 COMPD A
COMPD A
Compd A OR B IS GSK 2269557
Phosphatidylinositol 3-Kinase (PI3K)
PHASE 1….asthma & COPD
ASHTHMA COPD
DATA FOR COMPD A
6-(1H-indol-4-yl)-4-[5-[[4-(1-methylethyl)-1-piperazinyl]methyl]-2-oxazolyl]-1H-Indazole,
6-(1 H-lndol-4-yl)-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1 H- indazole
CAS 1254036-77-5 hcl salt
base 1254036-71-9, 440.54, C26 H28 N6 O
Formula C26H28N6O.HCl
EMAIL ME amcrasto@gmail.com
DATA FOR COMPD B
Methanesulfonamide, N-[5-[4-[5-[[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl]-2-oxazolyl]-1H-indazol-6-yl]-2-methoxy-3-pyridinyl]-,
N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide
1254036-66-2 CAS
C24 H28 N6 O5 S, 512.58
Compound B may be prepared according to known procedures, such as those disclosed in international patent application PCT/EP2010/055666 (publication number WO02010/125082)
EMAIL ME amcrasto@gmail.com
Phosphoinositide 3ΌΗ kinases (hereinafter PI3Ks) are a family of signal transducer enzymes which are involved in various cellular functions including cell growth, proliferation and differentiation. A wide variety of retroviruses and DNA-based viruses activate the PI3K pathway as a way of preventing host cell death during viral infection and ultimately exploiting the host cell synthesis machinery for its replication (Virology 344(1) p. 131-8 (2006) by Vogt et al.; and Nat. Rev. Microbiol. 6(4) p. 265-75 (2008) by Buchkovich et al). It has therefore been postulated that PI3K inhibitors may have potential therapeutic benefit in the treatment of viral infections such as influenza virus infection, in addition to the more established treatment of cancer and inflammatory diseases.
The Influenza NS1 protein activates Class la PI3Ks by binding to their regulatory subunit p85beta but not to other Class la regulatory subunits such as p85alpha. The recent crystal structure of the NS1-p85beta complex (Hale et al. Proc. Natl. Acad. Sci. U S A. 107(5) p.1954-1959 (2010)) is also suggestive of an interaction with the p110 kinase subunit providing a mechanism for catalytic activation of the kinase domain. This observation provides a rationale for isoform specificity not only with the p85 regulatory subunit but also potentially with the p110 catalytic subunit too. The function of PI3K during influenza virus infection has also been investigated by, for example, Ehrhardt et al. (Cell. Microbiol. 8(8) p. 1336-1348 (2006)), and the role of PI3K5 signalling in morbidity and lung pathology induced by influenza virus infection has been reported in WO 2010/083163.
There remains a need to provide compounds which are inhibitors of the activity or function of PI3K5 which may be useful in the treatment or prevention of influenza virus infection.
GSK 2269557 is an inhaled phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor in early clinical trials at GlaxoSmithKline for the treatment of patients with asthma and also for the treatment of chronic obstructive pulmonary disease (COPD) in patients who smoke cigarettes.
- 18 Nov 2014GlaxoSmithKline plans a phase II trial in Chronic obstructive pulmonary disease in Belgium, Denmark, the Netherlands and Russia (NCT02294734)
- 01 Jun 2014Phase-II clinical trials in Chronic obstructive pulmonary disease in Germany (Inhalation)
- 01 May 2014GlaxoSmithKline plans a phase II trial for Chronic obstructive pulmonary disease in Germany (NCT02130635)
| Study ID |
Status |
Title |
Patient Level Data |
| 115117 |
Completed |
A single-centre. double-blind, placebo controlled three part study to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of single and repeat doses of nebulised GSK2269557 in healthy male subjects |
|
| 115119 |
Active not recruiting |
A Double-Blind, Placebo Controlled, Randomised, Parallel Group Study to Evaluate the Safety, Tolerability and Pharmacokinetics of Multiple Doses of GSK2269557 Administered as a Dry Powder to COPD Patients |
|
| 116617 |
Completed |
A Single-Centre, Double-Blind, Placebo Controlled Two Part Study to Evaluate the Safety, Tolerability and Pharmacokinetics of Single and Repeat Doses of GSK2269557 as a Dry Powder in Healthy Subjects who Smoke Cigarettes |
|
| 116678 |
Not yet recruiting |
A Randomised, Double-blind (Sponsor Unblinded), Placebo-controlled, Parallel-group, Multicentre Study to Evaluate the Efficacy and Safety of GSK2269557 Administered in Addition to Standard of Care in Adult Subjects Diagnosed With an Acute Exacerbation of Chronic Obstructive Pulmonary Disease |
EMAIL ME amcrasto@gmail.com
CLICK ON IMAGES TO VIEW SIMILAR ROUTES FOR COMPD A AND B


