








| Patent | Submitted | Granted |
|---|---|---|
| Neoadjuvant treatment of Breast Cancer [US2008318880] | 2008-12-25 | |
| Selective androgen receptor modulators for treating diabetes [US2007281906] | 2007-12-06 | |
| Nuclear receptor binding agents [US8158828] | 2007-11-15 | 2012-04-17 |
| Treatment of hormone-refractory prostate cancer [US2004220281] | 2004-11-04 | |
| METABOLITES OF SELECTIVE ANDROGEN RECEPTOR MODULATORS AND METHODS OF USE THEREOF [US8003689] | 2010-01-07 | 2011-08-23 |
| Treatment of metastatic breast cancer with anthracyclines, and taxanes [US2006089317] | 2006-04-27 | |
| Serm reduction of lipid profiles [US2007135407] | 2007-06-14 | |
| TREATMENT OF HORMONE-UNRESPONSIVE METASTATIC PROSTATE CANCER [EP0737067] | 1996-10-16 | 2003-09-10 |
| Use of a combination of dppe with other chemotherapeutic agents for the treatment of breast cancer [US2006142287] | 2006-06-29 | |
| Neoadjuvant treatment of breast cancer [US2006160755] | 2006-07-20 |
Home » Posts tagged 'PHASE 3' (Page 10)
Tag Archives: PHASE 3
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GS 9883, Bictegravir an HIV-1 integrase inhibitor
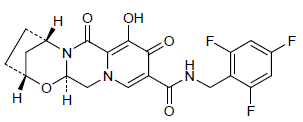
GS 9883, bictegravir
CAS 1611493-60-7
PHASE 3
HIV-1 integrase inhibitor
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
2,5-Methanopyrido(1′,2′:4,5)pyrazino(2,1-b)(1,3)oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-((2,4,6-trifluorophenyl)methyl)-, (2R,5S,13aR)-
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluoroheoctahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide
MF C21H18F3N3O5,
| MW | 449.37993 g/mol |
|---|
UNII-8GB79LOJ07; 8GB79LOJ07

BICTEGRAVIR
- 16 Nov 2015 Phase-III clinical trials in HIV-1 infections (Combination therapy, Treatment-naive) in USA (PO) (Gilead Pipeline, November 2015)
- 01 Jul 2015 Gilead Sciences completes a phase I trial in HIV-1 infections in USA and New Zealand (NCT02400307)
- 01 Apr 2015 Phase-I clinical trials in HIV-1 infections (In volunteers) in New Zealand (PO) (NCT02400307)
UPDATE Biktarvy (bictegravir/emtricitabine/tenofovir alafenamide); Gilead; For the treatment of HIV-1 infection in adults, Approved February 2018
Human immunodeficiency virus infection and related diseases are a major public health problem worldwide. Human immunodeficiency virus type 1 (HIV-1) encodes three enzymes which are required for viral replication: reverse transcriptase, protease, and integrase. Although drugs targeting reverse transcriptase and protease are in wide use and have shown effectiveness, particularly when employed in combination, toxicity and development of resistant strains have limited their usefulness (Palella, et al. N. Engl. J Med. (1998) 338:853-860; Richman, D. D. Nature (2001) 410:995-1001). Accordingly, there is a need for new agents that inhibit the replication of HIV and that minimize PXR activation when co-administered with other drugs.
Certain polycyclic carbamoylpyridone compounds have been found to have antiviral activity, as disclosed in PCT/US2013/076367. Accordingly, there is a need for synthetic routes for such compounds.
SYNTHESIS
WO 2014100323

PATENTS
xample 42
Preparation of Compound 42
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorohe
octahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide

42

Step 1
l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l ,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesuffhnic acid (0.195 mL, 3 mmol) and placed in a 75 deg C bath. The reaction mixture was stirred for 7 h, cooled and stored at -10 °C for 3 days and reheated to 75 °C for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (lR,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85 °C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HQ (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichloimethane:hexanes afforded the desired intermediate 42 A: LC S-ESI (m/z): [M+H]+ calculated for Ci5Hi7N206: 321.1 1 ; found: 321.3.
Step 3
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethyiammo)- V,A/-dimethyi(3H-[l ,2,3]triazolo[4,5-&]pyridm~3-yiox.y)methammimum hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LfJMS-ESlT (m/z): [M+H calculated for (\ ,l l.,, i \\:0< : 464.14; found: 464.2.
Step 4
To the crude reaction mixture of the previous step was added MgBr2
(0.258 g, 1.40 mmol). The reaction mixture was stirred at 50 °C for 10 minutes, acidified with 10% aqueous HC1, and extract twice with dichloromethane. The combined organic phases were dried over MgS04, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN H2O with 0.1 % TFA modifier) to afford compound 42: 1H~ M (400 MHz, DMSO-</6) δ 12.43 (s, 1H), 10.34 (t, J = 5.7 Hz, IH), 8.42 (s, 1H), 7.19 (t, J = 8.7 Hz, 2H), 5.43 (dd, ./’ 9.5, 4.1 Hz, I H), 5.08 (s, i l l ). 4.66 (dd, ./ 12.9, 4.0 Hz, IH), 4.59 (s, 1 1 1 ). 4.56 4.45 (m, 2H), 4.01 (dd, J = 12.7, 9.7 Hz, IH), 1.93 (s, 4H), 1.83 (d, J —— 12.0 Hz, I H),
1.56 (dt, J = 12.0, 3.4 Hz, I H). LCMS-ESI+ (m/z): [M+H]+ calculated for { · Ί ί ] ΝΓ :Χ.¾ϋ : 450.13; found: 450.2.
PATENT
WO2015177537
PATENT
WO2015196116
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015196116&redirectedID=true
PATENT
WO2015196137
PATENT
http://www.google.com/patents/US20140221356
Example 42 Preparation of Compound 42 (2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
Step 1
-
1-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-1,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesulfonic acid (0.195 mL, 3 mmol) and placed in a 75 deg C. bath. The reaction mixture was stirred for 7 h, cooled and stored at −10° C. for 3 days and reheated to 75° C. for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
-
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (1R,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85° C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HCl (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichlormethane:hexanes afforded the desired intermediate 42A: LCMS-ESI+ (m/z): [M+H]+ calculated for C15H17N2O6: 321.11; found: 321.3.
Step 3
-
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethylamino)-N,N-dimethyl(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yloxy)methaniminium hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LCMS-ESI+ (m/z): [M+H]+ calculated for C22H21F3N3O5: 464.14; found: 464.2.
Step 4
-
To the crude reaction mixture of the previous step was added MgBr2 (0.258 g, 1.40 mmol). The reaction mixture was stirred at 50° C. for 10 minutes, acidified with 10% aqueous HCl, and extract twice with dichloromethane. The combined organic phases were dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN/H2O with 0.1% TFA modifier) to afford compound 42: 1H-NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 10.34 (t, J=5.7 Hz, 1H), 8.42 (s, 1H), 7.19 (t, J=8.7 Hz, 2H), 5.43 (dd, J=9.5, 4.1 Hz, 1H), 5.08 (s, 1H), 4.66 (dd, J=12.9, 4.0 Hz, 1H), 4.59 (s, 1H), 4.56-4.45 (m, 2H), 4.01 (dd, J=12.7, 9.7 Hz, 1H), 1.93 (s, 4H), 1.83 (d, J=12.0 Hz, 1H), 1.56 (dt, J=12.0, 3.4 Hz, 1H). LCMS-ESI+ (m/z): [M+H]+ calculated for C21H19F3N3O5: 450.13; found: 450.2.
PATENT
General Scheme I:

General Scheme II:



General Scheme II
General Scheme III:


General Scheme III
General Scheme IV:


G-1
General Scheme V:

II
EXAMPLES
In order for this invention to be more fully understood, the following examples are set forth. These examples are for the purpose of illustrating embodiments, and are not to be construed as limiting the scope of this disclosure in any way. The reactants used in the examples below may be obtained either as described herein, or if not described herein, are themselves either commercially available or may be prepared from commercially available materials by methods known in the art.
In one embodiment, a multi-step synthetic method for preparing a compound of Formula I is provided, as set forth below. In certain embodiments, each of the individual steps of the Schemes set forth below is provided. Examples and any combination of two or more successive steps of the below Examples are provided.
A. Acylation and amidation of Meldrum ‘s acid to form C-la:

[0520] In a reaction vessel, Meldrum’s acid (101 g, 1.0 equivalent) and 4-dimethylaminopyridine (1.8 g, 0.2 equivalents) were combined with acetonitrile (300 mL). The resulting solution was treated with methoxyacetic acid (6.2 mL, 1.2 equivalents). Triethylamine (19.4 mL, 2.0 equivalents) was added slowly to the resulting solution, followed by pivaloyl chloride (9.4 mL, 1.1 equivalents). The reaction was then heated to about 45 to about 50 °C and aged until consumption of Meldrum’s acid was deemed complete.
A separate reaction vessel was charged with acetonitrile (50 mL) and J-la (13.4 g, 1.2 equivalents). The resulting solution was treated with trifluoroacetic acid (8.0 mL, 1.5 equivalents), and then this acidic solution was added to the acylation reaction in progress at about 45 to about 50 °C.
The reaction was allowed to age for at least 18 hours at about 45 to about 50 °C, after which time the solvent was removed under reduced pressure. The crude residue was dissolved in ethyl acetate (150 mL), and the organic layer was washed with water. The combined aqueous layers were extracted with ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate solution, and the combined bicarbonate washes were back extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified twice via silica gel chromatography to yield C-la.
lH NMR (400 MHz, CDC13): δ 7.12 (br, 1H), 6.66 (app t, J= 8.1 Hz, 2H), 4.50 (app d, J= 5.7 Hz, 2H), 4.08 (s, 2H), 3.44 (s, 2H), 3.40 (s, 3H). 13C NMR (100 MHz, CDC13): δ 203.96, 164.90, 162.37 (ddd, J= 250.0, 15.7, 15.7 Hz), 161.71 (ddd, J = 250.3, 14.9, 10.9 Hz), 110.05 (ddd, J= 19.7, 19.7, 4.7 Hz), 100.42 (m), 77.58, 59.41, 45.71, 31.17 (t, J= 3.5 Hz). LCMS, Calculated: 275.23, Found: 275.97 (M).
I l l
B. Alkylation of C-la to form E-la:

A solution of C-la (248 mg, 1.0 equivalent) and 2-methyl tetrahydrofuran (1.3 niL) was treated with N,N-dimethylformamide dimethylacetal (0.1 mL, 1.1 equivalent) and stirred at room temperature overnight (~14 hours). The reaction was treated with aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalents), and was allowed to age for about 2 hours, and then was quenched via the addition of 2 Ν HC1
(1.5 mL).
The reaction was diluted via the addition of ethyl acetate, and phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield E-la.
1H NMR (400 MHz, CDC13): δ 10.85 (s, 1H), 9.86 (s, 1H), 8.02 (d, J= 13.1 Hz, 1H), 6.65 (dd, J= 8.7, 7.7 Hz, 2H), 4.53 (d, J= 3.9 Hz, 2H), 4.40 (t, J= 5.1 Hz, 1H), 4.18 (s, 2H), 3.42 (s, 6H), 3.39 (m, 2H), 3.37 (s, 3H). 13C MR (100 MHz, CDC13): δ 193.30, 169.15, 162.10 (ddd, J= 248.9, 15.5, 15.5 Hz), 161.7 (ddd, J =
250.0, 14.9, 1 1.1 Hz), 161.66, 1 11.08 (ddd J= 19.9, 19.9, 4.7 Hz) 103.12, 100.29 (ddd, J= 28.1, 17.7, 2.3 Hz), 76.30, 58.83, 54.98, 53.53, 51.57, 29.89 (t, J= 3.3 Hz). LCMS, Calculated: 390.36, Found: 390.92 (M).
c. Cyclization of E-la to form F-la:

E-1a F-1a
] E-la (0.2 g, 1.0 equivalent), dimethyl oxalate (0.1 g, 2.5 equivalents) and methanol (1.5 mL) were combined and cooled to about 0 to about 5 °C. Sodium methoxide (0.2 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly while keeping the internal temperature of the reaction below about 10 °C throughout the addition. After the addition was completed the reaction was heated to about 40 to about 50 °C for at least 18 hours.
After this time had elapsed, the reaction was diluted with 2 N HC1 (1.5 mL) and ethyl acetate (2 mL). The phases were separated, and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and solvent was removed under reduced pressure. The resulting crude oil was purified via silica gel chromatography to afford F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
D. Alkylation and cyclization of C-la to form F-la:
1 . DMFDMA

C-1a NaOMe, MeOH, 40 °C F-1a
To a reaction vessel were added C-la (245 mg, 1.0 equivalent) and N,N-dimethylformamide dimethylacetal (0.5 mL, 4.3 equivalent). The reaction mixture was agitated for approximately 30 minutes. The reaction was then treated with 2-methyl tetrahydrofuran (2.0 mL) and aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalent). The reaction was allowed to age for several hours and then solvent was removed under reduced pressure.
The resulting material was dissolved in methanol and dimethyl oxalate was added (0.3 g, 2.5 equivalents). The reaction mixture was cooled to about 0 to about 5 °C, and then sodium methoxide (0.4 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly. After the addition was completed the reaction was heated to about 40 to about 50 °C.
After this time had elapsed, the reaction was cooled to room temperature and quenched via the addition of 2 Ν HC1 (1.5 mL). The reaction was then diluted with ethyl acetate, and the resulting phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
E. Condensation of F-la with N-la to form G-la:

K2C03, MeCN, 75 °C
To a reaction vessel were added F-la (202 mg, 1.0 equivalent) and acetonitrile (1.4 mL). The resulting solution was treated with glacial acetic acid (0.2 mL, 6.0 equivalents) and methane sulfonic acid (0.01 mL, 0.3 equivalents). The reaction was then heated to about 70 to about 75 °C.
After 3 hours, a solid mixture of N-la (0.128g, 1.5 equivalents) and potassium carbonate (0.2 g, 2.7 equivalents) was introduced to the reaction at about 70 to about 75 °C. After the addition was completed, the reaction was allowed to progress for at least about 1 hour.
After this time had elapsed, water (1.4 mL) and dichloromethane (1.4 mL) were introduced to the reaction. The phases were separated, and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over magnesium sulfate, then were filtered and concentrated under reduced pressure. The resulting crude material was purified via silica gel chromatography to obtain G-la.
lR NMR (400 MHz, CDC13): δ 10.23 (t, J= 5.5 Hz, 1H), 8.39 (s, 1H), 6.60 (t, J= 8.1 Hz, 2H), 5.29 (dd, J= 9.5, 3.7 Hz, 2H), 4.57 (d, J= 5.4 Hz, 3H), 4.33 (dd, J = 12.8, 3.8 Hz, 1H), 4.02 – 3.87 (m, 1H), 3.94 (s, 3H), 2.06 – 1.88 (m, 4H), 1.78 (dd, J = 17.2, 7.5 Hz, 1H), 1.55 – 1.46 (m, 1H). 13C MR (100 MHz, CDC13): δ 174.53, 163.75, 162.33 (dd, J= 249.4, 15.7, 15.7 Hz), 161.86 (ddd, J= 250.4, 14.9, 10.9 Hz), 154.18, 154.15, 142.44, 129.75, 1 18.88, 1 10.58 (ddd, J= 19.8, 4.7, 4.7 Hz), 100.42 (m), 77.64, 74.40, 61.23, 54.79, 51.13, 38.31, 30.73, 29.55, 28.04. LCMS, Calculated: 463.14, Found: 464.15 (M+H).
Γ. Deprotection of G-la to form a compound of Formula la:
G-la (14 g) was suspended in acetonitrile (150 mL) and dichloromethane (150 mL). MgBr2 (12 g) was added. The reaction was heated to 40 to 50 °C for approximately 10 min before being cooled to room temperature. The reaction was poured into 0.5M HC1 (140 mL) and the layers separated. The organic layer was washed with water (70 mL), and the organic layer was then concentrated. The crude product was purified by silica gel chromatography (100% dichloromethane up to 6% ethanol/dichloromethane) to afford la.
REFERENCES
| Patent | Submitted | Granted |
|---|---|---|
| POLYCYCLIC-CARBAMOYLPYRIDONE COMPOUNDS AND THEIR PHARMACEUTICAL USE [US2014221356] | 2013-12-19 | 2014-08-07 |
| US9216996 | Dec 19, 2013 | Dec 22, 2015 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydropyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepines and methods for treating viral infections |
see full gravir series at…………..http://medcheminternational.blogspot.in/p/ravir-series.html
//////////
C1CC2CC1N3C(O2)CN4C=C(C(=O)C(=C4C3=O)O)C(=O)NCC5=C(C=C(C=C5F)F)F
OR
c1c(cc(c(c1F)CNC(=O)c2cn3c(c(c2=O)O)C(=O)N4[C@H]5CC[C@H](C5)O[C@@H]4C3)F)F
![]()

BICTEGRAVIR, NEW PATENT, WO 2018005328, CONCERT PHARMA
WO2018005328) DEUTERATED BICTEGRAVIR
CONCERT PHARMACEUTICALS, INC.
TUNG, Roger, D.; (US)

Concert CEO Roger Tung
Novel deuterated forms of bictegravir is claimed. Gilead Sciences is developing the integrase inhibitor bictegravir as an oral tablet for the treatment of HIV-1 infection.
This invention relates to deuterated forms of bictegravir, and pharmaceutically acceptable salts thereof. In one aspect, the invention provides a compound of Formula (I) or a pharmaceutically acceptable salt thereof, wherein each of Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11b is independently hydrogen or deuterium; provided that if each Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, and Y11 is hydrogen, then Y11b is deuterium.

Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the
CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect. Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at http://www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme’s activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug’s metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res 1985, 14:1-40 (“Foster”); Kushner, DJ et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9:101-09 (“Fisher”)). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p.35 and Fisher at p.101).
[8] The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem.1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
Exemplary Synthesis
[72] Deuterium-modified analogs of bictegravir can be synthesized by means known in the art of organic chemistry. For instance, using methods described in US Patent No.9,216,996 (Haolun J. et al., assigned to Gilead Sciences, Inc. and incorporated herein by reference), using deuterium-containing reagents provides the desired deuterated analogs.
[73] Such methods can be carried out utilizing corresponding deuterated and optionally, other isotope-containing reagents and/or intermediates to synthesize the compounds delineated herein, or invoking standard synthetic protocols known in the art for introducing isotopic atoms to a chemical structure.
[74] A convenient method for synthesizing compounds of Formula I is depicted in the Schemes below.
 [75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[76] In a manner analogous to the procedure described in US 9,216,996, acetal deprotection of carboxylic acid 7 followed by cyclization with appropriately deuterated aminocyclopentanol 9 provides carboxylic acid intermediate 10. Amide coupling with appropriately deuterated benzylamine 11 followed by deprotection of the methyl ether ultimately affords a compound of Formula I in eight overall steps from compound 1.
[77] Use of appropriately deuterated reagents allows deuterium incorporation at the Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11bpositions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and/or Y11b.
[78] Appropriately deuterated intermediates 2a and 2b, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 2 below.
S h 2 S th i f C d 2 d 2b

[79] Synthesis of compound 2a (wherein Y3=H) by acetal formation of N,N-dimethylformamide (DMF) with dimethylsulfate has been described in Mesnard, D. et. al. J. Organomet. Chem.1989, 373, 1-10. Replacing DMF with N,N-dimethylformamide-d1 (98-99 atom % D; commercially available from Cambridge Isotope Laboratories) in this reaction would thereby provide compound 2b (wherein Y3=D).
[80] Use of appropriately deuterated reagents allows deuterium incorporation at the Y3 position of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at Y3.
[81] Appropriately deuterated intermediates 4a-4d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 3 below.

[82] As described in Malik, M. S. et. al. Org. Prep. Proc. Int.1991, 26, 764-766, acetaldehyde is converted to alkylhalide 14a via reaction with chlorine gas and subsequent acetal protection with CaCl2 in methanol. As described in CN 103739506, reaction of 14a with aqueous ammonia and then sodium hydroxide provides primary amine 4a (wherein Y9=Y10a=Y10b=H). Replacing acetaldehyde with acetaldehyde-d1, acetaldehyde-2,2,2-d3, or acetaldehyde-d4 (all commercially available from CDN Isotopes with 98-99 atom % D) in the sequence then provides access to compounds 4b (Y9=D, Y10a=Y10b=H), 4c (Y9=H,
Y10a=Y10b=D) and 4d (Y9=Y10a=Y10b=D) respectively (Schemes 3b-d).
[83] Use of appropriately deuterated reagents allows deuterium incorporation at the Y9, Y10a, and Y10b positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y9, Y10a, and/or Y10b.
[84] Appropriately deuterated intermediates 9a-9d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 4 below.
 [85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[86] As depicted in Scheme 4b, aminocyclopentanol 9b (Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b= Y8=D) is obtained through an analogous synthetic sequence using cyclopentadiene-d6 and performing the penultimate hydrogenation with D2 in place of H2. Cyclopentadiene-d6 is prepared according to the procedure described in Cangoenuel, A. et. al. Inorg. Chem.2013, 52, 11859-11866.
[87] Alternatively, as shown in Scheme 4c, the meso-diol obtained in Scheme 4a is oxidized to the diketone following the procedure reported by Rasmusson, G.H. et. al. Org. Syn.1962, 42, 36-38. Subsequent mono-reduction with sodium borodeuteride and CeCl3 then affords the D1-alcohol in analogy to the method described in WO 2001044254 for the all-protio analog using sodium borohydride. Reduction of the remaining ketone using similar conditions provides the meso-D2-diol in analogy to the method reported in Specklin, S. et. al. Tet. Lett.2014, 55, 6987-6991 for the all protio analog using sodium borohydride. The meso-D2-diol is then converted to 9c (Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=H, Y6=Y8=D) following the same procedures outlined in Scheme 4a.
[88] Likewise, the meso-diol obtained in Scheme 4b may be converted to 9d
(Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=D, Y6=Y8=H) in an analogous manner as depicted in Scheme 4d. The use of deuterated solvents such as D2O or MeOD may be considered to reduce the risk of D to H exchange for ketone containing intermediates.
[89] Use of appropriately deuterated reagents allows deuterium incorporation at the Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and Y8 positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and/or Y8.
[90] Appropriately deuterated intermediates 11a-11d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 5 below.
Scheme 5. Synthesis of Benzylamines 11a-11d

//////////////////
TEVA’S CEP 1347, KT 7515 a MAP3K11 (MLK3) inhibitor potentially for the treatment of Parkinson’s disease.
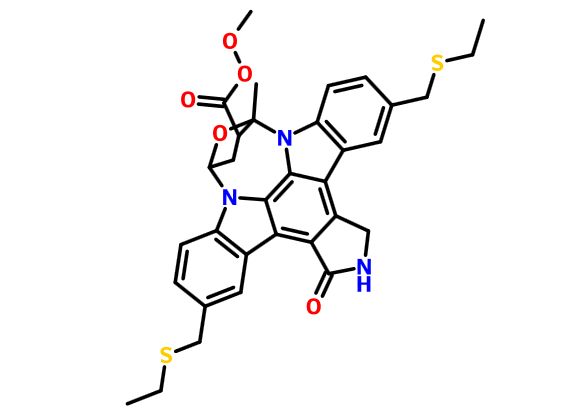

CEP-1347; KT-7515
(9S,10R,12R)-5-16-Bis[(ethylthio)methyl]-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester
3,9-Bis(etsm)-K-252a; CEP1347; 3,9-Bis((ethylthio)methyl)-K-252a; AC1L31ZX
3,9-bis[(ethylthio)methyl]-K-252a
Phase III
A MAP3K11 (MLK3) inhibitor potentially for the treatment of Parkinson’s disease.
![]()
MW 615.76, MF C33H33N3O5S2
Inhibitor of c-jun N-terminal kinase (JNK) signaling. Rescues motor neurons undergoing apoptosis (EC50 = 20 nM). Blocks Aβ-induced cortical neuron apoptosis (EC50 ~51 nM). Does not inhibit ERK1 activity. Neuroprotective.

Scheme 1 a
a (a) Ac2O, DMAP, THF, room temperature, 93%; (b) Cl2CHOCH3, TiCl4, CH2Cl2, 66%; (c) NaBH4 CH3OH, CHCl3, 65%; (d) NaOCH3, CH3OH, ClCH2CH2Cl, room temperature, 90%; (e) ROH, CSA, CH2Cl2; (f) RSH, CSA, CH2Cl2.
Inhibitor of c-jun N-terminal kinase (JNK) signaling. Rescues motor neurons undergoing apoptosis (EC50 = 20 nM). Blocks Aβ-induced cortical neuron apoptosis (EC50 ~51 nM). Does not inhibit ERK1 activity. Neuroprotective.
Apoptosis has been proposed as a mechanism of cell death in Alzheimer’s, Huntington’s and Parkinson’s diseases and the occurrence of apoptosis in these disorders suggests a common mechanism.
Events such as oxidative stress, calcium toxicity, mitochondria defects, excitatory toxicity, and deficiency of survival factors are all postulated to play varying roles in the pathogenesis of the diseases.
However, the transcription factor c-jun may play a role in the pathology and cell death processes that occur in Alzheimer’s disease.
Parkinson’s disease (PD) is also a progressive disorder involving the specific degeneration and death of dopamine neurons in the nigrostriatal pathway. In Parkinson’s disease, dopaminergic neurons in the substantia nigra are hypothesized to undergo cell death by apoptotic processes.
The commonality of biochemical events and pathways leading to cell death in these diseases continues to be an area under intense investigation.
The current therapy for PD and AD remains targeting replacement of lost transmitter, but the ultimate objective in neurodegenerative therapy is the functional restoration and/or cessation of progression of neuronal loss.
a novel approach for the treatment of neurodegenerative diseases through the development of kinase inhibitors that block the active cell death process at an early transcriptional independent step in the stress activated kinase cascade.
In particular, preclinical data will be presented on the c-Jun Amino Kinase pathway inhibitor, CEP-1347/KT-7515, with respect to it’s properties that make it a desirable clinical candidate for treatment of various neurodegenerative diseases.
CEP-1347 is also known as KT-7515 and is being developed by Cephalon and Kyowa Hakko for treatment of Parkinson’s disease and cognitive disorders.
It is believed to be a JNK-MAP kinase inhibitor. CEP-1347 has the chemical name 9alpha,12alpha-Epoxy-5,16-bis(ethylsulfanylmethyl)-10beta-hydroxy-9-methyl-1-oxo-2,3,9,10,11,12alpha-hexahydro-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4- i][1,6]benzodiazocine-10-carboxylic acid methyl ester and has the chemical structure as depicted in Formula 7.

PATENT
https://google.com/patents/WO2005082920A1?cl=en
The compound with the structure outlined below is presently in clinical trials for Parkinson’s disease (Idrugs, 2003, 6(4), 377-383).

This compound is in the following referred to as Compound I. The chemical name of Compound I is [9S-(9α,10β,12α)]-5,16-Rw[(ethylthio)methyl]-2,3,9,10,l l,12-hexahydro- 10-hydroxy-9-methyl- 1 -oxo-9, 12-epoxy- 1 H-diindolo[l ,2,3 -fg:3 ‘,2’, 1 ‘-kl]ρyrrolo[3,4- i][l,6]benzodiazocine-10-carboxylic acid methyl ester.
The following references relate to Compound I, in particular to methods for its preparation [J.Med. Chem. 1997, 40(12), 1863-1869; Curr. Med. Chem. – Central Nervous System Agents, 2002, 2(2), 143-155] and its potential medical uses, mainly in diseases in the central nervous system (CNS), in particular for treatment of neurodegenerative diseases, e.g. Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, peripheral neuropathy, AIDS dementia, and ear injuries such as noise-induced hearing loss [Progress in Medicinal Chemistry (2002), 40, 23-62; Bioorg. Med. Chem. Lett. 2002,12(2), 147-150; Neuroscience, Oxford, 1998, 86(2), 461-472; J. Neurochemistry (2001), 77(3), 849-863; J. Neuroscience (2000), 20(1), 43-50; J. Neurochemistry (2002), 82(6), 1424-1434; Hearing Research, 2002, 166(1-2), 33-43].
The following patent documents relate to Compound I, including its medical use and synthesis: WO 9402488, WO9749406, US 5621100, EP 0651754 and EP 112 932. By the known methods, Compound I is synthesized in a solid amorphous form. The inventors have now discovered 5 crystalline forms of Compound I (named alpha, beta, gamma, delta and epsilon) thereby providing an opportunity to improve the manufacturing process of Compound I and its pharmaceutical use. There exists a need for crystalline forms, which may exhibit desirable and beneficial chemical and physical properties. There also exists a need for reliable and reproducible methods for the manufacture, purification, and formulation of Compound I to permit its feasible commercialisation.
EXAMPLES
In the following the starting material ” Compound I” may, e.g., be prepared as described by Kaneko M. et al in J. Med. Chem. 1997, 40, 1863-1869.
Example 1. Preparation of crystalline alpha form of Compound I
Method I):
6.0 g amorphous Compound I was dissolved in 30 ml acetone. 0,6 g potassium carbonate was added and the suspension was stirred at room temperature for 1 hour before it was filtered to remove potential minor insoluble impurities and inorganic salts. The filter cake was washed with acetone. The filtrate was then evaporated on a rotary evaporator under reduced pressure at 60°C to a final volume of 10 ml to which 100 ml methanol was added slowly. The product separated as an oil, which almost dissolved on heating to reflux. Subsequently the residual insoluble impurities were removed by filtration. The filtrate was left with stirring at room temperature. A crystalline solid separated and was isolated by filtration. The filter cake was washed with methanol and dried in vacuo at 60°C overnight. Yield 2,83 g (47%), mp=182.4°C (DSC onset value), Weight loss by heating: 0.5%, Elemental analysis: 6.71%N, 63.93%C, 5.48%H, theoretical values corrected for 0.5% H2O: 6.79%N, 64.05%C, 5.43%H. XRPD analysis conforms with the alpha form. Method II):
5 g amorphous Compound I was dissolved in 25 ml acetone by gentle heating. 10 ml Methanol was added very slowly until the solution got turbid. The solution was allowed to cool to room temperature by natural cooling. The suspension was filtered and the filter-cake discarded. During filtration more material precipitated in the filtrate. The filtrate was heated until all material redissolves. Cold methanol was then added to the solution until precipitation was observed. The slightly turbid solution was then heated until all material was in solution. The solution was allowed to cool to room temperature, and the precipitate was removed by filtration. The second filter-cake was discarded. During the filtration some material separated in the filtrate. Heating redissolved the beginning crystallisation in the filtrate. Cold methanol was then added to the solution until precipitation was observed. The suspension was heated until a clear solution was obtained. The solution was allowed to reach room temperature by natural cooling. After a short period of time (15 min) precipitation begun. The precipitated pale yellow product was isolated by filtration and dried in vacuo at 50°C overnight. mp=188.9°C (DSC onset value), Weight loss by heating: 0.3%>, Elemental analysis: 6.53%N, 64.33%C, 5.43%H, theoretical values: 6.82%N, 64.37%C, 5.37%H. XRPD analysis conforms with the alpha form. Method III:
0.5g Compound I in a mixture of isopropyl acetate (10 mL) and water (0.6 mL) was heated to reflux with stirring. The compound was not completely dissolved so isopropyl acetate (10 mL) and water (0.6 mL) were added and heated to reflux. Stirring was stopped and the experiment was allowed to cool to room temperature. The crystalline product obtained were isolated by filtration and dried in vacuo at 40° C. Yield = 0.25g, mp = 183.7°C (DSC onset value). XRPD analysis conforms with the alpha form. Method IV: 0.5g Compound I in a mixture of ethyl acetate (10 mL) and water (0.4 mL) was heated to 70° C with stirring. The experiment was allowed to cool to room temperature. The crystalline product obtained were isolated by filtration and dried in vacuo at 40° C. XRPD analysis conforms with the alpha form.
PATENT
https://www.google.com/patents/US20050261762
PATENT
http://www.google.co.ug/patents/EP2004158A2?cl=en
CEP-1347 (KT7515) (Maroney et al. 1998; Roux et al. 2002).
PAPER
Neurotrophic 3,9-bis[(alkylthio)methyl]- and -bis(alkoxymethyl)-K-252a derivatives
J Med Chem 1997, 40(12): 1863
http://pubs.acs.org/doi/full/10.1021/jm970031d


The synthesis of the title compound used as the starting material was the indolocarbazole alkaloid K-252A (I). Compound (I) was protected as the diacetyl derivative (II) by treatment with Ac2O and DMAP. Formylation of (II) with dichloromethyl methyl ether in the presence of TiCl4 afforded dialdehyde (III), which was further reduced to diol (IV) using NaBH4 in MeOH-CHCl3. Condensation of diol (IV) with ethanethiol in the presence of camphorsulfonic acid furnished the bis-sulfanyl compound (V). The acetyl protecting groups of (V) were finally removed by treatment with sodium methoxide. Alternatively, diol (IV) was first deacetylated by treatment with NaOMe, and the deprotected bis(hydroxymethyl) compound (VI) was then condensed with ethanethiol to produce the title bis-sulfayl compound 8.
3,9-Bis[(ethylthio)methyl]-K-252a (8):
mp 163−165 °C;
IR (KBr) 1725, 1680 cm-1; FAB-MSm/z 615(M+);
1H-NMR (400 MHz, DMSO-d6) δ 1.23 (t, 6H, J = 7.3 Hz), 1.99 (dd, 1H, J = 4.8, 14.1 Hz), 2.132 (s, 3H), 2.489 (q, 2H, J = 7.3 Hz), 2.505 (q, 2H, J = 7.3 Hz), 3.37 (dd, 1H, J = 7.6, 14.1 Hz), 3.92 (s, 3H), 3.94 (s, 2H), 3.98 (s, 2H), 4.95 (d, 1H, J = 17.6 Hz), 5.02 (d, 1H, J = 17.6 Hz), 6.32 (s, 1H), 7.10 (dd, 1H, J = 4.8, 7.6 Hz), 7.450 (m, 2H), 7.84 (d, 1H, J = 8.5 Hz), 7.88 (d, 1H, J = 8.8 Hz), 7.95 (d, 1H, J = 1.0 Hz), 8.60 (s, 1H), 9.13 (d, 1H, J = 0.7 Hz);
HRFAB-MS calcd for C33H33N3O5S2 615.1862, found 615.1869. Anal. (C33H33N3O5S2·0.5H2O) C, H, N.
References
Maroney et al (1998) Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J.Neurosci. 18 104. PMID: 9412490.
Saporito et al (1998) Preservation of cholinergic activity and prevention of neuron death by CEP-1347/KT-7515 following excitotoxic injury of the nucleus basalis magnocellularis. Neuroscience 86 461. PMID: 9881861.
Bozyczko-Coyne et al (2001) CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J.Neurochem. 77 849. PMID: 11331414.
| WO1994002488A1 * | Jul 26, 1993 | Feb 3, 1994 | Cephalon Inc | BIS-STAUROSPORINE AND K-252a DERIVATIVES |
| Reference | ||||
|---|---|---|---|---|
| 1 | * | KANEKO M ET AL: “Neurotrophic 3,9-Bis[(alkylthio)methyl]- and -Bis(alkoxymethyl)-K-252a Derivatives” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 40, no. 12, 1997, pages 1863-1869, XP002128804 ISSN: 0022-2623 cited in the application | ||
//////////CEP 1347, KT 7515 ,
CCSCC1=CC2=C(C=C1)N3C4CC(C(O4)(N5C6=C(C=C(C=C6)CSCC)C7=C8CNC(=O)C8=C2C3=C75)C)C(=O)OOC
MELOGLIPTIN

Melogliptin
Phase III
A DP-IV inhibitor potentially for treatment of type II diabetes.
![]()
EMD-675992; GRC-8200
CAS No. 868771-57-7
4-fluoro-1-[2-[[(1R,3S)-3-(1,2,4-triazol-1-ylmethyl)cyclopentyl]amino]acetyl]pyrrolidine-2-carbonitrile
4(S)-Fluoro-1-[2-[(1R,3S)-3-(1H-1,2,4-triazol-1-ylmethyl)cyclopentylamino]acetyl]pyrrolidine-2(S)-carbonitrile
4(S)-Fluoro-1-[2-[(1R,3S)-3-(1H-1,2,4-triazol-1-ylmethyl)cyclopentylamino]acetyl]pyrrolidine-2(S)-carbonitrile
Note………The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
MELOGLIPTIN

GRC-8200, a dipeptidyl peptidase IV inhibitor (DPP-IV), is currently undergoing phase II clinical trials at Glenmark Pharmaceuticals and Merck KGaA for the treatment of type 2 diabetes. In 2006, the compound was licensed by Glenmark Pharmaceuticals to Merck KGaA in Europe, Japan and N. America for the treatment of type 2 diabetes, however, these rights were reaquired by Glenmark in 2008.


