PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
General information Recombinant von Willebrand Factor (rVWF) is co-expressed with recombinant Factor VIII (rFVIII) in Chinese hamster ovary (CHO) cells as part of the ADVATE (Centrally authorised product) manufacturing process. The rVWF protein is separated from the FVIII and further purified.
Vonicog alfa is expressed as a 2813 amino acid pro-VWF molecule. The pro-VWF is composed of A, B, C and D repeats, which contain various functional domains that have been identified. The mature VWF monomer is a 2050 amino acid protein. Every monomer contains a number of specific domains with a specific function. Elements of note are: • The D’/D3 domain, which binds to Factor VIII • The A1 domain, which binds to: Platelet gp1b-receptor, Heparin, Collagen • The A3 domain, which binds to collagen • The C1 domain, in which the RGD domain binds to platelet integrin αIIbβ3 when this is activated • The “cysteine knot” domain Monomers of pro-VWF are subsequently N-glycosylated, arranged into dimers via a C-terminal disulfide bond in the endoplasmic reticulum and into multimers by crosslinking of N-terminal cysteine residues via disulfide bonds.
Figure 1. Structure of Von Willebrand Factor Monomer/Dimer
After reduction of disulfide bonds in electrophoretic analysis, rVWF appears as a single predominant band having an apparent molecular weight of approximately 260 kDa. In low resolution agarose gel electrophoresis, rVWF shows a characteristic ladder of bands also known as multimers. In this analysis, rVWF contains as many distinct bands as VWF detectable in normal human plasma or VWF isolated from human plasma but in addition, has a zone with unresolved bands in the ultra-high molecular weight range. Highresolution electrophoresis shows a single band for all multimer levels without any satellite bands, as rVWF has never been exposed to ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) cleavage.
Vonicog alfa should not be used in the treatment of Hemophilia A.[4]
In the UK it is available only via a named patient access program.[7]
Vonicog alfa was approved for medical use in the United States in December 2015, in the European Union in August 2018, and in Australia in April 2020.[3][5][4][8] It was granted orphan drug designations in both the United States and the European Union.[4][1]
Medical uses
Vonicog alfa is indicated in adults with von Willebrand Disease (VWD), when desmopressin (DDAVP) treatment alone is ineffective or not indicated for the
The following side effects may occur during treatment with vonicog alfa: hypersensitivity (allergic) reactions, thromboembolic events (problems due to the formation of blood clots in the blood vessels), development of inhibitors (antibodies) against von Willebrand factor, causing the medicine to stop working and resulting in a loss of bleeding control.[4] The most common side effects with vonicog alfa (which may affect up to 1 in 10 patients) are dizziness, vertigo (a spinning sensation), dysgeusia (taste disturbances), tremor, rapid heartbeat, deep venous thrombosis (blood clot in a deep vein, usually in the leg), hypertension (high blood pressure), hot flush, vomiting, nausea (feeling sick), pruritus (itching), chest discomfort, sensations like numbness, tingling, pins and needles at the site of infusion, and an abnormal reading on the electrocardiogram (ECG).[4]
Singal M, Kouides PA: Recombinant von Willebrand factor: a first-of-its-kind product for von Willebrand disease. Drugs Today (Barc). 2016 Dec;52(12):653-664. doi: 10.1358/dot.2016.52.12.2570978. [PubMed:28276537]
Brown R: Recombinant von Willebrand factor for severe gastrointestinal bleeding unresponsive to other treatments in a patient with type 2A von Willebrand disease: a case report. Blood Coagul Fibrinolysis. 2017 Oct;28(7):570-575. doi: 10.1097/MBC.0000000000000632. [PubMed:28379876]
Gill JC, Castaman G, Windyga J, Kouides P, Ragni M, Leebeek FW, Obermann-Slupetzky O, Chapman M, Fritsch S, Pavlova BG, Presch I, Ewenstein B: Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015 Oct 22;126(17):2038-46. doi: 10.1182/blood-2015-02-629873. Epub 2015 Aug 3. [PubMed:26239086]
Lenting PJ, Christophe OD, Denis CV: von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood. 2015 Mar 26;125(13):2019-28. doi: 10.1182/blood-2014-06-528406. Epub 2015 Feb 23. [PubMed:25712991]
Chung MC, Popova TG, Jorgensen SC, Dong L, Chandhoke V, Bailey CL, Popov SG: Degradation of circulating von Willebrand factor and its regulator ADAMTS13 implicates secreted Bacillus anthracis metalloproteases in anthrax consumptive coagulopathy. J Biol Chem. 2008 Apr 11;283(15):9531-42. doi: 10.1074/jbc.M705871200. Epub 2008 Feb 8. [PubMed:18263586]
Molecular Formula: C13H16N2O2Molecular Weight: 232.28Percent Composition: C 67.22%, H 6.94%, N 12.06%, O 13.78%
Literature References: A hormone of the pineal gland, also produced by extra-pineal tissues, that lightens skin color in amphibians by reversing the darkening effect of MSH, q.v. Melatonin has been postulated as the mediator of photic-induced antigonadotrophic activity in photoperiodic mammals and has also been shown to be involved in thermoregulation in some ectotherms and in affecting locomotor activity rhythms in sparrows. Isoln from the pineal glands of beef cattle: Lerner et al.,J. Am. Chem. Soc.80, 2587 (1958); Wurtman et al.,Science141, 277 (1963). Structure: Lerner et al.,J. Am. Chem. Soc.81, 6084 (1959). Crystal and molecular structure: A. Wakahara, Chem. Lett.1972, 1139. Synthesis from 5-methoxyindole as starting material by two different routes: Szmuszkovicz et al.,J. Org. Chem.25, 857 (1960). Biochemical role of melatonin: Chem. Eng. News45, 40 (May 1, 1967). Pharmacological studies: Barchas et al.,Nature214, 919 (1967). Identification of antigonadal action sites in mouse brain: J. D. Glass, G. R. Lynch, Science214, 821 (1981). Binding studies in human hypothalamus: S. M. Reppert et al.,Science242, 78 (1988). Efficacy in control of estrus in red deer: G. W. Asher, Anim. Reprod. Sci.22, 145 (1990). Reviews: M. K. Vaughn, Int. J. Rev. Physiol.24, 41-95 (1981); D. C.Klein et al.,Life Sci.28, 1975-1986 (1981). Book: Advan. Biosci.vol. 29, N. Birau, W. Schlott, Eds. (Pergamon Press, New York, 1981) 420 pp. Review of etiological role in clinical disease: A. Miles, D. Philbrick, Crit. Rev. Clin. Lab. Sci.25, 231-253 (1987); in psychiatric disorders: eidem,Biol. Psychiatry23, 405-425 (1988).Properties: Pale yellow leaflets from benzene, mp 116-118°. uv max: 223, 278 nm (e 27550, 6300).Melting point: mp 116-118°Absorption maximum: uv max: 223, 278 nm (e 27550, 6300)Therap-Cat-Vet: Control of estrus.
Melatonin is a hormone primarily released by the pineal gland that regulates the sleep–wake cycle.[3][4] As a dietary supplement, it is often used for the short-term treatment of insomnia, such as from jet lag or shift work, and is typically taken by mouth.[5][6][7] Evidence of its benefit for this use, however, is not strong.[8] A 2017 review found that sleep onset occurred six minutes faster with use, but found no change in total time asleep.[6] The melatonin receptor agonist medication ramelteon may work as well as melatonin supplements,[6] at greater cost but with different adverse effects, for some sleep conditions.[9]
Melatonin was discovered in 1958.[3] It is sold over the counter in Canada and the United States;[10][13] in the United Kingdom, it is a prescription-only medication.[7] It is not approved by the US Food and Drug Administration (FDA) for any medical use.[10] In Australia and the European Union, it is indicated for difficulty sleeping in people over the age of 54.[20][11] In the European Union, it is indicated for the treatment of insomnia in children and adolescents.[12] It was approved for medical use in the European Union in 2007.[11]
Chemical Synthesis of Melatonin The methods for the chemical synthesis of melatonin are generally not so complicated and do not involve more than three steps of conversion. Three synthesis reactions of melatonin from primary literatures are shown below;
Reaction 1
In 1958 melatonin was first isolated and characterised by A.B.Lerner. It was know as one of a substituted 5-hydroxyindole derivative in the pineal gland that could lighten pigment cells. It had not been know to exist in biological tissue although it had been isolated as a urinary excretion product in rats after administration of 5-hydroxytryptamine. Melatonin or N-acetyl-5-methoxytryptamine (40 mg) was prepared by reducing 100 mg of 5-methoxyindole-3-acetonitrile with 160 mg of sodium and 2 ml of ethanol. Then the product was acetylated with 4 ml of both glacial acetic acid and acetic anhydride at 100 oC for 1 minute. Purification was achieved by countercerrent distribution and silicic acid chromatography.
Reaction 2
5-Methoxytryptamine hydrochloride (1g, 4.75 mmole) was dissolved in pyridine (10 ml) and acetic anhydride (10 ml) and kept overnight at 20 oC. The solution was poured onto iced, neutralised with dilute hydrochloric acid and extracted with chloroform (2×25 ml). The combined extracts were washed with water, dried in MgSO4 and evaporated to afford a liquid of N,N diacetyltryptamine derivative. The liquid was then poured into water (50 ml) and extracted with chlroform (2×25 ml). The combined organic layers were washed with water (25 ml), dried in MgSO4 and evaporated to dryness. The residual solid crystallised from benzene to afford melatonin 819 mg, 80% yield.
Reaction 3
The more reactive indoles (1a-1d) were alkylated at the 3 position by reaction with nitroethene generated in situ by thermolysis of nitroethyl acetate. The nitroethyl acetate used for this purpose was prepared by acetylation of nitroethanol with acetic anhydride using NaOAc as a catalyst. These conditions constitute a substantial improvement of the overal yield of the reation. Reduction of the nitroethylated indoles (2a-d) by hydrogenation over PtO2, followed by acetylation fo the resluting tryptamines with acetic anhydride-pyridine completed the synthesis of melatonin and its derivatives (4a-d).
Biological Synthesis and Metabolism of Melatonin
The biosynthesis of melatonin (Fig.1) is initiated by the uptake of the essential amino acid tryptophan into pineal parenchymal cells. Tryptophan is the least abundant of essential amino acids in normal diets. It is converted to another amino acid, 5-hydroxytryptophan, through the action of the enzyme tryptopahn hydroxylase and then to 5-hydroxytryptamine (serotonin) by the enzyme aromatic amino acid decarboxylase. Serotonin concentrations are higher in the pineal than in any other organ or in any brain region. They exhibit a striking diurnal rhythm remaining at a maximum level during the daylight hours and falling by more than 80% soon after the onset of darkness as the serotonin is converted to melatonin, 5-hydroxytryptophol and other methoxyindoles. Serotonin’s conversion to melatonin involves two enzymes that are characteristic of the pineal : SNAT (serotonin-N-acetyltransferase) which converts the serotonin to N-acetylserotonin, and HIOMT (hydroxyindole-O-methyltrasferase) which trasfers a methyl group from S-adenosylmethionine to the 5-hydroxyl of the N-acetylserotonin. The activities of both enzymes rise soon after the onset of darkness because of the enhanced release of norepinephrine from sympathetic neurons terminating on the pineal parenchymal cells. Another portion of the serotonin liberated from pineal cells after the onset of darkness is deaminated by the enzyme monoamine oxidase (MAO) and then either oxidized to form 5-hydroxyindole acetic acid or reduced to form 5-hydroxytryptophol (Fig.1). Both of these compounds are also substrates for HIOMT and can thus be converted in the pineal to 5-methoxyindole acetic acid 5-methoxytryptophol (Fig.1). The level of this latter indole, like that of melatonin, rises markedly in the pineal with the onset of darkness. Since 5-methoxytryptophol synthesis does not require the acetylation of serotonin, the nocturnal increase in pineal SNAT activity cannot be the trigger that causes pineal methoxyindole levels to rise. More likely, a single unexplained process- the intraparenchymal release of stored pineal serotonin, which then becomes accessible to both SNAT and MAO. This process ultimately controls the rates at which all three major pineal methoxyindoles are synthesized and generates the nocturnal increases in pineal melatonin and 5-methoxytryptophol. The proportion of available serotonin acetylated at any particular time of day or night depends on the relative activities of pineal SNAT and MAO at that time. The rates of methylation of all three 5-hydroxyindoles formed from pinela serotonin depends on HIOMT activity.Fig.1 Biosynthesis of pineal methoxyindoles from serotonin
Serotonin may be either acetylated to form N-acetylserotonin through the action of the enzyme serotonin-N-acetyltransferase (SNAT), or oxidatively deaminated by monoamine oxidase (MAO) to yield an unstable aldehyde. This compound is then either oxidized to 5-hydroxyindole acetic acid by the enzyme aldehyde dehydrogenase (ADH), or reduced to from 5-hydroxytryptophol by aldehyde reductase (AR). Each of these 5-hydroxyindole derivatives of serotonin is a substrate for hydroxyindole-O-methyltrasferase (HIMOT). The enzymatic trasfer of a methyl group from S-adenosylmethionine to these hydroxyindoles yields melatonin (5-hydroxy-N-acetyltryptamine), 5-methoxyindole acetic acid and 5-methoxytryptophol respectively. Pineal serotonin is synthesized from the essential amino acid tryptophan by 5-hydroxylation folloed by decarboxylation. The first step in ths enzymic sequence is catalysed by tryptophan hydroxylase. The second step is catalysed by aromatic L-amino acid decarboxylase.
Medical uses
In the European Union it is indicated for the treatment of insomnia in children and adolescents aged 2–18 with autism spectrum disorder (ASD) and / or Smith–Magenis syndrome, where sleep hygiene measures have been insufficient[12] and for monotherapy for the short-term treatment of primary insomnia characterized by poor quality of sleep in people who are aged 55 or over.[11]
Sleep disorders
Positions on the benefits of melatonin for insomnia are mixed.[8] An Agency for Healthcare Research and Quality (AHRQ) review from 2015 stated that evidence of benefit in the general population was unclear.[8] A review from 2017, found a modest effect on time until onset of sleep.[3] Another review from 2017 put this decrease at six minutes to sleep onset but found no difference in total sleep time.[6] Melatonin may also be useful in delayed sleep phase syndrome.[3] Melatonin appears to work as well as ramelteon but costs less.[6]
A 2020 Cochrane review found no evidence that melatonin helped sleep problems in people with moderate to severe dementia due to Alzheimer’s disease.[26] A 2019 review found that while melatonin may improve sleep in minimal cognitive impairment, after the onset of Alzheimer’s it has little to no effect.[27] Melatonin may, however, help with sundowning.[28]
Jet lag and shift work
Melatonin is known to reduce jet lag, especially in eastward travel. If the time it is taken is not correct, however, it can instead delay adaption.[29]
Melatonin appears to have limited use against the sleep problems of people who work shift work.[30] Tentative evidence suggests that it increases the length of time people are able to sleep.[30]
Adverse effects
Melatonin appears to cause very few side effects as tested in the short term, up to three months, at low doses.[clarification needed] Two systematic reviews found no adverse effects of exogenous melatonin in several clinical trials and comparative trials found the adverse effects headaches, dizziness, nausea, and drowsiness were reported about equally for both melatonin and placebo.[31][32] Prolonged-release melatonin is safe with long-term use of up to 12 months.[33] Although not recommended for long term use beyond this, low-dose melatonin is generally safer, and a better alternative, than many prescription and over the counter sleep aids if a sleeping medication must be used for an extended period of time. Low-doses of melatonin are usually sufficient to produce a hypnotic effect in most people. Higher doses do not appear to result in a stronger effect, but instead appear to cause drowsiness for a longer period of time.[34]
In those taking warfarin, some evidence suggests there may exist a potentiating drug interaction, increasing the anticoagulant effect of warfarin and the risk of bleeding.[41]
Functions
When eyes receive light from the sun, the pineal gland’s production of melatonin is inhibited and the hormones produced keep the human awake. When the eyes do not receive light, melatonin is produced in the pineal gland and the human becomes tired.
Circadian rhythm
In animals, melatonin plays an important role in the regulation of sleep–wake cycles.[42] Human infants’ melatonin levels become regular in about the third month after birth, with the highest levels measured between midnight and 8:00 am.[43] Human melatonin production decreases as a person ages.[44] Also, as children become teenagers, the nightly schedule of melatonin release is delayed, leading to later sleeping and waking times.[45]
Antioxidant
Melatonin was first reported as a potent antioxidant and free radical scavenger in 1993.[46] In vitro, melatonin acts as a direct scavenger of oxygen radicals and reactive nitrogen species including OH•, O2−•, and NO•.[47][48] In plants, melatonin works with other antioxidants to improve the overall effectiveness of each antioxidant.[48] Melatonin has been proven to be twice as active as vitamin E, believed to be the most effective lipophilic antioxidant.[49] Via signal transduction through melatonin receptors, melatonin promotes the expression of antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, glutathione reductase, and catalase.[50][51]
Melatonin occurs at high concentrations within mitochondrial fluid which greatly exceed the plasma concentration of melatonin.[52][53][54] Due to its capacity for free radical scavenging, indirect effects on the expression of antioxidant enzymes, and its significant concentrations within mitochondria, a number of authors have indicated that melatonin has an important physiological function as a mitochondrial antioxidant.[50][52][53][54][55]
The melatonin metabolites produced via the reaction of melatonin with reactive oxygen species or reactive nitrogen species also react with and reduce free radicals.[51][55] Melatonin metabolites generated from redox reactions include cyclic 3-hydroxymelatonin, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), and N1-acetyl-5-methoxykynuramine (AMK).[51][55]
Immune system
While it is known that melatonin interacts with the immune system,[56][57] the details of those interactions are unclear. An antiinflammatory effect seems to be the most relevant. There have been few trials designed to judge the effectiveness of melatonin in disease treatment. Most existing data are based on small, incomplete trials. Any positive immunological effect is thought to be the result of melatonin acting on high-affinity receptors (MT1 and MT2) expressed in immunocompetent cells. In preclinical studies, melatonin may enhance cytokine production,[58] and by doing this, counteract acquired immunodeficiences. Some studies also suggest that melatonin might be useful fighting infectious disease[59] including viral, such as HIV, and bacterial infections, and potentially in the treatment of cancer.
In bacteria, protists, fungi, and plants, melatonin is synthesized indirectly with tryptophan as an intermediate product of the shikimate pathway. In these cells, synthesis starts with D-erythrose 4-phosphate and phosphoenolpyruvate, and in photosynthetic cells with carbon dioxide. The rest of the synthesising reactions are similar, but with slight variations in the last two enzymes.[62][63]
It has been hypothesized that melatonin is made in the mitochondria and chloroplasts.[64]
Mechanism
Mechanism of melatonin biosynthesis
In order to hydroxylate L-tryptophan, the cofactor tetrahydrobiopterin (THB) must first react with oxygen and the active site iron of tryptophan hydroxylase. This mechanism is not well understood, but two mechanisms have been proposed:
1. A slow transfer of one electron from the THB to O2 could produce a superoxide which could recombine with the THB radical to give 4a-peroxypterin. 4a-peroxypterin could then react with the active site iron (II) to form an iron-peroxypterin intermediate or directly transfer an oxygen atom to the iron.
2. O2 could react with the active site iron (II) first, producing iron (III) superoxide which could then react with the THB to form an iron-peroxypterin intermediate.
Iron (IV) oxide from the iron-peroxypterin intermediate is selectively attacked by a double bond to give a carbocation at the C5 position of the indole ring. A 1,2-shift of the hydrogen and then a loss of one of the two hydrogen atoms on C5 reestablishes aromaticity to furnish 5-hydroxy-L-tryptophan.[65]
A decarboxylase with cofactor pyridoxal phosphate (PLP) removes CO2 from 5-hydroxy-L-tryptophan to produce 5-hydroxytryptamine.[66] PLP forms an imine with the amino acid derivative. The amine on the pyridine is protonated and acts as an electron sink, enabling the breaking of the C-C bond and releasing CO2. Protonation of the amine from tryptophan restores the aromaticity of the pyridine ring and then imine is hydrolyzed to produce 5-hydroxytryptamine and PLP.[67]
It has been proposed that histidine residue His122 of serotonin N-acetyl transferase is the catalytic residue that deprotonates the primary amine of 5-hydroxytryptamine, which allows the lone pair on the amine to attack acetyl-CoA, forming a tetrahedral intermediate. The thiol from coenzyme A serves as a good leaving group when attacked by a general base to give N-acetylserotonin.[68]
N-acetylserotonin is methylated at the hydroxyl position by S-adenosyl methionine (SAM) to produce S-adenosyl homocysteine (SAH) and melatonin.[67][69]
Regulation
In vertebrates, melatonin secretion is regulated by activation of the beta-1 adrenergic receptor by norepinephrine.[70] Norepinephrine elevates the intracellular cAMP concentration via beta-adrenergic receptors and activates the cAMP-dependent protein kinase A (PKA). PKA phosphorylates the penultimate enzyme, the arylalkylamine N-acetyltransferase (AANAT). On exposure to (day)light, noradrenergic stimulation stops and the protein is immediately destroyed by proteasomalproteolysis.[71] Production of melatonin is again started in the evening at the point called the dim-light melatonin onset.
