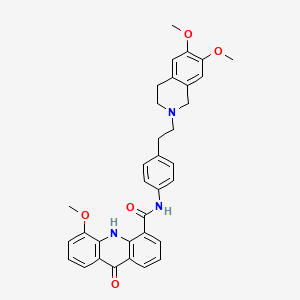
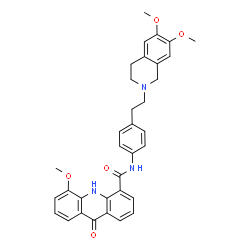
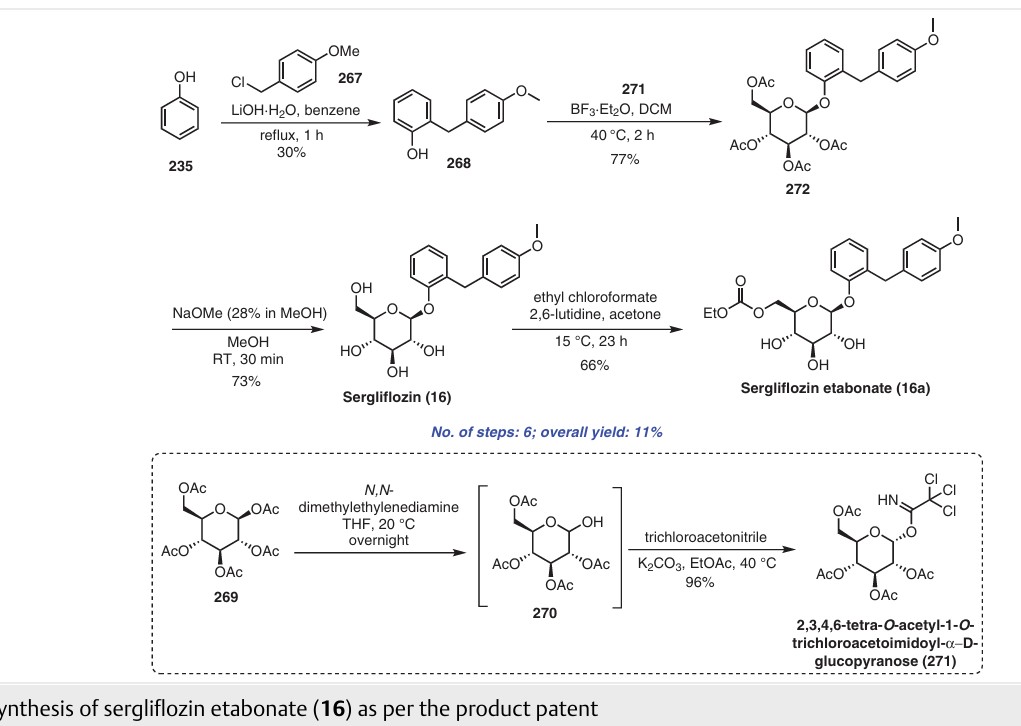
Elacridar
C34H33N3O5, 563.6 g/mol
依克立达;gw0918
UNII-N488540F94
143851-84-7 (maleate salt(1:1))
143851-98-3 (monoHCl)
4-Acridinecarboxamide, N-[4-[2-(3,4-dihydro-6,7-dimethoxy-2(1H)-isoquinolinyl)ethyl]phenyl]-9,10-dihydro-5-methoxy-9-oxo-[ACD/Index Name]
7582
AR7621300
N-[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]phenyl]-5-methoxy-9-oxo-10H-acridine-4-carboxamide
GF120918
Elacridar (GF120918)
GF-120918
GG-918
GW-120918
GW-918
GF-120918A (HCl)