ALTERNATE……….

SEE..http://apisynthesisint.blogspot.in/2015/12/melogliptin.html
See more at: http://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html
see all gliptins……….http://organicsynthesisinternational.blogspot.in/p/gliptin-series-22.html

DISCLAIMER…….The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
/////////
MK 7655, RELEBACTAM, a β-Lactamase inhibitor
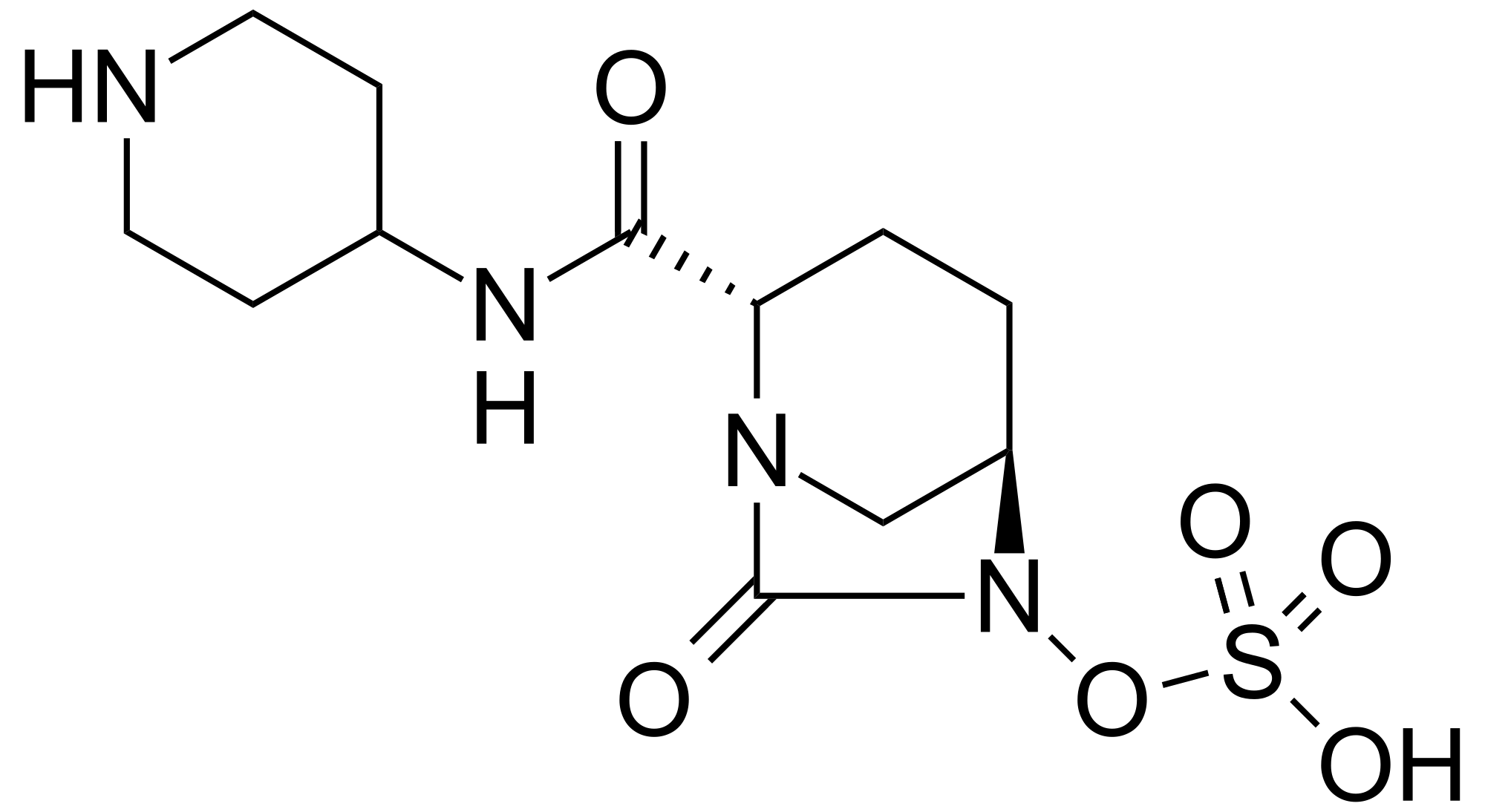
MK 7655, RELEBACTAM
(1R,2S,5R)-7-Oxo-N-(4-piperidinyl)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
| MF C12H22N4O7S | |
| MW | 366.39068 g/mol |
|---|
CAS 1174020-13-3
β-Lactamase inhibitor
MK-7655 is a beta-lactamase inhibitor in phase III clinical studies at Merck & Co for the treatment of serious bacterial infections…….See clinicaltrials.gov, trial identifier numbers NCT01505634 and NCT01506271.
In 2014, Qualified Infectious Disease Product (QIDP) and Fast Track designations were assigned by the FDA for the treatment of complicated urinary tract infections, complicated intra-abdominal infections and hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia.

PAPER
A concise synthesis of a beta-lactamase inhibitor
Org Lett 2011, 13(20): 5480
http://pubs.acs.org/doi/abs/10.1021/ol202195n
http://pubs.acs.org/doi/suppl/10.1021/ol202195n/suppl_file/ol202195n_si_001.pdf

MK-7655 (1) is a β-lactamase inhibitor in clinical trials as a combination therapy for the treatment of bacterial infection resistant to β-lactam antibiotics. Its unusual structural challenges have inspired a rapid synthesis featuring an iridium-catalyzed N–H insertion and a series of late stage transformations designed around the reactivity of the labile bicyclo[3.2.1]urea at the core of the target.
H NMR (400 MHz, DMSO-d6): δ 8.30 (br s, 2H), 8.20 (d, J = 7.8 Hz, 1H), 4.01 (s, 1H), 3.97-3.85 (m, 1H), 3.75 (d, J = 6.5 Hz, 1H), 3.28 (dd, J = 12.9, 2.5 Hz, 2H), 3.05-2.93 (m, 4H), 2.08-1.97 (m, 1H), 1.95-1.79 (m, 3H), 1.73-1.59 (m, 4H);
13C NMR (DMSO-d6, 100 MHz) δ 169.7, 166.9, 59.8, 58.3, 46.9, 44.3, 42.9, 28.5, 28.3, 20.8, 18.9;
HRMS calculated for C12H20N4O6S (M+H): 349.1182, found: 349.1183.
[α]D 25 = -23.3 (c = 1.0, CHCl3)
PATENT
WO 2009091856
http://www.google.com/patents/WO2009091856A2?cl=en
EXAMPLE IA
(2S ,5 R)-7-Oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo [3.2.1 ]octane-2-carboxamide

Step 1 : tert-butyl 4-({[(2S,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]oct-2- yljcarbonyl } amino)piperidine- 1 -carboxylate : To a solution of (2S,5R)-6-(phenylmethoxy)-7-oxo-l,6-diazabicyclot3.2.1]octane-
2-carboxylic acid (1.484 g, 5.37 mmol) in dry dichloromethane (60 ml) was added triethylamine (1.88 ml, 13.49 mmol), 2-chloro-l-methylpyridinium iodide (1.60 g, 6.26 mmol), and 4-amino-l- BOC-piperidine (1.30 g, 6.49 mmol) sequentially at room temperature under nitrogen. The reaction was then heated to 500C for 1 hour. The reaction mixture was concentrated under vacuum and purified by silica gel chromatography on an Isco Combiflash (40 g silica gel, 40 mL/min, 254 nM, 15% to 100% EtOAc/hexane over 14 column volumes then 100% EtOAc for 4 column volumes; title compuond eluted at 65% ethyl acetate/hexane) to afford the title compound as a pale orange solid.
Step 2: tert-butyl 4-({[(2S,5R)-6-hydroxy-7-oxo-l ,6-diazabicyclo[3.2.1]oct-2- yl] carbonyl } amino)piρeridine- 1 -carboxylate:
Palladium on carbon (394 mg; 10% Pd/C) was added to a solution of the product of step 1 (1.81 g, 3.95 mmol) in methanol (50.6 mL) and the resulting mixture was stirred under hydrogen (balloon) overnight. LC/MS analysis indicated the reaction was not complete. Acetic acid (6 drops) and additional catalyst (159 mg of 10% Pd/C) were added to the reaction and the resulting mixture was stirred under hydrogen (balloon) for an additional 90 minutes. Additional catalyst (0.2085 g of 10% Pd/C) was added to the reaction and stirring under hydrogen was continued for an additional 2.5 hours at which time the reaction was judged complete by LC-MS analysis. The reaction was filtered through a celite pad and the collected solid was washed well wtih MeOH. The filtrate was concentrated under vacuum to afford the title compound as a colorless oil which was used without purification in the next step.
Step 3 : tert-butyl-4-({ [(2S,5R)-7-oxo-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]oct-2- yl] carbonyl } amino)ρiperidine- 1 -carboxylate:
To a solution of the product of step 2 (1.455 g, 3.95 mmol; theoretical yield of step 2) in dry pyridine (30 mL) was added sulfur trioxide pyridine complex (3.2 g, 20.11 mmol) at room temperature under nitrogen. The resulting thick mixture was stirred over the weekend.
The reaction was filtered and the white insoluble solids were washed well with dichloromethane. The filtrate was concentrated in vacuo. The residue was further azeotroped with toluene to remove excess pyridine to afford the title compound which was used without purification in the next step.
Step 4: (2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2- carboxamide:
To a mixture of the product of step 3 (1.772 g, 3.95 mmol; theoretical yield of step 3) in dry dichloromethane (30 ml) at 00C under nitrogen was slowly added trifluoroacetic acid (6.1 ml, 79 mmol). Immediately the reaction became a solution. After 1 hour, additional trifluoroacetic acid (8 ml) was added to the reaction. The reaction was stirred at 00C until judged complete by LC-MS analysis then concentrated in vacuo. The residue was triturated with ether (3X) to remove excess TFA and organic impurities. The resulting white insoluble solid was collected via centrifugation, dried in vacuo, then purified by preparative HPLC (250X21.2 mm Phenomenex Synergi Polar-RP 80A column; 10 micron; 35 mL/min.; 210 nM; 0% to 30% methanol/water over 15 minutes; title compound eluted at 10% methanol/water). Fractions containing the title compound were combined and Iyophilized overnight to afford the title compound as a white solid. LC-MS (negative ionization mode) m/e 347 (M-H).
PAPER
Discovery of MK-7655, a beta-lactamase inhibitor for combination with Primaxin
Bioorg Med Chem Lett 2014, 24(3): 780
http://www.sciencedirect.com/science/article/pii/S0960894X13014856

PATENT
WO 2014200786
http://www.google.dj/patents/WO2014200786A1?cl=en


![]()




Exemplary Scheme

– 50% isolated yield overall from 1 to 5

O via crystallization
XAMPLE 1
(2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide 
Preparation of (15′,45)-5-((2-nitrophenyl)sulfonyl)-2-oxa-5-azabicyclo[2.2.2]octan-3 one (2)
To a reactor (R-1) equipped with an additional funnel, nitrogen inlet and agitator was charged (2S,5S)-5-hydroxypiperidine-2-carboxylic acid (77.3 wt%) (50.0 g, 344 mmol), and water (150 mL). Agitation was begun, the pH adjusted to 10-11 by addition of 10 N NaOH (~ 46.5 mL) and the reactor charged with acetone (50.0 mL).
In a separate reactor (R-2) equipped with an agitator and nitrogen inlet was charged 2-nitrobenzene-l-sulfonyl chloride (97%) (106.0 g, 478 mmol) and acetone (80 mL). The contents of R-2 were transferred to R-1 at 23-30 °C while the pH of the solution was maintained at 10-11 by simultaneously addition of 10 N NaOH. After 15 to 30 min, the pH was adjusted to about 6 by addition of 12 N HC1. The solution was charged with EtOAc (500 mL) and the pH adjusted to 3.0 by addition of 12 N HC1. The layers were separated and the aqueous back-extracted with EtOAc (150 mL x 2).
To a separate reactor (R-3) was charged product la in the combined organic layers, 2-nitrobenzene-l-sulfonyl chloride (73.0 g, 329 mmol), and triethylamine (130 mL). The batch in R-3 was agitated at 20-28°C for 30 min. The solution was charged with water (100 mL), the layers separated, and the aqueous back extracted with EtOAc (150 mL x 2). The combined EtOAc layer was washed with 10% NaHC03 (100 mL) and brine (100 mL). The organic phase was concentrated to 150 mL upon which a crystalline slurry was formed. The concentrated solution was agitated at 13-18°C for 2-3 hours followed by filtration of crystalline solids. The resulting wet cake was washed with EtOAc (60 mL) and then dried under vacuum oven at 25-30°C to afford 2 (65.6 g, 79% yield), m.p. 126.0-126.7 °C. 1H NMR (CDC13, 400 MHz) δ: 8.02 (m, 1 H), 7.80-7.71 (m, 2 H), 7.66 (m, 1 H), 4.88 (m, 1 H), 4.55 (dd, J= 3.8, 2.7 Hz, 1 H), 3.78 (dt, J= 11.2, 3.0 Hz, 1 H), 3.66 (dd, J = 11.2, 1.1 Hz, 1 H), 2.44 (m, 1 H), 2.11 (m, 2 H), 1.91 (m, 1 H); 13C NMR (CDC13, 100 MHz) δ: 168.4, 148.3, 134.4, 132.1, 131.0, 130.7, 124.2, 73.5, 51.4, 48.0, 25.1, 23.2
Preparation oftert-butyl 4-((25*,55)-l-((2-nitrophenyl)sulfonyl)-5-(((2- nitrophenyl)sulfony l)oxy)piperidine-2-carboxamido)piperidine- 1 -carboxylate (3)
To a reactor (R-l) was charged lactone 2 (65.5 g, 210 mmol), THF (131 mL) and tert-butyl 4-aminopiperidine-l -carboxylate (44.5 g, 222 mmol). The stirred solution was heated to reflux (typical temperature 72 °C) for ~18 hr. The reaction was cooled to 25-35 °C and then charged with THF (325 mL) and 4-dimethylaminopyridine (40.1 g, 328 mmol) followed by agitation for 30 minutes.
To a separate reactor (R-2) was charged 2-nitrobenzene-l-sulfonyl chloride (60.9 g,
275 mmol) and THF (200 mL). The contents of R-2 were added to R-l over the course of 45 to 75 minutes maintaining batch temperature of 20 to 30°C. The batch in R-l was agitated for 2 to 4 hours at a temperature of 20 to 30°C.
To a separate reactor (R-3) was charged water (600 mL) and methanol (600 mL). The contents of R-3 were charged to the main batch over the course of 45 to 75 minutes with agitation while maintaining temperature of 20 to 30°C. The batch was cooled to 5 to -5°C and then agitated at 5 to -5°C for at least 4 hours. The solids were filtered and then washed twice with methanol (130 mL x 2). The wet cake was dried in a vacuum oven at 40 to 50°C to afford 3 (144.0 g, 98% yield), m.p. 131.8-133.1 °C. 1H NMR (CDC13, 400 MHz) δ: 8.14 (m, 2 H), 7.83-7.74 (m, 6 H), 6.50 (d, J= 7.9 Hz, 1 H), 4.69 (m, 1 H), 4.43 (s, 1H), 4.11 (dd, , J= 13.7, 4.9 Hz, 1H), 3.95 (m, 2H), 3.83 (m, 1H), 3.47 (s, 1H), 3.10 (dd, J= 13.7, 11.0 Hz, 1H), 2.81 (m, 2H), 2.51 (m, 1H), 2.12 (m, 1H), 1.85-1.72 (m, 4H), 1.45 (s, 9H), 1.26 (m, 1H); 13C NMR (CDC13, 100 MHz) δ: 166.9, 154.6, 148.2, 147.6, 135.2, 134.8, 132.6, 132.5, 131.9, 131.6, 131.4, 129.7, 124.9, 124.7, 79.8, 76.5, 55.0, 47.1, 46.0, 31.8, 31.5, 28.4, 27.3, 24.4.
Preparation of N-4-nitrobenzene sulfonyl-O-benzylhydroxylamine
![]()
To a reactor (R-l) was charged O-benzylhydroxylamine hydrochloride (61.0g, 382 mmol) and pyridine (400 mL). The solution cooled to 5 to -5°C.
To a separate reactor (R-2) was charged 4-nitrobenzenesulfonyl chloride (89.0 g, 402 mmol) and pyridine (200 mL). The contents of R-2 were transferred to R-l at a rate to maintain temperature range of -5 to -5°C. The batch in R-l was agitated at 5 to -5 °C for 15 to 45 minutes then warmed to 20 to 30°C for 45 to 75 minutes. Water (250 mL) was then added at a rate to maintain 20 to 30°C and agitated 5 to 15 minutes. The solids were filtered and the wet cake washed with water (100 mL x 3). The wet cake was dried in vacuum oven at 50°C to afford N-4-nitrobenzenesulfonyl-O-benzylhydroxylamine (113.3 g, 96% yield), m.p. 128.4-130.0 °C. 1H NMR (CDCls, 400 MHz) δ: 8.36 (d, J = 8.9 Hz, 2 H), 8.11 (d, J = 8.9 Hz, 2 H), 7.36 (m, 5H), 7.11 (s, 1H), 5.02 (s, 2H); 13C NMR (CDC13, 100 MHz) δ: 151.0, 142.5, 134.9, 130.2, 129.7, 129.3, 128.9, 124.5, 80.2.
Step C. Preparation of tert-butyl 4-((2S,5R)-5-((benzyloxy)amino)piperidine -2-carboxamido)piperidine- 1 -carboxylate (4)
Boc
To a reactor (R-l) was charged tert-butyl 4-((2R,5R)-l-((2-nitrophenyl)sulfonyl)-5-(((2-nitrophenyl)sulfonyl)oxy)piperidine-2-carboxamido)piperidine-l -carboxylate (3) (110 g, 158 mmol), N-4-nitrobenzene sulfonyl-O-benzylhydroxylamine (58 g, 188 mmol), potassium carbonate (25.9 g, 187 mmol) and dimethylacetamide (440 mL). The stirred solution was heated to 60 to 70°C for 24 – 32 hours. The batch was cooled to 20 to 30°C and charged with toluene (660 mL). The batch was extracted with 1 N sodium hydroxide (3×220 mL) then washed with water (220 mL).
The toluene solution was azotropically distilled at ~50°C to about 1/3 volume. The solution was solvent-switched to MeOH at 45-55°C, adjusted to 237 mL.
The batch was cooled to 20-25°C, charged with thioglycolic acid (57.9 g, 629 mmol) at 10 °C, and then charged with K2CO3 anhydrous (172.0 g, 1225 mmol). The batch was agitated at 10-15°C for 0.5 h, warmed to 20-25°C, agitated at 20-25°C for 10-15 h, and heated at 48-53°C for 3-6 h.
The batch was charged with 10 wt% sodium chloride (1.10 L) and toluene (880 mL) at about 40°C. The layers were separated and the aq. layer back-extracted with toluene (3 x440 mL). The combined organic layer was washed with 10% NaHC03 (2 x220 mL). The batch was concentrated at 40-50°C to 165 mL, then cooled to 35-40°C. The batch was charged with seed (50 mg) and agitated for 1 h at 35-40°C. The batch was charged with heptanes (110 mL) at 35-40°C over 1 h, then slowly cooled to 15-20°C over 1 h. The batch was agitated for 3 h and the solids filtered. The wet cake was washed with toluene/heptanes (137.5 mL) then dried in vacuum oven at 30 °C for 3-8 h to affored 4. (47.3 g, 70% overall yield from 3), m.p. 117.5-118.0 °C. 1H NMR (CDC13, 500 MHz) δ: 7.37-7.29 (m, 5 H), 6.64 (d, J= 8.2 Hz, 1 H), 5.36 (brs, 1 H), 4.67 (s, 2 H), 4.00 (m, 2 H), 3.90 (m, 1 H), 3.28 (ddd, J= 11.8, 4.0, 1.7 Hz, 1 H), 3.12 (dd, J= 10.2, 3.2 Hz, 1 H), 2.95 (m, 1 H), 2.86 (m, 2 H), 2.46 (dd, J= 11.8, 9.5 Hz, 1 H), 2.10 (m, 1 H), 1.93-1.83 (m, 3 H), 1.58 (brs, 1 H), 1.45 (s, 9 H), 1.41 (m, 1 H), 1.35-1.23 (m, 3 H); 13C NMR (CDC13, 125 MHz) δ: 172.8, 154.7, 137.7, 128.4 (4 C), 127.9, 79.6, 76.9, 59.8, 57.0, 49.2, 46.1, 42.8 (br, 2 C), 32.0 (2 C), 28.4 (3 C), 28.3, 27.2.
Step D: Preparation of tert-butyl 4-((lR,2S,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1 ]octane-2-carboxamido)piperidine- 1 -carboxylate (5)
To a reactor (R-l) was charged tert-butyl 4-((2S,5R)-5-((benzyloxy)amino)piperidine-2-carboxamido)piperidine-l-carboxylate (4) (46.3 g, 107 mmol), dichloromethane (463 mL), and Hunig’s base (58.0 mL). The batch was cooled to -18°C and then charged with triphosgene in four portions (25.1 g total; 85 mmol) at <-8°C. The batch was agitated at -5 to 0°C for 0.5 h then charged with 11.4 wt% aqueous H3P04 at -5 to 0 °C (347 g, 3541 mmol). The batch was agitated at 20-25°C for 15-20 h then phase cut. The aqueous layer was back-extracted with dichloromethane (138 mL). The combined organic layer was washed with 10% NaHC03 (115 mL), then water (115 mL). The organic solution was concentrated at atmospheric pressure to ~80
mL, then charged with MTBE (347 mL) at 35-45 °C over 0.5 h, then concentrated at 35-45 °C to 231 mL two times to form a slurry.
The slurry was charged with heptanes (139 mL) at 35-45 °C over 2 h, then slowly cooled to 15-20°C over 1 h. The batch was agitated at 15-20°C for 6-8 h. Solids were filtered and the wet cake washed with MTBE/heptanes (1.4 : 1 , 185 mL) then dried under vacuum at 25-30°C for 5-10 hours to afford 5 (43.7 g, 92% yield), m.p. 161.3-161.8 °C. 1H NMR (CDC13, 500 MHz) δ: 7.45-7.32 (m, 5 H), 6.55 (d, J= 8.2 Hz, 1 H), 5.05 (d, J= 11.6 Hz, 1 H), 4.90 (d, J= 11.6 Hz, 1 H), 4.02 (m, 2 H), 3.90 (m, 2 H), 3.30 (m, 1 H), 2.99 (dt, J= 11.7, 1.1 Hz, 1 H), 2.86 (m, 2 H), 2.64 (d, J = 11.7 Hz, 1 H), 2.37 (dd, J= 14.6, 6.9 Hz, 1 H), 2.04-1.82 (m, 4 H), 1.58 (m, 1 H), 1.45 (s, 9 H), 1.30 (m, 2 H); 13C NMR (CDC13, 125 MHz) δ: 168.3, 167.5, 154.7, 135.6, 129.2 (2 C), 128.8, 128.6 (2 C), 79.7, 78.3, 60.4, 57.8, 47.5, 46.8, 42.5 (br, 2 C), 32.0, 31.7, 28.4 (3 C), 20.8, 17.2.
Step E: Preparation of tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1“|octane- 2-carboxamido) iperidine- 1 -carboxylate
tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-l -carboxylate (9.2 g, 20.1 mmol) was charged to a glass bottle, and the solids were dissolved in THF (150 mL). The solution was then charged to a hydrogenation reactor along with Pd/Al203 (10 wt%, 1.5 g). The reaction was purged three times with hydrogen and then set to a hydrogen pressure of 50 psi. The reaction temperature was adjusted to 25°C and the reaction was allowed to agitate for 22 hours. After the reaction was complete as determined by HPLC analysis, the solution was filtered through SOLKA-FLOC® (Interational Fiber Corporation, North Tonawanda, NY) to remove the catalyst and the filter cake was washed with THF. The filtrate and washes were then solvent switched by vacuum distillation to iPrOAc to a final volume of 40 mL. The resulting iPrOAc slurry was aged at room temperature for 1 hour. The solids were then filtered and washed with iPrOAc (20 mL) and dried under vacuum and N2 at 40°C to afford the title product (6.62 g., 17.97 mmol, 90% isolated yield). Spectral data matched the reference compound.
Preparation of (2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide
tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-l-carboxylate (20 g, 54.3 mmol), THF (200 mL), 2-picoline (10.9 mL, 309 mmol) and pyridine-S03 complex (30.2 g, 190 mmol) were charged to a flask under nitrogen. The heterogeneous mixture was allowed to stir overnight (~15 h). The reaction mixture was cooled to -10°C then DCM (200 mL) was added. 0.5 M K2HP04 (168 mL, 84 mmol) was added over 10 minutes. Bu4NHS04 (19.4 g, 57 mmol) was then added over 10 minutes. The biphasic mixture was stirred for 30 minutes, phase cut and the water layer was back extracted with 40 ml of DCM. The combined DCM solution was washed with water (120 ml), phase cut and the organic solution was solvent-switched to MeCN (320 ml) by vacuum distillation with 3 bed volumes of MeCN (total 1.0 L) and used as is in the next step. The solution of Bu4N+ OSO3 salt 7 in MeCN solution was used with an assumed yield of 100% (37.5 g, 54.3 mmol). The reaction mixture was cooled in an ice bath, and TMSI (10.26 ml, 70.7 mmol) was added via addition funnel over 30 minutes between 0°C and 5°C. The resulting mixture was agitated for 1-2 h and then quenched with H20:MeCN (1 :1, 6 ml) to afford a slurry. The slurry was warmed to room temperature and agitated for 12 h and after this time the pH of the supernatant was about 3.0. Tetrabutylammonium acetate (13.6 ml, 13.59 mmol) was slowly added over 30 min. The slurry was agitated for 1 h and pH of the supernatant was about 4.0. Solids were collected by filtration. The solid was washed with 60 mL of aqueous MeCN to afford 19.5 g of the crude product 8 in a 93% isolated yield from compound 6 .
At this stage, all byproducts (including hydro lyzation products of TMS-carbonate) and impurities were soluble in the organic phase.
The product was dissolved back into 140 ml of MeCN:H20 (1 :2) at room temperature. 1-Butanol (390 ml) as antisolvent was slowly added into the solution to afford a slurry. The slurry was agitated overnight. The white crystalline solid was filtered and washed with 3:1 IPA: water (40 ml) and dried under vacuum and nitrogen at room temperature to afford the title product in the form of a crystalline hydrate. (Yield = 16.3 g, 82%). Spectral data matched reference compound.
Preparation of (2S,5R)-7-oxo-2-(piperidin- 1 -ium-4-ylcarbamoyl)- 1 ,6-diazabicyclo[3.2.1 ]octan-6-yl sulfate (1).
tert-Butyl 4-( {[(25*,5i?)-6-hydroxy-7-oxo- 1 ,6-diazabicyclo[3.2.1 ]oct-2-yl]carbonyl}amino)piperidine-l-carboxylate 16 (0.54 g, 1.5 mmol), THF (5.4 mL), 2-picoline (0.29 mL, 2.9 mmol) and pyridine-S03 complex (0.70 g, 4.4 mmol) were charged to a vial under nitrogen. The heterogeneous mixture was allowed to stir overnight (~15 hr). The reaction mixture was cooled to -10°C then dichloromethane (5.4 mL) was added. 0.5 M K2HPO4 (4.5 mL, 2.3 mmol) was added over 10 minutes. BU4NHSO4 (0.53 g, 1.54 mmol) was then added over 10 min. The biphasic mixture was stirred for 30 min, phase cut and the water layer was back extracted with 1 ml of DCM. The combined DCM solution was washed with water (2.0 mL), phase cut and the organic solution was solvent-switched to MeCN (3.2 mL) by vacuum distillation with 3 bed volumes of MeCN. The product was used as is in the next step (water content less than 1000 ppm).
The solution of Bu4N+S04~~ salt 8 in MeCN solution was used with an assumed yield of 100% (1.0 g, 1.47 mmol). The reaction mixture was cooled in an ice bath, and Ν,Ο-bis(trimethylsilyl)trifluoroacetamide (BSTFA) (0.4 lg, 1.59 mmol) was added into the reaction and was allowed to stir for 10 min. TMSI (0.06g, 0.27 mmol) was added between 0°C and 5°C. The resulting mixture was allowed to agitate for 2 hr and then quenched with H2O (0.07g, 4.1 mmol) and acetic acid (0.08g, 1.5 mmol) to afford a slurry. The slurry was warmed to room temperature and agitated for 12 hr. Filter to collect the solid. The solid was washed with MeCN/water (94:6, 1 mL X 4) to afford the crystalline product 1 (0.38 g) in a 75% yield.
If NO-bis(trimethylsilyl)acetamide (BSA) (0.32g, 1.59 mmol) was applied, the reaction needed 24 hr to achieve full conversion.
Patent
WO2015033191
Scheme 1.

