



TIRUPATI, INDIA
 .
.
Home » Posts tagged 'INDIA' (Page 4)
Tag Archives: INDIA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
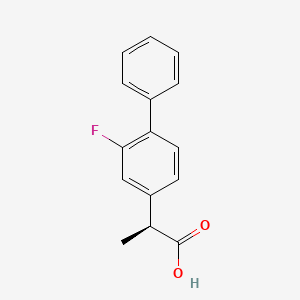
(1S)-(-)-beta-Pinene
(1S)-(1)-beta-Pinene, (1S)-(-)-beta-Pinene

 .
.
 .
.

13C NMR
 .
.
 .
.
APT
 .
.
DEPT
 .
.
COSY
 .
.
HETCOR

IR
MASS
 .
.
 .
.
RAMAN

|
take a tour
Amalner, Jalgaon, Maharashtra, India
Amalner – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Amalner
Amalner, India is a city and a municipal council in Jalgaon district in the state of Maharashtra, India, situated on the bank of the Bori River. Amalner is the …




 10000 devout Hindus were present for the Hindu Dharmajagruti Sabha at Amalner, Maharashtra
10000 devout Hindus were present for the Hindu Dharmajagruti Sabha at Amalner, Maharashtra




end of amalner…………
Daulatabad Fort Market
India / Maharashtra / Aurangabad /Daulatabad, Maharashtra – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Daulatabad,_Maharashtra
Daulatabad also known as Devagiri is a town which includes the Devagiri-Daulatabad fort It carries the distinction of remaining undefeated in battle.


Marketplace

Market place and Hotel/Dhaba
Nearby cities: Aurangabad, New Aurangabad, CIDCO. , Gangapur
Coordinates: 19°56’36″N 75°13’17″E






//////////////
Pirarubicin Hydrochloride
 Pirarubicin Hydrochloride
Pirarubicin Hydrochloride 
(7S,9S)-7-((2R,4S,5S,6S)-4-amino-6-methyl-5-((R)-tetrahydro-2H-pyran-2-yloxy)-tetrahydro-2H-pyran-2-yloxy)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione HCl
(CAS 95343-20-7)
THP Hydrochloride
(7S,9S)-7-((2R,4S,5S,6S)-4-amino-6-methyl-5-((R)-tetrahydro-2H-pyran-2-yloxy)-tetrahydro-2H-pyran-2-yloxy)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione HCl
MF C32H38ClNO12
MW 664.1
BASE 72496-41-4
Pirarubicin
or Pinorubicin
or Therarubicin
or (8S,10S)-10-(((2R,4S,5S,6S)-4-Amino-6-methyl-5-(((R)-tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H-pyran-2-yl)oxy)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione
or Pirarubicin
Pirarubicin Hcl is an analogue of the anthracycline anti-neoplastic doxorubicin, which is an inhibitor of Topo II.
Target: Topoisomerase
Pirarubicin is an anthracycline drug. An analogue of the anthracycline antineoplastic antibiotic doxorubicin. Pirarubicin intercalates into DNA and interacts with topoisomerase II, thereby inhibiting DNA replication and repair and RNA and protein synthesis. This agent is less cardiotoxic than doxorubicin and exhibits activity against some doxorubicin-resistant cell lines.

Pirarubicin (THP-adriamycin or THP-doxorubicin) was found during a search of new anthracycline antibiotics among 4′-O-substituted compounds having less toxicities than other anthracycline anticancer drugs in 1979 by Umezawa et al. In its preclinical studies, this compound possessed almost similar antitumor efficacies to doxorubicin, but was effective against doxorubicin-resistant P388 and other murine tumor cell lines. This compound was rapidly incorporated into tumor cells, inhibiting DNA polymerase alpha and subsequently DNA synthesis.
Inhibition of RNA synthesis was also noted. In the clinical studies, clinical responses were established against head and neck cancer, breast cancer, urogenital cancers, ovarian cancer, uterine cancer, acute leukemia, and malignant lymphoma, showing a wide antitumor spectrum clinically. Among the side effects, cardiac toxicity, alopecia and disturbance of the digestive organs were mild. From these results, THP-adriamycin seems to be a useful clinical drug for human solid tumors.
Pirarubicin (INN) is an anthracycline drug. An analogue of the anthracycline antineoplastic antibiotic doxorubicin. Pirarubicin intercalates into DNA and interacts with topoisomerase II, thereby inhibiting DNA replication and repair and RNA and protein synthesis. This agent is less cardiotoxic than doxorubicin and exhibits activity against some doxorubicin-resistant cell lines
 .
.
EP 0014853
https://www.google.com/patents/EP0014853B1?cl=en
")
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3S)-3-glycoloyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl 3-amino-2,3,6-trideoxy-4-O-[(2R)-tetrahydro-2H-pyran-2-yl]-α-L–lyxo-hexopyranoside | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
|
|
| Identifiers | |
| 72496-41-4 |
|
| L01DB08 | |
| PubChem | CID 3033521 |
| ChemSpider | 2298189 |
| UNII | D58G680W0G |
| KEGG | D01885 |
| ChEMBL | CHEMBL1398373 |
| Synonyms | (9S)-7-[(2R,4S,5S,6S)-4-amino-6-methyl-5-[(2R)-oxan-2-yl]oxyoxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione |
| Chemical data | |
| Formula | C32H37NO12 |
| 627.63 g/mol | |
TAKE A TOUR
Bijapur, Karnataka, INDIA
-
Bijapur – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/BijapurVijayapur city, formerly Bijapur, is the district headquarters of Bijapur District of Karnataka state. It is also the headquarters for Bijapur Taluka. Bijapur city is well …
.
 .
.







 GOLCONDA
GOLCONDA



Badami Cave Temple, near Bijapur





//////////
India’s Strides to buy Aspen’s Australian generic pharmaceutical business
![]()
India’s Strides to buy Aspen’s Australian generic pharmaceutical business
India-based Strides Arcolab has signed an agreement with subsidiaries of South African drugmaker Aspen Pharmacare Holdings to acquire its generic pharmaceutical business in Australia and certain branded pharmaceutical assets for around A$380m ($300m).
see
About Strides
-
 Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on development and manufacture of IP-led niche generics and bio-pharmaceuticals. It is also among the world’s largest manufacturers of specialty soft gelatin capsules. With world-class manufacturing facilities, an innovative R&D hub in Bangalore and a strong commercial platform to market branded and commodity generics globally, Strides has earned a reputation for building and scaling profitable businesses in a short span of time.
Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on development and manufacture of IP-led niche generics and bio-pharmaceuticals. It is also among the world’s largest manufacturers of specialty soft gelatin capsules. With world-class manufacturing facilities, an innovative R&D hub in Bangalore and a strong commercial platform to market branded and commodity generics globally, Strides has earned a reputation for building and scaling profitable businesses in a short span of time.

.
Chandos Street, St Leonards

///////////
S-flurbiprofen (TT-063)
Cas 51543-39-6,
MW 244.26,
MF C15 H13 F O2
[1,1′-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (αS)-
- [1,1′-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (S)-
- (+)-(S)-Flurbiprofen
- (+)-Flurbiprofen
- (2S)-2-(2-Fluoro-1,1′-biphenyl-4-yl)propanoic acid
- (2S)-2-(2-Fluoro-4-biphenyl)propanoic acid
- (S)-Flurbiprofen
- Dexflurbiprofen
- Esflurbiprofen
- S-(+)-Flurbiprofen
- d-Flurbiprofen
On October 20, 2014, Taisho filed for manufacturing and marketing approval for TT-063 from the Ministry of Health, Labour and Welfare as a new drug candidate that will follow the Type 2 diabetes treatment Lusefi®, which was launched in May 2014. TT-063 is a patch formulation that has been co-developed by Taisho and TOKUHON Corporation with the aim of obtaining an indication for osteoarthritis. In Phase 3 clinical trials comparing TT-063 with therapeutic drugs already on the market, TT-063 has been found to be more effective than the control drugs in patients with osteoarthritis of the knee joint (January 16, 2014 announcement ).
Furthermore, Taisho is also preparing to file for approval from the Ministry of Health, Labour and Welfare for CT-064, an oral formulation of the osteoporosis treatment agent Bonviva launched in August 2013. Taisho has confirmed the effectiveness of CT-064 for osteoporosis patients through Phase 3 clinical trials (September 22, 2014 announcement).

In the central nervous system field, TS-091 transitioned from Phase 1 to Phase 2 in Japan in May 2014. Clinical trials of TS-091 have commenced to confirm the effectiveness of this drug in patients with central disorders of hypersomnolence. In addition, Phase 1 clinical trials of TS-091 have commenced overseas. TS-111 and TS-121 are undergoing Phase 1 clinical trials overseas with the aim of obtaining an indication for depression.
Faced with intensifying competition in new drug discovery, we will jointly implement R&D activities with research institutions outside the Taisho Group, and with companies in Japan and overseas, as we work to enhance our drug development pipeline (lineup of drugs in development). Our goal is to discover many more new drugs, primarily in our priority fields.
| Company | Taisho Pharmaceutical Holdings Co. Ltd. |
| Description | Topical anti-inflammatory analgesic patch containing S-flurbiprofen |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase III |
| Standard Indication | Osteoarthritis |
| Indication Details | Treat osteoarthritis (OA) and scapulohumeral periarthritis |
| Regulatory Designation | |

Scheme 2.
Reagents and conditions: (a) THF, EDC, Et3N; (b) TFA; (c) 0.5 equiv 2,5-dimethoxybenzoquinone, EtOH, 50–80 °C for 3–5 h; (d) 1 equiv naphthoquinone, MeOH, rt, overnight.
http://www.sciencedirect.com/science/article/pii/S0960894X13011773
……………………………………………
2-(6-methoxynaphthalen-2-yl) propanoic acid By way of illustration, chemically, flurbiprofen is 2-(2-fluoro-4-biphenylyl) propionic acid and is described in US Patent No. 3,755,427. NSAIDs, such as flurbiprofen, are usually supplied as a racemate. However, recently there has been renewed interest in the separate enantiomers of flurbiprofen, i.e. S-flurbiprofen and R-flurbiprofen.

R-Flurbιprofen

S-Flurtιprofen
Flurbiprofen is a potent inhibitor of cyclooxygenase (both COX-I and COX-2) in humans and it is understood that the inhibitory effect lies predominantly in the S- enantiomer.
Flurbiprofen is generally produced in the form of a racemic compound. It is known that from the racemic compound, flurbiprofen having a high optical purity can be produced by an optical resolution method using, for example, an optically active amine compound, such as α-phenylethylamine, as an optical resolution agent, as is described in US Patent No. 5,599,969. In addition, whether dealing with racemic, S- or R- 2-aryl propionic acid, there is also a need to make the synthetic process as efficient as possible.
Example 2 – Ibuprofen
Example 2.1 Resolution procedure
Racemic ibuprofen (530g) is dissolved in toluene (1335ml) and methanol (900ml).
The mixture is heated to dissolve the solid. S-1-Phenylethylamine (247g) is dissolved in toluene (200ml) and the solution is added with stirring at 600C over about 3 hours while the temperature is maintained at about 65-700C. The mixture is cooled gradually to 0 to 50C to induce crystallisation and stirred at this temperature for 1 hour. The crystals are filtered off, washed with toluene (600ml) and dried in a Vacuum oven at 550C to form crude S-ibuprofen / S-1-phenylethylamine salt (635g).
Crude S-ibuprofen / S-1-phenylethylamine salt (635g) is stirred with toluene (1930ml) and methanol (800ml) and the mixture is heated to 6O0C to dissolve the solid. The solution is cooled gradually to 0 to 5°C to induce crystallisation. The crystals are filtered off and dried in a vacuum oven at 55°C to form pure S-ibuprofen / S-I- phenylethylamine salt (510g). This recrystallisation of the S-ibuprofen / S-I- phenylethylamine salt may be repeated if necessary to upgrade the enantiomeric purity if required.
Pure S-ibuprofen / S-1-phenylethylamine salt (485g) is mixed with toluene (1700ml) with stirring. Water (300ml) and concentrated hydrochloric acid (17Og) are added and
÷ibe mixture is stirred at 600C. The lower aqueous layer is separated off and the upper organic layer is retained. The hydrochloric acid wash is repeated, then the toluene solution is washed with water. Water (370ml) and 47% sodium hydroxide
(118g) are added and the solution is heated to 600C and allowed to settle. The lower aqueous layer is separated and the upper toluene layer is washed with water. The aqueous phases are combined and heptane (420ml) is added. Hydrochloric acid
(130g) is added and the mixture is heated to 600C, stirred and settled. The organic layer is separated off and washed with water. The solution is cooled to -100C to induce crystallisation and the crystals are separated off by filtration, washed with heptane and dried under vacuum to yield (S)-ibuprofen (28Og) at an enantiomeric purity of over 99%.
Example 2.2 Racemisation procedure
Toluene/methanol mother liquors from the filtration of crude S-ibuprofen / S-I- phenylethylamine salt in the resolution procedure (2400ml, containing an estimated 130g of ibuprofen) is charged into a 3 L 3 necked round bottomed flask and methanol and toluene are distilled out at atmospheric pressure (volume removed approximately 1400 ml). The batch is then cooled to around 60°C and washed twice with hydrochloric acid (20 ml concentrated hydrochloric acid in 200 ml of water), and then twice with water (200 ml). Toluene is charged (80 ml) followed by methanol (200 ml) and caustic soda solution (45Og of 28% w/w solution, 5 molar equivalents). The mixture is heated to reflux for about 6 hours. Solvent is then removed at atmospheric pressure until the vapour temperature reaches approximately 85°C. The mixture is cooled to around 60°C and concentrated hydrochloric acid is charged at about 60 to 70°C until the pH of the mixture is 1 or less. The layers are allowed to separate and the bottom aqueous layer removed. The organic layer is washed with water (200 ml) and then azeotroped to dryness using a Dean and Stark trap. A solution of racemic ibuprofen in toluene remains.
…………………………………………
PATENT
http://www.google.com/patents/CN104478703A?cl=en
Preparation of R – (+) _ flurbiprofen:
The racemic flurbiprofen as a starting material, to obtain an intermediate product of formula I as shown and then the ester prepared as shown in Formula II with 5-isosorbide monobenzyl ether, ester hydrolysis after obtained R – (+) – flurbiprofen;

