
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


DARUNAVIR
206361-99-1 CAS NO
[(1S,2R)-3-[[(4-Aminophenyl)sulfonyl] (2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester
M. P.:- 72-74 °C (dec)
MW: 547.66
Darunavir and processes for its preparation are disclosed in EP0715618, W09967417, EP1725566 and Bioorganic & Medicinal Chemistry Letters (2004), 14(4), 959-963.
J Med Chem. 2013 May 23;56(10):4017-27. doi: 10.1021/jm400231v
US20050250845 discloses various pseudopolymorphs of darunavir and processes for their preparation. According to this application, “pseudopolymorph” is defined as a crystalline form of a compound in which solvent molecules are incorporated in the lattice structure. The Form B disclosed in the patent application is a pseudopolymorph wherein water is used as solvent. The thermogravimetric experiments of the Form B shows weight loss of 3.4% in the temperature range 25-78°C (water), 5.1% in the temperature range 25-1 10°C (ethanol and water) and further 1.1% weight loss (ethanol) in temperature range 110-200° C. Further at the drying step the Form B showed about 5.6% weight loss. The obtained dried product was hygroscopic and it adsorbed up to 6.8% water at high relative humidity. Amorphous form of darunavir is disclosed in US20050250845 and the publication in J.Org. Chem. 2004, 69, 7822 – 7829.
US 7700645 patent disclosed amorphous Darunavir, various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. Journal of Organic Chemistry 2004, 69, 7822-7829 disclosed amorphous Darunavir is obtained by purification with column chromatography in 2% methanol in chloroform as eluent. PCT publication WO2010086844A1 disclosed crystalline dimethylsulfoxide solvate and crystalline tetrahydrofuran solvate of darunavir. The publication also disclosed the amorphous darunavir having the IR spectrum with characteristic peaks at about 1454 and 1365 cm“1
PCT publication WO201 1083287A2 disclosed crystalline darunavir hydrate substantially free of any non aqueous solvent.
Drug information:- Darunavir is an Anti-microbial drug further classified as anti-viral agent of the class protease inhibitor. It is used either single or in combination with other drugs for the treatment of human immunodeficiency virus.
Darunavir (brand name Prezista, formerly known as TMC114) is a drug used to treat HIV infection. It is in the protease inhibitor class. Prezista is an OARAC recommended treatment option for treatment-naïve and treatment-experienced adults and adolescents.Developed by pharmaceutical company Tibotec, darunavir is named after Arun K. Ghosh, the chemist who discovered the molecule at the University of Illinois at Chicago. It was approved by the Food and Drug Administration (FDA) on June 23, 2006.[2]
Darunavir is a second-generation protease inhibitor (PIs), designed specifically to overcome problems with the older agents in this class, such as indinavir. Early PIs often have severe side effects and drug toxicities, require a high therapeutic dose, are costly to manufacture, and show a disturbing susceptibility to drug resistant mutations. Such mutations can develop in as little as a year of use, and effectively render the drugs useless.
Darunavir was designed to form robust interactions with the protease enzyme from many strains of HIV, including strains from treatment-experienced patients with multiple resistance mutations to PIs.
Darunavir received much attention at the time of its release, as it represents an important treatment option for patients with drug-resistant HIV. Patient advocacy groups pressured developer Tibotec not to follow the previous trend of releasing new drugs at prices higher than existing drugs in the same class. Darunavir was priced to match other common PIs already in use, such as the fixed-dose combination drug lopinavir/ritonavir.
PREZISTA (darunavir) is an inhibitor of the human immunodeficiency virus (HIV-1) protease.
PREZISTA (darunavir), in the form of darunavir ethanolate, has the following chemical name: [(1S,2R)-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]-carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester monoethanolate. Its molecular formula is C27H37N3O7S • C2H5OH and its molecular weight is 593.73. Darunavir ethanolate has the following structural formula:
 |
Darunavir ethanolate is a white to off-white powder with a solubility of approximately 0.15 mg/mL in water at 20°C.