Formula (V)
Formula (VI)

Formula (I)
Scheme – 1
Example -1
Preparation of (2S, 5R)-Sulfuric acid mono-{2-[N’-(4-aminopiperidinyl)-carbonyl]-7-oxo- l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester (I).
Step-1: Preparation of (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (IV):
To a 250 ml round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-sodium 6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carboxylate (11.1 gm, 0.037 mol, prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) in water (180 ml) followed by l-tert-butoxycarbonyl-4-amino-piperidine (7.8 gm, 0.039 mol), EDC hydrochloride (11 gm, 0.055 mol) and 1 -hydro ybenzotriazole (4.8 gm, 0.037 mol) at 30°C successively under stirring. The reaction mixture was stirred for 24 hours at 30°C to provide a suspension. The suspension was filtered under suction and washed with 45°C warm water (40 ml) to provide (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 12.7 gm quantity in 74% yield after drying under vacuum.
Analysis
NMR: (CDC13,) = 7.36-7.44 (m, 5H), 6.56 (d,lH), 5.06 (d,lH), 4.91 (d, 1H), 4.03 (br s, 1H), 3.88-3.97 (m, 2H), 3.29 (s, 1H), 3.00 (d, 1H), 2.86 (t, 2H), 2.64 (d, 1H), 2.37 (dd, 1H), 1.85-2.01 (m, 4H), 1.54-1.62 (m, 2H), 1.45 (s, 9H), 1.25-1.36 (m, 2H).
MS (ES+) C24H34N405 = 459.5 (M+l).
Step-2: Preparation of (2S, 5R)-tert-butyl { (6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (V):
To a 100 ml single neck round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (9 g, 19.5 mmol) in methanol (90 ml) followed by 10% palladium on carbon (2.7 g) at 35°C. The reaction mixture was stirred under 1 atm hydrogen pressure at 35°C for 2 hours. The catalyst was removed by filtering the reaction mixture under suction over a celite bed. The celite bed was washed with dichloromethane (50 ml). The combined filtrate was evaporated under vacuum below 35°C to provide (2S, 5R)-tert-butyl {(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 8.45 g quantity; it was used as such for the next reaction.
Analysis
NMR: (CDC13,) = 6.60 (d, 1H), 3.88-4.10 (m, 4H), 3.78 (s, 1H), 3.20 (d, 1H), 3.90 (t, 2H), 2.80 (d, 1H), 2.46 (dd, 1H), 2.1-2.2 (m, 1H), 2.85-2.20 (m, 4H), 1.70-1.80 (m, 1H), 2.47 (s, 9H), 1.30-1.41 (m, 3H).
MS (ES+) C17H28N405 = 369.4 (M+l).
Step-3: Preparation of Tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (VI):
To a 100 ml single neck round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-tert-butyl {(6-hydroxy-7-oxo-l,6-diaza-bicyclo [3.2.1 ]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (6.40 g, 7.6 mmol) in dichloromethane (90 ml), triethyl amine (9.3 ml), followed by pyridine – sulfur trioxide complex (5.4 g, 34.2 mmol) at 35°C under stirring. The reaction mixture was stirred for additional 4 hours at 35°C. The solvent was evaporated under vacuum below 40°C to provide a residue. The residue was stirred with 0.5N aqueous potassium dihydrogen phosphate solution (90 ml) for 1 hour. The resulting solution was extracted with dichloromethane (2 x 100 ml) to remove impurities. To the aqueous layer was added tetrabutyl ammonium hydrogen sulfate (6.9 g, 20.52 mmol) and the reaction mixture was stirred for 14 hours at 35°C. It was extracted with dichloromethane (3 x 30 ml). Combined organic layer was dried over sodium sulfate and evaporated under vacuum to provide tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 8.0 g quantity in 62% yield.
Analysis
NMR: (CDC13,) – 6.64 (d, 1H), 4.36 (br s, 1H), 4.05(br s, 2H), 3.90-4.00 (m, 1H), 3.87 (d, 1H), 2.28-3.34 (m, 10H), 3.80-3.95 (m, 2H), 3.74 (d, 1H), 2.42 (dd, 1H), 2.15-2.24 (m, 1H), 1.82-1.97 (m, 4H), 1.61-1.74 (m, 14 H), 1.41-1.52 (m, 10 H), 1.02 (t, 12H).
MS (ES-) C17H27N408S. N(C4H9)4 = 447.4 (M-l) as a free sulfonic acid.
Step-4: Synthesis of (2S, 5R)- Sulfuric acid mono-{ [(4-aminopiperidin-4-yl) carbonyl]-7-oxo-l,6-diaza-bicyclo[3.2.1]-oct-6-yl} ester (I):
To a 100 ml round bottom flask equipped with magnetic stirrer was charged a solution of tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (6.0 g) in dichloromethane (15 ml). The solution was cooled to -10°C under stirring and to it was added trifluoro acetic acid (15 ml) drop wise. The reaction mixture was stirred at -10°C for 1 hour. Solvents were evaporated under vacuum below 30°C to its 1/3 volume to provide a thick residue. The thick residue was stirred twice with diethyl ether (60 ml each time) to provide a precipitation. The solid obtained was filtered at suction and suspended in acetone (90 ml). To the suspension was added 10% solution of sodium-2-ethyl-hexanoate in acetone to adjust pH between 4.5 to 5.5. The suspension was stirred for 10 minutes and filtered under suction. The wet cake was washed with acetone and dried under vacuum below 40°C to provide 3 gm crude compound. The crude compound was stirred with aqueous isopropanol (3ml water: 21 ml iospropanol) for overnight to purify further. The resulting suspension was filtered under suction and washed with aqueous isopropanol (1 ml water: 7 ml IPA mixture). Finally the cake was dried under vacuum below 40°C to provide the title compound as a off-white solid in 1.8 g quantity in 65% yield.
Analysis
H1NMR (DMSO-d6, D20 exchange) = 8.19 (d, exchanges with D20), 3.99 (s, 1H), 3.82-3.92 (m, 1H), 3.72 (d, 1H), 2.24 (br d, 3H), 2.90-3.04 (m, 5H), 1.96-2.06 (m, 1H), 1.80-1.94 (m, 3H), 1.58-1.72 (m, 4H).
MS (ES+) C12H20N4O6S = 349.2 (M+l) as a free sulfonic acid;
Purity by HPLC: 99.2%
Specific rotation: [a] D -45.25 °, (c 0.3%, water)
SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
//////
C1CC(N2CC1N(C2=O)OS(=O)(=O)O)C(=O)NC3CCNCC3.O
UPDATE,,,,,,,,,,
Improved Preparation of a Key Hydroxylamine Intermediate for Relebactam: Rate Enhancement of Benzyl Ether Hydrogenolysis with DABCO
Jianguo Yin, Mark Weisel, Yining Ji, Zhijian Liu  , Jinchu Liu, Debra J. Wallace, Feng Xu , Benjamin D. Sherry, and Nobuyoshi Yasuda*
, Jinchu Liu, Debra J. Wallace, Feng Xu , Benjamin D. Sherry, and Nobuyoshi Yasuda*
, Jinchu Liu, Debra J. Wallace, Feng Xu , Benjamin D. Sherry, and Nobuyoshi Yasuda* Process R&D Department, MRL, Merck & Co., Inc., Rahway, New Jersey 07065, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00381
Publication Date (Web): February 1, 2018
Copyright © 2018 American Chemical Society
*E-mail: nobuyoshi_yasuda@merck.com

Previous methods to prepare a bicyclic N-hydroxyl urea intermediate in the synthesis of the potent β-lactamase inhibitor relebactam were effective, but deemed unsuitable for long-term use. Therefore, we developed an in situ protection protocol during hydrogenolysis and a robust deprotection/isolation sequence of this unstable intermediate employing a reactive crystallization. During the hydrogenation studies, we discovered a significant rate enhancement of O-benzyl ether hydrogenolysis in the presence of organic amine bases, especially DABCO. The broader utility of the application of organic bases on the hydrogenolysis of a range of O– and N-benzyl-containing substrates was demonstrated.

5 could be isolated by concentrating the filtrate and storing the solution at 5 °C overnight. 1H NMR (500 MHz, CDCl3): δ 6.58 (d, J = 7.9 Hz, 1H), 4.10–3.86 (m, 4H), 3.55 (bs, 1H), 3.14 (bd, J = 11.5 Hz, 1H), 2.86 (bt, J = 12.0 Hz, 2H), 2.76 (d, J = 11.5 Hz, 1H), 2.36 (dd, J = 15.1, 7.1 Hz, 1H), 2.12 (m, 1H), 2.00–1.82 (m, 3H), 1.66 (m, 1H), 1.44 (s, 9H), 1.31 (m, 2H), 0.25 (S, 9H). 13C NMR (125 MHz, CDCl3): δ 169.2, 168.3, 154.8, 79.8, 60.7, 60.0, 47.3, 46.9, 42.6 (br, 2C), 32.2, 31.9, 28.5 (3C), 20.5, 17.5, −0.75 (3C). (+)-ESI HRMS: calcd for C20H36N4NaO3Si (M + Na)+, 463.2347; found, 463.2348.
Tesmilifene , Antagonist of intracellular histamine

Tesmilifene
BMS-217380; BMY-33419; DPPE
CAS No. 98774-23-3(Tesmilifene), 92981-78-7(Tesmilifene hydrochloride)
Tesmilifene
CAS 98774-23-3
N,N-Diethyl-2-[4-(phenylmethyl)phenoxy]ethanamine
DPPE
MFC19H25NO
MW 283.41
Percent Composition: C 80.52%, H 8.89%, N 4.94%, O 5.65%

Hydrochloride
CAS 92981-78-7
BMS-217380-01; BMY-33419
MF C19H25NO.HCl
MF 319.87
Percent Composition: C 71.34%, H 8.19%, N 4.38%, O 5.00%, Cl 11.08%
Properties: White crystals from isopropanol + acetone (3:1), mp 156-158°. pKa 10.9.
Melting point: mp 156-158°
pKa: pKa 10.9
Therap-Cat: Antineoplastic adjunct (chemosensitizer).
Tesmilifene is a novel potentiator of chemotherapy which, when added to doxorubicin, achieved an unexpected and very large survival advantage over doxorubicin alone in a randomized trial in advanced breast cancer.
PHASE 23 FOR An estrogen receptor antagonist potentially for the treatment of advanced breast cancer, gastric cancer
Tesmilifene is a novel agent that augments cytotoxicity of various chemotherapeutic agents both in vitro and in vivo. It binds selectively to the high-affinity microsomal antiestrogen binding site (Ki=50nm) but has no affinity for estrogen receptors. Inhibits concanavalin-A-induced histamine release in mast cells and acts as a novel antagonist of intracellular histamine.
US 4803227


The target product can be prepared by reacting para-benzylphenol (I) with 2-diethylaminoethylchloride hydrochloride (II) either by means of NaOH in H2O or with K2CO3 in DMF/acetone (at 60 C in both cases), followed by treatment with HCl to obtain the corresponding hydrochloride salt.
| EP 0153160; JP 1985190742; US 4803227 |
US 4803227
http://www.google.com/patents/US4803227
Tesmilifene is a small molecule chemopotentiator under development by YM BioSciences, a Candian pharmaceutical company that specialises in the development of cancer treatments. It is indicated for use in combination with standard cytotoxic drugs, such as taxanes and anthracyclines, which are widely used in the treatment of metastatic disease – when cancers spread to distant sites in the body.
Tesmilifene, the company’s lead investigational compound, is currently in phase III development for patients with metastatic breast cancer. At the end of January 2007, an independent safety monitoring board advised the company that its ongoing registration trial should be stopped; it was considered unlikely that significant differences in overall survival (primary endpoint) between treatment arms would emerge over time. The company had hoped that the addition of tesmilifene to standard epirubicin/cyclophosphamide therapy would confer a survival benefit similar to that seen in its earlier phase III trial.
In light of these disappointing results, YM BioSciences plans a detailed analysis of its phase III data in advanced breast cancer to see if it can identify why tesmilifene failed to add clinical benefit in this trial.
DRUG RESISTANCE LIMITS EFFECTIVENESS OF CHEMOTHERAPY
Cytotoxic drugs have proved potent weapons in the fight against malignant tumours and are considered first-line therapy for the treatment of many cancers. However, while patients often respond well to a first course of chemotherapy over time the response to drug treatment diminishes and the tumour may eventually become drug resistant. In some cases resistance can develop across several classes of anti-cancer drugs, leading to multidrug resistance. The development of drug resistance limits the effectiveness of many anti-cancer agents and is an important contributor to cancer deaths.
The development of agents that can overcome drug resistance is seen as one of the most important areas of cancer research and for which there is significant unmet need. Various approaches are being explored to boost the use of cytotoxic agents including chemopotentiators, chemoprotectants and liposomal formulations.
Clearly any agent that can prevent or reverse drug resistance would have a major impact on treatment strategies, enhancing the benefits of standard cytotoxic drugs.
TESMILIFENE MAY BOOST CYTOTOXIC EFFECTS OF ANTHRACYCLINES
Anthracyclines are a class of cytotoxic agents with proven efficacy in the treatment of breast cancer. They include agents such as doxorubicin and epirubicin among others. Because patients with metastatic breast cancer may have received anthracycline therapy for earlier stage breast cancer (adjuvant therapy) or following disease recurrence, there is a risk that they will fail to respond to continued treatment.
A phase III trial in 305 patients with advanced breast cancer has shown that when tesmilifene is combined with doxorubicin it appears to improve survival over treatment with doxorubicin alone. In this trial approximately half the patients were treated with both tesmilifene and doxorubicin, while the other half received doxorubicin alone. Although there were no significant differences in tumour response rates, progression-free survival, or average duration of response between treatment arms at endpoint, overall survival was significantly improved in the combination arm. Among patients treated with tesmilifene and doxorubicin overall survival was 23.6 months compared with 15.6 months for those treated with doxorubicin alone.
Researchers have suggested that tesmilifene may enhance the anti-tumour effects of anthracyclines in several ways:
- Reducing the cancer cell’s ability to become resistant
- Decreasing the metabolism or “break-down” of doxorubicin
- Disrupting the cancer cell’s energy source
TESMILIFENE REGISTRATION TRIAL
In March 2004 YM BioSciences began its pivotal international phase III trial of tesmilifene in metastatic breast cancer. By September 2005, 723 patients had been enrolled in the trial, which was designed once again to compare the efficacy and safety of tesmilifene and an antrhacycline (epirubicin) with epirubicin alone.
“At the end of January 2007, an independent safety monitoring board advised the company that its ongoing registration trial should be stopped.”
Given the survival benefit seen in the earlier trial, which was carried out by the Canadian National Cancer Institute, the company was optimistic about outcome in its pivotal registration trial. However, an interim analysis of 351 events suggested that significant differences in overall survival were unlikely to be seen between the two treatment arms as the data matured and the trial was brought to a premature end.
In addition to its work on anthracyclines, YM BioSciences has also been exploring the potential of tesmilifene to enhance the efficacy of taxanes, also standard chemotherapy for metastatic breast cancer. Other potential applications include:
- Adjuvant therapy for breast cancer, i.e. immediately post-surgery and before the cancer has recurred or metastasised
- Hormone-refractory prostate cancer
- Lung cancer
- Non-Hodgkin’s lymphoma
MARKETING COMMENTARY
Although there have been major advances in the treatment of breast cancer in the last 10 to 15 years, it remains a disease for which improved treatments are still urgently needed. Estimates from the WHO suggest that metastatic breast cancer will claim the lives of over 40,000 patients a year.
Current treatments for metastatic breast cancer are rarely curative but can nonetheless do much to improve patients’ quality of life or duration of survival. . By boosting the cytotoxic effects of standard chemotherapy agents such as anthracyclines, while protecting healthy cells, tesmilifene was thought to have potential to extend the benefits of cytotoxic therapy to more patients. This is now in doubt following premature ending of its pivotal registration trial in advanced breast cancer.
Literature References: Intracellular histamine antagonist with chemopotentiating and cytoprotective activity. Structurally similar to tamoxifen, q.v., although binds anti-estrogen binding site (AEBS) with no affinity for the estrogen receptor.
Prepn: L. J. Brandes, M. W. Hermonat, Biochem. Biophys. Res. Commun. 123, 724 (1984); and use as antineoplastic: eidem, US 4803227 (1989 to Univ. Manitoba); and study of binding affinity: M. Poirot et al., Bioorg. Med. Chem. 8, 2007 (2000). Spectral analysis of interaction with P450 isozymes: L. J. Brandes et al., Cancer Chemother. Pharmacol. 45, 298 (2000).
Clinical evaluation in combination with cyclophosphamide in prostate cancer: L. J. Brandes et al., J. Clin. Oncol. 13, 1398 (1995); in combination with doxorubicin in breast cancer: L. Reyno et al., J. Clin. Oncol. 22, 269 (2004).
Bioorg Med Chem 2000,8(8),2007
Product Literature References
Enhancement of cytotoxicity of natural product drugs against multidrug resistant variant cell lines of human head and neck squamous cell carcinoma and breast carcinoma by tesmilifene.: P. J. Ferguson, et al.; Cancer Lett. 274, 279 (2009), Abstract;
Phase III study of N,N-diethyl-2-[4-(phenylmethyl) phenoxy]ethanamine (BMS-217380-01) combined with doxorubicin versus doxorubicin alone in metastatic/recurrent breast cancer: National Cancer Institute of Canada Clinical Trials Group St: L. Reyno, et al.; J. Clin. Oncol. 22, 269 (2004), Abstract;
Synergy between tamoxifen and cisplatin in human melanoma cells is dependent on the presence of antiestrogen-binding sites.: J.A. Jones, et al.; Cancer Res. 57, 2657 (1997), Abstract;
Influence of DPPE on histamine release from isolated rat mast cells.: N. Grosman; Agents Actions 41, 1 (1994), Abstract;
Histamine is an intracellular messenger mediating platelet aggregation.: S.P: Saxena, et al.; Science 243, 1596 (1989), Abstract;
///////Tesmilifene, Antineoplastic Adjunct, Chemosensitizer, PHASE 3, Tesmilifene hydrochloride, BMY-33419, BMS-217380, DPPE, N,N-DPPE, Antagonist of intracellular histamine
CCN(CC)CCOC1=CC=C(C=C1)CC2=CC=CC=C2
see……….http://apisynthesisint.blogspot.in/2015/12/tesmilifene-antagonist-of-intracellular.html
Mirogabalin


Mirogabalin, A-2000700, DS-5565
1138245-13-2, C12H19NO2, 209.28
[(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
2-[(1R,5S,6S)-6-(aminomethyl)-3-ethyl-6-bicyclo[3.2.0]hept-3-enyl]acetic acid
UNII-S7LK2KDM5U
| Originator |
Daiichi Sankyo
|
|---|---|
| Therapeutic Claim |
Treatment of fibromyalgia
|
Phase III clinical trials at Daiichi Sankyo for the treatment of pain associated with fibromyalgia
| Class |
Analgesic drugs (small molecules)
|
|---|---|
| Mechanism of action |
CACNA2D1 protein modulators
|
SYNTHESIS
 SEE
SEE
[(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid benzenesulfonatee

DESIRED
[(1S,5R,6R)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid , optical isomer of the compound
 UNDESIRED
UNDESIRED
Mirogabalin (DS-5565) is a drug developed by Daiichi Sankyo and related to drugs such as gabapentin and pregabalin. Similarly to these drugs, mirogabalin binds to the α2δ calcium channels (1 and 2), but with significantly higher potency than pregabalin. It has shown promising results in Phase II clinical trials for the treatment of diabetic peripheral neuropathic pain,[1][2] and is currently in Phase III trials.
Mirogabalin, a voltage-dependent calcium channel subunit alpha-2/delta-1 ligand, is in phase III clinical trials at Daiichi Sankyo for the treatment of pain associated with fibromyalgia. The company is also conducting phase III clinical studies for the treatment of chronic pain and pain associated with diabetic peripheral neuropathy.
Mirogabalin besylate
cas 1138245-21-2
UNII: 01F4FRP8YL
C12-H19-N-O2.C6-H6-O3-S, 367.4635
Bicyclo(3.2.0)hept-3-ene-6-acetic acid, 6-(aminomethyl)-3-ethyl-, (1R,5S,6S)-, benzenesulfonate (1:1)
SEE
Tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate…..http://www.google.com/patents/US20140094623?cl=zh
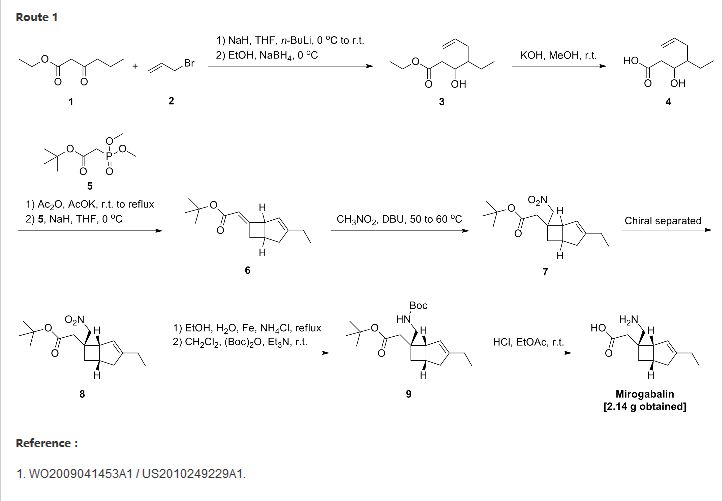
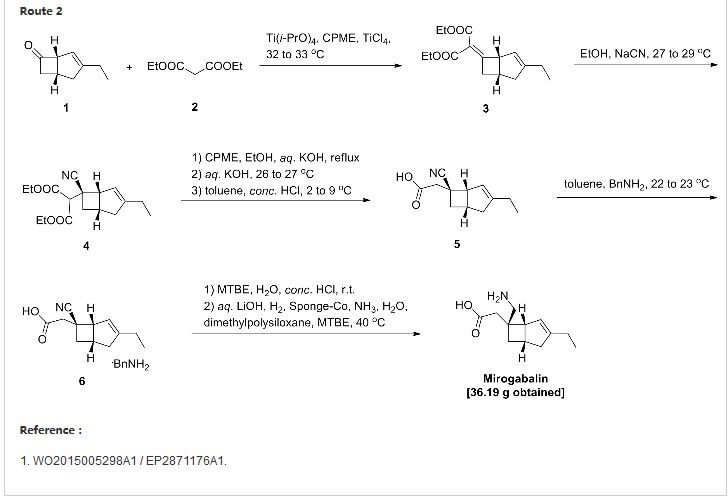
PATENT
WO 2009041453
https://www.google.co.in/patents/EP2192109A1
(Example 21) [(1S,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid (exemplary compound No: 8, optically active form of the compound of Example 8)
(21-a) Resolution of tert-butyl (±)-[(1R,5S,6S)-3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate
Tert-butyl (±)-[(1R,5S,6S)-3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (230 g, 778 mmol) was resolved using Chiralpak IC (N-Hex:EtOH=98:2, 1.0 mL/min, 40°C) manufactured by Daicel Chemical Industries, Ltd. to respectively obtain 115 g of a peak 1 (retention time: 5.2 min) and 93.7 g of a peak 2 (retention time: 6.3 min).
(21-b) Tert-butyl ([(1R,5S,6S)-6-(tert-butoxycarbonylamino)methyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate
Tert-butyl [(1R,5S,6S)-3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (peak 1, 7.0 g, 23.7 mmol) was dissolved in ethanol (60 mL) and water (21 mL). To the solution, iron powder (13.27 g, 237 mmol) and ammonium chloride (628.1 mg, 11.9 mmol) were added, and the mixture was stirred for 5.5 hours under heating to reflux. The mixture was allowed to cool, then diluted with saturated saline, a saturated aqueous solution of sodium bicarbonate, and ethyl acetate, and filtered through Celite to remove insoluble matter. The filtrate was separated into organic and aqueous layers. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate. Then, the solvent was distilled off under reduced pressure to obtain a pale yellow oil substance (7.02 g). This substance was dissolved in dichloromethane (200 mL). To the solution, (Boc)2O (5.25 g, 25 mmol) and triethylamine (5.01 g, 50 mmol) were added, and the mixture was stirred overnight at room temperature. The solvent was distilled off under reduced pressure, and the residue was then purified by silica gel chromatography to obtain the title compound of interest as a pale yellow oil substance (8.82 g, <100%). (21-c) [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
A 4 N hydrochloric acid-ethyl acetate solution (100 mL) was added to tert-butyl (1R,5S,6S)-[6-(tert-butoxycarbonylaminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate (9.82 g, 23.7 mmol), and the mixture was stirred at room temperature for 1 hour. Then, the solvent was distilled off under reduced pressure. The residue was dissolved in dichloromethane. To the solution, triethylamine was added dropwise, and the resulting powder was collected by filtration, then washed with dichloromethane, and then dried to obtain 4.02 g of a white powder. This powder was washed with ethanol and ethyl acetate to obtain the title compound of interest as a white powder (2.14 g, 43%).
(Example 31) [(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid benzenesulfonate (exemplary compound No: 8, optically active benzenesulfonate)
(1R,5S,6S)-6-(aminomethyl)-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid (4.50 g, 20.6 mmol) was dissolved by heating in a 1 M aqueous solution (22.7 mL) of benzenesulfonic acid monohydrate, and the solution was then allowed to cool to room temperature. The resulting solid was collected by filtration. The solid was washed with water (15 mL) and then dried using a vacuum pump to obtain the compound of interest as a colorless solid (6.45 g, 77%).
PATENT
JP 2010241796
PATENT
WO 2012169475
-
 Reference Example 1[6-Aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
Reference Example 1[6-Aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid(1-a) Ethyl 4-ethyl-3-hydroxyhept-6-enoateSodium hydride (>63% oil, 2.09 g, 55 mmol) was added to a solution of ethyl 3-oxohexanoate (7.91 g, 50 mmol) in tetrahydrofuran (50 mL) under ice cooling, and the mixture was stirred in this state for 10 minutes. To the reaction solution, n-butyllithium (1.58 M solution in hexane, 34.8 mL, 55 mmol) was added dropwise, and the mixture was further stirred for 10 minutes under ice cooling. Then, allyl bromide (4.7 mL, 55 mmol) was added thereto, and the mixture was stirred in this state for 1 hour and then further stirred at room temperature for 4 hours. To the reaction solution, 1 N hydrochloric acid and a saturated aqueous solution of ammonium chloride were added, followed by extraction with n-pentane. The organic layer was washed with saturated saline and dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was dissolved in ethanol (80 mL). To the solution, sodium borohydride (1.51 g, 40 mmol) was added under ice cooling, and the mixture was stirred in this state for 2 hours. 1 N hydrochloric acid (50 mL) was added thereto, and the mixture was stirred for 30 minutes. Then, saturated saline was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography to obtain the compound of interest as a pale yellow oil substance (3.64 g, 37%, mixture of diastereomers).
(1-b) 4-Ethyl-3-hydroxyhept-6-enoic acid
(1-c) Tert-butyl 3-ethylbicyclo[3.2.0]hept-3-en-6-ylideneacetate
-
1H-NMR (400 MHz, CDCl3): δ ppm:
-
Major isomer: 1.06 (3H, t, J=7.4 Hz), 1.45 (9H, s), 2.07-2.22 (3H, m), 2.59-2.70 (2H, m), 2.87-2.96 (1H, m), 3.30 (1H, ddt, J=8.6, 18.4, 2.7 Hz), 3.86-3.88 (1H, m), 5.22-5.23 (1H, m), 5.45-5.47 (1H, m).
-
Minor isomer: 1.08 (3H, t, J=7.3 Hz), 1.49 (9H, s), 2.07-2.21 (3H, m), 2.43-2.47 (1H, m), 2.59-2.70 (1H, m), 2.75-2.85 (1H, m), 2.87-2.96 (1H, m), 4.28-4.31 (1H, m), 5.35-5.38 (1H, m), 5.45-5.47 (1H, m).
(1-d) Tert-butyl [3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate
(1-e) [6-Aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetic acid
1H-NMR (400 MHz, CDCl3): δ ppm: 0.91 (3H, t, J=7.5 Hz), 1.28 (3H, t, J=7.2 Hz), 1.43-1.55 (2H, m), 1.98-2.28 (2H, m), 2.45-2.48 (2H, m), 2.88-2.93 (1H, m), 4.07-4.10 (1H, m), 4.10-4.20 (2H, m), 5.01-5.09 (2H, m), 5.75-5.86 (1H, m).Ethyl 4-ethyl-3-hydroxyhept-6-enoate (3.64 g, 18.2 mmol) was dissolved in a 2 N solution of potassium hydroxide in methanol (120 mL), and the solution was stirred overnight at room temperature. From the reaction solution, the solvent was distilled off under reduced pressure. To the residue, a 1 N aqueous sodium hydroxide solution (200 mL) was then added, followed by extraction with diethyl ether. The aqueous layer was made acidic by the addition of concentrated hydrochloric acid under ice cooling, followed by extraction with diethyl ether again. The organic layer was washed with saturated saline and dried over anhydrous magnesium sulfate. Then, the solvent was distilled off under reduced pressure to obtain the compound of interest as a pale yellow oil substance (3.14 g, <100%, mixture of diastereomers).1H-NMR (400 MHz, CDCl3): δ ppm: 0.91-0.96 (3H, m), 1.39-1.52 (3H, m), 2.01-2.28 (2H, m), 2.52-2.55 (2H, m), 4.05-4.15 (2H, m), 5.03-5.10 (2H, m), 5.74-5.86 (1H, m).4-Ethyl-3-hydroxyhept-6-enoic acid (3.13 g, 18.2 mmol) was dissolved in acetic anhydride (15 mL). To the solution, potassium acetate (4.27 g, 43.6 mmol) was added, and the mixture was stirred at room temperature for 100 minutes. The reaction solution was heated to reflux and stirred for 3.5 hours to form “3-ethylbicyclo[3.2.0]hept-6-en-6-one” in the reaction solution. To the reaction solution, ice water and toluene were then added, and this mixture was stirred overnight at room temperature. The mixture was separated into aqueous and organic layers by the addition of saturated saline (50 mL) and toluene (20 mL). Then, the organic layer was washed with a 1 N aqueous sodium hydroxide solution and saturated saline in this order, then dried over anhydrous magnesium sulfate, and filtered. The filtrate was added to a reaction solution prepared by adding sodium hydride (>65% oil, 761.9 mg, 20 mmol) to a solution of tert-butyl dimethoxyphosphorylacetate (4.48 g, 20 mmol) in tetrahydrofuran (50 mL) under ice cooling, and the mixture was further stirred for 1 hour. The reaction solution was separated into aqueous and organic layers by the addition of a saturated aqueous solution of ammonium chloride and saturated saline. The aqueous layer was subjected to extraction with ethyl acetate. The organic layers were combined, then washed with saturated saline, and then dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography to obtain the compound of interest as a pale yellow oil substance (1.32 g, 31%, E/Z mixture).Tert-butyl [3-ethylbicyclo[3.2.0]hept-3-en-6-ylideneacetate (1.32 g, 5.63 mmol) was dissolved in nitromethane (7 mL). To the solution, 1,8-diazabicyclo[5.4.0]undec-7-ene (1.2 mL, 7.3 mmol) was added, and the mixture was heated with stirring at 50 to 60° C. for 7 hours. The mixture was allowed to cool, and a saturated aqueous solution of potassium dihydrogen phosphate was then added thereto, followed by extraction with ethyl acetate. Then, the organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography to obtain the compound of interest as a colorless oil substance (1.39 g, 84%).1H-NMR (400 MHz, CDCl3): δ ppm: 1.09 (3H, t, J=7.4 Hz), 1.46 (9H, s), 1.52 (1H, dd, J=7.6, 13.2 Hz), 2.06 (1H,d, 16.6 Hz), 2.14 (2H, q, J=7.4 Hz), 2.30 (1H, ddd, J=2.4, 7.6, 13.2 Hz), 2.47 (2H, s), 2.49 (1H, dd, J=7.6,16.6 Hz), 2.86 (1H, quint, J=7.6 Hz), 3.21-3.22 (1H, m), 4.75 (1H, d, J=11.7 Hz), 4.84 (1H, d, J=11.7 Hz), 5.27 (1H, s).Tert-butyl [3-ethyl-6-(nitromethyl)bicyclo[3.2.0]hept-3-en-6-yl]acetate (1.09 g, 4.71 mmol) was dissolved in ethanol (10 mL) and water (5 mL). To the solution, iron powder (1.32 g, 23.5 mmol) and ammonium chloride (249.6 mg, 4.71 mmol) were added, and the mixture was stirred for 2 hours under heating to reflux. The mixture was allowed to cool, then diluted with saturated saline, a saturated aqueous solution of sodium bicarbonate, and ethyl acetate, and filtered through Celite to remove insoluble matter. The filtrate was separated into organic and aqueous layers. The organic layer was washed with saturated saline and then dried over anhydrous magnesium sulfate, and the solvent was then distilled off under reduced pressure. To the residue, a 4 N solution of hydrochloric acid in ethyl acetate (20 mL) was added, and the mixture was stirred at room temperature for 1 hour. Then, the solvent was distilled off under reduced pressure. The residue was suspended in dichloromethane. To the suspension, triethylamine was added dropwise, and the resulting powder was collected by filtration, then washed with dichloromethane, and then dried to obtain the compound of interest as a white powder (425.1 mg, 43%).1H-NMR (400 MHz, CD3OD): δ ppm: 1.10 (3H, t, J=7.4 Hz), 1.48 (1H, dd, J=7.5, 12.5 Hz), 2.03-2.08 (2H, m), 2.14 (2H, q, J=7.4 Hz), 2.46 (1H, d, J=16.2 Hz), 2.46-2.53 (1H, m), 2.51 (1H, d, J=16.2 Hz), 2.85 (1H, quint, J=7.5 Hz), 3.09-3.10 (1H, m), 3.14 (1H, d, J=13.0 Hz), 3.18 (1H, d, J=13.0 Hz), 5.38 (1H, dd, J=1.7, 3.7 Hz).(Step of Performing Optical Resolution from Diastereomeric Mixture)- Reference Example 2Tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate
-
 Acetonitrile (4.7 L, 8.6 v/w) was added to tert-butyl [6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate (627.0 g, net: 543.6 g, 2.05 mol, 85:15 diastereomeric mixture), and the mixture was stirred at 40° C. To the reaction solution, D-mandelic acid (116.3 g, 0.76 mmol, 0.37 eq.) was added, and the mixture was stirred at 40° C. for 1 hour and then allowed to cool slowly to 3° C. After stirring at 3° C. for 1 hour, the resulting crystal was collected by filtration. Then, the crystal was dried under reduced pressure under the condition of 40° C. to obtain tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate as a white powder (251.2 g, yield: 29.4%, 97.6% ee, 99.6% de).
Acetonitrile (4.7 L, 8.6 v/w) was added to tert-butyl [6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate (627.0 g, net: 543.6 g, 2.05 mol, 85:15 diastereomeric mixture), and the mixture was stirred at 40° C. To the reaction solution, D-mandelic acid (116.3 g, 0.76 mmol, 0.37 eq.) was added, and the mixture was stirred at 40° C. for 1 hour and then allowed to cool slowly to 3° C. After stirring at 3° C. for 1 hour, the resulting crystal was collected by filtration. Then, the crystal was dried under reduced pressure under the condition of 40° C. to obtain tert-butyl [(1R,5S,6S)-6-aminomethyl-3-ethylbicyclo[3.2.0]hept-3-en-6-yl]acetate D-mandelate as a white powder (251.2 g, yield: 29.4%, 97.6% ee, 99.6% de). -
1H-NMR (400 MHz, DMSO-d6) δ ppm: 1.04 (3H, t, J=7.6 Hz), 1.28-1.35 (1H, m), 1.39 (9H, s), 1.96-2.11 (4H, m), 2.28 (1H, d, J=15.6 Hz), 2.33 (1H, d, J=15.6 Hz), 2.36-2.40 (1H, m), 2.72 (1H, quint, J=7.6 Hz), 3.00 (1H, d, J=13.2 Hz), 3.03 (1H, d, J=13.2 Hz), 3.31 (1H, br s), 4.54 (1H, s), 5.21-5.23 (1H, m), 7.13-7.25 (3H, m), 7.35-7.37 (2H, m).
-
[α]20 D −104.4° (C=0.108, MeOH).
-
Anal. calcd for C24H35NO5: C, 69.04; H, 8.45; N, 3.35; Found C, 69.15; H, 8.46; N, 3.46.
-



PATENT
WO 2012169474
PATENT
[Step D-2]
a compound having the formula (Va) (and its enantiomers), and to carry out optical resolution by chloride with optically active organic amine, and is a process for preparing a compound having the general formula (VIa) .
[Formula 19] The solvent used in this step, MTBE, CPME, ethers such as THF; aromatic hydrocarbons such as toluene; esters such as ethyl acetate; EtOH, alcohols such as diisopropyl alcohol CH; s three nitriles such as CN; ketones such as acetone; or is a mixed solvent of these solvents and water, preferably toluene, ethyl acetate, CH 3 CN, are MTBE, More preferably, toluene, MTBE. Optically active organic amine used in this step, preferably, (1R, 2R) -trans-1- amino-2-indanol, (S) -2- phenylglycinol, (R) -1- ( p- tolyl) ethylamine, (1R, 2S) -2- amino-1,2-diphenyl ethanol, (S) -1- (2- naphthyl) ethylamine, (R) -1- (4- bromophenyl) ethylamine, (1S, 2R) – (+) – 1- amino-2-indanol is a L- phenylalaninol, etc., more preferably, (1R, 2R) -trans-1- amino-2-indanol, (S ) -2-phenylglycinol. Equivalent of the optically active organic amine to be used have the general formula (Va) compound having a relative (and its enantiomers) are 0.5-1.1 equivalents. The reaction temperature of this step is such as about 0-50 ℃, preferably, after aging the crystals at about 10-30 ℃, is obtained by filtering the compound of formula (VIa). The time required to chloride present step is not particularly limited, but is usually 4 to about 48 hours. In this step, (1) with respect to (Va) compound with (and its enantiomers), directly to a compound of formula (VIa) with the desired configuration by the action of the above-mentioned optically active amine How to get, or, with respect to (2) compounds having formula (Va) (or its enantiomer), first, quinine, (1S, 2S) -trans-1- amino-2-indanol, (R) -2- by the action of an optically active amine such as phenylglycinol, it allowed to temporarily deposit the enantiomer having the unnecessary configuration, after removing the precipitate by filtration, against followed by compound obtained from the filtrate, (1R, 2R ) -trans-1- amino-2-indanol, by the action of optically active amines such as (S) -2- phenylglycinol, to precipitate the salt of the compound of formula (VIa) with the desired configuration How to get Te, one of the methods is used.
 Known compounds having the general formula (Va) which are used in the above Step D-1 or step D-2, which can be prepared according to step A-C, as otherwise, it is disclosed in Patent Document 5 It can be prepared by method (the following scheme).
Known compounds having the general formula (Va) which are used in the above Step D-1 or step D-2, which can be prepared according to step A-C, as otherwise, it is disclosed in Patent Document 5 It can be prepared by method (the following scheme).
[Formula 20] specific production method according to the present method will be described later as a reference example.

a compound having the formula (Va) (and its enantiomers), and to carry out optical resolution by chloride with optically active organic amine, and is a process for preparing a compound having the general formula (VIa) .
[Formula 19] The solvent used in this step, MTBE, CPME, ethers such as THF; aromatic hydrocarbons such as toluene; esters such as ethyl acetate; EtOH, alcohols such as diisopropyl alcohol CH; s three nitriles such as CN; ketones such as acetone; or is a mixed solvent of these solvents and water, preferably toluene, ethyl acetate, CH 3 CN, are MTBE, More preferably, toluene, MTBE. Optically active organic amine used in this step, preferably, (1R, 2R) -trans-1- amino-2-indanol, (S) -2- phenylglycinol, (R) -1- ( p- tolyl) ethylamine, (1R, 2S) -2- amino-1,2-diphenyl ethanol, (S) -1- (2- naphthyl) ethylamine, (R) -1- (4- bromophenyl) ethylamine, (1S, 2R) – (+) – 1- amino-2-indanol is a L- phenylalaninol, etc., more preferably, (1R, 2R) -trans-1- amino-2-indanol, (S ) -2-phenylglycinol. Equivalent of the optically active organic amine to be used have the general formula (Va) compound having a relative (and its enantiomers) are 0.5-1.1 equivalents. The reaction temperature of this step is such as about 0-50 ℃, preferably, after aging the crystals at about 10-30 ℃, is obtained by filtering the compound of formula (VIa). The time required to chloride present step is not particularly limited, but is usually 4 to about 48 hours. In this step, (1) with respect to (Va) compound with (and its enantiomers), directly to a compound of formula (VIa) with the desired configuration by the action of the above-mentioned optically active amine How to get, or, with respect to (2) compounds having formula (Va) (or its enantiomer), first, quinine, (1S, 2S) -trans-1- amino-2-indanol, (R) -2- by the action of an optically active amine such as phenylglycinol, it allowed to temporarily deposit the enantiomer having the unnecessary configuration, after removing the precipitate by filtration, against followed by compound obtained from the filtrate, (1R, 2R ) -trans-1- amino-2-indanol, by the action of optically active amines such as (S) -2- phenylglycinol, to precipitate the salt of the compound of formula (VIa) with the desired configuration How to get Te, one of the methods is used.
Known compounds having the general formula (Va) which are used in the above Step D-1 or step D-2, which can be prepared according to step A-C, as otherwise, it is disclosed in Patent Document 5 It can be prepared by method (the following scheme).[Formula 20] specific production method according to the present method will be described later as a reference example.
[Step E]
Formula (V) or a compound having the general formula (VI) from (and / or its enantiomer) is a process for preparing a compound of formula (VII) (and / or its enantiomer), the general formula (V) is a compound having (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst, reduction with a solvent, or a compound having the general formula (VI) (and / or its enantiomer) solution compounds having the general formula (V) obtained by salt (and / or its enantiomer) solution, under a hydrogen atmosphere to carry out a reduction reaction in the presence of a metal catalyst, by a compound of formula (VII) This is a method of manufacturing a.
Formula 21] (1) Kaishio step formula compound with a (VI) (and / or its enantiomer) is suspended in an organic solvent, washed with an aqueous solution obtained by adding an acid, by liquid separation and the organic layer , compounds having general formula (V) (and / or its enantiomer) solution containing it can get. The solvent used in this step include aromatic hydrocarbons such as toluene, ethers such as MTBE, an ester such as ethyl acetate, and the like, preferably toluene, or is MTBE. Acid used in this step is not particularly limited, hydrochloric acid, sulfuric acid, phosphoric acid, citric acid, malonic acid can be used.

