
Home » Posts tagged 'fda' (Page 5)
Tag Archives: fda
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Italian API Manufacturer Receives FDA Warning Letter for Data Integrity Issues
Italian API Manufacturer Receives FDA Warning Letter for Data Integrity Issues
On July 7th the US FDA issued a Warning Letter to Trifarma S.p.A. for violating Good Manufacturing Standards at their facility in Rozzano, Italy. The company produces APIs and had been inspected early this year.
Read more about this Warning Letter here
On July 7th the US FDA issued a Warning Letter to Trifarma S.p.A. for violating Good Manufacturing Standards at their facility in Rozzano, Italy. The company produces APIs and had been inspected early this year. As a result of the inspection and the response of the company to the GMP findings the FDA decided to issue a Warning Letter.
While so far mainly Indian Manufacturers have been blamed by FDA and EU Inspectors for data integrity issues, now also an European API manufacturer has been cited for that problem. According to the Warning Letter the firm deleted all electronic raw data supporting the companies high performance liquid chromatography (HPLC) testing. Moreover, Trifarma failed to retain basic chromatographic information such as injection sequence, instrument method or integration method for the tests.
In a response to the FDA the firm explained that it has been researching backup systems since July 2013 and will have a backup system online by the third quarter of 2014. But FDA is not satisfied with this answer. Some interim actions such as storing backup data on each computer, including the integration method as part of that data are not sufficient. The FDA expects to see backups of the injection sequence, the instrument method and audit trails. According to the FDA the firm does not address how it will ensure that electronic files are not deleted prematurely from local computers.
In addition further basic GMP provisions are not met in the lab. There are no proper controls in place to prevent the unauthorized manipulation of the raw electronic data. All persons in the lab were able to delete and/or adulterate data because all lab employees were granted full privileges to the computer systems. Some equipment in place in the lab such as the HPLC and the GC lacked active audit trail functions to record changes to data, including information on original results, the identity of the person making the change, and the date of the change.
The FDA also expected to see electronic raw data supporting cleaning, method and process validations but the company was not able to provide these data. Another critical deviation referred to the fact that the company did not document any training of production employees on the production operations they perform. The company did change an SOP on how to perform training at the manufacturing site in July 2013 in order to include on-the-job training but Trifarma is not following it’s own procedures.
Interestingly the US FDA has used the information gathered in a previous inspection of another production site of the company to check the compliance in the Rozzano site. Trifarma received a 483 form on similar deficiencies for it’s Ceriano Laghetto plant but did not take the necessary actions to check if similar problems exist also at other manufacturing sites. From this the FDA concluded that there is not robust quality system is in place. The FDA also references the ICH Q7 Guide GMP for APIs and expects form API manufacturers to meet the requirements stated in that Guide.
Source. FDA Warning Letter for Trifarma S.p.A.
|
email me amcrasto@gmail.com |
FDA Approves Beleodaq (belinostat) for Peripheral T-Cell Lymphoma

Belinostat (PXD101)
FAST TRACK FDA , ORPHAN STATUS
July 3, 2014 — The U.S. Food and Drug Administration today approved Beleodaq (belinostat) for the treatment of patients with peripheral T-cell lymphoma (PTCL), a rare and fast-growing type of non-Hodgkin lymphoma (NHL). The action was taken under the agency’s accelerated approval program.
- PDX101
- PX 105684
- PXD-101
- PXD101
- UNII-F4H96P17NZ
Belinostat (PXD101) is a novel HDAC inhibitor with IC50 of 27 nM, with activity demonstrated in cisplatin-resistant tumors.
CLINICAL TRIALS…http://clinicaltrials.gov/search/intervention=Belinostat+OR+PXD101

| Identifiers | |
|---|---|
| CAS | 414864-00-9 |
| PubChem | 6918638 |
| ChemSpider | 5293831 |
| UNII | F4H96P17NZ |
| ChEBI | CHEBI:61076 |
| ChEMBL | CHEMBL408513 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C15H14N2O4S |
| Molar mass | 318.35 g mol−1 |
Belinostat inhibits the growth of tumor cells (A2780, HCT116, HT29, WIL, CALU-3, MCF7, PC3 and HS852) with IC50 from 0.2-0.66 μM. PD101 shows low activity in A2780/cp70 and 2780AD cells. Belinostat inhibits bladder cancer cell growth, especially in 5637 cells, which shows accumulation of G0-G1 phase, decrease in S phase, and increase in G2-M phase. Belinostat also shows enhanced tubulin acetylation in ovarian cancer cell lines. A recent study shows that Belinostat activates protein kinase A in a TGF-β signaling-dependent mechanism and decreases survivin mRNA.
PTCL comprises a diverse group of rare diseases in which lymph nodes become cancerous. In 2014, the National Cancer Institute estimates that 70,800 Americans will be diagnosed with NHL and 18,990 will die. PTCL represents about 10 to 15 percent of NHLs in North America.
Beleodaq works by stopping enzymes that contribute to T-cells, a type of immune cell, becoming cancerous. It is intended for patients whose disease returned after treatment (relapsed) or did not respond to previous treatment (refractory).
“This is the third drug that has been approved since 2009 for the treatment of peripheral T-cell lymphoma,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Today’s approval expands the number of treatment options available to patients with serious and life-threatening diseases.”
The FDA granted accelerated approval to Folotyn (pralatrexate) in 2009 for use in patients with relapsed or refractory PTCL and Istodax (romidepsin) in 2011 for the treatment of PTCL in patients who received at least one prior therapy.
The safety and effectiveness of Beleodaq was evaluated in a clinical study involving 129 participants with relapsed or refractory PTCL. All participants were treated with Beleodaq until their disease progressed or side effects became unacceptable. Results showed 25.8 percent of participants had their cancer disappear (complete response) or shrink (partial response) after treatment.
The most common side effects seen in Beleodaq-treated participants were nausea, fatigue, fever (pyrexia), low red blood cells (anemia), and vomiting.
The FDA’s accelerated approval program allows for approval of a drug based on surrogate or intermediate endpoints reasonably likely to predict clinical benefit for patients with serious conditions with unmet medical needs. Drugs receiving accelerated approval are subject to confirmatory trials verifying clinical benefit. Beleodaq also received orphan product designation by the FDA because it is intended to treat a rare disease or condition.
Beleodaq and Folotyn are marketed by Spectrum Pharmaceuticals, Inc., based in Henderson, Nevada. Istodax is marketed by Celgene Corporation based in Summit, New Jersey.
| MW 318.07 | |
| MF | C15H14N2O4S |
414864-00-9 cas no
866323-14-0
(2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]acrylamide
A novel HDAC inhibitor
…………………………
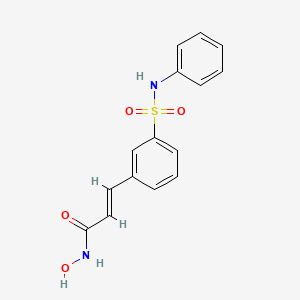
Belinostat (PXD101) is experimental drug candidate under development byTopoTarget for the treatment of hematological malignancies and solid tumors. It is a histone deacetylase inhibitor.[1]
A hydroxamate-type inhibitor of histone deacetylase.
NCI: A novel hydroxamic acid-type histone deacetylase (HDAC) inhibitor with antineoplastic activity. Belinostat targets HDAC enzymes, thereby inhibiting tumor cell proliferation, inducing apoptosis, promoting cellular differentiation, and inhibiting angiogenesis. This agent may sensitize drug-resistant tumor cells to other antineoplastic agents, possibly through a mechanism involving the down-regulation of thymidylate synthase
In 2007 preliminary results were released from the Phase II clinical trial of intravenous belinostat in combination with carboplatin and paclitaxel for relapsedovarian cancer.[2] Final results in late 2009 of a phase II trial for T cell lymphomawere encouraging.[3] Belinostat has been granted orphan drug and fast trackdesignation by the FDA.[4]

The study of inhibitors of histone deacetylases indicates that these enzymes play an important role in cell proliferation and differentiation. The inhibitor Trichostatin A (TSA) (Yoshida et al., 1990a) causes cell cycle arrest at both G1 and G2 phases (Yoshida and Beppu, 1988), reverts the transformed phenotype of different cell lines, and induces differentiation of Friend leukaemia cells and others (Yoshida et al., 1990b). TSA (and SAHA) have been reported to inhibit cell growth, induce terminal differentiation, and prevent the formation of tumours in mice (Finnin et al., 1999).
Trichostatin A (TSA)

Suberoylanilide Hydroxamic Acid (SAHA)

Cell cycle arrest by TSA correlates with an increased expression of gelsolin (Hoshikawa et al., 1994), an actin regulatory protein that is down regulated in malignant breast cancer (Mielnicki et al., 1999). Similar effects on cell cycle and differentiation have been observed with a number of deacetylase inhibitors (Kim et al., 1999). Trichostatin A has also been reported to be useful in the treatment of fibrosis, e.g., liver fibrosis and liver cirrhosis. See, e.g., Geerts et al., 1998.
Recently, certain compounds that induce differentiation have been reported to inhibit histone deacetylases. Several experimental antitumour compounds, such as trichostatin A (TSA), trapoxin, suberoylanilide hydroxamic acid (SAHA), and phenylbutyrate have been reported to act, at least in part, by inhibiting histone deacetylase (see, e.g., Yoshida et al., 1990; Richon et al., 1998; Kijima et al., 1993). Additionally, diallyl sulfide and related molecules (see, e.g., Lea et al., 1999), oxamflatin (see, e.g., Kim et al., 1999), MS-27-275, a synthetic benzamide derivative (see, e.g., Saito et al., 1999; Suzuki et al., 1999; note that MS-27-275 was later re-named as MS-275), butyrate derivatives (see, e.g., Lea and Tulsyan, 1995), FR901228 (see, e.g., Nokajima et al., 1998), depudecin (see, e.g., Kwon et al., 1998), and m-carboxycinnamic acid bishydroxamide (see, e.g., Richon et al., 1998) have been reported to inhibit histone deacetylases. In vitro, some of these compounds are reported to inhibit the growth of fibroblast cells by causing cell cycle arrest in the G1 and G2 phases, and can lead to the terminal differentiation and loss of transforming potential of a variety of transformed cell lines (see, e.g., Richon et al, 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988). In vivo, phenybutyrate is reported to be effective in the treatment of acute promyelocytic leukemia in conjunction with retinoic acid (see, e.g., Warrell et al., 1998). SAHA is reported to be effective in preventing the formation of mammary tumours in rats, and lung tumours in mice (see, e.g., Desai et al., 1999).
The clear involvement of HDACs in the control of cell proliferation and differentiation suggest that aberrant HDAC activity may play a role in cancer. The most direct demonstration that deacetylases contribute to cancer development comes from the analysis of different acute promyelocytic leukaemias (APL). In most APL patients, a translocation of chromosomes 15 and 17 (t(15;17)) results in the expression of a fusion protein containing the N-terminal portion of PML gene product linked to most of RARσ (retinoic acid receptor). In some cases, a different translocation (t(11 ;17)) causes the fusion between the zinc finger protein PLZF and RARα. In the absence of ligand, the wild type RARα represses target genes by tethering HDAC repressor complexes to the promoter DNA. During normal hematopoiesis, retinoic acid (RA) binds RARα and displaces the repressor complex, allowing expression of genes implicated in myeloid differentiation. The RARα fusion proteins occurring in APL patients are no longer responsive to physiological levels of RA and they interfere with the expression of the RA- inducible genes that promote myeloid differentiation. This results in a clonal expansion of promyelocytic cells and development of leukaemia. In vitro experiments have shown that TSA is capable of restoring RA-responsiveness to the fusion RARα proteins and of allowing myeloid differentiation. These results establish a link between HDACs and oncogenesis and suggest that HDACs are potential targets for pharmaceutical intervention in APL patients. (See, for example, Kitamura et al., 2000; David et al., 1998; Lin et al., 1998).
BELINOSTAT
Furthermore, different lines of evidence suggest that HDACs may be important therapeutic targets in other types of cancer. Cell lines derived from many different cancers (prostate, coloreetal, breast, neuronal, hepatic) are induced to differentiate by HDAC inhibitors (Yoshida and Horinouchi, 1999). A number of HDAC inhibitors have been studied in animal models of cancer. They reduce tumour growth and prolong the lifespan of mice bearing different types of transplanted tumours, including melanoma, leukaemia, colon, lung and gastric carcinomas, etc. (Ueda et al., 1994; Kim et al., 1999).
Psoriasis is a common chronic disfiguring skin disease which is characterised by well-demarcated, red, hardened scaly plaques: these may be limited or widespread. The prevalence rate of psoriasis is approximately 2%, i.e., 12.5 million sufferers in the triad countries (US/Europe/Japan). While the disease is rarely fatal, it clearly has serious detrimental effects upon the quality of life of the patient: this is further compounded by the lack of effective therapies. Present treatments are either ineffective, cosmetically unacceptable, or possess undesired side effects. There is therefore a large unmet clinical need for effective and safe drugs for this condition. Psoriasis is a disease of complex etiology. Whilst there is clearly a genetic component, with a number of gene loci being involved, there are also undefined environmental triggers. Whatever the ultimate cause of psoriasis, at the cellular level, it is characterised by local T-cell mediated inflammation, by keratinocyte hyperproliferation, and by localised angiogenesis. These are all processes in which histone deacetylases have been implicated (see, e.g., Saunders et al., 1999; Bernhard et al, 1999; Takahashi et al, 1996; Kim et al , 2001 ). Therefore HDAC inhibitors may be of use in therapy for psoriasis. Candidate drugs may be screened, for example, using proliferation assays with T-cells and/or keratinocytes.
………………………………………………………………………..
PXD101/Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.


…………………………………..
GENERAL SYNTHESIS
IGNORE 10

ENTRY 45 IS BELINOSTAT
Scheme 1

By using amines instead of aniline, the corresponding products may be obtained. The use of aniline, 4-methoxyaniline, 4-methylaniline, 4-bromoaniline, 4-chloroaniline, 4-benzylamine, and 4-phenethyamine, among others, is described in the Examples below.
In another method, a suitable amino acid (e.g., ω-amino acid) having a protected carboxylic acid (e.g., as an ester) and an unprotected amino group is reacted with a sulfonyl chloride compound (e.g., RSO2CI) to give the corresponding sulfonamide having a protected carboxylic acid. The protected carboxylic acid is then deprotected using base to give the free carboxylic acid, which is then reacted with, for example, hydroxylamine 2-chlorotrityl resin followed by acid (e.g., trifluoroacetic acid), to give the desired carbamic acid.
One example of this approach is illustrated below, in Scheme 2, wherein the reaction conditions are as follows: (i) RSO2CI, pyridine, DCM, room temperature, 12 hours; (ii) 1 M LiOH or 1 M NaOH, dioxane, room temperature, 3-48 hours; (iii) hydroxylamine 2-chlorotrityl resin, HOAt, HATU, DIPEA, DCM, room temperature, 16 hours; and (iv) TFA/DCM (5:95, v/v), room temperature, 1.5 hours.
Scheme 2

Additional methods for the synthesis of compounds of the present invention are illustrated below and are exemplified in the examples below.
Scheme 3A

Scheme 3B

Scheme 4


Scheme 8

Scheme 9

……………………………………………………………………..
SYNTHESIS
Example 1
3-Formylbenzenesulfonic acid, sodium salt (1)

Oleum (5 ml) was placed in a reaction vessel and benzaldehyde (2.00 g, 18.84 mmol) was slowly added not exceeding the temperature of the reaction mixture more than 30°C. The obtained solution was stirred at 40°C for ten hours and at ambient temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate. The aqueous phase was treated with CaC03 until the evolution of C02 ceased (pH~6-7), then the precipitated CaSO4was filtered off and washed with water. The filtrate was treated with Na2CO3 until the pH of the reaction medium increased to pH 8, obtained CaCO3 was filtered off and water solution was evaporated in vacuum. The residue was washed with methanol, the washings were evaporated and the residue was dried in desiccator over P2Oβ affording the title compound (2.00 g, 51%). 1H NMR (D20), δ: 7.56-8.40 (4H, m); 10.04 ppm (1 H, s).
Example 2 3-(3-Sulfophenyl)acrylic acid methyl ester, sodium salt (2)

