

Graphical abstract

WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » Posts tagged 'PFIZER' (Page 4)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PALBOCICLIB
Mechanism of action: selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6
Indication: Estrogen receptor-positive (ER+), HER2-negative (HER2 -) breast cancer
February 3, 2015
syn……….https://newdrugapprovals.org/2014/01/05/palbociclib/
The U.S. Food and Drug Administration today granted accelerated approval to Ibrance (palbociclib) to treat advanced (metastatic) breast cancer.
Breast cancer in women is the second most common type of cancer in the United States. It forms in the breast tissue and in advanced cases, spreads to surrounding normal tissue. The National Cancer Institute estimates that 232,670 American women were diagnosed with breast cancer and 40,000 died from the disease in 2014.
Ibrance works by inhibiting molecules, known as cyclin-dependent kinases (CDKs) 4 and 6, involved in promoting the growth of cancer cells. Ibrance is intended for postmenopausal women with estrogen receptor (ER)-positive, human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer who have not yet received an endocrine-based therapy. It is to be used in combination with letrozole, another FDA-approved product used to treat certain kinds of breast cancer in postmenopausal women.
“The addition of palbociclib to letrozole provides a novel treatment option to women diagnosed with metastatic breast cancer,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The FDA is committed to expediting marketing approval of cancer drugs through our accelerated approval regulations.”
syn……….https://newdrugapprovals.org/2014/01/05/palbociclib/
The FDA granted Ibrance breakthrough therapy designation because the sponsor demonstrated through preliminary clinical evidence that the drug may offer a substantial improvement over available therapies. It also received a priority review, which provides for an expedited review of drugs intended to provide a significant improvement in safety or effectiveness in the treatment of a serious condition or meet an unmet medical need. Ibrance is being approved more than two months ahead of the prescription drug user fee goal date of April 13, 2015, the date when the agency was scheduled to complete its review of the application.
Ibrance is being approved under the FDA’s accelerated approval program, which allows approval of a drug to treat a serious or life-threatening disease based on clinical data showing the drug has an effect on a surrogate endpoint reasonably likely to predict clinical benefit to patients. This program provides earlier patient access to promising new drugs while the company conducts confirmatory clinical trials.
The drug’s efficacy was demonstrated in 165 postmenopausal women with ER-positive, HER2-negative advanced breast cancer who had not received previous treatment for advanced disease. Clinical study participants were randomly assigned to receive Ibrance in combination with letrozole or letrozole alone. Participants treated with Ibrance plus letrozole lived about 20.2 months without their disease progressing (progression-free survival), compared to about 10.2 months seen in participants receiving only letrozole. Information on overall survival is not available at this time.
The most common side effects of the drug were a decrease in infection-fighting white blood cells called neutrophils (neutropenia), low levels of white blood cells (leukopenia), fatigue, low red blood cell counts (anemia), upper respiratory infection, nausea, inflammation of the lining of the mouth (stomatitis), hair loss (alopecia), diarrhea, low blood platelet counts (thrombocytopenia), decreased appetite, vomiting, lack of energy and strength (asthenia), damage to the peripheral nerves (peripheral neuropathy) and nosebleed (epistaxis). Healthcare professionals should inform patients of these risks.
It is recommended that treatment begin with a 125 milligram dose for 21 days, followed by seven days without treatment. Healthcare professionals are advised to monitor complete blood count prior to start of therapy and at the beginning of each cycle, as well as on Day 14 of the first two cycles, and as clinically indicated.
Ibrance is marketed by New York City-based Pfizer, Inc.
see synthesis……….https://newdrugapprovals.org/2014/01/05/palbociclib/
New York City-based Pfizer, Inc.


Pfizer World Headquarters building in New York City. Zoetis, based in Madison, N.J., traces its roots back to 1952 as a Pfizer unit and has made at least 10 …
 Pfizer’s NYC headquarters
Pfizer’s NYC headquarters


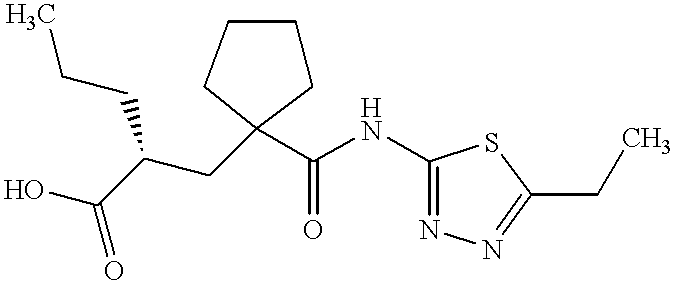
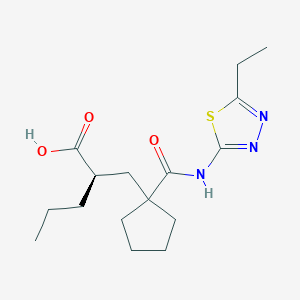
UK-414,495
Molecular Formula: C16H25N3O3S
Molecular Weight: 339.453
UK 414495
CAS 388630-36-2
OF
(-)-(2R)-2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid;
AND
Cyclopentanepropanoic acid, 1-[[(5-ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]-α-propyl-, (αR)-
((R)-2-({1-[(5-ethyl-1,3,4-thiadiazol-2-yl) carbamoyl]cyclopentyl}methyl) valeric acid)
(2R)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
…………………………………………………
Cas 337962-93-3 RACEMIC…………2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid

…………………………………………………………………..
ITS ENANTIOMER
(+)-(2S)-2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid……………337962-74-0
CAS SUMMARY
| Cas number | 388630-36-2 337962-74-0 (enantiomer) 337962-93-3 (racemate) 388630-59-9 (sodium salt) |

 desired
desired
UK-414,495 is a drug developed by Pfizer for the treatment of female sexual arousal disorder.[1] UK-414,495 acts as a potent, selective inhibitor of the enzyme neutral endopeptidase, which normally serves to break down the neuropeptide VIP. The consequent increase in VIP activity alters blood flow to the genital region leading to increased lubrication and muscle relaxation.[2][3][4]

A female equivalent of Viagra could soon be available to help women increase their sexual arousal, scientists claim.
For years they have endeavoured to create an alternative for women that mimics the effects of the male Viagra pill.
Now, the pharmaceutical company behind the original pill has created a prototype which increases blood flow to the genitalia in a similar way to Viagra.

Pfizer have come up with a prototype version of the female equivalent of Viagra
More than half of women experience sexual dysfunction at some point in their lives.
They may suffer a lack of desire, emotional or mental health problems and physical problems that mean they avoid having sex.
Pharmaceutical giant Pfizer has developed a drug, so far called only UK-414,495, which is supposed to increase sexual arousal, but will not affect desire, mood or emotional problems.
Some women take Viagra with mixed results and the drug has been used in fertility treatment to increase blood flow to the pelvis and encourage an embryo to implant in the womb.
But this is the first pill that claims to be an equivalent of the male Viagra.
The research, which involved animals, is published by the British Journal of Pharmacology, though Pfizer say they won’t develop the drug and warn that the chemical may not work the same way in humans, according to the Telegraph.
Chris Wayman, the lead researcher, said: ‘Before this work, we knew surprisingly little about the processes that control all of these changes.

Pfizer claim the tablets may help overcome female sexual arousal disorder
‘Now that we are beginning to establish the pathways involved in sexual arousal, scientists may be able to find ways of helping women who would like to overcome female sexual arousal disorder.
‘While the particular chemical compound in this research did not prove appropriate for further developments, the implications of the research could lead to the development of a product in the future.’
Viagra was originally developed as a treatment for high blood pressure and the heart condition angina, but men who took part in early trials realised the drug had an interesting side effect.
Clinical trials suggested the drug had little effect on angina and instead induced erections in men.
The drug first went on sale in 1998 and has since been prescribed to 25million men, creating a multi-billion pound global market.
The name Viagra has become so associated with men’s erectile problems that many cures are marketed as ‘herbal viagra’.
It is known by many nicknames, including Vitamin V and the Blue Pill.
Read more: http://www.dailymail.co.uk/health/article-1265842/Female-Viagra-help-women-increase-sexual-arousal.html#ixzz39lkmpSik
…………………………………

