
Home » Posts tagged 'gsk' (Page 2)
Tag Archives: gsk
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GSK 2126458, Omipalisib, PI3K/mTOR inhibitor

GSK 2126458
CAS 1086062-66-9
OMipalisib;GSK2126458;GSK-2126458;GSK2126458 (GSK458);GSK212;
2,4-Difluoro-N-[2-methoxy-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl]benzenesulfonamide;
2,4-Difluoro-N-[2-Methoxy-5-[4-(pyridazin-4-yl)quinolin-6-yl]pyridin-3-yl]benzenesulfonaMide
2,4-Difluoro-N-[2-methoxy-5-[4-(4-pyridazinyl)quinolin-6-yl]pyridin-3-yl]benzenesulfonamide
phosphoinositide 3 kinase inhibitor
idiopathic pulmonary fibrosis
PHASE 1
MW 505.49598
MF C25H17F2N5O3S
GSK…….http://www.gsk.com/media/280387/product-pipeline-2014.pdf
![]()
Omipalisib (GSK2126458): Omipalisib, also known as GSK2126458, is a small-molecule pyridylsulfonamide inhibitor of phosphatidylinositol 3-kinase (PI3K) with potential antineoplastic activity. PI3K inhibitor GSK2126458 binds to and inhibits PI3K in the PI3K/mTOR signaling pathway, which may trigger the translocation of cytosolic Bax to the mitochondrial outer membrane, increasing mitochondrial membrane permeability and inducing apoptotic cell death. Bax is a member of the proapoptotic Bcl2 family of proteins. PI3K, often overexpressed in cancer cells, plays a crucial role in tumor cell regulation and survival.
GlaxoSmithKline (GSK) is developing omipalisib (GSK-2126458), a phosphoinositide 3-kinase/mammalian target of rapamycin (PI3K/mTOR) inhibitor as well as mTOR complex 1 and 2 inhibitor, for the potential oral treatment of cancer and idiopathic pulmonary fibrosis
MEDKOO

![]()
![]()
![]()
![]()
|
Certificate of Analysis: |
|
|
QC data: |
GSK2126458 is a highly potent PI3K and mTOR inhibitor. In vivo, GSK2126458 showed anti-tumor activity in both pharmacodynamic and tumor growth efficacy models. GSK2126458 reduced the phosphorylated AKT, p70S6K contents in a dose and time dependent way. The IC50 of GSK2126458 is 2 nM for pAKT in the HCC1954 breast carcinoma cell line. In various human tumor cells, GSK2126458 had a width of inhibitory activity for potent cell growth and induced cell death. Notably, GSK2126458 acted mainly by not induction of apoptosis but cell cycle arrest, particularly in G1-phase
GlaxoSmithKline (GSK) is developing omipalisib (GSK-2126458), a phosphoinositide 3-kinase/mammalian target of rapamycin (PI3K/mTOR) inhibitor as well as mTOR complex 1 and 2 inhibitor, for the potential oral treatment of cancer and idiopathic pulmonary fibrosis
GSK-2126458 is a phosphatidylinositol 3-Kinase (PI3K) inhibitor in early clinical development for the oral treatment of solid tumors and for the oral treatment of lymphoma. Early clinical studies are ongoing for the treatment of idiopathic pulmonary fibrosis. The compound is being developed b GlaxoSmithKline.
In August 2009, a phase I trial began for solid tumors and lymphoma . In April 2012, phase Ib co-clinical trials in advanced prostate cancer (PC) were underway . In March 2013, a phase I trial was initiated in the UK in patients with idiopathic pulmonary fibrosis
In April 2014, a phase I, open-label, multicenter, dose-escalation study (study number P3K113794) and safety data were presented at the 105th AACR meeting in San Diego, CA. Advanced solid tumor patients (n = 69) received oral continuous GSK-2126458 or intermittent GSK-2126458 bid + trametinib. For GSK-2126458 and trametinib, the MTD in QD cohort was 2 and 1 mg, respectively, and also 1 and 1.5 mg, respectively
PAPER ![]()
![]()
![]()
![]()
![]()
![]()
![]()
Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rampamycin
ACS Med Chem Lett 2010, 1(1): 39

Phosphoinositide 3-kinase α (PI3Kα) is a critical regulator of cell growth and transformation, and its signaling pathway is the most commonly mutated pathway in human cancers. The mammalian target of rapamycin (mTOR), a class IV PI3K protein kinase, is also a central regulator of cell growth, and mTOR inhibitors are believed to augment the antiproliferative efficacy of PI3K/AKT pathway inhibition. 2,4-Difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3-pyridinyl}benzenesulfonamide (GSK2126458, 1) has been identified as a highly potent, orally bioavailable inhibitor of PI3Kα and mTOR with in vivo activity in both pharmacodynamic and tumor growth efficacy models. Compound 1 is currently being evaluated in human clinical trials for the treatment of cancer.

synthesis![]()
![]()
![]()
![]()
![]()


………………..
PATENT
WO 2008144463
http://www.google.co.in/patents/WO2008144463A1?cl=en
Example 345
2,4-difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3- pyridinyl } benzenesulf onamide
a) 6-bromo-4-(4-pyridazinyl)quinoline
Dissolved 6-bromo-4-iodoquinoline (17.43 g, 52.2 mmol), 4- (tributylstannanyl)pyridazine (19.27 g, 52.2 mmol), and PdC12(dppf)-CH2C12 (2.132 g, 2.61 mmol) in 1,4-dioxane (200 mL) and heated to 105 °C. After 3 h, added more palladium catalyst and heated for 6 h. Concentrated and dissolved in methylene chloride/methanol. Purified by column chromatography (combiflash) with 2% MeOH/EtOAc to 5% MeOH/EtOAc to give the crude title compound. Trituration with EtOAc furnished 6-bromo-4-(4-pyridazinyl)quinoline (5.8 g, 20.27 mmol, 38.8 % yield). MS(ES)+ m/e 285.9, 287.9 [M+H]+.
b) 2,4-difluoro-N-{2-(methyloxy)-5-[4-(4-pyridazinyl)-6-quinolinyl]-3- pyridinyl } benzenesulf onamide A slurry of 6-bromo-4-(4-pyridazinyl)quinoline (4.8 g, 16.78 mmol), bis(pinacolato)diboron (4.69 g, 18.45 mmol) , PdC12(dppf)-CH2C12 (530 mg, 0.649 mmol) and potassium acetate (3.29 g, 33.6 mmol) in anhydrous 1,4-dioxane (120 ml) was heated at 100 °C for 3 h. The complete disappearance of the starting bromide was observed by LCMS. The reaction was then treated with N-[5-bromo-2- (methyloxy)-3-pyridinyl]-2,4-difluorobenzenesulfonamide (6.68 g, 17.61 mmol) and another portion of PdC12(dppf)-CH2C12 (550 mg, 0.673 mmol), then heated at 110 °C for 16 h. The reaction was allowed to cool to room temperature, filtered, and concentrated. Purification of the residue by chromatography (Analogix; 5% MeOH / 5% CH2C12 / 90% EtOAC) gave 6.5 g (76%) desired product. MS(ES)+ m/e 505.9 [M+H]+.
INTERMEDIATES:
Intermediate 1 Similar but not same
Scheme A:

Conditions: a) Tributyl(vinyl)tin, Pd(PPh3)4, dioxane, reflux; b) OsO4, NaIO4, 2,6- lutidine, r-BuOH, dioxane, H2O, rt; c) (4-pyridyl)boronic acid, Pd(PPh3)4, 2 M K2CO35 DMF, 100 DC.
4-(4-pyridinyl)-6-quinolinecarbaldehydeSimilar but not same

a) 4-chloro-6-ethenylquinoline
A mixture of 6-bromo-4-chloroquinoline (6.52 g, 26.88 mmol; see J. Med. Chem., H 268 (1978) ), tributyl(vinyl)tin (8.95 g, 28.22 mmol), and tetrakistriphenylphospbine palladium (0) (0.62 g, 0.54 mmol) in 1,4-dioxane (150 mL) was refluxed for 2.0 h, cooled to room temperature, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (0-4% MeOH:CH2Cl2) to give the title compound (5.1 g) as a pale yellow solid. MS (ES)+ m/e 190 [M+H]+. This material was used directly in the next step.
b) 4-chloro-6-quinolinecarbaldehyde
A mixture of 4-chloro-6-ethenylquinoline (5.1 g, 26.88 mmol), 2,6-lutidine
(5.76 g, 53.75 mmol), sodium (meta) periodate (22.99 g, 107.51 mmol), and osmium tetroxide (5.48 g of a 2.5% solution in tert-butanol, 0.538 mmol) in l,4-dioxane:H2θ (350 mL of 3: 1 mixture) was stirred for 3.5 h at room temperature and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (CH2Cb) to give the title compound (4.26 g, 83% for 2 steps) as a pale yellow solid. MS (ES)+ m/e 192 [M+H]+.
c) 4-(4-pyridmyl)-6-qumolinecarbaldehyde
A mixture of 4-chloro-6-quinolinecarbaldehyde (3.24 g, 16.92 mmol), A- pyridylboronic acid (3.12 g, 25.38 mmol), tetrakistriphenylphosphine palladium (0) (0.978 g, 0.846 mmol), and 2M aqueous K2CO3 (7.02 g, 50.76 mmol, 25.4 mis of 2M solution) in DMF (100 mL) was heated at 100 °C for 3.0 h and cooled to room temperature. The mixture was filtered through Celite and the Celite was washed with EtOAc. The filtrate was transferred to a separatory funnel, washed with water and saturated NaCl, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (5% MeOH:CH2Cl2) to give the title compound (2.03 g, 51%) as a tan solid. MS (ES)+ m/e 235 [M+H]+.
Intermediate 2
Preparation of 2-amino-5 -bromo-N,N-dimethyl-3 -pyridinesulfonamideSimilar but not same

a) 2-ammo-5-bromo-3-pyridinesulfonyl chloride
To a cooled (0 °C) solution of chlorosulfonic acid (58 mL) under vigorous stirring was added 5-bromo-2-pyridinamine (86.7 mmol) portionwise. The reaction mixture was then heated at reflux for 3 hrs. Upon cooling to room temperature, the reaction mixture was poured over ice (-100 g) with vigorous stirring. The resulting yellow precipitate was collected by suction filtration, washing with cold water and petroleum ether to provide the title compound as an orange-yellow solid (18.1 g, 77% yield). MS(ES)+ m/e 272.8 [M+H]+.
* Other sulfonyl chlorides can be prepared using this procedure by varying the choice of substituted aryl or heteroaryl.
b) 2-amino-5-bromo-N,N-dimethyl-3-pyridinesulfonamide
To a cold (0 DC) suspension of 2-amino-5-bromo-3-pyridinesulfonyl chloride (92.1 mmol) in dry 1,4-dioxane (92 mL) was added pyridine (101.3 mmol) followed by a 2M solution of dimethylamine in THF (101.3 mmol). The reaction was allowed to warm to rt for 2 h, heated to 50 DC for 1 h, then cooled to rt. After standing for 2 h, the precipitate was collected by filtration and rinsed with a minimal amount of cold water. Drying the precipitate to constant weight under high vacuum provided 14.1 g (55%) of the title compound as a white solid. MS(ES)+ m/e 279.8, 282.0 [M+H]+.
Intermediate 3
Preparation of 2-amino-N,N-dimethyl-5-(4,4,5,5-tetramethyl-l,3.2-dioxaborolan-2- yl)-3 -pyridinesulfonamideSimilar but not same

c) To a solution of 2-amino-5-bromo-N,N-dimethyl-3 -pyridinesulfonamide (7.14 mmol) in 1,4-dioxane (35 mL) was added 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi-l,3,2- dioxaborolane (7.86 mmol), potassium acetate (28.56 mmol) and [1,1 ‘- bis(diphenylphosphmo)-ferrocene] dichloropalladium(II) dichloromethane complex (1 :1) (0.571 mmol). The reaction mixture was stirred at 100 °C for 18 h. The reaction was concentrated in vacuo, re-dissolved in ethyl acetate (50 mL) and purified on silica using 60% ethyl acetate/hexanes to yield the title compound as a tan solid (86 %). IH ΝMR (400 MHz, DMSOd6) δ ppm 8.41 (d, 1 H, J =1.52), 7.92 (d, 1 H, J = 1.77), 2.68 (s, 6 H), 1.28 (s, 12 H).
* Other boronate or boronic acids can be prepared using this procedure by varying the choice of aryl or heteroaryl bromide. Scheme 17:

Conditions: a) NaO(Rl), (Rl)OH, O 0C to room temperature; b) SnCl2-2H2O, ethyl acetate, reflux; c) (R2)SO2C1, pyridine, O 0C to room temperature.
Intermediate 4
Preparation of N-r5-bromo-2-(methyloxy)-3-pyridinyll-2,4- difluorobenzenesulfonamide
N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4- difluorobenzenesulfonamide a) 5-bromo-2-(methyloxy)-3-nitropyridine

To a cooled (0 °C) solution of 5-bromo-2-chloro-3-nitropyridine (50 g, 211 mmol) in methanol (200 mL) was added dropwise over 10 minutes 20% sodium methoxide (50 mL, 211 mmol) solution. The reaction, which quickly became heterogeneous, was allowed to warm to ambient temperature and stirred for 16 h. The reaction was filtered and the precipitate diluted with water (200 mL) and stirred for 1 h. The solids were filtered, washed with water (3 x 100 mL) and dried in a vac oven (40 °C) to give 5-bromo-2-(methyloxy)-3-nitropyridine (36 g, 154 mmol, 73.4 % yield) as a pale yellow powder. The original filtrate was concentrated in vacuo and diluted with water (150 mL). Saturated ammonium chloride (25 mL) was added and the mixture stirred for 1 h. The solids were filtered, washed with water, and dried in a vac oven (40 °C) to give a second crop of 5-bromo-2-(methyloxy)-3- nitropyridine (9 g, 38.6 mmol, 18.34 % yield). Total yield = 90%. MS(ES)+ m/e 232.8, 234.7 [M+H]+.
b) 5-bromo-2-(methyloxy)-3-pyridinamine

