
Home » 2013 (Page 10)
Yearly Archives: 2013
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Argatroban

Argatroban
Molecular Formula: C23H36N6O5S
Formula Weight: 508.63
CAS No.: 74863-84-6
(2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2-
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl]
sulfonylamino]pentanoyl]-4-methyl-piperidine-2-
carboxylic acid
PATENT
US 7,589,106, 7,687,516, EP 0008746; US 4258192, US 4201863
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in the liver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).

Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.
……………………………………………………….
Argatroban monohydrate

Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5-[(aminoiminomethyl) amino]-1-oxo2[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4-methyl-2- piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. cas 141396-28-3
Argipidine, Argatroban monohydrate, GN1600, DK-7419, MDI-805 Acova, Slonnon, Novastan
Mitsubishi Chemical (Originator), Encysive Pharmaceuticals (Licensee), Mitsubishi Pharma (Distributor), Daiichi Pharmaceutical (Codevelopment), GlaxoSmithKline (Codevelopment), Mitsubishi Pharma (Codevelopment), Sanofi-SynthLabo (Codevelopment)
Antithrombocytopenic, CARDIOVASCULAR DRUGS, Cerebrovascular Diseases, Treatment of, HEMATOLOGIC DRUGS, Hematopoiesis Disorders Therapy, Ischemic Stroke, Treatment of, NEUROLOGIC DRUGS, Peripheral Vascular Disease, Treatment of, Stroke, Treatment of, Treatment of Peripheral Obstructive Vascular Disease, Thrombin Inhibitors
Synthesis of argatroban on the method reported in the literature there are two synthetic routes, patent EP8746, US4258192, US4201863, JP8115267 relates to a route is: with 4 – methyl-piperidine as a starting material was prepared first intermediate body (2R, 4R) -4 – methyl-2 – ethyl-piperidine, and the first and a t-BOC protected amino nitro-L-arginine condensation, and then the 3 – methyl – 8 – quinoline sulfonyl chloride condensation after hydrolysis, hydrogenation, hydration be argatroban. This entry route synthesis process complicated procedure to be carried out under the protection of nitrogen, the raw material is highly toxic gas phosgene, the operation more difficult.
US4117127, JP02-212473, EP823430, EP8746, JC S Perk Transl 1981 (5), JP02-212473 relates to an alternative route is: nitro L-arginine prior to the 3 – methyl-8 – subsequent condensation quinoline sulfonyl chloride Intermediate (2R, 4R) -4 – methyl-2 – piperidinecarboxylate condensation, and then after hydrolysis, hydrogenation, hydration be argatroban. This synthetic route despite the relatively simple process method, to obtain raw materials, but this method using reagents such as phosphorus oxychloride, phosphorus trichloride has a pungent odor, easy to absorb moisture in humid air, intense smoke, environmental pollution, greater stimulation of the body’s respiratory tract, can cause eye and skin irritation and burning, and the use of this method, complex operation, low yield, high cost.
Patent CN100586946C Argatroban discloses a method for separating optically active isomeric compounds, the feedstock argatroban mixed solvent of alcohol and water was heated to reflux 5-10 hours, cooled and allowed to stand, and filtered to give White crystalline product, dried, repeated 2-6 times. [0008] Patent CN101033223A discloses a Argatroban is the main by-product (2R, 4R)-l_ [N2-(3_ methyl-8 – quinolinesulfonyl)-L-arginyl] -4 – methyl-2 – carboxylic acid, argatroban, and the byproducts are difficult to isolate, argatroban two diastereomeric isomer 21 (S) and 21 (R) separation of work attracted a lot of research persons. Because both physical and chemical properties are very similar, so separation is very difficult. 1993 Rawson, Thomas E.; VanGorp, Kimmie A.; Yang, Janet so first by high pressure liquid chromatography and column chromatography separation to obtain a single 21 (S) and 21 (R) argatroban [ Journal of Pharmaceutical Sciences vol. 82, No. 6,672]; Thibaudeau Karen et al. reported Protein A chromatographic separation [US6440417]. However, due to the separation of these methods a small amount of low efficiency, so there is no practical value industrialization. 2006 China Tianjin Weijie Technology Co., Ltd. Song Honghai et al. Reported using recrystallization Separation 21 (S) and 21 (R) argatroban way [0 With 951,936 it], so that the mass 21 (5) Aga music classes as possible, but the law of low yield, complicated operation, high cost, and a large amount of a small amount of 21 (S) of 21 (R)-product argatroban, from the viewpoint of industrial production, is still a ideal method. [0009] These methods can be effectively prepared argatroban, but the purity of the desired product is not high, poor color, content is low, affecting the quality of the results of its preparation.
U.S. Pat. No. 4,201,863 (6 May 1980) and EP 8746 (filed on 22 Aug. 1979 with priority based on the application for the cited US patent) describe a class of N2-arylsulphonyl-L-argininamide drugs, with anti-thrombotic activity, and the processes for obtaining them. Of these, the compound 4-methyl-1-[N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid (argatroban, isomers mixture) is described. The described process comprises the synthesis of an intermediate NG-substituted-N2-quinolinesulphonyl-L-argininamide from which the desired compound is obtained by catalyzed hydrogenolysis or acidolysis and catalyzed hydrogenation. The general conditions provided for the hydrogenolysis and hydrogenation reaction are: i) inert solvents (methanol, ethanol, tetrahydrofuran or dioxane); ii) presence of a catalyst (Raney nickel, palladium, platinum, ruthenium, rhodium); iii) hydrogen atmosphere at a pressure between 1 and 100 kg/cm2 and preferably between 5 and 50 kg/cm2; iv) temperature between 0° C. and 200° C. and preferably between 50° C. and 150° C.; v) reaction temperature from 2 hours to 120 hours. The crude product obtained is then purified by trituration or by re-crystallization from diethyl ether-tetrahydrofuran, diethyl ether-methanol or from water-methanol or by chromatography. No example is given of this purification step. In particular, both U.S. Pat. No. 4,210,863 and EP 8746 in example 1(E) describe the preparation of argatroban, isomers mixture. This compound is obtained in amorphous form by hydrogenation of [NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid in ethanol in the presence of Pd/C with hydrogen pressure of 10 kg/cm2 at 100° C. for 8 hours. The catalyst is removed by filtration of the ethanol solution which is then evaporated without further purification and/or re-crystallization steps. In the US patent at issue as indeed in patent application EP 8746, no mention is made of polymorphic forms of the compounds and, for the obtained compound, the following characteristics are reported: Amorphous solid, I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.01; H 6.98; N 16.61.
U.S. Pat. No. 4,258,192 (24 Mar. 1981) (continuation-in-part of the aforesaid patent application U.S. Pat. No. 4,201,863) and the same patent application EP 8746 describe the stereoisomers and the preparation thereof, including argatroban used as an active principle in medicaments, i.e. the stereoisomer (2R,4R)-4methyl-[4N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid, with the following characteristics: melting point (m.p.). 188-191° C.; I.R. (KBr) (cm−1) 3400, 1620, 1460, 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.05; H 6.94; N 16.65. The compound is prepared according to the description given in examples 1(E) in U.S. Pat. No. 4,258,192 and 2(E) and 3 in EP 8746 respectively by hydrogenation of (2R,4R) 1-[NG-nitro-N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid in ethanol in presence of acetic acid catalyzed by Pd/C. After filtering the mass to remove the catalyst, the solvent is evaporated and the residue suspended in chloroform, the solution treated with a saturated sodium bicarbonate solution or 1N sodium hydroxide solution and after washing, the solvent is evaporated. The compound is then re-crystallized from ethanol. Again in this case, no reference is made to the obtainment of monohydrate polymorphic forms.
Said polymorphic forms are described instead in the publication Biochem. Biophys. Res. Comm. 1981, 101, 440-446 in the context of stereoisomer preparation. The monohydrate polymorph of the (2R,4R) stereoisomer is prepared by re-crystallization from ethanol/water and the reported characteristics are: m.p. 176-180° C.; [α]D 27 +76.1° (c 1, 0.2N HCl).
U.S. Pat. No. 5,925,760 (20 Jul. 1999) and EP 0823430 (filed 4 Aug. 1997) subsequently describe a new method for preparing argatroban by means of a new intermediate N2-(3-methyl-8-quinolinesulphonyl)-NG-nitro-L-arginine. In particular the patent makes reference to the preparation of a crystalline monohydrate form of argatroban, referring back to examples (D) and (E) of Japanese patent publication No. (Hei)-2-31055/1990 and generically to an I.R. spectrum identical to that of the commercially available argatroban compound. The relevant example in the cited patent publication is example (E), while example (D) concerns the preparation of (2R,4R)-1-[NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid. This compound represents the starting compound for argatroban preparation by catalytic reduction in the presence of Pd/C. The crude argatroban obtained is then purified by extraction with chloroform, treatment with a saturated sodium bicarbonate solution and, after solvent evaporation, re-crystallization from ethanol or from 15% alcohol in water. It should be noted however that the Japanese patent makes no mention of the monohydrate form of argatroban being obtained and that for the compound the following characteristics are reported: m.p. 188-191° C.; molecular composition (theoretical/found) (%): C 54.31/54.01; H 7.13/6.98; N 16.52/16.61; I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380. These analytical data, with the exception of the unreported melting point, are the same as those indicated in the cited patent documents describing a mixture of (2R,4R)-4methyl-[4N2-(3S-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid and (2R,4R)-4methyl-1-[N2-(3R-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid isomers of argatroban, but do not correspond to the melting point given in the publication, being the only document that identifies the monohydrate form of argatroban.
More recently, patent application CN 1,951,937 (filing date 10 Nov. 2006) described a method for preparing hydrated argatroban by treating argatroban with large quantities of water (more than 60 and up to 80 volumes of distilled water per gram of argatroban) at a temperature of 80-100° C. for a time of 0.5-1 hour and crystallization by cooling. The water content reported is comprised between 3.3 and 3.8% and the ratio of dextroisomer R to levoisomer S is R:S=63-67: 37-33.
Argatroban is a compound of wide therapeutic use, for which reason the need still exists to provide a compound of pharmaceutically acceptable quality obtained by easily industrialized and economically convenient methods. With regard to the monohydrate, this form is preferable for the applicative purpose since the anhydrous form is unstable and tends to become hydrated and/or wet. Moreover it crystallizes only with difficulty at the correct ratio between the diastereoisomers.

