agonist (anxiolytic and antidepressant activity) and 5-HT7 antagonist (temperature, circadian rhythms, learning and memory, sleep) . Currently, in the United States and Europe as an adjunctive treatment of severe depression (MDD) Phase III clinical trials; III clinical trials the treatment of schizophrenia in the United States, Europe and Japan, meanwhile, is still the United States II Adult ADHD clinical trials.
Wherein, X is fluorine, chlorine, bromine or iodine; R 1 are the same as defined in the compounds illustrated and R are as defined above for formula I. The present invention also provides processes for preparing key intermediates Bray prazosin or a salt thereof, the method as shown in Scheme 6:
14- (3-chloro-2-carboxaldehyde-phenyl-1 -) – Reference Example Synthesis of piperazine-1-carboxylic acid tert-butyl ester
A mixture of 2-chloro-6-fluorobenzaldehyde (500mg, 3.15mmol), piperazine-1-carboxylate (646mg, 3.47mmol) was dissolved in N, N- dimethylformamide (5mL), and nitrogen at, at room temperature was added potassium carbonate (2.18g, 15.77mmol), the mixture was stirred for 4 hours at 80 ℃, cooled and filtered, water (20mL), ethyl acetate (3 × 5mL) was extracted, dried over anhydrous sodium sulfate, filtered The desiccant was concentrated to give a solid, after with petroleum ether (50mL) beating 1h, filtered to give a pale yellow solid (750mg, 75% yield).
1 HNMR (400 MHz, CDCl 3 ): δ10.37 (s, 1H), 7.40 (t, 1H), 7.01 (d, 1H), 6.99 (d, 1H), 3.20 (m, 4H), 3.00 (s, 4H), 1.47 (s, 9H) ESI: [M + 1] + = 325.8.
Reference Example 21- carboxylic acid (3-chloro-2-carboxaldehyde-phenyl-1 -) – -4- piperazine
A mixture of 2-chloro-6-fluorobenzaldehyde (500mg, 3.15mmol), 1- formyl piperazine (396mg, 3.47mmol) was dissolved in DMF (5mL), and under nitrogen at room temperature was added potassium carbonate (2.18g, 15.77mmol). The mixture was stirred for 4 hours at 80 ℃, cooled water (20mL), ethyl acetate (3 × 5mL) was extracted, dried over anhydrous sodium sulfate, and concentrated to give a solid with petroleum ether (50mL) After beating 1h, filtered to give a pale yellow solid (588mg, yield 70%).
1 HNMR (400 MHz, CDCl 3 ): [delta] 10.45 (s, 1H), 8.13 (s, 1H), 7.44 (t, 1H), 7.18 (d, 1H), 7.02 (d, 1H), 3.80 (s, 2H), 36.4 (s, 2H), 3.10 (m, 4H) ESI: [M + 1] + = 253.1.
Acetyl-31- (3-chloro-2-carboxaldehyde-phenyl-1 -) – 4- Reference piperazine
A mixture of 2-chloro-6-fluorobenzaldehyde (500mg, 3.15mmol), 1- acetyl-piperazine (444mg, 3.47mmol) was dissolved in DMF (5mL), and under nitrogen at room temperature was added potassium carbonate (2.18g, 15.77 mmol), the mixture was stirred at 80 ℃ 4 hours, cooled and filtered, water (20mL), ethyl acetate (3 × 5mL) was extracted, dried over anhydrous sodium sulfate, and concentrated to give a solid, after with petroleum ether (50mL) beating 1h, filtered to give a pale yellow solid (588mg, yield 70%).
1 HNMR (400 MHz, CDCl 3 ): [delta] 10.44 (s, 1H), 7.44 (t, 1H), 7.17 (d, 1H), 7.03 (d, 1H), 3.79 (bs, 4H), 3.10 (m, 4H), 2.18 (s, 3H) ESI: [M + 1] + = 267.1.
Piperazine-1-carboxylic acid tert-butyl ester – 14- (2-ethoxycarbonyl phenyl and thien-4-yl) Example
Under nitrogen, to N, was added the product (1.0g, 3.08mmol) of Reference Example 1 N- dimethylformamide (5mL) at room temperature within, ethyl thioglycolate (388mg, 3.20mmol), potassium carbonate (1.38 g, 10mmol), the mixture was stirred for 4 hours at 80 ℃, cooled and filtered, water (20mL), ethyl acetate (3x5mL) was extracted, dried over anhydrous sodium sulfate, the drying agent filtered, and concentrated to give a solid with petroleum ether (50mL ) after beating 1h, filtered to give a pale yellow solid (900mg, 75% yield).
1 HNMR (400 MHz, CDCl 3 ): [delta] 8.40 (s, 1H), 7.58 (d, 1H), 7.37 (t, 1H), 6.95 (d, 1H), 4.44 (q, 2H), 3.64 (m, 4H), 3.15 (m, 4H) ESI: [M + 1] + = 391.1.
Example 24- (2-carboxy-benzothiophen-4-yl) – piperazine-1-carboxylate Synthesis of
Of the product (1.0g, 2.5mmol) in Example 1 was dissolved into 1,4-dioxane (5mL), was added 4N aqueous sodium hydroxide solution (1.8mL, 7.2mmol), the mixture was stirred for 3h at 80 ℃, cooled to room temperature, water (5mL) and ethyl acetate (10mL), separated and the aqueous phase with 1N HCl at 0 ℃ pH was adjusted to about 4.0, the resulting solid was filtered, dried to give a pale yellow solid.
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 7.98 (s, 1H), 7.64 (d, 1H), 7.42 (t, 1H), 6.95 (d, 1H), 3.53 (bs, 4H), 3.035 ( bs, 4H) ESI: [M-1] – = 361.1.
34- (benzothiophen-4-yl) Example – Synthesis of piperazine-1-carboxylic acid tert-butyl ester
The product of Example 2 (20g, 54mmol) will be implemented, cuprous oxide (1g, 7mmol) was dissolved in quinoline (50mL) inside, heated to 140 ℃ overnight. After cooling and filtration, the filtrate was added water, extracted with ethyl acetate, the organic phase was washed with 1N HCl to slightly acidic, saturated aqueous sodium bicarbonate solution, purified by silica gel column chromatography, the concentrated solid slurried with petroleum ether to give an off-white solid (13g, yield 70%).
1 HNMR (400 MHz, CDCl 3 ): [delta] 7.57 (d, 1H), 7.41 (s, 2H), 7.27 (t, 1H), 6.88 (d, 1H), 3.66 (m, 4H), 3.01 (m, 4H), 1.50 (s, 9H) ESI: [M + 1] + = 319.1.
44- (benzothiophene-4-yl) Example – Synthesis of piperazine-1-carboxylic acid tert-butyl ester
The product of Example 2 (500mg, 1.35mmol) will be implemented, silver carbonate (40mg, 0.135mmol) and acetic acid (8mg) was dissolved in dimethylsulfoxide (5mL) inside, heated to 120 ℃, the reaction overnight, cooled and filtered, and the filtrate Water was added, extracted with ethyl acetate, and concentrated by column chromatography to obtain the target substance.
1 HNMR (400 MHz, CDCl 3 ): [delta] 7.57 (d, 1H), 7.41 (s, 2H), 7.27 (t, 1H), 6.88 (d, 1H), 3.66 (m, 4H), 3.01 (m, 4H), 1.50 (s, 9H) ESI: [M + 1] + = 319.1.