scheme
http://www.google.com/patents/US20020052370




| Ex | Prec | n | Y | Data |
| 43 | Prep 37 | 0 |
 |
1H NMR (CDCl3, 400 MHz) δ: 0.92 (t, 3H), 1.35 (t, 3H), 1.25-1.80 (m, 11H), 2.20-2.50 (m, 4H), 2.95 (q, 2H), 12.10 (bs, 1H); LRMS: m/z 339.8 (MH+) Anal. Found: C, 56.46; H, 7.46; N, 12.36. C16H25N3O3S requires C, 56.62; H, 7.44; N, 12.37%. |
Example 29 (2R)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
[0354]
desired[0355] and
Example 30 (2S)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
[0356]
 undesired
undesired[0357] The acid from Example 4 (824 mg) was further purified by HPLC using an AD column and using hexane:iso-propanol:trifluoroacetic acid (85:15:0.2) as eluant to give the title compound of example 29 as a white foam, 400 mg, 99.5% ee, 1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.36 (m, 6H), 1.50-1.80 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.30 (bs, 1H), LRMS: m/z 338 (MH−), [α]D=−9.0°(c=0.1, methanol),
and
the title compound of example 30 as a white foam, 386 mg, 99% ee, 1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H);
[0358] LRMS: m/z 338 (MH−); and [α]D=+3.8°(c=0.1, methanol).
[0359] Alternatively, Example 29 may be prepared as follows:
[0360] To a solution of the product from Preparation 51a (574 g, 1.45 mol) in dichloromethane (2.87 L) was added trifluoroacetic acid (1.15 L) over a period of 50 minutes with cooling at 10° C. After addition was complete, the reaction was allowed to warm to ambient temperature with stirring under a nitrogen atmosphere for 24 hours. Deionised water (2.6 L) was then added. The reaction mixture was then washed with deionised water (3×2.6 L). The dichloromethane layer was concentrated to a volume of approximately 1 L to give the crude title compound (439 g, 1.29 mol, 96% yield) as a solution in dichloromethane. A purified sample of the title compound was obtained using the following procedure. To a dichloromethane solution (2.34 L) of the crude product, that had been filtered to remove any particulate contamination, was added isopropyl acetate (1.38 L). The resultant mixture was distilled at atmospheric pressure whilst being simultaneously replaced with isopropyl acetate until the solution temperature reached 87° C. The heating was stopped and the solution was allowed to cool to ambient temperature with stirring for 14 hours to give a cloudy brown solution. The agitation rate was then increased and crystallisation commenced. The suspension was then allowed to granulate for 12 hours at ambient temperature. The resultant suspension was then cooled to 0° C. for 3.5 hours and the solid was then collected by filtration. The filter cake was then washed with isopropyl acetate (2×185 ml, then 2×90 ml) and the solid was dried under vacuum at 40-45° C. for 18 hours to give the title compound (602 g, 0.18 mol, 70% yield) as a cream coloured, crystalline solid;
m.p.: 130-136° C.;
LRMS (negative APCI): m/z [M−H]− 338;
1H-NMR (CDCl3, 300 MHz) δ: 0.92 (t, 3H), 1.27-1.52 (m, 7H), 1.52-1.89 (m, 8H), 2.11-2.27 (m, 1H), 2.27-2.37 (m, 1H), 2.42-2.55 (m, 1H), 2.65 (dd, 2H), 3.00 (q, 2H), 12.25 (bs, 1H).
[0361] Example 29 may be purified as follows:
[0362] The title product from Example 29 was disolved in methanol. To this solution was added sodium methoxide (1 equivalent) in methanol (1 ml/g of Example 29) and the mixture was stirred at room temperature for 20 minutes. The solvent was removed in vacuo and the residue was azeotoped with ethyl acetate to give a brown residue. Ethyl acetate was added and the solution filtered to give a brown solid which was washed with tert-butylmethyl ether to give the crude sodium salt of Example 29. This crude product (35 g) was partitioned between water (200 ml) and ethyl acetate (350 ml). Concentrated hydrochloric acid (˜7 ml) was added until the pH of the aqueous layer was pH2. The aqueous phase was washed with ethyl acetate (2×100 ml). The combined layers were dried using magnesium sulphate. The solvent was removed in vacuo to give a light brown solid (31 g). Ethyl acetate (64 ml, 2 ml/g) and diisopropyl ether (155 ml, 5 ml/g) were added and the mixture heated to 68° C. until a clear solution was obtained (˜30 min). Upon cooling to room temperature, crystallisation of the free acid occurred. After 30 minutes stirring at room temperature the product was collected by filtration and washed with diisopropyl ether. The product was dried in a vacuum oven at 50° C. overnight. (20.2 g, 61% recovery from the sodium salt.); m.p. 135 degC (determined using a Perkin Elmer DSC7 at a heating rate of 20° C./minute).
[0372] The title compound of Example 29 metabolysed to form (2R)-1-(2-{[(5-ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}pentyl)cyclopentanecarboxylic acid.

[0373] This compound was prepared as follows:
[0374] The product from Preparation 102 (430 mg, 1 mmol) was taken up in ethanol (5 mls) and methanol (1 ml) and hydrogenated at 30 psi hydrogen pressure at room temperature for 2 h. The mixture was then filtered through a plug of Arbocel®) and evaporated to a yellow oil. This oil was purified by column chromatography using firstly 19:1, then 9:1 DCM:MeOH as eluant to provide the product as a clear oil (120 mg, 35%); 1HNMR (400 MHz, CDCl3) 0.88 (t, 3H), 1.20-1.88 (m, 13H), 1.90-2.03 (m, 1H), 2.24-2.38 (m, 1H), 2.43-2.72 (m, 2H), 2.95 (q, 2H); LRMS m/z 340.2 (M+H).
Example 31 (R)-2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid
[0375] and

Example 32 (S)-2-{[1-({[2-(Hydroxymethyl)-2.3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid
[0376]

[0377] 2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid (WO 9110644, Example 8) was further purified by HPLC using an AD column and hexane:isopropanol:trifluoroacetic acid (90:10:0.1) as eluant, to give the title compound of Example 31, 99% ee, [α]D=+10.40 (c=0.067, ethanol) and the title compound of Example 32, 99% ee, [α]D=−10.9° (c=0.046, ethanol).
………………..
http://www.google.com/patents/US6734186
Example 7 (+)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl)methyl]pentanoic Acid (F63)

The acid from Preparation 18 (18/ex4) (824 mg) was further purified by HPLC using an AD column and using hexane:iso-propanol:trifluoroacetic acid (85:15:0.2) as eluant to give the title compound of example 7 as a white foam, 386 mg, 99% ee,1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H); LRMS: m/z 338 (MH-); and [α]D=+3.80°(c=0.1, methanol)
Novel selective inhibitors of neutral endopeptidase for the treatment of female sexual arousal disorder. Synthesis and activity of functionalized glutaramides
J Med Chem 2006, 49(14): 4409

Female sexual arousal disorder (FSAD) is a highly prevalent sexual disorder affecting up to 40% of women. We describe herein our efforts to identify a selective neutral endopeptidase (NEP) inhibitor as a potential treatment for FSAD. The rationale for this approach, together with a description of the medicinal chemistry strategy, lead compounds, and SAR investigations are detailed. In particular, the strategy of starting with the clinically precedented selective NEP inhibitor, Candoxatrilat, and targeting low molecular weight and relatively polar mono-carboxylic acids is described. This led ultimately to the prototype development candidate R–13, for which detailed pharmacology and pharmacokinetic parameters are presented.
ACID ENTRY 13
…………………………………………..
WO 2002002513
http://www.google.com/patents/WO2002002513A1?cl=en
…………………..
WO 2002003995
http://www.google.com/patents/WO2002003995A2?cl=en
Scheme 12
LiAIHψ THF, 6hr at reflux




Example 1
( f?)-2-r(1 r(5-ethyl-1.3.4-thiadiazol-2-yl)aminolcarbonyl)cvclopentyl) methyllpentanoic acid
and
Example 2
( S)-2-r(1-fr(5-Ethyl-1.3.4-thiadiazol-2-vnaminolcarbonyl)cvclopentyl)- methyllpentanoic acid
The title product from stage c) below (824mg) was further purified by HPLC using an AD column and using hexane:/sσ-propanol:trifluoroacetic acid (85:15:0.2) as elutant to give the title product from Example 1 , 400mg, 99.5% ee, 1H NMR (CDCI3, 400MHz) δ: 0.90 (t, 3H), 1.36 (m, 6H), 1.50-1.80 (m, 9H), 2.19 (m, 1 H), 2.30 (m, 1 H), 2.44 (m, 1 H), 2.60 (m, 1 H), 2.98 (q, 2H), 12.10-12.30 (bs, 1 H), LRMS : m/z 338 (MH“ ), [α]D = -9.0° (c = 0.1 , methanol), and the title product from Example 2, 386mg, 99% ee, 1H NMR (CDCl3, 400MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 ( , 1 H), 2.30 ( , 1H), 2.44 (m, 1 H), 2.60 (m, 1 H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H); LRMS: m/z 338 (MH“); and [α]D = +3.8° (c = 0.1 , methanol)
Preparation of Starting Materials a) 1 -r2-(tø/t-Butoxycarbonyl)-4-pentvπ-cvclopentane carboxylic acid
A mixture of 1 -[2-(tø t-butoxycarbonyl)-4-pentenyl]-cyclopentane carboxylic acid (EP 274234) (23g, 81.5mmol) and 10% palladium on charcoal (2g) in dry ethanol (200ml) was hydrogenated at 30psi and room temperature for 18 hours. The reaction mixture was filtered through Arbocel®, and the filtrate evaporated under reduced pressure to give a yellow oil. The crude product was purified by column chromatography on silica gel, using ethyl acetate:pentane (40:60) as the eluant, to provide the desired product as a clear oil, 21 g, 91%; 1H NMR (CDCI3, 0.86 (t, 3H), 1.22-1.58 (m, 15H), 1.64 (m, 4H), 1.78 (dd, 1H), 2.00-2.18 ( , 3H), 2.24 ( , 1H); LRMS : m/z 283 (M-HV b) tert-Butyl 2-1Ϊ1 -flT5-ethyl-1.3.4-thiadiazol-2-vnaminolcarbonyl)- cvclopentvDmethyllpentanoate.
1 -(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.21 mmol), 1 – hydroxybenzotriazole hydrate (0.2mmol), N-methylmorpholine (0.31 mmol) and 2-amino-5-ethyl-1 ,3,4-thiadiazole (0.22mmol) were added to a solution of the product from stage a) above (150mg, 0.53mmol) in N,N- dimethylformamide (3ml), and the reaction stirred at 90°C for 18 hours. The cooled solution was diluted with ethyl acetate (90ml), washed with water
(3x25ml), and brine (25ml), then dried (MgSO ) and evaporated under reduced pressure. The crude product was purified by chromatography on silica gel, using ethyl acetate:pentane (30:70) as the eluant to afford the title compound, 92%; 1H NMR (CDCI3, 300MHz) δ: 0.82 (t, 3H), 1.20-1.80 (m, 22H), 1.84 (m, 1 H), 2.20 (m, 4H), 3.04 (q, 2H), 9.10 (bs, 1 H); LRMS : m/z
396.2 (MH+).
c) . 2-r(1-H,(5-ethyl-1.3.4-thiadiazol-2-yl)amino1carbonyl)cvclopentyl) methyllpentanoic acid.
Trifluoroacetic acid (5ml) was added to a solution of the title product from stage b) above (0.31 mmol) in dichloromethane (5ml), and the solution stirred at room temperature for 4 hours. The reaction mixture was concentrated under reduced pressure and the residue azeotroped with toluene and dichloromethane to afford the title compound as a clear oil, 81 %, 1H NMR
(CDCI3, 400MHz) δ: 0.92 (t, 3H), 1.35 (t, 3H), 1.25-1.80 (m, 11 H), 2.20-2.50 (m, 4H), 2.95 (q, 2H), 12.10 (bs, 1 H); LRMS : m/z 339.8 (MH+); Anal. Found: C, 56.46; H, 7.46; N, 12.36. Cι6H25N3O3S requires C, 56.62; H, 7.44; N, 12.37%.
………………………………………