wherein, in formula I, X is Cl or Br;
(2) by the R – (+) _ flurbiprofen obtained (RS) – flurbiprofen:
The R _ (+) _ flurbiprofen 200mg, potassium hydroxide 150mg, 0. 5mL water into IOmL reaction flask and heated to 120 ° C and held for 2h, then water was added 15mL, cooled to room temperature, the resulting stirring the mixed solution with 10% hydrochloric acid to pH = 0. 5, extracted with ethyl acetate, combined several layers, washed with water until neutral, the organic solvent is recovered, the resulting residue was added at 60~90 ° C under an appropriate amount of petroleum ether by recrystallization, obtained (RS) – flurbiprofen 100mg, 50% yield.
(3) Preparation of (S) -⑴- flurbiprofen:
In 25mL single-necked flask, followed by adding (RS) – flurbiprofen 123mg, Portugal TOA 29. 8mg, isopropanol lmL, the mixture was stirred at reflux until clear, half the amount of the solvent evaporated under reduced pressure except , set the refrigerator overnight. The precipitate was collected by suction filtration as white crystals, after washing a small amount of isopropanol, which was dissolved in water, washed with 10% aqueous sodium hydroxide (10% NaOH mean mass fraction) adjusted pH = 13, the sheet-like precipitate was filtered off Portuguese octylamine white crystals. The resulting filtrate was added dropwise with stirring 10% hydrochloric acid to pH = 1, extracted with ethyl acetate, the organic layer was washed with water to recover the solvent, the resulting residue was purified by an appropriate amount of petroleum ether and recrystallized at 60~90 ° C. The product was collected by filtration, and dried in vacuo to give a white (S) – (+) _ flurbiprofen needle crystal 45. 3mg, 65% yield, mp 102~103 ° C, [α] = + 44 ° (C = 1, methanol), ee value of 92.6% (ee value measurement method: (S) – (+) – flurbiprofen after chiral amine derivatization reagents, by HPLC analysis).
wherein in step (3) is a byproduct eleven R _ (+) _ flurbiprofen, its follow step (1) of racemic reused.
Step (1) of the specific operation is as follows:
(la) 1:. Synthesis of 2,6-sorbitol dehydration -D- -5- benzyl ether: 4: 3
250ml volumetric flask isosorbide 18. 25g (125mmol), lithium hydroxide monohydrate 5. 25g (125mmol) and 60ml of dimethyl sulfoxide (DMSO), heated to 90 ° C, stirred for 30min, constant pressure equalizing dropping funnel was added dropwise benzyl chloride 14. 4ml (125mmol), 90 ° C the reaction 19-20h, reaction mixture was adjusted to pH 1 with 2M hydrochloric acid, extracted with ethyl acetate (50ml * 3), the organic layers combined, washed with water ( 30ml * 2), dried over anhydrous sodium sulfate overnight, filtered and concentrated residue Cheng baby gel column chromatography (petroleum ether: ethyl acetate = 5: 1) to give a cream solid, that is 1: 4: 3: 2,6 Dehydration -D- sorbitol -5- benzyl ether 24. 5g, m.p. 59 ~61 ° C.
(Ib) · 2- (2- fluoro-4-biphenylyl) propionyl chloride Synthesis
50ml vial before racemic flurbiprofen was added 2. 44g (IOmmol), anhydrous toluene 20ml, freshly distilled thionyl chloride was added dropwise 0. 8ml (Ilmmol), N, N- dimethylformamide amide (DMF) 2 dropwise, stirred at room temperature 2h, the solvent was distilled off under reduced pressure to give a pale yellow gum, i.e., 2- (2-fluoro-4-biphenylyl) propionyl chloride, it was used directly in the reaction without isolation.
(lc). R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester synthesis
The (Ib) resulting acid chloride was dissolved in 20ml of dry toluene was added dropwise at room temperature, dimethyl amine 3. 5ml, solid precipitation, stirred for about Ih, ice salt bath, a bath temperature of minus 10-15Ό, stirred at this temperature IOmin so, and then the constant pressure dropping funnel (Ia) 5 isosorbide monobenzyl ether (2. 83g, 12mmol) in toluene, keeping the reaction temperature, stirring 8h. The ice bath was removed and the reaction mixture under reduced pressure to remove the solvent, the residue was extracted with ethyl acetate. The extract was washed with water, dried over anhydrous sodium sulfate overnight, ethyl acetate was removed under reduced pressure, the residue was a white gel, recrystallized from petroleum ether to give a white solid that R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester 3. 65g (7. 88mmol), in order to put the racemic flurbiprofen yield based on 78.8%.
(ld) R – Synthesis of flurbiprofen – (+)
Under ice bath (Ic) obtained R-2- (2- fluoro-4-biphenylyl) propionic acid monobenzyl ether isosorbide 5- ester 2. 3Ig (5mmol) was dissolved in 20ml of acetone / water (1/1) was added Iml hydrochloric acid to adjust pH to 3, stirred for 3-4h, the reaction solution was extracted with ethyl acetate (20ml * 2), sash organic layer was washed with ice (10ml * 2), dried over anhydrous sodium sulfate overnight , filtration, and the filtrate was concentrated, the residue was recrystallized from ether to give white crystals, i.e. L-flurbiprofen 1.02g (4 18mmol.), yield 83.5%, optical purity 93% (HPLC method); input-racemic flurbiprofen dollars, the total yield of 78.8% * 83.5% = 65.8%.
Step (1) reaction of the formula:

FLURBIPROFEN RACEMIC
3-Fluoro-4-phenyl-α-methylphenylacetic acid 1
M.p. 110-113°C (lit.3d 111-113.5°C).
1 H NMR (CDCl3, δ ppm) 7.51-7.55 (m, 2H), 7.49-7.37 (m, 4H), 7.21-7.16 (m, 2H), 3.85-3.78 (q, 1H, J = 7.1 Hz, CH), 1.60-1.57 (d, 3H, J = 7.1 Hz, CH3);
13C NMR (CDCl3 δ ppm) 180.4 (COOH), 161.3 & 158.0 (3-Ar-C), 140.9 & 140.8, 135.4, 130.9 & 130.8 (5-Ar-C), 128.9, 128.4, 128.2 & 128.0 (4-Ar-C), 127.7 (4′-Ar-C), 123.7 & 123.7 (6-Ar-C), 115.5 & 115.2 (2-Ar-C), 44.8 (CH), 18.0 (CH3).
(d) Sagami Chemical Research Center. Jpn. Kokai Tokkyo Koho JP 8216840, 1982 (Chem. Abstr. 1982, 97: 5996s).

 |
| RACEMIC |
|
Flurbiprofen
CAS : 5104-49-4
: 2-Fluoro-a-methyl[1,1¢-biphenyl]-4-acetic acid
Additional Names: 2-(2-fluoro-4-biphenylyl)propionic acid; 3-fluoro-4-phenylhydratropic acid
Manufacturers’ Codes: BTS-18322; U-27182
Trademarks: Adfeed (Lead Chem.); Ansaid (Pfizer); Antadys (Thamex); Cebutid (Boots); Froben (Boots); Flurofen (Boots); Ocufen (Allergan); Stayban (Boots); Zepolas (Mikasa)
Molecular Formula: C15H13FO2
Molecular Weight: 244.26
Percent Composition: C 73.76%, H 5.36%, F 7.78%, O 13.10%
Literature References: Prepn: FR M5737; Adams et al., US 3755427 (1968, 1973 both to Boots Co., Ltd.). Pharmacology: Chalmers et al., Ann. Rheum. Dis. 31, 319 (1972); ibid. 32, 58 (1973); Glenn et al., Agents Actions 3, 210 (1973); Nishizawa et al.,Thromb. Res. 3, 577 (1973). HPLC determn in urine and plasma: J. M. Hutzler et al., J. Chromatogr. B 749, 119 (2000). Symposium on pharmacokinetics and clinical efficacy in pain management: Am. J. Med. 80, Suppl. 3A, 1-157 (1986).
Properties: Crystals from petr ether, mp 110-111°. Slightly sol in water (pH 7.0); readily sol in most polar solvents.
Melting point: mp 110-111°
Therap-Cat: Anti-inflammatory; analgesic.
|
racemic


s form


| Patent | Submitted | Granted |
|---|---|---|
| Methods to accelerate the isolation of novel cell strains from pluripotent stem cells and cells obtained thereby [US2008070303] | 2006-11-21 | 2008-03-20 |
| Herpes Virus-Based Compositions and Methods of Use in the Prenatal and Perinatal Periods [US2008226601] | 2006-06-05 | 2008-09-18 |
| METHOD OF REDUCING ABETA42 AND TREATING DISEASES [US2008021085] | 2007-06-21 | 2008-01-24 |
| METHODS TO ACCELERATE THE ISOLATION OF NOVEL CELL STRAINS FROM PLURIPOTENT STEM CELLS AND CELLS OBTAINED THEREBY [US2010184033] | 2009-07-16 | 2010-07-22 |
| Pyridyl Amide T-Type Calcium Channel Antagonists [US2011112064] | 2011-05-12 | |
| PROCESS FOR THE MANUFACTURE OF RACEMIC 2-ARYL-PROPIONIC ACID [US2011172460] |
| Patent | Submitted | Granted |
|---|---|---|
| Nitroxyderivatives having antinflammatory, analgesic and antithrombotic activity [US6613784] | 2003-09-02 | |
| Global method for mapping property spaces [US6675136] | 2004-01-06 | |
| Method of reducing Abeta42 and treating diseases [US2006004086] | 2006-01-05 | |
| 11-Beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia [US7179802] | 2004-06-03 | 2007-02-20 |
| 11-BETA-HYDROXYSTEROID DEHYDROGENASE 1 INHIBITORS USEFUL FOR THE TREATMENT OF DIABETES, OBESITY AND DYSLIPIDEMIA [US6730690] | 2004-03-11 | 2004-05-04 |
| Process for producing optically active flurbiprofen [US7214820] | 2006-06-22 | 2007-05-08 |
| Pyridyl Amide T-Type Calcium Channel Antagonists [US7875636] | 2009-11-05 | 2011-01-25 |
| METHOD FOR PRODUCING OPTICALLY ACTIVE ESTER AND METHOD FOR PRODUCING OPTICALLY ACTIVE CARBOXYLIC ACID [US8115008] | 2010-09-16 | 2012-02-14 |
| DRUG SUBSTANCE PREPARATIONS, PHARMACEUTICAL COMPOSITIONS AND DOSAGE FORMS [US2010087538] | 2010-04-08 | |
| (R)-2-(3-Benzoylphenyl)propionic acid salts and pharmaceutical preparations containing them [EP0935961] | 1999-08-18 | 2008-04-02 |
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

Taisho Pharmaceutical Co., Ltd. (大正製薬株式会社 Taishō Seiyaku Kabushiki-gaisha?) (TYO: 4535) is a Japanese pharmaceutical company based in Tokyo.

.
////////////
| Tirupati తిరుపతి |
|
|---|---|
| City | |

Clockwise from top: Tirumala Venkateswara Temple, Tirumala ghat road, City skyline and Chandragiri fort
|
|
|
Tirupati
Location in Andhra Pradesh, India |
|
| Coordinates: 13.65°N 79.42°ECoordinates: 13.65°N 79.42°E | |
| Country | India |
| State | Andhra Pradesh |
| Region | Rayalaseema |
| District | Chittoor |
| Government | |
| • Member of Parliament | Varaprasad Rao Velagapalli |
| Area | |
| • City | 24 km2 (9 sq mi) |
| Elevation | 161 m (528 ft) |
| Population (2011)[1] | |
| • City | 287,035 |
| • Density | 12,000/km2 (31,000/sq mi) |
| • Metro[2] | 459,985 |
| Languages | |
| • Official | Telugu |
| Time zone | IST (UTC+5:30) |
| PIN | 517501 |
| Telephone code | +91–877 |
| Vehicle registration | AP 03 |
| Website | Tirupati Mucnicipal Corporation |

 .
.
 .
.
Kapila Theertham in Tirupati








Food Service During Tirumala Tirupati Devastanam’s ‘Srinivasa Kalyanam Utsavam’ at MARG Swarnabhoomi



Methyl (S)-aminobutyrate hydrochloride…..Levetiracetam intermediate
Methyl (S)-aminobutyrate hydrochloride…..Levetiracetam intermediate
 |
- (s)-2-aminobutyric Acid Methyl Ester
- CAS No.: 15399-22-1
PATENT
http://www.google.im/patents/WO2003014080A2?cl=en
(S)-amino butyric acid

Step 1 – Synthesis of methyl (S)-aminobutyrate hydrochloride


……………………………(23) …………………………………………………….(24)
5.0g of (S)-amino butyric acid (23) was suspended in 50 ml of methanol and stirred at 0-5°C. 6.35g of thionyl chloride was added dropwise over 45 min to form a clear solution. After stirring for 20 hours at room temperature, the reaction was concentrated under reduced pressure to dryness and the almost colourless residue solidified to give the required product which was dried in an oven at 50°C under vacuum (7.6g; 102% crude yield). The same reaction was scaled-up from 200g of the amino acid and provided 296g (99.5% yield) of product (24). Analysis gave the following results:
1H NMR (DMSO-de) : d 0.94 (3H, t) 1.88 (2H, q) 3.75 (3H, s) 3,9 (1H, m) 8,8
(3H, m). m.p. : 107°C-110°C IR : 2876 cm 1, 1742 cm 1.
TLC : Si02, 20%MeOH/80%EtOAc/ l%NH OH, UV & IR. (TLC is an abbreviation for thin layer chromatography).

