PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
2-{(E)-2-[(3E)-2-(4-{2-[(4-{[(2-Amino-4-oxo-3,4-dihydro-6-pteridinyl)methyl]amino}benzoyl)amino]-2-carboxyethyl}phenoxy)-3-{(2E)-2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene ]ethylidene}-1-cyclohexen-1-yl]vinyl}-3,3-dimethyl-1-(4-sulfobutyl)-3H-indolium-5-sulfonateOTL-38Tyrosine, N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-O-[(6E)-6-[(2E)-2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[(E)-2-[3,3-dimethy l-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt
2-(2-(2-(4-((2S)-2-(4-(((2-amino-4-oxo-3,4-dihydropteridin-6-yl)methyl)amino)benzamido)-2-carboxyethyl)phenoxy)-3-(2-(3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene)ethylidene)cyclohex-1-en-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium inner salt,sodium salt (1:4)
3H-Indolium, 2-(2-(2-(4-((2S)-2-((4-(((2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl)amino)benzoyl)amino)-2-carboxyethyl)phenoxy)-3-(2-(1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene)ethylidene)-1-cyclohexen-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1 (4-sulfobutyl)-, inner salt,sodium salt (1:4)
Intraoperative Imaging and Detection of Folate Receptor Positive Malignant Lesions
Pafolacianine, sold under the brand name Cytalux, is an optical imaging agent.[1][2]
The most common side effects of pafolacianine include infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity.[2]
It was approved for medical use in the United States in November 2021.[2][3]
Pafolacianine is a fluorescent drug that targets folate receptor (FR).[1]
Medical uses
Pafolacianine is indicated as an adjunct for intraoperative identification of malignant lesions in people with ovarian cancer.[1][2]
History
The safety and effectiveness of pafolacianine was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.[2] Of the 134 women (ages 33 to 81 years) who received a dose of pafolacianine and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.[2]
In another aspect of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0029] In a fourth embodiment of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0030]
[0032] wherein C is any carbon isotope. In this embodiment, the amino acid linker is selected from a group consisting of methyl 2-di-tert-butyl dicarbonate-amino-3-(4-phenyl)propanoate, 3-(4-hydroxyphenyl)-2-(di-tert-butyl-dicarbonate methylamino)propanoic acid, 2-amino-4-(4-hydroxyphenyl)butanoic acid, and Tert-butyl (2-di-tert-butyl dicarbonate- amino)-3-(4-hydroxyphenyl)propanoate . In a particular embodiment, the aqueous base is potassium hydroxide (KOH). The method of this embodiment may also further include purifying the compound by preparatory HPLC.
EXAMPLE 1 : General synthesis of Pte – L Tyrosine – S0456 (OTL-0038)
[0088] Scheme:
C33H37CIF3N
Reactants for Step I:
[0089] A 500 mL round bottom flask was charged with a stirring bar, pteroic acid
equiv) and HATU (13.45 g, 35.28 mmol, 1 .2 equiv) then DMF (147 mL) was added to give a brown suspension [suspension A]. DIPEA (20.48 mL, 1 17.62 mmol, 4.0 equiv) was added slowly to suspension A at 23 °C, over 5 minutes. The suspension turned in to a clear brown solution within 10 minutes of addition of DIPEA. The reaction was stirred at 23 °C for 2.5 h. Reaction was essentially complete in 30 minutes as judged by LC/MS but was stirred further for 2.5 h. The formation of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI (Figure 12) was confirmed by LC/MS showing m/z 409→m/z 684. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column . The reaction mixture was cannulated as a steady stream to a stirred solution of aq. HCI (2.0 L, 0.28 M) over the period of 30 minutes to give light yellow precipitate of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The precipitated Pte_N 10(TFA)_L_Tyr(- OfBu)-OfBu HCI was filtered using sintered funnel under aspirator vacuum, washed with water (8 * 300 mL) until the pH of the filtrate is between 3 and 4. The wet solid was allowed to dry under high vacuum for 12 hours on the sintered funnel. In a separate batch, where this wet solid (3) was dried under vacuum for 48 hours and then this solid was stored at -20 0 C for 48 h. However, this brief storage led to partial decomposition of 3. The wet cake (58 g) was transferred to a 500 mL round bottom flask and was submitted to the next step without further drying or purification.
Reactants for Step II:
The wet solid (58 g) was assumed to contain 29.40 mmol of the desired compound (3) (i. e. quantitative yield for the step I ).
[0090] A 500 mL round bottom flask was charged with a stirring bar, Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI as a wet cake (58 g, 29.40 mmol, 1 equiv). A solution of TFA:TIPS:H20 (95:2.5:2.5, 200 mL) was added at once to give a light brown suspension. The reaction content was stirred at 23°C for 1 .5 hours and was monitored by LC/MS. The suspension became clear dull brown solution after stirring for 5 minutes. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column. The formation of Pte_TFA_L_Tyr (Figure 12) was confirmed by showing m/z 684→m/z 572. Reaction time varies from 30 min to 1 .5 hours depending on the water content of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The reaction mixture was cannulated as a steady stream to a stirred MTBE (1 .8 L) at 23 °C or 100 °C to give light yellow precipitate of Pte_TFA_L_Tyr. The precipitated Pte_TFA_L_Tyr was filtered using sintered funnel under aspirator vacuum, washed with MTBE (6 * 300 mL) and dried under high vacuum for 8 hours to obtain Pte_TFA_L_Tyr (14.98 g, 83.98% over two steps) as a pale yellow solid. The MTBE washing was tested for absence of residual TFA utilizing wet pH paper (pH between 3-4). The yield of the reaction was between 80-85% in different batches. The deacylated side product was detected in 3.6% as judged by LC/MS. For the different batches this impurity was never more than 5%.
Reactants for Step III:
[0091] A 200 mL round bottom flask was charged with a stirring bar and Pte_TFA_L_Tyr (13.85 g, 22.78 mmol, 1 equiv), then water (95 mL) was added to give a yellow suspension [suspension B]. A freshly prepared solution of aqueous 3.75 M NaOH (26.12 mL, 97.96 mmol, 4.30 equiv), or an equivalent base at a corresponding temperature using dimethylsulfoxide (DMSO) as a solvent (as shown in Table 1 ), was added dropwise to suspension B at 23 °C, giving a clear dull yellow solution over 15 minutes [solution B]. The equivalence of NaOH varied from 3.3 to 5.0 depending on the source of 4 (solid or liquid phase synthesis) and the residual TFA. Trianion 5 (Figure 12) formation was confirmed by LC/MS showing m/z 572→m/z 476 while the solution pH was 9-10 utilizing wet pH paper. The pH of the reaction mixture was in the range of 9-10. This pH is crucial for the overall reaction completion. Notably, pH more than 10 leads to hydrolysis of S0456. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. The presence of hydrolysis by product can be visibly detected by the persistent opaque purple/blue to red/brown color.
TABLE 1 : Separate TFA deprotection via trianion formation; S0456
[0092] The precipitated OTL-0038 product could also be crashed out by adding the reaction solution steady dropwise to acetone, acetonitrile, isopropanol or ethyl acetate/acetone mixture. Acetone yields optimal results. However, viscous reactions could be slower due to partial insolubility and/or crashing out of S0456. In this reaction, the equivalence of the aqueous base is significant. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. This solution phase synthesis provides Pte_N10(TFA)_Tyr-OH »HCI salt and desires approximately 4.1 to approximately 4.8 equiv base as a source to hydrolyze the product. Particularly, precipitation of Pte_Tyr_S0456 was best achieved when 1 mL of reaction mixture is added dropwise to the stirred acetone (20 mL). Filtration of the precipitate and washing with acetone (3 x10 mL) gave the highest purity as judged from LC/MS chromatogram.
[0093] During experimentation of this solution-phase synthesis of Pte – L Tyrosine -S0456 (OTL-0038) at different stages, some optimized conditions were observed:
Mode of addition: Separate TFA deprotection via trianion formation; S0456 @ 23 °C; reflux.
Stability data of Pte – L Tyrosine – S0456 (OTL-0038):
Liquid analysis: At 40 °C the liquid lost 8.6% at 270 nm and 1 % at 774 nm. At room temperature the liquid lost about 1 .4% at 270 nm and .5% at 774 nm. At 5 °C the
270 nm seems stable and the 774 nm reasonably stable with a small degradation purity.
Source Purity Linker S0456 Base Solvent Duration % Conversion
4.3-4.6
Solution 0.95
95% 1 equiv equiv H20 15 min 100% phase equiv
K2C03
PATENT
US 20140271482
FDA approves pafolacianine for identifying malignant ovarian cancer lesions
On November 29, 2021, the Food and Drug Administration approved pafolacianine (Cytalux, On Target Laboratories, LLC), an optical imaging agent, for adult patients with ovarian cancer as an adjunct for interoperative identification of malignant lesions. Pafolacianine is a fluorescent drug that targets folate receptor which may be overexpressed in ovarian cancer. It is used with a Near-Infrared (NIR) fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
Efficacy was evaluated in a single arm, multicenter, open-label study (NCT03180307) of 178 women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer scheduled to undergo primary surgical cytoreduction, interval debulking, or recurrent ovarian cancer surgery. All patients received pafolacianine. One hundred and thirty-four patients received fluorescence imaging evaluation in addition to standard of care evaluation which includes pre-surgical imaging, intraoperative palpation and normal light evaluation of lesions. Among these patients, 36 (26.9%) had at least one evaluable ovarian cancer lesion detected with pafolacianine that was not observed by standard visual or tactile inspection. The patient-level false positive rate of pafolacianine with NIR fluorescent light with respect to the detection of ovarian cancer lesions confirmed by central pathology was 20.2% (95% CI 13.7%, 28.0%).
The most common adverse reactions (≥1%) occurring in patients were nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, pruritus, and hypersensitivity.
The recommended pafolacianine dose is 0.025 mg/kg administered intravenously over 60 minutes, 1 to 9 hours before surgery. The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of pafolacianine.
USFDA approves new drug to help identify cancer lesions
This drug is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery.By The Health Master -December 2, 2021
The U.S. Food and Drug Administration (USFDA) has approved Cytalux (pafolacianine), an imaging drug intended to assist surgeons in identifying ovarian cancer lesions. The drug is designed to improve the ability to locate additional ovarian cancerous tissue that is normally difficult to detect during surgery.
Cytalux is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery. The drug is a diagnostic agent that is administered in the form of an intravenous injection prior to surgery.
Alex Gorovets, M.D., deputy director of the Office of Specialty Medicine in the FDA’s Center for Drug Evaluation and Research said, “The FDA’s approval of Cytalux can help enhance the ability of surgeons to identify deadly ovarian tumors that may otherwise go undetected.
By supplementing current methods of detecting ovarian cancer during surgery, Cytalux offers health care professionals an additional imaging approach for patients with ovarian cancer.”
The American Cancer Society estimates there will be more than 21,000 new cases of ovarian cancer and more than 13,000 deaths from this disease in 2021, making it the deadliest of all female reproductive system cancers.
Conventional treatment for ovarian cancer includes surgery to remove as many of the tumors as possible, chemotherapy to stop the growth of malignant cells or other targeted therapy to identify and attack specific cancer cells.
Ovarian cancer often causes the body to overproduce a specific protein in cell membranes called a folate receptor. Following administration via injection, Cytalux binds to these proteins and illuminates under fluorescent light, boosting surgeons’ ability to identify the cancerous tissue.
Currently, surgeons rely on preoperative imaging, visual inspection of tumors under normal light or examination by touch to identify cancer lesions. Cytalux is used with a Near-Infrared fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
The safety and effectiveness of Cytalux was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.
Of the 134 women (ages 33 to 81 years) who received a dose of Cytalux and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.
The most common side effects of Cytalux were infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity. Cytalux may cause fetal harm when administered to a pregnant woman.
The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of Cytalux. There is a risk of image interpretation errors with the use of Cytalux to detect ovarian cancer during surgery, including false negatives and false positives.
A hydrolase enzyme that converts L-asparagine and water to L-aspartate and NH3.
NCI: Asparaginase Erwinia chrysanthemi. An enzyme isolated from the bacterium Erwinia chrysanthemi (E. carotovora). Asparagine is critical to protein synthesis in leukemic cells, which cannot synthesize this amino acid due to the absence of the enzyme asparagine synthase. Asparaginase hydrolyzes L-asparagine to L-aspartic acid and ammonia, thereby depleting leukemic cells of asparagine and blocking protein synthesis and tumor cell proliferation, especially in the G1 phase of the cell cycle. This agent also induces apoptosis in tumor cells. The Erwinia-derived product is often used for those patients who have experienced a hypersensitivity reaction to the E. Coli formulation. (NCI Thesaurus)
The present invention concerns a conjugate of a protein having substantial L-asparagine aminohydrolase activity and polyethylene glycol, particularly wherein the polyethylene glycol has a molecular weight less than or equal to about 5000 Da, particularly a conjugate wherein the protein is a L-asparaginase from Erwinia, and its use in therapy.Proteins with L-asparagine aminohydrolase activity, commonly known as L- asparaginases, have successfully been used for the treatment of Acute Lymphoblastic Leukemia(ALL) in children for many years. ALL is the most common childhood malignancy (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393).[0003] L-asparaginase has also been used to treat Hodgkin’s disease, acute myelocytic leukemia, acute myelomonocytic leukemia, chronic lymphocytic leukemia, lymphosarcoma, reticulosarcoma, and melanosarcoma (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).The anti-tumor activity of L-asparaginase is believed to be due to the inability or reduced ability of certain malignant cells to synthesize L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669). These malignant cells rely on an extracellular supply of L-asparagine. However, the L-asparaginase enzyme catalyzes the hydrolysis of L-asparagine to aspartic acid and ammonia, thereby depleting circulating pools of L-asparagine and killing tumor cells which cannot perform protein synthesis without L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0004] L-asparaginase from E. coli was the first enzyme drug used in ALL therapy and has been marketed as Elspar® in the USA or as Kidrolase® and L-asparaginase Medac® in Europe. L- asparaginases have also been isolated from other microorganisms, e.g., an L-asparaginase protein from Erwinia chrysanthemi, named crisantaspase, that has been marketed as Erwinase® (Wriston Jr., J.C. (1985) “L-asparaginase” Meth. Enzymol. 113, 608-618; Goward, CR. et al. (1992) “Rapid large scale preparation of recombinant Erwinia chrysanthemi L-asparaginase”, Bioseparation 2, 335-341). L-asparaginases from other species of Erwinia have also been identified, including, for example, Erwinia chrysanthemi 3937 (Genbank Accession#AAS67028), Erwinia chrysanthemi NCPPB 1125 (Genbank Accession #CAA31239), Erwinia carotovora (Genbank Accession #AAP92666), and Erwinia carotovora subsp. Astroseptica (Genbank Accession #AAS67027). These Erwinia chrysanthemi L-asparaginases have about 91-98% amino acid sequence identity with each other, while the Erwinia carotovora L- asparaginases have approximately 75-77% amino acid sequence identity with the Erwinia chrysanthemi L-asparaginases (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0005] L-asparaginases of bacterial origin have a high immunogenic and antigenic potential and frequently provoke adverse reactions ranging from mild allergic reaction to anaphylactic shock in sensitized patients (Wang, B. et al. (2003) “Evaluation of immunologic cross reaction of anti- asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),Leukemia 17, 1583-1588). E. coli L-asparaginase is particularly immunogenic, with reports of the presence of anti-asparaginase antibodies to E. coli L-asparaginase following i.v. or i.m. administration reaching as high as 78% in adults and 70% in children (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0006] L-asparaginases from Escherichia coli and Erwinia chrysanthemi differ in their pharmacokinetic properties and have distinct immunogenic profiles, respectively (Klug Albertsen, B. et al. (2001) “Comparison of intramuscular therapy with Erwinia asparaginase and asparaginase Medac: pharmacokinetics, pharmacodynamics, formation of antibodies and influence on the coagulation system” Brit. J. Haematol. 115, 983-990). Furthermore, it has been shown that antibodies that developed after a treatment with L-asparaginase from E. coli do not cross react with L-Asparaginase from Erwinia (Wang, B. et al., Leukemia 17 (2003) 1583-1588). Thus, L-asparaginase from Erwinia (crisantaspase) has been used as a second line treatment of ALL in patients that react to E. coli L-asparaginase (Duval, M. et al. (2002) “Comparison of Escherichia co/z-asparaginase with £Vwzm‘α-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment ofCancer, Children’s Leukemia Group phase 3 trial” Blood 15, 2734-2739; Avramis and Panosyan,Clin. Pharmacokinet. (2005) 44:367-393).[0007] In another attempt to reduce immunogenicity associated with administration of microbial L-asparaginases, an E. coli L-asparaginase has been developed that is modified with methoxy- polyethyleneglycol (mPEG). This method is commonly known as “PEGylation” and has been shown to alter the immunological properties of proteins (Abuchowski, A. et al. (1977) “Alteration of Immunological Properties of Bovine Serum Albumin by Covalent Attachment of Polyethylene Glycol,” J.Biol.Chem. 252 (11), 3578-3581). This so-called mPEG-L- asparaginase, or pegaspargase, marketed as Oncaspar® (Enzon Inc., USA), was first approved in the U.S. for second line treatment of ALL in 1994, and has been approved for first- line therapy of ALL in children and adults since 2006. Oncaspar® has a prolonged in vivo half-life and a reduced immunogenicity/antigenicity.[0008] Oncaspar® is E. coli L-asparaginase that has been modified at multiple lysine residues using 5 kDa mPEG-succinimidyl succinate (SS-PEG) (U.S. Patent No. 4,179,337). SS-PEG is aPEG reagent of the first generation that contains an instable ester linkage that is sensitive to hydro lysis by enzymes or at slightly alkaline pH values (U.S. Patent No. 4,670,417; Makromol. Chem. 1986, 187, 1131-1144). These properties decrease both in vitro and in vivo stability and can impair drug safety.[0009] Furthermore, it has been demonstrated that antibodies developed against L-asparaginase from E. coli will cross react with Oncaspar® (Wang, B. et al. (2003) “Evaluation of immunologic cross-reaction of anti-asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),” Leukemia 17, 1583-1588). Even though these antibodies were not neutralizing, this finding clearly demonstrated the high potential for cross-hypersensitivity or cross-inactivation in vivo. Indeed, in one report 30-41% of children who received pegaspargase had an allergic reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0010] In addition to outward allergic reactions, the problem of “silent hypersensitivity” was recently reported, whereby patients develop anti-asparaginase antibodies without showing any clinical evidence of a hypersensitivity reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588). This reaction can result in the formation of neutralizing antibodies to E. coli L-asparaginase and pegaspargase; however, these patients are not switched to Erwinia L-asparaginase because there are not outward signs of hypersensitivity, and therefore they receive a shorter duration of effective treatment (Holcenberg, J., J. Pediatr. Hematol. Oncol. 26 (2004) 273-274).[0011] Erwinia chrysanthemi L-asparaginase treatment is often used in the event of hypersensitivity to E. co/z-derived L-asparaginases. However, it has been observed that as many as 30-50% of patients receiving Erwinia L-asparaginase are antibody-positive (Avramis andPanosyan, Clin. Pharmacokinet. (2005) 44:367-393). Moreover, because Erwinia chrysanthemi L-asparaginase has a significantly shorter elimination half-life than the E. coli L-asparaginases, it must be administered more frequently (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393). In a study by Avramis et al., Erwinia asparaginase was associated with inferior pharmacokinetic profiles (Avramis et al., J. Pediatr. Hematol. Oncol. 