
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TEDIZOLID (torezolid)
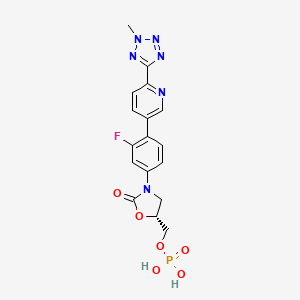
TEDIZOLID PHOSPHATE
[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxo-5-oxazolidinyl]methyl]phosphate,
DA 7157
THERAPEUTIC CLAIM Treatment of complicated skin and skin structure infections
CHEMICAL NAMES
1. 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5- [(phosphonooxy)methyl]-, (5R)-
2. [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxooxazolidin-5- yl]methyl hydrogen phosphate
http://www.ama-assn.org/resources/doc/usan/tedizolid-phosphate.pdf
MOLECULAR FORMULA C17H16FN6O6P
MOLECULAR WEIGHT 450.3
TRADEMARK None as yet
SPONSOR Trius Therapeutics
CODE DESIGNATION TR-701 FA
CAS REGISTRY NUMBER 856867-55-5
Note: This adoption statement supersedes the USAN torezolid phosphate (N09/81), which is hereby rescinded and replaced by the USAN tedizolid phosphate (N10/118).\
……………………………..
Tedizolid, 856866-72-3
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
(5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone,
TR 700
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701″, while the active ingredient is called “TR-700″.[4][5]

Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.
…………………………………………………….
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistance. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in the discovery of antibacterial agents.
Several antibacterial agents have been described in the prior art (for example, see PCT International Application Nos. PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178 and PCT/US2009/041200). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.
Various oxazolidinone-containing compounds have been disclosed for use asantibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog
active drug of Formula I is (5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone, i.e.,

These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010).
US Patent Publication No. 20070155798, recently disclosed a series of potently anti-bacterial oxazolidinones including

wherein R═H, PO(OH)2, and PO(ONa)2.
Cubist Announces Submission of New Drug Application for Investigational Antibiotic Tedizolid for Treatment of Serious Skin Infections
LEXINGTON, Mass.–(BUSINESS WIRE)– Cubist Pharmaceuticals, Inc. today announced that it has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of its investigational antibiotic tedizolid phosphate (TR-701). Cubist is seeking approval of tedizolid phosphate for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Tedizolid phosphate is a once daily oxazolidinone being developed for both intravenous (I.V.) and oral administration for the treatment of serious Gram-positive infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
http://www.drugs.com/nda/tedizolid_131023.html
…………………………………………………………..
Espinoza-González NA, Welsh O, de Torres NW, Cavazos-Rocha N, Ocampo-Candiani J, Said-Fernandez S, Lozano-Garza G, Choi SH, Vera-Cabrera L.
Molecules. 2008 Jan 11;13(1):31-40.
…………………………………………..
imp patents
|
12-3-2010
|
OXAZOLIDINONE CONTAINING DIMER COMPOUNDS, COMPOSITIONS AND METHODS TO MAKE AND USE
|
|
|
10-20-2010
|
Oxazolidinone derivatives
|
|
|
7-31-2009
|
NOVEL OXAZOLIDINONE DERIVATIVES
|
…………………………………………..
TEDIZOLID disodium salt
59 nos in
http://www.google.com/patents/US20130102523

 38 nos
38 nos
Tedizolid (formerly known as torezolid or TR-700) is the active hydroxymethyl oxazolidinone having the following formula:

Pharmaceutical prodrugs such as tedizolid phosphate (also referred to as TR-701, torezolid phosphate, and TR-701 “free acid” or FA) have the following formula:

The disodium salt of tedizolid phosphate, has the following structure:

Example 1 Preparation of the Phosphate Monohydrogen Diester, Formula III
In this and the following Examples, “Formula III” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00024
and M=OH.
A 1-L, three-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula Ia below (16.0 g, 0.0499 mol], THF (320 mL, 20 vol) and Et3N (21.9 g, 0.216 mol, 5.0 equiv.).
Figure US20100305069A1-20101202-C00025
POCl3 (3.31 g, 0.0216 mol, 0.5 equiv.) was added dropwise via syringe over 5 minutes. The reaction temperature was maintained below 25° C. The batch was aged for 16 hours at room temperature at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction vessel was then immersed in an ice-water bath and a 500-mL addition funnel charged with 320 mL of H2O was attached to the reaction vessel. When the temperature of the reaction reached 2.7° C., H2O was added drop wise over 30 minutes. The temperature of the reaction was maintained below 10° C. Upon completion of the H2O addition, the ice-water bath was removed and the batch was aged for 3 hours. The solution was transferred to a 2-L round-bottom flask and concentrated under reduced pressure on a rotary evaporator. After removal of most of the THF from the solution, the aqueous mixture was extracted with 5 1-L portions of CH2Cl2:MeOH (9:1). The CH2Cl2 layers were combined and concentrated to a dark oil. This crude material was purified on 200 g of silica gel, eluting with 10% MeOH/CH2Cl2 to 20% 2 N NH3 in MeOH/CH2Cl2. Fractions containing mostly the bis-ester (as judged by TLC Rf=0.3 eluting with 20% 2 N NH3 in MeOH/CH2Cl2) were combined and concentrated under reduced pressure on a rotary evaporator, during which time a white precipitate was observed. The flask containing the slurry was removed from the rotary evaporator and equipped with a magnetic stir bar and allowed to stir while cooling to room temperature over 3 hours, during which time the slurry thickened. The solid was filtered and dried in a vacuum oven at 45° C. for 16 hours to give 3.55 g of bis-ester as an off-white solid (20% yield). HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min. This reaction was repeated and the combined lots of the compound of Formula III (6.7 g) were slurried in 100 mL of MeOH (15 vol). The slurry was heated to 40° C. for 30 minutes and then allowed to cool to room temperature over 1 hour. The off-white solid was filtered and dried in a vacuum oven at 40° C. for 16 hours to give 6.15 g of the compound of Formula III (92% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min.
Example 2 Preparation of the Diphosphate Dihydrogen Diester, Formula IV
In Examples 2-5, “Formula IV” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00026
n=0 and M=O-imidazolium salt.
A 250-mL 3-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula IIa below (5.0 g, 11.1 mmol), carbonyldiimidazole (890 mg, 5.55 mmol, 0.5 equiv.) and DMF (100 mL, 20 vol).
Figure US20100305069A1-20101202-C00027
The suspension was heated to 50° C. and held at that temperature for 4 hours at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction was filtered at 50° C. and dried in a vacuum oven at 50° C. for 24 hours to give 5.15 g of the imidazolium salt (i.e., the compound of Formula IV) as an off-white solid (98% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 94.5% (AUC), tR=14.6 min.
TABLE 1
Method A (Waters XBridge C18 Column)
Time (min) Flow (mL/min) % A % B
0.0 1.0 98.0 2.0
15.0 1.0 5.0 95.0
25.0 1.0 5.0 95.0
27.0 1.0 98.0 2.0
30.0 1.0 98.0 2.0
A = 87% 25 mM ammonium bicarbonate solution in water/13% Acetonitrile
B = Acetonitrile
Wavelength = 300 nm
 disodium salt is TR 701
disodium salt is TR 701
……………………………………
Various oxazolidinone-containing compounds have been disclosed for use as antibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog,
These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010). The latter two applications are assigned to the same assignee as in the present application
………………………………………………………………………………………………………………………..
SYNTHESIS

DESCRIPTION OF COMPDS
10,
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)

………………………………………………………………………………………………………………………….
18
Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
………………………………………………………………………………………………………………………………………………………….
33
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
…………………………………………………………………………………………………………………………………………..
59
(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)

………………………………………………………………………………………………………………………………………………………
72
mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72)

