
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Saracatinib, AZD0530 in phase 3 for Ovary Cancer,

Saracatinib
NCGC00241099, cas 379231-04-6
893428-71-2 (trihydrate)
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methyl-1-piperazinyl)ethoxy]-5-[(tetrahydro-2H-pyran-4-yl)oxy]-4-quinazolinamine
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydropyran-4-yloxy)quinazolin-4-amine
4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline
4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline
AZD0530
C27H32ClN5O5
542.03
AstraZeneca Pharmaceuticals LP
| Astrazeneca Ab, Astrazeneca Uk Ltd, |
Saracatinib (AZD0530) is a highly selective, orally available, dual-specific Src/Abl kinase inhibitor with IC50 of 2.7 and 30 nM for c-Src and Abl kinase, respectively.Saracatinib (AZD0530) demonstrated potent antimigratory and antiinvasive effects in vitro, and inhibited metastasis in a murine model of bladder cancer. Antiproliferative activity of AZD0530 in vitro varied between cell lines (IC50=0.2 ~10 mM).
c-Src, Bcr–Abl, Yes1, Lck.target
AZD0530 is orally available 5-, 7-substituted anilinoquinazoline with anti-invasive and anti-tumor activities. AZD0530 is a dual-specific inhibitor of Src and Abl, protein tyrosine kinases that are overexpressed in chronic myeloid leukemia cells. This agent binds to and inhibits these tyrosine kinases and their effects on cell motility, cell migration, adhesion, invasion, proliferation, differentiation, and survival. Specifically, AZD0530 inhibits Src kinase-mediated osteoclast bone resorption.
AZD-0530 is a highly selective, dual-specific small molecule Src/Abl kinase inhibitor currently in phase II/III clinical trials at AstraZeneca for the treatment of ovarian cancer. Phase II clinical trials are also under way at the company for the treatment of solid tumors and hematological neoplasms. The Mayo Clinic is developing AZD-0530 in phase II clinical studies for the treatment of metastatic pancreas cancer.
Additional phase II trials are under way at the National Cancer Institute (NCI) for the treatment of colorectal cancer, prostate cancer, breast cancer, lung cancer, stomach cancer, soft tissue sarcoma, stage II or IV melanoma and thymic malignancies. A phase II trial for pancreatic cancer has been suspended. Src and Abl kinase are highly expressed in various human tumor types. No recent development has been reported for research into the treatment of head and neck cancer.
Phase II study of Saracatinib (AZD0530) for for the treatment of patients with hormone receptor-negative metastatic breast cancer : Nine patients were treated on study. After a median of 2 cycles (range 1-3), no patient had achieved CR, PR, or SD >6 months. The median time to treatment failure was 82 days (12-109 days).The majority (89%) of patients discontinued saracatinib because of disease progression. One patient acquired potentially treatment-related grade 4 hypoxia with interstitial infiltrates and was removed from the study. Common adverse events included fatigue, elevated liver enzymes, nausea, hyponatremia, dyspnea, cough, and adrenal insufficiency. CONCLUSIONS: These efficacy results were not sufficiently promising to justify continued accrual to this study. Based on this series, saracatinib does not appear to have significant single-agent activity for the treatment of patients with ER(-)/PR(-) MBC. (source: Clin Breast Cancer. 2011 Oct;11(5):306-11.)
Phase II study of Saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: Saracatinib has insufficient activity as a single agent in patients with advanced gastric adenocarcinoma to warrant further investigation. Further development in gastric cancer would require rational drug combinations or identification of a tumor phenotype sensitive to Src inhibition. (source: Invest New Drugs. 2011 Mar 12. [Epub ahead of print]).
Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Nine patients were enrolled. All patients had received prior radiotherapy and six patients had received prior chemotherapy for recurrent or metastatic disease. The most common adverse event was fatigue. Eight patients had progression of disease by response evaluation criteria in solid tumors (RECIST) within the first eight-week cycle and one patient was removed from the study after 11 days due to clinical decline with stable disease according to the RECIST criteria. Median overall survival was six months. The study was closed early due to lack of efficacy according to the early stopping rule. CONCLUSION: Single-agent saracatinib does not merit further study in recurrent or metastatic HNSCC. (source: Anticancer Res. 2011 Jan;31(1):249-53.)
893428-72-3 Saracatinib difumarate
893428-73-4 also
Saracatinib (AZD0530) is a Src inhibitor for c-Src with IC50 of 2.7 nM.
…………………………….

http://www.google.com/patents/WO2001094341A9?cl=en

………………….
WO 2006064217
http://www.google.fm/patents/EP1871769A2?cl=en
4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline which compound is disclosed as Compound No. 73 within the Table in Example 14 of International Patent Application WO 01/94341. That compound is described herein by way of the Formula I

and as AZD0530, the code number by which the compound is known.
AZD0530 is an inhibitor of the Src family of non-receptor tyrosine kinase enzymes and, thereby, is a selective inhibitor of the motility of tumour cells and a selective inhibitor of the dissemination and invasiveness of mammalian cancer cells leading to inhibition of metastatic tumour growth. In particular, the compound AZD0530 is an inhibitor of c-Src non-receptor tyrosine kinase and should be of value as an anti-invasive agent for use in the containment and/or treatment of solid tumour disease in the human or animal body. The route for preparing the compound of the Formula I that is disclosed in International Patent Application WO 01/94341 involves the reaction of the compound 4-(6-chloro-2,3-methylenedioxyanilino)-7-hydroxy-5-tetrahydropyran-4-yloxyquinazoline with an alkylating agent to form the 2-(4-methylpiperazin-l-yl)ethoxy side-chain at the 7-position. The product of the reaction is disclosed in WO 01/94341 in the form of a dihydrochloride salt and in the form of a free base.
Example 14 4-(6-chloro-2,3-methylenedioxyaniIino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline (route 4)
Under an atmosphere of nitrogen gas, l-(2-hydroxyethyl)-4-methylpiperazine (13.93 g) was added to a stirred mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-fluoro- 5-tetrahydropyran-4-yloxyquinazoline (12.9 g), sodium te/t-pentoxide (9.87 g) and 1 ,2-diethoxyethane (37.5 ml). Water (1.34 g) and 1,2-diethoxyethane (25 ml) were added and the resultant reaction mixture was stirred and heated to 86°C for 18 hours. The reaction mixture was cooled to 5O0C and, under vacuum distillation at approximately 60 millibar pressure, approximately 50 ml of reaction solvent was distilled off. The reaction mixture was neutralised to pH 7.0 to 7.6 by the addition of a mixture of concentrated aqueous hydrochloric acid (36%, 10 ml) and water (84 ml) at a rate that kept the temperature of the reaction mixture at a maximum of 6O0C. With the temperature of the reaction mixture being kept at 6O0C, the reaction mixture was extracted with ethyl acetate (225 ml). The organic solution was washed with water (50 ml). Water (25 ml) was added and, with the temperature being kept at 6O0C, the mixture was stirred for 10 minutes, then allowed to stand for 30 minutes and the aqueous layer was separated. The organic layer was concentrated to a volume of about 100 ml by distillation of solvent at about 9O0C under atmospheric pressure. The residual mixture was cooled during 1 hour to 450C and held at that temperature for 2 hours to allow crystallisation of product. The mixture was warmed briefly to 550C and then cooled during 4 hours to 180C and held at that temperature for 1 hour. The crystalline precipitate was isolated by filtration and washed in turn with water (17 ml) and with tø’t-butyl methyl ether (17 ml). There was thus obtained 4-(6-chloro-2,3-πiethylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline as a trihydrate (11 g; 88% purity by HPLC using Method B, retention time 7.3 minutes); NMR Spectrum: (CDCl3) 1.65 (br s, 3H), 1.9-2.05 (m, 2H), 2.2-2.3 (m, 2H), 2.31 (s, 3H), 2.4-2.8 (m, 8H), 2.9 (m, 2H), 3.6-3.7 (m, 2H), 3.95-4.05 (m, 2H), 4.2-4.25 (m, 2H), 4.8 (m,lH), 6.05 (s, 2H), 6.55 (s, IH), 6.75 (d, IH), 6.85 (s, IH), 7.0 (d, IH), 8.55 (s, IH), 9.25 (s, IH).
A portion (10 g) of the material so obtained was placed on a filter and dried at ambient temperature in a stream of dry nitrogen gas. The resultant material was dissolved at 6O0C in dry isopropanol (140 ml) whilst maintaining a dry nitrogen atmosphere. The solution was allowed to cool to ambient temperature and to stand under a dry nitrogen atmosphere for 2 days. The resultant crystalline solid was isolated by filtration under a dry nitrogen atmosphere. The material (8 g) so obtained was a crystalline anhydrous form of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l -yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline, m.p. 142 to 1440C.
Example 15
4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline difumarate salt
A mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline trihydrate (27.1 g), isopropanol (200 ml) and water (10 ml) was heated to 75°C. A mixture of fumaric acid (12.8 g), isopropanol (200 ml) and water (40 ml) was heated to 😯0C. A portion (80 ml) of the warmed solution of the quinazoline compound was added to the fumaric acid solution whilst the temperature was maintained at 750C. The resultant mixture was stirred at 750C for 75 minutes. The remainder of the quinazoline compound solution was added during 1 hour whilst the temperature was maintained at 750C. Isopropanol (50 ml) was added and the resultant mixture was stirred at 750C for 7 hours. The mixture was cooled slowly over at least 25 minutes to 5O0C and was stirred at that temperature for 6 hours. The mixture was cooled slowly over at least 20 minutes to 2O0C and was stirred at that temperature for 18.5 hours. The crystalline solid was isolated by filtration, washed twice with a 10:1 mixture of isopropanol and water (50 ml and 100 ml respectively) and dried in vacuo at 450C to constant weight. There was thus obtained 4-(6-chloro- 2,3-methylenedioxyanilino)-7-[2-(4-methylρiperazin-l-yl)ethoxy]-5-tetrahydropyran- 4-yloxyquinazoline difumarate salt (37.0 g); m.p. 233-2370C; NMR Spectrum: (DMSOd6) 1.76-1.88 (m, 2H), 2.1-2.17 (m, 2H)5 2.33 (s, 3H), 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H)3 3.83-3.9 (m, 2H), 4.24 (t, 2H)5 4.98-5.07 (m, IH), 6.07 (s, 2H)3 6.6 (s, 4H)5 6.83 (d5 IH)3 6.84 (d, IH)5 6.91 (d3 IH)5 7.05 (d, IH)3 8.33 (s, IH)3 9.18 (s, IH).
Example 16
4-(6-chloro-2,3-methyIenedioxyaniIino)-7-[2-(4-methyIpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yIoxyquinazolme difumarate salt
A mixture of 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline trihydrate (27.1 g), isopropanol (210 ml) and water (30 ml) was heated to 4O0C and the mixture was filtered. The filter was washed with isopropanol (20 ml) and the washings were added to the warm filtrate. The resultant solution was warmed to 75°C.
A mixture of fumaric acid (12.8 g), isopropanol (200 ml) and water (20 ml) was heated to 700C and the resultant mixture was filtered. A portion (110 ml) of the fumaric acid solution was added to the warmed solution of 4-(6-chloro-2,3-methylenedioxyanilino)- 7-[2-(4-methylpiperazin- 1 -yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline whilst the temperature was maintained at 75°C. Seed crystals of 4-(6-chloro-
253-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]-5-tetrahydropyran- 4-yloxyquinazoline difumarate salt (0.02 g) were added and the resultant mixture was stirred at 750C for 1 hour. The remainder of the fumaric acid solution was added during 1 hour whilst the temperature was maintained at 750C and the resultant mixture was stirred at 750C for 14 hours.
The mixture was cooled slowly over at least 2 hours to 200C and was stirred at that temperature for 1 hour. The crystalline solid was isolated by filtration, washed twice with a 10:1 mixture of isopropanol and water (50 ml and 100 ml respectively) and dried in vacuo at 450C to constant weight. There was thus obtained 4-(6-chloro-253-methylenedioxyanilino)- 7-[2-(4-methylpiperazin-l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline difumarate salt (35.8 g); m.p. 234-237°C; NMR Spectrum: (DMSOd6) 1.76-1.88 (m, 2H)5 2.1-2.17 (m5 2H)5 2.33 (s5 3H)5 2.6 (br s, 8H), 2.78 (t, 2H), 3.51-3.6 (m, 2H), 3.83-3.9 (m, 2H), 4.24 (t, 2H)5 4.98-5.07 (m, IH), 6.07 (s, 2H)5 6.6 (s, 4H), 6.83 (d, IH)5 6.84 (d, IH)5 6.91 (d, IH)5 7.05 (d, IH)5 8.33 (s5 IH)5 9.18 (s5 IH).
Example 17 4-(6-chloro-2,3-methylenedioxyanilino)-7-[2-(4-methyIpiperazin-l-yI)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline sesquifumarate salt
A mixture of 4-(6-chloro-253-methylenedioxyanilino)-7-[2-(4-methylpiperazin- l-yl)ethoxy]-5-tetrahydropyran-4-yloxyquinazoline difurnarate (0.15 g) and water (20 ml) was warmed using a heat gun to obtain a solution. The sample was allowed to evaporate slowly at ambient temperature to a volume of about 3 ml under a flow of air for 24 hours whereupon a precipitate had started to form. The mixture was placed in a refridgerator at 4°C for 2 days. The resultant precipitate was isolated by filtration and washed with water. There was thus obtained 4-(6-chloro-253-methylenedioxyanilino)-7-[2-(4-methylpiperazin-l-yl)ethoxy]- 5-tetrahydropyran-4-yloxyquinazoline as a sesquifumarate tetrahydrate salt (0.084 g) which was characterised using XRPD5 DSC5 TGA5 FTIR and solution NMR techniques.
………………..
A simplified process for the manufacture of AZD0530, a potent SRC kinase inhibitor
Org Process Res Dev 2011, 15(3): 688
http://pubs.acs.org/doi/abs/10.1021/op100161y

