
Home » Posts tagged 'fda' (Page 26)
Tag Archives: fda
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GSK’s Votrient meets primary objective in Phase III ovarian cancer trial

pazopanib
GlaxoSmithKline’s (GSK) Votrient (pazopanib) has met the primary objective of a statistically significant improvement in the time to disease progression or death that is the progression-free survival (PFS) against placebo in Phase III ovarian cancer..
Pazopanib (trade name Votrient) is a potent and selective multi-targeted receptortyrosine kinase inhibitor of VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-a/β, and c-kit that blocks tumor growth and inhibits angiogenesis. It has been approved for renal cell carcinoma and soft tissue sarcoma by the U.S. Food and Drug Administration.Pazopanib may also be active in ovarian cancer Pazopanib also appears effective in the treatment of non-small cell lung carcinoma.
Flamel Technologies today announced that the U.S. Food and Drug Administration (FDA) has approved the company’s New Drug Application (NDA) for Bloxiverz (neostigmine methylsulfate)

Neostigmine
Flamel Technologies Announces FDA Approval of Bloxiverz
06/03/2013 — Flamel Technologies today announced that the U.S. Food and Drug Administration (FDA) has approved the company’s New Drug Application (NDA) for Bloxiverz (neostigmine methylsulfate), a drug used intravenously in the operating room for the reversal of the effects of non-depolarizing neuromuscular blocking agents after surgery. Flamel expects to launch Bloxiverz in July 2013 in 0.5 and 1.0 mg/mL strengths
Chemistry
Neostigmine, N,N,N-trimethyl-meta-(dimethylcarbomoyloxy)-phenylammonium methylsulfonate, which can be viewed as a simplified analog of physostigmine, is made by reacting 3-dimethylaminophenol with N-dimethylcarbamoyl chloride, which forms the dimethylcarbamate, and its subsequent alkylation using dimethylsulfate forming the desired compound. ![]()
- J.A. Aeschlimann, U.S. Patent 1,905,990 (1933).
Neostigmine shows notable UV/VIS absorption at 261nm, 267nm, and 225nm.
Neostigmine’s 1H NMR Spectroscopy reveals shifts at: 7.8, 7.7, 7.4, 7.4, 3.8, and 3.1 parts per million. The higher shifts are due to the aromatic hydrogens. The lower shifts at 3.8ppm and 3.1ppm are due to the electronic withdrawing nature of the tertiary and quarterary nitrogen, respectively.
by WORLD DRUG TRACKER
DR ANTHONY CRASTO
FDA Approves Tafinlar (dabrafenib mesylate, GSK 2118436) for Advanced Melanoma

DABRAFENIB
1195765-45-7
Benzenesulfonamide, N-[3-[5-(2-amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-4-thiazolyl]-2-fluorophenyl]-2,6-difluoro-
MW 519.56
MF C23 H20 F3 N5 O2 S2
- Dabarefenib
- Dabrafenib
- GSK 2118436
- Tafinlar
Dabrafenib (trade name Tafinlar) is a drug for the treatment of cancers associated with a mutated version of the gene BRAF. Dabrafenib acts as an inhibitor of the associated enzyme B-Raf, which plays a role in the regulation of cell growth. Dabrafenib has clinical activity with a manageable safety profile in clinical trials of phase 1 and 2 in patients with BRAF(V600)-mutated metastatic melanoma.[1][2]
The Food and Drug Administration approved dabrafenib as a single agent treatment for patients with BRAF V600E mutation-positive advanced melanoma on May 30, 2013.[3] Clinical trial data demonstrated that resistance to dabrafinib and other BRAF inhibitors occurs within 6 to 7 months.[4] To overcome this resistance, the BRAF inhibitor dabrafenib was combined with the MEK inhibitor trametinib.[4] As a result of this research, on January 8, 2014, the FDA approved the combination of dabrafenib and trametinib for the treatment of patients with BRAF V600E/K-mutant metastatic melanoma.[5]