To a solution of 5-bromo-2-(methyloxy)-3-nitropyridine (45 g, 193 mmol) in ethyl acetate (1 L) was added tin(II) chloride dihydrate (174 g, 772 mmol). The reaction mixture was heated at reflux for 4 h. LC/MS indicated some starting material remained, so added 20 mol% tin (II) chloride dihydrate and continued to heat at reflux. After 2 h, the reaction was allowed to cool to ambient temperature and concentrated in vacuo. The residue was treated with 2 N sodium hydroxide and the mixture stirred for 1 h. The mixture was then with methylene chloride (1 L), filtered through Celite, and washed with methylene chloride (500 mL). The layers were separated and the organics dried over magnesium sulfate and concentrated to give 5-bromo-2-(methyloxy)-3-pyridinamine (23 g, 113 mmol, 58.7 % yield). The product was used crude in subsequent reactions. MS(ES)+ m/e 201.9, 203.9 [M+H]+.
c) N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4-difluorobenzenesulfonamide
To a cooled (0 °C) solution of 5-bromo-2-(methyloxy)-3-pyridinamine (20.3 g, 100 mmol) in pyridine (200 mL) was added slowly 2,4-difluorobenzenesulfonyl chloride (21.3 g, 100 mmol) over 15 min (reaction became heterogeneous). The ice bath was removed and the reaction was stirred at ambient temperature for 16 h, at which time the reaction was diluted with water (500 mL) and the solids filtered off and washed with copious amounts of water. The precipitate was dried in a vacuum oven at 50 °C to give N-[5-bromo-2-(methyloxy)-3-pyridinyl]-2,4- difluorobenzenesulfonamide (12 g, 31.6 mmol, 31.7 % yield) MS(ES)+ m/e 379.0, 380.9 [M+H]+.
References
|
1: Zhang Y, Xue D, Wang X, Lu M, Gao B, Qiao X. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. Mol Med Rep. 2014 Jan;9(1):83-90. doi: 10.3892/mmr.2013.1781. Epub 2013 Nov 7. PubMed PMID: 24213221.
2: Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, Fukunaba-Kalabis M, Wubbenhorst B, Chen TY, Liu Q, Sproesser K, DeMarini DJ, Gilmer TM, Martin AM, Marmorstein R, Schultz DC, Speicher DW, Karakousis GC, Xu W, Amaravadi RK, Xu X, Schuchter LM, Herlyn M, Nathanson KL. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013 Sep 26;4(6):1090-9. doi: 10.1016/j.celrep.2013.08.023. Epub 2013 Sep 19. PubMed PMID: 24055054; PubMed Central PMCID: PMC3956616.
3: Kim HG, Tan L, Weisberg EL, Liu F, Canning P, Choi HG, Ezell SA, Wu H, Zhao Z, Wang J, Mandinova A, Griffin JD, Bullock AN, Liu Q, Lee SW, Gray NS. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem Biol. 2013 Oct 18;8(10):2145-50. doi: 10.1021/cb400430t. Epub 2013 Aug 13. PubMed PMID: 23899692; PubMed Central PMCID: PMC3800496.
4: Khalili JS, Yu X, Wang J, Hayes BC, Davies MA, Lizee G, Esmaeli B, Woodman SE. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012 Aug 15;18(16):4345-55. doi: 10.1158/1078-0432.CCR-11-3227. Epub 2012 Jun 25. PubMed PMID: 22733540; PubMed Central PMCID: PMC3935730.
5: Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012 Apr;11(4):909-20. doi: 10.1158/1535-7163.MCT-11-0989. Epub 2012 Mar 2. PubMed PMID: 22389471.
6: Wang M, Gao M, Miller KD, Sledge GW, Zheng QH. [11C]GSK2126458 and [18F]GSK2126458, the first radiosynthesis of new potential PET agents for imaging of PI3K and mTOR in cancers. Bioorg Med Chem Lett. 2012 Feb 15;22(4):1569-74. doi: 10.1016/j.bmcl.2011.12.136. Epub 2012 Jan 10. PubMed PMID: 22297110.
7: Schenone S, Brullo C, Musumeci F, Radi M, Botta M. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011;18(20):2995-3014. Review. PubMed PMID: 21651476.
8: Leung E, Kim JE, Rewcastle GW, Finlay GJ, Baguley BC. Comparison of the effects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells. Cancer Biol Ther. 2011 Jun 1;11(11):938-46. Epub 2011 Jun 1. PubMed PMID: 21464613; PubMed Central PMCID: PMC3127046.
Sun Pharma to acquire GSK’s Australian opiates business
![]()
Sun Pharma to acquire GSK’s Australian opiates business
India-based Sun Pharmaceutical Industries has agreed to acquire GlaxoSmithKline’s (GSK) opiates business in Australia.
The agreement has been signed by wholly-owned subsidiaries of both firms, with the financial terms not disclosed.
Sun Pharma API business executive vice-president Iftach Seri said: “The global opiates market holds good potential and the addition of GSK’s Opiates business will strengthen our positioning further.”
Sun Pharmaceutical Industries Ltd(SUN.NS), India’s largest drugmaker by sales, said on Tuesday it has agreed to buy GlaxoSmithKline’s(GSK.L) opiates business in Australia to strengthen its pain management portfolio.
The business consists of analgesics made from raw materials found in opium poppy plants, and includes two manufacturing sites in the states of Tasmania and Victoria.
Financial details of the deal were not disclosed. A Sun Pharma spokesman declined to comment. Glaxo did not immediately respond to a request seeking comment.
Glaxo supplies a quarter of the world’s medicinal opiate needs from poppies grown by farmers in Tasmania, according to the company website. The company’s Australian opiates business brought in revenue of A$89 million ($69.63 million) in 2013.
Australia’s poppy industry is the world’s largest legal supplier of pharmaceutical grade opiates for painkillers, and Glaxo is one of three firms that control the crop and production in Tasmania.
The other two are Johnson & Johnson’s (JNJ.N) unit Tasmanian Alkaloids, and privately-held TPI Enterprises.
Glaxo’s decision to part with the opiates business comes as Tasmania’s poppy industry is facing a tough crop and the United Nations is expected to cut the state’s poppy crop area this week.
Glaxo said the deal would allow it to “focus on delivering its innovative product portfolio” in Australia.
“The opiates business has been an important part of our Australian business for many years, but as our portfolio transitions, we believe now is the right time to hand this business over to someone else,” Steve Morris, general manager of GSK Opiates, said in a statement.
The business employs 185 staff, including 155 in Victoria state and 30 in Tasmania state. Sun Pharma said it would hire all employees from both sites.
“The acquisition is a part of our strategy towards building our portfolio of opiates and accessing strong capabilities in this segment,” said Iftach Seri, executive vice president of the active pharmaceuticals ingredients business at Sun Pharma.
Both companies said they expect to close the deal by August.
Sun Pharma shares closed 1.93 percent higher on Tuesday, while the broader Nifty rose 0.44 percent.
($1 = 1.2781 Australian dollars)
Lupin and Celon Pharma partner for generic version of GSK’s Advair Diskus

Vinita Gupta, 43, Group President and CEO, Lupin Pharmaceuticals and Director, Lupin
India-based drugmaker Lupin has signed an agreement with Polish biopharmaceutical firm Celon Pharma to develop a fluticasone / salmeterol dry powder inhaler (DPI).
Under the deal, Lupin will take the responsibility for commercialisation of the product, which is a generic version of GlaxoSmithKline’s (GSK) Advair Diskus.
Lupin CEO Vinita Gupta said: “We are very pleased to partner with Celon given their experience in the development and manufacturing of fluticasone/salmeterol DPI in Europe…………..http://www.pharmaceutical-technology.com/news/newslupin-celon-pharma-partner-generic-version-gsks-advair-diskus-4514718?WT.mc_id=DN_News
![]()
Vinita Gupta, 43, Group President and CEO, Lupin Pharmaceuticals and Director, Lupin, is based in the United States, but has been in India a lot in the past one year.

With an expanding role in Lupin’s universe, Vinita has been spending more time outside the US, at times taking her six-year-old son, Krish with her. “He is getting exposure at a much younger age,” she says. Gupta herself was exposed to business at the age of 11 by her father Desh Bandhu Gupta, Lupin’s founder and Chairman.
“We almost had a family board at home, discussing work,” she says. Currently work goes well indeed, with Gupta taking new initiatives in India and also making the business more global. “I am focusing on drivers for growth in our business for the next five years,” she says.
Gupta is married to US-based businessman Brij Sharma.

DB Gupta (centre) Chairman, Vinita Gupta (right) CEO and Nilesh Gupta

Epelsiban being developed by GlaxoSmithKline for the treatment of premature ejaculation in men.

Epelsiban
557296
GSK-557296
GSK-557296-B
(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
(3R,6R)-6-[(2S)-butan-2-yl]-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-morpholin-4-yl-2-oxoethyl]piperazine-2,5-dione
(3R, 6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[(1R)- 1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1-methylpropyl]-2,5- piperazinedione
Glaxo Group Limited INNOVATOR
Epelsiban (GSK-557,296-B)[1][2] is an oral drug which acts as a selective, sub-nanomolar (Ki=0.13 nM) oxytocin receptor antagonist with >31000-fold selectivity over the related vasopressin receptors and is being developed by GlaxoSmithKline for the treatment of premature ejaculation in men.[3][4]


benzenesulfonic acid;(3R,6R)-6-[(2S)-butan-2-yl]-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-morpholin-4-yl-2-oxoethyl]piperazine-2,5-dione,CAS 1159097-48-9
UNII-H629P9T4UN, GSK557296B, Epelsiban besylate (USAN), Epelsiban besylate [USAN], 1159097-48-9, H629P9T4UN
GSK-557296 is being developed in early clinical studies at GlaxoSmithKline for enhancement of embryo and or blastocyst implantation in women undergoing IVF treatment. The product has been in phase II clinical development for the treatment of premature ejaculation.
Preterm labor is a major clinical problem leading to death and disability in newborns and accounts for 10% of all births and causes 70% of all infant mortality and morbidity.
Oxytocin (OT) is a potent stimulant of uterine contractions and is responsible for the initiation of labor via the interaction with the OT receptors in the mammalian uterus. OT antagonists have been shown to inhibit uterine contractions and delay preterm delivery. So there is increasing interest in OT antagonists because of their potential application in the prevention of preterm labor. Although several tocolytics have already been approved in clinical practice, they have harmful maternal or fetal side effects.
The first clinically tested OT antagonist atosiban has a much more tolerable side effect profile and has recently been approved for use in Europe. However, atosiban is a peptide and a mixed OT/vasopressin V1a receptor antagonist that has to be given by iv infusion and is not suitable for long-term maintenance treatment, as it is not orally bioavailable.
Hence there has been considerable interest in overcoming the shortcomings of the peptide OT antagonists by identifying orally active nonpeptide OT antagonists with a higher degree of selectivity toward the vasopressin receptors (V1a, V1b, V2) with good oral bioavailability. Although several templates have been investigated as potential selective OT antagonists, few have achieved the required selectivity for the OT receptor vs the vasopressin receptors combined with the bioavailability and physical chemical properties required for an efficacious oral drug.
Therefore our objective was to design a potent, orally active OT antagonist with high levels of selectivity over the vasopressin receptor with good oral bioavailability in humans that would delay labor safely by greater than seven days and with improved infant outcome, as shown by a reduced combined morbidity score.
| Patent | Submitted | Granted |
|---|---|---|
| Compounds [US7919492] | 2010-12-02 | 2011-04-05 |
| Piperazinediones as Oxytocin Receptor Antagonists [US7550462] | 2007-11-01 | 2009-06-23 |
| Compounds [US8202864] | 2011-06-23 | 2012-06-19 |
| Novel compounds [US2009247541] | 2009-10-01 |
………………………………………
PATENT
https://www.google.com/patents/US7919492
Example 3
Method A