Process research and development of a synthetic route towards a novel SRC kinase inhibitor is described. The Medicinal Chemistry route was very long and suffered from extensive use of chlorinated solvents and chromatography. A number of steps in the Medicinal Chemistry route were also unattractive for large-scale use for a variety of reasons. The route was modified to produce a shorter synthetic scheme that started from more readily available materials. By using the modified route, the title compound was manufactured on kilogram scale without recourse to chromatography and in significantly fewer steps. The scaled synthesis required two Mitsunobu couplings, which were developed and scaled successfully. An interesting hydrazine impurity was identified in the second Mitsunobu coupling; a mechanism for its formation is proposed, and a method for its control is described. The formation and control of some other interesting impurities are also described.
N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine Difumarate (AZD0530 Difumarate)
Final Purification of N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine Difumarate (AZD0530 Difumarate)
AZD0530 difumarate (4.234 kg at 89% w/w, 4.87 mol) was refluxed in a mixture of IPA (10.L) and water (10.L, Fresenius). A solution was not obtained, so further IPA (450 mL) and water (450 mL, Fresenius) were added, and the mixture was refluxed. The resulting solution was cooled to 68 °C and screened over 3.5 min through a 20 μm in-line filter into a vessel preheated to 65 °C. IPA(20.4 L) at 65 °C was added via the first vessel and in-line filter, and the resulting solution was stirred at 65 °C for 2 h. Crystallisation was evident after 20 min. The mixture was allowed to self-cool to ambient temperature overnight before filtering and washing the cake with a mixture (prescreened through a 20 μm membrane) of water (640 mL) and IPA (5.76 L). The cake was washed with IPA (6.4 L, prescreened) and MTBE (6.4 L, prescreened) and dried to constant weight under reduced pressure at 50 °C to give AZD0530 difumarate (2.865 kg, at 95.2% w/w, 3.52 mol, 72% yield). Spectroscopic analysis was in agreement with the reported data…………Ford, J. G.; McCabe, J. F.; O’Kearney-McMullan, A.; O’Keefe, P.; Pointon, S. M.; Powell, L.; Purdie, M.; Withnall, J. WO/2006/064217, 2006.
……………………….
SEE…………N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor
J Med Chem 2006, 49(22): 6465

…………..
1: Hannon RA, Finkelman RD, Clack G, Iacona RB, Rimmer M, Gossiel F, Baselga J, Eastell R. Effects of Src kinase inhibition by saracatinib (AZD0530) on bone turnover in advanced malignancy in a Phase I study. Bone. 2012 Jan 8. [Epub ahead of print] PubMed PMID: 22245630.
2: Gucalp A, Sparano JA, Caravelli J, Santamauro J, Patil S, Abbruzzi A, Pellegrino C, Bromberg J, Dang C, Theodoulou M, Massague J, Norton L, Hudis C, Traina TA. Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin Breast Cancer. 2011 Oct;11(5):306-11. Epub 2011 May 3. PubMed PMID: 21729667; PubMed Central PMCID: PMC3222913.
3: Mackay HJ, Au HJ, McWhirter E, Alcindor T, Jarvi A, Macalpine K, Wang L, Wright JJ, Oza AM. A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: a trial of the PMH phase II consortium. Invest New Drugs. 2011 Mar 12. [Epub ahead of print] PubMed PMID: 21400081.
4: Fury MG, Baxi S, Shen R, Kelly KW, Lipson BL, Carlson D, Stambuk H, Haque S, Pfister DG. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011 Jan;31(1):249-53. PubMed PMID: 21273606.
5: Renouf DJ, Moore MJ, Hedley D, Gill S, Jonker D, Chen E, Walde D, Goel R, Southwood B, Gauthier I, Walsh W, McIntosh L, Seymour L. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs. 2010 Dec 18. [Epub ahead of print] PubMed PMID: 21170669.
6: Dalton RN, Chetty R, Stuart M, Iacona RB, Swaisland A. Effects of the Src inhibitor saracatinib (AZD0530) on renal function in healthy subjects. Anticancer Res. 2010 Jul;30(7):2935-42. PubMed PMID: 20683035.
7: Arcaroli JJ, Touban BM, Tan AC, Varella-Garcia M, Powell RW, Eckhardt SG, Elvin P, Gao D, Messersmith WA. Gene array and fluorescence in situ hybridization biomarkers of activity of saracatinib (AZD0530), a Src inhibitor, in a preclinical model of colorectal cancer. Clin Cancer Res. 2010 Aug 15;16(16):4165-77. Epub 2010 Aug 3. PubMed PMID: 20682712.
8: Morrow CJ, Ghattas M, Smith C, Bönisch H, Bryce RA, Hickinson DM, Green TP, Dive C. Src family kinase inhibitor Saracatinib (AZD0530) impairs oxaliplatin uptake in colorectal cancer cells and blocks organic cation transporters. Cancer Res. 2010 Jul 15;70(14):5931-41. Epub 2010 Jun 15. PubMed PMID: 20551056; PubMed Central PMCID: PMC2906706.
9: Hannon RA, Clack G, Rimmer M, Swaisland A, Lockton JA, Finkelman RD, Eastell R. Effects of the Src kinase inhibitor saracatinib (AZD0530) on bone turnover in healthy men: a randomized, double-blind, placebo-controlled, multiple-ascending-dose phase I trial. J Bone Miner Res. 2010 Mar;25(3):463-71. PubMed PMID: 19775203.
10: Rajeshkumar NV, Tan AC, De Oliveira E, Womack C, Wombwell H, Morgan S, Warren MV, Walker J, Green TP, Jimeno A, Messersmith WA, Hidalgo M. Antitumor effects and biomarkers of activity of AZD0530, a Src inhibitor, in pancreatic cancer. Clin Cancer Res. 2009 Jun 15;15(12):4138-46. Epub 2009 Jun 9. PubMed PMID: 19509160.
11: Chen Y, Guggisberg N, Jorda M, Gonzalez-Angulo A, Hennessy B, Mills GB, Tan CK, Slingerland JM. Combined Src and aromatase inhibition impairs human breast cancer growth in vivo and bypass pathways are activated in AZD0530-resistant tumors. Clin Cancer Res. 2009 May 15;15(10):3396-405. PubMed PMID: 19451593.
12: Lara PN Jr, Longmate J, Evans CP, Quinn DI, Twardowski P, Chatta G, Posadas E, Stadler W, Gandara DR. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium study. Anticancer Drugs. 2009 Mar;20(3):179-84. PubMed PMID: 19396016; PubMed Central PMCID: PMC3225398.
13: Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM, Wilkinson RW, Elvin P, Boyer B, Carragher N, Plé PA, Bermingham A, Holdgate GA, Ward WH, Hennequin LF, Davies BR, Costello GF. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009 Jun;3(3):248-61. Epub 2009 Feb 7. PubMed PMID: 19393585.
14: de Vries TJ, Mullender MG, van Duin MA, Semeins CM, James N, Green TP, Everts V, Klein-Nulend J. The Src inhibitor AZD0530 reversibly inhibits the formation and activity of human osteoclasts. Mol Cancer Res. 2009 Apr;7(4):476-88. PubMed PMID: 19372577.
15: Schweppe RE, Kerege AA, French JD, Sharma V, Grzywa RL, Haugen BR. Inhibition of Src with AZD0530 reveals the Src-Focal Adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009 Jun;94(6):2199-203. Epub 2009 Mar 17. PubMed PMID: 19293266; PubMed Central PMCID: PMC2690419.
16: Purnell PR, Mack PC, Tepper CG, Evans CP, Green TP, Gumerlock PH, Lara PN, Gandara DR, Kung HJ, Gautschi O. The Src inhibitor AZD0530 blocks invasion and may act as a radiosensitizer in lung cancer cells. J Thorac Oncol. 2009 Apr;4(4):448-54. PubMed PMID: 19240653; PubMed Central PMCID: PMC2716757.
17: Gwanmesia PM, Romanski A, Schwarz K, Bacic B, Ruthardt M, Ottmann OG. The effect of the dual Src/Abl kinase inhibitor AZD0530 on Philadelphia positive leukaemia cell lines. BMC Cancer. 2009 Feb 13;9:53. PubMed PMID: 19216789; PubMed Central PMCID: PMC2654659.
18: Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008 Oct 23;27(49):6365-75. Epub 2008 Aug 4. PubMed PMID: 18679417.
Src inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo.
Simpkins et al. Clin Cancer Res. 2012 Aug 15. PMID: 22896656.
Saracatinib (AZD0530) is a potent modulator of ABCB1-mediated multidrug resistance in vitro and in vivo.
Liu et al. Int J Cancer. 2012 May 24. PMID: 22623106.
Common PIK3CA mutants and a novel 3′ UTR mutation are associated with increased sensitivity to saracatinib.
Arcaroli et al. Clin Cancer Res. 2012 May 1;18(9):2704-14. PMID: 22553375.
Phase I study of saracatinib (AZD0530) in combination with paclitaxel and/or carboplatin in patients with solid tumours.
Kaye et al. Br J Cancer. 2012 May 22;106(11):1728-34. PMID: 22531637.
Timely post on litigation and polymorphism: Case study with Celgene’s Revlimid
Hi there, folks. It has been a while since I blogged. I decided that I would continue once again and share some of the references that I came across while perusing the journals, while using the free blog-hosting services that are available. This particular post was something I came across while looking at some stocks online. I was searching taking a look at Celgene (CELG) stock and came across this discussion regarding the tricky world of patent litigation and more specifically, its application to matters concerning crystal form and polymorphism. Having worked both sides of generic and brand-name pharma, I am interested in how this case will work out.
I know that this article’s subject matter is on the periphery of what is involved in developing a process. I have worked on crystallizations and been concerned with polymorph control, so this is a textbook case, that is current, about patent…
View original post 256 more words
Taiho’s Colon Cancer Drug Ups OS in Phase 3