Piperazine hydrochloride – 51- (benzothiophen-4-yl) Example
At room temperature, the product of Example 3 will be implemented (2g, 6.2mmol) was dissolved in dioxane (6mL) was added 4N HCl / dioxane (6mL), stirred 3h, concentrated to dryness, the residue was beating ethyl acetate, filtered to obtain the target substance (1.3g, 95% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 7.75 (d, 1H), 7.69 (d, 1H), 7.53 (t, 1H), 7.31 (t, 1H), 6.97 ( t, 1H), 3.30 (bs, 8H) ESI: [M + 1] + = 219.2.
Example 61- formyl-4- (2-ethoxycarbonyl phenyl and thien-4-yl) – piperazine Synthesis
In N 2 protected, at room temperature was added the product of Reference Example 2 to DMF (5mL) inside (1.0g, 3.7mmol), ethyl thioglycolate (410mg, 3.80mmol), potassium carbonate (1.38g, 10mmol), the mixture 80 ℃ stirred for 4 hours. Cooling water was added (20mL), ethyl acetate (3 × 5mL) was extracted, dried over anhydrous sodium sulfate, and concentrated to give a solid with petroleum ether (50mL) After beating 1h, filtered to give a pale yellow solid (1.0 g, yield 83%).
1 HNMR (400 MHz, CDCl3): [delta] 8.15 (d, 2H), 7.59 (d, 1H), 7.41 (t, 1H), 6.94 (d, 1H), 4.44 (q, 2H), 3.85 (t, 2H ), 3.68 (t, 2H), 3.21-3.15 (m, 4H), 1.44 (t, 3H) ESI: [M + 1] + = 319.1.
Example 71- formyl-4- (2-carboxy-benzothiophen-4-yl) – piperazine
The product (1.0g, 3.1mmol) of Example 6 was dissolved in methanol (5mL) and water (2mL) the addition of lithium hydroxide (420mg, 10mmol), the mixture was stirred at room temperature for 5h, was added water (5mL) and acetic acid ethyl ester (10mL), extracted, the aqueous phase was collected, the pH was adjusted to about 4.0 at 0 ℃ with 1N HCl solution, the precipitated solid was filtered and dried to give a pale yellow solid (510mg, 56% yield).
Example 81- formyl (benzothiophen-4-yl) -4 – piperazine
The product (1.0g, 3.4mmol) Example 7 will be implemented, cuprous oxide (50mg) was dissolved in quinoline (5mL) inside, heated to 140 ℃ overnight. After cooling and filtration, water was added, extracted with ethyl acetate, the organic phase was washed with aqueous 1N HCl to slightly acidic, then with saturated aqueous sodium bicarbonate solution, and concentrated by silica gel column chromatography, the resulting solid was slurried with petroleum ether to give white solid (520mg, 62% yield).
1 HNMR (400 MHz, CDCl 3 ): [delta] 8.15 (s, 1H), 7.62 (d, 1H), 7.42 (m, 2H), 7.31 (t, 1H), 6.04 (d, 1H), 3.82 (t, 2H), 3.63 (t, 2H), 3.19-3.12 (m, 4H) ESI: [M + 1] + = 247.1.
Example 91- (benzothiophen-4-yl) – piperazine hydrochloride
A mixture of the product of Example 8 (500mg) was dissolved in dioxane (2mL) was added 4N HCl / dioxane (3mL), stirred 3h, concentrated to dryness, slurried with ethyl acetate, filtered to give the target (470mg, yield 90%).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 7.75 (d, 1H), 7.69 (d, 1H), 7.53 (t, 1H), 7.31 (t, 1H), 6.97 ( t, 1H), 3.30 (bs, 8H) ESI: [M + 1] + = 219.2.
Example 10 1-Acetyl-4- (2-ethoxycarbonyl phenyl and thien-4-yl) – piperazine Synthesis
Under the protection of N2, at room temperature was added the product of Reference Example 3 (1.0g, 3.74mmol) to DMF (5mL) inside, ethyl thioglycolate (388mg, 3.20mmol), potassium carbonate (1.38g, 10mmol), the mixture was 80 ℃ stirred for 4 hours, cooled water was added (20mL), ethyl acetate (3 × 5mL) was extracted, dried over anhydrous sodium sulfate, and concentrated to give a solid with petroleum ether (50mL) beating 1h, filtered to give a pale yellow solid (863mg, yield 70%).
1 HNMR (400 MHz, CDCl 3 ): [delta] 8.17 (s, 1H), 7.60 (d, 1H), 7.42 (t, 1H), 7.01 (d, 1H), 4.44 (q, 2H), 3.94 (br, 2H), 3.80 (br, 2H), 3.21 (br, 4H), 2.19 (s, 3H), 1.44 (t, 3H) ESI: [M + 1] + = 333.3.
Example 11 1-Acetyl-(2-carboxy-benzothiophen-4-yl) -4 – piperazine
The product (1.0g, 3.0mmol) from Example 10 was dissolved in methanol (5mL) and water (2mL) the addition of lithium hydroxide (300mg, 7.2mmol), the mixture was stirred at rt for 3h, water was added (5mL) and ethyl acetate ester (10mL), separated and the aqueous phase was collected, the pH adjusted with aqueous 1N HCl at 0 ℃ to about 4.0, and the precipitated solid was filtered, dried to give a pale yellow solid (820mg, yield 90%).
Example 12 1-Acetyl-4- (benzothiazol-4-yl) – piperazine
The product of Example 11 (1.0g, 3.2mmol), cuprous oxide (50mg) was dissolved in quinoline (5mL) inside, heated to 140 ℃ overnight. After cooling and filtration, water was added, extracted with ethyl acetate, washed with 1N HCl solution was added to a weak acid, a saturated aqueous solution of sodium bicarbonate, silica gel column chromatography, and concentrated to give a solid slurried with petroleum ether to give a white solid (600mg , yield 70%).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 7.95 (s, 1H), 7.65 (d, 1H), 7.41 (t, 1H) 6.95 (d, 1H), 3.69 (q, 4H), 3.10 (t , 2H), 3.02 (t, 2H), 2.06 (s, 3H) ESI: [M + 1] + = 261.1.
EXAMPLE 131- (benzothiophen-4-yl) – piperazine hydrochloride
A mixture of the product of Example 12 (1g, 3.8mmol) was dissolved in dioxane (6mL) was added 4N HCl / dioxane (6mL), stirred 3h, concentrated dry, with ethyl beating, filtered to give the product (870mg, yield 90%).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 7.75 (d, 1H), 7.69 (d, 1H), 7.53 (t, 1H), 7.31 (t, 1H), 6.97 ( t, 1H), 3.30 (bs, 8H) ESI: [M + 1] + = 219.2.
141- (benzothiophen-4-yl) Example – piperazine
The product of (500mg, 1.38mmol) of Example 2 was dissolved in quinoline (3mL) the addition of cuprous oxide (20mg), the reaction temperature was raised to 140 ℃ After 2h, the reaction continues to heat up to 240 ℃ 3h, cooled to room temperature, filtered , water was added, extracted with ethyl acetate, washed with saturated aqueous sodium bicarbonate, silica gel column chromatography, and concentrated to give the desired product. 1 HNMR (300 MHz, DMSO-d 6 ): [delta] 8.74 (bs, 1H), 7.75 (d, 1H), 7.69 (d, 1H), 7.51 (d, 1H), 7.31 (t, 1H), 6.95 ( d, 1H), 3.24 (m, 8H) ESI: [M + 1] + = 219.2.
