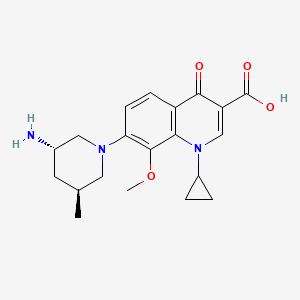
Graphical abstract

WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » Posts tagged 'QIDP' (Page 2)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
WCK 5222
DEC2015
Wockhardt has received Qualified Infectious Disease Product (QIDP) status for its new drug WCK 5222, a product from its breakthrough New Drug Discovery program in Anti Infectives from the US Food and Drug Administration (FDA).
This is the fourth product from the company to receive this coveted status. During last year, the company has received approval for WCK 771 & WCK 2349 and in early this year approval was received for WCK 4873. The only company globally to receive QIDP status for 4 drugs from US FDA.
Wockhardt is one of the few companies with end to end integrated capabilities for its products, starting with the manufacture of the oral and sterile API’s, the dose forms and marketing through wholly owned subsidiary in the US, enabling the company to capture maximum value.
Ten compounds generally represented by a general Formula (I) were used and are as follows:
(a) Sodium salt of ir ns-7-oxo-6-sulphooxy-l ,6-diazabicyclo[3.2.1]-octane-2-carbonitrile (Compound A);

(b) trans-sulphuric acid mono-[2-(5-carboxamido)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound B);

(c) trans-sulphuric acid mono-[2-(5-(piperidin-4-yl)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound C);

(d) trans-sulphuric acid mono-[2-(5-azetidin-3-ylmethyl-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound D);

(e) (25,5i?)-7-Oxo-6-sulphooxy-2-[N’-((i?)-piperidine-3-carbonyl)-hydrazinocarbonyl] -1,6-diaza-bicyclo[3.2.1]octane (Compound E);

(f) (25, 5i?)-7-Oxo-N-[(25)-pyrrolidin-2-ylmethoxy]-6-(sulfooxy)-l,6-diaza bicyclo [3.2.1] octane-2-carboxamide (Compound F);

(g) (25,5i?)-7-Oxo-6-sulphooxy-2-[N’-((i?)-pyrrolidine-3-carbonyl)-hydrazinocarbonyl]-l ,6-diaza -bicyclo[3.2.1]octane (Compound G);

(h) (25,5i?)-7-Oxo-N-[(25)-piperidine-2-ylmethyloxy]-6-(sulfooxy)-l ,6-diazabicyclo
octane-2-carboxamide (Compound H);

(i) trans-sulphuric acid mono-[2-(5-((5)-l-amino-ethyl)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound I); and

j) trans-sulphuric acid mono-[2-(5-((5)-pyrrolidin-2-yl)-[l,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound J).

////
 . CH3SO3H
. CH3SO3H
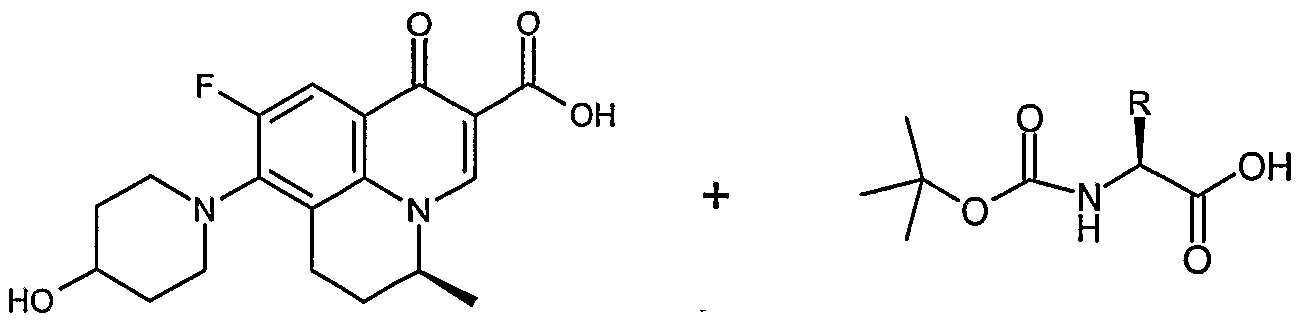
8-{4-[2(S)-Amino-propionyloxy] piperidine-l-yl}-9-fluoro-5 (S)-methyl-ό, 7-dihydro-l- oxo-lH, 5H-benzo[i,j]quinolizine-2-carboxylic acid of structural Formula I can be used to treat bacterial Gram-positive, Gram-negative and anaerobic infections; especially infections caused by resistant Gram-positive organism and Gram-negative organism, mycobacterial infections and emerging nosocomial pathogen infections.
Formula I
U.S. Patent Nos. 6,750,224 and 7,247,642 describes optically pure S-(-)-benzoquinolizine carboxylic acids, their derivatives, salts, pseudopolymorphs, polymorphs and hydrates thereof, their processes of preparation and their pharmaceutical compositions.
WO 2007102061
http://www.google.co.in/patents/WO2007102061A2?cl=en

Scheme 1
Experimental:
(S)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[ij] quinolizine-2-carboxylic acid was prepared as per procedure described in Chem. Pharm. Bull. 1996, 44(4), 642-645.
Example-l
Preparation of (2’S,5S)-9-fluoro-6,7-dihydro-8-(4-(N-tert-butoxycarbonyI-L-aIaninyl- oxy)-piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid:
Method-1 : To a mixture of N-tert-butoxycarbonyl-L-alanine (473 g) in dichloromethane (2 L), dicyclohexylcarbodiimide (515 g) dissolved in dichloromethane (2 L) was charged at -10 to 0 0C to provide a turbid suspension. To the turbid suspension, 300 g of (S)-9-fluoro-6,7- dihydro-8-(4-hydroxy-piperidin- 1 -yl)-5-methyl- 1-oxo- lH,5H-benzo[i,j]quinolizine-2- carboxylic acid was added followed by 4-N,N-dimethylamino pyridine (58 g) and the reaction mixture was stirred at -10 to 5 °C temperature over a period of 2 h. Suspension was filtered and solid was washed with 500 ml of dichloromethane. The filtrate was washed with water. Filtrate was dried over anhydrous sodium sulfate. Dried organic layer was then concentrated to its half volume where upon solid was precipitated. The solid was filtered and washed with 300 ml of dichloromethane. Clear organic filtrate was concentrated to dryness to provided an oily mass. Oily mass was triturated with diethyl ether (4 L) to provide white solid. The solid was filtered under suction and washed with diethyl ether (1 L) to provide title compound in 415 g (94%) quantity.
Method-2: To a mixture of triethylamine (98.0 ml) and N-tert-butoxycarbonyl-L-alanine (110 g) in tetrahydrofuran (1050 ml) and N,N-dimethyl formamide (350 ml) mixture, was added 2,4,6-trichlorobenzoyl chloride (100 ml). The resultant mixture was stirred at a temperature -5 to 0 °C for 5 h. To the > reaction mixture 4-N,N-dimethylamino pyridine (24g) and (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[i,j]quinolizine-2-carboxylic acid (70 g) was added. The reaction mixture was stirred for additional 7 h at -5 to 0 0C temperature. The suspension was filtered at room temperature and the filtrate was extracted with ethyl acetate after addition of water. The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 85 g quantity.
Method-3: To a solution N-tert-butoxycarbonyl-L-alanine (7.9 g) in tetrahydrofuran (75 ml) and N,N-dimethyl formamide (25 ml) mixture at -10 to 0°C was added methanesulfonyl chloride (2.42 ml) dropwise. To the above solution triethylamine (8.7 ml) was added dropwise over 5 min. the reaction was stirred for 1.5 h maintaining the temperature between at -10 to 0 0C. To the reaction mixture (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l- yl)-5-methyl-l-oxo-lH,5H-benzo[ij]quinolizine-2-carboxylic acid (5.01 g) and 4-N5N- dimethylamino pyridine (1.70 g) was added. The reaction mixture was stirred for additional 1 h at -5 to 0 °C temperature. The suspension was filtered at room temperature and the filtrate was diluted with water (300 ml) and extracted with ethyl acetate (150 ml x 2). The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 6.38 g (86%) quantity.
Example-2
Preparation of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-L-alaninyl-oxy-piperidin-l-yl)-5-methyl- l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonic acid salt:
To a mixture of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-N-tert-butoxycarbonyl-L-alaninyloxy- piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid (415 g) in acetone (4.5 L) was charged methanesulfonic acid (66 ml). Reaction mixture was stirred at 65-67 °C temperature for overnight. The suspension was filtered at 40-45 0C. Solid was washed with acetone (1.5 L) followed by diethyl ether (1.5 L). Off white solid was dried under 40 to 45 mm vacuum at 55-60 °C temperature over the period of 3-4 h. Title compound was obtained as a free flowing off white material 383.0 g (93%).
For MF: C23H30FN3O8S, MS (ES+) m/z 432 (obtained as free base for MF: C22H26FN3O5);
M.P. 278.50 0C by DSC


The tablets may optionally be coated with film forming agents and/or pharmaceutically acceptable excipients. Particularly suitable for use are commercially available coating compositions comprising film-forming polymers marketed under various trade names, such as Opadry® and Eudragit®. The coating layers over the tablet may be applied as solution/dispersion of coating ingredients using conventional techniques known in the art.
The present invention is further illustrated by the following examples which are provided merely to be exemplary of the invention and do not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Example 1 :
Table 1 provides the composition of batches of the present invention.
Table 1

Procedure: The compound of Formula I or pharmaceutically acceptable salts, esters or products thereof, lactose and croscannellose sodium were sifted and dry mixed in a rapid mixer granulator. The above mass was granulated by spraying aqueous solution of povidone. The granules were dried in a fluidized bed drier, sifted and oversize granules were milled in a Quadra mill. The resultant granules were mixed with talc, croscarmellose sodium, microcrystalline cellulose and sodium stearyl fumarate in a double cone blender. The lubricated granules were compressed into tablets using suitable tooling. Tablets were coated with aqueous dispersion of opadry.
Table 2 provides the dissolution data for the compound of formula I or pharmaceutically acceptable salts, esters or products thereof tablets prepared as per the formula given in Table 1. For determination of drug release rate, USP Type 2 Apparatus (rpm 50) was used wherein 0.1 N hydrochloric acid (900 ml) was used as a medium. Table 2: Dissolution data




| WO1991012815A1 * | Feb 25, 1991 | Sep 5, 1991 | Squibb Bristol Myers Co | COMPOSITIONS AND METHODS FOR TREATING INFECTIONS CAUSED BY ORGANISMS SENSITIVE TO β-LACTAM ANTIBIOTICS |
| WO2000068229A2 * | May 8, 2000 | Nov 16, 2000 | S K Agarwal | (s)-benzoquinolizine carboxylic acids and their use as antibacterial agents |
| WO2001085095A2 * | May 3, 2001 | Nov 15, 2001 | Shiv Kumar Agarwal | Chiral fluoroquinolizinone arginine salt forms |
| WO2002009758A2 * | Jul 31, 2001 | Feb 7, 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| EP2062582A1 * | Aug 14, 2007 | May 27, 2009 | Tianjin Hemey Bio-Tech Co., Ltd. | The antibiotics composition comprising beta-lactam antibiotics and buffers |
| US4524073 * | Jul 20, 1983 | Jun 18, 1985 | Beecham Group P.1.C. | β-Lactam compounds |
| US6465428 * | Aug 25, 2000 | Oct 15, 2002 | Aventis Pharma S.A. | Pharmaceutical combinations based on dalfopristine and quinupristine, and on cefepime |
| US20040254381 * | Aug 15, 2003 | Dec 16, 2004 | Day Richard A. | Antibiotic compositions and methods of using the same |
| US20050148571 * | Nov 29, 2002 | Jul 7, 2005 | Nancy Niconovich | Method of treating bacterial infections using gemifloxacin or a salt thereof and a betha-Lactam antibiotic |
| US20090148512 * | Apr 17, 2008 | Jun 11, 2009 | Lannett Co Inc | Novel uses of chloramphenicol and analogous thereof |
| US20090232744 * | Feb 26, 2009 | Sep 17, 2009 | Pari Pharma Gmbh | Macrolide compositions having improved taste and stability |
| WO2002009758A2 * | 31 Jul 2001 | 7 Feb 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| US6750224 | 17 Aug 2000 | 15 Jun 2004 | Wockhardt Limited | Antibacterial optically pure benzoquinolizine carboxylic acids, processes, compositions and methods of treatment |


Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.

///////////keywords USFDA, Qualified Infectious Disease Product status, Wockhardt, drugs, WCK 2349, QIDP
 STEREOCENTERS SHOWN
STEREOCENTERS SHOWN
There is an urgent medical need for novel antibacterial agents to treat hospital infections, specially those caused by multidrug-resistant Gram-positive pathogens. The need may also be fulfilled by either exploring antibacterial agents having new mechanism of action or expanding known classes of antibacterial drugs. The paper describes a new chemical entity, compound 21, derived from hitherto little known “floxacin”. The choice of the entity was made from a series of synthesized prodrugs and salts of the active chiral benzoquinolizine carboxylic acid, S-(−)-nadifloxacin. The chemistry, physicochemical characteristics, and essential bioprofile of 21 qualifies it for serious consideration as a novel drug entity against hospital infections of multi-drug-resistant Staphylococcus aureus, and its progress up to clinical phase I trials in humans is described.
S-(−)-Nadifloxacin is S-(−)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-1-yl)-5-methyl-1-oxo-1H,5H-benzo[i,j] quinolizine-2-carboxylic acid (1). Prodrugs and aqueous soluble salts of 1were synthesized and explored for possible use in parenteral or oral formulations………….De Souza, N. J.; Agarwal, S. K.; Patel, M. V.; Bhawsar, S. B.; Beri, R. K.; Yeole, R. D.; Shetty, N.; Khorakiwala, H. F. Chiral Fluoroquinolone Arginine Salt Form. US patent 6,514,986, 2003.
quinolones has now grown to four generations, the first generation to nalidixic acid is represented as the representative of the second generation to PPA, only the Gram-negative bacteria effectively, the third generation is the development of these drugs the peak period, there has been a lot of drugs, and is a broad-spectrum antibiotic, which to norfloxacin, ciprofloxacin and other representatives. The fourth-generation quinolone antibiotics is in the third generation on the basis of a broad spectrum of antibacterial spectrum further expanded to make it available against mycoplasma and chlamydia infections.
[0003] R & D has been relatively popular domestic antibiotics, the most widely used on the market today is the third generation fluoroquinolones. Nadifloxacin developed by the Japanese company Otsuka, belongs to the third-generation quinolone antibacterial drugs, topical treatment of acne and folliculitis. 1993 for the first time in Japan (trade name: Acuatim), 2004 in the German market (trade name: Nadixa), 2005 in China listed (trade name: By Union, ointment).
[0004] nadifloxacin irritation due to its absorption and vascular problems, only made of topical formulations for in vitro Propionibacterium acnes (propionibacterium acnes) caused by acne. Wherein the S-(-) – that is the main role difloxacin isomer, the antibacterial activity of the R-isomer of 64 to 256 times that of racemic 2 times.
[0005] fine that gatifloxacin is S-(-) _ nadifloxacin salt on the basis of the system.Significantly improved solubility nadifloxacin well absorbed by the body, so it retains nadifloxacin broad spectrum antimicrobial, antibacterial activity, especially methicillin-sensitive Staphylococcus aureus and methicillin-resistant Staphylococcus aureus Effective characteristics (Antimicrobial Agents and Chemotherapy, 2004,3188 ~ 31920; J. Med. Chem. 2005 (48), 5232 ~ 5242). Pre-clinical tests prove that the product on the market anti-methicillin-resistant Staphylococcus aureus Antibiotic better compare the efficacy, including vancomycin, trovafloxacin, quinupristin + dalfopristin, linezolid amine.
[0006] fine molecular structure that gatifloxacin following formula:
[0007]