Inhibitor of BRAF(V600) mutants
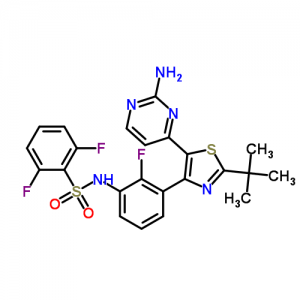
May 29, 2013 — GlaxoSmithKline plc announced today that the U.S. Food and Drug Administration (FDA) has approved Tafinlar (dabrafenib). Tafinlar is indicated as a single-agent oral treatment for unresectable melanoma (melanoma that cannot be removed by surgery) or metastatic melanoma (melanoma which has spread to other parts of the body) in adult patients with BRAF V600E mutation. Tafinlar is not indicated for the treatment of patients with wild-type BRAF melanoma. The mutation must be detected by an FDA-approved test, such as the companion diagnostic assay from bioMérieux S.A., THxID™-BRAF.
Among those with metastatic melanoma, approximately half have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread
Tafinlar is approved for patients with the BRAF V600E mutation, which accounts for approximately 85 percent of all BRAF V600 mutations in metastatic melanoma.
GSK will be making Tafinlar available for prescription no later than in the early third quarter of 2013.
In 2010, GSK entered a collaboration with bioMérieux to develop a companion diagnostic test to detect BRAF V600 (V600E and V600K) gene mutations found in several cancers, including melanoma. bioMérieux has received FDA pre-market approval of THxID™-BRAF. Currently, it is the only FDA-approved test that detects the V600K mutation.
The primary outcome measure was the estimation of the overall intracranial response rate (OIRR) in each cohort. The OIRR for Cohort A was 18 percent (95% CI: 9.7, 28.2). For Cohort B, the OIRR was also 18 percent (95% CI: 9.9, 30.0). The median duration of response was 4.6 months (95% CI: 2.8, Not Reached) and 4.6 months (95% CI: 1.9, 4.6) in Cohort A and Cohort B, respectively.
Melanoma is the most serious and deadly form of skin cancer. According to statistics from the National Cancer Institute, in 2013 there will be an estimated 9,480 deaths resulting from melanoma in the United States. When melanoma spreads in the body, the disease is called metastatic melanoma.Approximately half of all people with metastatic melanoma have a BRAF mutation, which is an abnormal change in a gene that can enable some melanoma tumours to grow and spread. One in two patients worldwide with metastatic melanoma is expected to survive for a year after diagnosis, while in the U.S., the five-year survival rate was 16 percent (2003-2009).The median age of a newly diagnosed metastatic melanoma patient is almost a decade younger than other cancers.
Tafinlar (dabrafenib) is now approved for the treatment of adult patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. Limitation of use: Tafinlar is not recommended for use in patients with wild-type BRAF melanoma.
Tafinlar is not approved or licensed in Europe and may not be approved in other parts of the world for the treatment of patients with BRAF V600 mutation-positive unresectable melanoma or metastatic melanoma.
Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2•CH4O3S and a molecular weight of 615.68. Dabrafenib mesylate has the following chemical structure:
 |
Dabrafenib mesylate is a white to slightly colored solid with three pKas: 6.6, 2.2, and -1.5. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
TAFINLAR (dabrafenib) capsules are supplied as 50-mg and 75-mg capsules for oral administration. Each 50-mg capsule contains 59.25 mg dabrafenib mesylate equivalent to 50 mg of dabrafenib free base. Each 75-mg capsule contains 88.88 mg dabrafenib mesylate equivalent to 75 mg of dabrafenib free base.
The inactive ingredients of TAFINLAR are colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose. Capsule shells contain hypromellose, red iron oxide (E172), and titanium dioxide (E171).
Dabrafenib mesylate
1195768-06-9 cas of mesylate
N-[3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl]-2,6-difluorobenzenesulfonamide;methanesulfonic acid
Chemical structure

………………….
PATENT
http://www.google.com/patents/WO2009137391A2?cl=en
WO 2009137391
Example 58a: Λ/-{3-r5-(2-Amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Following a procedure analogous to the procedure described in Example 51, Step B using Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (196 mg, 0.364 mmol) and ammonia in methanol 7M (8 ml, 56.0 mmol) and heating to 90 0C for 24 h, the title compound, Λ/-{3- [5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide was obtained (94 mg, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 10.83 (s, 1 H), 7.93 (d, J=5.2 Hz, 1 H), 7.55 – 7.70 (m, 1 H), 7.35 –
7.43 (m, 1 H), 7.31 (t, J=6.3 Hz, 1 H), 7.14 – 7.27 (m, 3 H), 6.70 (s, 2 H), 5.79 (d, J=5.13 Hz, 1 H), 1.35 (s, 9 H). MS (ESI): 519.9 [M+H]+.
Example 58b: Λ/-{3-r5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
19.6 mg of Λ/-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (may be prepared in accordance with example 58a) was combined with 500 μl_ of ethyl acetate in a 2-mL vial at room temperature. The slurry was temperature-cycled between 0-400C for 48 hrs. The resulting slurry was allowed to cool to room temperature and the solids were collected by vacuum filtration. The solids were analyzed by Raman, PXRD, DSC/TGA analyses, which indicated a crystal form different from the crystal form resulting from Example 58a, above. Example 58c: Λ/-{3-r5-(2-amino-4-pyrimidinylV2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