TAS-102 (nonproprietary names: trifluridine and tipiracil hydrochloride)
Taiho’s Colon Cancer Drug Ups OS in Phase 3
Taiho Pharmaceutical Co. Ltd. announced results from its global Phase 3 RECOURSE trial on its oral combination anticancer drug TAS-102 in refractory metastatic colorectal cancer (mCRC). Read more…
FULL STORY
TAS-102 is an anti-cancer drug under development for colorectal cancer.[1]
Clinical trials
A phase II trial reported in 2011[2] and a phase III trial is due to end in 2014.[1][3]
Mechanism
TAS-102 consists of the cytotoxin trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil.[4] Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.[1]
References
- “New Drug for Colorectal Cancer Shows Promise in Phase II Trial”. 28 Aug 2012.
- “Novel Drug TAS-102 Makes Headway in Refractory Colorectal Cancer”. 4 Oct 2011.
- “Phase II study of TAS-102 for pretreated metastatic colorectal cancer”. 29 Aug 2012.
- “A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA.”. Sep 2004.

Trifluridine
Trifluridine (also called trifluorothymidine or TFT) is an anti-herpesvirus antiviral drug, used primarily on the eye. It was sold under the trade name, Viroptic, by Glaxo Wellcome, now merged into GlaxoSmithKline. The brand is now owned by Monarch Pharmaceuticals, which is wholly owned by King Pharmaceuticals.
It is a nucleoside analogue, a modified form of deoxyuridine, similar enough to be incorporated into viral DNA replication, but the -CF3 group added to the uracil component blocks base pairing.
It is a component of the experimental anti-cancer drug TAS-102.
A Cochrane Systematic Review showed that trifluridine was a more effective treatment than idoxuridine or vidarabine, significantly increasing the relative number of successfully healed eyes in 14 days.[1]
References
- Wilhelmus KR (2010). “Antiviral treatment and other therapeutic interventions for herpes simplex virus epithelial keratitis”. Cochrane Database Syst Rev 12: CD002898. doi:10.1002/14651858.CD002898.pub4. PMID 21154352.
External links
- Costin D, Dogaru M, Popa A, Cijevschi I (2004). “Trifluridine therapy in herpetic in keratitis”. Rev Med Chir Soc Med Nat Iasi 108 (2): 409–12. PMID 15688823.
- Kuster P, Taravella M, Gelinas M, Stepp P (1998). “Delivery of trifluridine to human cornea and aqueous using collagen shields.”. CLAO J 24 (2): 122–4. PMID 9571274.
- O’Brien W, Taylor J (1991). “Therapeutic response of herpes simplex virus-induced corneal edema to trifluridine in combination with immunosuppressive agents.”. Invest Ophthalmol Vis Sci 32 (9): 2455–61. PMID 1907950.
- “Trifluridine Ophthalmic Solution, 1%” (PDF). Retrieved 2007-03-24.


Fig 4. Open Babel bond-line chemical structure with annotated hydrogens. Click to toggle size.
Spectrum Plot

Fig 5. 1H NMR spectrum of C10H11F3N2O5 in CDCL3 at 400 MHz

Tipiralacil, also known as TPI, is a thymidine phosphorylase inhibitor (TPI). Tipiracil is one of the active components in TAS-102, which is an anticancer drug candidate currently in clinical trials. TAS-102 consists of the cytotoxin Trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil. Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.
183204-72-0 (Tipiracil -HCl); 183204-74-2(Tipiracil ).

|
References |
1: Peters GJ, Bijnsdorp IV. TAS-102: more than an antimetabolite. Lancet Oncol. 2012 Dec;13(12):e518-9. doi: 10.1016/S1470-2045(12)70426-6. PubMed PMID: 23182191.
2: Yoshino T, Mizunuma N, Yamazaki K, Nishina T, Komatsu Y, Baba H, Tsuji A, Yamaguchi K, Muro K, Sugimoto N, Tsuji Y, Moriwaki T, Esaki T, Hamada C, Tanase T, Ohtsu A. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012 Oct;13(10):993-1001. doi: 10.1016/S1470-2045(12)70345-5. Epub 2012 Aug 28. PubMed PMID: 22951287.
3: Sobrero A. TAS-102 in refractory colorectal cancer: caution is needed. Lancet Oncol. 2012 Oct;13(10):959-61. doi: 10.1016/S1470-2045(12)70376-5. Epub 2012 Aug 28. PubMed PMID: 22951286.
4: Doi T, Ohtsu A, Yoshino T, Boku N, Onozawa Y, Fukutomi A, Hironaka S, Koizumi W, Sasaki T. Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012 Jul 24;107(3):429-34. doi: 10.1038/bjc.2012.274. Epub 2012 Jun 26. PubMed PMID: 22735906; PubMed Central PMCID: PMC3405214.
5: Suzuki N, Nakagawa F, Nukatsuka M, Fukushima M. Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp Ther Med. 2011 May;2(3):393-397. Epub 2011 Mar 21. PubMed PMID: 22977515; PubMed Central PMCID: PMC3440718.
6: Shintani M, Urano M, Takakuwa Y, Kuroda M, Kamoshida S. Immunohistochemical characterization of pyrimidine synthetic enzymes, thymidine kinase-1 and thymidylate synthase, in various types of cancer. Oncol Rep. 2010 May;23(5):1345-50. PubMed PMID: 20372850.
7: Temmink OH, Bijnsdorp IV, Prins HJ, Losekoot N, Adema AD, Smid K, Honeywell RJ, Ylstra B, Eijk PP, Fukushima M, Peters GJ. Trifluorothymidine resistance is associated with decreased thymidine kinase and equilibrative nucleoside transporter expression or increased secretory phospholipase A2. Mol Cancer Ther. 2010 Apr;9(4):1047-57. doi: 10.1158/1535-7163.MCT-09-0932. Epub 2010 Apr 6. PubMed PMID: 20371715.
8: Bijnsdorp IV, Kruyt FA, Fukushima M, Smid K, Gokoel S, Peters GJ. Molecular mechanism underlying the synergistic interaction between trifluorothymidine and the epidermal growth factor receptor inhibitor erlotinib in human colorectal cancer cell lines. Cancer Sci. 2010 Feb;101(2):440-7. doi: 10.1111/j.1349-7006.2009.01375.x. Epub 2009 Sep 29. PubMed PMID: 19886911.
9: Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M, Kruyt FA. Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared to 5-fluorouracil in colon cancer cells. Int J Cancer. 2010 May 15;126(10):2457-68. doi: 10.1002/ijc.24943. PubMed PMID: 19816940.
10: Bijnsdorp IV, Kruyt FA, Gokoel S, Fukushima M, Peters GJ. Synergistic interaction between trifluorothymidine and docetaxel is sequence dependent. Cancer Sci. 2008 Nov;99(11):2302-8. doi: 10.1111/j.1349-7006.2008.00963.x. Epub 2008 Oct 18. PubMed PMID: 18957056.
11: Overman MJ, Kopetz S, Varadhachary G, Fukushima M, Kuwata K, Mita A, Wolff RA, Hoff P, Xiong H, Abbruzzese JL. Phase I clinical study of three times a day oral administration of TAS-102 in patients with solid tumors. Cancer Invest. 2008 Oct;26(8):794-9. doi: 10.1080/07357900802087242. PubMed PMID: 18798063.
12: Overman MJ, Varadhachary G, Kopetz S, Thomas MB, Fukushima M, Kuwata K, Mita A, Wolff RA, Hoff PM, Xiong H, Abbruzzese JL. Phase 1 study of TAS-102 administered once daily on a 5-day-per-week schedule in patients with solid tumors. Invest New Drugs. 2008 Oct;26(5):445-54. doi: 10.1007/s10637-008-9142-3. Epub 2008 Jun 5. PubMed PMID: 18528634.
13: Temmink OH, Emura T, de Bruin M, Fukushima M, Peters GJ. Therapeutic potential of the dual-targeted TAS-102 formulation in the treatment of gastrointestinal malignancies. Cancer Sci. 2007 Jun;98(6):779-89. Epub 2007 Apr 18. Review. PubMed PMID: 17441963.
14: Temmink OH, Hoebe EK, van der Born K, Ackland SP, Fukushima M, Peters GJ. Mechanism of trifluorothymidine potentiation of oxaliplatin-induced cytotoxicity to colorectal cancer cells. Br J Cancer. 2007 Jan 29;96(2):231-40. PubMed PMID: 17242697; PubMed Central PMCID: PMC2360012.
Using molecular techniques, researchers improved diagnosis and treatment of cancer

The ABC Medical Center, located in Mexico City, implemented various molecular diagnostic methods that can detect the genetic alterations in several types of cancer, so they can select a personalized therapy for each patient and direct it against the mutated genes that cause disease.
For this the Laboratory of Molecular Pathology was created, thanks to a cancer patient who decided to make a donation to the hospital to acquire the necessary equipment, sensitive to the existing need in Mexico for a place where timely diagnosis of the disease is made.
“While the techniques of molecular biology have been applied to clinical diagnosis for over 20 years, they had just been established in the country. It is estimated that only about five years ago in some of the National Institutes of Health and universities, where it was first used in the field of research, “says Dr. César Lara Torres, head of…
View original post 419 more words
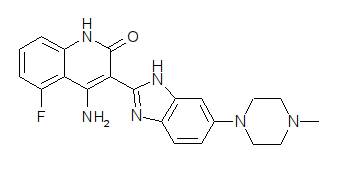
Dovitinib in phase 3 for Cancer, bladder (urothelial carcinoma)