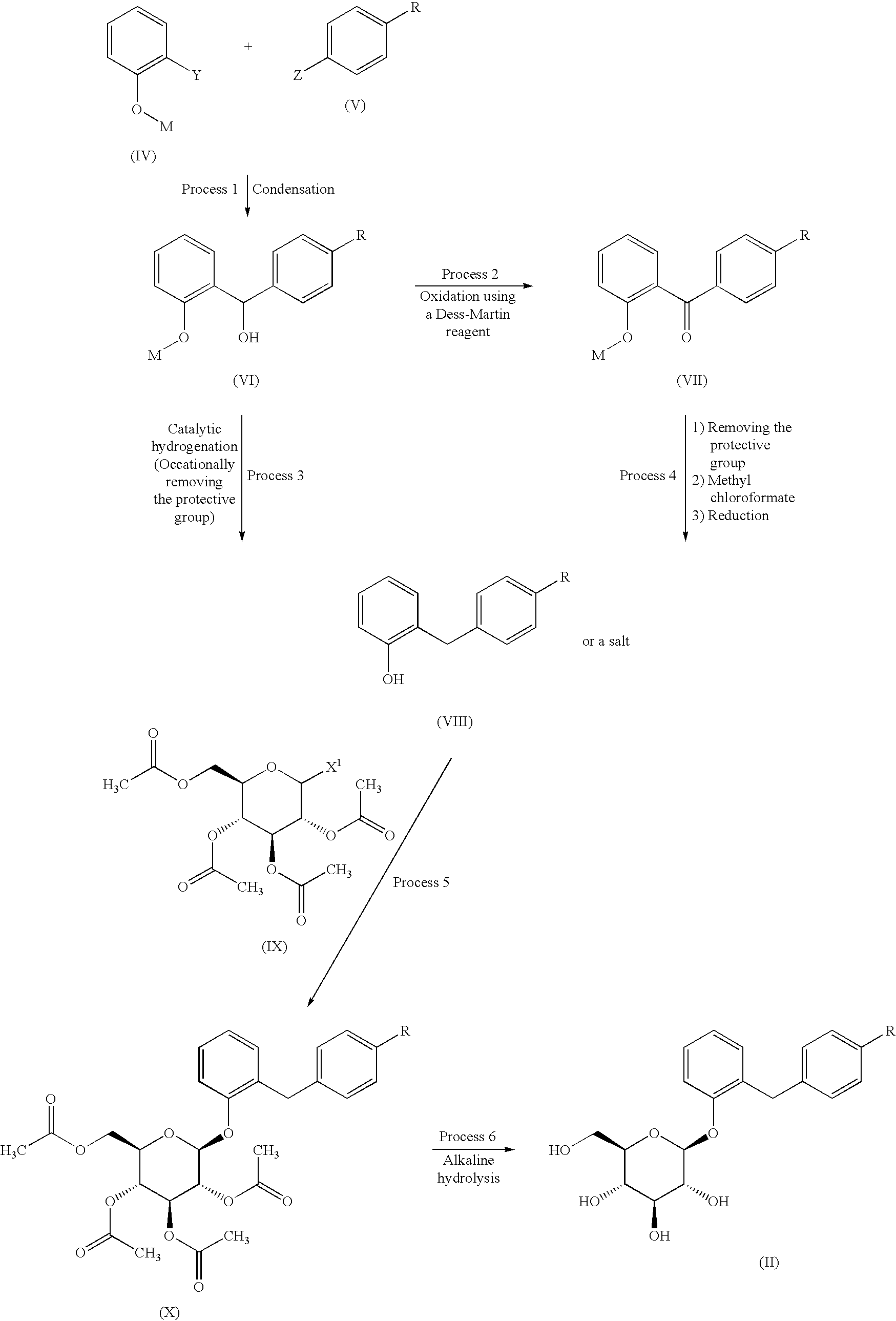
GlaxoSmithKline (previously Glaxo Wellcome ) was developing elacridar, an inhibitor of the multidrug resistance transporter BCRP (breast cancer resistant protein), as an oral bioenhancer for the treatment of solid tumors.
Elacridar is an oral bioenhancer which had been in early clinical trials at GlaxoSmithKline for the treatment of cancer, however, no recent development has been reported. It is a very potent inhibitor of P-glycoprotein, an ABC-transporter protein that has been implicated in conferring multidrug resistance to tumor cells.