Step A: methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate

Methyl 3-amino-2-fluorobenzoate (50 g, 1 eq) was charged to reactor followed by dichloromethane (250 ml_, 5 vol). The contents were stirred and cooled to ~15°C and pyridine (26.2 ml_, 1.1 eq) was added. After addition of the pyridine, the reactor contents were adjusted to ~15°C and the addition of 2,6-diflurorobenzenesulfonyl chloride (39.7 ml_, 1.0 eq) was started via addition funnel. The temperature during addition was kept <25°C. After complete addition, the reactor contents were warmed to 20-250C and held overnight. Ethyl acetate (150 ml.) was added and dichloromethane was removed by distillation. Once distillation was complete, the reaction mixture was then diluted once more with ethyl acetate (5 vol) and concentrated. The reaction mixture was diluted with ethyl acetate (10 vol) and water (4 vol) and the contents heated to 50- 55°C with stirring until all solids dissolve. The layers were settled and separated. The organic layer was diluted with water (4 vol) and the contents heated to 50-55° for 20-30 min. The layers were settled and then separated and the ethyl acetate layer was evaporated under reduced pressure to ~3 volumes. Ethyl Acetate (5 vol.) was added and again evaporated under reduced pressure to ~3 volumes. Cyclohexane (9 vol) was then added to the reactor and the contents were heated to reflux for 30 min then cooled to 0 0C. The solids were filtered and rinsed with cyclohexane (2 x 100 ml_). The solids were air dried overnight to obtain methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2- fluorobenzoate (94.1 g, 91 %).
Step B: Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide

Methyl 3-{[(2,6-difluorophenyl)sulfonyl]amino}-2-fluorobenzoate (490 g, 1 equiv.), prepared generally in accordance with Step A, above, was dissolved in THF (2.45 L, 5 vols) and stirred and cooled to 0-3 0C. 1 M lithium bis(trimethylsilyl)amide in THF (5.25 L, 3.7 equiv.) solution was charged to the reaction mixture followed addition of 2-chloro-4- methylpyrimidine (238 g, 1.3 equiv.) in THF (2.45 L, 5 vols). The reaction was then stirred for 1 hr. The reaction was quenched with 4.5M HCI (3.92 L, 8 vols). The aqueous layer (bootom layer) was removed and discarded. The organic layer was concentrated under reduced pressure to ~2L. IPAC (isopropyl acetate) (2.45L) was added to the reaction mixture which was then concentrated to ~2L. IPAC (0.5L) and MTBE (2.45 L) was added and stirred overnight under N2. The solids were filtered. The solids and mother filtrate added back together and stirred for several hours. The solids were filtered and washed with MTBE (~5 vol). The solids were placed in vacuum oven at 50 0C overnight. The solids were dried in vacuum oven at 30 0C over weekend to obtain Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (479 g, 72%).
Step C: Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide

To a reactor vessel was charged Λ/-{3-[(2-chloro-4-pyrimidinyl)acetyl]-2-fluorophenyl}- 2,6-difluorobenzenesulfonamide (30 g, 1 eq) followed by dichloromethane (300 ml_). The reaction slurry was cooled to ~10°C and N-bromosuccinimide (“NBS”) (12.09 g, 1 eq) was added in 3 approximately equal portions, stirring for 10-15 minutes between each addition. After the final addition of NBS, the reaction mixture was warmed to ~20°C and stirred for 45 min . Water (5 vol) was then added to the reaction vessel and the mixture was stirred and then the layers separated. Water (5 vol) was again added to the dichloromethane layer and the mixture was stirred and the layers separated. The dichloromethane layers were concentrated to -120 ml_. Ethyl acetate (7 vol) was added to the reaction mixture and concentrated to -120 ml_. Dimethylacetamide (270 ml.) was then added to the reaction mixture and cooled to -1O0C. 2,2-Dimethylpropanethioamide (1.3 g, 0.5 eq) in 2 equal portions was added to the reactor contents with stirring for -5 minutes between additions. The reaction was warmed to 20-25 0C. After 45 min, the vessel contents were heated to 75°C and held for 1.75 hours . The reaction mixture was then cooled to 5°C and water (270 ml) was slowly charged keeping the temperature below 300C. Ethyl acetate (4 vol) was then charged and the mixture was stirred and layers separated. Ethyl acetate (7 vol) was again charged to the aqueous layer and the contents were stirred and separated. Ethyl acetate (7 vol) was charged again to the aqueous layer and the contents were stirred and separated. The organic layers were combined and washed with water (4 vol) 4 times and stirred overnight at 20-250C. The organic layers were then concentrated under heat and vacuum to 120 ml_. The vessel contents were then heated to 500C and heptanes (120 ml.) were added slowly. After addition of heptanes, the vessel contents were heated to reflux then cooled to 0°C and held for -2 hrs. The solids were filtered and rinsed with heptanes (2 x 2 vol). The solid product was then dried under vacuum at 300C to obtain Λ/-{3-[5-(2-chloro-4-pyrimidinyl)- 2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (28.8 g, 80%).
Step D: Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide
In 1 gal pressure reactor, a mixture of Λ/-{3-[5-(2-chloro-4-pyrimidinyl)-2-(1 ,1- dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide (120 g) prepared in accordance with Step C, above, and ammonium hydroxide (28-30%, 2.4 L, 20 vol) was heated in the sealed pressure reactor to 98-103 0C and stirred at this temperature for 2 hours. The reaction was cooled slowly to room temperature (20 0C) and stirred overnight. The solids were filtered and washed with minimum amount of the mother liquor and dried under vacuum. The solids were added to a mixture of EtOAc (15 vol)/ water (2 vol) and heated to complete dissolution at 60-70 0C and the aqueous layer was removed and discarded. The EtOAC layer was charged with water (1 vol) and neutralized with aq. HCI to ~pH 5.4-5.5. and added water (1vol). The aqueous layer was removed and discarded at 60-70 0C. The organic layer was washed with water (1 vol) at 60-70 0C and the aqueous layer was removed and discarded. The organic layer was filtered at 60 0C and concentrated to 3 volumes. EtOAc (6 vol) was charged into the mixture and heated and stirred at 72 0C for 10 min , then cooled to 2O0C and stirred overnight. EtOAc was removed via vacuum distillation to concentrate the reaction mixture to ~3 volumes. The reaction mixture was maintained at -65-7O0C for ~30mins. Product crystals having the same crystal form as those prepared in Example 58b (and preparable by the procedure of Example 58b), above, in heptanes slurry were charged. Heptane (9 vol) was slowly added at 65-70 0C. The slurry was stirred at 65-70 0C for 2-3 hours and then cooled slowly to 0-50C. The product was filtered, washed with EtOAc/heptane (3/1 v/v, 4 vol) and dried at 45°C under vacuum to obtain Λ/-{3-[5-(2- amino-4-pyrimidinyl)-2-(1 , 1 -dimethylethyl)-1 ,3-thiazol-4-yl]-2-fluorophenyl}-2,6- difluorobenzenesulfonamide (102.3 g, 88%).
Example 58d: Λ/-{3-r5-(2-amino-4-pyrimidinvn-2-(1.1-dimethylethylV1.3-thiazol-4-yll-2- fluorophenyl}-2,6-difluorobenzenesulfonamide methanesulfonate
 MESYLATE
MESYLATETo a solution of Λ/-{3-[5-(2-amino-4-pyrimidinyl)-2-(1 ,1-dimethylethyl)-1 ,3-thiazol-4-yl]-2- fluorophenyl}-2,6-difluorobenzenesulfonamide (204 mg, 0.393 mmol) in isopropanol (2 ml_), methanesulfonic acid (0.131 ml_, 0.393 mmol) was added and the solution was allowed to stir at room temperature for 3 hours. A white precipitate formed and the slurry was filtered and rinsed with diethyl ether to give the title product as a white crystalline solid (210 mg, 83% yield).
1H NMR (400 MHz, DMSO-d6) δ ppm 10.85 (s, 1 H) 7.92 – 8.05 (m, 1 H) 7.56 – 7.72 (m, 1 H) 6.91 – 7.50 (m, 7 H) 5.83 – 5.98 (m, 1 H) 2.18 – 2.32 (m, 3 H) 1.36 (s, 9 H). MS (ESI): 520.0 [M+H]+.
…………………………………………………………………
PAPER
ACS Medicinal Chemistry Letters (2013), 4(3), 358-362.
http://pubs.acs.org/doi/abs/10.1021/ml4000063
http://pubs.acs.org/doi/suppl/10.1021/ml4000063/suppl_file/ml4000063_si_001.pdf

…………………………………………………………….
Patent
http://www.google.com/patents/WO2014158467A1?cl=en

Dara Phoenix (Dabrafenib) by the British GlaxoSmithKline (GSK) has developed Sisu threonine protein kinase (BRAF) inhibitor, as monotherapy ro ー kinds of clothes capsules for carrying BRAF V600E mutation surgical unresectable melanoma or metastatic melanoma treatment of adult patients, Dara Phoenix mesylate in May 2013 was approved by the US Food and Drug Administration (FDA), and is listed on the United States, the trade name Tafinlar (Da Feina). Since the European Medicines Agency (EMA) Committee for Medicinal Products for human use (CHMP) positive evaluation of Tafinlar, making the drug is expected to become after Roche’s Weiluofeini (Vemurafinib) to enter the European market, following a second BRAF inhibitors.
The chemical name Phoenix Dallas: N- [3- [5- (2- amino-4-pyrimidinyl) -2_ (tert-butyl) ~ ~ thiazol-4-yl] _2_ fluorophenyl] – 2,6_-difluorobenzenesulfonamide.

World Patent No. W02009137391, No. W02011047238 and W02012148588 number reported Dallas and Phoenix and its medicinal value synthesis method of the composition. According to the structural characteristics of Dara Phoenix and its analogues, the synthesis of such substances currently have A, B and C are three routes.

A more common route is the synthetic route, by reaction of 3-amino-2-fluorobenzoate (IX) first and 2,6_-difluorobenzene sulfonyl chloride (III) to amidation reaction occurs sulfonamide intermediate ( X); intermediate (X) with 2-chloro-4-methyl pyrimidine (XI) The condensation reaction occurs under the action of a strong base to give the intermediate (XII); intermediate (XII) to give the intermediate bromo
(XIII); intermediate (XIII) with 2,2_ dimethyl thiopropionamide (VI) to give the cyclized intermediate (XIV); and finally, the intermediate (XIV) by ammonolysis to afford the title compound Dallas Phoenix (I).