Dovitinib
(3E)-4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1,3-dihydrobenzimidazol-2-ylidene]quinolin-2-one, is one kind of white crystalline powder, odorless, little bitter taste.
4-Amino-5-fluoro-3-[6-(4-methyl-1-piperazinyl)-1H-benzimidazol-2-yl]-2(1H)-quinolinone 2-hydroxypropanoate
4-Amino-5-fluoro-3-[6-(4-methyl-1-piperazinyl)-1H-benzimidazol-2-yl]-2(1H)-quinolinone 2-hydroxypropanoate hydrate (1:1:1);
| CAS No. | 405169-16-6 (free base), 915769-50-5, 804551-71-1 of lactate | ||
| TKI-258; CHIR-258. | |||
| Formula | C21H21FN6O.C3H6O3.H2O | ||
| Molecular Weight | 500.53 |
for treatment of cancer
Novartis Ag, innovator
Dovitinib lactate is the orally bioavailable lactate salt of a benzimidazole-quinolinone compound with potential antineoplastic activity. Dovitinib strongly binds to fibroblast growth factor receptor 3 (FGFR3) and inhibits its phosphorylation, which may result in the inhibition of tumor cell proliferation and the induction of tumor cell death. In addition, this agent may inhibit other members of the RTK superfamily, including the vascular endothelial growth factor receptor; fibroblast growth factor receptor 1; platelet-derived growth factor receptor type 3; FMS-like tyrosine kinase 3; stem cell factor receptor (c-KIT); and colony-stimulating factor receptor 1; this may result in an additional reduction in cellular proliferation and angiogenesis, and the induction of tumor cell apoptosis. The activation of FGFR3 is associated with cell proliferation and survival in certain cancer cell types
Dovitinib (TKI258) is a highly potent, novel multitargeted growth factor receptor kinase inhibitor with IC50 of 1, 2, 10, 8, 27, 36 nM for FLT3, c-KIT, VEGFR1/2/3, PDGFRß and CSF-1R, respectively. It shows both antitumor and antiangiogenic activities in vivo. [1] It potently inhibits FGFR3 with an inhibitory concentration of 50% (IC50) of 5 nM in in vitro kinase assays and selectively inhibited the growth of B9 cells and human myeloma cell lines expressing wild-type (WT) or activated mutant FGFR3. Antiproliferative activity of Dovitinib (TKI258) against MV4;11 was ~24-fold greater compared with RS4;11, indicating more potent inhibition against cells with constitutively activated FLT3 ITD. [2][3]
References on Dovitinib (TKI258)
- [1] Clin Cancer Res 2005;11:3633-3641
- [2] Blood 2005;105: 2941-2948
- [3] Clin Cancer Res 2005;11:5281-5291
Dovitinib lactate is an angiogenesis inhibitor in phase III clinical trials at Novartis for the treatment of refractory advanced/metastatic renal cell cancer. Early clinical trials are also under way at the company for the oral treatment of several types of solid tumors, multiple myeloma and glioblastoma multiforme. Phase II trials are ongoing for the treatment of castration-resistant prostate cancer and for the treatment of Von-Hippel Lindau disease, for the treatment of non-small cell lung cancer (NSCLC) and for the treatment of colorectal cancer. Novartis and Seoul National University Hospital are conducting phase II clinical studies for the treatment of adenoid cystic carcinoma. Additional phase II clinical trials are ongoing at Asan Medical Center for the treatment of metastatic or advanced gastrointestinal stromal tumors (GIST). The University of Pennsylvania is conducting phase II clinical trials for the treatment of advanced malignant pheochromocytoma or paraganglioma. Phase II clinical studies are ongoing by Novartis for the treatment of advanced malignant pleural mesothelioma which has progressed following prior platinum-antifolate chemotherapy (DOVE-M) and for the oral treatment of hepatocellular carcinoma.
In 2009, Novartis discontinued development of dovitinib lactate for the treatment of acute myeloid leukemia (AML) based on the observation of time dependent drug accumulation. A phase I trial was also stopped for the same reason.
The drug candidate has been shown to inhibit multiple growth factor tyrosine kinases, including vascular endothelial growth factor receptor (VEGFR) tyrosine kinases VEGFR1 and VEGFR2, fibroblast growth factor receptor (FGFR) and platelet-derived growth factor receptor (PDGFR) tyrosine kinases. In previous studies, the benzimidazole-quinoline inhibited VEGF-mediated human microvascular endothelial cell (HMVEC) proliferation and demonstrated concentration-dependent antiangiogenic activity in in vitro assays, as well as potent antiproliferative activity against a subset of cancer cell lines.
In 2013, an orphan drug designation was assigned in the U.S. for the treatment of adenoid cystic carcinoma.
“Molecularly Targeted Agents for Renal Cell Carcinoma: The Next Generation”, C. Lance Cowey and Thomas E. Hutson -Clinical Advances in Hematology & Oncology, 2010, 8, 357.
Lee S. H.; Lopes de Menezes, D. Vora, J. Harris, A.; Ye, H. Nordahl, L.; Garrett, E.; Samara, E.; Aukerman, S. L.; Gelb, A. B. Heise, C. In Vivo Target Modulation and Biological Activity of CHIR-258, a Multitargeted Growth Factor Receptor Kinase Inhibitor, in Colon Cancer Models. Clin. Cancer Res. 2005, 11 (10), 3633–3641.
Lopes de Menezes, D. E.; Peng, J.; Garrett, E. N.; Louie, S. G.; Lee, S. H.; Wiesmann, M.; Tang, Y.; Shephard, L.; Goldbeck, C.; Oei, Y.; Ye, H.; Aukerman, S. L.; Heise, C. CHIR-258: A Potent Inhibitor of FLT3 Kinase in Experimental Tumor Xenograft Models of Human Acute Myelogenous Leukemia. Clin. Cancer Res. 2005, 11 (14), 5281–5291.
Trudel, S.; Li, Z. H.; Wei, E.; Wiesmann, M.; Chang, H.; Chen, C.; Reece, D.; Heise, C.; Stewart, A. K. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood 2005, 105 (7), 2941–2948.
Synthesis of Dovitinib
Tetrahedron Letters 47 (2006) 657–660
LHMDS mediated tandem acylation–cyclization of 2-aminobenzenecarbonitriles with 2-benzymidazol-2-yl acetates: a short and efficient route to the synthesis of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones
William R. Antonios-McCrea, Kelly A. Frazier, Elisa M. Jazan, Timothy D. Machajewski, Christopher M. McBride, Sabina Pecchi, Paul A. Renhowe, Cynthia M. Shafer and Clarke Taylor


cas 852433-84-2
…………………………..
852433-84-2
…………………………..
WO 2002022598 or https://www.google.com/patents/EP1317442A1?cl=en


………………………………
WO 2003087095 or http://www.google.fm/patents/US20030028018?cl=un

…………………..
WO 2005046589 or http://www.google.com/patents/EP1692085A2?cl=en
lactate salt of the compound of
Structure I or the tautomer thereof is administered to the subject and/or is used to prepare the medicament. [0062] In some embodiments, the compound of Structure I has the following formula

……………………
WO 2006125130
http://www.google.com/patents/WO2006125130A1?cl=en
formula IHB

HIB
Scheme 1



Example 4
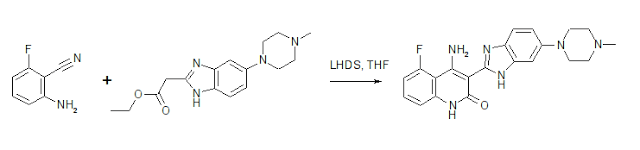
Synthesis of 4-Amino-5-fluoro-3-[6-(4-rnethyl-piperazin-1 -yl)-1 H-benzimidazol- 2-yl]-1 H-quinolin-2-one Procedure A

[0149] [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (250 g, 820 mmol) (dried with ethanol as described above) was dissolved in THF (3800 ml_) in a 5000 ml_ flask fitted with a condenser, mechanical stirrer, temperature probe, and purged with argon. 2-Amino-6-fluoro-benzonitrile (95.3 g, 700 mmol) was added to the solution, and the internal temperature was raised to 40°C. When all the solids had dissolved and the solution temperature had reached 4O0C, solid KHMDS (376.2 g, 1890 mmol) was added over a period of 5 minutes. When addition of the potassium base was complete, a heterogeneous yellow solution was obtained, and the internal temperature had risen to 62°C. After a period of 60 minutes, the internal temperature decreased back to 40°C, and the reaction was determined to be complete by HPLC (no starting material or uncyclized intermediate was present). The thick reaction mixture was then quenched by pouring it into H2O (6000 ml_) and stirring the resulting mixture until it had reached room temperature. The mixture was then filtered, and the filter pad was washed with water (1000 ml_ 2X). The bright yellow solid was placed in a drying tray and dried in a vacuum oven at 50°C overnight providing 155.3 g (47.9%) of the desired 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1 H-benzimidazol-2- yl]-1 H-quinolin-2-one.
Procedure B
[0150] A 5000 mL 4-neck jacketed flask was equipped with a distillation apparatus, a temperature probe, a N2 gas inlet, an addition funnel, and a mechanical stirrer. [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (173.0 g, 570 mmol) was charged into the reactor, and the reactor was purged with N2 for 15 minutes. Dry THF (2600 mL) was then charged into the flask with stirring. After all the solid had dissolved, solvent was removed by distillation (vacuum or atmospheric (the higher temperature helps to remove the water) using heat as necessary. After 1000 mL of solvent had been removed, distillation was stopped and the reaction was purged with N2. 1000 mL of dry THF was then added to the reaction vessel, and when all solid was dissolved, distillation (vacuum or atmospheric) was again conducted until another 1000 mL of solvent had been removed. This process of adding dry THF and solvent removal was repeated at least 4 times (on the 4thdistillation, 60% of the solvent is removed instead of just 40% as in the first 3 distillations) after which a 1 mL sample was removed for Karl Fischer analysis to determine water content. If the analysis showed that the sample contained less than 0.20% water, then reaction was continued as described in the next paragraph. However, if the analysis showed more than 0.20% water, then the drying process described above was continued until a water content of less than 0.20% was achieved.
[0151] After a water content of less than or about 0.20% was achieved using the procedure described in the previous paragraph, the distillation apparatus was replaced with a reflux condenser, and the reaction was charged with 2-amino-6- fluoro-benzonitrile (66.2 g, 470 mmol)( in some procedures 0.95 equivalents is used). The reaction was then heated to an internal temperature of 38-420C. When the internal temperature had reached 38-420C, KHMDS solution (1313 g, 1.32 mol, 20% KHMDS in THF) was added to the reaction via the addition funnel over a period of 5 minutes maintaining the internal temperature at about 38-50°C during the addition. When addition of the potassium base was complete, the reaction was stirred for 3.5 to 4.5 hours (in some examples it was stirred for 30 to 60 minutes and the reaction may be complete within that time) while maintaining the internal temperature at from 38-420C. A sample of the reaction was then removed and analyzed by HPLC. If the reaction was not complete, additional KHMDS solution was added to the flask over a period of 5 minutes and the reaction was stirred at 38-420C for 45-60 minutes (the amount of KHMDS solution added was determined by the following: If the IPC ratio is < 3.50, then 125 ml_ was added; if 10.0 >IPC ratio >3.50, then 56 mL was added; if 20.0 ≥IPC ratio >10, then 30 mL was added. The IPC ratio is equal to the area corresponding to 4-amino-5-fluoro-3-[6- (4-methyl-piperazin-1 -yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one) divided by the area corresponding to the uncyclized intermediate). Once the reaction was complete (IPC ratio > 20), the reactor was cooled to an internal temperature of 25- 300C, and water (350 mL) was charged into the reactor over a period of 15 minutes while maintaining the internal temperature at 25-35°C (in one alternative, the reaction is conducted at 400C and water is added within 5 minutes. The quicker quench reduces the amount of impurity that forms over time). The reflux condenser was then replaced with a distillation apparatus and solvent was removed by distillation (vacuum or atmospheric) using heat as required. After 1500 mL of solvent had been removed, distillation was discontinued and the reaction was purged with N2. Water (1660 mL) was then added to the reaction flask while maintaining the internal temperature at 20-300C. The reaction mixture was then stirred at 20-300C for 30 minutes before cooling it to an internal temperature of 5- 100C and then stirring for 1 hour. The resulting suspension was filtered, and the flask and filter cake were washed with water (3 x 650 mL). The solid thus obtained was dried to a constant weight under vacuum at 5O0C in a vacuum oven to provide 103.9 g (42.6% yield) of 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1H- benzimidazol-2-yl]-1H-quinolin-2-one as a yellow powder.
Procedure C