COMPLETE SYNTHESIS
Example 5
Preparation of 2-cyano-5-bromopyridine
In 1 L of dimethylformamide was dissolved 100 g of 2,5-dibromopyridine, 32 g of cupper cyanide and 17.8 g of sodium cyanide were added to the solution at room temperature and the solution was stirred at the temperature of 150° C. for 7 hours for reaction. After being cooled to room temperature, the reaction mixture was added with water and extracted with ethyl acetate. The organic layer was washed with brine, dehydrated, filtered and concentrated in vacuo. The title compound 54 g was obtained. Yield 70%.
1HNMR(CDCl3) δ 8.76(s,1H), 7.98(dd,1H), 7.58(dd,1H)
Example 6
Preparation of 2-(tetrazol-5-yl)-5-bromopyridine
10 g of 2-cyano-5-bromopyridine prepared in the Preparation example 5 was dissolved in 100 ml of dimethylformamide, 5.33 g of sodiumazide, and 4.4 g of ammonium chloride were added to the solution at room temperature, and the solution was stirred at the temperature of 110° C. for 3 hours for reaction. The reaction mixture was added with water and then was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated and concentrated in vacuo thereby to obtain 10.5 g of the title compound. Yield 85%.
Preparation Example 7 Preparation of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 2-(2-methyltetrazol-5-yl)-5-bromopyridine
10.5 g of 2-(tetrazol-5-yl)-5-bromopyridine prepared in the Preparation example 6 was dissolved in 100 ml of dimethylformamide. And then 6.5 g of sodium hydroxide was added to the solution and 9.3 g of iodomethane was slowly added to the solution at the temperature of 0° C. The solution was stirred for 6 hours at room temperature, added with water, extracted with ethyl acetate. And then the organic layer was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to obtain 4 g of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 5 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine.
1) 2-(1-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.77(t,1H), 8.23(dd,1H), 8.04(dd,1H), 4.46(s,3H)
2) 2-(2-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.80(t,1H), 8.13(dd,1H), 7.98(dd,1H), 4.42(s,3H)
Example 1
Preparation of N-Carbobenzyloxy-3-fluoroaniline
3-fluoroaniline 100 g was dissolved in 1 L of tetrahydrofuran (THF) and the solution was added with 150 g (1.8 mol) of sodium bicarbonate (NaHCO3). After being cooled to 0° C., the solution was slowly added with 154 ml of N-carbobenzyloxy chloride (CbzCl) for reaction. While the temperature was maintained at 0° C., the reaction mixture was let to react for 2 hours with stirring. Afterwards, the reaction was extracted with 0.5 L of ethyl acetate. The organic layer, after being separated, was washed with brine, dried over anhydrous magnesium sulfate (MgSO4) and concentrated in vacuo. The residue was washed twice with n-hexane to afford the title compound as white crystal. 132 g. Yield 85%.
Example 2
Preparation of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
132 g of N-carbobenzyloxy-3-fluoroaniline 132 g prepared in the Preparation example 1 was dissolved in 1.3 L of tetrahydrofuran and the solution was cooled to −78° C. 370 ml of n-buthyllitium (n-BuLi, 1.6M/n-hexane) was slowly added to the solution in a nitrogen atmosphere, followed by stirring for 10 min. And 84 ml of (R)-(−)-glycidylbuthylate was slowly added to the reaction mixture, stirred at the same temperature for 2 hours and allowed to react for 24 hours at room temperature. After completion of the reaction, the solution was added with ammonium chloride (HH4Cl) solution and extracted with 0.5 L of ethyl acetate at room temperature. The organic layer, thus separated, was washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was dissolved in 100 ml of ethyl acetate and washed with n-hexane to give white crystals, which were purified to the title compound. 80 g. Yield 70%.
1H NMR (DMSO-d6) δ 7.85(t,1H), 7.58(dd,1H), 7.23(dd,1H), 4.69(m,1H), 4.02 (t,1H), 3.80(dd,1H), 3.60(br dd,2H).
Example 3
Preparation of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 300 ml of acetonitryl was dissolved 30 g of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 2, and 46 g of trifluoroacetic acid silver salt (CF3COOAg) and 43 g of iodide were added to the solution. After being stirred for one day at room temperature, the solution was added with water and was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine and dehydrated. And then the residue was filtered, concentrated in vacuo and dried thereby to form the title compound 44 g. Yield 94%.
1H NMR (DMSO-d6) δ 7.77(t,1H), 7.56(dd,1H), 7.20(dd,1H), 5.20(m,1H), 4.70 (m,1H), 4.07(t,1H), 3.80(m,1H), 3.67(m,2H), 3.56(m,3H)
Example 4
Preparation of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 660 ml of 1,4-dioxan was dissolved 50 g of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 3, 52 g of hexabutylditin ((Bu3Sn)2) and 9.3 g of dichlorobistriphenylphosphinpalladium were added into the solution, and stirred for 2 hours. The solution was filtered using celite and concentrated in vacuo. The residue was purified by column chromatography and 45 g of the title compound was formed.
1H NMR (DMSO-d6) δ 7.74(m,3H), 5.20(t,1H), 4.71(m,1H), 4.08(t,1H), 3.82(dd,1H), 3.68(m,1H), 3.52(m,1H), 1.48(m, 6H), 1.24(m, 6H), 1.06(m,6H), 0.83(t,9H)
COMPD 10
Example 1 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)
In 150 ml of 1-methyl-2-pyrrolidone was dissolved 37 g of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine, 10.44 g of lithium chloride and 2.9 g of dichlorobistriphenylphospine palladium(II) at room temperature and then stirred at the temperature of 120° C. for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8 g of the title compound. Yield 26%.
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.70(m,2H), 7.49(dd,1H), 5.25(t,1H), 4.74(m,1H), 4.46(s,3H), 4.14(t,1H), 3.88(dd,1H), 3.68(m,1H), 3.58 (m,1H)
COMPD 18
Example 28 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
In 5 ml of methylenchloride was dissolved 100 mg of the compound 10. The solution was added with 43 mg of diethylaminosulfurtrifloride (DAST) and 0.078 ml of triethylamine and then stirred for 24 hours. After being concentrating, the reaction mixture was purified by column chromatography to obtain the title compound 75 mg. Yield 75%.
1H NMR (DMSO-d6) δ 8.91(s,1H), 8.19(m,2H), 7.74(t,1H), 7.66(dd,1H) 7.49 (dd,1H), 5.06(m,1H), 4.89(m,2H), 4.46(s,3H), 4.23(t,1H), 3.95(dd,1H)
COMPD 33
Example 37 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
In 10 ml of methanol was dissolved 400 mg of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methansulfonyloxymethyl oxazolidin-2-on prepared in the secondary step of the Example 24. The solution was added with 90 mg of sodium methoxide at room temperature and then stirred for one day at room temperature. The solution was extracted with ethyl acetate and the organic layer, thus separated, was washed with water and brine. The organic layer was dehydrated, filtered, concentrated in vacuo and purified by column chromatography to provide the title compound 200 mg. Yield 58%.
1H NMR(CDCl3) δ 8.90(s,1H), 8.29(d,1H), 8.04(d,1H), 7.61(dd,1H), 7.58 (t,1H), 7.38(dd,1H), 4.80(m,1H), 4.45(s,3H), 4.08(t,1H), 3.96(dd,1H), 3.67 (m,2H), 3.43(s,3H)
COMPD 59
Example 58 Preparation of mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) and (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)
1. The Primary Step
In 10 ml of mixture solvent (tetrahydrofuran:methylenchloride=1:1) was dissolved 1 g of compound 10. The solution was added with 0.6 g of tetrazole and 2.3 g of di-tetrabutyl diisoprophylphosphoamidite and stirred for 15 hours at room temperature. The reaction mixture was refrigerated to −78° C., added with 0.7 g of metachloroperbenzoic acid and stirred for 2 hours. After being cooling to −78° C., the reaction mixture was added with metachloroperbenzoic acid (0.7 g). When the reaction mixture was stirred for 2 hours, the temperature of the reaction mixture was raised to room temperature. The reaction mixture was then added with ethyl acetate. The organic layer, thus separated, was washed with sodium bisulfate, sodium bicarbonate and brine, dehydrated, filtered and concentrated in vacuo, followed by purification with column chromatography thereby to provide (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl phosphoric acid ditetrabuthylester (0.71 g, 71%).
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.74(t,1H), 7.68 (dd,1H), 7.49(dd,1H), 4.98(m,1H), 4.46(s,3H), 4.23(t,1H), 4.18(m,1H), 4.09(m,1H), 3.89 (dd,1H), 1.39(s,9H), 1.38(s,9H)
The crystal prepared the above method was dissolved in a mixture of methanol and chloroform. And then the solution added with 3.4 ml of sodium methoxide (0.3M methanol solution) at the room temperature and stirred for 10 hours. The reaction mixture was concentrated to prepare the residue. The residue was crystallized and filtered thereby to obtain the title compound (compound 59) 300 mg.
1H NMR (D2O) δ 8.27(s,1H), 7.56(dd,2H), 7.06(m,2H), 6.90(m,1H), 4.79 (m,1H), 4.63(s,3H), 3.90(m,4H)
COMPD 72
The Secondary Step
In 30 ml of methylenchloride was dissolved the compound (0.7 g) in the Primary Step. The solution was added with 15 ml of trifluoroacetic acid and then stirred for 1 hour at room temperature. The reaction mixture was concentrated in vacuo to prepare the residue. The residue was crystallized with ethanol and ethyl ether to obtain mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) 400 mg.
1H NMR (DMSO-d6) δ 8.92(s,1H), 8.20(m,2H), 7.74(t,1H), 7.66(dd,1H), 7.500(dd,1H), 4.95 (m,1H), 4.46(s,3H), 4.21(t,1H), 4.05(m,2H), 3.91(dd,1H)
………………………………………………………
IMPURITIES
| Organic Impurities in TR-701 FA Drug Substance | |
| Impurity | |
| ‘Name’ | Structure and Chemical Name |
| Rx600013 ‘Des-methyl TR- 701’ |
 |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(2H-1,2,3,4-tetrazol-5- | |
| yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5-yl)methyl | |
| phosphate | |
| Rx600024 ‘Pyrophosphate’ |
 |
| trihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl pyrophosphate | |
| Rx600014 ‘Ring opened’ |
 |
| dihydrogen 3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetraazol-5- | |
| yl)-3-pyridinyl]aniline}-2-hydroxypropyl phosphate | |
| Rx600023 ‘Me-isomer’ |