|
|
4-11-2012
|
METHODS FOR THE PREPARATION OF HEXAHYDROFURO[2,3-b]FURAN-3-OL
|
|
|
12-28-2011
|
Substituted Aminophenylsulfonamide Compounds as Hiv Protease Inhibitor
|
|
|
12-23-2011
|
POLYMORPHS OF DARUNAVIR
|
|
|
12-14-2011
|
METHODS FOR THE PREPARATION OF N-ISOBUTYL-N-(2-HYDROXY-3-AMINO-4-PHENYLBUTYL)-P-NITROBENZENESULFONYLAMIDE DERIVATIVES
|
|
|
11-30-2011
|
Protease inhibitor precursor synthesis
|
|
|
6-31-2011
|
PROCESS FOR THE PREPARATION OF (3R,3AS,6AR)-HEXAHYDROFURO [2,3-B] FURAN-3-YL (1S,2R)-3-[[(4-AMINOPHENYL) SULFONYL] (ISOBUTYL) AMINO]-1-BENZYL-2-HYDROXYPROPYLCARBAMATE
|
|
|
9-29-2010
|
Aminophenylsulfonamide Derivatives as Hiv Protease Inhibitor
|
|
|
8-11-2010
|
Process for the preparation of (3R,3aS,6aR)-hexahydrofuro [2,3-b] furan-3-yl (1S,2R)-3[[(4-aminophenyl) sulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate
|
|
|
7-30-2010
|
RELATING TO ANTI-HIV TABLET FORMULATIONS
|
|
|
7-30-2010
|
COMBINATION FORMULATIONS
|
|
7-2-2010
|
METHODS AND INTERMEDIATES USEFUL IN THE SYNTHESIS OF HEXAHYDROFURO [2,3-B]FURAN-3-OL
|
|
|
5-7-2010
|
METHODS AND COMPOSITIONS FOR TREATING HIV INFECTIONS
|
|
|
4-21-2010
|
Pseudopolymorphic forms of a hiv protease inhibitor
|
|
|
9-21-2007
|
Immunoassays, Haptens, Immunogens and Antibodies for Anti-HIV Therapeutics
|
|
|
6-23-2006
|
Method for treating HIV infection through co-administration of tipranavir and darunavir
|
|
|
6-3-2005
|
Combination of cytochome p450 dependent protease inhibitors
|
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010086844A1 | Dec 8, 2009 | Aug 5, 2010 | Mapi Pharma Hk Limited | Polymorphs of darunavir |
| WO2011048604A2 * | Sep 16, 2010 | Apr 28, 2011 | Matrix Laboratories Limited | An improved process for the preparation of darunavir |
| WO2011083287A2 | Oct 6, 2010 | Jul 14, 2011 | Cipla Limited | Darunavir polymorph and process for preparation thereof |
| CN102584844A * | Jan 11, 2011 | Jul 18, 2012 | 浙江九洲药业股份有限公司 | Darunavir crystal form and method for preparing same |
| US6248775 | Apr 8, 1999 | Jun 19, 2001 | G. D. Searle & Co. | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| US7700645 | May 16, 2003 | Apr 20, 2010 | Tibotec Pharmaceuticals Ltd. | Pseudopolymorphic forms of a HIV protease inhibitor |
| Reference | ||
|---|---|---|
| 1 | JOURNAL OF ORGANIC CHEMISTRY vol. 69, 2004, pages 7822 – 7829 | |
| 2 | * | VAN GYSEGHEM E ET AL: “Solid state characterization of the anti-HIV drug TMC114: Interconversion of amorphous TMC114, TMC114 ethanolate and hydrate“, EUROPEAN JOURNAL OF PHARMACEUTICAL SCIENCES, ELSEVIER, AMSTERDAM, NL, vol. 38, no. 5, 8 December 2009 (2009-12-08), pages 489-497, XP026764329, ISSN: 0928-0987, DOI: 10.1016/J.EJPS.2009.09.013 [retrieved on 2009-09-24] |
Virus-encoded proteases, which are essential for viral replication, are required for the processing of viral protein precursors. Interference with the processing of protein precursors inhibits the formation of infectious virions. Accordingly, inhibitors of viral proteases may be used to prevent or treat chronic and acute viral infections. Darunavir has HIV protease inhibitory activity and is particularly well suited for inhibiting HIV-I and HIV -2 viruses. Darunavir, chemically (1 S^R.S’R.S’aS.e’aRJ-fS’he ahydrofuro^.S-b ]furanyl-[3-( 4-aminobenzenesulfonyl)isobutylamino [- 1-benzyl-zhydroxypropyl]carbamate. Darunavir is represented by the following structure:
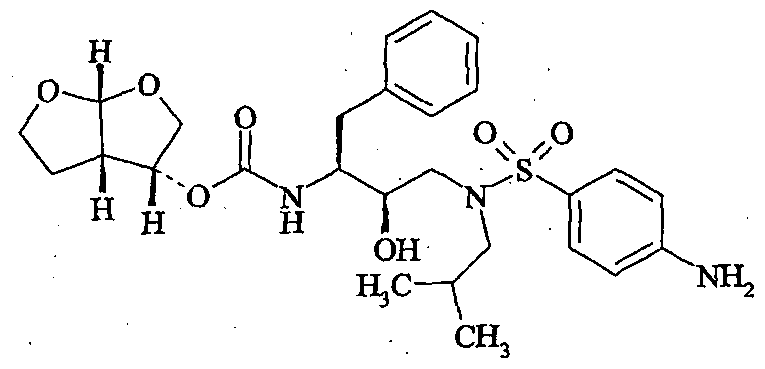
Darunavir and its pharmaceutically acceptable salts were disclosed in US 6248775 patent, wherein Darunavir is prepared by condensing 2R-hydroxy-3-[[(4-aminophenyl)sulfonyl](2- methylpropyl)amino]-1S(phenylmethyl)propylamine with hexahydro-furo[2,3-b]furan-3-ol in anhydrous acetonitrile in the presence of anhydrous pyridine and Ν,Ν’-disuccinimidyl carbonate at ambient temperature.