Formula (V) or a compound having the general formula (VI) from (and / or its enantiomer) is a process for preparing a compound of formula (VII) (and / or its enantiomer), the general formula (V) is a compound having (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst, reduction with a solvent, or a compound having the general formula (VI) (and / or its enantiomer) solution compounds having the general formula (V) obtained by salt (and / or its enantiomer) solution, under a hydrogen atmosphere to carry out a reduction reaction in the presence of a metal catalyst, by a compound of formula (VII) This is a method of manufacturing a.
Formula 21] (1) Kaishio step formula compound with a (VI) (and / or its enantiomer) is suspended in an organic solvent, washed with an aqueous solution obtained by adding an acid, by liquid separation and the organic layer , compounds having general formula (V) (and / or its enantiomer) solution containing it can get. The solvent used in this step include aromatic hydrocarbons such as toluene, ethers such as MTBE, an ester such as ethyl acetate, and the like, preferably toluene, or is MTBE. Acid used in this step is not particularly limited, hydrochloric acid, sulfuric acid, phosphoric acid, citric acid, malonic acid can be used.
(2) the reduction reaction step
compounds having the general formula (V) (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst was reduced in a solvent, a cyano group (or a nitro group) and an amino group It is converted into, and is a step for preparing a compound of formula (VII). This reaction is usually carried out in a neutral or basic conditions.
The solvent used in this step include aromatic hydrocarbons such as toluene, MTBE, ethers such as THF, alcohols of C1-C4, or is water, preferably toluene, MTBE, or water , and the Particularly preferred is water.
Metal catalyst used in this step, vinegar Sanskrit nickel, sponge cobalt, or palladium – is carbon, preferably, sponge nickel (eg, Kawaken Fine Chemicals Co., Ltd. of PL-9T, NDT-65, NDT- 90, NDHT-90M, NDHT-M3, and the like, or, Nikko Rika Co., Ltd. R-100, R-200, such as R-205, R-211, R-2311), or, sponge cobalt (for example, the river Research ODHT-60 manufactured by Fine Chemical Co., Ltd., OFT-55, or the like, or is a Nikko Rika Co., Ltd. R-400, R-400K, such as R-401, R-455, such as A-8B46 manufactured by Johnson Matthey) .
In this step, when carrying water as a solvent is usually added to the base. As the base used, preferably an inorganic base, particularly preferred are lithium hydroxide, sodium hydroxide, alkali metal hydroxides such as potassium hydroxide.
In this step, by the addition of aqueous ammonia, it is possible to improve the yield, it is not necessarily added aqueous ammonia.
In this step, by the addition of dimethyl polysiloxane, it is possible to suppress the generation of bubbles from the reaction liquid, it is not necessarily added dimethylpolysiloxane.
The reaction temperature in this step is about 20-60 ℃, preferably, is about 30-50 ℃.
The reaction time of this step, the raw material is not particularly limited as long as it is a time that is substantially consumed, it is usually 2 to about 12 hours.
In this step, after the completion of the reaction, the catalyst was removed by filtration, by adding an acid to the filtrate, by then crystallizing the compound of formula (VII), and filtering and washing the precipitate, pure products a you can get.
compounds having the general formula (V) (and / or its enantiomer), under a hydrogen atmosphere in the presence of a metal catalyst was reduced in a solvent, a cyano group (or a nitro group) and an amino group It is converted into, and is a step for preparing a compound of formula (VII). This reaction is usually carried out in a neutral or basic conditions.
The solvent used in this step include aromatic hydrocarbons such as toluene, MTBE, ethers such as THF, alcohols of C1-C4, or is water, preferably toluene, MTBE, or water , and the Particularly preferred is water.
Metal catalyst used in this step, vinegar Sanskrit nickel, sponge cobalt, or palladium – is carbon, preferably, sponge nickel (eg, Kawaken Fine Chemicals Co., Ltd. of PL-9T, NDT-65, NDT- 90, NDHT-90M, NDHT-M3, and the like, or, Nikko Rika Co., Ltd. R-100, R-200, such as R-205, R-211, R-2311), or, sponge cobalt (for example, the river Research ODHT-60 manufactured by Fine Chemical Co., Ltd., OFT-55, or the like, or is a Nikko Rika Co., Ltd. R-400, R-400K, such as R-401, R-455, such as A-8B46 manufactured by Johnson Matthey) .
In this step, when carrying water as a solvent is usually added to the base. As the base used, preferably an inorganic base, particularly preferred are lithium hydroxide, sodium hydroxide, alkali metal hydroxides such as potassium hydroxide.
In this step, by the addition of aqueous ammonia, it is possible to improve the yield, it is not necessarily added aqueous ammonia.
In this step, by the addition of dimethyl polysiloxane, it is possible to suppress the generation of bubbles from the reaction liquid, it is not necessarily added dimethylpolysiloxane.
The reaction temperature in this step is about 20-60 ℃, preferably, is about 30-50 ℃.
The reaction time of this step, the raw material is not particularly limited as long as it is a time that is substantially consumed, it is usually 2 to about 12 hours.
In this step, after the completion of the reaction, the catalyst was removed by filtration, by adding an acid to the filtrate, by then crystallizing the compound of formula (VII), and filtering and washing the precipitate, pure products a you can get.
[Step F]
compounds having the formula (VII) (and / or its enantiomer), to produce the presence of an organic acid and a solvent, a compound having formula (VIII) is allowed to form salts with (and / or its enantiomer) It is a method.
Chemical Formula 22] The solvent used in this step include water, anisole, aqueous acetone, water CH 3 CN, MTBE water – acetone, anisole – acetate, anisole – acetone, anisole – acetate – acetone, acetone – water -CH 3 CN single like, or it is a mixed solvent, preferably, water, anisole. The organic acid used in this step is an organic acid that is pharmacologically today preferably a benzenesulfonic acid. Equivalent of the organic acid used in this step is preferably a compound having the formula (VII) with respect to (and / or its enantiomer) is about 1.00-1.10 equivalents. This step is carried out in the range of usually about -15-50 ℃, preferably, after aging the crystals at a temperature of about -10-30 ℃, filtration, by washing, the general formula (VIII) a compound having a (and / or its enantiomers) get. The time required for chloride in this step is not particularly limited, but is usually 1 to about 24 hours.
 In the present invention, compounds having formula (IX) prepared via the process F from Step A (and / or its enantiomer) may be very produced as pure compounds. Compounds of formula (IX) which can be obtained by the present invention typically have a quality below.
In the present invention, compounds having formula (IX) prepared via the process F from Step A (and / or its enantiomer) may be very produced as pure compounds. Compounds of formula (IX) which can be obtained by the present invention typically have a quality below.
Chemical Formula 22] The solvent used in this step include water, anisole, aqueous acetone, water CH 3 CN, MTBE water – acetone, anisole – acetate, anisole – acetone, anisole – acetate – acetone, acetone – water -CH 3 CN single like, or it is a mixed solvent, preferably, water, anisole. The organic acid used in this step is an organic acid that is pharmacologically today preferably a benzenesulfonic acid. Equivalent of the organic acid used in this step is preferably a compound having the formula (VII) with respect to (and / or its enantiomer) is about 1.00-1.10 equivalents. This step is carried out in the range of usually about -15-50 ℃, preferably, after aging the crystals at a temperature of about -10-30 ℃, filtration, by washing, the general formula (VIII) a compound having a (and / or its enantiomers) get. The time required for chloride in this step is not particularly limited, but is usually 1 to about 24 hours.
In the present invention, compounds having formula (IX) prepared via the process F from Step A (and / or its enantiomer) may be very produced as pure compounds. Compounds of formula (IX) which can be obtained by the present invention typically have a quality below.The content of the diastereomer represented by the formula (X): 0.1% less than the
content of the enantiomers represented by the formula (XI): 1.0% less than
the formula (XII) and the double bond represented by the formula (XIII) The total content of regioisomers: less than 0.5%
(Note that each content is calculated from the area percentage of the free form of formula (IX) (VII) in the by test High Performance Liquid Chromatography)
[formula 23] [of 24]


content of the enantiomers represented by the formula (XI): 1.0% less than
the formula (XII) and the double bond represented by the formula (XIII) The total content of regioisomers: less than 0.5%
(Note that each content is calculated from the area percentage of the free form of formula (IX) (VII) in the by test High Performance Liquid Chromatography)
[formula 23] [of 24]
Next, the present invention is described by examples in detail, the present invention is, which however shall not be construed as limited thereto.
The internal standard substance in a magnetic resonance spectra (NMR), and using tetramethylsilane and abbreviations indicate the multiplicity, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and brs = It shows a broad singlet.
In the name of the compound, “R” and “S” indicate the absolute configuration at the asymmetric carbon. Furthermore, “RS” and “SR” indicates that the asymmetric carbon atom is racemic. In addition, “(1RS, and 5SR) -” if such a can shows the relative arrangement of the 1-position and the 5-position, as well shows only one of the diastereomers, its diastereomers are racemic We show that.
In the name of the compound, “E” and “Z” indicates the arrangement of positional isomers in the structure of the compound having a position isomerism.
The internal standard substance in a magnetic resonance spectra (NMR), and using tetramethylsilane and abbreviations indicate the multiplicity, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and brs = It shows a broad singlet.
In the name of the compound, “R” and “S” indicate the absolute configuration at the asymmetric carbon. Furthermore, “RS” and “SR” indicates that the asymmetric carbon atom is racemic. In addition, “(1RS, and 5SR) -” if such a can shows the relative arrangement of the 1-position and the 5-position, as well shows only one of the diastereomers, its diastereomers are racemic We show that.
In the name of the compound, “E” and “Z” indicates the arrangement of positional isomers in the structure of the compound having a position isomerism.
“EZ” and “ZE” indicates that it is a mixture of regioisomers. Way more notation, is in accordance with the conventions in this area of the normal.
(Example 1)
(2EZ)-3-ethoxy -2 – [(1R, 5S) -3- Echirubishikuro [3.2.0] hept-3-en-6-ylidene] -3-oxo-propanoic acid (2EZ) -3-Ethoxy-2 – [(1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] -3-Oxopropanoic acid [of 25] malonic acid mono ethyl ester (2.9 g, AlCl in THF (20 mL) solution of 22.0 mmol) 3 (3.9 g, after addition of 29.4 mmol) in -10 ° C, (1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en- 6-one (2.0 g, 14.7 mmol) was added and stirred for 25 h at -10 ° C. Under ice-cooling After stirring was added with water (10 mL) CPME and (10 mL), and the organic layer was separated and aqueous layer 1 1 25 ° C. The aqueous layer 1 was extracted with CPME (20 mL), the organic layer 2 was separated and the organic layer was combined with the organic layer one. After washing the combined organic layers with 1 N hydrochloric acid (6 mL), and concentrated under reduced pressure at an external temperature of 40 ° C, to give the title compound (4.8 g) as a crude product. 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.07 (3H, t, J = 7.6 Hz), 1.35 (1.5H, t, 7.2 Hz), 1.41 (1.5H, t, 7.2 Hz), 2.08- 2.16 (2H, m), 2.23-2.31 (1H, m), 2.67-2.75 (1H, m), 2.83-3.05 (2H, m), 3.40-3.48 (0.5H, m), 3.57-3.64 (0.5H , m), 4.27-4.41 (3H, m), 5.29 (0.5H, s), 5.50 (0.5H, s)

(Example 2) [(RS, 5SR)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] -3-oxo propanedioic acid dimethyl (racemic) Dimethyl [(RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] Propanedioate (Racemate) [of 26] THF for (3.2 mL), TiCl at 0 ° C 4 (0.175 mL, 1.60 mmol) a It was then added and stirred for 20 minutes. Subsequently (1RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (112 mg, 0.819 mmol), dimethyl malonate (113 μL, 0.989 mmol) was added and stirred for 50 min After, it was added pyridine (265 μL, 3.28 mmol). After 1 hour stirring at 0 ° C, and subjected to stirring overnight with warming to room temperature, quenched with water (6 mL), and extracted three times with toluene (6 mL). The toluene layer saturated aqueous sodium bicarbonate solution (6 mL), washed with saturated brine (6 mL), after distilling off the solvent, PTLC (hexane: ethyl acetate = 5: 1) and subjected to purification, the title compound as a colorless oil The resulting (135 mg, 65%). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.05 (3H, D, J = 7.6 Hz), 2.09 (2H, Q, J = 7.6 Hz), 2.21 (1H, dd, J = 16.8, 3.2 Hz ), 2.60-2.76 (2H, m), 2.91 (1H, quint, J = 7.2 Hz), 3.30 (1H, ddd, J = 19.1, 8.4, 3.6 Hz), 3.73 (3H, s), 3.78 (3H, . s), 4.29 (1H, M), 5.34 (1H, s) 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.2, 24.2, 32.6, 39.8, 42.7, 51.6, 51.7, 117.5, 120.9, 148.9 , 164.6, 164.9, 177.6.

(Example 7) [(1R, 5S)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] propane two acid diethyl Diethyl [(1R, 5S) -3-ethylbicyclo [3.2.0 ] hept-3-en-6-Ylidene] Propanedioate [of 31] to CPME (159 mL), 0 ° C with Ti (Oi-Pr) 4 (16.0 mL, 54.6 mmol) After addition of, TiCl 4 and stirred for 1 hour at (18.0 mL, 164 mmol) and over 8 minutes was added dropwise 0 ° C. Then diethyl malonate (25.72 g, 161 mmol), was added (1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (19.87 g, 146 mmol), 30-40 ° it was stirred for 4 hours at C. The reaction was quenched with water (100 mL), and extracted with toluene (40 mL). After the organic layer is concentrated under reduced pressure, to obtain a crude product of the title compound as a yellow oil (43.61 g).

(Example 8) [(RS, 5SR)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] propane diacid di -tert- butyl (racemic) Di-tert-butyl [( RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] Propanedioate (Racemate) [of 32] with respect to THF (30 mL), and TiCl at 0 ° C 4 and (1.6 mL, and the mixture was stirred for 30 minutes was added 14.7 mmol). Subsequently (1RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (1.00 g, 7.34 mmol), malonic acid di -tert- butyl (1.91 g, 8.81 mmol) was added After stirring for 1.5 hours, it was added pyridine (2.2 mL, 29.4 mmol). 0 ° 3.5 hours after stirring at C, and subjected to stirring overnight with warming to room temperature, quenched with water (10 mL), and extracted two times with toluene (10 mL). After washed with saturated brine (10 mL), the solvent was distilled off under reduced pressure, silica gel column chromatography (hexane: ethyl acetate = 20: 1) and subjected to purification to give the title compound (2.26 g, 92% ). 1 H NMR (CDCl 3 ) (500 MHz): delta = 1.07 (3H, t, J = 7.5 Hz), 1.47 (9H, s), 1.52 (9H, s), 2.06-2.14 (2H, M), 2.16 -2.24 (1H, m), 2.60-2.69 (2H, m), 2.90 (1H, quint, J = 7.0 Hz), 3.25 (1H, ddd, J = 18.6, 8.5, 3.5 Hz), 4.12-4.23 (1H , m), 5.36 (1H, s).

(Example 9) 5 – [(RS, 5SR)-3-Echirubishikuro [3.2.0] hept-3-en-6-ylidene] -2,2-dimethyl-1,3-dioxane -4-6- dione (racemic) 5 – [(RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] 2,2-dimethyl-1,3-dioxane-4-6-dione (Racemate) [of 33] THF for (80 mL), TiCl at 0 ° C 4 was stirred for 10 minutes was added (4.5 mL, 41 mmol). Subsequently (1RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one (2.81 g, 20.6 mmol), Meldrum’s acid (3.57 g, 24.8 mmol) was added and after stirring for 50 minutes , pyridine (6.53 g, 82.6 mmol) it was added. After 1.5 h stirring at 0 ° C, and subjected to stirring overnight with warming to room temperature, quenched with water (80 mL), and extracted three times with toluene (50 mL). The organic layers with saturated brine (50 mL), washed with 1 M HCl (10 mL), after distilling off the solvent, silica gel column chromatography (hexane: ethyl acetate = 9: 1-6: 1) to perform purification, as a white solid to give the title compound (4.51 g, 83.2%). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.05 (3H, t, J = 7.6 Hz), 1.69 (3H, s), 1.71 (3H, s), 2.11 (2H, Q, J = 7.6 Hz ), 2.20-2.35 (1H, m), 2.65-2.85 (1H, m), 2.92-3.13 (2H, m), 3.47-3.63 (1H, m), 4.45-4.59 (1H, m), 5.43 (1H , s). 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.1, 24.3, 27.59, 27.64, 34.1, 42.3, 42.8, 60.7, 104.4, 108.5, 119.4, 150.3, 160.1, 160.7.

(Example 10) [(1R, 5S, 6R)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane two acid dimethyl Dimethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate [of 34] Dimethyl [(1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en- 6-ylidene] propanedioate (517 mg, 1.66 mmol) was dissolved in MeOH (5.2 mL), was added sodium cyanide (90 mg, 1.84 mmol) at room temperature and stirred for 2 hours at room temperature. After quenching with 10% aqueous acetic acid (5 mL), and extracted three times with ethyl acetate (5 mL), the solvent was distilled off under reduced pressure to give the title compound as an oil (667 mg). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.08 (3H, t, J = 7.6 Hz), 1.80 (1H, dd, J = 12.4, 8.0 Hz), 2.01-2.22 (3H, M), 2.54 (1H, dd, J = 16.8, 7.6 Hz), 2.73 (1H, ddd, J = 12.8, 8.8, 2.8 Hz), 3.18 (1H, quint, J = 7.6 Hz), 3.67 (1H, s), 3.78 ( . 3H, s), 3.82 (3H, s), 5.16-5.28 (1H, M) 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.2, 24.4, 32.1, 37.5, 39.2, 42.5, 52.9, 53.0 , 54.6, 55.0, 118.8, 123.2, 153.9, 166.62, 166.63.

(Example 11) [(1R, 5S, 6R)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane two acid diethyl Diethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate [of 35] Diethyl obtained by the method shown in Example 7 [(1R, 5S) -3-ethylbicyclo [3.2 .0] hept-3-en-6-Ylidene] Propanedioate crude product (43.61 g, 146 mmol) was dissolved in EtOH (262 mL) and was added sodium cyanide (7.15 g, 146 mmol) at room temperature , it was stirred for 4 hours at room temperature. Acetate (8.76 g), after the reaction quenched with water (180 mL), the solvent it was concentrated to approximately 340 mL under reduced pressure. Water was added (80 mL), then extracted three times with ethyl acetate (150 mL), the solvent was distilled off under reduced pressure to give the title compound as an oil (HPLC quantitative value: 44.29 g, 96.3% (( 1R, 5S) -3-Ethylbicyclo [3.2.0] total yield from hept-3-en-6-one)). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.07 (3H, t, J = 7.6 Hz), 1.28 (3H, t, J = 7.2 Hz), 1.31 (3H, t, J = 7.2 Hz), 1.80 (1H, dd, J = 12.6, 7.6 Hz), 2.01-2.19 (3H, m), 2.53 (1H, dd, J = 16.8, 7.6 Hz), 2.72 (1H, ddd, J = 12.6, 9.2, 2.8 Hz), 3.16 (1H, quint, J = 7.6 Hz), 3.61 (1H, s), 3.67-3.82 (1H, M), 4.15-4.33 (4H, M), 5.21-5.26 (1H, M). 13 C NMR (CDCl 3 ) (100 MHz):. delta = 12.2, 14.0, 24.4, 32.2, 37.7, 39.3, 42.5, 55.0, 55.2, 62.00, 62.02, 119.0, 123.3, 153.7, 166.21, 166.23 (HPLC analysis conditions) Diethyl [(1R, 5S, 6R) -6-cyano-3-ethylbicyclo [3.2.0] hept-3-en-6-yl] propanedioate quantification method column: Cadenza CW-C18 (Imtakt, 3 μm, 4.6 mm × 150 mm), 40 ° Cdetection wavelength: UV 205 nm mobile phase: MeCN: 0.1% AcOH aqueous solution = 10: 90-80: 20 (gradient) (0-2 min: MeCN 10%, 2-17 min: MeCN 10 → 80%, 17-25 min: MeCN 80%, 25-30 min: MeCN 80 → 10%, 40 min: STOP) measurement time: 40 min flow rate: 1.0 mL / min retention time: Diethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate: 18.6 min, Diethyl [(1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3 en-6-ylidene] propanedioate: 19.7 min

(Example 12) [(1R, 5S, 6R)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane two acid diethyl Diethyl [(1R, 5S, 6R) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate [of 36] under a nitrogen atmosphere, Ti (Oi-Pr) 4 (25.1 g, 88.11 mmol) the CPME (210 In addition to mL), TiCl it over 1 hour at 10-30 ° C 4 was added dropwise (29.0 mL, 264 mmol). After stirring for 30 minutes at 25-30 ° C, was added diethyl malonate (38.8 g, 242 mmol) at 3-4 ° C, stirred for 30 minutes at 1-4 ° C, (1R, 5S) -3-Ethylbicyclo- [3.2.0] In addition hept-3-en-6-one a (30.0 g, 220 mmol) at 1-4 ° C, after which the mixture was stirred for 2.5 hours at 32-33 ° C, ice cold cold water (150 mL) was added thereto at the bottom, and the aqueous layer was removed at room temperature. After washing with the organic layer 1 N hydrochloric acid (60 mL), and concentrated under reduced pressure at an external temperature of 40-45 ° C up to 120 mL, Diethyl [(1R, 5S) -3-ethylbicyclo [3.2.0] hept- 3-en-6-ylidene] got CPME solution of propanedioate. Under a nitrogen atmosphere, after addition of EtOH (150 mL) to the above solution was added sodium cyanide (10.8 g, 220 mmol), and stirred for 4.5 h at 27-29 ° C. After cooling to 14 ℃, was added a solution prepared by diluting concentrated sulfuric acid (10.8 g) in water (60 mL), was added additional water and (150 mL). And the external temperature 35-45 ° C under reduced pressure concentrated to 240 mL, after removing the aqueous layer was added CPME (60 mL), the organic layer was washed with 20% brine (60 mL), CPME of the title compound solution was obtained (91.4%, HPLC quantitative value).

(Example 13) [(RS, 5SR, 6RS)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] propane diacid di -tert- butyl (racemic) Di tert-butyl [(RS, 5SR, 6RS) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] Propanedioate (Racemate) [of 37] Di-tert-butyl [( 1RS, 5SR) -3-ethylbicyclo [3.2.0] hept-3-en-6-ylidene] propanedioate (5.00 g, 14.9 mmol) was dissolved in DMAc (50 mL), and sodium cyanide at room temperature (586 mg , it was added 12.0 mmol), and stirred for 1 hour at room temperature. After quenching with 1 M HCl (30 mL), and extracted three times with ethyl acetate (50 mL), and the solvent was evaporated under reduced pressure. Silica gel column chromatography (hexane: ethyl acetate = 20: 1) to give to give the title compound as an oil (5.10 g, 94%). 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.06 (3H, t, J = 7.5 Hz), 1.46 (9H, s), 1.50 (9H, s), 1.78 (1H, dd, J = 12.3, 8.0 Hz), 2.00-2.18 (3H, m), 2.51 (1H, dd, J = 17.0, 7.5 Hz), 2.68 (1H, ddd, J = 12.6, 8.5, 3.0 Hz), 3.13 (1H, quint, J = 7.5 Hz), 3.40 (1H, s), 3.65-3.73 (1H, m), 5.24 (1H, s).

(Example 14) (RS, 5SR, 6RS)-6-(2,2-dimethyl-4,6-dioxo-1,3-dioxane-5-yl) -3-Echirubishikuro [3.2.0] hept – 3-en-6-carbonitrile (racemic) (RS, 5SR, 6RS)-6-(2,2-Dimethyl-4, 6-Dioxo-1,3-Dioxan-5-YL) -3-Ethylbicyclo [ 3.2.0] hept-3-ene-6-carbonitrile (Racemate) [of 38] 5 – [(RS, 5SR) -3-Ethylbicyclo [3.2.0] hept-3-en-6-Ylidene] -2, 2-dimethyl-1,3-dioxane-4,6-dione (100.8 mg, 0.384 mmol) was dissolved in EtOH (1.0 mL) and was added sodium cyanide (22.0 mg, 0.449 mmol) at room temperature, room temperature in it was stirred for 3 hours. After quenching with phosphate buffer (pH 7) (5 mL), and extracted three times with ethyl acetate (5 mL), the solvent was distilled off under reduced pressure, a white solid to give the title compound (23.6 mg, 21.2 %). 1 H NMR (CD 3 OD) (400 MHz): delta = 1.03 (3H, t, J = 7.6 Hz), 1.61 (3H, s), 1.92-2.25 (4H, M), 2.45 (1H, dd, J = 16.8, 7.2 Hz), 2.66-2.80 (1H, m), 3.00 (1H, quint, J = 7.6 Hz), 3.72-3.87 (1H, m), 4.85 (1H, s), 5.23-5.33 (1H, . M) 13 C NMR (CD 3 OD) (100 MHz): delta = 12.66, 12.69, 25.3, 34.1, 38.8, 39.4, 43.3, 57.0, 75.8, 102.9, 123.67, 123.70, 127.9, 150.5, 167.9.

(Example 15) [(RS, 5SR, 6SR)-3-ethyl-6- (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] ethyl acetate (racemic) Ethyl [(RS, 5SR, 6SR) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] acetate (Racemate) [of 39] Diethyl [(RS, 5SR) -3-Ethylbicyclo [ 3.2.0] hept-3-en-6-ylidene] propanedioate (256.0 mg, 0.920 mmol) was dissolved in toluene (2.5 mL), was added DBU (152 mL), nitromethane (55 mL), at room temperature for 17 time it was stirred. After quenching with 1 M HCl (5 mL), and extracted three times with ethyl acetate (5 mL), and the resulting ethyl acetate solution was washed with saturated brine (5 mL). The solvent was evaporated under reduced pressure, as a pale yellow oily substance Diethyl [(1RS, 5SR, 6SR) -3-ethyl-6- (nitromethyl) bicyclo- [3.2.0] hept-3-en-6-yl] propanedioate was obtained (336.9 mg). The resulting Diethyl [(RS, 5SR, 6SR) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] – Propanedioate a (336.9 mg) DMSO and (3.4 mL) It was dissolved in water (50 μL, 2.78 mmol), sodium chloride (64.8 mg, 1.11 mmol) was added, followed by 10 hours heated and stirred at 140 ° C. After cooling to room temperature, the reaction was quenched with 1 M HCl (5 mL), was extracted three times with ethyl acetate (5 mL), and the resulting ethyl acetate solution was washed with saturated brine (5 mL). The solvent was evaporated under reduced pressure to give the title compound as a brown oily substance (261.6 mg, 2 process overall yield 72.4%). Diethyl [(RS, 5SR, 6SR) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] Propanedioate 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.08 (3H, t, J = 7.6 Hz), 1.17-1.35 (6H, m), 1.73 (1H, dd, J = 13.2, 7.6 Hz), 2.05 (1H, d, J = 16.4 Hz), 2.05-2.22 (2H, m), 2.42-2.58 (2H, m), 2.75 (1H, quint, J = 7.6 Hz), 3.46 (1H, brs), 3.79 (1H, s), 4.09-4.27 (4H, m), 4.96 (2H, s), 5.27 (1H, s). 13 C NMR (CDCl 3 ) (100 MHz): delta = 12.3, 13.97, 14.04, 24.4, 31.6, 36.1, 42.5, 45.6, 53.6, 55.5, 61.49, 61.53, 80.1, 120.7, 152.0, 167.7, 167.8. Ethyl [(RS, 5SR, 6SR) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] acetate 1 H NMR (CDCl 3 ) (400 MHz): delta = 1.07 (3H, t, J = 7.6 Hz), 1.25 (3H, t, J = 7.6 Hz), 1.52 (1H, dd, J = 12.6, 7.2 Hz), 2.04 (1H, d, J = 16.4 Hz), 2.05-2.19 (2H, m), 2.23-2.35 (1H, m), 2.50 (1H, dd, J = 15.8, 7.6 Hz), 2.62 (2H, s) , 2.86 (1H, quint, J = 7.6 Hz), 3.21 (1H, brs), 4.12 (4H, q, J = 7.6 Hz), 4.76 (2H, d, J = 11.6 Hz), 4.83 (2H, d, J = 11.6 Hz), 5.24 (1H, s).

(Example 16) [(RS, 5SR, 6RS)-3-ethyl-6- (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] propane diacid di -tert- butyl (racemic ) Di-tert-butyl [(RS, 5SR, 6RS) -3-ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] Propanedioate (Racemate) [of 40] Di- tert-butyl [(1RS, 5SR) -3-ethylbicyclo [3.2.0] hept-3-en-6-ylidene] propanedioate a (2.55 g) was dissolved in toluene (26 mL), DBU (1.45 mL), nitromethane (1.05 mL) was added and stirred for 49 hours at room temperature. After quenching with 1 M HCl (50 mL), and extracted three times with ethyl acetate (50 mL), and the resulting ethyl acetate solution was washed with saturated brine (50 mL). The solvent was distilled off under reduced pressure to give the title compound as a pale yellow oil (2.36 g, 78% yield). 1 H NMR (CDCl 3 ) (500 MHz): delta = 1.09 (t, 3H, J = 7.4 Hz), 1,45 (s, 9H), 1.49 (s, 9H), 1.71 (dd, 1H, J = 12.9, 7.4 Hz), 2.03 (d, 1H, 16.7 Hz), 2.09-2.19 (m, 2H), 2.47 (dd, 2H, J = 16.7, 7.9 Hz), 2.59 (ddd, 1H, J = 11.7, 8.9 , 2.7 Hz), 2.67 (quint, 1H, J = 7.4 Hz), 3.52 (brs, 1H), 3.64 (s, 1H), 4.88 (d, 1H, J = 10.9 Hz), 4.95 (d, 1H, J = 10.9 Hz), 5.28 (m, 1H).