SYN

The condensation of 2-(4-nitrophenyl)ethyl bromide with 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline by means of K2CO3 and KI in DMF at 100 C gives 6,7-dimethoxy-2-[2-(4-nitrophenyl)ethyl]-1,2,3,4-tetrahydroisoquinoline,
Which is reduced with H2 over Pd/C in ethanol to yield the corresponding amine . Finally, this compound is condensed with 5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxylic acid by means of DCC and HOBt in DMF to afford the target carboxamide.
The intermediate 5-methoxy-9-oxo-9,10-dihydroacridine-4-carboxylic acidhas been obtained as follows: The condensation of 2-amino-3-methoxybenzoic acid with 2-bromobenzoic acid by means of K2CO3 and copper dust give the diphenylamine , which is cyclized to the target acridine Elacridar by means of POCl3 in refluxing acetonitrile.
PATENT
WO-2019183403
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019183403&tab=PCTDESCRIPTION&_cid=P11-K1LK8Y-65903-1
Deuterated analogs of elacridar as P-gp/BCRP inhibitor by preventing efflux useful for treating cancer.
Elacridar, previously referred to as GF120918, is a compound with the structure of 9,10-dihydro-5-methoxy-9-oxo-N-[4-[2-(1 ,2,3,4-tetrahydro- 6,7-dimethoxy-2-isoquinolinyl)ethyl] phenyl]-4-acridine-carboxamide or, as sometimes written, N-4-[2-(1 ,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy- 9-oxo-4-acridine carboxamide. Elacridar was originally described as a P-gp selective inhibitor but is now recognized as a dual P-gp/BCRP inhibitor. (Matsson P, Pedersen JM, Norinder U, Bergstrom CA, and Artursson P 2009 Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm Res 26:1816-1831 ).
003 Elacridar has been examined with some success both in vitro and in vivo as a P-gp and BCRP inhibitor. By way of example, in cancer patients, coadministration of elacridar with therapeutic agents such as paclitaxel (P-gp substrate) and topotecan (BCRP substrate) improved their oral absorption – presumably by preventing efflux into the intestinal lumen by P-gp/BCRP pumps located in the Gl tract. Similarly, in rodents, elacridar has been coadministered with some success with pump substrates such as morphine, amprenavir, imatinib, dasatinib, gefitinib, sorafenib, and sunitinib to increase drug levels in the brain (by blocking efflux mediated by P-gp and BCRP at the blood brain barrier). A summary of some of these studies can be found in a study report by Sane et al. (Drug Metabolism And Disposition 40:1612-1619, 2012).
004 Administration of elacridar has several limitations. By way of example, elacridar has unfavorable physicochemical properties; it is practically insoluble in water, making it difficult to formulate as, for example, either an injectable or oral dosage form. Elacridar’s poor solubility and high lipophilicity result in dissolution rate-limited absorption from the gut lumen.
005 A variety of approaches have been pursued in order to increase efficacy of elacridar. For example, United States Patent Application Publication 20140235631 discloses a nanoparticle formulation in order to increase oral bioavailability.
006 Sane et al. (Journal of Pharmaceutical Sciences, Vol. 102, 1343-1354 (2013)) report a micro-emulsion formulation of elacridar to try and overcome its dissolution-rate-limited bioavailability.
007 Sawicki et al. (Drug Development and Industrial Pharmacy, 2017 VOL. 43, NO. 4, 584-594) described an amorphous solid dispersion formulation of freeze dried elacridar hydrochloride-povidone K30-sodium dodecyl sulfate. However, when tested in healthy human volunteers, extremely high doses (e.g. 1000 mg) were required to achieve a Cmax of 326 ng/ml. (Sawicki et al. Drug Deliv. and Transl.
Res. Published online 18 Nov 2016).