Example 15 7- [4- (2-carboxy-4-yl-benzothiophene-1-piperazinyl) butoxy] -3,4-dihydro -2 (1H) – quinolinone Preparation of
7- [4- (2-ethoxycarbonyl-4-phenyl and thienyl-1-piperazinyl) butoxy] -3,4-dihydro -2 (1H) – quinolinone (300mg, 0.59 mmol) was dissolved in methanol (3mL) and water (1mL) was added lithium hydroxide (76mg, 1.8mmol), stirred at rt for 3h, ethyl acetate was added, the aqueous phase was adjusted with 1N dilute hydrochloric acid to about pH 4.0, using bis dichloromethane: methanol (10: 1) extraction, concentration did a white solid (210mg, 46% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 10.01 (s, 1H), 7.88 (s, 1H), 7.61 (d, 1H), 7.38 (t, 1H), 7.03 (q, 1H), 6.93 ( d, 1H), 6.48 (m, 2H), 3.92 (m, 4H), 3.35 (s, 4H), 2.84 (s, 4H), 2.77 (s, 2H), 2.62 (s, 2H), 1.72 (m , 4H), ESI: [M-1] – = 478.3.
EXAMPLE 167- [4- (benzothiazol-4-yl-1-piperazinyl) butoxy] -3,4-dihydro -2 (1H) – quinolinone Preparation of
The product (500mg, 1.04mmol) of Example 15 will be implemented, cuprous oxide (50mg) was dissolved in quinoline (5mL) inside, heated to 140 ℃ overnight. After cooling and filtration, water was added, extracted with ethyl acetate, washed with 1N HCl solution was added until pH = 4.0, dichloromethane: methanol (10: 1) extracted, dried over anhydrous sodium sulfate, and silica gel column chromatography to give a solid (320mg , yield 70%).
1 HNMR (400 MHz, DMSO-d 6 ): δ10.00 (s, 1H), 7.69 (d, 1H), 7.61 (d, 1H), 7.40 (d, 1H), 7.27 (t, 1H), 7.04 ( d, 1H), 6.89 (d, 1H), 6.50 (dd, 1H), 6.45 (d, 1H), 3.93 (t, 2H), 3.06 (br, 4H), 2.78 (t, 2H), 2.60 (br , 4H), 2.41 (t, 4H), 1.74 (t, 2H), 1.60 (t, 2H) ESI: [M + 1]+ = 436.3.
Preparation of piperazine-1-carboxylic acid tert-butyl ester – Example 17 4- (2-ethoxycarbonyl phenyl and thien-4-yl)
Under nitrogen at room temperature was added to ethanol (5mL) within the reference product (200mg, 0.62mmol) of Example 1, ethyl mercaptoacetate (0.081ml, 0.74mmol), potassium carbonate (342mg, 2.48mmol), the mixture was 85 ℃ stirred for 18 hours, concentrated, and column chromatography to obtain the target substance (100mg, 42% yield).
1 HNMR (400 MHz, CDCl 3 ): [delta] 8.40 (s, 1H), 7.58 (d, 1H), 7.37 (t, 1H), 6.95 (d, 1H), 4.44 (q, 2H), 3.64 (m, 4H), 3.15 (m, 4H) ESI: [M + 1] + = 391.1.
Preparation of piperazine-1-carboxylic acid tert-butyl ester – Example 18 4- (2-ethoxycarbonyl phenyl and thien-4-yl)
Under nitrogen, was added at room temperature to DMF (5mL) within the reference product (200mg, 0.62mmol) of Example 1, ethyl mercaptoacetate (0.081ml, 0.74mmol), DIPEA (342mg, 2.48mmol), the mixture was at 105 ℃ stirred for 18 hours, 1N HCl solution was added adjust the pH = 7, and extracted with methyl tert-butyl ether, the ether layer was then washed three times with saturated brine, dried over anhydrous sodium sulfate, the drying agent filtered, and concentrated by column chromatography to obtain the target (170mg, yield 71%).
1 HNMR (400 MHz, CDCl 3 ): [delta] 8.40 (s, 1H), 7.58 (d, 1H), 7.37 (t, 1H), 6.95 (d, 1H), 4.44 (q, 2H), 3.64 (m, 4H), 3.15 (m, 4H) ESI: [M + 1] + = 391.1.
Preparation of piperazine-1-carboxylic acid tert-butyl ester – Example 19 4- (2-ethoxycarbonyl phenyl and thien-4-yl)
Under nitrogen at room temperature was added the product of Reference Example 1 to ethanol (5mL) inside (200mg, 0.62mmol), ethyl mercaptoacetate (0.081ml, 0.74mmol), sodium hydroxide (100mg, 2.48mmol), the mixture 85 ℃ stirred for 6 hours, concentrated to column chromatography to obtain the target substance (70mg, 30% yield).
1 HNMR (400 MHz, CDCl 3 ): [delta] 8.40 (s, 1H), 7.58 (d, 1H), 7.37 (t, 1H), 6.95 (d, 1H), 4.44 (q, 2H), 3.64 (m, 4H), 3.15 (m, 4H) ESI: [M + 1] + = 391.1.
Piperazine hydrochloride – (2-carboxy-benzothiophen-4-yl) Example 201-
The product of Example 2 (200mg, 0.55mmol), was dissolved in THF (5mL), concentrated hydrochloric acid (0.5mL), 50 ℃ heated 6h. Cooling, methyl tert-butyl ether (5mL), filtered to give the target (130mg, yield 79%).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 8.04 (s, 1H), 7.69 (d, 1H), 7.43 (t, 1H), 7.00 (d, 1H), 3.30 ( bs, 8H) ESI: [M + 1] + = 262.9.
Piperazine hydrochloride – (benzothiophene-4-yl) Example 211-
The product of Example 20 (130mg, 0.43mmol) was added to diphenyl ether (3mL) in, 260 ℃ heating 0.5h. Cooled and filtered to give the object (60mg, 55% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 7.75 (d, 1H), 7.69 (d, 1H), 7.53 (t, 1H), 7.31 (t, 1H), 6.97 ( t, 1H), 3.30 (bs, 8H) ESI: [M + 1] + = 219.2.
Preparation of tert-butyl piperazine-1 – Example 224- (2-carboxy-benzothiophen-4-yl)
Under nitrogen, to N, at room temperature was added N- dimethylformamide (5mL) within the reference product (200g, 0.62mmol) of Example 1, thioglycolic acid (114mg, 1.23mmol), sodium methoxide (133mg, 2.45mmol ), and the mixture was stirred at 105 ℃ 18 hours. Cooling, water was added, extracted with ethyl acetate, separated and the aqueous phase was adjusted pH = 5 or so, the precipitated solid was filtered and dried to obtain the target substance (130mg, 58% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 7.98 (s, 1H), 7.64 (d, 1H), 7.42 (t, 1H), 6.95 (d, 1H), 3.53 (bs, 4H), 3.035 ( bs, 4H) ESI: [M-1] – = 361.1.