…..

the protection of 4-methylpiperidine (I) with (Boc)2O gives the carbamate (II), which is condensed with benzyl chloroformate by means of sec-butyl lithium and TMEDA in ethyl ether to yield (?-trans-1-(tert-butoxycarbonyl)-4-methylpiperidine-2-carboxylic acid benzyl ester (III). Deprotection of the NH group of (III) with HCl in ethyl acetate affords (?-trans-4-methylpiperidine-2-carboxylic acid benzyl ester (IV), which is condensed with the protected arginine derivative (V) by means of isobutyl chloroformate and TEA to provide the corresponding amide as a diastereomeric mixture. Resolution of this mixture by flash chromatography furnishes the desired diastereomer (VI), which is treated with HCl in ethyl acetate in order to remove the Boc-protecting group to yield compound (VII). Condensation of compound (VII) with 3-methylquinoline-8-sulfonyl chloride (VIII) by means of TEA in dichloromethane affords the expected sulfonamide (IX). Finally, this compound is submitted to hydrogenation with H2 over Pd/C in AcOH/ethanol in order to produce debenzylation, cleavage of the NO2 group and hydrogenation of the pyridine ring to yield argatroban.
………….
Argatroban, i.e., (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino) pentyl)-4-methyl-2-piperidine carboxylic acid, has two diastereoisomers: 21(R) and 21(S). Usually the ratio of 21(R) to 21(S) is 64-65: 36-35 (U.S. Pat. No. 6,440,417, Cossy. J., et al, Bioorganic & Medicine Chemistry Letters, 11 (2001), 1989-1992, Journal of pharmaceutical Sciences, Vol. 82, No. 6, 672 (1993)).
The structure formula of Argatroban is reported below:

-
- 21(S) Argatroban, X=CH3, Y═H;
- 21(R) Argatroban, X=H, Y=CH3;
- Argatroban, 21(S): 21 (R)=35:65.
The chemical names of the two diastereoisomers mentioned above are:
- 21(S) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3S)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-72-6); and
- 21(R) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3R)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-71-5).
In 1978, S. Akamoto et al from Japanese Mitsubishi Chemical Corporation first disclosed the anti-thrombin activity of Argatroban monohydrate (U.S. Pat. No. 4,101,653). In the next 20 years, numerous researchers had in-depth studies on Argatroban about its biological activity and medicine values. In 1981, S. Akamoto compared Argatroban with heparin in vivo (Okamoto, S. et al., Biochem. Biophys. Res. Commun. 101, 440 (1981)); T. Kumoto disclosed its three-dimensional selective activity (Kumada, T. et al., Thromb. Res. 24, 285 (1981)). In 1984, R. Kumato made a clinical evaluation of hemodialysis of Argatroban (Kikumoto, R. et al., Biochemistry 23, 85 (1984)), and in 1986, he further disclosed that Argatroban can inhibit the thrombin activity of mammals, and can be used as active ingredient to treat and prevent thrombosis and as an inhibitor of platelet aggregation. Argatroban monohydrate can be used as a selective anti-thrombosis agent for treatment of chronic arterial blockage and cerebral thrombosis, etc (JP 61-48829). In 1992 and 1993, Taparelli and Jakubowski separately disclosed the reversibility of Argatroban in anti-thrombin (Taparelli, C., Trends Pharmacol. Sci., 1993, 14, 366, Jakubowski, J. A. et al, Rep. Med. Chem., 1992, 27, 99). In 1990s, many researchers such as L. R. Buch reported other related research (Buch, L. R., Cadiosvasc. Drug Rev., 1991, 9, 247, Strupcnewski, J. D. et al., Academic: San Diego, 1991; Vol. 26, p 299, Brundish, D. et al., J. Med. Chem. 1999; 42, 4584, Shebuski, R. J., Academic: San Diego, 1999; Vol. 26, p 98). In 1992, Argatroban monohydrate was first approved as an anti-thrombin medicine in Japan (Hijikata-Okunomiya, A., et al, Thromb. Hemostasis, 1992, 18, 135).
Updated in sept 2015, reader there may be duplication
| Argatroban |
| AC1L99H9; AC-15185; TL8005144; C04931; | |
| Molecular Formula: | C23H36N6O5S |
|---|---|
| Molecular Weight: | 508.63414 g/mol |
(2R,4R)-1-[5-(diaminomethylideneamino)-2-[(3-methyl-1,2,3,4-tetrahydroquinolin-8-yl)sulfonylamino]pentanoyl]-4-methylpiperidine-2-carboxylic acid, cas 74863-84-6
| Patent | Submitted | Granted |
|---|---|---|
| Prodrugs of (2R)-2-Propyloctanoic Acid For the Treatment of Stroke [US7495029] | 2008-06-05 | 2009-02-24 |
Tomiya Mano, Jin Shiomura, “Argatroban preparations for ophthalmic use.” U.S. Patent US5506241, issued October, 1986.
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in the liver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).
Argatroban is a direct, selective thrombin inhibitor. The American College of Cardiologists (ACC) recommend using bivalirudin or argatroban in patients who have had, or at risk for, heparin induced thrombocytopenia (HIT) and are undergoing percutaneous coronary intervention. Argatroban is a non-heparin anticoagulant shown to both normalize platelet count in patients with HIT and prevent the formation of thrombi. Parental anticoagulants must be stopped and a baseline activated partial thromboplastin time must be obtained prior to administering argatroban.

Transitioning to warfarin in individuals with heparin induced thrombocytopenia
Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.

http://www.google.com/patents/WO2009124906A2?cl=en
………….
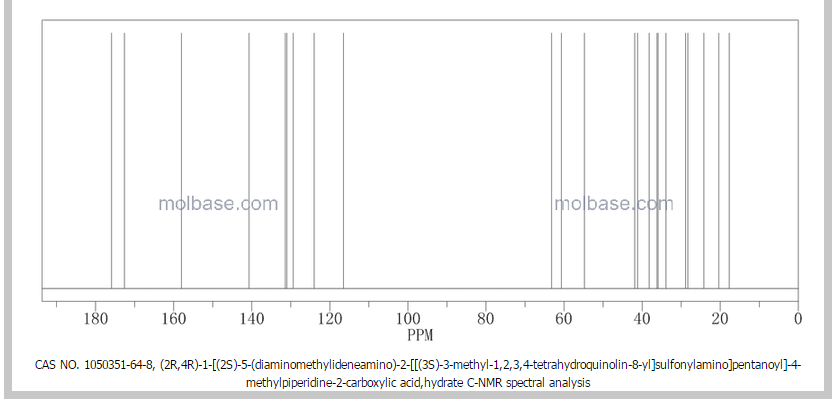
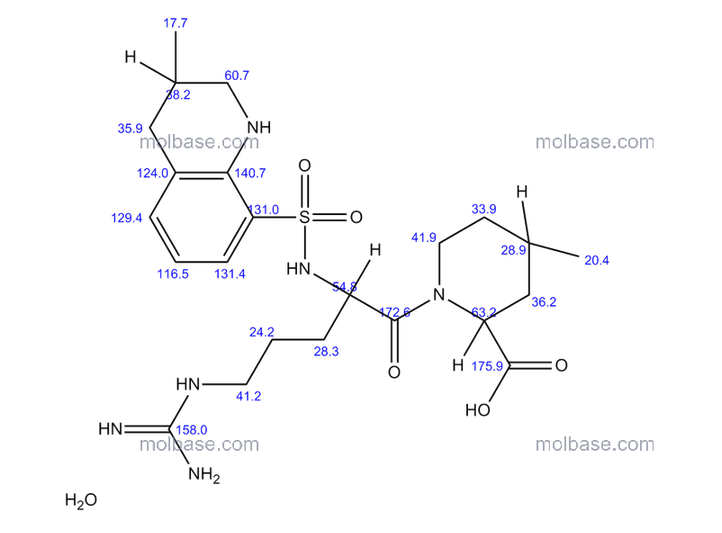
NMR paper
Complete 1H and 13C assignments of (21R) and (21S) diastereomers of argatroban
Article first published online: 20 DEC 2007
DOI: 10.1002/mrc.2122, http://onlinelibrary.wiley.com/doi/10.1002/mrc.2122/abstract