1H NMR PREDICT

13C PREDICT

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com
JALGAON, MAHARASHTRA, INDIA

.




 .
.



.
 MANUDEVI
MANUDEVI

(S)-2-amino-butanamide hydrochloride………. Key intermediate of Levetiracetam
(S)-2-amino-butanamide hydrochloride………. Key intermediate of Levetiracetam
(S)-2-amino-butanamide hydrochloride
Key intermediate of Levetiracetam

-
CAS Number 7682-20-4
-
Linear Formula CH3CH2CH(NH2)CONH2 · HCl

Stage B
(S)-2-aminobutyramide hydrochloride Preparation
Into the above (S)-2-aminobutyric acid methyl ester hydrochloride is added Isopropanol is then added, followed by the introduction of ammonia gas at a pressure about 60 psi (413 kPa) until the reaction is complete. After filtering to remove formed ammonium chloride, the solvent is partially evaporated and isopropanol hydrochloride is added. The mixture is stirred while solid product forms, then the solid is separated by filtration and washed with isopropanol.
The product was characterized by the following 1H NMR data (200 MHz, DMSO-d6): 0.9-1.0(t,3H), 1.8-1.9(Q,2H), 3.7-3.8(t, 1H), 7.5-7.7(Br,NH2), 8.0-8.2(Br,NH2)
1H NMR PREDICT

………..
13C NMR PREDICT

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
 COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR amcrasto@gmail.com
TIRUPATI, INDIA
.
| Tirupati తిరుపతి |
|
|---|---|
| City | |
|
Clockwise from top: Tirumala Venkateswara Temple, Tirumala ghat road, City skyline and Chandragiri fort
|
|

Tirupati
Location in Andhra Pradesh, India |
|
| Coordinates: 13.65°N 79.42°ECoordinates: 13.65°N 79.42°E | |
| Country | India |
| State | Andhra Pradesh |
| Region | Rayalaseema |
| District | Chittoor |
| Government | |
| • Member of Parliament | Varaprasad Rao Velagapalli |
| Area | |
| • City | 24 km2 (9 sq mi) |
| Elevation | 161 m (528 ft) |
| Population (2011)[1] | |
| • City | 287,035 |
| • Density | 12,000/km2 (31,000/sq mi) |
| • Metro[2] | 459,985 |
| Languages | |
| • Official | Telugu |
| Time zone | IST (UTC+5:30) |
| PIN | 517501 |
| Telephone code | +91–877 |
| Vehicle registration | AP 03 |
| Website | Tirupati Mucnicipal Corporation |
.
.
Kapila Theertham in Tirupati
Food Service During Tirumala Tirupati Devastanam’s ‘Srinivasa Kalyanam Utsavam’ at MARG Swarnabhoomi
Levetiracetam Green process construction

Dr. Rakeshwar Bandichhorl Director API – R&D,
Dr Reddys
LEVETIRACETAM GREEN PROCESS
An alternate synthesis of levetiracetam
Ravikumar Mylavarapu a , Ramasamy Vijaya Anand a , Golla China Mala Kondaiah a , Lekkala
Amarnath Reddy a , Gade Srinivas Reddy a , Arnab Roy a , Apurba Bhattacharya a , Kagga
Mukkanti b & Rakeshwar Bandichhor a
a Innovation Plaza, IPDO, R&D , Dr. Reddy’s Laboratories Ltd. , Survey Nos. 42, 45,46 & 54,
Bachupally, Qutubullapur, 500073, R.R. Dist, Andhra Pradesh, India
b Center for Environmental Science, Institute of Science and Technology , J.N.T. University ,
Kukatpally, Hyderabad, 500 072, Andhra Pradesh, India
Ravikumar Mylavarapu a , Ramasamy Vijaya Anand a , Golla China Mala Kondaiah a , Lekkala
Amarnath Reddy a , Gade Srinivas Reddy a , Arnab Roy a , Apurba Bhattacharya a , Kagga
Mukkanti b & Rakeshwar Bandichhor a
a Innovation Plaza, IPDO, R&D , Dr. Reddy’s Laboratories Ltd. , Survey Nos. 42, 45,46 & 54,
Bachupally, Qutubullapur, 500073, R.R. Dist, Andhra Pradesh, India
b Center for Environmental Science, Institute of Science and Technology , J.N.T. University ,
Kukatpally, Hyderabad, 500 072, Andhra Pradesh, India
Email: rakeshwarb@drreddys.com
Green Chemistry Letters and Reviews
Vol. 3, No. 3, September 2010, 225230
Vol. 3, No. 3, September 2010, 225230
Ravikumar Mylavarapu , Ramasamy Vijaya Anand , Golla China Mala Kondaiah , Lekkala Amarnath Reddy ,
Gade Srinivas Reddy , Arnab Roy , Apurba Bhattacharya , Kagga Mukkanti & Rakeshwar Bandichhor (2010)
Gade Srinivas Reddy , Arnab Roy , Apurba Bhattacharya , Kagga Mukkanti & Rakeshwar Bandichhor (2010)
An alternate
synthesis of levetiracetam, Green Chemistry Letters and Reviews, 3:3, 225-230, DOI: 10.1080/17518251003716568
To link to this article: http://dx.doi.org/10.1080/17518251003716568
synthesis of levetiracetam, Green Chemistry Letters and Reviews, 3:3, 225-230, DOI: 10.1080/17518251003716568
To link to this article: http://dx.doi.org/10.1080/17518251003716568
You might enjoy reading:
– See more at: http://organicsynthesisinternational.blogspot.in/#sthash.ruewyXXk.dpuf
Dr Rakeshwar Bandichhor

| Rakeshwar Bandichhor Associate Director, API, R&D Dr. Reddy’s Laboratories India |
 |
|||||||||||
 |
||||||||||||
| BiographyRakeshwar Bandichhor holds a doctorate in Chemistry from University of Lucknow/University of Regensburg, Germany and worked as Postdoctoral Fellow at University of Regensburg, Germany, University of Pennsylvania and Texas A&M University. Dr. Rakeshwar has more than 150 papers including patents and book chapters published/accepted in various International Journals and contributed to more than 60 academic national and international conferences. He has won the various awards in his career | ||||||||||||
| Dr. Rakeshwar has more than 80 papers including patents and book chapters published/accepted in various International Journals. Notably, in the area of Organic Chemistry, Dr. Rakeshwar has coauthored a chapter in the book entitled “Green Chemistry in Pharmaceutical industry”. He has won the various awards in his career e.g. Chairman Excellence Award in the category of individual functional excellence, Best Cost Leadership Award for the development of Lopinavir, Ritonavir & their components and Anveshan Award at Dr. Reddy’s. As a part of organizational building efforts, he also supervises master’s & Ph.D. students in their dissertations. He has been invited in several conferences e.g. IIT-Mumbai, IGCW-2009, BIT-Ranchi, BITS Pilani, 9th Heterocyclic Conference, University of Florida, JNTU-Hyderabad, ISCB-2011, Apollo Hospitals Educational & Research Foundation, Hyderabad etc. to deliver lectures. He is also currently acting as an Associate Editor of GERF Bulletin of Bioscience. Recently, he has become a member National Advisory Board of Indian Society of Chemists and Biologists. |
||||||||||||
|
||||||||||||

Innovation Plaza, IPDO, R&D , Dr. Reddy’s Laboratories Ltd.


 .
.
MAHABALIPURAM, INDIA
Mahabalipuram – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Mahabalipuram
Mahabalipuram, also known as Mamallapuram is a town in Kancheepuram district in the Indian state of Tamil Nadu. It is around 60 km south from the city of …Shore Temple – Seven Pagodas – Pancha Rathas –
.
.



Krishna’s Butter Ball in Mahabalipuram, India. The surface below the rock is …




http://www.weather-forecast.com/locations/Mamallapuram
 Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
 Moonraikers Restaurant, Mamallapuram
Moonraikers Restaurant, Mamallapuram


 Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
 .
.
A carving at the Varaha Temple, Mahabalipuram

/////////////
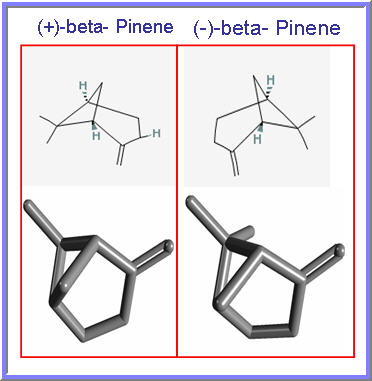
GSK 2269557 In Phase 1….Asthma , COPD, is it COMPD A OR B?
 COMPD A
COMPD A
Compd A OR B IS GSK 2269557
Phosphatidylinositol 3-Kinase (PI3K)
Glaxo Group Limited INNOVATOR
PHASE 1….asthma & COPD
ASHTHMA COPD
DATA FOR COMPD A
6-(1H-indol-4-yl)-4-[5-[[4-(1-methylethyl)-1-piperazinyl]methyl]-2-oxazolyl]-1H-Indazole,
6-(1 H-lndol-4-yl)-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1 H- indazole
CAS 1254036-77-5 hcl salt
base 1254036-71-9, 440.54, C26 H28 N6 O
Formula C26H28N6O.HCl
EMAIL ME amcrasto@gmail.com
DATA FOR COMPD B
Methanesulfonamide, N-[5-[4-[5-[[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl]-2-oxazolyl]-1H-indazol-6-yl]-2-methoxy-3-pyridinyl]-,
N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide
1254036-66-2 CAS
C24 H28 N6 O5 S, 512.58
Compound B may be prepared according to known procedures, such as those disclosed in international patent application PCT/EP2010/055666 (publication number WO02010/125082)
EMAIL ME amcrasto@gmail.com
Phosphoinositide 3ΌΗ kinases (hereinafter PI3Ks) are a family of signal transducer enzymes which are involved in various cellular functions including cell growth, proliferation and differentiation. A wide variety of retroviruses and DNA-based viruses activate the PI3K pathway as a way of preventing host cell death during viral infection and ultimately exploiting the host cell synthesis machinery for its replication (Virology 344(1) p. 131-8 (2006) by Vogt et al.; and Nat. Rev. Microbiol. 6(4) p. 265-75 (2008) by Buchkovich et al). It has therefore been postulated that PI3K inhibitors may have potential therapeutic benefit in the treatment of viral infections such as influenza virus infection, in addition to the more established treatment of cancer and inflammatory diseases.
The Influenza NS1 protein activates Class la PI3Ks by binding to their regulatory subunit p85beta but not to other Class la regulatory subunits such as p85alpha. The recent crystal structure of the NS1-p85beta complex (Hale et al. Proc. Natl. Acad. Sci. U S A. 107(5) p.1954-1959 (2010)) is also suggestive of an interaction with the p110 kinase subunit providing a mechanism for catalytic activation of the kinase domain. This observation provides a rationale for isoform specificity not only with the p85 regulatory subunit but also potentially with the p110 catalytic subunit too. The function of PI3K during influenza virus infection has also been investigated by, for example, Ehrhardt et al. (Cell. Microbiol. 8(8) p. 1336-1348 (2006)), and the role of PI3K5 signalling in morbidity and lung pathology induced by influenza virus infection has been reported in WO 2010/083163.
There remains a need to provide compounds which are inhibitors of the activity or function of PI3K5 which may be useful in the treatment or prevention of influenza virus infection.
GSK 2269557 is an inhaled phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor in early clinical trials at GlaxoSmithKline for the treatment of patients with asthma and also for the treatment of chronic obstructive pulmonary disease (COPD) in patients who smoke cigarettes.
- 18 Nov 2014GlaxoSmithKline plans a phase II trial in Chronic obstructive pulmonary disease in Belgium, Denmark, the Netherlands and Russia (NCT02294734)
- 01 Jun 2014Phase-II clinical trials in Chronic obstructive pulmonary disease in Germany (Inhalation)
- 01 May 2014GlaxoSmithKline plans a phase II trial for Chronic obstructive pulmonary disease in Germany (NCT02130635)
EMAIL ME amcrasto@gmail.com
CLICK ON IMAGES TO VIEW SIMILAR ROUTES FOR COMPD A AND B
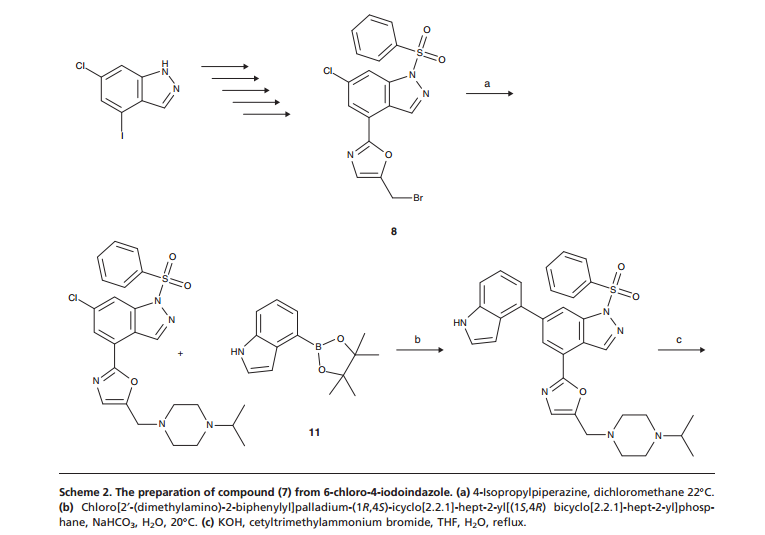
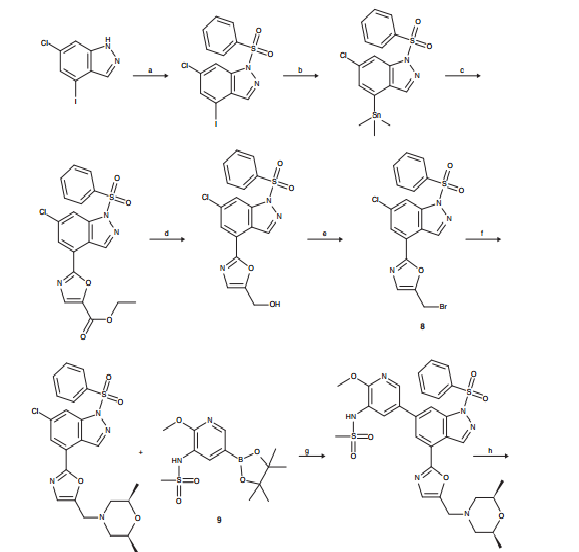
CLICK ON IMAGE TO VIEW
…………………………………………………………………….
COMPD A
WO 2012032065
http://www.google.com/patents/WO2012032065A1?cl=en
Example 68
6-(1 H-lndol-4-yl)-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1,3-oxazol-2-yl)-1 H- indazole