29 (2007) 239-247). E. coli L-asparaginase and pegaspargase therefore have been the preferred first-line therapies for ALL over Erwinia L-asparaginase.[0012] Numerous biopharmaceuticals have successfully been PEGylated and marketed for many years. In order to couple PEG to a protein, the PEG has to be activated at its OH terminus. The activation group is chosen based on the available reactive group on the protein that will bePEGylated. In the case of proteins, the most important amino acids are lysine, cysteine, glutamic acid, aspartic acid, C-terminal carboxylic acid and the N-terminal amino group. In view of the wide range of reactive groups in a protein nearly the entire peptide chemistry has been applied to activate the PEG moiety. Examples for this activated PEG-reagents are activated carbonates, e.g., p-nitrophenyl carbonate, succinimidyl carbonate; active esters, e.g., succinimidyl ester; and for site specific coupling aldehydes and maleimides have been developed (Harris, M., Adv. Drug – A -DeI. Rev. 54 (2002), 459-476). The availability of various chemical methods for PEG modification shows that each new development of a PEGylated protein will be a case by case study. In addition to the chemistry the molecular weight of the PEG that is attached to the protein has a strong impact on the pharmaceutical properties of the PEGylated protein. In most cases it is expected that, the higher the molecular weight of the PEG, the better the improvement of the pharmaceutical properties (Sherman, M. R., Adv. Drug Del. Rev. 60 (2008), 59-68; Holtsberg, F. W., Journal of Controlled Release 80 (2002), 259-271). For example, Holtsberg et al. found that, when PEG was conjugated to arginine deaminase, another amino acid degrading enzyme isolated from a microbial source, pharmacokinetic and pharmacodynamic function of the enzyme increased as the size of the PEG attachment increased from a molecular weight of 5000Da to 20,000 Da (Holtsberg, F.W., Journal of Controlled Release 80 (2002), 259-271).[0013] However, in many cases, PEGylated biopharmaceuticals show significantly reduced activity compared to the unmodified biopharmaceutical (Fishburn, CS. (2008) Review “The Pharmacology of PEGylation: Balancing PD with PK to Generate Novel Therapeutics” J. Pharm. Sd., 1-17). In the case of L-asparaginase from Erwinia carotovora, it has been observed that PEGylation reduced its in vitro activity to approximately 57% (Kuchumova, A.V. et al. (2007) “Modification of Recombinant asparaginase from Erwinia carotovora with Polyethylene Glycol 5000” Biochemistry (Moscow) Supplement Series B: Biomedical Chemistry, 1, 230-232). The L-asparaginase from Erwinia carotovora has only about 75% homology to the Erwinia chrysanthemi L-asparaginase (crisantaspase). For Oncaspar® it is also known that its in vitro activity is approximately 50% compared to the unmodified E. coli L-asparaginase.[0014] The currently available L-asparaginase preparations do not provide alternative or complementary therapies— particularly therapies to treat ALL— that are characterized by high catalytic activity and significantly improved pharmacological and pharmacokinetic properties, as well as reduced immunogenicity. L-asparaginase protein has at least about 80% homology or identity with the protein comprising the sequence of SEQ ID NO:1, more specifically at least about 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% homology or identity with the protein comprising the sequence of SEQ ID NO:1. SEQ ID NO:1 is as follows:ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGE QFSNMASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTV KSDKPVVFVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSA RYITKTNASTLDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKV DILYGYQDDPEYLYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY (SEQ ID NO:1) [0048] The term “comprising the sequence of SEQ ID NO:1” means that the amino-acid sequence of the protein may not be strictly limited to SEQ ID NO:1 but may contain additional amino-acids.ExamplesExample 1 : Preparation of Recombinant Crisantaspase [0100] The recombinant bacterial strain used to manufacture the naked recombinant Erwinia chrysanthemi L-asparaginase protein (also referred to herein as “r-crisantaspase”) was an E. coli BL21 strain with a deleted ansB gene (the gene encoding the endogenous E. coli type II L- asparaginase) to avoid potential contamination of the recombinant Erwinia chrysanthemi L- asparaginase with this enzyme. The deletion of the ansB gene relies on homologous recombination methods and phage transduction performed according to the three following steps:1) a bacterial strain (NMI lOO) expressing a defective lambda phage which supplies functions that protect and recombine electroporated linear DNA substrate in the bacterial cell was transformed with a linear plasmid (kanamycin cassette) containing the kanamycin gene flanked by an FLP recognition target sequence (FRT). Recombination occurs to replace the ansB gene by the kanamycin cassette in the bacterial genome, resulting in a ΛansB strain; 2) phage transduction was used to integrate the integrated kanamycin cassette region from the ΛansB NMI lOO strain to the ansB locus in BL21 strain. This results in an E. coli BL21 strain with a deleted ansB gene and resistant to kanamycin; 3) this strain was transformed with a FLP -helper plasmid to remove the kanamycin gene by homologous recombination at the FRT sequence. The genome of the final strain (BL21 ΛansB strain) was sequenced, confirming full deletion of the endogenous ansB gene.[0101] The E. co/z‘-optimized DNA sequence encoding for the mature Erwinia chrysanthemi L- asparaginase fused with the ENX signal peptide from Bacillus subtilis was inserted into an expression vector. This vector allows expression of recombinant Erwinia chrysanthemi L- asparaginase under the control of hybrid T5/lac promoter induced by the addition of Isopropyl β- D-1-thiogalactopyranoside (IPTG) and confers resistance to kanamycin.[0102] BL21 ΛansB strain was transformed with this expression vector. The transformed cells were used for production of the r-crisantaspase by feed batch glucose fermentation in Reisenberg medium. The induction of the cell was done 16h at 23°C with IPTG as inducer. After cell harvest and lysis by homogenization in 1OmM sodium phosphate buffer pH6 5mM EDTA (Buffer A), the protein solution was clarified by centrifugation twice at 1500Og, followed by 0.45μm and 0.22μm filtration steps. The recombinant Erwinia chrysanthemi L-asparaginase was next purified using a sequence of chromatography and concentration steps. Briefly, the theoretical isoelectric point of the Erwinia chrysanthemi L-asparaginase (7.23) permits the recombinant enzyme to adsorb to cation exchange resins at pH6. Thus, the recombinant enzyme was captured on a Capto S column (cation exchange chromatography) and eluted with salt gradient in Buffer A. Fractions containing the recombinant enzyme were pooled. The pooled solution was next purified on Capto MMC column (cation exchange chromatography) in Buffer A with salt gradient. . The eluted fractions containing Erwinia chrysanthemi L-asparaginase were pooled and concentrated before protein separation on Superdex 200pg size exclusion chromatography as polishing step. Fractions containing recombinant enzymes were pooled, concentrated, and diafiltered against 10OmM sodium phosphate buffer pH8. The purity of the final Erwinia chrysanthemi L-asparaginase preparation was evaluated by SDS-PAGE (Figure 1) and RP-HPLC and was at least 90%. The integrity of the recombinant enzyme was verified byN-terminal sequencing and LC-MS. Enzyme activity was measured at 37°C using Nessler’s reagent. The specific activity of the purified recombinant Erwinia chrysanthemi L-asparaginase was around 600 U/mg. One unit of enzyme activity is defined as the amount of enzyme that liberates lμmol of ammonia from L-asparagine per minute at 37°C. Example 2: Preparation of 10 kDa mPEG-L- Asparaginase Conjugates[0103] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration between 2.5 and 4 mg/mL, in the presence of 150 mg/mL or 36 mg/mL 10 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 10 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 10 kDa mPEG-L-asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) residues being conjugated corresponding to PEGylation of 78% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (39% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 50% of accessible amino groups (e.g., lysine residues and/or the N-terminus)) . SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 3: Preparation of 5 kDa mPEG-L-Asparaginase Conjugates[0104] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration of 4 mg/mL, in the presence of 150 mg/mL or 22.5 mg/mL 5 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 5 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 5 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) being conjugated corresponding to PEGylation of 84% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (36% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 43% of accessible amino groups (e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 4: Preparation of 2 kDa mPEG-L-Asparaginase Conjugates[0105] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer pH 8.0 at a protein concentration of 4 mg/mL in the presence of150 mg/mL or 22.5 mg/mL 2 kDa mPEG-NHS for 2 hours at 22°C. The resulting crude 2 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 2 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as reference, one corresponding to maximum PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N- terminus) being conjugated corresponding to PEGylation of 86% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (47% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 55% of accessible amino groups {e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 5: Activity of mPEG-r-Crisantaspase Conjugates[0106] L-asparaginase aminohydrolase activity of each conjugate described in the proceeding examples was determined by Nesslerization of ammonia that is liberated from L-asparagine by enzymatic activity. Briefly, 50μL of enzyme solution were mixed with 2OmM of L-asparagine in a 50 mM Sodium borate buffer pH 8.6 and incubated for 10 min at 37°C. The reaction was stopped by addition of 200μL of Nessler reagent. Absorbance of this solution was measured at 450 nm. The activity was calculated from a calibration curve that was obtained from Ammonia sulfate as reference. The results are summarized in Table 2, below:Table 2: Activity of mPEG-r-crisantaspase conjugates
* the numbers “40%” and “100%” indicate an approximate degree of PEGylation of respectively 40-55% and 100% of accessible amino groups (see Examples 2-4, supra).** the ratio mol PEG / mol monomer was extrapolated from data using TNBS assay, that makes the assumption that all amino groups from the protein (e.g., lysine residues and the N-terminus) are accessible.[0107] Residual activity of mPEG-r-crisantaspase conjugates ranged between 483 and 543 Units/mg. This corresponds to 78-87% of L-asparagine aminohydrolase activity of the unmodified enzyme. Example 6: L-Asparagine-Depleting Effect of Unmodified Crisantaspase
PAPER
Biotechnology and Applied Biochemistry (2019), 66(3), 281-289. |
Crisantaspase is an asparaginase enzyme produced by Erwinia chrysanthemi and used to treat acute lymphoblastic leukemia (ALL) in case of hypersensitivity to Escherichia coli l-asparaginase (ASNase). The main disadvantages of crisantaspase are the short half-life (10 H) and immunogenicity. In this sense, its PEGylated form (PEG-crisantaspase) could not only reduce immunogenicity but also improve plasma half-life. In this work, we developed a process to obtain a site-specific N-terminal PEGylated crisantaspase (PEG-crisantaspase). Crisantaspase was recombinantly expressed in E. coli BL21(DE3) strain cultivated in a shaker and in a 2-L bioreactor. Volumetric productivity in bioreactor increased 37% compared to shaker conditions (460 and 335 U L−1 H−1, respectively). Crisantaspase was extracted by osmotic shock and purified by cation exchange chromatography, presenting specific activity of 694 U mg−1, 21.7 purification fold, and yield of 69%. Purified crisantaspase was PEGylated with 10 kDa methoxy polyethylene glycol-N-hydroxysuccinimidyl (mPEG-NHS) at different pH values (6.5–9.0). The highest N-terminal pegylation yield (50%) was at pH 7.5 with the lowest poly-PEGylation ratio (7%). PEG-crisantaspase was purified by size exclusion chromatography and presented a KM value three times higher than crisantaspase (150 and 48.5 µM, respectively). Nonetheless, PEG-crisantaspase was found to be more stable at high temperatures and over longer periods of time. In 2 weeks, crisantaspase lost 93% of its specific activity, whereas PEG-crisantaspase was stable for 20 days. Therefore, the novel PEG-crisantaspase enzyme represents a promising biobetter alternative for the treatment of ALL.
Figure S1 – Amino acid sequence of the enzyme crisantaspase without the signal peptide and with the lysines highlighted in red (Swiss-Prot/TrEMBL accession number: P06608|22-348 AA).
……………………………………………………………………………………………………………………………..
As a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in patients who are allergic to E. coli-derived asparaginase products Press ReleaseFor Immediate Release:June 30, 2021
FDA Approves Component of Treatment Regimen for Most Common Childhood Cancer
Alternative Has Been in Global Shortage Since 2016
Today, the U.S. Food and Drug Administration approved Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) as a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in adult and pediatric patients who are allergic to the E. coli-derived asparaginase products used most commonly for treatment. The only other FDA-approved drug for such patients with allergic reactions has been in global shortage for years.
“It is extremely disconcerting to patients, families and providers when there is a lack of access to critical drugs for treatment of a life-threatening, but often curable cancer, due to supply issues,” said Gregory Reaman, M.D., associate director for pediatric oncology in the FDA’s Oncology Center of Excellence. “Today’s approval may provide a consistently sourced alternative to a pivotal component of potentially curative therapy for children and adults with this type of leukemia.”
Acute lymphoblastic leukemia occurs in approximately 5,700 patients annually, about half of whom are children. It is the most common type of childhood cancer. One component of the chemotherapy regimen is an enzyme called asparaginase that kills cancer cells by depriving them of substances needed to survive. An estimated 20% of patients are allergic to the standard E. coli-derived asparaginase and need an alternative their bodies can tolerate.
Rylaze’s efficacy was evaluated in a study of 102 patients who either had a hypersensitivity to E. coli-derived asparaginases or experienced silent inactivation. The main measurement was whether patients achieved and maintained a certain level of asparaginase activity. The study found that the recommended dosage would provide the target level of asparaginase activity in 94% of patients.
The most common side effects of Rylaze include hypersensitivity reactions, pancreatic toxicity, blood clots, hemorrhage and liver toxicity.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Health Canada, where the application review is pending.
Rylaze received Fast Track and Orphan Drug designations for this indication. Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fulfill an unmet medical need. Orphan Drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted approval of Rylaze to Jazz Pharmaceuticals.
DUBLIN, June 30, 2021 /PRNewswire/ — Jazz Pharmaceuticals plc (Nasdaq: JAZZ) today announced the U.S. Food and Drug Administration (FDA) approval of Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn) for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase.1Rylaze is the only recombinant erwinia asparaginase manufactured product that maintains a clinically meaningful level of asparaginase activity throughout the entire duration of treatment, and it was developed by Jazz to address the needs of patients and healthcare providers with an innovative, high-quality erwinia-derived asparaginase with reliable supply.
“We are excited to bring this important new treatment to patients who are in critical need, and we are grateful to FDA for the approval of Rylaze based on its established safety and efficacy profile. We are pleased Rylaze was approved before the trial is complete and are diligently working to advance additional clinical trial data. We are committed to quickly engaging with FDA to evolve the Rylaze product profile with additional dosing options and an IV route of administration,” said Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals. “Thank you to our collaborators within the Children’s Oncology Group, the clinical trial investigators, patients and their families, and all of the other stakeholders who helped us achieve this significant milestone.”
Rylaze was granted orphan drug designation for the treatment of ALL/LBL by FDA in June 2021. The Biologics Licensing Application (BLA) approval followed review under the Real-Time Oncology Review (RTOR) program, an initiative of FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The company expects Rylaze will be commercially available in mid-July.
“The accelerated development and approval of Rylaze marks an important step in bringing a meaningful new treatment option for many ALL patients – most of whom are children – who cannot tolerate E. coli-derived asparaginase medicine,” said Dr. Luke Maese, assistant professor at the University of Utah, Primary Children’s Hospital and Huntsman Cancer Institute. “Before the approval of Rylaze, there was a significant need for an effective asparaginase medicine that would allow patients to start and complete their prescribed treatment program with confidence in supply.”
Recent data from a Children’s Oncology Group retrospective analysis of over 8,000 patients found that patients who did not receive a full course of asparaginase treatment due to associated toxicity had significantly lower survival outcomes – regardless of whether those patients were high risk or standard risk, slow early responders.2
About Study JZP458-201 The FDA approval of Rylaze, also known as JZP458, is based on clinical data from an ongoing pivotal Phase 2/3 single-arm, open-label, multicenter, dose confirmation study evaluating pediatric and adult patients with ALL or LBL who have had an allergic reaction to E. coli-derived asparaginases and have not previously received asparaginase erwinia chrysanthemi. The study was designed to assess the safety, tolerability and efficacy of JZP458. The determination of efficacy was measured by serum asparaginase activity (SAA) levels. The Phase 2/3 study is being conducted in two parts. The first part is investigating the intramuscular (IM) route of administration, including a Monday-Wednesday-Friday dosing schedule. The second part remains active to further confirm the dose and schedule for the intravenous (IV) route of administration.
The FDA approval of Rylaze was based on data from the first of three IM cohorts, which demonstrated the achievement and maintenance of nadir serum asparaginase activity (NSAA) greater than or equal to the level of 0.1 U/mL at 48 hours using IM doses of Rylaze 25 mg/m2. The results of modeling and simulations showed that for a dosage of 25 mg/m2 administered intramuscularly every 48 hours, the proportion of patients maintaining NSAA ≥ 0.1 U/mL at 48 hours after a dose of Rylaze was 93.6% (95% CI: 92.6%, 94.6%).1
The most common adverse reactions (incidence >15%) were abnormal liver test, nausea, musculoskeletal pain, fatigue, infection, headache, pyrexia, drug hypersensitivity, febrile neutropenia, decreased appetite, stomatitis, bleeding and hyperglycemia. In patients treated with the Rylaze, a fatal adverse reaction (infection) occurred in one patient and serious adverse reactions occurred in 55% of patients. The most frequent serious adverse reactions (in ≥5% of patients) were febrile neutropenia, dehydration, pyrexia, stomatitis, diarrhea, drug hypersensitivity, infection, nausea and viral infection. Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received Rylaze. Adverse reactions resulting in permanent discontinuation included hypersensitivity (6%) and infection (3%).1
The company will continue to work with FDA and plans to submit additional data from a completed cohort of patients evaluating 25mg/m2 IM given on Monday and Wednesday, and 50 mg/m2 given on Friday in support of a M/W/F dosing schedule. Part 2 of the study is evaluating IV administration and is ongoing. The company also plans to submit these data for presentation at a future medical meeting.