US 7700645 patent disclosed amorphous Darunavir, various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. Journal of Organic Chemistry 2004, 69, 7822-7829 disclosed amorphous Darunavir is obtained by purification with column chromatography in 2% methanol in chloroform as eluent. PCT publication WO2010086844A1 disclosed crystalline dimethylsulfoxide solvate and crystalline tetrahydrofuran solvate of darunavir. The publication also disclosed the amorphous darunavir having the IR spectrum with characteristic peaks at about 1454 and 1365 cm“1
PCT publication WO201 1083287A2 disclosed crystalline darunavir hydrate substantially free of any non aqueous solvent.
Darunavir Ethanolate, has the chemical name: [(1 S, 2R)-3-[[(4-aminophenyl) sulfonyl](2- methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3/?, 3aS, 6a/?)- hexahydrofuro[2,3-i>]furan-3-yl ester monoethanolate and has the following structural formula:

Darunavir and its process are first disclosed in US 6248775, wherein 2 ?-hydroxy-3-[[(4- aminophenyl)sulfonyl](2-methylpropyl)amino]-1 S(phenylmethyl) propylamine (4) is reacted with (3R, 3aS, 6aR)-hexahydrofuro[2,3- >]furan-3-ol in anhydrous acetonitrile in the presence of N, W-disuccinimidyl carbonate, anhydrous pyridine at ambient temperature followed by workup to get Darunavir (Scheme A).
Scheme A

Darunavir
US 20050250845 disclosed the various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. The same application disclosed the amorphous Darunavir by Raman spectra without process details.
WO 2005063770 discloses process for the preparation of Darunavir ethanolate, wherein 2R-hydroxy-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-1 S-(phenylmethyl)propyl amine (4) is reacted with (3R, 3aS, 6a ?)-hexahydrofuro[2,3-b]furan-3-ol in the presence of N, /V-disuccinimidyl carbonate, triethylamine, 41% methylamine in ethanol in a mixture of ethyl acetate and acetonitrile followed by workup and crystallization from ethanol to get Darunavir ethanolate (Scheme B).
Scheme B

In the prior art process, compound of formula 4 condensed with (3/?, 3aS, 6aR)- hexahydrofuro[2,3-6]furan-3-ol in large excess of solvent or solvent mixture containing large excess of base or mixture of bases to get Darunavir. Further, the obtained products by the processes described in the prior art are not satisfactory, from purity point of view. We have repeated the Darunavir synthetic procedures as described in the prior art and found that relatively large amounts of impurities were obtained along with Darunavir (Table-1) which need repeated crystallizations in different solvents to get desired quality of the final product resulting in poor yields. Among other impurities, the carbonic acid [(1/?,2S)-1-{((4-amino-benzenesulfonyl)-isobutyl-amino)-methyl}-2-((3R,3aSI6aR)- hexahydro-furot2,3-/3]furan-3-yloxycarbonylamino)-3-phenyl-propylester (3R,3aS,6aR)- hexahydro-furo[2,3-ft]furan-3-yl ester (difuranyl impurity of formula 1) is identified.


Conditions:-
i. Phenyl magnesium bromide, Cuprous cyanide, tetrahydrofuran, 23 °C, 1 h,
ii. t-Butyl hydroperoxide, titanium tetraisopropoxide, diethyl D-tartrate, dichloromethane, -22 °C, 24 h,
iii. Azidotrimethylsilane, titanium tetraisopropoxide, Benzene, reflux, 25 min,
iv. 2-Acetoxyisobutyryl chloride, Chloroform, 23 °C, 8 h,
v. Isobutyl amine, isopropanol, 80 °C, 12 h,
vi 4-aminobenzenesulfonyl chloride, aq. Sodium bicarbonate, dichloromethane, 23 °C, 12 h,
vii. 10% palladium on carbon, hydrogen gas (50 psi), methanol, acetic acid, tetrahydrofuran, room temperature, 2 h,
viii. [3R, 3aS,6aS]-3-hydroxyhexahydrofuro[2,3-b]-furan, disuccanamidyl carbonate, triethylamine, acetonitrile, 23 °C, 12 h
Schematic Representation for Synthesis of Darunavir
Preparation of Darunavir is described in US patent 05,158,713, and also in WO9967417 and WO9967254. Accordingly, 2-vinyloxirane 1 on reacting with phenyl magnesium bromide in presence of tetrahydrofuran solvent and cuprous cyanide catalyst give 4-phenylbut-2-ene-1-ol 2. Oxidizing 2 with t-Butyl hydroperoxide in presence of titanium tetraisopropoxide and diethyl D-tartrate using dichloromethane as solvent give [(3S)-3-benzyloxiran-2-yl]methanol 3.