(Example 17) [(RS, 5SR, 6SR)-3-ethyl-6- (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] optical resolution of acetic acid [(1RS, 5SR, 6SR ) -3-Ethyl-6- (nitromethyl) bicyclo [3.2.0] optical resolution of hept-3-en-6-YL] acetic acid [of 41] [(RS, 5SR, 6SR) -3-Ethyl-6 – (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] acetic acid (0.2 g, 0.84 mmol) and CH 3 CN (3.0 mL) to dissolve the table of the optically active organic amine of the following (0.42 mmol) was at room temperature stirred with, precipitated filtered crystals selectivity and dried to determine yield. The results I shown in the table below.[Table 1] * (1S, 5R, 6R) – the body is the main product ** (1R, 5S, 6S) – the body is the main product

(HPLC optical analysis condition)
Column: CHIRALPAK AD-RH 4.6 × 250 mm
mobile phase: 10 mM pH 2.0 phosphate buffer / MeCN = 25/75 (isocratic)
flow rate: 1.0 mL / min
Column temperature: 40 ° C
Detection wavelength: UV 210 nm
analysis time: 80 minutes
retention time: (1S, 5R, 6R) – Body: 35.2 min, (1R, 5S, 6S) – Body: 42.1 min
Column: CHIRALPAK AD-RH 4.6 × 250 mm
mobile phase: 10 mM pH 2.0 phosphate buffer / MeCN = 25/75 (isocratic)
flow rate: 1.0 mL / min
Column temperature: 40 ° C
Detection wavelength: UV 210 nm
analysis time: 80 minutes
retention time: (1S, 5R, 6R) – Body: 35.2 min, (1R, 5S, 6S) – Body: 42.1 min
(Example 18) [(1R, 5S, 6S)-3-ethyl-6- (nitromethyl) bicyclo [3.2.0] hept-3-en-6-yl] acetic acid [(1R, 5S, 6S) -3 -Ethyl-6 (Nitromethyl) bicyclo [3.2.0] hept-3-en-6-YL] acetic acid [of 42] quinine (5.97 g, 18.4 mmol) was dissolved in acetone (300 mL), [( RS, 5SR, 6SR) -3-Ethyl-6 (Nitromethyl) -bicyclo [3.2.0] hept-3-en-6-YL] acetic acid (10.0 g, I was added 33.4 mmol). After stirring 20 hours at room temperature, it was carried out 5 hours of stirring it was cooled to 0 ° C. After filtering off the solid, washed with cold acetone, the combined filtrate and washing was concentrated under reduced pressure, further CH 3 CN were added and again concentrated to the concentration residue (6.4 g, ee 65.2%) was obtained. The resulting residue (6.4 g, ee 65.2%) and CH 3 was dissolved in CN (43 mL), (S) – it was added phenylglycinol (1.37 g, 1 eq minute) – (+). After stirring for 20 hours at room temperature and stirred for 5 hours and cooled to 0 ° C. The precipitated crystals were collected by filtration, and added to dilute hydrochloric acid and ethyl acetate was dissolved by liquid separation, and dried under reduced pressure after the organic layer was concentrated to give the title compound (1.39 g, 14%, ee 92.0%). 1 H NMR (400 MHz, CDCl 3 ): delta = 1.09 (t, 3H, J = 7.6 Hz), 1.47-1.57 (M 2H), 2.06-2.17 (M, 3H), 2.27-2.33 (M, 1H) , 2.49-2.55 (m, 1H), 2.66 (s, 2H),, 2.88 (quint, 1H, J = 7.6 Hz), 3.17 (bs, 1H), 4.78 (d, 1H, J = 11.5 Hz), 4.86 (d, 1H, J = 11.5Hz), 5.27-5.28 (m, 1H)

(Example 28) [(1R, 5S, 6S)-6-cyano-3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid benzyl amine salt Benzylammonium [(1R, 5S, 6S) -6-cyano-3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetate [of 52] Diethyl obtained by the method of Example 12 [(1RS, 5SR, 6RS) -6-cyano -3-Ethylbicyclo [3.2.0] Hept- 3-en-6-YL] After the addition of EtOH (390 mL) to CPME solution of propanedioate, heating under reflux, 8 N aqueous solution of potassium hydroxide (6.9 mL, 55.07 mmol ) after adding a total of 5 times every 1 hour, refluxed for 5 hours and returned to room temperature. The addition of water (60 mL) and 8N aqueous potassium hydroxide (24 mL) to the above EtOH solution, and after stirring for 2 h at 26-27 ° C, under reduced pressure at an external temperature of 40-45 ° C until 150 mL It was concentrated. To remove the organic layer by water (180 mL) and toluene (90 mL) was added for liquid separation. The resulting aqueous solution Toluene (150 mL) added, cooled to, was added concentrated hydrochloric acid 42.5 mL at 2-9 ° C, the pH was adjusted to 1.4. By separation to remove the aqueous layer was added toluene (300 mL) benzylamine (23.6 g, 220.28 mmol) and. After stirring for 30 minutes at 44-46 ° C make the inoculation, and concentrated under reduced pressure until 300 mL at 44-46 ° C. After stirring overnight at 22-23 ° C, and crystals were filtered off. And vacuum dried at 40 ° C, was obtained as a white crystalline title compound 54.4 g (79.2% from (1R, 5S) -3-Ethylbicyclo [3.2.0] hept-3-en-6-one) a.

(Example 33) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid [(1R, 5S, 6S) – 6 (aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid [of 57] Benzylammonium [(1R, 5S, 6S) -6-cyano-3-Ethylbicyclo [3.2. 0] hept-3-en-6-yl] acetate (40.0 g) in toluene (200 mL), was added 2 mol / L hydrochloric acid (100 mL) at room temperature and dissolved. And allowed to stand the solution to drain the aqueous layer to obtain an organic layer. To the stirred addition of 10% aqueous sodium chloride solution (about 100 mL), and the aqueous layer was removed after standing. The solution of water (100 mL) was added to, was adjusted to 10.0 to pH added 8 mol / L aqueous potassium hydroxide solution (about 15.7 mL), the organic layer was removed to standing. The solution to the sponge cobalt (10 g), 28% aqueous ammonia (13 mL), 2% dimethylpolysiloxane / toluene solution (2 mL) was added and warmed to 40 ° C in a hydrogen gas pressure (0.45 MPa) It was stirred for 8 hours.After cooling to room temperature, filtering the reaction mixture to remove the sponge cobalt. The sponge cobalt on the filter it was washed with water (80 mL). The resulting solution was stirred for 0.5 hours added the activated carbon (4 g), to remove the charcoal by filtration. The activated carbon on the filter it was washed with water (60 mL). The solution I was adjusted to about pH 6.0 with concentrated hydrochloric acid (about 32.7g) a. Then, after stirring for 0.5 hours was added potassium chloride (55.0 g), and cooled to 0 ° C. The resulting was filtered and crystals were washed with 20% brine cooled to about 0 ° C (80 mL), and dried overnight in vacuum at 50 ° C to give the title compound as white crystals (26.9 g, content 88.3 %, 88.7% content in terms of yield).

(Example 34) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid [(1R, 5S, 6S) – 6 (aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid [of 58] (R) -Phenylethanaminium [(1R, 5S, 6S) -6-cyano-3 ethylbicyclo [3.2.0] hept-3-en-6-yl] acetate (35.9g, 99.2 mmol, 95.7% de, ee 99.2%) in toluene (120 mL) and 1 mol / L hydrochloric acid (150 mL) was added , it was stirred. After removing the aqueous layer, the organic layer was washed twice with water (120 mL), and concentrated. The obtained residue in MTBE to (150 mL) and sponge nickel (10.1 g) was added, under hydrogen pressure (approximately 4 atm) and stirred for 3 hours at room temperature. The reaction of 2 mol / L aqueous potassium hydroxide solution (72 mL) was added, After stirring for 30 minutes, a sponge nickel was filtered off. It was washed with a filtration sponge nickel 2 mol / L potassium hydroxide solution (12 mL). After combining the filtrate and washings, the organic layer was removed to obtain an aqueous layer. The organic layer was re-extracted with 2M aqueous potassium hydroxide solution. The matched aqueous layer was cooled, after adjusting the pH adding concentrated hydrochloric acid (about 12 mL) to 7.5, and the mixture was stirred at 0 ° C for about 3 hours. Filtered the precipitated crystals were washed with ice-cold water (24 mL), and dried under reduced pressure at 50 ° C, to give the title compound (18.3g, 88%, 99.8% de) and.

(Example 35) [(1R, 5S, 6S)-6-(aminomethyl) -3-Echirubishikuro [3.2.0] hept-3-en-6-yl] acetic acid one benzenesulfonate [(1R, 5S, 6S)-6-(aminomethyl) -3-Ethylbicyclo [3.2.0] hept-3-en-6-YL] acetic acid Monobenzenesulfonate [of 59] MTBE (83 mL), acetone (4.0 mL), water ( with respect to a mixture of 0.98 mL), at 0 ° C [(1R, 5S, 6S) -6- (Aminomethyl) -3-ethylbicyclo [3.2.0] hept-3-en-6-yl] acetic acid ( 4.07 g, 19.5 mmol) was added and stirred to form a slurry solution. This BsOH (3.08 g, 19.5 mmol) it was added acetone (10.1 mL) solution of. 0 ° After stirring for 1 hour at C, and stirred for 2 hours and allowed to warm to room temperature. Over 1 hour and gradually cooled to -10 ° C, and stirred for 2.5 hours. The resulting was filtered crystals, after washing with acetone and cooled to 0 ° C (12 mL), and by vacuum-dried at 40 ° C, as white crystals of the title compound was obtained (6.44 g, 90.1% ). Various spectrum data of the obtained title compound was almost (extent the structure can be identified) coincides with (described in Patent Documents 5 and 6) the known information. (Purity measurement method -1) column: Cadenza CW-C18 (Imtakt, 3 μm, 4.6 mm × 150 mm), 40 ° C detection wavelength: UV 205 nm mobile phase: MeCN: 5 mM ammonium hydrogen carbonate aqueous solution = ten ninety -80: 20 (gradient) (0-12 min: MeCN 10%, 12-27 min: MeCN 10 → 80%, 27-45 min: MeCN 80%, 45-50 min: MeCN 80 → 10%, 50- 60 min: MeCN 10%, 60 min: STOP) measurement time: 60 min flow rate: 1.0 mL / min infusion sample concentration: 5mg / mL sample injection volume: 2μL retention time: the title compound (as free form): 12.5 min diastereoisomers Marr (Compound X): 13.5 min double bond position isomer (compound XII or XIII): 9.4 min, 9.6 min, 11.4 min

| Patent | Submitted | Granted |
|---|---|---|
| Bicyclic [gamma]-amino acid derivative [US7947738] | 2010-09-30 | 2011-05-24 |
| Optical Resolution Methods for Bicyclic Compounds Using Enzymes [US2015038738] | 2014-10-10 | 2015-02-05 |
| WO2015005298A1 * | Jul 8, 2014 | Jan 15, 2015 | Daiichi Sankyo Company,Limited | METHOD FOR PRODUCING OPTICALLY ACTIVE BICYCLIC γ-AMINO ACID DERIVATIVE |
CONSTRUCTION
References
- Vinik A, Rosenstock J, Sharma U, Feins K, Hsu C, Merante D, et al. Efficacy and safety of mirogabalin (DS-5565) for the treatment of diabetic peripheral neuropathic pain: a randomized, double-blind, placebo- and active comparator-controlled, adaptive proof-of-concept phase 2 study. Diabetes Care. 2014 Dec;37(12):3253-61. doi: 10.2337/dc14-1044. PMID 25231896
- Vinik A, Sharma U, Feins K, Hsu C, Merante D. DS-5565 for the Treatment Of Diabetic Peripheral Neuropathic Pain: Randomized, Double-Blind, Placebo- And Active Comparator-Controlled Phase II Study (S20.004) Neurology April 8, 2014; 82(10): Supplement S20.004
Tokyo, Japan – (February 4, 2015) – Daiichi Sankyo Company, Limited (hereafter, Daiichi Sankyo) today announced enrollment of the first patients in large-scale, multi-national clinical programs evaluating the safety and efficacy of investigational mirogabalin (DS-5565), the first preferentially selective alpha-2 delta ligand. The phase 3 clinical program across Asia includes the REDUCER (An Asian, phase 3, multicenter, RandomizEd, Double-blind, placebo-controlled 14-week stUdy of DS-5565 in patients with diabetiC pEripheral neuRopathic pain followed by a 52-week open-label extension) study and the NEUCOURSE (An AsiaN, phasE 3, mUltiCenter, randomized, dOUble-blind, placebo-contRolled 14-week study of DS-5565 in patientS with postherpetic neuralgia followed by a 52-week open-label Extension) study which will evaluate investigational mirogabalin for the treatment of diabetic peripheral neuropathic pain (DPNP) and postherpetic neuralgia (PHN), respectively. The phase 3 global ALDAY (A Randomized, Double-Blind, Placebo- and Active-Controlled Study of DS-5565 in Patients with Pain Associated with Fibromyalgia) clinical program is ongoing and will evaluate mirogabalin for the treatment of pain associated with fibromyalgia in three identical studies.
“Pain associated with the neurologic conditions of diabetic peripheral neuropathic pain, postherpetic neuralgia and fibromyalgia can be debilitating,” said Lesley Arnold, MD, Professor of Psychiatry and Behavioral Neuroscience and Director of the Women’s Health Research Program, University of Cincinnati and lead investigator of the ALDAY program. “New treatment options are needed to help people living with these neurologic conditions relieve and manage their chronic pain and hopefully, improve their function and quality of life.”
“We are pleased that our global clinical development program evaluating the efficacy and safety of mirogabalin continues to move forward and has progressed into phase 3,” said Mahmoud Ghazzi, MD, PhD, Executive Vice President and Global Head of Development for Daiichi Sankyo. “Daiichi Sankyo is committed to identifying and studying new medicines that could help improve the management of chronic pain for people with diabetic peripheral neuropathy, postherpetic neuralgia and pain associated with fibromyalgia.”
About the REDUCER and NEUCOURSE Phase 3 Clinical Studies
The REDUCER study will last 14 weeks and is being conducted at approximately 200 centers in Japan, Taiwan and Korea. The NEUCOURSE study will also last 14 weeks and is being conducted at approximately 200 centers in Japan, Taiwan, Korea, Singapore, Malaysia and Thailand. The studies will include about 750 patients each with either diabetic peripheral neuropathic pain or postherpetic neuralgia, respectively. The objectives of the double-blind studies are to evaluate safety and efficacy of mirogabalin by comparing change in the average daily pain score (ADPS) from baseline to Week 14 in patients receiving a total daily dose of either 15 mg, 20 mg or 30 mg of mirogabalin versus placebo. Both studies will be followed by one-year open-label extension studies to assess long-term safety and efficacy of mirogabalin. For more information on the REDUCER study in patients with diabetic peripheral neuropathic pain, please visit
https://www.clinicaltrials.gov/ct2/show/NCT02318706?term=Mirogabalin&rank=3.
For more information on the NEUCOURSE study in patients with postherpetic neuralgia, please visithttps://www.clinicaltrials.gov/ct2/show/NCT02318719?term=Mirogabalin&rank=1.
About the ALDAY Phase 3 Clinical Program
The ALDAY program is a large clinical phase 3 program evaluating mirogabalin for the treatment of pain associated with fibromyalgia, and includes three, randomized, double-blind, placebo- and active-controlled studies, and an open label safety study that will be carried out over the next three years. Approximately 4,000 patients with pain associated with fibromyalgia will be enrolled at approximately 800 clinical centers at more than 40 countries worldwide. The primary objective of the studies in the ALDAY program is to compare change in weekly ADPS from baseline to Week 13 in patients receiving a total daily dose of either 15 mg or 30 mg of mirogabalin versus placebo. Weekly ADPS is based on daily pain scores reported by the patient that best describes his or her worst pain over the previous 24 hours. The primary objective of the phase 3 open-label extension study is to assess the long-term safety of a total daily dose of mirogabalin 15 mg or mirogabalin 30 mg in patients with pain associated with fibromyalgia. For more information on the studies in the ALDAY program, please visit
https://clinicaltrials.gov/ct2/show/NCT02187471?term=DS5565&rank=1
https://clinicaltrials.gov/ct2/show/NCT02187471?term=ds-5565&rank=2
https://clinicaltrials.gov/ct2/show/NCT02146430?term=ds-5565&rank=3
For more information on the open-label extension study, please visithttps://clinicaltrials.gov/ct2/show/NCT02234583?term=ds-5565&rank=4
For patient recruitment or additional clinical study information, please visit http://www.aldaystudy.com/.
About Diabetic Peripheral Neuropathic Pain
Diabetic peripheral neuropathy is a disorder that causes nerve damage to the extremities and is one of the most common long-term complications of diabetes.1 Symptoms include sharp pains or increased sensitivity, numbness, loss of balance and coordination, tingling, burning, or prickling sensations, which typically worsen at night.1 Up to 50 percent of people with diabetes have peripheral neuropathy2 and it is estimated that between 11 and 26 percent of people with diabetes experience diabetic peripheral neuropathic pain (DPNP).3-6 However, DPNP is often undertreated and underreported.2
About Postherpetic Neuralgia
Postherpetic neuralgia is pain that occurs after recovering from shingles, an infection that is caused by the herpes zoster (chickenpox) virus. Pain from postherpetic neuralgia can range in severity, and is typically described as burning, sharp, or stabbing.7 Other symptoms include sensitivity to touch, itching, numbness, and in rare cases, muscle weakness or paralysis can occur.7 The risk of developing postherpetic neuralgia increases with age and it mainly affects people older than 60.7 Studies have shown that only half of all patients affected with the condition will be relieved from pain within a year.8 Most people will require more than one treatment to help ease the pain.7
About Fibromyalgia
Fibromyalgia is a chronic disorder that causes widespread muscle pain, generalized tender points and fatigue.9 Other common symptoms include sleep disturbances, morning stiffness, memory and thinking problems (sometimes called fibro fog), tingling in the hands and feet and headaches.9 Fibromyalgia is often misdiagnosed and suboptimally treated.10-17 The overall estimated prevalence of fibromyalgia is approximately two to three percent in the general population, with a higher prevalence in women.18-22 Pain that occurs with fibromyalgia has a substantial impact on the patient, and can be associated with societal and economic burdens.23-29
About Mirogabalin
Mirogabalin is an investigational drug that is currently being studied for the treatment of DPNP, PHN and pain associated with fibromyalgia. Mirogabalin is preferentially selective in regards to how it binds to α2δ-1 subunit, a protein that may help to regulate how the brain processes pain signals. It has a unique binding profile and long duration of action.30*,31
About Daiichi Sankyo
Daiichi Sankyo Group is dedicated to the creation and supply of innovative pharmaceutical products to address the diversified, unmet medical needs of patients in both mature and emerging markets. While maintaining its portfolio of marketed pharmaceuticals for hypertension, dyslipidemia and bacterial infections used by patients around the world, the Group has also launched treatments for thrombotic disorders and is building new product franchises. Furthermore, Daiichi Sankyo research and development is focused on bringing forth novel therapies in oncology and cardiovascular-metabolic diseases, including biologics. The Daiichi Sankyo Group has created a “Hybrid Business Model,” to respond to market and customer diversity and optimize growth opportunities across the value chain. For more information, please visit: www.daiichisankyo.com.
| trial(s) |
|
|---|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(1R,5S,6S)-6-(aminomethyl)-3-ethyl-bicyclo(3.2.0)hept-3-ene-6-acetic acid
|
|
| Identifiers | |
| CAS Registry Number | 1138245-21-2 |
| PubChem | CID: 49802951 |
| ChemSpider | 32701007 |
| Chemical data | |
| Formula | C12H19NO2 |
| Molecular mass | 209.285 g/mol |
/////////
1138245-13-2, CCC1=C[C@@H]2[C@H](C1)C[C@@]2(CC(=O)O)CN
CCC1=CC2C(C1)CC2(CC(=O)O)CN
smiles besylate……CCC1=C[C@@H]2[C@H](C1)C[C@@]2(CC(=O)O)CN.c1ccc(cc1)S(=O)(=O)O
see
ATAGABALIN ALS0
SEE ……..SERIES………http://apisynthesisint.blogspot.in/p/gabalin-series.html
крисаборол , كريسابورول , Crisaborole, AN 2728
Crisaborole
Treatment for Inflammatory Skin Diseases, including Atopic Dermatitis and Psoriasis
C14H10BNO3, Average mass251.045 Da
4-[(1-Hydroxy-1,3-dihydro-2,1-benzoxaborol-5-yl)oxy]benzonitrile ,
4-((1-Hydroxy-1,3-dihydrobenzo(c)(1,2)oxaborol-6-yl)oxy)benzonitrile
CAS 906673-24-3, AN-2728
Benzonitrile, 4-[(1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy]-
1,3-Dihydro-1-hydroxy-5-(4-cyanophenoxy)-2,1-benzoxaborole
5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole
crisaborol, crisaborole, Crisaborole, crisaborolum
UNII-Q2R47HGR7P
крисаборол
كريسابورول
In phase 3 for treatment of mild to moderate atopic dermatitis……Anacor Pharmaceuticals, Inc.
Psoriasis is a chronic skin disorder caused by inflammatory cell infiltration into the dermis and epidermis, and is accompanied by keratinocyte hyperproliferation. Once triggered, a strong T-cell response is mounted, and a cascade of cytokine and chemokine production is induced.
Down-regulation of certain cytokines and chemokines is considered to be a good approach to treatment, and indeed, the biologics targeting TNF-α demonstrate the effectiveness of this approach.However, biologics have intrinsic challenges, such as limited administration route, side effects, quality control and production cost.
Small molecule approaches to treat psoriasis include systemic or topical steroids, cyclosporine, psoralen plus UVA (PUVA), retinoids, methotrexete, and vitamin D3 analogs.Atopic dermatitis is an allergic skin disorder, which is typically treated with topical steroids, antihistamines, and calcineurin inhibitors.
However, there is still a need for new treatment with improved safety profile. Recently phosphodiesterase 4 (PDE4) inhibitors have been in development for such skin diseases. CC-10004 is in development as an oral treatment for psoriasis and atopic dermatitis. AWD-12-281 was, until recently, in development for the topical treatment of atopic dermatitis. In addition, roflumilast is under Phase 1 development for both diseases.

Figure 1.
PDE4 inhibitors aiming at skin inflammatory diseases.

Anacor’s lead product candidate is crisaborole, an investigational non-steroidal topical PDE-4 inhibitor in development for the potential treatment of mild-to-moderate atopic dermatitis and psoriasis
crisaborole is an investigational topical antiinflammatory drug in phase III clinical development by Anacor Pharmaceuticals for the treatment of mild to moderate atopic dermatitis and in phase II clinical trials in mild to moderate psoriasis
A novel boron-containing small molecule, Crisaborole inhibits the release of pro-inflammatory cytokines including TNF-alpha, IL-12, and IL-23, known mediators of the inflammation associated with psoriasis.
Synthesis
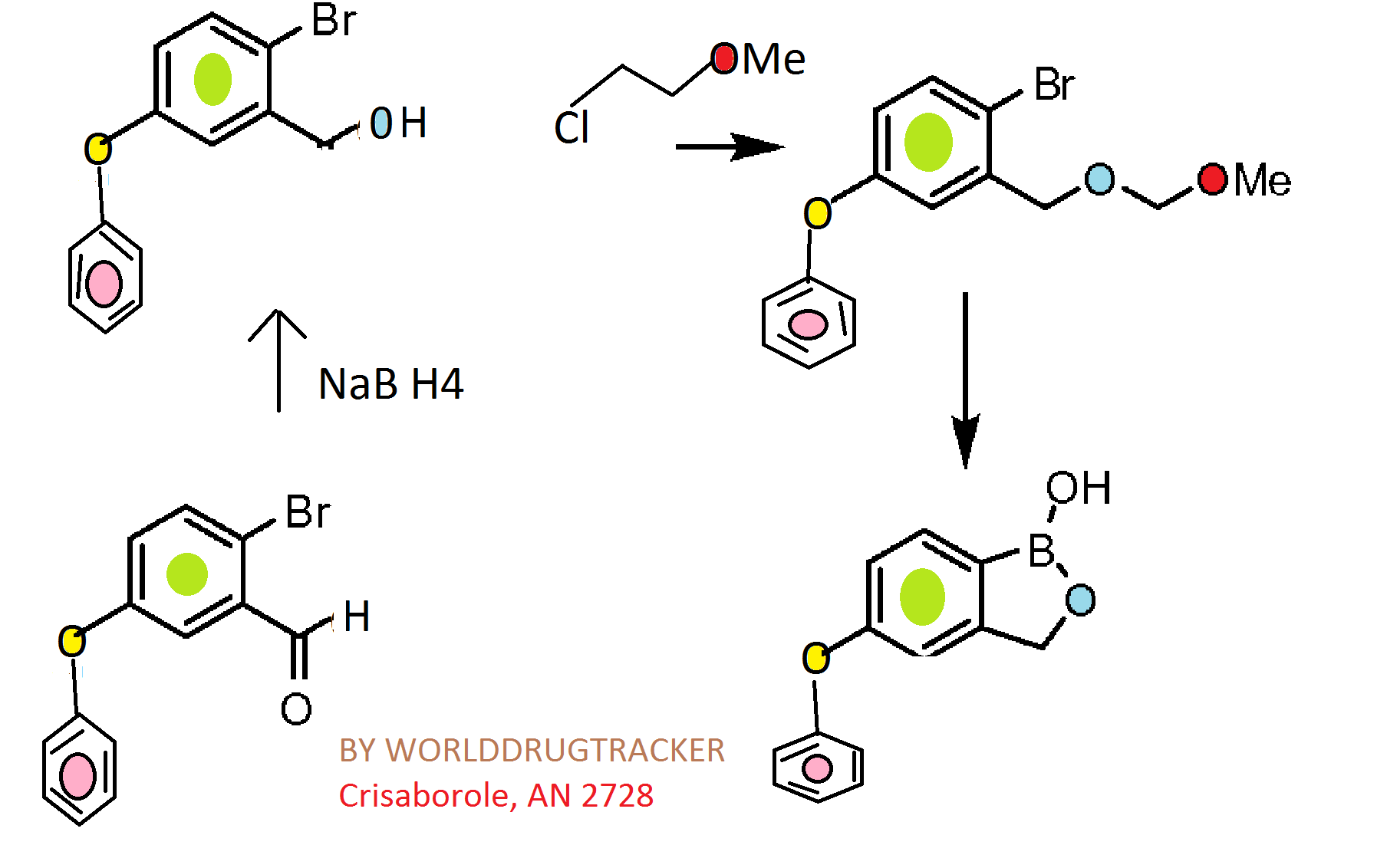
CKICK ON IMAGE FOR CLEAR VIEW
| Originator | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Therapeutic Claim | |||||||||||||||||||||
| Class | |||||||||||||||||||||
| Mechanism of action | |||||||||||||||||||||
| WHO ATC code(s) | |||||||||||||||||||||
| EPhMRA code(s) | |||||||||||||||||||||
| Clinical trial(s) |
|
PAPER
Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis
Bioorg Med Chem Lett 2009, 19(8): 2129
http://www.sciencedirect.com/science/article/pii/S0960894X09002996
- Anacor Pharmaceuticals, Inc., 1020 E. Meadow Circle, Palo Alto, CA 94303, USA
A series of phenoxy benzoxaboroles were synthesized and screened for their inhibitory activity against PDE4 and cytokine release. 5-(4-Cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2728) showed potent activity both in vitro and in vivo. This compound is now in clinical development for the topical treatment of psoriasis and being pursued for the topical treatment of atopic dermatitis


Scheme 1.
Reagents and conditions: (a) ethylene glycol, p-TsOH, toluene, reflux, 6 h (quant.); (b) K2CO3, DMF, 100 °C, overnight (82–96%); (c) 3 M HCl, THF, reflux, 2 h (80–100%); (d) NaBH4, MeOH, rt, 1 h (quant.); (e) 3,4-dihydro-2H-pyran, camphorsulfonic acid, CH2Cl2, rt, 2 h (quant.); (f) (i-PrO)3B, n-BuLi, THF, −78 °C to rt, 3 h; (g) 6 M HCl, THF, rt, 3 h (37–44%); (h) 6 M NaOH, MeOH, 1,4-dioxane, reflux, 6 days (79%); (i) diethylamine (for 5f) or morpholine (for 5g), EDCI, HOBt, DMAP, DMF, rt, overnight (41–70%).
PATENT
http://www.google.co.in/patents/WO2006089067A2?cl=en
4.2. q 5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole (C17) [0264] 1H-NMR (300 MHz,
δ ppm 4.95 (s, 2H), 7.08 (dd, J= 7.9, 2.1 Hz, IH), 7.14 (d, J= 8.8 Hz, IH), 7.15 (d, J= 2.1 Hz, IH), 7.78 (d, J= 7.9 Hz, IH), 7.85 (d, J= 9.1 Hz, 2H), 9.22 (s, IH).
PATENT
EXAMPLE 15
http://www.google.com/patents/WO2007095638A2?cl=en
4-(4-Cvanophenoxy)phenylboronic acid (C97)

(a) (4-cyanophenyl) (4-bromophenyl) ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 1000C for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with IN NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4- bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-de): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p.l94-198°C. MS: m/z = 239 (M+), 240 (M+ 1) (ESI+) and m/z = 238 (M-I) (ESI-). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6 + D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.
FURTHER METHOD

2-Bromo-5-(4-cvanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J= 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
PATENT
http://www.google.im/patents/EP1976536A2?cl=en
2.2.a 2-Bromo-5-(4-cyanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J- 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
2.2.b 2-Bromo-4-(4-cyanophenoxγ)benzyl Alcohol
1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.58 (d, IH), 7.39 (d, IH), 7.18 (dd, IH), 7.11- (d, 2H), 5.48 (t, IH) and 4.50 (d, 2H) ppm.
2.2.c 5- (4-Cyanophenoxy) -1 -Indanol
M.p.50-53°C. MS (ESI+): m/z = 252 (M+l). HPLC: 99.7% purity at 254 nm and 99.0% at 220 nm. 1H NMR (300 MHz, DMSOd6): δ 7.80 (d, 2H), 7.37 (d, IH), 7.04 (d, 2H), 6.98-6.93 (m, 2H), 5.27 (d, IH)5 5.03 (q, IH), 2.95-2.85 (m, IH), 2.75-2.64 (m, IH), 2.39-2.29 (m, IH) and 1.85-1.74 (m, IH) ppm.
2.2. d 2-Bromo-5-(tert-butyldimethylsiloxy)benzyl Alcohol [0429] 1H-NMR (300 MHz, CDCl3) δ (ppm) 0.20 (s, 6H), 0.98 (s, 9H), 4.67 (br s,lH), 6.65 (dd, J= 8.2, 2.6 Hz, IH), 6.98 (d, J= 2.9 Hz, IH), 7.36 (d, J= 8.8 Hz, IH).
3.2.k 2-Bromo-5-(2-cyanophenoχy)-l-(methoxymethoxymethyl)benzene [0443] 1H-NMR (300 MHz, CDCl3) δ (ppm) 3.41 (s, 3H), 4.64 (s, 2H), 4.76 (s, 2H), 6.8-6.9 (m, 2H), 7.16 (td, J= 7.6, 0.9 Hz, IH), 7.28 (d, J= 2.9 Hz, IH), 7.49 (ddd, J= 8.8, 7.6, 1.8 Hz, IH)5 7.56 (d, J= 8.5 Hz, IH), 7.67 (dd, J= 7.9, 1.8 Hz, IH).
EXAMPLE 32
Alternative Preparation of C17 -Intermediate

The procedure described in Example II I was followed for 1H NMR characterization of the current alcohol-borate intermediate. 1H NMR determination indicated there were 72.7 mol% of the desired alcohol-borate intermediate [2-bromo- 5-(4-cyanophenoxy)benzyl] diisopropyl borate, 20.7 mol% of an unknown intermediate and 6.5 mol% of unreacted alcohol. 1H NMR (CDCl3, 300 MHz) of [2- bromo-5-(4-cyanophenoxy)benzyl] diisopropyl borate: δ= 7.61 (d, J= 9.0 Hz, 2H), 7.52 (d, J= 8.4 Hz, IH), 7.15 (d, J= 3.0 Hz, IH), 7.03 (d, J= 8.7 Hz, 2H), 6.84 (dd, J= 8.7 Hz, J= 3.0 Hz, IH), 4.85 (s, 2H), 4.35 (septet, J= 6.1 Hz, 2H), 1.11 (d, J= 6.1 Hz, 12H) ppm.
PATENT
http://www.google.com/patents/US20090291917
- Example 154-(4-Cyanophenoxy)phenylboronic acid (C97)
-

-
(a) (4-cyanophenyl)(4-bromophenyl)ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 100° C. for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with 1N NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4-bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
-
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p. 194-198° C. MS: m/z=239 (M+), 240 (M+1) (ESI+) and m/z=238 (M−1) (ESI−). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6+D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.