Sodium salt of 3-formylbenzenesulfonic acid (1) (1.00 g, 4.80 mmol), potassium carbonate (1.32 g, 9.56 mmol), trimethyl phosphonoacetate (1.05 g, 5.77 mmol) and water (2 ml) were stirred at ambient temperature for 30 min., precipitated solid was filtered and washed with methanol. The filtrate was evaporated and the title compound (2) was obtained as a white solid (0.70 g, 55%). 1H NMR (DMSO- dβl HMDSO), δ: 3.68 (3H, s); 6.51 (1 H, d, J=16.0 Hz); 7.30-7.88 (5H, m).
Example 3 3-(3-Chlorosulfonylphenyl)acrylic acid methyl ester (3)

To the sodium salt of 3-(3-sulfophenyl)acrylic acid methyl ester (2) (0.670 g, 2.53 mmol) benzene (2 ml), thionyl chloride (1.508 g, 0.9 ml, 12.67 mmol) and 3 drops of dimethylformamide were added and the resultant suspension was stirred at reflux for one hour. The reaction mixture was evaporated, the residue was dissolved in benzene (3 ml), filtered and the filtrate was evaporated to give the title compound (0.6’40 g, 97%).
Example 4 3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a)

A solution of 3-(3-chlorosulfonylphenyl)acrylic acid methyl ester (3) (0.640 g, 2.45 mmol) in dichloromethane (2 ml) was added to a mixture of aniline (0.465 g, 4.99 mmol) and pyridine (1 ml), and the resultant solution was stirred at 50°C for one hour. The reaction mixture was evaporated and the residue was partitioned between ethyl acetate and 10% HCI. The organic layer was washed successively with water, saturated NaCl, and dried (Na2S0 ). The solvent was removed and the residue was chromatographed on silica gel with chloroform-ethyl acetate (7:1 , v/v) as eluent. The obtained product was washed with diethyl ether to give the title compound (0.226 g, 29%). 1H NMR (CDCI3, HMDSO), δ: 3.72 (3H, s); 6.34 (1H, d, J=16.0 Hz); 6.68 (1 H, br s); 6.92-7.89 (10H, m).
Example 5 3-(3-Phenylsulfamoylphenyl)acrylic acid (5a)

3-(3-Phenylsulfamoylphenyl)acrylic acid methyl ester (4a) (0.220 g, 0.69 mmol) was dissolved in methanol (3 ml), 1N NaOH (2.08 ml, 2.08 mmol) was added and the resultant solution was stirred at ambient temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The aqueous layer was acidified with 10% HCI and stirred for 30 min. The precipitated solid was filtered, washed with water and dried in desiccator over P2Os to give the title compound as a white solid (0.173 g, 82%). Example 6 3-(3-Phenylsulfamoylphenyl)acryloyl chloride (6a)

To a suspension of 3-(3-phenylsulfamoylphenyl)acrylic acid (5a) (0.173 g, 0.57 mmol) in dichloromethane (2.3 ml) oxalyl chloride (0.17 ml, 1.95 mmol) and one drop of dimethylformamide were added. The reaction mixture was stirred at 40°C for one hour and concentrated under reduced pressure to give crude title compound (0.185 g).
Example 7
N-Hydroxy-3-(3-phenylsulfamoylphenyl)acrylamide (7a) (PX105684) BELINOSTAT

To a suspension of hydroxylamine hydrochloride (0.200 g, 2.87 mmol) in tetrahydrofuran (3.5 ml) a saturated NaHCOβ solution (2.5 ml) was added and the resultant mixture was stirred at ambient temperature for 10 min. To the reaction mixture a 3-(3-phenylsulfamoylphenyl)acryloyl chloride (6a) (0.185 g) solution in tetrahydrofuran (2.3 ml) was added and stirred at ambient temperature for one hour. The reaction mixture was partitioned between ethyl acetate and 2N HCI. The organic layer was washed successively with water and saturated NaCl, the solvent was removed and the residue was washed with acetonitrile and diethyl ether.
The title compound was obtained as a white solid (0.066 g, 36%), m.p. 172°C. BELINOSTAT

1H NMR (DMSO-d6, HMDSO), δ: 6.49 (1 H, d, J=16.0 Hz); 7.18-8.05 (10H, m); 9.16 (1 H, br s); 10.34 (1 H, s); 10.85 ppm (1 H, br s).
HPLC analysis on Symmetry C18column: impurities 4% (column size 3.9×150 mm; mobile phase acetonitrile – 0.1 M phosphate buffer (pH 2.5), 40:60; sample concentration 1 mg/ml; flow rate 0.8 ml/ min; detector UV 220 nm).
Anal. Calcd for C15Hι4N204S, %: C 56.59, H 4.43, N 8.80. Found, %: C 56.28, H 4.44, N 8.56.
……………………………………………………………………….
SYNTHESIS
US20100286279

…………………………………………………….
SYNTHESIS AND SPECTRAL DATA
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (28, belinostat, PXD101).
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
The methyl ester (27) (8.0 g) was prepared according to reported synthetic route,
(Watkins, C. J.; Romero-Martin, M.-R.; Moore, K. G.; Ritchie, J.; Finn, P. W.; Kalvinsh, I.;
Loza, E.; Dikvoska, K.; Gailite, V.; Vorona, M.; Piskunova, I.; Starchenkov, I.; Harris, C. J.;
Duffy, J. E. S. Carbamic acid compounds comprising a sulfonamide linkage as HDAC
inhibitors. PCT Int. Appl. WO200230879A2, April 18, 2002.)
but using procedure D (Experimental Section) or method described for 26 to convert the methyl ester to crude
hydroxamic acid which was further purified by chromatography (silica, MeOH/DCM = 1:10) to
afford 28 (PXD101) as off-white or pale yellow powder (2.5 g, 31%).
LC–MS m/z 319.0 ([M +H]+).
1H NMR (DMSO-d6) 12–9 (very broad, 2H), 7.90 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.70 (d, J
= 7.8 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d,
J = 7.8 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.50 (d, J = 15.8 Hz, 1H);
13C NMR (DMSO-d6) 162.1,
140.6, 138.0, 136.5, 135.9, 131.8, 130.0, 129.2, 127.1, 124.8, 124.1, 121.3, 120.4.
Anal.
(C15H14N2O4S) C, H, N
………………………………………………..
SYNTHESIS
PXDIOI / Belinostat®
(E)-N-hydroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide, also known as PXD101 and Belinostat®, shown below, is a well known histone deacetylate (HDAC) inhibitor. It is being developed for treatment of a range of disorders mediated by HDAC, including proliferative conditions (such as cancer and psoriasis), malaria, etc.

PXD101 was first described in WO 02/30879 A2. That document describes a multi-step method of synthesis which may conveniently be illustrated by the following scheme.
Scheme 1
Not isolated

ed on (A)
on (D)

d on (H)

There is a need for alternative methods for the synthesis of PXD101 and related compounds for example, methods which are simpler and/or employ fewer steps and/or permit higher yields and/or higher purity product.
Scheme 5

DMAP, toluene



Synthesis 1 3-Bromo-N-phenyl-benzenesulfonamide (3)

To a 30 gallon (-136 L) reactor was charged aniline (2) (4.01 kg; 93.13 g/mol; 43 mol), toluene (25 L), and 4-(dimethylamino)pyridine (DMAP) (12 g), and the mixture was heated to 50-600C. 3-Bromobenzenesulfonyl chloride (1) (5 kg; 255.52 g/mol; 19.6 mol) was charged into the reactor over 30 minutes at 50-600C and progress of the reaction was monitored by HPLC. After 19 hours, toluene (5 L) was added due to losses overnight through the vent line and the reaction was deemed to be complete with no compound (1) being detected by HPLC. The reaction mixture was diluted with toluene (10 L) and then quenched with 2 M aqueous hydrochloric acid (20 L). The organic and aqueous layers were separated, the aqueous layer was discarded, and the organic layer was washed with water (20 L), and then 5% (w/w) sodium bicarbonate solution (20 L), while maintaining the batch temperature at 45-55°C. The batch was then used in the next synthesis.
Synthesis 2 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrylic acid ethyl ester (5)

To the batch containing 3-bromo-N-phenyl-benzenesulfonamide (3) (the treated organic layer obtained in the previous synthesis) was added triethylamine (2.97 kg; 101.19 g/mol; 29.4 mol), tri(o-tolyl)phosphine (119 g; 304.37 g/mol; 0.4 mol), and palladium (II) acetate (44 g; 224.51 g/mol; 0.2 mol), and the resulting mixture was degassed four times with a vacuum/nitrogen purge at 45-55°C. Catalytic palladium (0) was formed in situ. The batch was then heated to 80-900C and ethyl acrylate (4) (2.16 kg; 100.12 g/mol; 21.6 mol) was slowly added over 2.75 hours. The batch was sampled after a further 2 hours and was deemed to be complete with no compound (3) being detected by HPLC. The batch was cooled to 45-55°C and for convenience was left at this temperature overnight.
The batch was then reduced in volume under vacuum to 20-25 L, at a batch temperature of 45-55°C, and ethyl acetate (20 L) was added. The batch was filtered and the residue washed with ethyl acetate (3.5 L). The residue was discarded and the filtrates were sent to a 100 gallon (-454 L) reactor, which had been pre-heated to 600C. The 30 gallon (-136 L) reactor was then cleaned to remove any residual Pd, while the batch in the 100 gallon (-454 L) reactor was washed with 2 M aqueous hydrochloric acid and water at 45-55°C. Once the washes were complete and the 30 gallon (-136 L) reactor was clean, the batch was transferred from the 100 gallon (-454 L) reactor back to the 30 gallon (-136 L) reactor and the solvent was swapped under vacuum from ethyl acetate/toluene to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it. The batch was then cooled to 0-100C and held at this temperature over the weekend in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). A sample of the wet-cake was taken for Pd analysis. The Pd content of the crude product (5) was determined to be 12.9 ppm.
The wet-cake was then charged back into the 30 gallon (-136 L) reactor along with ethyl acetate (50 L) and heated to 40-500C in order to obtain a solution. A sparkler filter loaded with 12 impregnated Darco G60® carbon pads was then connected to the reactor and the solution was pumped around in a loop through the sparkler filter. After 1 hour, a sample was taken and evaporated to dryness and analysed for Pd content. The amount of Pd was found to be 1.4 ppm. A second sample was taken after 2 hours and evaporated to dryness and analysed for Pd content. The amount of Pd had been reduced to 0.6 ppm. The batch was blown back into the reactor and held at 40-500C overnight before the solvent was swapped under vacuum from ethyl acetate to toluene while maintaining a batch temperature of 45-55°C (the volume was reduced to 20-25 L). At this point, the batch had precipitated and heptanes (10 L) were added to re-dissolve it and the batch was cooled to 0-100C and held at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with heptanes (5 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum for 25 hours. A first lot of the title compound (5) was obtained as an off-white solid (4.48 kg, 69% overall yield from 3-bromobenzenesulfonyl chloride (1)) with a Pd content of 0.4 ppm and a purity of 99.22% (AUC) by HPLC.
Synthesis 3 (E)-3-(3-Phenylsulfamoyl-phenyl)-acrvlic acid (6)

To the 30 gallon (-136 L) reactor was charged the (E)-3-(3-phenylsulfamoyl-phenyl)- acrylic acid ethyl ester (5) (4.48 kg; 331.39 g/mol; 13.5 mol) along with 2 M aqueous sodium hydroxide (17.76 L; -35 mol). The mixture was heated to 40-50°C and held at this temperature for 2 hours before sampling, at which point the reaction was deemed to be complete with no compound (5) being detected by HPLC. The batch was adjusted to pH 2.2 using 1 M aqueous hydrochloric acid while maintaining the batch temperature between 40-500C. The product had precipitated and the batch was cooled to 20-300C and held at this temperature for 1 hour before filtering and washing the cake with water (8.9 L). The filtrate was discarded. The batch was allowed to condition on the filter overnight before being charged back into the reactor and slurried in water (44.4 L) at 40-500C for 2 hours. The batch was cooled to 15-20°C, held for 1 hour, and then filtered and the residue washed with water (8.9 L). The filtrate was discarded. The crude title compound (6) was transferred to an oven for drying at 45-55°C under vacuum with a slight nitrogen bleed for 5 days (this was done for convenience) to give a white solid (3.93 kg, 97% yield). The moisture content of the crude material was measured using Karl Fischer (KF) titration and found to be <0.1% (w/w). To the 30 gallon (-136 L) reactor was charged the crude compound (6) along with acetonitrile (47.2 L). The batch was heated to reflux (about 80°C) and held at reflux for 2 hours before cooling to 0-10°C and holding at this temperature overnight in order to precipitate the product. The batch was filtered and the residue was washed with cold acetonitrile (7.9 L). The filtrate was discarded and the residue was dried under vacuum at 45-55°C for 21.5 hours. The title compound (6) was obtained as a fluffy white solid (3.37 kg, 84% yield with respect to compound (5)) with a purity of 99.89% (AUC) by HPLC.
Synthesis 4 (E)-N-Hvdroxy-3-(3-phenylsulfamoyl-phenyl)-acrylamide (PXD101) BELINOSTAT

To the 30 gallon (-136 L) reactor was charged (E)-3-(3-phenylsulfamoyl-phenyl)-acrylic acid (6) (3.37 kg; 303.34 g/mol; 11.1 mol) and a pre-mixed solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in isopropyl acetate (IPAc) (27 g in 30 L; 152.24 g/mol; 0.18 mol). The slurry was stirred and thionyl chloride (SOCI2) (960 mL; density ~1.631 g/mL; 118.97 g/mol; -13 mol) was added to the reaction mixture and the batch was stirred at 20-300C overnight. After 18.5 hours, the batch was sampled and deemed to be complete with no compound (6) being detected by HPLC. The resulting solution was transferred to a 100 L Schott reactor for temporary storage while the
30 gallon (-136 L) reactor was rinsed with isopropyl acetate (IPAc) and water. Deionized water (28.9 L) was then added to the 30 gallon (-136 L) reactor followed by 50% (w/w) hydroxylamine (6.57 L; -1.078 g/mL; 33.03 g/mol; -214 mol) and another charge of deionized water (1.66 L) to rinse the lines free of hydroxylamine to make a 10% (w/w) hydroxylamine solution. Tetrahydrofuran (THF) (6.64 L) was then charged to the
30 gallon (-136 L) reactor and the mixture was stirred and cooled to 0-100C. The acid chloride solution (from the 100 L Schott reactor) was then slowly charged into the hydroxylamine solution over 1 hour maintaining a batch temperature of 0-10°C during the addition. The batch was then allowed to warm to 20-300C. The aqueous layer was separated and discarded. The organic layer was then reduced in volume under vacuum while maintaining a batch temperature of less than 300C. The intention was to distill out 10-13 L of solvent, but this level was overshot. A larger volume of isopropyl acetate (IPAc) (16.6 L) was added and about 6 L of solvent was distilled out. The batch had precipitated and heptanes (24.9 L) were added and the batch was held at 20-30°C overnight. The batch was filtered and the residue was washed with heptanes (6.64 L). The filtrate was discarded and the residue was dried at 45-55°C under vacuum with a slight nitrogen bleed over the weekend. The title compound (PXD101) was obtained as a light orange solid (3.11 kg, 89% yield with respect to compound (6)) with a purity of 99.25% (AUC) by HPLC.
The title compound (PXD101) (1.2 kg, 3.77 mol) was dissolved in 8 volumes of 1:1 (EtOH/water) at 600C. Sodium bicarbonate (15.8 g, 5 mol%) was added to the solution. Water (HPLC grade) was then added at a rate of 65 mL/min while keeping the internal temperature >57°C. After water (6.6 L) had been added, crystals started to form and the water addition was stopped. The reaction mixture was then cooled at a rate of 10°C/90 min to a temperature of 0-10cC and then stirred at ambient temperature overnight. The crystals were then filtered and collected. The filter cake was washed by slurrying in water (2 x 1.2 L) and then dried in an oven at 45°C for 60 hours with a slight nitrogen bleed. 1.048 kg (87% recovery) of a light orange solid was recovered. Microscopy and XRPD data showed a conglomerate of irregularly shaped birefringant crystalline particles. The compound was found to contain 0.02% water.
As discussed above: the yield of compound (5) with respect to compound (1) was 69%. the yield of compound (6) with respect to compound (5) was 84%. the yield of PXD101 with respect to compound (6) was 89%.
……………….
FORMULATION
Formulation Studies
These studies demonstrate a substantial enhancement of HDACi solubility (on the order of a 500-fold increase for PXD-101) using one or more of: cyclodextrin, arginine, and meglumine. The resulting compositions are stable and can be diluted to the desired target concentration without the risk of precipitation. Furthermore, the compositions have a pH that, while higher than ideal, is acceptable for use.