[0008] S-(-) _ nadifloxacin (C19H21FN2O4) with L-arginine salt, the further improve the play a major role in antibacterial s-(-) – nadifloxacin isomer content, and improved oral bioavailability, so that it can develop an oral or injectable preparations.
[0009] the literature (J. Med. Chem. 2005 (48), 5232 ~ 5242) discloses the synthesis of S_ (_) _ Nadifloxacin-L-arginine salt, S-(-) _ that fluoride gatifloxacin and L-arginine salt in the reaction solvent system, which solvent system is mainly methanol – water system, according to the paper reported in S-(-) – Nadifloxacin-L-arginine salt, yields were and related substances are not high enough.
Example 1
[0026] In equipped with oil bath, magnetic stirrer, thermometer, reflux condenser flask at 25 ° C was added (S) – (-) – nadifloxacin (100. 0g, 278mmol), dioxane ring (300ml), and the reaction solution was added dropwise to the L-arginine 4g, 278mmol) in distilled water (250ml) was added. Then heated to 50_60 ° C stirred 1.5 hours, and then adding activated carbon (3. Og) for 5 minutes, filtered hot, and then added dropwise at 55-60 ° C dioxane (700ml), and the natural cooling to 30 -35 ° C for 2 hours crystallization. The solid was collected by filtration and acetone (IOOml) wash. Dried at room temperature M hours. To give a white solid 137g, yield: 92%.
Example 1
Preparation of the single crystal of S-(- -9-fluoro-6,7-dihvdro-8-(4-hvdroxypiperidin-l-ylV5- methyl-l-oxo-lH,5H-benzo[“i,ι‘lquinolizine-2-carboxylic acid L-arginine salt terahvdrate.
S-(-)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[i,j]quinolizine-2-carboxylic acid L-arginine salt (1.0 g) was dissolved in a mixture of acetone (40 ml) and water (10 ml) by heating the suspension at 70 °C for 15 minutes. The clear solution thus obtained was left for slow evaporation at room temperature in a beaker covered with a perforated aluminum foil. The crystal formation started after 2 days. Finally the single crystal was selected for X-ray crystal analysis from a cluster left after complete evaporation of the solvent. The ORTEP diagrams are described in Figures 1 and 2.
EXAMPLE 1
S-(-)-9-Fluoro-6,7-dihvdro-8-(4-hvdroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo Ti l quinolizine-2-carboxylic acid arginine salt Synthesis of SubstantiaUy CrystaUine product A solution of L-(+)-arginine (48.372 g, 0.278 mole) in distilled water (600 ml) was added dropwise over a period of 30 min to the stirred solution/suspension of finely powdered S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo [ij] quinolizine-2-carboxylic acid (100 g, 0.278 mole) in acetone (1250 ml). The obtained clear solution was stirred for 30 min and concentrated on a water bath in vacuum (175 mbar) at 80°C. When product started solidifying, the concentration was carried out in vacuum (50 mbar) at 80°C up to dryness. Hexane (1 liter) was added, the reaction mixture was stirred for 4 hr, the solid thus separated was filtered and dried in vacuum (0.7 mbar) for 12 hrs at 70 °C. Yield 145 g (96.9%), m.p. 238-242 °C, and solubility 6 mg/ml (pH 9.5 buffer solution).
The substantially crystalline S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5- methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid arginine salt prepared according to Example 1 possesses the following properties: a) Crystalline form, with a degree of crystallinity as determined by X-ray powder diffraction and as shown in Fig. 1. , b) A thermogram as determined by Differential scanning calorimetry and as shown in Fig. 3. c) Particle size measured as mean mass diameter (MMD) of 83.92 μm, as determined by laser diffraction technique. d) Density of 0.51 g/cm3 (untapped) and 0.7 g/cm3 (tapped). e) Hygroscopicity of 0% increase of weight upon storage for 14 days up to 22% relative atmospheric humidity as determined gravimetricaUy. f) A content of moisture water of 0.1 % by weight as determined by titration according to Karl Fischer. g) A content of acetone of 0.014 % by weight as determined by gas chromatography
Example 1
S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyI-l-oxo-lH,5H-benzo [ij] quinolizine-2-car boxy lie acid anhydrate
Method A
S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yI)-5-methyl-l-oxo-lH,5H-benzo [ij] quinoIizine-2-carboxylic acid (3.0 g) obtained according to the process described in literature [K Hashimoto et al., Chem.Pharm.Bull.44, 642-5(1996)] was dissolved in acetonitrile (250 ml) at 85 °C. The resulting clear solution was filtered (to remove if any fibrous material is in suspension). The filtrate was concentrated to 125 ml and left at room temperature for crystallization. The crystals thus separated were filtered and dried in a drying cabinet at 40 °C for 2 hr in vacuum at 50 mm of Hg to obtain constant weight. Yield 2.6 g (86%).
Method B:
S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyI-l-oxo-lH,5H-benzo [ij] quinolizine-2-carboxyIic acid (2.0 g) obtained according to the process described in literature [K.Hashimoto etal., Chem.Pharm.Bull.44, 642-5(1996)] was dissolved in ethyl alcohol (95 %; 200 ml) at 80 °C. The obtained clear solution was filtered (to remove if any fibrous material is in suspension), concentrated to 100 ml and left for crystallization. The separated solid was Altered and dried in a drying cabinet at 40 °C for 3 hr in vacuum at 50 mm of Hg to obtain constant weight. Yield 1.7 g (85 %).
M.p.258-62 °C, moisture content 0 % (by Karl Fisher method) [CXJD 26 -299°, HPLC purity 99.8%
Example 8
S-(-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyI-l-oxo-lH,5H-benzo [ij] quinolizine-2-carboxylic acid, L-arginine salt 0.75 hydrate
L-(+)-Arginine (0.958 g., 5.5 mmoles) was added in portions to a suspension solution of S- (-)-9-fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo [ij] quinoIizine-2-carboxyIic acid 0.2 hydrate (2.0 g., 5.5 mmole) in methanol (400 ml). The obtained solution was concentrated in vacuum to give the desired product as a yellow solid, which was dried at 50 °C at 50 mm/Hg for 5 hours. Yield 3.0 g. (100%), m.p. 220- 223 °C (dec), m/z 535 (M+H), moisture content 2.3% (by Karl Fisher, required 2.46%), [CIJD 25 -144 ° (1% methanol c=l), solubility 93 mg/ml.
……………………………..
https://www.jstage.jst.go.jp/article/cpb1958/44/4/44_4_642/_article
(S)-(-)-Nadifloxacin [(S)-(-)-9-fluoro-6, 7-dihydro-8-(4-hydroxy-1-piperidyl)-5-methyl-1-oxo-1H, 5H-benzo[i, j]quinolizine-2-carboxylic acid, (S)-(-)-OPC-7251], an antibacterial agent, was synthesized from (S)-(-)-5, 6-difluoro-2-methyl-1, 2, 3, 4-tetrahydroquinoline (DFTQ), which was prepared by the optical resolution of recemic DFTQ with 2, 3-di-O-benzoxyl-L-tartaric acid. Racemization of the undesired enantiomer [(R)-(+)-DFTQ] was studied in the presence of various acids and the best result was obtained in the case of methanesulfonic acid. The absolute configuration of (-)-nadifloxacin was determined as S by X-ray crystallographic analysis.
https://www.jstage.jst.go.jp/article/cpb1958/44/4/44_4_642/_pdf ………..FREE PDF

Ishikawa, H.; Tabusa, F.; Miyamoto, H.; Kano, M.; Ueda, H.; Tamaoka, H.; Nakagawa, K. Studies on antibacterial agents. I. Synthesis of substituted 6,7-dihydro-1-oxo-1H,5H-benzo[i,j]-quinolizine-2-carboxylic acids. Chem. Pharm. Bull. 1989, 37, 2103-2108.
(b) Kurokawa, I.; Akamatsu, H.; Nishigima, S.; Asada, Y.; Kawabata, S. Clinical and Bacteriologic Evaluation of OPC-7251 in Patients with Acne: A Double Blind Group Comparison Study vs Cream Base. J. M. Acad. Dermatol. 1991, 25, 674−81.
(c) Morita, S.; Otsubo, K.; Matsubara, J.; Ohtnai, T.; Uchida, M. An Efficient Synthesis of a Key Intermediate towards (S)-(−)-Nadifloxacin. Tetrahedron: Asymmetry 1995, 6 (1), 245−254.
(7) (a) Patel, M. V.; Gupte, S. V.; Sreenivas, K.; Chugh, Y.; Agarwal, S. K.; De Souza, N. J. S-(−)-Nadifloxacin: Oral Bioavailbility and Bioefficacy in Mouse Model of Staphylococcal Septicemia. Abstract of Papers, 40th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Diego, CA, September 2000; American Society for Microbiology: Washington, DC, 2000; Poster F-558.
 A Chiral Benzoquinolizine-2-carboxylic acid Arginine Salt Active against Vancomycin Intermediate Staphylococcus aureus (VISA). Abstract of Papers, 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, September 2003;American Society for Microbiology: Washington, DC, 2003; Poster F-430
A Chiral Benzoquinolizine-2-carboxylic acid Arginine Salt Active against Vancomycin Intermediate Staphylococcus aureus (VISA). Abstract of Papers, 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, September 2003;American Society for Microbiology: Washington, DC, 2003; Poster F-430Some quinolones introduced for clinical use.

Radezolid
869884-78-6 cas no
http://www.ama-assn.org/resources/doc/usan/radezolid.pdf
Rib-X Pharmaceuticals
Phase II completed
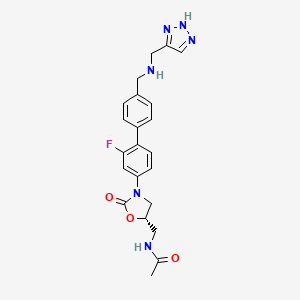
N-{[(5S)-3-(2-fluoro-4′-{[(1H-1,2,3-triazol-5-ylmethyl)amino]methyl}biphenyl-4-yl)-2-oxo-1,3-oxazolidin-5-yl]methyl}acetamide
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide
Rib-X Pharmaceuticals has completed two Phase II clinical trials of radezolid for the treatment of pneumonia and uncomplicated skin infections. The trial completion dates were in 2008 and 2009, but to date the Phase III trials have not been initiated [1-6].
Radezolid (INN, codenamed RX-1741) is a novel oxazolidinone antibiotic being developed by Rib-X Pharmaceuticals, Inc. for the treatment of serious multi-drug–resistant infections. Radezolid has completed two phase-II clinical trials. One of these clinical trials was for uncomplicated skin and skin-structure infections (uSSSI) and the other clinical trial was for community acquired pneumonia (CAP).
Oxazolidinone antibiotics are a relatively new class of antibacterial agents with activity against a broad spectrum of gram-positive pathogens. The first member of this new class to be commercialized, linezolid, was approved in 2000. Since that time the development of linezolid resistant organisms has prompted efforts to discover more effective members of the oxazolidinone class.
A new family of biaryl oxazolidinone antibacterials with activity against both linezolid-susceptible and -resistant Gram-positive bacteria, as well as certain Gram-negative bacteria has been reported (see Bioorganic & Medicinal Chemistry Letters, 2008, 18, 6175-6178, and PCT Patent Publication WO 2005/019211).
Among the known biaryloxazolidinones is N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-bipheny- l-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide, more commonly known as radezolid (RX-1741), currently being developed for multi-drug-resistant infections.
Although a monohydrochloride salt of radezolid was disclosed in PCT Patent Publication WO 2006/133397, there is a continuing need for new salts and polymorphs thereof having improved properties such as solubility to optimize bioavailability on therapeutic administration.
Synthesis 1
http://www.google.co.il/patents/WO2005019211A2?hl=iw&cl=en
Scheme A

Scheme B

Scheme C

Scheme D

Scheme E


Scheme G


Scheme I

Scheme J

producing compounds of the present invention. Known iodoaryl oxazolidinone intermediate 50 (see U.S. Patent Nos. 5,523,403 and 5,565,571) is coupled to a substituted aryl boronic acid (the Suzuki reaction) to produce biaryl alcohol 51. Mesylate 52, azide 53, and amine 54 are then synthesized using chemistry well known to those skilled in the art. Scheme 1

NaN3, DMF, 70 °C


NO 2
http://www.google.com/patents/US20100234615
| TABLE 1 | |
| Compound | |
| Number | Structure |
| 1 |
 |
Example 1 Synthesis of Compound 1
Compound 1 and its hydrochloride salt are synthesized according to the following Scheme:


4-Methoxybenzyl Azide
1001.
A solution of 4-methoxybenzyl chloride 1000 (51.8 g, 331.0 mmol) in anhydrous DMF (200 mL) was treated with solid sodium azide (21.5 g, 331.0 mmol, 1.0 equiv) at 25° C., and the resulting mixture was stirred at 25° C. for 24 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was quenched with H2O (400 mL) and ethyl acetate (EtOAc, 400 mL) at room temperature.
The two layers were separated, and the aqueous layer was extracted with EtOAc (200 mL). The combined organic extracts were washed with H2O (2×200 mL) and saturated NaCl aqueous solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude 4-methoxybenzyl azide (51.2 g, 53.95 g theoretical, 94.9% yield) was obtained as colorless oil, which by HPLC and 1H NMR was found to be essentially pure and was directly used in the subsequent reaction without further purifications. For 4-methoxybenzyl azide 1001:
1H NMR (300 MHz, CDCl3) δ 3.84 (s, 3H, ArOCH3), 4.29 (s, 2H, Ar—CH2), 6.96 (d, 2H, J=8.7 Hz), 7.28 (d, 2H, J=7.8 Hz).
C-[1-(4-Methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-Methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine
(1003 and 1004).
A solution of 4-methoxybenzyl azide 1001 (61.2 g, 375.5 mmol) in toluene (188 mL) was heated with propargylamine 1002 (commercially available, 30.97 g, 38.6 mL, 563.0 mmol, 1.5 equiv) at 25° C., and the resulting reaction mixture was warmed up to gentle reflux at 100-110° C. for 21 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was cooled down to room temperature before being concentrated in vacuo to remove the excess amount of propargylamine and solvent.
The oily residue was then treated with 30% ethyl acetate-hexane (v/v, 260 mL), and the resulting mixture was warmed up to reflux and stirred at reflux for 30 min before being cooled down to room temperature for 1 h. The pale-yellow solids were then collected by filtration, washed with 30% ethyl acetate-hexane (v/v, 2×100 mL), and dried in vacuo at 40° C. for overnight to afford the crude, cycloaddition product (78.8 g, 81.75 g theoretical, 96.4%) as a mixture of two regioisomers, C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004), in a ratio of 1.2 to 1 by 1H NMR.
The crude cycloaddition product was found to be essentially pure and the two regioisomers were not separated before being used directly in the subsequent reaction without further purification. For 1003 and 1004:
1H NMR (300 MHz, DMSO-d6) δ 1.82 (br. s, 2H, NH2), 3.72 and 3.73 (two s, 3H, Ar—OCH3), 5.47 and 5.53 (two s, 2H, ArCH2), 6.89 and 6.94 (two d, 2H, J=8.7 Hz, Ar—H), 7.17 and 7.29 (two d, 2H, J=8.7 Hz, Ar—H), 7.58 and 7.87 (two br. s, 1H, triazole-CH); C11H14N4O, LCMS (EI) m/e 219 (M++H) and 241 (M++Na).
4-({tert-Butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-Butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009).
Method A. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.0 g, 91.74 mmol) in 1,2-dichloroethane (DCE, 280 mL) was treated with 4-formylphenylboronic acid 1005 (commercially available, 12.39 g, 82.57 mmol, 0.9 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.2 g, 137.6 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h.
When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture was concentrated in vacuo. The residue, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with tetrahydrofuran (THF, 100 mL) and water (H2O, 100 mL).
The resulting solution was subsequently treated with solid potassium carbonate (K2CO3, 37.98 g, 275.2 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.02 g, 91.74 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL). The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo.
The crude, regioisomeric 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 35.98 g, 37.32 g, 96.4%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo.
This crude material was directly used in the subsequent reaction without further purification. For 1008 and 1009:
1H NMR (300 MHz, DMSO-d6) δ 1.32 and 1.37 (two br. s, 9H, COOC(CH3)3), 3.70, 3.73 and 3.74 (three s, 3H, Ar—OCH3), 4.07-4.39 (m, 4H), 5.49 and 5.52 (two s, 2H), 6.70-8.04 (m, 9H, Ar—H and triazole-CH); C23H29BN4O5, LCMS (EI) m/e 453 (M++H) and 475 (M++Na).
Method B. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.06 g, 92.0 mmol) in tetrahydrofuran (THF, 300 mL) was treated with 4-formylphenylboronic acid (13.11 g, 87.4 mmol, 0.95 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.25 g, 138.0 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h.
When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with water (H2O, 200 mL).
The resulting aqueous solution was subsequently heated with solid potassium carbonate (K2CO3, 38.0 g, 276 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.08 g, 92 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL).
The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude, 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 38.45 g, 39.50 g, 97.3%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo. This crude material was found to be essentially identical in every comparable aspect as the material obtained from Method A and was directly used in the subsequent reaction without further purification.
(5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester
(1011 and 1012).
A suspension of the crude regioisomeric mixture of 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 37.62 g, 83.23 mmol) and N-[3-(3-fluoro-4-iodo-phenyl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1010, 28.32 g, 74.9 mmol, 0.90 equiv) in toluene (150 mL) was treated with powder K2CO3 (34.45 g, 249.7 mol, 3.0 equiv), EtOH (50 mL), and H2O (50 mL) at 25° C.,
and the resulting mixture was degassed three times under a steady stream of Argon at 25° C. Pd(PPh3)4 (866 mg, 0.749 mmol, 0.01 equiv) was subsequently added to the reaction mixture, and the resulting reaction mixture was degassed three times again under a stead stream of Argon at 25° C. before being warmed up to gentle reflux for 18 h. When TLC and HPLC/MS showed the coupling reaction was complete, the reaction mixture was cooled down to room temperature before being treated with H2O (100 mL) and ethyl acetate (100 mL). The two layers were then separated, and the aqueous layer was extracted with EtOAc (100 mL).
The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×150 mL), H2O (100 mL), and the saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The residual oil was solidified upon standing at room temperature in vacuo to afford the crude, (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-y]methyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester (1011) and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1012) as a regioisomeric mixture.
This crude product (43.36 g, 49.28 g theoretical, 88%) was used directly in the subsequent reaction without further purification. For the mixture of 1011 and 1012: 1H NMR (300 MHz, DMSO-d6) δ 1.35 and 1.38 (two br. s, 9H, COO(CH3)3), 1.85 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.73 and 3.76 (two s, 3H, Ar—OCH3), 3.79 (dd, 1H, J=6.6, 9.1 Hz), 4.18 (t, 1H, J=9.1 Hz), 4.35-4.43 (m, 4H), 4.73-4.81 (m, 1H), 5.50 (br. s, 2H), 6.90 and 6.98 (two d, 2H, J=8.7 Hz), 7.28 and 7.32 (two d, 2H, J=8.7 Hz), 7.35 (dd, 2H, J=2.2, 8.6 Hz), 7.42 (dd, 1H, J=2.2, 8.6 Hz), 7.49-7.63 (m, 4H, aromatic-H), 7.90 and 7.99 (two br. s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3); C35H39FN6O6, LCMS (EI) m/e 659 (M++H) and 681 (M++Na).
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1013)
and
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H–[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1014).
A solution of a regioisomeric mixture of (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1011 and 1012, 37.28 g, 56.65 mmol) in ethyl acetate (EtOAc, 150 mL) and methanol (MeOH, 30 mL) was treated with a solution of 4 N hydrogen chloride in 1,4-dioxane (113.3 mL, 453.2 mmol, 8.0 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 12 h. When TLC and HPLC/MS showed that the N-BOC deprotection reaction was complete,
the solvents were removed in vacuo. The residue was then suspended in 250 mL of 5% methanol (MeOH) in acetonitrile (CH3CN), and the resulting slurry was stirred at room temperature for 1 h. The solids were then collected by filtration, washed with toluene (2×100 mL) and 5% methanol in acetonitrile (2×50 mL), and dried in vacuo to afford a regioisomeric mixture of the crude, (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 30.0 g, 33.68 g theoretical, 89.1% yield) as off-white crystals in a ratio of 1.2 to 1.
This material was found by 1H NMR and HPLC/MS to be essentially pure and was directly used in the subsequent reactions without further purification. For the regioisomeric mixture of 1013 and 1014:
1H NMR (300 MHz, DMSO-d6) δ 1.84 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.71 and 3.74 (two s, 3H, Ar—OCH3), 3.80 (dd, 1H, J=6.6, 9.1 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.23-4.30 (m, 4H), 4.73-4.80 (m, 1H), 5.58 and 5.70 (two s, 2H), 6.88 and 6.93 (two d, 2H, J=8.7 Hz), 7.15 and 7.32 (two d, 2H, J=8.7 Hz), 7.43 (dd, 2H, J=2.2, 8.6 Hz), 7.52-7.62 (m, 6H, aromatic-H), 8.28 (s, 1H, triazole-CH), 8.32 (t, 1H, J=5.8 Hz, NHCOCH3), 9.91 and 10.32 (two br. s, 2H, ArCH2N+H2); C30H31FN6O4, LCMS (EI) m/e 559 (M++H) and 581 (M++Na).
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide hydrochloride (1 hydrochloride salt).
A solution of the crude regioisomeric mixture of (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-1H-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 29.17 g, 49.07 mmol) in trifluoroacetic acid(TFA, 150 mL) was warmed up to 65-70° C., and the resulting reaction mixture was stirred at 65-70° C. for 12 h. When TLC and HPLC/MS showed that the deprotection reaction was complete, the solvents were removed in vacuo.
The residual solids were then treated with ethyl acetate (EtOAc, 100 mL) and H2O (150 mL) before being treated with a saturated aqueous solution of sodium carbonate (30 mL) at room temperature. The resulting mixture was then stirred at room temperature for 1 h before the solids were collected by filtration, washed with EtOAc (2×50 mL) and H2O (2×50 mL), and dried in vacuo at 40-45° C. to afford the crude, (5S)-N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl)-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1 as the free base, 18.9 g, 21.49 g theoretical, 87.9%) as off-white powders, which by HPLC/MS and 1H NMR was found to be one pure regioisomer and this regioisomer was found to be identical as the material obtained from deprotection of 1013 alone by the same method.
For 1 as the free base: 1H NMR (300 MHz, DMSO-d6) δ 1.85 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.74 (s, 2H), 3.77 (s, 2H), 3.79 (dd, 1H, J=6.4, 9.2 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.72-4.81 (m, 1H), 7.39-7.62 (m, 7H, aromatic-H), 7.73 (s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH); C22H23FN6O3, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
A suspension of 1 free base (18.0 g, 41.1 mmol) in ethyl acetate (EtOAc, 80 mL), and methanol (MeOH, 20 mL) was treated with a solution of 4.0 N hydrogen chloride in 1,4-dioxane (41.1 mL, 164.4 mmol, 4.0 equiv) at room temperature, and the resulting mixture was stirred at room temperature for 8 h. The solvents were then removed in vacuo, and the residue was further dried in vacuo before being treated with a mixture of 10% methanol in acetonitrile (80 mL). The solids were collected by filtration, washed with 10% MeOH/acetonitrile (2×40 mL), and dried in vacuo to afford 1 hydrochloride salt (18.13 g, 19.50 g theoretical, 93% yield) as off-white crystals.
The crude 1 hydrochloride salt can be recrystallized from acetonitrile and water, if necessary, according to the following procedure: A suspension of the crude 1 hydrochloride salt (50.0 g) in acetonitrile (1250 mL) was warmed up to reflux before the distilled water (H2O, 280 mL) was gradually introduced to the mixture. The resulting clear yellow to light brown solution was then stirred at reflux for 10 min before being cooled down to 45-55° C. The solution was then filtered through a Celite bed at 45-55° C., and the filtrates were gradually cooled down to room temperature before being further cooled down to 0-5° C. in an ice bath for 1 h. The solids were then collected by filtration, washed with acetonitrile (2×50 mL), and dried in vacuo at 40° C. for 24 h to afford the recrystallized 1 hydrochloride salt (42.5 g, 50.0 g theoretical, 85% recovery) as off-white crystals.
For 1: 1H NMR (300 MHz, DMSO-d6) δ 1.86 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.84 (dd, 1H, J=6.4, 9.2 Hz), 4.19 (t, 1H, J=9.1 Hz), 4.24 (br. s, 2H), 4.31 (br. s, 2H), 4.74-4.79 (m, 1H), 7.44 (dd, 1H, J=2.2, 8.6 Hz), 7.57-7.66 (m, 6H, aromatic-H), 8.17 (s, 1H, triazole-CH), 8.30 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH);
13C NMR (75 MHz, DMSO-d6) δ 22.57, 40.69, 41.50, 47.36, 49.23, 71.85, 105.70 (d, J=28.5 Hz), 114.14 (d, J=2.9 Hz), 122.29 (d, J=13.3 Hz), 128.82 (d, J=3.0 Hz), 130.70, 130.94, 131.0, 131.22, 135.30, 137.92 (br. s), 139.66 (d, J=11.2 Hz), 154.11, 159.13 (d, J=243.5 Hz), 170.19;
C22H23FN6O3—HCl, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
……………………………..
http://www.sciencedirect.com/science/article/pii/S0960894X0801192X

| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6969726 * | Jun 2, 2004 | Nov 29, 2005 | Rib X Pharmaceuticals Inc | Biaryl heterocyclic compounds and methods of making and using the same |
| US20050043317 * | Jun 2, 2004 | Feb 24, 2005 | Jiacheng Zhou | Biaryl heterocyclic compounds and methods of making and using the same |
|
9-17-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
|
|
|
9-17-2010
|
Process for the synthesis of triazoles
|
|
|
4-28-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
|
|
|
11-26-2008
|
Biaryl heterocyclic compounds and methods of making and using the same
|
|
|
10-26-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
12-13-2006
|
Biaryl heterocyclic compounds and methods of making and using the same
|
|
|
11-30-2005
|
Biaryl heterocyclic compounds and methods of making and using the same
|

October 10, 2012
Rib-X Pharmaceuticals announced that the FDA designated radezolid as a Qualified Infectious Disease Product (QIDP) for the indications of acute bacterial skin and skin structure infections (ABSSSI) and community-acquired bacterial pneumonia (CABP).
The QIDP designation will enable Rib-X to benefit from certain incentives for the development of new antibiotics, including an additional five years of market exclusivity, priority review and eligibility for fast-track status, provided under the new Generating Antibiotic Incentives Now (GAIN) program. GAIN was included in the FDA Safety and Innovation Act (FDASIA), formerly known as PDUFA V, which received bipartisan Congressional support and was signed into law by President Obama in July 2012.
Radezolid has completed two Phase 2 clinical trials with an oral formulation in uncomplicated skin and skin structure infections (uSSSI) and in CABP. A Phase 1 study with an IV formulation was recently completed in healthy subjects. Rib-X recently announced data from a positive Phase 1 IV dosing study conducted in healthy subjects and an in vivo long-term safety study vs. linezolid (Zyvox; Pfizer).
Radezolid is a next-generation oxazolidinone with a safety profile permitting long-term treatment of resistant infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
For more information call (203) 624-5606 or visit www.rib-x.com



FINAFLOXACIN
(S-cyano-1-cyclopropyl-ό-fluoro-T-^aS, 7aS)-hexahydropyrrolo [3,4- b]-1,4-oxazin-6(2H)-yl]-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid)
7-[(4aS,7aS)-3,4,4a,5,7,7a-hexahydro-2H-pyrrolo[3,4-b][1,4]oxazin-6-yl]-8-cyano-1-cyclopropyl-6-fluoro-4-oxoquinoline-3-carboxylic acid |
BAY-35-3377
BY-377
CAS Registry Number: 209342-40-5
HYD SALT
(-)-(4aS,7aS)-8-Cyano-1-cyclopropyl-6-fluoro-4-oxo-7-(perhydropyrrolo[3,4-b]-1,4-oxazin-6-yl)-1,4-dihydroquinoline-3-carboxylic acid hydrochloride
209342-41-6,
| C20 H19 F N4 O4 . Cl H | |
| MW | 434.849 |
Synonyms: Finafloxacin, UNII-D26OSN9Q4R,
MerLion Pharmaceuticals (Singapore)…POSTER…….http://www.merlionpharma.com/sites/default/files/file/PPS/F1-2036_Wohlert.pdf
H. pylori, Broad-Spectrum
Finafloxacin is a novel fluoroquinolone being developed by MerLion Pharmaceuticals. Under neutral pH conditions (pH 7.2–7.4), the compound has shown in vitro activity equivalent to that of ciprofloxacin. However, under slightly acidic pH5.8 the compound shows enhanced potency.
Other marketed fluoroquinolones, such as ciprofloxacin, levofloxacin and moxifloxacin, exhibit reduced activity at slightly acidic pH 5.0–6.5. This feature of finafloxacin makes the compound suitable for use in the treatment of infections in acidic foci of infections such as urinary tract infections
Finafloxacin hydrochloride, a novel highly potent antibiotic, is in phase III clinical trials at Alcon for the treatment of ear infections. MerLion Pharmaceuticals is evaluating the product in phase II clinical trials at for the treatment of Helicobacter pylori infection and for the treatment of lower uncomplicated urinary tract infections in females.
A quinolone, finafloxacin holds potential for the treatment of Helicobacter pylori infection and urinary tract infection. Unlike existing antibiotics, finafloxacin demonstrates a unique acid activated activity whereby it becomes increasingly active under acidic conditions.
In 2009, a codevelopment agreement was signed between Chaperone Technologies and MerLion Pharmaceuticals. In 2011, finafloxacin hydrochloride was licensed to Alcon by MerLion Pharmaceuticals in North America for the treatment of ear infections.
MerLion Pharmaceuticals has announced that the FDA has granted a Qualified Infectious Disease Product Designation and Fast Track Status for finafloxacin. The company is currently recruiting patients for the Phase II clinical trial of the compound for the treatment of complicated urinary tract infections (cUTI) and/or acute pyelonephritis compared to ciprofloxacin
Finafloxacin and derivatives thereof can be synthesized according to the methods described in U.S. Patent No. 6,133,260 to Matzke et al., the contents of which are herein incorporated by reference in their entirety. The compositions of the invention are particularly directed toward treating mammalian and human subjects having or at risk of having a microbial tissue infection. Microbial tissue infections that may be treated or prevented in accord with the method of the present invention are referred to in J. P. Sanford et al., “The Sanford Guide to Antimicrobial Therapy 2007” 37 Edition (Antimicrobial Therapy, Inc.). Particular microbial tissue infections that may be treatable by embodiments of the present invention include those infections caused by bacteria, protozoa, fungi, yeast, spores, and parasites.
SYNTHESIS
WO1998026779A1
http://www.google.sc/patents/WO1998026779A1 COPY PASTE ON BROWSER
8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-di-azabicyclo [4.3.0] non-8-yl)-l, 4-dihydro-4-oxo-3-quinolinecarboxylic acid.
The compounds, which are suitable for use in the invention are known already to some extent in EP-A-0350733, EP-A-0550903 as well as from DE-A-4329600 or can be prepared according to the processes described in .
If, for example 9,10-difluoro-3 ,8-dimethyl-7-oxo-2 ,3-dihydro-7H-pyrido [l ,2,3-d, e] [l, 3,4] benzoxadiazine-6 -carboxylic acid and 2-oxa-5 ,8-diazabicyclo [4.3.0] nonane, the reaction can be represented by the following equation:

The 7-halo-quinolonecarboxylic acid derivatives used for preparing the compounds of Fomel (I) of the invention are known or can be prepared by known methods. Thus, the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid, or of the 7-chloro-8-cyano-l-cyclopropyl-6-fluoro- l been ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester described in EP-A-0 276 700th The corresponding 7-fluoro derivatives can be, for example, via the following reaction sequence to build:

An alternative process for preparing the intermediate compound 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride as the starting material for the preparation of 7-chloro-
8-cyano-1-cyclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is used (EP-A-0276700) and in the 3-cyano-2 ,4,5-trifluoro- benzoyl can be converted, is based on 5-fluoro-l ,3-xylene, 5-fluoro-l ,3-xylene, in the presence of a catalyst under ionic conditions in the nucleus disubstituted to 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene, and this is subsequently chlorinated chlorinated under free radical conditions in the side chains of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methylbenzene. This is the 2,4-dichloro-5-fluoro-3-dichloromethyl-benzoic acid to give 2,4-dichloro-5-fluoro-3-formyl-benzoic acid, and then hydrolyzed to 2,4-dichloro-5-fluoro-3 N-hydroxyiminomethyl acid implemented. By treatment with thionyl chloride, 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride is obtained, which can still be ,4,5-trifluoro-ben-zoylfluorid converted by a chlorine / fluorine exchange on-3-cyano-2 .



The amines used for the preparation of compounds of formula (I) according to the invention are known from EP-A-0550903, EP-A-0551653 as well as from DE-A-4 309 964th
An alternative to the synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-dihydro-drobromid or the free base 1 S, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0 ] nonane and the corresponding IR, 6R enantiomer provides the following path represents:
Starting material for this synthesis is the cis-l ,4-dihydroxy-2-butene, which is converted to the bis-mesylate with mesylation tosylamide for 1-tosylpyrrolidine. This is converted into the epoxide m-chloroperbenzoic. The ring opening of the epoxide by heating in isopropanol with ethanolamine to trans-3-hydroxy-4 – (2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl)-pyrrolidine in 80% yield. Tetrahydrofuran is then in pyridine / reacted with tosyl chloride, with cooling to Tris-tosylate, which as a crude product in a mixture with some tetra-tosyl derivative with basichen reaction conditions to give the racemic trans-5 ,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane is cylisiert. At this stage occurs with high selectivity of a chromatographic resolution kieselgelgebundenem poly (N-methacryloyl-L-leucine-d menthylamide) as the stationary phase. The desired enantiomer, (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane, is of a purity of
> 99% ee. Cleavage of the p-tosyl protecting groups is carried out with HBr-acetic acid to the lS, 6S-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide, the one with a base such as sodium or potassium hydroxide or with the aid of ion exchanger can be converted into the free base. The analogous sequence may be used for the preparation of lR, 6R-2-Oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide.
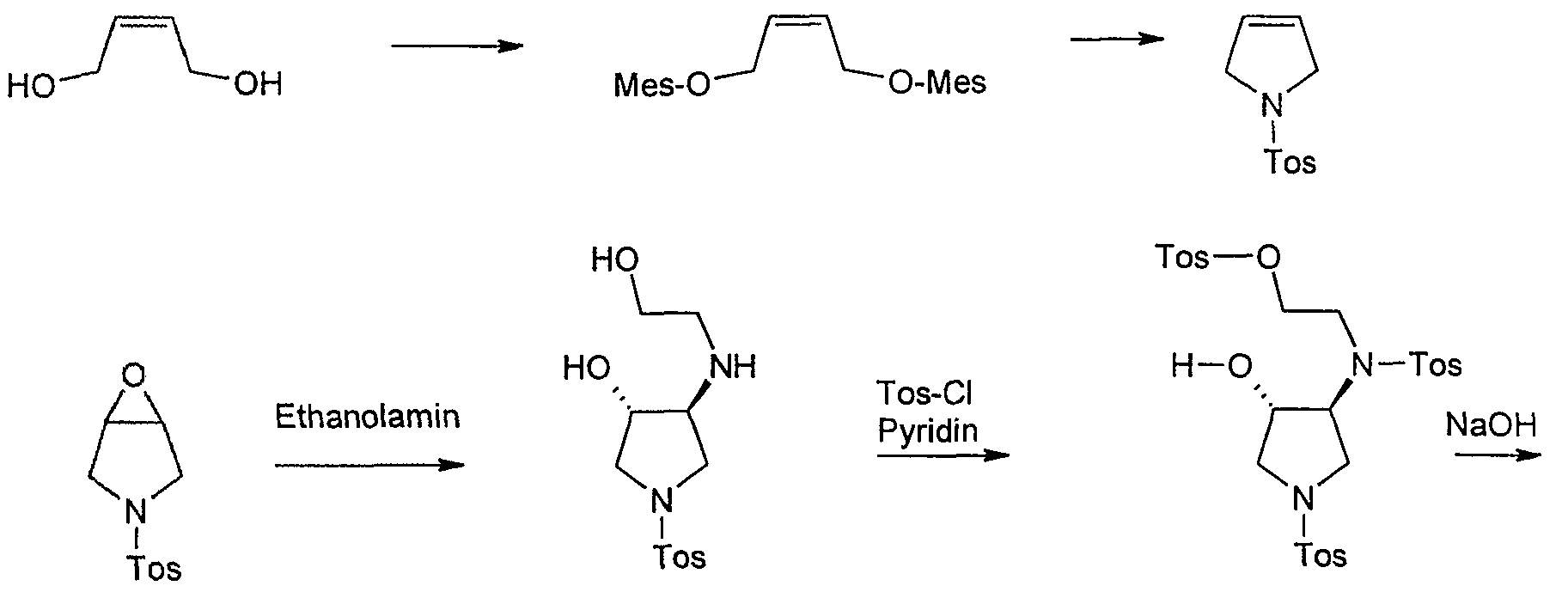

HBr / AcOH

Synthesis of lS, 6S-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Examples of compounds of the invention are mentioned in addition to the compounds listed in the preparation examples, the compounds listed in Table 1 below, which can be used both in racemic form as well as enantiomerically pure or diastereomerically pure compounds. Table 1:


Example 1 Z
8-cyano-1-cyclopropyl-6 ,7-difluoro-1 ,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester

a 3-bromo-2 ,4,5-trifluoro-benzoate
To a mixture of 1460 ml of methanol and 340 g of triethylamine, 772 g of 3-bromo-2 ,4,5-trifluoro-benzoyl fluoride was added dropwise under ice cooling. There is one
Stirred for an hour at room temperature. The Reaktionsgemsich is concentrated, the residue dissolved in water and methylene chloride, and the aqueous phase was extracted with methylene chloride. After drying the organic phase over sodium sulfate, concentrated, and the residue was distilled in vacuum. This gives 752.4 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester of boiling point 122 ° C/20 mbar.
b. 3-Cyano-2 ,4,5-trifluoro-benzoic acid methyl ester:
269 g of 3-bromo-2 ,4,5-trifluoro-benzoic acid methyl ester and 108 g of copper cyanide are heated to reflux in 400 ml of dimethylformamide for 5 hours. , All volatile components of the reaction mixture are then distilled off in vacuo. The distillate was then fractionated on a column. This gives 133 g of 3-cyano-2 ,4,5-trifluoro-benzoate of boiling point 88-89 ° C / 0.01 mbar.
c. 3-Cyano-2 ,4,5-trifluoro-benzoic acid
A solution of 156 g of 3-cyano-2 ,4,5-trifluoro-benzoate in 960 ml of glacial acetic acid, 140 ml of water and 69 ml concentrated sulfuric acid is heated for 8 hours under reflux. Then the acetic acid is distilled off under vacuum and the residue treated with water. Of failed-ne solid is filtered off, washed with water and dried. Obtained
118.6 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid as a white solid, mp 187-190 ° C.
d 3-cyano-2 ,4,5-trifluoro-benzoyl chloride:
111 g of 3-cyano-2 ,4,5-trifluoro-benzoic acid and 84 g of oxalyl chloride are stirred in 930 ml of dry methylene chloride with the addition of a few drops of dimethylformamide for 5 hours at room temperature. The methylene chloride is evaporated and the residue distilled in vacuo. This gives 117.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl chloride as a yellow oil.
e 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester:
To a solution of 36.5 g of 3-dimethylamino-acrylate and 26.5 g of triethylamine in 140 ml toluene, a solution of 55 g 3-cyano-2, 4,5 – trifluoro-benzoyl chloride are added dropwise in 50 ml of toluene so that the temperature 50-55 ° C remains. Then stirred for 2 hours at 50 ° C.
The reaction mixture is concentrated in vacuo and used without further
Processing used in the next step. f 2 – (3-cyano-2 ,4,5-trifluoro-benzoyl)-3-cyclopropylamino-acrylic acid ethyl ester:
To the reaction product of step e 30 g of glacial acetic acid are added dropwise at 20 ° C. A solution of 15.75 g of cyclopropyl amine in 30 ml of toluene is added dropwise. The mixture is stirred at 30 ° C for 1 hour. Are then added 200 ml of water, stirred 15 minutes, the organic phase is separated off and shakes it again with 100 ml of water. The organic phase is dried over sodium sulfate and concentrated in vacuo. The crude product thus obtained is a set-without further purification in the next step.
g 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester:
The reaction product from stage f and 27.6 g of potassium carbonate are stirred in 80 ml dimethylformamide for 16 hours at room temperature. The reaction mixture is then poured into 750 ml ice water, the solid filtered off with suction and washed with 80 ml cold methanol. After drying, 47 g of 8 – cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinoline carboxylic acid ethyl ester, mp 209-211 ° C.
Example 2 Z
2,4-dichloro-5-fluoro-l ,3-dimethylbenzene

a solvent-free
In 124 g of 3,5-dimethyl-fluorobenzene 1 g of anhydrous iron (III) chloride are pre-loaded and launched with the speed of chlorine (about 4 h), with which the reaction. This is initially slightly exothermic (temperature increase from 24 to 32 ° C) and is maintained by cooling below 30 ° C. After addition of 120 g of chlorine, the mixture is determined. According to GC analysis are 33.4% monochloro compound, formed 58.4% desired product and 5%> overchlorinated connections. The hydrogen chloride is removed and the reaction mixture is then distilled in a column in a water jet vacuum:
In the run 49 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene obtained at 72-74 ° C/22 mbar. After 5 g of an intermediate fraction proceed at 105 ° C/22 mbar 75 g of 2,4 – dichloro-5-fluoro-l ,3-dimethylbenzene via, Melting range: 64 – 65 ° C.
b in 1,2-dichloroethane
1 kg of 3,5-dimethyl-fluorobenzene and 15 g of anhydrous iron (III) chloride are placed in 1 1 1 ,2-dichloroethane and chlorine is introduced in the same extent as the reaction proceeds (about 4 h). The reaction is initially exothermic (temperature rise from 24 to 32 ° C) and is kept below 30 ° C by cooling. After the introduction of 1200 g of chlorine are according to GC analysis 4% monochloro compound, 81.1% and 13.3% desired product overchlorinated connections emerged. After distilling off the solvent and the hydrogen chloride is distilled in a column in a water jet vacuum:
In the run 40 g of 2-chloro-5-fluoro-l ,3-dimethylbenzene receive. After some intermediate run going at 127-128 ° C/50 mbar 1115 g of 2,4-dichloro-5-fTuor-l ,3-dimethyl-ethylbenzene over.
Example 3 Z
2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloromethylbenzene

In a photochlorination using chlorine inlet and outlet for the hydrogen chloride to a scrubber and a light source in the vicinity of the chlorine inlet tube, 1890 g of 2,4-dichloro-5-fluoro-l ,3-dimethylbenzene pre-loaded and at 140 to 150 ° C. Chlorine metered. Within 30 hours 3850 g of chlorine are introduced. The content of the desired product according to GC analysis is 71.1% and the proportion of connections minderchlorierten 27.7%. The DestiUaton a 60 cm column with Wilson spirals provides a flow of 1142 g, which can be reused in the chlorination. The main fraction at 160-168 ° C / 0.2 mbar gives 2200 g of 2,4-dichloro-5-fluoro-3-dichloromethyl-l-trichloro-methyl benzene having a melting range of 74-76 ° C. After one recrystallization
Sample from methanol, the melting point 81-82 ° C.
Example Z 4
2,4-dichloro-5-fluoro-3-formyl-benzoic acid

In a 2500 ml stirred apparatus with gas discharge are presented 95% sulfuric acid at 70 ° C. and under stirring, 500 g of molten added dropwise 2,4-dichloro-5-fluoro-3-dichloromethyl-1 trichloromethylbenzene. It is after a short while hydrochloric development. Is metered during a 2 h and stirred until the evolution of gas after. After cooling to 20 ° C., the mixture is discharged ice to 4 kg and the precipitated solid is filtered off with suction. The product is after-washed with water and dried.
Yield: 310 g, melting range: 172-174 ° C
Example Z 5
2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl-benzoic acid

In a stirred reactor 80 g of hydroxylamine hydrochloride in 500 ml of ethanol are charged and added dropwise 200 ml of 45% strength sodium hydroxide solution and then with 40 – 200 g of 2,4-dichloro-5-fluoro-3-formyl-benzoic acid added 45.degree.The reaction is slightly exothermic and it is stirred for 5 h at 60 ° C. After cooling to
Room temperature is provided by the dropwise addition of hydrochloric acid to pH <3, the product taken up in tert-butyl methyl ether, the organic phase separated and the solvent distilled off. The residue obtained 185 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid, melting range: 190 – 194 ° C.
Example No. 6
2,4-dichloro-3-cyano-5-benzoyl-fιuor

In a stirred vessel with metering and gas outlet via a reflux condenser to a scrubber 600 ml of thionyl chloride are introduced and registered at 20 ° C. 210 g of 2,4-dichloro-5-fluoro-3-N-hydroxyiminomethyl benzoic acid in the proportion as hydrochloric developed and sulfur dioxide. After the addition the mixture is heated until the gas evolution under reflux. Mixture is then distilled, and boiling in the range of 142-145 ° C/10 mbar, 149 g of 2,4-dichloro-3-cyano-5-fluoro-benzoyl chloride (98.1% purity by GC) Melting range: 73-75 ° C.
Example No. 7
3-Cyano-2 ,4,5-trifluoro-benzoyl

50 g of potassium fluoride are suspended in 120 ml of tetramethylene sulfone and at 15 mbar for drying distilled (ca. 20 mL).Then, 50.4 g of 2,4 – dichloro-3-cyano-5-fluoro-benzoyl chloride was added and stirred at an internal temperature with exclusion of moisture for 12 hours at 180 ° C. Are removed by vacuum distillation to 32.9 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride in the boiling range of 98 –
Obtain 100 ° C/12 mbar.
Example No. 8
3-Cyano-2 ,4,5-trifluoro-benzoyl chloride

76.6 g of 3-cyano-2 ,4,5-trifluoro-benzoyl fluoride together with 1 g of anhydrous
Aluminum chloride introduced at 60-65 ° C and then added dropwise 25 g of silicon tetrachloride gas in the course of development. After the evolution of gas at 65 ° C is distilled in a vacuum. Boiling range 120-122 ° C/14 mbar, 73.2 g of 3 – cyano-2 ,4,5-trifluoro-benzoyl chloride over.
Example No. 9
1 – (toluene-4-sulfonyl-pyrroline

In a 20 1 HC4-HWS boilers are 2.016 kg (17.6 mol)
Submitted methanesulfonyl chloride in dichloromethane and 12 1 at -10 ° C internal temperature under strong cooling (-34 ° C) solution of 705 g (8.0 mol) of 2-butene-l ,4-diol in 1.944 kg (2.68 1 , 19.2 mol) of triethylamine was added dropwise over 30 minutes. A yellow suspension stirred for 1 hour at -10 ° C and then treated with 4 1 of water, the temperature rises to 0 ° C.The suspension is warmed to room temperature, stirred for 10 minutes at room temperature and then fed in a 30 1 separating funnel. The phases are stirred separately (good phase separation) and the aqueous phase extracted with 2 1 of dichloromethane. The combined dichloromethane phases are presented in a pre-cooled 20 1 HC4 vessel and kept at 0 ° C.
In another 20-1 HC4 boiler distillation 1.37 kg (8.0 mol) toluenesulfonamide be submitted in 6 1 toluene. It is mixed with 3.2 kg of 45% sodium hydroxide solution, 0.8 1 of water and 130.5 g Tetrabutylammomiimhydrogensulfat, heated to 40 ° C maximum temperature inside and creates a vacuum. Then, the previously obtained
Dichloromethane (15.2 1) was added dropwise over 1.5 hours while the dichloromethane was removed by distillation at 450 mbar (bath temperature: 60 ° C). During the distillation is foaming. In the end, a solution is available at an internal temperature of 33-40 ° C. After the addition of dichloromethane is distilled off, until barely distillate is (duration: about 85 minutes; internal temperature 40 ° C at 60 ° C bath temperature at the end). The vessel contents will be warm transferred to a separating funnel and rinsed the tank with water and 5 1 2 1 toluene at 50 ° C. Before phase separation, the solids are extracted in the intermediate phase and washed with 0.5 1 of toluene. The organic phase is extracted with 2.4 1 of water, separated and evaporated to dryness on a rotary evaporator. The solid residue (1758 g) is suspended in 50 ° C bath temperature in 1.6 1 of methanol, the suspension is transferred into a 10 1-flanged flask and the flask rinsed with diisopropyl 2,4 1. The mixture is heated to reflux temperature (59 ° C) and stirred for 30 minutes under reflux. The suspension is cooled to 0 ° C., stirred at 0 ° C for 1 hour and extracted with 0.8 1 of a cold mixture of ether Methanol/Diisopropyl-: washed (1 1.5). The crystals are dried under a nitrogen atmosphere at 50 ° C/400 mbar.
Yield: 1456 g (81.5% of theory)
Example Z 10
3 – (toluene-4-sulfonylV6-oxa-3-aza-bicvclo [3.1.0] hexane
o “|” h “CH3
334.5 g (1.5 mol) of l-(toluene-4-sulphonyl)-pyrroline are dissolved in 1.5 1 of dichloromethane at room temperature and over 15 minutes with a suspension of 408 g (approx. 1.65 to 1, 77 mol) of 70-75% m-chloroperbenzoic acid in 900 ml of dichloromethane (cools added in manufacturing from). The mixture is heated under reflux for 16 hr (test for
Peroxide with KI / starch paper shows yet to peroxide), the suspension was cooled to 5 ° C, sucks the precipitated m-chlorobenzoic acid and washed with 300 ml of dichloromethane (peroxide with Precipitation: negative; precipitate was discarded). The filtrate is to destroy excess peroxide with 300 ml of 10% sodium sulfite solution, washed twice (test for peroxide runs now negative), extracted with 300 ml of saturated sodium bicarbonate solution, washed with water, dried with sodium sulfate and about a quarter of the volume evaporated. Again on test peroxide: negative. The mixture is concentrated and the solid residue is stirred with ice cooling, 400 ml of isopropanol, the precipitate filtered off and dried at 70 ° C in vacuum.
Yield: 295 g (82.3%),
Mp: 136-139 ° C,
TLC (dichloromethane methanol 98:2): 1 HK (Jodkammer)
Example CLOSED
trans-3-Hydroxy-4-(2-hydroxy-ethylamino-l-(‘toluene-4-sulfonyl’) pyrrolidine

643.7 g (2.65 mol) 3 – (Toluoι-4-sulfonyl)-6-oxa-3-aza-bicyclo [3.1.0] hexane to 318.5 ml with ethanolamine in 4 1 of isopropanol at reflux for 16 hours cooked. After TLC monitoring, further 35.1 ml (total 5.86 mol) of ethanolamine added to the mixture and boiled again until the next morning. The mixture is filtered hot with suction and the filtrate concentrated on a rotary evaporator to 3.5 ltr. After seeding and stirring at room temperature for 3.5 1 diisopropyl ether are added, and stirred at 0 ° C for 6 hours. The precipitated crystals are filtered off, with 250 ml of a mixture of isopropanol / diisopropyl ether (1: 1) and washed 2 times with 300 ml of diisopropyl ether and dried overnight under high vacuum.
Yield: 663.7 g (83% of theory), content: 96.1% (area% by HPLC). Example Z 12
trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl)-pyrrolidin-3-yl] – ftoluol-4-sulfonyl)-amino]-ethyl ester)

552 g (1.837 mol) of trans-3-hydroxy-4-(2-hydroxy-ethylamino)-l-(toluene-4-sulfonyl) – pyrrolidine are dissolved under argon in 1.65 1 tetrahydrofuran and 0.8 1 of pyridine dissolved and at -10 ° C in portions 700 g (3.675 mol) p-toluenesulfonyl chloride are added thereto. The mixture is then stirred at this temperature for 16 hours. The work is done by adding 4.3 18.5 1% aqueous hydrochloric acid, extraction twice with dichloromethane (3 1, 2 1), washing the combined organic phases with saturated Natriurnhydrogencarbonatlösung (3 1, 2 1), drying over sodium sulfate, extracting and distilling off the solvent in vacuo. The residue is dried overnight at the oil pump and crude in the next reaction. There were 1093 g as a hard foam (content [area% by HPLC]: 80% Tris-tosyl-product and 13% tetra-tosyl-product, yield see next step). Example Z 13
rac. trans-5 ,8-bis-tosyl-2-oxa-5 .6-diazabicyclor4 .3.01 nonane

1092 g of crude trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l-(toluene-4-sulfonyl) – pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino]-ethyl} were dissolved in tetrahydrofuran and 9.4 1 at 0-3 ° C with 1.4 1 of a 1.43 molar solution of sodium hydroxide in
Methanol reacted. After half an hour at this temperature, 2.1 1 of water and 430 ml of diluted (2:1) was added to the mixture and acetic acid with previously isolated crystals of trans-toluene-4-sulfonic acid {2 – [[4-hydroxy-l – (toluene-4-sulfo-phenyl)-pyrrolidin-3-yl] – (toluene-4-sulfonyl)-amino] ethyl}-seeded. The suspension is stirred overnight at 0 to -4 ° C. The next morning, the crystals are filtered off, washed twice with 400 ml of cold mixture of tetrahydrofuran / water (4:1) and dried at 3 mbar at 50 ° C overnight.
Yield: 503 g of white crystals (62.7%> of theory over 2 steps), content: 99.7% (area% by HPLC). Example Z 14
Preparative chromatographic resolution of racemic rac. trans-5.8-bis-tosyl-2-oxa-5.6-diazabicyclor4.3.0] nonane
The chromatography of the racemate at room temperature in a column (inner diameter 75 mm), which with 870 g of a chiral stationary phase (kie-selgelgebundenes poly (N-methacryloyl-L-leucine-d menthylamide) based on the mer captomodifizierten silica Polygosil 100 , 10 microns; see EP-A 0 379 917) is filled (bed height: 38 cm). Detection is carried out using a UV detector at 254 nm
For the sample application using a solution of a concentration of 100 g of rac. trans-5 ,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 3000 ml of tetrahydrofuran. A Trenncyclus is carried out under the following conditions: with the aid of a pump is required for 2 min at a flow of 50 ml / min, a part of the sample solution and the same time at a flow rate of 50 ml / min, pure n-heptane to the column.
Thereafter eluted at a flow rate of 100 ml / min 18 minutes with a mixture of n-Heptan/Tetrahydrofuran (3/2 vol / vol). This is followed for 3 minutes at a flow of 100 ml / min elution with pure tetrahydrofuran. Thereafter, further eluted with n-Heptan/Tetrahydro-furan (3/2 vol / vol). This cycle is repeated several times.
The first eluted enantiomer is the (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo-[4.3.0] nonane, which is isolated by concentration. The eluate of the more retarding enantiomers is largely evaporated in vacuo, and the precipitated crystals are filtered off with suction and dried. From the separation of 179 g of racemate in this
As 86.1 g (96.2% of theory) of the enantiomer (lS, 6S) -5,8-bis-tosyl-2-oxa-5, 6 – diazabicyclo [4.3.0] nonane having a purity of> 99 % ee. Example Z 15
(LR, 6R-2-oxa-5.6-diazabicvclo [4.3.0] nonane dihydrobromide

38.3 g (87 mmol) of (lS, 6R) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] nonane in 500 ml of 33 -% HBr / glacial acetic acid 10 g added anisole and heated for 4 hours at 60 ° C (bath). After standing overnight, the suspension is cooled, the precipitate filtered, with
100 ml of abs. Ethanol and dried at 70 ° C under high vacuum.
Yield: 23.5 g (93%) of white solid product, mp 309-310 ° C (dec.), DC (dichloromethane/methanol/17% aq ammonia 30:8:1.): 1 HK
[Α] D: + 0.6 ° (c = 0.53, H 2 O) (fluctuating).
Example Z 16
(LS.6S-2-oxa-5.6-diazabicvclor4.3.01nonan-Dihvdrobromid

Z is analogous to Example 15 from (lS, 6S) -5,8-bis-tosyl-2-oxa-5 ,6-diazabicyclo [4.3.0] no-nan (1S, 6S)-2-oxa-5, 6-diazabicyclo [4.3.0] nonane dihydrobromide receive. Example Z 17
(1 R.6R-2-oxa-5.8-diazabicvclo [4.3.Olnonan

1 Method: 5,8 g (20 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydro-drobromid are suspended in 100 ml of isopropanol at room temperature with 2.4 g ( 42.9 mmol) and powdered potassium hydroxide while leaving about 1 hour in an ultrasonic bath. The suspension is cooled in an ice bath, filtered, washed with isopropanol and the undissolved salt, the filtrate was concentrated and distilled in a Kugelrohr oven at 150-230 ° C oven temperature and 0.7 mbar. Obtained 2.25 g (87.9% of theory) of a viscous oil which crystallizes. [Α] D -21.3 ° (c = 0.92, CHC1 3) Accordingly, this reaction can be carried out in ethanol.
2 Method: A homosexual genie catalyzed mixture of (lR, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane dihydrobromide and 620 mg (11 mmol) of powdered potassium hydroxide is dry in a Kugelrohr apparatus at 0.2 mbar and increasing oven temperature to 250 ° C distilled. Obtained 490 mg (76.6% of theory) of (lR, 6R) -2 – oxa-5 ,8-diazabicyclo [4.3.0] nonane as a viscous oil which slowly crystallized.
3 Method: 100 g of moist, pretreated cation exchanger (Dowex 50WX, H + – form, 100-200 mesh, capacity: 5.1 meq / g of dry or 1.7 meq / mL) are charged into a column with about 200 ml 1 N HC1 activated and washed neutral with water 3 1. A solution of 2.9 g (10 mmol) of (lS, 6R)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane
Dihydrobromide in 15 ml of water is added to the ion exchanger, and then washed with 2 1 water, and eluted with approximately 1 1 1 N ammonia solution. The eluate is evaporated. concentrated. Yield: 1.3 g of a viscous oil (quantitative), DC (dichloromethane/methanol/17% NH 3 30:8:1): 1 HK, GC: 99.6% (area).
Example Z 18
(LS.6SV2-oxa-5.8-diazabicvclor4.3.01nonan

Z is analogous to Example 17 from (lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane-di-hydrobromide the free base (lS, 6S)-2-oxa-5 ,8-diazabicyclo [ 4.3.0] nonane made.
Example Z 19
2 – (2,4-dichloro-3-cyano-5-fluoro-benzoyl)-3-dimethylamino-acrylic acid ethyl ester

To a solution of 626 g (4.372 mol) of 3-dimethylamino-acrylate and 591 g (4.572 mol) of ethyl-diisopropyl-amine (Hunigs base) in 1060 ml of dichloromethane, a solution of 1075 g starting at room temperature 2,4-dichloro -3-cyano-5-fluoro-benzoyl chloride (94% pure, corresponding to 1010.5 g = 4.00 mol) was dropped in 850 ml of dichloromethane. The temperature rises to 50-55 ° C (dropwise addition about 90 minutes). Then stirred for 2 hours at 50 ° C and the reaction mixture was used without further purification in the next step.
Example Z 20
2 – (2,4-dichloro-3-Cyano-5-fluoro-benzoyl-3-cvclopropylamino-acrylate

