
Home » Posts tagged 'Janssen Research & Development'
Tag Archives: Janssen Research & Development
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
JNJ-54861911, Atabecestat , атабецестат , أتابيسيستات ,
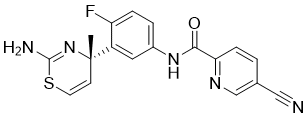
Atabecestat, JNJ-54861911
367.40, C18 H14 F N5 O S
Atabecestat is a beta-secretase inhibitor drug candidate.
(S)-N-(3-(2-amino-4-methyl-4H-1,3-thiazin-4-yl)-4-fluorophenyl)-5-cyanopicolinamide
WO 2017111042, 1H-NMR (CDCl3) δ: 1.71 (3H, s), 4.06 (3H, s), 6.29 (2H, d, J = 2.4 Hz), 7.07 (1H, dd, J = 11.3, 8.8 Hz), 7.65 (2H, dd, J = 6.8, 2.8 Hz), 7.86 (1H, ddd, J = 8.8, 4.1, 2.8 Hz), 8.19 (1H, dd, J = 8.1, 2.0 Hz), 8.43 (1H, d, J = 8.1 Hz), 8.89 (1H, d, J = 2.0 Hz), 9.81 (1H, s).
[α]D -11.8±1.0° (DMSO, 23°C, c=0.518)
![]()

Presented by: Yuji Koriyama, associate director at Shionogi & Co.
Target: β-site amyloid presursor protein cleaving enzyme 1 (BACE1), an enzyme whose buildup is implicated in Alzheimer’s disease
Disease: Alzheimer’s disease
Reporter’s notes: Presented by Koriyama, who told the audience he was attending the ACS National Meeting for the first time, JNJ-5486911 joins dozens of clinical candidates from many companies in Phase II and III trials to treat Alzheimer’s disease. Researchers started with a hit that inhibited BACE1 with approximately 2,600 nM affinity and advanced the program until finally reaching a compound with roughly 1 nM affinity. The compound is being jointly developed by Shionogi & Co. and Janssen Pharmaceuticals.
- Originator Shionogi
- Developer Janssen Research & Development
- Class Antidementias; Small molecules
- Mechanism of Action Amyloid precursor protein secretase inhibitors
Highest Development Phases
- Phase II/III Alzheimer’s disease
Most Recent Events
- 16 Jul 2017 Pharmacodynamics data from preclinical trials in Alzheimer’s disease presented at the Alzheimer’s Association International Conference (AAIC-2017)
- 15 Dec 2016 Biomarkers information updated
- 01 Jun 2016 Janssen Research & Development completes a phase I pharmacokinetic interaction trial in Healthy volunteers in Germany (PO) (NCT02611518)

SYNTHESIS


PATENTS
| Applicants: | SHIONOGI & CO., LTD. [JP/JP]; 1-8, Doshomachi 3-chome, Chuo-ku, Osaka-shi, Osaka 5410045 (JP) (For All Designated States Except US). HORI, Akihiro [JP/JP]; (JP) (For US Only). YONEZAWA, Shuji [JP/JP]; (JP) (For US Only). FUJIKOSHI, Chiaki [JP/JP]; (JP) (For US Only). MATSUMOTO, Sae [JP/JP]; (JP) (For US Only). KOORIYAMA, Yuuji [JP/JP]; (JP) (For US Only). UENO, Tatsuhiko [JP/JP]; (JP) (For US Only). KATO, Terukazu [JP/JP]; (JP) (For US Only) |
| Inventors: | HORI, Akihiro; (JP). YONEZAWA, Shuji; (JP). FUJIKOSHI, Chiaki; (JP). MATSUMOTO, Sae; (JP). KOORIYAMA, Yuuji; (JP). UENO, Tatsuhiko; (JP). KATO, Terukazu; (JP) |
PATENT
PATENT


PATENT
WO 2017111042




Scheme 1-D
[Chem. 27]

Example 1-4
Preparation of Compound 15
[Chem. 31]