[0152] [6-(4-Methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-acetic acid ethyl ester (608 g, 2.01 mol) (dried) and 2-amino-6-fluoro-benzonitrile (274 g, 2.01 mol) were charged into a 4-neck 12 L flask seated on a heating mantle and fitted with a condenser, mechanical stirrer, gas inlet, and temperature probe. The reaction vessel was purged with N2, and toluene (7.7 L) was charged into the reaction mixture while it was stirred. The reaction vessel was again purged with N2 and maintained under N2. The internal temperature of the mixture was raised until a temperature of 630C (+/- 3°C) was achieved. The internal temperature of the mixture was maintained at 63°C (+/- 30C) while approximately 2.6 L of toluene was distilled from the flask under reduced pressure (380 +/- 10 torr, distilling head t = 40°C (+/- 1O0C) (Karl Fischer analysis was used to check the water content in the mixture. If the water content was greater than 0.03%, then another 2.6 L of toluene was added and distillation was repeated. This process was repeated until a water content of less than 0.03% was achieved). After a water content of less than 0.03% was reached, heating was discontinued, and the reaction was cooled under N2 to an internal temperature of 17-19°C. Potassium t-butoxide in THF (20% in THF; 3.39 kg, 6.04 moles potassium t-butoxide) was then added to the reaction under N2 at a rate such that the internal temperature of the reaction was kept below 20°C. After addition of the potassium t-butoxide was complete, the reaction was stirred at an internal temperature of less than 2O0C for 30 minutes. The temperature was then raised to 25°C, and the reaction was stirred for at least 1 hour. The temperature was then raised to 30°C, and the reaction was stirred for at least 30 minutes. The reaction was then monitored for completion using HPLC to check for consumption of the starting materials (typically in 2-3 hours, both starting materials were consumed (less than 0.5% by area % HPLC)). If the reaction was not complete after 2 hours, another 0.05 equivalents of potassium t-butoxide was added at a time, and the process was completed until HPLC showed that the reaction was complete. After the reaction was complete, 650 mL of water was added to the stirred reaction mixture. The reaction was then warmed to an internal temperature of 50°C and the THF was distilled away (about 3 L by volume) under reduced pressure from the reaction mixture. Water (2.6 L) was then added drop wise to the reaction mixture using an addition funnel. The mixture was then cooled to room temperature and stirred for at least 1 hour. The mixture was then filtered, and the filter cake was washed with water (1.2 L), with 70% ethanol (1.2 L), and with 95% ethanol (1.2 L). The bright yellow solid was placed in a drying tray and dried in a vacuum oven at 50°C until a constant weight was obtained providing 674 g (85.4%) of the desired 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1-yl)-1 H- benzimidazol-2-yl]-1 H-quinolin-2-one.
Preparation of Lactic Acid salt of 4-Amino-5-fluoro-3-[6-(4-methyl-piperazin-1- yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one

D,L-Lactic Acid

[0154] A 3000 ml_ 4-necked jacketed flask was fitted with a condenser, a temperature probe, a N2 gas inlet, and a mechanical stirrer. The reaction vessel was purged with N2 for at least 15 minutes and then charged with 4-amino-5-fluoro- 3-[6-(4-methyl-piperazin-1-yl)-1 H-benzimidazol-2-yl]-1 H-quinoiin-2-one (484 g, 1.23 mol). A solution of D,L-Lactic acid (243.3 g, 1.72 mol of monomer-see the following paragraph), water (339 mL), and ethanol (1211 mL) was prepared and then charged to the reaction flask. Stirring was initiated at a medium rate, and the reaction was heated to an internal temperature of 68-720C. The internal temperature of the reaction was maintained at 68-72°C for 15-45 minutes and then heating was discontinued. The resulting mixture was filtered through a 10-20 micron frit collecting the filtrate in a 12 L flask. The 12 L flask was equipped with an internal temperature probe, a reflux condenser, an addition funnel, a gas inlet an outlet, and an overhead stirrer. The filtrate was then stirred at a medium rate and heated to reflux (internal temperature of about 780C). While maintaining a gentle reflux, ethanol (3,596 mL) was charged to the flask over a period of about 20 minutes. The reaction flask was then cooled to an internal temperature ranging from about 64-700C within 15-25 minutes and this temperature was maintained for a period of about 30 minutes. The reactor was inspected for crystals. If no crystals were present, then crystals of the lactic acid salt of 4-amino-5-fluoro-3-[6-(4-methyl- piperazin-1-yl)-1 H-benzimidazol-2-yl]-1 H-quinolin-2-one (484 mg, 0.1 mole %) were added to the flask, and the reaction was stirred at 64-7O0C for 30 minutes before again inspecting the flask for crystals.
[0155] Once crystals were present, stirring was reduced to a low rate and the reaction was stirred at 64-700C for an additional 90 minutes. The reaction was then cooled to about 00C over a period of about 2 hours, and the resulting mixture was filtered through a 25-50 micron fritted filter. The reactor was washed with ethanol (484 ml_) and stirred until the internal temperature was about 00C. The cold ethanol was used to wash the filter cake, and this procedure was repeated 2 more times. The collected solid was dried to a constant weight at 500C under vacuum in a vacuum oven yielding 510.7 g (85.7%) of the crystalline yellow lactic acid salt of 4-amino-5-fluoro-3-[6-(4-methyl-piperazin-1 -yl)-1 H-benzimidazol-2-yl]-1 H-quinolin- 2-one. A rubber dam or inert conditions were typically used during the filtration process. While the dry solid did not appear to be very hygroscopic, the wet filter cake tends to pick up water and become sticky. Precautions were taken to avoid prolonged exposure of the wet filter cake to the atmosphere.
[0156] Commercial lactic acid generally contains about 8-12% w/w water, and contains dimers and trimers in addition to the monomeric lactic acid. The mole ratio of lactic acid dimer to monomer is generally about 1.0:4.7. Commercial grade lactic acid may be used in the process described in the preceding paragraph as the monolactate salt preferentially precipitates from the reaction mixture.
[0157] It should be understood that the organic compounds according to the invention may exhibit the phenomenon of tautomerism. As the chemical structures within this specification can only represent one of the possible tautomeric forms at a time, it should be understood that the invention encompasses any tautomeric form of the drawn structure. For example, the compound having the formula NIB is shown below with one tautomer, Tautomer INBa:

HIB

Tautomer HIBa
Other tautomers of the compound having the formula NIB, Tautomer INlBb and Tautomer IHBc, are shown below:

Tautomer IIIBb

Tautomer IIIBc
……………………….
WO 2006127926
……………………..
Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: A novel class of receptor tyrosine kinase inhibitors
J Med Chem 2009, 52(2): 278
………………………………
WO 2003087095
…………………..
WO 2005046589
……………………
WO 2006125130
……………………….
WO 2006127926
……………………..
Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: A novel class of receptor tyrosine kinase inhibitors
J Med Chem 2009, 52(2): 278
Qingqi Chen’s Book……”Anticancer Drug Research Guide.” New drugs in development for cancers

Chen Qingqi,
http://chen.medkoo.com/Anticancer-NewDrug.htm
Qingqi Chen’s Book
“Anticancer Drug Research Guide.”
New drugs in development for cancers

Ezatiostat……….designed to stimulate the production of blood cells in the bone marrow

Ezatiostat
168682-53-9 (Ezatiostat); 286942-97-0 (Ezatiostat HCl salt)
gamma-Glu-S-BzCys-PhGly diethyl ester

Ezatiostat hydrochloride
Target: glutathione S-transferase P1-1 (GSTP1-1) inhibitor
Pathway: hsa00480 Glutathione metabolism
Activity: Treatment of disorders of bone marrow cellular growth and differentiation
see http://www.ncbi.nlm.nih.gov/pubmed?term=TLK-199&cmd=search
| Telik, Inc. |
innovator
TLK199; TLK-199; TLK 199; Brand name: TELINTRA®。ethyl (2R)-[(4S)-4-amino-5-ethoxy-5-oxopentanoyl]-S-benzyl-L-cysteinyl-2- phenylglycinate.
ethyl (2S)-2-amino-5-[[(2R)-3-benzylsulfanyl-1-[[(1R)-2-ethoxy-2-oxo-1-phenylethyl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoate.
IUPAC/Chemical name:
(S)-ethyl 2-amino-5-(((R)-3-(benzylthio)-1-(((S)-2-ethoxy-2-oxo-1-phenylethyl)amino)-1-oxopropan-2-yl)amino)-5-oxopentanoate
C27H35N3O6S
Exact Mass: 529.2246
nmr.http://www.medkoo.com/Product-Data/Ezatiostat/ezatiostat-QC-CRB40225web.pdf
Telintra is a small molecule product candidate designed to stimulate the production of blood cells in the bone marrow. Many conditions are characterized by depleted bone marrow, including myelodysplastic syndrome, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the 3 major blood elements (white blood cells, red blood cells and platelets). A reduction in blood cell levels is also a common, toxic effect of many standard chemotherapeutic drugs.
Ezatiostat is a liposomal small-molecule glutathione analog inhibitor of glutathione S-transferase (GST) P1-1 with hematopoiesis-stimulating activity. After intracellular de-esterification, the active form of ezatiostat binds to and inhibits GST P1-1, thereby restoring Jun kinase and MAPK pathway activities and promoting MAPK-mediated cellular proliferation and differentiation pathways. This agent promotes the proliferation and maturation of hematopoietic precursor cells, granulocytes, monocytes, erythrocytes and platelets
Phase II trial myelodysplastic syndrome (MDS): Cancer. 2012 Apr 15;118(8):2138-47.
Phase I trial myelodysplastic syndrome (MDS): J Hematol Oncol. 2012 Apr 30;5:18. doi: 10.1186/1756-8722-5-18; Blood. 2009 Jun 25;113(26):6533-40; J Hematol Oncol. 2009 May 13;2:20.
Ezatiostat hydrochloride is the hydrochloride acid addition salt of ezatiostat. Ezatiostat, also known as TLK199 or TER 199, is a compound of the formula:

Ezatiostat has been shown to induce the differentiation of HL-60 promyelocyte leukemia cells in vitro, to potentiate the activity of cytotoxic agents both in vitro and in vivo, and to stimulate colony formation of all three lineages of hematopoietic progenitor cells in normal human peripheral blood.
In preclinical testing, ezatiostat has been shown to increase white blood cell production in normal animals, as well as in animals in which white blood cells were depleted by treatment with cisplatin or fluorouracil. Similar effects may provide a new approach to treating myelodysplastic syndrome (MDS).
Many conditions, including MDS, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the three major blood elements (white blood cells, red blood cells, and platelets), are characterized by depleted bone marrow. Myelosuppression, which is characterized by a reduction in blood cell levels and in a reduction of new blood cell generation in the bone marrow, is also a common, toxic effect of many standard chemotherapeutic drugs.
Ezatiostat hydrochloride in a liposomal injectable formulation was studied in a clinical trial for the treatment of MDS, and results from this trial, reported by Raza et al., J Hem. One, 2:20 (published online 13 May 2009), demonstrated that administration of TLK199 was well tolerated and resulted in multi-lineage hematologic improvement.
Ezatiostat hydrochloride in a tablet formulation has been evaluated in a clinical trial for the treatment of MDS, as reported by Raza et al., Blood, 113:6533-6540 (prepublished online 27 April 2009) and a single-patient report by Quddus et al., J Hem. One, 3:16 (published online 23 April 2010), and is currently being evaluated in clinical trials for the treatment of MDS and for severe chronic idiopathic neutropenia.
When used for treating humans, it is important that a crystalline therapeutic agent like ezatiostat hydrochloride retains its polymorphic and chemical stability, solubility, and other physicochemical properties over time and among various manufactured batches of the agent. If the physicochemical properties vary with time and among batches, the administration of a therapeutically effective dose becomes problematic and may lead to toxic side effects or to ineffective therapy, particularly if a given polymorph decomposes prior to use, to a less active, inactive, or toxic compound.
Therefore, it is important to choose a form of the crystalline agent that is stable, is manufactured reproducibly, and has physicochemical properties favorable for its use as a therapeutic agent.
Ezatiostat hydrochloride (USAN) has the molecular weight of 566.1, the trademark of Telintra®, and the CAS registry number of 286942-97-0. Ezatiostat hydrochloride has been evaluated for the treatment of myelodysplastic syndrome (MDS), in a Phase I-IIa study using a liposomal formulation (U.S. Pat. No. 7,029,695), as reported at the 2005 Annual Meeting of the American Society for Hematology (Abstract #2250) and by Raza et al. in Journal of Hematology & Oncology, 2:20 (published online on 13 May 2009); and in a Phase I study using a tablet formulation, as reported at the 2007 Annual Meeting of the American Society for Hematology (Abstract #1454) and by Raza et al. in Blood, 113:6533-6540 (prepublished online on 27 Apr. 2009), and in a single patient case report by Quddus et al. in Journal of Hematology & Oncology, 3:16 (published online on 23 Apr. 2010).
…………………………………………………………………
http://www.google.com/patents/US20110301376
Preparation of Ezatiostat Hydrochloride
In another aspect, this invention provides a process comprising the steps of contacting a compound of formula:

or a salt thereof with a compound of formula:

or a salt thereof and an activating agent under conditions which provide a compound of formula:

In one embodiment, the process further comprises deprotecting the compound of formula:

under conditions which provide a compound of formula:

or a salt thereof. In another embodiment, the compound provided is ezatiostat hydrochloride.
In another aspect, this invention provides a process comprising contacting a compound of formula:

or a salt thereof with an ethylating agent under conditions which provide a compound of formula:

In another embodiment, the process further comprises debenzylating the compound of formula:

under conditions which provide a compound of formula:

or a salt thereof.
In another aspect, this invention provides a process comprising the steps of contacting a compound of formula:

or a salt thereof having a t-butoxycarbonyl group with an activating agent and a compound of formula:

or a salt thereof under conditions which provide a compound of formula:

In another embodiment, the process further comprises deprotecting the tertiarybutyloxycarboyl (Boc) group under conditions to provide a compound of formula:

or a salt thereof.
Certain preferred embodiments of this invention are illustrated in the reaction scheme and described below. In the peptide coupling the amino acid reagents are used generally at a 1:1 molar ratio, and the activating reagent (isobutyl chloroformate) and the base (NMM) are used in slight excess over the amino acid reagents; while in the esterification of the N-BOC-L-glutamic acid γ-benzyl ester the esterifying agent (diethyl sulfate) and base are used in about 1.4-fold excess.