CLICK ON IMAGE TO VIEW

…………………………………………………………………….
COMPD A
WO 2012032065
http://www.google.com/patents/WO2012032065A1?cl=en
Example 68
6-(1 H-lndol-4-yl)-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1 H- indazole
Method A
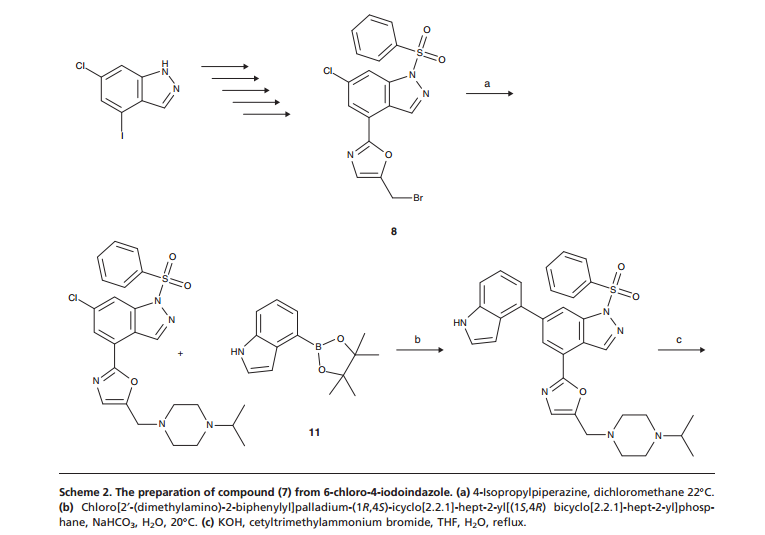
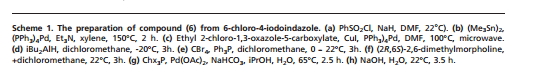
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1/-/-indazole (97 mg, 0.194 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (61.3 mg, 0.252 mmol, available from Frontier Scientific Europe), chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (10.87 mg, 0.019 mmol) and potassium phosphate tribasic (124 mg, 0.582 mmol) were dissolved in 1 ,4-dioxane (1 ml) and water (0.1 ml) and heated in a Biotage Initiator microwave at 100°C for 30 min. Additional 4-(4,4,5,5- tetramethyl-1 ,3,2-dioxabotolan-2-yl)-1 H-indole (61.3 mg, 0.252 mmol) and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (5 mg) were added and the reaction heated at 1 10°C for 30 min, then 140°C for 30 min. The solvent was removed in vacuo and the residue purified by silica gel chromatography, eluting with 0-25% methanol in dichloromethane. The appropriate fractions were combined and concentrated to give a brown solid which was dissolved in MeOH:DMSO (1 ml, 1 : 1 , v/v) and purified by MDAP (method H). The appropriate fractions were concentrated in vacuo to give the title compound as a white solid (30 mg).
LCMS (Method A): Rt 0.57 mins, MH+ 441.
Method B
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1 H-indazole (75.17 g, 150 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (73.1 g, 301 mmol), sodium bicarbonate (37.9 g, 451 mmol), and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (8.43 g, 15.03 mmol) were suspended in nitrogen purged 1 ,4-dioxane (1200 ml_) and water (300 ml_). The reaction vessel was placed under alternating vacuum and nitrogen five times with overhead stirring, then finally placed under a nitrogen atmosphere and heated to 120°C for 2.5 h.
The reaction mixture was cooled to 45°C and then treated with 2M aqueous sodium hydroxide (376 ml_, 752 mmol). After stirring at 45°C overnight (~ 13h), the mixture was cooled to RT and DCM (600 ml) and water (400 ml) were added. The layers were separated and the aqueous re-extracted with DCM: 1 ,4-dioxane (1 : 1). Brine was added and the mixture filtered through Celite, washing with DCM: 1 ,4-dioxane (1 : 1). The layers were separated and 2M HCI (1000 ml) added to the organic. The mixture was again filtered through Celite washing with 500 ml 2M HCI keeping the washings separate. The filtrate layers were then separated and the organic layer was washed with the acid washings from the Celite. Layers were separated and the acidic aqueous combined. This was then back-washed with 2×500 ml of DCM; each wash requiring a Celite filtration. The acidic aqueous was then given a final filtration through Celite washing the Celite pad with 150 ml of 2M HCI.
The acidic aqueous was transfered to a beaker (5000 ml) and with vigorous stirring 2M NaOH was added to basify the mixture to pH 10-11. The mixture was then extracted using 1 ,4-dioxane: DCM (1 : 1) (5 x 500 ml). The combined organics were washed with brine, dried over magnesium sulphate, filtered and evaporated to yield a brown foam that was dried in vacuo at 50°C overnight. This material was split into three batches and each was purified by reverse phase column chromatography (3x 1.9 kg C18 column), loading in DMF/TFA (1 : 1 , 30 ml) then eluting with 3-40% MeCN in Water + 0.25% TFA (Note: Columns 2 & 3 used a different gradient starting with 10% MeCN).
Appropriate fractions were combined, the acetotnitrile removed in vacuo and the acidic aqueous basified to pH10 by addition of saturated aqueous sodium carbonate solution to the stirred solution. The resultant solid was collected by filtration, washed with water then dried in vacuo at 65°C overnight to give the title compound (28.82 g) as a pale brown foam.
LCMS (Method A): Rt 0.68 mins, MH+ 441.