UV Absorbance
The ultraviolet (UV absorbance E\ value for PXD-101 was determined by plotting a calibration curve of PXD-101 concentration in 50:50 methanol/water at the λmax for the material, 269 nm. Using this method, the E1i value was determined as 715.7.
Methanol/water was selected as the subsequent diluting medium for solubility studies rather than neat methanol (or other organic solvent) to reduce the risk of precipitation of the cyclodextrin.
Solubility in Demineralised Water
The solubility of PXD-101 was determined to be 0.14 mg/mL for demineralised water. Solubility Enhancement with Cvclodextrins
Saturated samples of PXD-101 were prepared in aqueous solutions of two natural cyclodextrins (α-CD and γ-CD) and hydroxypropyl derivatives of the α, β and Y cyclodextrins (HP-α-CD, HP-β-CD and HP-γ-CD). All experiments were completed with cyclodextrin concentrations of 250 mg/mL, except for α-CD, where the solubility of the cyclodextrin was not sufficient to achieve this concentration. The data are summarised in the following table. HP-β-CD offers the best solubility enhancement for PXD-101.

Phase Solubility Determination of HP-β-CD
The phase solubility diagram for HP-β-CD was prepared for concentrations of cyclodextrin between 50 and 500 mg/mL (5-50% w/v). The calculated saturated solubilities of the complexed HDACi were plotted against the concentration of cyclodextrin. See Figure 1.
………………………..

- Plumb, Jane A.; Finn, Paul W.; Williams, Robert J.; Bandara, Morwenna J.; Romero, M. Rosario; Watkins, Claire J.; La Thangue, Nicholas B.; Brown, Robert (2003). “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101”. Molecular Cancer Therapeutics 2 (8): 721–728. PMID 12939461.
- “CuraGen Corporation (CRGN) and TopoTarget A/S Announce Presentation of Belinostat Clinical Trial Results at AACR-NCI-EORTC International Conference”. October 2007.
- Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma, December 2009
- “Spectrum adds to cancer pipeline with $350M deal.”. February 2010.
- Helvetica Chimica Acta, 2005 , vol. 88, 7 PG. 1630 – 1657, MP 172
- WO2009/40517 A2, ….
- WO2006/120456 A1, …..
- Synthetic Communications, 2010 , vol. 40, 17 PG. 2520 – 2524, MP 172
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 PG. 4694 – 4720, NMR IN SUP INFO
| US2008274120 | 11-7-2008 | Histone Deacetylase (Hdac) Inhibitors (Pxd101) for the Treatment of Cancer Alone or in Combination With Chemotherapeutic Agent |
| US2008227845 | 9-19-2008 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US2008213399 | 9-5-2008 | Combination Therapies Using Hdac Inhibitors |
| US2008194690 | 8-15-2008 | Pharmaceutical Formulations Of Hdac Inhibitors |
| US7407988 | 8-6-2008 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US7402603 | 7-23-2008 | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| US7183298 | 2-28-2007 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US2005107445 | 5-20-2005 | Carbamic acid compounds comprising a sulfonamide linkage as HDAC inhibitors |
| US6888027 | 5-4-2005 | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising asulfonamide linkage as hdac inhibitors |
| US7973181 | 7-6-2011 | HYDROXAMIC ACID DERIVATIVES AS INHIBITORS OF HDAC ENZYMATIC ACTIVITY |
| US7928081 | 4-20-2011 | Combined Use of Prame Inhibitors and Hdac Inhibitors |
| US2011077305 | 3-32-2011 | 5-LIPOXYGENASE INHIBITORS |
| US2011003777 | 1-7-2011 | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
| US2010286279 | 11-12-2010 | Methods of Synthesis of Certain Hydroxamic Acid Compounds |
| US2010190694 | 7-30-2010 | Methods for identifying patients who will respond well to cancer treatment |
| US2010010010 | 1-15-2010 | HDAC INHIBITORS |
| US2009312311 | 12-18-2009 | COMBINATION OF ORGANIC COMPOUNDS |
| US2009192211 | 7-31-2009 | CYCLOOXYGENASE-2 INHIBITOR/HISTONE DEACETYLASE INHIBITOR COMBINATION |
| US7557140 | 7-8-2009 | CARBAMIC ACID COMPOUNDS COMPRISING A SULFONAMIDE LINKAGE AS HDAC INHIBITORS |
| WO1998038859A1 * | Mar 4, 1998 | Sep 11, 1998 | Thomas E Barta | Sulfonyl divalent aryl or heteroaryl hydroxamic acid compounds |
| WO1999024399A1 * | Nov 12, 1998 | May 20, 1999 | Darwin Discovery Ltd | Hydroxamic and carboxylic acid derivatives having mmp and tnf inhibitory activity |
| WO2000056704A1 * | Mar 22, 2000 | Sep 28, 2000 | Duncan Batty | Hydroxamic and carboxylic acid derivatives |
| WO2000069819A1 * | May 12, 2000 | Nov 23, 2000 | Thomas E Barta | Hydroxamic acid derivatives as matrix metalloprotease inhibitors |
| WO2001038322A1 * | Nov 22, 2000 | May 31, 2001 | Methylgene Inc | Inhibitors of histone deacetylase |
| EP0570594A1 * | Dec 7, 1992 | Nov 24, 1993 | SHIONOGI & CO., LTD. | Hydroxamic acid derivative based on aromatic sulfonamide |
| EP0931788A2 * | Dec 16, 1998 | Jul 28, 1999 | Pfizer Inc. | Metalloprotease inhibitors |
| GB2312674A * | Title not available |
| WO2002030879A2 | Sep 27, 2001 | Apr 18, 2002 | Prolifix Ltd | Carbamic acid compounds comprising a sulfonamide linkage as hdac inhibitors |
| WO2005063806A1 | Dec 30, 2003 | Jul 14, 2005 | Council Scient Ind Res | Arginine hydrochloride enhances chaperone-like activity of alpha crystallin |
| US4642316 | May 20, 1985 | Feb 10, 1987 | Warner-Lambert Company | Parenteral phenytoin preparations |
| WO2008090585A2 * | Jan 25, 2008 | Jul 31, 2008 | Univ Roma | Soluble forms of inclusion complexes of histone deacetylase inhibitors and cyclodextrins, their preparation processes and uses in the pharmaceutical field |
| WO2009109861A1 * | Mar 6, 2009 | Sep 11, 2009 | Topotarget A/S | Methods of treatment employing prolonged continuous infusion of belinostat |
| WO2010048332A2 * | Oct 21, 2009 | Apr 29, 2010 | Acucela, Inc. | Compounds for treating ophthalmic diseases and disorders |
| WO2011064663A1 | Nov 24, 2010 | Jun 3, 2011 | Festuccia, Claudio | Combination treatment employing belinostat and bicalutamide |
| US20110003777 * | Mar 6, 2009 | Jan 6, 2011 | Topotarget A/S | Methods of Treatment Employing Prolonged Continuous Infusion of Belinostat |
………………………..
SPECTRUM
Tiny Biotech With Three Cancer Drugs Is More Alluring Takeover Bet Now
Forbes
The drug is one of Spectrum’s two drugs undergoing phase 3 clinical trials. Allergan paid Spectrum $41.5 million and will make additional payments of up to $304 million based on achieving certain milestones. So far, Raj Shrotriya, Spectrum’s chairman, …
……………………………..


FDA Guidance for Industry: Electronic Source Data in Clinical Investigations
FDA Guidance for Industry: Electronic Source Data in Clinical Investigations
The FDA published its new Guidance for Industry (GfI) – “Electronic Source Data in Clinical Investigations” in September 2013. The Guidance defines the expectations of the FDA concerning electronic source data generated in the context of clinical trials. Find out more about this Guidance.
|
FDA Guidance for Industry: Electronic Source Data in Clinical Investigations |
|
After more than 5 years and two draft versions, the final version of the Guidance for Industry (GfI) – “Electronic Source Data in Clinical Investigations” was published in September 2013. This new FDA Guidance defines the FDA’s expectations for sponsors, CROs, investigators and other persons involved in the capture, review and retention of electronic source data generated in the context of FDA-regulated clinical trials. In an effort to encourage the modernization and increased efficiency of processes in clinical trials, the FDA clearly supports the capture of electronic source data and emphasizes the agency’s intention to support activities aimed at ensuring the reliability, quality, integrity and traceability of this source data, from its electronic source to the electronic submission of the data in the context of an authorization procedure. The Guidance addresses aspects as data capture, data review and record retention. When the computerized systems used in clinical trials are described, the FDA recommends that the description not only focus on the intended use of the system, but also on data protection measures and the flow of data across system components and interfaces. In practice, the pharmaceutical industry needs to meet significant requirements regarding organisation, planning, specification and verification of computerized systems in the field of clinical trials. The FDA also mentions in the Guidance that it does not intend to apply 21 CFR Part 11 to electronic health records (EHR). Author: Source: |
FDA grants orphan drug designation to Insys Therapeutics’ pharmaceutical cannabidiol


| Systematic (IUPAC) name | |
|---|---|
| 2-[(1R,6R)-6-isopropenyl-3-methylcyclohex-2-en-1-yl]-5-pentylbenzene-1,3-diol | |
| Clinical data | |
| Trade names | Epidiolex |
| AHFS/Drugs.com | International Drug Names |
| Legal status | Schedule I (US)Schedule II (Can)(THC – Schedule/Level I; THC and CBD two main chemicals in cannabis) |
| Pharmacokinetic data | |
| Bioavailability | 13-19% (oral),[1] 11-45% (mean 31%; inhaled)[2] |
| Half-life | 9 h[1] |
| Identifiers | |
| CAS number | 13956-29-1 |
| ATC code | None |
| PubChem | CID 644019 |
| ChemSpider | 24593618 |
| UNII | 19GBJ60SN5 |
| Chemical data | |
| Formula | C21H30O2 |
| Mol. mass | 314.4636 |
| Physical data | |
| Melt. point | 66 °C (151 °F) |
| Boiling point | 180 °C (356 °F) (range: 160–180 °C)[3] |
FDA grants orphan drug designation to Insys Therapeutics’ pharmaceutical cannabidiol – Pharmaceutical Technology
US-based specialty pharmaceutical company Insys Therapeutics has obtained orphan drug designation from the US Food and Drug Administration (FDA) for its pharmaceutical cannabidiol for treatment of Lennox-Gastaut Syndrome.
Insys Therapeutics president and CEO Michael Babich said: “With no cure and persistence of seizures with current antiepileptic medications, the orphan drug designation recognises the significant, unmet need that exists among children with this severe form of epilepsy and the teams who provide their care.
“We have the unique opportunity to test a controlled pharmaceutical CBD product for Lennox-Gastaut Syndrome, and our company is committed to advancing cannabinoid therapies that have the potential to provide significant medical benefits to patients across multiple indications.
“We expect to file an investigational new drug application (IND) for CBD in the second half of 2014.”
Cannabidiol (CBD) is one of at least 60 active cannabinoids identified in cannabis.[4] It is a major phytocannabinoid, accounting for up to 40% of the plant’s extract.[5] CBD is considered to have a wider scope of medical applications than tetrahydrocannabinol(THC).[5] An orally-administered liquid containing CBD has received orphan drug status in the US, for use as a treatment for dravet syndrome under the brand name, Epidiolex.[6]
Clinical applications

The bud of a Cannabis sativa flower coated with trichomes
Antimicrobial actions
CBD absorbed transcutaneously may attenuate the increased sebum production at the root of acne, according to an untested hypothesis.[7]
Neurological effects
A 2010 study found that strains of cannabis containing higher concentrations of cannabidiol did not produce short-term memory impairment vs. strains with similar concentrations of THC, but lower concentrations of CBD. The researchers attributed this attenuation of memory effects to CBD’s role as a CB1 antagonist. Transdermal CBD is neuroprotective in animals.[8]
Cannabidiol’s strong antioxidant properties have been shown to play a role in the compound’s neuroprotective and anti-ischemiceffects.[9]
- Parkinson’s disease
It has been proposed that CBD may help people with Parkinson’s disease, but promising results in animal experiments were not confirmed when CBD was trialled in humans.[10]
Psychotropic effect
CBD has anti-psychotic effects and may counteract the potential psychotomimetic effects of THC on individuals with latentschizophrenia;[5] some reports show it to be an alternative treatment for schizophrenia that is safe and well-tolerated.[11] Studies have shown CBD may reduce schizophrenic symptoms due to its apparent ability to stabilize disrupted or disabled NMDA receptor pathways in the brain, which are shared and sometimes contested by norepinephrine and GABA.[11][12] Leweke et al. performed a double blind, 4 week, explorative controlled clinical trial to compare the effects of purified cannabidiol and the atypical antipsychoticamisulpride on improving the symptoms of schizophrenia in 42 patients with acute paranoid schizophrenia. Both treatments were associated with a significant decrease of psychotic symptoms after 2 and 4 weeks as assessed by Brief Psychiatric Rating Scale andPositive and Negative Syndrome Scale. While there was no statistical difference between the two treatment groups, cannabidiol induced significantly fewer side effects (extrapyramidal symptoms, increase in prolactin, weight gain) when compared to amisulpride.[13]
Studies have shown cannabidiol decreases activity of the limbic system[14] and decreases social isolation induced by THC.[15] Cannabidiol has also been shown to reduce anxiety in social anxiety disorder.[16][17] However, chronic cannabidiol administration in rats was recently found to produce anxiogenic-like effects, indicating that prolonged treatment with cannabidiol might incite anxiogenic effects.[18]
Cannabidiol has demonstrated antidepressant-like effects in animal models of depression.[19][20][21]
Cancer
The American Cancer Society says: “There is no available scientific evidence from controlled studies in humans that cannabinoids can cure or treat cancer.”[22] Laboratory experiments have been performed on the potential use of cannabinoids for cancer therapy but as of 2013 results have been contradictory and knowledge remains poor.[23] Cannabinoids have been recommended for cancer pain but the adverse effects may make them a less than ideal treatment; two cannabinoid-based medicines have been approved as a backup remedy for nausea associated withchemotherapy.[4]
Dravet syndrome
Dravet syndrome is a rare form of epilepsy that is difficult to treat. Dravet syndrome, also known as Severe Myoclonic Epilepsy of Infancy (SMEI), is a rare and catastrophic form of intractable epilepsy that begins in infancy. Initial seizures are most often prolonged events and in the second year of life other seizure types begin to emerge.[24] While high profile and anecdotal reports have sparked interest in treatment with cannabinoids,[25] there is insufficient medical evidence to draw conclusions about their safety or efficacy.[25][26]
CBD-enhanced cannabis
Decades ago, selective breeding by growers in US dramatically lowered the CBD content of cannabis; their customers preferred varietals that were more mind-altering due to a higher THC, lower CBD content.[27] To meet the demands of medical cannabis patients, growers are currently developing more CBD-rich strains.[28]
In November 2012, Tikun Olam, an Israeli medical cannabis facility announced a new strain of the plant which has only cannabidiol as an active ingredient, and virtually no THC, providing some of the medicinal benefits of cannabis without the euphoria.[29][30] The researchers said the cannabis plant, enriched with CBD, “can be used for treating diseases like rheumatoid arthritis, colitis, liver inflammation, heart disease and diabetes”. Research on CBD enhanced cannabis began in 2009, resulting in Avidekel, a cannabis strain that contains 15.8% CBD and less than 1% THC. Raphael Mechoulam, a cannabinoid researcher, said “…Avidekel is thought to be the first CBD-enriched cannabis plant with no THC to have been developed in Israel”.[31]
Pharmacology
Pharmacodynamics
Cannabidiol has a very low affinity for CB1 and CB2 receptors but acts as an indirect antagonist of their agonists.[9] While one would assume that this would cause cannabidiol to reduce the effects of THC, it may potentiate THC’s effects by increasing CB1 receptor density or through another CB1-related mechanism.[32] It is also an inverse agonist of CB2receptors.[9][33] Recently, it was found to be an antagonist at the putative new cannabinoid receptor, GPR55, a GPCR expressed in the caudate nucleus and putamen.[34]Cannabidiol has also been shown to act as a 5-HT1A receptor agonist,[35] an action which is involved in its antidepressant,[19][36] anxiolytic,[36][37] and neuroprotective[38][39]effects. Cannabidiol is an allosteric modulator of μ and δ-opioid receptors.[40] Cannabidiol’s pharmacologial effects have also been attributed to PPAR-γ receptor agonism andintracellular calcium release.[5]
Pharmacokinetic interactions
There is some preclinical evidence to suggest that cannabidiol may reduce THC clearance, modestly increasing THC’s plasma concentrations resulting in a greater amount of THC available to receptors, increasing the effect of THC in a dose-dependent manner.[41][42] Despite this the available evidence in humans suggests no significant effect of CBD on THC plasma levels.[43]
Pharmaceutical preparations
Nabiximols (USAN, trade name Sativex) is an aerosolized mist for oral administration containing a near 1:1 ratio of CBD and THC. The drug was approved by Canadian authorities in 2005 to alleviate pain associated with multiple sclerosis.[44][45][46]
Isomerism