 |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl phosphate | |
| Rx600025 ‘Overalkylated- phosphorylated impurity’ |
  |
| (R)-1-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-3- | |
| hydroxypropan-2-yl dihydrogen phosphate; | |
| (R)-3-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-2- | |
| hydroxypropyl dihydrogen phosphate | |
| Rx600020 ‘Dimer impurity’ |
 |
| dihydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl- | |
| 2H-1,2,3,4-tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3- | |
| oxazolidin-5-yl]methyl pyrophosphate | |
| Rx600026 “Chloro” |
 |
| (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H- | |
| tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one | |
| Rx600001 TR-700 |
 |
| 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)- | |
| pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one | |
| Rx600022 ‘Bis phosphate’ |
 |
| hydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4- | |
| tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl | |
| phosphate | |
| Rx600042 |
 |
| 3-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-2-hydroxypropyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
| Rx600043 |
 |
| 2-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-1-hydroxyethyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
……………………………………………..
| US4128654 | 10 Feb 1978 | 5 Dec 1978 | E. I. Du Pont De Nemours And Company | 5-Halomethyl-3-phenyl-2-oxazolidinones |
| US4250318 | 9 Aug 1978 | 10 Feb 1981 | Delalande S.A. | Novel 5-hydroxymethyl oxazolidinones, the method of preparing them and their application in therapeutics |
| US4340606 | 23 Oct 1980 | 20 Jul 1982 | E. I. Du Pont De Nemours And Company | 3-(p-Alkylsulfonylphenyl)oxazolidinone derivatives as antibacterial agents |
| US4461773 | 5 Jan 1984 | 24 Jul 1984 | E. I. Dupont De Nemours And Company | P-Oxooxazolidinylbenzene compounds as antibacterial agents |
| US4476136 | 24 Feb 1982 | 9 Oct 1984 | Delalande S.A. | Aminomethyl-5 oxazolidinic derivatives and therapeutic use thereof |
| US4948801 | 29 Jul 1988 | 14 Aug 1990 | E. I. Du Pont De Nemours And Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| US5523403 | 22 May 1995 | 4 Jun 1996 | The Upjohn Company | Tropone-substituted phenyloxazolidinone antibacterial agents |
| US5565571 | 28 Apr 1994 | 15 Oct 1996 | The Upjohn Company | Substituted aryl- and heteroaryl-phenyloxazolidinones |
| US5652238 | 27 Sep 1994 | 29 Jul 1997 | Pharmacia & Upjohn Company | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| US5688792 | 16 Aug 1994 | 18 Nov 1997 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US6365751 | 17 Apr 2001 | 2 Apr 2002 | Zeneca Ltd. | Antibiotic oxazolidinone derivatives |
| US6627646 * | 17 Jul 2001 | 30 Sep 2003 | Sepracor Inc. | Norastemizole polymorphs |
| US6689779 | 18 May 2001 | 10 Feb 2004 | Dong A Pharm. Co., Ltd. | Oxazolidinone derivatives and a process for the preparation thereof |
| US7129259 | 1 Dec 2004 | 31 Oct 2006 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141583 | 23 Apr 2001 | 28 Nov 2006 | Astrazeneca Ab | Oxazolidinone derivatives with antibiotic activity |
| US7144911 | 24 Dec 2003 | 5 Dec 2006 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| US7202257 | 6 Jul 2004 | 10 Apr 2007 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US7396847 | 9 Sep 2002 | 8 Jul 2008 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7462633 | 29 Jun 2004 | 9 Dec 2008 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US7473699 | 25 Feb 2003 | 6 Jan 2009 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7498350 | 24 Nov 2003 | 3 Mar 2009 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US7816379 | 17 Dec 2004 | 19 Oct 2010 | Dong-A Pharm. Co., Ltd. | Oxazolidinone derivatives |
| US20020115669 | 29 Aug 2001 | 22 Aug 2002 | Wiedeman Paul E. | Oxazolidinone chemotherapeutic agents |
| US20030166620 | 18 May 2001 | 4 Sep 2003 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| US20040180906 | 24 Dec 2003 | 16 Sep 2004 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20050038092 | 29 Jun 2004 | 17 Feb 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20050107435 | 9 Sep 2002 | 19 May 2005 | Gravestock Michael B. | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US20050288286 | 6 Jul 2004 | 29 Dec 2005 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20060116386 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US20060116400 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline derivatives as antibacterial agents |
| US20060270637 | 24 Feb 2004 | 30 Nov 2006 | Astrazeneca Ab | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents |
| US20070155798 | 17 Dec 2004 | 5 Jul 2007 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20070185132 | 29 Jun 2004 | 9 Aug 2007 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereo |
| US20070191336 | 23 Dec 2004 | 16 Aug 2007 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070203187 | 22 Jan 2007 | 30 Aug 2007 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20070208062 | 24 May 2005 | 6 Sep 2007 | Astrazeneca Ab | 3-(4-(2-dihydroisoxazol-3-ylpyridin-5-yl)phenyl)-5-triazol-1-ylmethyloxazolidin-2-one derivatives as mao inhibitors for the treatment of bacterial infections |
| US20080021012 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-[4-{6-Substituted Alkanoyl Pyridin-3-Yl}-3-Phenyl]-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones As Antibacterial Agents |
| US20080021071 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-{4-(Pyridin-3-Yl) Phenyl}-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20080064689 | 24 May 2004 | 13 Mar 2008 | Astrazeneca Ab | 3-[4-(6-Pyridin-3-Yl)-3-Phenyl] -5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20090018123 | 19 Jun 2006 | 15 Jan 2009 | Milind D Sindkhedkar | Oxazolidinones Bearing Antimicrobial Activity Composition and Methods of Preparation |
| US20090192197 | 16 Sep 2008 | 30 Jul 2009 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20100093669 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| US20100227839 | 3 Feb 2010 | 9 Sep 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin- 5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| AU2004299413A1 | Title not available | |||
| AU2009200606A1 | Title not available | |||
| CA2549062A1 | 17 Dec 2004 | 30 Jun 2005 | Chong Hwan Cho | Novel oxazolidinone derivatives |
| CN101982468A | 17 Dec 2004 | 2 Mar 2011 | 东亚制药株式会社 | Novel oxazolidinone derivatives and pharmaceutical compositions comprising the derivatives |
| EP0312000A1 | 12 Oct 1988 | 19 Apr 1989 | The Du Pont Merck Pharmaceutical Company | Aminomethyl oxooxazolidinyl aroylbenzene derivatives useful as antibacterial agents |
| EP0352781A2 | 27 Jul 1989 | 31 Jan 1990 | The Du Pont Merck Pharmaceutical Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| EP1699784A1 | 17 Dec 2004 | 13 Sep 2006 | Dong-A Pharmaceutical Co., Ltd. | Novel oxazolidinone derivatives |
| EP2305657A2 | 17 Dec 2004 | 6 Apr 2011 | Dong-A Pharmaceutical Co., Ltd. | Oxazolidinone derivatives |
| EP2435051A1 | 27 May 2010 | 4 Apr 2012 | Trius Therapeutics | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
| IN236862A1 | Title not available | |||
| JPS5799576A | Title not available | |||
| KR20110071107A | Title not available | |||
| NZ547928A | Title not available | |||
| NZ575842A | Title not available | |||
| WO1993009103A1 | 5 Oct 1992 | 13 May 1993 | Upjohn Co | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| WO1993023384A1 | 21 Apr 1993 | 25 Nov 1993 | Michael Robert Barbachyn | Oxazolidinones containing a substituted diazine moiety and their use as antimicrobials |
| WO1995007271A1 | 16 Aug 1994 | 16 Mar 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO1995014684A1 | 27 Sep 1994 | 1 Jun 1995 | Michel R Barbachyn | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| WO2001094342A1 | 18 May 2001 | 13 Dec 2001 | Cho Jong Hwan | Novel oxazolidinone derivatives and a process for the preparation thereof |
| WO2002081470A1 | 3 Apr 2002 | 17 Oct 2002 | Astrazeneca Ab | Oxazolidinones containing a sulfonimid group as antibiotics |
| WO2003022824A1 | 9 Sep 2002 | 20 Mar 2003 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| WO2003035648A1 | 23 Oct 2002 | 1 May 2003 | Astrazeneca Ab | Aryl substituted oxazolidinones with antibacterial activity |
| WO2003047358A1 | 2 Dec 2002 | 12 Jun 2003 | Vaughan Leslie Crow | Cheese flavour ingredient and method of its production |
| WO2003072575A1 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring) methyl-oxazolidinone derivatives and their use as antibacterial agents |
| WO2003072576A2 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | Oxazolidinone derivatives, processes for their preparation, and pharmaceutical compositions containing them |
| WO2004048350A2 | 24 Nov 2003 | 10 Jun 2004 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| WO2004083205A1 | 16 Mar 2004 | 30 Sep 2004 | Astrazeneca Ab | Antibacterial 1, 3- oxazolidin -2- one derivatives |
| WO2005005398A2 | 29 Jun 2004 | 20 Jan 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| WO2005051933A1 | 23 Nov 2004 | 9 Jun 2005 | Vijay Kumar Kaul | An improved process for the synthesis of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester, a key intermediate for oxazolidinone antimicrobials and compounds prepared thereby |
| WO2005058886A1 | 17 Dec 2004 | 30 Jun 2005 | Dong A Pharm Co Ltd | Novel oxazolidinone derivatives |
| WO2005116017A1 | 24 May 2005 | 8 Dec 2005 | Astrazeneca Ab | Process for the preparation of aryl substituted oxazolidinones as intermediates for antibacterial agents |
| WO2006038100A1 | 6 Oct 2005 | 13 Apr 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007023507A2 | 19 Jun 2006 | 1 Mar 2007 | Milind D Sindkhedkar | Oxazolidinones bearing antimicrobial activity composition and methods of preparation |
| WO2007138381A2 | 13 Oct 2006 | 6 Dec 2007 | Delorme Daniel | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| WO2010042887A2 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| WO2010091131A1 | 3 Feb 2010 | 12 Aug 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| WO2010138649A1 | 27 May 2010 | 2 Dec 2010 | Trius Therapeutics, Inc. | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
……………………………………………………………………………………….