To the reaction mixture from the above step 306 g (5.1 mol) of glacial acetic acid are added dropwise under cooling at about 15 ° C. Then, with further cooling at 10-15 ° C. 267.3 g (4.68 mol) of cyclopropyl amine is added dropwise. Immediately after which the reaction mixture is mixed with 1300 ml of water under ice-cooling and 15 minutes stirred well. The dichloromethane layer was separated and used in the next step.
Example 21 Z
7-chloro-8-cyano-1-cyclopropyl-6-fluoro-1.4-dihydro-4-oxo-3-chinolincarbonsäureethyl ester

To a heated to 60-70 ° C suspension of 353 g (2.554 mol) of potassium carbonate in 850 ml of N-methylpyrrolidone, the dichloromethane phase is dropped from the precursor (about 90 minutes). During the addition of the dichloromethane at the same time
Reaction mixture was distilled off. Then the reaction mixture for 5 Vz hours at 60-70 ° C is well stirred. The mixture is cooled to about 50 ° C. and distilled under a vacuum of about 250 mbar residual dichloromethane from. At room temperature is added dropwise 107 ml 30% hydrochloric acid under ice cooling, then to obtain a pH of 5-6 is set. Then, 2,200 ml of water are added under ice cooling. The reaction mixture is thoroughly stirred for 15 minutes, the solid was then filtered off and washed on the filter twice with 1000 ml of water and extracted three times with 1000 ml of ethanol and then dried in a vacuum oven at 60 ° C.
Yield: 1200 g (89.6% of theory).
This product can be purified, if desired by, the solid is stirred in 2000 ml of ethanol for 30 minutes at reflux. You filtered hot with suction, washed with 500 ml of ethanol and dried at 60 ° C in vacuum. Melting point: 180-182 ° C.
Η-NMR (400 MHz, CDC1 3): d = 1.2 to 1.27 (m, 2H), 1.41 (t, 3H), 1.5-1.56 (m, 2H), 4, 1 to 4.8 (m, 1H), 4.40 (q, 2H), 8.44 (d, J = 8.2 Hz, H), 8.64 (s, 1H) ppm.
Example Z 22
7-chloro-8-cyano-1-cvclopropyl-6-fluoro-1 ,4-dihydro-4-oxo-3-quinolinecarboxylic acid

33.8 g (0.1 mol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylate dissolved in a mixture of 100 ml of acetic acid, 20 ml water and 10 ml concentrated sulfuric acid was heated for 3 hours under reflux. After cooling, the mixture is poured onto 100 ml of ice water, the precipitate filtered off, washed with water and ethanol and dried at 60 ° C in vacuum.
Yield: 29.6 g (96% of theory),
Mp 216-21 C. (with decomposition)
Example 1

A 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS.6S-2-oxa-5.8-diazabicvclo [4.3.0] non-8-yl – 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid
1.00 g (3.26 mmol) of 7-chloro-8-cyano-l-cyclopropyl-6-fluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are heated with 501 mg (3.91 mmol) of ( lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] nonane and 0.9 ml of triethylamine in 30 ml of acetonitrile was stirred at 40-45 ° C under argon for 25 hours. All volatile components in vacuo. removed and the residue recrystallized from ethanol. Yield: 1.22 g (94%)
Melting point: 294 ° C. (with decomposition)
B) 8-Cyano-l-cyclopropyl-6-fluoro-7-(‘(lS.6S-2-oxa-5 ,8-diazabicvclo [4.3.01nonan-8-YLV 1.4-dihydro-4-oxo-3- quinoline carboxylic acid Hvdrochlorid
200 mg (0.63 mmol) of 8-cyano-l-cyclopropyl-6 ,7-difluoro-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid ethyl ester to be 97 mg (0.75 mmol) of (lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] nonane and 0.17 ml of triethylamine in 3 ml of acetonitrile was stirred at 40-45 ° C for 2 hours under argon. All volatile components in vacuo. removed, the residue treated with water, insolubles filtered off and the filtrate was extracted with dichloromethane. The organic phase is dried over sodium sulfate and then concentrated under reduced pressure. a. The resulting residue is dissolved in 6 ml of tetrahydrofuran and 2 ml of water and 30 mg (0.72 mmol) of lithium hydroxide monohydrate was added. After 16 hours of stirring at room temperature, acidified with dilute hydrochloric acid and the resulting precipitate was filtered off with suction and dried. Yield: 155 mg (57%) Melting point:> 300 ° C
C) 8-Cyano-l-cvclopropyl-6-fluoro-7-((lS, 6S-2-oxa-5.8-diazabicvclo [4.3.01non-8 yiyi.4-dihydro-4-oxo-3-quinolinecarboxylic acid hydrochloride
1 g (2.5 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5 ,8-diazabicyclo [4.3.0] non-8-yl )-l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid is suspended in 20 ml of water was added to the suspension, 10 ml hydrochloric acid and stirred for In at room temperature for 3 hours. The resulting precipitate is filtered off, washed with ethanol and dried at 80 ° C under high vacuum.
Yield: 987 mg (90.6% of theory), Melting point: 314-316 ° C. (with decomposition).
D) 8-Cyano-l-cvclopropyl-6-fluoro-7-(iS, 6S)-2-oxa-5.8-diazabicyclo [4.3.0] non-8-YLV 1 ,4-dihydro-4-oxo-3 -quinoline carboxylic acid hydrochloride
86.4 g (217 mmol) of 8-cyano-l-cyclopropyl-6-fluoro-7-((lS, 6S)-2-oxa-5, 8 – diazabicyclo [4.3.0] non-8-yl) – l ,4-dihydro-4-oxo-3-quinolinecarboxylic acid are dissolved at room temperature in 963 ml of water and 239 ml of 1 N aqueous sodium hydroxide solution. After filtration and washing with 200 ml of water is added to 477 ml in aqueous hydrochloric acid and the precipitated crystals placed at 95 ° C to 100 ° C in solution. The solution is cooled overnight, the precipitated crystals are filtered off with suction and washed three times with 500 ml of water and dried in vacuum.
Yield 90 g (94.7% of theory), content:> 99% (area% by HPLC) 99.6% ee. [] D 23: -112 ° (c = 0.29, N NaOH).
……………….
Tetrahedron Lett 2009, 50(21): 2525
A novel approach to Finafloxacin hydrochloride (BAY35-3377)Pages 2525-2528 |
||
Finafloxacin hydrochloride, an important clinical compound was synthesized by a novel synthetic approach. An active intermediate ethyl 7-chloro-8-cyano-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate 19 was prepared by a new route. The chiral (S,S′)-N-Boc 10 was derived from protected pyrrolidine and the absolute stereochemistry was established by X-ray analysis.
http://www.sciencedirect.com/science/article/pii/S0040403909005875
……………….



| WO2011003091A1 * | 2 Jul 2010 | 6 Jan 2011 | Alcon Research, Ltd. | Compositions comprising finafloxacin and methods for treating ophthalmic, otic, or nasal infections |
| US7723524 | 29 Sep 2004 | 25 May 2010 | Daiichi Pharmaceutical Co., Ltd. | 8-cyanoquinolonecarboxylic acid derivative |
| US8536167 | 2 Jul 2010 | 17 Sep 2013 | Alcon Research, Ltd. | Methods for treating ophthalmic, otic, or nasal infections |
| DE4329600A1 * | 2 Sep 1993 | 9 Mar 1995 | Bayer Ag | Pyrido [1,2,3-d,e] [1,3,4] benzoxadiazinderivate |
| EP0276700A1 * | 15 Jan 1988 | 3 Aug 1988 | Bayer Ag | 8-Cyano-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids, process for their preparation, and antibacterial agents containing them |
| EP0350733A2 * | 30 Jun 1989 | 17 Jan 1990 | Bayer Ag | 7-(1-Pyrrolidinyl)-3-quinolone- and -naphthyridone-carboxylic-acid derivatives, method for their preparation and for substituted mono- and bi-cyclic pyrrolidine intermediates, and their antibacterial and feed additive compositions |
| EP0550903A1 * | 28 Dec 1992 | 14 Jul 1993 | Bayer Ag | Quinolone- and naphthyridone carboxylic acid derivatives as antibacterial agents |
| EP0603887A2 * | 23 Dec 1993 | 29 Jun 1994 | Daiichi Pharmaceutical Co., Ltd. | Bicyclic amine derivatives |
| EP0676199A1 * | 23 Mar 1995 | 11 Oct 1995 | Pfizer Inc. | Use of trovafloxacin or derivatives thereof for the manufacture of a medicament for the treatment of H. pylori infections |
| GB2289674A * | Title not available |


Nemonoxacin 奈诺沙星
378746-64-6 CAS
TG-873870
WARNER CHILCOTT ORIGINATOR
CLINICAL TRIALS http://clinicaltrials.gov/search/intervention=Nemonoxacin
(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
7-[3(S)-Amino-5(S)-methylpiperidin-1-yl]-1-cyclopropyl-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
Taigexyn has been approved in Taiwan IN 2014
TaiGen Biotechnology Receives Marketing Approval from the Taiwan Food and Drug Administration for Taigexyn in Taiwan
TAIPEI, March 13, 2014 /PRNewswire/ — TaiGen Biotechnology Company, Limited (“TaiGen”) today announced that the Taiwan Food and Drug Administration (TFDA) has approved the new drug application (NDA) of Taigexyn® (nemonoxacin) oral formulation (500 mg) for the treatment of community-acquired bacterial pneumonia (CAP). With this NDA approval, Taiwan is the first region to grant marketing approval to Taigexyn®. An NDA for Taigexyn® was also submitted to China FDA (CFDA) in April 2013 and is currently under review.
Nemonoxacin is a novel non-fluorinated quinolone antibiotic undergoing clinical trials.
TaiGen Biotechnology announced that the FDA has granted nemonoxacin (Taigexyn) Qualified Infectious Disease Product (QIDP) and Fast Track designations for community-acquired bacterial pneumonia (CAP) and acute bacterial skin and skin structure infections (ABSSSI).
Nemonoxacin is a novel non-fluorinated quinolone broad spectrum antibiotic available in both oral and intravenous formulations. Nemonoxacin demonstrates activity against gram-positive and gram-negative bacteria and atypical pathogens. Nemonoxacin also possesses activities against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant pathogens.
Nemonoxacin is a novel non-flourinated quinolone antibiotic registered in Taiwan for the oral treatment of community-acquired pneumonia. Clinical trials are in development at TaiGen Biotechnology for the treatment of diabetic foot infections and for the treatment of moderate to severe community-acquired pneumonia with an intravenous formulation. The drug is thought to accomplish its antibacterial action through topoisomerase inhibition.
Originally developed at Procter & Gamble, nemonoxacin was the subject of a strategic alliance formed in January 2005 between P&G and TaiGen to further the development and commercialization of nemonoxacin. In 2012, the product was licensed by TaiGen Biotechnology to Zhejiang Medicine in China for manufacturing, sales and marketing. In 2014, TaiGen out-licensed the exclusive rights of the product in Russian Federation, Commonwealth Independent States and Turkey to R-Pharm.
TaiGen has completed two Phase 2 clinical studies, one in CAP and the other in diabetic foot infections with demonstrated efficacy and safety. In the clinical trials conducted to date, nemonoxacin has shown activity against drug-resistant bacteria such as MRSA, quinolone-resistant MRSA, as well as quinolone-resistant Streptococcus pneumoniae.

Malate salt
Nemonoxacin malate anhydrous
951163-60-3 CAS NO, MW: 505.5209
Nemonoxacin malate hemihydrate
951313-26-1, MW: 1029.0566
Chemical structure of nemonoxacin as a malate salt (C20H25N3O4·C4H6O5·H2O). Nemonoxacin is the free base, and its molecular mass is 371.44 g/mol. The molecular mass of the salt, nemonoxacin malate, is 514.53 g/mol.


……………………..
isomeric compounds are:

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD1…….DESIRED

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
Example 1
Malate salts of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and (3S,5R)-7- [3-ammo-5-methyl-piperidinyl]- 1 -cyclopropyl- 1 ,4-dihydro-8-methoxy-4-oxo-3- quinolinecarboxylic acid (Compound 1′) were synthesized as follows:
(A) Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9) and (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (Compound 9′): Compound 9′ was synthesized as shown in Scheme 1 below:
Scheme 1

3 4 Boc

A 50-L reactor was charged with Compound 2 (5.50 kg, 42.60 mol), methanol (27 L) and cooled to 10-150C. Thionyl chloride (10.11 kg, 2.0 equiv.) was added via an addition funnel over a period of 65 min, with external cooling to keep temperature below 30°. The resulting solution was stirred at 250C for 1.0 hour, after which methanol was removed under reduced pressure. The oily residue was azeotroped with ethyl acetate (3 x 2.5 L) to remove residual methanol, dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by slow addition of triethylamine (3.6 kg) below 3O0C. The resulting suspension was filtered to remove triethylamine hydrochloride.
The filtrate was charged to a 50 L reactor, along with DMAP (0.53 kg). Di- fert-butyl dicarbonate (8.43 kg) was added via hot water heated addition funnel, over a period of 30 min at a temperature of 20-300C. The reaction was complete after 1 hour as determined by TLC analysis. The organic phase was washed with ice cold IN HCl (2 x 7.5 L), saturated sodium bicarbonate solution (1 x 7.5 L), dried over magnesium sulfate, and filtered. After ethyl acetate was removed under reduced pressure, crystalline slurry was obtained, triturated with MTBE (10.0 L), and filtered to afford Compound 3 as a white solid (5.45 kg, 52.4%).
Anal. Calcd for CHHI7NO5 : C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for CHHI8NO5, [M+H] 244.1185. Found
244.1174; 1H NMR (CDCl3, 500 MHz):δ=4.54 (dd, J= 3.1, 9.5 Hz, IH), 3.7 (s, 3H), 2.58-2.50 (m, IH), 2.41 (ddd, IH, J= 17.6, 9.5, 3.7), 2.30-2.23 (m, IH), 1.98-1.93 (m, IH), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5. Mp 70.20C.
A 50-L reactor was charged with Compound 3 (7.25 kg, 28.8 mol), DME (6.31 kg), and Bredereck’s Reagent (7.7 kg, 44.2 mole). The solution was agitated and heated to 750C + 50C for three hours. The reaction was cooled to O0C over an hour, during which time a precipitate formed. The mixture was kept at O0C for an hour, filtered, and dried in a vacuum oven for at least 30 hours at 3O0C + 50C to give compound 4 as a white crystalline solid (6.93 kg, 77.9%).
Anal. Calcd for Ci4H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for Ci4H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR (CDCl3, 499.8 MHz) δ = 7.11 (s, IH), 4.54 (dd, IH, J= 10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, IH), 3.00 (s, 6H), 2.97-2.85 (m,lH), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ = 172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. MP 127.90C. A 10-gallon Pfaudler reactor was charged with ESCAT 142 (Engelhard Corp.
N.J, US) 5% palladium powder on carbon (50% wet, 0.58 kg wet wt), Compound 4 (1.89 kg, 6.33 mol), and isopropanol (22.4 Kg). After agitated under a 45-psi hydrogen atmosphere at 450C for 18 hrs, the reaction mixture was cooled to room temperature and filtered though a bed of Celite (0.51 kg). The filtrate was evaporated under reduced pressure to give a thick oil, which was solidified on standing to afford Compound 5 (1.69 kg, 100%) as a 93:7 diastereomeric mixture.
A sample of product mixture was purified by preparative HPLC to give material for analytical data. Anal. Calcd for Ci2Hi9NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for Ci2Hi9NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ = 4.44 (m, IH), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, IH), 1.43 (s, 9H), 1.20 (d, j = 6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ = 175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.90C.
A 50-L reactor was charged with Compound 5 (3.02 kg, 11.7 mol), absolute ethanol (8.22 kg), and MTBE (14.81 kg). Sodium borohydride (1.36 kg, 35.9 mol) was added in small portions at 00C + 50C. A small amount of effervescence was observed. The reaction mixture was warmed to 1O0C + 50C and calcium chloride dihydrate (2.65 kg) was added in portions at 1O0C + 50C over an hour. The reaction was allowed to warm to 2O0C + 50C over one hour and agitated for an additional 12 hours at 200C + 50C. After the reaction was cooled to -50C + 50C, ice-cold 2N HCl (26.9 kg) was added slowly at of O0C + 50C. Agitation was stopped. The lower aqueous phase was removed. The reactor was charged with aqueous saturated sodium bicarbonate (15.6 kg) over five minutes under agitation. Agitation was stopped again and the lower aqueous phase was removed. The reactor was charged with magnesium sulfate (2.5 kg) and agitated for at leastlO minutes. The mixture was filtered though a nutsche filter, and concentrated under reduced pressure to afford Compound 6 (1.80 kg, 66%). Anal. Calcd for CnH23NO4: C, 56.6 H, 9.94; N, 6.00. Found C, 56.0; H, 9.68;
N, 5.96; HRMS (ESI+) Expected for CnH24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz) δ = 6.34 (d, J= 8.9 Hz, IH, NH), 4.51 (t, J= 5.8, 5.3 Hz, IH, NHCHCH2OH), 4.34 (t, J= 5.3, 5.3 Hz, IH, OBCHCH2OH), 3.46-3.45, (m, IH, NHCH), 3.28 (dd, J= 10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J= 10.2, 5.8 Hz , IH, CH3CHCHHOH), 3.16 (dd, J = 10.2, 6.2 Hz, IH, NHCHCHHOH), 3.12 (dd, J= 10.6, 7.1 Hz , IH, CH3CHCHHOH), 1.53-1.50 (m, IH, CH3CHCHHOH), 1.35 (s, 9H, 0(CHB)3, 1.30 (ddd, J = 13.9, 10.2, 3.7 Hz, IH, NHCHCHHCH), 1.14 (ddd, J= 13.6, 10.2, 3.4 Hz, IH, NHCHCHHCH), 0.80 (d, J= 6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.10C. A 50 L reactor was charged with a solution of Compound 6 (5.1 kg) in isopropyl acetate (19.7 kg). The reaction was cooled to 150C + 5°C and triethylamine (7.8 kg) was added at that temperature. The reactor was further cooled to O0C + 50C and methanesulfonyl chloride (MsCl) (6.6 kg) was added. The reaction was stirred for a few hours and monitored for completion by HPLC or TLC. The reaction was quenched by saturated aqueous bicarbonate solution. The organic phase was isolated and washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase was dried, filtered, and concentrated in vacuo below 550C + 50C to afford compound 7 as a solid/liquid slurry, which was used in the subsequent reaction without further purification.
After charged with 9.1 kg of neat benzylamine, a 50 L reactor was warmed to 550C, at which temperature, a solution of compound 7 (8.2 kg) in 1,2- dimethoxyethane (14.1 kg) was added. After the addition, the reaction was stirred at 6O0C + 50C for several hours and monitored for completion by TLC or HPLC. The reaction was cooled to ambient temperature and the solvent was removed under vacuum. The residue was diluted with 11.7 kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture was obtained upon standing. The upper organic layer was collected. The isolated middle layer was extracted twice again with 11.7 kg portions of 15% (v/v) ethyl acetate/hexanes solution. The combined organic layers were concentrated under vacuum to give an oily residue. The residue was then purified by chromatography to afford Compound 8 as an oil. A 40 L pressure vessel was charged with 0.6 kg 50% wet, solid palladium on carbon (ElOl, 10 wt. %) under flow of nitrogen. A solution of Compound 8 (3.2 kg) in 13.7 kg of absolute ethanol was then added to the reactor under nitrogen. The reactor was purged with nitrogen and then pressurized with hydrogen at 45 psi. The reaction was then heated to 45°C. It was monitored by TLC or LC. Upon completion, the reaction was cooled to ambient temperature, vented, and purged with nitrogen. The mixture was filtered through a bed of Celite and the solid was washed with 2.8 kg of absolute ethanol. The filtrate was concentrated under vacuum to afford Compound 9 as a waxy solid.
TLC R/(Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain) = 0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, IH), 3.80-3.68 (m, IH), 2.92 (d, J=I 1.4 Hz,
IH), 2.77 (AB quart, JAB=12.0 Hz, v=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, IH), 1.82-1.68 (m, 2H), 1.54 (br s, IH), 1.43 (s, 9H), 1.25-1.15 (m, IH), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H). Similarly, (3S,5R)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester
(Compound 9′) was synthesized as shown in Scheme 2.
Scheme 2