Preparation of piperazine-1-carboxylic acid tert-butyl ester – (2-carboxy-benzothiophen-4-yl) Example 234-
Under nitrogen, to N, at room temperature was added N- dimethylformamide (5mL) within the reference product (200g, 0.62mmol) of Example 1, thioglycolic acid (114mg, 1.23mmol), sodium hydroxide (99mg, 2.45 mmol), the mixture was stirred at 105 ℃ 18 hours. Cooling, water was added, extracted with ethyl acetate, separated and the aqueous phase was adjusted pH = 5 or so, the precipitated solid was filtered and dried to obtain the target substance (180mg, yield 81%).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 7.98 (s, 1H), 7.64 (d, 1H), 7.42 (t, 1H), 6.95 (d, 1H), 3.53 (bs, 4H), 3.035 ( bs, 4H) ESI: [M-1] – = 361.1.
Example 24 7- [4- (2-ethoxycarbonyl-4-phenyl and thienyl-1-piperazinyl) butoxy] -2 (1H) – quinolinone Preparation of
2-chloro-6- (4- (4 – ((2-oxo-1,2-dihydro-quinolin-7-yl) oxy) butyl) piperazin-1-yl) benzaldehyde (80mg , 0.18mmol) was dissolved in DMF (5mL) was added DIPEA (94mg, 0.73mmol) and ethyl mercaptoacetate (0.024mL, 0.22mmol), 110 ℃ stirred for 16 hours.Cooling, water was added, extracted with ethyl acetate, the ethyl acetate layer was washed with saturated brine, dried over anhydrous sodium sulfate, and silica gel column chromatography to give a solid (40mg, 46% yield).
1 HNMR (400 MHz, DMSO-d 6 ): δ11.69 (s, 1H), 11.24 (s, 1H), 8.09 (s, 1H), 7.81 (d, 1H), 7.74 (d, 1H), 7.57 ( d, 1H), 7.48 (t, 1H), 7.04 (d, 1H), 6.82 (m, 2H), 6.30 (d, 1H), 4.32 (m, 4H), 4.06 (t, 2H), 3.67-3.16 (m, 8H), 1.96 (m, 2H), 1.84 (m, 2H), 1.32 (t, 3H) ESI: [M + 1] + = 506.4.
Example 25 7- [4- (2-carboxy-4-yl-benzothiophene-1-piperazinyl) butoxy] -3,4-dihydro -2 (1H) – quinolinone Preparation of
7- [4- (2-ethoxycarbonyl-4-phenyl and thienyl-1-piperazinyl) butoxy] -3,4-dihydro -2 (1H) – quinolinone (100mg, 0.19 mmol) was dissolved in acetic acid (3mL) and concentrated hydrochloric acid (0.5mL), 100 ℃ stirred for 10 hours. The reaction mixture was poured into ice water, stirred for 10min after filtration, to obtain the target substance (40mg, 43% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 10.01 (s, 1H), 7.88 (s, 1H), 7.61 (d, 1H), 7.38 (t, 1H), 7.03 (q, 1H), 6.93 ( d, 1H), 6.48 (m, 2H), 3.92 (m, 4H), 3.35 (s, 4H), 2.84 (s, 4H), 2.77 (s, 2H), 2.62 (s, 2H), 1.72 (m , 4H), ESI: [M-1] – = 478.3.
Example 26 7- [4- (benzothiazol-4-yl-1-piperazinyl) butoxy] -3,4-dihydro -2 (1H) – quinolinone Preparation of
The product (400mg, 0.83mmol) of Example 25 will be implemented, silver carbonate (46mg, 0.16mmol) was dissolved in DMSO (5mL) and the acetic acid was heated to 120 ℃ overnight. Cooling, water was added, extracted with ethyl acetate, the ethyl acetate layer was washed with saturated aqueous sodium bicarbonate and brine again each wash, dried over anhydrous sodium sulfate, and silica gel column chromatography to give a solid (80mg, yield 22%).
1 HNMR (400 MHz, DMSO-d 6 ): δ10.00 (s, 1H), 7.69 (d, 1H), 7.61 (d, 1H), 7.40 (d, 1H), 7.27 (t, 1H), 7.04 ( d, 1H), 6.89 (d, 1H), 6.50 (dd, 1H), 6.45 (d, 1H), 3.93 (t, 2H), 3.06 (br, 4H), 2.78 (t, 2H), 2.60 (br , 4H), 2.41 (t, 4H), 1.74 (t, 2H), 1.60 (t, 2H) ESI: [M + 1]+ = 436.3.
Example 27 7- [4- (2-carboxy-4-yl-benzothiophene-1-piperazinyl) butoxy] -2 (1H) – quinolinone Preparation of
2-chloro-6- (4- (4 – ((2-oxo-1,2-dihydro-quinolin-7-yl) oxy) butyl) piperazin-1-yl) benzaldehyde (80mg , 0.18mmol) was dissolved in DMF (5mL) was added sodium hydroxide (29mg, 0.73mmol) and thioglycolic acid (0.025mL, 0.36mmol), 120 ℃ stirred for 16 hours. Cooling, water was added, adjusted with 1N HCl aqueous solution is about pH = 5, extracted with ethyl acetate, the ethyl acetate layer was washed with saturated brine, dried over anhydrous sodium sulfate, and silica gel column chromatography to give a solid (40mg, yield 46 %).
ESI: [M + 1] + = 478.0.
Piperazine hydrochloride – (2-carboxy-benzothiophen-4-yl) Example 28 1-
The product of Example 17 (100mg, 0.25mmol) was dissolved in acetic acid (3mL) and concentrated hydrochloric acid (0.5 mL) in, 100 ℃ stirred for 10 hours. The reaction mixture was poured into ice water, stirred for 10min after suction filtration to give the object (38mg, 50% yield).
1 HNMR (400 MHz, DMSO-d 6 ): [delta] 9.46 (bs, 2H), 8.04 (s, 1H), 7.69 (d, 1H), 7.43 (t, 1H), 7.00 (d, 1H), 3.30 ( bs, 8H) ESI: [M + 1] + = 262.9.
uUpdate july 2015
On July 10, the U.S. Food and Drug Administration approved Rexulti (brexpiprazole) tablets to treat adults with schizophrenia and as an add-on treatment to an antidepressant medication to treat adults with major depressive disorder (MDD).
Schizophrenia is a chronic, severe, and disabling brain disorder affecting about one percent of Americans. Typically, symptoms are first seen in adults younger than 30 years of age and include hearing voices; believing other people are reading their minds or controlling their thoughts; and being suspicious or withdrawn.
MDD, commonly referred to as depression, is also a severe and disabling brain disorder characterized by mood changes and other symptoms that interfere with a person’s ability to work, sleep, study, eat, and enjoy once-pleasurable activities. Episodes of depression often recur throughout a person’s lifetime, although some may experience a single occurrence. Other signs and symptoms of MDD include loss of interest in usual activities; significant change in weight or appetite; insomnia or excessive sleeping (hypersomnia); restlessness/pacing (psychomotor agitation); increased fatigue; feelings of guilt or worthlessness; slowed thinking or impaired concentration; and suicide attempts or thoughts of suicide. Not all people with MDD experience the same symptoms.
“Schizophrenia and major depressive disorder can be disabling and can greatly disrupt day-to-day activities,” said Mitchell Mathis, M.D., director of the Division of Psychiatry Products in the FDA’s Center for Drug Evaluation and Research. “Medications affect everyone differently so it is important to have a variety of treatment options available for patients with mental illnesses.”
The effectiveness of Rexulti in treating schizophrenia was evaluated in 1,310 participants in two 6-week clinical trials. Rexulti was shown to reduce the occurrence of symptoms of schizophrenia compared to placebo (inactive tablet).