………………………………..
Original Research Article
SEE
The discovery of small molecule inhibitors of neutral endopeptidase. Structure-activity studies on functionalized glutaramides
Chem Biol Drug Des 2006, 67(1): 74
Optimization of oral pharmacokinetics in the discovery of clinical candidates for the treatment of sexual dysfunction
237th ACS Natl Meet (March 22-26, Salt Lake City) 2009, Abst MEDI 173
Novel selective inhibitors of neutral endopeptidase for the treatment of female sexual arousal disorder. Synthesis and activity of functionalized glutaramides
J Med Chem 2006, 49(14): 4409
Bioorganic & Medicinal Chemistry (2007), 15(1), 142-159
Journal of Medicinal Chemistry (2007), 50(24), 6165-6176.
|
5-7-2004
|
Treatment of sexual dysfunction
|
|
|
11-15-2002
|
Treatment of sexual dysfunction
|
|
|
5-3-2002
|
Cyclopentyl-substituted glutaramide derivatives as inhibitors of neutral endopeptidase
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (R)-2-({1-[(5-ethyl-1,3,4-thiadiazol-2-yl)carbamoyl]cyclopentyl}methyl)valeric acid | |
| Clinical data | |
| Legal status | ? |
| Identifiers | |
| CAS number | 337962-93-3 |
| ATC code | ? |
| PubChem | CID 9949799 |
| Chemical data | |
| Formula | C16H25N3O3S |
| Mol. mass | 339.452 g/mol |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6734186 * | Nov 8, 2000 | May 11, 2004 | Pfizer Inc. | Phosphodiesterase 2 inhibitor |
| US7956195 * | Dec 21, 2007 | Jun 7, 2011 | Abbott Laboratories | reacting arylboronic acids with a cycloalkanone, in the presence of a rhodium catalyst or BINAP, to form a substituted arylcycloalkanone, then formin of a hydantoin, alkylation of the hydantoin, resolution, hydrolysis of the hydantoin to the amino acids and esterification of acids; chemical intermediates |
| WO2005007166A1 * | Jul 12, 2004 | Jan 27, 2005 | Alasdair Mark Naylor | Treatment of sexual dysfunction |
Female Sexual Response
The female sexual response phase of arousal is not easily distinguished from the phase of desire until physiological changes begin to take place in the vagina and clitoris as well as other sexual organs. Sexual excitement and pleasure are accompanied by a combination of vascular and neuromuscular events which lead to engorgement of the clitoris, labia and vaginal wall, increased vaginal lubrication and dilatation of the vaginal lumen (Levin, 1980; Ottesen, 1983; Levin, 1991; Levin, 1992; Sjoberg, 1992; Wagner, 1992; Schiavi et al., 1995; Masters et al., 1996; Berman et al., 1999).
Vaginal engorgement enables transudation to occur and this process is responsible for increased vaginal lubrication. Transudation allows a flow of plasma through the epithelium and onto the vaginal surface, the driving force for which is increased blood flow in the vaginal capillary bed during the aroused state. In addition engorgement leads to an increase in vaginal length and luminal diameter, especially in the distal ⅔ of the vaginal canal. The luminal dilatation of the vagina is due to a combination of smooth muscle relaxation of its wall and skeletal muscle relaxation of the pelvic floor muscles. Some sexual pain disorders such as vaginismus are thought to be due, at least in part, by inadequate relaxation preventing dilatation of the vagina; it has yet to be ascertained if this is primarily a smooth or skeletal muscle problem. (Levin, 1980; Oltesen, 1983; Levin, 1991; Levin, 1992; Sjoberg, 1992; Wagner, 1992; Schiavi et al., 1995; Master et al., 1996; Berman et al., 1999).
The vasculature and micro vasculature of the vagina are innervated by nerves containing neuropeptides and other neurotransmitter candidates. These include calcitonin gene-related peptide (CGRP), neuropeptide Y (NPY; Sequence No. 4), nitric oxide synthase (NOS), substance P and vasoactive intestinal peptide (VIP; Sequence No. 8) (Hoyle et al., 1996). Peptides that are present in the clitoris are discussed infra. Nitric oxide synthase, which is often colocalised with VIP (Sequence No. 8), displays a greater expression, immunologically, in the deep arteries and veins rather than in the blood vessels of the propria (Hoyle et al., 1996).
Female Sexual Dysfunction
It is known that some individuals can suffer from female sexual dysfunction (FSD). FSD is best defined as the difficulty or inability of a woman to find satisfaction in sexual expression. FSD is a collective term for several diverse female sexual disorders (Leiblum, 1998, Berman et al., 1999). The woman may have lack of desire, difficulty with arousal or orgasm, pain with intercourse or a combination of these problems. Several types of disease, medications, injuries or psychological problems can cause FSD.
Studies investigating sexual dysfunction in couples reveals that up to 76% of women have complaints of sexual dysfunction and that 30-50% of women in the USA experience FSD.
Sub-types of FSD include hypoactive sexual desire disorder, female sexual arousal disorder, orgasmic disorder and sexual desire disorder.
Treatments in development are targeted to treat specific subtypes of FSD, predominantly desire and arousal disorders.
The categories of FSD are best defined by contrasting them to the phases of normal female sexual response: desire, arousal and orgasm (Leiblum 1998). Desire or libido is the drive for sexual expression—and manifestations often include sexual thoughts either when in the company of an interested partner or when exposed to other erotic stimuli. In contrast, sexual arousal is the vascular response to sexual stimulation, an important component of which is vaginal lubrication and elongation of the vagina. Thus, sexual arousal, in contrast to sexual desire, is a response relating to genital (e.g. vaginal and clitoral) blood flow and not necessarily sensitivity. Orgasm is the release of sexual tension that has culminated during arousal. Hence, FSD typically occurs when a woman has an inadequate or unsatisfactory response in any of these phases, usually desire, arousal or orgasm. FSD categories include hypoactive sexual desire disorder, sexual arousal disorder, orgasmic disorders and sexual pain disorders.
Hypoactive sexual desire disorder is present if a woman has no or little desire to be sexual, and has no or few sexual thoughts or fantasies. This type of FSD can be caused by low testosterone levels, due either to natural menopause or to surgical menopause. Other causes include illness, medications, fatigue, depression and anxiety.
Female sexual arousal disorder (FSAD) is characterised by inadequate genital response to sexual stimulation. The genitalia (e.g. the vagina and/or the clitoris) do not undergo the engorgement that characterises normal sexual arousal. The vaginal walls are poorly lubricated, so that intercourse is painful. Orgasms may be impeded. Arousal disorder can be caused by reduced oestrogen at menopause or after childbirth and during lactation, as well as by illnesses, with vascular components such as diabetes and atherosclerosis. Other causes result from treatment with diuretics, antihistamines, antidepressants eg SSRIs or antihypertensive agents. FSAD is discussed in more detail infra.
Sexual pain disorders (which include dyspareunia and vaginismus) are characterised by pain resulting from penetration and may be caused by medications which reduce lubrication, endometriosis, pelvic inflammatory disease, inflammatory bowel disease or urinary tract problems.
The prevalence of FSD is difficult to gauge because the term covers several types of problem, some of which are difficult to measure, and because the interest in treating FSD is relatively recent. Many women’s sexual problems are associated either directly with the female ageing process or with chronic illnesses such as diabetes and hypertension.
There are wide variations in the reported incidence and prevalence of FSD, in part explained by the use of differing evaluation criteria, but most investigators report that a significant proportion of otherwise healthy women have symptoms of one or more of the FSD subgroups. By way of example, studies comparing sexual dysfunction in couples reveal that 63% of women had arousal or orgasmic dysfunction compared with 40% of men have erectile or ejaculatory dysfunction (Frank et al., 1978).
However, the prevalence of female sexual arousal disorder varies considerably from survey to survey. In a recent National Health and Social Life Survey 19% of women reported lubrication difficulties whereas 14% of women in an outpatient gynaecological clinic reported similar difficulties with lubrication (Rosen et al., 1993).
Several studies have also reported dysfunction with sexual arousal in diabetic women (up to 47%), this included reduced vaginal lubrication (Wincze et al., 1993). There was no association between neuropathy and sexual dysfunction.
Numerous studies have also shown that between 11-48% of women overall may have reduced sexual desire with age. Similarly, between 11-50% of women report problems with arousal and lubrication, and therefore experience pain with intercourse. Vaginismus is far less common, affecting approximately 1% of women.
Studies of sexually experienced women have detailed that 5-10% have primary anorgasmia. Another 10% have infrequent orgasms and a further 10% experience them inconsistently (Spector et al., 1990).
Because FSD consists of several subtypes that express symptoms in separate phases of the sexual response cycle, there is not a single therapy. Current treatment of FSD focuses principally on psychological or relationship issues. Treatment of FSD is gradually evolving as more clinical and basic science studies are dedicated to the investigation of this medical problem. Female sexual complaints are not all psychological in pathophysiology, especially for those individuals who may have a component of vasculogenic dysfunction (eg FSAD) contributing to the overall female sexual complaint. There are at present no drugs licensed for the treatment of FSD. Empirical drug therapy includes oestrogen administration (topically or as hormone replacement therapy), androgens or mood-altering drugs such as buspirone or trazodone. These treatment options are often unsatisfactory due to low efficacy or unacceptable side effects.
Since interest is relatively recent in treating FSD pharmacologically, therapy consists of the following:- psychological counselling, over-the-counter sexual lubricants, and investigational candidates, including drugs approved for other conditions. These medications consist of hormonal agents, either testosterone or combinations of oestrogen and testosterone and more recently vascular drugs, that have proved effective in male erectile dysfunction. None of these agents has been demonstrated to be very effective in treating FSD.
Female Sexual Arousal Disorder (FSAD)
The sexual arousal response consists of vasocongestion in the pelvis, vaginal lubrication and expansion and swelling of the external genitalia. The disturbance causes marked distress and/or interpersonal difficulty. Studies investigating sexual dysfunction in couples reveals that there is a large number of females who suffer from sexual arousal dysfunction; otherwise known as female sexual arousal disorder (FSAD).
The Diagnostic and Statistical Manual (DSM) IV of the American Psychiatric Association defines Female Sexual Arousal Disorder (FSAD) as being:
“a persistent or recurrent inability to attain or to maintain until completion of the sexual activity adequate lubrication-swelling response of sexual excitement. The disturbance must cause marked distress or interpersonal difficulty.”
FSAD is a highly prevalent sexual disorder affecting pre-, peri- and post menopausal (±HRT) women. It is associated with concomitant disorders such as depression, cardiovascular diseases, diabetes and UG disorders.
The primary consequences of FSAD are lack of engorgement/swelling, lack of lubrication and lack of pleasurable genital sensation. The secondary consequences of FSAD are reduced sexual desire, pain during intercourse and difficulty in achieving an orgasm.
It has recently been hypothesised that there is a vascular basis for at least a proportion of patients with symptoms of FSAD (Goldstein et al., 1998) with animal data supporting this view (Park et al., 1997).
Drug candidates for treating FSAD, which are under investigation for efficacy, are primarily erectile dysfunction therapies that promote circulation to the male genitalia. They consist of two types of formulation, oral or sublingual medications (Apomorphine, Phentolamine, Sildenafil), and prostaglandin (PGE1-Alprostadil) that are injected or administered transurethrally in men, and topically to the genitalia in women.
The present invention seeks to provide an effective means of treating FSD, and in particular FSAD.
SUMMARY
The present invention is based on findings that FSAD is associated with reduced genital blood flow—in particular reduced blood flow in the vagina and/or the clitoris. Hence, treatment of women with FSAD can be achieved by enhancement of genital blood flow with vasoactive agents. In our studies, we have shown that cAMP mediates vaginal and clitoral vasorelaxation and that genital (e.g. vaginal and clitoral) blood flow can be enhanced/potentiated by elevation of cAMP levels. This is a seminal finding.
In this respect, no one has previously proposed that FSAD can be treated in such a way—i.e. by direct or indirect elevation of cAMP levels. Moreover, there are no teachings in the art to suggest that FSAD was associated with a detrimental modulation of cAMP activity and/or levels or that cAMP is responsible for mediating vaginal and clitoral vasorelaxation. Hence, the present invention is even further surprising.
In addition, we have found that by using agents of the present invention it is possible to increase genital engorgement and treat FSAD—e.g. increased lubrication in the vagina and increased sensitivity in the vagina and clitoris. Thus, in a broad aspect, the present invention relates to the use of a cAMP potentiator to treat FSD, in particular FSAD.
The present invention is advantageous as it provides a means for restoring a normal sexual arousal response—namely increased genital blood flow leading to vaginal, clitoral and labial engorgement. This will result in increased vaginal lubrication via plasma transudation, increased vaginal compliance and increased genital (e.g. vaginal and clitoral) sensitivity. Hence, the present invention provides a means to restore, or potentiate, the normal sexual arousal response.
More particularly, the present invention relates to:
A pharmaceutical composition for use (or when in use) in the treatment of FSD, in particular FSAD; the pharmaceutical composition comprising an agent capable of potentiating cAMP in the sexual genitalia of a female suffering from FSD, in particular FSAD; wherein the agent is optionally admixed with a pharmaceutically acceptable carrier, diluent or excipient.
The use of an agent in the manufacture of a medicament (such as a pharmaceutical composition) for the treatment of FSD, in particular FSAD; wherein the agent is capable of potentiating cAMP in the sexual genitalia of a female suffering from FSD, in particular FSAD.
A method of treating a female suffering from FSD, in particular FSAD; the method comprising delivering to the female an agent that is capable of potentiating cAMP in the sexual genitalia; wherein the agent is in an amount to cause potentiation of cAMP in the sexual genitalia of the female; wherein the agent is optionally admixed with a pharmaceutically acceptable carrier, diluent or excipient.
An assay method for identifying an agent that can be used to treat FSD, in particular FSAD, the assay method comprising: determining whether an agent can directly or indirectly potentiate cAMP; wherein a potentiation of cAMP in the presence of the agent is indicative that the agent may be useful in the treatment of FSD, in particular FSAD.