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007
Orphan Drugs: NPS Pharmaceuticals’ Natpara Receives EC Designation
NPS Pharmaceuticals, a New Jersey-based biopharmaceutical company developing treatments for rare diseases, announces January 3, 2014, that the European Commission (EC) grants Orphan Drug Designation (ODD) to Natpara (Recombinant Human Parathyroid Hormone (rhPTH[1-84]), for the treatment of Hypoparathyroidism. Natpara is a bioengineered replacement for endogenous Parathyroid Hormone (PTH) – “a hormone replacement therapy for the underlying cause of Hypoparathyroidism”.
Hypoparathyroidism is the decreased function of the parathyroid glands resulting in the under production of PTH. PTH’s functions are :
• Modulation of serum calcium and phosphate
• Regulation of renal excretion of phosphate and calcium
• Activation of Vitamin D
• Maintenance of normal bone turnover.
Natpara Regulatory Activities
• In 2007, receives FDA ODD Designation
• In October 2013, submission of US Biologic License Application (BLA) to FDA
• In January 2014, receives EC ODD.
NPS Pharmaceuticals also makes orphan drug Gattex, an injectable treatment for rare disease Short Bowel Syndrome…
View original post 33 more words
TOSEDOSTAT ….An aminopeptidase inhibitor with antineoplastic activity.