Compound 12 (3.0 g, 20.3 mmol) was dissolved in N-methylpyrrolidone (18 mL), and the solution was cooled to 5°C. Thionyl chloride (3.1 g, 26.1 mmol) was added to obtain a solution of Compound 13.
To a suspension of Compound 11 (5.0 g, 16.8 mmol) in ethyl acetate (50 mL) were added sodium bicarbonate (3.5 g, 42.0 mmol) and water (50 mL), and the mixture was stirred for 5 min at 20°C.
The layers were separated, and the organic layer was concentrated to 10 g under reduced pressure. N-Methylpyrrolidone (5 mL) and 35% hydrochloric acid (0.9 g) were added, and the mixture was cooled to 3°C. The solution of Compound 13 and N-methylpyrrolidone (1.5 mL) were added to obtain a solution of Compound 15.
The solution of Compound 15 was added to a mixture of water (15 mL) and ethyl acetate (10 mL). After stirring the mixture for 1 hour, triethylamine (14.8 g, 14.6 mmol), N-methylpyrrolidone (1.5 mL) and water (5 mL) were added and further stirred for 1 hour. Water (45 mL) was added, and the mixture was stirred for 1 hour, filtered and dried to obtain crystals of Compound 15 (Crystalline Form I, 5.71 g, 92.4%).
Compound 15
1H-NMR (CDCl3) δ: 1.71 (3H, s), 4.06 (3H, s), 6.29 (2H, d, J = 2.4 Hz), 7.07 (1H, dd, J = 11.3, 8.8 Hz), 7.65 (2H, dd, J = 6.8, 2.8 Hz), 7.86 (1H, ddd, J = 8.8, 4.1, 2.8 Hz), 8.19 (1H, dd, J = 8.1, 2.0 Hz), 8.43 (1H, d, J = 8.1 Hz), 8.89 (1H, d, J = 2.0 Hz), 9.81 (1H, s).
[α]D -11.8±1.0° (DMSO, 23°C, c=0.518)
Example 1-5
To a suspension of Compound 11 (1831 g, 6.2 mol) in ethyl acetate (18L) were added sodium bicarbonate (1293 g, 15.4 mol) and water (18L), and the mixture was stirred for 5 min at 20°C. The layers were separated, and the organic layer was concentrated to 3.8 kg under reduced pressure to obtain a concentrated solution of Compound 14.
Compound 12 (912 g, 6.2 mol) was dissolved in N-methylpyrrolidone (64L), and the solution was cooled to 4°C. Thionyl chloride (951 g, 8.0 mol) was added, and the mixture was stirred for 30 min. The concentrated solution of Compound 14 was added to obtain a solution of Compound 15.
The solution of Compound 15 and N-methylpyrrolidone (1.6 L) were added to water (18 L), and the mixture was stirred for 40 min at 25°C. 24% sodium hydroxide in water (5 kg), sodium bicarbonate (259 g, 3.1 mmol) and water (2.7 L) were added to the mixture. The mixture was stirred for 1 hour, filtered and dried to obtain crystals (metastable Form II) of Compound 15 (1.93 kg, 85.4%).
Example 1-3
Preparation of Compound 11
[Chem. 30]