click on image for clear view
1H NMR PREDICT


13C NMR PREDICT
References
1 Di Nisio M, Middeldorp S, Buller HR. Direct thrombin inhibitors. N Engl J Med 2005;353:1028-40. PMID 16148288
2http://www.pharmatimes.com/Article/12-07-03/UK_launch_for_Mitsubishi_s_blood_thinner_Exembol.aspx
3Dhillon S. Argatroban: A Review of its Use in the Management of Heparin-Induced Thrombocytopenia. Am J Cardiovasc Drugs 2009; 9 (4): 261-82. Link text
- Hursting MJ, Lewis BE, Macfarlane DE. (2005). “Transitioning from argatroban to warfarin therapy in patients with heparin-induced thrombocytopenia.”. Clin Appl Thromb Hemost 11 (3): 279–87. doi:10.1177/107602960501100306. PMID 16015413.
External links
| EP0823430A1 * | Aug 4, 1997 | Feb 11, 1998 | Mitsubishi Chemical Corporation | Method for preparing n2-arylsulfonyl-l-argininamides |
| US4201863 * | Aug 31, 1978 | May 6, 1980 | Mitsubishi Chemical Industries, Limited | N2 -Arylsulfonyl-L-argininamides and the pharmaceutically acceptable salts thereof |
| Reference | ||||
|---|---|---|---|---|
| 1 | None | |||
| 2 | * | OKAMOTO S ET AL: “Potent inhibition of thrombin by the newly synthesized arginine derivative No. 805. The importance of stereo-structure of its hydrophobic carboxamide portion” BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 101, no. 2, 30 July 1981 (1981-07-30), pages 440-446, XP024844713 ISSN: 0006-291X [retrieved on 1981-07-30] cited in the application | ||
| 3 | * | SONG H: “Method for preparing argatroban monohydrate in pure water” CASREACT,, 25 April 2007 (2007-04-25), XP002493272 & CN 1 951 937 A (TIANJIN WEIJIE TECHNOLOGY CO L [CN]) 25 April 2007 (2007-04-25) | ||
| Citing Patent | Filing date | Publication date | Applicant | Title |
| WO2012136504A1 | Mar 26, 2012 | Oct 11, 2012 | Lundbeck Pharmaceuticals Italy S.P.A. | Method for the preparation of process intermediates for the synthesis of argatroban monohydrate |
| CN102408468A * | Sep 20, 2011 | Apr 11, 2012 | 海南灵康制药有限公司 | Argatroban compound and preparation method thereof |
| EP2752412A1 | Mar 26, 2012 | Jul 9, 2014 | Lundbeck Pharmaceuticals Italy S.p.A. | Intermediates for the synthesis of Argatroban monohydrate |
Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5[(aminoiminomethyl)amino]1-oxo-2-[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4methyl-2-piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. The structural formula is:
|
Argatroban Injection is a sterile, non-pyrogenic, clear, colorless to pale yellow isotonic solution. It is supplied in a single use polyolefin bag containing 250 mg of argatroban in 250 mL sodium chloride solution (1 mg/mL). Each mL contains 1 mg argatroban, 9 mg sodium chloride, USP, and 3 mg sorbitol, NF in water for injection, USP. The pH of the solution is between 3.2 to 7.5.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2-
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl] sulfonylamino]pentanoyl]-4-methyl-piperidine-2- carboxylic acid |
|
| Clinical data | |
| Trade names | Argatroban |
| AHFS/Drugs.com | monograph |
| Routes of administration |
intravenous |
| Pharmacokinetic data | |
| Bioavailability | 100% (intravenous) |
| Protein binding | 54% |
| Metabolism | hepatic |
| Biological half-life | 39 and 51 minutes |
| Identifiers | |
| CAS Registry Number | 74863-84-6 |
| ATC code | B01AE03 |
| PubChem | CID: 440542 |
| DrugBank | DB00278 |
| ChemSpider | 389444 |
| UNII | OCY3U280Y3 |
| KEGG | C04931 |
| ChEMBL | CHEMBL1166 |
| Chemical data | |
| Formula | C23H36N6O5S |
| Molecular mass | 508.635 g/mol |
//////
EMA introduces new fee reductions for orphan drugs aimed at big companies
December 04, 2013 | By Márcio Barra
The European medicines Agency (EMA) announced yesterday that, starting January 2014, there will be greater fee-reduction rates for orphan medicines in 2014 for big companies, something that was previously only in place for micro, small or medium-sized enterprises (SMEs).
View original post 266 more words
DRUG SPOTLIGHT …… DOXOFYLLINE

DOXOFYLLINE
LAUNCHED 1987, Istituto Biologico Chemioterapico ABC
69975-86-6 CAS NO
7-(1,3-dioxolan-2-ylmethyl)-1,3-dimethylpurine-2,6-dione
1H-Purine-2,6-dione, 3,7-dihydro-7-(1,3-dioxolan-2-ylmethyl)-1,3-dimethyl- (9CI)
7-(1,3-Dioxolan-2-ylmethyl)-3,7-dihydro-1,3-dimethyl-1H-purine-2,6-dione; 7-[1,3-(Dioxolan-d4)-2-ylmethyl)]theophylline; 2-(7�-Theophyllinemethyl)-1,3- dioxolane; ABC 12/3; ABC 1213; Ansimar; Dioxyfilline; Doxophylline; Maxivent; Ventax;
Synonyms
-
2-(7′-Teofillinmetil)-1,3-diossolano
-
2-(7′-Teofillinmetil)-1,3-diossolano [Italian]
-
2-(7′-Theophyllinemethyl)-1,3-dioxolane
-
5-26-14-00120 (Beilstein Handbook Reference)
-
7-(1,3-Dioxolan-2-ylmethyl)theophylline
| Formula | C11H14N4O4 |
|---|---|
| Mol. mass | 266.25 g/mol |
- ABC 12/3
- Ansimar
- BRN 0561195
- Dioxyfilline
- Doxofilina
- Doxofilina [INN-Spanish]
- Doxofylline
- Doxofyllinum
- Doxofyllinum [INN-Latin]
- Doxophylline
- EINECS 274-239-6
- Maxivent
- UNII-MPM23GMO7Z
- Ventax
Doxofylline (INN), (also known as doxophylline) is a xanthine derivative drug used in the treatment of asthma.[1]
Doxofylline is a xanthine molecule that appears to be both bronchodilator and anti-inflammatory with an improved therapeutic window over conventional xanthines such as Theophylline and the evidence supporting the effects of Doxofylline in the treatment of lung diseases
It has antitussive and bronchodilator[2] effects, and acts as aphosphodiesterase inhibitor.[3]
In animal and human studies, it has shown similar efficacy to theophylline but with significantly fewer side effects.[4]
Unlike other xanthines, doxofylline lacks any significant affinity for adenosine receptorsand does not produce stimulant effects. This suggests that its antiasthmatic effects are mediated by another mechanism, perhaps its actions on phosphodiesterase.[1]
Doxofylline, [7-(1, 3-dioxolan-2-ylmethyl)-3, 7-dihydro-1, 3-dimethyl-1H-purine-2, 6-dione] is a new bronchodilator xanthine based drug which differs from theophylline by the presence of dioxalane group at position 7. It is used in the treatment of bronchial asthma, chronic obstructive pulmonary disease (COPD), and chronic bronchitis . The mechanism of action is similar to that of theophylline in that it inhibits phosphodiesterase (PDE-IV), thereby preventing breakdown of cyclic adenosine monophosphate (cAMP). Increase in cAMP inhibits activation of inflammatory cells resulting in bronchodilating effect [52]. In contrast to theophylline, doxofylline has very low affinity towards adenosine A1 and A2 receptors which explain its better safety profile
Doxofylline (7-(l,3-dioxalan-2-ylmethyl)-theophylline) is a drug derived from theophylline which is used in therapy as a bronchodilator, with anti-inflammatory action, in reversible airway obstruction. It is commonly administered in doses ranging from 800 to 1200 mg per day, orally, according to a dosage which provides for the intake of two to three dosage units per day in order to maintain therapeutically effective haematic levels. The doxofylline tablets commercially available generally contain 400 mg of active ingredient and release almost all the drug within one hour from intake. The half- life of the drug is around 6-7 hours and for this reason several administrations are required during the 24-hour period.
Obviously a drop in haematic concentration of the drug in an asthmatic patient or patient suffering from COPD (chronic obstructive pulmonary disease) can result in serious consequences, in which case the patient must have recourse to rescue medication, such as salbutamol inhalers.
Pharmaceutical techniques for obtaining the modified release of drugs have been known for some time, but no modified release formulation of doxofylline is known. In fact the present inventors have observed that there are significant difficulties in the production of a doxofylline formula that can be administered only once a day and in particular have encountered problems correlated with bioequivalence.
Various attempts to formulate doxofylline in modified release systems, with different known polymers, have not provided the desired results, i.e. a composition that can be administered once a day, bio equivalent to the plasmatic concentration obtained with the traditional compositions currently on sale. In fact currently, dosage units containing 400 mg of active ingredient are currently administered two/three times a day for a daily average of approximately 1000 mg of active ingredient, a dosage considered necessary to maintain the therapeutic haematic levels of doxofylline.
Such a dosage unit is currently marketed by Dr. Reddy’s Laboratories Ltd as DOXOBID and has the following quali-quantitative composition: doxofylline (400 mg), colloidal silicon dioxide (13 mg), corn starch (63 mg), mannitol (40 mg), povidone (7 mg), microcrystalline cellulose (64 mg), talc (30 mg), magnesium stearate (3 mg) and water (0.08 ml).
Xanthine is a dioxypurine that is structurally related to uric acid. Xanthine can be represented by the following structure:

Caffeine, theophylline and theobromine are methylated xanthines. Methylated xanthines such as caffeine and theophylline are typically used for their bronchodilating action in the management of obstructive airways diseases such as asthma. The bronchodilator effects of methylxanthines are thought to be mediated by relaxation of airway smooth muscle. Generally, methylxanthines function by inhibiting cyclic nucleotide phosphodiesterases and antagonizing receptor-mediated actions of adenosine.
Theophylline can be represented by the following structure:

However, when administered intravenously or orally, theophylline has numerous undesired or adverse effects that are generally systemic in nature. It has a number of adverse side effects, particularly gastrointestinal disturbances and CNS stimulation. Nausea and vomiting are the most common symptoms of theophylline toxicity. Moderate toxicity is due to relative epinephrine excess, and includes tachycardia, arrhythmias, tremors, and agitation. Severe toxicity results in hallucinations, seizures, dysrhythmias and hypotension. The spectrum of theophylline toxicity can also include death.
Furthermore, theophylline has a narrow therapeutic range of serum concentrations above which serious side effects can occur. The pharmacokinetic profile of theophylline is dependent on liver metabolism, which can be affected by various factors including smoking, age, disease, diet, and drug interactions.
Generally, the solubility of methylxanthines is low and is enhanced by the formation of complexes, such as that between theophylline and ethylenediamine (to form aminophylline). The formation of complex double salts (such as caffeine and sodium benzoate) or true salts (such as choline theophyllinate) also enhances aqueous solubility. These salts or complexes dissociate to yield the parent methylxanthine when dissolved in aqueous solution. Although salts such as aminophylline have improved solubility over theophylline, they dissociate in solution to form theophylline and hence have similar toxicities.
Dyphylline is a covalently modified derivative of xanthine (1,3, -dimethyl-7-(2,3-dihydroxypropl)xanthine. Because it is covalently modified, dyphylline is not converted to free theophylline in vivo. Instead, it is absorbed rapidly in therapeutically active form. Dyphylline has a lower toxicity than theophylline. Dyphylline can be represented by the following structure:

Dyphylline is an effective bronchodilator that is available in oral and intramuscular preparations. Generally, dyphylline possesses less of the toxic side effects associated with theophylline.
U.S. Pat. No. 4,031,218 (E1-Antably) discloses the use of 7-(2,3-dihydroxypropyl)-1,3-di-n-propylxanthine, a derivative of theophylline, as a bronchodilator. U.S. Pat. No. 4,341,783 (Scheindlin) discloses the use of dyphylline in the treatment of psoriasis and other diseases of the skin by topical administration of dyphylline. U.S. Pat. No. 4,581,359 (Ayres) discloses methods for the management of bronchopulmonary insufficiency by administering an N-7-substituted derivative of theophylline, including dyphylline, etophylline, and proxyphylline.
At present, domestic synthetic Doxofylline composed of two main methods: one is by the condensation of theophylline prepared from acetaldehyde and ethylene glycol, but this method is more complex synthesis of acetaldehyde theophylline, require high periodate oxidation operation. Another is a halogenated acetaldehyde theophylline and ethylene glycol is prepared by reaction of an organic solvent, the method were carried out in an organic solvent, whereby the product Theophylline caused some pollution, conducive to patients taking.
current domestic Doxofylline synthetic methods reported in the literature are: 1, CN Application No. 94113971.9, the name “synthetic drugs Doxofyllinemethod” patents, the patent is determined by theophylline with a 2 – (halomethyl) -1,3 – dimethoxy-dioxolane in a polar solvent, with a base made acid absorbent,Doxofylline reaction step. 2, CN Application No. 97100911.2, entitled “Synthesis of Theophylline,” the patent, the patent is obtained from 7 – (2,2 – dialkoxy-ethyl) theophylline with ethylene glycol in N, N-dimethylformamide solvent with an alkali metal carbonate to make the condensing agent, p-toluenesulfonic acid catalyst in the condensation Doxofylline.
Doxofylline of xanthine asthma drugs, and its scientific name is 7 – (1,3 – dioxolan – ethyl methyl) -3,7 – dihydro-1,3 – dimethyl-1H – purine-2 ,6 – dione. The drug developed by the Italian Roberts & Co. in 1988, listed its tablet tradename Ansimar. This product is compared with similar asthma drugs, high efficacy, low toxicity, oral LD50 in mice is 1.5 times aminophylline, non-addictive. Adenosine and its non-blocking agents, it does not produce bronchial pulmonary side effects, no aminophylline like central and cardiovascular system. U.S. patent (US4187308) reported the synthesis of doxofylline, theophylline and acetaldehyde from ethylene glycol p-toluenesulfonic acid catalyst in the reaction of benzene as a solvent Doxofylline. Theophylline acetaldehyde by the method dyphylline derived reaction with a peroxy periodate or 7 – (2,2 – dialkoxy-ethyl) ammonium chloride aqueous solution in the decomposition of theophylline converted to acetaldehyde theophylline . Former method is relatively complex, and the high cost of using periodic acid peroxide, low yield after France. And theophylline acetaldehyde and ethylene glycol solvent used in the reaction of benzene toxicity, harm to health, and the yield is low, with an average around 70%, not suitable for industrial production.
SYN 1

Theophylline-7-acetaldehyde (I) could react with ethylene glycol (II) in the presence of p-toluenesulfonic acid in refluxing benzene to produce Doxofylline.
SYN 2