Heating 3 with azidotrimethylsilane in presence of titanium tetraisopropoxide using benzene as solvent give (2S,3S)-3-azido-4-phenyl-butane-1,2-diol 4. The 1,2-dipl compound 4 underwent cyclization when treated with 2-acetoxyisobutyryl chloride in chloroform give (2S)-2-[(1S)-1-azido-2-phenyl-ethyl]oxirane 5, which was further heating with isobutylamine and isopropanol at higher temperature give (2R,3S)-3-azido-1-(isobutylamino)-4-phenyl-butan-2-ol 6. Compound 6 was reacted with 4-aminobenzenesulfonyl chloride in presence of aq. Sodium bicarbonate as base and dichloromethane as solvent resulting in to 4-amino-N-[(2R,3S)-3-azido-2-hydroxy-4-phenyl-butyl]-N-isobutyl-benzenesulfonamide 7.
Hydrogenating 7 with 10% palladium on carbon catalyst using hydrogen gas (50 psi) in methanol and tetrahydrofuran solvent in presence of small amount of acetic acid at ambient temperature resulted in to 4-amino-N-[(2R,3S)-3-amino-2-hydroxy-4-phenyl-butyl]-N-isobutyl-benzenesulfonamide 8. The final step involves reacting 8 with [3R,3aS,6aS]-3-hydroxyhexahydrofuro[2,3-b]-furan and disuccanamidyl carbonate in presence of triethylamine base and acetonitrile as solvent afford [(1S,2R)-3-[[(4-Aminophenyl)sulfonyl] (2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester also called Darunavir 9.
…………………………
http://www.google.com/patents/WO2013114382A1?cl=en
process for the preparation of amorphous Darunavir is as

Process for the preparation of intermediate 2 is as shown in below scheme.

Examples
Example -1 : Preparation of [(1S, 2S)-3-chloro-2-hydroxy-1-(phenyl methyl) propyl] carbamic acid tert-butyl ester (5).
The solution of (3S)-3-(tert-butoxycarbonyl) amino-1-chloro-4-phenyl-2-butanone (Chloromethyl ketone 6,100 g) and aluminium isopropoxide (35 g) in isoprpylalcohol was heated to mild reflux and maintained for 3 hours. After completion of reaction distilled off isopropyl alcohol up to 50 % under vacuum and the resultant mass was cooled to 25-35°C. Water was added to the distillate, pH was adjusted to 3.0-4.0 with acetic acid and maintained the stirring for 2 hours at 25-35°C. The obtained solid was filtered and washed with water. The wet cake was taken into isopropyl alcohol (400mL) and heated to reflux for 60minutes, the mass was cooled to 25-35°C again maintain the stirring for 60minutes, the obtained solid was filtered and washed with isopropyl alcohol. The wet product was dried under normal drying to get title compound 5 (yield 80 g). Example -2: Preparation of [(1 S, 2R)-3-[(2-methylpropyl) amino]-2-hydroxy-1- (phenylmethyl) propyl] carbamic acid tert-butyl ester (4).
The mixture of [(1S, 2S)-3-chloro-2-hydroxy-1-(phenylmethyl) propyl] carbamic acid tert-butyl ester (5,100 g), isobutyl amine (294 g), sodium carbonate (31.3 g) and water was heated to 60 – 65°C and maintained for 3hours. After completion of reaction water (200 mL) was added and distilled out excess isobutyl amine under vacuum at below 75°C. Water (800 mL) was added to the distillate, cooled to 25-35°C and stirred for 2 hours. The obtained solid was filtered and washed with water to get title compound 4 (yield 105 g).
Example -3: Preparation of [(1S, 2R)-3-[[(4-nitrophenyl) sulfonyl] (2-methylpropyl) amino]- 2-hydroxy-1-(phenylmethyl) propyl] carbmic acid tert-butylester (3).