TOSEDOSTAT
An aminopeptidase inhibitor with antineoplastic activity.
- CHR 2797
- CHR-2797
- Tosedostat
- UNII-KZK563J2UW
- BB-76163Vernalis (Originator)
| CAS No. | 238750-77-1 |
| Chemical Name: | Tosedostat |
| Synonyms: | BB-76163;Chr-2797;tosedostat;CHR2797 (Tosedostat);Tosedostat (CHR2797);α-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]-benzeneaceticacidcyclopentlyester;alpha-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]benzeneacetic acid cyclopentyl ester;Benzeneacetic acid, alpha-(((2R)-2-((1S)-1-hydroxy-2-(hydroxyamino)-2-oxoethyl)-4-methyl-1-oxopentyl)amino)-, cyclopentyl ester, (alphas)- |
| Molecular Formula: | C21H30N2O6 |
| Formula Weight: | 406.47 |
CHR-2797 is an oral, once-daily experimental cancer therapy in phase II clinical development at Chroma Therapeutics for the oral treatment of refractory acute myeloid leukemia in elderly patients. It is also in early clinical development for the treatment of refractory solid tumors alone or in combination with chemotherapy.
No recent development has been reported for phase I/II studies evaluating CHR-2797 as monotherapy in hematologic/blood cancer. A phase I/II clinical trial of the compound in combination with erlotinib for non-small cell lung cancer was terminated in 2010 due to very poor recruitment of patients to the study.
Cell Therapeutics is also conducting phase II clinical trials of the compound for the treatment of myelodysplasia and acute myeloid leukemia.
CHR- 2797 is an inhibitor of aminopeptidases and has demonstrated strong preclinical efficacy as monotherapy in addition to demonstrating strong synergy with a number of leading cancer therapies in a range of cancer cells. It was originally licensed from Vernalis, where it was being evaluated for its potential in treating multiple sclerosis; however development in this indication has been discontinued.
In 2008, orphan drug designation was assigned to CHR-2797 in the U.S. for the treatment of acute myeloid leukemia. In 2011, the compound was licensed to Cell Therapeutics by Chroma Therapeutics in Central America, North America and South America for exclusive marketing and codevelopment for the oral treatment of blood-related cancers and other cancers.
In corporate news, biopharmaceutical company Cell Therapeutics, Inc. (CTIC) was up more than 6% and near 52 week highs after saying Thursday that the U.S. FDA has removed the partial clinical hold on tosedostat and all studies underway have been allowed to continue. Tosedostat is under development for the treatment of blood-related cancers. It is currently being studied in Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia and high-risk myelodysplastic syndromes.