(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
as a white lyophilisate (88 mg, 23%) after freeze-drying from 1,4-dioxane
HPLC Rt=2.70 minutes (gradient 2); m/z [M+H]+=519
1H NMR (CDCl3) δ 7.49 (d, 1H), 7.27-7.15 (m, 4H), 7.10 (d, 1H), 6.68 (s, 1H), 6.40 (d, 1H), 4.10 (dd, 1H), 4.01 (d, 1H), 3.74-3.52 (m, 5H), 3.28-3.07 (m, 5H), 2.97-2.84 (m, 2H), 2.79-2.71 (m, 1H), 2.62 (s, 3H), 2.59 (s, 3H), 1.65-1.53 (m, 1H), 0.98-0.80 (m, 2H), 0.70 (t, 3H), 0.45 (d, 3H).
Example 3
Method B

(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione
A suspension of {(3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-6-[(1S)-1-methylpropyl]-2,5-dioxo-1-piperazinyl}(2,6-dimethyl-3-pyridinyl)acetic acid hydrochloride (5.0 g, 10.3 mmol) (intermediate 5) in dry dichloromethane (50 ml) was treated with 1,1-carbonyldiimidazole (2.6 g, 16 mmol) and the reaction mixture was stirred under nitrogen for 18 hours. Morpholine (4.8 ml, 55 mmol) was added and the resultant solution was left to stand under nitrogen for 18 hours. The solvent was removed in vacuo and the residue was separated between ethyl acetate and water. The organic phase was washed with brine and dried over anhydrous magnesium sulphate. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. This was applied to a basic alumina cartridge (240 g) and eluted using a gradient of 0-7.5% methanol in diethyl ether (9CV), 7.5-10% methanol in diethyl ether (1CV) and 10% methanol in diethyl ether (1CV). The required fractions were combined and evaporated in vacuo to give (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione as a white solid (2.4 g, 45%).
HPLC Rt=2.72 minutes (gradient 2); m/z [M+H]+=519
………………………………………
WO 2011051814
http://www.google.com/patents/WO2011051814A1?cl=en
This invention relates to novel crystalline forms of (3R, 6R)-3-(2,3-dihydro-1 H- inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1 S)-1 – methylpropyl]-2,5-piperazinedione benzenesulfonate salt, processes for their preparation, pharmaceutical compositions containing them and to their use in medicine. The benzenesulfonate salt of Compound A is represented by the following structure:

In one aspect, the present invention provides a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form provides an X-ray powder diffraction pattern substantially in accordance with Figure 1 .
In another aspect, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:

In an additional aspect, the invention includes a crystalline form of {3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2.
In certain aspects, the invention encompasses a crystalline form of (3R, 6R)-3- (2,3-dihydro-1 H-inden-2-yl)-1 -[(1 R)-1 -(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2- oxoethyl]-6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said compound is characterized by an X-ray powder diffraction pattern substantially in accordance with Figure 2 In one aspect, the invention also provides a crystalline form of {3R, 6R)-3-(2,3- dihydro-1 H-inden-2-yl)-1-[(1 R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]- 6-[(1 S)-1 -methylpropyl]-2,5-piperazinedione benzenesulfonate hydrate, wherein said crystalline form is characterized by an X-ray powder diffraction pattern comprising the peaks:

Experimental
Process Scheme

Stage 4
Acetone / Water Recrystallisation
Compound A-form I Ste8e 5 Besylate salt
MW 676.83 Acetone / Water
Recrystallisation MW 676.83 Process description for isolation of Compound A-Form 1
Stage 0
methyl d-alloisoleucinate hydrochloride (Compound 2) was charged to ethyl acetate. A solution of potassium carbonate in water was then added. The mixture was then stirred vigorously at room temperature for 1 hour. The two layers were separated and the aqueous layer further extracted with ethyl acetate. The organic layers were combined and washed with brine. The organic layers were then concentrated in vacuo and filtered to yield methyl D-alloisoleucinate (Compound 3) as a pale yellow oil.
Stage 1
2,6-dimethyl-3-pyridinecarbaldehyde (Compound 4) in methanol at ambient temperature was treated with D-alloisoleucinate (Compound 3) in methanol followed by 2,2,2- trifluoroethanol and the reaction mixture was warmed to 40°C. When formation of the intermediate imine (methyl A/-[(2,6-dimethyl-3-pyridinyl)methylidene]-D-alloisoleucine) was complete Compound 5 was added followed by 1-isocyano-2- [(phenylmethyl)oxy]benzene (Compound 6) and the reaction mixture was stirred at 40°C until formation of Compound 7 was deemed complete.
Stage 2
Palladium on carbon catalyst was treated with a solution of Compound 7 in methanol and 2,2,2-trifluoroethanol and diluted with acetic acid. The vessel was purged with nitrogen and the reaction mixture warmed to 50°C and hydrogenated at 4.0-4.5 barg. When the reaction was deemed complete it was cooled to ambient temperature and the catalyst removed by filtration and washed through with methanol. The organic solution of 2- {(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1-piperazinyl}- 2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) was concentrated at reduced pressure and then diluted with /‘so-propyl acetate and concentrated at reduced pressure.
The residue was diluted with /‘so-propyl acetate and washed with aqueous ammonia. The aqueous phase was separated and extracted into another portion of /‘so-propyl acetate. The combined organic phases were washed with water, concentrated by distillation at reduced pressure, diluted with /‘so-propyl acetate and concentrated by distillation at reduced pressure, to leave a concentrated solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8). The product was finally dissolved in 1 ,4-dioxane for the next stage and stored into drums.
Stage 3 Solution of 2-{(3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-6-[(1 S)-1 -methylpropyl]-2,5-dioxo-1 – piperazinyl}-2-(2,6-dimethyl-3-pyridinyl)-/\/-(2-hydroxyphenyl)acetamide (Compound 8) in 1 ,4-dioxane was treated with 1 ,1 ‘-carbonyl diimidazole at ambient temperature to form a solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1 -[1-(2,6-dimethyl-3-pyridinyl)- 2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1 -methylpropyl]-2,5- piperazinedione (Compound 9).
In a separate vessel morpholine in 1 ,4-dioxane was heated to 80-85°C. The solution containing (3R,6R)-3-(2,3-dihydro-1 H-inden-2-yl)-1-[1 – (2,6-dimethyl-3-pyridinyl)-2-oxo-2-(2-oxo-1 ,3-benzoxazol-3(2H)-yl)ethyl]-6-[(1 S)-1- methylpropyl]-2,5-piperazinedione (Compound 9) was slowly added to the morpholine in 1 ,4-dioxane. The reaction mixture was stirred for one hour at 80-85°C and cooled before concentration by distillation at reduced pressure.
The concentrated solution of Compound A was diluted with /‘so-propyl acetate and washed with aqueous sodium hydroxide followed by water. The /so-propyl acetate solution of COMPOUND A was then concentrated by distillation at reduced pressure and cooled to ambient temperature. The concentrated solution of Compound A was then diluted with acetone and treated with benzenesulfonic acid and seed crystals were added and the reaction mixture stirred until crystallisation occurred. The slurry of Compound A besylate was heated to 50°C, a temperature cycle was performed, and finally the slurry was cooled to -10°C and isolated by filtration. The filter cake was washed with cold acetone (-10°C) to give Compound A besylate (intermediate grade) as a wet cake.
Yield: 44% from Compound 5
39% from Compound 5
Stage 4
Compound A besylate (intermediate grade wet cake, Compound A besylate ) was suspended in acetone (17.4 vol including acetone content of wet cake) and heated to 55- 60°C. Water (0.66 vol) was added until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (3.2 vol). The temperature of the reaction mixture was adjusted to 45-50°C before the addition of seed crystals (0.00025wt). When crystallisation was complete the reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins.
The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was heated to 45-50°C and stirred at 45-50°C for 30mins. The reaction mixture was cooled to -3-2°C over 4.5 h and stirred for at least 1 h before the product was isolated by filtration. The wet cake was washed with acetone at 0°C (3 x 3.1 vol) and blown dry before being unloaded. COMPOUND A besylate was dried at 50°C under vacuum for 3 days. Compound A besylate was then milled. Yield: 66% Stage 5
Compound A besylate (OBU-D-02) was suspended in acetone (8 vol) and water (1 .1 vol) and heated to 48-52°C until dissolution was observed. The reaction mixture was then filtered into another vessel and the lines washed through with acetone (2 vol). The reaction mixture was cooled to 20-25°C before the addition of Form 1 seed crystals (0.0025wt). When crystallisation was complete the reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins. The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to 0-5°C over 1 h and stirred at 0-5°C for 30mins.
The reaction mixture was heated to 20-25°C and stirred at 20-25°C for 30mins. The reaction mixture was cooled to -12— 8°C over 3.5 h and stirred for 15 h before the product was isolated by filtration. The wet cake was washed with acetone at -10°C (2 x 3 vol) and blown dry before being unloaded. Compound A besylate was dried at ambient temperature under vacuum for 6 days with a wet nitrogen bleed to afford Form 1 . Compound A besylate was then milled. Yield: 67%
Recrystallisation of Compound A besylate anhydrate (Form 2)