see
http://www.google.co.in/patents/WO2006089067A2?cl=en
see
http://www.google.com/patents/US20090291917
| US5688928 * | Jun 7, 1995 | Nov 18, 1997 | Prolinx, Inc. | Phenylboronic acid complexing reagents derived from aminosalicylic acid |
| US5880188 * | May 26, 1995 | Mar 9, 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US5962498 * | Dec 2, 1994 | Oct 5, 1999 | Procyon Pharmaceuticals, Inc. | Protein kinase C modulators. C. indolactam structural-types with anti-inflammatory activity |
| US6369098 * | Oct 4, 2000 | Apr 9, 2002 | Bethesda Pharmaceuticals, Inc. | Dithiolane derivatives |
| US20030032673 * | Jul 19, 2002 | Feb 13, 2003 | Isis Innovation Limited | Therapeutic strategies for prevention and treatment of alzheimer’s disease |
| US20050239170 * | Jul 16, 2001 | Oct 27, 2005 | Hedley Mary L | Alpha-MSH related compounds and methods of use |
| US20060009386 * | May 12, 2005 | Jan 12, 2006 | The Brigham And Women’s Hospital, Inc. | Use of gelsolin to treat infections |
|
Methods of treating anti-inflammatory conditions through the use of boron- containing small molecules are disclosed.
|
|
… Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … Francisco, CA Mar. 6-10, 2009. 7 , “AN2728 … Kyoto, Japan, May 14-18, 2008. 10, “AN2728 …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3- dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
… Dermatology Annual Meeting, San Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … 7, “AN2728 … Francisco, CA May 6-10, 2009. 10, “AN2728 …
|
|
… from the group consisting of AN-2728, AN-2898, CBS- 3595, apremilast, ELB- 353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, …
|
|
“AN2728” is the compound 4-(l-hydroxy-l,3-dihydro-2 … GSK256066, oglemilast, tetomilast, apremilast, AN2728, Compound A, Compound B, …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
85.用于治疗疼痛的UK-500,001。 85. for the treatment of pain UK-500,001. 86.用 于治疗疼痛的AN2728。 86. for the treatment of pain AN2728.
|
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
///////////crisaborole, AN 2728, PHASE 3, Anti-inflammatory, Phosphodiesterase, Oxaborole, Psoriasis, Atopic dermatitis, borole
Ribociclib, рибоциклиб , ريبوسيكليب , 瑞波西利
Ribociclib
рибоциклиб , ريبوسيكليب , 瑞波西利
Ribociclib (LEE 011)
CAS: 1211441-98-3
Chemical Formula: C23H30N8O
Exact Mass: 434.25426
7-Cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide
FDA UNII
-
TK8ERE8P56
Current developer: Novartis /Astex Pharmaceuticals.
Novartis Ag, Astex Therapeutics Ltd.
NMR.http://file.selleckchem.com/downloads/nmr/S744002-LEE011-2-HNMR-Selleck%20.pdf
http://file.selleckchem.com/downloads/hplc/S744002-LEE011-2-HPLC-Selleck.pdf
Ribociclib (LEE011) is an orally available, and highly specific CDK4/6 inhibitor. Phase 3.
CDK4 AND 6
(Cell-free assay)Product Ingredients
NOW FDA APPROVED 2017 since the blog post was written
| Kisqali | FDA 3/13/2017 | To treat postmenopausal women with a type of advanced breast cancer Drug Trials Snapshot |

| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Ribociclib hydrochloride | 63YF7YKW7E | 1211443-80-9 | JZRSIQPIKASMEV-UHFFFAOYSA-N |
| Ribociclib succinate | BG7HLX2919 | 1374639-75-4 | NHANOMFABJQAAH-UHFFFAOYSA-N |
RIBOCICLIB SUCCINATE
STRUCTURE ….LINK
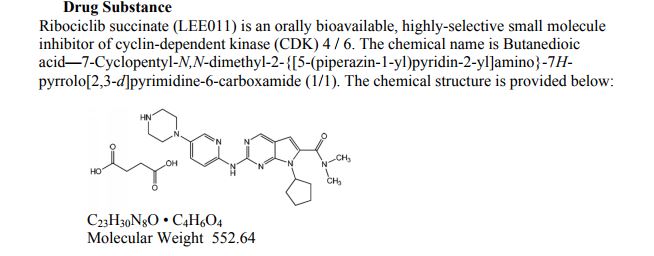
Ribociclib is in phase III clinical trials by Novatis for the treatment of postmenopausal women with advanced breast cancer.
Phase II clinical trials are also in development for the treatment of liposarcoma, ovarian cancer, fallopian tube cancer, peritoneum cancer, endometrial cancer, and gastrointestinal cancer.
Ribociclib, also known as LEE011, is an orally available cyclin-dependent kinase (CDK) inhibitor targeting cyclin D1/CDK4 and cyclin D3/CDK6 cell cycle pathway, with potential antineoplastic activity. CDK4/6 inhibitor LEE011 specifically inhibits CDK4 and 6, thereby inhibiting retinoblastoma (Rb) protein phosphorylation. Inhibition of Rb phosphorylation prevents CDK-mediated G1-S phase transition, thereby arresting the cell cycle in the G1 phase, suppressing DNA synthesis and inhibiting cancer cell growth. Overexpression of CDK4/6, as seen in certain types of cancer, causes cell cycle deregulation
Orally bioavailable CDK4/6-selective inhibitor that has been tested in Phase III clinical trials for treatment of advanced breast cancer.
CDK full name of cyclin-dependent kinases, there are many other subtypes CDK1-11, capable of binding to cell cycle proteins regulate the cell cycle. Pfizer Palbociclib been submitted for FDA review under phase II clinical data, Novartis Ribociclib (LEE011), Lilly Abemaciclib (LY2835219) the three CDK4 / 6 inhibitors have entered late stage development for the treatment of breast cancer
SYNTHESIS

WO2010020675
US20120115878
WO2010020675
http://www.google.co.in/patents/WO2010020675A1?cl=en
Example 74
7-Cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide

Following Buchwald Method B, then General Procedure A, 2-chloro-7-cyclopentyl-7H- pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide (300 mg, 1.02 mmol) and 5-piperazin-1- yl-pyridin-2-ylamine (314 mg, 1.13 mmol) gave 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2- ylamino)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide (142 mg, 36%). MS(ESI) m/z 435.3 (M+H)+
POSTER

SYNTHESIS
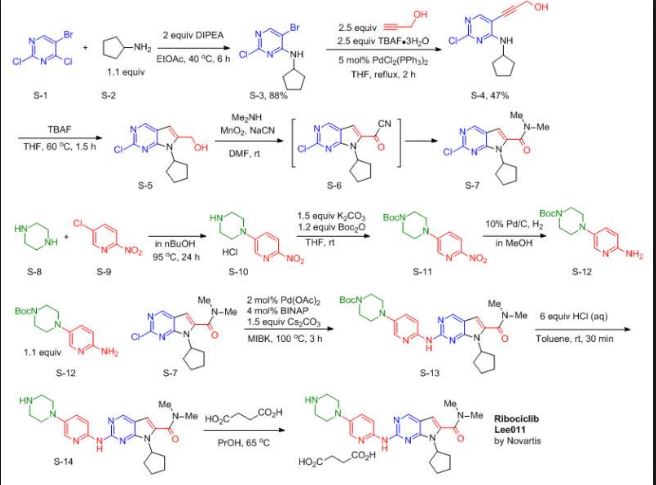


TAKEN FROM ….http://www.joygooo.com/news_71.htm?pageNum=21
PCT Int Appl, WO2012061156.
US Pat Appl Publ, US20120115878
PCT Int Appl, WO2011130232 5) Brain, Christopher Thomas et al; Preparation of pyrrolopyrimidine Derivatives for Use as CDK4 / 6 inhibitors;. PCT Int Appl, WO2011101409.
PCT Int Appl, WO2011101417. 7) Besong, Gilbert et al;.
PCT Int Appl, WO2010020675.
PCT Int Appl, WO2007140222.
Route 1


Reference:1. WO2012064805A1 / US20120115878A1.
2. WO2010020675A1 / US8415355B2.
3. WO2011130232A1 / US20130035336A1.
Clinical Trial Information( data from http://clinicaltrials.gov, updated on 2015-10-17)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT02571829 | Not yet recruiting | Liposarcoma|Soft Tissue Sarcoma | Hadassah Medical Organization | December 2015 | Phase 2 |
| NCT02524119 | Not yet recruiting | Hepatocellular Carcinoma | University of Texas Southwestern Medical Center|Novartis …more | November 2015 | Phase 2 |
| NCT02494921 | Recruiting | Prostate Cancer | Rahul Aggarwal|University of California, San Francisco | September 2015 | Phase 1|Phase 2 |
| NCT02420691 | Recruiting | Gastrointestinal Cancer | M.D. Anderson Cancer Center|Novartis | August 2015 | Phase 2 |
| NCT02431481 | Not yet recruiting | Normal Renal Function|Impaired Renal Function | Novartis Pharmaceuticals|Novartis | August 2015 | Phase 1 |
Protocols from literature
|
In vitro protocol:: |
Pharmacologic growth inhibition: Clin Cancer Res. 2013 Nov 15;19(22):6173-82. Cell-cycle analysis: Clin Cancer Res. 2013 Nov 15;19(22):6173-82. Senescence and apoptosis assays: Clin Cancer Res. 2013 Nov 15;19(22):6173-82. |
|
In vivo protocol: |
Xenograft therapeutic trials: Clin Cancer Res. 2013 Nov 15;19(22):6173-82 Immunohistochemistry of xenografted neuroblastomas.Clin Cancer Res. 2013 Nov 15;19(22):6173-82 |
Ribociclib (LEE011) is a Me-Too version of palbociclib. Their structures are compared side-by-side as the following:
 |
Ribociclib (LEE011) is currently being developed by Novartis and Astex. According its Novartis’s website, LEE011 is a novel, orally available, selective inhibitor of CDK4/6 kinases, which induces complete dephosphorylation of Rb and G1 arrest in cancer cells. In preclinical in vitro and in vivo tumor models, LEE011 has been shown active in cancers harboring aberrations that increase CDK4/6 activity, including those directly linked to the kinases as well as activating alterations in the upstream regulators. First-in-human study of LEE011 in patients with solid tumors and lymphoma is currently ongoing. (source: http://www.novartisoncology.us/research/pipeline/lee011.jsp).
Treatment with LEE011 significantly reduced proliferation in 12 of 17 human neuroblastoma-derived cell lines by inducing cytostasis at nanomolar concentrations (mean IC50 = 307 ± 68 nmol/L in sensitive lines). LEE011 caused cell-cycle arrest and cellular senescence that was attributed to dose-dependent decreases in phosphorylated RB and FOXM1, respectively. In addition, responsiveness of neuroblastoma xenografts to LEE011 translated to the in vivo setting in that there was a direct correlation of in vitro IC50 values with degree of subcutaneous xenograft growth delay. Although our data indicate that neuroblastomas sensitive to LEE011 were more likely to contain genomic amplification of MYCN (P = 0.01), the identification of additional clinically accessible biomarkers is of high importance. LEE011 is active in a large subset of neuroblastoma cell line and xenograft models, and supports the clinical development of this CDK4/6 inhibitor as a therapy for patients with this disease. (Clin Cancer Res. 2013 Nov 15;19(22):6173-82)
|
References |
1. Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, Li Y, Carpenter EL, Attiyeh EF, Diskin SJ, Kim S, Parasuraman S, Caponigro G, Schnepp RW, Wood AC, Pawel B, Cole KA, Maris JM. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013 Nov 15;19(22):6173-82. doi: 10.1158/1078-0432.CCR-13-1675. Epub 2013 Sep 17. PubMed PMID: 24045179; PubMed Central PMCID: PMC3844928.
2. Caponigro, Giordano; Stuart, Darrin; Kim, Sunkyu; Loo, Alice; Delach, Scott. Pharmaceutical combinations of a CDK4/6 inhibitor and a B-RAF inhibitor for treatment of proliferative diseases such as cancer. PCT Int. Appl. (2014), WO 2014018725 A1 20140130.
3. Kim, Sunkyu; Doshi, Shivang; Haas, Kristy; Kovats, Steven; Huang, Alan Xizhong; Chen, Yan. Combination therapy comprising a cyclin dependent kinase 4/6 (CDK4/6) inhibitor and a phosphatidylinositol 3-kinase (PI3K) inhibitor for use in the treatment of cancer. PCT Int. Appl. (2013), WO 2013006532 A1 20130110
4. Kim, Sunkyu; Doshi, Shivang; Haas, Kristy; Kovats, Steven. Combination of cyclin dependent kinase 4/6 (CDK4/6) inhibitor and fibroblast growth factor receptor (FGFR) kinase inhibitor for the treatment of cancer. PCT Int. Appl. (2013), WO 2013006368 A1 20130110
5. Calienni, John Vincent; Chen, Guang-Pei; Gong, Baoqing; Kapa, Prasad Koteswara; Saxena, Vishal. Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof. U.S. Pat. Appl. Publ. (2012), US 20120115878 A1 20120510.
6. Borland, Maria; Brain, Christopher Thomas; Doshi, Shivang; Kim, Sunkyu; Ma, Jianguo; Murtie, Josh; Zhang, Hong. Combination comprising a cyclin dependent kinase 4 or cyclin dependent kinase (cdk4/6) inhibitor and an Mtor inhibitor for treating cancer. PCT Int. Appl. (2011), WO 2011130232 A1 20111020
7. Besong, Gilbert; Brain, Christopher Thomas; Brooks, Clinton A.; Congreve, Miles Stuart; Dagostin, Claudio; He, Guo; Hou, Ying; Howard, Steven; Li, Yue; Lu, Yipin; et al. Preparation of pyrrolopyrimidine compounds as CDK inhibitors. PCT Int. Appl. (2010), WO 2010020675 A1 20100225.
CLIP
Cyclin-dependent kinase inhibitors (14 compounds) under clinical evaluation.
LEE-011 is one of the most selective inhibitors for CDK4 and CDK6 [59] and is being developed by Astex Pharmaceuticals™ and Novartis. In January 2014 this inhibitor entered phase III clinical trials for the treatment of breast cancer [60]. Due to encouraging results LEE-011 has now become the main competing drug-candidate with Pfizer’s PD0332991 (palbociclib), see Figure 3 [59].

Figure 3. Comparison of Astex/Novartis’ LEE-011 and Pfizer’s PD0332991 structures.
Upon comparison of the chemical structure of Novartis’ LEE-011 and Pfizer’s PD0332991, the similarity is evident. The major difference lies in the bicyclic core since LEE-011 possesses a pyrrolo-pyrimidine and PD0332991 a pyridopyrimidine. The “east” part of the structure is also modified. The structural similarities make their analogous CDKs inhibition profiles (high selectivity for CDK4 and CDK6) quite obvious Moreover, both derivatives are orally administered which is pretty advantageous compared with dinaciclib, which is also in phase III clinical trials but is administered intravenously.
http://www.mdpi.com/1420-3049/19/9/14366/htm
- Kurt, S. LEE011 CDK Inhibitor Showing Early Promise in Drug-Resistant Cancers. Oncol. Times 2014, 36, 39–40. [Google Scholar]
- Macmillan Publishers Limited. CDK inhibitors speed ahead. Nat. Rev. Drug Discov. 2014, 13, 323. [Google Scholar] [CrossRef]
Sources:
1)Rader, JulieAnn et al.;Dual CDK4/CDK6 Inhibition Induces Cell-Cycle Arrest and Senescence in Neuroblastoma;Clinical Cancer Research (2013), 19(22), 6173-6182
2)Tavares, Francis X. and Strum, Jay C.;Preparation of pyrazinopyrrolopyrimidine derivatives and analogs for use as CDK inhibitors;PCT Int. Appl., WO2012061156
3)Calienni, John Vincent et al.;Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof;U.S. Pat. Appl. Publ., US20120115878
4)Borland, Maria et al;Combination comprising a cyclin dependent kinase 4 or cyclin dependent kinase (cdk4/6) inhibitor and an Mtor inhibitor for treating cancer;PCT Int. Appl., WO2011130232
5)Brain, Christopher Thomas et al;Preparation of pyrrolopyrimidine derivatives for use as CDK4/6 inhibitors;PCT Int. Appl., WO2011101409
6)Brain, Christopher Thomas and Perez, Lawrence Blas; Preparation of deuterated pyrrolopyrimidine compounds as inhibitors of CDK4/6 for treating cancer; PCT Int. Appl., WO2011101417
7)Besong, Gilbert et al.;Preparation of pyrrolopyrimidine compounds as CDK inhibitors;PCT Int. Appl., WO2010020675
8)Brain, Christopher Thomas et al.;Preparation of pyrrolopyrimidine compounds as protein kinase inhibitors; PCT Int. Appl., WO2007140222
9)A Randomized Double-blind, Placebo-controlled Study of LEE011 in Combination With Letrozole for the Treatment of Postmenopausal Women With Hormone Receptor Positive, HER2 Negative, Advanced Breast Cancer Who Received no Prior Therapy for Advanced Disease;ClinicalTrials.gov Identifier: NCT01958021
/////////Ribociclib, novartis, LEE011, astex, phase 3, CDK inhibitors
CN(C)C(=O)c1cc2cnc(nc2n1C3CCCC3)Nc4ccc(cn4)N5CCNCC5
Etelcalcetide, AMG 416, KAI-4169, velcalcetide
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2

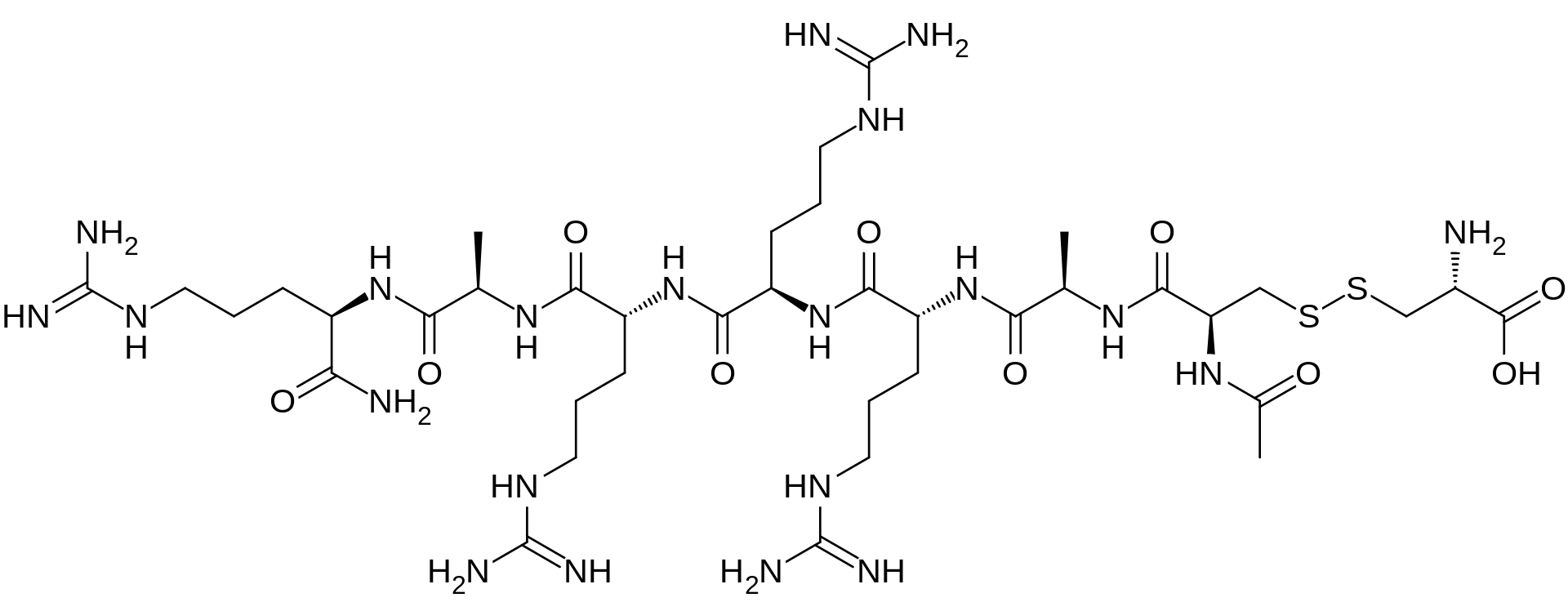
AMG 416 IS (Ac-D-Cys(L-Cys-OH)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2)
Etelcalcetide (AMG 416, KAI-4169, velcalcetide)
The main chain has 7 amino acids, all in the D-configuration. The side-chain cysteine residue is in the L-configuration. The molecular formula of AMG 416 (free base) is C38H73N21O10S2, and has a calculated average molecular mass of 1048.3 Da.
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine, hydrochloride (1:?)
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
http://www.amgenpipeline.com/pipeline/
WO 2011/014707. , the compound may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The main chain has 7 amino acids, all in the D-configuration and the side-chain cysteine residue is in the L-configuration. The amino terminal is acetylated and the carboxyl-terminal is amidated. This compound (“AMG-416”) has utility for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis patients. A liquid formulation comprising AMG-416 may be administered to a subject intravenously. The hydrochloride salt of AMG-416 may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
Therapeutic peptides pose a number of challenges with respect to their formulation. Peptides in general, and particularly those that contain a disulfide bond, typically have only moderate or poor stability in aqueous solution. Peptides are prone to amide bond hydrolysis at both high and low pH.
Disulfide bonds can be unstable even under quite mild conditions (close to neutral pH). In addition, disulfide containing peptides that are not cyclic are particularly prone to dimer formation. Accordingly, therapeutic peptides are often provided in lyophilized form, as a dry powder or cake, for later reconstitution.
A lyophilized formulation of a therapeutic peptide has the advantage of providing stability for long periods of time, but is less convenient to use as it requires the addition of one or more diluents and there is the potential risk for errors due to the use of an improper type or amount of diluent, as well as risk of contamination. In addition, the lyophilization process is time consuming and costly.
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
Generic Name:Etelcalcetide
Synonym:KAI-4169
CAS Number:1262780-97-1
N-acetyl-D-cysteinyl-S-(L-cysteine disulfide)-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide
Mechanism of Action:Activates calcium sensing receptor on parathyroid glands reducing PTH synthesis and secretion
Indication: secondary hyperparathyroidism associated with chronic kidney disease
Development Stage: Phase III
Developer:KAI Pharmaceuticals/Amgen Inc.
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
HYDROCHLORIDE
Generic Name:Etelcalcetide Hydrochloride
AMG 416, KAI-4169, previously also known as velcalcetide hydrochloride
CAS :1334237-71-6
Chemical Name:N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
Mechanism of Action:Activates calcium sensing receptor on parathyroid glands reducing PTH synthesis and secretion
Indication: secondary hyperparathyroidism associated with chronic kidney disease
Development Stage: Phase III
Developer:KAI Pharmaceuticals/Amgen Inc.
Method for preparing etelcalcetide and its salts, particularly hydrochloride. See WO2014210489, for a prior filing claiming stable liquid formulation of etelcalcetide. Amgen, following its acquisition of KAI Pharmaceuticals, and Japanese licensee Ono Pharmaceuticals are developing etelcalcetide, a long-acting iv isozyme-selective peptide-based protein kinase C epsilon inhibitor and agonist of the calcium-sensing receptor, for treating secondary hyperparathyroidism (SHPT) in patients with end-stage renal disease receiving dialysis.
In August 2015, an NDA was submitted seeking approval of the drug for SHPT in patients with chronic kidney disease (CKD) on hemodialysis (HD) in the US.
In September 2015, Amgen filed an MAA under the centralized procedure in the EU for the approval of etelcalcetide for treating SHPT in patients with CKD on HD therapy.
KAI is also investigating a transdermal patch formulation of the drug for treating primary HPT.
Secondary hyperparathyroidism in patients with chronic kidney disease receiving dialysis
AMG 416 is a peptide agonist of the human cell surface calcium-sensing receptor (CaSR). It is being investigated as a treatment for secondary hyperparathyroidism in patients with chronic kidney disease receiving dialysis.
Etelcalcetide is a novel calcimimetic agent that suppresses the secretion of parathyroid hormone and is in clinical development for the treatment of SHPT in patients with CKD on hemodialysis. Etelcalcetide is administered intravenously three times per week at the end of each dialysis session. It acts by binding to and activating the calcium-sensing receptor on the parathyroid gland, thereby causing decreases in parathyroid hormone (PTH). Sustained elevations in PTH are known to be associated with significant clinical consequences for patients with CKD.
The submission includes data from three Phase 3 studies, all of which met the primary endpoints, including two pooled placebo-controlled trials in more than 1,000 patients and a head-to-head study evaluating etelcalcetide compared with cinacalcet.
About Secondary HyperparathyroidismSHPT is a common and serious condition that is often progressive among patients with CKD, and it affects many of the approximately two million people throughout the world who are receiving dialysis, including 450,000 people in the U.S. The disorder develops early in the course of CKD and usually manifests as increased levels of PTH as a result of increased production from the parathyroid glands (four small glands in the neck). Patients with end stage renal disease who require maintenance dialysis often have substantial elevations of PTH that are commonly associated with abnormal calcium and phosphorus levels and an increased risk of significant clinical consequences.
About Etelcalcetide (AMG 416)Etelcalcetide is a novel calcimimetic agent in clinical development for the treatment of SHPT in CKD patients on hemodialysis that is administered intravenously at the end of the dialysis session. Etelcalcetide binds to and activates the calcium-sensing receptor on the parathyroid gland, thereby decreasing PTH levels.
About Sensipar® (cinacalcet)Sensipar® (cinacalcet) is the first oral calcimimetic agent approved by the FDA for the treatment of SHPT in adult patients with CKD on dialysis. Sensipar is not indicated for use in adult patients with CKD who are not on dialysis because of an increased risk of hypocalcemia. The therapy is also approved in the U.S. for treatment of hypercalcemia in adult patients with parathyroid carcinoma and hypercalcemia in adult patients with primary HPT for whom parathyroidectomy would be indicated on the basis of serum calcium levels, but who are unable to undergo parathyroidectomy. Sensipar binds to the calcium-sensing receptor, resulting in a drop in PTH levels by inhibiting PTH synthesis and secretion. In addition, the reductions in PTH lower serum calcium and phosphorus levels.
Milestones
- 25 Aug 2015 Preregistration for Secondary hyperparathyroidism in USA (IV)
- 29 May 2015 Pooled analysis efficacy and adverse events data from two phase III trials in secondary hyperparathyroidism released by Amgen
- 21 Apr 2015 Amgen plans to submit Biological License Application to USFDA and Marketing Authorisation Application to EMA for Secondary hyperparathyroidism
PATENT
WO2011014707
http://www.google.com/patents/WO2011014707A2?cl=en
PATENT
WO 2015154031
The hydrochloride salt of AMG 416 has the chemical structure:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
(SEQ ID NO:l)
The main chain has 7 amino acids, all in the D-configuration. The side-chain cysteine residue is in the L-configuration. The molecular formula of AMG 416 (free base) is C38H73N21O10S2, and has a calculated average molecular mass of 1048.3 Da.
AMG 416 and a method for its preparation are described in International Pat. Publication No. WO 2011/014707, which is incorporated herein by reference for any purpose. As described in International Pat. Publication No. WO 2011/014707, AMG 416 may be assembled by solid-phase synthesis from the corresponding Fmoc-protected D-amino acids. After cleavage from the resin, the material may be treated with Boc-L-Cys(NPyS)-OH to form the disulfide bond. The Boc group may then be removed with trifluoroacetate (TFA) and the resulting product purified by reverse-phase high pressure liquid chromatography (HPLC) and isolated as the TFA salt form by lyophilization. The TFA salt can be converted to a pharmaceutically acceptable salt by carrying out a subsequent salt exchange procedure. Such procedures are well known in the art and include, e.g., an ion exchange technique, optionally followed by purification of the resultant product (for example by reverse phase liquid chromatography or reverse osmosis).
There is a need for an efficient method of producing AMG 416, or a pharmaceutically acceptable salt thereof (e.g., AMG 416 HC1), and particularly one appropriate for commercial scale manufacturing.
In a first aspect, provided is a method for preparing AMG 416, the method comprising: providing a resin-bound peptide having a structure selected from the group consisting of Fmoc-D-Cys(Trt)-D-Ala-D- Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:2) and Ac-D-Cys(Trt)-D-Ala-D- Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:3); cleaving the peptide from the solid support; and activating the side chain of the D-Cys residue of the cleaved peptide.
In a second aspect, provided is a method for preparing AMG 416, the method comprising: providing a peptide having a structure of Ac-D-Cys(SPy)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 (SEQ ID NO:4); and contacting the peptide with L-Cys to produce a conjugated product.
In yet a third aspect provided is a method for preparing AMG 416, the method comprising: providing a resin-bound peptide having a structure selected from the group consisting of Fmoc-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:2) and Ac-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:3); cleaving the peptide from the solid support, i.e., to provide an unsupported peptide, and activating the side chain of the D-Cys residue of the unsupported peptide to generate an AMG 416 SPy intermediate (where SPy is 2-pyridinesulfenyl or S-Pyr), dissolving the AMG 416 SPy intermediate in an aqueous 0.1% TFA (trifluoroacetic acid solution), and purifying the AMG 416 SPy derivative by HPLC.
The term “AMG 416”, also known as etelcalcetide, formerly known as velcalcetide or KAI-4169, refers to a compound having the chemical name: N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-arginamide disulfide with L-cysteine, which has the following structural formula:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
Reference to AMG 416, or to any compound or AMG 416 fragment, intermediate, or precursor as described herein, is intended to encompass neutral, uncharged forms thereof, as well as pharmaceutically acceptable salts, hydrates and solvates thereof.
The terms “AMG 416 hydrochloride” and “AMG 416 HC1” are interchangeable and refer to a hydrochloride salt form of AMG 416 having the following structural formula:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · xHCl
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1 shows the chemical structure of AMG 416 (Ac-D-Cys(L-Cys-OH)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2) (SEQ ID NO: l).
FIG. 2 shows the chemical structure of Rink Amide AM resin and Ac-D-Cys(Trt)- D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-Resin (SEQ ID NO:3).
FIG. 3 shows a reaction scheme in which the SPy intermediate product (Ac-D-Cys(SPy)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2) (SEQ ID NO:4) is formed from the peptidyl-resin (Ac-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-NH-Resin) (SEQ ID NO:3).
FIG. 4 shows a reaction scheme in which a TFA salt of AMG 416 is formed from the SPy intermediate (AA1_7(SPy)).
FIG. 5 shows a reaction scheme in which the HC1 salt of AMG 416 is formed from the TFA salt of AMG 416.
FIG. 6 shows a reaction scheme in which Boc-D-Arg(Pbf)-OH is formed from Boc-D-Arg-OH.
FIG. 7 shows a reaction scheme in which D-Arg(Pbf)-OH is formed from Boc-D-Arg(Pbf)-OH.
EXAMPLE 5
Purification of the SPy Intermediate and Production of AMG 416 HC1
An alternative method for preparation of AMG 416 HC1 salt is described here. As described in Example 2 above, the SPy intermediate product was dried at 20°C under full vacuum after cleavage from the resin, precipitation and filtration. The precipitate was then dissolved in a 0.1% TFA aqueous solution and loaded onto a C-18 column for HPLC purification. The column was run at <60 bar and the solution temperature was 15-25 °C throughout. The eluents were 0.1% TFA in acetonitrile and 0.1% TFA in water. The fractions were stored at 5°C, they were sampled and then fractions were pooled. The combined pools from two runs were diluted and a concentration/purification run was performed using the same HPLC column to decrease the total volume and remove additional impurities. The fractions were stored at 5°C.
The fractions containing the AMG 416 SPy intermediate were subjected to azeotropic distillation to change the solvent from the 0.1% TFA to a 15% water in IPA solution, charging with IPA as needed. To the resultant AMG 416 SPy intermediate in IPA solution was then added L-Cysteine 1.15 eq and the reaction was allowed to proceed at room temperature for conjugation to occur and to form the AMG 416 TFA salt as described above in Example 4. The AMG 416 TFA solution was added to a solution of 12M aqueous HC1, 0.27 L/kg and IPA 49.4 L/kg over 3 hours via subsurface addition, resulting in direct precipitation of the AMG 416 4.5 HC1 salt. The batch was aged for 3 hours and sampled for analysis.
The material was filtered and slurry washed with 96 wt% IPA, 10 L/kg. The cake was then re-slurried for 4 hours in 10 L/kg of 96% wt% IPA. The material was filtered and further slurry washed with 96% IPA, 10 L/kg and then IPA 10 L/kg. The material was dried under full vacuum at 25°C. The dry cake was dissolved in water 8 L/kg and the batch was concentrated via distillation to remove residual IPA and achieve the desired concentration. The solution temperature was kept below 25 °C throughout the distillation.
PATENT
WO2014210489
SEE
EXAMPLE 1
Solubility of AMG 416 in Succinate Buffered Saline
In this study, the solubility of AMG 416 in succinate buffered-saline was investigated. AMG 416 HC1 (103 mg powder, 80 mg peptide) was dissolved in 200 iL of sodium succinate buffered saline (25 mM succinate, 0.9% saline, pH 4.5). After briefly vortexing, a clear solution was obtained with a nominal concentration of 400 mg/mL. Because expansion of the solution volume was not determined, the solubility of AMG 416 can be conservatively stated as at least 200 mg/mL. Although the maximal solubility was not determined in this experiment, AMG 416 is soluble in pH 4.5 succinate buffered saline to concentrations of at least 200 mg/mL.
REFERENCES
- “Amgen Submits New Drug Application For Novel Intravenous Calcimimetic Etelcalcetide (AMG 416)”
- “Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects
- “Evidence for Chronic Kidney Disease-Mineral and Bone Disorder Associated With Metabolic Pathway Changes”
KAI-4169, a novel calcium sensing receptor agonist, decreases serum iPTH, FGF-23 and improves serum bone markers in a phase 2 study in hemodialysis subjects with chronic kidney disease-mineral and bone disorder
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO054
KAI-4169, a novel peptide agonist of the calcium sensing receptor, attenuates PTH and soft tissue calcification and restores parathyroid gland VDR levels in uremic rats
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
Long term safety and efficacy of velcalcetide (AMG 416), a calcium-sensing receptor (CaSR) agonist, for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis (HD) patients
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
Preclinical PK and PD relationship for KAI-4169, a novel calcimimetic
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
KAI-4169, a novel calcimimetic for the treatment of secondary hyperparathyroidism
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
Characterization of KAI-4169, a novel peptide for the treatment of chronic kidney disease – Mineral and bone disorder, in a phase I study in healthy males
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
| WO2011014707A2 | Jul 29, 2010 | Feb 3, 2011 | Kai Pharmaceuticals, Inc. | Therapeutic agents for reducing parathyroid hormone levels |
//////////////Etelcalcetide, AMG 416, KAI-4169, velcalcetide, peptide drugs
Ravidasvir, PPI-668, BI 238630

Ravidasvir dihydrochloride
C42H50N8O6.2(HCl), 835.83
CAS 1303533-81-4
Phase II/IIIHepatitis C