| 7 double bond isomers and their 30 stereoisomers | ||||||||
|---|---|---|---|---|---|---|---|---|
| Formal numbering | Terpenoid numbering | Number of stereoisomers | Natural occurrence | Convention on Psychotropic SubstancesSchedule | Structure | |||
| Short name | Chiral centers | Full name | Short name | Chiral centers | ||||
| Δ5-cannabidiol | 1 and 3 | 2-(6-isopropenyl-3-methyl-5-cyclohexen-1-yl)-5-pentyl-1,3-benzenediol | Δ4-cannabidiol | 1 and 3 | 4 | No | unscheduled | -5-pentyl-1,3-benzenediol.png) |
| Δ4-cannabidiol | 1, 3 and 6 | 2-(6-isopropenyl-3-methyl-4-cyclohexen-1-yl)-5-pentyl-1,3-benzenediol | Δ5-cannabidiol | 1, 3 and 4 | 8 | No | unscheduled | -5-pentyl-1,3-benzenediol.png) |
| Δ3-cannabidiol | 1 and 6 | 2-(6-isopropenyl-3-methyl-3-cyclohexen-1-yl)-5-pentyl-1,3-benzenediol | Δ6-cannabidiol | 3 and 4 | 4 | ? | unscheduled | -5-pentyl-1,3-benzenediol.png) |
| Δ3,7-cannabidiol | 1 and 6 | 2-(6-isopropenyl-3-methylenecyclohex-1-yl)-5-pentyl-1,3-benzenediol | Δ1,7-cannabidiol | 3 and 4 | 4 | No | unscheduled | -5-pentyl-1,3-benzenediol.png) |
| Δ2-cannabidiol | 1 and 6 | 2-(6-isopropenyl-3-methyl-2-cyclohexen-1-yl)-5-pentyl-1,3-benzenediol | Δ1-cannabidiol | 3 and 4 | 4 | Yes | unscheduled | -5-pentyl-1,3-benzenediol.png) |
| Δ1-cannabidiol | 3 and 6 | 2-(6-isopropenyl-3-methyl-1-cyclohexen-1-yl)-5-pentyl-1,3-benzenediol | Δ2-cannabidiol | 1 and 4 | 4 | No | unscheduled | -5-pentyl-1,3-benzenediol.png) |
| Δ6-cannabidiol | 3 | 2-(6-isopropenyl-3-methyl-6-cyclohexen-1-yl)-5-pentyl-1,3-benzenediol | Δ3-cannabidiol | 1 | 2 | No | unscheduled | -5-pentyl-1,3-benzenediol.png) |
See also: Tetrahydrocannabinol#Isomerism, Abnormal cannabidiol.
Chemistry
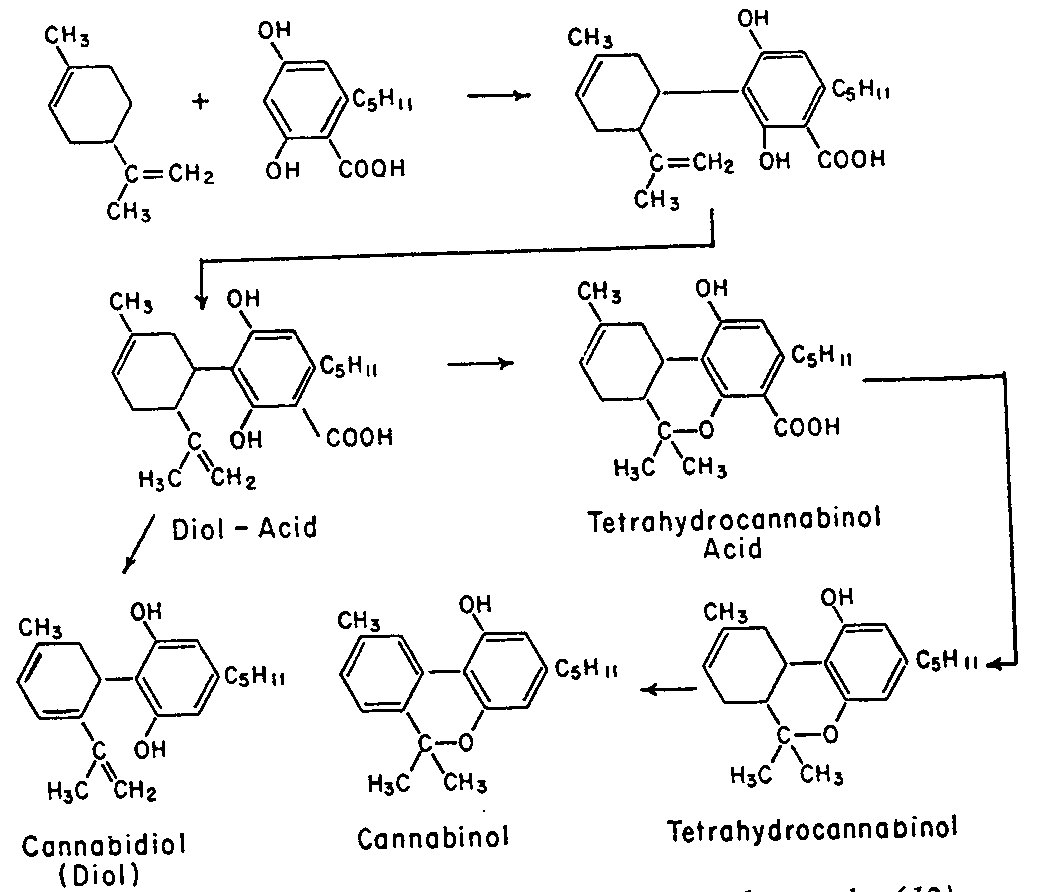
Cannabidiol is insoluble in water but soluble in organic solvents, such as pentane. At room temperature it is a colorless crystalline solid.[47] In strongly basic medium and the presence of air it is oxidized to a quinone.[48] Under acidic conditions it cyclizes to THC.[49] The synthesis of cannabidiol has been accomplished by several research groups.[50][51][52]

http://pubs.rsc.org/en/content/articlelanding/2005/ob/b416943c#!divAbstract
https://www.unodc.org/unodc/en/data-and-analysis/bulletin/bulletin_1964-01-01_4_page005.html