Different [0009] B is the first route by reaction of 3-amino-2-fluorobenzoate (IX) amino group protection, and thus condensation, cyclization, and bromo; then be obtained by deprotection of the amino group and the sulfonamide Intermediate (XIV); similarly, the intermediate
(XIV) obtained by ammonolysis target compound Dara Phoenix (I).

c route design features that first aminolysis reaction, and then give the desired product by deprotection and amino sulfonamide reaction. Clearly, this design is suitable for the route of these substituted amino ー aminolysis reaction, and for compounds such as Dallas Phoenix having pyrimidinylamino structure is not applicable. The reason is that if there are two aromatic amino groups will make the final sulfonamide ー reaction step to lose selectivity.

Example IV: the reaction flask was added N- [3- (5- formyl-2-t-butyl-ko -4_ thiazolyl) -2_ fluorophenyl] -2,6_ difluoro benzenesulfonamide (VIII) (5.4g, 11.5mmol), N, N- dimethylformamide dimethyl acetal (DMF-DMA) (2.74g, 23mmol) and xylene 50mL, heated to 140 ° C. About every four hours methanol was distilled out of the resulting reaction system, the reaction takes about 24 hours in total, the end of the reaction was detected by TLC. Cool, add hexane 40mL, have produced a yellow solid, filtered, and dried solids obtained after January nitrate melon (1.36,11.5mmol), sodium hydroxide (0.46g, 11.5mmol) and n-Ding enjoy 5OmL, warmed to 120 ° C, The reaction for 12 inches, TLC the reaction was complete. Cooling, with a crystal precipitated crystallized slowly for 3 inches, and filtered. The filter cake starched water, filtered and dried to yield an off-white solid Dara Phoenix (I) 3.58g, yield 60%.
References
- Gibney, G. T.; Zager, J. S. (2013). “Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies”. Expert Opinion on Drug Metabolism & Toxicology 9 (7): 1. doi:10.1517/17425255.2013.794220. PMID 23621583.
- Huang, T.; Karsy, M.; Zhuge, J.; Zhong, M.; Liu, D. (2013). “B-Raf and the inhibitors: From bench to bedside”. Journal of Hematology & Oncology 6: 30. doi:10.1186/1756-8722-6-30. PMC 3646677. PMID 23617957.
- “GSK melanoma drugs add to tally of U.S. drug approvals”. Reuters. May 30, 2013.
- “Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations” 367 (18). New England Journal of Medicine. November 1, 2012. pp. 1694–703. doi:10.1056/NEJMoa1210093. PMC 3549295. PMID 23020132.
“Dabrafenib/Trametinib Combination Approved for Advanced Melanoma”. OncLive. January 9, 2013.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| N-{3-[5-(2-aminopyrimidin-4-yl)-2-tert-butyl-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzenesulfonamide | |
| Clinical data | |
| Trade names | Tafinlar |
|
|
| Legal status | |
| Identifiers | |
| CAS number | 1195765-45-7 |
| ATC code | L01XE23 |
| PubChem | CID 44462760 |
| ChemSpider | 25948204 |
| ChEBI | CHEBI:75045 |
| ChEMBL | CHEMBL2028663 |
| Chemical data | |
| Formula | C23H20F3N5O2S2 |
| Molecular mass | 519.56 g/mol |
Roche wins FDA approval for novel diabetes Dx
 Roche’s next-generation diabetes diagnostic works on the Cobas Integra 800 analyzer. –Courtesy of Roche
Roche’s next-generation diabetes diagnostic works on the Cobas Integra 800 analyzer. –Courtesy of Roche
Read more: Roche wins FDA approval for novel diabetes Dx – FierceMedicalDevices

The FDA has opened the inside track to Novartis’ experimental lung cancer drug, LDK378, which gained “Breakthrough Therapy” designation

The FDA has opened the inside track to Novartis’ experimental lung cancer drug, which gained “Breakthrough Therapy” designation that speeds the development and review schedules for new treatments. The Swiss drug giant plans to file for approval the drug, now in mid-stage clinical trials, in early 2014. Since clinical development began in 2011, the program has advanced with lightning speed compared with those that take 10 years or so to trial before submitted for approval.
While there are no guarantees of an FDA approval for Novartis’ compound, code-named LDK378, the “breakthrough” tag provides an early nod to the potential of the candidate to improve treatment for patients with metastatic non-small cell lung cancer with anaplastic lymphoma kinase (ALK) mutations.
The “breakthrough” designation is also important because Novartis’ compound and others with the coveted status have a shot to be approved by the FDA without completing all three phases of clinical trials typically required before an approval decision.
Novartis’ LDK378 joined the “breakthrough” club after showing an 80% response rate in patients studied in Phase I trial of 88 subjects with advanced cases of ALK-positive NSCLC. The company has already begun a pair of Phase II studies of the compound for patients with the same kind of ALK-positive cancers, which account for about 3% to 8% of cases of NSCLC. And plans call for kicking off Phase III development of the new drug later this year.
“LDK378 is a strong example of our research approach, which focuses on identifying the underlying cause of disease pathways,” said Alessandro Riva, Novartis’ global head of oncology development, in a statement. “This Breakthrough Therapy designation will allow us to collaborate more closely with the FDA and potentially to expedite the availability of an important new treatment option for patients with ALK+ NSCLC.”
Intravenous formulation of Melphalan, which is in a Phase III trial for use as a conditioning treatment prior to autologous stem cell transplant for patients with multiple myeloma