Besylate salt ………………………………………………………………Besylate salt
C30H38 4O4■ C6H603S C30H38 4O4■
MW 676.83 MW 676.83
COMPOUND A besylate is charged to the vessel and treated with methyl ethyl ketone (MEK) (8vol) and water (0.35vol) and the solution heated until dissolution is observed (ca. 55-60°C). The solution is then filtered and recharged to the vessel. Pressure is then reduced to 650mbar and the reaction mixture heated further to distil out solvent. MEK is added at the same rate as solvent is removed by distillation keeping the reaction mixture volume constant. After 4 volumes of MEK have been added the reaction mixture is treated with Form 2 seed crystals (2%wt) and the distillation continued in the same manner until another 7 volumes of MEK has been added. The vacuum is then released to an atmospheric pressure of nitrogen and the temperature of the reaction mixture adjusted to 65°C. The reaction mixture is then filtered and washed with pre heated MEK (2vol at 65°C). The purified COMPOUND A besylate anhydrate is then sucked dry and dried further in a vacuum oven at 65°C at l OOmbar with a nitrogen bleed. Yield 89%
NMR data is the same for Forms 1 and 2.
1 H NMR (500MHz, DMSO-d6) 5ppm 0.71-0.80(m, 6H) 0.87-0.98(m, 1 H) 1 .31 (br. S, 1 H) 1.69(br. S, 1 H) 2.68(s, 3H) 2.69(s, 3H) 2.72-2.79(m, 1 H) 2.80-2.87(m, 1 H) 2.88-3.01 (m, 3H) 3.18-3.25(m, 1 H) 3.27-3.33(m, 1 H) 3.38-3.46(m, 1 H) 3.47-3.52(m, 1 H)3.53-3.57(m, 1 H) 3.60-3.71 (m, 3H) 3.83(dd, J=9.46,3.15 Hz, 1 H) 3.89 (br. S, 1 H)6.10(br. S, 1 H) 7.1 1 – 7.14(m, 2H) 7.19-7.23(m, 2H) 7.30-7.35(m, 3H)7.59-7.63(m, 2H) 7.67(d, J=7.25Hz, 1 H) 8.12(br. S, 1 H) 8.50(d, J=3.78Hz, 1 H)
………………………………………..
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm201287w

A six-stage stereoselective synthesis of indanyl-7-(3′-pyridyl)-(3R,6R,7R)-2,5-diketopiperazines oxytocin antagonists from indene is described. SAR studies involving mono- and disubstitution in the 3′-pyridyl ring and variation of the 3-isobutyl group gave potent compounds (pKi > 9.0) with good aqueous solubility. Evaluation of the pharmacokinetic profile in the rat, dog, and cynomolgus monkey of those derivatives with low cynomolgus monkey and human intrinsic clearance gave 2′,6′-dimethyl-3′-pyridyl R–sec-butyl morpholine amide Epelsiban (69), a highly potent oxytocin antagonist (pKi = 9.9) with >31000-fold selectivity over all three human vasopressin receptors hV1aR, hV2R, and hV1bR, with no significant P450 inhibition. Epelsiban has low levels of intrinsic clearance against the microsomes of four species, good bioavailability (55%) and comparable potency to atosiban in the rat, but is 100-fold more potent than the latter in vitro and was negative in the genotoxicity screens with a satisfactory oral safety profile in female rats.
(3R,6R)-3-(2,3-Dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione (69 EPELSIBAN)
References
- Borthwick AD, Liddle J, Davies DE, Exall AM, Hamlett C, Hickey DM, Mason AM, Smith IE, Nerozzi F, Peace S, Pollard D, Sollis SL, Allen MJ, Woollard PM, Pullen MA, Westfall TD, Stanislaus DJ (January 2012). “Pyridyl-2,5-diketopiperazines as potent, selective, and orally bioavailable oxytocin antagonists: synthesis, pharmacokinetics, and in vivo potency”. Journal of Medicinal Chemistry 55 (2): 783–96. doi:10.1021/jm201287w. PMID 205501.
- 2 Borthwick, A. D.; Liddle, J. (January 2013). “Retosiban and Epelsiban: Potent and Selective Orally available Oxytocin Antagonists”. In Domling, A. Methods and Principles in Medicinal Chemistry: Protein-Protein Interactions in Drug Discovery. Weinheim: Wiley-VCH. pp. 225–256. ISBN 978-3-527-33107-9.
- 3 World Health Organization (2011). “International Nonproprietary Names for Pharmaceutical Substances (INN): Proposed INN: List 105”. WHO Drug Information 25 (2): 179.
- 4 USAN Council (2011). “Statement on a Nonproprietary Name Adopted by the USAN Council” (PDF). Retrieved 2011-10-28.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3R,6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethylpyridin-3-yl)-2-(morpholin-4-yl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]piperazine-2,5-dione | |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | 872599-83-2 1159097-48-9 (besylate) |
| ATC code | None |
| PubChem | CID 11634973 |
| ChemSpider | 9809717 |
| KEGG | D10117 |
| Chemical data | |
| Formula | C30H38N4O4 |
| Molecular mass | 518.6 g/mol |
| Cited Patent | Filing date | Publication date | Applicant | Title | |
|---|---|---|---|---|---|
| WO2003053443A1 | Dec 20, 2002 | Jul 3, 2003 | Glaxo Group Ltd | Substituted diketopiperazines as oxytocin antagonists | |
| WO2006000399A1 | Jun 21, 2005 | Jan 5, 2006 | Glaxo Group Ltd | Novel compounds | |
| EP2005006760W | Title not available | ||||
| US6914160 | Jul 31, 2003 | Jul 5, 2005 | Pfizer Inc | Oxytocin inhibitors | |
| US20070254888 | Jun 21, 2005 | Nov 1, 2007 | Glaxo Group Limited | Piperazinediones as Oxytocin Receptor Antagonists |
| US8202864 * | Feb 25, 2011 | Jun 19, 2012 | Glaxo Group Limited | Compounds |
| US8716286 | Oct 28, 2010 | May 6, 2014 | Glaxo Group Limited | Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione |
| US8742099 | May 20, 2013 | Jun 3, 2014 | Glaxo Group Limited | Compounds |
| US8815856 | Mar 18, 2014 | Aug 26, 2014 | Glaxo Group Limited | Crystalline forms of (3R, 6R)-3-(2,3-dihydro-1H-inden-2-yl)-1-[(1R)-1-(2,6-dimethyl-3-pyridinyl)-2-(4-morpholinyl)-2-oxoethyl]-6-[(1S)-1-methylpropyl]-2,5-piperazinedione |
| US20120202811 * | Apr 19, 2012 | Aug 9, 2012 | Glaxo Group Limited | Novel compounds |
Tafenoquine…..GSK Launches Phase 3 Malaria Drug Trials