http://pubs.rsc.org/en/content/articlelanding/2005/ob/b416943c#!divAbstract
Biosynthesis
Cannabis produces CBD-carboxylic acid through the same metabolic pathway as THC, until the last step, where CBDA synthase performs catalysis instead of THCA synthase.[53]
Legal status
Cannabidiol is not scheduled by the Convention on Psychotropic Substances.
Cannabidiol is a Schedule II drug in Canada.[54]
Cannabidiol’s legal status in the United States:
The DEA Drug Schedule classifies synthetic THC (Tetrahydrocannabinol) as a schedule III substance (eg Marinol); while the natural marijuana plant is listed as Schedule I. Cannabidiol is not named specifically on the list.[55] However the CSA does mention all natural Phytocannabinoids in Schedule 1 Code 7372, which would include CBD.[55]
Marijuana (along with all of its cannabinoids) is defined by 21 U.S.C. §802(16), which is part of the Controlled Substances Act.[56][57][58] There is an exemption for certain Hemp products produced abroad. Under this exception, what are known as industrial hemp-finished products are legally imported into the United States each year. Hemp finished products which meet the specific definitions including hemp oil which may contain cannabidiol are legal in the United States but aren’t used for getting high.[59]
Some cannabidiol oil is derived from marijuana and therefore contains higher levels of THC.[60] This type of cannabidiol oil would be considered a Schedule I as a result of the THC present.[60]
US patent
In October 2003, U.S. patent #6630507 entitled “Cannabinoids as antioxidants and neuroprotectants” was assigned to “The United States Of America As Represented By The Department Of Health And Human Services.” The patent was filed in April 1999 and listed as the inventors: Aidan J. Hampson, Julius Axelrod, and Maurizio Grimaldi, who all held positions at the National Institute of Mental Health (NIMH) in Bethesda, MD, which is part of the National Institutes of Health (NIH), an agency of the United States Department of Health and Human Services (HHS). The patent mentions cannabidiol’s ability as an antiepileptic, to lower intraocular pressure in the treatment of glaucoma, lack of toxicity or serious side effects in large acute doses, its neuroprotectant properties, its ability to prevent neurotoxicity mediated by NMDA, AMPA, or kainate receptors; its ability to attenuate glutamate toxicity, its ability to protect against cellular damage, its ability to protect brains from ischemic damage, its anxiolytic effect, and its superior antioxidant activity which can be used in the prophylaxis and treatment of oxidation associated diseases.[61]
| “ | “Oxidative associated diseases include, without limitation, free radical associated diseases, such as ischemia, ischemic reperfusion injury, inflammatory diseases, systemic lupus erythematosus, myocardial ischemia or infarction, cerebrovascular accidents (such as a thromboembolic or hemorrhagic stroke) that can lead to ischemia or an infarct in the brain, operative ischemia, traumatic hemorrhage (for example a hypovolemic stroke that can lead to CNS hypoxia or anoxia), spinal cord trauma, Down’s syndrome, Crohn’s disease, autoimmune diseases (e.g. rheumatoid arthritis or diabetes), cataract formation, uveitis, emphysema, gastric ulcers, oxygen toxicity, neoplasia, undesired cellular apoptosis, radiation sickness, and others. The present invention is believed to be particularly beneficial in the treatment of oxidative associated diseases of the CNS, because of the ability of the cannabinoids to cross the blood brain barrier and exert their antioxidant effects in the brain. In particular embodiments, the pharmaceutical composition of the present invention is used for preventing, arresting, or treating neurological damage in Parkinson’s disease, Alzheimer’s disease and HIV dementia; autoimmune neurodegeneration of the type that can occur in encephalitis, and hypoxic or anoxic neuronal damage that can result from apnea, respiratory arrest or cardiac arrest, and anoxia caused by drowning, brain surgery or trauma (such as concussion or spinal cord shock).”[61] | ” |
On November 17, 2011, the Federal Register published that the National Institutes of Health of the United States Department of Health and Human Services was “contemplating the grant of an exclusive patent license to practice the invention embodied in U.S. Patent 6,630,507” to the company KannaLife based in New York, for the development and sale of cannabinoid and cannabidiol based therapeutics for the treatment of hepatic encephalopathy in humans.[62][63][64]
References
- Mechoulam R, Parker LA, Gallily R (November 2002). “Cannabidiol: an overview of some pharmacological aspects”. J Clin Pharmacol (Review) 42 (11 Suppl): 11S–19S.doi:10.1177/0091270002238789. PMID 12412831.
- Scuderi C, Filippis DD, Iuvone T, Blasio A, Steardo A, Esposito G (May 2009). “Cannabidiol in medicine: a review of its therapeutic potential in CNS disorders”.Phytother Res (Review) 23 (5): 597–602. doi:10.1002/ptr.2625. PMID 18844286.
- McPartland JM, Russo EB (2001). “Cannabis and cannabis extracts: greater than the sum of their parts?”. Journal of Cannabis Therapeutics 1(3/4): 103–132. doi:10.1300/J175v01n03_08.
- Borgelt LM, Franson KL, Nussbaum AM, Wang GS (February 2013). “The pharmacologic and clinical effects of medical cannabis”. Pharmacotherapy (Review) 33(2): 195–209. doi:10.1002/phar.1187. PMID 23386598.
- Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimarães FS (December 2012). “Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders”. Philos. Trans. R. Soc. Lond., B, Biol. Sci.(Review) 367 (1607): 3364–78. doi:10.1098/rstb.2011.0389. PMC 3481531.PMID 23108553.
- Wilner, AN (25 March 2014). “Marijuana for Epilepsy: Weighing the Evidence”.Medscape Neurology. WebMD. Retrieved 2 April 2014.
- Russo EB (August 2011). “Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects”. Br. J. Pharmacol. (Review) 163 (7): 1344–64. doi:10.1111/j.1476-5381.2011.01238.x. PMC 3165946. PMID 21749363.
- Liput, D. J.; Hammell, D. C.; Stinchcomb, A. L.; Nixon, K (2013). “Transdermal delivery of cannabidiol attenuates binge alcohol-induced neurodegeneration in a rodent model of an alcohol use disorder”. Pharmacology Biochemistry and Behavior 111: 120–7.doi:10.1016/j.pbb.2013.08.013. PMID 24012796.
- Mechoulam R, Peters M, Murillo-Rodriguez E, Hanus LO (August 2007). “Cannabidiol–recent advances”. Chem. Biodivers. (Review) 4 (8): 1678–92.doi:10.1002/cbdv.200790147. PMID 17712814.
- Iuvone T, Esposito G, De Filippis D, Scuderi C, Steardo L (2009). “Cannabidiol: a promising drug for neurodegenerative disorders?”. CNS Neurosci Ther 15 (1): 65–75.doi:10.1111/j.1755-5949.2008.00065.x. PMID 19228180.
- Zuardi AW, Crippa JA, Hallak JE, Moreira FA, Guimarães FS (April 2006).“Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug”. Braz. J. Med. Biol. Res. (Review) 39 (4): 421–9. doi:10.1590/S0100-879X2006000400001.PMID 16612464.
- Long, L. E.; Malone, D. T.; Taylor, D. A. (2005). “Cannabidiol Reverses MK-801-Induced Disruption of Prepulse Inhibition in Mice”. Neuropsychopharmacology 31 (4): 795–803. doi:10.1038/sj.npp.1300838. PMID 16052245.
- Leweke, FM; Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, Klosterkötter J, Hellmich M and Koethe D. (2012). “Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia”. Translational Psychiatry 2 (3): e94–.doi:10.1038/tp.2012.15. PMC 3316151. PMID 22832859.
- José Alexandre de Souza Crippa, Antonio Waldo Zuardi, Griselda E J Garrido, Lauro Wichert-Ana, Ricardo Guarnieri, Lucas Ferrari, Paulo M Azevedo-Marques, Jaime Eduardo Cecílio Hallak, Philip K McGuire and Geraldo Filho Busatto (October 2003). “Effects of Cannabidiol (CBD) on Regional Cerebral Blood Flow”.Neuropsychopharmacology 29 (2): 417–426. doi:10.1038/sj.npp.1300340.PMID 14583744.
- Daniel Thomas Malone, Dennis Jongejana and David Alan Taylora (August 2009). “Cannabidiol reverses the reduction in social interaction produced by low dose Δ9-tetrahydrocannabinol in rats”. Pharmacology Biochemistry and Behavior 93 (2): 91–96.doi:10.1016/j.pbb.2009.04.010. PMID 19393686.
- Mateus M Bergamaschi, Regina Helena Costa Queiroz, Marcos Hortes Nisihara Chagas, Danielle Chaves Gomes de Oliveira, Bruno Spinosa De Martinis, Flávio Kapczinski, João Quevedo, Rafael Roesler, Nadja Schröder, Antonio E Nardi, Rocio Martín-Santos, Jaime Eduardo Cecílio (May 2011). “Cannabidiol Reduces the Anxiety Induced by Simulated Public Speaking in Treatment-Naïve Social Phobia Patients”.Neuropsychopharmacology 36 (6): 1219–1226. doi:10.1038/npp.2011.6.PMC 3079847. PMID 21307846.
- Crippa JA, Derenusson GN, Ferrari TB, Wichert-Ana L, Duran FL, Martin-Santos R, Simões MV, Bhattacharyya S, Fusar-Poli P, Atakan Z, Santos Filho A, Freitas-Ferrari MC, McGuire PK, Zuardi AW, Busatto GF, Hallak JE. (January 2011). “Neural basis of anxiolytic effects of cannabidiol (CBD) in generalized social anxiety disorder: a preliminary report”. J Psychopharmacol. 25 (1): 121–130.doi:10.1177/0269881110379283. PMID 20829306.
- ElBatsh, MM; Assareh, N; Marsden, CA; Kendall, DA (May 2012). “Anxiogenic-like effects of chronic cannabidiol administration in rats”. Psychopharmacology 221 (2): 239–247. doi:10.1007/s00213-011-2566-z. PMID 22083592.
- Zanelati, T; Biojone, C; Moreira, F; Guimarães, F; Joca, S (January 2010).“Antidepressant-like effects of cannabidiol in mice: possible involvement of 5-HT1A receptors”. British Journal of Pharmacology 159 (1): 122–8. doi:10.1111/j.1476-5381.2009.00521.x. PMC 2823358. PMID 20002102.
- Réus, GZ; Stringari, RB; Ribeiro, KF; Luft, T; Abelaira, HM; Fries, GR; Aguiar, BW; Kapczinski, F; Hallak, JE; Zuardi, AW; Crippa JA; Quevedo, J (October 2011). “Administration of cannabidiol and imipramine induces antidepressant-like effects in the forced swimming test and increases brain-derived neurotrophic factor levels in the rat amygdala”. Acta Neuropsychiatrica 23 (5): 241–248. doi:10.1111/j.1601-5215.2011.00579.x.
- El-Alfy, AT; Ivey, K; Robinson, K; Ahmed, S; Radwan, M; Slade, D; Khan, I; ElSohly, M; Ross, S (June 2010). “Antidepressant-like effect of Δ9-tetrahydrocannabinol and other cannabinoids isolated from Cannabis sativa L”. Pharmacology Biochemistry and Behavior 95 (4): 434–442. doi:10.1016/j.pbb.2010.03.004. PMC 2866040.PMID 20332000.
- Young, Saundra (15 Jan 2014), 3-year-old is focus of medical marijuana battle, CNN, retrieved 2014-01-16
- Cridge BJ, Rosengren RJ (2013). “Critical appraisal of the potential use of cannabinoids in cancer management”. Cancer Manag Res 5: 301–13.doi:10.2147/CMAR.S36105. PMC 3770515. PMID 24039449.
- http://www.dravetfoundation.org/dravet-syndrome/what-is-dravet-syndrome#sthash.jAC0bZ89.dpuf What is Dravet Syndrome?
- Melville, Nancy A. (14 Aug 2013), Seizure Disorders Enter Medical Marijuana Debate, Medscape Medical News, retrieved 2014-01-14
- Gloss D, Vickrey B (13 June 2012). “Cannabinoids for epilepsy”. Cochrane Database Syst Rev (Review) 6: CD009270. doi:10.1002/14651858.CD009270.pub2.PMID 22696383.
- Romney, Lee (13 September 2012). “On the frontier of medical pot to treat boy’s epilepsy”. Los Angeles Times.
- Jump up^ Good, Alastair (26 October 2010). “Growing marijuana that won’t get you high”. The Daily Telegraph (London).
- Jump up^ Sidner, Sara (8 November 2012). Medical marijuana without the high (video). CNN. “An Israeli company has cultivated a new type of medical marijuana.”
- Jump up^ Solon, Olivia (5 July 2012). “Medical Marijuana Without the High”. Wired.com
- Jump up^ Lubell, Maayan (3 July 2012). “What a drag, Israeli firm grows ‘highless’ marijuana”.Reuters. Retrieved 31 Jan 2014.
- Jump up^ Hayakawa, K.; Mishima, K.; Hazekawa, M.; Sano, K.; Irie, K.; Orito, K.; Egawa, T.; Kitamura, Y.; Uchida, N.; Nishimura, R.; Egashira, N.; Iwasaki, K.; Fujiwara, M. (2008). “Cannabidiol potentiates pharmacological effects of Δ9-tetrahydrocannabinol via CB1 receptor-dependent mechanism”. Brain Research 1188: 157–164.doi:10.1016/j.brainres.2007.09.090. PMID 18021759.
- Jump up^ Pertwee, R. G. (2008). “The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin”.British Journal of Pharmacology 153 (2): 199–215. doi:10.1038/sj.bjp.0707442.PMC 2219532. PMID 17828291.
- Jump up^ Ryberg E, Larsson N, Sjögren S, et al. (2007). “The orphan receptor GPR55 is a novel cannabinoid receptor”. British Journal of Pharmacology 152 (7): 1092–101.doi:10.1038/sj.bjp.0707460. PMC 2095107. PMID 17876302.
- Jump up^ Russo EB, Burnett A, Hall B, Parker KK (August 2005). “Agonistic properties of cannabidiol at 5-HT1a receptors”. Neurochemical Research 30 (8): 1037–43.doi:10.1007/s11064-005-6978-1. PMID 16258853.
- ^ Jump up to:a b Resstel LB, Tavares RF, Lisboa SF, Joca SR, Corrêa FM, Guimarães FS (January 2009). “5-HT1A receptors are involved in the cannabidiol-induced attenuation of behavioural and cardiovascular responses to acute restraint stress in rats”. British Journal of Pharmacology 156 (1): 181–8. doi:10.1111/j.1476-5381.2008.00046.x.PMC 2697769. PMID 19133999.
- Jump up^ Campos AC, Guimarães FS (August 2008). “Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats”. Psychopharmacology 199 (2): 223–30. doi:10.1007/s00213-008-1168-x.PMID 18446323.
- Jump up^ Mishima K, Hayakawa K, Abe K, et al. (May 2005). “Cannabidiol prevents cerebral infarction via a serotonergic 5-hydroxytryptamine1A receptor-dependent mechanism”.Stroke; a Journal of Cerebral Circulation 36 (5): 1077–82.doi:10.1161/01.STR.0000163083.59201.34. PMID 15845890.
- Jump up^ Hayakawa K, Mishima K, Nozako M, et al. (March 2007). “Repeated treatment with cannabidiol but not Delta9-tetrahydrocannabinol has a neuroprotective effect without the development of tolerance”. Neuropharmacology 52 (4): 1079–87.doi:10.1016/j.neuropharm.2006.11.005. PMID 17320118.
- Jump up^ Kathmann, Markus; Flau, Karsten; Redmer, Agnes; Tränkle, Christian; Schlicker, Eberhard (2006). “Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors”. Naunyn-Schmiedeberg’s Archives of Pharmacology 372 (5): 354–361.doi:10.1007/s00210-006-0033-x. PMID 16489449.
- Jump up^ Bornheim, LM; Kim, KY; Li, J; Perotti, BY; Benet, LZ (August 1995). “Effect of cannabidiol pretreatment on the kinetics of tetrahydrocannabinol metabolites in mouse brain”. Drug Metabolism and Disposition 23 (8): 825–831. PMID 7493549.
- Jump up^ Klein, C; Karanges, E; Spiro, A; Wong, A; Spencer, J; Huynh, T; Gunasekaran, N; Karl, T; Long, LE; Huang, XF; Liu, K; Arnold, JC; McGregor, IS (November 2011). “Cannabidiol potentiates Δ⁹-tetrahydrocannabinol (THC) behavioural effects and alters THC pharmacokinetics during acute and chronic treatment in adolescent rats”.Psychopharmacology 218 (2): 443–457. doi:10.1007/s00213-011-2342-0.PMID 21667074.
- Jump up^ Hunt, CA; Jones, RT; Herning, RI; Bachman, J (June 1981). “Evidence that Cannabidiol Does Not Significantly Alter the Pharmacokinetics of Tetrahydrocannabinol in Man”.Journal of Pharmacokinetics and Biopharmaceutics 9 (3): 245–260.doi:10.1007/BF01059266. PMID 6270295.
- Jump up^ United States Adopted Names Council: Statement on a nonproprietary name
- Jump up^ “Fact Sheet – Sativex”. Health Canada. Retrieved 16 May 2013.
- Jump up^ GWPharma- Welcome
- Jones PG, Falvello L, Kennard O, Sheldrick GM Mechoulam R (1977). “Cannabidiol”.Acta Cryst. B33 (10): 3211–3214. doi:10.1107/S0567740877010577.
- Mechoulam R, Ben-Zvi Z (1968). “Hashish—XIII On the nature of the beam test”.Tetrahedron 24 (16): 5615–5624. doi:10.1016/0040-4020(68)88159-1. PMID 5732891.
- Gaoni Y, Mechoulam R (1966). “Hashish—VII The isomerization of cannabidiol to tetrahydrocannabinols”. Tetrahedron 22 (4): 1481–1488. doi:10.1016/S0040-4020(01)99446-3.
- Petrzilka T, Haefliger W, Sikemeier C, Ohloff G, Eschenmoser A (1967). “Synthese und Chiralität des (-)-Cannabidiols”. Helv. Chim. Acta 50 (2): 719–723.doi:10.1002/hlca.19670500235. PMID 5587099.
- Gaoni Y, Mechoulam R (1985). “Boron trifluoride etherate on alumuna – a modified Lewis acid reagent. An improved synthesis of cannabidiol”. Tetrahedron Letters 26 (8): 1083–1086. doi:10.1016/S0040-4039(00)98518-6.
- Kobayashi Y, Takeuchi A, Wang YG (2006). “Synthesis of cannabidiols via alkenylation of cyclohexenyl monoacetate”. Org. Lett. 8 (13): 2699–2702.doi:10.1021/ol060692h. PMID 16774235.
- Marks, M.; Tian, L.; Wenger, J.; Omburo, S.; Soto-Fuentes, W.; He, J.; Gang, D.; Weiblen, G.; Dixon, R. (2009). “Identification of candidate genes affecting Δ9-tetrahydrocannabinol biosynthesis in Cannabis sativa”. Journal of Experimental Botany60 (13): 3715–3726. doi:10.1093/jxb/erp210. PMC 2736886. PMID 19581347.
- Controlled Drugs and Substances Act – Schedule II
- CSA Schedule, List of drugs by schedule.
- Definition of marijuana under the Controlled Substances Act.
- Title 21 US Code Controlled Substances Act, text of the CSA.
- Hemp Industries Assn., v. Drug Enforcement Admin., 9th Circuit Court of Appeals case involving industrial hemp.
- Hemp, Many definitions of common terms associated with hemp, including the history of hemp use.
- Cannabidiol: The side of marijuana you don’t know
- US patent 6630507, Hampson, Aidan J.; Axelrod, Julius; Grimaldi, Maurizio, “Cannabinoids as antioxidants and neuroprotectants”, issued 2003-10-07
- “Federal Register | Prospective Grant of Exclusive License: Development of Cannabinoid(s) and Cannabidiol(s) Based Therapeutics To Treat Hepatic Encephalopathy in Humans”. Federalregister.gov. November 17, 2011. Retrieved August 13, 2013.
- “KannaLife Sciences, Inc. Signs Exclusive License Agreement With National Institutes Of Health Office Of Technology Transfer (NIH-OTT)”. thestreet.com. Retrieved 2012-07-09.
- “KannaLife in R&D Collaboration for Cannabinoid-Based Drugs”. Genengnews.com. Retrieved 2013-04-04.
External links
- Project CBD Non-profit educational service dedicated to promoting and publicizing research into the medical utility of cannabidiol.
OLD CUT PASTE
LONDON, Nov. 15, 2013
GW Pharmaceuticals plc (AIM: GWP, Nasdaq: GWPH, “GW”) announced today that the U.S. Food and Drug Administration (FDA) has granted orphan drug designation for Epidiolex(R), our product candidate that contains plant-derived Cannabidiol (CBD) as its active ingredient, for use in treating children with Dravet syndrome, a rare and severe form of infantile-onset, genetic, drug-resistant epilepsy syndrome. Epidiolex is an oral liquid formulation of a highly purified extract of CBD, a non-psychoactive molecule from the cannabis plant. Following receipt of this orphan designation, GW anticipates holding a pre-IND meeting with the FDA in the near future to discuss a development plan for Epidiolex in Dravet syndrome.
Dravet syndrome is a rare pediatric epilepsy syndrome with a distinctive but complex electroclinical presentation. Onset of Dravet syndrome occurs during the first year of life with clonic and tonic-clonic seizures in previously healthy and developmentally normal infants. Prognosis is poor and patients typically develop intellectual disability and life-long ongoing seizures. There are approximately 5,440 patients with Dravet in the United States and an estimated 6,710 Dravet patients in Europe. These figures may be an underestimate as this syndrome is reportedly underdiagnosed.
In addition to GW’s clinical development program for Epidiolex in Dravet syndrome, which is expected to commence in 2014, GW has also made arrangements to enable independent U.S. pediatric epilepsy specialists to treat high need pediatric epilepsy cases with Epidiolex immediately. To date in 2013, a total of seven “expanded access” INDs have been granted by the FDA to U.S. clinicians to allow treatment with Epidiolex of approximately 125 children with epilepsy. These children suffer from Dravet syndrome, Lennox-Gastaut syndrome, and other pediatric epilepsy syndromes. GW is aware of further interest from additional U.S. and ex-U.S. physicians to host similar INDs for Epidiolex. GW expects data generated under these INDs to provide useful observational data during 2014 on the effect of Epidiolex in the treatment of a range of pediatric epilepsy syndromes.
“I, together with many colleagues in the U.S. who specialize in the treatment of childhood epilepsy, very much welcome the opportunity to investigate Epidiolex in the treatment of Dravet syndrome. The FDA’s timely approval of the orphan drug designation for Epidiolex in Dravet syndrome is a key milestone that comes after many years of reported clinical cases that suggest encouraging evidence of efficacy for CBD in this intractable condition,” stated Dr. Orrin Devinsky, Professor of Neurology, Neurosurgery and Psychiatry in New York City. “With GW now making plans to advance Epidiolex through an FDA development program, we have the prospect for the first time of fully understanding the science of CBD in epilepsy with a view to making an appropriately tested and approved prescription medicine available in the future for children who suffer from this debilitating disease.”
“GW is proud to be at the forefront of this important new program to treat children with Dravet Syndrome and potentially other forms of intractable childhood epilepsy. For families in these circumstances, their lives are significantly impacted by constant and often times very severe seizures in children where all options to control these seizures have been exhausted,” stated Dr. Stephen Wright, GW’s R&D Director. “GW intends to advance a full clinical development program for Epidiolex in Dravet syndrome as quickly as possible, whilst at the same time helping families in the short term through supporting physician-led INDs to treat intractable cases. Through its efforts, GW aims to provide the necessary evidence to confirm the promise of CBD in epilepsy and ultimately enabling children to have access to an FDA-approved prescription CBD medicine.”
“This orphan program for Epidiolex in childhood epilepsy is an important corporate strategic priority for GW. Following receipt of today’s orphan designation, GW now intends to commence discussions with the FDA regarding the U.S. regulatory pathway for Epidiolex,” stated Justin Gover, GW’s Chief Executive Officer. “GW intends to pursue this development in-house and retains full commercial rights to Epidiolex.”
About Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition — generally a disease or condition that affects fewer than 200,000 individuals in the U.S. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication.
About GW Pharmaceuticals plc
Founded in 1998, GW is a biopharmaceutical company focused on discovering, developing and commercializing novel therapeutics from its proprietary cannabinoid product platform in a broad range of disease areas. GW commercialized the world’s first plant-derived cannabinoid prescription drug, Sativex(R), which is approved for the treatment of spasticity due to multiple sclerosis in 22 countries. Sativex is also in Phase 3 clinical development as a potential treatment of pain in people with advanced cancer. This Phase 3 program is intended to support the submission of a New Drug Application for Sativex in cancer pain with the U.S. Food and Drug Administration and in other markets around the world. GW has established a world leading position in the development of plant-derived cannabinoid therapeutics and has a deep pipeline of additional clinical-stage cannabinoid product candidates targeting epilepsy (including an orphan pediatric epilepsy program), Type 2 diabetes, ulcerative colitis, glioma and schizophrenia. For further information, please visit http://www.gwpharm.com.
Cannabidiol (CBD) is one of at least 85 cannabinoids found in cannabis.It is a major constituent of the plant, second totetrahydrocannabinol (THC), and represents up to 40% in its extracts. Compared with THC, cannabidiol is not psychoactive in healthy individuals, and is considered to have a wider scope of medical applications than THC, including to epilepsy, multiple sclerosis spasms, anxiety disorders, bipolar disorder,schizophrenia,nausea, convulsion and inflammation, as well as inhibiting cancer cell growth. There is some preclinical evidence from studies in animals that suggests CBD may modestly reduce the clearance of THC from the body by interfering with its metabolism.Cannabidiol has displayed sedative effects in animal tests. Other research indicates that CBD increases alertness. CBD has been shown to reduce growth of aggressive human breast cancer cells in vitro, and to reduce their invasiveness.