HN Boc HN Boc
NaBH4,EtOH w – “ MsCI1TEA . „ _. – – _. „ Benzyl Amine
THF EA1CoId

(B) Synthesis of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-l,4-dihydro-quinoline-3- carboxylic acid (Compound 10): Compound 10 was prepared according to the method described in U.S. Patent
6,329,391.
(C) Synthesis of borone ester chelate of l-Cyclopropyl-7-fluoro-8-methoxy-4-oxo- l,4-dihydro-quinoline-3-carboxylic acid (Compound 11):
Scheme 3

Toluene, tert-Butylmethyl ether 20-500C, filter
A reactor was charged with boron oxide (2.0 kg, 29 mol), glacial acetic acid (8.1 L, 142 mol), and acetic anhydride (16.2 L, 171 mol). The resulting mixture was refluxed at least 2 hours, and then cooled to 400C, at which temperature, 7- fluoroquinolone acid compound 10 (14.2 kg, 51 mol) was added. The mixture was refluxed for at least 6 hours, and then cooled to about 900C. Toluene (45 L) was added to the reaction. At 5O0C, terϊ-butylmethyl ether (19 L) was added to introduce precipitation. The mixture was then cooled to 200C and filtered to isolate the precipitation. The isolated solid was then washed with teτt-butylmethyl ether (26 L) prior to drying in a vacuum oven at 4O0C (50 torr) to afford Compound 11 in a yield of 86.4%. Raman (cm 1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, IH), 8.38-8.33 (m, IH), 7.54 (t, J=9.8 Hz, IH), 4.38-4.35 (m, IH), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 6θA, 200 μm), Mobile Phase: 1 :1 (v/v) CH3CN : 0.5N NaCl (aq), UV (254/366 nm) visualization; R^O.4-0.5. (D) Synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidmyl]-l- cyclopropyl-l,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1) and malate salt of (3S,5R)-7-[3-amino-5-methyl-piperidmyl]-l-cyclopropyl-l,4- dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (Compound 1′)
Compound 1 was synthesized from compound 9 as shown in Scheme 4 below:
Scheme 4

5O0C 3 d
a 6 0 N HCI (aq) CH2CI2 35°40°C 12 h t> Extract pH ad]ust to ~7-8 50″-65″C filter



A reactor was charged with Compound 11 (4.4 kg, 10.9 mol), Compound 9 (2.1 kg, 9.8 mol), triethylamine (TEA) (2.1 L, 14.8 mol), and acetonitrile (33.5 L, 15.7 L/kg). The resulting mixture was stirred at approximately 500C till completion of the reaction, as monitored by HPLC or reverse phase TLC. It was cooled to approximately 35°C and the reaction volume was reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. After 28.2 kg of 3.0 N NaOH (aq) solution was added, the reaction mixture was warmed to approximately 4O0C, distilled under vacuum until no further distillates were observed, and hydro lyzed at room temperature. Upon completion of hydrolysis, which was monitored by HPLC or reverse phase TLC, 4-5 kg of glacial acetic acid was added to neutralize the reaction mixture.
The resulting solution was extracted 3 times with 12.7 kg (9.6 L) of dichloromethane. The organic layers were combined and transferred to another reactor. The reaction volume was reduced to approximately a half by evaporation at 400C. After 20.2 Kg 6.0N HCl (aq) solution was added, the reaction mixture was stirred for at least 12 hours at 35°C. After the reaction was completed as monitored by HPLC or reverse phase TLC, agitation was discontinued to allow phase separation. The organic phase was removed and the aqueous layer was extracted with 12.7 kg (9.6 L) of dichloromethane. The aqueous layer was diluted with 18.3 kg distilled water and warmed to approximately 500C. Dichloromethane was further removed by distillation under vacuum (100-400 torr).
The pH of the aqueous solution was then adjusted to 7.8-8.1 by adding about 9.42 kg of 3.0 N NaOH (aq) below 65°C. The reaction mixture was stirred at 500C for at least an hour and then cooled to room temperature. The precipitate was isolated by suction filtration, washed twice with 5.2 kg of distilled water, and dried with suction for at least 12 hours and then in a convection oven at 55°C for additional 12 hours. Compound 12 (3.2 kg, 79%) was obtained as a solid.
A reactor was charged with 3.2 kg of Compound 12 and 25.6 kg of 95% ethanol. To the reactor was added 1.1 kg of solid D,L-malic acid. The mixture was refluxed temperature (~80°C). Distilled water (-5.7 L) was added to dissolve the precipice and 0.2 kg of activated charcoal was added. The reaction mixture was passed through a filter. The clear filtrate was cooled to 45°C and allowed to sit for at least 2 hours to allow crystallization. After the reaction mixture was further cooled to 5°C, the precipitate was isolated by suction filtration, washed with 6.6 kg of 95% ethanol, and dried with suction for at least 4 hours. The solid was further dried in a convection oven at 450C for at least 12 hours to afford 3.1 kg of Compound 1 (yield: 70%). NEMONOXACIN
NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, IH), 7.37 (d, J=9.0 Hz, IH), 7.05 (d, J=9.0 Hz, IH), 4.23-4.18 (m, IH), 4.10-3.89 (m, IH), 3.66 (br s, IH), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, IH), 3.34 (d, J=9.3 Hz, IH), 3.16 (d, J=12.9 Hz, IH), 2.65 (dd, J=16.1, 4.1 Hz, IH), 2.64-2.53 (m, IH), 2.46 (dd, J=16.1, 8.0 Hz, IH), 2.06 (br s, IH), 1.87 (d, J=14.4 Hz, IH), 1.58-1.45 (m, IH), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H), 0.85-0.78 (m, 2H).
Similarly, Compound 1′ was synthesized from Compound 9′ as shown in Scheme 5 below:
Scheme 5

(3S,5R)-7-[3-amino-5-methyl-piperidinyl]-l-cyclopropyl-l,4-dihydro-8- methoxy-4-oxo-3 -quinolinecarboxylic acid
COMPD 1’….NOT DESIRED
…………………
US2007/232650 A1,
malate salts of

(3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (hereinafter Compound I, see also intermediate (23) in Section D, of Detailed Description of the Invention).
EXAMPLES Example 1 Synthesis of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid and malate salt thereof A. Synthesis of (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8)

(2S)-1-(1,1-Dimethylethyl)-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester, (2). A 50-L reactor is charged with compound (1) (5.50 Kg, 42.60 mol), methanol (27 L) and cooled to 10-15° C. Thionyl chloride (10.11 Kg, 2.0 equiv.) is added via addition funnel over a period of 65 min, with external cooling to maintain temperature at <30°. The resulting solution is stirred at 25° C.+5° C. for 1.0 hour, after which the methanol is distilled off under reduced pressure. The resulting thick oil is azeotroped with ethyl acetate (3×2.5 L) to remove residual methanol. The residue is dissolved in ethyl acetate (27.4 L), charged into a 50 L reactor, and neutralized by the addition of triethylamine (3.6 Kg) from an addition funnel over 30 minutes. The temperature of the neutralization is maintained below 30° C. via external cooling. The resulting suspension of triethylamine hydrochloride is removed by filtration, and the clarified mother liquor solution is charged to a 50 L reactor, along with DMAP (0.53 Kg). Di-tert-butyl dicarbonate (8.43 Kg) is added via hot water heated addition funnel, over a period of 30 min with external cooling to maintain temperature at about 20-30° C. The reaction is complete after 1 hour as determined by TLC analysis. The organic phase is washed with ice cold 1N HCl (2×7.5 L), saturated sodium bicarbonate solution (1×7.5 L), and dried over magnesium sulfate. The mixture is filtered through a nutsche filter and ethyl acetate is removed under reduced pressure to yield a crystalline slurry that is triturated with MTBE (10.0 L) and filtered to afford intermediate (2) as a white solid (5.45 Kg, 52.4%). Anal. Calcd for C11H17NO5: C, 54.3; H, 7.04; N, 5.76. Found: C, 54.5; H, 6.96; N, 5.80. HRMS (ESI+) Expected for C11H18NO5, [M+H] 244.1185. Found 244.1174; 1H NMR (CDCl3, 500 MHz): δ=4.54 (dd, J=3.1, 9.5 Hz, 1H), 3.7 (s, 3H), 2.58-2.50 (m, 1H), 2.41 (ddd, 1H, J=17.6, 9.5, 3.7), 2.30-2.23 (m, 1H), 1.98-1.93 (m, 1H), 1.40 (s, 9H); 13C NMR (CDCl3, 125.70 MHz) δ 173.3, 171.9, 149.2, 83.5, 58.8, 52.5, 31.1, 27.9, 21.5; Mp 70.2° C.
(2S,4E)-1-(1,1-Dimethylethyl)-4-[(dimethylamino)methylene]-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (3). A 50-L reactor is charged with intermediate (2) (7.25 Kg, 28.8 mol), DME (6.31 Kg), and Bredereck’s Reagent (7.7 Kg, 44.2 mole). The solution is agitated and heated to 75° C.±5° C. for at least three hours. The progress of the reaction is monitored by HPLC. The reaction is cooled to 0° C.±5° C. over on hour during which time a precipitate forms. The mixture is held at 0° C.±5° C. for one hour and filtered though a nutsche filter and the product dried in a vacuum oven for at least 30 hours at 30° C.±5° C. to give intermediate (3) as a white crystalline solid (6.93 Kg, 77.9%). Anal. Calcd for C14H22N2O5: C, 56.4; H, 7.43; N, 9.39. Found C, 56.4; H, 7.32; N, 9.48; HRMS (ESI+) Expected for C14H22N2O5, [M+H] 299.1607. Found 299.1613; 1H NMR(CDCl3, 499.8 MHz)δ=7.11 (s, 1H), 4.54 (dd, 1H, J=10.8, 3.6), 3.74 (s, 3H), 3.28-3.19 (m, 1H), 3.00 (s, 6H), 2.97-2.85 (m, 1H), 1.48 (s, 9H); 13C NMR (CDCl3, 125.7 MHz) δ=172.6, 169.5, 150.5, 146.5, 90.8, 82.2, 56.0, 52.3, 42.0, 28.1, 26.3. Mp 127.9° C.
(2S,4S)-1-(1,1-Dimethylethyl)-4-methyl-5-oxo-1,2-pyrrolidinedicarboxylic acid-2-methyl ester (4). A 10-gallon Pfaudler reactor is inerted with nitrogen and charged with ESCAT 142 5% palladium powder on carbon (50% wet, 0.58 Kg wet wt.), intermediate (3) (1.89 Kg, 6.33 mol) and isopropanol (22.4 Kg). The reaction mixture is agitated under a 45-psi hydrogen atmosphere at 45° C. for 18 hrs. The reaction mixture is then cooled to room temperature and filtered though a bed of Celite (0.51 Kg) in a nutsche filter to remove catalyst. The mother liquor is evaporated under reduced pressure to give a thick oil that crystallizes on standing to afford 4 (1.69 Kg, 100%) as a 93:7 diastereomeric mixture. A sample of product mixture is purified by preparative HPLC to give material for analytical data. Anal. Calcd for C12H19NO5: C, 56.0; H, 7.44; N, 5.44. Found C, 55.8; H, 7.31; N, 5.44; MS (ESI+) Expected for C12H19NO5, [M+H] 258.1342. Found 258.1321; 1H NMR (CDCl3, 499.8 MHz) δ=4.44 (m, 1H), 3.72 (s, 3H), 2.60-2.48 (m, 2H), 1.59-1.54 (m, 1H), 1.43 (s, 9H), 1.20 (d, j=6.8 Hz,3H); 13C NMR (CDCl3, 125.7 MHz) δ=175.7, 172.1, 149.5, 83.6, 57.4, 52.5, 37.5, 29.8, 27.9, 16.2. Mp 89.9° C.
(1S,3S)-(4-Hydroxyl-1-hydroxymethyl-3-methyl-butyl)-carbamic acid tert-butyl ester (5). A 50-L reactor is charged with intermediate (4) (3.02 Kg, 11.7 mol), absolute ethanol (8.22 Kg), and MTBE (14.81 Kg). The solution is agitated and cooled to 0° C.±5° C. and sodium borohydride (1.36 Kg, 35.9 mol) is added in small portions so as to maintain reaction temperature at 0° C.±5° C. A small amount of effervescence is observed. The reaction mixture is warmed to 10° C.±5° C. and calcium chloride dihydrate (2.65 Kg) is added portion wise at a slow rate over an hour so as to maintain a reaction temperature of 10° C.±5° C. The reaction is allowed to warm to 20° C.±5° C. over one hour and agitated for an additional 12 hours at 20° C.±5° C. The reaction is cooled to −5° C.±5° C., ice-cold 2N HCl (26.9 Kg) is added at a rate to maintain a reaction temperature of 0° C.±5° C. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=1) is removed. The reactor is charged with aqueous saturated sodium bicarbonate (15.6 Kg) over five minutes. Agitation is stopped to allow phases to separate. The lower aqueous phase (pH=8) is removed. The reactor is charged with magnesium sulfate (2.5 Kg) and agitated for at least 10 minutes. The mixture is filtered though a nutsche filter, and condensed under reduced pressure to afford intermediate (5) (1.80 Kg, 66%). Anal. Calcd for C11H23NO4: C, 56.6; H, 9.94; N, 6.00. Found C, 56.0; H, 9.68; N, 5.96; HRMS (ESI+) Expected for C11H24NO4, [M+H] 234.1705. Found 234.1703; 1H NMR (CDCl3, 500 MHz)δ=6.34(d, J=8.9 Hz, 1H, NH), 4.51 (t, J=5.8, 5.3 Hz, 1H, NHCHCH2OH), 4.34 (t, J=5.3, 5.3 Hz, 1H, CH3CHCH2OH), 3.46-3.45, (m, 1H, NHCH), 3.28 (dd, J=10.6, 5.3 Hz, NHCHCHHOH), 3.21 (dd, J=10.2, 5.8 Hz, 1H, CH3CHCHHOH), 3.16 (dd, J=10.2, 6.2 Hz, 1H, NHCHCHHOH), 3.12 (dd, J=10.6, 7.1 Hz, 1H, CH3CHCHHOH), 1.53-1.50 (m, 1H, CH3CHCHHOH), 1.35 (s, 9H, O(CH 3)3, 1.30 (ddd, J=13.9, 10.2, 3.7 Hz, 1H, NHCHCHHCH), 1.14 (ddd, J=13.6, 10.2, 3.4 Hz, 1H, NHCHCHHCH), 0.80 (d, J=6.6 Hz, 3H, CH3); 13C NMR (CDCl3, 125.7 MHz) δ 156.1, 77.9, 50.8, 65.1, 67.6, 65.1, 35.6, 32.8, 29.0, 17.1. Mp 92.1° C.
(2S,4S)-Methanesulfonic acid 2-tert-butoxycarbonylamino-5-methanesulfonyloxy-4-methyl-pentyl ester (6). A 50 L reactor is charged with a solution of intermediate (5) (5.1 Kg) in isopropyl acetate (i-PrOAc) 11.8 Kg followed by a rinse with an additional 7.9 Kg i-PrOAc. The reaction is cooled to 15° C.±5° C. and triethylamine (TEA) (7.8 Kg) is added while maintaining the set temperature. The reactor is further cooled to 0° C.±5° C. and methanesulfonyl chloride (MsCl) (6.6 Kg) is added to the reaction solution while maintaining the set temperature. The reaction is stirred for a few hours and monitored for completion by HPLC or TLC. The reaction is quenched by the addition of a saturated aqueous bicarbonate solution and the resulting isolated organic phase is washed successively with cold 10% aqueous triethylamine solution, cold aqueous HCl solution, cold saturated aqueous bicarbonate solution, and finally saturated aqueous brine solution. The organic phase is dried, filtered, and concentrated in vacuo below 55° C.±5° C. until a solid/liquid slurry containing intermediate (6) is obtained. The slurry is used crude in subsequent reaction without further characterization.
(3S,5S)-(1-Benzyl-5-methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (7). A 50 L reactor is charged with 9.1 Kg of neat benzylamine. The reactor is brought to 55° C. and a solution of intermediate (6) (8.2 Kg) in 1,2-dimethoxyethane (DME) (14.1 Kg) is added to the reactor while maintaining a temperature of 60° C.±5° C. After complete addition of this solution, the reaction is stirred at 60° C.±5° C. for several hours and monitored for completion by TLC or HPLC. The reaction is cooled to ambient temperature and volatiles (DME) are removed by rotary evaporation under vacuum. The residue is diluted with 11.7 Kg of 15% (v/v) ethyl acetate/hexanes solution and treated, while agitating, with 18.7 Kg of 20% (wt) aqueous potassium carbonate solution. A triphasic mixture is obtained upon settling. The bottom aqueous phase is removed and the middle phase is set aside. The upper organic phase is collected and held for combination with extracts from additional extractions. The isolated middle phase is extracted twice again with 11.7 Kg portions of 15% (v/v) ethyl acetate/hexanes solution, each time combining the extracts with original organic phase. The combined organic extracts are transferred into a rotary evaporator and solvent is removed under vacuum until an oily residue remains. The residue is then purified via large-scale preparative chromatography to afford purified intermediate (7) as an oil.
(3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8). A 40 L pressure vessel is charged with 0.6 Kg 50% wet, solid palladium on carbon (E101, 10 wt. %) under flow of nitrogen. A solution of 3.2 Kg intermediate (7) in 13.7 Kg of absolute ethanol is then charged to the reactor under nitrogen. The reactor is purged with nitrogen and is then pressurized with hydrogen at 45 psi. The reaction is then heated to 45° C. while maintaining a hydrogen pressure of 45 psi. The reaction is monitored by TLC or LC until complete. The reaction is cooled to ambient temperature, vented, and purged with nitrogen. The reactor contents are filtered through a bed of Celite and the solids are washed with 2.8 Kg of absolute ethanol. The filtrate is concentrated by rotary evaporation under vacuum until a waxy solid is obtained to afford intermediate (8): TLC Rf (Silica F254, 70:30 v/v ethyl acetate-hexanes, KMnO4 stain)=0.12; 1H NMR (300 MHz, CDCl3) δ 5.31 (br s, 1H), 3.80-3.68 (m, 1H), 2.92 (d, J=11.4 Hz, 1H), 2.77 (AB quart, JAB=12.0 Hz, Δν=50.2 Hz, 2H), 2.19 (t, J=10.7 Hz, 1H), 1.82-1.68 (m, 2H), 1.54 (br s, 1H), 1.43 (s, 9H), 1.25-1.15 (m, 1H), 0.83 (d, J=6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 155.3, 78.9, 54.3, 50.8, 45.3, 37.9, 28.4, 27.1, 19.2; MS (ESI+) m/z 215 (M+H), 429 (2M+H).
B. Synthesis of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (19)