[(1 S, 2R)-3-[(2-methylpropyl) amino]-2-hydroxy-1 -(phenyl methyl) propyl] carbamic acid tert-butyl ester (4, 100 gm) and triethylamine (39.04 g) was added to methylenedichloride (1200 mL) and the temperature was raised to 40°C. p-nitro benzene sulfonyl chloride solution (72.3g of p-NBSC dissolve in 300mL methylenedichloride) was added slowly at 40-45°C for 2-3 hrs. The reaction was maintained for 3hours at 40 – 45°C. After completion of the reaction, water (500 mL) was added, separated the organic layer and distilled out methylene dichloride at atmospheric pressure. Finally, strip out the methylene dichloride by using isopropyl alcohol (200 mL). Isopropyl alcohol (1000 mL) was added to the distillate and maintained the stirring for 60 minutes at 70- 80°C. Cooled the mass to 30 – 35°C, filtered and washed with Isopropyl alcohol to get title compound 3 (yield 145 g). Example – 4: Preparation of 4-Amino-N-(2R, 3S) (3-amino-2-hydroxy-4-phenylbutyl)-N- isobutyl-benzene sulfonamide (1).
(1S, 2R)-{1-benzyl-2-hydroxy-3-[isobutyl-(4-nitro-benzenesulfonyl)-amino]-propyl}-carbamic acid tert-butyl ester (3, 100g), 10% palladium carbon (10gm) and triethanolamine (2gm) were suspended in isopropyl alcohol. The reaction was heated to 40 – 45°C and maintained under 4 – 6kg/cm2 of hydrogen pressure for 3 hours. After completion of reaction, the mass was filtered and hydrochloric acid (70mL) was added to the filtered mass. The solution was heated to reflux and maintained for 2-3hours. After completion of reaction the mass was cooled to 25-35°C, the reaction mass pH was adjusted to 6.0 – 7.0 with 20% sodium hydroxide solution and distilled out isopropyl alcohol under vacuum at below 55°C. Ethanol (200mL) and water (400mL) was added to the distillate, the mass pH was adjusted to 9.0 – 10.0 with 20% sodium hydroxide solution at 25-35°C and maintained the stirring for 2 hours at 25-35°C. The mass was cooled to 0 – 5°C, filtered and wash with water. The wet product was taken into ethanol (350mL), maintained the stirring for 30minutes at reflux temperature. The mass was cooled to 2 – 4°C, stirred for 2 hours, filtered and washed with ethanol (50 mL). The wet product was dried under normal drying to get title compound 1 (Yield 60 g).
Example-5: Preparation of ethyl-2-(4,5-dihydrofuran-3-yl)-2-oxoacetate (VI).
2, 3-Dihydrofuran (250 g) was taken in toluene (2000 mL) and triethyl amine (505 g) was added to above solution. Ethyl oxalyl chloride (536.5 g) was slowly added to the above mixture by maintaining temperature at 25-30°C and maintained the stirring for 5 hours. After completion of reaction separated the organic layer, washed the organic layer with 8% sodium bicarbonate solution (2x500mL). Organic layer was distilled completely under vacuum to get title compound VI (Yield 560g).
1 H NMR : 1.38 (t, 3H), 2.93 (t, 2H), 4.34 (q, 2H), 4.63 (t, 2H), 8.02 (s, 1 H).
Example-6: Preparation of ethyl-2-(3-bromo-2-ethoxytetrahydrofuran-3-yl)-2-oxoacetate (V).
Ethyl-2-(4,5-dihydrofuran-3-yl)-2-oxoacetate (Vl, 100g) was dissolved in dichloromethane (500ml) and Ethanol (150mL) was added. The reaction mass was cooled to 5 to 10°C. N- bromosuccinimide (1 15 g) was added lot wise by maintaining temp below 10°C. Reaction mass was then stirred at 20-30°C till completion of reaction. Reaction mass was washed with sodium bicarbonate solution (2%, 3x400mL) and the organic layer was used for the next step.
Example-7: Preparation of hexahydrofuro [2, 3-b] furan-3-ol (IV).
To the solution of Ethyl-2-(3-bromo-2-ethoxy tetra hydrofuran-3-yl)-2-oxoacetate in dichloromethane (V, 500mL) as prepared in above example, sodium sulphite solution (225g was dissolved in 1700mL of water) was added at 25-35°C. Reaction mass was stirred for 5-8hours at the same temperature and separated the organic and aqueous layers. Organic layer was washed with water (340mL). Distilled out the solvent completely get ethyl-2-(2-ethoxy tetra hydrofuran-3- yl)-2-oxoacetate. Sodium borohydride (35.5g)was dissolved in ethanol (400mL) under nitrogen atmosphere, ethyl-2-(2-ethoxytetra hydrofuran-3-yl)-2-oxoacetate was dissolved in ethanol (100mL) and slowly added to above solution at 15-30°C. Reaction mass was heated to 30-45X, maintained for 5-8 hours, the reaction mass temperature was raised to 55°C and stirred for 8 hours. The reaction mass was cooled to 20-30°C, ammonium chloride solution (1 5g in 200mL water) was slowly added and stirred for 1-2hours. The reaction mass was filtered and filtrate was distilled out under vacuum to get residue. Dichloromethane (600mL) was added to residue and cooled to -10°C. Hydrochloric acid (85mL) was added slowly drop wise in 2 hours by maintaining temp -5 to 0°C, reaction mass was stirred for 60minutes at -5 to 0°C and distilled the solvent completely. The obtained residue was stripped out with isopropyl alcohol (2x200mL, 1x100mL), ethyl acetate (500mL) was added to the resultant residue, stirred for 30-60minutes and cooled to 10-15°C. The solution was filtered and filtrate was concentrated to get title compound IV (yield 56 g).