Investor Webcast The company will host an investor webcast on the Rylaze approval in July. Details will be announced separately.
About Rylaze™(asparaginase erwinia chrysanthemi (recombinant)-rywn) Rylaze, also known as JZP458, is approved in the U.S. for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase. Rylaze has orphan drug designation for the treatment of ALL/LBL in the United States. Rylaze is a recombinant erwinia asparaginase that uses a novel Pseudomonas fluorescens expression platform. JZP458 was granted Fast Track designation by the U.S. Food and Drug Administration (FDA) in October 2019 for the treatment of this patient population. Rylaze was approved as part of the Real-Time Oncology Review program, an initiative of the FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
RYLAZE should not be given to people who have had:
Serious allergic reactions to RYLAZE
Serious swelling of the pancreas (stomach pain), serious blood clots, or serious bleeding during previous asparaginase treatment
RYLAZE may cause serious side effects, including:
Allergic reactions (a feeling of tightness in your throat, unusual swelling/redness in your throat and/or tongue, or trouble breathing), some of which may be life-threatening
Swelling of the pancreas (stomach pain)
Blood clots (may have a headache or pain in leg, arm, or chest)
Bleeding
Liver problems
Contact your doctor immediately if any of these side effects occur.
Some of the most common side effects with RYLAZE include: liver problems, nausea, bone and muscle pain, tiredness, infection, headache, fever, allergic reactions, fever with low white blood cell count, decreased appetite, mouth swelling (sometimes with sores), bleeding, and too much sugar in the blood.
RYLAZE can harm your unborn baby. Inform your doctor if you are pregnant, planning to become pregnant, or nursing. Females of reproductive potential should use effective contraception (other than oral contraceptives) during treatment and for 3 months following the final dose. Do not breastfeed while receiving RYLAZE and for 1 week after the final dose.
Tell your healthcare provider if there are any side effects that are bothersome or that do not go away.
These are not all the possible side effects of RYLAZE. For more information, ask your healthcare provider.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088 (1-800-332-1088).
About ALL ALL is a cancer of the blood and bone marrow that can progress quickly if not treated.3 Leukemia is the most common cancer in children, and about three out of four of these cases are ALL.4 Although it is one of the most common cancers in children, ALL is among the most curable of the pediatric malignancies due to recent advancements in treatment.5,6 Adults can also develop ALL, and about four of every 10 cases of ALL diagnosed are in adults.7 The American Cancer Society estimates that almost 6,000 new cases of ALL will be diagnosed in the United States in 2021.7 Asparaginase is a core component of multi-agent chemotherapeutic regimens in ALL.8 However, asparaginase treatments derived from E. coli are associated with the potential for development of hypersensitivity reactions.9
AboutLymphoblastic Lymphoma LBL is a rare, fast-growing, aggressive subtype of Non-Hodgkin’s lymphoma, most often seen in teenagers and young adults.8 LBL is a very aggressive lymphoma – also called high-grade lymphoma – which means the lymphoma grows quickly with early spread to different parts of the body.10,11
About Jazz Pharmaceuticals plc Jazz Pharmaceuticals plc (NASDAQ: JAZZ) is a global biopharmaceutical company whose purpose is to innovate to transform the lives of patients and their families. We are dedicated to developing life-changing medicines for people with serious diseases – often with limited or no therapeutic options. We have a diverse portfolio of marketed medicines and novel product candidates, from early- to late-stage development, in neuroscience and oncology. We actively explore new options for patients including novel compounds, small molecules and biologics, and through cannabinoid science and innovative delivery technologies. Jazz is headquartered in Dublin, Ireland and has employees around the globe, serving patients in nearly 75 countries. For more information, please visit www.jazzpharmaceuticals.com and follow @JazzPharma on Twitter.
About The Children’s Oncology Group (COG) COG (childrensoncologygroup.org), a member of the NCI National Clinical Trials Network (NCTN), is the world’s largest organization devoted exclusively to childhood and adolescent cancer research. COG unites over 10,000 experts in childhood cancer at more than 200 leading children’s hospitals, universities, and cancer centers across North America, Australia, and New Zealand in the fight against childhood cancer. Today, more than 90% of the 14,000 children and adolescents diagnosed with cancer each year in the United States are cared for at COG member institutions. Research performed by COG institutions over the past 50 years has transformed childhood cancer from a virtually incurable disease to one with a combined 5-year survival rate of 80%. COG’s mission is to improve the cure rate and outcomes for all children with cancer.
Caution Concerning Forward-Looking Statements This press release contains forward-looking statements, including, but not limited to, statements related to Jazz Pharmaceuticals’ belief in the potential of Rylaze to provide a reliable therapeutic option for adult and pediatric patients to maximize their chance for a cure, plans for a mid-July 2021 launch of Rylaze, the availability of a reliable supply of Rylaze and other statements that are not historical facts. These forward-looking statements are based on Jazz Pharmaceuticals’ current plans, objectives, estimates, expectations and intentions and inherently involve significant risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in such forward-looking statements as a result of these risks and uncertainties, which include, without limitation, effectively launching and commercializing new products; obtaining and maintaining adequate coverage and reimbursement for the company’s products; delays or problems in the supply or manufacture of the company’s products and other risks and uncertainties affecting the company, including those described from time to time under the caption “Risk Factors” and elsewhere in Jazz Pharmaceuticals’ Securities and Exchange Commission filings and reports (Commission File No. 001-33500), including Jazz Pharmaceuticals’ Annual Report on Form 10-K for the year ended December 31, 2020 and future filings and reports by Jazz Pharmaceuticals. Other risks and uncertainties of which Jazz Pharmaceuticals is not currently aware may also affect Jazz Pharmaceuticals’ forward-looking statements and may cause actual results and the timing of events to differ materially from those anticipated. The forward-looking statements herein are made only as of the date hereof or as of the dates indicated in the forward-looking statements, even if they are subsequently made available by Jazz Pharmaceuticals on its website or otherwise. Jazz Pharmaceuticals undertakes no obligation to update or supplement any forward-looking statements to reflect actual results, new information, future events, changes in its expectations or other circumstances that exist after the date as of which the forward-looking statements were made.
Jazz Media Contact: Jacqueline Kirby Vice President, Corporate Affairs Jazz Pharmaceuticals plc CorporateAffairsMediaInfo@jazzpharma.com Ireland, +353 1 697 2141 U.S. +1 215 867 4910
Jazz Investor Contact: Andrea N. Flynn, Ph.D. Vice President, Head, Investor Relations Jazz Pharmaceuticals plc investorinfo@jazzpharma.com Ireland, +353 1 634 3211
References
Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) injection, for intramuscular use Prescribing Information. Palo Alto, CA: Jazz Pharmaceuticals, Inc.
Gupta S, Wang C, Raetz EA et al. Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children’s Oncology Group. J Clin Oncol. 2020 Jun 10;38(17):1897-1905. doi: 10.1200/JCO.19.03024
Salzer W, Bostrom B, Messinger Y et al. 2018. Asparaginase activity levels and monitoring in patients with acute lymphoblastic leukemia. Leukemia & Lymphoma. 59:8, 1797-1806, DOI: 10.1080/10428194.2017.1386305.
Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57(4):748–757. DOI: 10.3109/10428194.2015.1101098.
Indication:Ovarian cancer; Breast cancer; Non small cell lung cancer (NSCLC)лурбинектединلوربينيكتيدين芦比替定(1R,1’R,2’R,3’R,11’S,12’S,14’R)-5′,12′-Dihydroxy-6,6′-dimethoxy-7′,21′,30′-trimethyl-27′-oxo-2,3,4,9-tetrahydrospiro[β-carboline-1,26′-[17,19,28]trioxa[24]thia[13,30]diazaheptacyclo[12.9.6.13,11. 02,13.04,9.015,23.016,20]triaconta[4,6,8,15,20,22]hexaen]-22′-yl acetate [ACD/IUPAC Name]2CN60TN6ZS497871-47-3[RN]9397
Lurbinectedin is in phase III clinical development for the treatment of platinum refractory/resistant ovarian cancer.
Phase II clinical trials are also ongoing for several oncology indications: non-small cell lung cancer, breast cancer, small cell lung cancer, head and neck carcinoma, neuroendocrine tumors, biliary tract carcinoma, endometrial carcinoma, germ cell tumors and Ewing’s family of tumors.
Lurbinectedin, sold under the brand name Zepzelca, is a medication for the treatment of adults with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.[1][2][3]
The most common side effects include leukopenia, lymphopenia, fatigue, anemia, neutropenia, increased creatinine, increased alanine aminotransferase, increased glucose, thrombocytopenia, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.[1][2][3]
Lurbinectedin is a synthetic tetrahydropyrrolo [4, 3, 2-de]quinolin-8(1H)-one alkaloid analogue with potential antineoplastic activity.[4] Lurbinectedin covalently binds to residues lying in the minor groove of DNA, which may result in delayed progression through S phase, cell cycle arrest in the G2/M phase and cell death.[4]
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Structure
Lurbinectedin is structurally similar to trabectedin, although the tetrahydroisoquinoline present in trabectedin is replaced with a tetrahydro β-carboline which enables lurbinectedin to exhibit increased antitumor activity compared with trabectedin.[7]
Biosynthesis
Lurbinectedin a marine agent isolated from the sea squirt species Ecteinascidia turbinata. Synthetic production is necessary because very small amounts can be obtained from sea organisms. For example, one ton (1000 kg) of sea squirts are required to produce one gram of trabectedin, which is analogue of lurbinectedin. Complex synthesis of lurbinectedin starts from small, common starting materials that require twenty-six individual steps to produce the drug with overall yield of 1.6%.[8][9]
Mechanism of action
According to PharmaMar,[10] lurbinectedin inhibits the active transcription of the encoding genes. This has two consequences. On one hand, it promotes tumor cell death, and on the other it normalizes tumor microenvironment. Active transcription is the process by which there are specific signal where information contained in the DNA sequence is transferred to an RNA molecule. This activity depends on the activity of an enzyme called RNA polymerase II. Lurbinectedin inhibits transcription through a very precise mechanism. Firstly, lurbinectedin binds to specific DNA sequences. It is at these precise spots that slides down the DNA to produce RNA polymerase II that is blocked and degraded by lurbinectedin. Lurbinectedin also has important role in tumor microenvironment. The tumor cells act upon macrophages to avoid them from behaving like an activator of the immune system. Literally, macrophages work in any tumor’s favor. Macrophages can contribute to tumor growth and progression by promoting tumor cell proliferation and invasion, fostering tumor angiogenesis and suppressing antitumor immune cells.[11][12] Attracted to oxygen-starved (hypoxic) and necrotic tumor cells they promote chronic inflammation. So, not only that macrophages inhibit immune system avoiding the destruction of tumor cells, but they also create tumor tissue that allows tumor growth. However, macrophages associated with tumors are cells that are addicted to the transcription process. Lurbinectedin acts specifically on the macrophages associated with tumors in two ways: firstly, by inhibiting the transcription of macrophages that leads to cell death and secondly, inhibiting the production of tumor growth factors. In this way, lurbinectedin normalizes the tumor microenvironment.
History
Lurbinectedin was approved for medical use in the United States in June 2020.[5][1][2][3][6]
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 participants with metastatic SCLC who had disease progression on or after platinum-based chemotherapy.[3][6] Participants received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.[3] The trial was conducted at 26 sites in the United States, Great Britain, Belgium, France, Italy, Spain and Czech Republic.[6]
The U.S. Food and Drug Administration (FDA) granted the application for lurbinectedin priority review and orphan drug designations and granted the approval of Zepzelca to Pharma Mar S.A.[3][13]
Research
Clinical Trials
Lurbinectedin can be used as monotherapy in the treatment of SCLC. Lurbinectedin monotherapy demonstrated the following clinical results in relapsed extensive stage SCLC:
For sensitive disease (chemotherapy-free interval of ≥ 90 days) overall response rate (ORR) was 46.6% with 79.3% disease control rate and median overall survival (OS) being increased to 15.2 months.[14]
For resistant disease (chemotherapy-free interval of < 90 days) overall response rate (ORR) was 21.3% with 46.8% disease control rate and 5.1 months median overall survival (OS).[14]
Lurbinectedin is also being investigated in combination with doxorubicin as second-line therapy in a randomized Phase III trial.[medical citation needed] While overall survival in this trial is not yet known, response rates at second line were
91.7% in sensitive disease with median progression-free survival of 5.8 months, and
33.3% in resistant disease with median progression-free of 3.5 months.[15]
Lurbinectedin is available in the U.S. under Expanded Access Program (EAP).[15][16]
Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01 /771 15; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 1 1456- 1 1460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001 , Curr. Med. Chem. Anti-Cancer Agents, 1 , pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731 , ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641 , and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources.
At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to
specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum- sensitive ovarian cancer in combination with pegylated liposomal doxorubicin.
ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula
It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought.
The first synthetic process for producing ecteinascidin compounds was described in US Patent 5,721 ,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence.
An improvement in the preparation of one intermediate used in such process was disclosed in US Patent 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale.
A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens.
Cyanosafracin B
An improvement in such hemisynthetic process was disclosed in
EP 1.287.004.
To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600.
WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula:
Total synthesis strategies for the synthesis of the pentacyclic core -743 are overviewed in Figure I.
X = OH or CI
R = Protecting Group
WO2007087220 JOC 2008, 73, 9594-9600
EXAMPLE 3: SYNTHESIS OF COMPOUND 17.
Scheme X above provides an example of the synthesis of compound 17 from intermediate 10.
Compounds 16 and 17 are obtainable from intermediate 15 using the same procedures than those previously described in WO03/014127.
The ecteinascidins are exceedingly potent antitumour agents isolated from the marine tunicate Ecteinascidia turbinata. Several ecteinascidins have been reported previously in the patent and scientific literature. See, for example:
U.S. Patent No 5.256.663, which describes pharmaceutical compositions comprising matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins, and the use of such compositions as antibacterial, antiviral, and/ or antitumour agents in mammals.
U.S. Patent No 5.089.273, which describes novel compositions of matter extracted from the tropical marine invertebrate, Ecteinascidia turbinata, and designated therein as ecteinascidins 729, 743, 745, 759A, 759B and 770. These compounds are useful as antibacterial and/or antitumour agents in mammals.
U.S. Patent No 5.149.804 which describes Ecteinascidins 722 and 736 (Et’s 722 and 736) isolated from the Caribbean tunicate Ecteinascidia turbinata and their structures. Et’s 722 and 736 protect mice in vivo at very low concentrations against P388 lymphoma, B 16 melanoma, and Lewis lung carcinoma.
U.S. Patent No 5.478.932, which describes ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX- 1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.654.426, which describes several ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo protection against P388 lymphoma, B 16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX-1 human lung and MX- 1 human mammary carcinoma xenografts.
U.S. Patent No 5.721.362 which describes a synthetic process for the formation of ecteinascidin compounds and related structures.
U.S. Patent No 6.124.292 which describes a series of new ecteinascidin- like compounds.
WO 0177115, WO 0187894 and WO 0187895, which describe new synthetic compounds of the ecteinascidin series, their synthesis and biological properties.
See also: Corey, E.J., J. Am. Chem. Soc, 1996, 118 pp. 9202-9203; Rinehart, et al., Journal of Natural Products, 1990, “Bioactive Compounds from Aquatic and Terrestrial Sources”, vol. 53, pp. 771- 792; Rinehart et al., Pure and Appl. Chem., 1990, “Biologically active natural products”, vol 62, pp. 1277- 1280; Rinehart, et al., J. Org. Chem., 1990, “Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent Antitumour Agents from the Caribbean Tunicate Ecteinascidia tuminata”, vol. 55, pp. 4512-4515; Wright et al., J. Org. Chem., 1990, “Antitumour Tetrahydroisoquinoline Alkaloids from the Colonial ascidian Ecteinascidia turbinata”, vol. 55, pp. 4508-4512; Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional anitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 1 1456- 1 1460; Science 1994, “Chemical Prospectors Scour the Seas for Promising Drugs”, vol. 266, pp.1324; Koenig, K.E., “Asymmetric Synthesis”, ed. Morrison, Academic Press, Inc., Orlando, FL, vol. 5, 1985, p. 71; Barton, et al., J. Chem Soc. Perkin Trans., 1 , 1982, “Synthesis and Properties of a Series of Sterically Hindered Guanidine bases”, pp. 2085; Fukuyama et al., J. Am. Chem. Soc, 1982, “Stereocontrolled Total Synthesis of (+)-Saframycin B”, vol. 104, pp. 4957; Fukuyama et al., J. Am. Chem. Soc, 1990, “Total Synthesis of (+) – Saframycin A”, vol. 112, p. 3712; Saito, et al., J. Org. Chem., 1989, “Synthesis of Saframycins. Preparation of a Key tricyclic Lactam Intermediate to Saframycin A”, vol. 54, 5391; Still, et al., J Org. Chem., 1978, “Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution”, vol. 43, p. 2923; Kofron, W.G.; Baclawski, L.M., J. Org. Chem., 1976, vol. 41, 1879; Guan et al., J. Biomolec Struc & Dynam., vol. 10, pp. 793-817 (1993); Shamma et al., “Carbon- 13 NMR Shift Assignments of Amines and Alkaloids”, p. 206 (1979); Lown et al., Biochemistry, 21, 419-428 (1982); Zmijewski et al., Chem. Biol. Interactions, 52, 361-375 (1985); Ito, CRC Crit. Rev. Anal. Chem., 17, 65- 143 (1986); Rinehart et al., “Topics in Pharmaceutical Sciences 1989”, pp. 613-626, D. D. Breimer, D. J. A. Cromwelin, K. K. Midha, Eds., Amsterdam Medical Press B. V., Noordwijk, The Netherlands (1989); Rinehart et al., “Biological Mass Spectrometry”, 233-258 eds. Burlingame et al., Elsevier Amsterdam (1990); Guan et al., Jour. Biomolec. Struct. & Dynam., vol. 10 pp. 793-817 (1993); Nakagawa et al., J. Amer. Chem. Soc, 11 1 : 2721-2722 (1989);; Lichter et al., “Food and Drugs from the Sea Proceedings” (1972), Marine Technology Society, Washington, D.C. 1973, 117- 127; Sakai et al., J. Amer. Chem. Soc, 1996, 1 18, 9017; Garcϊa-Rocha et al., Brit. J. Cancer, 1996, 73: 875-883; and pommier et al., Biochemistry, 1996, 35: 13303- 13309;
In 2000, a hemisynthetic process for the formation of ecteinascidin compounds and related structures such as phthalascidin starting from natural bis(tetrahydroisoquinoline) alkaloids such as the saframycin and safracin antibiotics available from different culture broths was reported; See Manzanares et al., Org. Lett., 2000, “Synthesis of Ecteinascidin ET-743 and Phthalascidin Pt-650 from Cyanosafracin B”, Vol. 2, No 16, pp. 2545-2548; and International Patent Application WO 00 69862.