Tafenoquine
N-[2,6-dimethoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinolin-8-yl]pentane-1,4-diamine
Medicines for Malaria Venture
Walter Reed Army Institute (Originator)
April 28, 2014
GlaxoSmithKline (GSK) and Medicines for Malaria Venture (MMV) announced the start of a Phase 3 global program to evaluate the efficacy and safety of tafenoquine, an investigational medicine which is being developed for the treatment and relapse prevention (radical cure) of Plasmodium vivax (P. vivax) malaria.
P. vivax malaria, a form of the disease caused by one of several species of Plasmodium parasites known to infect humans, occurs primarily in South and South East Asia, Latin America and the horn of Africa. Severe anemia, malnutrition and respiratory distress are among the most serious consequences described to be caused by the infection.
The Phase 3 program includes two randomized, double-blind treatment studies to investigate tafenoquine in adult patients with P. vivax malaria. The DETECTIVE study (TAF112582) aims to evaluate the efficacy, safety and tolerability of tafenoquine as a radical cure for P. vivax malaria, co-administered with chloroquine, a blood stage anti-malarial treatment. The GATHER study (TAF116564) aims to assess the incidence of hemolysis and safety and efficacy of tafenoquine compared to primaquine, the only approved treatment currently available for the radical cure of P. vivax malaria.
Tafenoquine is not yet approved or licensed for use anywhere in the world.
“P. vivax malaria can affect people of all ages and is particularly insidious because it has the potential to remain dormant within the body in excess of a year, and causes some patients to experience repeated episodes of illness after the first mosquito bite,” said Nicholas Cammack, head, Tres Cantos Medicines Development Center for Diseases of the Developing World. “Our investigation of tafenoquine for the treatment of P. vivax malaria is part of GSK’s efforts to tackle the global burden of malaria. Working with our partners, including MMV, we are determined to stop malaria in all its forms.”
“One of the big challenges we face in tackling malaria is to have new medicines to prevent relapse, caused by dormant forms of P. vivax,” said Dr. Timothy Wells, MMV’s chief scientific officer. “The Phase 3 program is designed to build upon the promising results of the Phase 2b study which showed that treatment with tafenoquine prevented relapses. If successful, tafenoquine has the potential to become a major contributor to malaria elimination. It’s a great privilege to be working with GSK on this project; they have a clear commitment to changing the face of public health in the countries in which we are working.”


Tafenoquine succinate, Etaquine, SB-252263, WR-238605
in phase 2
Medicines for Malaria Venture
Walter Reed Army Institute (Originator)
Tafenoquine is an 8-aminoquinoline drug manufactured by GlaxoSmithKline that is being investigated as a potential treatment for malaria, as well as for malaria prevention.[1][2]
The proposed indication for tafenoquine is for treatment of the hypnozoite stages of Plasmodium vivax (and also Plasmodium ovale) that are responsible for relapse of these malaria species even when the blood stages are successfully cleared. This is only now achieved by administration of daily primaquine for 14 days. The main advantage of tafenoquine is that it has a long half-life (2–3 weeks) and therefore a single treatment may be sufficient to clear hypnozoites. The shorter regimen has been described as an advantage.[3]
Like primaquine, tafenoquine causes haemolysis in people with G-6-P deficiency.[1] Indeed the long half-life of tafenoquine suggests that particular care should be taken to ensure that individuals with severe deficiency do not receive the drug.
The dose of tafenoquine has not been firmly established, but for the treatment of Plasmodium vivax malaria, a dose of 800 mg over three days has been used.[4]
Synonyms

………………..
US 4431807

Nitration of 1,2-dimethoxybenzene (XXIX) with HNO3/AcOH gives 4,5-dimethoxy-1,2-dinitrobenzene (XXX), which is treated with ammonia in hot methanol to yield 4,5-dimethoxy-2-nitroaniline (XXXI). Cyclization of compound (XXXI) with buten-2-one (XXXII) by means of H3PO4 and H3AsO4 affords 5,6-dimethoxy-4-methyl-8-nitroquinoline (XXXIII), which is selectively mono-demethylated by means of HCl in ethanol to provide 5-hydroxy-6-methoxy-4-methyl-8-nitroquinoline (XXXIV). Reaction of quinoline (XXXIV) with POCl3 gives the corresponding 5-chloro derivative (XXXV), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH to yield the diaryl ether (XXXVI). Finally, the nitro group of (XXXVI) is reduced by means of H2 over PtO2 in THF or H2 over Raney nickel.

Nitration of 2-fluoroanisole (XXXVII) with HNO3/Ac2O gives 3-fluoro-4-methoxynitrobenzene (XXXVIII), which is reduced to the corresponding aniline (XXXIX) with SnCl2/HCl. Reaction of compound (XXXIX) with Ac2O yields the acetanilide (XL), which is nitrated with HNO3 to afford 5-fluoro-4-methoxy-2-nitroacetanilide (XLI). Hydrolysis of (XLI) with NaOH provides 5-fluoro-4-methoxy-2-nitroaniline (XLII), which is cyclized with buten-2-one (XXXII) by means of As2O5 and H3PO4 to furnish 5-fluoro-6-methoxy-4-methyl-8-nitroquinoline (XLIII). Condensation of quinoline (XLIII) with 3-(trifluoromethyl)phenol (IV) by means of K2CO3 gives the diaryl ether (XXXIV), which is finally reduced by means of H2 over PtO2 in THF.
………………..
US 4617394

Reaction of 8-amino-6-methoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinoline (XIV) with phthalic anhydride (XV) affords the phthalimido derivative (XVI), which is oxidized with MCPBA to yield the quinoline N-oxide (XVII). Treatment of compound (XVII) with neutral alumina gives the quinolone derivative (XVIII), which by reaction with POCl3 in refluxing CHCl3 provides the 2-chloroquinoline derivative (XIX). Alternatively, reaction of the quinoline N-oxide (XVII) with POCl3 as before also gives the 2-chloroquinoline derivative (XIX) The removal of the phthalimido group of compound (XIX) by means of hydrazine in refluxing ethanol gives the chlorinated aminoquinoline (XX), which is finally treated with MeONa in hot DMF.
……………….
US 6479660; WO 9713753

Chlorination of 6-methoxy-4-methylquinolin-2(1H)-one (I) with SO2Cl2 in hot acetic acid gives the 5-chloro derivative (II), which is nitrated with HNO3 in H2SO4 to yield the 8-nitroquinolinone (III). Condensation of compound (III) with 3-(trifluoromethyl)phenol (IV) by means of KOH in NMP provides the diaryl ether (V), which is treated with refluxing POCl3 to afford the 2-chloroquinoline (VI). Reaction of compound (VI) with MeONa in refluxing methanol results in the 2,6-dimethoxyquinoline derivative (VII), which is reduced with hydrazine over Pd/C to give the 8-aminoquinoline derivative (VIII). Condensation of aminoquinoline (VIII) with N-(4-iodopentyl)phthalimide (IX) by means of diisopropylamine in hot NMP yields the phthalimido precursor (X), which is finally cleaved with hydrazine in refluxing ethanol.

Reaction of 1,4-dibromopentane (XI) with potassium phthalimide (XII) gives N-(4-bromopentyl)phthalimide (XIII), which is then treated with NaI in refluxing acetone.