008 Montesinos et al. (Mol Pharm. 2015 Nov 2; 12(11 ):3829-38) attempted several PEGylated liposome formulations of elacridar which resulted in a partial increase in half life, but without an increase in efficacy when co-administered with a therapeutic agent.
009 Because of the great unpredictability in the art and poor correlations in many cases between animal and human data, the value of such formulation attempts await clinical trial.
0010 Studies of the whole body distribution of a microdose of 11C elacridar after intravenous injection showed high level accumulation in the liver (Bauer et al. J Nucl Med. 2016;57:1265-1268). This has led some to suggest that systemic levels of elacridar are also substantially limited by clearance in the liver.
0011 A potentially attractive strategy for improving metabolic stability of some drugs is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the rate of formation of inactive metabolites by replacing one or more hydrogen atoms with deuterium atoms.
Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the absorption, distribution, metabolism, excretion and/or toxicity (‘ADMET’) properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
0012 Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, M I et al, J Pharm Sci, 1975, 64:367-91 ; Foster, A B, Adv Drug Res 1985, 14:1 -40 (“Foster”); Kushner, D J et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, M B et al, Curr Opin Drug Discov Devel, 2006, 9:101 -09 (“Fisher”)). The results have been variable and unpredictable. For some compounds, deuteration indeed caused decreased metabolic clearance in vivo. For others, no change in metabolism was observed. Still others demonstrated increased metabolic clearance. The great unpredictability and variability in deuterium effects has led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting metabolism (see Foster at p. 35 and Fisher at p. 101 ).
0013 The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem. 1991 , 34, 2871 -76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