Ecteinascidin 736 was first discovered by Rinehart and features a tetrahydro-β-carboline unit in place of the tetrahydroisoquinoline unit more usually found in the ecteinascidin compounds isolated from natural sources; See for example Sakai et al., Proc. Natl. Acad. Sci. USA 1992, “Additional antitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo”, vol. 89, 11456-11460.
Et-736
WO 9209607 claims ecteinascidin 736, as well as ecteinascidin 722 with hydrogen in place of methyl on the nitrogen common to rings C and D of ecteinascidin 736 and O-methylecteinascidin 736 with methoxy in place of hydroxy on ring C of ecteinascidin 736.
Despite the positive results obtained in clinical applications in chemotherapy, the search in the field of ecteinascidin compounds is still open to the identification of new compounds with optimal features of cytotoxicity and selectivity toward the tumour and with a reduced systemic toxicity and improved pharmacokinetic properties.
Ecteinascidins is a group of naturally occurring marine compounds and analogs thereof, which are well identified and structurally characterized, and are disclosed to have antibacterial and cytotoxic properties. See for example, European Patent 309.477; WO 03/66638; WO 03/08423; WO 01/77115; WO 03/014127; R. Sakai et al., 1992, Proc. Natl. Acad. Sci. USA 89, pages 11456-11460; R. Menchaca et al., 2003, J. Org. Chem. 68(23), pages 8859-8866; and I. Manzanares et al., 2001, Curr. Med. Chem. Anti–Cancer Agents, 1, pages 257-276; and references therein. Examples of ecteinascidins are provided by ET-743, ET-729, ET-745, ET-759A, ET-759B, ET-759C, ET-770, ET-815, ET-731, ET-745B, ET-722, ET-736, ET-738, ET-808, ET-752, ET-594, ET-552, ET-637, ET-652, ET-583, ET-597, ET-596, ET-639, ET-641, and derivatives thereof, such as acetylated forms, formylated forms, methylated forms, and oxide forms.
[0003] The structural characterizations of such ecteinascidins are not given again explicitly herein because from the detailed description provided in such references and citations any person of ordinary skill in this technology is capable of obtaining such information directly from the sources cited here and related sources.
[0004] At least one of the ecteinascidin compounds, ecteinascidin 743 (ET-743), has been extensively studied, and it will be referred to specifically herein to illustrate features of this invention. ET-743 is being employed as an anticancer medicament, under the international nonproprietary name (INN) trabectedin, for the treatment of patients with advanced and metastatic soft tissue sarcoma (STS), after failure of anthracyclines and ifosfamide, or who are unsuited to receive such agents, and for the treatment of relapsed platinum-sensitive ovarian cancer in combination with pegylated liposomal doxorubicin.
[0005] ET-743 has a complex tris(tetrahydroisoquinoline) structure of formula
[0006] It was originally prepared by isolation from extracts of the marine tunicate Ecteinascidia turbinata. The yield was low, and alternative preparative processes had been sought.
[0007] The first synthetic process for producing ecteinascidin compounds was described in U.S. Pat. No. 5,721,362. This process employed sesamol as starting material and yielded ET-743 after a long and complicated sequence of 38 examples each describing one or more steps in the synthetic sequence.
[0008] An improvement in the preparation of one intermediate used in such process was disclosed in U.S. Pat. No. 6,815,544. Even with this improvement, the total synthesis was not suitable for manufacturing ET-743 at an industrial scale.
[0009] A hemisynthetic process for producing ecteinascidin compounds was described in EP 1.185.536. This process employs cyanosafracin B as starting material to provide ET-743. Cyanosafracin B is a pentacyclic antibiotic obtained by fermentation from the bacteria Pseudomonas fluorescens.
[0010] An improvement in such hemisynthetic process was disclosed in EP 1.287.004.
[0011] To date four additional synthetic process (2 total and 2 formal synthesis) have been disclosed in patent applications JP 2003221395, WO 2007/045686, and WO 2007/087220 and in J. Org. Chem. 2008, 73, pages 9594-9600.
[0012] WO 2007/045686 also relates to the synthesis of Ecteinascidins-583 and 597 using intermediate compounds of formula:
[0013] Total synthesis strategies for the synthesis of the pentacyclic core of ET-743 are overviewed in FIG. 1.
PAPER
Angewandte Chemie, International Edition (2019), 58(12), 3972-3975.
An efficient and scalable approach is described for the total synthesis of the marine natural product Et‐743 and its derivative lubinectedin, which are valuable antitumor compounds. The method delivers 1.6 % overall yield in 26 total steps from Cbz‐protected (S)‐tyrosine. It features the use of a common advanced intermediate to create the right and left parts of these compounds, and a light‐mediated remote C−H bond activation to assemble a benzo[1,3]dioxole‐containing intermediate.
“Lurbinectedin”. Drug Information Portal. U.S. National Library of Medicine.
“Lurbinectedin”. NCI Dictionary of Cancer Terms. National Cancer Institute.
Clinical trial number NCT02454972 for “Clinical Trial of Lurbinectedin (PM01183) in Selected Advanced Solid Tumors” at ClinicalTrials.gov
FDA grants accelerated approval to lurbinectedin for metastatic small cell lung cancer
On June 15, 2020, the Food and Drug Administration granted accelerated approval to lurbinectedin(ZEPZELCA, Pharma Mar S.A.) for adult patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy.
Efficacy was demonstrated in the PM1183-B-005-14 trial (Study B-005; NCT02454972), a multicenter open-label, multi-cohort study enrolling 105 patients with metastatic SCLC who had disease progression on or after platinum-based chemotherapy. Patients received lurbinectedin 3.2 mg/m2 by intravenous infusion every 21 days until disease progression or unacceptable toxicity.
The main efficacy outcome measures were confirmed overall response rate (ORR) determined by investigator assessment using RECIST 1.1 and response duration. Among the 105 patients, the ORR was 35% (95% CI: 26%, 45%), with a median response duration of 5.3 months (95% CI: 4.1, 6.4). The ORR as per independent review committee was 30% (95% CI: 22%, 40%) with a median response duration of 5.1 months (95% CI: 4.9, 6.4).
The most common adverse reactions (≥20%), including laboratory abnormalities, were myelosuppression, fatigue, increased creatinine, increased alanine aminotransferase, increased glucose, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhea.
The recommended lurbinectedin dose is 3.2 mg/m2 every 21 days.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this application, a modified Project Orbis was undertaken because of the timing of submission to other regulatory agencies. FDA is collaborating with the Australian Therapeutic Goods Administration (TGA). FDA approved this application 2 months ahead of the goal date. The review is ongoing for the Australian TGA.
1: Calvo E, Moreno V, Flynn M, Holgado E, Olmedo ME, Lopez Criado MP, Kahatt C, Lopez-Vilariño JA, Siguero M, Fernandez-Teruel C, Cullell-Young M, Soto Matos-Pita A, Forster M. Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: results from a phase I study. Ann Oncol. 2017 Oct 1;28(10):2559-2566. doi: 10.1093/annonc/mdx357. PubMed PMID: 28961837.
2: Erba E, Romano M, Gobbi M, Zucchetti M, Ferrari M, Matteo C, Panini N, Colmegna B, Caratti G, Porcu L, Fruscio R, Perlangeli MV, Mezzanzanica D, Lorusso D, Raspagliesi F, D’Incalci M. Ascites interferes with the activity of lurbinectedin and trabectedin: Potential role of their binding to alpha 1-acid glycoprotein. Biochem Pharmacol. 2017 Nov 15;144:52-62. doi: 10.1016/j.bcp.2017.08.001. Epub 2017 Aug 4. PubMed PMID: 28782526.
3: Belgiovine C, Bello E, Liguori M, Craparotta I, Mannarino L, Paracchini L, Beltrame L, Marchini S, Galmarini CM, Mantovani A, Frapolli R, Allavena P, D’Incalci M. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br J Cancer. 2017 Aug 22;117(5):628-638. doi: 10.1038/bjc.2017.205. Epub 2017 Jul 6. PubMed PMID: 28683469; PubMed Central PMCID: PMC5572168.
4: Jimeno A, Sharma MR, Szyldergemajn S, Gore L, Geary D, Diamond JR, Fernandez Teruel C, Soto Matos-Pita A, Iglesias JL, Cullell-Young M, Ratain MJ. Phase I study of lurbinectedin, a synthetic tetrahydroisoquinoline that inhibits activated transcription, induces DNA single- and double-strand breaks, on a weekly × 2 every-3-week schedule. Invest New Drugs. 2017 Aug;35(4):471-477. doi: 10.1007/s10637-017-0427-2. Epub 2017 Jan 20. PubMed PMID: 28105566.
5: Paz-Ares L, Forster M, Boni V, Szyldergemajn S, Corral J, Turnbull S, Cubillo A, Teruel CF, Calderero IL, Siguero M, Bohan P, Calvo E. Phase I clinical and pharmacokinetic study of PM01183 (a tetrahydroisoquinoline, Lurbinectedin) in combination with gemcitabine in patients with advanced solid tumors. Invest New Drugs. 2017 Apr;35(2):198-206. doi: 10.1007/s10637-016-0410-3. Epub 2016 Nov 21. PubMed PMID: 27873130.
6: Harlow ML, Maloney N, Roland J, Guillen Navarro MJ, Easton MK, Kitchen-Goosen SM, Boguslawski EA, Madaj ZB, Johnson BK, Bowman MJ, D’Incalci M, Winn ME, Turner L, Hostetter G, Galmarini CM, Aviles PM, Grohar PJ. Lurbinectedin Inactivates the Ewing Sarcoma Oncoprotein EWS-FLI1 by Redistributing It within the Nucleus. Cancer Res. 2016 Nov 15;76(22):6657-6668. doi: 10.1158/0008-5472.CAN-16-0568. Epub 2016 Oct 3. PubMed PMID: 27697767; PubMed Central PMCID: PMC5567825.
7: Céspedes MV, Guillén MJ, López-Casas PP, Sarno F, Gallardo A, Álamo P, Cuevas C, Hidalgo M, Galmarini CM, Allavena P, Avilés P, Mangues R. Lurbinectedin induces depletion of tumor-associated macrophages, an essential component of its in vivo synergism with gemcitabine, in pancreatic adenocarcinoma mouse models. Dis Model Mech. 2016 Dec 1;9(12):1461-1471. Epub 2016 Oct 20. PubMed PMID: 27780828; PubMed Central PMCID: PMC5200894.
8: Santamaría Nuñez G, Robles CM, Giraudon C, Martínez-Leal JF, Compe E, Coin F, Aviles P, Galmarini CM, Egly JM. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol Cancer Ther. 2016 Oct;15(10):2399-2412. Epub 2016 Sep 14. PubMed PMID: 27630271.
9: Metaxas Y, Cathomas R, Mark M, von Moos R. Combination of cisplatin and lurbinectedin as palliative chemotherapy in progressive malignant pleural mesothelioma: Report of two cases. Lung Cancer. 2016 Dec;102:136-138. doi: 10.1016/j.lungcan.2016.07.012. Epub 2016 Jul 14. PubMed PMID: 27440191.
10: Lima M, Bouzid H, Soares DG, Selle F, Morel C, Galmarini CM, Henriques JA, Larsen AK, Escargueil AE. Dual inhibition of ATR and ATM potentiates the activity of trabectedin and lurbinectedin by perturbing the DNA damage response and homologous recombination repair. Oncotarget. 2016 May 3;7(18):25885-901. doi: 10.18632/oncotarget.8292. PubMed PMID: 27029031; PubMed Central PMCID: PMC5041952.
11: Takahashi R, Mabuchi S, Kawano M, Sasano T, Matsumoto Y, Kuroda H, Kozasa K, Hashimoto K, Sawada K, Kimura T. Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary. PLoS One. 2016 Mar 17;11(3):e0151050. doi: 10.1371/journal.pone.0151050. eCollection 2016. PubMed PMID: 26986199; PubMed Central PMCID: PMC4795692.
12: Pernice T, Bishop AG, Guillen MJ, Cuevas C, Aviles P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J Pharm Biomed Anal. 2016 May 10;123:37-41. doi: 10.1016/j.jpba.2016.01.043. Epub 2016 Jan 21. PubMed PMID: 26871278.
13: Elez ME, Tabernero J, Geary D, Macarulla T, Kang SP, Kahatt C, Pita AS, Teruel CF, Siguero M, Cullell-Young M, Szyldergemajn S, Ratain MJ. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin Cancer Res. 2014 Apr 15;20(8):2205-14. doi: 10.1158/1078-0432.CCR-13-1880. Epub 2014 Feb 21. PubMed PMID: 24563480.
14: Romano M, Frapolli R, Zangarini M, Bello E, Porcu L, Galmarini CM, García-Fernández LF, Cuevas C, Allavena P, Erba E, D’Incalci M. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Int J Cancer. 2013 Nov;133(9):2024-33. doi: 10.1002/ijc.28213. Epub 2013 May 25. PubMed PMID: 23588839.
15: Vidal A, Muñoz C, Guillén MJ, Moretó J, Puertas S, Martínez-Iniesta M, Figueras A, Padullés L, García-Rodriguez FJ, Berdiel-Acer M, Pujana MA, Salazar R, Gil-Martin M, Martí L, Ponce J, Molleví DG, Capella G, Condom E, Viñals F, Huertas D, Cuevas C, Esteller M, Avilés P, Villanueva A. Lurbinectedin (PM01183), a new DNA minor groove binder, inhibits growth of orthotopic primary graft of cisplatin-resistant epithelial ovarian cancer. Clin Cancer Res. 2012 Oct 1;18(19):5399-411. doi: 10.1158/1078-0432.CCR-12-1513. Epub 2012 Aug 15. PubMed PMID: 22896654.
FDA 11/25/2020, Imcivree, To treat obesity and the control of hunger associated with pro-opiomelanocortin deficiency, a rare disorder that causes severe obesity that begins at an early age Drug Trials Snapshot, 10MG/ML, SOLUTION;SUBCUTANEOUS, Orphan
update Imcivree EMA APPROVED 2021/7/16
DESCRIPTION
IMCIVREE contains setmelanotide acetate, a melanocortin 4 (MC4) receptor agonist. Setmelanotide is an 8 amino acid cyclic peptideanalog of endogenous melanocortin peptide α-MSH (alpha-melanocyte stimulating hormone).
The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-Lhistidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C49H68N18O9S2 (anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide is:
IMCIVREE injection is a sterile clear to slightly opalescent, colorless to slightly yellow solution. Each 1 mL of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with 2 to 4 molar equivalents of acetate, and the following inactive ingredients: 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-glycero-3phosphoethanolamine sodium salt, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 11 mg mannitol, 5 mg phenol, 10 mg benzyl alcohol, 1 mg edetate disodium dihydrate, and Water for Injection. The pH of IMCIVREE is 5 to 6.
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor. Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects. Setmelanotide increases resting energy expenditure in both obese animals and humans. Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).
Setmelanotide, sold under the brand name Imcivree, is a medication for the treatment of obesity.[1]
The most common side effects include injection site reactions, skin hyperpigmentation (skin patches that are darker than surrounding skin), headache and gastrointestinal side effects (such as nausea, diarrhea, and abdominal pain), among others.[1] Spontaneous penile erections in males and adverse sexual reactions in females have occurred with treatment.[1] Depression and suicidal ideation have also occurred with setmelanotide.[1]
SYN
WO 2011060355
Medical uses
Setmelanotide is indicated for chronic weight management (weight loss and weight maintenance for at least one year) in people six years and older with obesity due to three rare genetic conditions: pro-opiomelanocortin (POMC) deficiency, proprotein subtilisin/kexin type 1 (PCSK1) deficiency, and leptin receptor (LEPR) deficiency confirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes considered pathogenic (causing disease), likely pathogenic, or of uncertain significance.[1] Setmelanotide is the first FDA-approved treatment for these genetic conditions.[1]
Setmelanotide is not approved for obesity due to suspected POMC, PCSK1, or LEPR deficiency with variants classified as benign (not causing disease) or likely benign or other types of obesity, including obesity associated with other genetic syndromes and general (polygenic) obesity.[1]
Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).[5] (19.6-fold selectivity for MC4 over MC3, the second target of highest activity.)