1 H NMR (400MHz ,DMSO-d6) d = 13.41 (br. s., 1 H), 11.35 (br. s., 1 H), 8.59 (br. s., 1 H), 8.07 (d, J = 1.5 Hz, 1 H), 7.90 (br. s., 1 H), 7.51 – 7.44 (m, 2 H), 7.32 (s, 1 H), 7.27 – 7.21 (m, 2 H), 6.61 – 6.58 (m, 1 H), 3.73 (br. s., 2 H), 2.64 – 2.36 (m, 9 H), 0.97 – 0.90 (m, 6 H)
Method C
Potassium hydroxide (145.6 g) was added to a suspension of 6-(1 H-indol-4-yl)-4-(5-{[4-(1- methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)-1 H-indazole (300.7 g) and cetyltrimethylammonium bromide (9.3 g) in tetrahydrofuran (6.0 L) and water (30 ml) stirring under nitrogen at ambient temperature. The mixture was heated at reflux for 17 hours and was then cooled to 20-25°C. Ethyl acetate (3.0 L) and water (3.0 L) were added, stirred for 10 minutes and then separated. The organic layer was extracted with hydrochloric acid (1 M, 1 x 3.0 L, 2 x 1.5L) and the acidic extracts combined and basified to ~pH 8 by the addition of saturated sodium carbonate solution (2.1 L). After ageing for 30 minutes the resultant suspension was filtered, washed with water (300 ml) and the solid dried under vacuum at 65°C to give the title compound as a pale yellow solid (127.9 g).
LCMS (Method B): Rt 2.44 min, MH+ 441.
…………………………………………………………………………
WO 2010125082
http://www.google.co.in/patents/WO2010125082A1?cl=en
Example 6
6-(1 H-lndol-4-yl)-4-(5-{[4-(1 -methylethyl)-1 -piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1 H- indazole
Method A
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1H-indazole (97 mg, 0.194 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (61.3 mg, 0.252 mmol, available from Frontier Scientific Europe), chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (10.87 mg, 0.019 mmol) and potassium phosphate tribasic (124 mg, 0.582 mmol) were dissolved in 1 ,4-dioxane (1 ml) and water (0.1 ml) and heated in a Biotage Initiator microwave at 1000C for 30 min. Additional 4-(4, 4,5,5- tetramethyl-1 ,3,2-dioxabotolan-2-yl)-1 H-indole (61.3 mg, 0.252 mmol) and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (5 mg) were added and the reaction heated at 1 1O0C for 30 min, then 14O0C for 30 min. The solvent was removed in vacuo and the residue purified by silica gel chromatography, eluting with 0-25% methanol in dichloromethane. The appropriate fractions were combined and concentrated to give a brown solid which was dissolved in MeOH:DMSO (1 ml, 1 :1 , v/v) and purified by MDAP (method A). The appropriate fractions were concentrated in vacuo to give the title compound as a white solid (30 mg).
LCMS (Method A): Rt 0.57 mins, MH+ 441.
Method B
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1 H-indazole (75.17 g, 150 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (73.1 g, 301 mmol), sodium bicarbonate (37.9 g, 451 mmol), and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (8.43 g, 15.03 mmol) were suspended in nitrogen purged 1 ,4-dioxane (1200 ml.) and water (300 ml_). The reaction vessel was placed under alternating vacuum and nitrogen five times with overhead stirring, then finally placed under a nitrogen atmosphere and heated to 1200C for 2.5 h.
The reaction mixture was cooled to 45°C and then treated with 2M aqueous sodium hydroxide (376 ml_, 752 mmol). After stirring at 450C overnight (~ 13h), the mixture was cooled to RT and DCM (600 ml) and water (400 ml) were added. The layers were separated and the aqueous re-extracted with DCM: 1 ,4-dioxane (1 :1 ). Brine was added and the mixture filtered through Celite, washing with DCM: 1 ,4-dioxane (1 :1 ). The layers were separated and 2M HCI (1000 ml) added to the organic. The mixture was again filtered through Celite washing with 500 ml 2M HCI keeping the washings separate. The filtrate layers were then separated and the organic layer was washed with the acid washings from the Celite. Layers were separated and the acidic aqueous combined. This was then back-washed with 2×500 ml of DCM; each wash requiring a Celite filtration. The acidic aqueous was then given a final filtration through Celite washing the Celite pad with 150 ml of 2M HCI.