A suspension of Compound 9 (20.0 g, 29.0 mmol) in N,N-dimethylacetamide (30 mL) was cooled to 5°C. 1,8-diazabicyclo(5,4,0)-7-undecene (39.7 g, 260.8 mmol) was added, and the mixture was stirred for 22 hours. Water (70 mL) was added to afford a solution of Compound 10.
To a mixture of ethyl acetate (200 mL), water (40 mL) and 62% sulfuric acid (12.7 g) was added the solution of Compound 10, and the mixture was cooled to 10°C. 15% sulfuric acid (3.7 g) was added, and the mixture was warmed to 20°C. The layers were separated, and the organic layer was washed with 5% sodium chloride in water (95 g). The layers were separated, and the organic layer was concentrated in vacuo to 42 mL. Ethyl acetate (20 mL) and 50% potassium carbonate in water (20 g) were added, and the mixture was warmed to 40°C. 4-chlorobenzenethiol (6.29 g, 43.5 mmol) and ethyl acetate (11 mL) were added, and the mixture was stirred for 1 hour. After cooling to 20°C, ethyl acetate (100 mL), water (68 mL) and 15% hydrochloric acid (42.6 g) were added. The layers were separated, and ethyl acetate (149 mL) and 20% potassium carbonate in water (40.5 g) were added to the aqueous layer. The layers were separated, and the organic layer was washed with water (100 mL). The layers were separated, and the organic layer was concentrated to 20 mL. Acetic acid (1.7 g, 29.0 mmol) was added, and the mixture was cooled to 5°C and stirred for 90 min, filtered and dried to afford 7.19 g of crystals of Compound 11 (yield: 83.4%, optical purity of (S)-isomer: 100%).
Compound 11
1H-NMR (DMSO-d6) δ: 6.74 (1H, dd, J=11.86, 8.56 Hz), 6.62 (1H, dd, J=6.97, 2.93 Hz), 6.35-6.40 (2H, m), 6.11 (1H, dd, J=9.60, 4.71 Hz), 1.90 (3H, s), 1.49 (3H, s).
The optical purity was determined as follows.
(Sample Preparation)
25 mg of Compound 11 was weighed and dissolved in a solvent to prepare a 50 mL sample solution.
(Method)
Using liquid chromatography, the peak area was determined by automatic integration method for each of (R)- and (S)-isomers of Compound 11.
(Conditions)
Detector: ultraviolet absorptiometer (wave length: 230 nm)
Column: CHIRALCEL OD-RH, φ4.6×150 mm, 5 μm, (Daicel Corporation)
Column Temp.: constant at around 40°C
Mobile Phase: water/acetonitrile (LC grade)/methanol (LC grade)/triethylamine (1320:340:340:1)
Flow Rate: 1.0 mL/min (retention time of Compound 11: about 8 min for (R)-isomer, about 9 min for (S)-isomer)
Time span of measurement: over 15 min from the sample injection
Injection Volume: 10 μL
Sample Cooler Temp.: constant at around 25°C
Autoinjector Rinse Solution: water/acetonitrile (1:1)

//////////////JNJ-54861911, Atabecestat , атабецестат , أتابيسيستات ,Phase III , Alzheimer’s disease, DEMENTIA, Shionogi, Developer, Janssen Research & Development
C[C@]1(C=CSC(N)=N1)c3cc(NC(=O)c2ccc(C#N)cn2)ccc3F
J and J Submits Leukemia Drug, Ibrutinib for Approval