Tosedostat is a proprietary orally bioavailable inhibitor of the M1 family of aminopeptidases with potential antineoplastic activity.
Tosedostat is converted intracellularly into a poorly membrane-permeable active metabolite (CHR-79888) which inhibits the M1 family of aminopeptidases, particularly puromycin-sensitive aminopeptidase (PuSA), and leukotriene A4 (LTA4) hydrolase; inhibition of these aminopeptidases in tumor cells may result in amino acid deprivation, inhibition of protein synthesis due to a decrease in the intracellular free amino acid pool, an increase in the level of the proapoptotic protein Noxa, and cell death.
Noxa is a member of the BH3 (Bcl-2 homology 3)-only subgroup of the proapoptotic Bcl-2 (B-cell CLL/lymphoma 2) protein family
Cell Therapeutics announced that it has received notification from the U.S. Food and Drug Administration (FDA) that the partial clinical hold on tosedostat (IND 075503) has been removed and all studies underway may continue. Tosedostat is a first-in-class selective inhibitor of aminopeptidases, which are required by tumor cells to provide amino acids necessary for growth and tumor cell survival, and is under development for the treatment of blood-related cancers.
Tosedostat is currently being studied in the United States and European Union in investigator-sponsored and cooperative group-sponsored Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia (AML) and high-risk myelodysplastic syndromes (MDS).
“We are pleased that the FDA has responded favorably to the tosedostat clinical trial data provided and removed the partial clinical hold to allow further development of tosedostat in ongoing and future studies,” said John Pagel, MD, PhD, Associate Member, Clinical Research Division, Fred Hutchinson Cancer Research Center; Associate Professor, Medical Oncology Division, University of Washington School of Medicine; and Principal Investigator in the tosedostat first-line AML/MDS trial.
Recently, WO 93/20047 disclosed a class of hydroxamic acid based MMP inhibitors which also are active in inhibiting TNF production.
As mentioned above, MMP inhibitors have been proposed with hydroxamic acid or carboxylic acid zinc binding groups. The following patent publications disclose hydroxamic acid-based MMP inhibitors:
US 4599361 (Searle) EP-A-0236872 (Roche) EP-A-0274453 (Bellon) WO 90/05716 (British Bio-technology) WO 90/05719 (British Bio-technology) WO 91/02716 (British Bio-technology) EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) EP-A-0497192 (Roche) WO 92/13831 (British Bio-technology) WO 92/17460 (SmithKline Beecham) WO 92/22523 – (Research Corporation Technologies) WO 93/09090 (Yamanouchi) WO 93/09097 (Sankyo) WO 93/20047 (British Bio-technology) WO 93/24449 (Celltech) WO 93/24475 (Celltech) EP-A-0574758 (Roche) The following patent publications disclose carboxylic acid-based MMP inhibitors:
EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) WO 93/24449 (Celltech) WO 93/24475 (Celltech)
|
|
|
TOSEDOSTAT
| WO1996033166A1 * | 17 Apr 1996 | 24 Oct 1996 | Du Pont Merck Pharma | Hydroxamic and carboxylic acids as metalloprotease inhibitors |
| WO1998011063A1 * | 8 Sep 1997 | 19 Mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| GB2268934A * | Title not available |
| US5652262 * | 14 mar 1994 | 29 lug 1997 | British Biotech Pharmaceutical, Ltd. | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| US5821262 * | 4 ott 1994 | 13 ott 1998 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as inhibitors of cytokine production |
| US5861436 * | 29 apr 1997 | 19 gen 1999 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| EP0423943A2 | 19 set 1990 | 24 apr 1991 | Beecham Group p.l.c. | Use of collagenase inhibitors in the treatment of demyelinating diseases, in particular multiple sclerosis |
| JPH03157372A | Titolo non disponibile | |||
| WO1997049674A1 | 20 giu 1997 | 31 dic 1997 | Francesca Abrate | Matrix metalloproteinase inhibitors |
| WO1998011063A1 | 8 set 1997 | 19 mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| WO1999040910A1 | 27 gen 1999 | 19 ago 1999 | Andrew Paul Ayscough | Anti-inflammatory agents |
| WO1999044602A1 | 5 mar 1999 | 10 set 1999 | British Biotech Pharm | Inflammatory cell inhibitors |
| WO1999046241A1 | 12 mar 1998 | 16 set 1999 | British Biotech Pharm | Cytostatic agents |
| WO2000044373A1 * | Jan 27, 2000 | Aug 3, 2000 | Raymond Paul Beckett | Antibacterial hydroxamic acid derivatives |
| US6545051 | Jan 27, 2000 | Apr 8, 2003 | British Biotech Pharmaceuticals, Ltd. | Antibacterial hydroxamic acid derivatives |
Drugs Fut 2009, 34(2): 115
PLoS One (2013), 8(2), e57641.
WO 1999046241
WO 1995019956
WO 1998011063
US 6462023
US 20100260674
WO 2000044373
WO 9940910
NMR
http://file.selleckchem.com/downloads/nmr/S152202-CHR-2797-NMR-Selleck.pdf
Anti-Metastatic and Anti-Invasive Agents Compounds which have the property of inhibiting the action of the metalioproteinase enzymes involved in connective tissue breakdown and remodelling, such as fibroblast collagenase (Type 1 ), PMN-collagenase, 72 kDa-gelatinase, 92 kDa- gelatinase, stromelysin, stromelysin-2 and PUMP-1 (known as “matrix metalloproteinases”, and herein referred to as MMPs) have been proposed and are being tested in the clinic for the treatment of solid tumours. Cancer cells are particularly adept at utilising the MMPs to achieve rapid remodelling of the extracellular matrix, thereby providing space for tumour expansion and permitting metastasis. MMP inhibitors should minimise these processes and thus slow or prevent cancer progression.
In view of the rapid emergence of multidrug-resistant bacteria, the development of antibacterial agents with novel modes of action that are effective against the growing number of resistant bacteria, particularly the vancomycin resistant enterococci and β-lactam antibiotic-resistant bacteria, such as methicillin-resistant Staphylocccus aureus, is of utmost importance.
The natural antibiotic actinonin (see for example J. C. S Perkin I, 1975, 819) is a hydroxamic acid derivative of Structure (A):

In ddition to actinonin, various structural analogues of actinonin have also been shown to have antibacterial activity (see for example Broughton et al. (Devlin et al. Journal of the Chemical Society. Perkin Transactions 1 (9):830-841, 1975; Broughton et al. Journal of the Chemical Society. Perkin Transactions 1 (9):857-860, 1975).
The matlystatin group of compounds, share a number of structural similarities with actinonin. Both are peptidic molecules with functional hydroxamic acid metal binding groups (Ogita et al., J. Antibiotics. 45(11):1723-1732; Tanzawa et al., J. Antibiotics. 45(11):1733-1737; Haruyama et al., J. Antibiotics. 47(12):1473-1480; Tamaki et al., J. Antibiotics. 47(12):1481-1492).
………………………………………………………….
EXAMPLE 44 2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl-pentanoylamine]-2-phenyl-ethanoic acid cyclopentyl ester