Intermediate (12): A reactor is charged with a solution of intermediate (11) (1.2 Kg, 7.7 mol, 1.0 eq) in anhydrous toluene (12 L) followed by ethylene glycol (1.8 L, 15.7 mol, 4.2 eq) and solid p-toluenesulfonic acid (120 g, 10 wt. %). The reaction mixture is stirred at ambient temperature for at least 30 minutes and then heated to reflux, collecting the water/toluene azeotrope in a Dean Stark type trap apparatus until the reaction is complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to ambient temperature and poured into an aqueous solution of sodium bicarbonate (6 L). The organic toluene phase was removed and washed with saturated sodium bicarbonate solution (6 L), distilled water (2×6 L), and saturated aqueous brine (6 L). The organic phase was removed and dried over MgSO4, filtered, and evaporated under reduced pressure to afford intermediate (12) as an oil (1.3 Kg, 86%). The material is used without further purification in subsequent reaction steps.
Intermediate (13): A reactor is charged with a solution of intermediate (12) (1.2 Kg, 6.0 mol, 1.0 eq) in anhydrous tetrahydrofuran (12 L) and n-butyllithium (2.5M in hexanes, 2.6 L, 6.6 mol, 1.1 eq) is added at −40° C., while maintaining this temperature throughout the addition. The reaction is stirred for at least one hour at −40° C. and trimethylborate (0.9 L, 7.8 mol, 1.3 eq) is added to the mixture while maintaining the temperature at or below −40° C. The reaction mixture is stirred for at least one hour at −40° C. until complete as determined by TLC analysis (30% EtOAc/Hexanes v/v). The reaction is warmed slightly to −30° C. and acetic acid (3 L) is added slowly. Upon complete addition, water is added (0.5 L) to the reaction and the mixture is allowed to quickly warm to ambient temperature while stirring overnight. Organic solvent is removed from the reaction by distillation under reduced pressure at 45° C. To the reaction residue is added 3-4 volumes of water (6 L) and 30% hydrogen peroxide (0.7 L, 1.0 eq) slowly at ambient temperature with cooling provided to control the exotherm. The reaction is stirred for at least an hour at ambient temperature until complete as determined by TLC (15% EtOAc/Hexanes v/v). The reaction mixture is cooled to 0-5° C. and excess peroxide is quenched with the addition of 10% aqueous sodium bisulfite solution (2 L). The mixture is tested to ensure a negative peroxide result and the reaction is acidified by the addition of 6N HCl (aq) (1.2 L). The reaction is stirred until the hydrolysis reaction is complete as determined by TLC or NMR analysis. The resulting solids are collected by suction filtration to afford intermediate (13) as a yellow solid (1.0 Kg, 79%).
Intermediate (14): A reactor is charged with intermediate (13) (0.53 Kg, 3.0 mol, 1.0 eq) and dissolved in dry toluene (2.7 Kg, 3.1 L). To this solution is added dimethylsulfate (0.49 Kg, 3.9 mol, 1.30 eq) followed by solid potassium carbonate (0.58 Kg, 4.2 mol, 1.4 eq). The reaction mixture is heated to reflux and held for at least 1 hour until complete as determined by HPLC. During this time, vigorous gas evolution is observed. The reaction is then cooled to ambient temperature and diluted with distilled water (3.2 L) along with 30% NaOH (aq) (0.13 Kg, 0.33 eq). The aqueous phase is separated and the remaining toluene phase is extracted twice more with distilled water (3.2 L) combined with 30% NaOH (aq) (0.13 Kg, 0.33 eq), removing the aqueous phase each time. The organic upper phase is concentrated by distillation in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature, checked for quality and yield by HPLC, and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (14) assumed, 0.56 Kg).
Intermediate (15a,b): A reactor is charged with 1.8 Kg (2.1 L) anhydrous toluene along with sodium hydride (0.26 Kg, 6.6 mol, 2.20 eq) as a 60 wt. % dispersion in mineral oil. To this mixture is added (0.85 Kg, 7.2 mol, 2.4 eq) diethylcarbonate as the reaction mixture is heated to 90° C. over 1 hour. A solution of intermediate (14) (˜1.0 eq) in toluene from the previous step is added to the reaction while maintaining a temperature of 90° C.±5° C. Gas evolution can be observed during this addition. After complete addition, the reaction is stirred for at least 30 minutes or until complete as determined by HPLC analysis. Upon completion, the mixture is cooled to ambient temperature and diluted with 10 wt. % aqueous sulfuric acid (3.8 Kg, 3.9 mol, 1.3 eq) with agitation. The phases are allowed to separate and the lower aqueous phase is removed. The remaining organic phase is concentrated in vacuo (<100 mbar) at approximately 40° C. until a concentrated toluene solution is achieved. The resulting solution is cooled to ambient temperature and carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (15a,b) assumed, 0.85 Kg).
Intermediate (16a,b; 17a,b): A reactor is charged with a solution of intermediate (15a,b) (0.85 Kg, ˜3.0 mol, ˜1.0 eq) in toluene from the previous step. To the reactor is then added dimethylformamide-dimethylacetal (0.54 Kg, 4.5 mol, 1.5 eq) and the resulting solution is heated to reflux temperature (˜95-105° C.). The lower boiling solvent (methanol from reaction) is allowed to distill off while the temperature is maintained at ≧90° C. Heating is continued for at least 1 hour or until complete as determined by HPLC analysis. Upon completion, the reaction containing the mixture of intermediate (16a,b), is cooled to ambient temperature and toluene (1.8 Kg, 2.1 L) along with cyclopropylamine (0.21 Kg, 3.6 mol, 1.2 eq) are added to the reaction. The reaction is stirred at ambient temperature for at least 30 minutes until complete as determined by HPLC. Upon completion, the reaction is diluted with 10 wt. % aqueous sulfuric acid (2.9 Kg, 3.0 mol, 1.0 eq) with agitation, and the phases are then allowed to separate. The aqueous phase is removed and the organic phase is concentrated under reduced pressure (<100 mbar) at approximately 40° C. by distillation. When the desired concentration is achieved, the solution is cooled to ambient temperature and the toluene solution containing the mixture of intermediate (17a,b) is carried forward to the next step in the synthesis without further purification (theoretical yield for intermediate (17a,b) assumed, ˜1.1 Kg).
Intermediate (18): A reactor is charged with a solution of the mixture of intermediate (17a,b) (˜4.7 Kg, ˜3.0 mol) at ambient temperature. To the reactor is added N,O-bis(trimethylsilyl)acetamide (0.61 Kg, 3.0 mol, 1.0 eq) and the reaction is heated to reflux temperature (˜105-115° C.) for at least 30 minutes or until complete as determined by HPLC analysis. If not complete, an additional amount of N,O-bis(trimethylsilyl)acetamide (0.18 Kg, 0.9 mol, 0.3 eq) is added to the reaction to achieve completion. Upon completion, the reaction is cooled to below 40° C. and organic solvent is removed under reduced pressure (<100 mbar) at approximately 40° C. by distillation until a precipitate is formed. The reaction is cooled to ambient temperature and the precipitated solids are isolated by suction filtration and washed with distilled water twice (1×1.8 L, 1×0.9 L). The solid is dried to afford intermediate (18) as a white solid (0.76 Kg, 82%). The material is used without further purification in the next reaction step.
Intermediate (19): A reactor is charged with solid intermediate (18) (0.76 Kg, ˜2.5 mol, ˜1.0 eq) at ambient temperature followed by ethanol (5.3 Kg, 6.8 L) and 32 wt. % aqueous hydrochloric acid (1.1 Kg, 10 mol). The reaction mixture is brought to reflux temperature (76-80° C.) during which time the mixture first becomes homogeneous and later becomes heterogeneous. The mixture is heated at reflux for at least 5 hours or until complete as determined by TLC analysis (15% EtOAc/Hexanes v/v). Upon completion, the reaction is cooled to 0° C.±5° C. and the precipitated solid is isolated by filtration and washed with distilled water (1.7 Kg) followed by ethanol (1.7 Kg). The isolated solid is dried to afford intermediate (19) as a white solid (0.65 Kg, ˜95%). 1H NMR (CDCl3, 300 MHz) δ (ppm): 14.58 (s, 1H), 8.9 (s, 1H), 8.25 (m, 1H), 7.35 (m, 1H), 4.35 (m, 1H), 4.08 (s, 3H), 1.3 (m, 2H), 1.1 (m, 2H) 19F NMR (CDCl3+CFCl3, 292 MHz) δ (ppm): −119. HPLC: 99.5% by area.
C. Synthesis of borone ester chelate of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20)

A reactor is charged with boron oxide (2.0 Kg, 29 mol) followed by dilution with glacial acetic acid (8.1 L, 142 mol) and acetic anhydride (16.2 L, 171 mol). The resulting mixture is heated to reflux temperature for at least 2 hours. The reaction contents are cooled to 40° C. and the solid 7-fluoroquinolone acid intermediate (19) (14.2 Kg, 51 mol) is added to the reaction mixture. The mixture is again heated to reflux temperature for at least 6 hours. Reaction progress is monitored by HPLC and NMR. The mixture is cooled to approximately 90° C. and toluene (45 L) is added to the reaction. The reaction is further cooled to 50° C. and tert-butylmethyl ether (19 L) is added to the reaction mixture to bring about precipitation of the product. The mixture is then cooled to 20° C. and the solid product 19 is isolated by filtration. The isolated solids are then washed with tert-butylmethyl ether (26 L) prior to drying in a vacuum oven at 40° C. (50 torr). The product yield obtained for intermediate (20) in this reaction is 86.4%. Raman (cm−1): 3084.7, 3022.3, 2930.8, 1709.2, 1620.8, 1548.5, 1468.0, 1397.7, 1368.3, 1338.5, 1201.5, 955.3, 653.9, 580.7, 552.8, 384.0, 305.8. NMR (CDCl3, 300 MHz) δ (ppm): 9.22 (s, 1H), 8.38-8.33 (m, 1H), 7.54 (t, J=9.8 Hz, 1H), 4.38-4.35 (m, 1H), 4.13 (s, 3H), 2.04 (s, 6H), 1.42-1.38 (m, 2H), 1.34-1.29 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization; Rf=0.4-0.5.
D. Coupling of 1-Cyclopropyl-7-fluoro-8-methoxy-4-oxo-1,4-dihydro-quinoline-3-carboxylic acid (20) to (3S,5S)-(5-Methyl-piperidin-3-yl)-carbamic acid tert-butyl ester (8), and synthesis of malate salt of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid (25)

A reactor is charged with solid intermediate (20) (4.4 Kg, 10.9 mol) followed by dilution with a solution of triethylamine (TEA) (2.1 L, 14.8 mol) and piperidine side chain intermediate (8) (2.1 Kg, 9.8 mol) in acetonitrile (33.5 L, 15.7 L/Kg) at room temperature. The resulting mixture is warmed to approximately 50° C. until reaction is judged complete. Reaction progress is monitored by HPLC or reverse phase TLC. When complete, the reaction is cooled to approximately 35° C. and reaction volume is reduced to approximately half by distillation of acetonitrile under vacuum between 0-400 torr. The reactor is then charged with 28.2 Kg of 3.0N NaOH (aq) solution and the temperature is raised to approximately 40° C. Distillation under vacuum is continued between 1-4 hours or until no further distillates are observed. The reaction is then cooled to room temperature and the hydrolysis reaction is monitored by HPLC or reverse phase TLC. Upon completion, the reaction mixture is neutralized to a pH of between 6-8 by adding ˜4-5 Kg of glacial acetic acid. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The extraction process is repeated two additional times using 12.7 Kg (9.6 L) of dichloromethane, collecting the lower, organic phase each time. The aqueous phase is discarded and the organic extracts are combined in a single reactor. The reactor contents are heated to 40° C. and the reaction volume is reduced to approximately one half by distillation. The reactor is then charged with 20.2 Kg 6.0N HCl (aq) solution, the temperature is adjusted to 35° C., and agitation is allowed for at least 12 hours to permit the Boc deprotection reaction to occur. The reaction is monitored by HPLC or reverse phase TLC. When complete, agitation is discontinued and the phases are allowed to separate. The lower, organic phase is removed and set aside. The reactor is then charged with 12.7 Kg (9.6 L) of dichloromethane as an extraction solvent, the mixture is agitated, phases are allowed to separate, and the organic dichloromethane phase is removed. The organic extracts are combined and discarded. The remaining aqueous phase is diluted with 18.3 Kg distilled water and the temperature is raised to approximately 50° C. Distillation under vacuum (100-400 torr) is performed to remove residual dichloromethane from the reaction. The pH of the reaction is then adjusted to between 7.8-8.1 using about 9.42 Kg of 3.0N NaOH (aq) solution while keeping the temperature of the reaction below 65° C. The reaction is cooled to 50° C. and the precipitated solids are aged for at least an hour prior to cooling the mixture to room temperature. The solids are isolated by suction filtration and washed twice with 5.2 Kg portions of distilled water. The solids are dried for at least 12 hours with suction and then for an additional 12 hours in a convection oven at 55° C. The yield achieved for intermediate (23) in this example is 3.2 Kg (79%). A reactor is charged with 3.2 Kg solid intermediate (23) and the solids are suspended in 25.6 Kg of 95% ethanol as solvent. To the reactor is then added 1.1 Kg of solid D,L-malic acid (24), and the mixture is heated to reflux temperature (˜80° C.). Distilled water (˜5.7 L) is added to the reaction until a complete solution is achieved and 0.2 Kg of activated charcoal is added. The reaction mixture is passed through a filter to achieve clarification, cooled to 45° C. and held for a period of at least 2 hours to allow crystallization to occur. The reaction mixture is further cooled to 5° C. and the suspended solids are isolated by suction filtration. The solids are then washed with 6.6 KG of 95% ethanol and dried for at least 4 hours with suction under vacuum. The solids are then further dried in a convection oven for at least 12 hours at 45° C. to afford 3.1 Kg of intermediate (24) (70%). NMR (D2O, 300 MHz) δ (ppm): 8.54 (s, 1H), 7.37 (d, J=9.0 Hz, 1H), 7.05 (d, J=9.0 Hz, 1H), 4.23-4.18 (m, 1H), 4.10-3.89 (m, 1H), 3.66 (br s, 1H), 3.58 (s, 3H), 3.45 (d, J=9.0 Hz, 1H), 3.34 (d, J=9.3 Hz, 1H), 3.16 (d, J=12.9 Hz, 1H), 2.65 (dd, J=16.1, 4.1 Hz, 1H), 2.64-2.53 (m, 1H), 2.46 (dd, J=16.1, 8.0 Hz, 1H), 2.06 (br s, 1H), 1.87 (d, J=14.4 Hz, 1H), 1.58-1.45 (m, 1H), 1.15-0.95 (m, 2H), 0.91 (d, J=6.3 Hz, 3H); 0.85-0.78 (m, 2H). TLC (Whatman MKC18F Silica, 60 Å, 200 μm), Mobile Phase: 1:1 (v/v) CH3CN:0.5N NaCl (aq), UV (254/366 nm) visualization. HPLC: Mobile Phase H2O with 0.1% formic acid/Acetonitrile with 0.1% formic acid, gradient elution with 88% H2O/formic acid to 20% H2O/formic acid, Zorbax SB-C8 4.6 mm×150 mm column, Part No. 883975.906, 1.5 ml/min rate, 20 min run time, 292 nm, Detector Model G1314A, S/N JP72003849, Quat Pump Model G1311A, S/N US72102299, Auto Sampler Model G1313A, S/N DE14918139, Degasser Model G1322A, S/N JP73007229; approximate retention time for intermediate (19): 13.0 min; approximate retention time for intermediate (20): 11.6 min; approximate retention time for intermediate (21): 16.3 min; approximate retention time for intermediate (22): 18.2 min; approximate retention time for intermediate (23): 8.6 min; approximate retention time for compound (25): 8.6 min.
………………..
REF
A. ARJONA ET AL: “Nemonoxacin“, DRUGS OF THE FUTURE, vol. 34, no. 3, 1 January 2009 (2009-01-01), page 196, XP55014485, ISSN: 0377-8282, DOI: 10.1358/dof.2009.034.03.1350294
| 2 | * | ANONYMOUS: “TaiGen Announces Positive Data From the Phase II Study of Nemonoxacin (TG-873870) in Community-Acquired Pneumonia“, INTERNET CITATION, [Online] 7 April 2008 (2008-04-07), page 1, XP007919900, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#16> [retrieved on 2011-12-12] |
| 3 | * | ANONYMOUS: “TaiGen Biotechnology Initiates Phase II Trial Of Nemonoxacin For Treatment Of Adult Community Acquired Pneumonia (CAP)“, 20070108, [Online] 8 January 2007 (2007-01-08), page 1, XP007919910, Retrieved from the Internet: URL:http://www.taigenbiotech.com/news.html#11> [retrieved on 2011-12-12] |
| 4 | * | ANONYMOUS: “TaiGen Initiates Phase 1B Trial of a Novel Quinolone Antibiotic“, 20050618, 18 June 2005 (2005-06-18), pages 1-2, XP007919904, |
| 5 | * | See also references of WO2010002415A1 |
| WO2007110834A2 * | Mar 26, 2007 | Oct 4, 2007 | Procter & Gamble | Malate salts, and polymorphs of (3s,5s)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid |
| WO2009023473A2 * | Aug 5, 2008 | Feb 19, 2009 | Chi-Hsin Richard King | Antimicrobial parenteral formulation |
| WO2010009014A2 * | Jul 10, 2009 | Jan 21, 2010 | Taigen Biotechnology Co., Ltd. |
|
7-4-2012
|
TREATMENT OF ANTIBIOTIC-RESISTANT BACTERIA INFECTION
|
|
|
4-18-2012
|
Coupling Process For Preparing Quinolone Intermediates
|
|
|
10-19-2011
|
Malate salts, and polymorphs of (3S,5S)-7-[3-amino-5-methyl-piperidinyl]-1-cyclopropyl-1,4-dihydro-8-methoxy-4-oxo-3-quinolinecarboxylic acid
|
|
|
6-18-2010
|
STEREOSELECTIVE SYNTHESIS OF PIPERIDINE DERIVATIVES
|
|
|
2-19-2010
|
PNEUMONIA TREATMENT
|
|
|
5-6-2009
|
Hydride reduction process for preparing quinolone intermediates
|
|
|
2-13-2009
|
ANTIMICROBIAL PARENTERAL FORMULATION
|
|
|
11-26-2008
|
Coupling process for preparing quinolone intermediates
|
| US8158798 | Oct 27, 2008 | Apr 17, 2012 | Taigen Biotechnology Co., Ltd. | Coupling process for preparing quinolone intermediates |
| US8211909 | Sep 8, 2008 | Jul 3, 2012 | Taigen Biotechnology Co., Ltd. | Treatment of antibiotic-resistant bacteria infection |
| WO2010002965A2 * | Jul 1, 2009 | Jan 7, 2010 | Taigen Biotechnology Co., Ltd. | Pneumonia treatmen |
WO 2007110834
WO 2007110835
WO 2007110836
WO 1999014214
WO 2010077798

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.