1236408-39-1
C19 H19 N5 O2
US 20100197591
| Inventores | Gary E. Aspnes, Robert L. Dow, Michael J. Munchhof |
| Beneficiário Original | Pfizer Inc |
1-(4-(4-Amino-2-methoxy-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl)phenyl)cyclobutanecarbonitrile
1-[4-(4-amino-7,8-dihydro-2-methoxy-5-oxopyrido[4,3-d]pyrimidin-6(5H)-yl)phenyl]-Cyclobutanecarbonitrile,
nmr……http://pubs.acs.org/doi/suppl/10.1021/op400215h/suppl_file/op400215h_si_001.pdf
Enzyme acyl-CoA:diacylglycerol acyltransferase-1 (DGAT-1) catalyzes the rate-limiting step in triglyceride synthesis. It has recently emerged as an attractive target for therapeutic intervention in the treatment of Type II diabetes and obesity.
It is estimated that somewhere between 34 and 61 million people in the US are obese and, in much of the developing world, incidence is increasing by about 1% per year. Obesity increases the likelihood of death from all causes by 20%, and more specifically, death from coronary artery disease and stroke are increased by 25% and 10%, respectively. Key priorities of anti-obesity treatments are to reduce food intake and/or hyperlipidemia. Since the latter has been suggested to provoke insulin resistance, molecules developed to prevent the accumulation of triglyceride would not only reduce obesity but they would also have the additional effect of reducing insulin resistance, a primary factor contributing to the development of diabetes. The therapeutic activity of leptin agonists has come under scrutiny through their potential to reduce food intake and, also, to reverse insulin resistance; however, their potential may be compromised by leptin-resistance, a characteristic of obesity. Acyl coenzyme A:diacylglycerol acyltransferase 1 (DGAT-1) is one of two known DGAT enzymes that catalyze the final step in mammalian triglyceride synthesis and an enzyme that is tightly implicated in both the development of obesity and insulin resistance. DGAT-1 deficient mice are resistant to diet-induced obesity through a mechanism involving increased energy expenditure. US researchers have now shown that these mice have decreased levels of tissue triglycerides, as well as increased sensitivity to insulin and to leptin. Importantly, DGAT-1 deficiency protects against insulin resistance and obesity in agouti yellow mice, a model of severe leptin resistance. Thus, DGAT-1 may represent a useful target for the treatment of insulin and leptin resistance and hence human obesity and diabetes. Chen, H. C., et al., J Clin Invest, 109(8), 1049-55 (2002).
Although studies show that DGAT-1 inhibition is useful for treating obesity and diabetes, there remains a need for DGAT-1 inhibitors that have efficacy for the treatment of metabolic disorders (e.g., obesity, Type 2 diabetes, and insulin resistance syndrome (also referred to as “metabolic syndrome”)).

………………………………..
US 20100197591

Scheme II outlines the general procedures one could use to provide compounds of the general Formula (II).


Scheme IV outlines a general procedure for the preparation of compounds of the general Formula VI.