Ravidasvir
PPI-668 free base; BI 238630;
CAS:1242087-93-9
C42H50N8O6, 762.38
Chemical Name:methyl N-[(1S)-1-({(2S)-2-[5-(6-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]- 3- methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl}naphthalen-2-yl) -1H- benzimidazol- 2-yl]pyrrolidin-1-yl}carbonyl)-2-methylpropyl]carbamate
Mechanism of Action:NS5A Inhibitor
Indication: hepatitis C
Development Stage: Phase II
Developer:Presidio Pharmaceuticals, Inc
- OriginatorXTL Biopharmaceuticals
- Developer Pharco Corporation; Presidio Pharmaceuticals
- Class Antivirals; Benzimidazoles; Carbamates; Naphthalenes; Pyrrolidines; Small molecules
- Mechanism of Action Hepatitis C virus NS 5 protein inhibitors; Hepatitis C virus replication inhibitors
- 31 Aug 2015 Ascletis plans to initiate the phase II EVEREST trial for Hepatitis C (Combination therapy; Treatment-naive) in Taiwan
- 31 Aug 2015 Taiwan Food and Drug Administration approves Clinical Trial Application to initiate a phase II trial for interferon free regimen comprising danoprevir and ravidasvir in Hepatitis C
- 24 Jun 2015 Efficacy data from a phase IIa trial in Hepatitis C released by Ascletis
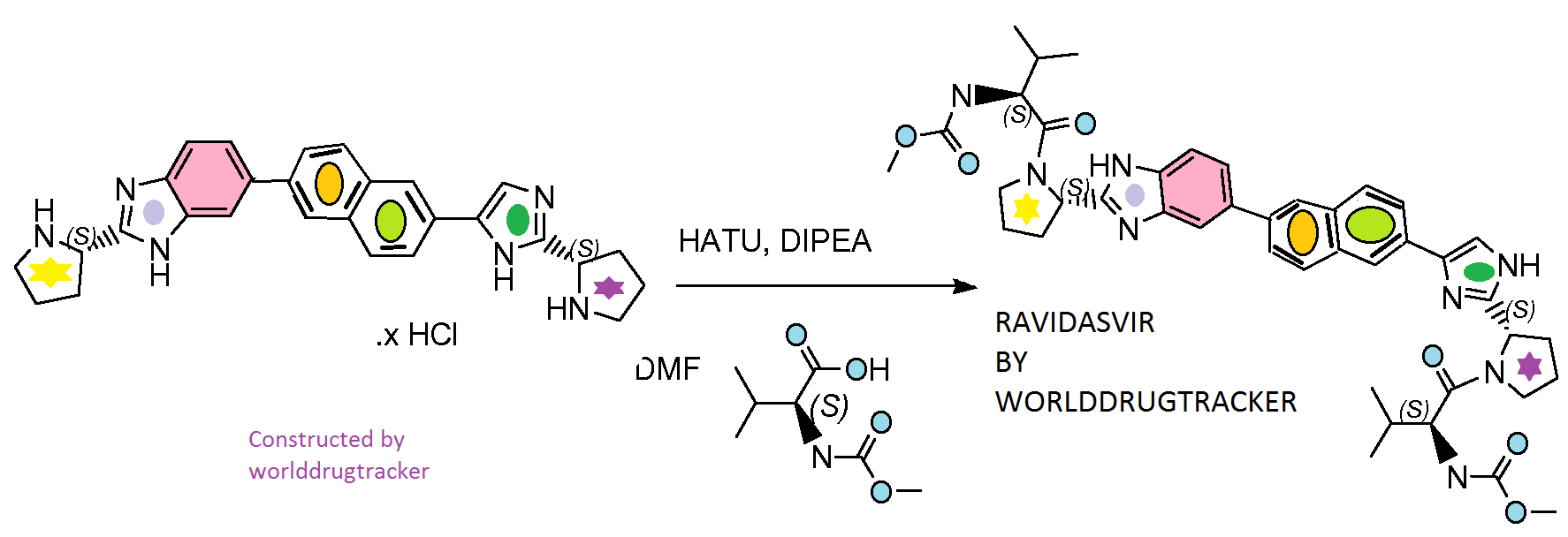
Ravidasvir [Methyl N-[(1S)-1-({(2S)-2-[5-(6-{2-[(2S)-1-{(2S)-2-[(methoxycarbonyl)amino]- 3- methylbutanoyl}pyrrolidin-2-yl]-1H-imidazol-4-yl}naphthalen-2-yl) -1H- benzimidazol- 2-yl]pyrrolidin-1-yl}carbonyl)-2-methylpropyl]carbamate] is an Nonstructural protein 5A (NS5A) inhibitor. It is an antiviral agent that is being developed as a potential treatment for hepatitis C virus infection.
PPI-668, a non-structural 5A (NS5A) protein of hepatitis C virus (HCV) inhibitor, is in phase II clinical studies at Presidio Pharmaceuticals for the treatment of chronic genotype 1 hepatitis C virus infection.
Ravidasvir has 50% inhibitory concentrations (EC50s) values of 0.02-1.3 nM in replicon assays for HCV genotypes 1-7 (gt1-gt7).
Ravidasvir was developed by Presidio Pharmaceuticals Inc, later Ascletis licensed it. Ravidasvir is in Phase II clinical trials proving interferon (IFN)-free regimen to treat chronic hepatitis C (CHC). Ascletis is now the first Chinese company to file clinical trial applications in China for an IFN-free regimen.
In 2014, Ascletis acquired rights for development and commercialization in Greater China and Pharco in Egypt for the treatment of hepatitis C.
Hepatitis C virus infection is a major health problem worldwide and no vaccine has yet been developed against this virus. The standard therapy of pegylated-interferon and ribavirin induces serious side effects and provides viral eradication in less than 50% of patients. Combination therapy of HCV including ribavirin and interferon are currently is the approved therapy for HCV. Unfortunately, such combination therapy also produces side effects and is often poorly tolerated, resulting in major clinical challenges in a significant proportion of patients. The combination of direct acting agents can also result in drug-drug interactions. To date, no HCV therapy has been approved which is interferon free. There is therefore a need for new combination therapies which have reduced side effects, and interferon free, have a reduced emergence of resistance, reduced treatment periods and/or and enhanced cure rates.
Nonstructural protein 5A (NS5A) is a zinc-binding and proline-rich hydrophilic phosphoprotein that plays a key role in Hepatitis C virus RNA replication.
A number of direct-acting antiviral agents (DAAs) are under development for the treatment of chronic HCV infection. These agents block viral production by directly inhibiting one of several steps of the HCV lifecycle. several viral proteins involved in the HCV lifecycle, such as the non-structural (NS)3/4A serine protease, the NS5B RNA-dependent RNA polymerase (RdRp), and the NS5A protein, have been targeted for drug development. Two NS3/4A protease inhibitors already approved for clinical use, numerous other protease inhibitors are being developed as well as inhibitors of viral replication, including nucleoside/nucleotide analogue inhibitors of HCV RdRp, non-nucleoside inhibitors of RdRp, cyclophilin inhibitors, and NS5A inhibitors.
Inhibition of NS5A at picomolar concentrations has been associated with significant reductions in HCV RNA levels in cell culture-based models, which makes these agents among the most potent antiviral molecules yet developed.
Activity:
This NS5A inhibitor has been shown to possess high efficacy against HCV genotype 1, with up to 3.7 log10 mean HCV RNA reductions, in a Phase Ib clinical trial. Activity was demonstrated against variants harbouring the L31M substitution. In an added genotype-2/3 cohort, the first 2 patients achieved mean 3.0 log10 RNA level reductions [1].
Results from the Phase IIa study involving a combination therapy with Faldaprevir and Deleobuvir plus Ravidasvir came with positive news where the said combination cured 92 percent of those with genotype 1a of hepatitis C virus (HCV) when given with ribavirin. The results presented at the 49th annual meeting of the European Association for the Study of the Liver (EASL) in London [2, 3].
The 36 study participants were randomly dived into three even cohorts of 12 each: The first received 600 mg of Deleobuvir twice a day as well as once-daily doses of Faldaprevir (120 mg), Ravidasvir and Ribavirin. The second group received the same regimen except the Faldaprevir dose was 400 mg. The third group took the regimen with the higher dose of Faldaprevir, but without Ribavirin. All participants were treated for 12 weeks with follow up for next 24 weeks.
Ninety-two percent of the first and second cohorts (11 out of 12 in both cases) achieved a sustained virologic response 12 weeks after completing therapy (SVR12, considered a cure). In the end, 14 participants were required for the third cohort, because one was incarcerated early on during treatment and another experienced viral rebound at week eight as a result of not adhering to the treatment regimen. Of the other 12 participants, eight, or two-thirds, have achieved an SVR12, while one more participant stopped taking the therapy at week eight but has since achieved an SVR8.
PATENT
WO 2011054834
http://www.google.co.in/patents/WO2011054834A1?cl=en
Scheme 1

GOING TO PRODUCT USING STRUCTURES FROM PATENT
 IIa
IIa
 IIIa one of side chain
IIIa one of side chain
DO NOT MISS OUT synthesis of XIIIa or XIII’a, this is needed in one of side chain
 L-boc-prolinol
L-boc-prolinol
 Z-boc-prolinal
Z-boc-prolinal
 XXIV
XXIV
 XIIIa
XIIIa
or

 XVIb
XVIb


MY CONSTRUCTION of 3

Compound 3 was prepared following the procedure reported for the synthesis of compound 1 using intermediate XVIIIb instead of intermediate XVIIIa. see my construction below

Compound 3. BASE
1H NMR (400 MHz, DMSO-d6) δ ppm 8.34 (2 H, s), 8.21 (1 H, s), 8.19
(1 H, d, J=8.69 Hz), 8.06 – 8.11 (2 H, m), 8.00 (1 H, dd, J=8.88, 1.61 Hz), 7.88 – 7.96
(2 H, m), 7.86 (1 H, d, J=8.48 Hz), 7.32 (1 H, d, J=8.48 Hz), 7.34 (1 H, d, J=8.53 Hz), 5.27 (1 H, dd, J=8.17, 5.33 Hz), 5.17 (1 H, t, J=7.00 Hz), 4.15 (2 H, t, J=7.95 Hz), 3.84
– 3.96 (4 H, m), 3.56 (6 H, s), 2.38 – 2.47 (2 H, m), 1.95 – 2.30 (8 H, m), 0.86 (3 H, d,
J=6.70 Hz), 0.85 (3 H, d, J=6.70 Hz), 0.81 (6 H, d, J=6.63 Hz).
[a] 2°= -148.98 0 (c 0.3336 w/v %, MeOH)
Alternative preparation of compound 3 and the corresponding HC1 salt
N-methoxycarbonyl-L- Valine (3.09 g, 17.7 mmol, 2.1 equiv) was dissolved in dichloro- methane (300 mL). Triethylamine (11.7 mL, 84.1 mmol, 10 equiv) and (l-cyano-2- ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluoro- phosphate were added (7.57 g, 17.7 mmol, 2.1 eq). The reaction mixture was stirred at room temperature for 5 minutes, after which XVIIIb was added (5 g, 8.41 mmol in case x.HCl equals 4 HC1). Stirring was continued for 30 minutes. HC1 in iPrOH (6N) was added to the mixture (until pH = 2), and the resulting mixture was stirred for 5 minutes. The solution was then washed with saturated aqueous sodium carbonate (2 x 200 mL) and once with brine (200 mL). The organic layer was separated, dried on magnesium sulphate and filtrated. After removal of the solvent in vacuum, the obtained residue was further dried in vacuum to afford an orange powder (6.84 g)
The powder was purified by silica gel column chromatography using gradient elution with 0 to 10 % MeOH (7N NH3) in dichloromethane, resulting in compound 3 (2.81 g) as a foam.
Compound 3 was dissolved in iPrOH (40 mL) and HC1 (6N in iPrOH, 10 mL) was added. The volatiles were removed in vacuum. Then, iPrOH (30 mL) was added and the mixture was heated at reflux. The solution was cooled to room temperature and stirred at room temperature for 4 days. tBuOMe (100 mL) was added to the solution, resulting in white precipitation, which was filtered, washed immediately with tBuOMe (3 x 10 mL) under nitrogen atmosphere and dried under vacuum at 40°C. The residue was mixed with acetonitrile and evaporated to dryness (2x). The residue was stirred in acetonitrile (150 mL) and the mixture was sonicated for 10 minutes. The precipitate was filtered under nitrogen atmosphere, washed twice with acetonitrile (50 mL) and dried in vacuum at 40°C, resulting in a slightly yellow powder (4 g).
HCL salt of compound 3:
[a] *° = -110.02 ° (589 nm, 20 °C, c 0.429 w/v%, MeOH)
1H NMR (600 MHz, DIMETHYLFORMAMIDE- y, 280K) δ ppm 0.86 (d, J=6.6 Hz, 6 H), 0.95 (d, J=7.0 Hz, 6 H), 2.03 – 2.20 (m, 2 H), 2.26 – 2.37 (m, 3 H), 2.39 – 2.61 (m, 5 H), 3.61 – 3.63 (m, 6 H), 3.93 – 4.01 (m, 2 H), 4.23 – 4.32 (m, 2 H), 4.32 – 4.39 (m, 2 H), 5.49 (t, J=7.5 Hz, 1 H), 5.52 (dd, J=8.3, 5.3 Hz, 1 H), 7.22 (d, J=8.8 Hz, 1 H), 7.27 (d, J=8.8 Hz, 1 H), 7.98 (d, J=8.6 Hz, 1 H), 8.01 (dd, J=8.6, 1.1 Hz, 1 H), 8.03 (dd, J=8.8, 1.8 Hz, 1 H), 8.09 (d, J=8.8 Hz, 1 H), 8.19 (d, J=8.8 Hz, 1 H), 8.22 (dd, J=8.4, 1.8 Hz, 1 H), 8.25 (s, 1 H), 8.32 (s, 1 H), 8.41 (s, 1 H), 8.88 (s, 1 H).
Anal. Calcd for C42H5oN806 . 2 HCl . 4 H20: C 55.56, H 6.66 , N 12.34. Found: C 55.00, H 6.60, N 12.30
Going reverse…………………..
Intermediate XVIIIb
2.8 preparation of intermediate XVIIIb (A=

To a solution of XVIIb (960 mg, 1.48 mmol) in CH2C12 (25mL) was added HCI (5-6 M in isopropanol, 5 mL). The mixture was stirred at room temperature overnight. The solvent was evaporated, the obtained solid was dried in vacuum and used as such in the next step. 2.8a Alternative preparation of intermediate XVIIIb (A=
XVIIb (19.52 g, 30.1 mmol, 1.00 equiv.) was dissolved in dichloromethane (200 mL) and HCI in isopropanol (5-6 N, 300 mL) was added. The reaction mixture was stirred for 1 hour at room temperature. tBuOMe (1000 mL) was added to the suspension and the slurry was stirred at roomtemperature for 30 minutes. The filtered solid was rinced with tBuOMe (2x 100 mL) and dried under vacuum overnight to afford XVIIIb as a powder (15.2 g). 1H NMR (400 MHz, MeOD-d4) δ ppm 2.15 – 2.37 (m, 2 H), 2.37 – 2.52 (m, 2 H), 2.52 – 2.69 (m, 2 H), 2.69 – 2.88 (m, 2 H), 3.56 – 3.71 (m, 4 H), 5.19 – 5.41 (m, 2 H), 7.90 – 8.02 (m, 3 H), 8.05 (dd, J= 8.6, 1.6 Hz, 1 H), 8.10 – 8.25 (m, 4 H), 8.30 (d, J=1.4 Hz, 1 H), 8.47 (d, J=1.2 Hz, 1 H)
INTERMEDIATE XVIIb
2.7 reparation of intermediate XVIIb (A= PG= Boc)
To boronic ester XVIb (1.22 g, 2.26 mmol), bromide Xllla (1072 mg, 3.39 mmol), sodium bicarbonate (380 mg, 4.52 mmol), Pd(dppf)Cl2 (166 mg, 0.226 mmol) in toluene (50 mL), was added water (1 mL). The resulting mixture was heated at reflux overnight. The reaction mixture was filtered, evaporated to dryness and purified by column chromatography by gradient elution with heptane to ethyl acetate. The collected fractions containing the product were pooled and the volatiles were removed under reduced pressure. The residue (960 mg, 65 %) was used as such in the next reaction.
2.7a Alternative preparation of intermediate XVIIb (A= . PG= Boc)

XVIb (10 g, 18.5 mmol), Xlll’a (8.76 g, 24 mmol), NaHC03 (9.32 g, 111 mmol) and Pd(dppf)Cl2 (lg) were stirred in dioxane/water (140 mL, 6/1) under argon. The mixture was heated to 85 °C for 15 hours. Brine (100 mL ) was added and the mixture was extracted with CH2CI2, after drying on MgSC^, filtration and evaporation of the solvent, the residue was purified by column chromotography by gradient elution with CH2CI2 to EtOAc to afford XVIIb (7 g, 58 %).

To a stirred, deoxygenated solution of Vlllb (20.0 g, 45.2 mmol, 1.00 equiv.), Ilia (20.6 g, 49.7 mmol, 1.1 equiv.) and sodium bicarbonate (11.4 g, 136 mmol, 3.0 equiv.) in 1 ,4-dioxane/water (500 mL, 5: 1) under nitrogen, was added l.,.r-Bis(diphenyi~ phosphmo)ferrocene-paiIadium(]I)dichloride dichJoromethane complex (2.50 g, 4.52 mmol, 0.1 equiv.). The mixture was heated at 80°C under argon for 15 hours and cooled to room temperature. The reaction mixture was diluted with dichloromethane (500 mL) and washed with brine (2 x 150 mL) dried on magnesium sulphate; filtered and evaporated to dryness to afford a dark brown foam (43 g). The foam was purified using silicagel column chromatography (gradient elution with 0-6% MeOH in CH2CI2) to afford XVIIb (19.52 g, 65%) as an off-white powder.
INTERMEDIATE XVIb

Bromide XVb (1890 mg, 3.83 mmol), 4,4,4\4\5,5,5\5*-octamethyl-2,2′-bis(l,3,2- dioxaborolane) (2437 mg, 9.59 mmol), KF (390 mg; 6.71 mmol) and (dppf)PdCl2 (281 mg, 0.384 mmol) were dissolved in toluene (50 mL) and heated 3 days at reflux.
The solids were removed by filtration over dicalite and the filtrate was evaporated to dryness on silica. The residue was purified by column chromatography using a heptane to ethylacetate gradient. The fractions containing the product were pooled and the solvent was removed under reduced pressure. The residue (1.22 g, 59 %) was used as such in the next reaction
Under nitrogen, Ilia (25 g, 60.5 mmol), 6-bromonaphthalen-2-yl trifluoromethane- sulfonate (20 g, 56.7 mmol), K3P04 (36.65 g, 173 mmol) and (PPh3)4Pd (717 mg, 0.62 mmol) were stirred in THF (60 mL) and water (15 mL) with the heating mantle at 85 °C (reflux) for 2 hours. CH2CI2 (50 mL) was added and the water layer was separated. The organic layer was dried on MgS04 and after filtration, the filtrate was concentrated resulting in a sticky solid. The residue was purified by column
chromatography (petroleum ether/Ethyl acetate 15/1 to 1/1) to afford XVb (20 g;
40.6 mmol). Compound XVb (1 g, 2.0 mmol), potassium acetate (0.5 g, 5.0 mmol), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bis(l,3,2-dioxaborolane) (1.29 g, 5.0 mmol), and Pd(dppf)Cl2 (0. lg) were stirred in DMF (15 mL) under argon. The mixture was heated at 60°C for 5 hours. After cooling, CH2CI2 (50 mL) was added and the mixture was washed with saturated NaHC03. The water layer was separated and extracted with CH2CI2. The organic layers were combined and dried on MgSC^. After filtration the solvent was removed and the product was purified by column chromatography (gradient elution with petroleum ether/ethyl acetate 10/1 to 1/1) to give of XVIb (0.7 g,1.3 mmol, 65 %) as light yellow solid.
INTERMEDIATE XVb

2,6-Dibromonaphthalene (6.92 g, 24.2 mmol), boronic ester Ilia (2 g, 4.84 mmol), NaHC03 (813 mg, 9.68 mmol), (dppf)PdCl2(710 mg, 0.968 mmol) were dissolved in toluene (75 mL). Water (1 mL) was added and the mixture was heated for 7 hours at reflux. The solids were removed by filtration over dicalite and the filtrate was evaporated to dryness on silica. The residue was purified by column chromatography by gradient elution with heptane to ethylacetate. The appropriate fractions were pooled and the solvent was removed under reduced pressure. The residue (1.89 g, 79 %) was used as such in the next step.
1.2 Preparation of intermediate IIIa (PG= Boc)
IIIaTo a mixture of Ila (200 g, 546 mmol), potassium acetate (160.8 g, 1.64 mol) and 4,4,4*,4*,5,5,5*,5*-octamethyl-2,2,-bis(l,3,2-dioxaborolane) (416 g, 1.64 mol) in DMF (3L) was added Pd(dppf)Cl2 (20 g) under nitrogen gas. The reaction mixture was stirred at 85°C for 15 hours. The mixture was diluted with ethyl acetate, washed with water and brine, dried over magnesium sulfate, the solids removed by filtration, and the solvents of the filtrate were removed under reduced pressure. The residue was purified by silica column chromatography (petroleum ether : ethyl acetate 10: 1 to 2: 1) to afford 125 g of Ilia as a white solid (contains 15% of boronic acid).
INT IIa
1.1 preparation of intermediate Ila (PG= Boc; X= Br)
Ma
To a solution of Boc-Z-Proline (2669 mg, 12.4 mmol) in pyridine/DMF (30 mL, 1/1) was added di(lH-imidazol-l-yl)ketone (2205 mg, 13.6 mmol). The mixture was stirred at 45°C for 2 hours. 4-bromobenzene-l,2-diamine (2319 mg, 12.4 mmol) was added and the mixture was stirred at ambient temperature overnight. The solvent was removed and the residue heated in acetic acid (15 mL) at 100°C for 30 minutes. After
concentration of the residue, the mixture was partitioned between ethyl acetate and a saturated sodium bicarbonate solution. The organic phase was separated and washed with water, after drying over Na2SC”4, the mixture was filtrated and the filtrate was concentrated in vacuum. The obtained residue was purified by flash chromatography using CH2Cl2/EtOAc 90/10 to 50/50, resulting in compound Ila (3.146 g, 69 %).
DO NOT MISS OUT synthesis of XIIIa or XIII’a, this is needed in one of side chain
2.1 preparation of L-boc-prolinol
Borane-methyl sulfide complex (180 mL, 1.80 mol) was added dropwise to a solution of N-Boc- L-Proline (300 g, 1.39 mol) in anhydrous THF (3.0 L) which was cooled to 0°C. When gas evolution ceased, the ice bath was removed and the solution was stirred at 10°C for 18 hours. Thin layer chromatography (TLC) showed that no starting material remained and that the desired product was formed. The solution was cooled to 0°C and methanol (2.4 L) was slowly added. The solvents were removed under reduced pressure. The residue was reconstituted in dichloromethane (1 L), washed with
NaHC03 (500 mL, saturated, aqueous) and brine (500 mL), dried over MgS04, the solids were removed via filtration, and the solvents of the filtrate were removed under reduced pressure to afford a white solid, 260 g (93%), used in the next step without further purification.
2.2 preparation of Z-boc-prolinal
To a solution of Z-boc-prolinol (100 g, 500 mmol) in CH2CI2 (1.5 L) at 0°C were added successively, under vigorous stirring, 2,2,6,6-tetramethylpiperidine-l-oxyl (TEMPO; 1.56 g, 10 mmol) and NaBr (5.14 g, 50 mmol). To the resulting mixture was added dropwise a solution of NaHC03 (6.3 g, 75 mmol) and 6% NaCIO in active chlorine (750 mL, 750 mmol) at 0°C over a period of 1 hour. TLC showed no starting material remained and that the desired product was formed. The mixture was rapidly extracted with dichloromethane (2 x 1.5 L). The organic layers were combined, washed with NaHS04 (10%, 1 L) and KI (4%, 200 mL), then with Na2S203 (10%, 1 L) and brine (1.5 L), dried over MgS04, the solids were removed via filtration, and the solvents evaporated to afford a yellow oil, Z-boc-prolinal, (89 g, 92%>), used in the next step without further purification.
2.3 preparation of intermediate XXIV
ammonia
XXIV
Aqueous ammonia (25~28%>, 200 mL) was added dropwise to a solution of L-boc- prolinal (89 g, 0.44 mol) and glyoxal (183 mL of 40% in water) in methanol (1 L). The reaction mixture was sealed and reacted at 10°C. After 16 hours, additional glyoxal (20 mL) and aqueous ammonia (20 mL) were added and reacted for an additional 6 hours. The solvents were removed under reduced pressure, and the crude was reconstituted in ethyl acetate (1.0 L), washed with water and brine, dried over MgSC^, the solids were removed via filtration and the solvents were removed under reduced pressure. The crude was purified by column chromatography (silica gel, dichloromethane to methanol/dichloromethane 1 :70) to obtain 73 g (70%) intermediate XXIV as a white solid.
1H NMR: (CD3OD 400 MHz) δ 6.95 (s, 2H), 4.82-4.94 (m, 1H), 3.60-3.70 (m, 1H), 3.41-3.50 (m, 1H), 2.20-2.39 (m, 1H), 1.91-2.03 (m, 3H), 1.47 (s, 3H), 1.25 (s, 6H)
2.4 preparation of intermediate XHIa (PG= Boc)
XXIV Xllla
N-Bromosuccinimide (47.2 g, 0.26 mol) was added portion wise over 1 hour to a cooled (ice-ethanol bath, -10 °C) solution of XXIV (63.0 g, 0.26 mol) in CH2C12 (1.5 L) and stirred at similar temperature for 2 hours. The reaction mixture was concentrated in vacuum and the residue was purification by preparatory HPLC to provide 25.3 g (30%) of Xllla as a pale yellow solid.
1H NMR: CD3OD 400Mhz
δ 6.99-7.03 (s,lH), 4.77-4.90 (m, 1H), 3.61-3.68 (m, 1H), 3.42-3.50 (m, 1H), 2.20-2.39 (m, 1H), 1.89-2.05 (m, 3H), 1.47 (s, 3H), 1.27 (s, 6H).
2.4a preparation of intermediate XHI’a (PG= Boc)
To a solution of iodine (43.3 g, 170.5 mmol, 2 eq) in chloroform (210 mL) in a round bottomed flask (1L) a suspension of XXIV (20 g, 84.3 mmol) in an aqueous NaOH solution (2M, 210 mL) was added. The mixture was stirred at room temperature for 15 hours. To the resulting reaction mixture was added a saturated aqueous Na2S2C”3 solution (100 mL) and the organic layer was separated. The aqueous layer was extracted with chloroform (4x 150 mL). The organic layers were combined, washed with water and dried on magnesium sulphate. The solids were filtered and the solution was evaporated to dryness to afford diiodide (38.61 g, 89 %).
The above obtained intermediate diiodide (2.24 g, 4.58 mmol) and sodium sulfite (4.82 g, 38 mmol) were placed in a round bottomed flask (100 mL) and suspended in 30% EtOH/water (80 mL). The resulting mixture was refluxed for 40 hours. The solvent was removed and after addition of H20 (20 mL), the mixture was stirred at room temperature overnight. The solids were filtered, washed with water and dried in a vacuum oven to afford compound XHI’a (1.024 g, 61 %).
1H NMR (400 MHz, DMSO-d6) δ ppm 1.16 and 1.38 (2x br. s., 9 H), 1.68 – 2.02 (m, 3 H), 2.02 – 2.27 (m, 1 H), 3.18 – 3.38 (m, 1 H), 3.38 – 3.59 (m, 1 H), 4.53 – 4.88 (m, 1 H), 6.81 (m, -0.1 H), 7.05 – 7.28 (m, -0.9 H), 11.90 – 12.20 (m, -0.9 H), 12.22 – 12.40 (m, -0.1 H)
PATENT
WO 2011149856
http://www.google.co.in/patents/WO2011149856A1?cl=en
1st scheme

IN ABOVE SCHEME CONVERSION OF f to g N-methoxycarbonyl-L-Val-OH is used,
USE R =H IN LAST STEP TO GET RAVIDASVIR
EXAMPLE 1 – Synthesis of compounds of Formula lie
Scheme 1-1 describes preparation of target molecules and their analogs with symmetrical and non-symmetrical functionalized ends.
[0341] Step a. To a solution of 2-bromonaphthane a (62.0 g, 300 mmol) in DCM (1 L) was added A1C13 (44.0 g, 330 mmol) and 2-chloroacetyl chloride (34.0 g, 330 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h and then H20 added (500 mL) and extracted. The organic layer was washed with H20, dried over anhydrous Na2S04, evaporated under reduced pressure to give 80 g crude product, which was purified by re-crystallization from 10% EtOAc- hexane (v/v) to yield b (28 g, 36% yield) as a white solid: JH NMR (500 MHz, CDC13) δ 8.44 (s, 1H), 8.07 (s, 1H), 8.04 (d, J= 11.0 Hz, 1H), 7.84 (d, J= 8.5 Hz, 2H), 7.66 (d, J= 8.5 Hz, 1H), 4.81 (s, 2H) ppm; LCMS (ESI) m/z 282.9 (M + H)+.
Step b. To a solution of b (28.0 g, 100 mmol) in DCM (500 mL) was added N-Boc- L-Pro-OH (24.7 g, 115 mmol) and Et3N (70.0 mL, 500 mmol) and the mixture was stirred at rt for 2 h. The mixture was concentrated under reduced pressure to afford crude c which was used for the next step without further purification. LC-MS (ESI) m/z 462.1 (M + H)+.
Step c. To a solution of c (46.0 g, 100 mmol) in toluene (500 mL) was added
NH4OAc (77 g, 1.0 mol) and the mixture was stirred at 110 °C overnight, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (petroleum ether/EtOAc l :l(v/v)) to afford d (30 g, 68% yield) as a yellow solid: LC-MS (ESI) m/z 442 A (M + H)+.
Step d. To a solution of d (10.0 g, 23.0 mmol) in anhydrous DME (200 mL) and equal molar of boronate e was added PPh3 (1.2 g, 4.6 mmol), Pd(PPh3)4 (1.6 g, 2.3 mmol), and 2.0 M Na2C03 solution. The mixture was refluxed under argon overnight. The organic solvent was removed under reduced pressure and the residue was treated with H20, extracted with EtOAc (2 x 200 mL). The combined organic phase was dried, filtered, and concentrated in vacuo to give a residue, which was purified by silica gel column chromatography (petroleum
ether/EtOAc 3: l(v/v)) to afford f (10 g, 96% yield) as a yellow solid. LC-MS (ESI): m/z 709.3 (M+H)+.
Step e. To a stirred solution of f (150 mg, 0.29 mmol) in dioxane (3 mL) was added 4.0 N HCl in dioxane (3 mL) dropwise. The mixture was stirred at rt for 4 h, and then
concentrated to yield a yellowish solid (134 mg), which was used directly for the next step. The residue (134 mg, 0.290 mmol) was suspended in THF (5 mL) and DIPEA (0.32 mL) was added and followed by addition of N-methoxycarbonyl-L-Val-OH (151 mg, 0.860 mmol). After stirring for 15 min, HATU (328 mg, 0.860 mmol) was added and the mixture was stirred at rt for another 2 h and then concentrated. The residue was purified by prep-HPLC to obtain g (40 mg, 19% yield).
2nd scheme

SCHEME SIMILAR UPTO PENULTIMATE STEP
Note 9 is not final product pl ignore it
Step a. Referring to Scheme 1-2, to a solution of compound 3 (2.0 g, 4.5 mmol) in dioxane (25 mL) was added 4.0 N HCl in dioxane (25 mL). After stirring at rt for 4 h, the reaction mixture was concentrated and the residue was dried in vacuo to give a yellowish solid (2.1 g), which was used directly for the next step without further purification.
[0347] Step b. To the residue of step a (4.5mmol) was added DMF (25 mL), followed by adding HATU (2.1 g, 5.4 mmol), DIPEA (3.7 mL, 22.5 mmol) and N-methyl carbamate-L-valine (945 mg, 5.4 mmol). After stirring at rt for 15 min, the reaction mixture was added slowly to H20 (400 mL). A white solid precipitated was filtered and dried to give compound 6 (2.2 g, 98% yield). LC-MS (ESI): m/z 499.1 (M+H)+.
[0348] Step c. To a mixture of compound 6 (800 mg, 1.6 mmol), compound 7 (718 mg, 1.6 mmol), and NaHC03 (480 mg, 5.7 mmol) in 1 ,2-dimethoxyethane (15mL) and H20 (5mL) was added Pd(dppf)Cl2 (59 mg, 0.08 mmol). After stirring at 80°C overnight under an atmosphere of N2, the reaction mixture was concentrated. The residue was partitioned between 20%
methanol/CHCl3 (100 mL) and H20 (100 mL). The organic phase was separated and the aqueous phase was extracted with 20% methanol/CHCl3 (100 mL) again. The combined organic phase was consequently washed with brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (Petroleum
ether/EtOAc=15: l(v/v)) to give compound 8 (1.0 g, 85% yield) as a yellow solid. LC-MS (ESI): m/z 732.4 (M+H)+.
Step d. To a solution of compound 8 (200 mg, 0.27 mmol) in dioxane (3.0 mL) was added 4 N HCl in dioxane (3.0 mL). After stirring at rt for 2 h, the reaction mixture was concentrated and the residue was dried in vacuo to give an HCl salt in quantitative yield, which was used directly for the next step without further purification…………..CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
CAUTION SIMILAR BUT NOT SAME……..Step e. To a solution of the salt (0.27 mmol) in DMF (5.0 mL) was added DIPEA (0.47mL, 2.7 mmol), followed by adding N,N-dimethyl-D-phenyl glycine (59 mg, 0.33 mmol) and HATU (125 mg, 0.33 mmol). After stirring at rt for lh, the reaction mixture was partitioned between H20 and DCM. The organic phase was washed successively with H20 and brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by prep-HPLC to give compound 9……..CAUTION SIMILAR BUT NOT SAME. LC-MS (ESI): m/z 793.4 (M+H)+.
3rd scheme

SCHEME SIMILAR UPTO PENULTIMATE STEP
15 NOT THE COMPD PL IGNORE IT IF YOU NEED RAVIDASVIR
Step a. To a mixture of compound 3 (3.2 g, 7.2 mmol), bis(pinacolato)diboron (3.86 g, 15.2 mmol), and KOAc (1.85g, 18.8mmol) in 1,4-dioxane (100 mL) was added Pd(dppf)Cl2 (440 mg, 0.6 mmol). After stirring at 80 °C for 3 h under an atmosphere of N2, the reaction mixture was concentrated. The residue was purified with silica gel column chromatography (Petroleum ether/EtOAc=2/l(v/v)) to give compound 11 (2.8 g, 80% yield) as a white solid. LC- MS (ESI): m/z 490.3 (M+H)+.
[0352] Step b. To a mixture of compound 11 (626 mg, 1.27 mmol), compound 12 (570 mg, 1.27 mmol), and NaHC03 (420 mg, 4.99 mmol) in 1, 2-dimethoxyethane (30 mL) and H20 (10 mL) was added Pd(dppf)Cl2 (139 mg, 0.19 mmol). After stirring at 80°C overnight under an atmosphere of N2, the reaction mixture was concentrated. The residue was partitioned between 20% methanol/CHCl3 (100 mL) and H20 (100 mL). The aqueous phase was extracted with 20% methanol/CHCl3 (100 mL) again. The combined organic phase was consequently washed with brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (Petroleum ether/EtOAc=2/l(v/v)) to give compound 13 (635 mg, 68% yield) as a yellow solid. LC-MS (ESI): m/z 732.4 (M+H)+.
Step c. To a solution of compound 13 (200 mg, 0.27 mmol) in dioxane (3.0 mL) was added 4 N HC1 in dioxane (3.0 mL). After stirring at rt for 2 h, the reaction mixture was concentrated and the residue was dried in vacuo to yield the HC1 salt of compound 14 in quantitative yield, which was used directly for the next step without further purification…..CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
CAUTION SIMILAR BUT NOT SAME………Step d. To a solution of the salt (0.27 mmol) in DMF (5.0 mL) was added DIPEA (0.47 mL, 2.7 mmol), followed by adding N,N-dimethyl-D-phenyl glycine (59 mg, 0.33 mmol) and HATU (125 mg, 0.33 mmol). After stirring at rt for lh, the reaction mixture was partitioned between H20 and DCM. The organic phase was consequently washed with H20 and brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by prep-HPLC to give compound 15..CAUTION SIMILAR BUT NOT SAME. LC-MS (ESI): m/z 793.4 (M+H)+.
4 th scheme