Doxofylline can be prepared by N-alkylation of theophylline (I) with bromoacetaldehyde ethylene glycol acetal (II) using Na2CO3 in refluxing H2O (1).
.…………………………………….
Synthesis
EXAMPLE
A mixture of 15 g of theophyllineacetic aldehyde, 30 ml of ethylene glycol and 1.5 g of p-toluenesulphonic acid in 600 ml of benzene is heated under reflux in a flask provided with a Marcusson apparatus.
After two hours the separation of the water is complete.
The reaction mixture is washed with 200 ml of a 3.5% aqueous solution of sodium bicarbonate.
The organic phase is dried and concentrated to dryness under reduced pressure, to leave a product residue which is taken up in ethyl ether, separated by filtration and purified by ethanol.
2-(7′-theophyllinemethyl)-1,3-dioxolane is obtained.
M.P. 144
Average yield 70%
Analysis: C.sub.11 H.sub.14 N.sub.4 O.sub.4 : M.W. 266.26: Calculated: C%, 49.62; H%, 5.30; N%, 21.04. Found: C%, 49.68; H%, 5.29; N%, 21.16.
………………………………..
the reaction is:
a, anhydrous theophylline and bromoacetaldehyde ethylene glycol as the basic raw material, purified water as a solvent with anhydrous sodium carbonate as acid-binding agent;
Doxofylline
UV (95% C2H5OH, nm) λmax273 (ε9230); λmin244 (ε2190)
IR (KBr, cm-1) 1134 (CO); 1233 (CN) ; 1547 (C = N); 1656 (C = C); 1700 (C = O); 2993 (CH)
1H-NMR [CDCl3, δ (ppm)] 3.399 (s, 3H, N-CH3); 3.586 (S, 3H, N-CH3); 3.815-3.885 (m, 4H, OCH2 × 2); 4.581 (d, 2H, CH2); 5.211 (t, 1H, CH ); 7.652 (S, 1H, CH = N)
13C-NMR [CDCL3, δ (ppm)] 27.88 (CH3); 29.69 (CH3); 47.87 (CH2); 65.37 ( OCH2); 100.76 (CH); 107.26 (C = C); 142.16 (CH = N); 148.22 (C = C); 151.59 (C = O); 155.25 ( C
……………………………
HPLC
http://www.scipharm.at/download.asp?id=1401
…………………..
- Cirillo R, Barone D, Franzone JS (1988). “Doxofylline, an antiasthmatic drug lacking affinity for adenosine receptors”. Arch Int Pharmacodyn Ther 295: 221–37.PMID 3245738.
- Poggi R, Brandolese R, Bernasconi M, Manzin E, Rossi A (October 1989). “Doxofylline and respiratory mechanics. Short-term effects in mechanically ventilated patients with airflow obstruction and respiratory failure”. Chest 96 (4): 772–8.doi:10.1378/chest.96.4.772. PMID 2791671.
- Dini FL, Cogo R (2001). “Doxofylline: a new generation xanthine bronchodilator devoid of major cardiovascular adverse effects”. Curr Med Res Opin 16 (4): 258–68.doi:10.1185/030079901750120196. PMID 11268710.
- Sankar J, Lodha R, Kabra SK (March 2008). “Doxofylline: The next generation methylxanthine”. Indian J Pediatr 75 (3): 251–4. doi:10.1007/s12098-008-0054-1.PMID 18376093.
- Dali Shukla, Subhashis Chakraborty, Sanjay Singh & Brahmeshwar Mishra. Doxofylline: a promising methylxanthine derivative for the treatment of asthma and chronic obstructive pulmonary disease. Expert Opinion on Pharmacotherapy. 2009; 10(14): 2343-2356, DOI 10.1517/14656560903200667, PMID 19678793
- Farmaco, Edizione Scientifica, 1981 , vol. 36, 3 pg. 201 – 219, mp 144 – 144.5 °C
- Drugs Fut 1982, 7(5): 301
| US6313131 | 16 feb 2000 | 6 nov 2001 | Upsher-Smith Laboratories, Inc. | Method of kidney treatment |
| US6348470 * | 20 maart 1998 | 19 feb 2002 | Korbonits Dezsoe | Antitussive compositions |
| US6423719 | 16 feb 2000 | 23 juli 2002 | Upsher-Smith Laboratories, Inc. | Method for treating benign prostate hyperplasia |
| CN101647776B | 2 sept 2009 | 20 april 2011 | 吴光彦 | Doxofylline venous injection with small volume as well as preparation method and quality control method thereof |
| DE3114130A1 * | 8 april 1981 | 28 jan 1982 | Abc Ist Biolog Chem Spa | Neue theophyllinylmethyldioxolan-derivate, verfahren zu ihrer herstellung und sie enthaltende pharmazeutische ansaetze |
| EP0272596A2 * | 16 dec 1987 | 29 juni 1988 | ISTITUTO BIOLOGICO CHEMIOTERAPICO “ABC” S.p.A. | Theophyllinemethyldithiolan and theophyllinemethyldithianyl derivates, a method for their preparation and pharmaceutical compositions in which they are included |
| WO2011146031A1 | 16 mei 2011 | 24 nov 2011 | Bilgic Mahmut | Pharmaceutical composition comprising n- acetylcysteine and a xanthine |
| WO2013055302A1 | 14 mei 2012 | 18 april 2013 | Mahmut Bilgic | Effervescent composition comprising n- acetylcysteine and doxophylline or theophylline |
………………………………………………………………………………………..
I n case Images are blocked on your computer, VIEW AT
14-chapter 4.pdf – Shodhganga
Although various bioanalytical methods for estimation of doxofylline in …. 1H and 13C-NMR spectra of doxofylline and its degradation products were recorded by….. CLICK ABOVE
SPECTRAL DATA

The ESI mass spectrum exhibited a protonated molecular ion peak at m/z 267 in positive ion mode indicating the molecular weight of 266. The tandem mass spectrum showed the fragment ions m/z 223, 181.2, 166.2, 138.1, 124.1 and 87.1.
The FT-IR spectrum, two strong peaks at 1697cm-1 and 1658cm-1 indicated presence of two carbonyl groups. A strong peak at frequency 1546cm-1 indicated presence of C=N stretch. A medium peak at 1232cm-1 was due to C-O stretch
FT IR
1H and 13C-NMR spectra of doxofylline and its degradation products were recorded by using Bruker NMR 300MHz instrument with a dual broad band probe and z-axis gradients. Spectra were recorded using DMSO-d6 as a solvent and tetramethylsilane as an internal standard.
4.2.6 Validation
1H NMR
13 C NMR
COMPARISONS
Drug spotlight- Zafirlukast