IBRUTINIB
1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidin-1-yl]prop-2-en-1-one
New Drug Application Submitted to U.S. FDA for Ibrutinib in the Treatment of Two B-Cell Malignancies
If approved, ibrutinib will address a high unmet need in relapsed/refractory chronic lymphocytic leukemia and relapsed/refractory mantle cell lymphoma
RARITAN, N.J., July 10, 2013
Janssen Research & Development, LLC announced the submission of a New Drug Application for ibrutinib to the U.S. Food and Drug Administration (FDA) for its use in the treatment of previously treated patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), and for its use in the treatment of previously treated patients with mantle cell lymphoma (MCL). The regulatory submission for ibrutinib is supported by data from two pivotal Phase 2 studies, one in relapsed/refractory CLL/SLL (PCYC-1102) and one in relapsed/refractory MCL (PCYC-1104), both of which were published in The New England Journal of Medicine online on June 19, 2013. Ibrutinib is a novel Bruton’s tyrosine kinase (BTK) inhibitor being jointly developed by Janssen and Pharmacyclics, Inc. for the treatment of B-cell malignancies.
If approved, ibrutinib would be the first in a class of oral BTK inhibitors and is one of the first medicines to file for FDA approval via the new Breakthrough Therapy Designation pathway. Ibrutinib will be co-commercialized in the U.S. by Janssen Biotech, Inc. and Pharmacyclics.
“The FDA submission is another important milestone for ibrutinib since we formed our strategic partnership with Pharmacyclics just 18 months ago,” said Peter F. Lebowitz, M.D., Ph.D., Global Oncology Head, Janssen. “Both companies recognize that there is great unmet need among these patient populations, and together in close collaboration with the FDA, as part of its Breakthrough Therapy Designation pathway, we have been able to accelerate the ibrutinib development program for the benefit of patients.”
About Chronic Lymphocytic Leukemia
Chronic Lymphocytic Leukemia (CLL) is a slow-growing blood cancer that starts in the white blood cells (lymphocytes), most commonly from B-cells. CLL is the second most common adult leukemia. Approximately 16,000 patients in the US are diagnosed each year with CLL. The prevalence of CLL is approximately 113,000 in the US. The disease is a chronic disease of the elderly with an average survival of about 5 years. Patients commonly receive multiple lines of treatment over the course of their disease.
In CLL the genetic mutation 17p deletion occurs when the short arm of chromosome 17 is missing. Del 17p is associated with abnormalities of a key tumor suppressor gene, TP53, which results in poor response to chemoimmunotherapy and worse treatment outcomes. It occurs in about 7% of treatment naive CLL patients and is estimated to be approximately 20% to 40% of relapsed or refractory patients harboring the mutation.
About Ibrutinib
Ibrutinib , previously publicly known as PCI-32765, is an experimental drug candidate for the treatment of various types of cancer. It was first synthesized at Celera Genomics as a selective inhibitor of Bruton’s tyrosine kinase (Btk).It was later discovered to have anti-lymphoma properties in vivo by scientists at Pharmacyclics, Inc.Ibrutinib is currently under development by Pharmacyclics, Inc and Johnson & Johnson‘sJanssen Pharmaceutical division for chronic lymphocytic leukemia, mantle cell lymphoma,diffuse large B-cell lymphoma, and multiple myeloma. It also has potential effects against autoimmune arthritis.
Janssen Biotech, Inc. and Pharmacyclics entered a collaboration and license agreement in December 2011 to co-develop and co-commercialize ibrutinib. Ibrutinib was designed to specifically target and selectively inhibit an enzyme called Bruton’s tyrosine kinase (BTK). BTK is a key mediator of at least three critical B-cell pro-survival mechanisms occurring in parallel – regulation of apoptosis, adhesion, and cell migration and homing. Through these multiple signals, BTK regulation helps to direct malignant B-cells to lymphoid tissues, thus allowing access to a micro environment necessary for survival.
The effectiveness of ibrutinib alone or in combination with other treatments is being studied in several B-cell malignancies, including chronic lymphocytic leukemia/small lymphocytic lymphoma, mantle cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, Waldenstrom’s macroglobulinemia and multiple myeloma. To date five Phase III trials have been initiated with ibrutinib and a total of 26 trials are currently registered on www.clinicaltrials.gov.
About Pharmacyclics
Pharmacyclics® is a clinical-stage biopharmaceutical company focused on developing and commercializing innovative small-molecule drugs for the treatment of cancer and immune mediated diseases. Our mission and goal is to build a viable biopharmaceutical company that designs, develops and commercializes novel therapies intended to improve quality of life, increase duration of life and resolve serious unmet medical healthcare needs; and to identify promising product candidates based on scientific development and administrational expertise, develop our products in a rapid, cost-efficient manner and pursue commercialization and/or development partners when and where appropriate.
Presently, Pharmacyclics has three product candidates in clinical development and several preclinical molecules in lead optimization. The Company is committed to high standards of ethics, scientific rigor, and operational efficiency as it moves each of these programs to viable commercialization.
The Company is headquartered in Sunnyvale, California and is listed on NASDAQ under the symbol PCYC. To learn more about how Pharmacyclics advances science to improve human healthcare visit at http://www.pharmacyclics.com.