The acidic aqueous was transfered to a beaker (5000 ml) and with vigorous stirring 2M NaOH was added to basify the mixture to pH 10-11. The mixture was then extracted using 1 ,4-dioxane:DCM (1 :1 ) (5 x 500 ml). The combined organics were washed with brine, dried over magnesium sulphate, filtered and evaporated to yield a brown foam that was dried in vacuo at 500C overnight.
This material was split into three batches and each was purified by reverse phase column chromatography (3x 1.9 kg C18 column), loading in DMF/TFA (1 :1 , 30 ml) then eluting with 3-40% MeCN in Water + 0.25% TFA (Note: Columns 2 & 3 used a different gradient starting with 10% MeCN).
Appropriate fractions were combined, the acetotnitrile removed in vacuo and the acidic aqueous basified to pH10 by addition of saturated aqueous sodium carbonate solution to the stirred solution. The resultant solid was collected by filtration, washed with water then dried in vacuo at 65°C overnight to give the title compound (28.82 g) as a pale brown foam.
LCMS (Method A): Rt 0.68 mins, MH+ 441. 1H NMR (400MHz ,DMSOd6) d = 13.41 (br. s., 1 H), 11.35 (br. s., 1 H), 8.59 (br. s., 1 H), 8.07 (d, J = 1.5 Hz, 1 H), 7.90 (br. s., 1 H), 7.51 – 7.44 (m, 2 H), 7.32 (s, 1 H), 7.27 – 7.21 (m, 2 H), 6.61 – 6.58 (m, 1 H), 3.73 (br. s., 2 H), 2.64 – 2.36 (m, 9 H), 0.97 – 0.90 (m, 6 H)
EMAIL ME amcrasto@gmail.com
COMPD B
WO2010125082
http://www.google.co.in/patents/WO2010125082A1?cl=en
Example 1
Λ/-[5-[4-(5-{[(2/?,6S)-2,6-Dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-
6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide
Method A
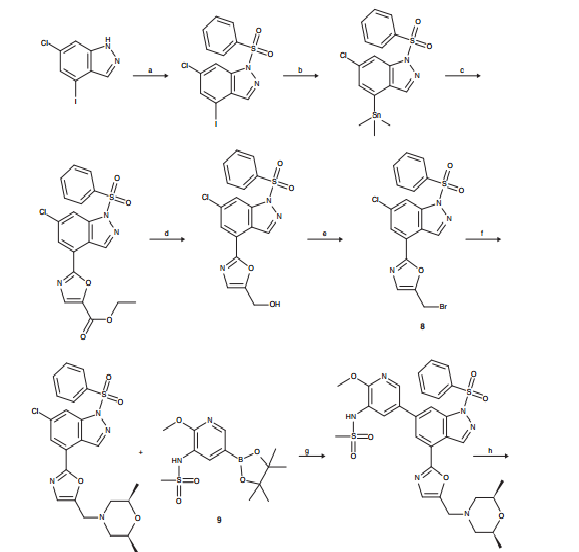
To a solution of 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1 ,3-oxazol-2- yl)-1-(phenylsulfonyl)-1 H-indazole (0.20 g, 0.411 mmol) and N-[2-(methoxy)-5-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3-pyridyl]methanesulfonamide (0.175 g, 0.534 mmol) in 1 ,4-dioxane (2 ml) was added chloro[2′-(dimethylamino)-2-biphenylyl]palladium- 1 (1 /?,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4/?)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol), potassium phosphate tribasic (0.262 g, 1.23 mmol) and water (0.2 ml). The reaction mixture was heated and stirred at 12O0C under microwave irradiation for 1 h. Additional chloroP’^dimethylamino^-biphenylyOpalladium-^I R^S^bicycloP^.ilhept^- yl[(1 S,4/?)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol) and potassium phosphate tribasic (80 mg) were added and the reaction heated to 12O0C under microwave irradiation for 1 h. Additional potassium phospate tribasic (80 mg) was added and the reaction heated under the same conditions for a further 1 h. The reaction mixture was filtered through a silica SPE and eluted with methanol. The solvent was removed in vacuo and the residue partitioned between dichloromethane (5 ml) and water (5 ml). The layers were separated and the aqueous extracted with further dichloromethane (2x 2 ml). The combined organics were concentrated under a stream of nitrogen and the residue dissolved in MeOH:DMSO (3ml, 1 :1 , v/v) and purified by MDAP (method A) in 3 injections. The appropriate fractions were combined and concentrated to give a white solid which was dissolved in MeOH:DMSO (1 ml, 1 :1 , v/v) and further purified by MDAP (method B). The appropriate fractions were basified to pH 6 with saturated sodium bicarbonate solution and extracted with ethyl acetate (2x 25 ml). The combined organics were dried and evaporated in vacuo to give a white solid which was further dried under nitrogen at 4O0C for 3 h to give the title compound as a white solid (26 mg). LCMS (Method A): Rt 0.53 mins, MH+ 513.
Method B N-[2-(Methyloxy)-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinyl]methanesulfonamide (101 g, 308 mmol), 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4- morpholinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)-1 H-indazole (83.3 g, 154 mmol) and sodium bicarbonate (38.8 g, 462 mmol) were suspended in 1 ,4-dioxane (1840 ml) and water (460 ml) under nitrogen and heated to 800C. Chloro[2′-(dimethylamino)-2- biphenylyl]palladium-1 (1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)-bicyclo[2.2.1]hept-2- yl]phosphane (8.63 g, 15.40 mmol) was added and the mixture stirred overnight at 800C.