ZAFIRLIKAST 
cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate 107753-78-6
Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 (1990);
U. S. Patent No. 4,859,692;
U. S. Patent No. 5,993,859;
http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020547s031lbl.pdf
Zafirlukast is an oral leukotriene receptor antagonist (LTRA) for the maintenance treatment of asthma, often used in conjunction with an inhaled steroid and/or long-acting bronchodilator. It is available as a tablet and is usually dosed twice daily. Another leukotriene receptor antagonist is montelukast (Singulair), taken once daily. Zileuton (Zyflo), also used in the treatment of asthma via its inhibition of 5-lipoxygenase, is taken four times per day.
Zafirlukast blocks the action of the cysteinyl leukotrienes on the CysLT1 receptors, thus reducing constriction of the airways, build-up of mucus in the lungs andinflammation of the breathing passages.
Zafirlukast is marketed by Astra Zeneca with the brand names Accolate, Accoleit, and Vanticon. It was the first LTRA to be marketed in the USA and is now approved in over 60 countries, including the UK, Japan, Taiwan, Italy, Spain, Canada, Brazil, China and Turkey
Healthy young men who received a single oral 40 mg dose attained peak plasma zafirlukast concentrations that averaged 607 μg/L at 3.4 hours. The elimination half-life ranged from 12 to 20 hours. In another study involving a 20 mg single oral dose in healthy men, the elimination half-life averaged 5.6 hours.[1][2]
A letter was submitted to the FDA by Zeneca Pharmaceuticals on July 22, 1997, notifying them of a change in product labeling that includes the following potential reaction in patients undergoing a dosage reduction of oral steroids who are currently taking zafirlukast:
PRECAUTIONS-Eosinophilic Conditions: The reduction of the oral steroid dose, in some patients on ACCOLATE therapy, has been followed in rare cases by the occurrence of eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy sometimes presenting as Churg–Strauss syndrome, a systemic eosinophilic vasculitis. Although a causal relationship with ACCOLATE has not been established, caution is required when oral steroid reduction is being considered.1
NDA..020547 26/09/1996, ACCOLATE, ASTRAZENECA, 20MG TABLET
| US Patent No | Expirey Date | patent use code |
|---|---|---|
| 5482963 | Jan 9, 2013 | |
| 5612367 | Mar 18, 2014 | U-189 |
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | R03DC01 | C 31 H 33 N 3 O 6 S | 575.69 g / mol | 107753-78-6 |
| monohydrate | R03DC01 | C 31 H 33 N 3 O 6 S · H 2 O | 593.70 g / mol | 143052-93-1 |
| calcium (2: 1) | R03DC01 | C 62 H 64 CaN 6 O 12 S 2 | 1189.43 g / mol | 107753-86-6 |
Application
-
antihistamine effect
-
LTD4-antagonist
Classes of substances
-
Benzenesulfonamide (s -imidy), as well as their derivatives
-
Esters of carbamic acid
-
Cyclopentanes
-
Hydroxybenzoic acid amides, and hydroxy acids alkoksibenzoynyh
-
Indoles
-
-
-
-
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), with the chemical name 4(5-cyclopentyloxy-carbonylamino-1-methyl-indol-3ylmethyl)-3-methoxy-N-o-tolylsulfonylbenzamide. The molecular weight of zafirlukast is 575.7 and the structural formula is:

Zafirlukast, a fine white to pale yellow amorphous powder, is practically insoluble in water. It is slightly soluble in methanol and freely soluble in tetrahydrofuran, dimethylsulfoxide, and acetone.The empirical formula is: C31H33N3O6S
- Fischer JD, Song MH, Suttle AB, Heizer WD, Burns CB, Vargo DL, Brouwer KL. Comparison of zafirlukast (Accolate) absorption after oral and colonic administration in humans. Pharmaceut. Res. 17: 154-159, 2000.
- Bharathi DV, Naidu A, Jagadeesh B, Laxmi KN, Laxmi PR, Reddy PR, Mullangi R. Development and validation of a sensitive LC-MS/MS method with electrospray ionization for quantitation of zafirlukast, a selective leukotriene antagonist in human plasma: application to a clinical pharmacokinetic study. Biomed. Chromatogr. 22: 645-653, 2008.
- Zafirlukast (U.S. National Library of Medicine)
- Zafirlukast (patient information)

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate | |
| Clinical data | |
| Trade names | Accolate |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a697007 |
| Pregnancy cat. | B1 (Australia), B (United States) |
| Legal status | POM (UK) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | Unknown |
| Protein binding | 99% |
| Metabolism | Hepatic (CYP2C9-mediated) |
| Half-life | 10 hours |
| Excretion | Biliary |
| Identifiers | |
| CAS number | 107753-78-6 |
| ATC code | R03DC01 |
| PubChem | CID 5717 |
| IUPHAR ligand | 3322 |
| DrugBank | DB00549 |
| ChemSpider | 5515 |
| UNII | XZ629S5L50 |
| KEGG | D00411 |
| ChEBI | CHEBI:10100 |
| ChEMBL | CHEMBL603 |
| Chemical data | |
| Formula | C31H33N3O6S |
| Mol. mass | 575.676 g/mol |
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| United Kingdom | Akkolat | AstraZeneca |
| Italy | Akkoleit | – “- |
| Zafirst | Chiesi | |
| Japan | Akkolat | AstraZeneca |
| USA | – “- | Zeneca |
| Ukraine | No | No |
Formulations
-
Tablets of 20 mg, 40 mg

is a first anti-asthmatic leukotriene antagonist (Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 ‘(1990); U. S. Patent No. 4,859,692 and The Merck Index, 12th Edition, 10241). Methods for the preparation of Zafirlukast are described in J. Med. Chem., v. 33, 1781 (1990), U. S. Patent 4,859,692 and U.S. Patent 5,993,859 starting from methyl 3-methoxy-4-(l-methyl-5-nitroindol-3-ylmethyl)benzoate [la]



in the presence of an equivalent quantity of silver(I) oxide,

The above process has serious disadvantages in the isolation of the product [4] in step (b) which is due to the fact that alkylation of indole, that is unsubstituted at positions 1-, 2- and 3-, at the 3-position, is accompanied by the undesired process of poly alkylation, to form polysubstituted indoles of formula [7] and/or formula [8] :

while at the same time some quantity of the starting unreacted indole remains in the reaction mixture. Most common methods for the separation of alkyl (indol-3-ylmethyl)benzoate of formula [4] from by-products of polyalkylation and starting unreacted indole, which are all covalent compounds with similar physical properties, include column chromatography that is an unpractical method for industrial scale applications.
Formula (I) compound for the synthesis of an important intermediate of zafirlukast.Reported in the patent EP199543 synthesized compound (I) of the conventional method, the following formula:

(A) (I)
In this method, Intermediate A and 5 – nitro-indole silver oxide in the presence of a catalyst, for docking composite formula (I) compound. Reported only 45% of the reaction yield, the reaction is difficult to complete the reaction and post-treatment using chromatographic methods, resulting in product purification more difficult. And the use of more expensive silver oxide catalysts, high cost.
W00246153 reported a catalyst for the above reaction to zinc bromide, Compound (I), after treatment of the compound (I) with sodium hydroxide hydrolysis of the intermediate (B), separating the product and raw materials purification products.