1-[4-(4-amino-2-methoxy-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl)phenyl]cyclobutanecarbonitrilePotassium nitrate (7.88 g, 77.0 mmol) was suspended in sulfuric acid (45 mL) at 0° C. and stirred for 30 minutes until a clear and colorless solution was obtained (NOTE—a blast shield is highly recommended). An addition funnel was charged with 1-phenylcyclobutanecarbonitrile (11.40 g, 72.5 mmol), and this neat starting material was added drop wise at such a rate that the internal reaction temperature did not exceed 10° C. Upon completion of the addition (which required 90 min), the mixture was poured onto 300 g of ice and stirred vigorously for 30 minutes. The resulting suspension was filtered, and the solid was washed with water and dried under vacuum to afford give 1-(4-nitrophenyl)cyclobutanecarbonitrile (13.53 g, 92%) as a light tan powder.
1H NMR (500 MHz, CHLOROFORM-d) δ ppm 2.11-2.21 (m, 1H) 2.47-2.58 (m, 1H) 2.66 (s, 2H) 2.88-2.96 (m, 2H) 7.63 (d, J=8.54 Hz, 2H) 8.29 (d, J=8.54 Hz, 2H).
A steel hydrogenation vessel was loaded with 1-(4-nitrophenyl)cyclobutanecarbonitrile (103.6 g, 0.51 mol), 10% palladium on activated carbon (10.3 g; contains ˜50% of water), and 2-methyltetrahydrofuran (1.3 L). The mixture was stirred under 30 psi of hydrogen gas at 45° C. for 4 h. The mixture was filtered through a pad of celite and filtrate concentrated. Heptane (1 L) was added to the obtained oil and the heterogeneous mixture was stirred while slowly cooled to room temperature, causing the product aniline to solidify. The solid was filtered off and dried in vacuum to give 1-(4-aminophenyl)cyclobutanecarbonitrile (86.6 g, 98%).
1H NMR (CHLOROFORM-d) δ ppm 7.12-7.25 (m, 2H), 6.61-6.76 (m, 2H), 3.68 (br. s., 2H), 2.68-2.88 (m, 2H), 2.48-2.64 (m, 2H), 2.30-2.45 (m, 1H), 1.94-2.14 (m, 1H)
A mixture of 1-(4-aminophenyl)cyclobutanecarbonitrile (42.2 g, 245 mmol), triethylamine (27.1 mL, 394 mmol), and ethyl acrylate (28.0 mL, 258 mmol) were combined in ethanol (27 mL) and heated to reflux for 24 hours. The mixture was concentrated to dryness and toluene (600 mL) added and concentrated to dryness to give ethyl N-[4-(1-cyanocyclobutyl)phenyl]beta-alaninate as brown oil, which was used without further purification.
1H NMR (CHLOROFORM-d) δ ppm 7.22 (d, 2H), 6.63 (d, 2H), 4.12-4.21 (m, 3H), 3.47 (q, J=6.3 Hz, 2H), 2.74-2.83 (m, 2H), 2.53-2.66 (m, 4H), 2.33-2.45 (m, 1H), 2.00-2.11 (m, 1H), 1.28 (t, 3H)
Ethyl N-[4-(1-cyanocyclobutyl)phenyl]-beta-alaninate was combined with cyanoacetic acid (22.9 g, 270 mmol) and 4-dimethylaminopyridine (2.30 g, 18.8 mmol) in N,N-dimethylformamide (400 mL) and cooled to 0° C. Diisopropylcarbodiimide (41.7 mL, 270 mmol) was then added drop wise over 30 minutes. Once addition was complete, the reaction was slowly warmed up to room temperature and stirred for 16 hours. Reaction was then poured into saturated aqueous sodium bicarbonate (600 mL) and stirred for 30 mintues. Ethyl acetate (1 L) was added and the mixture was filtered to remove the insoluble diisopropylurea. The phases of the filtrate were separated, and the organic phase was washed with brine and dried over sodium sulfate and concentrated to give ethyl N-(cyanoacetyl)-N-[4-(1-cyanocyclobutyl)phenyl]-beta-alaninate as yellow oil that was used with out further purification in the following step.
ethyl N-(cyanoacetyl)-N-[4-(1-cyanocyclobutyl)phenyl]-beta-alaninate and 1,8-diazabicyclo[5.4.0]undec-7-ene (350 mmol) were combined in methanol (400 mL) and heated to 70° C. for 30 minutes. The mixture was concentrated to dryness then partitioned between water (400 mL) and 2:1 ethyl acetate:heptane (400 mL). The aqueous phase was separated and acidified to pH 2 by the addition of 1M hydrochloric acid (400 mL). The precipitate was filtered off and washed with water (300 mL) and 2:1 ethyl acetate:heptane (300 mL) give 1-(4-(1-cyanocyclobutyl)phenyl)-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (31.7 g, 44% over 3 steps) as an off-white solid.
1H NMR (DMSO-d6) δ ppm 7.39-7.45 (m, 2H), 7.31 (d, 2H), 3.78 (t, J=6.7 Hz, 2H), 2.79 (t, 2H), 2.66-2.75 (m, 2H), 2.53-2.64 (m, 2H), 2.16-2.31 (m, 1H), 1.91-2.04 (m, 1H)
m/z (M+1)=294.4
1-(4-(1-Cyanocyclobutyl)phenyl)-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (50.0 g, 170 mmol) and N,N-dimethylformamide (0.66 mL, 8.5 mmol) in dichloromethane (350 mL) was cooled to 0° C. Oxalyl chloride (18.0 mL, 203 mmol) was added over 15 minutes. The mixture was warmed to room temperature over 2 hours. Methanol (300 mL) was then added as a steady stream, and the mixture was heated at 45° C. for 16 hours. The mixture was cooled to room temperature and concentrated to get rid of most of the dichloromethane. Methanol (200 mL) was added and the thick slurry was stirred for 2 hours. The solid was filtered and dried under vacuum to give 1-(4-(1-cyanocyclobutyl)phenyl)-4-methoxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (48.3 g, 92%) as an off-white powder.
1H NMR (400 MHz, DMSO-d6) δ ppm 1.91-2.03 (m, 1H) 2.18-2.31 (m, 1H) 2.54-2.63 (m, 2H) 2.67-2.75 (m, 2H) 3.03 (t, J=6.73 Hz, 2H) 3.85 (t, J=6.73 Hz, 2H) 4.01 (s, 3H) 7.33 (d, J=8.78 Hz, 2H) 7.44 (d, J=8.78 Hz, 2H)
m/z (M+1)=308.4
1-(4-(1-Cyanocyclobutyl)phenyl)-4-methoxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (12.04 g, 37.9 mmol) and cyanamide (1.64 g, 41.0 mmol) were suspended in methanol (200 mL) at room temperature. A solution of 25% sodium methoxide in methanol (45.0 mmol) was then added drop wise over 10 minutes to obtain a clear homogeneous solution of the intermediate cyanamide adduct. In one portion, sulfuric acid (5.06 mL, 94.9 mmol) was added, and the mixture was heated to 50° C. for 16 hours. The mixture was then cooled to room temperature and basified to pH 10-11 by the addition of 1N sodium hydroxide, and the thick suspension was stirred for 20 minutes. The solid was filtered, washed with cold methanol and water, and dried under vacuum to obtain the crude product as a mixture contaminated with the vinylogous amide (4-amino-1-[4-(1-cyanocyclobutyl)phenyl]-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile). This solid mixture was heated to reflux in methanol (150 mL) for 3 hours then cooled to room temperature and filtered. The solid collected was then dissolved in a minimal amount of acetic acid (30 mL) at 60° C. to obtain a clear yellow solution. Water was then added drop wise at 60° C. until the cloudiness persisted, and the mixture was allowed to return to room temperature. Another 50 mL of water was added and the fine suspension was filtered, washed with water, and dried under vacuum to afford the title compound (4A) (6.80 g, 51%) as a light yellow solid.
1H NMR (500 MHz, DMSO-d6) δ ppm 1.97-2.06 (m, 1H) 2.23-2.34 (m, 1H) 2.59-2.67 (m, 2H) 2.71-2.79 (m, 2H) 2.96 (t, J=6.71 Hz, 2H) 3.86 (s, 3H) 3.91 (t, J=6.71 Hz, 2H) 7.39-7.44 (d, J=8.54, 2H) 7.47-7.51 (d, J=8.54, 2H) 7.81 (br. s., 1H) 8.35 (br. s., 1H).
m/z (M+1)=350.4
………………………..
paper
http://pubs.acs.org/doi/abs/10.1021/op400215h

Obesity and overweight are generally defined by body mass index (BMI), which is correlated with total body fat and estimates the relative risk of disease. BMI is calculated by weight in kilograms divided by height in meters squared (kg/m2). Overweight is typically defined as a BMI of 25-29.9 kg/m2, and obesity is typically defined as a BMI of 30 kg/m2. See, e.g., National Heart, Lung, and Blood Institute, Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults, The Evidence Report, Washington, D.C.: U.S. Department of Health and Human Services, NIH publication no. 98-4083 (1998).
Another aspect of the invention is for the treatment or delaying the progression or onset of diabetes or diabetes-related disorders including Type 1 (insulin-dependent diabetes mellitus, also referred to as “IDDM”) and Type 2 (noninsulin-dependent diabetes mellitus, also referred to as “NIDDM”) diabetes, impaired glucose tolerance, insulin resistance, hyperglycemia, and diabetic complications (such as atherosclerosis, coronary heart disease, stroke, peripheral vascular disease, nephropathy, hypertension, neuropathy, and retinopathy).
Yet another aspect of the invention is the treatment of diabetes- or obesity-related co-morbidities, such as metabolic syndrome. Metabolic syndrome includes diseases, conditions or disorders such as dyslipidemia, hypertension, insulin resistance, diabetes (e.g., Type 2 diabetes), weight gain, coronary artery disease and heart failure. For more detailed information on Metabolic Syndrome, see, e.g., Zimmet, P.Z., et al., “The Metabolic Syndrome: Perhaps an Etiologic Mystery but Far From a Myth —Where Does the International Diabetes Federation Stand?,” Diabetes & Endocrinology, 7(2), (2005); and Alberti, K. G., et al., “The Metabolic Syndrome —A New Worldwide Definition,” Lancet, 366, 1059-62 (2005). Administration of the compounds of the invention may provide a statistically significant (p<0.05) reduction in at least one cardiovascular disease risk factor, such as lowering of plasma leptin, C-reactive protein (CRP) and/or cholesterol, as compared to a vehicle control containing no drug. The administration of compounds of the invention may also provide a statistically significant (p<0.05) reduction in glucose serum levels.
In yet another aspect of the invention, the condition treated is impaired glucose tolerance, hyperglycemia, diabetic complications such as sugar cataracts, diabetic neuropathy, diabetic nephropathy, diabetic retinopathy and diabetic cardiomyopathy, anorexia nervosa, bulimia, cachexia, hyperuricemia, hyperinsulinemia, hypercholesterolemia, hyperlipidemia, dyslipidemia, mixed dyslipidemia, hypertriglyceridemia, nonalcoholic fatty liver disease, atherosclerosis, arteriosclerosis, acute heart failure, congestive heart failure, coronary artery disease, cardiomyopathy, myocardial infarction, angina pectoris, hypertension, hypotension, stroke, ischemia, ischemic reperfusion injury, aneurysm, restenosis, vascular stenosis, solid tumors, skin cancer, melanoma, lymphoma, breast cancer, lung cancer, colorectal cancer, stomach cancer, esophageal cancer, pancreatic cancer, prostate cancer, kidney cancer, liver cancer, bladder cancer, cervical cancer, uterine cancer, testicular cancer and ovarian cancer.
Pfizer’s XALKORI® Granted Regular FDA Approval
Standard of Care for Patients With Metastatic ALK-Positive Non-Small Cell Lung Cancer
NEW YORK, November 21, 2013–(BUSINESS WIRE)–Pfizer Inc. announced today that the U.S. Food and Drug Administration (FDA) has granted Pfizer’s XALKORI® (crizotinib) regular approval for the treatment of patients with metastatic ALK-positive non-small cell lung cancer (NSCLC) as detected by an FDA-approved test. XALKORI was previously granted accelerated approval in August 2011 due to the critical need for new agents for people living with ALK-positive NSCLC
read all at
http://www.pharmalive.com/pfizer%E2%80%99s-xalkori-granted-regular-fda-approval