0014 Considering elacridar’s challenging physicochemical and ADMET properties in humans, in spite of recent formulation advancements, there remains a need in the art for elacridar analogs that can achieve higher, less variable levels in the systemic circulation, at the blood-brain barrier, and elsewhere to optimize efflux inhibition.
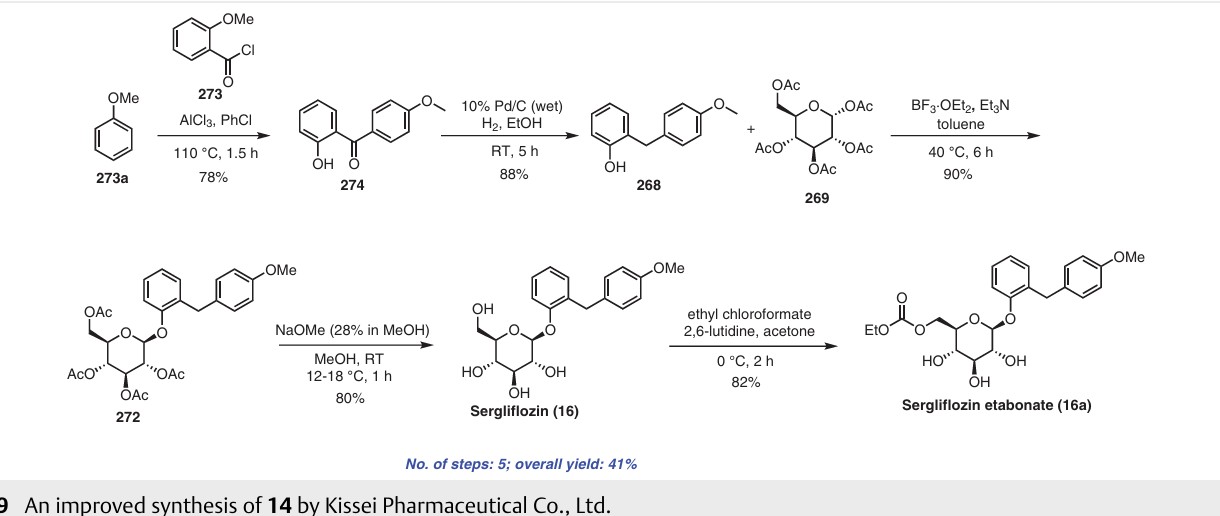
Example 1 : Synthesis of Instant Analogs and Compositions
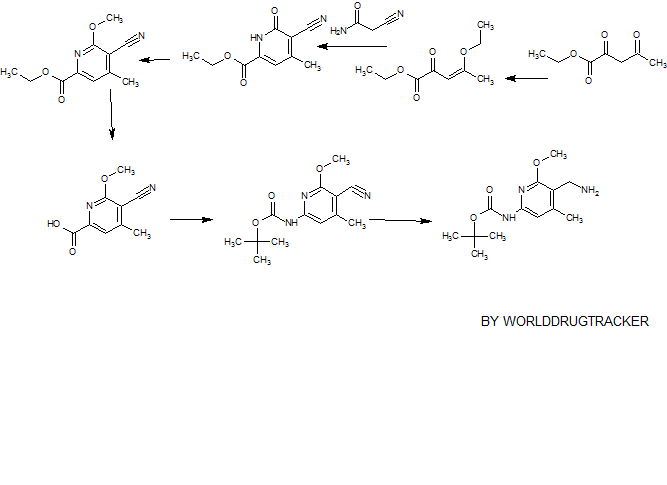
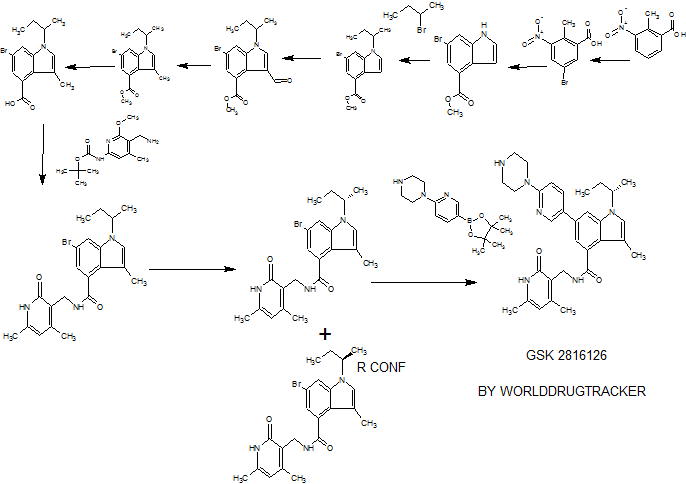
00179 This example demonstrates a synthetic method for making elacridar analogs, deuterium substitutions based upon the deuteration of the starting compounds. The synthesis and the analog numbers refer to Figure 4.
00180 Step 1
00181 A 12L three-neck flask was charged with compound 1 (270.5 g, 1.618 mol), compound 2 (357.8 g, 1.78 mol, 1.1 eq.), K2C03 (447 g, 3.236 mol, 2.0 eq), Cu (20.6 g, 0.324 mol, 0.2 eq.) and ethanol (2.7 L) and the resulting mixture was heated to reflux under nitrogen for 1 hour. The reaction mixture was cooled to room
temperature after the reaction progress was checked with LC-MS. Water (2.7 L) was added and the mixture was filtered through a pad of Celite. The Celite was washed with water (1.35L) and the combined filtrate was adjusted to pH~2 by addition of concentrated HCI (~410 mL) over 15 min. The resulting suspension was stirred at 10°C for 1.5 hours and the solid was filtered, washed with water (2.7 L) and dried at 45°C using a vacuum oven for 2 days to give compound 3 (465 g, ~100%) as a yellow solid.
00182 Step 2
00183 A suspension of compound 3 (498 g, 1.734 mol) in acetonitrile (4.0 L) was heated to reflux under stirring. To the suspension was added POCb (355.5 mL,
3.814 mol, 2.2 eq.) drop-wise over 2h. The mixture was heated at reflux for 2.5h and then cooled to 30 °C. To the mixture was slowly added water (3.0 L) and the resultant thick slurry was heated to reflux for 1 5h. The slurry was cooled to 10 °C and filtered. The solid was washed with water (2 X 1.0 L), acetonitrile (2 X 1.0 L) and dried using a vacuum oven overnight at 45 °C to afford compound 4 (426 g, 91.3%) as a yellow solid.