History
Setmelanotide was evaluated in two one-year studies.[1] The first study enrolled participants with obesity and confirmed or suspected POMC or PCSK1 deficiency while the second study enrolled participants with obesity and confirmed or suspected LEPR deficiency; all participants were six years or older.[1] The effectiveness of setmelanotide was determined by the number of participants who lost more than ten percent of their body weight after a year of treatment.[1]
The effectiveness of setmelanotide was assessed in 21 participants, ten in the first study and eleven in the second.[1] In the first study, 80 percent of participants with POMC or PCSK1 deficiency lost ten percent or more of their body weight.[1] In the second study, 46 percent of participants with LEPR deficiency lost ten percent or more of their body weight.[1]
The study also assessed the maximal (greatest) hunger in sixteen participants over the previous 24 hours using an eleven-point scale in participants twelve years and older.[1] In both studies, some, but not all, of participants’ weekly average maximal hunger scores decreased substantially from their scores at the beginning of the study.[1] The degree of change was highly variable among participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for setmelanotide orphan disease designation, breakthrough therapy designation, and priority review.[1] The FDA granted the approval of Imcivree to Rhythm Pharmaceutical, Inc.[1]
Synthesis of Example 1i.e., Ac-Arg-cyclo(Cys-D-Ala-His-D-Phe-Arg-Trp-Cys)-NH2
The title peptide having the above structure was assembled using Fmoc chemistry on an Apex peptide synthesizer (Aapptec; Louisville, Ky., USA). 220 mg of 0.91 mmol/g (0.20 mmoles) Rink Amide MBHA resin (Polymer Laboratories; Amherst, Mass., USA) was placed in a reaction well and pre-swollen in 3.0 mL of DMF prior to synthesis. For cycle 1, the resin was treated with two 3-mL portions of 25% piperidine in DMF for 5 and 10 minutes respectively, followed by 4 washes of 3-mL DMF—each wash consisting of adding 3 mL of solvent, mixing for 1 minute, and emptying for 1 minute. Amino acids stocks were prepared in NMP as 0.45M solutions containing 0.45M HOBT. HBTU was prepared as a 0.45M solution in NMP and DIPEA was prepared as a 2.73M solution in NMP. To the resin, 2 mL of the first amino acid (0 9 mmoles, Fmoc-Cys(Trt)-OH) (Novabiochem; San Diego, Calif., USA) was added along with 2 mL (0.9 mmoles) of HBTU and 1.5 mL (4.1 mmoles) of DIPEA. After one hour of constant mixing, the coupling reagents were drained from the resin and the coupling step was repeated. Following amino acid acylation, the resin was washed with two 3-mL aliquots of DMF for 1 minute. The process of assembling the peptide (deblock/wash/acylate/wash) was repeated for cycles 2-9 identical to that as described for cycle 1. The following amino acids were used: cycle 2) Fmoc-Trp(Boc)-OH (Genzyme; Cambridge, Mass., USA); cycle 3) Fmoc-Arg(Pbf)-OH (Novabiochem); cycle 4) Fmoc-DPhe-OH (Genzyme); cycle 5) Fmoc-His(Trt)-OH (Novabiochem); cycle 6) Fmoc-D-Ala-OH (Genzyme); cycle 7) Fmoc-Cys(Trt)-OH, (Novabiochem); and cycle 8) Fmoc-Arg(Pbf)-OH (Genzyme). The N-terminal Fmoc was removed with 25% piperidine in DMF as described above, followed by four 3-mL DMF washes for 1 minute. Acetylation of the N-terminus was performed by adding 0.5 mL of 3M DIPEA in NMP to the resin along with 1.45 mL of 0.45M acetic anhydride in NMP. The resin was mixed for 30 minutes and acetylation was repeated. The resin was washed with 3 mL of DMF for a total of 5 times followed with 5 washes with 5 mL of DCM each.
To cleave and deprotect the peptide, 5mL of the following reagent was added to the resin: 2% TIS/5% water/5% (w/v) DTT/88% TFA. The solution was allowed to mix for 3.5 hours. The filtrate was collected into 40 mL of cold anhydrous ethyl ether. The precipitate was pelleted for 10 minutes at 3500 rpm in a refrigerated centrifuge. The ether was decanted and the peptide was re-suspended in fresh ether. The ether workup was performed three times. Following the last ether wash, the peptide was allowed to air dry to remove residual ether.
The peptide was dissolved in 10% acetonitrile and analyzed by mass spectrometry and reverse-phase HPLC employing a 30×4.6 cm C18 column (Vydac; Hesperia, Calif., USA) with a gradient of 2-60% acetonitrile (0.1% TFA) over 30 minutes. This analysis identified a product with ˜53% purity. Mass analysis employing electrospray ionization identified a main product containing a mass of 1118.4 corresponding to the desired linear product. The crude product (˜100 mg) was diluted to a concentration of 2 mg/mL in 5% acetic acid. To this solution, 0.5M iodine/methanol was added dropwise with vigorous stirring until a pale yellow color was achieved. The solution was vigorously stirred for another 10 minutes. Excess iodine was then quenched by adding 1.0M sodium thiosulfate under continuous mixing until the mixture was rendered colorless. The peptide was re-examined by mass spectrometry analysis and HPLC. Mass spectrometry analysis identified a main species with a mass of 1116.4 which indicated successful oxidation to form the cyclic peptide. The peptide solution was purified on a preparative HPLC equipped with a C18 column using a similar elution gradient. The purified product was re-analyzed by HPLC for purity (>95%) and mass spectrometry (1116.9 which is in agreement with the expected mass of 1117.3) and subsequently lyophilized. Following lyophilization, 28 mg of purified product was obtained representing a 24% yield.
The other exemplified peptides were synthesized substantially according to the procedure described for the above-described synthetic process. Physical data for select exemplified peptides are given in Table 1.
The acetate salt of Example 1 (200 mg, 0.18 mmole) was dissolved in 10 mL of water. Sodium pamoate (155 mg, 0.36 mmole) was dissolved in 10 mL of water. The two solutions were combined and mixed well. The precipitates were collected by centrifugation at 3000 rpm for 20 minutes, washed for three times with water, and dried by lyophilization.
^ Jump up to:abc Lee EC, Carpino PA (2015). “Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014)”. Pharmaceutical Patent Analyst. 4 (2): 95–107. doi:10.4155/ppa.15.1. PMID25853469.
^ Jackson VM, Price DA, Carpino PA (August 2014). “Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies”. Expert Opinion on Investigational Drugs. 23 (8): 1055–66. doi:10.1517/13543784.2014.918952. PMID25000213. S2CID23198484.
The peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It is being researched by Rhythm Pharmaceuticals for the treatment of obesity and diabetes. In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity. As of December 2014, the drug is in phase II clinical trials for obesity and PWS.
1: Lee EC, Carpino PA. Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014). Pharm Pat Anal. 2015;4(2):95-107. doi: 10.4155/ppa.15.1. PubMed PMID: 25853469.
2: Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LH, Reitman ML, Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015 Apr;100(4):1639-45. doi: 10.1210/jc.2014-4024. Epub 2015 Feb 12. PubMed PMID: 25675384; PubMed Central PMCID: PMC4399297.
3: Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, Hofmann SM, Tschöp MH, Van der Ploeg LH, Müller TD. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015 Feb 4;7(3):288-98. doi: 10.15252/emmm.201404508. PubMed PMID: 25652173; PubMed Central PMCID: PMC4364946.
4: Jackson VM, Price DA, Carpino PA. Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies. Expert Opin Investig Drugs. 2014 Aug;23(8):1055-66. doi: 10.1517/13543784.2014.918952. Epub 2014 Jul 7. Review. PubMed PMID: 25000213.
5: Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013 Feb;62(2):490-7. doi: 10.2337/db12-0598. Epub 2012 Oct 9. PubMed PMID: 23048186; PubMed Central PMCID: PMC3554387.
6: Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA, Culler MD, Yang H, Dixit VD, Butler AA. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides. 2009 Oct;30(10):1892-900. doi: 10.1016/j.peptides.2009.07.012. Epub 2009 Jul 29. PubMed PMID: 19646498; PubMed Central PMCID: PMC2755620.
External links
“Setmelanotide”. Drug Information Portal. U.S. National Library of Medicine.
Tepezza (teprotumumab-trbw) is a fully human monoclonal antibody (mAb) and a targeted inhibitor of the insulin-like growth factor 1 receptor (IGF-1R) for the treatment of active thyroid eye disease (TED).
FDA Approves Tepezza (teprotumumab-trbw) for the Treatment of Thyroid Eye Disease (TED) – January 21, 2020
Today, the U.S. Food and Drug Administration (FDA) approved Tepezza (teprotumumab-trbw) for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis). Today’s approval represents the first drug approved for the treatment of thyroid eye disease.
“Today’s approval marks an important milestone for the treatment of thyroid eye disease. Currently, there are very limited treatment options for this potentially debilitating disease. This treatment has the potential to alter the course of the disease, potentially sparing patients from needing multiple invasive surgeries by providing an alternative, non surgical treatment option,” said Wiley Chambers, M.D., deputy director of the Division of Transplant and Ophthalmology Products in the FDA’s Center for Drug Evaluation and Research. “Additionally, thyroid eye disease is a rare disease that impacts a small percentage of the population, and for a variety of reasons, treatments for rare diseases are often unavailable. This approval represents important progress in the approval of effective treatments for rare diseases, such as thyroid eye disease.”
Thyroid eye disease is associated with the outward bulging of the eye that can cause a variety of symptoms such as eye pain, double vision, light sensitivity or difficulty closing the eye. This disease impacts a relatively small number of Americans, with more women than men affected. Although this condition impacts relatively few individuals, thyroid eye disease can be incapacitating. For example, the troubling ocular symptoms can lead to the progressive inability of people with thyroid eye disease to perform important daily activities, such as driving or working.
Tepezza was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive Tepezza or a placebo. Of the patients who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than 2 millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.
The most common adverse reactions observed in patients treated with Tepezza are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache. Tepezza should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for 6 months following the last dose of Tepezza.
The FDA granted this application Priority Review, in addition to Fast Track and Breakthrough Therapy Designation. Additionally, Tepezza received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases or conditions. Development of this product was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and efficacy of products for use in rare diseases or conditions.
The FDA granted the approval of Tepezza to Horizon Therapeutics Ireland DAC.
Teprotumumab (RG-1507), sold under the brand name Tepezza, is a medication used for the treatment of adults with thyroid eye disease, a rare condition where the muscles and fatty tissues behind the eye become inflamed, causing the eyes to be pushed forward and bulge outwards (proptosis).[1]
The most common adverse reactions observed in people treated with teprotumumab-trbw are muscle spasm, nausea, alopecia (hair loss), diarrhea, fatigue, hyperglycemia (high blood sugar), hearing loss, dry skin, dysgeusia (altered sense of taste) and headache.[1] Teprotumumab-trbw should not be used if pregnant, and women of child-bearing potential should have their pregnancy status verified prior to beginning treatment and should be counseled on pregnancy prevention during treatment and for six months following the last dose of teprotumumab-trbw.[1]
It is a human monoclonal antibody developed by Genmab and Roche. It binds to IGF-1R.
Teprotumumab was first investigated for the treatment of solid and hematologic tumors, including breast cancer, Hodgkin’s and non-Hodgkin’s lymphoma, non-small cell lung cancer and sarcoma.[2][3] Although results of phase I and early phase II trials showed promise, research for these indications were discontinued in 2009 by Roche. Phase II trials still in progress were allowed to complete, as the development was halted due to business prioritization rather than safety concerns.
Teprotumumab was subsequently licensed to River Vision Development Corporation in 2012 for research in the treatment of ophthalmic conditions. Horizon Pharma (now Horizon Therapeutics, from hereon Horizon) acquired RVDC in 2017, and will continue clinical trials.[4] It is in phase III trials for Graves’ ophthalmopathy (also known as thyroid eye disease (TED)) and phase I for diabetic macular edema.[5] It was granted Breakthrough Therapy, Orphan Drug Status and Fast Track designations by the FDA for Graves’ ophthalmopathy.[6]
In a multicenter randomized trial in patients with active Graves’ ophthalmopathy Teprotumumab was more effective than placebo in reducing the clinical activity score and proptosis.[7] In February 2019 Horizon announced results from a phase 3 confirmatory trial evaluating teprotumumab for the treatment of active thyroid eye disease (TED). The study met its primary endpoint, showing more patients treated with teprotumumab compared with placebo had a meaningful improvement in proptosis, or bulging of the eye: 82.9 percent of teprotumumab patients compared to 9.5 percent of placebo patients achieved the primary endpoint of a 2 mm or more reduction in proptosis (p<0.001). Proptosis is the main cause of morbidity in TED. All secondary endpoints were also met and the safety profile was consistent with the phase 2 study of teprotumumab in TED.[8] On 10th of July 2019 Horizon submitted a Biologics License Application (BLA) to the FDA for teprotumumab for the Treatment of Active Thyroid Eye Disease (TED). Horizon requested priority review for the application – if so granted (FDA has a 60-day review period to decide) it would result in a max. 6 month review process.[9]
Teprotumumab-trbw was approved for use in the United States in January 2020, for the treatment of adults with thyroid eye disease.[1]
Teprotumumab-trbw was approved based on the results of two studies (Study 1 and 2) consisting of a total of 170 patients with active thyroid eye disease who were randomized to either receive teprotumumab-trbw or a placebo.[1] Of the subjects who were administered Tepezza, 71% in Study 1 and 83% in Study 2 demonstrated a greater than two millimeter reduction in proptosis (eye protrusion) as compared to 20% and 10% of subjects who received placebo, respectively.[1]
Avapritinib, sold under the brand name Ayvakit, is a medication used for the treatment of tumors due to one specific rare mutation: It is specifically intended for adults with unresectable or metastatic ( y) gastrointestinal stromal tumor (GIST) that harbor a platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutation.[1]
Avapritinib was approved based on the results from the Phase I NAVIGATOR[2][3] clinical trial involving 43 patients with GIST harboring a PDGFRA exon 18 mutation, including 38 subjects with PDGFRA D842V mutation.[1] Subjects received avapritinib 300 mg or 400 mg orally once daily until disease progression or they experienced unacceptable toxicity.[1] The recommended dose was determined to be 300 mg once daily.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] For subjects harboring a PDGFRA exon 18 mutation, the overall response rate was 84%, with 7% having a complete response and 77% having a partial response.[1] For the subgroup of subjects with PDGFRA D842V mutations, the overall response rate was 89%, with 8% having a complete response and 82% having a partial response.[1] While the median duration of response was not reached, 61% of the responding subjects with exon 18 mutations had a response lasting six months or longer (31% of subjects with an ongoing response were followed for less than six months).[1]
4-Chloro-6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/] [l,2,4]triazine (180 mg, 0.770 mmol), (4-fluorophenyl)(2-(piperazin-l-yl)pyrimidin-5-yl)methanone, HC1 (265 mg, 0.821 mmol) and DIPEA (0.40 mL, 2.290 mmol) were stirred in 1,4-dioxane (4 mL) at room temperature for 18 hours. Saturated ammonium chloride was added and the products extracted into DCM (x2). The combined organic extracts were dried over Na2S04, filtered through Celite eluting with DCM, and the filtrate concentrated in vacuo. Purification of the residue by MPLC (25- 100% EtOAc-DCM) gave (4-fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methanone (160 mg, 0.331 mmol, 43 % yield) as an off-white solid. MS (ES+) C25H22FN90 requires: 483, found: 484 [M + H]+.
(S)-2-Methylpropane-2-sulfinamide (110 mg, 0.908 mmol), (4-fluorophenyl)(2-(4-(6-(l- methyl- lH-pyrazol-4-yl)pyrrolo[2,l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5- yl)methanone (158 mg, 0.327 mmol) and ethyl orthotitanate (0.15 mL, 0.715 mmol) were stirred in THF (3.2 mL) at 70 °C for 18 hours. Room temperature was attained, water was added, and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0- 10% MeOH-EtOAc) gave (5,Z)-N-((4-fluorophenyl)(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)methylene)-2- methylpropane-2-sulfinamide (192 mg, 0.327 mmol, 100 % yield) as an orange solid. MS (ES+) C29H3iFN10OS requires: 586, found: 587 [M + H]+.
Step 3: Synthesis of (lS’)-N-(l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- l)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-
(lS’,Z)-N-((4-Fluorophenyl)(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2,l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)methylene)-2-methylpropane-2-sulfinamide (190 mg, 0.324 mmol) was taken up in THF (3 mL) and cooled to 0 °C. Methylmagnesium bromide (3 M solution in diethyl ether, 0.50 mL, 1.500 mmol) was added and the resulting mixture stirred at 0 °C for 45 minutes. Additional methylmagnesium bromide (3 M solution in diethyl ether, 0.10 mL, 0.300 mmol) was added and stirring at 0 °C continued for 20 minutes. Saturated ammonium chloride was added and the products extracted into EtOAc (x2). The combined organic extracts were washed with brine, dried over Na2S04, filtered, and concentrated in vacuo while loading onto Celite. Purification of the residue by MPLC (0-10% MeOH-EtOAc) gave (lS’)-N-(l-(4-fluorophenyl)-l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2, l- ] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol, 61.5 % yield) as a yellow solid (mixture of diastereoisomers). MS (ES+) C3oH35FN10OS requires: 602, found: 603 [M + H]+. Step 4: Synthesis of l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4-yl)pyrrolo[2,l- f\ [ 1 ,2,4] triazin-4- l)piperazin- 1 -yl)pyrimidin-5-yl)ethanamine:
(S)-N- ( 1 – (4-Fluorophenyl)- 1 -(2- (4- (6-( 1 -methyl- 1 H-pyrazol-4-yl)pyrrolo [2,1- /] [l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethyl)-2-methylpropane-2-sulfinamide (120 mg, 0.199 mmol) was stirred in 4 M HCl in 1,4-dioxane (1.5 mL)/MeOH (1.5 mL) at room temperature for 1 hour. The solvent was removed in vacuo and the residue triturated in EtOAc to give l-(4-fluorophenyl)- l-(2-(4-(6-(l -methyl- lH-pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4- yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine, HCl (110 mg, 0.206 mmol, 103 % yield) as a pale yellow solid. MS (ES+) C26H27FN10requires: 498, found: 482 [M- 17 + H]+, 499 [M + H]+.
Step 5: Chiral separation of (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine and (5)-1-(4- fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4-yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin- 1 -yl)pyrimidin- -yl)ethanamine:
The enantiomers of racemic l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl- lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (94 mg, 0.189 mmol) were separated by chiral SFC to give (R)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH- pyrazol-4-yl)pyrrolo[2, l-/][l,2,4]triazin-4-yl)piperazin- l-yl)pyrimidin-5-yl)ethanamine (34.4 mg, 0.069 mmol, 73.2 % yield) and (lS,)-l-(4-fluorophenyl)- l-(2-(4-(6-(l-methyl-lH-pyrazol-4- yl)pyrrolo[2, l-/] [l,2,4]triazin-4-yl)piperazin-l-yl)pyrimidin-5-yl)ethanamine (32.1 mg, 0.064 mmol, 68.3 % yield). The absolute stereochemistry was assigned randomly. MS (ES+)
Wu CP, Lusvarghi S, Wang JC, et al. (July 2019). “Avapritinib: A Selective Inhibitor of KIT and PDGFRα that Reverses ABCB1 and ABCG2-Mediated Multidrug Resistance in Cancer Cell Lines”. Mol. Pharm. 16 (7): 3040–3052. doi:10.1021/acs.molpharmaceut.9b00274. PMID31117741.