Blue light, principally around 460–480 nm, suppresses melatonin biosynthesis,[72] proportional to the light intensity and length of exposure. Until recent history, humans in temperate climates were exposed to few hours of (blue) daylight in the winter; their fires gave predominantly yellow light.[citation needed] The incandescent light bulb widely used in the 20th century produced relatively little blue light.[73] Light containing only wavelengths greater than 530 nm does not suppress melatonin in bright-light conditions.[74] Wearing glasses that block blue light in the hours before bedtime may decrease melatonin loss. Use of blue-blocking goggles the last hours before bedtime has also been advised for people who need to adjust to an earlier bedtime, as melatonin promotes sleepiness.[75]
When used several hours before sleep according to the phase response curve for melatonin in humans, small amounts (0.3 mg[77]) of melatonin shift the circadian clock earlier, thus promoting earlier sleep onset and morning awakening.[78] Melatonin is rapidly absorbed and distributed, reaching peak plasma concentrations after 60 minutes of administration, and is then eliminated.[61] Melatonin has a half life of 35–50 minutes.[79] In humans, 90% of orally administered exogenous melatonin is cleared in a single passage through the liver, a small amount is excreted in urine, and a small amount is found in saliva.[5] The bioavalibility of melatonin is between 10 and 50%.[61]
Melatonin is metabolized in the liver by cytochrome P450 enzyme CYP1A2 to 6-hydroxymelatonin. Metabolites are conjugated with sulfuric acid or glucuronic acid for excretion in the urine. 5% of melatonin is excreted in the urine as the unchanged drug.[61]
Some of the metabolites formed via the reaction of melatonin with a free radical include cyclic 3-hydroxymelatonin, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), and N1-acetyl-5-methoxykynuramine (AMK).[51][55]
In 1958, dermatology professor Aaron B. Lerner and colleagues at Yale University, in the hope that a substance from the pineal might be useful in treating skin diseases, isolated the hormone from bovine pineal gland extracts and named it melatonin.[85] In the mid-70s Lynch et al. demonstrated that the production of melatonin exhibits a circadian rhythm in human pineal glands.[86]
The discovery that melatonin is an antioxidant was made in 1993.[87] The first patent for its use as a low-dose sleep aid was granted to Richard Wurtman at MIT in 1995.[88] Around the same time, the hormone got a lot of press as a possible treatment for many illnesses.[89]The New England Journal of Medicine editorialized in 2000: “With these recent careful and precise observations in blind persons, the true potential of melatonin is becoming evident, and the importance of the timing of treatment is becoming clear.”[90]
It was approved for medical use in the European Union in 2007.[11]
Other animals
In vertebrates, melatonin is produced in darkness, thus usually at night, by the pineal gland, a small endocrine gland[91] located in the center of the brain but outside the blood–brain barrier. Light/dark information reaches the suprachiasmatic nuclei from retinal photosensitive ganglion cells of the eyes[92][93] rather than the melatonin signal (as was once postulated). Known as “the hormone of darkness”, the onset of melatonin at dusk promotes activity in nocturnal (night-active) animals and sleep in diurnal ones including humans.
Many animals use the variation in duration of melatonin production each day as a seasonal clock.[94] In animals including humans,[95] the profile of melatonin synthesis and secretion is affected by the variable duration of night in summer as compared to winter. The change in duration of secretion thus serves as a biological signal for the organization of daylength-dependent (photoperiodic) seasonal functions such as reproduction, behavior, coat growth, and camouflage coloring in seasonal animals.[95] In seasonal breeders that do not have long gestation periods and that mate during longer daylight hours, the melatonin signal controls the seasonal variation in their sexual physiology, and similar physiological effects can be induced by exogenous melatonin in animals including mynah birds[96] and hamsters.[97] Melatonin can suppress libido by inhibiting secretion of luteinizing hormone and follicle-stimulating hormone from the anterior pituitary gland, especially in mammals that have a breeding season when daylight hours are long. The reproduction of long-day breeders is repressed by melatonin and the reproduction of short-day breeders is stimulated by melatonin.
During the night, melatonin regulates leptin, lowering its levels.
Cetaceans have lost all the genes for melatonin synthesis as well as those for melatonin receptors.[98] This is thought to be related to their unihemispheric sleep pattern (one brain hemisphere at a time). Similar trends have been found in sirenians.[98]
Plants
Until its identification in plants in 1987, melatonin was for decades thought to be primarily an animal neurohormone. When melatonin was identified in coffee extracts in the 1970s, it was believed to be a byproduct of the extraction process. Subsequently, however, melatonin has been found in all plants that have been investigated. It is present in all the different parts of plants, including leaves, stems, roots, fruits, and seeds, in varying proportions.[19][99] Melatonin concentrations differ not only among plant species, but also between varieties of the same species depending on the agronomic growing conditions, varying from picograms to several micrograms per gram.[63][100] Notably high melatonin concentrations have been measured in popular beverages such as coffee, tea, wine, and beer, and crops including corn, rice, wheat, barley, and oats.[19] In some common foods and beverages, including coffee[19] and walnuts,[101] the concentration of melatonin has been estimated or measured to be sufficiently high to raise the blood level of melatonin above daytime baseline values.
Although a role for melatonin as a plant hormone has not been clearly established, its involvement in processes such as growth and photosynthesis is well established. Only limited evidence of endogenous circadian rhythms in melatonin levels has been demonstrated in some plant species and no membrane-bound receptors analogous to those known in animals have been described. Rather, melatonin performs important roles in plants as a growth regulator, as well as environmental stress protector. It is synthesized in plants when they are exposed to both biological stresses, for example, fungal infection, and nonbiological stresses such as extremes of temperature, toxins, increased soil salinity, drought, etc.[63][102][103]
Occurrence
Dietary supplement
Melatonin is categorized by the US Food and Drug Administration (FDA) as a dietary supplement, and is sold over-the-counter in both the US and Canada.[5] FDA regulations applying to medications are not applicable to melatonin,[15] though the FDA has found false claims that it cures cancer.[104] As melatonin may cause harm in combination with certain medications or in the case of certain disorders, a doctor or pharmacist should be consulted before making a decision to take melatonin.[29] In many countries, melatonin is recognized as a neurohormone and it cannot be sold over-the-counter.[105]
Food products
Naturally-occurring melatonin has been reported in foods including tart cherries to about 0.17–13.46 ng/g,[106] bananas and grapes, rice and cereals, herbs, plums,[107] olive oil, wine[108] and beer. When birds ingest melatonin-rich plant feed, such as rice, the melatonin binds to melatonin receptors in their brains.[109] When humans consume foods rich in melatonin, such as banana, pineapple, and orange, the blood levels of melatonin increase significantly.[110]
Beverages and snacks containing melatonin were being sold in grocery stores, convenience stores, and clubs in May 2011.[111] The FDA considered whether these food products could continue to be sold with the label “dietary supplements”. On 13 January 2010, it issued a Warning Letter to Innovative Beverage, creators of several beverages marketed as drinks, stating that melatonin, while legal as a dietary supplement, was not approved as a food additive.[112] A different company selling a melatonin-containing beverage received a warning letter in 2015.[113]
Commercial availability
Immediate-release melatonin is not tightly regulated in countries where it is available as an over-the-counter medication. It is available in doses from less than half a milligram to 5 mg or more. Immediate-release formulations cause blood levels of melatonin to reach their peak in about an hour. The hormone may be administered orally, as capsules, gummies, tablets, or liquids. It is also available for use sublingually, or as transdermal patches.[medical citation needed]
Formerly, melatonin was derived from animal pineal tissue, such as bovine. It is now synthetic, which limits the risk of contamination or the means of transmitting infectious material.[15][114]
Melatonin is the most popular over-the-counter sleep remedy in the US, resulting in sales in excess of US$400 million during 2017.[115]
Research
A bottle of melatonin tablets. Melatonin is available in timed-release and in liquid forms.
Various uses and effects of melatonin have been studied. A 2015 review of studies of melatonin in tinnitus found the quality of evidence low, but not entirely without promise.[116]
Headaches
Tentative evidence shows melatonin may help reduce some types of headaches including cluster and hypnic headaches.[117][118]
Cancer
A 2013 review by the National Cancer Institutes found evidence for use to be inconclusive.[119] A 2005 review of unblinded clinical trials found a reduced rate of death, but that blinded and independently conducted randomized controlled trials are needed.[120]
Protection from radiation
Both animal[121] and human[122][123][124] studies have shown melatonin to protect against radiation-induced cellular damage. Melatonin and its metabolites protect organisms from oxidative stress by scavenging reactive oxygen species which are generated during exposure.[125] Nearly 70% of biological damage caused by ionizing radiation is estimated to be attributable to the creation of free radicals, especially the hydroxyl radical that attacks DNA, proteins, and cellular membranes. Melatonin has been described as a broadly protective, readily available, and orally self-administered antioxidant that is without known, major side effects.[126]
Epilepsy
A 2016 review found no beneficial role of melatonin in reducing seizure frequency or improving quality of life in people with epilepsy.[127]
Secondary dysmenorrhoea
A 2016 review suggested no strong evidence of melatonin compared to placebo for dysmenorrhoea secondary to endometriosis.[128]
Delirium
A 2016 review suggested no clear evidence of melatonin to reduce the incidence of delirium.[129]
Gastroesophageal reflux disease
A 2011 review said melatonin is effective in relieving epigastric pain and heartburn.[130]
Psychiatry
Melatonin might improve sleep in people with autism.[131] Children with autism have abnormal melatonin pathways and below-average physiological levels of melatonin.[132][133] Melatonin supplementation has been shown to improve sleep duration, sleep onset latency, and night-time awakenings.[132][134][135] However, many studies on melatonin and autism rely on self-reported levels of improvement and more rigorous research is needed.
While the packaging of melatonin often warns against use in people under 18 years of age, studies suggest that melatonin is an efficacious and safe treatment for insomnia in people with ADHD, including children. However, larger and longer studies are needed to establish long-term safety and optimal dosing.[136]
Melatonin in comparison to placebo is effective for reducing preoperative anxiety in adults when given as premedication. It may be just as effective as standard treatment with midazolam in reducing preoperative anxiety. Melatonin may also reduce postoperative anxiety (measured 6 hours after surgery) when compared to placebo.[137]
Some supplemental melatonin users report an increase in vivid dreaming. Extremely high doses of melatonin increased REM sleep time and dream activity in people both with and without narcolepsy.[138] Some evidence supports an antidepressant effect.[139]
^ Jump up to:abc Buscemi N, Vandermeer B, Pandya R, Hooton N, Tjosvold L, Hartling L, et al. (November 2004). “Melatonin for treatment of sleep disorders” (PDF). Evidence Report/Technology Assessment No. 108. (Prepared by the University of Alberta Evidence-based Practice Center, Under Contract No. 290-02-0023.) AHRQ Publication No. 05-E002-2. Rockville, MD: Agency for Healthcare Research and Quality. Agency for Healthcare Research and Quality (AHRQ), US Department of Health and Human Services (108): 1–7. doi:10.1037/e439412005-001. PMC4781368. PMID15635761. Retrieved 5 June2013.
^ Jump up to:abcdef Matheson E, Hainer BL (July 2017). “Insomnia: Pharmacologic Therapy”. American Family Physician. 96 (1): 29–35. PMID28671376.
^ Jump up to:abc Brasure M, MacDonald R, Fuchs E, Olson CM, Carlyle M, Diem S, et al. (2015). “Management of Insomnia Disorder[Internet]”. AHRQ Comparative Effectiveness Reviews. 15 (16): EHC027–EF. PMID26844312. Evidence for benzodiazepine hypnotics, melatonin agonists in the general adult population, and most pharmacologic interventions in older adults was generally insufficient
^ Adams, Katie S. (2014). “Melatonin agonists in the management of sleep disorders: A focus on ramelteon and tasimelteon”. Mental Health Clinician. 4 (2): 59–64. doi:10.9740/mhc.n190087. Retrieved 25 October 2020. However, the clinical relevance of this objective and therefore the author’s conclusion that these results support the potential use of ramelteon in circadian rhythm sleep disorders is questionable. … It is unclear whether Takeda Pharmaceuticals will pursue FDA indications for ramelteon for circadian rhythm disorders given these results.
^ Boutin JA, Audinot V, Ferry G, Delagrange P (August 2005). “Molecular tools to study melatonin pathways and actions”. Trends in Pharmacological Sciences. 26 (8): 412–9. doi:10.1016/j.tips.2005.06.006. PMID15992934.
^ Hardeland R (July 2005). “Antioxidative protection by melatonin: multiplicity of mechanisms from radical detoxification to radical avoidance”. Endocrine. 27 (2): 119–30. doi:10.1385/ENDO:27:2:119. PMID16217125.
^“Australian Public Assessment Report for Melatonin” (PDF). Australian Government Department of Health and Ageing Therapeutic Goods Administration. January 2011. pp. 2, 4. Retrieved 9 January 2019. Monotherapy for the short term treatment of primary insomnia characterised by poor quality of sleep in patients who are aged 55 or over.
^ Gao C, Scullin MK, Bliwise DL (2019). “Mild Cognitive Impairment and Dementia”. In Savard J, Ouellet MC (eds.). Handbook of Sleep Disorders in Medical Conditions. Academic Press. pp. 253–276. doi:10.1016/b978-0-12-813014-8.00011-1. ISBN978-0-12-813014-8.
^ Lyseng-Williamson KA (November 2012). “Melatonin prolonged release: in the treatment of insomnia in patients aged ≥55 years”. Drugs & Aging. 29 (11): 911–23. doi:10.1007/s40266-012-0018-z. PMID23044640. S2CID1403262.
^ Database of Abstracts of Reviews of Effects (DARE): Quality-assessed Reviews [Internet]. York (UK): Centre for Reviews and Dissemination (UK); 1995. Optimal dosages for melatonin supplementation therapy in older adults: a systematic review of current literature. 2014.
^ Morera AL, Henry M, de La Varga M (2001). “[Safety in melatonin use]” [Safety in melatonin use]. Actas Espanolas de Psiquiatria (in Spanish). 29 (5): 334–7. PMID11602091.
^ Ardura J, Gutierrez R, Andres J, Agapito T (2003). “Emergence and evolution of the circadian rhythm of melatonin in children”. Hormone Research. 59 (2): 66–72. doi:10.1159/000068571. PMID12589109. S2CID41937922.
^ Poeggeler B, Saarela S, Reiter RJ, Tan DX, Chen LD, Manchester LC, Barlow-Walden LR (November 1994). “Melatonin—a highly potent endogenous radical scavenger and electron donor: new aspects of the oxidation chemistry of this indole accessed in vitro”. Annals of the New York Academy of Sciences. 738 (1): 419–20. Bibcode:1994NYASA.738..419P. doi:10.1111/j.1749-6632.1994.tb21831.x. PMID7832450. S2CID36383425.
^ Pieri C, Marra M, Moroni F, Recchioni R, Marcheselli F (1994). “Melatonin: a peroxyl radical scavenger more effective than vitamin E”. Life Sciences. 55 (15): PL271-6. doi:10.1016/0024-3205(94)00666-0. PMID7934611.
^ Jump up to:abcdefgh Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G, et al. (September 2016). “Update on melatonin receptors: IUPHAR Review 20”. British Journal of Pharmacology. 173 (18): 2702–25. doi:10.1111/bph.13536. PMC4995287. PMID27314810. Hence, one melatonin molecule and its associated metabolites could scavenge a large number of reactive species, and thus, the overall antioxidant capacity of melatonin is believed to be greater than that of other well‐known antioxidants, such as vitamin C and vitamin E, under in vitro or in vivo conditions (Gitto et al., 2001; Sharma and Haldar, 2006; Ortiz et al., 2013).
^ Jump up to:abc Reiter RJ, Rosales-Corral S, Tan DX, Jou MJ, Galano A, Xu B (November 2017). “Melatonin as a mitochondria-targeted antioxidant: one of evolution’s best ideas”. Cellular and Molecular Life Sciences. 74 (21): 3863–3881. doi:10.1007/s00018-017-2609-7. PMID28864909. S2CID23820389. melatonin is specifically targeted to the mitochondria where it seems to function as an apex antioxidant … The measurement of the subcellular distribution of melatonin has shown that the concentration of this indole in the mitochondria greatly exceeds that in the blood.
^ Jump up to:abc Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L (October 2016). “Melatonin as an antioxidant: under promises but over delivers”. Journal of Pineal Research. 61 (3): 253–78. doi:10.1111/jpi.12360. PMID27500468. S2CID35435683. There is credible evidence to suggest that melatonin should be classified as a mitochondria-targeted antioxidant.
^ Jump up to:abc Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, et al. (November 2015). “Melatonin: an ancient molecule that makes oxygen metabolically tolerable”. Journal of Pineal Research. 59 (4): 403–19. doi:10.1111/jpi.12267. PMID26272235. S2CID24373303. While originally thought to be produced exclusively in and secreted from the vertebrate pineal gland [53], it is now known that the indole is present in many, perhaps all, vertebrate organs [54] and in organs of all plants that have been investigated [48, 55, 56]. That melatonin is not relegated solely to the pineal gland is also emphasized by the reports that it is present in invertebrates [57–59], which lack a pineal gland and some of which consist of only a single cell.
^ Carrillo-Vico A, Guerrero JM, Lardone PJ, Reiter RJ (July 2005). “A review of the multiple actions of melatonin on the immune system”. Endocrine. 27 (2): 189–200. doi:10.1385/ENDO:27:2:189. PMID16217132. S2CID21133107.
^ Arushanian EB, Beĭer EV (2002). “[Immunotropic properties of pineal melatonin]”. Eksperimental’naia i Klinicheskaia Farmakologiia (in Russian). 65 (5): 73–80. PMID12596522.
^ Carrillo-Vico A, Reiter RJ, Lardone PJ, Herrera JL, Fernández-Montesinos R, Guerrero JM, Pozo D (May 2006). “The modulatory role of melatonin on immune responsiveness”. Current Opinion in Investigational Drugs. 7 (5): 423–31. PMID16729718.
^ Tan DX, Manchester LC, Liu X, Rosales-Corral SA, Acuna-Castroviejo D, Reiter RJ (March 2013). “Mitochondria and chloroplasts as the original sites of melatonin synthesis: a hypothesis related to melatonin’s primary function and evolution in eukaryotes”. Journal of Pineal Research. 54 (2): 127–38. doi:10.1111/jpi.12026. PMID23137057. S2CID206140413.
^ Sumi-Ichinose C, Ichinose H, Takahashi E, Hori T, Nagatsu T (March 1992). “Molecular cloning of genomic DNA and chromosomal assignment of the gene for human aromatic L-amino acid decarboxylase, the enzyme for catecholamine and serotonin biosynthesis”. Biochemistry. 31 (8): 2229–38. doi:10.1021/bi00123a004. PMID1540578.
^ Hickman AB, Klein DC, Dyda F (January 1999). “Melatonin biosynthesis: the structure of serotonin N-acetyltransferase at 2.5 A resolution suggests a catalytic mechanism”. Molecular Cell. 3 (1): 23–32. doi:10.1016/S1097-2765(00)80171-9. PMID10024876.
^ Lerner AB, Case JD, Takahashi Y (July 1960). “Isolation of melatonin and 5-methoxyindole-3-acetic acid from bovine pineal glands”. The Journal of Biological Chemistry. 235: 1992–7. PMID14415935.
^ Poeggeler B, Reiter RJ, Tan DX, Chen LD, Manchester LC (May 1993). “Melatonin, hydroxyl radical-mediated oxidative damage, and aging: a hypothesis”. Journal of Pineal Research. 14 (4): 151–68. doi:10.1111/j.1600-079X.1993.tb00498.x. PMID8102180. S2CID23460208.
^US patent 5449683, Wurtman RJ, “Methods of inducing sleep using melatonin”, issued 12 September 1995, assigned to Massachusetts Institute of Technology
^ Arendt J (August 2005). “Melatonin: characteristics, concerns, and prospects”. Journal of Biological Rhythms. 20 (4): 291–303. doi:10.1177/0748730405277492. PMID16077149. S2CID19011222. There is very little evidence in the short term for toxicity or undesirable effects in humans. The extensive promotion of the miraculous powers of melatonin in the recent past did a disservice to acceptance of its genuine benefits.
^ Reiter RJ (May 1991). “Pineal melatonin: cell biology of its synthesis and of its physiological interactions”. Endocrine Reviews. 12 (2): 151–80. doi:10.1210/edrv-12-2-151. PMID1649044. S2CID3219721.
^ Richardson GS (2005). “The human circadian system in normal and disordered sleep”. The Journal of Clinical Psychiatry. 66 Suppl 9: 3–9, quiz 42–3. PMID16336035.
^ Perreau-Lenz S, Pévet P, Buijs RM, Kalsbeek A (January 2004). “The biological clock: the bodyguard of temporal homeostasis”. Chronobiology International. 21 (1): 1–25. doi:10.1081/CBI-120027984. PMID15129821. S2CID42725506.