00184 Step 3:
00185 A 12L three-neck flask was charged with compound 5 (475g, 2.065 mol), compound 6 (474.8g, 2.065 mol), K2C03 (314g, 2.273 mol), Kl (68.6g, 0.413 moL) and DMF (2.5L) and the resulting mixture was heated to 70 °C and stirred for 2.5 hours. After LC-MS showed that the reaction was complete, the mixture was cooled to 50 °C and methanol (620 ml_) was added. Then the mixture was cooled to 30 °C and water (4.75 L) was added. The resulting suspension was cooled to 10 °C and for 1 hour. The solid was filtered, washed with water (2 X 2.5 L) and air dried for 2 days to afford the compound 7 (630 g, 89.1 %) as a yellow solid.
00186 Step 4
00187 To a solution of compound 7 (630 g, 1.84 mol) in THF/ethanol (8 L at 1 :1 ) was added Pd/C (10%, 50% wet, 30 g). The mixture was stirred under an
atmosphere of hydrogen (1 atm, balloon) at 15-20 °C for 4h. The reaction mixture was filtered through a pad of Celite and the pad was washed with TFIF (1.0 L). The filtrate was concentrated to 3 volumes under vacuum and hexanes (4.0 L) was added. The resulting slurry was cooled to 0 °C and stirred for 1 h. The solid was filtered and washed with hexanes (2 X 500 ml_) and air dried overnight to afford the compound 8 (522 g, 90.8%) as an off -white solid.
00188 Step 5
00189 A 5L three-neck flask was charged with compound 4 (250 g, 0.929 mol, 1 eq.), compound 8 (290 g, 0.929mol, 1 eq.) and DMF (2.5 L) and the resulting mixture was stirred at room temperature until it became a clear solution. To the solution was added TBTU (328 g, 1.021 mol, 1.1 eq.), followed by triethylamine (272 ml_, 1.95 mol, 2.1 eq.) and the resulting mixture was stirred at room temperature under nitrogen overnight. The mixture was poured slowly into water (7.5 L) with stirring and the resulting suspension was stirred for 1 hour at room temperature. The solid was filtered and washed with water (2 X 7 L). The solid thus obtained was dried using a vacuum oven at 50 °C for two days and 509.0 g (97.3%) of compound 9 was obtained as yellow solid.
00190 Step 6
00191 300.0 g (0.532 mol) of compound 9 was suspended in acetic acid (1.2 L) and heated to 70 °C. The resultant solution was hot filtered and heated to 70°C again. Preheated ethanol (70 °C, 3.6 L) was then added. To this solution was added concentrated HCI (66.0 ml_, 0.792 mol, 1.5 eq.) dropwise over 30 min. The resulting solution was stirred at 70°C until crystallization commenced (~about 20 min). The suspension was cooled to room temperature over 3h, filtered, washed with ethanol (2 X 1.8 L) and dried using a vacuum oven at 60°C over the weekend to afford compound 10 (253.0 g, 79.2%) as a brown solid.
Example 2 Manufacture of a Deuterated Elacridar analog EE60.
00192 EE60 is synthesized by the procedure shown in Figure 4 and as continued in Figure 5.
00193 The structure of EE60 is confirmed as follows: Samples of 5 pi are measured using an LC system comprising an UltiMate 3000 LC Systems (Dionex, Sunnyvale, CA) and an 2996 UV diode array detector (Waters). Samples are injected on to a 100 x 2mm (ID) 3.5 pm ZORBAX Extend-C18 column (Agilent, Santa Clara, CA). Elution is done at a flow rate of 0.4 mL/min using a 5 minute gradient from 20% to 95% B (mobile phase A was 0.1 % FICOOFI in water (v/v) and mobile phase B was methanol). 95% B is maintained for 1 min followed by re-equilibration at 20% B. Chromeleon (v6.8) is used for data acquisition and peak processing.