Mephalan
15 march 2013
Spectrum Pharmaceuticals has licensed an investigational multiple myeloma drug from Ligand Pharmaceuticals in a deal that could be worth over $50 million.
The treatment in question is an intravenous formulation of melphalan, which is in a Phase III trial for use as a conditioning treatment prior to autologous stem cell transplant for patients with multiple myeloma. Spectrum is assuming the responsibility for the trial and hopes to file Captisol-enabled melphalan in the first half of 2014.
The Captisol technology used to reformulate melphalan allows for longer administration durations and slower infusion rates. It has been used with six US Food and Frug Administration-approved products, including Onyx Pharmaceuticals’ multiple myeloma drug Kyprolis (carfilzomib )and Pfizer’s antifungal Vfend (voriconazole).
Melphalan hydrochloride (trade name Alkeran) is a chemotherapy drug belonging to the class of nitrogen mustard alkylating agents.
An alkylating agent adds an alkyl group (CnH2n+1) to DNA. It attaches the alkyl group to the guanine base of DNA, at the number 7 nitrogen atom of the imidazole ring.
Otherwise known as L-Phenylalanine Mustard, or L-PAM, melphalan is a phenylalanine derivative of mechlorethamine.
Uses
It is used to treat multiple myeloma[1] and ovarian cancer, and occasionally malignant melanoma.
The agent was first investigated as a possible drug for use in melanoma. It was not found to be effective, but has been found to be effective in the treatment of myeloma.
Oral or intravenous; dosing varies by purpose and route of administration as well as patient weight.
Melphalan Prescribing Information: Alkeran[2]
Melphalan Patient Information: MedlinePlus[3]
Melphalan Material Safety Data Sheet (MSDS): Sequoia Research Products[4]
- Facon T, Mary JY, Hulin C, et al. (October 2007). “Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomised trial”. Lancet 370 (9594): 1209–18. doi:10.1016/S0140-6736(07)61537-2. PMID 17920916.
- celgene.com
- nlm.nih.gov
- seqchem.com
Biogen submits haemophilia A drug to FDA

Mar 14 2013
Biogen Idec has filed the first long-lasting Factor VIII treatment for haemophilia A with the US Food and Drug Administration.
The US biotech major has submitted recombinant factor VIII Fc fusion protein (rFVIIIFc), the first haemophilia A product candidate “in a new class of long-lasting clotting factor therapies being developed with the goal of reducing the burden of treatment for this condition”. If approved, rFVIIIFc will be the first major advance in haemophilia A treatment in more than two decades, Biogen claims.
The regulatory submission is based on results from A-LONG, the largest Phase III study in haemophilia A to date. Glenn Pierce, Biogen’s head of global medical affairs, noted that in that trial, patients were able to inject rFVIIIFc once-weekly to twice-weekly, “which creates the potential for those currently on prophylactic treatment to reduce injections by 50 to 100 per year”. Moreover, patients currently treating bleeding episodes could potentially dose once per week “and maintain significant protection from bleeding with about the same total number of injections each year they use to treat bleeding episodes today”, he added.
Earlier this month, the FDA accepted for review the company’s BLA for its factor IX candidate, rFIXFc, for use in patients with haemophilia B.
Links
Phase III trial of Merck’s Vytorin passes vital safety test
mar 13 2013
Merck & Co’s stock enjoyed a boost yesterday after it revealed it has been given permission to continue a late-stage trial of its cholesterol buster Vytorin.
The Whitehouse Station, New Jersey-based firm must have a breathed a sigh of relief when the Data Safety Monitoring Board issued a green light for the Phase III IMPROVE for a second time, having found no significant safety concerns raised by the data.
After an earlier planned review of data last year, the Board, rather unusually, said it would undertake a second interim analysis at a later date, which had led to some concerns that there may be issues that could lead to the trial being halted, according to media reports.
However, it seems these fears are unfounded at this point, as the18,000-plus patient study – which is designed to determine whether Vytorin is more effective at reducing the risk of heart attack, stroke and death in patients with heart disease than simvastatin alone – has been cleared to conclude.
The drugmaker said the trial should finish in September next year, and it will no doubt be hoping for a positive outcome to prove the benefits of Vytorin – a combination of the generic simvastatin and the still-patented Zetia (ezetimibe) – and breathe a little new life into its heart franchise.
Citi Investment Research analyst Andrew Baum, however, expressed doubt in a research note the final analysis will show Merck’s drug is more effective than generic competition, according to the Associated Press.
 |
|
|---|---|
 |
|
| Combination of | |
| Ezetimibe | via Niemann-Pick C1-Like 1 protein |
| Simvastatin | Statin HMG-CoA reductase inhibitor |
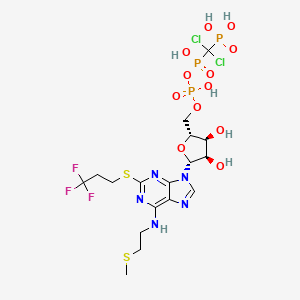
Cangrelor, AR-C69931MX Shows Improvement Over Plavix in Phase III Trial