Example-8: Preparation of Hexahydrofuro [2, 3-b] furan-3-yl acetate (III).
Hexahydrofuro [2, 3-b] furan-3-ol (IV, 60g) was dissolved in dichloromethane (300mL) and cooled to 0-5°C. To the cooled solution triethylamine (58.2 g), N, N-dimethylaminopyridine (1.12g) was added, acetic anhydride (56.5g) was added for 30-60 minutes at the same temperature, the mass temperature was raised to 25-35°C and stirred for 2-4hours. After completion of reaction the mass was cooled to 10-20°C, water (120mL) was added, stirred for 30minutes, separated the organic layer, washed with 10% sodium chloride solution (120mL) and distilled out dichloromethane to get title compound (yield 72g). Further, the product was purified by fractional distillation to get pure Hexahydrofuro [2, 3-b] furan-3-yl acetate III (yield 54g).
1 H NMR : 1.9-2.09(m, 2H), 2.10(s, 3H), 3.0-3.1 (m, 1 H), 3.86-4.03(m, 2H), 3.73(dd, 1 H), 4.10(dd, 1 H), 5.19(m, 1 H), 5.72 (d, 1 H)
Example-9: Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-yl acetate (II). To the buffer solution (104.3g of sodium dihydrogen orthophosphate dissolved in 530mL of water & pH adjusted to 6.0-6.5 with saturated sodium bicarbonate solution(68g in 680 mL water) solution) hexahydrofuro [2, 3-b] furan-3-yl acetate (111,115g) and CAL-B (17.25g) was added at 25-35°C, heated to 38-45°C and stirred for 24 hours. CAL-B (17.25g) was added stirred for 16 hours, again CAL- B (11.5g) was added at 38-45°C and stirred for 16 hours (pH should maintain 6.0-6.5). The reaction mass was cooled to 20-30°C, methylenedichloride (1 150mL) was added to the mass and stirred for 30 minutes. The reaction mass was filtered through hyflowbed then separated the organic layer and washed with 10%sodiumchloride solution (575mL). Organic layer was distilled completely under vacuum to get title compound II (yield 40. Og). Example-10: Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-ol (I).
(3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-yl acetate (II, 14.0g) was dissolved in methanol (42mL). Potassium carbonate (0.34g) was added and stirred at 25-35°C for 6-8hours. Methanol was distilled out completely under vacuum, to the distillate methylenedichloride (28mL) was added, stirred the mass for 30 minutes and again distilled the solvent to get residue. Dissolved the residue in dichloromethane (56mL), the resultant solution was treated with carbon and the solvent was completely distilled out get title compound I (yield 10.5g). Example-11 : Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b]-furan-3-yl-4-nitrophenyl carbonate (2).
To the solution of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-ol (l,100g) and Bis-nitrophenyl carbonate (257.2g) in methylene dichloride (1200mL), triethylamine solution (132 g in 300 mL of methylene dichloride) was added slowly at 20-30°C for 2-3hours. Maintained the reaction at the same temperature for 8-10hours, after completion of reaction water (500mL) was added for 30- 60minut.es and settled the reaction mass then separated the organic layer. Organic layer was washed with 10% acetic acid (100mL) and 10% sodium chloride solution (500mL), distilled the organic layer and co distilled with ethyl acetate (100mL). Ethyl acetate (300mL) was added to the distillate and heated to 50-55°C for 30-45minut.es to get clear solution, the solution was cooled to 5-10°C and maintained at the same temperature for 60 minutes. The obtained solid was filtered, washed with ethanol (100mL) and dried the wet material at 40-45°C for 10-14 hours to get title compound 2 (yield 160g). Example-12: Preparation of dimethylformamide solvate of Darunavir.