DARA BioSciences receives FDA orphan drug designation for KRN5500 (SPK 241) …..Antitumor agent

KRN5500
Antitumor agent
151276-95-8 cas
IUPAC/Chemical name:
(2E,4E)-N-(2-(((2R,3R,4R,5R,6S)-6-((7H-purin-6-yl)amino)-2-((S)-1,2-dihydroxyethyl)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)amino)-2-oxoethyl)tetradeca-2,4-dienamide
C28H43N7O7
Exact Mass: 589.32240
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-((((1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-, (E,E)-
L-glycero-beta-L-manno-Heptopyranosylamine, 4-deoxy-4-(((((2E,4E)-1-oxo-2,4-tetradecadienyl)amino)acetyl)amino)-N-1H-purin-6-yl-

-
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:

- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).
- EP 0525479; JP 1993186494; US 5461036; US 5631238

DARA BioSciences receives FDA orphan drug designation for KRN5500
DARA BioSciences has received orphan drug designation from the US Food and Drug Administration’s (FDA) Office of Orphan Products Development for KRN5500, for treating multiple myeloma
Multiple myeloma is a hematologic cancer or cancer of the blood.
KRN5500 is a non-opioid, non-narcotic compound that is currently being tested in Phase I clinical trial.
Earlier this year, KRN5500 received orphan status to be developed for the parenteral treatment of painful, chronic, chemotherapy-induced peripheral neuropathy (CCIPN) that is refractory to conventional analgesics in patients with cancer.
“We believe this myeloma-specific orphan designation enhances both the viability and the future market opportunity for this valuable pipeline product.”
DARA BioSciences MD, CEO and chief medical officer David J Drutz said: “It is noteworthy in this regard that up to 20% of myeloma patients have intrinsic peripheral neuropathy, an incidence that increases to the range of 75% in patients treated with neurotoxic drugs such as thalidomide or bortezomib.
KRN5500 is a semisynthetic derivative of the nucleoside-like antineoplastic antibiotic spicamycin, originally isolated from the bacterium Streptomyces alanosinicus. KRN 5500 inhibits protein synthesis by interfering with endoplasmic reticulum and Golgi apparatus functions. This agent also induces cell differentiation and caspase-dependent apoptosis.
KRN5500 is available as a solution for intravenous (IV) administration. KRN5500 was discovered in an effort to identify new agents that induced differentiation of myeloid leukemia cells.
Safety and efficacy data from Phase I trials have been leveraged to support DARA Therapeutics’ active IND and ongoing Phase 2a clinical trial. The objective of this Phase 2a feasibility study is to determine the potential of KRN5500 (a spicamycin analogue) to be a breakthrough medicine for the treatment of neuropathic pain in cancer patients.
Four clinical trials have been conducted in cancer patients, including one in Japan and 3 in the United States. Three of these studies are complete; the fourth was closed to patient accrual and treatment in December 2004.
A total of 91 patients with solid tumors have been treated with single IV KRN5500 doses of up to 21 mg/m2 and weekly doses of up to 42 mg/m2. While KRN5500 has not shown anti-cancer efficacy in any trial, its use in pain elimination is encouraging. (source: http://www.darabiosciences.com/krn5500.htm).

Chemical structures of KRN5500 and its known metabolites.
………………..
http://www.google.com/patents/EP0525479A1?cl=en
spk 241
- 6-[4′-N-(N’-trans,trans-2,4-tridecadienylglycyl)spicamynyl-amino]purine,
- (20) SPK241:


Example 52: Preparation of SPK241
-
[0214]To trans-2-dodecenal (4.5 g) dissolved in methylene chloride (80 ml) was added (carbomethoxymethylene)triphenylphosphorane (8.3 g), and the mixture was stirred for 2 hours. The reaction mixture was subjected to chromatography on a silica gel column with eluent systems of n-hexane- ethyl acetate (from 100:1 to 20:1) to give the methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g). Potassium hydroxide (6.5 g) was dissolved in a mixed solvent of ethanol-water (1:1) (100 ml). The methyl ester of trans,trans-2,4-tetradecadienoic acid (5.4 g) was added to the mixture, and the resulting mixture was stirred at 60 °C for 40 minutes. After the reaction mixture was cooled, it was adjusted to the weak acidic range of pH with citric acid and extracted with ethyl acetate. The ethyl acetate layer was dried over anhydrous sodium sulfate and concentrated to give trans,trans-2,4-tetradecadienoic acid (4.4 g). Hereafter, the title compound can be synthesized by the two methods described below.
-
[0215]In the first method, trans,trans-2,4-tetradecadienoic acid (4.3 g) is first dissolved in N,N-dimethylformamide (DMF, 50 ml). Para-nitrophenol (2.67 g) and N,N’-dicyclohexylcarbodiimide (3.9 g) were added to trans,trans-2,4-tetradecadienoic acid solution, and the mixture was stirred for 12 hours. After precipitates produced were removed by filtration and the solvent (DMF) was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of n-hexane-ethyl acetate (from 200:1 to 50:1) to give the active ester of trans,trans-2,4-tetradecadienoic acid (5.1 g). To the active ester (500 mg) dissolved in DMF (30 ml) were added 6-(4′-N-glycyl-spicamynyl-amino)purine hydrochloride (556 mg) and triethylamine (1.2 ml). The mixture was stirred for 12 hours. After the solvent was removed by distillation, the residue was subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 398 mg.
-
[0216]In the second method, trans,trans-2,4-tetradecadienoic acid (99.6 g) was dissolved in thionyl chloride (87 ml), and the mixture was stirred at room temperature. The excessive thionyl chloride was removed by distillation to give trans,trans-2,4-tetradecadienoic acid chloride (102.0 g). To glycine (66.8 g) dissolved in an aqueous 2N sodium hydroxide solution (540 ml) were added at the same time trans,trans-2,4-tetradecadienoic acid chloride (102.0 g) and 2N sodium hydroxide (270 ml) with 1/10 portions at a 3 minute interval. After the addition was completed, the mixture was warmed to room temperature, stirred for 15 minutes and acidified with concentrated hydrochloric acid (140 ml) under ice-cooling. Precipitates thus produced were collected by filtration and desiccated to give trans,trans-2,4-tetradecadienoyl glycine (75.0 g). To the solution of trans,trans-2,4-tetradecadienoyl glycine (4.7 g) and 6-(4′-N-glycyl-spicamynyl-amino)-purine (5.1 g) in N,N-dimethylformamide (DMF, 60 ml) was added N-hydroxysuccinimide (2.1 g), and the mixture was ice-cooled. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (3.4 g) dissolved in DMF (100 ml) was added dropwise to the mixture. After the addition was completed, the mixture was heated to room temperature and stirred for 12 hours. Water (500 ml) was added to the reaction mixture, and precipitates produced were collected by filtration and desiccated. Sodium methoxide (3.1 g) was added to a suspension of the precipitates in methanol (100 ml), and the mixture was stirred at room temperature, then ice-cooled and acidified by adding dropwise thereto a 10% methanolic hydrochloric acid solution. Precipitates produced were filtered, dried and subjected to chromatography on a silica gel column with eluent systems of chloroform-methanol (from 7:1 to 5:1) to give SPK241 in the yield of 5.00 g.
Physicochemical properties of SPK241
-
[0217]
- (1) Melting point: 182-183 °C,
- (2) Specific rotation [a]0 2S = 0 (c = 0.1, in methanol),
- (3) Elementary analysis:
- (4) FD mass spectrum (m/z): 590 (M + H) , C28 H4 3 N707
- (5) Infrared spectrum (KBr disc): 3250 cm-1, 1650 cm-1, 1620 cm-1,
- (6) Proton nuclear magnetic resonance spectrum (500 MHz, in CD30D) δH: 0.89 (3H, t, J = 7.3 Hz), 1.20-1.50 (14H, m), 2.18 (2H, dt, J = 7.3, 7.3 Hz), 3.6-3.8 (5H, m), 3.95 (1 H, d, J = 16.3 Hz), 3.98 (1H, d, J = 16.3 Hz), 4.00 (1H, dd, J = <1, 2.9 Hz), 4.15 (1H, dd, J = 10.8, 10.8 Hz), 5.66 (1 H, brs), 5.98 (1 H, d, J = 15.7 Hz), 6.12 (1 H, dt, J = 7.3, 15.7 Hz), 6.22 (1 H, dd, J = 10.0, 15.7 Hz), 7.17 (1 H, dd, J = 10.0, 15.7 Hz), 8.15 (1 H, s), 8.30 (1 H, s).

| DE3407979A1 * | Mar 3, 1984 | Sep 6, 1984 | Kirin Brewery | Spicamycin sowie verfahren zu seiner herstellung |
| JPS59161396A | Title not available | |||
| US3155647 | Jul 24, 1963 | Nov 3, 1964 | Olin Mathieson | Septaciding and derivatives thereof |
| WO1990015811A1 | Jun 14, 1990 | Dec 27, 1990 | Kirin Brewery | Spicamycin x and its use |
| EP1328236A2 * | Sep 20, 2001 | Jul 23, 2003 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| EP2305264A1 * | Sep 20, 2001 | Apr 6, 2011 | The General Hospital Corporation | Spicamycin derivatives for use in decreasing or preventing pain |
| EP2349285A2 * | Oct 9, 2009 | Aug 3, 2011 | Dara Biosciences, Inc. | Methods for treating or preventing pain using spicamycin derivatives |
| EP2597082A1 | Nov 24, 2011 | May 29, 2013 | Symrise AG | Compounds for masking an unpleasant taste |
| US5905069 * | Jan 26, 1998 | May 18, 1999 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin or derivatives thereof |
| US7196071 | Sep 20, 2001 | Mar 27, 2007 | The General Hospital Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
| US7375094 | Mar 15, 2007 | May 20, 2008 | The General Hospital Corporation | Produced via Streptomyces; antitumor agents; time-release agents; for opiod-resistant pain; drug screening |
| US7632825 | Apr 30, 2008 | Dec 15, 2009 | Bayer Pharmaceuticals Corporation | Methods of decreasing or preventing pain using spicamycin derivatives |
|
References 1: Mizumura Y. [Spicamycin derivative]. Nippon Rinsho. 2006 Feb;64(2):322-8. Review. Japanese. PubMed PMID: 16454188. 2: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2004 Apr;26(3):211-44. PubMed PMID: 15148527. 3: Borsook D, Edwards AD. Antineuropathic effects of the antibiotic derivative spicamycin KRN5500. Pain Med. 2004 Mar;5(1):104-8. PubMed PMID: 14996243. 4: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Dec;25(10):831-55. PubMed PMID: 14735233. 5: Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2003 Nov;25(9):747-71. PubMed PMID: 14685303. 6: Supko JG, Eder JP Jr, Ryan DP, Seiden MV, Lynch TJ, Amrein PC, Kufe DW, Clark JW. Phase I clinical trial and pharmacokinetic study of the spicamycin analog KRN5500 administered as a 1-hour intravenous infusion for five consecutive days to patients with refractory solid tumors. Clin Cancer Res. 2003 Nov 1;9(14):5178-86. PubMed PMID: 14613997. 7: Yamamoto N, Tamura T, Kamiya Y, Ono H, Kondoh H, Shirao K, Matsumura Y, Tanigawara Y, Shimada Y. Phase I and pharmacokinetic study of KRN5500, a spicamycin derivative, for patients with advanced solid tumors. Jpn J Clin Oncol. 2003 Jun;33(6):302-8. PubMed PMID: 12913085. 8: Kobierski LA, Abdi S, DiLorenzo L, Feroz N, Borsook D. A single intravenous injection of KRN5500 (antibiotic spicamycin) produces long-term decreases in multiple sensory hypersensitivities in neuropathic pain. Anesth Analg. 2003 Jul;97(1):174-82, table of contents. PubMed PMID: 12818962. 9: Gadgeel SM, Boinpally RR, Heilbrun LK, Wozniak A, Jain V, Redman B, Zalupski M, Wiegand R, Parchment R, LoRusso PM. A phase I clinical trial of spicamycin derivative KRN5500 (NSC 650426) using a phase I accelerated titration “2B” design. Invest New Drugs. 2003 Feb;21(1):63-74. PubMed PMID: 12795531. 10: Byrd JC, Lucas DM, Mone AP, Kitner JB, Drabick JJ, Grever MR. KRN5500: a novel therapeutic agent with in vitro activity against human B-cell chronic lymphocytic leukemia cells mediates cytotoxicity via the intrinsic pathway of apoptosis. Blood. 2003 Jun 1;101(11):4547-50. Epub 2003 Feb 20. PubMed PMID: 12595316. |
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US
 GADOBUTROL
GADOBUTROL
gadolinium(III) 2,2′,2”-(10-((2R,3S)-1,3,4-trihydroxybutan-2-yl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate
Gadobutrol, SH-L-562, Gadovist,138071-82-6
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US.
The agency has approved the new indication for Gadavist injection for intravenous use with magnetic resonance imaging of the breast to assess the presence and extent of malignant breast disease.
Approval is based on priority review of two Phase III studies with identical design (GEMMA-1 and GEMMA-2).
Bayer HealthCare’s Gadavist (gadobutrol)
Bayer’s Gadavist injection cleared for breast cancer evaluation
UPDATE……. Gadoteridol 279.3 mg/ml for injection , CDSCO INDIA 29.07.2021
For intravenous use in magnetic
reasonance imaging (MRI) in adults and
pediatric patients over 2 years of age for
whole body MRI including the head, neck,
liver, breast, musculoskeletal system and
soft tissue pathologies
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US.
GADOBUTROL
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Licence data | US FDA:link |
| Pregnancy cat. | C (US) |
| Legal status | POM (UK) ℞-only (US) |
| Routes | IV |
| Identifiers | |
| CAS number | 138071-82-6 |
| ATC code | V08CA09 |
| PubChem | CID 72057 |
| DrugBank | DB06703 |
| UNII | 1BJ477IO2L |
| KEGG | D07420 |
| Chemical data | |
| Formula | C18H31GdN4O9 |
| Mol. mass | 604.710 g/mol |
………………………..
Gadobutrol (INN) (Gd-DO3A-butrol) is a gadolinium-based MRI contrast agent (GBCA).
It received marketing approval in Canada[1] and in the United States.[2][3][4]
As of 2007, it was the only GBCA approved at 1.0 molar concentrations.[5]
Gadobutrol is marketed by Bayer Schering Pharma as Gadovist, and by Bayer HealthCare Pharmaceuticals as Gadavist.[6]




 WORLDCUP FOOTBALL WEEK 2014 BRAZIL
WORLDCUP FOOTBALL WEEK 2014 BRAZIL
……………………………………………….
http://www.google.com/patents/EP0988294B1?cl=en
-
This type of complexes with metal ions, in particular with paramagnetic metal ions; is used for the preparation of non-ionic contrast agents for the diagnostic technique known as magnetic resonance (MRI, Magnetic Resonance Imaging), among which are ProHance(R) (Gadoteridol, gadolinium complex of 10-(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid), and Gadobutrol (gadolinium complex of [10-[2,3-dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid).

-
[0003]Two different synthetic approaches are described in literature for the preparation of this kind of complexes, said approaches differing in the strategy taken to discriminate one of the four nitrogen atoms: the first one (Dischino et al., Inorg. Chem., 1991, 30, 1265 or EP 448191, EP 292689, EP 255471) is based on the selective protection of one of the nitrogen atoms by formation of the compound of formula (III), 5H,9bH-2a,4a,7-tetraazacycloocta[cd]pentalene, and on the subsequent hydrolysis to compound of formula (IV), 1-formyl-1,4,7,10-tetraazacyclododecane, followed by the carboxymethylation of the still free nitrogen atoms and by the deprotection and alkylation of the fourth nitrogen atom, according to scheme 1.