SCHEME SIMILAR UPTO PENULTIMATE STEP
5 NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
4 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
scheme ……..CAUTION SIMILAR BUT NOT SAME
EXAMPLE 2 – Synthesis of compounds of Formula Hie
Step a. Referring to Scheme 2-1, to a mixture of compound 1 (5.05 g, 13.8 mmol), bis(pinacolato)diboron (7.1 g, 27.9 mmol), and KOAc (3.2 g, 32.5 mmol) in 1,4-dioxane (100 mL) was added Pd(dppf)Cl2 (400 mg, 0.5 mmol). After stirring at 80 °C for 3 h under an atmosphere of N2, the reaction mixture was concentrated. The residue was purified by silica gel column chromatography (Petroleum ether/EtOAc=2/l(v/v)) to give compound 2 (3.0 g, 53% yield) as a gray solid. LC-MS (ESI): m/z 414.2 (M+H)+.
Step b. To a mixture of compound 2 (522 mg, 1.26 mmol), compound 3 (500 mg, 1.13 mmol), and NaHC03 (333 mg, 3.96 mmol) in 1, 2-dimethoxyethane (30 mL) and H20 (10 mL) was added Pd(dppf)Cl2 (74 mg, 0.1 mmol). After stirring at 80°C overnight under an atmosphere of N2, the reaction mixture was concentrated. The residue was partitioned between 20% methanol/CHCl3 (100 mL) and H20 (100 mL). The organic phase was separated and the aqueous phase was extracted with 20% methanol/CHCl3 (100 mL) again. The combined organic phase was consequently washed with brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (DCM/MeOH=50:l (v/v)) to give compound 4 (450 mg, 55% yield) as a yellow solid. LC-MS (ESI): m/z 649.3 (M+H)+.
Step c. To a stirred solution of compound 4 (160 mg, 0.25 mmol) in dioxane (2.0 mL) was added 4N HCl in dioxane (2.0 mL). After stirring at rt for 3h, the reaction mixture was concentrated and the residue was dried in vacuo to give an HCl salt in quantitative yield, which was used directly for the next step without further purification.4 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
SCHEME SIMILAR UPTO PENULTIMATE STEP
5 NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
scheme ……..CAUTION SIMILAR BUT NOT SAME
5 th scheme

SCHEME SIMILAR UPTO PENULTIMATE STEP
18NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
17 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
scheme ……..CAUTION SIMILAR BUT NOT SAME
Step a. Referring to Scheme 2-2, to a mixture of compound 2 (1.16 g, 2.32 mmol), compound 6 (1.40 g, 3.39 mmol), and NaHC03 (823 mg, 9.8 mmol) in 1, 2-dimethoxyethane (30 mL) and H20 (10 mL) was added Pd(dppf)Cl2 (103 mg, 0.14 mmol). After stirring at 80 °C over night under an atmosphere of N2, the reaction mixture was concentrated. The residue was partitioned between 20% methanol/CHCl3 (150 mL) and H20 (150 mL). The aqueous phase was extracted with 20% methanol/CHCl3 (150 mL) again. The combined organic phase was consequently washed with brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (Petroleum ether/acetone=1.5/l (v/v)) to give compound 16 (1.32g, 80% yield) as a yellow solid. LC-MS (ESI): m/z 706.4 (M + H)+.
tep b. To a solution of compound 16 (200 mg, 0.28 mmol) in dioxane (3.0 mL) was added 4 N HC1 in dioxane (3.0 mL). After stirring at rt for 2 h, the reaction mixture was concentrated and the residue was dried in vacuo to give the HC1 salt of compound 17 in quantitative yield, which was used directly for the next step…….17 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
6 th scheme
 scheme 2-3
scheme 2-3
SCHEME SIMILAR UPTO PENULTIMATE STEP
22NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
21 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
scheme ……..CAUTION SIMILAR BUT NOT SAME
Scheme 2-3
Step a. Referring to Scheme 2-3, to a solution of compound 1 (4.0 g, 10.9 mmol) in dioxane (40 mL) was added 4 N HC1 in dioxane (40 mL). After stirring at rt overnight, the reaction mixture was concentrated. The residue was washed with DCM, filtered, and dried in vacuo to afford a hydrochloride salt in quantitative yield, which was used for the next step without further purification.
Step b. To a solution of the salt (10.9 mmol) in DMF (30 mL) was added DIPEA (5.8 mL, 33.0 mmol), followed by adding N-methoxycarbonyl-L-valine (2.1 g, 12.1 mmol) and HATU (4.6 g, 12.1 mmol). After stirring at rt for lh, the reaction mixture was partitioned between H20 and DCM. The organic phase was consequently washed with H20 and brine, dried with anhydrous Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (DCM/Petroleum ether=4/l (v/v)) to give compound 19 (3.0 g, 65% yield). LC- MS (ESI): m/z 423.1 (M+H)+.
Step c. To a mixture of compound 11 (800 mg, 1.9 mmol), compound 19 (700 mg, 1.7 mmol), and NaHC03 (561 mg, 6.6 mmol) in 1, 2-dimethoxyethane (60 mL) and H20 (20 mL) was added Pd(dppf)Cl2 (183 mg, 0.25 mmol). After stirring at 80 °C overnight under an atmosphere of N2, the reaction mixture was concentrated. The residue was then partitioned between 20% methanol/CHCl3 (100 mL) and H20 (100 mL). The aqueous phase was extracted with 20% methanol/CHCl3(100 mL) again. The combined organic phase was consequently washed with brine, dried with Na2S04, filtered, and concentrated. The residue was purified by silica gel column chromatography (Petroleum ether/EtOAc=2/l(v/v)) to give compound 20 (600 mg, 52% yield) as a yellow solid. LC-MS (ESI): m/z 706.4 (M+H)+.
Step d. To a solution of compound 20 (200 mg, 0.28 mmol) in dioxane (3.0 mL) was added 4N HC1 in dioxane (3.0 mL). After stirring at rt for 2h, the reaction mixture was concentrated and the residue was dried in vacuo to yield the HC1 salt of compound 21 in quantitative yield, which was used directly for the next step without further purification.
21 CAN BE USED AS PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
7 th scheme

Scheme 6-2
SCHEME SIMILAR UPTO n-2 STEP in above scheme
84, 85 NOT THE COMPD, PL IGNORE IT IF YOU NEED RAVIDASVIR
83 CAN BE USED AS early PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
scheme ……..CAUTION SIMILAR BUT NOT SAME
Step a. Referring to Scheme 6-2, a solution of compound 78 (50.0 g, 0.30 mol) in THF (500 mL) and H20 (500 mL) was added K2C03 (83 g, 0.60 mol) and (Boc)20 (73. Og, 0.330 mol). After stirring at rt overnight, the reaction mixture was concentrated and the residue was extracted with EtOAc (250 mL x 3). The extracts were combined, washed with brine, and dried with anhydrous Na2S04. The solvent was removed and the residue was dried in vacuo to give crude compound 78 (62 g), which was used for the next step without further purification. LC-MS (ESI) m/z 230.1 (M + H)+.
[0453] Step b. To a solution of compound 78 (60.0 g, 260 mmol) in EtOH (1 L) was slowly added NaBH4 (50.0 g, 1.30 mol) at rt. After stirring at rt overnight, the reaction was quenched by adding acetone (10 mL). The resulting mixture was concentrated and the residue was diluted with EtOAc (500 mL). The mixture was washed with brined and dried in vacuo. The solvent was removed and the residue was purified by silica gel column chromatography (Petroleum ether/EtOAc = 1/1 (v/v)) to give compound 79 (42.0 g, 80% yield) as a white solid. LC-MS (ESI) m/z 202 A (M + H)+.
[0454] Step c. To a solution of compound 79 (30.0 g, 150 mmol) and DMSO (35.0 g, 450 mmol) in DCM (1 L) was added oxalyl chloride (28.0 g, 220 mmol) at -78 °C. After stirring at – 78 °C for 4 h, the reaction mixture was added Et3N (60.0 g, 600 mol) and the resulting mixture was stirred for another 1 h at -78 °C. Subsequently, the reaction was quenched by adding H20. The organic layer was separated and the aqueous layer was extracted with DCM (200mL x 2). The extracts were combined, washed with brine, and dried with Na2S04. The solvent was removed and the residue was dried in vacuo to give crude compound 80 (22.0 g) as a colorless oil, which was used immediately without further purification. LC-MS (ESI) m/z 200.1 (M + H)+.
[0455] Step d. A mixture of compound 80 (7.7 g, 38.5 mmol), 6-bromopyridine-2,3-diamine (8.0 g, 42.8 mmol) (PCT Intl. Appl. WO 2008021851) , and iodine (1.08 g, 4.28 mmol) in AcOH (30 mL) was stirred at rt overnight. The reaction mixture was neutralized by adding saturated aqueous NaHC03. The resulting mixture was extracted with EtOAc (200 mL x 3). The extracts were combined, washed with brine, and dried with anhydrous Na2S04. The solvent was removed and the residue was purified by silica gel column chromatography (DCM/MeOH = 80/1 (v/v)) to give compound 81 (7.8 g, 55% yield). LC-MS (ESI) m/z 367.1 (M + H)+.
[0456] Step e. A mixture of compound 82 (10.0 g, 20.1 mmol), bis(pinacolato)diboron (7.65 g, 30.1 mmol), potassium acetate (6.89 g, 70.3 mmol), and Pd(dppf)Cl2-CH2Cl2 (886 mg, 1.0 mmol) in 1,4-dioxane (200 mL) was stirred at 80 °C for 3 h under an atmosphere of N2. The reaction mixture was filtered through CELITE™ 545 and the filtered cake was washed with EtOAc (200 mL x 3). The filtrate was washed with brine and dried with anhydrous Na2S04. The solvent was removed and the residue was purified by silica gel column chromatography
(DCM/MeOH = 50/1 (v/v)) to give compound 83 (9.8 g, 89% yield) as a white solid: LC-MS (ESI) m/z 547.3 (M + H)+.83 CAN BE USED AS early PRECURSOR FOR RAVIDASVIR UPTO THIS POINT
PATENT
CN 102796084
http://www.google.com/patents/CN102796084A?cl=en
Step One: Formula (2) compounds strokes trichloride catalyst (AlCl3), chloroacetyl chloride (2-chloroacetylchloride) at room temperature to obtain a compound of formula (3),

(3);
wherein the reaction temperature is room temperature, the solvent is methylene chloride. Material I (i.e., formula (2) compound) and chloroacetyl chloride (2-chloroacetyl chloride) was slowly added, higher yields can be obtained. (3) The compound was recrystallized from ether to obtain.
In the present embodiment, the 20.5 g of formula (2) compound (0. Imol) and 26.2 g AlCl3 (0.2mol) was added to 200ml of dichloromethane, cooled to room temperature, stirring speed slowly was added 13.4 g of chloroacetyl chloride (I. 2mol), within three hours after the addition and then mixed by stirring maintained at room temperature for 3 hours. Was slowly added 50 ml of ice water, the precipitate was collected by filtration. The filter cake was washed with 10 ml of water and 10 ml petroleum ether (twice). The filtrate and the organic layer together with 50 ml of dichloromethane and extracted twice with 50 ml brine and then paint extraction solution, the extract was dried over magnesium sulfate, the solution was removed, the solid with 100 ml of diethyl ether and recrystallized to afford 20g (71% yield compounds) of formula (3).
Step II: Formula (3) with a compound of formula (4) compound under acidic conditions and chloroform (CCl3H) heating the reaction, and the reaction system reached reflux to give a compound of formula (5),

(5);
[0042] wherein, the formula (3) with a compound of formula (4) compound in acetonitrile (chloroform (CCl3H), the reaction system must be reached reflux, and must be reacted under acidic conditions to give the compound of formula (5). [0043] In this embodiment, the compound (3) (0. Imol) 28. 2 克 formula and the compound (4) (0. Imol) 21. 5 克 style with 3 g of trifluoroacetic acid was added to 200 ml of chloroform, in was stirred at reflux under nitrogen for 17 hours. After cooling to room temperature, spin-dry, to give 46. I g of a yellow solid of formula (5) compound (99% yield).
Step three: (5) the compound obtained in toluene (toluene) and ammonium acetate (NH4OAc) reflux (6) of
Thereof,

Compound of formula (5) is ammonium acetate with toluene under reflux conditions for ring closure.
In the present embodiment, the compound (0. Imol) and 10 g of ammonium acetate (NH4OAc) was added 46. I g of formula (5) to IJ 200ml of toluene, heated under reflux for 3 hours with stirring. Was slowly added 50 ml of ice water, filtered, washed with 100 ml of toluene and extracted twice with 50 ml brine and then paint extraction solution, the extract was dried over magnesium sulfate, the solution was removed, the solid with 100 ml of diethyl ether and recrystallized to afford 40g (89% compound yield) of the formula (6).
Step Four: (6) compound in the catalyst and the associated button pinacolato ester (Bis (pinacolato) diboron) reacting a compound of formula (7),

wherein, Pd (dppf) 2Cl2 can be replaced by another of a palladium catalyst, a palladium catalyst with the other, the same effect.
In the present embodiment, 44 g of the compound of formula (6) (0. Imol) and 3 g Pd (dppf) 2C12,25. 4 克 United pinacolato ester (0. Imol) and 8.4 g of sodium bicarbonate (0. Imol) was added to a 200 ml I. 4- dioxane, stirred at reflux for 24 hours. Diatomaceous earth filtration, spin dry. Spin-dry 100 ml of ethyl acetate dissolved. Anhydrous magnesium sulfate and spin dry. Recrystallization from ether to yield 40 g (82% yield) of a yellow solid of formula (7) compound.
Step Five: formula (7) under palladium catalyst compound and the compound (8) obtained by reacting the compound of formula (9),

wherein, Pd (dppf) 2Cl2 can be replaced by another of a palladium catalyst, a palladium catalyst with the other, the same effect.
In the present embodiment, 48.9 g of the compound of formula (7) (0. Imol) and 3 g Pd compound (8) (0. Imol) (dppf) 2C12,41. 3 and 8 克 style. 4 g of sodium hydrogen carbonate (0. Imol) was added to a 200 ml I. 4- dioxane, stirred at reflux for 24 hours. Diatomaceous earth filtration, spin dry. Spin-dry 100 ml of ethyl acetate dissolved. Anhydrous magnesium sulfate and spin dry. Recrystallized from ether to give compound 55 g (85% yield) of a yellow solid of formula (9).
[0056] Step Six: formula (9) compound deprotected under acidic conditions to give a compound of formula (10),
[0057]

In the present embodiment, the 64.8 grams of formula (9) compound (0. Imol) was added to 100 ml I. 4_ dioxane was stirred, 100 ml of 5M / L of I under nitrogen 4- dioxane solution of hydrochloric acid. Spin-dry for 24 hours later, get 52. I g of pale yellow solid formula (10) compound (99% yield).
Step 7: Formula (10) with a compound (11) in a condensing agent is 2- (7-azo BTA) -N, N, N ‘, N’- tetramethyluronium hexafluorophosphate phosphate (HATU) under condensation reaction conditions to give the final product compound C0S-101, i.e. the compound of formula (I):

In the present embodiment, the compound of formula 52. I g of (10) (0. Imol) was added to a 200 ml N, N- dimethylformamide (DMF) cooled to 0 ° with stirring, in a nitrogen atmosphere was added 20.2 g of triethylamine (0. 2mol) 0 After 10 minutes of stirring, was added 19 g of formula (11) compound (0. Ilmol) was added followed by 26 g HATU (0. 2mol), stirred at room temperature for 32 hours . Was slowly added 50 ml of ice water, the precipitate was collected by filtration. The filter cake was washed with 10 ml of water and 50 ml dichloromethane twice. Together with the filtrate and the organic layer was extracted 2 times 50 ml of dichloromethane, and then washed with 50 ml brine solution, the extract was dried over magnesium sulfate, the solution was removed, solid was recrystallized from 100 ml of ethanol, to give 50g (66% yield) The pale yellow compound C0S-101.
In summary this compound on C0S-101 non-structural protein 5A inhibitor, or a pharmaceutically acceptable salt thereof, the treatment of hepatitis C active substance. A compound of formula (3) Friedel-Crafts reaction occurs directly from 2-bromo-naphthalene chloride and chlorine. A compound of formula (3) with a compound of formula (4) condensing a compound of formula (5). The compound of formula (5) self-condensation of a compound of formula (6). Of formula (6) is reacted with boronic acid pinacol ester linking reaction of the compound of formula (7). A compound of formula (7) with a compound of formula (8) coupling reaction of a compound of formula (9). Off compound under acidic conditions (9) protect the compound of formula (10) and formula (10) compound condensation of the final product C0S-101, method of operation of the invention is simple, mild conditions, process maturity, yield and high purity suitable for industrial production.
PATENT
WO 2013123092
http://www.google.com/patents/WO2013123092A1?cl=en

Scheme 3

3-3 2HCI salt
Step 1. Referring to Scheme 3, compounds l-5a (1.3 kg , 1.0 eq.), 2-2a (975.0 g, 1.0 eq.), NaHCOs (860.0 g, 3.80 eq.), Pd(dppf)Cl2 (121.7 g, 0.05 eq.), purified water (5.2 L, 4.0 volume) and 1 ,2-dimethoxy ethane (DME) (24.7 L, 19.0 volume) were charged into a 50.0 L 4-necked round bottom flask under argon atmosphere. After being degassed using argon for a period of 30 min, the reaction mass was slowly heated to ~ 80 °C and stirred at this temperature for 12 – 14 hrs. HPLC analysis indicated that > 97% of compound 2-2a was consumed. Next, the reaction mass was concentrated to completely remove DME under vacuum (600 mmHg) at 40 – 45 °C and the residue was diluted with 20% (v/v) MeOH in DCM (13.0 L , 10 volume) and purified water (13.0 L, 10.0 volume) with stirring. The organic layer was separated and the aqueous layer was extracted with 20% (v/v) MeOH in DCM (6.5 L x 2, 10.0 volume). The combined organic extracts were washed twice with water (6.5 L x 2, 10.0 volume) and once with saturated brine (6.5 L, 5.0 volume) and dried over anhydrous Na2S04. The solvent was removed under vacuum (600 mmHg) and the residue was purified by flash column chromatography using silica gel with hexanes/EtOAc as eluent to give compound 3-1 (1.0 kg, 63% yield) as off white solid with a purity of > 98.0%> determined by HPLC analysis. LC-MS (ESI): m/z 649.3 [M + H]+. 1H NMR (400 MHz, d6– DMSO): δ 12.26 – 12.36 (m, 1H), 11.88 – 11.95 (m, 1H), 8.23 (s, 1H), 8.11 (s, 1H), 7.91 (m, 3H), 7.85 – 7.87 (m, 2H), 7.51 – 7.81 (m, 3H), 4.78 -4.99 (m, 2H), 3.55 – 3.59 (m, 2H), 3.35 – 3.44 (m, 2H), 2.30 – 2.47 (m, 2H), 1.85 – 2.01 (m, 6H), 1.39, 1.14, 1.04 (s, s, s, 18H) ppm. Alternatively, compound 3-1 can be obtained following the same procedure and using compounds l-4a and 2-3a instead of compounds l-5a and 2-2a as the Suzuki coupling components.
Step 2. Compound 3-1 (1.0 kg, 1.0 eq.) and IPA (7.0 L, 7.0 volume) were charged into a 20.0 L four-necked RB flask under nitrogen atm. The reaction mass was cooled to 18 – 20°C and 3.0 N HC1 in isopropyl alcohol (7.0 L, 7.0 volume) was added over a period of 90 – 120 min under nitrogen atmosphere. After stirring at 25 – 30 °C for 10 – 12 hrs under nitrogen atmosphere, HPLC analysis indicated that > 98%> compound 3-1 was consumed. Next, the reaction mass was concentrated to remove IPA under vacuum at 40 – 45 °C. The semi solid obtained was added to acetone (2.0 L, 2.0 volume) with stirring and the resulting suspension was filtered under nitrogen atmosphere. The solid was washed with acetone (2.0 L, 2.0 volume) and dried in a vacuum tray drier at 40 – 45 °C for 10 hrs to give compound 3- 2 (860 g, 94%o yield) as pale yellow solid with a purity of > 98.0%> determined by HPLC analysis. LC-MS (ESI): m/z 449.2 [M + H]+. 1H NMR (400 MHz, -DMSO): δ 10.49 – 10.59 (m, 2H), 10.10 and 9.75 (m, m, 2H), 8.60 (s, 1H), 8.31 (s, 2H), 8.15 (m, 1H), 8.13 – 8.15 (m, 2H), 7.96 – 8.09 (m, 2H), 7.82 (s, 2H), 5.08 (m, 2H), 3.39 – 3.53 (m, 4H), 2.47 – 2.54 (m, 3H), 2.37 (m, 1H), 2.14 – 2.21 (m, 2H), 2.08 (m, 2H) ppm.
Step 3. Compound 3-2 (2.2 kg, 1.0 eq.) was added to a four necked round bottom flask charged with DMF (4.4 L, 20.0 volume) under a nitrogen atmosphere. After stirring for 15 min, the mixture was added N-Moc-L-Valine (226.2 g, 3.52 eq.) in one lot at 25 – 30 °C. Next, the mixture was cooled to -20 to -15 °C, followed by adding HATU (372.9 g, 2.0 eq.) portion wise over 30 min. After stirring for 10 min, a solution of DIPEA (238.9 g, 5.0 eq.) in DMF (1.1 L, 5.0 volume) was added over 45 min. Subsequently, the reaction mass was warmed to 25 – 30 °C with stirring. After stirring for 1 hr, HPLC analysis indicated that > 99%) of compound 3-2 was consumed. The reaction mixture was poured into water (38.0 L) and the mixture was extracted with DCM (10.0 L x 3, 45.0 volume). The combined organic extracts were washed with water (10.0 L x 3, 45.0 volume) and saturated brine (10 L, 45.0 volume) and dried over anhydrous Na2S04. The solvent was removed at 40 – 45 °C under vacuum (600 mmHg) and the residue was purified by column chromatography on silica gel using DCM and MeOH as the eluent to give compound 3-3 (1.52 kg, 47% yield) as off white solid with a purity of > 97.0% determined by HPLC analysis. LC-MS (ESI): m/z 763.4 [M + H]+. 1H NMR (400 MHz, -DMSO): δ 8.60 (s, 1H), 8.29 (s, 1H), 8.20 (s, 1H), 8.09 – 8.14 (m, 2H), 7.99 – 8.05 (m, 2H), 7.86 – 7.95 (m, 3H), 7.20-7.21 (m, 2H), 5.24 – 5.33 (m, 2H), 4.06 – 4.18 (m, 4H), 3.83 (m, 2H), 3.53 (m, 6H), 2.26 – 2.55 (m, 10H), 0.85 (m, 6H), 0.78 (m, 6H) ppm. The transformation of 3-2 to 3-3 (Compound I) can be achieved via a range of conditions. One of these conditions is described below.
A reactor was charged with N-Moc-V aline (37.15 g, 0.211 mol), acetonitrile (750 mL) and DIPEA (22.5 g). The reaction mixture was agitated for 10 min and HOBT (35.3 g 0.361 mole) and EDCI (42.4 g, 0.221 mole) were added while keeping temperature < 2 °C. The reaction mixture was agitated for 30 min and DIPEA (22.5 g) and compound 3-2 (48.0 g, 0.092 mole) was added slowly to reactor over 30 min to keep temperature < 3 °C. The reaction mixture was agitated 4 hrs at 20 – 25 °C, and sample was submitted for reaction completion analysis by HPLC (IPC specification: < 1.0% area 3-2 remaining). At the completion of reaction as indicated by HPLC analysis, isopropyl acetate (750 mL) was added to the reactor and stirred for 10 min. The organic layer (product layer) was washed with brine (300 mL x 2) and 2% NaOH (200 mL). The organic solution was filtered through a silica gel pad to remove insoluble material. The silica gel pad was washed with isopropyl acetate and concentrated under vacuum (400 mm/Hg) to a minimum volume. The crude product was purified by column chromatography on silica gel using ethyl acetate and methanol as eluent to give compound 3-3 (38.0 g, 65%> yield) with purity of > 95 %>. LC-MS (ESI): m/z 763.4 [M + H]+.
Step 4. Compound 3-3 (132.0 g, 1.0 eq.) and ethanol (324.0 mL, 2.0 volume) were charged into a 10 L four-necked round bottom flask under nitrogen atmosphere. After stirring for 15 min, the suspension was cooled to 5 – 10 °C, to it was added 2.0 N HC1 in ethanol (190 mL, 1.5 volume) over 30 min. The resulting solution was allowed to warm to 25 – 30 °C. Acetone (3.96 L, 30.0 volume) was added over 90 min in to cause the slow precipitation. Next, the suspension was warmed to 60 °C and another batch of acetone (3.96 L, 30.0 volume) was added over 90 min. The temperature was maintained at 55 – 60 °C for 1 hr, and then allowed to cool to 25 – 30 °C. After stirring at 25 – 30 °C for 8 – 10 hrs, the mixture was filtered. The solid was washed with acetone (660.0 mL, 5.0 volume) and dried in a vacuum tray drier at 50 – 55 °C for 16 hrs to give the di-HCl salt of compound 3-3
(compound I) (101 g, 71% yield) as pale yellow solid with a purity of > 96.6% determined by HPLC analysis.
Preparation of N-Moc-L-Valine
N-Moc-L-Valine is available for purchase but can also be made. Moc-L-Valine was prepared by dissolving 1.0 eq of L-valine hydrochloride in 2-methyltetrahydrofuran (2- MeTHF) /water containing sodium hydroxide and sodium carbonate, and then treating with 1.0 eq of methyl chloroformate at 0 – 5°C for 6 hr. The reaction mixture was diluted with 2- MeTHF, acidified with HC1, and the organic layer was washed with water. The 2-MeTHF solution is concentrated and the compound is precipitated with n-heptane. The solid was rinsed with 2-MeTHF/ n-heptane and dried in vacuo to give N-Moc-L-Valine in 68% yield. Crystallization of Compound I to Yield Form A
Compound I Salt Formation and Crystallization, Example 1
Ethanol (3.19 L, 1.0 volume, 200 proof) was charged to the 230-L glass lined reactor under nitrogen atmosphere. Free base form of compound 3-3 (3.19 kg, 4.18 mol) was added to the flask with stirring, stir continued for an additional 20 to 30 min. To the thick solution of 3-3 in ethanol was added slowly 2.6 N HC1 in ethanol (3.19 L, 1.0 volume) to the above mass at 20 – 25 °C under nitrogen atmosphere. The entire mass was stirred for 20 min at rt, and then heated to 45 – 50 °C. Acetone (128.0 L, 40.0 volume) was added to the above reaction mass at 45 – 50 °C over a period of 3-4 hrs before it was cooled to ~25 °C and stirred for ~15 hrs. The precipitated solid was collected by filtration and washed with acetone (6.4 L x 2, 4.0 volume), suck dried for 1 hr and further dried in vacuum tray drier at 40 – 45 °C for 12 hrs. Yield: 2.5 kg (71.0% yield), purity by HPLC: 97.70%, XRPD: amorphous.
Isopropyl alcohol (7.5 L, 3.0 volume) was charged to a 50.0 L glass reactor protected under a nitrogen atmosphere. The amorphous di-HCl salt of 3-3 (2.5 kg) was added to the above reactor with stirring. The entire mass was heated to 60 – 65 °C to give a clear solution. Stir continued at 65 ± 2 °C for ~15 hrs, solid formation started during this time. The heating temperature was lowered to ~50 °C over a period of 3 hrs, methyl tertiary butyl ether (12.5 L, 5.0 volume) was added to the above mass slowly over a period of ~3 hrs with gentle agitation. The above reaction mass was further cooled to 25 – 30 °C over 2 – 3 hrs. The solid was collected by filtration, washed with 10.0% isopropyl alcohol in methyl tertiary butyl ether (6.25 L, 2.5 volume), suck dried for 1 hr and further dried in a tray drier at 45 – 50 °C under vacuum (600 mm/Hg) for 70 – 80 hrs. Yield: 2.13 kg (85.0% recovery, 61.0% yield based on the input of compound free base 3-3), purity by HPLC: 97.9%.
FIG. 1 : 1H NMR (500 MHz, -DMSO): δ 15.6 (bs, 2H), 14.7 (bs, 2H), 8.58 (s, 1H), 8.35 (s, 1H), 8.25 (s, 1H), 8.18 (d, J= 8.7 Hz, 1H), 8.13 (s, 1H), 8.06 (d, J= 8.6 Hz, 1H), 8.04 (s, 1H), 8.00 (s, 1H), 7.98 (d, J= 8.7 Hz, 1H), 7.91 (d, J= 8.6 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 7.33 (d, J= 8.6 Hz, 2H), 5.31 (m, 1H), 5.26 (m, 1H), 4.16 (d, J= 7.7 Hz, 1H), 4.04 (m, 2H), 3.87 (m, 2H), 3.55 (s, 6H), 2.42 (m, 2H), 2.22-2.26 (m, 4H), 2.07-2.14 (m, 4H), 0.86 (d, J= 2.6 Hz, 3H), 0.84 (d, J= 2.6 Hz, 3H), 0.78 (d, J= 2.2 Hz, 3H), 0.77 (d, J= 2.2 Hz, 3H), 3.06 (s, OMe of MTBE), 1.09 (s, t-Bu of MTBE), 1.03 (d, 2Me of IP A) ppm.
FIG. 2: 13C NMR (500 MHz, /-DMSO): δ 171.6, 171.5, 157.4, 156.1, 150.0, 138.2, 138.0, 133.5, 132.5, 131.3, 129.8, 129.4, 128.0, 127.0, 126.4, 125.6, 125.3, 124.4, 124.2, 115.8, 115.0, 112.5, 58.37, 58.26, 54.03, 53.34, 52.00 (2 carbons), 47.71 (2 carbons), 31.52, 31.47, 29.42 (2 carbons), 25.94, 25.44, 20.13, 20.07, 18.37, 18.36 ppm.
FIG. 3: FT-IR (KBr pellet): 3379.0, 2963.4, 2602.1, 1728.4, 1600.0, 1523.4, 1439.7, 1420.6, 1233.2, 1193.4, 1100.9, 1027.3 cm“1.
Elemental Analysis: Anal. Calcd for C42H52C12N806: C, 60.35; H, 6.27; N, 13.41; CI, 8.48. Found C, 58.63; H, 6.42; N, 12.65, CI, 8.2.
FIG. 1 is a representative 1H NMR spectrum of Compound I Form A.
FIG. 2 is a representative 13C NMR spectrum of Compound I Form A.
FIG. 3 is a representative FT-IR spectrum of Compound I Form A.
References:
1. Lalezari, J. P.; et. al. PPI-668, a potent new pan-genotypic HCV NS5A inhibitor: phase 1 efficacy and safety. Hepatology 2012, 56, 1065A-1066A.
- ClinicalTrials.govA Study of the Efficacy and Safety of PPI-668 (NS5A Inhibitor) Plus Sofosbuvir, With or Without Ribavirin, in Patients With Chronic Hepatitis C Genotype-4. NCT02371408(retrieved on 24-03-2015)
3. ClinicalTrials.gov Study of PPI-668, BI 207127 and Faldaprevir, With and Without Ribavirin, in the Treatment of Chronic Hepatitis C. NCT01859962 (retrieved on 15-09-2015)
4. Lalezari, J.; et. al. High rate of sustained virologic response in patients with hcv genotype-1a infection: a phase 2 trial of faldaprevir, deleobuvir and ppi-668, with and without ribavirin. EASL-The International Liver Congress 2014– 49th Annual Meeting of the European Association for the Study of the Liver London, United Kingdom April 9-13 (article here)
| US20070185175 * | 27 Jul 2006 | 9 Aug 2007 | Bristol-Myers Squibb Company | Benzothiazole and azabenzothiazole compounds useful as kinase inhibitors |
| US20080050336 * | 8 Aug 2007 | 28 Feb 2008 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| WO2012087976A2 * | 19 Dec 2011 | 28 Jun 2012 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| WO2013123092A1 * | 13 Feb 2013 | 22 Aug 2013 | Presidio Pharmaceuticals, Inc. | Solid forms comprising inhibitors of hcv ns5a, compositions thereof, and uses therewith |
| WO2013158776A1 * | 17 Apr 2013 | 24 Oct 2013 | Gilead Sciences, Inc. | Compounds and methods for antiviral treatment |
| US8765731 | 16 Nov 2012 | 1 Jul 2014 | Vertex Pharmaceuticals Incorporated | Benzimidazole analogues for the treatment or prevention of flavivirus infections |
| US8779156 | 24 Sep 2012 | 15 Jul 2014 | Vertex Pharmaceuticals Incorporated | Analogues for the treatment or prevention of flavivirus infections |
| US8809330 | 1 Nov 2013 | 19 Aug 2014 | Gilead Sciences, Inc. | Pyrazolo[1,5-A]pyrimidines for antiviral treatment |
| US8946238 | 20 Dec 2012 | 3 Feb 2015 | Gilead Sciences, Inc. | Pyrazolo[1,5-A]pyrimidines as antiviral agents |
| US8980878 | 17 Apr 2013 | 17 Mar 2015 | Gilead Sciences, Inc. | Compounds and methods for antiviral treatment |
| US20110274648 * | 4 Nov 2010 | 10 Nov 2011 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
////////////Phase III, Hepatitis C, RAVIDASVIR, PPI-668, BI 238630