The above compound was prepared using procedures similar to those described in example 8 using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1H, s), 5.2-5.14 (1H, m), 4.02 (1H, d, J=6.9 Hz), 2.94-2.85 (1H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1H, m) and 0.86 (6H, dd, J=6.5, 11 5 Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1, 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; 8 (MeOD), 7.33-7.19 (5H, m), 5.3 (1H, s), 5.11-5.06 (1H, m), 3.81 (1H, d, J=7.3 Hz), 2.83-2.74 (lH, m), 1.83-1.45 (10H, bm), 1.12-1.03 (lH, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3 Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2
Example 1
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

HO Ξ CONHOH
Prepared using procedures similar to those described in Preparative Example A using phenylglycine cyclopentyl ester.
Diastereoisomer A
Η-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d,
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, 14 dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2,
58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
Η-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d, J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.

tosedostat
http://www.google.it/patents/US6545051

42
| WO98/11063 | WO99/46241 ex 1b | WO 98/11063 analogy ex 8 |

43
| WO98/11063 | WO99/46241 ex 1a | WO 98/11063 analogy ex 8 |
……………………………………………………………………
entry 65 in http://www.google.com/patents/WO2000044373A1
……………………………………………………………………………………………………….
http://www.google.com/patents/WO1999044602A1
Example 43
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester
TC

HO Ξ CONHOH
Prepared using procedures similar to those described in example 8 of WO 98/11063, using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d, 34
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d,
J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and
0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3,
129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.
……………………………..
3R-isobutyl-4S-methoxy-dihydrofuran-2,5-dione (WO 97/02239)
…………………………………………………………………………..
2(S)-Amino(phenyl)ethanoic acid cyclopentyl ester

…………………………………………………………………..
2(R)-[2,2-Dimethyl-5-oxo-1,3-dioxolan-4(S)-yl]-4-methylpentanoic acid pentafluorophenyl ester

…………………………………………………………..
intermediates
238750-91-9
α-amino-, cyclopentyl ester Benzeneacetic acid,
……………….
cas 240489-34-3
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

…………………..
will be updated very soon… keep watching

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

TROPISETRON


TROPISETRON
89565-68-4
105826-92-4 (HCl)
Novartis (Originator)
Tropisetron (INN) is a serotonin 5-HT3 receptor antagonist used mainly as an antiemetic to treat nausea and vomiting following chemotherapy, although it has been used experimentally as an analgesic in cases of fibromyalgia.[1] The drug is available in a 5 mg oral preparation or in 2 mg intravenous form. It is marketed by Novartis in Europe, Australia, New Zealand, Japan, South Korea and the Philippines as Navoban, but is not available in the U.S. It is also available from Novell Pharmaceutical Laboratories and marketed in several Asian countries as Setrovel
Tropisetron is a 5-hydroxytryptamine receptor 3 (5-HT3) antagonist that was launched in 1992 by Novartis for the oral and injection treatment of chemotherapy-induced emesis. The drug has also been approved for the prophylaxis and treatment of post-operative nausea and vomiting, and is available in capsule and ampule formulations. In terms of clinical development, phase III trials were being carried out by the National Institute of Mental Health (NIHM) as an adjunct to risperidone therapy for the treatment of schizophrenia, but no recent development has been reported. Clinical trials for the treatment of fibromyalgia have also been conducted by Novartis, although recent progress reports on this indication have not been made available.
5-HT3 receptors are excitatory ligand-gated cation channel receptors that can be found in the presynaptic vagal afferents, the area postrema and the gastrointestinal tract. Stimulation of these receptors seems to be important in the emetic response and the gag reflex. It has also been shown that tropisetron shows agonistic effects on the 7 nicotinic acetylcholine receptor. This receptor is associated with auditory sensory gating, a neural mechanism believed to have important roles in information processing and cognition, both diminished in people with schizophrenia.
Tropisetron acts as both a selective 5-HT3 receptor antagonist and α7-nicotinic receptor agonist.[2][3]

Tropisetron is a well-tolerated drug with few side effects. Headache, constipation, and dizziness are the most commonly reported side effects associated with its use. Hypotension, transient liver enzyme elevation, immune hypersensitivity syndromes and extrapyramidal side effects have also been associated with its use on at least one occasion.There have been no significant drug interactions reported with this drug’s use. It is broken down by the hepatic cytochrome P450 system and it has little effect on the metabolism of other drugs broken down by this system.
As a biological stain and as trypanocide.
Tropisetron was originally developed by Novartis, and rights to the drug were subsequently acquired by Asta Medica (now, part of Meda). In December 1997, Novartis and Kyowa Hakko signed an agreement, pursuant to which the companies would copromote the product in Japan. Tropisetron is currently distributed in various countries worldwide, including Belgium, Germany, Italy, Japan, The Netherlands, Sweden and the U.K.
.
- Muller, W.; T. Stratz (2004). “Local treatment of tendinopathies and myofascial pain syndromes with the 5-HT3 receptor antagonist tropisetron”. Scand J Rheumatic Suppl. 119 (119): 44–48.PMID 15515413. Retrieved 2007-05-17.
- Macor JE, Gurley D, Lanthorn T, Loch J, Mack RA, Mullen G, Tran O, Wright N, Gordon JC (February 2001). “The 5-HT3 antagonist tropisetron (ICS 205-930) is a potent and selective alpha7 nicotinic receptor partial agonist”. Bioorganic & Medicinal Chemistry Letters11 (3): 319–21. doi:10.1016/S0960-894X(00)00670-3.PMID 11212100.
- Cui R, Suemaru K, Li B, Kohnomi S, Araki H (May 2009). “Tropisetron attenuates naloxone-induced place aversion in single-dose morphine-treated rats: role of alpha7 nicotinic receptors”.European Journal of Pharmacology 609 (1–3): 74–7.doi:10.1016/j.ejphar.2008.12.051. PMID 19374878.
-
Tropisetron hydrochloride (CAS 105826-92-4)

Application: A potent SR-3 antagonist CAS Number: 105826-92-4 Purity: ≥99% Molecular Weight: 320.82 Molecular Formula: C17H20N2O2 HCl - Navoban™ IS THE FORMULATION
Tropisetron hydrochloride is a potent SR (SR-3) antagonist. It is also a selective, partial agonist at AChR α 7 (α7 nicotinic receptors). Tropisetron hydrochloride is an inhibitor of HTR3.