Example 3: Manufacture of a Deuterated Elacridar analog EE59
00194 EE59 was synthesized by the procedure shown in Figure 6.
00195 The resulting yellowish brown precipitate was removed by filtration and the filter cake was dried overnight (72 mg). Analysis of the filter cake by LCMS indicated the presence of a single peak at multiple wavelengths (215 nm, 220 nm, 254 nm,
280 nm); each peak confirmed the presence of the desired product (LC retention time, 5.3 min; m/z = 575 [(M+FI)+]).
00196 1H NMR of EE598 revealed 1H NMR (400 MHz, DMSO-d6) d 12.3 ( s , 1H), 10.6 (s, 1H), 8.51-8.46 (m, 2H), 7.80 (d, J = 8.8 Hz, 1H), 7.66 (d, J = 7.6 Hz, 2H), 7.45-7.38 (m, 2H), 7.32-7.25 (m, 3H), 6.66 (d, J = 6.8 Hz, 2H), 3.62 (s, 2H), 2.86 (t, J = 6.8 Hz, 2H), 2.66 (m, 4H).
Example 4: Demonstration of superior properties of instant analogs and compositions: in vivo ADMET.
00197 Pharmacologic studies are performed according to Ward KW et al (2001 Xenobiotica 317783-797) and Ward and Azzarano (JPET 310:703-709, 2004).
Briefly, instant analogs are administered solutions in 10% aqueous polyethylene glycol-300 (PEG-300) or 6% Cavitron with 1 % dimethyl sulfoxide, or as well triturated suspensions in 0.5% aqueous HPMC containing 1 % Tween 80. Blood samples are collected at various times up to 48 h after drug administration; plasma samples are prepared and at “70°C until analysis.
00198 Mice. Instant analogs are administered to four groups of animals by oral gavage (10 ml/kg dose volume). Three groups receive instant analogs as a suspension at 3, 30, or 300 mg/kg, and the fourth group receive instant analogs as a solution in Cavitron at 3 mg/kg. Blood sampling in mice is performed via a tail vein at 0.5, 1 , 2, 4, 8, 24, and 32 h postdose.
00199 Rats. A total of seven groups of animals receive instant analogs by oral gavage (10 ml/kg). Three groups receive instant analogs as a suspension at 3, 30, or 300 mg/kg, and a fourth and fifth group each receive instant analogs as a solution in Cavitron or PEG-300, respectively, at 3 mg/kg. A sixth and seventh group of rats with indwelling hepatic portal vein catheters receive instant analogs by oral gavage (10 ml/kg) as a suspension at 3 or 30 mg/kg, respectively. Blood sampling in rats are performed via a lateral tail vein; samples are also obtained from the hepatic portal vein catheter. Blood samples are obtained before dosing and at 5, 15, 30, and 45 min, and 1 ,1.5, 2, 3, 4, 6, 8, 10, 24, and 32 h postdose.
00200 Dogs. Dogs receive instant analogs by lavage (4 ml/kg) on three separate occasions with dosages at 3 and 30 mg/kg as a suspension and 3 mg/kg as a solution in Cavitron. Blood samples are obtained from a cephalic vein and from the hepatic portal vein catheter before dosing and at 5, 15, 30, and 45 min and 1 , 1.5, 2, 3, 4, 6, 8, 10, 24, 32, and 48 h postdose.
00201 Monkeys. Monkeys receive instant analogs by oral gavage (8 ml/kg dose volume) on three separate occasions at dosages of 3 and 30 mg/kg as a suspension and 3 mg/kg as a solution in Cavitron. Blood samples are obtained from a femoral vein via an indwelling catheter and from the hepatic portal vascular access port
before dosing and at 5, 15, and 30 min and 1 , 1.5, 2, 4, 6, 8, 10, 24, 32, and 48 h postdose.
00202 Humans. Healthy volunteers receive instant analogs orally at doses ranging from 25 mg to 1000 mg. Blood samples are obtained and analyzed for analog concentrations at 0, 15 min, 30 min, 45 min, 60 min, 90 min, 120 min, 180 min, 2 hr, 4 hr, 6hr, 8 hr, 12 hr, 24 hr, and 48 h after administration .