^ Jump up to:ab Arendt J, Skene DJ (February 2005). “Melatonin as a chronobiotic”. Sleep Medicine Reviews. 9 (1): 25–39. doi:10.1016/j.smrv.2004.05.002. PMID15649736. Exogenous melatonin has acute sleepiness-inducing and temperature-lowering effects during ‘biological daytime’, and when suitably timed (it is most effective around dusk and dawn), it will shift the phase of the human circadian clock (sleep, endogenous melatonin, core body temperature, cortisol) to earlier (advance phase shift) or later (delay phase shift) times.
^ Chaturvedi CM (1984). “Effect of Melatonin on the Adrenl and Gonad of the Common Mynah Acridtheres tristis”. Australian Journal of Zoology. 32 (6): 803–09. doi:10.1071/ZO9840803.
^ Chen HJ (July 1981). “Spontaneous and melatonin-induced testicular regression in male golden hamsters: augmented sensitivity of the old male to melatonin inhibition”. Neuroendocrinology. 33 (1): 43–6. doi:10.1159/000123198. PMID7254478.
^ Paredes SD, Korkmaz A, Manchester LC, Tan DX, Reiter RJ (1 January 2009). “Phytomelatonin: a review”. Journal of Experimental Botany. 60 (1): 57–69. doi:10.1093/jxb/ern284. PMID19033551. S2CID15738948.
^ Bonnefont-Rousselot D, Collin F (November 2010). “Melatonin: action as antioxidant and potential applications in human disease and aging”. Toxicology. 278 (1): 55–67. doi:10.1016/j.tox.2010.04.008. PMID20417677.
^ Reiter RJ, Manchester LC, Tan DX (September 2005). “Melatonin in walnuts: influence on levels of melatonin and total antioxidant capacity of blood”. Nutrition. 21 (9): 920–4. doi:10.1016/j.nut.2005.02.005. PMID15979282.
^ Burkhardt S, Tan DX, Manchester LC, Hardeland R, Reiter RJ (October 2001). “Detection and quantification of the antioxidant melatonin in Montmorency and Balaton tart cherries (Prunus cerasus)”. Journal of Agricultural and Food Chemistry. 49 (10): 4898–902. doi:10.1021/jf010321. PMID11600041.
^ Lamont KT, Somers S, Lacerda L, Opie LH, Lecour S (May 2011). “Is red wine a SAFE sip away from cardioprotection? Mechanisms involved in resveratrol- and melatonin-induced cardioprotection”. Journal of Pineal Research. 50 (4): 374–80. doi:10.1111/j.1600-079X.2010.00853.x. PMID21342247. S2CID8034935.
^ Hattori A, Migitaka H, Iigo M, Itoh M, Yamamoto K, Ohtani-Kaneko R, et al. (March 1995). “Identification of melatonin in plants and its effects on plasma melatonin levels and binding to melatonin receptors in vertebrates”. Biochemistry and Molecular Biology International. 35(3): 627–34. PMID7773197.
^ Sae-Teaw M, Johns J, Johns NP, Subongkot S (August 2013). “Serum melatonin levels and antioxidant capacities after consumption of pineapple, orange, or banana by healthy male volunteers”. Journal of Pineal Research. 55 (1): 58–64. doi:10.1111/jpi.12025. PMID23137025. S2CID979886.
^ Rodriguez RR (13 January 2010). “Warning Letter”. Inspections, Compliance, Enforcement, and Criminal Investigations. U.S. Food and Drug Administration. Archived from the original on 12 January 2017.
^Bebida Beverage Company U.S. Food and Drug Administration 4 March 2015 (Accessed 8 December 2017)
^ Miroddi M, Bruno R, Galletti F, Calapai F, Navarra M, Gangemi S, Calapai G (March 2015). “Clinical pharmacology of melatonin in the treatment of tinnitus: a review”. European Journal of Clinical Pharmacology. 71 (3): 263–70. doi:10.1007/s00228-015-1805-3. PMID25597877. S2CID16466238.
^ Peres MF, Masruha MR, Zukerman E, Moreira-Filho CA, Cavalheiro EA (April 2006). “Potential therapeutic use of melatonin in migraine and other headache disorders”. Expert Opinion on Investigational Drugs. 15 (4): 367–75. doi:10.1517/13543784.15.4.367. PMID16548786. S2CID28114683.
^ Mills E, Wu P, Seely D, Guyatt G (November 2005). “Melatonin in the treatment of cancer: a systematic review of randomized controlled trials and meta-analysis”. Journal of Pineal Research. 39 (4): 360–6. doi:10.1111/j.1600-079X.2005.00258.x. PMID16207291. S2CID22225091.
^ Meltz ML, Reiter RJ, Herman TS, Kumar KS (March 1999). “Melatonin and protection from whole-body irradiation: survival studies in mice”. Mutation Research. 425 (1): 21–7. doi:10.1016/S0027-5107(98)00246-2. PMID10082913.
^ Reiter RJ, Herman TS, Meltz ML (December 1996). “Melatonin and radioprotection from genetic damage: in vivo/in vitro studies with human volunteers”. Mutation Research. 371 (3–4): 221–8. doi:10.1016/S0165-1218(96)90110-X. PMID9008723.
^ Reiter RJ, Herman TS, Meltz ML (February 1998). “Melatonin reduces gamma radiation-induced primary DNA damage in human blood lymphocytes”. Mutation Research. 397 (2): 203–8. doi:10.1016/S0027-5107(97)00211-X. PMID9541644.
^ Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ (January 2007). “One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species?”. Journal of Pineal Research. 42 (1): 28–42. doi:10.1111/j.1600-079X.2006.00407.x. PMID17198536. S2CID40005308.
^ Giannotti F, Cortesi F, Cerquiglini A, Bernabei P (August 2006). “An open-label study of controlled-release melatonin in treatment of sleep disorders in children with autism”. Journal of Autism and Developmental Disorders. 36 (6): 741–52. doi:10.1007/s10803-006-0116-z. PMID16897403. S2CID19724241.
^ Bendz LM, Scates AC (January 2010). “Melatonin treatment for insomnia in pediatric patients with attention-deficit/hyperactivity disorder”. The Annals of Pharmacotherapy. 44(1): 185–91. doi:10.1345/aph.1M365. PMID20028959. S2CID207263711.
The most common side effects include neutropenia (low levels of neutrophils, a type of white blood cell), infusion reactions, pneumonia (infection of the lungs), upper respiratory tract infection (such as nose and throat infections), diarrhoea and bronchitis (inflammation of the airways in the lungs).[3]
In the European Union it is indicated, in combination with pomalidomide and dexamethasone, for the treatment of adults with relapsed and refractory multiple myeloma (MM) who have received at least two prior therapies including lenalidomide and a proteasome inhibitor (PI) and have demonstrated disease progression on the last therapy.[3]
Researchers started a Phase I study with isatuximab in combination with pomalidomide and dexamethasone for the treatment of patients with multiple myeloma (MM). The results during the Phase I trial showed that 26 out of the 45 patients discontinued the treatment due to progression of the disease. The patients had already been heavily pretreated. The latter lead to a manageable safety profile where the dose of isatuximab in combination with pomalidomide and dexamethasone would be capped to the maximum of 10 mg/kg weekly every two weeks for future studies.[12]
Based on the remarkable findings during the Phase I trial, a Phase II trial was launched where researchers investigated isatuximab as a single agent in patients with MM. The heavily pretreated patients reacted well to the single administration of isatuximab during Phase II of the trial.[13]
A Phase III combination trial for plasma cell myeloma is comparing pomalidomide and dexamethasone with and without isatuximab is in progress with an estimated completion date of 2021.[medical citation needed]
Additionally, two Phase III trials were added in 2017. The first trial highlights whether there is an added value in the combination of isatuximab with bortezomib, lenalidomide and dexamethasone. The latter will be tested in patients with newly diagnosed MM who are not qualified for a transplant (IMROZ trial). The second trial evaluates the combinations of isatuximab with carfilzomib and dexamethasone compared to carfilzomib with dexamethasone. The second trial was designed for patients who were previously treated with one to three prior lines (IKEMA). There is currently[when?] no treatment for MM, however promising improvements have been made and the study is still ongoing.[14][15]
In March 2020, it was approved for medical use in the United States.[8][9][10]
The U.S. Food and Drug Administration (FDA) approved isatuximab-irfc in March 2020, based on evidence from a clinical trial (NCT02990338) of 307 subjects with previously treated multiple myeloma.[10] The trial was conducted at 102 sites in Europe, North America, Asia, Australia and New Zealand.[10]
The trial evaluated the efficacy and side effects of isatuximab-irfc in subjects with previously treated multiple myeloma.[10] Subjects were randomly assigned to receive either isatuximab-irfc (in combination with pomalidomide and low-dose dexamethasone) or active comparator (pomalidomide and low-dose dexamethasone).[10] Treatment was administered in both groups in 28-day cycles until disease progression or unacceptable toxicity.[10] Both subjects and health care providers knew which treatment was given.[10] The trial measured the time patients lived without the cancer growing (progression-free survival or PFS).[10]
It was approved for medical use in the European Union in May 2020.[3]
Structure and reactivity
The structure of isatuximab consists of two identical immunoglobulin kappa light chains and also two equal immunoglobulin gamma heavy chains. Chemically, isatuximab is similar to the structure and reactivity of daratumumab, hence both drugs show the same CD38 targeting. However, isatuximab shows a more potent inhibition of its ectozyme function. The latter gives potential for some non-cross reactivity. Isatuximab shows action of an allosteric antagonist with the inhibition of the CD38 enzymatic activity. Additionally, isatuximab shows potential where it can induce apoptosis without cross linking.[16] Lastly, Isatuximab reveals direct killing activity when a larger increase in apoptosis is detected in CD38 expressing cancer cells. Furthermore, isatuximab demonstrated a dose dependent inhibition of CD38 enzymatic activity. However, daratumumab with the same experimental conditions shows a more limited inhibition without a dose response.[17]
Reactions
Isatuximab binds uniquely to an epitope on the CD38 receptor and is the only CD38 antibody which can start apoptosis directly.[18] Isatuximab binds to a different CD38 epitopeamino-acid sequence than does the anti-CD38 monoclonal antibody daratumumab.[19] The binding with the CD38 receptor is mainly via the gamma heavy chains and are more potent than other CD38 antibodies such as daratumumab which can inhibit the enzymatic activity of CD38. Moreover, isatuximab inhibits the hydrolase activity of CD38.[medical citation needed]
The antibodies show signs of improving antitumor immunity by eliminating regulatory T cells, B cells and myeloid-derived suppressor cells. The difference in binding between isatuximab and daratumumab is in the recognition of the different amino acid groups. Isatuximab identifies 23 amino acids of CD38 to the contrary with daratumumab who has 27. The residue of Glu233 has a flexible sidechain and faces the N-terminal of Asp1 residue in the isatuximab light chain. The latter light chain of isatuximab is also flexible which makes the interaction between CD38/Glu233 and the Asp1 weaker than the other interactions between CD38 and isatuximab. The caspase-dependent apoptotic pathway and the lysosomal mediated cell death pathway in MM cells is induced by isatuximab. The MM cell death follows the downstream reactions of the lysosomal activation. The latter also activates the production of reactive oxygen species.[20]
Available forms
Isatuximab or isatuximab-irfc is available as a drug in an intravenous infusion form. Injection doses are 100 mg/5 mL (20 mg/mL) solution in single-dose vial or 500 mg/25 mL (20 mg/mL) solution in single-dose vial.[4]
Mechanism of action
Cancer of the blood that is distinguished by an overproduction of malignant plasma cells in the bone marrow is called multiple myeloma. The myeloma cells are marked with uniformed overexpression of CD38 surface glycoproteins. Although these proteins are also expressed on other myeloid and lymphoid cells, the extent is relatively minor compared to myeloma cells. The fact that CD38 glycoproteins carry out various important cellular functions, and that they are plentiful on the surface of myeloma cells, has made them an appealing target for multiple myeloma treatment.[21] CD38 was first described as an activation marker, but later the molecule displayed functions in adhesion to endothelial CD31 proteins, e.g. as an aiding component of the synapse complex, as well as an ectoenzyme implicated in the metabolism of extracellular NAD+ and cytoplasmic NADP. The tumour cells can evade the immune system, possibly due to adenosine, an immunosuppressive molecule that arises as a product of the ectoenzymatic activity of CD38.[22]
Isatuximab-irfc is an IgG1-derived monoclonal antibody that selectively binds to the CD38 that exists on the exterior of hematopoietic and multiple myeloma cells (as well as other tumor cells). This drug induces apoptosis of tumor cells and activates immune effector mechanisms such as complement dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cell-mediated cytotoxicity (ADCC). Isatuximab-irfc is able to stimulate natural killer (NK) cells in the absence of CD38-positive target tumor cells and blocks CD38-positive T-regulatory cells.[4] Furthermore, the NADase activity of CD38 is adjusted by isatuximab, similarly to other CD38 antibodies. Contrarily to daratumumab however, isatuximab can incite apoptosis directly without cross-linking, and in its binding epitope.[23] According to the FDA, isatuximab-irfc alone has reduced ADCC and direct tumor cell killing activity in vitro in comparison to when it is combined with pomalidomide. As well as increased anti-tumor activity as opposed to isatuximab-irfc or pomalidomide only in a human multiple myeloma xenograft model.[4]
Metabolism and toxicity
Metabolism
Isatuximab-irfc is likely to be metabolized through catabolic pathways into smaller peptides. When isatuximab is at a constant state it is expected that the ≥99% elimination will occur approximately two months after the last dose was administered. The clearance percentage diminished when the dosages were increased over time, as well as when multiple doses were administered. However, the elimination of isatuximab-irfc did not differ when applied as a single agent or as a combination therapy.[4]
Toxicity
A dose-limiting toxicity (DLT) has characterized been characterized as the development of any of the following: grade ≥ 3 non-hematologic toxicity; grade 4 neutropenia or grade 4 thrombocytopenia lasting more than 5 days; grade ≥ 2 allergic reactions or hypersensitivity (i.e., infusion reactions); or any other toxicity considered to be dose-limiting by the investigators or sponsor. Grade ≤ 2 infusion reactions were excluded from the DLT definition, because, with suitable care, patients that suffered a grade 2 infusion reaction prior to completion of the infusion were able to finalize isatuximab administration.[23]
There is no recommended reduced dose of isatuximab-irfc. In the eventuality of hematological toxicity it may be necessary to delay administration so that the blood count may be recovered.[4] Although there is no counteracting agent for isatuximab, clinical experience with overdoses is seemingly nonexistent as well. Overdose symptoms will probably be in line with the side effects attached to isatuximab. Therefore, infusion reactions, gastrointestinal disturbances and an elevated risk of infections may occur. It is necessary to carefully monitor the patient in case of an overdose and to employ clinically indicated symptomatic and supportive procedures.[21]
No studies have been conducted with isatuximab concerning carcinogenicity, genotoxicity or fertility.[4]
Pregnancy
When given to pregnant women isatuximab-irfc can cause fetal injury, due to the mechanism of action. It can precipitate depletion of immune cells as well as decreased bone density in the fetus. Pregnant women are therefore notified of the potential risks to a fetus, and women that are able to reproduce are advised to use effective contraceptives during treatment and at least five months subsequent to the last dose of isatuximab-irfc.
Furthermore, it is not recommended to combine isatuximab-irfc with pomalidomide in women that are carrying a child, because pomalidomide may cause birth defects and death of the unborn child.[4]
Indications
Isatuximab is indicated as a CD38-directed cytolytic antibody. By inhibiting the enzymatic activity of CD38.
The binding of isatuximab to CD38 on multiple myeloma (MM) cells leads to a trigger to several mechanisms leading to direct apoptosis of target cancer cells. The triggered pathways are the caspase-dependent apoptotic and the lysosome-mediated cell death pathway in MM cells.[24]
It is used in a combination with dexamethasone and pomalidomide. The drug is thus to treat patients with multiple myeloma. Restrictions for the use of isatuximab is that the patients have to be adults who have at least received two previous treatments with lenalidomide and a proteasome inhibitor.[4]
Isatuximab is currently[when?] also being tested in a Phase II trial as a monotherapy against refractory/recurrent systemic light-chain amyloidosis.[24]
Efficacy and side effects
Efficacy
A Phase III study of patients with refractory and relapsed MM, who were resistant to lenalidomide and a proteasome inhibitor, and could not have received daratumumab, another anti-CD38 monoclonal antibody was published in 2019 (ICARIA-MM). The addition of isatuximab to pomalidomide and dexamethasone improved progression free survival to 11.5 months compared to 6.5 months, with an overall response rate of 63%.[25]
Side effects
Adverse reactions to isatuximab-irfc may include neutropenia, infusion-related reactions and/or secondary primary malignancies.[4] Of these three the most commonly occurring ones are the infusion-related reactions.[24] Examples of the most frequent symptoms of infusion-related reactions are dyspnea, cough, chills, and nausea, while the severest signs and symptoms included hypertension and dyspnea.[4]
Effects on animals
The activity of isatuximab has been researched in mouse tumor models. It has been proven that isatuximab leads to antitumor activity in MM cells. Furthermore, the combination of isatuximab and pomalidomide will lead to an extra enhanced antitumor activity in MM cells. Thus, pomalidomide in vivo and in vitro leads to an increase of the activity of isatuximab.[24]
Animal studies in reproduction toxicity have not yet been carried out. So, the risks of birth defects and miscarriage risks are unknown.[4]
^ Orlowski RZ, Goldschmidt H, Cavo M, Martin TG, Paux G, Oprea C, Facon T (20 May 2018). “Phase III (IMROZ) study design: Isatuximab plus bortezomib (V), lenalidomide (R), and dexamethasone (d) vs VRd in transplant-ineligible patients (pts) with newly diagnosed multiple myeloma (NDMM)”. Journal of Clinical Oncology. 36 (15_suppl): TPS8055. doi:10.1200/JCO.2018.36.15_suppl.TPS8055.
“Isatuximab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02990338 for “Multinational Clinical Study Comparing Isatuximab, Pomalidomide, and Dexamethasone to Pomalidomide and Dexamethasone in Refractory or Relapsed and Refractory Multiple Myeloma Patients (ICARIA-MM)” at ClinicalTrials.gov
With the development of atomic science, radiation therapy such as cobalt hexahydrate, linear accelerator, and electron beam has become one of the main methods of cancer treatment. However, traditional photon or electron therapy is limited by the physical conditions of the radiation itself. While killing the tumor cells, it also causes damage to a large number of normal tissues on the beam path. In addition, due to the sensitivity of tumor cells to radiation, traditional radiation therapy For the more radiation-resistant malignant tumors (such as: glioblastoma multiforme, melanoma), the treatment effect is often poor.
In order to reduce the radiation damage of normal tissues around the tumor, the concept of target treatment in chemotherapy has been applied to radiation therapy; and for tumor cells with high radiation resistance, it is currently actively developing with high relative biological effects (relative Biological effectiveness, RBE) radiation sources, such as proton therapy, heavy particle therapy, neutron capture therapy. Among them, neutron capture therapy combines the above two concepts, such as boron neutron capture therapy, by the specific agglomeration of boron-containing drugs in tumor cells, combined with precise neutron beam regulation, providing better radiation than traditional radiation. Cancer treatment options.
Boron Neutron Capture Therapy (BNCT) is a high-capture cross-section of thermal neutrons using boron-containing ( 10 B) drugs, with 10 B(n,α) 7 Li neutron capture and nuclear splitting reactions. Two heavy charged particles of 4 He and 7 Li are produced. The average energy of the two charged particles is about 2.33 MeV, which has high linear energy transfer (LET) and short range characteristics. The linear energy transfer and range of α particles are 150 keV/μm and 8 μm, respectively, while the 7 Li heavy particles are For 175 keV/μm, 5 μm, the total range of the two particles is equivalent to a cell size, so the radiation damage caused to the organism can be limited to the cell level, when the boron-containing drug is selectively aggregated in the tumor cells, with appropriate The sub-radiation source can achieve the purpose of locally killing tumor cells without causing too much damage to normal tissues.
Since the effectiveness of boron neutron capture therapy depends on the concentration of boron-containing drugs in the tumor cell position and the number of thermal neutrons, it is also called binary cancer therapy; thus, in addition to the development of neutron sources, The development of boron-containing drugs plays an important role in the study of boron neutron capture therapy.
4-( 10 B)dihydroxyboryl-L-phenylalanine (4-( 10 B)borono-L-phenylalanine, L- 10 BPA) is currently known to be able to utilize boron neutron capture therapy (boron neutron capture therapy) , BNCT) An important boron-containing drug for the treatment of cancer.
Therefore, various synthetic methods of L-BPA have been developed. As shown in the following formula (A), the prior art L-BPA synthesis method includes two methods of forming a bond (a) and a bond (b):
Among them, the method for synthesizing L-BPA by forming the bond (a) is to try to introduce a substituent containing a dihydroxylboryl group or a borono group into the skeleton of the phenylalanine, thereby the pair of the amide substituent. The position forms a carbon-boron bond to produce L-BPA.
J. Org. Chem. 1998, 63, 8019 discloses a method for the cross-coupling reaction of (S)-4-iodophenylalanine with a diboron compound by palladium-catalyzed amine end treatment. Amine-protected (S)-4-iodophenylalanine (eg (S)-N-tert-butoxycarbonyl-4-iodophenylalanine ((S)-N-Boc-4-) Iodophenylalanine)) is prepared by cross-coupling with a diboron compound such as bis(pinacolato diboron) to give (S)-N-tert-butoxycarbonyl-4-pentanoylboryl phenylalanine The amine-terminated (S)-4-boranyl ester phenylalanine of the acid ((S)-N-Boc-4-pinacolatoborono phenylalanine); afterwards, the protecting group on the amine end and the boronic end are removed. The above substituents complete the preparation of L-BPA.