Gebreyohannes YK, Wozniak A, Zhai ME, et al. (January 2019). “Robust Activity of Avapritinib, Potent and Highly Selective Inhibitor of Mutated KIT, in Patient-derived Xenograft Models of Gastrointestinal Stromal Tumors”. Clin. Cancer Res. 25 (2): 609–618. doi:10.1158/1078-0432.CCR-18-1858. PMID30274985.
This new drug application provides for the use of KRINTAFEL (tafenoquine) tablets for the radical cure (prevention of relapse) of Plasmodium vivax malaria in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute P. vivax infection….https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/210795Orig1s000Ltr.pdf
Tafenoquine under the commercial name of Krintafel is an 8-aminoquinoline drug manufactured by GlaxoSmithKline that is being investigated as a potential treatment for malaria, as well as for malaria prevention.[2][3]
The proposed indication for tafenoquine is for treatment of the hypnozoite stages of Plasmodium vivax and Plasmodium ovale that are responsible for relapse of these malaria species even when the blood stages are successfully cleared. This is only now achieved by administration of daily primaquine for 14 days. The main advantage of tafenoquine is that it has a long half-life (2–3 weeks) and therefore a single treatment may be sufficient to clear hypnozoites. The shorter regimen has been described as an advantage.[4]
Like primaquine, tafenoquine causes hemolysis in people with G6PD deficiency.[2] Indeed, the long half-life of tafenoquine suggests that particular care should be taken to ensure that individuals with severe G6PD deficiency do not receive the drug.
The dose of tafenoquine has not been firmly established, but for the treatment of Plasmodium vivax malaria, a dose of 800 mg over three days has been used.[5]
In 2018 United States Food and Drug Administration (FDA) approved single dose tafenoquine for the radical cure (prevention of relapse) of Plasmodium vivax malaria[6].
Tafenoquine is used for the treatment and prevention of relapse of Vivax malaria in patients 16 years and older. Tafenoquine is not indicated to treat acute vivax malaria.[1]
Malaria is a disease that remains to occur in many tropical countries. Vivax malaria, caused by Plasmodium vivax, is known to be less virulent and seldom causes death. However, it causes a substantive illness-related burden in endemic areas and it is known to present dormant forms in the hepatocytes named hypnozoites which can remain dormant for weeks or even months. This dormant form produces ongoing relapses
FDA Approves Tafenoquine, First New P VivaxMalaria Treatment in 60 Years
JUL 23, 2018
The US Food and Drug Administration (FDA) has approved, under Priority Review, GlaxoSmithKline (GSK)’s tafenoquine (Krintafel), which is the first single-dose medicine for the prevention of Plasmodium vivax (P vivax) malaria relapse in patients over the age of 16 years who are receiving antimalarial therapy. This is the first drug to be approved for the treatment of P vivax in over 60 years.
“[The] approval of Krintafel, the first new treatment for Plasmodium vivax malaria in over 60 years, is a significant milestone for people living with this type of relapsing malaria.” Hal Barron, MD, chief scientific officer and president of research and development of GSK, said in the announcement, “Together with our partner, Medicines for Malaria Venture (MMV), we believe Krintafel will be an important medicine for patients with malaria and contribute to the ongoing effort to eradicate this disease.”
Tafenoquine is an 8-aminoquinoline derivative with activity against all stages of the P vivax lifecycle, including hypnozoites. It was first synthesized by scientists at the Walter Reed Army Institute of Research in 1978, and in 2008, GSK entered into a collaboration with MMV, to develop tafenoquine as an anti-relapse medicine.
After an infected mosquito bite, the P vivax parasite infects the blood and causes an acute malaria episode and can also lie dormant in the liver (in a form known as hypnozoite) from where it periodically reactivates to cause relapses, which can occur weeks, months, or years after the onset of the initial infection. The dormant liver forms cannot be readily treated with most anti-malarial treatments. Primaquine, an 8-aminoquinolone, has been the only FDA-approved medicine that targeted the dormant liver stage to prevent relapse; however, effectiveness only occurs after 14 days and the treatment has shown to have poor compliance.
“The US FDA’s approval of Krintafel is a major milestone and a significant contribution towards global efforts to eradicate malaria,” commented David Reddy, PhD, chief executive officer of MMV in a recent statement, “The world has waited decades for a new medicine to counter P vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance.”
Approval for tafenoquine was granted based on the efficacy and safety data gleaned from a comprehensive global clinical development program for P vivaxprevention of relapse which has been designed by GSK and MMV in agreement with the FDA. The program consisted of 13 studies assessing the safety of a 300 mg single-dose of tafenoquine, including 3 double-blind studies referred to as DETECTIVE Parts 1 and 2 and GATHER.
With the approval of tafenoquine, GSK has also been awarded a tropical disease priority review voucher by the FDA. Additionally, GSK is waiting for a decision from Australian Therapeutics Good Administration regarding the regulatory submission for the drug.
P vivax malaria has caused around 8.5 million clinical infections each year, primarily in South Asia, South-East Asia, Latin America, and the Horn of Africa, a peninsula in East Africa. Symptoms include fever, chills, vomiting, malaise, headache and muscle pain, and can lead to death in severe cases.
Tafenoquine should not be administered to: patients who have glucose-6-phosphate dehydrogenase (G6PD) deficiency or have not been tested for G6PD deficiency, patients who are breastfeeding a child known to have G6PD deficiency or one that has not been tested for G6PD deficiency, or patients who are allergic to tafenoquine or any of the ingredients in tafenoquine or who have had an allergic reaction to similar medicines containing 8-aminoquinolines
Stereochemistry
Tafenoquine contains a stereocenter and consists of two enantiomers. This is a mixture of (R) – and the (S) – Form:
Enantiomers of tafenoquine
(R)-Form
(S)-Form
CLIP
US 4431807
Nitration of 1,2-dimethoxybenzene (XXIX) with HNO3/AcOH gives 4,5-dimethoxy-1,2-dinitrobenzene (XXX), which is treated with ammonia in hot methanol to yield 4,5-dimethoxy-2-nitroaniline (XXXI). Cyclization of compound (XXXI) with buten-2-one (XXXII) by means of H3PO4 and H3AsO4 affords 5,6-dimethoxy-4-methyl-8-nitroquinoline (XXXIII), which is selectively mono-demethylated by means of HCl in ethanol to provide 5-hydroxy-6-methoxy-4-methyl-8-nitroquinoline (XXXIV). Reaction of quinoline (XXXIV) with POCl3 gives the corresponding 5-chloro derivative (XXXV), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH to yield the diaryl ether (XXXVI). Finally, the nitro group of (XXXVI) is reduced by means of H2 over PtO2 in THF or H2 over Raney nickel.
Nitration of 2-fluoroanisole (XXXVII) with HNO3/Ac2O gives 3-fluoro-4-methoxynitrobenzene (XXXVIII), which is reduced to the corresponding aniline (XXXIX) with SnCl2/HCl. Reaction of compound (XXXIX) with Ac2O yields the acetanilide (XL), which is nitrated with HNO3 to afford 5-fluoro-4-methoxy-2-nitroacetanilide (XLI). Hydrolysis of (XLI) with NaOH provides 5-fluoro-4-methoxy-2-nitroaniline (XLII), which is cyclized with buten-2-one (XXXII) by means of As2O5 and H3PO4 to furnish 5-fluoro-6-methoxy-4-methyl-8-nitroquinoline (XLIII). Condensation of quinoline (XLIII) with 3-(trifluoromethyl)phenol (IV) by means of K2CO3 gives the diaryl ether (XXXIV), which is finally reduced by means of H2 over PtO2 in THF.
CLIP
US 4617394
Reaction of 8-amino-6-methoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinoline (XIV) with phthalic anhydride (XV) affords the phthalimido derivative (XVI), which is oxidized with MCPBA to yield the quinoline N-oxide (XVII). Treatment of compound (XVII) with neutral alumina gives the quinolone derivative (XVIII), which by reaction with POCl3 in refluxing CHCl3 provides the 2-chloroquinoline derivative (XIX). Alternatively, reaction of the quinoline N-oxide (XVII) with POCl3 as before also gives the 2-chloroquinoline derivative (XIX) The removal of the phthalimido group of compound (XIX) by means of hydrazine in refluxing ethanol gives the chlorinated aminoquinoline (XX), which is finally treated with MeONa in hot DMF.
CLIP
US 6479660; WO 9713753
Chlorination of 6-methoxy-4-methylquinolin-2(1H)-one (I) with SO2Cl2 in hot acetic acid gives the 5-chloro derivative (II), which is nitrated with HNO3 in H2SO4 to yield the 8-nitroquinolinone (III). Condensation of compound (III) with 3-(trifluoromethyl)phenol (IV) by means of KOH in NMP provides the diaryl ether (V), which is treated with refluxing POCl3 to afford the 2-chloroquinoline (VI). Reaction of compound (VI) with MeONa in refluxing methanol results in the 2,6-dimethoxyquinoline derivative (VII), which is reduced with hydrazine over Pd/C to give the 8-aminoquinoline derivative (VIII). Condensation of aminoquinoline (VIII) with N-(4-iodopentyl)phthalimide (IX) by means of diisopropylamine in hot NMP yields the phthalimido precursor (X), which is finally cleaved with hydrazine in refluxing ethanol.
Reaction of 1,4-dibromopentane (XI) with potassium phthalimide (XII) gives N-(4-bromopentyl)phthalimide (XIII), which is then treated with NaI in refluxing acetone.
Reaction of 4-methoxyaniline (XXI) with ethyl acetoacetate (XXII) by means of triethanolamine in refluxing xylene gives the acetoacetanilide (XXIII), which is cyclized by means of hot triethanolamine and H2SO4 to yield 6-methoxy-4-methylquinolin-2(1H)-one (I), which is treated with refluxing POCl3 to provide 2-chloro-6-methoxy-4-methylquinoline (XXIV). Reaction of compound (XXIV) with SO2Cl2 in hot AcOH affords 2,5-dichloro-6-methoxy-4-methylquinoline (XXV), which is treated with MeONa in refluxing methanol to furnish 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Alternatively, the reaction of compound (XXIV) with MeONa as before gives 2,6-dimethoxy-4-methylquinoline (XXVII), which is treated with SO2Cl2 in hot AcOH to give the already described 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Nitration of compound (XXVI) with KNO3 and P2O5 gives the 8-nitroquinoline derivative (XXVIII), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH in hot NMP to yield the diaryl ether (VII). Finally, the nitro group of compound (VII) is reduced with hydrazine over Pd/C.
aResearch Center for Solar Energy Chemistry, Division of Chemical Engineering, Graduate School of Engineering Science, Osaka University, Toyonaka 560-8531, Japan E-mail:shiraish@cheng.es.osaka-u.ac.jp
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), Saitama 332-0012, Japan
Abstract
Tafenoquine (TQ), a fluorescent antimalarial drug, was used as a receptor for the fluorometric detection of hypochlorite (OCl−). TQ itself exhibits a strong fluorescence at 476 nm, but OCl−-selective cyclization of its pentan-1,4-diamine moiety creates a blue-shifted fluorescence at 361 nm. This ratiometric response facilitates rapid, selective, and sensitive detection of OCl− in aqueous media with physiological pH. This response is also applicable to a simple test kit analysis and allows fluorometric OCl− imaging in living cells.
Synthesis of the intermediate diazepinone (IV) is accomplished by a one-pot synthesis. Condensation of 2-chloro-3-aminopyridine (I) with the anthranilic ester (II) is effected in the presence of potassium tert-butoxide as a catalyst. The resulting anthranilic amide (III) is cyclized under the influence of catalytic amounts of sulfuric acid. Treatment of (IV) with chloroacetylchloride in toluene yields the corresponding choroacetamide (V). The side chain of AQ-RA 741 is prepared starting from 4-picoline, which is alkylated by reaction with 3-(diethylamino)propylchloride in the presence of n-butyllithium. Hydrogenation of (VIII) using platinum dioxide as a catalyst furnishes the diamine (IX), which is coupled with (V) in the presence of catalytic amounts of sodium iodide in acetone leading to AQ-RA 741 as its free base.
Shanks GD, Oloo AJ, Aleman GM et al. (2001). “A New Primaquine Analogue, Tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria”. Clin Infect Dis33 (12): 1968–74. doi:10.1086/324081. JSTOR4482936.PMID11700577.
Lell B, Faucher JF, Missinou MA et al. (2000). “Malaria chemoprophylaxis with tafenoquine: a randomised study”.Lancet355 (9220): 2041–5. doi:10.1016/S0140-6736(00)02352-7. PMID10885356.
Percent Composition: C 62.19%, H 6.09%, F 12.30%, N 9.07%, O 10.36%
Literature References: Analog of primaquine, q.v. Prepn: P. Blumbergs, M. P. LaMontagne, US4617394 (1986 to U.S. Sec. Army); M. P. LaMontagne et al.,J. Med. Chem.32, 1728 (1989). HPLC determn in blood and plasma: D. A. Kocisko et al.,Ther. Drug Monit.22, 184 (2000). Metabolism: O. R. Idowu et al.,Drug Metab. Dispos.23, 1 (1995). Clinical pharmacokinetics: M. D. Edstein et al.,Br. J. Pharmacol.52, 663 (2001). Clinical evaluation in prevention of malaria relapse: D. S. Walsh et al.,J. Infect. Dis.180, 1282 (1999); in malaria prophylaxis: B. Lell et al.,Lancet355, 2041 (2000); B. R. Hale et al.,Clin. Infect. Dis.36, 541 (2003).
Derivative Type: Succinate
CAS Registry Number: 106635-81-8
Trademarks: Etaquine (GSK)
Molecular Formula: C24H28F3N3O3.C4H6O4
Molecular Weight: 581.58
Percent Composition: C 57.83%, H 5.89%, F 9.80%, N 7.23%, O 19.26%
Properties: Crystals from acetonitrile, mp 146-149°. LD50 in male, female rats (mg/kg): 102, 71 i.p.; 429, 416 orally (LaMontagne).
April 28, 2014
GlaxoSmithKline (GSK) and Medicines for Malaria Venture (MMV) announced the start of a Phase 3 global program to evaluate the efficacy and safety of tafenoquine, an investigational medicine which is being developed for the treatment and relapse prevention (radical cure) of Plasmodium vivax (P. vivax) malaria.
P. vivax malaria, a form of the disease caused by one of several species of Plasmodium parasites known to infect humans, occurs primarily in South and South East Asia, Latin America and the horn of Africa. Severe anemia, malnutrition and respiratory distress are among the most serious consequences described to be caused by the infection.
The Phase 3 program includes two randomized, double-blind treatment studies to investigate tafenoquine in adult patients with P. vivax malaria. The DETECTIVE study (TAF112582) aims to evaluate the efficacy, safety and tolerability of tafenoquine as a radical cure for P. vivax malaria, co-administered with chloroquine, a blood stage anti-malarial treatment. The GATHER study (TAF116564) aims to assess the incidence of hemolysis and safety and efficacy of tafenoquine compared to primaquine, the only approved treatment currently available for the radical cure of P. vivax malaria.
Tafenoquine is not yet approved or licensed for use anywhere in the world.
“P. vivax malaria can affect people of all ages and is particularly insidious because it has the potential to remain dormant within the body in excess of a year, and causes some patients to experience repeated episodes of illness after the first mosquito bite,” said Nicholas Cammack, head, Tres Cantos Medicines Development Center for Diseases of the Developing World. “Our investigation of tafenoquine for the treatment of P. vivax malaria is part of GSK’s efforts to tackle the global burden of malaria. Working with our partners, including MMV, we are determined to stop malaria in all its forms.”
“One of the big challenges we face in tackling malaria is to have new medicines to prevent relapse, caused by dormant forms of P. vivax,” said Dr. Timothy Wells, MMV’s chief scientific officer. “The Phase 3 program is designed to build upon the promising results of the Phase 2b study which showed that treatment with tafenoquine prevented relapses. If successful, tafenoquine has the potential to become a major contributor to malaria elimination. It’s a great privilege to be working with GSK on this project; they have a clear commitment to changing the face of public health in the countries in which we are working.”
/////////////Tafenoquine, タフェノキン , Orphan, FDA 2018, KRINTAFEL, Priority Review, GlaxoSmithKline
Carglumic acid is a Carbamoyl Phosphate Synthetase 1 Activator. The mechanism of action of carglumic acid is as a Carbamoyl Phosphate Synthetase 1 Activator.
For the treatment of acute and chronic hyperammonaemia in patients with N-acetylglutamate synthase (NAGS) deficiency. This enzyme is an important component of the urea cycle to prevent build up of neurotoxic ammonium in the blood.
EMA
Carglumic acid exists as a white powder or colourless crystals. It is soluble in boiling water, slightly soluble in cold water and practically insoluble in organic solvents (cyclohexane, dichloromethane, ether). The water solubility of carglumic acid at pH 2.0 is 21.0 g/L. It increases rapidly between the pH 3.0 (28.2 g/L) and the pH 5.0 (440.9 g/L). The solubility of carglumic acid in water is stable between pH 6.0 (555.5 g/L) and pH 8.0 (553.9 g/L). Carglumic acid is prepared from L-glutamic acid. It exhibits stereoisomerism due to the presence of one chiral centre and has one optical isomer; N-carbamoyl-D-glutamic acid.
Percent Composition: C 37.90%, H 5.30%, N 14.73%, O 42.07%
Literature References: Metabolically stable analog of N-acetylglutamate, a physiological activator of the first enzyme of the urea cycle, carbamylphosphate synthetase (CAPS). Prepn: H. McIlwain, Biochem. J.33, 1942 (1939). Effect on blood urea and ammonia levels and potential clinical application: J.-E. O’Connor et al., Eur. J. Pediatr.143, 196 (1985). Evaluation in treatment of CAPS deficiency: G. Kuchler et al., J. Inher. Metab. Dis.19, 220 (1996); of N-acetylglutamate synthetase (NAGS) deficiency: B. Plecko et al., Eur. J. Pediatr.157, 996 (1998).
Properties: mp 174°.
Melting point: mp 174°
Therap-Cat: In treatment of inherited urea cycle disorders.