Analytical Methods
00203 Instant analogs are isolated from samples by precipitation with acetonitrile and quantified by LC/MS/MS coupled with an atmospheric pressure chemical ionization interface (475°C). Internal standards [in acetonitrile/10 mM ammonium formate, pH 3.0; 95:5 (v/v)] are added to 50 pi samples and vortexed and centrifuged for 30 min at 4000 rpm. The supernatants are injected onto the LC/MS/MS system using an HTS PAL autosampler (CTC Analytics, Zwingen, Switzerland) coupled to an Aria TX2 high-throughput liquid chromatographic system using turbulent flow technology (Cohesive Technologies, Franklin, MA) in focus mode. The mobile phase consists of a mixture of 0.1 % formic acid in water and 0.1 % formic acid in
acetonitrile. The turbulent flow column is a 0.5 X 50-mm Cyclone P column
(Cohesive Technologies) in series to a 2 X 20 mm, 4 pm Polar RP (Phenomenex, Torrance, CA) analytical column. Positive-ion multiple reaction monitoring is used for the detection of instant analogs and internal standard and the selected precursor and product ions are mlz 564 and 252, respectively. Using a (1/x) weighted linear regression analysis of the calibration curve, linear responses in analyte/internal standard peak area ratios are observed for instant analog concentrations ranging from 2 to 10,000 ng/ml.
00204 Alternatively, useful analytical methods to demonstrate the surprising and superior properties of the instant elacridar analogs are the methods as described by Stokvis et al, J Mass Spectr 2004: 39: 1122-1130.
PATENT
WO2014018932
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014018932&recNum=9&docAn=US2013052402&queryString=diabetes&maxRec=85830
claiming nano-particle composition comprising breast cancer resistance protein inhibitor (eg elacridar). Family member of the elacridar
PAPER
J Med Chem 1995, 38(13): 2418
PATENT
Product PATENT WO9212132
PATENT
US5604237
NMR includes d 2.60-2.95 (m,8H,CH2); 3.58 (s,2H,N–CH2 –Ph); 3.72 (s,6H,OMe); 4.05 (s,3H,OMe acridone); 6.78 (2s,2H,Ar.isoquinoline), 7.20-7.88 (m,8H,Ar.), 8.48 (t,2H,H1 and H8 acridone), 10.60 (s, 1H,CONH), 12.32 (s, 1H,NH acridone)
///////////Elacridar, GF-120918, GG-918 , GW-120918, GW-918, GF-120918A (HCl), solid tumors, GSK, GLAXO

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



















































 Japan
Japan
































 CARMEN
CARMEN





















 It’s a pleasure and a privilege to be interviewed, Bora.
It’s a pleasure and a privilege to be interviewed, Bora. By the time I discovered science blogs I knew my career goals were changing. I’d already been lucky enough to audit a science writing course at Princeton taught by Mike Lemonick from TIME, and thought that maybe science writing was a good choice for me. After reading chemistry blogs for a while I realized “Hey, I can do this!” and started my own blog, She Blinded Me with Science, in July 2006. It was the typical grad student blog, a mix of posts about papers I liked and life in the lab.
By the time I discovered science blogs I knew my career goals were changing. I’d already been lucky enough to audit a science writing course at Princeton taught by Mike Lemonick from TIME, and thought that maybe science writing was a good choice for me. After reading chemistry blogs for a while I realized “Hey, I can do this!” and started my own blog, She Blinded Me with Science, in July 2006. It was the typical grad student blog, a mix of posts about papers I liked and life in the lab.





{kind=link}
{kind=link}