1alphaH,5alphaH-Tropan-3alpha-yl Indole-3-carboxylate, could be produced through many synthetic methods.
Following is one of the synthesis routes: Indole 3-carbonyl chloride (I) is condensed with endo-8-methyl-8-azabicyclo[3.2.1]octan-3-ol (II) in the presence of butyllithium in THF, or Na2CO3 in the same solvent to produce the final product of Tropisetron.
IMPORTANT REFERENCES
Drugs Fut 1986, 11(2): 106
US 4789673
CN 102532128
CN 102887893
WO 2013123426
WO 2007099069
WO 2009033305
WO 2003032897
WO 2004054552
WO 2005105089
WO 2000048597
WO 2000048581
| CN101033225A | 2 Apr 2007 | 12 Sep 2007 | 北京成宇化工有限公司 | Process of preparing troipisetron |
| CN101787021A | 5 Mar 2010 | 28 Jul 2010 | 王明 | High-purified tropisetron hydrochloride compound |
| US4789673 | 10 Nov 1987 | 6 Dec 1988 | Peter Donatsch | Heterocyclic carboxylic acid amides and esters |
…………………………………………………………………………………………………………
5-HT3 receptor antagonists are a class of compounds which block 5-HT3receptors, and are also sometimes classified as serotonin M receptor antagonists. The 5-HT3 receptor antagonists comprise a defined and recognised class of pharmaceutically active compounds well known in the art and characterised, as their name implies, by their pharmacological activity. Various 5-HT3 receptor antagonist compounds are commercially available and clinically applied, e.g. in the treatment of emesis.
5-HT3 receptor antagonists from various sources have been published for a wide variety of uses, for example for the treatment of visceral pain, migraine, vascular and cluster headache, trigeminal neuralgia, arrhythmia, serotonin-induced gastro-intestinal disorders, including emesis induced by anti-cancer agents, anxiety, stress-related psychiatric disorders, depression, cognitive disorders, social withdrawal, panic attacks, agoraphobia, lung embolism, rhinitis or serotonin-induced nasal disorders, fibromyalgia and local treatment of pain caused by various non-inflammatory or inflammatory conditions. Some have been commercially introduced for the treatment of emesis.
In accordance with the present invention it has now surprisingly been found that 5-HT3 receptor antagonists are useful for the treatment of diseases caused or influenced by activation of thrombocytes.
Thrombocytes play a central role in blood coagulation (clotting) and are therefore also of high importance in the pathogenesis of cardiac infarction and stroke, furthermore also in thrombosis of the veins and inflammatory conditions in the development of atherosclerosis.
Activation of thrombocytes causing blood clotting is based on several mechanisms. It has ■ now been demonstrated that 5-HT3 receptors are present on platelets and that the number of these receptors at the platelet surface is increasing in a dose dependent fashion on addition of ADP (adenosine diphosphate) or TRAP (thrombin receptor activating peptide) known to stimulate thrombocyte activation. The increase of 5-HT3 receptors on addition of compounds inducing aggregation such as ADP and TRAP is proof that such receptors play a role in thrombotic processes. Platelet activity is also important in inflammatory processes in atherosclerotic conditions. Thrombocytes activated by thrombin may, as is already known, induce the production of I L- 1 β , IL-8, MCP (monocyte chemoattractant protein) and other inflammation mediators These are impeded by 5-HT3 receptor antagonists. This demonstrates that 5-HT3 receptor antagonists not only influence blood coagulation but also processes playing a role in the development of atherosclerosis.
Hence, the present invention relates to the use of a 5-HT3 receptor antagonist or of a pharmaceutically acceptable salt of such an antagonist for the manufacture of a pharmaceutical composition for the treatment of a disease caused or influenced by activation of thrombocytes, in particular myocardial infarction, stroke, thrombosis and atherosclerosis.
Any 5-HT3 receptor antagonist can be used in accordance with the invention. Preferred 5-HT3 receptor antagonists which may be employed in accordance with the present invention are ondansetron, 1 ,2,3,9-tetrahydro-9-methyl-3-[(2-methyl-1 H-imidazol-1-yl)- methyl]-4H-carbazol-4-one (cf. Merck Index, twelfth edition, item 6979), granisetron, ‘ endo-1-methyl-N-(9-methyl-9-aza-bicyclo[3.3.1]non-3-yl)-1 H-innidazole-3-carboxamide . (cf. loc. cit, item 4557), or dolasetron, 1 H-indole-3-carboxylic acid (2α,6α,8α,9αβ)- octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl ester, (cf. loc. cit., item 3471). ■•
Particular 5-HT3 receptor antagonists which may be employed in accordance with the ■ present invention are those of the formula 1 as defined in European Patent Publication EP O 189 002 B1, in particular tropisetron, indol-S-yl-carboxylic acid-endo-δ-methyl-δ-aza- bicyclo[3,2,1]-oct-3-yl-ester, (cf. loc. cit., item 9914), ramosetron, 4,5,6,7-tetrahydro-5- [(1-methyl-indol-3-yl)carbonyl]benzimidazole (U.S. Pat. No. 5,344,927), fabesetron, (+)- 10-methyl-7-(5-methyl-1 H-imidazol-4-ylmethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-6-one (EP 0 361 317), lintopride, N-(1-ethyl-2-imidazolin-2-y-methyl)-2-methoxy-4-amino-5- chlorobenzamide (Chem. Abstr. No. 107429-63-0), alosetron, 2,3,4,5-tetrahydro-5-methyl- 2-[(5-methyl-1 H-imidazol-4-yl)methyl]-1 H-pyrido[4,3-b]indol-1-one (EP 0 306 323), cilansetron, (-)-(R)-5,6,9,10-tetrahydro-10-[(2-methylimidazol-1-yl)methyl]-4H-pyrido- (3,2,1-jk)carbazol-11(8H)-one, palonosetron, 2-(3S)-1-azabicyclo[2.2.2]oct-3-yl-2,3,3a(S), 4,5,6-hexahydro-1 H-benz(de)isoquinolin-1-one, azasetron, N-(1-azabicyclo[2.2.2]octan-8- yl)-6-chloro-4-methyl-3-oxo-1 ,4-benzoxazine-8-carboxamide, and zatosetron, 5-chloro- 2,2-dimethyl-N-(8-methyl-8-azabicyclo[3.2.1]octan-3-yl)-3H-1-benzofuran-7-carboxamide.
5-HT3 receptor antagonists may be employed in accordance with the invention in free or in pharmaceutically acceptable salt form, e.g. as known in the art, for example, in the case of compounds mentioned above in pharmaceutically acceptable acid addition salt form, for example, in the case of ondansetron as the hydrochloride dihydrate, granisetron as the hydrochloride, dolasetron as the mesylate, tropisetron as the monohydrochloride, ramosetron, fabesetron, alosetron and cilansetron as the hydrochlorides, palonosetron as the monohydrochloride, azasetron as the hydrochloride, and zatosetron as the maleate.
References to 5-HT3 receptor antagonists collectively or individually throughout the present specification and claims are accordingly to be understood as embracing both free compounds and such pharmaceutically acceptable salt forms, e.g. as clinically employed, and further also solvates, e.g. hydrates, or specific crystal forms of any of these compounds or salts.
For use in accordance with the present invention tropisetron (especially in the formulation called Navoban™) is most preferred.
tropisetron hydrochloride is a peripheral neurons and central nervous system 5 – hydroxytryptamine 3 (5-ΗΤ3) receptors potent, highly selective competitive antagonist, mainly by selectively blocking peripheral neurons presynaptic 5-ΗΤ3 receptors inhibit vomiting reflex. For the prevention and treatment of child and adult cancer chemotherapy, radiotherapy and post-operative nausea and vomiting caused.
About tropisetron hydrochloride preparation of patent literature, including (for example): US4797406, US 4789673, CN 101033225, CN 101838266, CN 101787021. Tropisetron hydrochloride preparation generally is this: the firstsynthesis of acid chloride intermediates, and then through esterification acidification, refining and other steps, resulting tropisetron hydrochloride.
The preparation method has, in U.S. Patent (US3980668, US4789673, US4797406, US4803199) described with indole-3 – carboxylic acid with oxalyl chloride, treatment with methylene chloride and n-hexane to give 3 – methyl indole chloride, then with tropenol activation in n-butyl lithium produced under tropenol lithium reaction was treated to obtain tropisetron, and the final hydrochloride salt obtained tropisetron hydrochloride
in which the U.S. patent literature US 4797406 and US 4789673 introduces the indole-3 – carboxylic acid with oxalyl chloride to give 3 – indole chloride then with tropenol activation in n-butyl lithium in the reaction system tray alkyl granisetron. This method uses expensive n-butyl lithium and polluting chloride compound, and the multi-step, only about 20% yield.
Chinese patent literature described in CN 101033225 is improved indolesynthesis _3_ chloro acid as raw material of 1,3 – dimethyl-2 – chloro-imidazoline as the condensing agent, an organic base directly under conditions esterified with tropenol obtain the target product. Although this method avoids the acylation step, a condensing agent in preparing dimethyl-2-chloro-1,3 _ – chloro-imidazoline must use highly toxic phosgene, so there is an obvious lack of this route.
In the literature [tropisetron hydrochloride synthesis improvements, Huaihai Institute of Technology, 2003; 12 (4): 4 wide 43; tropisetron hydrochloride Improved synthesis of Hunan Normal University (Medical Sciences), 2006; 3 (4): 29 ^ 30], respectively, 50%, 52.8% obtained in a yield tropisetron hydrochloride, but still use of expensive n-butyl lithium as a condensation reaction activator.
………………………………………………………………………………………………………………
Example 2:
[0071] The indole-3 – carboxylic acid (20 g, 0. 124mol), benzenesulfonic acid (0. 791g, 0. 005mol), ethyl acetate (230ml), freshly activated 4A molecular sieves (0. 5-1 . Omm) (3 g) was added to a equipped with a thermometer, reflux condenser, 500ml three reaction flask, stir began to heat up, the temperature controlled at 75 ° C _77 ° C, and then began a slow drip Gato Decanter (19. 3 g, 0. 137mol), after the addition was complete the reaction refluxed for 11 hours.
[0072] The reaction was stopped, the organic layer was washed with IOOml a lmol / L of hydrochloric acid, extracting the product three times, the combined aqueous phase was then washed once with 50ml of ethyl acetate. Aqueous phase was 4mol / L sodium hydroxide aqueous solution was adjusted to pH = 9_10, precipitated yellow solid was suction filtered cake was washed with distilled water to neutral, dried under reduced pressure to obtain 33.1 Hector alkoxy granisetron crude. The crude product was dissolved at 60 ° C in 180ml of anhydrous ethanol was slowly added dropwise 12mol / L hydrochloric acid until the pH value 1_2, _5 ° C under crystallization 6 hours, filtered, the filter cake washed with ethanol to white . Drying get tropisetron hydrochloride concept .3 g, yield 71.1% (to indole-3 – carboxylic acid basis), as measured by liquid chromatography purity of 99.56%.
…………………………………………………………………………..
[0016] (3) tropisetron hydrochloride Preparation:
A solution of indole-3 – carbonyl chloride in THF was slowly added dropwise to sodium tropine THF solution, 35 ° C the reaction was stirred overnight, vacuum distillation recovery THF, and recrystallized from 95% ethanol to give a pale yellow solid adding 70mL ethanol, dissolved by heating, cooling pass into the HCl gas at room temperature, the reaction was stirred for 30min, filtered, tropisetron hydrochloride was crude. Recrystallization from absolute ethanol to give white crystalline product 21. 2g (Liquid chromatographic purity 99.91%), total yield of 53.29%, a melting point of view 3185, the product spectrum consistent with the literature.
…………………………………………………………………
Example 5 tropisetron hydrochloride refined
[0063] (a) The IOOg tropisetron hydrochloride was dissolved in 500ml water, then slowly adding 12% (g / ml) of sodium carbonate solution was stirred until the solution PH 8, resulting tropisetron sedimentation, filtration , 40 ° C under reduced pressure and dried to give tropisetron 84. lg.
[0064] (2) obtained in the above Step Tropisetron 84. Ig dissolved in 400ml ethanol, 5. 2g of activated charcoal, stirred at room temperature for 40 minutes, filtered decarbonization, collecting the filtrate.
[0065] (3) The use of the filtrate obtained in step purified by preparative chromatography separation tropisetron hydrochloride refined products, including the column using a mobile phase as the mobile phase volume accounted for 32% of total current volume of methylene chloride and with 68% aqueous hydrochloric acid to pH 1; stationary phase filler is silica; flow rate of 6. 2ml/min; column temperature: 25 ° C. The filtrate was collected, dried under reduced pressure, to obtain purified tropisetronhydrochloride 91. 7g, yield 91. 7%, HPLC purity of 99.9% method.
[0066] Elemental analysis = C17H21ClN2A
[0067] Theoretical value (%): C: 63. 65, H: 6. 60, N: 8. 73, Cl: 11. 05;
[0068] Found (%): C: 63. 62, H: 6. 64, N: 8. 74, Cl: 11. 06.
[0069] UV (MeOH) Amax: 214 (ε 38,222), 229 (ε 17,438), 282 (ε 13,405).
[0070] IR (KCl) CnT1: 3219,2496 (NH), 3103,748 (Ar CH), 2966,1428 (CH), 1692 (C = 0), 1580,1525 (Ar C = C), 1311, 1128 (CN), 1033 (CO).
[0071] 1HNMR (DMSO-D6) δ: 2. 10 (d, 2H, CH2), 2 · 32 (d, 4H, CH2), 2 · 52 (m, 2H, CH2), 2. 68 (s, 3H, CH3), 3. 88 (s, 2H,-CH), 5. 14 (s, H, CH), 8. 03 (m, H, CH), 8. 08 (d, H, CH), 10. 81 (s, H, HCl), 12. 10 (s, H, NH).
[0072] ESI-MS; EI m / z: 285 (M + H, 100%), 284 (M +, 21. 72%) ”
…………………………………………………………………………………………………………..
Example 1
[0048] equipped with stirrer, thermometer, condenser IOOOml reaction flask, add 80 grams of tropisetron hydrochloride (I Ci refined products) and 560ml of acetone-water (8:2) mixture, start stirring, heating heated to 60 ° C _65 ° C, until all dissolved clear, incubated for 30 minutes, filtered while hot. The filtrate was cooled to room temperature, and then incubated for 2 hours, crystalline precipitation, filtration, drying ie high purity tropisetron hydrochloride crystals, mp: 280.10C -281.1 ° C, purity 99.96% (HPLC normalization method), the solvent residue testing to meet the requirements.
[0049] Elementary analysis:
[0050] Found (calculated value), C: 63.64 (63.57), H: 6.60 (6.62), N: 8.73 (8.72),
[0051] Cl: 11.05 (11.01);
[0052] X-ray diffraction of the crystal shown in Figure 1. Instrument model and measurement conditions: Rigaku D / max 2500 type diffractometer; CuKa 40Kv 100mA; 2 Θ scan range: 0 – 50 °;
[0053] The infrared spectrum of this crystal is shown in Figure 2, as measured by KBr tablet.
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
Lorcaserin…Eisai Expands Marketing and Supply Agreement for Anti-obesity Agent Lorcaserin to Include Most Countries Worldwide

(1R)-8-chloro-1-methyl-2,3,4,5-tetrahydro-1H-3-benzazepine
Eisai Expands Marketing and Supply Agreement for Anti-obesity Agent Lorcaserin to Include Most Countries Worldwide
HATFIELD, England, November 8, 2013 /PRNewswire/ —
Eisai announces today that it has expanded the marketing and supply agreement between its U.S. subsidiary Eisai Inc. and U.S-based Arena Pharmaceuticals Inc.’s Swiss subsidiary, Arena Pharmaceuticals GmbH, for the anti-obesity agent lorcaserin hydrochloride (lorcaserin) (U.S. brand name: BELVIQ®). Whilst the existing agreement granted Eisai Inc. exclusive rights to market and distribute lorcaserin in 21 countries throughout the Americas, the expanded agreement now includes most countries and territories worldwide, most notably, the member states of the European Union, Japan and China (but excludes South Korea, Taiwan, Australia, New Zealand and Israel).http://www.pharmalive.com/eisai-expands-lorcaserin-marketing-and-supply-agreement
Lorcaserin (previously APD-356), a highly selective 5HT2C receptor agonist, is used for the treatment of obesity. It has been shown to reduce body weight and food intake in animal models of obesity…
View original post 4,225 more words