-
[0004]The step from 1,4,7,10-tetraazacyclododecane disulfate (a commercially available product) to compound (III) is effected according to the conventional method disclosed in US 4,085,106, followed by formation of the compound of formula (IV) in water-alcohol medium.
-
[0005]This intermediate is subsequently tricarboxymethylated with tert-butyl bromoacetate (TBBA) in dimethylformamide at 2.5°C and then treated with a toluene-sodium hydroxide diphasic mixture to give the compound of formula (V), 10-formyl-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic, tris(1,1-dimethylethyl) ester, which is subsequently hydrolysed to compound of formula (II) in acidic solution.
-
[0006]In the process described in WO 93/24469 for the synthesis of Gadobutrol, at first one of the nitrogen atoms is alkylated in conditions such as to minimize the formation of polyalkylated derivatives, then the monoalkylderivative is purified and carboxymethylated, according to scheme 2.

-
[0007]The alkylation of 1,4,7,1,0-tetraazacyclododecane with the epoxide of formula (VI), 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]octane, is carried out in anhydrous n-BuOH under reflux and the reaction mixture is extracted with water, evaporated to dryness and the residue is subsequently diluted with water and extracted with methylene chloride.
-
[0008]The aqueous phase containing the mono-alkylated product (65% yield in Example 7 which reports the procedure for the preparation of 5 kg of Gadobutrol) is directly carboxymethylated at 70°C with chloroacetic acid, keeping pH 9.5 by addition of NaOH. The reaction mixture is adjusted to pH 1, concentrated to dryness and dissolved in methanol to remove the undissolved salts. The filtrate is then concentrated under vacuum, dissolved in water, and loaded onto a cation exchanger in the H+ form to fix the product. The subsequent elution with ammonia displaces the desired product, which is concentrated to small volume and subsequently complexed with gadolinium oxide according to conventional methods, and the resulting complex is purified by means of ion exchange resins. The overall yield is 42%.
-
[0009]Although the first of these two processes could theoretically provide a higher yield, in that all the single steps (protection, carboxymethylation and deprotection) are highly selective, the complexity of the operations required to remove salts and solvents and to purify the reaction intermediates makes such theoretical advantage ineffective: the overall yield is in fact, in the case of Gadoteridol, slightly higher than 37%.
-
[0010]The preparation of Gadobutrol according to the alternative process (WO 93/24469) provides a markedly better yield (72%) only on laboratory scale (example 2): example 7 (represented in the above Scheme 2) actually evidences that, when scaling-up, the yield of this process also remarkably decreases (42%).
-
[0011]In addition to the drawback of an about 40% yield, both processes of the prior art are characterized by troublesome operations, which often involve the handling of solids, the use of remarkable amounts of a number of different solvents, some of them having undesirable toxicological or anyway hazardous characteristics.
-
[0012]Moreover, the synthesis described by Dischino makes use of reagents which are extremely toxic, such as tert-butyl bromoacetate, or harmful and dangerous from the reactivity point of view, such as dimethylformamide dimethylacetal.
-
[0013]An alternative to the use of dimethyl formamide dimethylacetal is suggested by J. Am. Chem. Soc. 102(20), 6365-6369 (1980), which discloses the preparation of orthoamides by means of triethyl orthoformate.
-
[0014]EP 0596 586 discloses a process for the preparation of substituted tetraazacyclododecanes, among them compounds of formula (XII), comprising:
- formation of the tricyclo[5.5.1.0] ring;
- alkylation with an epoxide;
- hydrolysis of the 10-formyl substituent;
- reaction with an acetoxy derivative bearing a leaving group at the alpha-position.
-
[0015]Nevertheless, this method requires quite a laborious procedure in order to isolate the product of step b).
-
[0016]It is the object of the present invention a process for the preparation of the complexes of general formula (XII)

wherein
- R1 and R2
- are independently a hydrogen atom, a (C1-C20) alkyl containing 1 to 10 oxygen atoms, or a phenyl, phenyloxy group, which can be unsubstituted or substituted with a (C1-C5) alkyl or hydroxy, (C1-C5) alkoxy, carbamoyl or carboxylic groups,
- Me3+
- is the trivalent ion of a paramagnetic metal;
comprising the steps represented in the following Scheme 3:






-
The process of the present invention keeps the high selectivity typical of the protection/deprotection strategy described by Dischino in the above mentioned paper, while removing all its drawbacks, thus providing for the first time a reproducible industrial process for the preparation of the concerned compounds in high yields and without use of hazardous substances.
-
[0019]The preparation of the gadolinium complex of 10-(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-tri-acetic) acid (Gadoteridol), according to scheme 4, is particularly preferred:

in which the synthetic steps a), b), c), d), e), and f) have the meanings defined above and the epoxide of formula (XI) in step d) is propylene oxide.
-
[0020]The preparation of the gadolinium complex of [10-[2,3-dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic) acid (Gadobutrol), according to the scheme 5, is also preferred.

in which the synthetic steps a), b), c), d), e), and f) have the meanings defined above and the epoxide of formula (XI) in step d) corresponds to the one of formula (VI), defined above.
-
[0021]On the other hand, step a) of the process of the present invention involves the use of triethyl orthoformate in the presence of an acid catalyst, instead of dialkylformamide-dialkylacetal.
-
[0022]Triethyl orthoformate can be added in amounts ranging from 105% to 200% on the stoichiometric value.
-
[0023]The reaction temperature can range from 110 to 150°C and the reaction time from 5 to 24 h.
-
[0024]The catalyst is a carboxylic acid having at least 3 carbon atoms, C3-C18, preferably selected from the group consisting of propionic, butyric and pivalic acids.
-
[0025]Triethyl orthoformate is a less toxic and less expensive product than N,N-dimethylformamide-dimethylacetal and does not involve the formation of harmful, not-condensable gaseous by-products. Moreover, triethyl orthoformate is less reactive than N,N-dimethylformamide-dimethylacetal, which makes it possible to carry out the loading procedures of the reactives as well as the reaction itself in utterly safe conditions even on a large scale, allows to better monitor the progress of the reaction on the basis of such operative parameters as time and temperature, without checking the progress by gas chromatography, and makes dosing the reactive less critical, in that it can be added from the very beginning without causing the formation of undesired by-products: all that rendering the process suitable for the production of compound (III) on the industrial scale in easily reproducible conditions.
-
[0026]The subsequent step b) involves the carboxymethylation of compound (III) in aqueous solution, using a haloacetic acid, to give compound (IX), i.e. the 10-formyl-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid salt with an alkali or alkaline-earth metal, the salts of compound (IX) with sodium, potassium or calcium being most preferred.




Example 2
-
[0065]

-
[0066]The procedure of Example 1 is followed until step C included, to obtain a solution of DO3A trisodium salt.
-
[0067]pH is adjusted to 12.3 with conc. HCl and 57.7 kg (0.4 kmol) of 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]-octane are added. After reaction for 4 h at 40°C and for 8 h at 80°C, the solution is cooled to 50°C, 120 kg of an aqueous solution containing 0.135 kmol of gadolinium trichloride are added. After 1 h the mixture is cooled at 17°C and acidified to pH 1.7 with conc. HCl, keeping this pH for 2 h. The solution is subsequently warmed to 50°C, pH is adjusted to 7 with sodium hydroxide, keeping these conditions for 1 h.
-
[0068]After that, the resulting crude Gadobutrol is purified repeating exactly the same process as in steps E and F of Example 1.

Recovery of the product (Gadobutrol)
-
[0069]The product-rich fraction is then thermally concentrated to a viscous residue and the residue is added with 350 kg of ethanol at 79°C.
-
[0070]The resulting suspension is refluxed for 1 h, then cooled, centrifuged and dried under reduced pressure to obtain 66.0 kg of Gadobutrol (0.109 kmol), HPLC assay 99.5% (A%).
Overall yield: 79.1% -
[0071]The IR and MS spectra are consistent with the indicated structure.

References
- Cheng, KT (2007). “Gadobutrol”. Molecular Imaging and Contrast Agent Database (MICAD) (Bethesda, MD: National Center for Biotechnology Information (NCBI)). PMID 20641787. NBK23589.
- http://bayerimaging.com/products/gadavist/index.php
- “FDA approves imaging agent for central nervous system scans” (Press release). U.S. Food and Drug Administration (FDA). March 15, 2011. Retrieved March 31, 2011.
- “U.S. FDA Approves Bayer’s Gadavist (Gadobutrol) Injection for MRI of the Central Nervous System” (Press release). Bayer HealthCare Pharmaceuticals. March 14, 2011. Retrieved March 31, 2011.
- “Gadobutrol 1.0-molar in Cardiac Magnetic Resonance Imaging (MRI) – Further Enhancing the Capabilities of Contrast-enhanced MRI in Ischaemic and Non-ischaemic Heart Disease?”
- “Gadavist full prescribing information”. Retrieved 2011-03-14.

EffRx Pharmaceuticals receives FDA orphan drug designation for EX404
EffRx Pharmaceuticals has received US Food and Drug Administration (FDA) orphan-drug designation for its proprietary metformin-based product, EX404, for treatment of paediatric polycystic ovary syndrome (PCOS).
Also known as Stein-Leventhal syndrome, PCOS is a heterogeneous disorder of chronic anovulation and hyperandrogenism.
The syndrome is believed to occur due to hormonal imbalance caused by increased levels of androgens and insulin in the body.
EffRx Pharmaceuticals chairman and CEO Christer Rosén said the FDA’s orphan drug designation of EX404 is a significant step forward in the clinical development programme.
For National Women’s Health Week, FDA Resources Help Women Make Informed Health Choices

For National Women’s Health Week, FDA Resources Help Women Make Informed Health Choices
By: Marsha B. Henderson, M.C.R.P. “Ask your mother.” In households throughout the country, women often make decisions about foods and medical products for themselves and their loved ones. As we celebrate National Women’s Health Week (May 11-17), I want to … Continue reading →http://blogs.fda.gov/fdavoice/index.php/2014/05/for-national-womens-health-week-fda-resources-help-women-make-informed-health-choices/?source=govdelivery&utm_medium=email&utm_source=govdelivery

Sanfilippo Syndrome: FDA Orphan Designations For Gene Therapy
SF is a genetic metabolism disorder that prohibits the proper breakdown of the body’s sugar molecules. There are 4 types of MPS III (MPS III A, MPS III B, MPS III C, and MPS III D), each with a deficiency in one of four lysosomal enzymes. The disease first affects the central nervous system, causing severe brain damage, and typically results in hearing loss, vision loss…
View original post 226 more words
The FDA’s Drug Review Process: Ensuring Drugs Are Safe and Effective

How Drugs are Developed and Approved
The mission of FDA’s Center for Drug Evaluation and Research (CDER) is to ensure that drugs marketed in this country are safe and effective. CDER does not test drugs, although the Center’s Office of Testing and Research does conduct limited research in the areas of drug quality, safety, and effectiveness.
CDER is the largest of FDA’s five centers. It has responsibility for both prescription and nonprescription or over-the-counter (OTC) drugs. For more information on CDER activities, including performance of drug reviews, post-marketing risk assessment, and other highlights, please see the CDER Update: Improving Public Health Through Human Drugs The other four FDA centers have responsibility for medical and radiological devices, food, and cosmetics, biologics, and veterinary drugs.
Some companies submit a new drug application (NDA) to introduce a new drug product into the U.S. Market. It is the responsibility of the company seeking to market a drug to test it and submit evidence that it is safe and effective. A team of CDER physicians, statisticians, chemists, pharmacologists, and other scientists reviews the sponsor’s NDA containing the data and proposed labeling.

The section below entitled From Fish to Pharmacies: The Story of a Drug’s Development, illustrates how a drug sponsor can work with FDA’s regulations and guidance information to bring a new drug to market under the NDA process.

From Fish to Pharmacies: A Story of Drug Development
Osteoporosis, a crippling disease marked by a wasting away of bone mass, affects as many as 2 million American, 80 percent of them women, at an expense of $13.8 billion a year, according to the National Osteoporosis Foundation., The disease may be responsible for 5 million fractures of the hip, wrist and spine in people over 50, the foundation says, and may cause 50,000 deaths. Given the pervasiveness of osteoporosis and its cost to society, experts say it is crucial to have therapy alternatives if, for example, a patient can’t tolerate estrogen, the first-line treatment.
Enter the salmon, which, like humans, produces a hormone called calcitonin that helps regulate calcium and decreases bone loss. For osteoporosis patients, taking salmon calcitonin, which is 30 times more potent than that secreted by the human thyroid gland, inhibits the activity of specialized bone cells called osteoclasts that absorb bone tissue. This enables bone to retain more bone mass.
Though the calcitonin in drugs is based chemically on salmon calcitonin, it is now made synthetically in the lab in a form that copies the molecular structure of the fish gland extract. Synthetic calcitonin offers a simpler, more economical way to create large quantities of the product.
FDA approved the first drug based on salmon calcitonin in an injectable. Since then, two more drugs, one injectable and one administered through a nasal spray were approved. An oral version of salmon calcitonin is in clinical trials now. Salmon calcitonin is approved only for postmenopausal women who cannot tolerate estrogen, or for whom estrogen is not an option.
How did the developers of injectable salmon calcitonin journey “from fish to pharmacies?”
After obtaining promising data from laboratory studies, the salmon calcitonin drug developers took the next step and submitted an Investigational New Drug (IND) application to CDER. The IND Web page explains the need for this application, the kind of information the application should include, and the Federal regulations to follow.
Once the IND application is in effect, the drug sponsor of salmon calcitonin could begin their clinical trials. After a sponsor submits an IND application, it must wait 30 days before starting a clinical trial to allow FDA time to review the prospective study. If FDA finds a problem, it can order a “clinical hold” to delay an investigation, or interrupt a clinical trial if problems occur during the study.
Clinical trials are experiments that use human subjects to see whether a drug is effective, and what side effects it may cause. The Running Clinical Trials Webpage provides links to the regulations and guidelines that the clinical investigators of salmon calcitonin must have used to conduct a successful study, and to protect their human subjects.
The salmon calcitonin drug sponsor analyzed the clinical trials data and concluded that enough evidence existed on the drug’s safety and effectiveness to meet FDA’s requirements for marketing approval. The sponsor submitted a New Drug Application (NDA) with full information on manufacturing specifications, stability and bioavailablility data, method of analysis of each of the dosage forms the sponsor intends to market, packaging and labeling for both physician and consumer, and the results of any additional toxicological studies not already submitted in the Investigational New Drug application. The NDA Web page provides resources and guidance on preparing the NDA application, and what to expect during the review process.

New drugs, like other new products, are frequently under patent protection during development. The patent protects the salmon calcitonin sponsor’s investment in the drug’s development by giving them the sole right to sell the drug while the patent is in effect. When the patents or other periods of exclusivity on brand-name drugs expire, manufacturers can apply to the FDA to sell generic versions. TheAbbreviated New Drug Applications (ANDA) for Generic Drug Products Webpageprovides links to guidances, laws, regulations, policies and procedures, plus other resources to assist in preparing and submitting applications.
Bringing Nonprescription Drug Products to the Market Under an OTC Monograph
OTC drugs can be brought to the market following the NDA process as described above or under an OTC monograph. Each OTC drug monograph is a kind of “recipe book” covering acceptable ingredients, doses, formulations, labeling, and, in some cases, testing parameters. OTC drug monographs are continually updated to add additional ingredients and labeling as needed. Products conforming to a monograph may be marketed without FDA pre-approval. The NDA and monograph processes can be used to introduce new ingredients into the OTC marketplace. For example, OTC drug products previously available only by prescription are first approved through the NDA process and their “switch” to OTC status is approved via the NDA process. OTC ingredients marketed overseas can be introduced into the U.S. market via a monograph under a Time and Extent Application (TEA) as described in 21 CFR 330.14. For a more thorough discussion of how OTC drug products are regulated visit FDA laws, regulations and guidances that affect small business. Information is also provided on financial assistance and incentives that are available for drug development.