Pfizer, GlaxoSmithKline and engineering giant Siemens have signed on as founding members of a new consortium set up by Singapore’s Agency for Science, Technology and Research (A*Star) to address challenges such as costs, regulatory compliance and processes to bring drugs from trials to markets.
READ ALL AT
The Agency for Science, Technology and Research (Abbreviation: A*STAR; Chinese: 新加坡科技研究局) is a statutory board under the Ministry of Trade and Industry of Singapore. The Agency was established in 1991 to foster scientific research and talent for a knowledge-based Singapore.
Established in 1991 as the former National Science and Technology Board (NSTB), A*STAR was established with the primary mission to raise the level of science and technology in Singapore.[1]
The current chairman of A*STAR is Mr. Lim Chuan Poh. He was formerly the Permanent Secretary (Education) and the Chief of Defence Force. Mr Lim took over the reins of A*STAR from Mr. Philip Yeo, who later became Chairman of SPRING Singapore, on 1 April 2007.[2]
The scientific leadership includes Tan Chorh Chuan, George Radda, Sydney Brenner, David Lane, Charles Zukoski and used to include Prof Low Teck Seng. Prof Low Teck Seng left A*Star on 19 July 2012 to join the National Research Foundation of the Prime Minister’s Office.
The agency is made up of:
The agency oversees 14 biomedical sciences, and physical sciences and engineering research institutes, and six consortia & centre, which are located in Biopolis and Fusionopolis, as well as their immediate vicinity.
A*STAR supports Singapore’s key economic clusters by providing intellectual, human and industrial capital to its partners in industry. It also supports extramural research in the universities, hospitals, research centres, and with other local and international partners.
Biomedical Research Council
The Biomedical Research Council (BMRC) oversees 7 research institutes and several other research units that focus on both basic as well as translational and clinical research to support the key industry clusters in Biomedical Sciences, pharmaceuticals, medical technology, biotechnology and healthcare services.
Having established a strong foundation in basic biomedical research capabilities, there is now an added focus on translating new knowledge and technologies created at the “benches” into new clinical applications for diagnosis and treatment that can one day be delivered at the “bedsides” of our hospitals and disease centres.
The research institutes and units under BMRC are:
The BMRC Research Institutes focus on building up core biomedical capabilities in the areas of bioprocessing; chemical synthesis; genomics and proteomics; molecular and cell biology; bioengineering and nanotechnology and computational biology. In addition, the Institute of Medical Biology (IMB) and Singapore Institute for Clinical Sciences (SICS) focus on translational and clinical research.
Science and Engineering Council
A*STAR’s Science and Engineering Research Council (SERC) promotes public sector research and development in the physical sciences & engineering.
SERC manages seven research institutes and several state-of-the art centres and facilities with core competencies in a wide range of fields including communications, data storage, materials, chemicals, computational sciences, microelectronics, advanced manufacturing and metrology to tackle global technological challenges and create future industries from its headquarters at Fusionopolis, Singapore’s iconic hub for science and technology research.
The research institutes and units under SERC are:
The seamless integration of the research institutes is key to addressing industry needs, which may span multiple disciplines. To this end, SERC’s broad range of capabilities are in a unique position to develop new technologies in areas such as automotives, aerospace, energy, electronic healthcare and medical technology, nanotechnology, photonics, sensors and sensor networks.
In July 2012, it was announced that A*STAR collaborates with Chinese language internet search provider Baidu to open a joint laboratory, called the Baidu-I2R Research Centre (BIRC), which aims to develop language processing technologies.[3]
Each year, the Agency gives out a number of scholarships and awards to young and aspiring scientists. These awards are meant to help Singapore achieve its goal of becoming a research hub by nurturing home-grown PhDs to serve both in the public sector and in industry. In 2008, a total of 101 scholarships were awarded to Bachelor of Science and PhD students who were to embark on their studies in overseas universities.[4] The administration of these awards are governed by the A*Star Graduate Academy, some of which are listed below:
CHEMICAL NAMES
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt)
http://www.ama-assn.org/resources/doc/usan/palbociclib.pdf FOR STRUCTURE AND DETAILS
Palbociclib, also known as PD0332991, is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity. PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin D1 kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation; inhibition of Rb phosphorylation prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the G1 phase), resulting in suppression of DNA replication and decreased tumor cell proliferation. PD 0332991 is a highly specific inhibitor of cyclin-dependent kinase 4 (Cdk4) (IC50 = 0.011 μmol/L) and Cdk6 (IC50 = 0.016 μmol/L), having no activity against a panel of 36 additional protein kinases. Check for active clinical trials or closed clinical trials using this agent. (NCI Thesaurus)
Date: April 10, 2013
Pfizer Inc. said that its experimental pill for advanced, often deadly breast cancer has been designated as a breakthrough therapy by the Food and Drug Administration.
The breakthrough designation, created under legislation enacted last summer to fund and improve operations of the FDA, is meant to speed up development and review of experimental treatments that are seen as big advances over existing therapies for serious diseases. Pfizer is working with the agency to determine exactly what research results it will need to apply for approval of the drug.
Palbociclib is being evaluated as an initial treatment for the biggest subgroup of postmenopausal women whose breast cancer is locally advanced or has spread elsewhere in the body. About 60% of women with such advanced breast cancer have tumors classified as ER+, or estrogen-receptor positive, but HER2-, or lacking an excess of the growth-promoting protein HER2.
Estrogen-receptor positive tumors have proteins inside and on the surface of their cells to which the estrogen hormone can attach and then fuel growth of cells. These tumors tend to grow slowly and can be fought with drugs that block estrogen’s effects.
Meanwhile, about 80% of breast cancer tumor cells are HER2 negative. That means that unlike HER2 positive tumors, they don’t produce too much of the HER2 protein, which makes tumors grow and spread more aggressively than in other breast cancer types.
New York-based Pfizer is currently running a late-stage study of palbociclib at multiple centers, comparing its effects when used in combination with letrozole with the effects of letrozole alone.
Letrozole, sold under the brand name Femara for about the past 15 years, is a pill that works by inhibiting aromatase. That’s an enzyme in the adrenal glands that makes estrogen.
According to Pfizer, palbociclib targets enzymes called cyclin dependent kinases 4 and 6. By inhibiting those enzymes, the drug has been shown in laboratory studies to block cell growth and suppress copying of the DNA of the cancer cells.
Pfizer, which has made research on cancer medicines a priority in recent years, also is testing palbociclib as a treatment for other cancers.
| Highlight of recent study using PD-0332991 |
Phase I study of PD-0332991: Forty-one patients were enrolled. DLTs were observed in five patients (12%) overall; at the 75, 125, and 150 mg once daily dose levels. The MTD and recommended phase II dose of PD 0332991 was 125 mg once daily. Neutropenia was the only dose-limiting effect. After cycle 1, grade 3 neutropenia, anemia, and leukopenia occurred in five (12%), three (7%), and one (2%) patient(s), respectively. The most common non-hematologic adverse events included fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for tumor response; 10 (27%) had stable disease for ≥4 cycles of whom six derived prolonged benefit (≥10 cycles). PD 0332991 was slowly absorbed (median T(max), 5.5 hours), and slowly eliminated (mean half-life was 25.9 hours) with a large volume of distribution (mean, 2,793 L). The area under the concentration-time curve increased linearly with dose. Using an E(max) model, neutropenia was shown to be proportional to exposure. CONCLUSIONS:
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
Phase I study of PD-0332991 in 3-week cycles (Schedule 2/1): Six patients had DLTs (18%; four receiving 200 mg QD; two receiving 225 mg QD); the MTD was 200 mg QD. Treatment-related, non-haematological adverse events occurred in 29 patients (88%) during cycle 1 and 27 patients (82%) thereafter. Adverse events were generally mild-moderate. Of 31 evaluable patients, one with testicular cancer achieved a partial response; nine had stable disease (≥10 cycles in three cases). PD 0332991 was slowly absorbed (mean T(max) 4.2 h) and eliminated (mean half-life 26.7 h). Volume of distribution was large (mean 3241 l) with dose-proportional exposure. Using a maximum effective concentration model, neutropenia was proportional to exposure. CONCLUSION: PD 0332991 was generally well tolerated, with DLTs related mainly to myelosuppression. The MTD, 200 mg QD, is recommended for phase II study. (source: Br J Cancer. 2011 Jun 7;104(12):1862-8)
| References |
1: Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568-76. doi: 10.1158/1078-0432.CCR-11-0509. Epub 2011 Nov 16. PubMed PMID: 22090362.
2: Smith D, Tella M, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in mouse plasma using automated micro-sample processing and microbore liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 Oct 1;879(27):2860-5. doi: 10.1016/j.jchromb.2011.08.009. Epub 2011 Aug 16. PubMed PMID: 21889427.
3: Katsumi Y, Iehara T, Miyachi M, Yagyu S, Tsubai-Shimizu S, Kikuchi K, Tamura S, Kuwahara Y, Tsuchiya K, Kuroda H, Sugimoto T, Houghton PJ, Hosoi H. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun. 2011 Sep 16;413(1):62-8. doi: 10.1016/j.bbrc.2011.08.047. Epub 2011 Aug 17. PubMed PMID: 21871868; PubMed Central PMCID: PMC3214763.
4: Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011 Jun 7;104(12):1862-8. doi: 10.1038/bjc.2011.177. Epub 2011 May 24. PubMed PMID: 21610706; PubMed Central PMCID: PMC3111206.
5: Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010 Nov 2;53(3):228-34. doi: 10.1016/j.jpba.2010.02.031. Epub 2010 Feb 26. PubMed PMID: 20236782.
6: Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. PubMed PMID: 19874578; PubMed Central PMCID: PMC2790859.
7: Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008 Jul 15;68(14):5519-23. doi: 10.1158/0008-5472.CAN-07-6404. PubMed PMID: 18632601.
8: Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427-38. PubMed PMID: 15542782.
Good News For Pfizer’s Orphan Drug Bosulif (Bosutinib) in Europe

mar 28,2013
Regulators in Europe have given a partial green light to Pfizer ‘s leukaemia drug Bosulif.
The European Commission has granted conditional marketing authorisation for Bosulif (bosutinib) for the treatment of adults with chronic, accelerated or blast phase Philadelphia chromosome-positive (Ph+) chronic myelogenous leukaemia (CML). The drug can be given to patients previously treated with one or more tyrosine kinase inhibitors ie Novartis’ Gleevec (imatinib) and Tasigna (nilotinib) or Bristol-Myers Squibb’s Sprycel (dasatinib).
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on January 17, 2013, adopts a positive opinion, recommending a conditional marketing authhorization for Pfizer’s orphan drug Bosulif (Bosutinib) for Chronic Leukemia (CML). Bosutinib receives orphan designation from the European Commission (EC) on August 4, 2010, for CML.
Pfizer receives FDA approval on September 4, 2012, for orphan drug Bosulif (Bosutinib) for CML. Pfizer receives on February 24, 2009, FDA Orphan Drug Designation (ODD) for Bosutinib for CML.