The reaction mixture was cooled to 450C, sodium hydroxide 2M aq. (770 ml, 1540 mmol) added and the reaction heated to 45 0C for 4 hours. The mixture was cooled to RT and diluted with water (610 ml_). Dichloromethane (920 ml.) was added, and the mixture was filtered twice through Celite (washed with 200 ml. 1 ,4-dioxane/DCM 2:1 each time). The phases were separated, and aqueous washed with 1 ,4-dioxane/DCM 2:1 (500 ml_). The aqueous phase was neutralised with hydrochloric acid to pH -7 and extracted with 1 ,4- dioxane/DCM 2:1 (1 L), then 1 ,4 dioxane/DCM 1 :1 (2×500 ml_). The organics were washed with brine (500 ml_), and filtered through Celite (washed with 200 ml. 1 ,4 dioxane/DCM 2:1 ), and evaporated to yield a dark black solid, which was purified in 4 batches:
Batch 1 : 28g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (14.78 g).
Batch 2: 3Og was dissolved in methanol and mixed with Fluorisil. The solvent was then removed by evaporation and the solid purified by column chromatography (1.5 kg silica column, solid sample injection module), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (9.44 g).
Batch 3: 31 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (17 g).
Batch 4: 29g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (21 g).
The mixed fractions from the 4 columns were combined and evaporated to yield 19 g which was dissolved in 200 ml. of Toluene/Ethanol/Ammonia 80:20:2 (+ additional 4ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (6.1 g).
All pure batches were combined (68 g) and recrystallised from ethanol (1200 ml_). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight. The resulting solid was then collected by filtration, washed sparingly with ethanol and dried under vacuum to give the title compound as an off-white solid (56 g). This material was recrystallised again from ethanol (1 100 ml_). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight with stirring. The resulting solid was collected by filtration and washed sparingly with ethanol. The solid was dried in vacuo at 600C for 5hrs to give the title compound as an off-white solid (45.51 g). LCMS (Method A): Rt 0.61 mins, MH+ 513.
The filtrate from the two recrystallisations was evaporated to yield -23 g of a solid residue that was dissolved in 200 ml. of Toluene/Ethanol/Ammonia 80:20:2 (+ additional 4ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give a further crop of the title compound as an off-white solid (18.5 g). This solid was then recrystallised from ethanol (370 ml_). The suspension was heated to reflux then the resulting solution stirred for 20 mins before being allowed to cool to room temperature naturally overnight. The solid was then dried in vacuo at 65°C overnight to give the title compound as an off-white solid (11.9O g). LCMS (Method A): Rt 0.62 mins, MH+ 513.
………………………………………..
http://www.google.co.in/patents/US8735390
Example 1N-[5-[4-(5-{[(2R,6S)-2,6-Dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide
Method A
To a solution of 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazole (0.20 g, 0.411 mmol) and N-[2-(methoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-pyridyl]methanesulfonamide (0.175 g, 0.534 mmol) in 1,4-dioxane (2 ml) was added chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol), potassium phosphate tribasic (0.262 g, 1.23 mmol) and water (0.2 ml). The reaction mixture was heated and stirred at 120° C. under microwave irradiation for 1 h. Additional chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol) and potassium phosphate tribasic (80 mg) were added and the reaction heated to 120° C. under microwave irradiation for 1 h. Additional potassium phospate tribasic (80 mg) was added and the reaction heated under the same conditions for a further 1 h. The reaction mixture was filtered through a silica SPE and eluted with methanol. The solvent was removed in vacuo and the residue partitioned between dichloromethane (5 ml) and water (5 ml). The layers were separated and the aqueous extracted with further dichloromethane (2×2 ml). The combined organics were concentrated under a stream of nitrogen and the residue dissolved in MeOH:DMSO (3 ml, 1:1, v/v) and purified by MDAP (method A) in 3 injections. The appropriate fractions were combined and concentrated to give a white solid which was dissolved in MeOH:DMSO (1 ml, 1:1, v/v) and further purified by MDAP (method B). The appropriate fractions were basified to pH 6 with saturated sodium bicarbonate solution and extracted with ethyl acetate (2×25 ml). The combined organics were dried and evaporated in vacuo to give a white solid which was further dried under nitrogen at 40° C. for 3 h to give the title compound as a white solid (26 mg).