CDER Small Business and Industry Assistance (CDER SBIA)
Drug sponsors which qualify as small businesses can take advantage of special offices and programs designed to help meet their unique needs. The CDER Small Business and Industry Assistance (CDER SBIA) Webpage provides links to FDA laws, regulations and guidances that affect small business. Information is also provided on financial assistance and incentives that are available for drug development.
| |
The path a drug travels from a lab to your medicine cabinet is usually long, and every drug takes a unique route. Often, a drug is developed to treat a specific disease. An important use of a drug may also be discovered by accident.
For example, Retrovir (zidovudine, also known as AZT) was first studied as an anti-cancer drug in the 1960s with disappointing results. Twenty years later, researchers discovered the drug could treat AIDS, and Food and Drug Administration approved the drug, manufactured by GlaxoSmithKline, for that purpose in 1987.
Most drugs that undergo preclinical (animal) testing never even make it to human testing and review by the FDA. The drugs that do must undergo the agency’s rigorous evaluation process, which scrutinizes everything about the drug–from the design of clinical trials to the severity of side effects to the conditions under which the drug is manufactured.



Investigational New Drug Application (IND)–The pharmaceutical industry sometimes seeks advice from the FDA prior to submission of an IND.
Sponsors–companies, research institutions, and other organizations that take responsibility for developing a drug. They must show the FDA results of preclinical testing in laboratory animals and what they propose to do for human testing. At this stage, the FDA decides whether it is reasonably safe for the company to move forward with testing the drug in humans.

Clinical Trials–Drug studies in humans can begin only after an IND is reviewed by the FDA and a local institutional review board (IRB). The board is a panel of scientists and non-scientists in hospitals and research institutions that oversees clinical research.
IRBs approve the clinical trial protocols, which describe the type of people who may participate in the clinical trial, the schedule of tests and procedures, the medications and dosages to be studied, the length of the study, the study’s objectives, and other details. IRBs make sure the study is acceptable, that participants have given consent and are fully informed of their risks, and that researchers take appropriate steps to protect patients from harm.

Phase 1 studies are usually conducted in healthy volunteers. The goal here is to determine what the drug’s most frequent side effects are and, often, how the drug is metabolized and excreted. The number of subjects typically ranges from 20 to 80.

Phase 2 studies begin if Phase 1 studies don’t reveal unacceptable toxicity. While the emphasis in Phase 1 is on safety, the emphasis in Phase 2 is on effectiveness. This phase aims to obtain preliminary data on whether the drug works in people who have a certain disease or condition. For controlled trials, patients receiving the drug are compared with similar patients receiving a different treatment–usually an inactive substance (placebo), or a different drug. Safety continues to be evaluated, and short-term side effects are studied. Typically, the number of subjects in Phase 2 studies ranges from a few dozen to about 300.

At the end of Phase 2, the FDA and sponsors try to come to an agreement on how large-scale studies in Phase 3 should be done. How often the FDA meets with a sponsor varies, but this is one of two most common meeting points prior to submission of a new drug application. The other most common time is pre-NDA–right before a new drug application is submitted.
Phase 3 studies begin if evidence of effectiveness is shown in Phase 2. These studies gather more information about safety and effectiveness, studying different populations and different dosages and using the drug in combination with other drugs. The number of subjects usually ranges from several hundred to about 3,000 people.

Postmarket requirement and commitment studies are required of or agreed to by a sponsor, and are conducted after the FDA has approved a product for marketing. The FDA uses postmarket requirement and commitment studies to gather additional information about a product’s safety, efficacy, or optimal use.

New Drug Application (NDA)–This is the formal step a drug sponsor takes to ask that the FDA consider approving a new drug for marketing in the United States. An NDA includes all animal and human data and analyses of the data, as well as information about how the drug behaves in the body and how it is manufactured

When an NDA comes in, the FDA has 60 days to decide whether to file it so that it can be reviewed. The FDA can refuse to file an application that is incomplete. For example, some required studies may be missing. In accordance with the Prescription Drug User Fee Act (PDUFA), the FDA’s Center for Drug Evaluation and Research (CDER) expects to review and act on at least 90 percent of NDAs for standard drugs no later than 10 months after the applications are received. The review goal is six months for priority drugs. (See “The Role of User Fees.”)
“It’s the clinical trials that take so long–usually several years,” says Sandra Kweder, M.D., deputy director of the Office of New Drugs in the CDER. “The emphasis on speed for FDA mostly relates to review time and timelines of being able to meet with sponsors during a drug’s development,” she says.
Drug Approval Process Infographic
Drug Review Steps Simplified
- Preclinical (animal) testing.
- An investigational new drug application (IND) outlines what the sponsor of a new drug proposes for human testing in clinical trials.
- Phase 1 studies (typically involve 20 to 80 people).
- Phase 2 studies (typically involve a few dozen to about 300 people).
- Phase 3 studies (typically involve several hundred to about 3,000 people).
- The pre-NDA period, just before a new drug application (NDA) is submitted. A common time for the FDA and drug sponsors to meet.
- Submission of an NDA is the formal step asking the FDA to consider a drug for marketing approval.
- After an NDA is received, the FDA has 60 days to decide whether to file it so it can be reviewed.
- If the FDA files the NDA, an FDA review team is assigned to evaluate the sponsor’s research on the drug’s safety and effectiveness.
- The FDA reviews information that goes on a drug’s professional labeling (information on how to use the drug).
- The FDA inspects the facilities where the drug will be manufactured as part of the approval process.
- FDA reviewers will approve the application or issue a complete response letter.
Supplemental Information About the Drug Approval Process
Reviewing Applications
Though FDA reviewers are involved with a drug’s development throughout the IND stage, the official review time is the length of time it takes to review a new drug application and issue an action letter, an official statement informing a drug sponsor of the agency’s decision.
Once a new drug application is filed, an FDA review team–medical doctors, chemists, statisticians, microbiologists, pharmacologists, and other experts–evaluates whether the studies the sponsor submitted show that the drug is safe and effective for its proposed use. No drug is absolutely safe; all drugs have side effects. “Safe” in this sense means that the benefits of the drug appear to outweigh the known risks.
The review team analyzes study results and looks for possible issues with the application, such as weaknesses of the study design or analyses. Reviewers determine whether they agree with the sponsor’s results and conclusions, or whether they need any additional information to make a decision.
Each reviewer prepares a written evaluation containing conclusions and recommendations about the application. These evaluations are then considered by team leaders, division directors, and office directors, depending on the type of application.
Reviewers receive training that fosters consistency in drug reviews, and good review practices remain a high priority for the agency.
Sometimes, the FDA calls on advisory committees, who provide FDA with independent opinions and recommendations from outside experts on applications to market new drugs, and on FDA policies. Whether an advisory committee is needed depends on many things.
“Some considerations would be if it’s a drug that has significant questions, if it’s the first in its class, or the first for a given indication,” says Mark Goldberger, M.D., a former director of one of CDER’s drug review offices. “Generally, FDA takes the advice of advisory committees, but not always,” he says. “Their role is just that–to advise.”Accelerated Approval
Traditional approval requires that clinical benefit be shown before approval can be granted. Accelerated approval is given to some new drugs for serious and life-threatening illnesses that lack satisfactory treatments. This allows an NDA to be approved before measures of effectiveness that would usually be required for approval are available.
Instead, less traditional measures called surrogate endpoints are used to evaluate effectiveness. These are laboratory findings or signs that may not be a direct measurement of how a patient feels, functions, or survives, but are considered likely to predict benefit. For example, a surrogate endpoint could be the lowering of HIV blood levels for short periods of time with anti-retroviral drugs.
Gleevec (imatinib mesylate), an oral treatment for patients with a life-threatening form of cancer called chronic myeloid leukemia (CML), received accelerated approval. The drug was also approved under the FDA’s orphan drug program, which gives financial incentives to sponsors for manufacturing drugs that treat rare diseases. Gleevec blocks enzymes that play a role in cancer growth. The approval was based on results of three large Phase 2 studies, which showed the drug could substantially reduce the level of cancerous cells in the bone marrow and blood.
Most drugs to treat HIV have been approved under accelerated approval provisions, with the company required to continue its studies after the drug is on the market to confirm that its effects on virus levels are maintained and that it ultimately benefits the patient. Under accelerated approval rules, if studies don’t confirm the initial results, the FDA can withdraw the approval.
Because premarket review can’t catch all potential problems with a drug, the FDA continues to track approved drugs for adverse events through a postmarketing surveillance program.
Bumps in the Road
If the FDA decides that the benefits of a drug outweigh the known risks, the drug will receive approval and can be marketed in the United States. But if there are problems with an NDA or if more information is necessary to make that determination, the FDA may issue a complete response letter.
Common problems include unexpected safety issues that crop up or failure to demonstrate a drug’s effectiveness. A sponsor may need to conduct additional studies–perhaps studies of more people, different types of people, or for a longer period of time.
Manufacturing issues are also among the reasons that approval may be delayed or denied. Drugs must be manufactured in accordance with standards called good manufacturing practices, and the FDA inspects manufacturing facilities before a drug can be approved. If a facility isn’t ready for inspection, approval can be delayed. Any manufacturing deficiencies found need to be corrected before approval.
“Sometimes a company may make a certain amount of a drug for clinical trials. Then when they go to scale up, they may lose a supplier or end up with quality control issues that result in a product of different chemistry,” says Kweder. “Sponsors have to show us that the product that’s going to be marketed is the same product that they tested.”
John Jenkins, M.D., director of CDER’s Office of New Drugs, says, “It’s often a combination of problems that prevent approval.” Close communication with the FDA early on in a drug’s development reduces the chance that an application will have to go through more than one cycle of review, he says. “But it’s no guarantee.”
The FDA outlines the justification for its decision in a complete response letter to the drug sponsor and CDER gives the sponsor a chance to meet with agency officials to discuss the deficiencies. At that point, the sponsor can ask for a hearing, correct any deficiencies and submit new information, or withdraw the application.
The Role of User Fees
Since PDUFA was passed in 1992, more than 1,000 drugs and biologics have come to the market, including new medicines to treat cancer, AIDS, cardiovascular disease, and life-threatening infections. PDUFA has allowed the Food and Drug Administration to bring access to new drugs as fast or faster than anywhere in the world, while maintaining the same thorough review process.
Under PDUFA, drug companies agree to pay fees that boost FDA resources, and the FDA agrees to time goals for its review of new drug applications. Along with supporting increased staff, drug user fees help the FDA upgrade resources in information technology. The agency has moved toward an electronic submission and review environment, now accepting more electronic applications and archiving review documents electronically.
The goals set by PDUFA apply to the review of original new human drug and biological applications, resubmissions of original applications, and supplements to approved applications. The second phase of PDUFA, known as PDUFA II, was reauthorized in 1997 and extended the user fee program through September 2002. PDUFA III, which extended to Sept. 30, 2007, was reauthorized in June 2002.
PDUFA III allowed the FDA to spend some user fees to increase surveillance of the safety of medicines during their first two years on the market, or three years for potentially dangerous medications. It is during this initial period, when new medicines enter into wide use, that the agency is best able to identify and counter adverse side effects that did not appear during the clinical trials.
On September 27, 2007, President Bush signed into law the Food and Drug Administration Amendments Act of 2007 which includes the reauthorization and expansion of the Prescription Drug User Fee Act. The reauthorization of PDUFA will significantly broaden and upgrade the agency’s drug safety program, and facilitate more efficient development of safe and effective new medications for the American public.
In addition to setting time frames for review of applications, PDUFA sets goals to improve communication and sets goals for specific kinds of meetings between the FDA and drug sponsors. It also outlines how fast the FDA must respond to requests from sponsors. Throughout a drug’s development, the FDA advises sponsors on how to study certain classes of drugs, how to submit data, what kind of data are needed, and how clinical trials should be designed.
The Quality of Clinical Data
The Food and Drug Administration relies on data that sponsors submit to decide whether a drug should be approved. To protect the rights and welfare of people in clinical trials, and to verify the quality and integrity of data submitted, the FDA’s Division of Scientific Investigations (DSI) conducts inspections of clinical investigators’ study sites. DSI also reviews the records of institutional review boards to be sure they are fulfilling their role in patient protection.
“FDA investigators compare information that clinical investigators provided to sponsors on case report forms with information in source documents such as medical records and lab results,” says Carolyn Hommel, a consumer safety officer in DSI.
DSI seeks to determine such things as whether the study was conducted according to the investigational plan, whether all adverse events were recorded, and whether the subjects met the inclusion/exclusion criteria outlined in the study protocol.
At the conclusion of each inspection, FDA investigators prepare a report summarizing any deficiencies. In cases where they observe numerous or serious deviations, such as falsification of data, DSI classifies the inspection as “official action indicated” and sends a warning letter or Notice of Initiation of Disqualification Proceedings and Opportunity to Explain (NIDPOE) to the clinical investigator, specifying the deviations that were found.
The NIDPOE begins an administrative process to determine whether the clinical investigator should remain eligible to receive investigational products and conduct clinical studies.
CDER conducts about 300-400 clinical investigator inspections annually. About 3 percent are classified in this “official action indicated” category.
The FDA has established an independent Drug Safety Oversight Board (DSOB) to oversee the management of drug safety issues. The Board meets monthly and has representatives from three FDA Centers and five other federal government agencies. The board’s responsibilities include conducting timely and comprehensive evaluations of emerging drug safety issues, and ensuring that experts–both inside and outside of the FDA–give their perspectives to the agency. The first meeting of the DSOB was held in June 2005.
Once the review is complete, the NDA might be approved or rejected. If the drug is not approved, the applicant is given the reasons why and what information could be provided to make the application acceptable. Sometimes the FDA makes a tentative approval recommendation, requesting that a minor deficiency or labeling issue be corrected before final approval. Once a drug is approved, it can be marketed.
Some approvals contain conditions that must be met after initial marketing, such as conducting additional clinical studies. For example, the FDA might request a postmarketing, or phase 4, study to examine the risks and benefits of the new drug in a different population or to conduct special monitoring in a high-risk population. Alternatively, a phase 4 study might be initiated by the sponsor to assess such issues as the longer term effects of drug exposure, to optimize the dose for marketing, to evaluate the effects in pediatric patients, or to examine the effectiveness of the drug for additional indications. Postmarketing surveillance is important, because even the most well-designed phase 3 studies might not uncover every problem that could become apparent once a product is widely used. Furthermore, the new product might be more widely used by groups that might not have been well studied in the clinical trials, such as elderly patients. A crucial element in this process is that physicians report any untoward complications. The FDA has set up a medical reporting program called Medwatch to track serious adverse events (1-800-FDA-1088). The manufacturer must report adverse drug reactions at quarterly intervals for the first 3 years after approval, including a special report for any serious and unexpected adverse reactions.
Recent Developments in Drug Approval
The Food and Drug Administration Modernization Act of 1997 (FDAMA) extended the use of user fees and focused on streamlining the drug approval process. In 1999, the 35 drugs approved by the FDA were reviewed in an average of 12.6 months, slightly more than the 12-month goal set by PDUFA. This act also increased patient access to experimental drugs and facilitated an accelerated review of important new medications. The law ended the ban on disseminating information to providers about non-FDA-approved uses of medications. A manufacturer can now provide peer-reviewed journal articles about an off-label indication of a product if the company commits to filing a supplemental application to establish the use of the unapproved indication. As part of this process, the company must still conduct its own phase 4 study. As a condition for an accelerated approval, the FDA can require the sponsor to carry out postmarketing studies to confirm a clinical benefit and product safety. Critics contend the 1997 act compromises public safety by lowering the standard of approval. Within a year after the law was passed, several drugs were removed from the market. Among these medications were mibefradil for hypertension, dexfenfluramine for morbid obesity, the antihistamine terfenadine, and bromfenac sodium for pain. More recently, additional drugs including troglitazone were removed from the market. Although the increase in recalls might reflect the dramatic increase in drugs approved and launched, others argue that several safety questions were ignored. Another concern was that many withdrawn drugs were me-too drugs which did not represent a noteworthy advance in therapy. Persons critical of the FDA believe changes in the approval process, such as allowing some new drugs to be approved based on only a single clinical trial, expanded use of accelerated approvals, and the use of surrogate end points, have created a dangerous situation. Proponents of the changes in the approval process argue that there is no evidence of increased risk from the legislative changes, and that these changes improve access to cancer patients and those with debilitating disease who were previously denied critical and lifesaving medications.
New drugs are an important part of modern medicine. Just a few decades ago, a disease such as peptic ulcers was a frequent indication for major surgery. The advent of new pharmacologic treatments has dramatically reduced the serious complications of peptic ulcer disease. Likewise, thanks to many new antiviral medications, the outlook for HIV-infected patients has improved dramatically. It is important that physicians understand the process of approving these new medications. Understanding the process can promote innovation, help physicians assess new products, underline the importance of reporting adverse drug events, and provide physicians with the information to educate patients about participating in a clinical trial.