However, since the selected 10 B-doped divaleryl diboron is not a commercially available compound, this method requires additional pretreatment of the preparation of the borating agent, resulting in a high process complexity and a long time consuming process. It is impossible to prepare a high yield of L-BPA. In addition, the carboxylic acid group of the protected (S)-4-iodophenylalanine at the amine end needs to be protected by a substituent to form a benzyl ester group to increase the process yield to 88%; however, The preparation of L-BPA in this manner also requires an additional step of deprotecting the carboxylic acid group, which in turn increases the process complexity of L-BPA.
Accordingly, the method provided in this document not only involves pre-treatment of the preparation of the borating agent, but also requires a large amount of process time and synthesis steps to complete the steps of protecting and deprotecting the carboxylic acid group, and is not advantageous as an industry. The main method of synthesizing L-BPA.
On the other hand, a method for synthesizing L-BPA by forming a bond (b) is a coupling reaction of an amino acid with a boron-containing benzyl fragment or a boron-containing benzaldehyde fragment. To synthesize L-BPA. Biosci. Biotech. Biochem. 1996, 60, 683 discloses an enantioselective synthesis of L-BPA which gives the hands of a cyclic ethers of boronic acid and L-proline The chiral derivatives from L-valine are subjected to a coupling reaction to produce L-BPA. However, this method requires the formation of a cyclic ether compound of boric acid from 4-boronobenzylbromide, followed by a coupling reaction with a chiral derivative of L-proline, and in the latter stage. The amino acid undergoes an undesired racemization in the synthesis step, so that the method requires an enzymatic resolution step to reduce the yield to obtain L-BPA having a certain optical purity.
Accordingly, the method provided in the literature still includes the steps of pretreatment of the preparation of the borating agent and post-treatment of the enzymatic resolution, so that the process involved in the method is complicated and takes a long time, and cannot be obtained. High yield of L-BPA.
In addition, L- 10 BPA (4-( 10 B)borono-L-phenylalanine, 4-( 10 B)dihydroxyboryl-L-phenylalanine) containing 10 boron is currently known to accumulate in tumor cells. The key factor is to use the thermal neutron beam to irradiate the boron element accumulated in the tumor cells to kill the tumor cells by capturing the high-energy particles generated by the reaction, thereby achieving the purpose of treating cancer. Therefore, 10 boron can promote the treatment of L- 10 BPA by boron neutron capture treatment.
However, the boron element present in nature contains about 19.9% of 10 boron and about 80.1% of 11 boron. Therefore, many researchers are still actively developing methods that can be applied to the synthesis of L-BPA, especially for the synthesis of 10- boron-rich L-BPA.
J.Org.Chem.1998,63,8019 additionally provides a method of synthesizing 10 boronated agents, since the method involves multiple steps, it is easy to greatly reduce the boron content of 1010 boron enriched material in the manufacturing process. Therefore, the method provided in this document is not suitable for the synthesis of 10- boron-rich L-BPA.
Another example is the Biosci.Biotech.Biochem.1996,60,683, before the enzymatic resolution step is not performed, the method provided by the articles could not be obtained with a certain L-BPA optical purity; 10 and the method for preparing boronated agents when also relates to multi-step, resulting in conversion of boron-rich material 10 occurs during the manufacturing process. Therefore, the method provided in this document is also not suitable for the synthesis of 10- boron-rich L-BPA.
Furthermore, Bull. Chem. Soc. Jpn. 2000, 73, 231 discloses the use of palladium to catalyze 4-iodo-L-phenylalanine with 4,4,5,5-tetramethyl-1,3,2 A method in which a dioxonium pentoxide (common name: pinacolborane) is subjected to a coupling reaction. However, this document does not mention how to prepare articles 10 boron enriched L-BPA using this method, and 4,4,5,5-tetramethyl-1,3,2-dioxaborolane not a commercial 10 The compounds available in the literature are not suitable for the synthesis of 10- boron-rich L-BPA.
In addition, Synlett. 1996, 167 discloses a method for coupling a iodophenylborate with a zinc derivative of L-serine zinc derivatives, which involves first preparing phenyl iodoborate. The ester and the preparation of a zinc derivative of L-type serine acid, etc., result in a lower yield of the produced L-BPA. In addition, since the 10- boron-rich triiodide 10 boron and 1,3-diphenylpropane-1,3-diol selected for this method are not commercially available compounds, the methods provided in this document are also provided. Still not suitable for the synthesis of 10- boron-rich L-BPA.
SYN
Repub. Korean Kongkae Taeho Kongbo, 2018060319,
PAPER
Research and Development in Neutron Capture Therapy, Proceedings of the International Congress on Neutron Capture Therapy, 10th, Essen, Germany, Sept. 8-13, 2002 (2002), 1-8.
PAPER
European Journal of Pharmaceutical Sciences (2003), 18(2), 155-163
Before preparing (S)-N-tert-butoxycarbonyl-4-dihydroxyborylphenylalanine from (S)-N-tert-butoxycarbonyl-4-iodophenylalanine, it is necessary to reveal Process for preparing (S)-N-tert-butoxycarbonyl-4-iodophenylalanine by using (S)-4-iodophenylalanine as a starting material and a process for preparing 10 tributyl borate with 10 boric acid.
1. Preparation of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine from (S)-4-iodophenylalanine
Please refer to the following reaction formula I, which is (S)-4-iodophenylalanine in a solvent of 1,4-dioxane (1,4-dioxane) and water (H 2 O) with hydrogen peroxide. Sodium (NaOH) and di-tert-butyl dicarbonate (Boc 2 O) are reacted to obtain a chemical reaction formula of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine.
In the preparation process, two reaction vessels were selected for the reaction.
The specific operation process is as follows:
1. Set up a reaction using a 3L three-neck bottle.
2. (S)-4-iodo-L-phenylalanine (200.00 g, 687.10 mmol, 1.00 eq) was added to the reaction system.
3. Add 1,4-dioxane (1.00 L) and water (1.00 L) to the reaction system, respectively.
4. Sodium hydroxide (68.71 g, 1.72 mol, 2.50 eq) was added to the reaction system, the solution gradually became clear, and the temperature rose slightly to 19 °C.
5. When the system is cooled to 0-10 ° C, di-tert-butyl dicarbonate (254.93 g, 1.17 mol, 268.35 mL, 1.70 eq) is added to the reaction system, and the temperature of the reaction system is naturally raised to 10 to 30 ° C and Stir at room temperature (about 30 ° C) for 8 hours.
6. The reaction was detected using high performance liquid chromatography (HPLC) until the starting of the reaction.
7. The temperature of the control system is less than 40 ° C, and the 1,4-dioxane in the reaction solution is concentrated.
8. The reaction system was lowered to room temperature (about 25 ° C), 100 mL of water was added, and the pH was adjusted to 1.8-2 with hydrochloric acid (2M (ie, molarity, M)).
9. Extract three times with ethyl acetate (2 L).
10. Combine the organic phases and wash twice with saturated brine (1 L).
11. The organic phase was dried over sodium sulfate (200 g).
12. Continue drying in an oven (40-45 ° C) to give (S)-N-tert-butoxycarbonyl-4-iodo-L-phenylalanine (250.00 g, 626.28 mmol, HPLC analysis, yield 93.00 %, purity 98%).
The prepared (S) -N- tert-butoxycarbonyl-4-iodo-phenylalanine was -L- Hydrogen 1 nuclear magnetic resonance spectrum analysis (1 HNMR) as follows:
Second, tributyl borate 10 was prepared from boronic acid 10
See the following reaction formulas II, 10 as boric acid (H 2 SO 4) is reacted with sulfuric acid in a solvent (butan-1-ol), and toluene (Toluene) in n-butanol, to obtain 10 tributyl borate (10 The chemical reaction formula of B(OBu) 3 ).
The specific operation process is as follows:
1. Set up a reaction device R1 using a 3L three-necked bottle, and configure a water separator on the device.
2. 10 boric acid (150.00 g, 2.46 mol, 1.00 eq) was added to the reaction R1 at room temperature (about 25 ° C).
3. Add n-butanol (1.00 L) to the reaction R1 at room temperature (about 25 ° C) and stir, and most of the boric acid cannot be dissolved.
4. Toluene (1.00 L) was added to the reaction R1 at room temperature (about 25 ° C) and stirred.
5. Concentrated sulfuric acid (4.82 g, 49.16 mmol, 2.62 mL, 0.02 eq) was added dropwise to the reaction at room temperature (about 25 ° C), at which time a large amount of solid remained undissolved.
6. The reaction system was heated to 130 ° C, and the water was continuously removed, stirred for 3.5 hours, and water (about 140 g) was formed in the water separator. The solids were all dissolved, and the solution changed from colorless to brown. .
8. Distill off most of the toluene at atmospheric pressure.
9. After most of the toluene is distilled off, the temperature of the system is lowered to 20 to 30 ° C, and the reaction liquids of the two reactions are combined, and the apparatus is changed for distillation.
10. Oil bath external temperature 108-110 ° C pump distillation under reduced pressure, Kelvin thermometer 45 ° C, distilled n-butanol.
11. Oil bath external temperature 108-110 ° C oil pump distillation under reduced pressure, the residual butanol was distilled off.
12. Oil bath external temperature 118-120 ° C oil pump vacuum distillation, Kelvin thermometer 55 ° C, began to produce products.
13. The temperature is raised to 135-140 ° C oil pump vacuum distillation, the product is completely distilled.
14. The product is obtained as a colorless liquid 10 tributyl borate (830.00g, 3.62mol, yield 73.58%).
The results of the 1 H NMR analysis of the obtained tributyl 10 borate were as follows:
Three, -N- tert-butoxycarbonyl-4-iodo-phenylalanine was prepared (S) of (S) -N- tert-butoxycarbonyl-4-hydroxy-10-yl -L- phenylalanine boron
Please refer to the following reaction formula III, which is (S)-N-tert-butoxycarbonyl-4-iodophenylalanine with tributyl 10 borate, t-butyl magnesium chloride (t-BuMgCl) and bis (2-A) yl aminoethyl) ether (BDMAEE) reaction, to produce (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine chemical reaction.
In the preparation process, two reaction vessels were selected for the reaction.
The specific operation process is as follows:
1. Set up a reaction using a 3L three-neck bottle.
2. Tributyl 10 borate (187.60 g, 87.98 mmol, 3.20 eq) was placed in the reaction system at room temperature (about 22 ° C).
3. Sodium hydride (20.45 g, 511.24 mmol, purity 60%, 2.00 eq) was added to the reaction system at room temperature (about 22 ° C). The reaction solution was a suspension and stirred at room temperature (about 22 ° C). 5 minutes.
4. Bis(2-methylaminoethyl)ether (327.73 g, 2.04 mol, 8.00 eq) was added to the reaction at room temperature (about 22 ° C).
5. N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (100.00 g, 255.62 mmol, 1.00 eq) was added to the reaction system at room temperature (about 22 ° C), and a large amount of solid was not dissolved.
6. Lower the temperature of the reaction system to 0-5 ° C, add t-butyl magnesium chloride (1.7 M, 1.20 L, 2.04 mol, 8.00 eq) to the reaction, control the temperature between 0-10 ° C, the dropping time is about It is 1.5 hours.
7. After the completion of the charging, the temperature of the reaction system was naturally raised to room temperature (20 to 30 ° C) and stirred at this temperature for 12 hours.
8. Using high performance liquid chromatography (HPLC) to detect about 9.00% of the remaining material.
9. When the temperature of the reaction system was lowered to -5 to 0 ° C, it was quenched by dropwise addition of 500 mL of water.
10. Lower the temperature of the system to 0-5 ° C, add methyl tert-butyl ether (500 mL) to the reaction system and adjust the pH to 2.9-3.1 (using a pH meter) with 37% HCl (about 500 mL). Exothermic, the temperature of the control system is between 0-15 °C.
11. The aqueous phase obtained by liquid separation was extracted once with methyl tert-butyl ether (500 mL), and the obtained organic phases were combined to give an organic phase of about 1.1 L.
12. Slowly add a sodium hydroxide aqueous solution (1 M, 400 mL) to the obtained organic phase, exotherm during the dropwise addition, and control the system temperature between 0-15 °C.
13. After the completion of the dropwise addition, the pH of the system was about 10, and the pH was adjusted to between 12.10 and 12.6 with an aqueous sodium hydroxide solution (4M). (measured with a pH meter)
14. Dispensing.
15. The aqueous phase 1 obtained after liquid separation was extracted once with n-butanol (500 ml) to obtain aqueous phase 2.
16. Combine the aqueous phase 2 of the two reaction vessels.
17. Adjust the pH of the aqueous phase to 2.9-3.1 with 37% HCl, stir for about 40 minutes, and precipitate a large amount of solid.
18. Filtration gave a white solid which was washed once with dichloromethane (50 mL).
19. At 25 ° C, the precipitated solid was slurried with dichloromethane (150 mL) and stirred for 10 min.
20. A white solid was filtered to give (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine (75.00g, 240.82mmol, by HPLC analysis, a yield of 47.11% , purity 99%).
The prepared (S) -N- tert-butoxycarbonyl group -4- (10 B) results dihydroxyboryl -L- phenylalanine 1 HNMR was as follows:
Preparation of L- 10 BPA from (S)-N-tert-Butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine
See the following reaction scheme IV, which is (S) -N- tert-butoxycarbonyl group -4- (10 B) of amine end dihydroxyboryl -L- phenylalanine deprotection of the chemical reaction, to obtain L- 10 BPA.
The specific operation process is as follows:
1. Set up a reaction using a 1L three-neck bottle.
2. room temperature (20-30 deg.] C) to (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxyboryl -L- phenylalanine (67.00g, 217.31mmol, 1.00eq) was added the reaction In the system.
3. room temperature (20-30 deg.] C) water (23.75mL) and acetone (Acetone, 420.00mL) were added dropwise to the reaction flask, stirred (S) -N- tert-butoxycarbonyl group -4- (10 B) dihydroxy Boronyl-L-phenylalanine.
4. Concentrated hydrochloric acid (23.93 g, 656.28 mmol, 23.46 mL, 3.02 eq) was added dropwise to the reaction system at room temperature (20-30 ° C). After the addition was completed, the reaction system was heated to 55-60 ° C and stirred for 4.5 hours.
5. HPLC detection until the reaction of the starting material is completed.
6. The temperature is controlled below 40 ° C, and the acetone in the reaction system is concentrated.
7. Lower the concentrated system to below 15 °C, adjust the pH of the system to about 1.5 with sodium hydroxide solution (4M) (pH meter detection), stir for 40 minutes and continue to adjust the pH of the system to 6.15 using sodium hydroxide solution (4M). ~6.25, a large amount of white solid precipitated, which was filtered to give a white solid, and rinsed with acetone (200mL).
8. Obtained as a white solid L- 10 BPA (36.00 g, 171.17 mmol, HPLC, yield 78.77%, purity 99%).
The analytical results obtained by the L- 10 BPA 1 HNMR are as follows:
Preparation of (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine from (S)-N-tert-butoxycarbonyl-4-iodophenylalanine
Please refer to the following reaction formula VII, which is a reaction of (S)-N-tert-butoxycarbonyl-4-iodophenylalanine with tributyl borate and t-butylmagnesium chloride (t-BuMgCl) to obtain (S The chemical reaction formula of -N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine.
The specific operation process is as follows:
1. Construct a reaction unit with a 250 mL three-neck bottle.
2. Tributyl borate (17.65 g, 76.68 mmol, 3.00 eq) was placed in a 250 mL reaction flask at 20-30 °C.
3. Sodium hydride (1.02 g, 25.56 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
4. (S)-N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (10.00 g, 25.56 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
5. Reduce the temperature of the reaction system to 0 ° C under nitrogen atmosphere, slowly add t-butyl magnesium chloride (1.7 M in THF, 120 mL, 8.00 eq) to the reaction, the dropping time is about 30 minutes, and the control temperature is 0. Between °C and 10 °C.
Stir at 20.20 ~ 30 ° C for 20 hours.
7. HPLC detection of the basic reaction of the raw materials, leaving only about 0.7% of the raw materials.
8. At a temperature of 0 ° C, 5 mL of water was added dropwise to the reaction to quench it. After complete quenching, stirring was continued for 10 minutes.
9. Cool down to 0 ° C, add methyl tert-butyl ether (50 mL) to the reaction and adjust the pH to 3 with 37% HCl (about 50 mL) (detected with a pH meter), adjust the pH during the process to exotherm, control the temperature at 0 Between °C and 15 °C.
12. The aqueous phase obtained by liquid separation was extracted once with methyl t-butyl ether (50 mL) and the organic phases were combined.
12. Add NaOH solution (1M, 55mL) to the obtained organic phase to adjust the pH to between 12.10-12.6. The process is exothermic and the temperature is controlled between 0 °C and 15 °C.
13. Liquid separation, the obtained aqueous phase was extracted once with n-butanol (50 mL), and most of the impurities were extracted and removed.
14. The aqueous phase obtained by liquid separation was adjusted to pH 3 with 37% HCl and stirred for about 30 minutes to precipitate a white solid.
15. Filtration gave a white solid which was washed once with dichloromethane (50 mL).
16. The precipitated solid was slurried with 25 mL of dichloromethane at 25 ° C and stirred for 10 minutes.
Please continue to refer to Reaction Scheme VII. The specific operation process is as follows:
1. Construct a reaction unit with a 250 mL three-neck bottle.
2. Tributyl borate (8.82 g, 38.34 mmol, 3.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
3. Sodium hydride (511.25 mg, 12.78 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
4. (S)-N-tert-Butoxycarbonyl-4-iodo-L-phenylalanine (5.00 g, 12.78 mmol, 1.00 eq) was added to a 250 mL reaction vial at 20-30 °C.
5. The temperature of the reaction system was lowered to 0 ° C under nitrogen atmosphere, and t-butyl magnesium chloride (1.7 M in THF, 60 mL, 8.00 eq) was added dropwise to the reaction, the dropwise addition time was about 30 minutes, and the control temperature was 0 ° C. -10 ° C between.
Stir at 6.20 ~ 30 ° C for 22 hours.
7. HPLC detection of the raw material reaction is completed.
8. At a temperature of 0 ° C, 2.5 mL of water was added dropwise to the reaction to quench it. After complete quenching, stirring was continued for 10 minutes.
9. Cool down to 0 ° C, add methyl tert-butyl ether (25 mL) to the reaction and adjust the pH to 3 with 37% HCl (about 25 mL) (detected with a pH meter), adjust the pH during the process to exotherm, control the temperature at 0 Between °C and 15 °C.
12. The aqueous phase obtained by liquid separation was extracted once with methyl t-butyl ether (25 mL) and the organic phases were combined.
12. Add NaOH solution (1M, 30mL) to the obtained organic phase to adjust the pH to between 12.10-12.6. The process is exothermic and the temperature is controlled between 0 °C and 15 °C.
13. Liquid separation, the obtained aqueous phase was extracted once with n-butanol (25 ml), and most of the impurities were extracted and removed.
14. The aqueous phase obtained by liquid separation was adjusted to pH 3 with 37% HCl and stirred for about 30 minutes to precipitate a white solid.
15. Filtration gave a white solid which was washed once with dichloromethane (25 mL).
16. The precipitated solid was slurried with 15 mL of dichloromethane at 25 ° C and stirred for 10 minutes.
17. Filtration gave (S)-N-tert-butoxycarbonyl-4-dihydroxyboryl-L-phenylalanine (3.4 g, obtained by HPLC, yield: 85.26%, purity 98%).
Bis(2-methylaminoethyl)ether is a complexing agent for Mg, which can reduce the occurrence of side reactions in the reaction. The reactions of Examples 6 and 7 were carried out without adding bis(2-methylaminoethyl)ether. The analysis showed that the iodine impurity in the reaction of Example 6 was about 17%, and the iodine impurity in the reaction of Example 7 was observed. About 28%. Therefore, it has been proved from the side that the addition of bis(2-methylaminoethyl)ether can protect the reaction from reducing iodine.
The BPA or 10 BPA obtained in the above examples were analyzed by chiral HPLC, and the ratio of the L-enantiomer to the D-enantiomer was 100:0.
The boron-containing drug L-BPA for neutron capture therapy disclosed in the present invention is not limited to the contents described in the above examples. The above-mentioned embodiments are only examples for convenience of description, and the scope of the claims should be determined by the claims.