The method reported in the literature a yield of 60%, but the actual operation is repeated only about 30% yield, and the operation is complicated, cumbersome and costly.
zaafirlukast is a selective and competitive receptor antagonist of leukotriene D4 and E4 (LTD4 and LTE4), components of slow-reacting substance of anaphylaxis (SRSA). Cysteinyl leukotriene production and receptor occupation have been correlated with the pathophysiology of asthma, including airway edema, smooth muscle constriction, and altered cellular activity associated with the inflammatory process, which contribute to the signs and symptoms of asthma.
The cysteinyl leukotrienes (LTC4 LTD4, LTE4) are the products of arachidonic acid metabolism and are various cells, including mast cells and eosinophills, these eicosinoids bind to cysteinyl leukotriene (CysLT) receptors. The CysLT type-1 (CysLT1) receptor is found in human airway and other pro-inflammatory cells. CysLTs have been correlated with the pathophysiology of asthma.
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), useful for the treatment of asthma and is commercially available in products sold under the brand name ACCOLATE™ as 10 and 20 mg tablets for oral administration. ACCOLATE™ is indicated for the prophylaxis and treatment of asthma in adults and children 5 years of age and older.
ACCOLATE™ film coated tablets contain amorphous zafirlukast as the active ingredient and the excipients croscarmellose sodium, lactose, magnesium stearate, microcrystalline cellulose, povidone, hypromellose, and titanium dioxide.
The greatest prevalence of asthma is in preschool children; however, the clinical utility of asthma therapy for this age group is limited by a narrow therapeutic index, long-term tolerability, and frequency and/or difficulty of administration. Asthma treatment requires an immediate perceivable effect. Inhalation therapy is a very common therapy prescribed for young children; inhalation therapy has the disadvantage of high dose variability.
 Process for the preparation of zafirlukast
Process for the preparation of zafirlukastUS 20040186300 A1



An Improved and Scalable Process for Zafirlukast: An Asthma Drug
Melting range: 142−145 °C; MS (m/z): 576 (M+ + H); IR (KBr, cm−1): 3326 (NH), 1679 (−C═O), 1H NMR (CDCl3) δ 7.0−8.0 (m, 11H), 3.7 (s, 3H), 4.0 (s, 2H), 3.9 (s, 3H), 2.6 (s, 3H), 1.45−1.8 (s, 9H). ……………………………………………………………….. US 20040186300 A1 http://www.google.com/patents/US20040186300 zafirlukast ethanolate as white powder with mp 132-133° C. (dec.) and 99.8% purity by HPLC. 1H NMR (CDCl3, δ, ppm): 1.22 (t, J 7.05 Hz, 3H), 1.45-1.87 (m, 8H), 2.66 (s, 3H), 3.67 (s, 3H), 3.73 (q, J 7.05 Hz, 4H), 3.79 (s, 3H), 3.98 (s, 2H), 5.08-5.23 (m, 1H), 6.58 (s, 1H), 6.73 (s, 1H), 7.01-7.51 (m, 9H), 8.23 (d, J 7.52 Hz, 1H), 9.67 (s, 1H).
Synthesis pathway
| Synthesis a) |
|---|
      |
| Synthesis of b) |
   |
-
Synthesis a)
-
US 4,859,692 (ICI; 08/22/1989; GB -prior. 4/17/1985; 17.10.1985).
-
EP 199 543 (ICI, Zeneca; appl. 16.4.1986; GB -prior. 4/17/1985).
-
-
Synthesis of b)
-
EP 490 649 (ICI, Zeneca; 11.12.1991; GB -prior. 12.12.1990).
-
Matassa, G. et al .: J. Med. Chem. (JMCMAR) 33, 1781 (1990).
-
Srinivas, K. et al .: Org. Process Res. Dev. (OPRDFK) 8 (6), 952 (2004).
-
added info Asthma is a disease that causes swelling and narrowing the airways of the lungs. Airways are air carriers to and from lungs. Swollen and narrower airways affect the air flow to and from the lungs and this lead to tightness of chest, wheezing, shortness of breath and cough. These symptoms are often occurs in early morning and in night. Asthma is caused by genetic and environmental factors, it was not curable completely but this can be controlled with good medical care. Leukotriene antagonists also known as leukast are the medicaments that are used to reduce leukotrienes, which are produced by several types of cells and causes inflammation in asthma and bronchitis. Leukotriene antagonists that are available in market are Montelukast, Zafirlukast and Pranlukast. Zafirlukast is the first leukast compound approved for management of Asthma. US FDA approved zafirlukast in the form of 10 mg and 20 mg tablet with the brand name of Accolate®.1 Subsequently this was approved and launched by innovator in few other countries. There are many synthetic routes for the preparation of Zafirlukast 4 is well documented in literature. Some of the key approaches are discussed here under. Scientists from ICI Americas Inc2 have reported process for the synthesis of 4, which starts with esterification of 3-methoxy-4-methyl benzoic acid 53 using methanol in presence of acetyl chloride PRODUCT PATENT ROUTE Allylic bromination of methyl ester 54 using bromine in presence of CCl4 resulted bromo compound 55, which was reacted with 5-nitro indole 124 using silver oxide as catalyst to obtain condensed compound 125. N-methylation of 125 utilizing methyl iodide in presence of NaH afforded N-methyl indole derivative 57. Thus obtained 57 was subjected to reduction using palladium carbon (Pd/C) in methanol followed by reacted with cyclopentyl chloroformate to obtain compound 59. Hydrolysis of 59 using LiOH.H2O subsequently reaction with o-toluene sulfonamide (OTSA) in presence of 1-[3-(dimethylamino)propyl]-3-ethyl carbodiimide hydrochloride (DMAPEC) and DMAP furnished zafirlukast 4. Matassa et al3 also reported similar procedure for the synthesis of Zafirlukast 4. 


Rare Disease Awareness Day: Phenylketonuria (PKU)
The National PKU Alliance (NPKUA) is celebrating December 3rd as National Phenylketonuria (PKU) Awareness Day to educate and spread awareness about the rare disease PKU. The organization has a webpage where there is a list of activities that one can participate in to help advocate for PKU.
A Norwegian doctor in 1934 discovers PKU. PKU is a rare, inherited metabolic disease where the body is not able to use the amino acid, Phenylalanine. The disease is caused by a deficiency of the liver produced enzyme Phenylalanine Hydroxylase (PAH). Without PAH, Phenylalanine builds up in the blood and poisons nerve cells in the brain. If PKU is not treated shortly after birth, it can be destructive to the nervous system, causing mental retardation. The disease is detectable after birth with appropriate blood testing during routine neonatal screening.
A mutation in a gene on chromosome 12 causes the disease. When this gene is…
View original post 275 more words