EXAMPLESAs relevant and unless otherwise noted, all operations were conducted under nitrogen purge and with stirring. Water was osmosis purified, and solvents were filtered. Unless otherwise stated, all temperatures are in degrees Celcius (° C.) and the following abbreviations have the following definitions:Et EthylHCl(g) HCl gasN-BOC or N-Boc N-tertiarybutyloxycarbonylL LiterKg KilogramNMM N-methylmorpholineMol Molew/w weight by weightExample 1Preparation of S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3)Without stirring, 45.1 Kg N-BOC-S-benzyl-L-cysteine (1) was added to a 600 L jacketed glass-lined reactor, followed by 45 L ethyl acetate. Stirring was started and the temperature was reduced to 13° C. NMM, 15.3 Kg, was added over 50 minutes, and rinsed in with 6 L ethyl acetate, and stirring stopped. Ethyl acetate, 315 L, was added to an 800 L cooled jacketed glass-lined reactor, followed by 20.7 Kg isobutyl chloroformate, rinsed in with 11 L ethyl acetate, and the mixture cooled to −10° C. The N-BOC-S-benzyl-L-cysteine NMM salt solution was added to the 800 L reactor over 5 hours, its reactor rinsed with 11 L ethyl acetate, and the rinse solution added to the 800 L reactor, while maintaining the temperature at (−10˜−7)° C. D-Phenylglycine ethyl ester hydrochloride, 31.2 Kg, was added in 8 portions over 50 minutes, followed by 15.3 Kg NMM in 8 portions over 1.3 hours, rinsed in with 2×5 L portions of ethyl acetate, allowing the mixture to warm to −1° C. by the end of the addition. The mixture was gradually warmed to 1° C. for 30 minutes, then to 20° C. over 2 hours, and maintained at (20˜25)° C. for 5 hours. The reaction mixture was washed twice with water: the first time adding 66 L water, stirring at room temperature for 40 minutes, allowing the phases to separate for 30 minutes, then removing the aqueous phase; the second time adding 68 L water, bringing the pH to 1.9 with the addition of 0.45 L 36% hydrochloric acid, stirring at room temperature for 35 minutes, allowing the phases to separate for 1 hour, then removing the aqueous phase. The organic phase was then heated to 38° C., and the pressure reduced to about 0.25 bar until no further gas was released, then to about (0.07-0.1) bar and solvents removed by distillation until 266 L of distillate had been removed. Four cycles of addition of 45 L ethyl acetate and removal of 45 L solvent by distillation were performed, and the water content of the remaining mixture was checked to ensure that it was below 0.1%. With the mixture at 36° C., 194 L heptanes was added, maintaining the temperature about 36° C., and held at that temperature for 2.3 hours. A further 194 L heptanes was added, allowing the temperature to cool to 30° C., and the temperature then reduced to −1° C. over 2.3 hours and then to −5° C. over 1 hour, and N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester recovered by filtration, washing twice with 30 L each of heptanes at −5° C., giving 85 Kg (63 Kg dry basis) N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester. Without stirring, the damp N-BOC-S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester was loaded into an 800 L jacketed glass-lined reactor, followed by 257 L ethyl acetate. Stirring was started and the temperature brought to 22° C., then the nitrogen purge stopped and 12.2 Kg hydrogen chloride gas was added through an immersion tube over 1.8 hours, allowing the temperature to increase to 38° C. The temperature was increased to 41° C., and the mixture held at that temperature for 9 hours. About 280 L of solvents were removed by distillation at that temperature and a pressure of (0.2˜0.1) bar over about 2 hours. Two cycles of addition of ethyl acetate and removal of solvent by distillation were performed, using 52 L in the first cycle and 77 L in the second cycle, and the viscous solution of S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3) in ethyl acetate, 148 Kg, was cooled to room temperature and filtered into a storage drum.Example 2Preparation of N-BOC-L-glutamic acid α-ethyl ester (6)Without stirring, 41 Kg N-BOC-L-glutamic acid γ-benzyl ester (4) was added to an 800 L jacketed glass-lined reactor, followed by 2.5 L water and 123 L ethyl acetate. The mixture was then stirred until the N-BOC-L-glutamic acid γ-benzyl ester completely dissolved, keeping the temperature below 15° C. Potassium carbonate fine powder, 23.4 Kg, was added in five batches, and the mixture then heated to 55° C. and maintained at that temperature for 40 minutes, giving a heterogeneous and completely fluid mixture. Diethyl sulfate, 26.2 Kg, was added over 2 hours, and rinsed in with 5 L ethyl acetate, with the temperature remaining at about 52° C. The nitrogen purge was stopped and a solution of 20 Kg ammonium chloride in 73 L water at room temperature added over 2 hours to the mixture, maintaining the temperature near 50° C., then rinsing in with 10 L water. Nitrogen purging was resumed, and the mixture was maintained at about 50° C. for 3 hours, then lowered to about 45° C., the stirring stopped, the phases allowed to separate for 30 minutes, and the lower, aqueous, phase removed. The organic phase, containing N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), was washed three times with water, each time adding 41 L water, stirring at room temperature for 30 minutes, allowing the phases to separate for 30 minutes, then removing the aqueous phase. The organic phase was heated to 35° C., and the pressure reduced, starting at about 0.2 bar and reducing as necessary until 82 Kg solvent had been removed by distillation, leaving about (70˜80) L of slightly opalescent solution. This solution was heated to 53° C., and 102 L heptanes was added, maintaining the same temperature. The solution was then filtered, rinsing with a further 13 L heptanes, then cooled to 32° C. to cause crystallization and maintained at that temperature for 1 hour. A further 66 L heptanes was added, and the mixture cooled to 22° C. and held for 1 hour, then cooled to −5° C. and held for another 1 hour. The mixture was then filtered to isolate the N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), which was washed twice, each time with 25 L heptanes cooled to (−5˜0)° C., and dried under vacuum at 40° C., giving 39.3 Kg N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5).A 4000 L hydrogenator was purged with nitrogen, then under nitrogen sweep and no stirring loaded with 39.2 Kg N-BOC-L-glutamic acid γ-benzyl α-ethyl ester (5), 2.0 Kg 5% palladium on carbon, and 432 L ethyl acetate, and purged (3 bar) and decompressed (0.2 bar) twice with nitrogen and twice with hydrogen. Stirring was begun and the mixture heated to (37±2)° C., hydrogenated at that temperature under 2.8 bar hydrogen pressure until no further hydrogen absorption occurred, then held under 2.8 bar hydrogen pressure for 12 hours. Completion of hydrogenation was confirmed by thin-layer chromatography of a sample. The mixture was cooled to 28° C., the hydrogen purged from the hydrogenator, and the hydrogenator purged (2 bar) and decompressed (0.2 bar) twice with nitrogen. The mixture was filtered through a filter precoated with 10 Kg powdered cellulose in 200 L ethyl acetate, then the filter washed with the ethyl acetate used to form the precoat, giving a total of 626 Kg of a dilute ethyl acetate solution containing 29.5 Kg N-BOC-L-glutamic acid α-ethyl ester (6). This was distilled at (35˜40)° C. and (0.16˜0.18) bar to give 67 L of concentrated solution, then 29 L of ethyl acetate added and the solution redistilled to again give 67 L of concentrated solution.Example 3Preparation of Ezatiostat HydrochlorideThe concentrated solution of N-BOC-L-glutamic acid α-ethyl ester (6), 61.2 Kg (containing 27.8 Kg N-BOC-L-glutamic acid α-ethyl ester), was added to a 600 L jacketed glass-lined reactor, rinsed in with 5 L ethyl acetate, then cooled to 14° C. NMM, 10.8 Kg, was added over 50 minutes and rinsed in with 5 L ethyl acetate, then stirring stopped, giving an ethyl acetate solution of N-BOC-L-glutamic acid α-ethyl ester NMM salt. Ethyl acetate, 475 L, was added to a 1300 L cooled jacketed glass-lined reactor, followed by 14.5 Kg isobutyl chloroformate, rinsed in with 2×10 L ethyl acetate, and the mixture cooled to −11° C. The N-BOC-L-glutamic acid α-ethyl ester NMM salt solution was added to the 1300 L reactor over 1.3 hours, its reactor rinsed with 10 L ethyl acetate, and the rinse solution added to the 1300 L reactor, then stirred for an additional 30 minutes, while maintaining the temperature at about −13° C.S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride (3) in ethyl acetate, 112 Kg (containing 41.3 Kg S-benzyl-L-cysteinyl-D-phenylglycine ethyl ester hydrochloride) was added in 4 portions over 45 minutes, and rinsed in with 5 L ethyl acetate, followed by 10.8 Kg NMM in 8 portions over 1.3 hours, rinsed in with 2×5 L portions of ethyl acetate, allowing the mixture to warm to −4° C. by the end of the addition. The mixture was gradually warmed to 30° C. over 2 hours, and maintained at (30˜35)° C. for 2 hours. The reaction mixture was washed twice with water: the first time adding 100 L water, heating to 41° C., allowing the phases to separate for 30 minutes, then removing the aqueous phase; the second time adding 100 L water, bringing the pH to 2.0 with the addition of 0.8 L 36% hydrochloric acid, stirring at 43° C. for 30 minutes, allowing the phases to separate for 1 hour, then removing the aqueous phase. The organic phase was then heated to 42° C., and the pressure reduced to about 0.25 bar until no further gas was released and solvents removed by distillation until 495 L of distillate had been removed. Four cycles of addition of 120 L ethyl acetate and removal of 120 L solvent by distillation were performed, and the water content of the remaining mixture was checked to ensure that it was below 0.1%. With the mixture at 42° C., 610 L of ethyl acetate was added, maintaining the temperature about 41° C., then heating to 58° C. to ensure dissolution. The solution was filtered, rinsing the filter with 18 L ethyl acetate, and the solution allowed to cool to 22° C. The nitrogen purge was stopped and 22.2 Kg hydrogen chloride gas was added through an immersion tube over 2 hours, then the mixture held at that temperature for 2 hours. The mixture was heated to 31° C. over 1.5 hours, and held at about that temperature for 15.5 hours. Solvents were removed by distillation at 33° C. and a pressure of about 0.13 bar over about 1.5 hours to give a volume of concentrated solution of about 630 L. Ethyl acetate, 100 L, was added, and the mixture cooled to 25° C. and held at that temperature for 30 minutes. The crude ezatiostat hydrochloride was recovered by filtration and washed with 30 L ethyl acetate, giving 113 Kg damp crude ezatiostat hydrochloride, which was dried at 40° C. under vacuum for 24 hours to give 52.8 Kg dry crude ezatiostat hydrochloride.Example 4Crystallization of Ezatiostat Hydrochloride to Form Pure Crystalline Ezatiostat Hydrochloride Ansolvate Form D61.5 Kg crude ezatiostat hydrochloride was added to a reactor at room temperature, followed by 399 liter (L) ethanol, and this mixture was heated to 68° C. to completely dissolve the ezatiostat hydrochloride, filtered, then allowed to cool to 65° C. and checked for clarity and the absence of crystallization. About 1.3 Kg of ezatiostat hydrochloride ansolvate form D was suspended in 9 L of ethyl acetate, and about one-half of this suspension was added to the ethanol solution. The mixture was cooled to 63° C. and the second half of the suspension added to the mixture. The resulting mixture was cooled gradually to 45° C., 928 L ethyl acetate was added, and the mixture was cooled to 26° C. and held at about that temperature for about 5 hours, then cooled to −2° C. The mixture, containing crystalline ezatiostat hydrochloride ansolvate, was filtered, and the residue washed twice with 65 L of chilled (0-5° C.) ethyl acetate. The crystalline ezatiostat hydrochloride ansolvate was dried at 30° C. for 48 hours, then cooled to room temperature and sieved. Analysis of the material by DSC and XRPD confirmed its identity as crystalline ezatiostat hydrochloride ansolvate, and Karl Fischer analysis showed a water content of 0.1%.Example 5Purifying Ezatiostat Hydrochloride Crystals to Form Pure Crystalline Ezatiostat Hydrochloride Ansolvate Form DCrude ezatiostat hydrochloride, 51.4 Kg, was added to a 600 L jacketed glass-lined reactor at room temperature, followed by 334 L of ethanol. The mixture was heated to 68° C. to completely dissolve the ezatiostat hydrochloride. The resulting solution was filtered into a 1300 L jacketed glass-lined reactor, and an additional 27 L ethanol warmed to 66° C. used to rinse the first reactor into the second reactor through the filter. The resulting solution in the second reactor was cooled to 63° C. and checked for complete dissolution; then 4 L of a seeding suspension of crystalline ezatiostat hydrochloride ansolvate in ethyl acetate was added, and the mixture cooled to 60° C. The remaining 4 L of the seeding suspension was added, and the mixture cooled to 47° C. over 2 hours. The solids in the mixture were shown by DSC to contain more than one form of ezatiostat hydrochloride, so the stages of heating to dissolution, cooling, and adding seeding suspension (this time 2×2 L), were repeated, then the mixture cooled to 41° C. This time the solids in the mixture were confirmed by DSC to be crystalline ezatiostat hydrochloride ansolvate. Ethyl acetate, 776 L, was added, and the mixture was cooled to 25° C. over 1.3 hours and further to 20° C. over an additional 5 hours, then cooled to −3° C. The mixture, containing crystalline ezatiostat hydrochloride ansolvate, was filtered and the solids washed twice with 54 L each of chilled (−5˜0)° C. ethyl acetate. The damp solids of crystalline ezatiostat hydrochloride ansolvate, 70 Kg, were dried in a vacuum oven at 25° C. for 16 hours, 35° C. for 7 hours, then at room temperature for 1 hour, then sieved. The crystalline ezatiostat hydrochloride ansolvate, 44.2 Kg, had a loss on drying at 40° C. under vacuum for 2 hours of 0.09%, and a water content by Karl Fischer analysis of 0.09%.

……………………………………………………………
U.S. Pat. No. 5,763,570
https://www.google.com/patents/US5763570
……………………………..
http://www.google.com/patents/WO2011156025A1?cl=en
Example 1. Preparation Of Ezatiostat Hydrochloride Ansolvate By Slurrying
[0082] Ezatiostat hydrochloride monohydrate was added to methyl tert-butyl ether at room temperature in excess, so that undissolved solids were present. The mixture was then agitated in a sealed vial at room temperature for 4 days, and the solids were then isolated by suction filtration. XRPD analysis of the solids established that the isolated solids were ezatiostat hydrochloride ansolvate.
[0083] Ezatiostat hydrochloride monohydrate was added to hexanes at 60 °C in excess, so that undissolved solids were present. The mixture was then agitated in a sealed vial at 60 °C for 4 days, and the solids were then isolated by suction filtration. XRPD analysis of the solids established that the isolated solids were ezatiostat hydrochloride ansolvate.
Example 2. Preparation Of Crystalline Ezatiostat Hydrochloride Ansolvate By Heating
[0084] DSC of crystalline ezatiostat hydrochloride monohydrate showed the pattern in FIG. 1, as discussed in paragraph above. Hot stage microscopy showed an initial melt followed by a recrystallization at 153 °C and a final melt at 166 °C. VT-XRPD, where XRPD patterns were obtained at 28 °C, 90 °C, and 160 °C during heating, and 28 °C after cooling of the formerly heated material, showed the presence of ezatiostat hydrochloride monohydrate at 28 °C and 90 °C during heating and of crystalline ezatiostat hydrochloride ansolvate at 160 °C and 28 °C after cooling of the formerly heated material. This confirmed that the transition at around 153/156 °C was a conversion of ezatiostat hydrochloride monohydrate form A to crystalline ezatiostat hydrochloride ansolvate form D and that the final DSC endothermic peak at about 177 °C (166 °C in the hot stage microscopy) was due to the melting of crystalline ezatiostat hydrochloride ansolvate. This was further confirmed by XRPD of the TG-IR material, where XRPD patterns obtained at room temperature both before and after heating to about 160 °C showed that the material before heating was form A and that the material after heating was form D ansolvate. DSC of crystalline ezatiostat hydrochloride ansolvate prepared by recrystallization showed the pattern in FIG. 5, with only the endothermic peak at about
177 °C followed by a broad endotherm at about (205 – 215) °C. Accordingly, the presence of the DSC endothermic peak at about 177 °C, for example at (177±2) °C, when measured under the conditions described above, is considered characteristic of crystalline ezatiostat hydrochloride ansolvate, and the substantial absence of thermal events at temperatures below this is considered indicative of the absence of other forms of ezatiostat hydrochloride
………………………………..
http://www.google.com/patents/WO2013082462A1?cl=en
Ezatiostat, also known as TLK199 or TER 199, is a compound of the formula:

[0003] Ezatiostat has been shown to induce the differentiation of HL-60 promyelocytic leukemia cells in vitro, to potentiate the activity of cytotoxic agents both in vitro and in vivo, and to stimulate colony formation of all three lineages of hematopoietic progenitor cells in normal human peripheral blood. In preclinical testing, ezatiostat has been shown to increase white blood cell production in normal animals, as well as in animals in which white blood cells were depleted by treatment with cisplatin or fluorouracil. Similar effects may provide a new approach to treating myelodysplasia syndrome (MDS).
[0004] Many conditions, including MDS, a form of pre-leukemia in which the bone marrow produces insufficient levels of one or more of the three major blood elements (white blood cells, red blood cells, and platelets), are characterized by depleted bone marrow.
Myelosuppression, which is characterized by a reduction in blood cell levels and in a reduction of new blood cell generation in the bone marrow, is also a common, toxic effect of many standard chemotherapeutic drugs.
[0005] Ezatiostat hydrochloride is the hydrochloride acid addition salt of ezatiostat.
Ezatiostat hydrochloride in a liposomal injectable formulation was studied in a clinical trial for the treatment of MDS, and results from this trial, reported by Raza et al., J. Hem. One, 2:20 (published online 13 May 2009), demonstrated that administration of TLK199 was well tolerated and resulted in multi-lineage hematologic improvement. Ezatiostat hydrochloride in a tablet formulation has been evaluated in a clinical trial for the treatment of MDS, as reported by Raza et al, Blood, 113:6533-6540 (prepublished online 27 April 2009) and a single-patient report by Quddus et al, J. Hem. One, 3:16 (published online 23 April 2010), and is currently being evaluated in clinical trials for the treatment of MDS and for severe chronic idiopathic neutropenia.
…………………………………..
http://www.google.com/patents/WO2013082462A1?cl=en
Example 1
[0048] 80 mg of crystalline ezatiostat hydrochloride was placed in a round bottom flask and dissolved in 25 mL of methanol. The solvent was then evaporated on a rotary evaporation apparatus under reduced pressure at 30 °C. After 30 minutes, the solid sample was removed from the round bottom flask and stored in a sealed vial at 2 °C in a refrigerator. Analysis of this sample was carried out within 24 hours of removing it from the rotary evaporation apparatus.
[0049] The resulting amorphous material was analyzed by 1H NMR, 13C NMR, DSC, and X-Ray powder diffraction experiments. The DSC conditions were 30 to 300 °C at 10 °C/ min using 7 mg of the amorphous material. The X-Ray powder diffraction was taken at 0-60 of 2theta. The crystalline ezatiostat hydrochloride was also analyzed.
Phase II trials: Ezatiostat is the first GSTP1-1 inhibitor shown to cause clinically significant and sustained reduction in RBC transfusions, transfusion independence, and multilineage responses in MDS patients. The tolerability and activity profile of ezatiostat may offer a new treatment option for patients with MDS. (source: Cancer. 2012 Apr 15;118(8):2138-47.)
|
References |
1: Galili N, Tamayo P, Botvinnik OB, Mesirov JP, Brooks MR, Brown G, Raza A. Prediction of response to therapy with ezatiostat in lower risk myelodysplastic syndrome. J Hematol Oncol. 2012 May 6;5:20. doi: 10.1186/1756-8722-5-20. PubMed PMID: 22559819; PubMed Central PMCID: PMC3407785.
2: Raza A, Galili N, Mulford D, Smith SE, Brown GL, Steensma DP, Lyons RM, Boccia R, Sekeres MA, Garcia-Manero G, Mesa RA. Phase 1 dose-ranging study of ezatiostat hydrochloride in combination with lenalidomide in patients with non-deletion (5q) low to intermediate-1 risk myelodysplastic syndrome (MDS). J Hematol Oncol. 2012 Apr 30;5:18. doi: 10.1186/1756-8722-5-18. PubMed PMID: 22546242; PubMed Central PMCID: PMC3416694.
3: Lyons RM, Wilks ST, Young S, Brown GL. Oral ezatiostat HCl (Telintra®, TLK199) and idiopathic chronic neutropenia (ICN): a case report of complete response of a patient with G-CSF resistant ICN following treatment with ezatiostat, a glutathione S-transferase P1-1 (GSTP1-1) inhibitor. J Hematol Oncol. 2011 Nov 2;4:43. doi: 10.1186/1756-8722-4-43. PubMed PMID: 22047626; PubMed Central PMCID: PMC3235963.
4: Raza A, Galili N, Smith SE, Godwin J, Boccia RV, Myint H, Mahadevan D, Mulford D, Rarick M, Brown GL, Schaar D, Faderl S, Komrokji RS, List AF, Sekeres M. A phase 2 randomized multicenter study of 2 extended dosing schedules of oral ezatiostat in low to intermediate-1 risk myelodysplastic syndrome. Cancer. 2012 Apr 15;118(8):2138-47. doi: 10.1002/cncr.26469. Epub 2011 Sep 1. PubMed PMID: 21887679.
5: Quddus F, Clima J, Seedham H, Sajjad G, Galili N, Raza A. Oral Ezatiostat HCl (TLK199) and Myelodysplastic syndrome: a case report of sustained hematologic response following an abbreviated exposure. J Hematol Oncol. 2010 Apr 23;3:16. doi: 10.1186/1756-8722-3-16. PubMed PMID: 20416051; PubMed Central PMCID: PMC2873355.
6: Steensma DP. Novel therapies for myelodysplastic syndromes. Hematol Oncol Clin North Am. 2010 Apr;24(2):423-41. doi: 10.1016/j.hoc.2010.02.010. Review. PubMed PMID: 20359635.
7: D’Alò F, Greco M, Criscuolo M, Voso MT. New treatments for myelodysplastic syndromes. Mediterr J Hematol Infect Dis. 2010 Aug 11;2(2):e2010021. doi: 10.4084/MJHID.2010.021. PubMed PMID: 21415972; PubMed Central PMCID: PMC3033133.
8: Raza A, Galili N, Callander N, Ochoa L, Piro L, Emanuel P, Williams S, Burris H 3rd, Faderl S, Estrov Z, Curtin P, Larson RA, Keck JG, Jones M, Meng L, Brown GL. Phase 1-2a multicenter dose-escalation study of ezatiostat hydrochloride liposomes for injection (Telintra, TLK199), a novel glutathione analog prodrug in patients with myelodysplastic syndrome. J Hematol Oncol. 2009 May 13;2:20. doi: 10.1186/1756-8722-2-20. PubMed PMID: 19439093; PubMed Central PMCID: PMC2694211.
9: Raza A, Galili N, Smith S, Godwin J, Lancet J, Melchert M, Jones M, Keck JG, Meng L, Brown GL, List A. Phase 1 multicenter dose-escalation study of ezatiostat hydrochloride (TLK199 tablets), a novel glutathione analog prodrug, in patients with myelodysplastic syndrome. Blood. 2009 Jun 25;113(26):6533-40. doi: 10.1182/blood-2009-01-176032. Epub 2009 Apr 27. PubMed PMID: 19398716.
| WO2013082462A1 * | Nov 30, 2012 | Jun 6, 2013 | Telik, Inc. | Amorphous ezatiostat ansolvate |
| US20120251496 * | Mar 20, 2012 | Oct 4, 2012 | Telik, Inc. | Ezatiostat for treating multiple myeloma |
……….

Known Yes1 kinase inhibitors, dasatinib and saracatinib.
| Compound name and NCGC ID | Structure | Clinical phase | Known targets | Yes1 IC50 (nM) |
|---|---|---|---|---|
| Dasatinib (1) NCGC00181129 |
 |
Approved | Lyn, PDGFR, KIT, Lck, BTK, Bcr–Abl, Fyn, Yes1, c-Src |
0.5 (<1.0)a |
| Saracatinib (2) NCGC00241099 |
|
Phase II/III | c-Src, Bcr–Abl, Yes1, Lck | 6.2 (0.70)a |
| AEE-788 (3) NCGC00263149 |
 |
Phase I/II | EGFR, HER-2, VEGFR-2 | 17.5 (13.1)a |
| Dovitinib (4) NCGC00249685 |
|
Phase III | FGFR, EGFR, PDGFR, VEGFR-1,2 | 31 (1.4)a |
| DCC-2036 (5) NCGC00263172 |
 |
Phase I/II | Bcr–Abl, Tie-2, Lyn, FLT3, VEGFR-2 | 2.5 (1.5)a |
| SGI-1776 (6) NCGC00263186 |
 |
Discontinued | Pim-1, FLT3 | 2670 (240)a |
| AMG-Tie-2-1 (7) NCGC00263199 |
 |
Preclinical | Tie-2 | 8.7 (22.0)a |
| AZ-23 (8) NCGC00250381 |
 |
Preclinical | Trk | 39.1 (3.0)a |
| Dorsomorphin (9) NCGC00165869 |
 |
Preclinical | AMPK, BMPR, TGFβ Receptor | 195.9 (29.8)a |
| AZ-628 (10) NCGC00250380 |
 |
Preclinical | Raf Kinase B,C | 348.3 (51.2)a |
- http://www.sciencedirect.com/science/article/pii/S0960894X13006677
- Data in parentheses were gathered by Reaction Biology Corp. using a [γ-33P]-ATP radiolabeled enzyme activity assay at an ATP concentration of 10 μM (www.reactionbiology.com).
SEE COMPILATION ON SIMILAR COMPOUNDS AT …………..http://drugsynthesisint.blogspot.in/p/nostat-series.html
Pharma Trends: Global Medicine Spending to Pass $1 Trillion in 2014
http://msg-latam-sfb.blogspot.in/2013/12/pharma-trends-global-medicine-spending.html

Very First Human Trials Using Cannbis To Treat Brain Cancer Are Under Way

The picture to your left is showing immunofluorescence of the human glioma cell line. (View more pictures here)
A European based pharmaceutical company called GW Pharmaceuticals is set to commence its first phase of clinical trials for the treatment of Glioblastoma Multiforme (GBM). It’s a bio-pharmaceutical company focused on discovering, developing and commercializing novel therapeutics from its proprietary cannabinoid product platform.
According to the New England Journal of Medicine, GBM accounts for approximately 50% of the 22,500 new cases of brain cancer diagnosed in the United States alone each year.(1) Treatment with regards to brain cancer are very limited which makes the study of cannabis and its effect on brain tumors crucial.