The preparation of the compound 3- (1- {3- [5- (1-Methyl-piperidin-4-ylmethoxy) -pyrimidin-2-yl] -benzyl} -6-oxo-1,6-dihydro-pyridazin-3 -yl) -benzonitrile (“A257”) takes place analogously to the following scheme
40.1 17.7 g (67.8 mmol) triphenyl are added to a suspension of 13.0 g (56.5 mmol) 3- (5-hydroxypyrimidin-2-yl) -benzoic acid methyl ester and 13.4 g (62.1 mmol) N-Boc-piperidinemethanol in 115 ml THF -phosphine and cooled to 5 ° C. To the suspension kept at this temperature, 13.3 ml (67.8 mmol) of diisopropylazodicarboxylate are added dropwise with stirring within 45 minutes. The reaction mixture is stirred for 1 hour at room temperature. Then a further 22.2 g (84.7 mmol) triphenylphosphine and 16.6 ml (84.7 mmol)
Diisopropyl azodicarboxylate added. The reaction mixture turns 18
Stirred for hours at room temperature and concentrated in vacuo. The resulting solid is filtered off with suction, washed with diethyl ether and chromatographed on a silica gel column with dichloromethane / methanol as the mobile phase: 4- [2- (3-methoxycarbonyl-phenyl) -pyrimidin-5-yloxymethyl] -piperidine-1-carboxylic acid tert .-butyl ester as lemon yellow crystals; 166 ° C .; ESI 428.
40.2 To a suspension of 1.71 g (3.99 mmol) of 4- [2- (3-methoxycarbonyl-phenyl) -pyrimidin-5-yloxymethyl] -piperidine-1-carboxylic acid tert-butyl ester in 20 ml of THF are added under nitrogen 25 ml (25 mmol) of a 1 M solution of diisobutylaluminum hydride in THF were added dropwise. The reaction mixture is stirred at room temperature for 1 hour, and 1 ml of a saturated sodium sulfate solution is added. The resulting precipitate is filtered off with suction and washed with THF and hot 2-propanol. The filtrate is evaporated and recrystallized from tert-butyl methyl ether: {3- [5- (1-Methyl-piperidin-4-ylmethoxy) -pyrimidin-2-yl] -phenyl} -methanol as beige crystals; Mp 175 ° C; ESI 314.
40.3 To a solution of 313 mg (1.00 mmol) {3- [5- (1-methyl-piperidin-4-ylmethoxy) -pyrimidin-2-yl] -phenyl} -methanol in 2 ml THF are successively added 264 mg (1.30 mmol) 3- (6-oxo-1, 6-dihydro-pyridazin-3-yl) benzonitrile and 397 mg (1.5 mmol) triphenylphosphine are added. The reaction mixture is cooled in an ice bath and 294 μl (1.5 mmol) of diisopropylazodicarboxylate are added dropwise with stirring. The
The reaction mixture is stirred for 18 hours at room temperature and evaporated. The residue is chromatographed on a silica gel column using dichloromethane / methanol. The product-containing fractions are combined, evaporated, the residue digested with tert-butyl methyl ether, filtered off with suction and dried in vacuo: 3- (1- {3- [5- (1-methylpiperidin-4-ylmethoxy) pyrimidine) -2-yl] -benzyl} -6-oxo-1,6-dihydro-pyridazin-3-yl) -benzonitrile as colorless crystals; M.p. 177 ° C; ESI 493; 1 H-NMR (de-DMSO): δ [ppm] = 1.33 (m, 2H), 1.75 (m, 3H), 1.89 (m, 2H), 2.17 (S, 3H), 2.80 (m, 2H), 4.05 (d, J = 6.1 Hz 1 2H), 5.45 (s, 2H) 1 7.16 (d, J = 10 Hz, 1 H), 7.49 (m, 2H), 7.73 (t, J = 7.8 Hz, 1H ), 7.93 (d, J = 7.8 Hz, 1H) 1 8.17 (d, J = 10 Hz, 1H), 8.24 (m, 2H), 8.38 (m, 2H), 8.64 (s, 2H).
The hemisulfate, citrate, tartrate, sulfate, succinate and hydrochloride are obtained from “A257” by salt formation.
Scheme 1. Reagents and conditions: a) PdCl2(PPh3)2, Na2CO3, ethanol/toluene/water, 90 °C, 8 h; b) SOCl2, CHCl3, reflux; c) SeO2, dioxane:H2O = 10:1, reflux, 12 h; d) NaOH, −30 °C; e) NaH, DMF/THF, 0 °C—room temperature, 12 h; f) dry ethanol, reflux; g) NaOH, DMF/H2O, 60 °C, 8 h, N2.
Scheme 2. Reagents and conditions: a) N,N-diisopropylethylamine, dry CH2Cl2, 0 °C—room temperature, 6 h; b) PdCl2(PPh3)2, Na2CO3, ethanol/toluene/water, 90 °C, 8 h; c) 10% aq. HCl, MeOH, reflux; d) K2CO3, dry DMF, 80 °C, 12 h; e) NaOH, DMF/H2O, 60 °C, 8 h, N2; f) PPh3, DIAD, THF, 0 °C—room temperature; g) SOCl2, CHCl3, reflux; h) 35% formaldehyde, NaBH4, MeOH.
Scheme 3. Reagents and conditions: a) PdCl2(PPh3)2, Na2CO3, ethanol/toluene/water, 90 °C, 8 h; b) NaBH4, MeOH, 0 °C—room temperature, 1 h; c) SOCl2, CHCl3, reflux; d) K2CO3, dry DMF, 80 °C, 12 h; e) 31a–31b: NaOH, DMF/H2O, 60 °C, 8 h, N2; f) 31c–31g: NaH, dry DMF, 0 °C—room temperature, 5 h.
Scheme 4. Reagents and conditions: a) K2CO3, dry DMF, 80 °C, 12 h; b) PdCl2(PPh3)2, Na2CO3, DME/DMF/water, 89 °C, 12 h; c) NaOH, DMF/H2O, 60 °C, 8 h, N2.
Scheme 5. Reagents and conditions: a) K2CO3, dry DMF, 80 °C, 12 h; b) PdCl2(PPh3)2, Na2CO3, DME/DMF/water, 89 °C, 12 h; c) NaOH, DMF/H2O, 60 °C, 8 h, N2.
Tirabrutinib (Velexbru®) is an orally administered, small molecule, Bruton’s tyrosine kinase (BTK) inhibitor being developed by Ono Pharmaceutical and its licensee Gilead Sciences for the treatment of autoimmune disorders and haematological malignancies. Tirabrutinib irreversibly and covalently binds to BTK in B cells and inhibits aberrant B cell receptor signalling in B cell-related cancers and autoimmune diseases. In March 2020, oral tirabrutinib was approved in Japan for the treatment of recurrent or refractory primary central nervous system lymphoma. Tirabrutinib is also under regulatory review in Japan for the treatment of Waldenström’s macroglobulinemia and lymphoplasmacytic lymphoma. Clinical development is underway in the USA, Europe and Japan for autoimmune disorders, chronic lymphocytic leukaemia, B cell lymphoma, Sjogren’s syndrome, pemphigus and rheumatoid arthritis. This article summarizes the milestones in the development of tirabrutinib leading to the first approval of tirabrutinib for the treatment of recurrent or refractory primary central nervous system lymphoma in Japan.
Cetuximab Sarotalocan Sodium is an antibody-drug-conjugate (molecular weight: 156,000-158,000) consisting of tetrasodium salt of Sarotalocan (6-({[3-({(OC-6-13)-bis({3-[bis(3-sulfopropyl)(3-sulfonatopropyl)azaniumyl]propyl}dimethylsilanolato-κO,κO‘)[(phtalocyaninato(2-)κN29,κN30,κN31,κN32)-1-yl]silicon}oxy)propoxy]carbonyl}amino)hexanoyl (C70H96N11O24S6Si3; molecular weight: 1,752.22)) attached to an average of 2-3 Lys residues of Cetuximab.
Originator Japan Tobacco Developer Japan Tobacco; JW Pharmaceutical Class Acetic acids; Amides; Antianaemics; Pyridones; Small molecules; Triazoles Mechanism of Action Hypoxia-inducible factor-proline dioxygenase inhibitors
Preregistration Anaemia
27 Dec 2019 Japan Tobacco and SalubrisBio enter into a development and marketing agreement for enarodustat (JTZ 951) in China, Hong Kong, Macau and Taiwan for Anaemia 29 Nov 2019 Preregistration for Anaemia in Japan (PO) 31 Oct 2019 Phase I development in Anaemia is ongoing in USA
Enarodustat is a potent and orally active factor prolyl hydroxylase inhibitor, with an EC50 of 0.22 μM. Enarodustat has the potential for renal anemia treatment
Inhibition of hypoxia inducible factor prolyl hydroxylase (PHD) represents a promising strategy for the discovery of a next generation treatment for renal anemia. We identified several 5,6-fused ring systems as novel scaffolds of the PHD inhibitor on the basis of pharmacophore analysis. In particular, triazolopyridine derivatives showed potent PHD2 inhibitory activities. Examination of the predominance of the triazolopyridines in potency by electrostatic calculations suggested favorable π–π stacking interactions with Tyr310. Lead optimization to improve the efficacy of erythropoietin release in cells and in vivo by improving cell permeability led to the discovery of JTZ-951 (compound 14), with a 5-phenethyl substituent on the triazolopyridine group, which increased hemoglobin levels with daily oral dosing in rats. Compound 14 was rapidly absorbed after oral administration and disappeared shortly thereafter, which could be advantageous in terms of safety. Compound 14 was selected as a clinical candidate.
Certain glycopyrronium salts and related compounds, as well as processes for making and methods of using these glycopyrronium salts and related compounds, are known. See, for example, US Patent No. 8,558,008, which issued to assignee Dermira, Inc. See also, for example, US Patent No. 2,956,062, which issued to assignee Robins Co Inc. A H. See also, for example, International Patent Application Publication Nos. WO 98/00132 Al and WO 2009/00109A1, both of which list applicant Sepracor, Inc., as well as US Patent Nos. 6,063,808 and 6,204,285, both of which issued to assignee Sepracor, Inc. Certain methods of treating hyperhidrosis using glycopyrronium salts and related compounds are known. See, for example GB 1,080,960. Certain forms of applying glycopyrrolate compounds to a subject are known. See, for example US Patent Nos. 6,433,003 and 8,618,160, both of which issued to assignee Rose U; also US Patent Nos. 7,060,289; 8,252,316; and 8,679,524, which issued to PurePharm, Inc.
[0004] One glycopyrronium salt which is useful in certain medical applications is the following compound:
[0005] As illustrated above, the absolute configuration at the three asymmetric chiral positions is 2R3’R1’RS. This means that the carbon indicated with the number, 2, has the stereochemical R configuration. The carbon indicated with the number, 3′, also has the stereochemical R configuration. The quatemary ammonium nitrogen atom, indicated with a positive charge, may have either the R or the S stereochemical configuration. As drawn, the compound above is a mixture of two diastereoisomers.
[0006] Certain processes for making glycopyrronium salts are known. However, these processes are not as safe, efficient, stereospecific, or stereoselective as the new processes disclosed herein, for example with respect to large-scale manufacturing processes. Certain publications show that higher anticholinergic activity is attributed to the 2R3’R configuration. However, to date, processes for making the 2R3’R isomers, as well as the 2R3’R1’R isomers are low yielding, involve too many reaction steps to be economically feasible, use toxic materials, and/or are not sufficiently stereospecific or stereoselective with respect to the products formed.
EXAMPLE 2
[0179] The below synthetic description refers to the numbered compounds illustrated in FIG. 2. Numbers which refer to these compounds in FIG. 2 are bolded and underlined in this Example.
[0180] Synthesis of R(-)-Cyclopentylmandelic acid (4)
[0181] R(-)-cyclopentylmandelic acid (compound 4) can be synthesized starting with
R(-)-mandelic acid (compound 1) according to Example 1.
[0182] Step 1 : Making Compound 2.
[0183] R(-)-mandelic acid (1) was suspended in hexane and mixed with pivaldehyde and a catalytic amount of trifluoromethanesulfonic acid at room temperature to form a mixture. The mixture was warmed to 36 °C and then allowed to react for about 5 hours. The mixture was then cooled to room temperature and treated with 8% aqueous sodium bicarbonate. The aqueous layer was removed and the organic layer dried over anhydrous sodium sulfate. After filtration and removal of the solvent under vacuum, the crude product was recrystallized to give (5R)-2-(tert-butyl)-5-phenyl-l,3-dioxolan-4-one (compound 2) in 88% yield (per S-enantiomer yield).
[0184] Step 2: Making Compound 3.
[0185] Compound 2 was reacted with lithium hexamethyl disilazide (LiHMDS) in hexane at -78 °C under stirring for one hour. Next, cyclopentyl bromide was added to the reaction mixture including compound 2 and LiHMDS . The reaction was kept cool for about four (4) hours and then slowly warmed to room temperature and allowed to react for at least twelve (12) more hours. The resulting mixture was then treated with 10% aqueous ammonium chloride. The aqueous layer was discarded and the organic layer dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue recrystallized from hexane to give pure product (5R)-2-(tert-butyl)-5-cyclopentyl-5-phenyl- l,3-dioxolan-4-one (3) in 63% yield (per S-enantiomer yield).
[0186] Step 3: Making Compound 4.
[0187] R(-)-cyclopentylmandelic acid (compound 4) was prepared by providing compound 3 in aqueous methanolic potassium hydroxide at 65 °C for four hours. After cooling this mixture to room temperature and removing the methanol under vacuum, the aqueous solution was acidified with aqueous hydrochloric acid. The aqueous solution was then extracted twice with ethyl acetate and the organic phase dried with anhydrous sodium sulfate. After removing the solvent and performing a recrystallization, pure R(-)- cyclopentylmandelic acid (compound 4) was obtained in 62% yield (based on S-enantiomer yield).
[0188] Next, a racemic mixture of l -methyl-3-pyrridinol (20) was provided:
[0189] Synthesis of 2R3 ‘R-glycopyrrolate base (8)
[0190] Step 4: Making Compound 8.
[0191] Enantiomerically pure R(-)-cyclopentylmandelic acid (4) was coupled to racemic l-methyl-3-pyrridinol (20) using 1, 1 -carbonyldiimideazole (CDI) activated esterification to make an enantiomerically pure mixture of the following erythro- and threo- glycopyrrolate bases (compounds 8 and 21, respectively):
[0192] The 2R3’R-glycopyrrolate base (compound 8) was then resolved using the 5- nitroisophthalate salt procedure in Finnish Patent 49713, to provide enantiomerically pure 2R3 Έ. {erythro) as well as pure 2R3 ‘S {threo). In this example, the 2R3 ‘S {threo) was discarded. The 2R3 Έ. {erythro) was separated as stereomerically pure compound 8.
[0193] Step 6: Making Compound 9.
[0194] The glycopyrrolate base, compound 8, was treated in dry acetonitrile with methyl bromoacetate at room temperature under stirring for three (3) hours. The crude product was dissolved in a small volume of methylene chloride and poured into dry ethyl ether to obtain a precipitate. This procedure was repeated three times to provide (3R)-3-((R)- 2-cyclopentyl-2-hydroxy-2-phenylacetoxy)-l -(2-ethoxy-2-oxoethyl)-l-methylpyrrolidin-l – ium bromide, also known as 3′(R)-[R-Cyclopentylphenylhydroxyacetoy]- -ethyl- l ‘methoxycarbonylpyrrolidinium bromide (compound 9) in 89% yield. Compound 9 included the following stereoisomers:
Delgocitinib, also known as LEO-124249 and JTE052, is a potent and selective JAK inhibitor. JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation.
Delgocitinib is a JAK inhibitor first approved in Japan for the treatment of atopic dermatitis in patients 16 years of age or older. Japan Tobacco is conducting phase III clinical trials for the treatment of atopic dermatitis in pediatric patients. Leo is developing the drug in phase II clinical trials for the treatment of inflammatory skin diseases, such as atopic dermatitis, and chronic hand eczema and for the treatment of discoid lupus erythematosus. Rohto is evaluating the product in early clinical development for ophthalmologic indications.
In 2014, the drug was licensed to Leo by Japan Tobacco for the development, registration and marketing worldwide excluding Japan for treatment of inflammatory skin conditions. In 2016, Japan Tobacco licensed the rights of co-development and commercialization in Japan to Torii. In 2018, Japan Tobacco licensed the Japanese rights of development and commercialization to Rohto for the treatment of ophthalmologic diseases.
(1) Optically active substance of 2-benzylaminopropan-1-ol
To a solution of (S)-(+)-2-aminopropan-1-ol (50.0 g) and benzaldehyde (74 ml) in ethanol (500 ml) was added 5% palladium carbon (5.0 g) at room temperature and normal pressure. Hydrogenated for 8 hours. The reaction mixture was filtered through celite and concentrated under reduced pressure to give the title compound (111.2 g). 1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.23-7.18 (1H, m), 4.53-4.47 (1H, m), 3.76 (1H, d, J = 13.5 Hz) , 3.66 (1H, d, J = 13.5 Hz), 3.29-3.24 (2H, m), 2.65-2.55 (1H, m), 1.99 (1H, br s), 0.93 (3H, d, J = 6.4 Hz) .
(2) Optically active substance of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester
To a mixture of optically active 2-benzylaminopropan-1-ol (111.2 g), potassium carbonate (111.6 g) and N, N-dimethylformamide (556 ml) cooled to 0 ° C., tert-butyl bromoacetate was added. Ester (109 ml) was added dropwise over 20 minutes and stirred at room temperature for 19.5 hours. The mixture was acidified to pH 2 by adding 2M aqueous hydrochloric acid and 6M aqueous hydrochloric acid, and washed with toluene (1000 ml). The separated organic layer was extracted with 0.1 M aqueous hydrochloric acid (300 ml). The combined aqueous layer was adjusted to pH 10 with 4M aqueous sodium hydroxide solution and extracted with ethyl acetate (700 ml). The organic layer was washed successively with water (900 ml) and saturated aqueous sodium chloride solution (500 ml). The separated aqueous layer was extracted again with ethyl acetate (400 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to give the title compound (160.0 g). 1 H-NMR (DMSO-D 6 ) δ: 7.37-7.26 (4H, m), 7.24-7.19 (1H, m), 4.26 (1H, dd, J = 6.9, 3.9 Hz), 3.76 (1H, d, J = 14.1 Hz), 3.68 (1H, d, J = 13.9 Hz), 3.45-3.39 (1H, m), 3.29-3.20 (1H, m), 3.24 (1H, d, J = 17.2 Hz), 3.13 ( 1H, d, J = 17.0 Hz), 2.84-2.74 (1H, m), 1.37 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
(3) Optically active substance of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester
(3)-(1) Optically active form of [benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester
To a solution of [benzyl- (2-hydroxy-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (160.0 g) cooled to 0 ° C. in chloroform (640 ml) was added thionyl chloride (50.0 ml). Was added dropwise and stirred at 60 ° C. for 2 hours. The reaction mixture was cooled to 0 ° C., saturated aqueous sodium hydrogen carbonate solution (1000 ml) and chloroform (100 ml) were added and stirred. The separated organic layer was washed with a saturated aqueous sodium chloride solution (500 ml), and the aqueous layer was extracted again with chloroform (450 ml). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain the title compound (172.9 g). 1 H-NMR (CDCl 3 ) δ: 7.40-7.22 (5H, m), 4.05-3.97 (0.4H, m), 3.93-3.81 (2H, m), 3.70-3.65 (0.6H, m), 3.44- 3.38 (0.6H, m), 3.29 (0.8H, s), 3.27 (1.2H, d, J = 2.4 Hz), 3.24-3.15 (0.6H, m), 3.05-2.99 (0.4H, m), 2.94 -2.88 (0.4H, m), 1.50 (1.2H, d, J = 6.4 Hz), 1.48 (3.6H, s), 1.45 (5.4H, s), 1.23 (1.8H, d, J = 6.8 Hz) .
(3)-(2) Optically active form of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester
[Benzyl- (2-chloro-1-methylethyl) -amino] acetic acid tert-butyl ester optically active substance (172.9 g) was dissolved in N, N-dimethylformamide (520 ml) and stirred at 80 ° C. for 140 minutes. did. The reaction mixture was cooled to 0 ° C., water (1200 ml) was added, and the mixture was extracted with n-hexane / ethyl acetate (2/1, 1000 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (400 ml), and the separated aqueous layer was extracted again with n-hexane / ethyl acetate (2/1, 600 ml). The combined organic layers were concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 50/1 to 40/1) to give the title compound (127.0 g ) 1 H-NMR (CDCl 3 ) δ: 7.37-7.29 (4H, m), 7.28-7.23 (1H, m), 4.05-3.97 (1H, m), 3.91 (1H, d, J = 13.5 Hz), 3.86 (1H, d, J = 13.7 Hz), 3.29 (2H, s), 3.03 (1H, dd, J = 13.9, 6.6 Hz), 2.91 (1H, dd, J = 13.9, 6.8 Hz), 1.50 (3H, d, J = 6.4 Hz), 1.48 (9H, s).