To a mixture of 4-amino-N-(2r,3S)(3-amino-2-hydroxy-4-phenylbutyl)-N-lsobutyl- benzenesulfonamide (1 ,25g) and N-methyl-2-pyrrolidinone (NMPO, 50mL), a solution of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]-furan-3-yl-4-nitrophenyl carbonate (2, 8.85g) and N-methyl- 2-pyrrolidinone (75mL) was added at -5 to 0°C for 2 to 3 hours under nitrogen atmosphere. The mass temperature was slowly raised to 25 to 30°C and stirred for 6 to 8 hours. The reaction mass was quenched in to the solution of methylene chloride (125mL) and water (250mL) at 25-35°C for 30 to 45 minutes. Separated the organic layer followed by washed with 10% sodium carbonate solution (150mL), 10% sodium chloride solution (150mL) and with water (6x150mL). Organic layer was dried over sodium sulphate and distill off the solvent under vacuum at below 50°C to obtain darunavir as a residue. To the residue Ν,Ν-dimethyl formamide (50mL) was added and cooled to 0 to -5°C, water (25mL) was added to the solution and maintained for 12hours at 0 to -5 °C, the obtained solid was filtered and washed with pre-cooled mixture of N,N-dimethyl formamide & water (25mL+25mL) to get dimethylformamide solvate of darunavir.
Example-13: Preparation of non-solvated crystalline Darunavir.
To a mixture of 4-amino-N-(2r,3S)(3-amino-2-hydroxy-4-phenylbutyl)-N-lsobutyl- benzenesulfonamide (1, 25g) and N-methyl-2-pyrrolidinone (NMPO, 50mL), a solution of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]-furan-3-yl-4-nitrophenyl carbonate (2, 18.85g) and N- methyl-2-pyrrolidinone (75mL) was added at -5 – 0°C for 2 to 3 hours under nitrogen atmosphere. The mass temperature was slowly raised to 25 – 30°C and stirred for 6 to 8 hours. The reaction mass was quenched in to the solution of methylene chloride (250mL) and water (250mL) at 25- 35°C for 30 – 45 minutes. Separated the organic layer followed by washed with 10% potassium carbonate solution (5x125mL), water (5x125mL), 20% sodium chloride solution (25mL), finally washed with 20% citric acid solution (125mL). The organic layer was treated with carbon and distilled off the solvent under vacuum at below 50°C to obtain darunavir as a residue. To the residue ethylacetate (250mL) was added and cooled to 0 to -5°C, to the cooled solution hexane (225mL) was added and maintained for 12hours at 0 to -5 °C, the obtained solid was filtered, washed with pre-cooled mixture of ethylacetate and hexane (25mL+25mL) and dried the compound to get non-solvated crystalline darunavir(yield 25g).
Example -14: Preparation of Amorphous Darunavir.
Darunavir (200g) as obtained in above example was dissolved in methylene dichloride (10L) and washed with water (3×1000 mL). Organic layer was taken into agitated thin film dryer (ATFD) feed tank. Applied initial temperature about 36 – 40°C and high vacuum (580mm/Hg) to the vessel. Slowly feed the solution to the Vessel (feed rate 5L r) over 1hour finally given the methylene chloride (3L) flushing. The material is collected in the material collecter. Dried at 58 -62°C for 40 hours to get amorphous darunavir (yield 160g).
………………..
http://www.google.com/patents/WO2011048604A2?cl=en

Preparation of Durumvir ethanolate
A solution of (3R,3aS,6a ?)-hexahydrofuro[2,3-D]furan-3-yl 4-nitrophenyl carbonate (5b, 75.4 g) in A -methyl-2-pyrrolidinone (300 mL) was added to a pre-cooled (-2 ± 2°C) solution of the compound of formula 4 (100 g) in W-methyl-2-pyrrolidinone (200 mL) at -4 to 0°C over a period of 2 h. The temperature of the reaction mass was slowly raised to 25 – 30°C and maintained for 8 h. After completion of the reaction (TLC monitoring), ethyl acetate (1000 mL) and purified water (500 mL) were added to the reaction mass. The layers were separated; organic layer was washed with sodium carbonate solution (2 X 500 mL) followed by sodium chloride solution. The organic layer was concentrated; ethanol (300 mL) was added, heated to 45 – 50°C, maintained for 1 h, filtered and washed with ethanol. The wet compound was taken into a mixture of ethyl acetate- ethanol (7:93, 600 mL), heated to reflux, charcoal was added and filtered. The resultant filtrate was cooled to 0 – 5°C, filtered the separated solid and washed with ethanol. The wet compound was dried at 45°C to obtain the in 124.3 g (yield-82.5%). The obtained Darunavir ethanolate had purity of 99.79% on area by HPLC and contained 0.08% on area by HPLC of the difuranyl impurity. Preparation of Amorphous Darunavir
Example – 4
A solution of Darunavir ethanolate (200 g) in dichloromethane (10 L) was taken into ATFD Feed tank. The solvent was evaporated by fed the solution slowly to the ATFD Vessel (feed rate 5 L /h) at 36 – 40°C and high vacuum (580 mm/Hg) over 2 h and then flushed with dichloromethane (3 L). The material is collected in the material collector in 160g with the HPLC Purity of 99.60% and particle size D50 of approximately 50 micrometers and Dgo of approximately 100 to 180 micrometers. Example-5
Darunavir Ethanolate (200 gm) was dissolved in Methylene chloride (1000 ml) and solvent was evaporated by applying vacuum followed by isolation of amorphous Darunavir as a solid as such or by charging n-Heptane or Isopropyl ether. Example – 6
Darunavir Ethanolate (10 g) was dissolved in ethyl acetate (50 mL). The solution was heated to 40 – 45°C and maintained for 30 min. Ethyl acetate was distilled off under vacuum completely to get residue in the form of semisolid. n-Heptane (50 mL) was added to the residue and stirred for 30 min. at ambient temperature. The separated solid was filtered, washed the wet cake with n-heptane (5 mL) and dried at 40 – 45°C under vacuum to get 8.0 g of amorphous Darunavir.