CADAZOLID, ACT-179811
1-Cyclopropyl-6-fluoro-7-[4-({2-fluoro-4-[(5R)-5-(hydroxymethyl)-2-oxo-1,3-oxazolidin-3-yl]phenoxy}methyl)-4-hydroxypiperidin-1-yl]-4-oxo-1,4-dihydroquinolin-3-carboxylic acid
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-(R)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid
| Formula | C29H29F2N3O8 |
|---|---|
| Mol. mass | 585.55 g/mol |
Actelion Pharmaceuticals Ltd / Actelion’s novel antibiotic cadazolid receives US FDA Qualified Infectious Disease Product designation for the treatment of Clostridium difficile-associated diarrhea .
ALLSCHWIL/BASEL, SWITZERLAND – 27 February 2014 – Actelion Ltd (six:ATLN) today announced that the US Food and Drug Administration (FDA) has designated cadazolid as both a Qualified Infectious Disease Product (QIDP) and a Fast Track development program for the treatment of Clostridium difficile-associated diarrhea (CDAD).
The QIDP designation for cadazolid means that – among other incentives – cadazolid would receive a nine-month priority review upon successful completion of the ongoing global Phase III IMPACT program. The Fast Track designation is intended to promote communication and collaboration between the FDA and the Company on the development of the drug.
The designations are based on the 2012 US Generating Antibiotic Incentives Now (GAIN) Act. The GAIN act is a legislative effort to incentivize the development of new antibiotic agents that target serious life-threatening infections.
Guy Braunstein, M.D. and Head of Clinical Development commented: “Clostridium difficile-associated diarrhea is a very serious and potentially life-threatening infection. There is a great need for an antibiotic that allows effective treatment of CDAD with low recurrence rates, particularly in infections caused by hypervirulent strains. The GAIN act highlights the importance of research in this area and we are very happy to receive the advantages that this designation for cadazolid will afford us.”
ABOUT THE IMPACT PROGRAM
IMPACT is an International Multi-center Program Assessing Cadazolid Treatment in patients suffering from Clostridium difficile-associated diarrhea (CDAD). The program comprises two Phase III studies comparing the efficacy and safety of cadazolid (250 mg administered orally twice daily for 10 days) versus vancomycin (125 mg administered orally four times daily for 10 days).
The IMPACT studies are designed to determine whether the clinical response after administration of cadazolid is non-inferior to vancomycin in subjects with CDAD, and whether administration of cadazolid is superior to vancomycin in the sustained clinical response. The program is expected to enroll approximately 1’280 subjects worldwide, and commenced enrollment in the fourth quarter of 2013.
ABOUT CADAZOLID
The novel antibiotic cadazolid is a strong inhibitor of Clostridium difficile protein synthesis leading to strong suppression of toxin and spore formation. In preclinical studies cadazolid showed potent in vitro activity against Clostridium difficile clinical isolates and a low propensity for resistance development. In a human gut model of CDAD, cadazolid had a very limited impact on the normal gut microflora.
Cadazolid absorption is negligible resulting in high gut lumen concentrations and low systemic exposure, even in severe cases of CDAD where the gut wall can be severely damaged and permeability to drugs potentially increased.
Cadazolid is an experimental antibiotic of the oxazolidinone class made by Actelion Pharmaceuticals Ltd. which is effective against Clostridium difficile, a major cause of drug resistant diarrhea in the elderly.[1] Current drug treatments for this infection involve orally delivered antibiotics, principally fidaxomicin, metronidazole and vancomycin; the last two drugs are the principal therapeutic agents in use, but fail in approximately 20 to 45% of the cases. The drug is presently in Phase III trials.[1] The drug works by inhibiting synthesis of proteins in the bacteria, thus inhibiting the production of toxins and the formation of spores.[2]
The chemical structure of cadazolid combines the pharmacophores of oxazolidinone and fluoroquinolone.[2]
In phase I tests, sixty four male patients reacted favourably to cadazolid which primarily acted and remained in the colon while displaying little toxicity even in regimes involving large doses.[1]
ABOUT CADAZOLID IN THE PHASE II STUDY
Cadazolid was studied in a Phase II multi-center, double-blind, randomized, active reference, parallel group, therapeutic exploratory study. The study evaluated the efficacy, safety and tolerability of a 10-day, twice daily oral administration of 3 doses (250 mg, 500 mg or 1,000 mg b.i.d.) of cadazolid in subjects with Clostridium difficile-associated diarrhea (CDAD). As the current standard of care for CDAD, oral vancomycin (125 mg qid for 10 days) was used as the active reference. The study was completed in December of 2012, after having enrolled 84 subjects with CDAD.
The results of the Phase II study indicate that the effect of all doses of cadazolid were numerically similar to, or better than vancomycin on key endpoints including CDAD clinical cure rates as well as sustained cure rates. Clinical cure rate was defined as the resolution of diarrhea and no further need for CDAD therapy at test-of-cure 24 to 72 hours after the last dose of treatment, while sustained cure rate was defined as clinical cure with no recurrence of CDAD up to 4 weeks post-treatment. Recurrence rates were numerically lower for all doses of cadazolid as compared to vancomycin. Cadazolid was safe and well tolerated.
ABOUT THE GAIN ACT (INCLUDING FAST TRACK DESIGNATION)
The Food and Drug Administration Safety and Innovation Act (FDASIA) was signed into law in July 2012. The GAIN Act is Title VIII to FDASIA. The purpose of the GAIN Act is to encourage pharmaceutical research of certain antibiotics by designation of products as QIDPs. These products are intended to treat serious or life-threatening infections and include those to treat certain specifically identified pathogens, which are listed in the GAIN Act. C. difficile is one such specifically identified pathogen and drugs to treat CDAD would be eligible for designation as a QIDP.
The GAIN Act also provides that qualifying drugs (QIDPs) are eligible for inclusion in the FDA’s Fast Track program. This program is intended to facilitate development and expedite review of new drugs and includes close early communication between the FDA and a drug’s sponsor.
ABOUT FAST TRACK DRUG DEVELOPMENT PROGRAMS
For further information regarding Fast Track Drug Development Programs, please refer to the FDA document “Guidance for Industry on Fast Track Drug Development Programs: Designation, Development, and Application Review”. This document is available on the Internet at:
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079736.pdf
ABOUT CLOSTRIDIUM DIFFICILE-ASSOCIATED DIARRHEA
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacterium that is the leading cause of nosocomial diarrhea. Clostridium difficile-associated diarrhea (CDAD or CDI for Clostridium difficile infection) can be a severe and life-threatening disease and results from the overgrowth in the colon of toxigenic strains of Clostridium difficile, generally during or after therapy with broad-spectrum antibiotics. CDAD is a major healthcare problem and a leading cause of morbidity in elderly hospitalized patients. The frequency and severity of CDAD in the western world has increased in recent years, and new hypervirulent and epidemic strains of Clostridium difficile have been discovered that are characterized by overproduction of toxins and other virulence factors, and by acquired resistance to fluoroquinolones such as moxifloxacin.
Current antibiotic therapy for CDAD includes vancomycin and metronidazole. While clinical cure rates are generally 85-90%, recurrences rates of 15-30 % with either drug are problematic as Clostridium difficile produces spores that are resistant to antibiotic treatment and routine disinfection. Spores surviving in the gut of patients and/or in the hospital environment may play a major role in re-infection and recurrence of CDAD after antibiotic treatment. Vancomycin and metronidazole are reported to promote spore formation in vitro at sub-inhibitory concentrations.
Actelion Ltd.
Actelion Ltd. is a leading biopharmaceutical company focused on the discovery, development and commercialization of innovative drugs for diseases with significant unmet medical needs.
Actelion is a leader in the field of pulmonary arterial hypertension (PAH). Our portfolio of PAH treatments covers the spectrum of disease, from WHO Functional Class (FC) II through to FC IV, with oral, inhaled and intravenous medications. Although not available in all countries, Actelion has treatments approved by health authorities for a number of specialist diseases including Type 1 Gaucher disease, Niemann-Pick type C disease, Digital Ulcers in patients suffering from systemic sclerosis, and mycosis fungoides in patients with cutaneous T-cell lymphoma.
Founded in late 1997, with now over 2,400 dedicated professionals covering all key markets around the world including the US, Japan, China, Russia and Mexico, Actelion has its corporate headquarters in Allschwil / Basel, Switzerland
…………………..
Preparation of the compound of formula II
The compound of formula II can be obtained by hydrogenation of the compound of formula VIII

VIII
over a noble metal catalyst such as palladium or platinum on charcoal in a solvent such as THF, MeOH or EA between 00C and 400C or by hydrolysis of in presence of a solution of HBr in water or AcOH between 00C and 800C in a solvent such as AcOH.
The compounds of formula III can be prepared as summarized in Scheme 1 hereafter.

IX VI IIIA: R1= H IIIS: ^ = SO2R5
Scheme 1
The compounds of formula V can be prepared as summarized in Scheme 2 hereafter.

II X XI

Scheme 2
The compounds of formula X can be prepared from the methylidene derivatives of formula XII as summarized in Scheme 3 hereafter.

Xc XII Xa: R1 = H

Scheme 3
Example 1:
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((/f)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid:
1 i. (R)-3-(3-fluoro-4-hydroxy-phenyl)-5-hydroxymethyl-oxazolidin-2-one:
A solution of (7?y)-3-(4-benzyloxy-3-fluoro-phenyl)-5-hydroxymethyl-oxazolidin-2-one (6.34 g, prepared according to WO 2004/096221) in THF/MeOH (1 :1; 200 ml) was hydrogenated over Pd/C 10% (1 g) overnight. The catalyst was filtered off, the filtrate evaporated under reduced pressure and the residue stirred in EA. The crystals were collected by filtration, affording 3.16 g (70% yield) of a colourless solid. 1H NMR (DMSOd6; δ ppm): 3.5 (m, IH), 3.64 (m, IH), 3.74 (dd, J = 8.8, 6.4, IH), 3.99 (t, J = 8.8, IH), 4.64 (m, IH), 5.16 (t, J = 5.6, IH), 6.93 (dd, J = 9.7, 8.8, IH), 7.08 (ddd, J = 8.8, 2.6, 1.2, IH), 7.45 (dd, J = 13.5, 2.6, IH), 9.66 (s, IH). MS (ESI): 228.1.
1. ii. 4-[2-fluoro-4- ((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]- 4-hydroxy-piperidine-l-carboxylic acid benzyl ester:
A solution of intermediate l.i (1.27 g) and l-oxa-6-aza-spiro[2.5]octane-6-carboxylic acid benzyl ester (1.60 g; prepared according to US 4244961) were dissolved in DMF (15 ml) and treated with Na2CO3 (1.16 g). The mixture was heated at 1000C overnight. The residue obtained after workup (DCM) was stirred in EA, and the solid was collected by filtration and sequentially washed with EA and Hex, affording 2.52 g (94.5% yield) of a beige solid.
1H NMR (DMSOd6; δ ppm): 1.57 (m, 4H), 3.14 (m, 2H), 3.54 (m, IH), 3.64 (m, IH), 3.79 (m, 5 H), 4.03 (t, J = 9.1, 1 H), 4.66 (m, 1 H), 4.78 (s, 1 H), 5.05 (s, 2 H), 5.16 (t,
J = 5.6, 1 H), 7.18 (m, 2 H), 7.32 (m, 5 H), 7.55 (d, J = 12, 1 H).
MS (ESI): 475.0.
1. iii. (R)-3-[3-fluoro-4-(4-hydroxy-piperidin-4-ylmethoxy)-phenyl]-5-hydroxymethyl- oxazolidin-2-one:
A suspension of intermediate l.ii (2.5 g) in EA/MeOH (1 :1; 100 ml) was hydrogenated over Pd/C for 48 h. The suspension was heated at 400C and the catalyst was filtered off.
The filtrate was evaporated under reduced pressure affording 1.61 g (89% yield) of a yellow powder.
1H NMR (DMSOd6; δ ppm): 1.4-1.63 (m, 4H), 2.67 (m, 2H), 2.83 (m, 2H), 3.53 (dd, J = 4.0, 12.0, IH); 3.66 (dd, J = 3.3, 12.0, IH), 3.71 (s, 2H); 3.80 (m, IH), 4.05 (t, J = 9.0,
IH), 4.48 (s, IH), 4.68 (m, IH), 5.20 (s, IH), 7.20 (m, 2H), 7.57 (d, IH).
MS (ESI): 341.5.
l.iv. l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid:
A solution of intermediate l.iii (200 mg), 7-chloro-l-cyclopropyl-6-fiuoro-l,4-dihydro- 4-0X0-3 -quinolinecarboxylic acid boron diacetate complex (241 mg; prepared according to WO 88/07998) and DIPEA (100 μl) in NMP (2 ml) was stirred at 85°C for 5 h. The reaction mixture was evaporated under reduced pressure and the residue was taken up in 5M HCl in MeOH (3 ml) and stirred. The resulting solid was collected by filtration and washed with MeOH to afford 230 mg (67% yield) of a yellow solid.
1H NMR (DMSOd6; δ ppm): 1.66-1.35 (m, 4H), 1.75 (d, J = 12.8, 2H), 1.95 (m, 2H), 3.33 (t broad, J = 11.0, 2H), 3.57 (m, 3H), 3.67 (dd, J = 12.3, 3.3, IH), 3.83 (m, 2H), 3.92 (s, 2H), 4.06 (t, J = 9.0, IH), 4.69 (m, IH), 7.24 (m, 2H), 7.60 (m, 2H), 7.90 (d, J = 13.3, IH), 8.66 (s, IH).
MS (ESI): 585.9.

Isavuconazole (BAL4815; trade name Cresemba) is a triazole antifungal drug. Its prodrug, isavuconazonium sulfate (BAL8557), was granted approval by the U.S. Food and Drug Administration (FDA) on March 6, 2015[1]
During its Phase III drug trials, Astellas partnered with Basilea Pharmaceutica, the developer of the drug, for rights to co-development and marketing of isavuconazole. [2]
On May 28, 2013, Basilea Pharmaceutica announced it had been granted orphan drug status by the FDA for treatment of aspergillosis.[3] Since then, it has also been granted orphan drug status for the treatment of invasive candidiasis.[4]

Isavuconazonium sulfate (BAL8557)—a prodrug of isavuconazole.
CLINICAL TRIALS…LINK
PATENTS
|
6-27-2012
|
Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole
|
|
|
11-18-2011
|
Antifungal Composition
|
|
|
9-29-2010
|
PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS
|
|
|
12-3-2008
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
3-14-2007
|
N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
11-3-2004
|
N-substituted carbamoyloxyalkyl-azolium derivatives
|
|
|
10-10-2001
|
Azoles for treatment of fungal infections
|
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.

Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:



……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
http://www.sciencedirect.com/science/article/pii/S0960894X02008922
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.


We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.

Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%).
Figure options

Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).
read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
There have been three phase III clinical trials of isavuconazole, ACTIVE, VITAL and SECURE. As of June 2015, SECURE and VITAL have been presented in abstract form and results from ACTIVE have not been released.[9]
The SECURE trial compared voriconazole and isavuconazole in invasive fungal infections due to aspergillus. Isuvaconazole was found to be non-inferior to voriconazole, anothertriazole antifungal, with all cause mortality at 18.6%, compared to 20.2% in the voriconazole group. It additionally demonstrated a similar side effect profile.[10]
Data from the VITAL study showed that isavuconazole could be used in treatment of invasive mucormycosis, but did not evaluate its clinical efficacy for this indication.[11]
The ACTIVE trial is a comparison of isuvaconazole and caspofungin for invasive candida infections and results are anticipated in the second half of 2015.[12][13]
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{2-[(1R,2R)-(2,5-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
|
|
| Clinical data | |
| Trade names | Cresemba (prodrug form) |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, intravenous |
| Identifiers | |
| ATC code | None |
| PubChem | CID: 6918485 |
| ChemSpider | 5293682 |
| UNII | 60UTO373KE |
| ChEBI | CHEBI:85979 |
| ChEMBL | CHEMBL409153 |
| NIAID ChemDB | 416566 |
| Chemical data | |
| Formula | C22H17F2N5OS |
| Molecular mass | 437.47 g/mol |


/////
Ceftazidime/avibactam Combination Receives Qualified Infectious Disease Product (QIDP) Designation from FDA
NEW YORK–(BUSINESS WIRE)–Sept. 12, 2013–Forest Laboratories, Inc. (NYSE:FRX), an international pharmaceutical manufacturer and marketer, today announced that the U.S. Food and Drug Administration (FDA) has designated its investigational drug, ceftazidime/avibactam, a qualified infectious disease product (QIDP). The QIDP designation was created by the Generating Antibiotic Incentives Now (GAIN) Act, which was part of the FDA Safety and Innovation Act (FDASIA), which was signed into law in 2012. The QIDP designation provides certain incentives for the development of new antibiotics, including priority review and eligibility for the FDA’s fast track program, and a five-year extension of exclusivity under the Hatch-Waxman Act………
READ ALL AT
http://www.pharmalive.com/fda-grants-forest-drug-combo-qidp-status