Method A
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1/-/-indazole (97 mg, 0.194 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (61.3 mg, 0.252 mmol, available from Frontier Scientific Europe), chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (10.87 mg, 0.019 mmol) and potassium phosphate tribasic (124 mg, 0.582 mmol) were dissolved in 1 ,4-dioxane (1 ml) and water (0.1 ml) and heated in a Biotage Initiator microwave at 100°C for 30 min. Additional 4-(4,4,5,5- tetramethyl-1 ,3,2-dioxabotolan-2-yl)-1 H-indole (61.3 mg, 0.252 mmol) and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (5 mg) were added and the reaction heated at 1 10°C for 30 min, then 140°C for 30 min. The solvent was removed in vacuo and the residue purified by silica gel chromatography, eluting with 0-25% methanol in dichloromethane. The appropriate fractions were combined and concentrated to give a brown solid which was dissolved in MeOH:DMSO (1 ml, 1 : 1 , v/v) and purified by MDAP (method H). The appropriate fractions were concentrated in vacuo to give the title compound as a white solid (30 mg).
LCMS (Method A): Rt 0.57 mins, MH+ 441.
Method B
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1 H-indazole (75.17 g, 150 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (73.1 g, 301 mmol), sodium bicarbonate (37.9 g, 451 mmol), and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 ,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4 )- bicyclo[2.2.1]hept-2-yl]phosphane (8.43 g, 15.03 mmol) were suspended in nitrogen purged 1 ,4-dioxane (1200 ml_) and water (300 ml_). The reaction vessel was placed under alternating vacuum and nitrogen five times with overhead stirring, then finally placed under a nitrogen atmosphere and heated to 120°C for 2.5 h.
The reaction mixture was cooled to 45°C and then treated with 2M aqueous sodium hydroxide (376 ml_, 752 mmol). After stirring at 45°C overnight (~ 13h), the mixture was cooled to RT and DCM (600 ml) and water (400 ml) were added. The layers were separated and the aqueous re-extracted with DCM: 1 ,4-dioxane (1 : 1). Brine was added and the mixture filtered through Celite, washing with DCM: 1 ,4-dioxane (1 : 1). The layers were separated and 2M HCI (1000 ml) added to the organic. The mixture was again filtered through Celite washing with 500 ml 2M HCI keeping the washings separate. The filtrate layers were then separated and the organic layer was washed with the acid washings from the Celite. Layers were separated and the acidic aqueous combined. This was then back-washed with 2×500 ml of DCM; each wash requiring a Celite filtration. The acidic aqueous was then given a final filtration through Celite washing the Celite pad with 150 ml of 2M HCI.
The acidic aqueous was transfered to a beaker (5000 ml) and with vigorous stirring 2M NaOH was added to basify the mixture to pH 10-11. The mixture was then extracted using 1 ,4-dioxane: DCM (1 : 1) (5 x 500 ml). The combined organics were washed with brine, dried over magnesium sulphate, filtered and evaporated to yield a brown foam that was dried in vacuo at 50°C overnight. This material was split into three batches and each was purified by reverse phase column chromatography (3x 1.9 kg C18 column), loading in DMF/TFA (1 : 1 , 30 ml) then eluting with 3-40% MeCN in Water + 0.25% TFA (Note: Columns 2 & 3 used a different gradient starting with 10% MeCN).
Appropriate fractions were combined, the acetotnitrile removed in vacuo and the acidic aqueous basified to pH10 by addition of saturated aqueous sodium carbonate solution to the stirred solution. The resultant solid was collected by filtration, washed with water then dried in vacuo at 65°C overnight to give the title compound (28.82 g) as a pale brown foam.
LCMS (Method A): Rt 0.68 mins, MH+ 441.
1 H NMR (400MHz ,DMSO-d6) d = 13.41 (br. s., 1 H), 11.35 (br. s., 1 H), 8.59 (br. s., 1 H), 8.07 (d, J = 1.5 Hz, 1 H), 7.90 (br. s., 1 H), 7.51 – 7.44 (m, 2 H), 7.32 (s, 1 H), 7.27 – 7.21 (m, 2 H), 6.61 – 6.58 (m, 1 H), 3.73 (br. s., 2 H), 2.64 – 2.36 (m, 9 H), 0.97 – 0.90 (m, 6 H)
Method C
Potassium hydroxide (145.6 g) was added to a suspension of 6-(1 H-indol-4-yl)-4-(5-{[4-(1- methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)-1 H-indazole (300.7 g) and cetyltrimethylammonium bromide (9.3 g) in tetrahydrofuran (6.0 L) and water (30 ml) stirring under nitrogen at ambient temperature. The mixture was heated at reflux for 17 hours and was then cooled to 20-25°C. Ethyl acetate (3.0 L) and water (3.0 L) were added, stirred for 10 minutes and then separated. The organic layer was extracted with hydrochloric acid (1 M, 1 x 3.0 L, 2 x 1.5L) and the acidic extracts combined and basified to ~pH 8 by the addition of saturated sodium carbonate solution (2.1 L). After ageing for 30 minutes the resultant suspension was filtered, washed with water (300 ml) and the solid dried under vacuum at 65°C to give the title compound as a pale yellow solid (127.9 g).
LCMS (Method B): Rt 2.44 min, MH+ 441.
…………………………………………………………………………
WO 2010125082
http://www.google.co.in/patents/WO2010125082A1?cl=en
Example 6
6-(1 H-lndol-4-yl)-4-(5-{[4-(1 -methylethyl)-1 -piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1 H- indazole

Method A
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1H-indazole (97 mg, 0.194 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (61.3 mg, 0.252 mmol, available from Frontier Scientific Europe), chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (10.87 mg, 0.019 mmol) and potassium phosphate tribasic (124 mg, 0.582 mmol) were dissolved in 1 ,4-dioxane (1 ml) and water (0.1 ml) and heated in a Biotage Initiator microwave at 1000C for 30 min. Additional 4-(4, 4,5,5- tetramethyl-1 ,3,2-dioxabotolan-2-yl)-1 H-indole (61.3 mg, 0.252 mmol) and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (5 mg) were added and the reaction heated at 1 1O0C for 30 min, then 14O0C for 30 min. The solvent was removed in vacuo and the residue purified by silica gel chromatography, eluting with 0-25% methanol in dichloromethane. The appropriate fractions were combined and concentrated to give a brown solid which was dissolved in MeOH:DMSO (1 ml, 1 :1 , v/v) and purified by MDAP (method A). The appropriate fractions were concentrated in vacuo to give the title compound as a white solid (30 mg).
LCMS (Method A): Rt 0.57 mins, MH+ 441.
Method B
6-Chloro-4-(5-{[4-(1-methylethyl)-1-piperazinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)- 1 H-indazole (75.17 g, 150 mmol), 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 H- indole (73.1 g, 301 mmol), sodium bicarbonate (37.9 g, 451 mmol), and chloro[2′- (dimethylamino)-2-biphenylyl]palladium-(1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)- bicyclo[2.2.1]hept-2-yl]phosphane (8.43 g, 15.03 mmol) were suspended in nitrogen purged 1 ,4-dioxane (1200 ml.) and water (300 ml_). The reaction vessel was placed under alternating vacuum and nitrogen five times with overhead stirring, then finally placed under a nitrogen atmosphere and heated to 1200C for 2.5 h.
The reaction mixture was cooled to 45°C and then treated with 2M aqueous sodium hydroxide (376 ml_, 752 mmol). After stirring at 450C overnight (~ 13h), the mixture was cooled to RT and DCM (600 ml) and water (400 ml) were added. The layers were separated and the aqueous re-extracted with DCM: 1 ,4-dioxane (1 :1 ). Brine was added and the mixture filtered through Celite, washing with DCM: 1 ,4-dioxane (1 :1 ). The layers were separated and 2M HCI (1000 ml) added to the organic. The mixture was again filtered through Celite washing with 500 ml 2M HCI keeping the washings separate. The filtrate layers were then separated and the organic layer was washed with the acid washings from the Celite. Layers were separated and the acidic aqueous combined. This was then back-washed with 2×500 ml of DCM; each wash requiring a Celite filtration. The acidic aqueous was then given a final filtration through Celite washing the Celite pad with 150 ml of 2M HCI.
The acidic aqueous was transfered to a beaker (5000 ml) and with vigorous stirring 2M NaOH was added to basify the mixture to pH 10-11. The mixture was then extracted using 1 ,4-dioxane:DCM (1 :1 ) (5 x 500 ml). The combined organics were washed with brine, dried over magnesium sulphate, filtered and evaporated to yield a brown foam that was dried in vacuo at 500C overnight.
This material was split into three batches and each was purified by reverse phase column chromatography (3x 1.9 kg C18 column), loading in DMF/TFA (1 :1 , 30 ml) then eluting with 3-40% MeCN in Water + 0.25% TFA (Note: Columns 2 & 3 used a different gradient starting with 10% MeCN).
Appropriate fractions were combined, the acetotnitrile removed in vacuo and the acidic aqueous basified to pH10 by addition of saturated aqueous sodium carbonate solution to the stirred solution. The resultant solid was collected by filtration, washed with water then dried in vacuo at 65°C overnight to give the title compound (28.82 g) as a pale brown foam.
LCMS (Method A): Rt 0.68 mins, MH+ 441. 1H NMR (400MHz ,DMSOd6) d = 13.41 (br. s., 1 H), 11.35 (br. s., 1 H), 8.59 (br. s., 1 H), 8.07 (d, J = 1.5 Hz, 1 H), 7.90 (br. s., 1 H), 7.51 – 7.44 (m, 2 H), 7.32 (s, 1 H), 7.27 – 7.21 (m, 2 H), 6.61 – 6.58 (m, 1 H), 3.73 (br. s., 2 H), 2.64 – 2.36 (m, 9 H), 0.97 – 0.90 (m, 6 H)
EMAIL ME amcrasto@gmail.com
COMPD B
http://www.google.co.in/patents/WO2010125082A1?cl=en
Example 1
Λ/-[5-[4-(5-{[(2/?,6S)-2,6-Dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-
6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide

Method A
To a solution of 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1 ,3-oxazol-2- yl)-1-(phenylsulfonyl)-1 H-indazole (0.20 g, 0.411 mmol) and N-[2-(methoxy)-5-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3-pyridyl]methanesulfonamide (0.175 g, 0.534 mmol) in 1 ,4-dioxane (2 ml) was added chloro[2′-(dimethylamino)-2-biphenylyl]palladium- 1 (1 /?,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4/?)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol), potassium phosphate tribasic (0.262 g, 1.23 mmol) and water (0.2 ml). The reaction mixture was heated and stirred at 12O0C under microwave irradiation for 1 h. Additional chloroP’^dimethylamino^-biphenylyOpalladium-^I R^S^bicycloP^.ilhept^- yl[(1 S,4/?)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol) and potassium phosphate tribasic (80 mg) were added and the reaction heated to 12O0C under microwave irradiation for 1 h. Additional potassium phospate tribasic (80 mg) was added and the reaction heated under the same conditions for a further 1 h. The reaction mixture was filtered through a silica SPE and eluted with methanol. The solvent was removed in vacuo and the residue partitioned between dichloromethane (5 ml) and water (5 ml). The layers were separated and the aqueous extracted with further dichloromethane (2x 2 ml). The combined organics were concentrated under a stream of nitrogen and the residue dissolved in MeOH:DMSO (3ml, 1 :1 , v/v) and purified by MDAP (method A) in 3 injections. The appropriate fractions were combined and concentrated to give a white solid which was dissolved in MeOH:DMSO (1 ml, 1 :1 , v/v) and further purified by MDAP (method B). The appropriate fractions were basified to pH 6 with saturated sodium bicarbonate solution and extracted with ethyl acetate (2x 25 ml). The combined organics were dried and evaporated in vacuo to give a white solid which was further dried under nitrogen at 4O0C for 3 h to give the title compound as a white solid (26 mg). LCMS (Method A): Rt 0.53 mins, MH+ 513.
Method B N-[2-(Methyloxy)-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3- pyridinyl]methanesulfonamide (101 g, 308 mmol), 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4- morpholinyl]methyl}-1 ,3-oxazol-2-yl)-1-(phenylsulfonyl)-1 H-indazole (83.3 g, 154 mmol) and sodium bicarbonate (38.8 g, 462 mmol) were suspended in 1 ,4-dioxane (1840 ml) and water (460 ml) under nitrogen and heated to 800C. Chloro[2′-(dimethylamino)-2- biphenylyl]palladium-1 (1 R,4S)-bicyclo[2.2.1]hept-2-yl[(1 S,4R)-bicyclo[2.2.1]hept-2- yl]phosphane (8.63 g, 15.40 mmol) was added and the mixture stirred overnight at 800C.
The reaction mixture was cooled to 450C, sodium hydroxide 2M aq. (770 ml, 1540 mmol) added and the reaction heated to 45 0C for 4 hours. The mixture was cooled to RT and diluted with water (610 ml_). Dichloromethane (920 ml.) was added, and the mixture was filtered twice through Celite (washed with 200 ml. 1 ,4-dioxane/DCM 2:1 each time). The phases were separated, and aqueous washed with 1 ,4-dioxane/DCM 2:1 (500 ml_). The aqueous phase was neutralised with hydrochloric acid to pH -7 and extracted with 1 ,4- dioxane/DCM 2:1 (1 L), then 1 ,4 dioxane/DCM 1 :1 (2×500 ml_). The organics were washed with brine (500 ml_), and filtered through Celite (washed with 200 ml. 1 ,4 dioxane/DCM 2:1 ), and evaporated to yield a dark black solid, which was purified in 4 batches:
Batch 1 : 28g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (14.78 g).
Batch 2: 3Og was dissolved in methanol and mixed with Fluorisil. The solvent was then removed by evaporation and the solid purified by column chromatography (1.5 kg silica column, solid sample injection module), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (9.44 g).
Batch 3: 31 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (17 g).
Batch 4: 29g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 ml.) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (21 g).
The mixed fractions from the 4 columns were combined and evaporated to yield 19 g which was dissolved in 200 ml. of Toluene/Ethanol/Ammonia 80:20:2 (+ additional 4ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (6.1 g).
All pure batches were combined (68 g) and recrystallised from ethanol (1200 ml_). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight. The resulting solid was then collected by filtration, washed sparingly with ethanol and dried under vacuum to give the title compound as an off-white solid (56 g). This material was recrystallised again from ethanol (1 100 ml_). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight with stirring. The resulting solid was collected by filtration and washed sparingly with ethanol. The solid was dried in vacuo at 600C for 5hrs to give the title compound as an off-white solid (45.51 g). LCMS (Method A): Rt 0.61 mins, MH+ 513.
The filtrate from the two recrystallisations was evaporated to yield -23 g of a solid residue that was dissolved in 200 ml. of Toluene/Ethanol/Ammonia 80:20:2 (+ additional 4ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give a further crop of the title compound as an off-white solid (18.5 g). This solid was then recrystallised from ethanol (370 ml_). The suspension was heated to reflux then the resulting solution stirred for 20 mins before being allowed to cool to room temperature naturally overnight. The solid was then dried in vacuo at 65°C overnight to give the title compound as an off-white solid (11.9O g). LCMS (Method A): Rt 0.62 mins, MH+ 513.
………………………………………..
http://www.google.co.in/patents/US8735390
Example 1N-[5-[4-(5-{[(2R,6S)-2,6-Dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide

Method A
To a solution of 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazole (0.20 g, 0.411 mmol) and N-[2-(methoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-pyridyl]methanesulfonamide (0.175 g, 0.534 mmol) in 1,4-dioxane (2 ml) was added chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol), potassium phosphate tribasic (0.262 g, 1.23 mmol) and water (0.2 ml). The reaction mixture was heated and stirred at 120° C. under microwave irradiation for 1 h. Additional chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (11.5 mg, 0.021 mmol) and potassium phosphate tribasic (80 mg) were added and the reaction heated to 120° C. under microwave irradiation for 1 h. Additional potassium phospate tribasic (80 mg) was added and the reaction heated under the same conditions for a further 1 h. The reaction mixture was filtered through a silica SPE and eluted with methanol. The solvent was removed in vacuo and the residue partitioned between dichloromethane (5 ml) and water (5 ml). The layers were separated and the aqueous extracted with further dichloromethane (2×2 ml). The combined organics were concentrated under a stream of nitrogen and the residue dissolved in MeOH:DMSO (3 ml, 1:1, v/v) and purified by MDAP (method A) in 3 injections. The appropriate fractions were combined and concentrated to give a white solid which was dissolved in MeOH:DMSO (1 ml, 1:1, v/v) and further purified by MDAP (method B). The appropriate fractions were basified to pH 6 with saturated sodium bicarbonate solution and extracted with ethyl acetate (2×25 ml). The combined organics were dried and evaporated in vacuo to give a white solid which was further dried under nitrogen at 40° C. for 3 h to give the title compound as a white solid (26 mg).
LCMS (Method A): Rt 0.53 mins, MH+ 513.
Method B
N-[2-(Methyloxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-pyridinyl]methanesulfonamide (101 g, 308 mmol), 6-chloro-4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazole (83.3 g, 154 mmol) and sodium bicarbonate (38.8 g, 462 mmol) were suspended in 1,4-dioxane (1840 ml) and water (460 ml) under nitrogen and heated to 80° C. Chloro[2′-(dimethylamino)-2-biphenylyl]palladium-1(1R,4S)-bicyclo[2.2.1]hept-2-yl[(1S,4R)-bicyclo[2.2.1]hept-2-yl]phosphane (8.63 g, 15.40 mmol) was added and the mixture stirred overnight at 80° C.
The reaction mixture was cooled to 45° C., sodium hydroxide 2M aq. (770 ml, 1540 mmol) added and the reaction heated to 45° C. for 4 hours. The mixture was cooled to RT and diluted with water (610 mL). Dichloromethane (920 mL) was added, and the mixture was filtered twice through Celite (washed with 200 mL 1,4-dioxane/DCM 2:1 each time). The phases were separated, and aqueous washed with 1,4-dioxane/DCM 2:1 (500 mL). The aqueous phase was neutralised with hydrochloric acid to pH ˜7 and extracted with 1,4-dioxane/DCM 2:1 (1 L), then 1,4 dioxane/DCM 1:1 (2×500 mL). The organics were washed with brine (500 mL), and filtered through Celite (washed with 200 mL 1,4 dioxane/DCM 2:1), and evaporated to yield a dark black solid, which was purified in 4 batches:
- Batch 1: 28 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (14.78 g).
- Batch 2: 30 g was dissolved in methanol and mixed with Fluorisil. The solvent was then removed by evaporation and the solid purified by column chromatography (1.5 kg silica column, solid sample injection module), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (9.44 g).
- Batch 3: 31 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (17 g).
- Batch 4: 29 g was dissolved in Toluene/Ethanol/Ammonia 80:20:2 (100 mL) and purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (21 g).
The mixed fractions from the 4 columns were combined and evaporated to yield 19 g which was dissolved in 200 mL of Toluene/Ethanol/Ammonia 80:20:2 (+additional 4 ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give the title compound as an off-white solid (6.1 g).
All pure batches were combined (68 g) and recrystallised from ethanol (1200 mL). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight. The resulting solid was then collected by filtration, washed sparingly with ethanol and dried under vacuum to give the title compound as an off-white solid (56 g). This material was recrystallised again from ethanol (1100 mL). The suspension was heated to reflux and a solution formed. The resulting solution was then cooled to room temperature overnight with stirring. The resulting solid was collected by filtration and washed sparingly with ethanol. The solid was dried in vacuo at 60° C. for 5 hrs to give the title compound as an off-white solid (45.51 g).
LCMS (Method A): Rt 0.61 mins, MH+ 513.
The filtrate from the two recrystallisations was evaporated to yield ˜23 g of a solid residue that was dissolved in 200 mL of Toluene/Ethanol/Ammonia 80:20:2 (+additional 4 ml of 0.88 NH3 to help solubility) then purified by column chromatography (1.5 kg silica column), eluting with Toluene/Ethanol/Ammonia 80:20:2 to give a further crop of the title compound as an off-white solid (18.5 g). This solid was then recrystallised from ethanol (370 mL). The suspension was heated to reflux then the resulting solution stirred for 20 mins before being allowed to cool to room temperature naturally overnight. The solid was then dried in vacuo at 65° C. overnight to give the title compound as an off-white solid (11.90 g).
LCMS (Method A): Rt 0.62 mins, MH+ 513.
Method C
10M Sodium hydroxide solution (0.70 ml) was added to a stirred suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (1.17 g) in water (5.8 ml). The resulting mixture was stirred at room temperature for 3.75 hours and was then washed with ethyl acetate (2×6 ml). The layers were separated and the aqueous phase was acidified to pH 6 with 2M hydrochloric acid (0.8 ml). The acidified aqueous layer was extracted twice with ethyl acetate (11 ml then 5 ml). The combined ethyl acetate extracts were dried by azeotropic distillation and diluted with further ethyl acetate (11 ml). The misture was stirred at room temperature for 112 hours. The slurry was seeded and then stirred at room temperature for 48 hours. The resultant suspension was filtered, washed with ethyl acetate (2×2 ml) and the solid dried under vacuum at 40° C. to give the title compound as a pale yellow solid (0.58 g).
LCMS (Method B): Rt 1.86 min, MH+ 513.
Method D
To a suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (596.5 g, 0.91 mol) in water (3.8 L) is added 5M sodium hydroxide (715 ml, 3.56 mol) over 20 mins at <25° C. The mixture is stirred at 20±3° C. for 2 h 45 min then washed with EtCN (3 L). The pH of the basic aqueous phase is adjusted to pH 6.6 using 2M hydrochloric acid (1.4 L), maintaining the temperature below 30° C. The mixture is then extracted with MeTHF (2×4.8 L), and the combined MeTHF extracts are washed with water (1.2 L). The mixture is concentrated to approx 2.4 L and EtOAc (3 L) is added. This put and take distillation is repeated a further 3 times. The mixture is adjusted to 60±3° C. and seeded twice (2×3 g) 35 mins apart. The resultant is aged for 1 h 10 mins then cooled over 2 h to 20-25° C., and aged for a further 15 h 50 min. The slurry is filtered, washed with EtOAc (2×1.2 L) and dried in vacuo at 45±5° C. for approx 3 day to give the title compound.
Preparation of Polymorphs of Compound A
Form (II)
Ethyl acetate (15 ml) was added to N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (2.1 g) and was stirred at ambient conditions overnight. The resultant slurry was filtered and dried under vacuum at 50° C. to give a new solid state form (91 ckw/w).
1H NMR (400 MHz, DMSO d6) d=13.49 (br s, 1H), 9.39 (s, 1H), 8.58 (s, 1H), 8.42 (d, J=2.2 Hz, 1H), 7.99 (d, J=2.2 Hz, 1H), 7.93 (d, J=1.2 Hz, 1H), 7.88 (s, 1H), 7.35 (s, 1H), 4.00 (s, 3H), 3.74 (s, 2H), 3.58 (m, 2H), 3.11 (s, 3H), 2.80 (d, J=10.3 Hz, 2H), 1.78 (t, J=10.3 Hz, 2H), 1.05 (d, J=6.4 Hz, 6H)
SODIUM SALT OF COMPD B
http://www.google.com/patents/US20140256721
Method D
To a suspension of N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1-(phenylsulfonyl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (596.5 g, 0.91 mol) in water (3.8 L) is added 5M sodium hydroxide (715 ml, 3.56 mol) over 20 mins at <25° C. The mixture is stirred at 20±3° C. for 2 h 45 min then washed with EtCN (3 L). The pH of the basic aqueous phase is adjusted to pH 6.6 using 2M hydrochloric acid (1.4 L), maintaining the temperature below 30° C. The mixture is then extracted with MeTHF (2×4.8 L), and the combined MeTHF extracts are washed with water (1.2 L). The mixture is concentrated to approx 2.4 L and EtOAc (3 L) is added. This put and take distillation is repeated a further 3 times. The mixture is adjusted to 60±3° C. and seeded twice (2×3 g) 35 mins apart. The resultant is aged for 1 h 10 mins then cooled over 2 h to 20-25° C., and aged for a further 15 h 50 min. The slurry is filtered, washed with EtOAc (2×1.2 L) and dried in vacuo at 45±5° C. for approx 3 day to give the title compound.
http://www.google.com/patents/US20140256721
Preparation of Salts of Compound ASodium Salt
Methanol (2 ml) was added to N-[5-[4-(5-{[(2R,6S)-2,6-dimethyl-4-morpholinyl]methyl}-1,3-oxazol-2-yl)-1H-indazol-6-yl]-2-(methyloxy)-3-pyridinyl]methanesulfonamide (0.3 g) followed by aqueous sodium hydroxide (0.129 ml) to give a solution. Tert-butylmethylether (4 ml) was added to the solution followed by seed crystals of the sodium salt and this suspension was stirred overnight at ambient conditions. The suspension was filtered, washed with tert-butylmethylether (2 ml) and air dried to give the sodium salt (0.2312 g) as a hydrate.
NMR: Consistent with salt formation
1H NMR (400 MHz, DMSO d6) d=13.35 (br s, 1H), 8.53 (s, 1H), 7.90 (d, J=1.2 Hz, 1H), 7.73 (s, 1H), 7.65 (d, J=2.5 Hz, 1H), 7.62 (d, J=2.2 Hz, 1H), 7.33 (s, 1H), 4.00 (s, 3H), 3.80 (s, 3H), 3.59 (m, 2H). 2.83 (d, J=10.3, 2H), 2.61 (s, 3H), 1.78 (t, J=10.5 Hz, 2H), 1.05 (d, J=6.1 Hz, 6H)
EMAIL ME amcrasto@gmail.com
EMAIL ME amcrasto@gmail.com
| US20100280029 * | 28 Apr 2010 | 4 Nov 2010 | Julie Nicole Hamblin | Novel compounds |
| WO2010125082A1 | 28 Apr 2010 | 4 Nov 2010 | Glaxo Group Limited | Oxazole substituted indazoles as pi3-kinase inhibitors |
| US20140256721 * | 14 Apr 2014 | 11 Sep 2014 | Glaxosmithkline Intellectual Property Development Limited | Novel Polymorphs and Salts |
| WO2012032065A1 | 6 Sep 2011 | 15 Mar 2012 | Glaxo Group Limited | Indazole derivatives for use in the treatment of influenza virus infection |
| WO2012032067A1 | 6 Sep 2011 | 15 Mar 2012 | Glaxo Group Limited | Polymorphs and salts of n- [5- [4- (5- { [(2r,6s) -2, 6 – dimethyl – 4 -morpholinyl] methyl} – 1, 3 – oxazol – 2 – yl) – 1h- inda zol-6-yl] -2- (methyloxy) – 3 – pyridinyl] methanesulfonamide |
| WO2012055846A1 | 25 Oct 2011 | 3 May 2012 | Glaxo Group Limited | Polymorphs and salts of 6-(1h-indol-4-yl)-4-(5- { [4-(1-methylethyl)-1-pi perazinyl] methyl} -1,3-oxazol-2-yl)-1h-indazole as pi3k inhibitors for use in the treatment of e.g. respiratory disorders |
| WO2012064744A2 * | 8 Nov 2011 | 18 May 2012 | Lycera Corporation | Tetrahydroquinoline and related bicyclic compounds for inhibition of rorϒ activity and the treatment of disease |
| WO2013088404A1 | 14 Dec 2012 | 20 Jun 2013 | Novartis Ag | Use of inhibitors of the activity or function of PI3K |
| WO2014068070A1 | 31 Oct 2013 | 8 May 2014 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for preventing antiphospholipid syndrome (aps) |
| US8524751 | 5 Mar 2010 | 3 Sep 2013 | GlaxoSmithKline Intellecutual Property Development | 4-oxadiazol-2-YL-indazoles as inhibitors of P13 kinases |
| US8536169 | 3 Jun 2009 | 17 Sep 2013 | Glaxo Group Limited | Compounds |
| US8575162 | 28 Apr 2010 | 5 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8580797 | 28 Apr 2010 | 12 Nov 2013 | Glaxo Smith Kline Intellectual Property Development Limited | Compounds |
| US8586583 | 2 Oct 2012 | 19 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8586590 | 2 Oct 2012 | 19 Nov 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8609657 | 2 Oct 2012 | 17 Dec 2013 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| US8658635 | 3 Jun 2009 | 25 Feb 2014 | Glaxosmithkline Intellectual Property Development Limited | Benzpyrazol derivatives as inhibitors of PI3 kinases |
| US8735390 | 6 Sep 2011 | 27 May 2014 | Glaxosmithkline Intellectual Property Development Limited | Polymorphs and salts |
| US8765743 | 3 Jun 2009 | 1 Jul 2014 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
…..
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
JALGAON, MAHARASHTRA, INDIA
.
.
.
MANUDEVI
Isradipine