The effectiveness of Rexulti as an add-on treatment for MDD was evaluated in two 6-week trials that compared Rexulti plus an antidepressant to placebo plus an antidepressant in 1,046 participants for whom an antidepressant alone did not adequately treat their symptoms. The participants taking Rexulti reported fewer symptoms of depression than those taking the placebo.
Rexulti and other drugs used to treat schizophrenia have a Boxed Warning alerting health care professionals about an increased risk of death associated with the off-label use of these drugs to treat behavioral problems in older people with dementia-related psychosis. No drug in this class is approved to treat patients with dementia-related psychosis.
The Boxed Warning also alerts health care professionals and patients to an increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants. Patients should be monitored for worsening and emergence of suicidal thoughts and behaviors. Rexulti must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
The most common side effects reported by participants taking Rexulti in clinical trials included weight gain and an inner sense of restlessness, such as feeling the need to move.
Rexulti is manufactured by Tokyo-based Otsuka Pharmaceutical Company Ltd.
update………….
4-Chlorobenzo[b]thiophene a key intermediate in brexpiprazole synthesis
We established an improved synthetic route to 4-chlorobenzo[b]thiophene, a key intermediate in brexpiprazole synthesis, via a practical decarboxylation process in three steps. Thermal analysis demonstrated that the coexistence of the decarboxylated product with DBU should be avoided and that removal of the product outside the reactor was vital. Our process yields the target compound by distillation under reduced pressure and is safe, highly batch efficient, cost-effective, and high yielding. Furthermore, manufacturing on a pilot scale was also accomplished through our approach.

4-Chlorobenzo[b]thiophene-2-carboxylic Acid (4)
4 as a white solid
mp 260 °C.
1H NMR (300 MHz, DMSO-
d6) δ 7.54 (d, 1H,
J = 11.6, 7.7 Hz), 7.56 (dd, 1H,
J = 17.8, 7.7 Hz), 8.03 (d, 1H,
J = 0.7 Hz), 8.07 (td, 1H,
J = 7.6, 0.9 Hz), 13.19 (brs, 1H).
13C NMR (75 MHz, DMSO-
d6) δ 122.21, 125.01, 126.84, 127.99, 128.96, 136.50, 136.57, 142.55, 163.10.
Elemental analysis calcd for C: 50.83%, H: 2.37%, found C: 50.84%, H: 2.21%.
2,3,4,6,7,8,9,10-Octahydropyrimido[1,2-a]azepin-1-ium 4-Chlorobenzo[b]thiophene-2-carboxylate 5
5 as a white solid
Mp 182.5 °C.
1H NMR (300 MHz, CDCl3) δ 1.66 (m, 6H), 1.80–1.75 (m, 2H), 2.98–2.94 (m, 2H), 3.45–3.39 (m, 4H), 3.55–3.51 (m, 2H), 7.31–7.20 (m, 2H), 7.69 (dd, 1H,
J = 3.9, 0.6 Hz), 7.97 (s, 1H), 13.19 (brs, 1H).
13C NMR (75 MHz, CDCl3) δ 19.51, 24.03, 26.70, 28.88, 31.99, 38.01, 48.36, 53.94, 121.04, 123.00, 125.17, 126.61, 128.98, 138.21, 142.43, 146.85, 165.91, 167.20.