Cangrelor, AR-C69931MX
[dichloro-[[[(2R,3S,4R,5R)-3,4-dihydroxy-5-[6-(2-methylsulfanylethylamino)-2-(3,3,3-trifluoropropylsulfanyl)purin-9-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]methyl]phosphonic acid
N-[2-(Methylthio)ethyl]-2-[(3,3,3-trifluoropropyl)thio]-5¢-adenylic acid monoanhydride with (dichloromethylene)bis[phosphonic acid]
163706-06-7 cas no
 Cangrelor
CangrelorUPDATE
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Company:
Approval Status:
Approved June 2015
Specific Treatments:
For reducing periprocedural thrombotic events
Therapeutic Areas
Kengreal (cangrelor)
MAR 09, 2013
The Medicines Company said yesterday it will pursue marketing approvals for its anti-clotting drug candidate Cangrelor after it met its primary efficacy endpoint in a Phase III clinical trial of improvement compared with Plavix (clopidogrel).
The intravenous small molecule antiplatelet agent reduced by 22% the likelihood of patients experiencing death, myocardial infarction, ischemia-driven revascularization, or stent thrombosis within 48 hours of taking it—to 4.7% from 5.9% of subjects randomized during CHAMPION PHOENIX. The Phase III trial compared Cangrelor to oral Plavix in 11,145 patients undergoing percutaneous coronary intervention.
Cangrelor also showed a 38% reduction (0.8% compared with 1.4%) over Plavix in the likelihood of patients experiencing the key secondary endpoint, incidence of stent thrombosis at 48 hours.
Cangrelor is designed to prevent platelet activation and aggregation that leads to thrombosis in acute care settings, including in patients undergoing percutaneous coronary intervention. During CHAMPION PHOENIX, Cangrelor made its best showing in patients with Q-wave myocardial infarction (QMI), lowering by 39% (to 0.2% compared with 0.3%) the incidence of QMI. Cangelor’s most disappoint showing was its inability to lower the odds of death compared with Clopidogrel; both drugs showed a likelihood of 0.3%.
“Our next step is to submit for market approvals in the U.S. and Europe. We anticipate submitting these data for a new drug application to the U.S. Food and Drug Administration in the second quarter with findings of prior trials, including the BRIDGE trial in patients awaiting open heart surgery,” Simona Skerjanec, PharmD, senior vp and innovation leader for antiplatelet therapies at The Medicines Company, said in a statement.
Cangrelor is a P2Y12 inhibitor under investigation as an antiplatelet drug[1] for intravenous application. Some P2Y12 inhibitors are used clinically as effective inhibitors of adenosine diphosphate-mediated platelet activation and aggregation.[1] Unlike clopidogrel (Plavix), which is a prodrug, cangrelor is an active drug not requiring metabolic conversion.
Poor interim results led to the abandonment of the two CHAMPION clinical trials in mid 2009.[2] The BRIDGE study, for short term use prior to surgery, continues.[3] The CHAMPION PHOENIX trial was a randomized study of over 11,000 patients published in 2013. It found usefulness of cangrelor in patients getting cardiac stents. Compared with clopidogrel given around the time of stenting, intravenous ADP-receptor blockade with cangrelor significantly reduced the rate of stent thrombosis and myocardial infarction.[4] Reviewers have questioned the methodology of the trial.[5]

One particularly preferred example of a reversible, short-acting P2Y12 inhibitor is cangrelor. Cangrelor is a potent, direct, and reversible antagonist of the platelet P2Y12 receptor. Cangrelor has a half-life of approximately less than 10 minutes, allowing for a return to normal platelet function in a very short period of time upon discontinuation of the drug. By reducing the need for a compound to be metabolized for activity, and by having a relatively short half-life, reversible, short-acting P2Y12 inhibitors are considered “reversible,” meaning that full platelet functionality may return rather quickly as compared to thienopyridines.
The binding of cangrelor to the P2Y12 receptor inhibits platelet activation as well as aggregation when mediated in whole or in part via this receptor. Cangrelor can be derived completely from synthetic materials, and is an analogue of adenosine triphosphate (ATP). ATP is a natural antagonist of the P2Y12 receptor sites and is found in humans.
The chemical structure for cangrelor is depicted below as Formula I.