Isradipine
CAS Registry Number: 75695-93-1
CAS Name: 4-(4-Benzofurazanyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylic acid methyl 1-methylethyl ester
Additional Names: isopropyl 4-(2,1,3-benzoxadiazol-4-yl)-1,4-dihydro-5-methoxycarbonyl-2,6-dimethyl-3-pyridinecarboxylate; 4-(2,1,3-benzoxadiazol-4-yl)-2,6-dimethyl-1,4-dihydro-3-isopropyloxycarbonylpyridine-5-carboxylic acid methyl ester; isrodipine
Manufacturers’ Codes: PN-200-110
Trademarks: Clivoten (Lifepharma); DynaCirc (Novartis); Esradin (Sigma-Tau); Lomir (Novartis); Prescal (Novartis)
Molecular Formula: C19H21N3O5
Molecular Weight: 371.39
Percent Composition: C 61.45%, H 5.70%, N 11.31%, O 21.54%
Properties: mp 168-170°.
Melting point: mp 168-170°
Derivative Type: S(+)-Form
Manufacturers’ Codes: PN-205-033
Properties: Crystals from ether + hexane, mp 142°. [a]D20 +6.7° (c = 1.5 in ethanol).
Melting point: mp 142°
Optical Rotation: [a]D20 +6.7° (c = 1.5 in ethanol)
Derivative Type: R(-)-Form
Manufacturers’ Codes: PN-205-034
Properties: Crystals from ether + hexane, mp 140°. [a]D20 -6.7° (c = 1.67 in ethanol).
Melting point: mp 140°
Optical Rotation: [a]D20 -6.7° (c = 1.67 in ethanol)
Keywords: Antianginal; Antihypertensive; Dihydropyridine Derivatives; Calcium Channel Blocker; Dihydropyridine Derivatives.
Isradipine (tradenames DynaCirc, Prescal) is a calcium channel blocker of the dihydropyridine class. It is usually prescribed for the treatment of high blood pressure in order to reduce the risk of stroke and heart attack. More recent research in animal models suggests that isradipine may have potential uses for treating Parkinson’s disease Chan et al. 2007.
Isradipine is given as either a 2.5mg or 5mg capsule. [1]
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html

Isradipine is a drug used to lower blood pressure but recently it was found by a team from Nortwestern University, that this molecule can also slow the progression of Parkinson’s disease, and restore the dopamine neurons (In animals tests). Isradipine is a calcium channel blocker of the 1,4-dihydropyridine class with a benzoxadiazole moiety in position 4.
The synthesis of the 1,4-dihydropyridine ring is quite classic, the first step consists in a Knoevenagel reaction of methyl acetoacetate on the benzoxadiazole 4-carboxaldehyde using piperidine and acetic acid as catalyst and diisopropylether as solvent in a 61% yield (this is the first time I see a Knoevenagel reaction in an ether!!!???? DCM, Toluene OK, but maybe I am wrong). The second step of this synthesis is the condensation of the acrylate obtained with the isopropyl aminocrotonate in ethanol to give the desire 1,4-dihydropyridine Isradipine in 67% yield after recrystallisation.
(WO/2005/005437) An improved Process for the Manufacture of Isradipine (Shasun Chemical & Drugs Limited)
The synthesis of the 1,4-dihydropyridine ring is quite classic, the first step consists in a Knoevenagel reaction of methyl acetoacetate on the benzoxadiazole 4-carboxaldehyde using piperidine and acetic acid as catalyst and diisopropylether as solvent in a 61% yield (this is the first time I see a Knoevenagel reaction in an ether!!!???? DCM, Toluene OK, but maybe I am wrong). The second step of this synthesis is the condensation of the acrylate obtained with the isopropyl aminocrotonate in ethanol to give the desire 1,4-dihydropyridine Isradipine in 67% yield after recrystallisation.

(WO/2005/005437) An improved Process for the Manufacture of Isradipine (Shasun Chemical & Drugs Limited)
Isradipine is 4-(4-Benzofi–razanyl)-l,4-d–hydro-2,6-dimethyl-3,5- pyiicϊ-nedicarboxylic acid med yl 1-methylethyl ester having die chemical structure of formula (I).

( I ) Isradipine is therapeutically indicated for treating cardiovascular diseases.
The cardiovascular diseases include angina, pectoris, hypertension and congestive heart failure. It is also used to treat high blood pressure. Isradipine was disclosed in the German specification DE 2949491 and US patent Nos. 4466972 and 4567271. DE 2949491 describes the general procedure to prepare 1,4-dihydropyridine derivatives. US 4466972, GBQ2103203A, LU 0088342A9, EP 0000150A1, EP 0000150B1, AU 0538515B2 and od er related patents describe the general mediod for d e preparation of Benzoxadiazoles and dieir derivatives of general formula (EL). These references in its entirety is hereby incorporated by reference into this application.

( II ) Where in Ri is -CH3 and -R2 is — CH(CH3)2 it refers to Isradipine of formula ( I ). When Ri and R2 are not identical the general procedures described in diese patent specifications produces a mixture of isomers of formula ( II ). These procedures for the preparation of Isradipine is characteristic of formation of the ‘ isomeric impurities, 1) 4-(4-Benzoi-urazanyl)-l,4-α^ydro-2,6-climedιyl-3,5- pyridinedicarboxylic acid di-methyl ester of formula ( III ) and 2) 4-(4- Benzofurazanyl)-l,4-α^ydro-2,6-d–methyl-3,5-pyrid–nedicarboxylic acid di-1- med ylethyl ester of formula ( IV) along with Isradipine. The US patent 4466972 describes the preparation of compounds of general formula (II) by refluxing 2, 1, 3-benzoxadiazole-4-carboxaldehyde, keto ester and concentrated ammonia or a β-amino ester in presence of ethanol, followed by evaporation and purification by chromatography.
H

( HI )H

( I V ) Tl ese symmetrical ester isomers ( III ) and ( IN ) are difficult to separate from the Isradipine and the separation is effected only by a chromatographic purifications. The drawback witii the procedures described in these patents is that it is very difficult to produce die product in commercial quantities as it involves d e purification of the product by chromatographic separations. A single step process for the preparation of Isradipine was described in CH 661270. This procedure involves first reacting 2,l,3-benzoxadiazole-4- carboxaldehyde with isopropyl acetoacetate in the presence of catalytic quantities of acetic acid and piperidine in refluxing toluene, and further reacting it widi mediyl-β-aminocrotonate. The Isradipine formed in d e reaction mixture was dien- separated by toluene distillation followed by cyclohexane treatment. The crude product obtained was dien crystallised from etiianol to get Isradipine. When we have repeated this process in our laboratory we got the Isradipine with substantially higher amount of symmetrical ester isomers ( III ) and ( IV ) are present in d e product. Removal of these symmetrical ester isomers is very difficult even after several repurifications from ethanol.

Stap 2

Example 4
Preparation of Isradipine using crude 2-acetyl-3-benzofurazan-4-yl-acrylic acid methyl ester Dissolved the crude 2-acetyl-3-benzofurazan-4-yl-acrylic acid methyl ester ‘ obtained in example – 1 (25 g, 0.10 mol) in absolute edianol (375 ml) and added in to the solution isopropyl-β-aminocrotonate (13.15 ml, 0.09 mol). Stirred the reaction mixture under nitrogen atmosphere at 25-28 °C for 7 hr. Removed sample from d e reaction mixture and analysed die sample by qualitative HPLC. Distilled ethanol from the reaction mixture- under vacuum at 50°C. Dissolved d e residue in ethyl acetate (235 ml) and washed twice widi water (90 ml). Dried the organic layer over sodium sulphate and distillation under vacuum at 50 °C. Dissolved the concentrate in ethanol (65 ml) at 70°C and slowly cooled to 5°C to get die product crystallised. Filtered the product and washed with pre cooled ethanol (25 ml). Recrystauised the product from ethanol (60 ml) and dried at 70°C under vacuum to obtain Isradipine (yield = 20 g, purity = 98.2% and Impurity III = 0.64%, Impurity IV = 0.51% by HPLQ
Example 5 Preparation of Isradipine using purified 2-acetyl-3-benzofurazan-4-yl-acrylic acid methyl ester Dissolved 2-acetyl-3-benzo–urazan-4-yl-acrylic acid methyl ester (25 g, 0.10 mol) in absolute ethanol (375 ml) and added in to the solution isopropyl-β- aminocrotonate (13.15 ml, 0.09 mol). Stirred the reaction mixture under nitrogen atmosphere at 25-28 °C for 5 hr. Removed sample from the reaction mixture and analysed the sample by qualitative HPLC. Distilled ethanol from the reaction mixture under vacuum at 50°C. Dissolved the residue in ethyl acetate (235 ml) and washed twice wid w?ater (90 ml). Dried die organic layer over sodium sulphate and distillation under vacuum at 50 °C. Dissolved the concentrate in ethanol (65 ml) at 70°C and slowly cooled to 5°C to get the product crystallised. Filtered the product. and washed with pre cooled ethanol (25 ml). Recrystallised the product from edianol (60 ml) and dried at 70°C under vacuum to obtain 25 g Isradipine (yield = 67%, purity 99.5%, Impurity III = 0.20%, and Impurity IV = 0.12% by HPLC)
Example 6 Preparation of Isradipine using purified 2-acetyl-3-benzofurazan-4-yl-acrylic acid methyl ester Dissolved the purified 2-acetyl-3-benzofurazan-4-yl-acrylic acid mediyl ester, obtained in example — 3 (25 g, 0.10 mol) in absolute ethanol (375 ml) and added in to the solution isopropyl-β-aminocrotonate (13.15 ml, 0.09 mol). Stirred the reaction mixture under nitrogen atmosphere at 25-28 °C for 5 hr. Removed sample from the reaction mixture and analysed the sample by qualitative HPLC. Distilled ethanol from die reaction mixture under vacuum at 50°C. Dissolved die residue in ethyl acetate (235 ml) and washed twice with water (90 ml). Dried the organic layer over sodium sulphate and distillation under vacuum at 50 °C. Dissolved the concentrate in ethanol (65 ml) at 70°C and slowly cooled to 5°C to get the product crystallised. Filtered the product and washed with pre cooled ethanol (25 ml) and dried at 70°C under vacuum to obtain 30g Isradipine (purity = 99.4%, Impurity III = 0.22%, and Impurity IV = 0.11% by HPLC). Throughout this application, various publications are referenced.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
Side effects
Common side effects include: [2]
- Dizziness
- Warmth, redness, or tingly feeling under your skin
- Headache
- Weakness, tired feeling
- Nausea, vomiting, diarrhea, upset stomach
- Skin rash or itching
Serious side effects include: [2]
- Lightheadedness or fainting
- Shortness of breath, especially from minimal physical activity
- Swelling in the hands and feet
- Rapid and/or heavy heartbeat
- Chest pain
If you experience one or more of these serious side effects, contact your health care provider immediately.
Significant drug interactions
There are other interactions beyond those listed below. Make sure to speak with a Pharmacist or Doctor if you have any concerns.
Three major interactions are listed below.
1. It is advised that those using Isradipine not take Anzemet (Dolasetron), as both agents can cause a dose-dependent PR intervaland QRS complex prolongation. [3]
2. Onmel/Sporanox (Itraconazole) exhibits a negative inotropic effect on the heart and thus could spur an additive effect when used concomitantly with Isradipine. Onmel/Sporanox also inhibits an important cytochrome liver enzyme (CYP 450 3A4) which is needed to metabolize Isradipine and other Calcium Channel Blockers. This will increase plasma levels of Isradipine and could cause an unintentional overdose of the medication. Caution is advised when administering both agents together. [4]
3. Zanaflex (Tizanidine) demonstrates anti-hypertensive effects and should be avoided in patients taking Isradipine due to the possibility of synergism between both medications. [5]
4. The anti-biotic Rifadin (Rifampin) lowered plasma concentrations of Isradipine to below detectable limits. [1]
5. Tagamet (Cimetidine) increased Isradipine mean peak plasma levels. A downward dose adjustment may be necessary with this particular instance of polypharmacy. [1]
6. Severe hypotension was reported with Duragesic (Fentanyl) anesthesia when it was combined with other Calcium Channel Blockers. Even though Isradipine, another Calcium Channel Blocker, has not been used in conjunction with Fentanyl anesthesia in any studies, caution is advised. [1]
Note: There was no significant interaction between Isradipine and Warfarin (Coumadin), Isradipine and Microzide Hydrochlorothiazide, Isradipine and Lanoxin (Digoxin), and Isradipine and Nitrostat (Nitroglycerin).
Overdose
Symptoms of an Isradipine overdose include: [1]
- Lethargy
- Sinus tachycardia
- Transient hypotension
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
…………………
US 4466972
http://www.google.com.na/patents/US4466972
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
………………………………….
http://www.google.com.ar/patents/CN101768153B?cl=en
isradipine is a class Benzofurazan dihydropyridine class of compounds, the synthesis is more complex, especially in the purified material is particularly difficult.
US Patent US4466972 and PCT application W02005 / 00437 respectively disclose two synthetic isradipine
Methods. However, the product prepared by conventional methods contain a certain amount of a formula homologues impurity, the impurity is structured as follows:

wherein, R1, R2 simultaneously or separately as methyl, ethyl and isopropyl. The homologue of the impurities with isradipine extremely difficult to separate, resulting in ineffective purification isradipine.
In addition, conventional processes for preparing key intermediates involved in 4-methyl benzofurazan. However, the preparation method of the key intermediates is still unsatisfactory. For example, the Chinese Patent 200 510 125 267 (Publication No. CN1847233A) in
Discloses an intermediate 4-methyl benzofurazan preparation methods, the preparation process is as follows:

It is clear that the above method steps long, dangerous operation, poor control, and cause a lot of pollution.
Accordingly, there is an urgent need to develop new, efficient and simple isradipine important intermediates for preparing 4-methyl benzofurazan method.
[0019]

[0020] (b) in an inert solvent, such that 4-methyl benzofurazan oxide reduction to form 4-methyl benzofurazan, i.e. a compound of formula 3;
[0021]

[0028]

[0029] to form isradipine.

Example 7 β – amino crotonic acid isopropyl ester (Compound 8)
The isopropyl acetoacetate (72.0g, 0 5mol.), Ammonium acetate (57. 8g, 0 75mol.) And tert-butanol / ethanol (1: 1,600ml) were mixed in IOOOml flask, 300 mesh sieve (50g), heated to reflux, TLC plate monitor. After the reaction is substantially completed, cooled to room temperature, filtered and the filtrate was concentrated until no liquid was distilled off, the residual liquid was distilled under reduced pressure, to collect 110-120 ° C fraction (degree of vacuum of 0. IMPa) to give compound 8 (66. Og). Y = 92. 4%.
1H-WR (CDCl3):…… 5 02-4 95 (1H, m), 4 48 (1H, s), 1 88 (3H, d), 1 23-1 21 (6H , d)
Example 8 isradipine (Isradipine)
In the protection teams, three-necked flask Compound 6 (3.0g, 20mmol), i3- amino crotonic acid isopropyl ester (Compound 8) (2. 7g, 16. 6mmol), methyl acetoacetate (3. 50g, 30mol), Ac2O (2. 05g, 20mmol), conc. H2SO4 (0. 4g, 4mmol) and tert-butanol / ethanol (1: 1,65ml) mixing the liquid phase monitoring, when the remainder is less than 3 Compound 6 When the 7% to terminate the reaction. The reaction was concentrated, the residue was dissolved CH2Cl2 (55ml), washed with water (45ml X Magic, dried, concentrated, drain pump, to give 6. 7g end yellow foam-like solid. Ethanol QOml) dissolved by heating, stirring crystallization (overnight) to give a pale yellow powder isradipine (4. 3g) (HPLC purity> 99.8%, impurity content homologues thereof are less than 0.1), yield 66.8%.
1H-WR (CDCl3):…… 7 62-7 60 (lH, m), 7 31-7 26 (2H, m), 5 46 (lH, s), 4 92-4 . 86 (1H, m), 3. 57 (3H, s), 2. 32-2. 30 (6H, m), 1. 21-1. 19 (3H, d), 0. 95-0. 94 (3H, d)
Comparative Example 1
isradipine (Isradipine) prepared by the United States Patent US4466972:
In the protection teams, three-necked flask Compound 6 (3.0g, 20mmol), i3- amino crotonic acid isopropyl ester (Compound 8) (2. 7g, 16. 6mmol), methyl acetoacetate (3. 50g, 30mol), Ac2O (2. 05g, 20mmol), conc. H2SO4 (0. 4g, 4mmol) and ethanol (65ml) were mixed and stirred, the liquid phase monitoring, when the compound 6 is less than 3.7% remaining, the reaction is stopped. The reaction was concentrated, the residue was dissolved CH2Cl2 (55ml), washed with water (45ml X Magic, dried, concentrated, drain pump, to give 6. 3g end yellow foam-like solid. Ethanol OOml) was dissolved by heating, stirring crystallization (overnight) to give a pale yellow powder isradipine (4. Ig) (HPLC purity: 99.0%, impurity content was homologues greater than 0.3%), a yield of 63.7%.
Compared with Comparative Example 1 (homolog impurity content was greater than 0.3%), was the content of impurities isradipine homologs prepared in Example 1-8 is less than 0.1% by the embodiment of the present invention.
The 10 cases of isradipine
Example 8 was repeated, except that, with t-butanol / ethanol (1: 2,70ml) or t-butanol / ethanol O: 1, 70ml) replaces t-butanol / ethanol (1: 1,65ml) 0
The results showed that isradipine yield of about 62%, the test substance impurity content of less than 0.1% homologous.
Further reading and references
- “”Isradipine: Brands, Medical Use, Clinical Data””.
- “Isradipine Side Effects”.
- “”Isradipine and Anzemet Drug Interactions””.
- “”Isradipine and Onmel Drug Interactions””.
- “”Isradipine and Zanaflex Drug Interactions””.
- Hattori T, Wang P (2006). “Calcium antagonist isradipine-induced calcium influx through nonselective cation channels in human gingival fibroblasts.”. Eur J Med Res 11 (3): 93–6. PMID 16751108.
- Ganz M, Mokabberi R, Sica D (2005). “Comparison of blood pressure control with amlodipine and controlled-release isradipine: an open-label, drug substitution study.”. J Clin Hypertens (Greenwich) 7 (4 Suppl 1): 27–31. doi:10.1111/j.1524-6175.2005.04450.x. PMID 15858400.
- Johnson B, Roache J, Ait-Daoud N, Wallace C, Wells L, Dawes M, Wang Y (2005). “Effects of isradipine, a dihydropyridine-class calcium-channel antagonist, on d-methamphetamine’s subjective and reinforcing effects.”. Int J Neuropsychopharmacol 8 (2): 203–13. doi:10.1017/S1461145704005036. PMID 15850499.
- Fletcher H, Roberts G, Mullings A, Forrester T (1999). “An open trial comparing isradipine with hydralazine and methyl dopa in the treatment of patients with severe pre-eclampsia.”. J Obstet Gynaecol 19 (3): 235–8. doi:10.1080/01443619964977. PMID 15512286.
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ (2007). “‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease.”.Nature 447 (3): 1081–1086. doi:10.1038/nature05865. PMID 17558391.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
External links
- MedlinePlus DrugInfo medmaster-a693048
- DDB 30003
- Drug offers hope for Parkinson’s – BBC News, 11 June 2007.
- [1] – Commentary of Chan et al. publication
- [2] – ArsTechnica – Why neurons die in Parkinson’s patients
…………
CN1847233A21 Nov 200518 Oct 2006圣玛精细化工有限责任公司Method for preparing 4-formoxylbenzofuran
US446697219 Mar 198221 Aug 1984Sandoz Ltd.Hypotensive, antiischemic, antispasmodic agents
WO2005005437A115 Jul 200420 Jan 2005Radhakrishnan Selvar MullaiyurAn improved process for the manufacture of isradipine.
References: Dihydropyridine calcium channel blocker. Prepn: P. Neumann, DE 2949491; idem, US 4466972 (1980, 1984 both to Sandoz). Prepn of enantiomers: A. Vogel, DE 3320616 (1983 to Sandoz), C.A. 101, 7162s (1984). Comparative study of in vitro effects on human and canine cerebral arteries: E. Müller-Schweinitzer, P. Neumann, J. Cereb. Blood Flow Metab.3, 354 (1983). Effect on a-adrenoceptor mediated vasoconstriction in rats: K. Jie et al., Arch. Int. Pharmacodyn. 278, 72 (1985). Pharmacokinetics: F. L. S. Tee, J. M. Jaffe, Eur. J. Clin. Pharmacol. 32, 361 (1987). Clinical evaluation in angina and coronary artery disease: C. E. Handler, E. Sowton, ibid. 27, 415 (1984); in hypertension: E. B. Nelson et al., Clin. Pharmacol. Ther. 40, 694 (1986). Comparison of hemodynamic effects of enantiomers: R. P. Hof et al., J. Cardiovasc. Pharmacol. 8, 221 (1986). Series of articles on pharmacology and clinical use: Am. J. Med. 86, 1-146 (1989).
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 3-methyl 5-propan-2-yl 4-(2,1,3-benzoxadiazol-4-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate | |
| Clinical data | |
| Trade names | DynaCirc |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a693048 |
|
|
| Legal status | |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 15-24% |
| Protein binding | 95% |
| Metabolism | 100% Hepatic |
| Half-life | 8 hours |
| Excretion | 70% Renal, 30% Fecal |
| Identifiers | |
| CAS number | 75695-93-1 |
| ATC code | C08CA03 |
| PubChem | CID 3784 |
| DrugBank | DB00270 |
| ChemSpider | 3652 |
| UNII | YO1UK1S598 |
| KEGG | D00349 |
| ChEMBL | CHEMBL1648 |
| Chemical data | |
| Formula | C19H21N3O5 |
| Molecular mass | 371.387 g/mol |
…….
Alleppey kerala INDIA…..Alappuzha
Alappuzha – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Alappuzha
pronunciation (help·info)), also known as Alleppey, is the administrative headquarters of Alappuzha District of Kerala state of southern India. Alappuzha is the …


Table in restaurant after eating fish, Alleppey, Kerala, India, South Asia,
PAGODA RESORTS ALLEPPEY KERALA INDIA

////////////
CILNIDIPINE 西尼地平

Cilnidipine
西尼地平
CAS 132203-70-4
- (E) – (±) 1 ,4 a dihydro-2 ,6 – dimethyl-4 – (3 – nitrophenyl) -3,5 – pyridinedicarboxylic acid, 2 – methoxy- ethyl butylester 3 – phenyl – 2 – propenyl ester FRC-8653 Cinalong
- More FRC 8653 1,4-Dihydro-2 ,6-dimethyl-4-(3-nitrophenyl) 3 ,5-pyridinedicarboxylic acid 2-methoxyethyl (2E)-3-phenyl-2-propenyl ester
- Molecular formula:C 27 H 28 N 2 O 7
- Molecular Weight:492.52
CAS Name: 1,4-Dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid 2-methoxyethyl (2E)-3-phenyl-2-propenyl ester
Additional Names: (±)-(E)-cinnamyl 2-methoxyethyl 1,4-dihydro-2,6-dimethyl-4-(m-nitrophenyl)-3,5-pyridinedicarboxylate
Cinnamyl 2-methoxyethyl 4-(3-nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate
Manufacturers’ Codes: FRC-8653
Trademarks: Atelec (Morishita); Cinalong (Fujirebio); Siscard (Boehringer, Ing.)
Percent Composition: C 65.84%, H 5.73%, N 5.69%, O 22.74%
Properties: Crystals from methanol, mp 115.5-116.6°. LD50 in male, female mice, rats (mg/kg): ³5000, ³5000, ³5000, 4412 orally;³5000 all species s.c.; 1845, 2353, 441, 426 i.p. (Wada).
Melting point: mp 115.5-116.6°
Toxicity data: LD50 in male, female mice, rats (mg/kg): ³5000, ³5000, ³5000, 4412 orally; ³5000 all species s.c.; 1845, 2353, 441, 426 i.p. (Wada)
Ajinomoto (INNOVATOR)
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
Antihypertensive; Dihydropyridine Derivatives; Calcium Channel Blocker; Dihydropyridine Derivatives.