CARBAGLU®
(carglumic acid) Tablet for Oral Suspension
DESCRIPTION
CARBAGLU tablets for oral suspension, contain 200 mg of carglumic acid. Carglumic acid, the active substance, is a Carbamoyl Phosphate Synthetase 1 (CPS 1) activator and is soluble in boiling water, slightly soluble in cold water, and practically insoluble in organic solvents.
Chemically carglumic acid is N-carbamoyl-L-glutamic acid or (2S)-2-(carbamoylamino) pentanedioic acid, with a molecular weight of 190.16.
The structural formula is:
Molecular Formula: C6H10N2O5
The inactive ingredients of CARBAGLU are croscarmellose sodium, hypromellose, microcrystalline cellulose, silica colloidal anhydrous, sodium lauryl sulfate, sodium stearyl fumarate.
Carglumic acid is an orphan drug used for the treatment of hyperammonaemia in patients with N-acetylglutamate synthase deficiency. This rare genetic disorder results in elevated blood levels of ammonia, which can eventually cross the blood–brain barrier and cause neurologic problems, cerebral edema, coma, and death. Carglumic acid was approved by the U.S. Food and Drug Administration (FDA) on 18 March 2010.
Carglumic Acid is an orally active, synthetic structural analogue of N-acetylglutamate (NAG) and carbamoyl phosphate synthetase 1 (CPS 1) activator, with ammonia lowering activity. NAG, which is formed by the hepatic enzyme N-acetylglutamate synthase (NAGS), is an essential allosteric activator of the enzyme carbamoyl phosphate synthetase 1 (CPS 1). CPS 1 plays an essential role in the urea cycle and converts ammonia into urea. Upon oral administration, carglumic acid can replace NAG in NAGS deficient patients and activates CPS 1, which prevents hyperammonaemia.
Carglumic acid is an orphan drug and a derivative of N-acetylglutamate that activates the first enzyme in the urea cycle that is responsible for removal and detoxification of ammonia, making this drug a valuable agent for therapy of hyperammonemia caused by rare forms of urea cycle defects. Clinical experience with carglumic acid is limited, but it has not been linked to significant serum enzyme elevations during therapy or to instances of clinically apparent acute liver injury.
Carglumic acid is an orphan drug, marketed by Orphan Europe under the trade name Carbaglu. Carglumic acid is used for the treatment of hyperammonaemia in patients with N-acetylglutamate synthase deficiency.[1][2] The initial daily dose ranges from 100 to 250 mg/kg, adjusted thereafter to maintain normal plasma levels of ammonia.
The US FDA approved it for treatment of hyperammonaemia on March 18, 2010. Orphan Drug exclusivity expired on March 18, 2017.[3]
Carbaglu (carglumic acid) Tablets 200 mg, is a white elongated tablet with three score marks on both sides engraved C’s on one side. It is a dispersible tablet designed to be dispersed in of water and ingested or administered through a syringe via a nasogastric tube. It is indicated for treatment of acute hyperammonemia in patients with NAGS deficiency.
The drug substance, carglumic acid, is an allosteric activator of a critical urea cycle enzyme, carbamoyl phosphate synthetase (CPS). It is a close analog of the naturally occurring activator, N-acetyl glutamate (NAG). Carglumic acid is a urea-like derivative of the amino acid L-glutamate and contains one chiral center. The drug substance solid form is the neutral dicarboxylic acid and is a white crystalline powder. The water solubility of the drug substance depends on the . polymorphic solid form has been found.
The drug substance is manufactured by .
The facility was found to have acceptable cGMP status during an inspection by
the Agency in November 2009. The synthesis of carglumic acid consists of a
Regarding characterization, the drug substance structure was determined by
NMR, MS, IR and Regarding impurities, two potential
impurities are possible due to
hydantoin-5-proprionic acid (HPA) and diaza-1,3-dione-2,4-carboxy-7-
cycloheptane (Diaza). Only the has been detected at
batch release and it increases in amount during storage at elevated temperatures
but not at room temperature. This impurity also increases during drug product
storage at room temperature but not at refrigerated temperatures, see above
discussion. The starting materials, , were not
detected in several batches and therefore routine testing is not required.
Regarding drug substance specification, identity testing is by IR and HPLC.
Other tests include optical rotation, melting point, pH of 0.5% solution, loss on
drying, residue on ignition, heavy metals, assay and impurities by HPLC.
Regarding chiral purity, the observed specific optical rotation is small and
therefore not a very precise method for determination of chiral purity. Although
a chiral HPLC method was developed, since the r was not detected in
any samples (the limit of detection was 0.1%) during the development, originally
the sponsor did not propose to implement the test in the specification. However,
the Agency recommended that the chiral HPLC method be included in the
specification to assure chiral purity, and the sponsor agreed to do so with the limit
for the NMT
Batch release data were provided that justified the proposed acceptance limits. In
general, measured total impurities were low in the drug substance, about .
Appropriate in-house reference standards were established.
Stability results for 3 batches stored at 25°C/60%RH for 36 months remained
within the tight specification limits. A re-test period of for the drug
substance stored in its original packaging at room temperature is granted.
B. Description of How the Drug Product is Intended to be Used
The drug product tablets may be dispersed in a minimum amount of water
mL per tablet) and ingested immediately or administered through a syringe via a
nasogastric tube. The suspension has a slightly acidic taste.
21 April 2017 EMA/CHMP/404487/2017 Committee for Medicinal Products for Human Use (CHMP) Assessment report Ucedane International non-proprietary name: carglumic acid Procedure No. EMEA/H/C/004019/0000
Carglumic acid (also called N-carbamyl-L-glutamate, or carbamylglutamate) is an orally active deacylaseresistant synthetic structural N-acetylglutamate (NAG) analogue. NAG, which is formed by the hepatic enzyme N-acetylglutamate synthase (NAGS), is an essential allosteric activator of the enzyme carbamoyl phosphate synthetase 1 (CPS-1). CPS-1 plays an essential role in the urea cycle and converts ammonia into urea which prevents hyperammonaemia. Despite a lower affinity of carbamoyl phosphate synthetase for carglumic acid than for N-acetylglutamate, carglumic acid has been shown in vivo to stimulate carbamoyl phosphate synthetase and to be much more effective than N-acetylglutamate in protecting against ammonia intoxication in rats.
Carglumic acid was first authorised in the EU as Carbaglu dispersible tablets in January 2003. At the time of approval Carbaglu was indicated for the treatment of hyperammonaemia associated with N-acetylglutamate synthase deficiency. Subsequently, the approved indications for Carbaglu have been extended and is now also authorised for the treatment of hyperammonaemia due to, isovaleric acidaemia, methymalonic acidaemia, or propionic acidaemia. Ucedane is indicated in treatment of hyperammonaemia due to N-acetylglutamate synthase primary deficiency. Proposed posology and method of administration for Ucedane
The chemical name of the active substance, carglumic acid, is N-Carbamyl-L-glutamic acid corresponding to the molecular formula C6H10N2O5. It has a relative molecular mass 190.16 g/mol and the following structure:
Carglumic acid exists as a white powder or colourless crystals. It is soluble in boiling water, slightly soluble in cold water and practically insoluble in organic solvents (cyclohexane, dichloromethane, ether). The water solubility of carglumic acid at pH 2.0 is 21.0 g/L. It increases rapidly between the pH 3.0 (28.2 g/L) and the pH 5.0 (440.9 g/L). The solubility of carglumic acid in water is stable between pH 6.0 (555.5 g/L) and pH 8.0 (553.9 g/L). Carglumic acid is prepared from L-glutamic acid. It exhibits stereoisomerism due to the presence of one chiral centre and has one optical isomer; N-carbamoyl-D-glutamic acid.
Adverse effects
The most common adverse effects include vomiting, abdominal pain, fever, and tonsillitis.[4]
Jump up^Caldovic L, Morizono H, Daikhin Y, Nissim I, McCarter RJ, Yudkoff M, Tuchman M (2004). “Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate”. J Pediatr. 145 (4): 552–4. doi:10.1016/j.jpeds.2004.06.047. PMID15480384.
Jump up^Elpeleg O, Shaag A, Ben-Shalom E, Schmid T, Bachmann C (2002). “N-acetylglutamate synthase deficiency and the treatment of hyperammonemic encephalopathy”. Ann Neurol. 52 (6): 845–9. doi:10.1002/ana.10406. PMID12447942.
CONTROLLED RELEASE PHARMACEUTICAL COMPOSITION WITH RESISTANCE AGAINST THE INFLUENCE OF ETHANOL EMPLOYING A COATING COMPRISING A POLYMER MIXTURE AND EXCIPIENTS
CONTROLLED RELEASE PHARMACEUTICAL COMPOSITION WITH RESISTANCE AGAINST THE INFLUENCE OF ETHANOL EMPLOYING A COATING COMPRISING NEUTRAL VINYL POLYMERS AND EXCIPIENTS
RECOMBINANT L-N-CARBAMOYLASE DERIVED FROM ARTHROBACTER AURESCENS, AND A METHOD FOR PRODUCING L-AMINO ACIDS BY USING THE SAME RECOMBINANT L-N-CARBAMOYLASE DERIVED FROM ARTHROBACTER AURESCENS, AND A METHOD FOR PRODUCING L-AMINO ACIDS BY USING THE SAME
Company:Swedish Orphan Biovitrum AB (SOBI) (Originator)
Nitisinone is a synthetic reversible inhibitor of 4-hydroxyphenylpyruvate dioxygenase. It is used in the treatment of hereditary tyrosinemia type 1. It is sold under the brand name Orfadin.
Nitisinone was first approved by the U.S. Food and Drug Administration (FDA) on January 18, 2002, then approved by the European Medicines Agency (EMA) on February 21, 2005. It was developed and marketed as Orfadin® bySwedish Orphan Biovitrum AB (SOBI) in the US .
The mechanism of action of nitisinone involves reversibile inhibition of 4-Hydroxyphenylpyruvate dioxygenase(HPPD). It is indicated for use as an adjunct to dietary restriction of tyrosine and phenylalanine in the treatment of hereditary tyrosinemia type 1 (HT-1).
Orfadin® is available as capsule for oral use, containing 2, 5 or 10 mg of free Nitisinone. The recommended initial dose is 1 mg/kg/day divided into two daily doses. Maximum dose is 2 mg/kg/day.
Nitisinone was launched in 2002 by Swedish Orphan (now Swedish Orphan Biovitrum) in a capsule formulation as an adjunct to dietary restriction of tyrosine and phenylalanine in the treatment of hereditary tyrosinemia type I. In 2015, this product was launched in Japan for the same indication. The same year, an oral suspension formulation for pediatric patients was registered in the E.U., and launch took place in the United Kingdom shortly after. This formulation was approved in 2016 in the U.S. for the same indication. In 2016, nitisinone tablet formulation developed by Cycle Pharmaceuticals was approved in Canada (this formulation is also available also in the U.S.).
Indication
Used as an adjunct to dietary restriction of tyrosine and phenylalanine in the treatment of hereditary tyrosinemia type 1.
Product name, strength, pharmaceutical form: Orfadin • Marketing authorisation holder: Swedish Orphan Biovitrum International AB • Date of authorisation: 21/02/2005
Procedure No. EMEA/H/C/004281/0000
During the meeting on 22 June 2017, the CHMP, in the light of the overall data submitted and the scientific discussion within the Committee, issued a positive opinion for granting a Marketing authorisation to Nitisinone MendeliKABS.
The chemical name of nitisinone is 2-[2-Nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione corresponding to the molecular formula C14H10F3NO5. It has a relative molecular mass of 329.23 g/mol and the following structure: Figure 1. Structure of nitisinone.
Nitisinone appears as off-white to yellowish non-hygroscopic fine crystalline powder. It is practically insoluble in unbuffered water. It is freely soluble in dichloromethane, sparingly soluble in ethyl alcohol, slightly soluble in isopropyl alcohol and 70% aqueous isopropyl alcohol and in pH 6.8 phosphate buffer, very slightly soluble in pH 4.5 acetate buffer and practically insoluble at pH 1.1. Solubility in acidified aqueous media depends on the acid counter ion. Solubility increases with increasing pH. Its pKa was found to be around 10. Nitisinone is achiral and does not show polymorphism.
Nitisinone is a white to yellowish-white crystalline powder poorly soluble in water. The active substance is a weak acid and it is highly soluble in the pH range 4.5-7.2 in phosphate buffer solutions. Nitisinone has the chemical name 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione. It does not show polymorphism.
Nitisinone (INN), also known as NTBC (an abbreviation of its full chemical name) is a medication used to slow the effects of hereditary tyrosinemia type 1. Since its first use for this indication in 1991, it has replaced liver transplantation as the first-line treatment for this rare condition. It is also being studied in the related condition alkaptonuria. It is marketed under the brand name Orfadin by the company Swedish Orphan Biovitrum (Sobi); it was first brought to market by Swedish Orphan International. It was originally developed as a candidate herbicide.
Since its first use for this indication in 1991, it has replaced liver transplantation as the first-line treatment for this rare condition.[4] I It is marketed under the brand name Orfadin.
It has been demonstrated that treatment with nitisinone can reduce urinary levels of homogentisic acid in alkaptonuria patients by 95%.[5] A series of clinical trials run by DevelopAKUre to determine whether nitisinone is effective at treating the ochronosis suffered by patients with alkaptonuria are ongoing.[6] If the trials are successful, DevelopAKUre will try to get nitisinone licensed for use by alkaptonuria patients.[7]
Alkaptonuria is caused when an enzyme called homogentisic dioxygenase (HGD) is faulty, leading to a buildup of homogenisate.[11]Alkaptonuria patients treated with nitisinone produce far less HGA than those not treated (95% less in the urine),[5] because nitisinone inhibits HPPD, resulting in less homogenisate accumulation. Clinical trials are ongoing to test whether nitisinone can prevent ochronosisexperienced by older alkaptonuria patients.[6]
Adverse effects
Nitisinone has several negative side effects; these include but are not limited to: bloated abdomen, dark urine, abdominal pain, feeling of tiredness or weakness, headache, light-colored stools, loss of appetite, weight loss, vomiting, and yellow-colored eyes or skin.[12]
Research
Nitisinone is being studied as a treatment for alkaptonuria.[13]
Research at the National Institutes of Health (NIH) has demonstrated that nitisinone can reduce urinary levels of HGA by up to 95% in patients with alkaptonuria. The primary parameter of the NIH trial was range of hip motion, for which the results were inconclusive.[citation needed]
Research done using alkaptonuric mice has shown that mice treated with nitisinone experience no ochronosis in knee joint cartilage. In contrast, all of the mice in the untreated control group developed ochronotic knee joints.[14]
The efficacy of Nitisinone is now being studied in a series international clinical trials called DevelopAKUre.[15] The studies will recruit alkaptonuria patients in Europe.[16] A larger number of patients will be recruited in these trials than in the previous NIH trial.[17] The trials are funded by the European Commission.[18]
Nitisinone has been shown to increase skin and eye pigmentation in mice, and has been suggested as a possible treatment for oculocutaneous albinism.[19][20]
History
Nitisinone was discovered as part of a program to develop a class of herbicides called HPPD inhibitors. It is a member of the benzoylcyclohexane-1,3-dione family of herbicides, which are chemically derived from a natural phytotoxin, leptospermone, obtained from the Australian bottlebrush plant (Callistemon citrinus).[21]HPPD is essential in plants and animals for catabolism, or breaking apart, of tyrosine.[22] In plants, preventing this process leads to destruction of chlorophyll and the death of the plant.[22] In toxicology studies of the herbicide, it was discovered that it had activity against HPPD in rats[23] and humans.[24]
In Type I tyrosinemia, a different enzyme involved in the breakdown of tyrosine, fumarylacetoacetate hydrolase is mutated and doesn’t work, leading to very harmful products building up in the body.[1] Fumarylacetoacetate hydrolase acts on tyrosine after HPPD does, so scientists working on making herbicides in the class of HPPD inhibitors hypothesized that inhibiting HPPD and controlling tyrosine in the diet could treat this disease. A series of small clinical trials attempted with one of their compounds, nitisinone, were conducted and were successful, leading to nitisinone being brought to market as an orphan drug Swedish Orphan International,[8] which was later acquired by Swedish Orphan Biovitrum (Sobi).
Sobi is now a part of the DevelopAKUre consortium. They are responsible for drug supply and regulatory support in the ongoing clinical trials that will test the efficiacy of nitisinone as a treatment for alkaptonuria.[25] It is hoped that if the trials are successful, nitisinone could also be licensed for treatment of alkaptonuria.[7]
Generic versions
There is no generic version of Orfadin in G7 countries. Prior to the market authorization of MDK-Nitisinone in Canada, the only Nitisinone product available globally was Orfadin.[26]Until recently, Nitisinone was not approved in Canada where it was distributed for over 20 years via a Health Canada Special Access Program. In September 2016, MendeliKABS was granted approval of a Priority New Drug Submission (PNDS) by Health Canada for a bioequivalent generic version of Orfadin capsules (MDK-Nitisinone). In November 2016 Cycle Pharma was also granted approval of a PNDS by Health Canada for Nitisinone tablets that are bioequivalent to Orfadin capsules.[27] SOBI was granted approval of a PNDS in December 2016.[28]
PAPER
1H NMR, 13C NMR, and Computational DFT Studies of the Structure of 2-Acylcyclohexane-1,3-diones and Their Alkali Metal Salts in Solution
Faculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warszawa, Poland
J. Org. Chem., 2006, 71 (12), pp 4636–4641
DOI: 10.1021/jo060583g
1H and 13C NMR spectra of 2-acyl-substituted cyclohexane-1,3-diones (acyl = formyl, 1; 2-nitrobenzoyl, 2; 2-nitro-4-trifluoromethylbenzoyl, 3) and lithium sodium and potassium salts of 1have been measured. The compound 3, known as NTBC, is a life-saving medicine applied in tyrosinemia type I. The optimum molecular structures of the investigated objects in solutions have been found using the DFT method with B3LYP functional and 6-31G** and/or 6-311G(2d,p) basis set. The theoretical values of the NMR parameters of the investigated compounds have been calculated using GIAO DFT B3LYP/6-311G(2d,p) method. The theoretical data obtained for compounds 1−3 have been exploited to interpret their experimental NMR spectra in terms of the equilibrium between different tautomers. It has been found that for these triketones an endo-tautomer prevails. The differences in NMR spectra of the salts of 1 can be rationalized taking into account the size of the cation and the degree of salt dissociation. It seems that in DMSO solution the lithium salt exists mainly as an ion pair stabilized by the chelation of a lithium cation with two oxygen atoms. The activation free energy the of formyl group rotation for this salt has been estimated to be 51.5 kJ/mol. The obtained results suggest that in all the investigated objects, including the free enolate ions, all atoms directly bonded to the carbonyl carbons lie near the same plane. Some observations concerning the chemical shift changes could indicate strong solvation of the anion of 1 by water molecules. Implications of the results obtained in this work for the inhibition mechanism of (4-hydroxyphenyl) pyruvate dioxygenase by NTBC are commented upon.