LCMS (Method A): Rt 0.53 mins, MH+ 513.
Method B
N-[2-(Methyloxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-pyridinyl]methanesulfonamide (101 g, 308 mmol), 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazole (83.3 g, 154 mmol) and sodium bicarbonate (38.8 g, 462 mmol) were suspended in 1,4-dioxane (1840 ml) and water (460 ml) under nitrogen and heated to 80° C. Chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (8.63 g, 15.40 mmol) was added and the mixture stirred overnight at 80° C.
The reaction mixture was cooled to 45° C., sodium hydroxide 2M aq. (770 ml, 1540 mmol) added and the reaction heated to 45° C. for 4 hours. The mixture was cooled to RT and diluted with water (610 mL). Dichloromethane (920 mL) was added, and the mixture was filtered twice through Celite (washed with 200 mL 1,4-dioxane/DCM 2:1 each time). The phases were separated, and aqueous washed with 1,4-dioxane/DCM 2:1 (500 mL). The aqueous phase was neutralised with hydrochloric acid to pH ˜7 and extracted with 1,4-dioxane/DCM 2:1 (1 L), then 1,4 dioxane/DCM 1:1 (2×500 mL). The organics were washed with brine (500 mL), and filtered through Celite (washed with 200 mL 1,4 dioxane/DCM 2:1), and evaporated to yield a dark black solid, which was purified in 4 batches:
- Batch 1: 28 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (14.78 g).
- Batch 2: 30 g was dissolved in methanol and mixed with Fluorisil. The solvent was then removed by evaporation and the solid purified by column chromatography (1.5 kg silica column, solid sample injection module), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (9.44 g).
- Batch 3: 31 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (17 g).
- Batch 4: 29 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (21 g).
The mixed fractions from the 4 columns were combined and evaporated to yield 19 g which was dissolved in 200 mL of Toluene/Ethanol/Ammonia 80:20:2 (+additional 4 ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (6.1 g).
All pure batches were combined (68 g) and recrystallised from ethanol (1200 mL). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight. The resulting solid was then collected by filtration, washed sparingly with ethanol and dried under vacuum to give the title compound as an off-white solid (56 g). This material was recrystallised again from ethanol (1100 mL). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight with stirring. The resulting solid was collected by filtration and washed sparingly with ethanol. The solid was dried in vacuo at 60° C. for 5 hrs to give the title compound as an off-white solid (45.51 g).
LCMS (Method A): Rt 0.61 mins, MH+ 513.
The filtrate from the two recrystallisations was evaporated to yield ˜23 g of a solid residue that was dissolved in 200 mL of Toluene/Ethanol/Ammonia 80:20:2 (+additional 4 ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give a further crop of the title compound as an off-white solid (18.5 g). This solid was then recrystallised from ethanol (370 mL). The suspension was heated to reflux then the resulting solution stirred for 20 mins before being allowed to cool to room temperature naturally overnight. The solid was then dried in vacuo at 65° C. overnight to give the title compound as an off-white solid (11.90 g).
LCMS (Method A): Rt 0.62 mins, MH+ 513.
Method C