(4) Optically active substance of 1-benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester
To a solution of [benzyl- (2-chloropropyl) -amino] acetic acid tert-butyl ester optically active substance (60.0 g) cooled to −72 ° C. and hexamethylphosphoramide (36.0 ml) in tetrahydrofuran (360 ml), Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 242 ml) was added dropwise over 18 minutes, and the temperature was raised to 0 ° C. over 80 minutes. A saturated aqueous ammonium chloride solution (300 ml) and water (400 ml) were sequentially added to the reaction mixture, and the mixture was extracted with ethyl acetate (500 ml). The organic layer was washed successively with water (700 ml) and saturated aqueous sodium chloride solution (500 ml), and the separated aqueous layer was extracted again with ethyl acetate (300 ml). The combined organic layers were dried over anhydrous sodium sulfate, concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography (developing solvent: n-hexane / ethyl acetate = 50/1 to 4/1). To give the title compound (50.9 g). 1 H-NMR (CDCl 3 ) δ: 7.34-7.21 (5H, m), 3.75 (1H, d, J = 12.6 Hz), 3.70-3.67 (1H, m), 3.58 (1H, d, J = 12.6 Hz ), 3.05-3.01 (1H, m), 2.99-2.95 (1H, m), 2.70-2.59 (1H, m), 1.41 (9H, s), 1.24 (3H, d, J = 7.1 Hz).
(5) Optically active substance of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester
1-Benzyl-3-methylazetidine-2-carboxylic acid tert-butyl ester optically active substance (43.5 g) and di-tert-butyl dicarbonate (38.2 g) in tetrahydrofuran / methanol (130 ml / 130 ml) solution 20% Palladium hydroxide carbon (3.5 g) was added thereto, and hydrogenated at 4 atm for 2 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (48.0 g). 1 H-NMR (DMSO-D 6 ) δ: 4.44 (1H, d, J = 8.8 Hz), 3.99-3.77 (1H, m), 3.45-3.37 (1H, m), 3.00-2.88 (1H, m) , 1.45 (9H, s), 1.40-1.30 (9H, m), 1.02 (3H, d, J = 7.2 Hz).
(6) Optically active substance of 3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester
Optically active substance (48.0 g) of 3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester cooled to -69 ° C. and 1-bromo-3-methyl-2-butene (25.4 ml) Lithium hexamethyldisilazide (1.0 M tetrahydrofuran solution, 200 ml) was added to a tetrahydrofuran solution (380 ml). The reaction mixture was warmed to −20 ° C. in 40 minutes and further stirred at the same temperature for 20 minutes. A saturated aqueous ammonium chloride solution (200 ml) and water (300 ml) were successively added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1 / 1,500 ml). The separated organic layer was washed successively with water (200 ml) and saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: n-hexane / ethyl acetate = 15/1 to 8/1) to give the titled compound (44.5 g). 1 H-NMR (CDCl 3 ) δ: 5.29-5.21 (1H, m), 3.77-3.72 (1H, m), 3.49-3.44 (1H, m), 2.73-2.52 (3H, m), 1.76-1.74 ( 3H, m), 1.66-1.65 (3H, m), 1.51 (9H, s), 1.43 (9H, s), 1.05 (3H, d, J = 7.3 Hz).
(7) Optically active substance of 3-methyl-2- (2-oxoethyl) azetidine-1,2-dicarboxylic acid di-tert-butyl ester
3-methyl-2- (3-methyl-but-2-enyl) -azetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (44.5 g) in chloroform / cooled to −70 ° C. An ozone stream was passed through the methanol solution (310 ml / 310 ml) for 1 hour. To this reaction mixture, a solution of triphenylphosphine (44.7 g) in chloroform (45 ml) was added little by little, and then the mixture was warmed to room temperature. To this mixture were added saturated aqueous sodium thiosulfate solution (200 ml) and water (300 ml), and the mixture was extracted with chloroform (500 ml). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure to obtain the title compound (95.0 g). This product was subjected to the next step without further purification. 1 H-NMR (DMSO-D 6 ) δ: 9.65 (1H, t, J = 2.6 Hz), 3.79-3.74 (1H, m), 3.45-3.40 (1H, m), 2.99-2.80 (3H, m) , 1.46 (9H, s), 1.34 (9H, s), 1.06 (3H, d, J = 7.2 Hz).
(8) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester
To a solution of the residue (95.0 g) obtained in (7) in tetrahydrofuran (300 ml) was added benzylamine (34 ml) at room temperature, and the mixture was stirred for 2 hours. The mixture was cooled to 0 ° C., sodium triacetoxyborohydride (83.3 g) was added, and the mixture was stirred at room temperature for 1.5 hours. Water (300 ml) was added to the reaction mixture, and the mixture was extracted with n-hexane / ethyl acetate (1/3, 600 ml). The separated organic layer was washed with water (300 ml) and saturated aqueous sodium chloride solution (200 ml), and then extracted twice with 5% aqueous citric acid solution (300 ml, 200 ml) and three times with 10% aqueous citric acid solution (250 ml × 3). . The combined aqueous layers were basified to pH 10 with 4M aqueous sodium hydroxide solution and extracted with chloroform (300 ml). The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure to obtain the title compound (46.9 g). 1 H-NMR (DMSO-D 6 ) δ: 7.34-7.26 (4H, m), 7.22-7.17 (1H, m), 3.74-3.65 (2H, m), 3.61 (1H, t, J = 7.8 Hz) , 3.28 (1H, t, J = 7.5 Hz), 2.76-2.66 (2H, m), 2.57-2.45 (1H, m), 2.15 (1H, br s), 2.05-1.89 (2H, m), 1.42 ( 9H, s), 1.27 (9H, s), 0.96 (3H, d, J = 7.1 Hz).
(9) Optically active substance of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride
2- (2-Benzylaminoethyl) -3-methylazetidine-1,2-dicarboxylic acid di-tert-butyl ester optically active substance (46.5 g), 4M hydrochloric acid 1,4-dioxane (230 ml) and water (4.1 ml) was mixed and stirred at 80 ° C. for 2 hours. The mixture was concentrated under reduced pressure, azeotroped with toluene, and then slurry washed with n-hexane / ethyl acetate (1/1, 440 ml) to give the title compound (30.1 g). 1 H-NMR (DMSO-D 6 ) δ: 10.24 (1H, br s), 9.64 (2H, br s), 8.90 (1H, br s), 7.58-7.53 (2H, m), 7.47-7.41 (3H , m), 4.21-4.10 (2H, m), 4.02-3.94 (1H, m), 3.46-3.37 (1H, m), 3.20-3.10 (1H, m), 2.99-2.85 (2H, m), 2.69 -2.54 (2H, m), 1.10 (3H, d, J = 7.2 Hz).
(10) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one
To a solution of 2- (2-benzylaminoethyl) -3-methylazetidine-2-dicarboxylic acid dihydrochloride optically active substance (29.1 g) and N, N-diisopropylethylamine (65 ml) in chloroform (290 ml), At room temperature, O- (7-azabenzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (41.3 g) was added and stirred for 4 hours. To this reaction mixture were added saturated aqueous sodium hydrogen carbonate solution (200 ml) and water (100 ml), and the mixture was extracted with chloroform (200 ml). The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 20/1 to 10/1) to give the titled compound (21.3 g). 1 H-NMR (DMSO-D 6 ) δ: 7.38-7.31 (2H, m), 7.30-7.22 (3H, m), 4.52 (1H, d, J = 14.8 Hz), 4.29 (1H, d, J = 14.8 Hz), 3.35-3.27 (2H, m), 3.22-3.17 (1H, m), 3.05 (2H, dd, J = 9.5, 4.0 Hz), 2.77-2.66 (1H, m), 2.16-2.10 (1H , m), 1.96-1.87 (1H, m), 0.94 (3H, d, J = 7.1 Hz).
(11) Optically active substance of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester
Concentrated sulfuric acid (4.8 ml) was slowly added dropwise to a suspension of lithium aluminum hydride (6.8 g) in tetrahydrofuran (300 ml) under ice cooling, and the mixture was stirred for 30 minutes. To this mixture was added dropwise a solution of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octan-5-one optically active substance (21.3 g) in tetrahydrofuran (100 ml) at the same temperature. Stir for 45 minutes. Water (7.0 ml), 4M aqueous sodium hydroxide solution (7.0 ml) and water (14.0 ml) were sequentially added to the reaction mixture, and the mixture was stirred as it was for 30 minutes. To this mixture was added anhydrous magnesium sulfate and ethyl acetate (100 ml), and the mixture was stirred and filtered through celite. Di-tert-butyl dicarbonate (23.4 g) was added to the filtrate at room temperature and stirred for 3 hours. The mixture was concentrated under reduced pressure to a half volume and washed twice with a saturated aqueous ammonium chloride solution (200 ml × 2). N-Hexane (200 ml) was added to the separated organic layer, and the mixture was extracted 5 times with a 10% aqueous citric acid solution. The separated aqueous layer was basified with 4M aqueous sodium hydroxide solution and extracted with chloroform. The organic layer was washed with a saturated aqueous sodium chloride solution (200 ml), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: chloroform / methanol = 40/1 to 20/1) to give the titled compound (15.6 g). 1 H-NMR (DMSO-D 6 ) δ: 7.34-7.27 (4H, m), 7.26-7.21 (1H, m), 3.84-3.69 (1H, m), 3.62-3.47 (2H, m), 3.19- 3.05 (1H, m), 3.02-2.92 (1H, m), 2.76-2.69 (1H, m), 2.47-2.24 (4H, m), 1.95-1.77 (1H, m), 1.36 (9H, s), 1.03 (3H, d, J = 7.0 Hz).
(12) Optically active substance of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester
20% of optically active form of 6-benzyl-3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (10.0 g) in tetrahydrofuran / methanol (50 ml / 50 ml) solution Palladium hydroxide on carbon (2.0 g) was added and hydrogenated at 4 atm for 24 hours. The mixture was filtered through Celite, and the filtrate was concentrated under reduced pressure to give the title compound (7.3 g). 1 H-NMR (DMSO-D 6 ) δ: 3.88-3.71 (1H, m), 3.44-3.06 (2H, m), 3.02-2.64 (4H, m), 2.55-2.38 (1H, m), 2.31- 2.15 (1H, m), 1.81-1.72 (1H, m), 1.37 (9H, s), 1.07 (3H, d, J = 7.0 Hz).
(13) Optical activity of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester body
The optically active substance (6.9 g) of 3-methyl-1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester was converted into 4-chloro-7H-pyrrolo [2,3-d] pyrimidine ( 4.3 g), potassium carbonate (7.7 g) and water (65 ml) and stirred for 4 hours at reflux. The mixture was cooled to room temperature, water (60 ml) was added, and the mixture was extracted with chloroform / methanol (10/1, 120 ml). The organic layer was washed successively with water, saturated aqueous ammonium chloride solution and saturated aqueous sodium chloride solution, and dried over anhydrous sodium sulfate. To this mixture, silica gel (4 g) was added, stirred for 10 minutes, filtered through celite, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / ethyl acetate = 1/1, then chloroform / methanol = 50/1 to 20/1) to give the title compound (10.0 g). Obtained. 1 H-NMR (DMSO-D 6 ) δ: 11.59 (1H, br s), 8.09 (1H, s), 7.12-7.09 (1H, m), 6.64-6.59 (1H, m), 4.09-3.66 (5H , m), 3.39-3.21 (1H, m), 2.64-2.44 (2H, m), 2.27-2.06 (1H, m), 1.36 (3H, s), 1.21 (6H, s), 1.11 (3H, d , J = 6.5 Hz).
(14) Optically active form of 4- (3-methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride
Optically active form of 3-methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] octane-1-carboxylic acid tert-butyl ester (9 0.5 g), 4M hydrochloric acid 1,4-dioxane (50 ml), chloroform (50 ml) and methanol (100 ml) were mixed and stirred at 60 ° C. for 30 minutes. The mixture was concentrated under reduced pressure and azeotroped with toluene to give the title compound (9.3 g). 1 H-NMR (DMSO-D 6 ) δ: 12.91 (1H, br s), 9.97-9.64 (2H, m), 8.45-8.35 (1H, m), 7.58-7.47 (1H, m), 7.04-6.92 (1H, m), 4.99-4.65 (1H, m), 4.32-3.21 (7H, m), 3.04-2.90 (1H, m), 2.46-2.31 (1H, m), 1.27 (3H, d, J = 6.0 Hz).
(15) 3- [3-Methyl-6- (7H-pyrrolo [2,3-d] pyrimidin-4-yl) -1,6-diazaspiro [3.4] oct-1-yl] -3-oxo Optically active form of propionitrile
4- (3-Methyl-1,6-diazaspiro [3.4] oct-6-yl) -7H-pyrrolo [2,3-d] pyrimidine dihydrochloride optically active substance (8.8 g) was converted to 1- The mixture was mixed with cyanoacetyl-3,5-dimethylpyrazole (6.8 g), N, N-diisopropylethylamine (20 ml) and 1,4-dioxane (100 ml) and stirred at 100 ° C. for 1 hour. The mixture was cooled to room temperature, saturated aqueous sodium hydrogen carbonate solution was added, and the mixture was extracted with chloroform / methanol (10/1). The separated organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: chloroform / methanol = 30/1 to 9/1). The residue obtained by concentration under reduced pressure was slurry washed with n-heptane / ethanol (2/1, 90 ml) to obtain a solid (7.3 g). The solid was slurried again with n-heptane / ethanol (5/1, 90 ml) to give the title compound as crystals 1 (6.1 g). 1 H-NMR (DMSO-D 6 ) δ: 11.60 (1H, br s), 8.08 (1H, s), 7.11 (1H, dd, J = 3.5, 2.4 Hz), 6.58 (1H, dd, J = 3.4 , 1.9 Hz), 4.18-4.14 (1H, m), 4.09-3.93 (3H, m), 3.84-3.73 (1H, m), 3.71 (1H, d, J = 19.0 Hz), 3.66 (1H, d, J = 18.7 Hz), 3.58 (1H, dd, J = 8.2, 6.0 Hz), 2.70-2.58 (2H, m), 2.24-2.12 (1H, m), 1.12 (3H, d, J = 7.1 Hz). [Α] D = + 47.09 ° (25 ° C., c = 0.55, methanol)
1-Butanol (39 ml) was added to the obtained crystal 1 (2.6 g), and the mixture was heated and stirred at 100 ° C. After complete dissolution, the solution was cooled to room temperature by 10 ° C. every 30 minutes and further stirred at room temperature overnight. The produced crystals were collected by filtration, washed with 1-butanol (6.2 ml), and dried under reduced pressure to give crystals 2 (2.1 g) of the title compound.
Janus kinase (JAK) inhibitors are of current interest for the treatment of various diseases including autoimmune diseases, inflammatory diseases, and cancer. To date, two JAK inhibitors have been approved by the U.S. Food & Drug Administration (FDA). Ruxolitinib has been approved for the treatment of primary myelofibrosis and polycythemia vera (PV), and tofacitinib has been approved for the treatment of rheumatoid arthritis. Other JAK inhibitors are in the literature. The compound 3-((3S,4R)-3-methyl-6-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1,6-diazaspiro[3.4]octan-1-yl)-3-oxopropanenitrile (Compound A) (see structure below) is an example of a spirocyclic JAK inhibitor reported in U.S. Pat. Pub. Nos. 2011/0136778 and International Pat. Pub. No. PCT/JP2016/070046.
[Chem. 1]
Step A. Preparation of S-MABB-HC (Compound [5])
[Chem. 2] Step 1
[Chem. 3] S-BAPO [1] (35.0 g, 212 mmol) was added to water (175 mL) at room temperature under nitrogen atmosphere. To the resulting suspension were added toluene (53 mL) and potassium carbonate (32.2 g, 233 mmol) at room temperature. To the resulting solution was added dropwise TBBA (434.4 g, 223 mmol) at room temperature, and then the used dropping funnel was washed with toluene (17 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 65°C for 21 hours, and then cooled to room temperature. After toluene (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (175 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of S-BBMO [2] (74.0 g, 212 mmol in theory). The given toluene solution of S-BBMO was used in the next step, assuming that the yield was 100 %.
A crude product of S-BBMO which was prepared by the same process was evaporated to dryness and then measured about NMR and MS. 1H-NMR (DMSO-d 6) δ: 7.36-7.13 (5H, m), 4.26 (1H, dd, J = 6.8, 3.9 Hz), 3.72 (2H, dd, J = 14.2, 6.8 Hz), 3.47-3.38 (1H, m), 3.30-3.08 (3H, m), 2.79 (1H, sext, J = 6.8 Hz), 1.35 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
MS: m/z = 280 [M+H] +
[0134]
Step 2
[Chem. 4] To the toluene solution of S-BBMO [2] (74.0 g, 212 mmol) were added toluene (200 mL), tetrahydrofuran (35 mL), and then triethylamine (25.7 g, 254 mmol) at room temperature under nitrogen atmosphere. To the mixture was added dropwise methanesulfonyl chloride (26.7 g, 233 mmol) at 0°C, and then the used dropping funnel was washed with toluene (10 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours and further at 65°C for 22 hours, and then cooled to room temperature. After sodium bicarbonate water (105 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with water (105 mL), aqueous layer was removed, and then the solvent was removed out of the organic layer in vacuo. Toluene (105 mL) was added to the residue, and the toluene solution was concentrated. The operation was repeated two more times to give a toluene solution of R-BCAB [3] (75.3 g, 212 mmol in theory). The given toluene solution of R-BCAB was used in the next step, assuming that the yield was 100 %.
A crude product of R-BCAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS. 1H-NMR (DMSO-d 6) δ: 7.28-7.11 (5H, m), 4.24-4.11 (1H, m), 3.80 (2H, d, J = 3.6 Hz), 3.24 (2H, d, J = 3.6 Hz), 2.98-2.78 (2H, m), 1.46-1.37 (12H, m).
MS: m/z = 298 [M+H] +
[0135]
Step 3
[Chem. 5] To the toluene solution of R-BCAB [3] (75.3 g, 212 mmol) were added tetrahydrofuran (88.0 mL) and 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (42.0 mL) at room temperature under nitrogen atmosphere. To the resulting solution was added dropwise a solution of lithium bis(trimethylsilyl)amide /tetrahydrofuran (195 mL, 233 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (17.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then warmed to room temperature. After water (175 mL) and toluene (175 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with aqueous ammonium chloride (175 mL) and then water (175 mL), and the solvent was removed out of the organic layer in vacuo. Ethyl acetate (175 mL) was added to the residue and the ethyl acetate solution was concentrated. The operation was repeated two more times to give an ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory). The given ethyl acetate solution of S-MABB was used in the next step, assuming that the yield was 100 %.
A crude product of S-MABB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS. 1H-NMR (DMSO-d 6) δ: 7.28-7.25 (10H, m), 3.75 (1H, d, J = 12.7 Hz), 3.68 (1H, d, J = 1.4 Hz), 3.66 (1H, d, J = 6.7 Hz), 3.46 (2H, d, J = 12.7 Hz), 3.30-3.17 (2H, m), 2.95 (1H, dd, J = 6.2, 1.2 Hz), 2.77 (1H, dd, J = 6.1, 2.2 Hz), 2.65-2.55 (1H, m), 2.48-2.40 (2H, m), 1.35 (9H, s), 1.35 (9H, s), 1.12 (3H, d, J = 7.2 Hz), 1.09 (3H, d, J = 6.2 Hz).
MS: m/z = 262 [M+H] +
[0136]
Step 4
[Chem. 6] To the ethyl acetate solution of S-MABB [4] (66.5 g, 212 mmol in theory) were added ethyl acetate (175 mL) and active carbon (3.5 g) under nitrogen atmosphere, and then the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the residue on the filter was washed with ethyl acetate (175 mL). The washings were added to the filtrate. To the solution was added S-MABB-HC crystal (17.5 mg) that was prepared according to the method described herein at 0°C, and then 4 M hydrogen chloride/ethyl acetate (53.0 mL, 212 mmol) was dropped thereto at 0°C. The reaction mixture was stirred at 0°C for 17 hours, and then the precipitated solid was collected on a filter, and washed with ethyl acetate (70 mL). The resulting wet solid was dried in vacuo to give S-MABB-HC [5] (48.3 g, 162 mmol, yield: 76.4 %).
S-MABB-HC which was prepared by the same process was measured about NMR, MS, and Cl-content. 1H-NMR (DMSO-d 6) δ: 11.08 (1H, br s), 10.94 (1H, br s), 7.52-7.42 (10H, m), 5.34 (1H, t, J = 8.4 Hz), 4.90 (1H, br s), 4.45-4.10 (5H, m), 3.92-3.49 (3H, br m), 3.10-2.73 (2H, br m), 1.35 (9H, s), 1.29 (9H, s), 1.24 (3H, d, J = 6.7 Hz), 1.17 (3H, d, J = 7.4 Hz).
MS: m/z = 262 [M+H-HCl] +
Cl content (ion chromatography): 11.9 % (in theory: 11.9 %).
[0137]
Step B. Preparation of S-MACB-HC (Compound [6])
[Chem. 7] To a solution of S-MABB-HC [5] (5.0 g, 16.8 mmol) in methanol (15.0 mL) was added 5 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., PH type, 54.1 % water-content 1.0 g) at room temperature under nitrogen atmosphere. The reaction vessel was filled with hydrogen, the reaction mixture was stirred at hydrogen pressure of 0.4 MPa at room temperature for 12 hours, the hydrogen in the reaction vessel was replaced with nitrogen, and then the 5 % palladium carbon was removed by filtration. The reaction vessel and the 5 % palladium carbon were washed with methanol (10 mL). The washings were added to the filtrate to give a methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory). The given methanol solution of S-MACB-HC was used in the next step, assuming that the yield was 100 %.