Bosutinib (rINN/USAN; codenamed SKI-606, marketed under the trade name Bosulif) is atyrosine kinase inhibitor undergoing research for use in the treatment of cancer. [1] [2]Originally synthesized by Wyeth, it is being developed by Pfizer.
Some commercial stocks of bosutinib (from sources other than the Pfizer material used for clinical trials) have recently been found to have the incorrect chemical structure, calling the biological results obtained with them into doubt.[3]
Bosutinib received US FDA approval on September 5, 2012 for the treatment of adult patients with chronic, accelerated, or blast phase Philadelphia chromosome-positive (Ph+)chronic myelogenous leukemia (CML) with resistance, or intolerance to prior therapy.[4][5][6]

Publication date: 25 January 2013
Author: Pfizer
Pfizer Inc. announced today that the U.S. Food and Drug Administration (FDA) has granted approval for the expansion of the company’s pneumococcal conjugate vaccine, Prevnar 13®* (Pneumococcal 13-valent Conjugate Vaccine [Diphtheria CRM197 Protein]), for use in older children and adolescents aged 6 years through 17 years for active immunization for the prevention of invasive disease caused by the 13 Streptococcus pneumoniae serotypes contained in the vaccine. For this age group, Prevnar 13 is administered as a one-time dose to patients who have never received Prevnar 13.1
“As a global leader in pneumococcal disease prevention, extending the impact of Prevnar 13 to older children and adolescents aged 6 through 17 years is a reflection of our dedication to improving public health worldwide,”said Susan Silbermann, president, vaccines, Pfizer. “We continue to work tirelessly to make this vaccine available to people at risk for invasive pneumococcal disease.”
The FDA approval followed submission and review of a Phase 3, open-label trial of Prevnar 13 in 592 older children and adolescents, including those with asthma.2 The study met all endpoints, demonstrating immunogenicity and establishing a safety profile in children aged 6 years through 17 years consistent with the safety profile established in previous trials in infants and young children.2
About Prevnar 13
Prevnar 13 was first introduced for use in infants and young children in December 2009 in Europe and in February 2010 in the U.S., and it is now approved for such use in nearly 120 countries worldwide. It is the most widely used pneumococcal conjugate vaccine in the world, and more than 500 million doses of Prevnar/Prevnar 13 have been distributed worldwide. Currently, Prevnar 13 is included as part of a national or regional immunization program in more than 60 countries, offering coverage against invasive pneumococcal disease to nearly 30 million children per year.3
Prevnar 13 is also approved for use in adults 50 years of age and older in more than 80 countries and it is the first and only pneumococcal vaccine to be granted World Health Organization prequalification in the adult population.3
About Pneumococcal Disease
Pneumococcal disease (PD) is a group of illnesses caused by the bacterium Streptococcus pneumoniae (S. pneumoniae), also known as pneumococcus.4 PD is associated with significant morbidity and mortality.4 Invasive manifestations of the disease include bacteremia (bacteria in the blood) and meningitis (infection of the tissues surrounding the brain and spinal cord).4 Invasive pneumococcal disease can affect people of all ages, although older adults and young children are at heightened risk.4,5,6
us fda data
STN#: 125324
Proper Name: Pneumococcal 13-valent Conjugate Vaccine (Diphtheria CRM197 Protein)
Tradename: Prevnar 13
Manufacturer: Wyeth Pharmaceuticals, Inc, License #0003
Indications:
Publication date: 4 March 2013
Author: Pfizer
Pfizer Inc. (NYSE:PFE) presented today the results from a Phase 3 study demonstrating the immunogenicity, tolerability and safety of Prevnar 13®(Pneumococcal 13-valent Conjugate Vaccine [Diphtheria CRM197 Protein])in adults infected with human immunodeficiency virus (HIV). The results were presented at the 20th Conference on Retroviruses and Opportunistic Infections (CROI) in Atlanta, Ga.
These data support planned regulatory submissions seeking to include data on HIV-infected immunocompromised adults in the Prevnar 13 label in the United States, the European Union, and other countries around the world.

DESVENLAFAXINE
read at
5 march 2013
Alembic has announced that it has received an NDA approval for extended release version of Pfizer’s anti depressant drug Pristiq. Pristiq sell approximately $550m in the US. Alembic has outlicensed rights to Ranbaxy for marketing in the US. The company will start marketing the product immediately.
Alembic will manufacture and supply the drug to Ranbaxy for marketing in the US. Vadodara-based pharma player, Alembic Pharmaceuticals Limited has received the approval from the US Food and Drug Administration (USFDA) for a bioequivalent version of Pristiq by Pfizer.

Xalkori, crizotinib,
(PF-02341066)
3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine
Crizotinib; 877399-52-5; Xalkori; PF-2341066; PF-02341066; (R)-crizotinib; 877399-52-5
| Molecular Formula: | C21H22Cl2FN5O |
|---|---|
| Molecular Weight: | 450.336683 g/mol |
Crizotinib an inhibitor of receptor tyrosine kinase for the treatment of non-small cell lung cancer (NSCLC). Verification of the presence of ALK fusion gene is done by Abbott Molecular’s Vysis ALK Break Apart FISH Probe Kit. This verification is used to select for patients suitable for treatment. FDA approved in August 26, 2011.
Crizotinib (1), an anaplastic lymphoma kinase (ALK) receptor tyrosine kinase inhibitor approved by the U.S. Food and Drug Administration in 2011, is efficacious in ALK and ROS positive patients
Feb 25, 2013
Pfizer has been granted China approval for Xalkori (crizotinib), an innovative treatment for patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) that is anaplastic lymphoma kinase (ALK) positive. The ALK-positive variation, which comprises between 3% and 5% of all NSCLC tumors, must be proved by a biomarker test. Pfizer said China’s approval came just eleven months after it submitted a new drug application to the SFDA for Xalkori
Crizotinib (trade name Xalkori,[1] Pfizer), is an anti-cancer drug acting as an ALK (anaplastic lymphoma kinase) and ROS1 (c-ros oncogene 1) inhibitor, approved for treatment of some non-small cell lung carcinoma (NSCLC) in the US and some other countries, and undergoing clinical trials testing its safety and efficacy in anaplastic large cell lymphoma, neuroblastoma, and other advanced solid tumors in both adults and children.[2]
Crizotinib the core structure is a substituted pyridine, the 3 – position of the ether as a chiral center adjacent, so with Mitsunobu reaction to complete, as is a typical Mitsunobu SN2 reaction, the reaction chiral center occurs in reverse, so easy to control, no racemization occurs. Pyridine substituted at position 5 by Suzuki reaction constructed.
Compound 1 The activation of the hydroxyl groups of methanesulfonyl chloride, and then with a 4 – iodopyrazole reaction 2 , 2 to 4 Suzuki reaction conversion can be used, but will generate a large quantity of the reaction product of their coupling, the first 2 converted to a Grignard reagent, and then with a boronic acid ester of 3 reaction 4 .

……………………
http://www.specchemonline.com/articles/view/biocatalyst-breakthroughs#.VTcW9yxabEs
http://www.google.com/patents/WO2014020467A2?cl=en
(R)-3-[l-(2,6-Dichloro-3-fluoro-phenyl)-ethoxy]-5-(l-piperidin-4-yl-lH-py- razol-4-yl)-pyridin-2-ylamine, also known as Crizotinib, is represented by the Formula (I):

Formula (I)
Crizotinib is a potent small-molecule inhibitor of c-Met/HGFR (hepatocyte growth factor receptor) kinase and ALK (anaplastic lymphoma kinase) activity. Enantiomerically pure compound of formula I was first disclosed in US Patent No. 7,858,643. Additionally, the racemate of compound of formula I was disclosed in U.S. patent application 2006/0128724, both of these references discloses similar methods for the synthesis of Compound of Formula I.
Conventionally, the compounds of formula I are prepared by reacting Bis(pinacolato)diboron with protected 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-pyridin-2-ylamine in the presence of Pd catalyst. The obtained product after deprotection is reacted with N- protected 4-(4-bromo-pyrazol-l-yl)-piperidine in the presence of Pd Catalyst. The obtained product is filtered through celite pad and purified by Column Chromatography. The final product of formula I was obtained by deprotection of the purified compound by using HCl/dioxane. US Patent No. 7,858,643 provides enantiomerically pure aminoheteroaryl compounds, particularly aminopyridines and aminopyrazines, having protein tyrosine kinase activity. More particularly, US 7,858,643 describes process for the preparation of 3-[(lR)-l-(2,6- dichloro-3-fluorophenyl)ethoxy]-5-(l-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine. The Scheme is summarized below in Scheme- 1 :