10M Sodium hydroxide solution (0.70 ml) was added to a stirred suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (1.17 g) in water (5.8 ml). The resulting mixture was stirred at room temperature for 3.75 hours and was then washed with ethyl acetate (2×6 ml). The layers were separated and the aqueous phase was acidified to pH 6 with 2M hydrochloric acid (0.8 ml). The acidified aqueous layer was extracted twice with ethyl acetate (11 ml then 5 ml). The combined ethyl acetate extracts were dried by azeotropic distillation and diluted with further ethyl acetate (11 ml). The misture was stirred at room temperature for 112 hours. The slurry was seeded and then stirred at room temperature for 48 hours. The resultant suspension was filtered, washed with ethyl acetate (2×2 ml) and the solid dried under vacuum at 40° C. to give the title compound as a pale yellow solid (0.58 g).
LCMS (Method B): Rt 1.86 min, MH+ 513.
Method D
To a suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (596.5 g, 0.91 mol) in water (3.8 L) is added 5M sodium hydroxide (715 ml, 3.56 mol) over 20 mins at <25° C. The mixture is stirred at 20±3° C. for 2 h 45 min then washed with EtCN (3 L). The pH of the basic aqueous phase is adjusted to pH 6.6 using 2M hydrochloric acid (1.4 L), maintaining the temperature below 30° C. The mixture is then extracted with MeTHF (2×4.8 L), and the combined MeTHF extracts are washed with water (1.2 L). The mixture is concentrated to approx 2.4 L and EtOAc (3 L) is added. This put and take distillation is repeated a further 3 times. The mixture is adjusted to 60±3° C. and seeded twice (2×3 g) 35 mins apart. The resultant is aged for 1 h 10 mins then cooled over 2 h to 20-25° C., and aged for a further 15 h 50 min. The slurry is filtered, washed with EtOAc (2×1.2 L) and dried in vacuo at 45±5° C. for approx 3 day to give the title compound.
Preparation of Polymorphs of Compound A
Form (II)
Ethyl acetate (15 ml) was added to N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (2.1 g) and was stirred at ambient conditions overnight. The resultant slurry was filtered and dried under vacuum at 50° C. to give a new solid state form (91 ckw/w).
1H NMR (400 MHz, DMSO d6) d=13.49 (br s, 1H), 9.39 (s, 1H), 8.58 (s, 1H), 8.42 (d, J=2.2 Hz, 1H), 7.99 (d, J=2.2 Hz, 1H), 7.93 (d, J=1.2 Hz, 1H), 7.88 (s, 1H), 7.35 (s, 1H), 4.00 (s, 3H), 3.74 (s, 2H), 3.58 (m, 2H), 3.11 (s, 3H), 2.80 (d, J=10.3 Hz, 2H), 1.78 (t, J=10.3 Hz, 2H), 1.05 (d, J=6.4 Hz, 6H)
SODIUM SALT OF COMPD B
http://www.google.com/patents/US20140256721
Method D
To a suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (596.5 g, 0.91 mol) in water (3.8 L) is added 5M sodium hydroxide (715 ml, 3.56 mol) over 20 mins at <25° C. The mixture is stirred at 20±3° C. for 2 h 45 min then washed with EtCN (3 L). The pH of the basic aqueous phase is adjusted to pH 6.6 using 2M hydrochloric acid (1.4 L), maintaining the temperature below 30° C. The mixture is then extracted with MeTHF (2×4.8 L), and the combined MeTHF extracts are washed with water (1.2 L). The mixture is concentrated to approx 2.4 L and EtOAc (3 L) is added. This put and take distillation is repeated a further 3 times. The mixture is adjusted to 60±3° C. and seeded twice (2×3 g) 35 mins apart. The resultant is aged for 1 h 10 mins then cooled over 2 h to 20-25° C., and aged for a further 15 h 50 min. The slurry is filtered, washed with EtOAc (2×1.2 L) and dried in vacuo at 45±5° C. for approx 3 day to give the title compound.
http://www.google.com/patents/US20140256721
Preparation of Salts of Compound ASodium Salt
Methanol (2 ml) was added to N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (0.3 g) followed by aqueous sodium hydroxide (0.129 ml) to give a solution. Tert-butylmethylether (4 ml) was added to the solution followed by seed crystals of the sodium salt and this suspension was stirred overnight at ambient conditions. The suspension was filtered, washed with tert-butylmethylether (2 ml) and air dried to give the sodium salt (0.2312 g) as a hydrate.
NMR: Consistent with salt formation
1H NMR (400 MHz, DMSO d6) d=13.35 (br s, 1H), 8.53 (s, 1H), 7.90 (d, J=1.2 Hz, 1H), 7.73 (s, 1H), 7.65 (d, J=2.5 Hz, 1H), 7.62 (d, J=2.2 Hz, 1H), 7.33 (s, 1H), 4.00 (s, 3H), 3.80 (s, 3H), 3.59 (m, 2H). 2.83 (d, J=10.3, 2H), 2.61 (s, 3H), 1.78 (t, J=10.5 Hz, 2H), 1.05 (d, J=6.1 Hz, 6H)