Reaction of 4-methoxyaniline (XXI) with ethyl acetoacetate (XXII) by means of triethanolamine in refluxing xylene gives the acetoacetanilide (XXIII), which is cyclized by means of hot triethanolamine and H2SO4 to yield 6-methoxy-4-methylquinolin-2(1H)-one (I), which is treated with refluxing POCl3 to provide 2-chloro-6-methoxy-4-methylquinoline (XXIV). Reaction of compound (XXIV) with SO2Cl2 in hot AcOH affords 2,5-dichloro-6-methoxy-4-methylquinoline (XXV), which is treated with MeONa in refluxing methanol to furnish 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Alternatively, the reaction of compound (XXIV) with MeONa as before gives 2,6-dimethoxy-4-methylquinoline (XXVII), which is treated with SO2Cl2 in hot AcOH to give the already described 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Nitration of compound (XXVI) with KNO3 and P2O5 gives the 8-nitroquinoline derivative (XXVIII), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH in hot NMP to yield the diaryl ether (VII). Finally, the nitro group of compound (VII) is reduced with hydrazine over Pd/C.
//////////////////////
J Med Chem 1989,32(8),1728-32

Synthesis of the intermediate diazepinone (IV) is accomplished by a one-pot synthesis. Condensation of 2-chloro-3-aminopyridine (I) with the anthranilic ester (II) is effected in the presence of potassium tert-butoxide as a catalyst. The resulting anthranilic amide (III) is cyclized under the influence of catalytic amounts of sulfuric acid. Treatment of (IV) with chloroacetylchloride in toluene yields the corresponding choroacetamide (V). The side chain of AQ-RA 741 is prepared starting from 4-picoline, which is alkylated by reaction with 3-(diethylamino)propylchloride in the presence of n-butyllithium. Hydrogenation of (VIII) using platinum dioxide as a catalyst furnishes the diamine (IX), which is coupled with (V) in the presence of catalytic amounts of sodium iodide in acetone leading to AQ-RA 741 as its free base.
- Shanks GD, Oloo AJ, Aleman GM et al. (2001). “A New Primaquine Analogue, Tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria”. Clin Infect Dis 33 (12): 1968–74. doi:10.1086/324081. JSTOR 4482936.PMID 11700577.
- Lell B, Faucher JF, Missinou MA et al. (2000). “Malaria chemoprophylaxis with tafenoquine: a randomised study”.Lancet 355 (9220): 2041–5. doi:10.1016/S0140-6736(00)02352-7. PMID 10885356.
- Elmes NJ, Nasveld PE, Kitchener SJ, Kocisko DA, Edstein MD (November 2008). “The efficacy and tolerability of three different regimens of tafenoquine versus primaquine for post-exposure prophylaxis of Plasmodium vivax malaria in the Southwest Pacific”. Transactions of the Royal Society of Tropical Medicine and Hygiene 102 (11): 1095–101.doi:10.1016/j.trstmh.2008.04.024. PMID 18541280.
- Nasvelda P, Kitchener S. (2005). “Treatment of acute vivax malaria with tafenoquine”. Trans R Soc Trop Med Hyg 99 (1): 2–5. doi:10.1016/j.trstmh.2004.01.013. PMID 15550254.
- Peters W (1999). “The evolution of tafenoquine–antimalarial for a new millennium?”. J R Soc Med 92 (7): 345–352.PMID 10615272.
- J Med Chem 1982,25(9),1094
|
8-3-2007
|
Methods and compositions for treating diseases associated with pathogenic proteins
|
|
|
12-6-2006
|
Process for the preparation of quinoline derivatives
|
|
|
3-14-2002
|
PROCESS FOR THE PREPARATION OF ANTI-MALARIAL DRUGS
|
|
|
4-2-1998
|
MULTIDENTATE METAL COMPLEXES AND METHODS OF MAKING AND USING THEREOF
|
|
|
4-18-1997
|
PROCESS FOR THE PREPARATION OF ANTI-MALARIAL DRUGS
|
|
|
12-20-1996
|
MULTIDENTATE METAL COMPLEXES AND METHODS OF MAKING AND USING THEREOF
|
|
|
12-15-1993
|
Use of interferon and a substance with an antimalarial activity for the treatment of malaria infections
|
|
|
10-15-1986
|
4-methyl-5-(unsubstituted and substituted phenoxy)-2,6-dimethoxy-8-(aminoalkylamino) quinolines
|
MEPOLIZUMAB….GSK to file severe asthma drug by year end
The first non-inhaled treatment for a difficult-to-treat form of severe asthma is getting closer to market after GlaxoSmithKline said it would initiate global filings for the drug at the end of this year, on the back of strong late-stage clinical data.
Mepolizumab – a monoclonal antibody that inhibits interleukin 5 – is being investigated as a treatment for severe eosinophilic asthma in patients who experience exacerbations despite high-dose oral or inhaled corticosteroids (ICS) and an additional controller such as long-acting beta-2 agonist.
Read more at: http://www.pharmatimes.com/Article/14-03-13/GSK_to_file_severe_asthma_drug_by_year_end.aspx#ixzz2vuANtYaK
Follow us: @PharmaTimes on Twitter
Mepolizumab (proposed trade name Bosatria) is a humanized monoclonal antibody that recognizes interleukin-5 (IL-5), and is used to treat certain kinds of asthma and white blood cell diseases.
 IL 5
IL 5
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | IL-5 |
Recent studies have concluded that mepolizumab may improve exacerbations in patients with severe eosinophilic asthma, an adult-onset asthma which represents less than 5% of all asthma.
IL-5 is a chemical messenger in the immune system that stimulates the growth of eosinophils. In eosinophilic asthma, eosinophils are present in the lungs. When mepolizumab was given to people with eosinophilic asthma, it eliminated eosinophils from the bloodstream,and reduced eosinophils in the lungs and bone marrow. Mepolizumab also reduced the number of asthma exacerbations, and reduced the need for corticosteroids.[1]Mepolizumab improved the quality of life, but the improvement was “not clinically meaningful,” according to a reviewer.[2] [3]
In a recent multi-centre, double-blinded, randomised, controlled trial study of Mepolizumab in severe eosinophilic asthma, Mepolizumab reduced the number of clinically significant exacerbations compared to a placebo. Additionally Mepolizumab reduced sputum and blood eosinophil counts and was shown to be safe for up to 12 months.[4]

Mepolizumab is also in development for the management of hypereosinophilic syndrome by GlaxoSmithKline (GSK) and has received orphan drug designation by the FDA.[5] Mepolizumab has been shown to reduce the need for corticosteroids and improve symptoms in FIP1L1/PDGFRA negative hypereosinophilic syndrome.[6]
UK pharma giant GlaxoSmithKline (LSE: GSK) says that a pivotal Phase III study of mepolizumab, an investigational IL-5 antagonist monoclonal antibody, met its primary endpoint of reduction in the frequency of exacerbations, in patients with severe eosinophilic asthma.
Mepolizumab could add £400 million ($668 million) to GSK’s revenue by 2021, according to estimates from Barclays reported by The Wall Street Journal. Analysts from Deutsche Bank forecast £300 million in mepolizumab sales by 2018 for the company, already a leader in the asthma treatment sector.