A crude product of S-MACB-HC which was prepared by the same process was evaporated to dryness and then measured about NMR and MS. 1H-NMR (DMSO-d 6) δ: 9.60 (br s, 1H), 4.97 (d, 1H, J = 9.2 Hz), 4.61 (d, 1H, J = 8.4 Hz), 4.01 (dd, 1H, J = 10.0, 8.4 Hz), 3.78-3.74 (m, 1H), 3.54 (dd, 1H, J = 9.6, 8.4 Hz), 3.35 (dd, 1H, J = 10.0, 6.0 Hz), 3.15-3.03 (m, 1H), 3.00-2.88 (m, 1H), 1.49 (s, 9H), 1.47 (s, 9H), 1.22 (d, 3H, J = 6.8 Hz), 1.14 (d, 3H, J = 7.2 Hz).
MS: m/z = 172 [M+H] + (free form)
[0138]
Step C. Preparation of S-ZMAB (Compound [7])
[Chem. 8] To the methanol solution of S-MACB-HC [6] (24.8 g, 16.8 mmol in theory) was added dropwise N,N-diisopropylethylamine (4.8 g, 36.9 mmol) at room temperature under nitrogen atmosphere, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. To the resulting reaction mixture was added dropwise benzyl chloroformate (3.0 g, 17.6 mmol) at 0°C, and then the used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C for 1 hour, and then the solvent was removed in vacuo. After toluene (25.0 mL) and an aqueous solution of citric acid (25.0 mL) was added to the residue and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with sodium bicarbonate water (25.0 mL) and then water (25.0 mL), and the solvent in the organic layer was removed out of the organic layer in vacuo. Toluene (15.0 mL) was added to the residue and the toluene solution was concentrated. The operation was repeated one more time to give a toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol in theory). The given toluene solution of S-ZMAB was used in the next step, assuming that the yield was 100 %.
A crude product of S-ZMAB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS. 1H-NMR (CDCl 3) δ: 7.38-7.28 (m, 10H), 5.16-5.04 (m, 4H), 4.60 (d, 1H, J = 9.2 Hz), 4.18-4.12 (m, 2H), 4.04 (t, 1H, J = 8.6 Hz), 3.66 (dd, 1H, J = 7.6, 7.2 Hz), 3.50 (dd, 1H, J = 8.0, 5.2 Hz), 3.05-2.94 (m, 1H), 2.60-2.50 (m, 1H), 1.43 (br s, 18H), 1.33 (d, 3H, J = 6.5 Hz), 1.15 (d, 3H, J = 7.2 Hz).
MS: m/z = 328 [M+Na] +.
[0139]
Step D. Preparation of RS-ZMBB (Compound [8])
[Chem. 9] To the toluene solution of S-ZMAB [7] (6.9 g, 16.8 mmol) was added tetrahydrofuran (15.0 mL) at room temperature under nitrogen atmosphere. A solution of lithium bis(trimethylsilyl)amide/tetrahydrofuran (14.7 mL, 17.6 mmol) was added dropwise to the toluene solution at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 6 hours, and then a solution of TBBA (3.4 g, 17.6 mmol) in tetrahydrofuran (2.5 mL) was added dropwise to the reaction mixture at -70°C. The used dropping funnel was washed with tetrahydrofuran (2.5 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at -70°C for 1 hour, and then warmed to room temperature. To the reaction mixture were added an aqueous ammonium chloride (25 mL) and toluene (25 mL) and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with an aqueous solution of citric acid (25 mL, x 2), sodium bicarbonate water (25 mL), and then water (25 mL), and then the solvent was removed out of the organic layer in vacuo. Acetonitrile (15 mL) was added to the residue and the acetonitrile solution was concentrated. The operation was repeated two more times. Acetonitrile (15 mL) and active carbon (0.25 g) were added to the residue, the mixture was stirred at room temperature for 2 hours. The active carbon was removed by filtration, and the reaction vessel and the residue on the filter was washed with acetonitrile (10 mL). The washings were added to the filtration, and then the filtration was concentrated in vacuo to give an acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory). The given acetonitrile solution of RS-ZMBB was used in the next step, assuming that the yield was 100 %.
A crude product of RS-ZMBB which was prepared by the same process was evaporated to dryness and then measured about NMR and MS. 1H-NMR (DMSO-d 6) δ: 7.38-7.29 (m, 5H), 5.09-4.96 (m, 2H), 3.91 (t, 0.4H, J = 8.0 Hz), 3.79 (t, 0.6H, J = 8.0 Hz), 3.55 (t, 0.4H, J = 7.2 Hz), 3.46 (t, 0.6H, J = 7.5 Hz), 3.14-3.04 (m, 1H), 2.83-2.72 (m, 2H), 1.38 (br s, 9H), 1.37 (br s, 3.6H), 1.34 (br s, 5.4H), 1.12-1.09 (m, 3H).
MS: m/z = 420 [M+H] +.
[0140]
Step E. Preparation of RS-ZMAA-DN.2H2O (Compound [9])
[Chem. 10] To the acetonitrile solution of RS-ZMBB [8] (13.2 g, 16.8 mmol in theory) was added acetonitrile (15 mL) at room temperature under nitrogen atmosphere. p-Toluenesulfonic acid mono-hydrate (6.4 g, 33.6 mmol) was added to the solution at room temperature. The reaction mixture was stirred at 50°C for 12 hours, and then cooled to room temperature, and water (7.5 mL) was added dropwise to the reaction mixture. The reaction mixture was cooled to 0°C, and then 4 mol/L aqueous sodium hydroxide (17.6 mL, 70.5 mmol) was added dropwise thereto. After stirring the reaction mixture at room temperature for 1 hour, acetonitrile (75 mL) was added dropwise thereto at room temperature, and the reaction mixture was stirred for 3 hours. The precipitated solid was collected on a filter, and washed with a mixture of acetonitrile : water = 4 : 1 (10 mL) and then acetonitrile (10 mL). The resulting wet solid was dried in vacuo to give RS-ZMAA-DN .2H 2O [9] (5.2 g, 13.4 mmol, yield: 85.4 %).
RS-ZMAA-DN .2H 2O which was prepared by the same process was measured about NMR, MS, Na-content, and water-content. 1H-NMR (DMSO-d 6) δ: 7.32-7.22 (m, 5H), 4.97 (d, 1H, J = 12.7 Hz), 4.84 (d, 1H, J = 12.7 Hz), 3.79 (t, 1H, J = 8.0 Hz), 3.29 (d, 1H, J = 14.8 Hz), 3.16-3.12 (m, 1H), 2.17-2.09 (m, 2H), 1.07 (d, 3H, J = 6.9 Hz).
MS: m/z = 352 [M+H] + (anhydrate)
Na content (ion chromatography): 13.3 % (after correction of water content)(13.1 % in theory)
Water content (Karl Fischer’s method): 9.8 % (9.3 % in theory)
[0141]
Step F. Preparation of RS-ZMAA (Compound [10])
[Chem. 11] To 1 mol/L hydrochloric acid (180 mL) were added RS-ZMAA-DN .2H 2O [9] (30 g, 77.5 mmol) and acetonitrile (60 mL), and the mixture was stirred at room temperature for about 15 minutes. After ethyl acetate (240 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The organic layer was washed with 10 % brine (60 mL x 2). The organic layer was stirred with magnesium sulfate (6 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with ethyl acetate (60 mL). The filtrate and the washings are combined, and the solvent was removed out in vacuo. Tetrahydrofuran (240 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated two more times. Tetrahydrofuran (60 mL) was added to the residue to give a tetrahydrofuran solution of RS-ZMAA [10]. The given tetrahydrofuran solution of RS-ZMAA was used in the next step, assuming that the yield was 100 %.
RS-ZMAA which was prepared by the same process was measured about NMR and MS. 1H-NMR (DMSO-D 6) δ: 7.35-7.28 (m, 5H), 5.06-4.94 (m, 2H), 3.86 (dt, 1H, J = 48.4, 7.9 Hz), 3.50 (dt, 1H, J = 37.9, 7.4 Hz), 3.16-3.02 (br m, 1H), 2.91-2.77 (br m, 2H), 1.08 (d, 3H, J = 6.9 Hz)
MS: m/z = 308 [M+H] +.
[0142]
Step G. Preparation of RS-ZMOO (Compound [11])
[Chem. 12] To the tetrahydrofuran solution of RS-ZMAA [10] (25.8 mmol in theory) was added tetrahydrofuran (50 mL) under nitrogen atmosphere. Boron trifluoride etherate complex (4.40 g) was added dropwise thereto at 0°C to 5°C. The used dropping funnel was washed with tetrahydrofuran (5 mL) and the washings were added to the reaction mixture. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (43.0 mL) at 0°C to 5°C, and the reaction mixture was stirred at 0°C to 5°C for about 30 minutes, and then further stirred at room temperature overnight. To the reaction mixture was added dropwise 1.2 mol/L borane-tetrahydrofuran complex (21.1 mL) at 0°C to 5°C, and then the reaction mixture was stirred at room temperature overnight. After stirring, water (40 mL) was added dropwise to the reaction mixture at 0°C to 15°C. To the reaction mixture was added sodium bicarbonate (5.42 g) at 0°C to 15°C. The sodium bicarbonate left in the vessel was washed with water (10 mL), and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 hours, and then toluene (50 mL) was added thereto and the reaction mixture was further stirred. The organic layer was separated out. The resulting organic layer was washed with 10 % brine (20 mL x 1), a mixture (x 3) of 5 % sodium bicarbonate water (20 mL) and 10 % brine (20 mL), a mixture (x 1) of 5 % aqueous potassium hydrogensulfate (10 mL) and 10 % brine (10 mL), and then 10 % brine (20 mL x 2). The organic layer was stirred with magnesium sulfate (8.9 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (20 mL). The washings were added to the filtration, and then the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (80 mL). The solution was concentrated in vacuo, and toluene (15 mL) was added thereto to give a toluene solution of RS-ZMOO [11]. The given toluene solution of RS-ZMOO was used in the next step, assuming that the yield was 100 %.
RS-ZMOO which was prepared by the same process was measured about NMR and MS. 1H-NMR (CDCl 3) δ: 7.39-7.30 (m, 5H), 5.10 (s, 2H), 4.15-4.01 (br m, 2H), 3.83-3.73 (br m, 3H), 3.48 (dd, 1H, J = 8.3, 6.4 Hz), 2.59-2.50 (br m, 1H), 2.46-2.40 (br m, 1H), 2.07-1.99 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 280 [M+H]+.
[0143]
Step H. Preparation of RS-ZMSS (Compound [12])
[Chem. 13] To the toluene solution of RS-ZMOO [11] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, triethylamine (5.27 g) was added dropwise thereto at -10°C to 10°C, and the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. To this reaction mixture was added dropwise methanesulfonyl chloride (5.69 g) at -10°C to 10°C, and then the used dropping funnel was washed with toluene (1.8 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 0°C to 10°C for about 2 hours, and then water (28 mL) was added dropwise thereto at 0°C to 20°C. The reaction mixture was stirred at 0°C to 20°C for about 30 minutes, and then, the organic layer was separated out. The resulting organic layer was washed twice with 10 % brine (18 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration, and the residue on the filter was washed with toluene (18 mL). The washings were added to the filtrate, and then the solvent was removed from the filtrate in vacuo. To the concentrated residue was added toluene up to 18 mL to give a toluene solution of RS-ZMSS [12]. The given toluene solution of RS-ZMSS was used in the next step, assuming that the yield was 100 %.
RS-ZMSS which was prepared by the same process was measured by NMR and MS. 1H-NMR (DMSO-D 6) δ: 7.37-7.27 (br m, 5H), 5.10-4.98 (m, 2H), 4.58-4.22 (br m, 4H), 3.84 (dt, 1H, J = 45.6, 8.1 Hz), 3.48-3.33 (br m, 1H), 3.17-3.10 (m, 6H), 2.81-2.74 (br m, 1H), 2.22-2.12 (m, 2H)
MS: m/z = 436 [M+H] +.
[0144]
Step I. Preparation of SR-ZMDB (Compound [13])
[Chem. 14] To a toluene solution of RS-ZMSS [12] (23.7 mmol in theory) was added toluene (55 mL) under nitrogen atmosphere. And, benzylamine (17.8 g) was added dropwise thereto at room temperature, and the used dropping funnel was washed with toluene (9.2 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at room temperature for about 1 hour, at 55°C to 65°C for about 3 hours, and then at 70°C to 80°C for 6 hours. After the reaction mixture was cooled to room temperature, 10 % NaCl (28 mL) was added dropwise thereto, and the reaction mixture was stirred at room temperature for about 30 minutes. After toluene (37 mL) was added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The resulting organic layer was washed with a mixture (x 2) of 10 % brine (18 mL) and acetic acid (2.84 g), and then 10 % brine (11 mL, x 1). The solvent of the organic layer was removed in vacuo to a half volume, and acetic anhydride (1.45 g) was added to the concentrated residue at room temperature. The mixture was stirred for about 3 hours. To the reaction mixture were added dropwise a solution of potassium hydrogensulfate (3.87 g) and water (92 mL) at room temperature. The reaction mixture was stirred, and then the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (18 mL), and toluene (73 mL) and then sodium bicarbonate (6.56 g) were added to the aqueous layer at room temperature, and the mixture was stirred. The organic layer was separated out, and washed with 10 % brine (11 mL). The organic layer was stirred with magnesium sulfate (2.75 g), the magnesium sulfate was removed by filtration. The residue on the filter was washed with toluene (18 mL), and the washings were added to the filtrate, and then the filtrate was concentrated in vacuo. Toluene (44 mL) was added to the concentrated residue to give a toluene solution of SR-ZMDB [13]. The given toluene solution of SR-ZMDB was used in the next step, assuming that the yield was 100 %. 1H-NMR (CDCl 3) δ: 7.35-7.20 (m, 10H), 5.08 (d, 2H, J = 23.6 Hz), 3.94 (q, 1H, J = 7.9 Hz), 3.73-3.42 (br m, 2H), 3.30-3.23 (m, 1H), 3.05 (dd, 1H, J = 19.7, 9.5 Hz), 2.79 (dt, 1H, J = 69.6, 6.1 Hz), 2.57-2.32 (br m, 4H), 1.96-1.89 (m, 1H), 1.09 (d, 3H, J = 6.9 Hz)
MS: m/z = 351 [M+H] +.
[0145]
Step J. Preparation of SR-MDOZ (Compound [14])
[Chem. 15] To a solution of 1-chloroethyl chloroformate (3.72 g) in toluene (28 mL) was added dropwise the toluene solution of SR-ZMDB [13] (23.7 mmol in theory) at 0°C to 10°C under nitrogen atmosphere, and then the used dropping funnel was washed with toluene (4.6 mL) and the washings were added to the reaction mixture. To the reaction mixture was added triethylamine (718 mg) at 0°C to 10°C, and the reaction mixture was stirred at 15°C to 25°C for about 2 hours. Then, methyl alcohol (46 mL) was added to the reaction mixture, and the mixture was stirred at 50°C to 60°C for additional about 2 hours. The solvent of the reaction mixture was removed in vacuo to a volume of about less than 37 mL. To the concentrated residue was added dropwise 2 mol/L hydrochloric acid (46 mL) at 15°C to 20°C, and the mixture was stirred, and the aqueous layer was separated out. The resulting aqueous layer was washed with toluene (28 mL, x 2). To the aqueous layer were added 20 % brine (46 mL) and tetrahydrofuran (92 mL), and then 8 mol/L aqueous sodium hydroxide (18 mL) was added dropwise thereto at 0°C to 10°C. The organic layer was separated out from the reaction mixture, washed with 20 % brine (18 mL, x 2), and then the solvent of the organic layer was removed in vacuo. To the concentrated residue was added tetrahydrofuran (92 mL), and the solution was concentrated in vacuo. The operation was repeated one more time. The concentrated residue was dissolved in tetrahydrofuran (92 mL). The solution was stirred with magnesium sulfate (2.75 g), and the magnesium sulfate was removed by filtration. The residue on the filter was washed with tetrahydrofuran (28 mL), the washings were added to the filtrate, and the filtrate was concentrated in vacuo. The volume of the concentrated residue was adjusted to about 20 mL with tetrahydrofuran to give a tetrahydrofuran solution of SR-MDOZ [14] (net weight: 4.01 g, 15.4 mol, yield: 65.0 %).
SR-MDOZ which was prepared by the same process was evaporated to dryness and then measured about NMR and MS. 1H-NMR (CDCl 3) δ: 7.37-7.28 (m, 5H), 5.08 (dd, 2H, J = 16.8, 12.8 Hz), 4.00 (dd, 1H, J = 17.1, 8.3 Hz), 3.40-3.31 (m, 1H), 3.24 (d, 1H, J = 12.7 Hz), 3.00 (dd, 1H, J = 54.9, 12.4 Hz), 2.87-2.57 (m, 3H), 2.47-2.27 (m, 1H), 1.91-1.80 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 261 [M+H] +.
[0146]
Step K. Preparation of SR-MDOZ-OX (Compound [15])
[Chem. 16] Under nitrogen atmosphere, oxalic acid (761 mg) was dissolved in tetrahydrofuran (40 mL), and the tetrahydrofuran solution of SR-MDOZ [14] (3.84 mmol in theory) was added dropwise to the solution of oxalic acid at room temperature. To the solution was added SR-MDOZ-OX crystal (1 mg) that was prepared according to the method described herein at room temperature, and the mixture was stirred at room temperature for about 3.5 hours to precipitate the crystal. To the slurry solution was added dropwise the tetrahydrofuran solution of SR-MDOZ (3.84 mmol) at room temperature, and the mixture was stirred at room temperature for about 1 hour. The slurry solution was heated, and stirred at 50°C to 60°C for about 2 hours, and then stirred at room temperature overnight. The slurry solution was filtrated, and the wet crystal on the filter was washed with tetrahydrofuran (10 mL), dried in vacuo to give SR-MDOZ-OX [15] (2.32 g, 6.62 mol, yield: 86.2 %).
SR-MDOZ-OX which was prepared by the same process was measured about NMR, MS, and elementary analysis. 1H-NMR (DMSO-D 6) δ: 7.37-7.30 (m, 5H), 5.15-5.01 (m, 2H), 3.92 (dt, 1H, J = 43.5, 8.4 Hz), 3.48-3.12 (br m, 5H), 2.67-2.56 (m, 1H), 2.46-2.35 (m, 1H), 2.12-2.05 (m, 1H), 1.13 (d, 3H, J = 6.9 Hz)
MS: m/z = 261 [M+H] +
elementary analysis: C 58.4wt % , H 6.4wt % , N 7.9 % wt % (theoretically, C 58.3wt % , H 6.3wt % , N 8.0wt % )
[0147]
Step L. Preparation of SR-MDPZ (Compound [16])
[Chem. 17] To SR-MDOZ-OX [15] (12.0 g, 34.2 mmol) were added ethanol (36 mL), water (72 mL), CPPY [20] (5.36 g, 34.9 mmol), and then K 3PO 4 (21.8 g, 103 mmol) under nitrogen atmosphere. The reaction mixture was stirred at 80°C for 5 hours, and then cooled to 40°C. Toluene (120 mL) was added thereto at 40°C, and the organic layer was separated out. The resulting organic layer was washed with 20 % aqueous potassium carbonate (48 mL), followed by washing twice with water (48 mL). The solvent of the organic layer was then removed in vacuo. tert-butanol (60 mL) was added to the residue and the tert-butanol solution was concentrated. The operation was repeated two more times. tert-Butanol (36 mL) was added to the concentrated residue to give a solution of SR-MDPZ [16] in tert-butanol (61.1 g, 34.2 mmol in theory). The given tert-butanol solution of SR-MDPZ was used in the next step, assuming that the yield was 100 %.
SR-MDPZ which was prepared by the same process was isolated as a solid from a mixture of ethyl acetate and n-heptane, and then measured about NMR and MS. 1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.41-7.26 (br m, 3H), 7.22-7.08 (br m, 3H), 6.64-6.51 (br m, 1H), 5.07-4.91 (br m, 2H), 4.09-3.67 (br m, 5H), 3.47-3.32 (br m, 1H), 2.67-2.55 (br m, 2H), 2.21-2.15 (br m, 1H), 1.11 (d, 3H, J = 6.9 Hz).
MS: m/z = 378 [M+H] +
[0148]
Step M. Preparation of SR-MDOP (Compound [17])
[Chem. 18] To the solution of SR-MDPZ [16] in tert-butanol (34.2 mmol in theory) were added ammonium formate (10.8 g, 171 mmol), water (60 mL), and 10 % palladium carbon (made by Kawaken Fine Chemicals Co., Ltd., M type, 52.6 % water-content, 1.20 g) under nitrogen atmosphere. The reaction mixture was stirred at 40°C for 13 hours, and then cooled to room temperature, and the resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tert-butanol (24 mL), the washings was added to the filtrate, and 8 M aqueous sodium hydroxide (25.7 mL, 205 mmol) and sodium chloride (13.2 g) were added to the filtrate. The reaction mixture was stirred at 50°C for 2 hours, and then toluene (84 mL) was added thereto at room temperature, and the organic layer was separated out. The resulting organic layer was washed with 20 % brine (60 mL), stirred with anhydrous sodium sulfate, and then the sodium sulfate was removed by filtration. The residue on the filter was washed with a mixture of toluene : tert-butanol = 1 : 1 (48 mL), the washings was added to the filtrate, and the filtrate was concentrated in vacuo. To the concentrated residue was added toluene (60 mL), and the solution was stirred at 50°C for 2 hours, and then the solvent was removed in vacuo. To the concentrated residue was added toluene (60 mL) again, and the solution was concentrated. To the concentrated residue was added toluene (48 mL), and the solution was stirred at room temperature for 1 hour, and then at ice temperature for 1 hour. The precipitated solid was collected on a filter, and washed with toluene (24 mL). The resulting wet solid was dried in vacuo to give SR-MDOP [17] (7.07 g, 29.1 mmol, yield: 84.8 %).