EMAIL ME amcrasto@gmail.com
EMAIL ME amcrasto@gmail.com
| US20100280029 * |
28 Apr 2010 |
4 Nov 2010 |
Julie Nicole Hamblin |
Novel compounds |
| WO2010125082A1 |
28 Apr 2010 |
4 Nov 2010 |
Glaxo Group Limited |
Oxazole substituted indazoles as pi3-kinase inhibitors |
| US20140256721 * |
14 Apr 2014 |
11 Sep 2014 |
Glaxosmithkline Intellectual Property Development Limited |
Novel Polymorphs and Salts |
| WO2012032065A1 |
6 Sep 2011 |
15 Mar 2012 |
Glaxo Group Limited |
Indazole derivatives for use in the treatment of influenza virus infection |
| WO2012032067A1 |
6 Sep 2011 |
15 Mar 2012 |
Glaxo Group Limited |
Polymorphs and salts of n- [5- [4- (5- { [(2r,6s) -2, 6 – dimethyl – 4 -morpholinyl] methyl} – 1, 3 – oxazol – 2 – yl) – 1h- inda zol-6-yl] -2- (methyloxy) – 3 – pyridinyl] methanesulfonamide |
| WO2012055846A1 |
25 Oct 2011 |
3 May 2012 |
Glaxo Group Limited |
Polymorphs and salts of 6-(1h-indol-4-yl)-4-(5- { [4-(1-methylethyl)-1-pi perazinyl] methyl} -1,3-oxazol-2-yl)-1h-indazole as pi3k inhibitors for use in the treatment of e.g. respiratory disorders |
| WO2012064744A2 * |
8 Nov 2011 |
18 May 2012 |
Lycera Corporation |
Tetrahydroquinoline and related bicyclic compounds for inhibition of rorϒ activity and the treatment of disease |
| WO2013088404A1 |
14 Dec 2012 |
20 Jun 2013 |
Novartis Ag |
Use of inhibitors of the activity or function of PI3K |
| WO2014068070A1 |
31 Oct 2013 |
8 May 2014 |
INSERM (Institut National de la Santé et de la Recherche Médicale) |
Methods for preventing antiphospholipid syndrome (aps) |
| US8524751 |
5 Mar 2010 |
3 Sep 2013 |
GlaxoSmithKline Intellecutual Property Development |
4-oxadiazol-2-YL-indazoles as inhibitors of P13 kinases |
| US8536169 |
3 Jun 2009 |
17 Sep 2013 |
Glaxo Group Limited |
Compounds |
| US8575162 |
28 Apr 2010 |
5 Nov 2013 |
Glaxosmithkline Intellectual Property Development Limited |
Compounds |
| US8580797 |
28 Apr 2010 |
12 Nov 2013 |
Glaxo Smith Kline Intellectual Property Development Limited |
Compounds |
| US8586583 |
2 Oct 2012 |
19 Nov 2013 |
Glaxosmithkline Intellectual Property Development Limited |
Compounds |
| US8586590 |
2 Oct 2012 |
19 Nov 2013 |
Glaxosmithkline Intellectual Property Development Limited |
Compounds |
| US8609657 |
2 Oct 2012 |
17 Dec 2013 |
Glaxosmithkline Intellectual Property Development Limited |
Compounds |
| US8658635 |
3 Jun 2009 |
25 Feb 2014 |
Glaxosmithkline Intellectual Property Development Limited |
Benzpyrazol derivatives as inhibitors of PI3 kinases |
| US8735390 |
6 Sep 2011 |
27 May 2014 |
Glaxosmithkline Intellectual Property Development Limited |
Polymorphs and salts |
| US8765743 |
3 Jun 2009 |
1 Jul 2014 |
Glaxosmithkline Intellectual Property Development Limited |
Compounds |
…..
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.


COCK WILL TEACH YOU NMR

COCK SAYS MOM CAN TEACH YOU NMR

Join me on twitter

 amcrasto@gmail.com
amcrasto@gmail.com
JALGAON, MAHARASHTRA, INDIA

.
![]()
![Image result for jalgaon railway station]()


![]()


 .
.





.

 MANUDEVI
MANUDEVI





















































 .
. .
.
 .
. .
. .
. .
. .
.
 .
. .
.


















![N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide](https://i0.wp.com/pic3.molbase.net/molpic/02/47/2473420.png)

![N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide NMR spectra analysis, Chemical CAS NO. 739366-20-2 NMR spectral analysis, N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-28/002/473/2473420_1h.png)
![N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide NMR spectra analysis, Chemical CAS NO. 739366-20-2 NMR spectral analysis, N-[2-[[2-[(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl]amino]-2-methylpropyl]-2-methylpyrazolo[1,5-a]pyrimidine-6-carboxamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-28/002/473/2473420_13c.png)







 LATUR AIRPORT
LATUR AIRPORT











 The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the







 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

































 Location in Madhya Pradesh
Location in Madhya Pradesh










{kind=link}