The study (MEA115588) evaluated the efficacy of two-dose regimens of mepolizumab in the treatment of patients with severe eosinophilic asthma. Patients remained on their current asthma maintenance therapy throughout the study and were randomized to receive either mepolizumab 75mg intravenous (IV), 100mg subcutaneous (SC), or placebo every four weeks.
For the primary end point, both mepolizumab treatment arms showed statistically significant reductions in the frequency of clinically significant exacerbations of asthma compared to placebo (75mg IV, 47%, p<0.001; 100mg SC, 53%, p<0.001).
Adverse events reported in the study were similar across all treatment groups. The most common reported adverse events across all treatment groups were nasopharyngitis, headache, upper respiratory tract infection and asthma. The frequency of adverse events was 83% in the placebo group, 84% in the mepolizumab 75mg IV and 78% in the mepolizumab 100mg SC group. The frequency of serious adverse events was 14% in the placebo group, 7% in the mepolizumab 75mg IV and 8% in the mepolizumab 100mg SC group.
Backs up earlier studies; regulatory filing mooted at year end
Dave Allen, head of GSK Respiratory Therapy Area Unit, R&D, said: “We are really pleased to have generated further positive data on mepolizumab, consistent with the findings from our earlier exacerbation study. We now have two studies showing a reduction in exacerbations in a specific group of patients with a severe form of asthma who continue to exacerbate despite treatment with high doses of their current maintenance therapies. This is very positive news for patients. For GSK it is exciting that this is the first non-inhaled treatment for severe asthma and we will be progressing towards global filings at the end of the year.”
In addition, a second Phase III study (MEA115575) designed to evaluate the use of mepolizumab 100mg SC, every four weeks in comparison to placebo in reducing daily oral corticosteroid use while maintaining asthma control also met its primary endpoint. The study showed that patients on mepolizumab 100mg SC were able to achieve greater reductions in their maintenance oral corticosteroid dose during weeks 20-24 compared to patients on placebo (p =0.008), while maintaining asthma control.
In this study adverse events were similar across treatment groups. The most common reported adverse events in the two treatment groups were headache, nasopharyngitis, bronchitis, sinusitis, fatigue and asthma. The frequency of adverse events was 92% in the placebo and 84% in the mepolizumab treatment group. Frequency of serious adverse events was 18% in the placebo group and 1% in the mepolizumab group.
Mepolizumab Useful in Refractory Eosinophilic Asthma, a Rare Subtype of Asthma
Eosinophil.
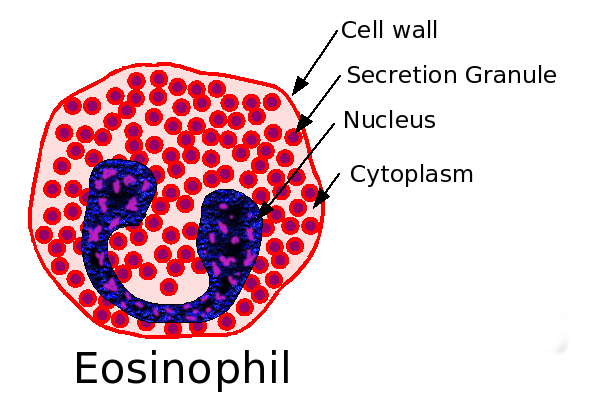
Eosinophil.Eeosinophilic form of asthma represents less than 5% of cases of adult-onset asthma and is difficult to treat.
Crystal structure of human IL-5. .
Mepolizumab reduced the number of blood and sputum eosinophils and allowed prednisone sparing in patients who had asthma with sputum eosinophilia despite prednisone treatment.
Mepolizumab therapy reduced exacerbations by 43% and improved Asthma Quality of Life Questionnaire (AQLQ) scores in patients with refractory eosinophilic asthma.
Eosinophils may have a role as important effector cells in the pathogenesis of severe exacerbations of asthma in patients with eosinophilic asthma.
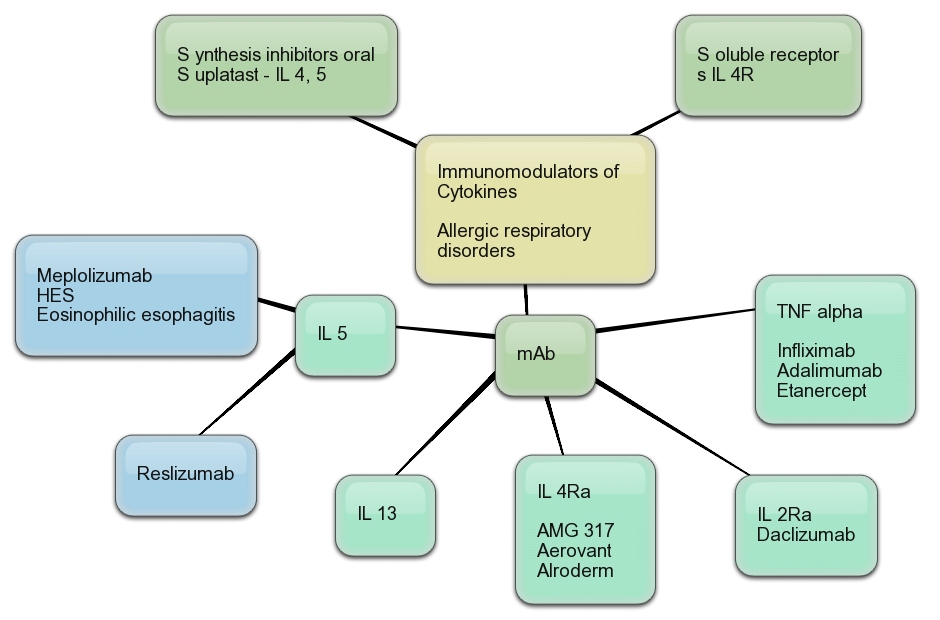
Cytokine targets for immunomodulators for allergic disorders.
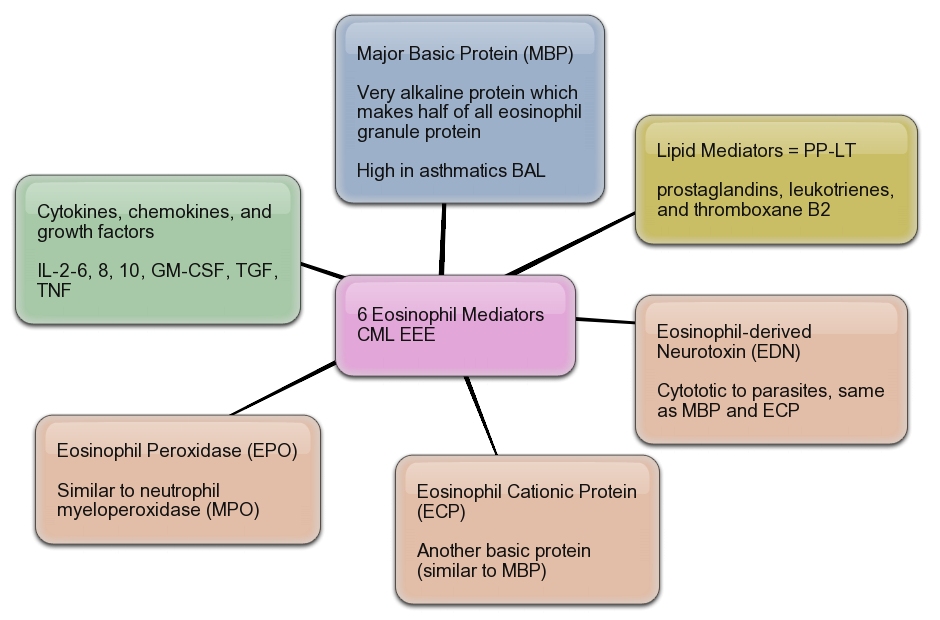
Mediators from Eosinophils
References
- Haldar P, Brightling CE, Hargadon B, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009 Mar 5;360(10):973-84.
- Nair P, Pizzichini MM, Kjarsgaard M, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009 Mar 5;360(10):985-93.
- Eosinophils in asthma – closing the loop or opening the door? Sally E. Wenzel, N Engl J Med. 2009 Mar 5;360(10):1026-7.
- Pavord, Ian D; Korn, Stephanie; Howarth, Peter; Bleecker, Eugene R; Buhl, Roland; Keene, Oliver N; Ortega, Hector; Chanez, Pascal (August 2012). “Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial”. The Lancet 380 (9842): 651–659. doi:10.1016/S0140-6736(12)60988-X.
- Phase III study of Bosatria (mepolizumab) showed disease control with reduced corticosteroid use in hypereosinophilic syndrome
- http://content.nejm.org/cgi/content/abstract/358/12/1215 Rothenberg et al 2008

GSK obtains FDA approval for bird flu vaccine
GlaxoSmithKline (GSK) has received approval from the US Food and Drug Administration (FDA) for the first adjuvanted vaccine to prevent H5N1 influenza, also known as bird flu.
GSK obtains FDA approval for bird flu vaccine http://www.pharmaceutical-technology.com/news/newsgsk-obtains-fda-approval-bird-flu-vaccine?WT.mc_id=DN_News
26 November 2013

GlaxoSmithKline (GSK) has received approval from the US Food and Drug Administration (FDA) for the first adjuvanted vaccine to prevent H5N1 influenza, also known as bird flu.
The FDA cleared the pandemic Influenza A (H5N1) virus monovalent vaccine, adjuvanted (also referred to as Q-Pan H5N1 influenza vaccine), for use in people aged 18 and older who are at increased risk of exposure to the virus.
The vaccine is composed of monovalent, inactivated, split A/H5N1 influenza virus antigen and GSK’s AS03 adjuvant.
The company said that in clinical studies, the adjuvanted formulation stimulated the required immune response while using a smaller amount of antigen as compared with a formulation without adjuvant.
GSK and Genmab seek FDA approval for ofatumumab combination therapy for CLL first-line treatment
GlaxoSmithKline (GSK) and Genmab have submitted a supplemental Biologics License Application (sBLA) to the US Food and Drug Administration (FDA) seeking the use of Arzerra (ofatumumab) in combination with an alkylator-based therapy in patients with chronic lymphocytic leukaemia (CLL) who have not received prior treatment.
READ AT
GSK and Genmab seek FDA approval for ofatumumab combination therapy for CLL first-line treatment

{kind=link}