Elemental analysis calcd for C: 59.25%, H: 5.80%, N: 7.68%, found C: 59.10%, H: 5.44%, N: 7.53%.
4-Chlorobenzo[b]thiophene (2)
1H NMR (300 MHz, CDCl3) δ 7.26 (t, 1H, J = 7.8 Hz), 7.36 (dd, 1H, J = 7.8, 0.9 Hz), 7.50 (d, 1H, J = 5.7 Hz), 7.52 (d, 1H, J = 5.7 Hz), 7.76 (d, 1H, J = 7.8 Hz).
13C NMR (75 MHz, CDCl3) δ 121.12, 122.40, 125.02, 127.43, 128.93, 138.06, 141.07.
Elemental analysis calcd for C: 56.98%, H: 2.99%, found C: 56.76%, H: 2.94%.
SEE
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00340
http://pubs.acs.org/doi/suppl/10.1021/acs.oprd.5b00340/suppl_file/op5b00340_si_001.pdf
Safe and Efficient Decarboxylation Process: A Practical Synthetic Route to 4-Chlorobenzo[b]thiophene
Bulk Pharmaceutical Chemicals Department, Second Tokushima Factory, Production Headquarters, Otsuka Pharmaceutical Co., Ltd., 224-18, Hiraishi Ebisuno, Kawauchi-cho, Tokushima 771-0182, Japan
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00340

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

































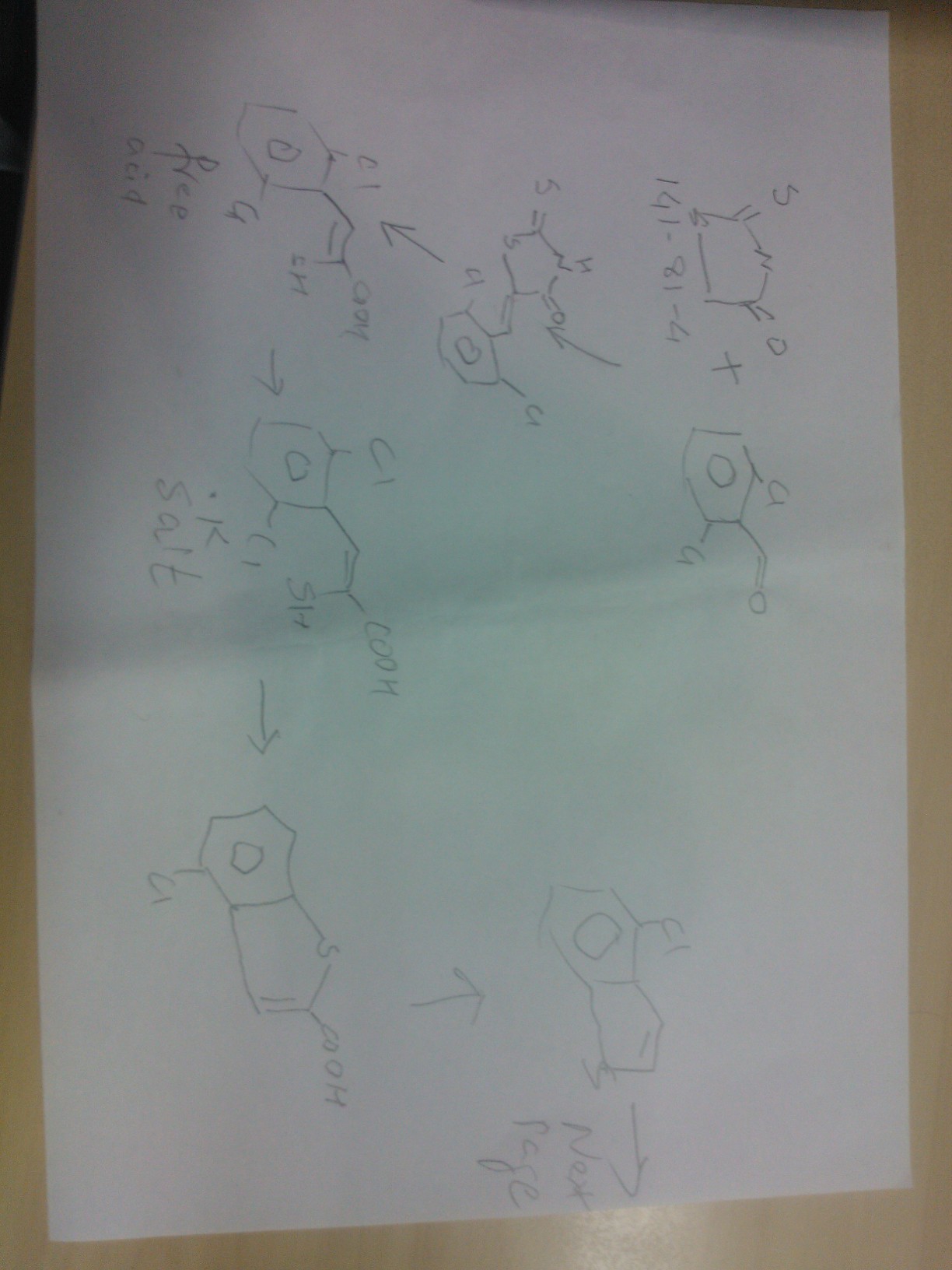
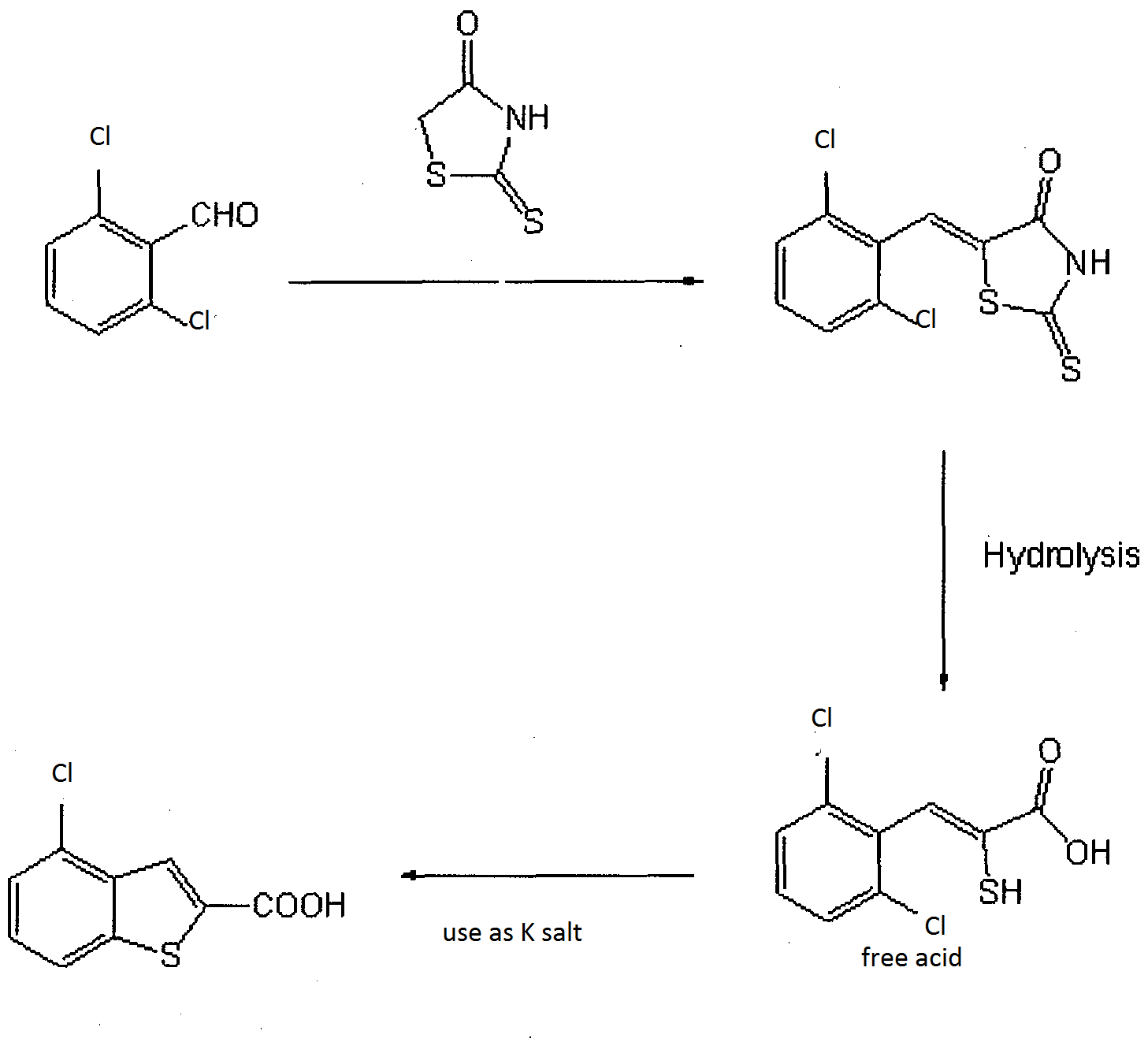
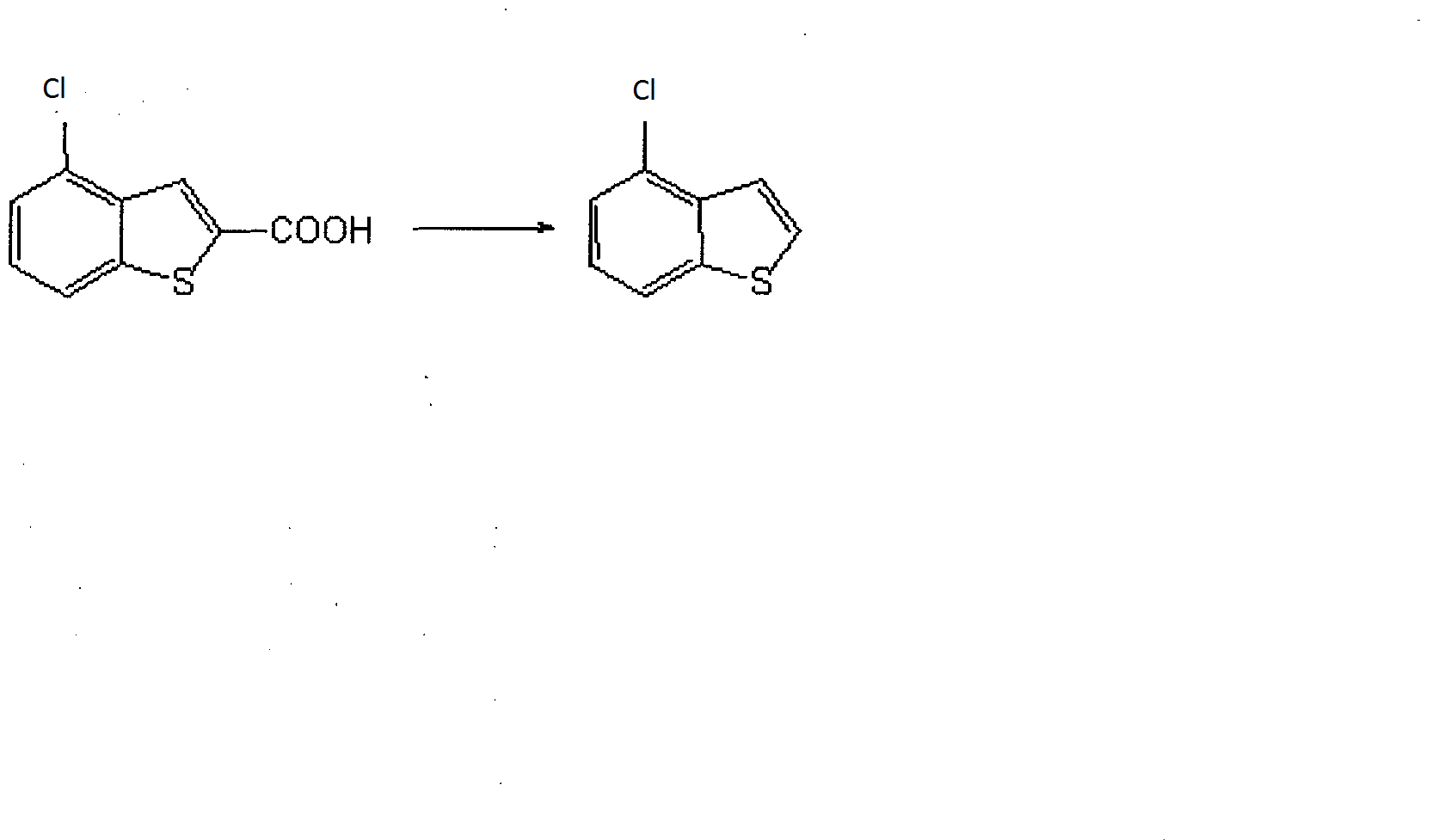
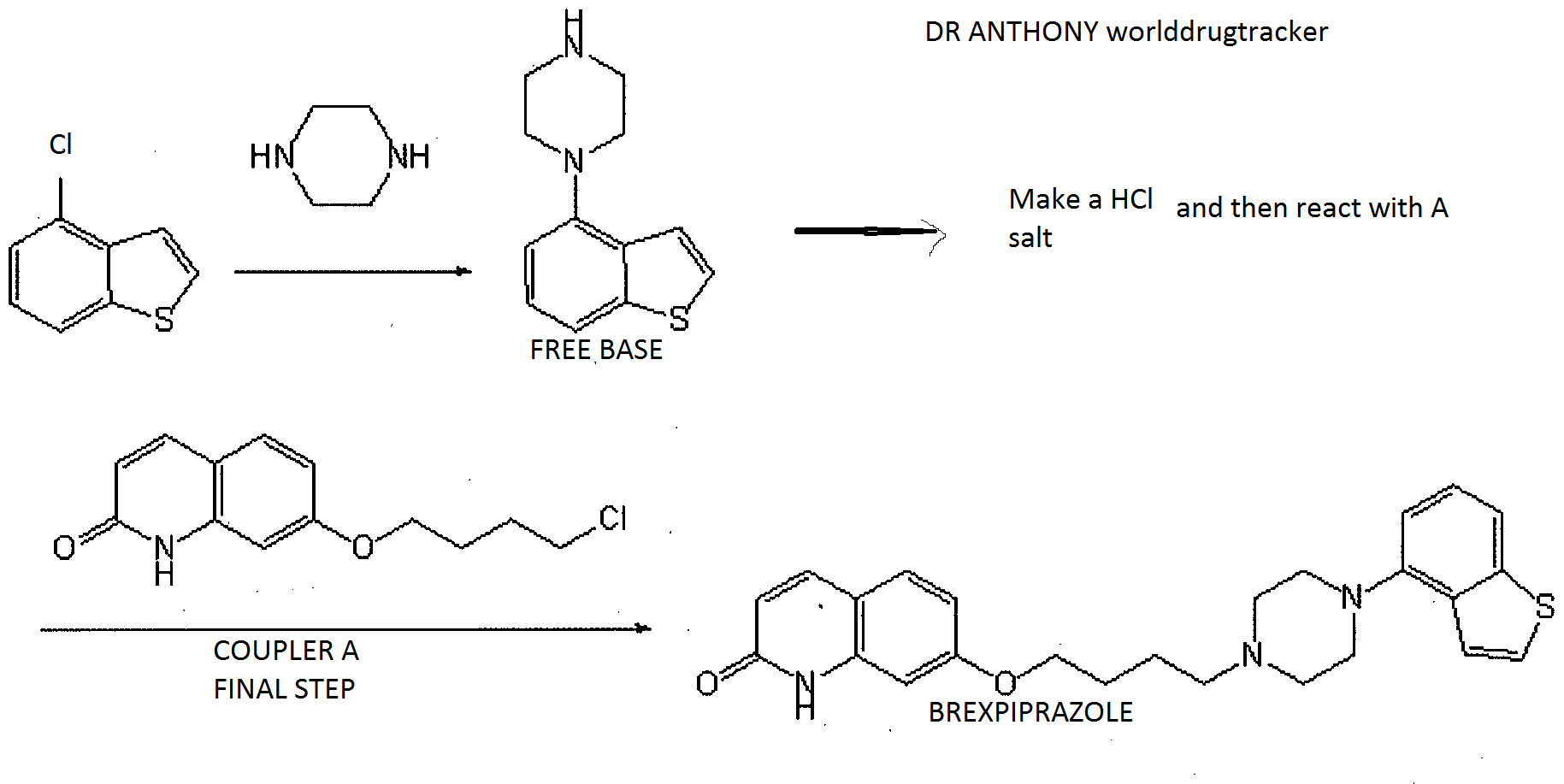

















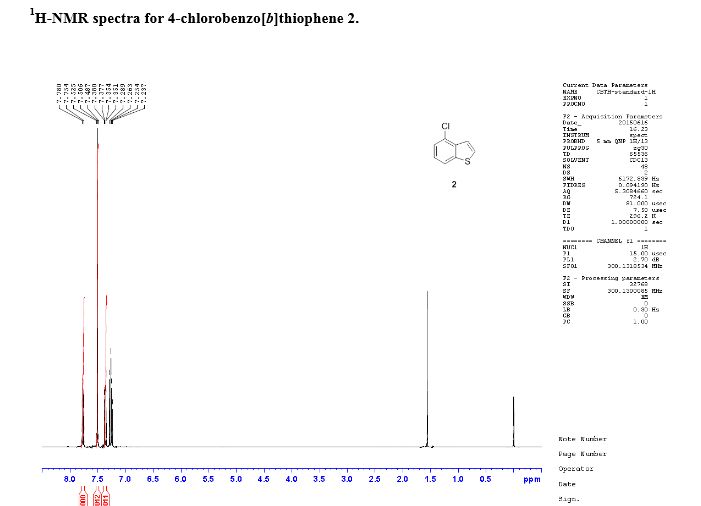
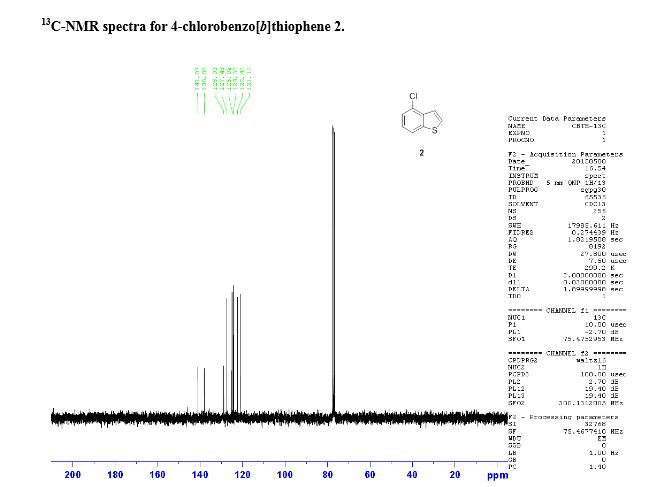



 DR ANTHONY CRASTO
DR ANTHONY CRASTO















 .