Example – 7
Darunavir Ethanolate (10 g) was placed into a dry round bottom flask and heated to 110 – 120°C to melt and maintained under vacuum for 4 h. The reaction mass was slowly cooled to 25 – 35°C. The obtained glass type crystal was broken into powder to afford 8.5 g of amorphous Darunavir.
Example – 8
Darunavir Ethanolate (5.0 g) was suspended into glycerol (25 g), heated to 110 – 120°C under vacuum and maintained for 30min. Water (50 mL) was added to the cooled reaction mass at 25 – 35°C under stirring and the obtained suspension was stirred for 30 min at 25 – 35°C. The separated solid was filtered and dried at 40 – 45°C under vacuum to yield 3.5 g of amorphous Darunavir. Example – 9
Carbonic acid [(1 R,2S)-1-{((4-amino-benzenesulfonyl)-isobutyl-amino)-methyl}-2- ((3/?,3aS,6aR)-hexahydro-furo[2,3-ft]furan-3-yloxycarbonylamino)-3-phenyl-propylester (3R,3aS,6a ?)-hexahydro-furo[2,3- )]furan-3-yl ester (difuranyl impurity, 1).
The difuranyl impurity (1) isolated from the mother liquor by preparative HPLC using a mixture of formic acid-water (1 :99) as eluent. The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.

1H-NMR (DMSO-cfe, 300 MHz, ppm) – δ 0.79 (d, J=6.6 Hz, 6H, 15 & 15′), 1.14-1.20 (m, 1 H, 20Ha), 1.34-1.42 (m, 1 H, 20Hb), 1.75-1.85 (m, 2H, 20’Ha & 14), 1.94-2.01(m, 1 H, 20’Hb), 2.54-2.64 (m, 2H, 8Ha & 13Ha), 2.74-2.89 (m, 3H, 8Hb, 13Hb & 19), 3.00-3.11 (m, 2H, 5Ha & 19′), 3.34-3.39 (m, 1H, 5Hb), 3.54-2.63 (m, 3H, 21 Ha & 17Ha), 3.65-3.74 (m, 3H, 21’Ha, 21 Hb &17Hb), 3.81-3.89 (m, 2H, 21’Hb & 17’Ha), 3.94-4.04 (m, 2H, 7 & 17’Hb), 4.81-4.88 (m, 1 H, 6), 4.92-4.96 (m, 1 H, 18′), 5.03-5.10 (m, 1 H, 18), 5.11 (d, J=5.4 Hz, 1 H, 22′), 5.61 (d, J=5.1 Hz, 1 H, 22), 6.03 (brs, 2H, NH2, D20 exchangeable), 6.63 (d, J=8.7 Hz, 2H, 2 & 2″), 7.15-7.28 (m, 5H, 10H, 10Ή, 11 H, 11′ & 12), 7.40 (d, J=8.7 Hz, 2H, 3 & 3′), 7.55 (d, J=9.3 Hz, 1 H, NH, D20 exchangeable).
“H-NMR (DMSO-d6, 75 MHz, ppm)- δ 19.56 & 19.81 (15C & 15’C), 25.42 (20 ), 25.47 (20C), 26.28 (14C), 35.14 (8C), 44.45(19’C), 45.01 (19C), 49.21 (5C), 53.39 (7C), 57.55 (13C), 68.70 (21 ‘C), 68.74 (21C), 69.95 (17’C), 70.20(17C), 72.65 (6C), 76.27 (18C), 79.59 (18’C), 108.70 (22’C), 108.75 (22C), 112.69 (2C), 122.56 (4C), 126.12 (12C), 128.04 (11 C & 11’C), 129.03 (10C & 10’C), 129.08 (3C), 138.03 (9C), 152.99 (1C), 153.55 (16’C), 155.32 (16C).
DIP MS: m/z (%) 1108 [M+Hf, 1131 [M+Naf
……………
http://www.google.com/patents/US20130244297

According to the present invention Darunavir having the below impurity not more than 0.1, preferably 0.05%.

………….
DARUNAVIR