Cilnidipine (INN) is a calcium channel blocker. It is sold as Atelec in Japan, asCilaheart, Cilacar in India, and under various other trade names in East Asian countries.
Cilnidipine is a dual blocker of L-type voltage-gated calcium channels in vascular smooth muscle and N-type calcium channels in sympathetic nerve terminals that supply blood vessels. However, the clinical benefits of cilnidipine and underlying mechanisms are incompletely understood.
Clinidipine is the novel calcium antagonist accompanied with L-type and N-type calcium channel blocking function. It was jointly developed by Fuji Viscera Pharmaceutical Company, Japan and Ajinomoto, Japan and approved to come into market for the first time and used for high blood pressure treatment in 1995. in india j b chemicals & pharmaceuticals ltd and ncube pharmaceutical develope a market of cilnidipine.
Hypertension is one of the most common cardiovascular disease states, which is defined as a blood pressure greater than or equal to 140/90 mm Hg. Recently, patients with adult disease such as hypertension have rapidly increased. Particularly, since damages due to hypertension may cause acute heart disease or myocardial infarction, etc., there is continued demand for the development of more effective antihypertensive agent.
Meanwhile, antihypertensive agents developed so far can be classified into Angiotensin II Receptor Blocker (ARB), Angiotensin-Converting Enzyme Inhibitor (ACEI) or Calcium Chanel Blocker (CCB) according to the mechanism of actions. Particularly, ARB or CCB drugs manifest more excellent blood pressure lowering effect, and thus they are more frequently used.
However, these drugs have a limit in blood pressure lowering effects, and if each of these drugs is administered in an amount greater than or equal to a specific amount, various side-effects may be caused. Therefore, there have been many attempts in recent years to obtain more excellent blood pressure lowering effect by combination therapy or combined preparation which combines or mixes two or more drugs.
Particularly, since side-effect due to each drug is directly related to the amount or dose of a single drug, there have been active attempts to combine or mix two or more drugs thereby obtaining more excellent blood pressure lowering effect through synergism of the two or more drugs while reducing the amount or dose of each single drug.
For example, US 20040198789 discloses a pharmaceutical composition for lowering blood pressure combining lercanidipine, one of CCB, and valsartan, irbesartan or olmesartan, one of ARB, etc. In addition, a combined preparation composition which combines or mixes various blood pressure lowering drugs or combination therapy thereof has been disclosed.
cilnidipine Compared with other calcium antagonists, clinidipine can act on the N-type calcium-channel that existing sympathetic nerve end besides acting on L-type calcium-channel that similar to most of the calcium antagonists. Due to its N-type calcium-channel blocking properties, it has more advantages compared to conventional calcium-channel blockers. It has lower incidence of Pedal edema, one of the major adverse effects of other calcium channel blockers. Cilnidipine has similar blood pressure lowering efficacy as compared to amlodipine. One of the distinct property of cilnidipine from amlodipine is that it does not cause reflex tachycardia.
In recent years, cardiovascular disease has become common, the incidence increased year by year, about a patient of hypertension in China. 3-1. 500 million, complications caused by hypertension gradually increased, and more and more young patients with hypertension technology. In recent years, antihypertensive drugs also have great development, the main first-line diuretic drug decompression 3 – blockers, calcium channel blockers, angiotensin-converting enzyme inhibitors, ar blockers and vascular angiotensin II (Ang II) receptor antagonist.
In the anti-hypertensive drugs, calcium antagonists are following a – blockers after another rapidly developing cardiovascular drugs, has been widely used in clinical hypertension, angina and other diseases, in cardiovascular drugs in the world, ranked first.
Cilnidipine for the long duration of the calcium channel blockers, direct relaxation of vascular smooth muscle, dilation of peripheral arteries, the peripheral resistance decreased, with lower blood pressure, heart rate without causing a reflex effect.
Cilnidipine is a dihydropyridine CCB as well as an antihypertensive. Cilnidipinehas L- and N-calcium channel blocking actions. Though many of the dihydropyridine CCBs may cause an increase in heart rate while being effective for lowering blood pressure, it has been confirmed that cilnidipine does not increase the heart rate and has a stable hypotensive effect. (Takahiro Shiokoshi, “Medical Consultation & New Remedies” vol. 41, No. 6, p. 475-481)
- http://www.mcyy.com.cn/e-product2.asp
- Löhn M, Muzzulini U, Essin K, et al. (May 2002). “Cilnidipine is a novel slow-acting blocker of vascular L-type calcium channels that does not target protein kinase C”. J. Hypertens.20 (5): 885–93. PMID12011649.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
Cilnidipine (CAS NO.: 132203-70-4), with its systematic name of (+-)-(E)-Cinnamyl 2-methoxyethyl 1,4-dihydro-2,6-dimethyl-4-(m-nitrophenyl)-3,5-pyridinedicarboxylate, could be produced through many synthetic methods.
Following is one of the synthesis routes: By cyclization of 2-(3-nitrobenzylidene)acetocetic acid cinnamyl ester (I) with 2-aminocrotonic acid 2-methoxyethyl ester (II) by heating at 120 °C.
………………..
http://www.google.com/patents/EP0161877A2?cl=en
AN EXAMPLE
Example 1
-
3.51 g (10 mM) of 2-(3-nitrobenzylidene) acetoacetic acid cinnamyl ester were mixed with 1.38 g (12 mM) of 3-aminocrotonic acid methyl ester, and heated at 120°C for 3 hours. The reaction mixture was separated by silica gel column chromatography, and 3.00 g of cinnamyl methyl 4-(3-nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (trans) were obtained (yield 67%). This derivative was recrystallized once from methanol.
-
Elemental Analysis; C25H24N206
- Calcd. (%) C: 66.95, H: 5.39, N: 6.25
- Found (%) C: 67.03, H: 5.31, N: 6.20
(trans)
-
- m.p.; 143.5-144.5°C
- IR (cm-1); vNH 3370, νCO 1700, νNO2 1530, 1350
- NMR δCDCl3; 2.34(s,6H), 3.60(s,3H), 4.69(d,2H), 5.13(s,lH), 6.14(tt,lH), 6.55(d,lH), 7.1-8.1(m,9H)
(cis)
-
- m.p.; 136-137°C
- IR (cm-1); vNH 3360, νCO 1700, 1650, νNO2 1530, 1350
- NMR δCDCl3; 2.30(s,6H), 3,60(s,3H), 4.80(d,lH), 5.10(s,1H), 5.77(tt,lH), 6.56(d,1H), 6.64(bs,1H), 7.1-8.1(m,9H)
EXAMPLE 13
-
Example 13 Cinnamyl 2-methoxyethyl 4-(3-nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate
-
Elemental Analysis; C27H28N2O7
- Calcd. (%) C: 65.84, H: 5.73, N: 5.69
- Found (%) C: 65.88, H: 5.70, N: 5.66
- m.p.; 115.5-116.5°C
- IR (cm-1); vNH 3380, νCO 1710, 1680, νNO2 1530, 1350
- NMR δCDCl3; 2.34(s,6H), 3.25(s,3H), 3.50(t,2H), 4.15(t,2H), 4.68(d,2H), 5.15(s,lH), 5.9-6.9(m,3H), 7.1-8.2(m,9H)

cyclization of 2-(3-nitrobenzylidene)acetocetic acid cinnamyl ester (I) with 2-aminocrotonic acid 2-methoxyethyl ester (II) by heating at 120 C.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
MORE
NMR
CARBOHYDRATE POLYMERS 90 PG 1719-1724 , YR2012
Numerous peaks were found in the spectrum of cilnidipine: 2.3555 (3H, s, CH3), 2.3886(3H, s, CH3), 3.2843(CD3OD), 3.3292(3H, s, OCH3), 3.5255–3.5623(2H, m, CH3OCH2CH2 ), 4.1224–4.1597(2H, m, CH3OCH2CH2 ), 4.6695–4.7293(2H, m, CH2 CH CH ), 4.8844(D2O), 5.1576(1H, s, CH), 6.2609(1H, dt, CH2 CH CH ), 6.5518(1H, d, CH2 CH CH ), 7.2488–7.3657(6H, m, ArH), 7.7002(1H, dd, ArH), 7.9805(1H, dd, ArH), 8.1548(1H, s, ArH)
References:
Dihydropyridine calcium channel blocker. Prepn: T. Kutsuma et al., EP 161877; eidem, US 4672068(1985, 1987 both to Fujirebio).
Pharmacology: K. Ikeda et al., Oyo Yakuri 44, 433 (1992).
Mechanism of action study: M. Hosonoet al., J. Pharmacobio-Dyn. 15, 547 (1992).
LC-MS determn in plasma: K. Hatada et al., J. Chromatogr. 583, 116 (1992). Clinical study: M. Ishii, Jpn. Pharmacol. Ther. 21, 59 (1993).
Acute toxicity study: S. Wada et al., Yakuri to Chiryo 20, Suppl. 7, S1683 (1992), C.A. 118, 32711 (1992).
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
U.S Patent No. 4,572,909 discloses amlodipine;
U.S Patent No. 4,446,325 discloses aranidipine;
U.S Patent No. 4,772,596 discloses azelnidipine;
U.S Patent No. 4,220,649 discloses barnidipine;
U.S Patent No. 4,448,964 discloses benidipine;
U.S Patent No. 5,856,346 discloses clevidipine;
U.S Patent No. 4,466,972 discloses isradipine;
U.S Patent No. 4,885,284 discloses efonidipine; and
U.S Patent No. 4,264,61 1 discloses felodipine.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
-
Planar chemical structures of these calcium blockers of formula (I) are shown below.









-
Amlodipine is 2-(2-aminoethoxymethyl)-4-(2-chlorophenyl)-3-ethoxycarbonyl-5-methoxycarbonyl-6-methyl-1,4-dihydropyridine disclosed in USP 4,572,909, Japanese patent publication No. Sho 58-167569 and the like.
-
Aranidipine is 3-(2-oxopropoxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(2-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,446,325 and the like.
-
Azelnidipine is 2-amino-3-(1-diphenylmethyl-3-azetidinyloxycarbonyl)-5-isopropoxycarbonyl-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,772,596, Japanese patent publication No. Sho 63-253082 and the like.
-
Barnidipine is 3-(1-benzyl-3-pyrrolidinyloxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,220,649, Japanese patent publication No. Sho 55-301 and the like.
-
Benidipine is 3-(1-benzyl-3-piperidinyloxycarbonyl)-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine and is described in the specifications of U.S. Patent No. 4,501,748, Japanese patent publication No. Sho 59-70667 and the like.
-
Cilnidipine is 2,6-dimethyl-5-(2-methoxyethoxycarbonyl)-4-(3-nitrophenyl)-3-(3-phenyl-2-propenyloxycarbonyl)-1,4-dihydropyridine disclosed in USP 4,672,068, Japanese patent publication No. Sho 60-233058 and the like.
-
Efonidipine is 3-[2-(N-benzyl-N-phenylamino)ethoxycarbonyl]-2,6-dimethyl-5-(5,5-dimethyl-1,3,2-dioxa-2-phosphonyl)-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,885,284, Japanese patent publication No. Sho 60-69089 and the like.
-
Elgodipine is 2,6-dimethyl-5-isopropoxycarbonyl-4-(2,3-methylenedioxyphenyl)-3-[2-[N-methyl-N-(4-fluorophenylmethyl)amino]ethoxycarbonyl]-1,4-dihydropyridine disclosed in USP 4,952,592, Japanese patent publication No. Hei 1-294675 and the like.
-
Felodipine is 3-ethoxycarbonyl-4-(2,3-dichlorophenyl)-2,6-dimethyl-5-methoxycarbonyl-1,4-dihydropyridine disclosed in USP 4,264,611, Japanese patent publication No. Sho 55-9083 and the like.
-
Falnidipine is 2,6-dimethyl-5-methoxycarbonyl-4-(2-nitrophenyl)-3-(2-tetrahydrofurylmethoxycarbonyl)-1,4-dihydropyridine disclosed in USP 4,656,181, Japanese patent publication (kohyo) No. Sho 60-500255 and the like.
-
Lemildipine is 2-carbamoyloxymethyl-4-(2,3-dichlorophenyl)-3-isopropoxycarbonyl-5-methoxycarbonyl-6-methyl-1,4-dihydropyridine disclosed in Japanese patent publication No. Sho 59-152373 and the like.
-
Manidipine is 2,6-dimethyl-3-[2-(4-diphenylmethyl-1-piperazinyl)ethoxycarbonyl]-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,892,875, Japanese patent publication No. Sho 58-201765 and the like.
-
Nicardipine is 2,6-dimethyl-3-[2-(N-benzyl-N-methylamino)ethoxycarbonyl]-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,985,758, Japanese patent publication No. Sho 49-108082 and the like.
-
Nifedipine is 2,6-dimethyl-3,5-dimethoxycarbonyl-4-(2-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,485,847 and the like.
-
Nilvadipine is 2-cyano-5-isopropoxycarbonyl-3-methoxycarbonyl-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,338,322, Japanese patent publication No. Sho 52-5777 and the like.
-
Nisoldipine is 2,6-dimethyl-3-isobutoxycarbonyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 4,154,839, Japanese patent publication No. Sho 52-59161 and the like.
-
Nitrendipine is 3-ethoxycarbonyl-2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-1,4-dihydropyridine disclosed in USP 3,799,934, Japanese patent publication (after examination) No. Sho 55-27054 and the like.
-
Pranidipine is 2,6-dimethyl-5-methoxycarbonyl-4-(3-nitrophenyl)-3-(3-phenyl-2-propen-1 -yloxycarbonyl)-1,4-dihydropyridine disclosed in USP 5,034,395, Japanese patent publication No. Sho 60-120861 and the like.
read more on dipine series………http://organicsynthesisinternational.blogspot.in/p/dipine-series.html
MAHABALIPURAM, INDIA
Mahabalipuram – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Mahabalipuram
Mahabalipuram, also known as Mamallapuram is a town in Kancheepuram district in the Indian state of Tamil Nadu. It is around 60 km south from the city of …Shore Temple – Seven Pagodas – Pancha Rathas –
.
.
Krishna’s Butter Ball in Mahabalipuram, India. The surface below the rock is …
http://www.weather-forecast.com/locations/Mamallapuram
Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
Moonraikers Restaurant, Mamallapuram
Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
.
A carving at the Varaha Temple, Mahabalipuram
/////////////

{kind=link}