.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..




 dalat city
dalat city hanoi
hanoi



























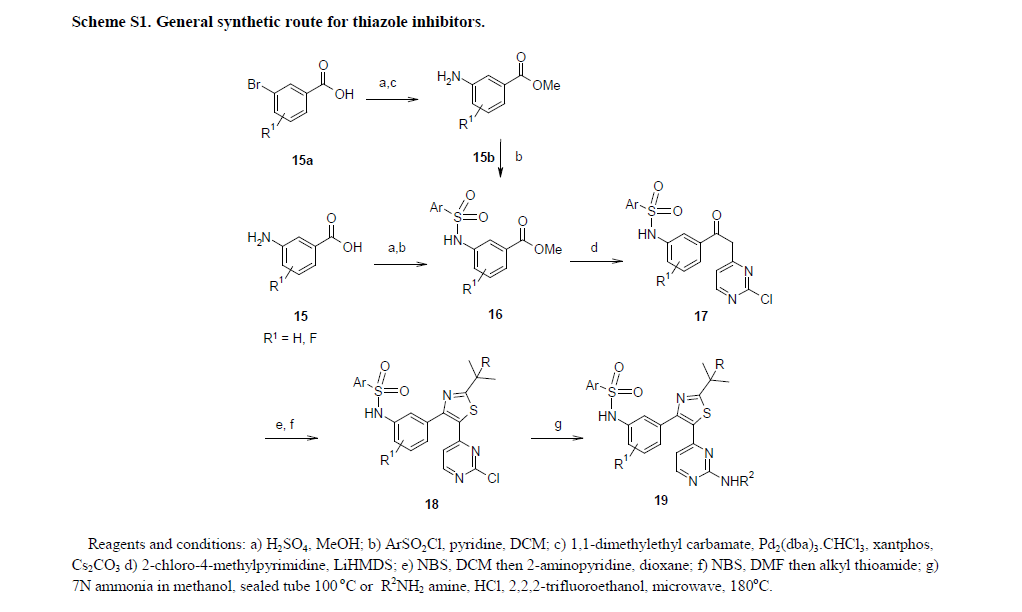



























![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_1h.png)
![N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide NMR spectra analysis, Chemical CAS NO. 1195765-45-7 NMR spectral analysis, N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2015-01-11/001/566/1566739_13c.png)

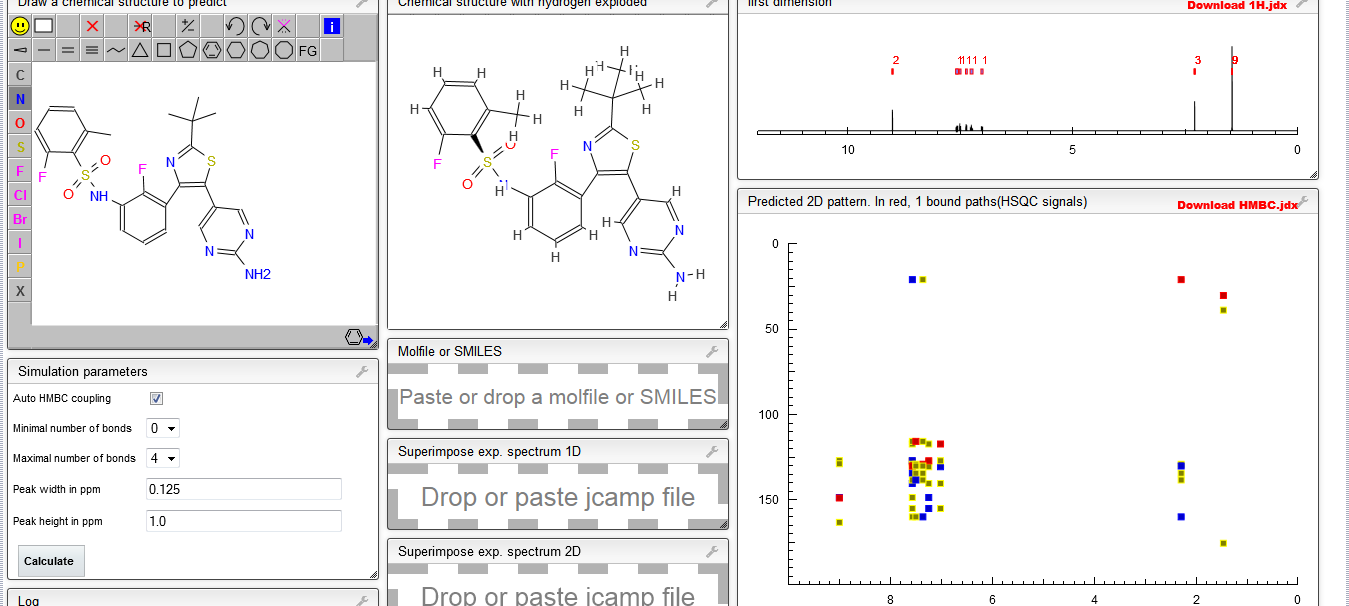
 Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate;Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, 1195768-23-0
Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate;Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, 1195768-23-0 methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester;
methyl 3-bromo-2-fluorobenzylate; PC3663; fluorobromobenzoic acid methyl ester; methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3
methyl 3-amino-2-fluorobenzoate, CAS No. 1195768-18-3 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, CAS No. 1195768-19-4
Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate, CAS No. 1195768-19-4 CAS No. 1042055-86-6 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate
CAS No. 1042055-86-6 Methyl 3-(tert-butoxycarbonylamino)-2-fluorobenzoate 2-fluoro-3-bromobenzoic acid;3-Bromo-2-fluoro-benzoic acid, CAS No. 161957-56-8
2-fluoro-3-bromobenzoic acid;3-Bromo-2-fluoro-benzoic acid, CAS No. 161957-56-8  DABARAFENIB
DABARAFENIB


 CARMEN
CARMEN





















 It’s a pleasure and a privilege to be interviewed, Bora.
It’s a pleasure and a privilege to be interviewed, Bora. By the time I discovered science blogs I knew my career goals were changing. I’d already been lucky enough to audit a science writing course at Princeton taught by Mike Lemonick from TIME, and thought that maybe science writing was a good choice for me. After reading chemistry blogs for a while I realized “Hey, I can do this!” and started my own blog,
By the time I discovered science blogs I knew my career goals were changing. I’d already been lucky enough to audit a science writing course at Princeton taught by Mike Lemonick from TIME, and thought that maybe science writing was a good choice for me. After reading chemistry blogs for a while I realized “Hey, I can do this!” and started my own blog, 




{kind=link}