2-(2-Nitro-4-trifluoromethylbenzoyl)cyclohexane-1,3-dione (NTBC; 3). The compound was prepared in the same manner as 2. The synthesis of an appropriate benzoic acid derivative was started from the transformation of commercially available 2-nitro-4-trifluoromethylaniline into benzonitrile by the classical Sandmeyer method. Then the nitrile was hydrolyzed in 65% sulfuric acid to give 2-nitro-4-trifluoromethylbenzoic acid.13 The obtained triketone 3 had a mp of 140−142 °C (lit.14 141−143 °C). For NMR data, see Supporting Information….. https://pubs.acs.org/doi/suppl/10.1021/jo060583g/suppl_file/jo060583gsi20060420_080852.pdf
NMR data for 2-(2-nitro-4-trifluoromethylbenzoyl)cyclohexane-1,3-dione, 3, in CDCl3
The condensation of cyclohexane-1,3-dione (I) with 2-nitro-4-(trifluoromethyl)benzoyl chloride by means of TEA in dichloromethane gives the target Nitisinone.EP 0186118
JP 1986152642, US 4774360, US 4780127
Nitisinone
Synonyms:NTBC, SC 0735
ATC:A16AX04
Use:treatment of inherited tyrosinemia type I
Chemical name:2-[2-nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione
NTBC is a drug marketed by Swedish Orphan Biovitrum International AB under the brand name Orfadin® and it is used to slow the effects of hereditary tyrosinemia type 1 (HT-1) in adult and pediatric patients. It has been approved by FDA and EMA in January 2002 and February 2005 respectively.
HT-1 disease is due to a deficiency of the final enzyme of the tyrosine catabolic pathway fumarylacetoacetate hydrolase. NTBC is a competitive inhibitor of 4-hydroxyphenylpyruvate dioxygenase (HPPD), an enzyme which precedes fumarylacetoacetate hydrolase. By inhibiting the normal catabolism of tyrosine in patients with HT-1, NTBC prevents the accumulation of the toxic intermediates maleylacetoacetate and fumarylacetoacetate, that in patients with HT-1 are converted to the toxic metabolites succinylacetone and succinylacetoacetate, the former inhibiting the porphyrin synthesis pathway leading to the accumulation of 5-aminolevulinate.
Usefulness of NTBC in the treatment of further diseases has also been documented. A non-comprehensive list is reported hereinafter.
Effectiveness of Orfadin® in the treatment of diseases where the products of the action of HPPD are involved (e.g., HT-1) has been described notably in EP0591275B1 corresponding to U.S. Pat. No. 5,550,165B1. Synthesis of NTBC is also described in this patent.
WO2011106655 reports a method for increasing tyrosine plasma concentrations in a subject suffering from oculocutaneous/ocular albinism, the method comprising administering to the subject a pharmaceutically acceptable composition comprising NTBC in the range of between about 0.1 mg/kg/day to about 10 mg/kg/day.
U.S. Pat. No. 8,354,451B2 reports new methods of combating microbial infections due to fungi or bacteria by means of administration to a subject of a therapeutically active amount of NTBC.
WO2010054273 discloses NTBC-containing compositions and methods for the treatment and/or prevention of restless leg syndrome (RLS).
EP1853241B1 claims the use of NTBC in the treatment of a neurodegenerative disease, notably Parkinson disease.
Introne W. J., et al., disclosed usefulness of nitisinone in the treatment of alkaptonuria (Introne W. J., et al., Molec. Genet. Metab., 2011, 103, 4, 307). The key step of the synthesis reported in EP0591275B1 (now propriety of Swedish Orphan Biovitrum International AB, SE), involves the reaction of 2-nitro-4-trifluromethylbenzoyl chloride and cyclohexane-1,3-dione in the presence of triethylamine and then use of acetone cyanohydrin in order to promote the rearrangement of the key intermediate enol ester. After washing and extraction from CH2Cl2, the crude product is recrystallized from ethyl acetate to get the desired 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione as a solid having a melting point of 88-94° C.
Another patent (U.S. Pat. No. 4,695,673) filed in name of Stauffer Chemical Company disclosed a process of synthesis of acylated 1,3-dicarbonyl compounds in which the intermediate enol ester is isolated prior to its rearrangement into the final product, said rearrangement making use of a cyanohydrin compound derived from alkali metal, methyl alkyl ketone, benzaldehyde, cyclohexanone, C2-C5aliphatic aldehyde, lower alkyl silyl or directly by using hydrogen cyanide.
Yet another patent (U.S. Pat. No. 5,006,158) filed in name of ICI Americas Inc. disclosed a process similar to the one disclosed in U.S. Pat. No. 4,695,673 wherein the intermediate enol ester was isolated prior to its rearrangement into the final product by use of potassium cyanide. Said reaction can optionally be done by concomitant use of a phase transfer catalyst such as Crown ethers. The preferred solvent for conducting such a reaction is 1,2-dichloroethane.
Still a further patent (EP0805791) filed in name of Zeneca Ltd disclosed an alternative synthesis of nitisinone involving the reaction of 1,3-cyclohexanedione and variously substituted benzoyl chloride in the presence of sodium or potassium carbonate in CH3CN or DMF. Best yields were obtained using CH3CN as solvent and sodium carbonate as the base. Reaction was performed at 55-57° C. in 17 hours.
It is well known that one of the problems of the actual drug formulation (i.e., Orfadin® capsules) is its chemical instability. Indeed, even if Orfadin® has to be stored in a refrigerator at a temperature ranging from 2° C. to 8° C., its shelf life is of only 18 months. After first opening, the in-use stability is a single period of 2 months at a temperature not above 25° C., after which it must be discarded. It will be evident that such storage conditions have an impact in the distribution chain of the medicine, in terms of costs and also in terms of logistics for the patient. Therefore, there is an urgent need of more stable formulations, both from a logistic supply chain point of view, and from the patient compliance point of view. Since the formulation of Orfadin® contains only the active ingredient and starch as excipient, relative instability may be attributed to the active pharmaceutical ingredient itself; in other words it can derive from the way it is synthesized and/or the way it is extracted from the reaction mixture, and/or the way it is finally crystallized. Furthermore, some impurities may contribute to render the final product less stable overtime. Consequently, it is of major importance to identify a process of synthesis and/or a crystallization method that enable the reliable production of a highly pure and stable product.
Impurities as herein-above mentioned can derive either from the final product itself (through chemical degradation) or directly from the starting materials/solvents used in the process of synthesis. Regarding the latter option, it is therefore primordial to ascertain that at each step, impurities are completely removed in order not to get them at the final stage, also considering that some of them could potentially be cyto/genotoxic.
The impurities correlated to nitisinone can be either derived from the starting materials themselves (i.e., impurities 1 and 2) or obtained as side products during the process of synthesis and/or under storage conditions (i.e., impurities 3 to 5) and are the following:
2-nitro-4-(trifluoromethyl) benzoic acid (Impurity no 1),
1,3-cyclohexanedione (CHD) (Impurity no 2),
4-(trifluoromethyl)salicylic acid (Impurity no 3),
2-[3-nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione (Impurity no 4), and
6-trifluoromethyl-3,4-dihydro-2H-xanthene-1,9-dione (Impurity no 5).
Impurity-2, impurity-3, and impurity-5 have been previously reported in WO2015101794. Strangely, impurity-4 has never been reported, even if it is an obvious side-product which can easily be formed during the coupling reaction between 1,3-cyclohexanedione and 2-nitro-4-(trifluoromethyl) benzoic acid, the latter being not 100% pure but containing some amount of regioisomer 3-nitro-4-(trifluoromethyl) benzoic acid.
Potential genotoxicity of impurity no 4 which possesses an aromatic nitro moiety was assessed using in-silico techniques and resulted to be a potential genotoxic impurity. According to the FDA ICH M7 guidelines, daily intake of a mutagenic impurity (Threshold of Toxicological Concern, TTC) in an amount not greater than 1.5 μg per person is considered to be associated with a negligible risk to develop cancer over a lifetime of exposure. Consequently, assuming a daily dose of 2 mg/kg, for a person weighing 70 kg, the maximum tolerated impurity content of such a compound would be of about 11 ppm, as calculated according to the equation underneath.
concentration limit ( ppm ) = T T C ( µg / day ) Dose ( g / day )
It is therefore of paramount importance to ensure that the process of synthesis of nitisinone and the purification steps of the same give rise to an API devoid of such impurity no 4, or at least far below the threshold of 11 ppm as indicated above. The skilled person will understand that total absence of said impurity is highly desirable.
It is well known in the pharmaceutical field that investigation of potential polymorphism of a solid API is of crucial importance and is also recommended by major regulatory authorities such as FDA.
Notwithstanding the fact that nitisinone has been used for years to treat HT-1 patients, it appears that no NTBC formulation fully satisfies the requisites of stability and/or compliance standard for the patients. Therefore, there is an unmet medical need of long-term pure and stable formulations.
Example 1
Thionyl chloride (162 g, 1.36 mol) was added dropwise into a suspension of 2-nitro-4-trifluoromethylbenzoic acid (228 g, 0.97 mol) in toluene (630 g) at 80° C. The thus obtained solution was kept under stirring at 80° C. for 20 hours, and then cooled to 50° C. The volatiles were removed under reduced pressure in order to get the expected 2-nitro-4-trifluoromethylbenzoyl chloride as an oil. The latter, cooled to 25° C. was added dropwise to a suspension of 1,3-cyclohexanedione (109 g, 0.97 mol) and potassium carbonate (323 g, 2.33 mol) in CH3CN (607 g). After 18 h the mixture was diluted with water (500 ml) and slowly acidified to about pH=1 with HCl 37%. The mixture was then warmed to about 55° C. and the phases were separated. The organic layer was washed with a 10% aqueous solution of sodium chloride and then, concentrated under reduced pressure at a temperature below 55° C. to reach a volume of 380 ml. The thus obtained mixture was stirred at 55° C. for 1 h and then cooled to 0° C. in 16 to 18 h. The resulting solid was filtered and rinsed several times with pre-cooled (0° C.) toluene. The wet solid was dried at 60° C. under vacuum for 6 h to provide nitisinone (164 g) as a white to yellowish solid with a purity of 98.4% as measured by HPLC and a content of potentially genotoxic impurity no 4 of 6.1 ppm measured by HPLC/MS.
Example 2
Nitisinone as obtained from example 1 (164 g) was added to a 3/1 (w/w) mixture of CH3CN/toluene (volume of solvent: 638 ml). The mixture was warmed gently to about 55° C. under stirring until solids were completely dissolved. The solution was then concentrated under reduced pressure maintaining the internal temperature below 50° C. to reach a volume of 290 ml. Then, more toluene (255 g) was added and the solution was concentrated again under reduced pressure until the residual volume reached 290 ml. The solution was heated to about 55° C. for 1 h and successively cooled slowly in 10 to 12 h to 10° C. The resulting solid was filtered and rinsed several times with pre-cooled (0° C.) toluene. The wet solid was dried at about 60° C. under vacuum for 4 h to provide nitisinone (136 g) as a white to yellowish solid, with a purity of 99.94% and a 99.8% assay measured by HPLC and a d(90) particle size between 310 and 350 μm. The content of potential genotoxic impurity no 4 resulted below 1 ppm.
^ Jump up to:abLock, E. A.; Ellis, M. K.; Gaskin, P.; Robinson, M.; Auton, T. R.; Provan, W. M.; Smith, L. L.; Prisbylla, M. P.; Mutter, L. C.; Lee, D. L. (1998). “From toxicological problem to therapeutic use: The discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), its toxicology and development as a drug”. Journal of Inherited Metabolic Disease. 21 (5): 498–506. doi:10.1023/A:1005458703363. PMID9728330.
Jump up^Kavana, Michael; Moran, Graham R. (2003). “Interaction of (4-Hydroxyphenyl)pyruvate Dioxygenase with the Specific Inhibitor 2-[2-Nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione†”. Biochemistry. 42 (34): 10238–45. doi:10.1021/bi034658b. PMID12939152.
Jump up^Phornphutkul, Chanika; Introne, Wendy J.; Perry, Monique B.; Bernardini, Isa; Murphey, Mark D.; Fitzpatrick, Diana L.; Anderson, Paul D.; Huizing, Marjan; Anikster, Yair; Gerber, Lynn H.; Gahl, William A. (2002). “Natural History of Alkaptonuria”. New England Journal of Medicine. 347 (26): 2111–21. doi:10.1056/NEJMoa021736. PMID12501223.
Jump up^Preston, A. J.; Keenan, C. M.; Sutherland, H.; Wilson, P. J.; Wlodarski, B.; Taylor, A. M.; Williams, D. P.; Ranganath, L. R.; Gallagher, J. A.; Jarvis, J. C. (2013). “Ochronotic osteoarthropathy in a mouse model of alkaptonuria, and its inhibition by nitisinone”. Annals of the Rheumatic Diseases. 73 (1): 284–9. doi:10.1136/annrheumdis-2012-202878. PMID23511227.
Jump up^G. Mitchell, D.W. Bartlett, T.E. Fraser, T.R. Hawkes, D.C. Holt, J.K. Townson, R.A. Wichert Mesotrione: a new selective herbicide for use in maize Pest Management Science, 57 (2) (2001), pp. 120–128
CAS Name: 2-[2-Nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione
Additional Names: NTBC
Trademarks: Orfadin (Swedish Orphan )
Molecular Formula: C14H10F3NO5
Molecular Weight: 329.23
Percent Composition: C 51.07%, H 3.06%, F 17.31%, N 4.25%, O 24.30%
Literature References: Herbicidal triketone that inhibits 4-hydroxyphenylpyruvate dioxygenase (HPPD), an enzyme involved in plastoquinone biosynthesis in plants and in tyrosine catabolism in mammals. Prepn: C. G. Carter, EP186118 (1986 to Stauffer); idem,US5006158 (1991 to ICI). Inhibition of HPPD in plants: M. P. Prisbylla et al.,Brighton Crop Prot. Conf. – Weeds1993, 731; in rats: M. K. Ellis et al.,Toxicol. Appl. Pharmacol.133, 12 (1995). LC determn in plasma: M. Bielenstein et al.,J. Chromatogr. B730,177 (1999). Clinical evaluation in hereditary tyrosinemia type I: S. Lindstedt et al.,Lancet340, 813 (1992). Review of toxicology and therapeutic development: E. A. Lock et al,J. Inherited Metab. Dis.21, 498-506 (1998); of clinical experience: E. Holme, S. Lindstedt, ibid. 507-517.
Properties: Solid, mp 88-94°.
Melting point: mp 88-94°
Therap-Cat: In treatment of inherited tyrosinemia type I.
////////////////Nitisinone, ニチシノン , Orfadin, FDA 2002, NTBC , SC-0735 , SYN-118 , JAPAN 2015, JAP 2015, EU 2005, Priority, Orphan

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....












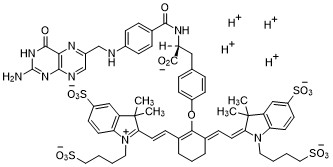
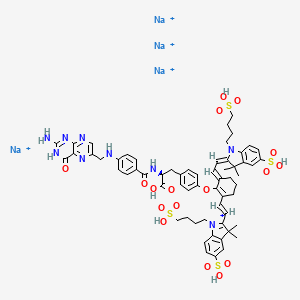




































-Tafenoquin_Structural_Formula_V1.svg)
-Tafenoquin_Structural_Formula_V1.svg)
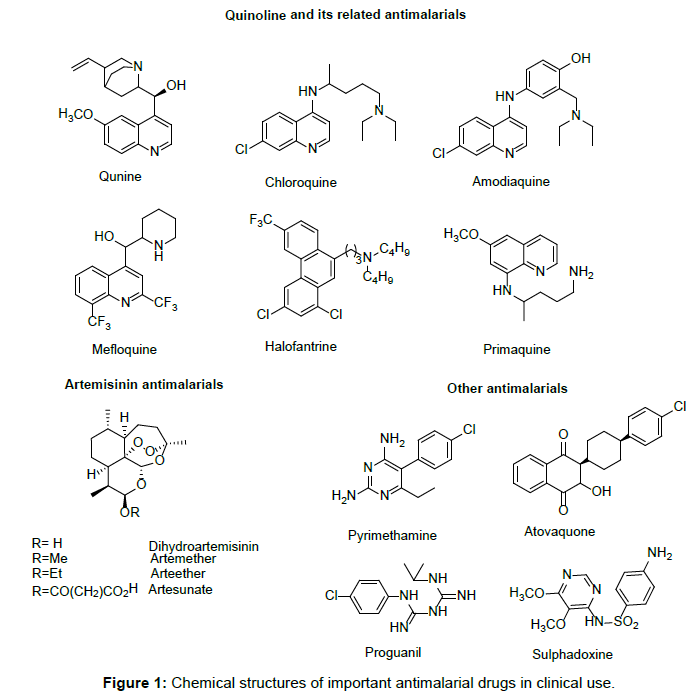
 CLIP
CLIP
















-Tafenoquin_Structural_Formula_V1.svg)







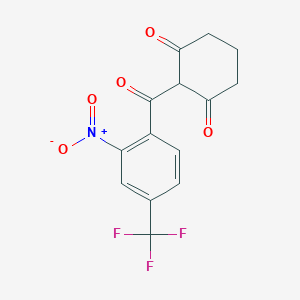
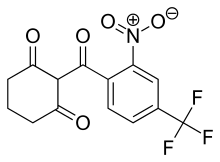











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}