Scheme-1
wherein, “Boc” means tert-butoxycarbonyl; and a) (Boc)2, DMF, Dimethylaminopyridine b) Pd(dppf)Cl2, KOAc, Dichloromethane; c) HC1, Dioxane, Dichloromethane; d) Pd(PPh3)2Cl2, Na2C03, DME/H20; e) 4M HCl/Dioxane, Dichloromethane
A similar process has been disclosed in the U.S. patent application 2006/0128724 for the preparation of Crizotinib. J. Jean Cui et. al. in J. Med. Chem. 2011, 54, 6342-6363, also provides a similar process for the preparation of Crizotinib and its derivatives.
However, above mentioned synthetic process requires stringent operational conditions such as filtration at several steps through celite pad. Also column chromatography is required at various steps which is not only tedious but also results in significant yield loss. Another disadvantage of above process involves extensive use of palladium catalysts, hence metal scavengers are required to remove palladium content from the desired product at various steps which makes this process inefficient for commercial scale.
Yet another disadvantage of above process is the cost of Bis(pinacolato)diboron. This reagent is used in excess in the reaction mixture resulting in considerable cost, especially during large-scale syntheses.
US Patent No. 7,825,137 also discloses a process for the preparation of Crizotinib where Boc protected 4-(4-iodo-pyrazol-l-yl)-piperidine is first reacted with Bis(pinacolato)diboron in the presence of Pd catalyst. The reaction mixture is filtered through a bed of celite and the obtained filtrate is concentrated and purified by silica gel chromatography to give to form tert-butyl-4-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]piperidine-l- carboxylate. To this compound, 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]- pyridin-2-ylamine is added in the presence of a Pd catalyst. The reaction mixture is stirred for 16h at 87°C. The reaction mixture is filtered through celite pad and the concentrated filtrate is purified on silica gel column to obtain (4-{6-amino-5-[(R)-l-(2,6-dichloro-3-fluoro- phenyl)-ethoxy]-pyri- din-3-yl}-pyrazol-l-yl)-piperidine-l-carboxylic acid tert-butyl ester of 95% purity. To the solution of resulting compound in dichloromethane 4N HCl/Dioxane is added and thereby getting the reaction suspension is filtered in Buchner funnel lined with filter paper. The obtained solid is dissolved in HPLC water and pH is adjusted to 10 with the addition of Na2C03 Compound is extracted using dichloroform and is purified on a silica gel column by eluting with CH2Cl2 MeOH/NEt3 system to obtain Crizotinib. The scheme is summarized below in scheme 2:

Formula (i) Formula (ii)

Formula (iii) Formula (ii) ula (iv)

Formula (v) Formula (I)
Scheme-2
Preparation of Crizotinib:
To a stirred solution of Tert-butyl 4-(4-{ 6-amino-5-[(li?)-l-(2,6-dichloro-3- fluorophenyl)ethoxy]pyridin-3 -yl } – lH-pyrazol- 1 -yl)piperidine- 1 -carboxylate (material obtained in Example 3) (l.Og, 0.00181 moles) in dichloromethane (-13 ml) at 0°C was added 4.0 M dioxane HQ (6.7 ml, 0.0272 moles). Reaction mixture was stirred at room temperature for 4h. After the completion of reaction monitored by TLC, solid was filtered and washed with dichloromethane (10 ml). The obtained solid was dissolved in water (20 ml); aqueous layer was extracted with dichloromethane (10×2). The pH of aqueous layer was adjusted to 9-10 with Na2C03 and compound was extracted with dichloromethane (10 x 3), combined organic layers were washed with water (20 ml), evaporated under vacuum to get solid product. The solid was stirred with ether (10 ml), filtered off, washed well with ether, dried under vacuum to get Crizotinib.
Yield: 0.45g (55 %)
HPLC Purity: 99.35 %
1HNMR (400 MHz, CDC13) δ: 7.76 (d, J = 1.6 Hz, 1H), 7.56 (s, 1H), 7.49 (s, 1H), 7.30 (dd, J = 9.2 Hz), 7.0 (m, 1H), 6.86 (d, J = 1.6 Hz, 1H), 6.09 ( q, J= 6.8 Hz, 1H), 4.75 (brs, 1H), 4.19 (m, 1H), 3.25 (m, 2H), 2.76 (m, 2H), 2.16 (m, 2H), 1.92 (m, 2H), 1.85 (d, J= 6.8 Hz, 3H), 1.67 (brs, 1H)
…………………………
http://www.sciencedirect.com/science/article/pii/S0040403914000872
A novel approach for the synthesis of Crizotinib (1) is described. In addition, new efficient procedures have been developed for the preparation of (S)-1-(2,6-dichloro-3-fluorophenyl)ethanol (2) and tert-butyl 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)piperidine-1-carboxylate (4), the key intermediates required for the synthesis of Crizotinib.

4-(4-Iodo-1H-pyrazol-1-yl)piperidine is a key intermediate in the synthesis of Crizotinib. We report a robust three-step synthesis that has successfully delivered multi-kilogram quantities of the key intermediate. The process includes nucleophilic aromatic substitution of 4-chloropyridine with pyrazole, followed by hydrogenation of the pyridine moiety and subsequent iodination of the pyrazole which all required optimization to ensure successful scale-up.

……………………

A robust six-step process for the synthesis of crizotinib, a novel c-Met/ALK inhibitor currently in phase III clinical trials, has been developed and used to deliver over 100 kg of API. The process includes a Mitsunobu reaction, a chemoselective reduction of an arylnitro group, and a Suzuki coupling, all of which required optimization to ensure successful scale-up. Conducting the Mitsunobu reaction in toluene and then crystallizing the product from ethanol efficiently purged the reaction byproduct. A chemoselective arylnitro reduction and subsequent bromination reaction afforded the key intermediate 6. A highly selective Suzuki reaction between 6 and pinacol boronate 8, followed by Boc deprotection, completed the synthesis of crizotinib 1.
3-[(1R)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-amine 1
crizotinib1 (20.7 kg, 80%) as a white solid.
Mp 192 °C;
1H NMR (400 MHz, CDCl3) δ: 7.78 (d, J = 1.8 Hz, 1H), 7.58 (s, 1H), 7.52 (s, 1H), 7.31 (dd, J = 9.0, 4.9 Hz, 1H), 7.06 (m, 1H), 6.89 (d, J = 1.7 Hz, 1H), 6.09 (q, 1H), 4.79 (br s, 2H), 4.21 (m, 1H), 3.26 (m, 2H), 2.78 (m, 2H), 2.17 (m, 2H), 1.90 (m, 2H), 1.87 (d, J = 6.7 Hz, 3H), 1.63 (br s, 1H).
13C NMR (100.6 MHz, CDCl3) δ: 157.5 (d, J = 250.7 Hz), 148.9, 139.8, 137.0, 135.7, 135.6, 129.9, 129.0 (d, J = 3.7 Hz), 122.4, 122.1 (d, J = 19.0 Hz), 119.9, 119.3, 116.7 (d, J = 23.3 Hz), 115.0, 72.4, 59.9, 45.7, 34.0, 18.9.
LC-MS: found m/z 450.0, 451.0, 452.0, 453.0, 454.0, 455.0.
Anal. Calcd for C21H22Cl2FN5O: C, 56.01; H, 4.92; N, 15.55. Found: C, 56.08; H, 4.94; N, 15.80.
Cui, J. J.; Botrous, I.; Shen, H.; Tran-Dube, M. B.; Nambu, M. D.; Kung, P.-P.; Funk, L. A.; Jia, L.; Meng, J. J.; Pairish, M. A.; McTigue, M.; Grodsky, N.; Ryan, K.; Alton, G.; Yamazaki, S.; Zou, H.; Christensen, J. G.; Mroczkowski, B.Abstracts of Papers; 235th ACS National Meeting, New Orleans, LA, United States, April 6–10, 2008.


![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-1h.png)
![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-13c.png)
| WO2006021881A2 * | 15 Aug 2005 | 2 Mar 2006 | Pfizer | Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
| WO2006021884A2 * | 15 Aug 2005 | 2 Mar 2006 | Pfizer | Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors |
| WO2013181251A1 * | 29 May 2013 | 5 Dec 2013 | Ratiopharm Gmbh | Crizotinib hydrochloride salt in crystalline |
| EP2620140A1 * | 26 Jan 2012 | 31 Jul 2013 | ratiopharm GmbH | Crizotinib containing compositions |
| WO2010048131A1 * | Oct 20, 2009 | Apr 29, 2010 | Vertex Pharmaceuticals Incorporated | C-met protein kinase inhibitors |
| WO2011042389A2 * | Oct 4, 2010 | Apr 14, 2011 | Bayer Cropscience Ag | Phenylpyri(mi)dinylazoles |
| US7825137 | Nov 23, 2006 | Nov 2, 2010 | Pfizer Inc. | Method of treating abnormal cell growth |
| US7858643 | Aug 26, 2005 | Dec 28, 2010 | Agouron Pharmaceuticals, Inc. | Crizotinib, a c-Met protein kinase inhibitor anticancer agent; 3-[(R)-1-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-5-(1-piperidin-4-yl-1H-pyrazol-4-yl)-pyridin-2-ylamine is crizotinib |
| US20060128724 | Aug 26, 2005 | Jun 15, 2006 | Agouron Pharmaceuticals, Inc. | Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
| 1 | J. JEAN CUI J. MED. CHEM. vol. 54, 2011, pages 6342 – 6363 | |
| 2 | ORG. PROCESS RES. DEV. vol. 15, 2011, pages 1018 – 1026 | |
| 3 | * | PIETER D. DE KONING ET AL: “Fit-for-Purpose Development of the Enabling Route to Crizotinib (PF-02341066)“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 15, no. 5, 16 September 2011 (2011-09-16), pages 1018-1026, XP055078841, ISSN: 1083-6160, DOI: 10.1021/op200131n |