SR-MDOP which was prepared by the same process was measured about NMR and MS. 1H-NMR (DMSO-d 6) δ: 11.57 (br s, 1H), 8.07 (s, 1H), 7.10 (d, 1H, J = 3.2 Hz), 6.58 (d, 1H, J = 3.2 Hz), 3.92-3.59 (br m, 4H), 3.49 (dd, 1H, J = 8.3, 7.2 Hz), 2.93 (dd, 1H, J = 7.2, 6.1 Hz), 2.61-2.53 (m, 2H), 2.12-2.01 (br m, 2H), 1.10 (d, 3H, J = 6.9 Hz).
MS: m/z = 244 [M+H] +.
[0149]
Step N. Preparation of Compound A mono-ethanolate (Compound [18])
[Chem. 19] Under nitrogen atmosphere, acetonitrile (60 mL) and triethylamine (416 mg, 4.11 mmol) were added to SR-MDOP [17] (5.00 g, 20.5 mmol), and to the solution was added dropwise a solution of DPCN [21] (3.69 g, 22.6 mmol) in acetonitrile (35 mL) at 45°C, and then the used dropping funnel was washed with acetonitrile (5.0 mL) and the washings were added to the reaction mixture. The reaction mixture was stirred at 45°C for 3 hours, and then cooled to room temperature. After 5 % sodium bicarbonate water (25 mL), 10 % brine (25 mL), and ethyl acetate (50 mL) were added to the reaction mixture and then the mixture was stirred, the organic layer was separated out. The solvent of the organic layer was removed out in vacuo. Tetrahydrofuran (50 mL) was added to the residue and the tetrahydrofuran solution was concentrated. The operation was repeated three more times. To the concentrated residue was added tetrahydrofuran (50 mL), and water was added the solution to adjust the water content to 5.5 %. The resulting precipitate was removed by filtration. The reaction vessel and the residue on the filter were washed with tetrahydrofuran (15 mL), the washings were added to the filtrate, and the solvent was removed out of the filtrate in vacuo. To the concentrated residue were added ethanol (50 mL) and Compound A crystal (5.1 mg) that was prepared according to the method described in the following Example 15. The mixture was stirred at room temperature for 1 hour, and concentrated in vacuo. To the residue was ethanol (50 mL), and the solution was concentrated again. To the concentrated residue was added ethanol (15 mL), and the solution was stirred at room temperature for 1 hour. The precipitated solid was collected on the filter, and washed with ethanol (20 mL). The resulting wet solid was dried in vacuo to give Compound A mono-ethanolate [18] (6.26 g, 17.6 mmol, yield: 85.5 %).
Compound A mono-ethanolate which was prepared by the same process was measured by NMR and MS. 1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.34 (t, 1H, J = 5.1 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.92 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 3.44 (dq, 2H, J = 6.7, 5.1 Hz), 2.69-2.60 (m, 2H), 2.23-2.13 (br m, 1H), 1.12 (d, 3H, J = 7.1 Hz), 1.06 (t, 3H, J = 6.7 Hz).
MS: m/z = 311 [M+H] +
[0150]
Step O. Purification of Compound A (Compound [19])
[Chem. 20] Compound A mono-ethanolate [18] (4.00 g, 11.2 mmol) and n-butanol (32 mL) were mixed under nitrogen atmosphere, and the mixture was dissolved at 110°C. The mixture was cooled to 85°C, and Compound A crystal (4.0 mg) that was prepared according to the method described herein was added thereto, and the mixture was stirred at 85°C for 2 hours, at 75°C for 1 hour, and then at room temperature for 16 hours. The precipitated solid was collected on a filter, and washed with n-butanol (8.0 mL) and then ethyl acetate (8.0 mL). The resulting wet solid was dried in vacuo to give Compound A [19] (3.18 g, 10.2 mmol, yield: 91.3 %).
Compound A which was prepared by the same process was measured by NMR and MS. 1H-NMR (DMSO-d 6) δ: 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.5 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.69-2.59 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H] +
[0151]
Using Compound A, which was prepared by the same method, the single crystal X-ray analysis was carried out.
(1) Preparation of Single crystal
To 10 mg of Compound A in a LaPha ROBO Vial(R) 2.0 mL wide-mouthed vial was added 0.5 mL of chloroform. The vial was covered with a cap, in which Compound A was completely dissolved. In order to evaporate the solvent slowly, a hole was made on the septum attached in the cap with a needle of a TERUMO(R) syringe, and the vial was still stood at room temperature. The resulting single crystal was used in the structural analysis.
(2) Measuring instrument
Beam line: SPring-8 BL32B2
Detector: Rigaku R-AXIS V diffractometer
(3) Measuring method
The radiant light of 0.71068Å was irradiated to the single crystal to measure X-ray diffraction data.
(4) Assay method
Using the X-ray anomalous scattering effect of the chlorine atom in the resulting Compound A chloroform-solvate, the absolute configuration of Compound A was identified as (3S,4R). Based on the obtained absolute configuration of Compound A, the absolute configurations of each process intermediate were identified.
REFERENCES
1: Nakagawa H, Nemoto O, Yamada H, Nagata T, Ninomiya N. Phase 1 studies to assess the safety, tolerability and pharmacokinetics of JTE-052 (a novel Janus kinase inhibitor) ointment in Japanese healthy volunteers and patients with atopic dermatitis. J Dermatol. 2018 Jun;45(6):701-709. doi: 10.1111/1346-8138.14322. Epub 2018 Apr 17. PubMed PMID: 29665062; PubMed Central PMCID: PMC6001687.
2: Nakagawa H, Nemoto O, Igarashi A, Nagata T. Efficacy and safety of topical JTE-052, a Janus kinase inhibitor, in Japanese adult patients with moderate-to-severe atopic dermatitis: a phase II, multicentre, randomized, vehicle-controlled clinical study. Br J Dermatol. 2018 Feb;178(2):424-432. doi: 10.1111/bjd.16014. Epub 2018 Jan 15. PubMed PMID: 28960254.
3: Tanimoto A, Shinozaki Y, Yamamoto Y, Katsuda Y, Taniai-Riya E, Toyoda K, Kakimoto K, Kimoto Y, Amano W, Konishi N, Hayashi M. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. Exp Dermatol. 2018 Jan;27(1):22-29. doi: 10.1111/exd.13370. Epub 2017 Jul 3. PubMed PMID: 28423239.
4: Nomura T, Kabashima K. Advances in atopic dermatitis in 2015. J Allergy Clin Immunol. 2016 Dec;138(6):1548-1555. doi: 10.1016/j.jaci.2016.10.004. Review. PubMed PMID: 27931536.
5: Amano W, Nakajima S, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. JAK inhibitor JTE-052 regulates contact hypersensitivity by downmodulating T cell activation and differentiation. J Dermatol Sci. 2016 Dec;84(3):258-265. doi: 10.1016/j.jdermsci.2016.09.007. Epub 2016 Sep 13. PubMed PMID: 27665390.
6: Tanimoto A, Shinozaki Y, Nozawa K, Kimoto Y, Amano W, Matsuo A, Yamaguchi T, Matsushita M. Improvement of spontaneous locomotor activity with JAK inhibition by JTE-052 in rat adjuvant-induced arthritis. BMC Musculoskelet Disord. 2015 Nov 6;16:339. doi: 10.1186/s12891-015-0802-0. PubMed PMID: 26546348; PubMed Central PMCID: PMC4636776.
7: Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, Dainichi T, Honda T, Otsuka A, Kimoto Y, Yamamoto Y, Tanimoto A, Matsushita M, Miyachi Y, Kabashima K. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. 2015 Sep;136(3):667-677.e7. doi: 10.1016/j.jaci.2015.03.051. Epub 2015 Jun 24. PubMed PMID: 26115905.
8: Tanimoto A, Ogawa Y, Oki C, Kimoto Y, Nozawa K, Amano W, Noji S, Shiozaki M, Matsuo A, Shinozaki Y, Matsushita M. Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflamm Res. 2015 Jan;64(1):41-51. doi: 10.1007/s00011-014-0782-9. Epub 2014 Nov 12. PubMed PMID: 25387665; PubMed Central PMCID: PMC4286029.
References
“Anzupgo EPAR”. European Medicines Agency. 25 July 2024. Retrieved 25 July 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
“Anzupgo PI”. Union Register of medicinal products. 23 September 2024. Retrieved 27 September 2024.
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:
Synthesis of Delgocitinib
Dec 26,2023
Synthesis of Delgocitinib
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:
A stereocontrolled kilogram scale synthesis of delgocitinib has been disclosed, beginning with an SN2 reaction involving bromolactone 128 and benzyl amine to provide α-amino lactone 129, which was isolated as the HCl salt after precipitation from hydrochloric acid in ethyl acetate. Amine 129 was then acylated with enantiomerically pure acid chloride 131 (prepared by thionyl chloride treatment of commercial acid 130) to furnish lactone 132. In the crucial spirocyclic ring ringforming sequence of the synthesis, lactone 132 was treated with LHMDS to form an enolate that underwent SN2 displacement of the chloride, forming the spirolactone 133 and establishing both stereocenters with 98:2 dr and 96% ee.
The lactone ring of 133 was then opened by an attack of potassium phthalimide on the γ- carbon, and the resulting carboxylic acid was converted to the ethyl ester by treatment with ethyl iodide. Finally, treatment with diethylenetriamine released phthalimide, providing a free amine for subsequent cyclization to spirolactam 134 via the corresponding ethyl ester intermediate. This sequence took place in 80% yield over four steps and provided the spirolactam in >99% de after recrystallization.
The carbonyl groups within spirolactam 134 were then reduced with lithium aluminum hydride and aluminum chloride in THF, and the resulting diamine 135 was crystallized as a succinic acid salt in 86% yield. The SNAr reaction of 135 with chloropyrrolopyrimidine 136 followed by hydrogenative removal of the benzyl protecting group provided amine 137 in 92% yield over 2 steps. Finally, amine 137 was acylated with cyanoacetyl pyrazole 138 and recrystallized from n-butanol with 3 wt % BHT to provide delgocitinib in 86% yield, >99% ee, and >99% de.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....














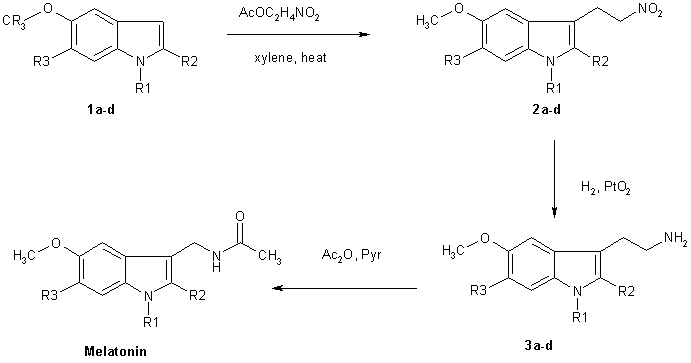
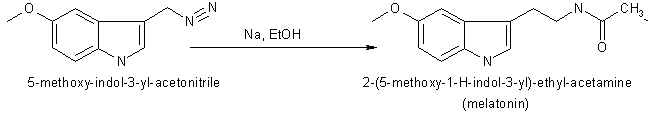
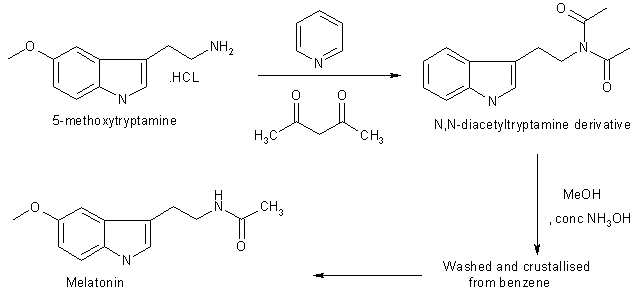
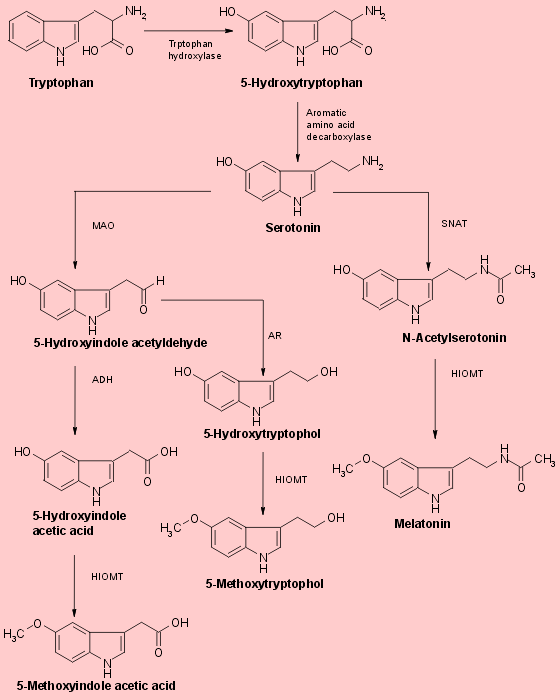 Fig.1 Biosynthesis of pineal methoxyindoles from serotonin
Fig.1 Biosynthesis of pineal methoxyindoles from serotonin




.png)









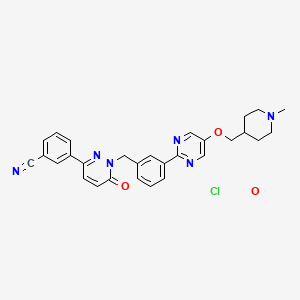
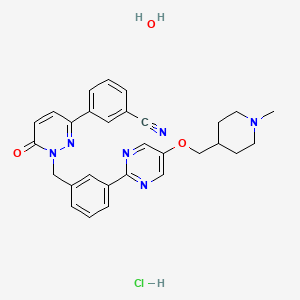
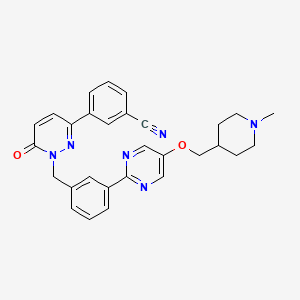


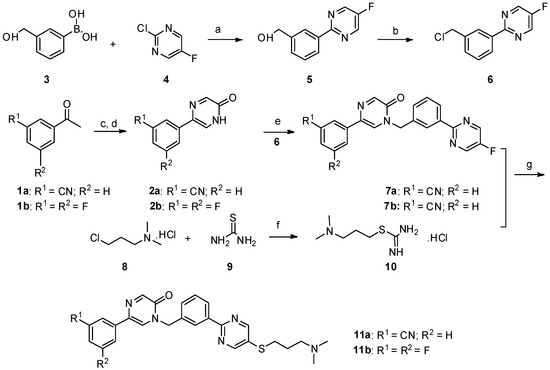
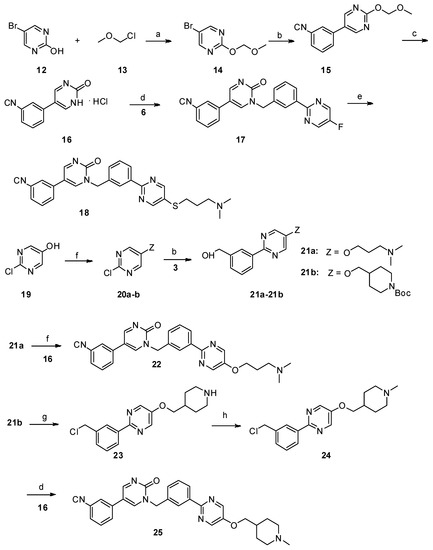
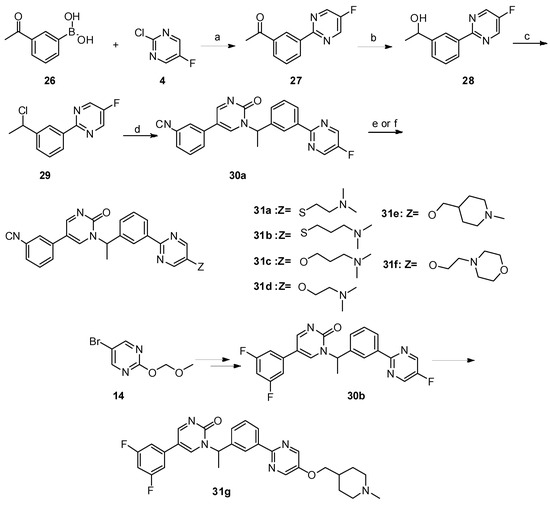
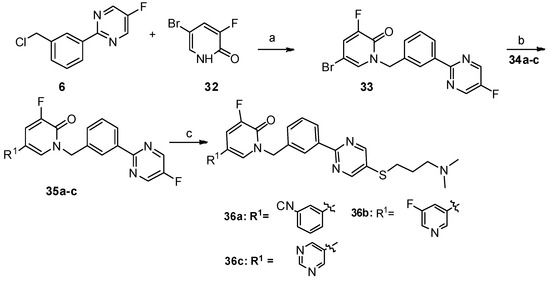
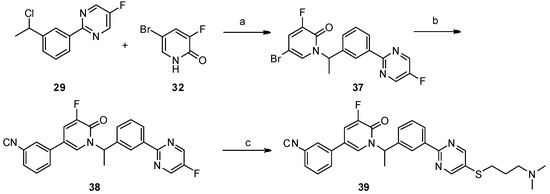
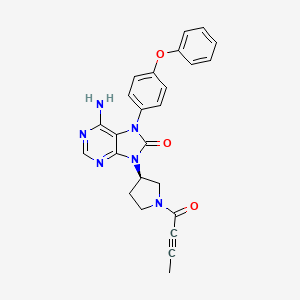



.png)
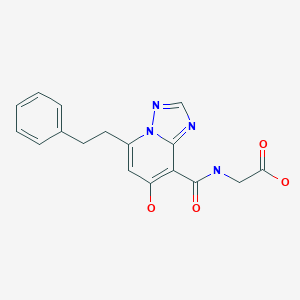
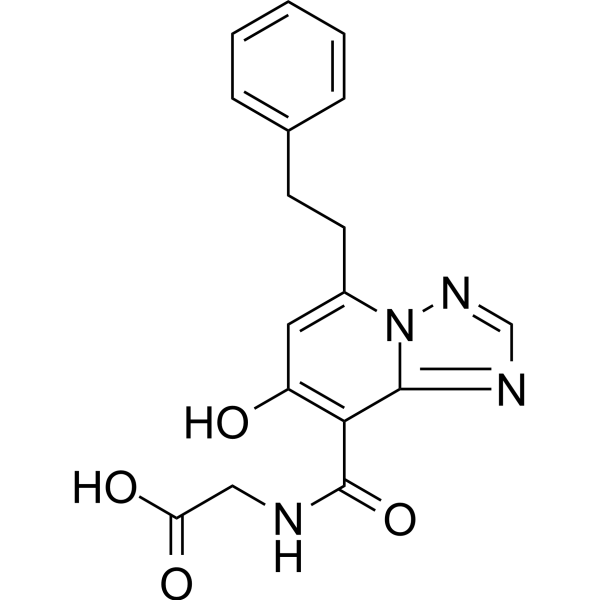


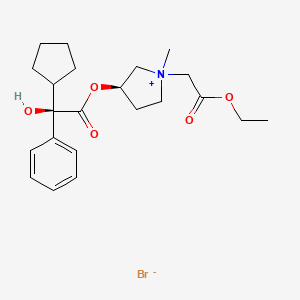
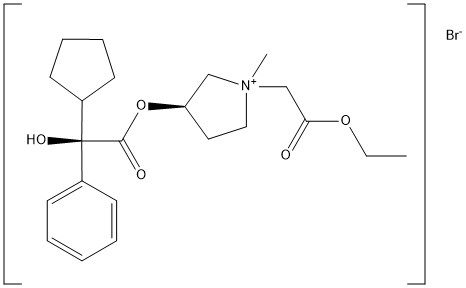
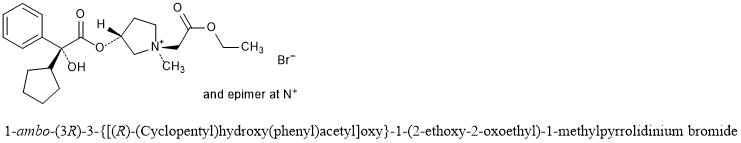






























{kind=link}
{kind=link}
{kind=link}
{kind=link}