Cangrelor is clinically well tolerated and safe and has no drug-drug interaction with aspirin, heparin or nitroglycerin. Unlike orally dosed thienopyridines, cangrelor can be administered intravenously and binds directly to P2Y12 receptor sites of platelets. In each of the embodiments of the present invention, the term “cangrelor” encompasses the compound of Formula I as well as tautomeric, enantiomeric and diastereomeric forms thereof, and racemic mixtures thereof, other chemically active forms thereof, and pharmaceutically acceptable salts of these compounds, including a tetrasodium salt. These alternative forms and salts, processes for their production, and pharmaceutical compositions comprising them, are well known in the art and set forth, for example, in U.S. Pat. No. 5,721,219. Additional disclosure relevant to the production and use of cangrelor may be found in U.S. Pat. Nos. 5,955,447, 6,130,208 and 6,114,313, as well as in U.S. Appln. Publication Nos. 2006/0270607 and 2011/0112030.
Invasive procedures means any technique where entry to a body cavity is required or where the normal function of the body is in some way interrupted by a medical procedure and/or treatment that invades (enters) the body, usually by cutting or puncturing the skin and/or by inserting instruments into the body. Invasive procedures can include coronary artery bypass grafting (CABG), orthopedic surgeries, urological surgeries, percutaneous coronary intervention (PCI), other general invasive procedures, such as endarterectomy, renal dialysis, cardio-pulmonary bypass, endoscopic procedures or any medical, surgical, or dental procedure that could result in excessive bleeding or hemorrhage to the patient.
Perioperative means the period of a patient’s invasive procedure which can occur in hospitals, surgical centers or health care providers’ offices. Perioperative includes admission, anesthesia, surgery, to recovery.
Thrombosis is the formation of a blood clot (thrombus) inside a blood vessel obstructing the flow of blood through the circulatory system. When a blood vessel is injured, the body uses platelets and fibrin to form a blood clot to prevent blood loss. Some examples of the types of thrombosis include venous thrombosis which includes deep vein thrombosis, portal vein thrombosis, renal vein thrombosis, jugular vein thrombosis, Budd-Chiari syndrome, Paget-Schroetter disease, cerebral venous sinus thrombosis, cerebral venous sinus thrombosis and arterial thrombosis which includes stroke and myocardial infarction.
The compound cangrelor from the Medicines Company is represented by the structure

 OR
ORRN: 163706-36-3



…………
J. Med. Chem., 1999, 42 (2), pp 213–220
http://pubs.acs.org/doi/full/10.1021/jm981072s
10l (AR-C69931MX)
N6–(2-Methylthioethyl)-2-(3,3,3-trifluoropropylthio)-5‘-adenylic Acid, Monoanhydride withDichloromethylenebis(phosphonic acid) (10l). Prepared as the triammonium salt in 4% yield from 3l: 1H NMR δ(D2O) 8.30 (1H, s, H8), 5.97 (1H, d, J = 5.5 Hz, H1‘), 4.65 (1H, m, H2‘), 4.47 (1H, m, H3‘), 4.28 (1H, m, H4‘), 4.17 (2H, m, H5‘a and H5‘b), 3.67 (br s, NHCH2), 3.21 (2H, t, J = 7.6 Hz, SCH2), 2.72 (2H, t, J = 6.6 Hz, SCH2CH2CF3), 2.58 (2H, m, NCH2CH2), 2.04 (3H, s, SCH3);31P NMR δ(D2O) 8.80 (d, 1P, J = 18.6 Hz, Pγ), 0.42 (dd, 1P, J1 = 18.9 Hz, J2 = 28.9 Hz, Pβ), −9.41 (d, 1P, J = 29.0 Hz, Pα). Anal. (C17H34Cl2F3N8O12P3S2·3H2O) H, N, S; C: calcd, 23.16; found, 23.66.
References
- Cangrelor Attenuates Coated-Platelet Formation
- CHAMPION Trials With Cangrelor Stopped for Lack of Efficacy
- What Cangrelor Failure Means to Medicines
- Effect of Platelet Inhibition with Cangrelor during PCI on Ischemic Events (2013) Bhatt, DL etal. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMoa1300815 (published initially online).
- The Duel between Dual Antiplatelet Therapies (2013) Lange, RA and Hillis, LD. New England Journal of Medicine March 10, 2013 DOI: 10.1056/NEJMe1302504
- 15th European Federation for Medicinal Chemistry International Symposium on Medicinal Chemistry (Sept 6 1998, Edinburgh)1998,:Abst P.281
- Specific P2Y12 purinoceptor antagonist; inhibits ADP-induced platelet aggregation. Prepn: A. H. Ingall et al., WO 9418216 (1994 to Fisons); eidem, US 5721219 (1998 to Astra); and in vivo antithrombotic activity: idem et al., J. Med. Chem. 42, 213 (1999).
- In vivo antithrombotic effects in canine arterial thrombosis: J. Huang et al., J. Pharmacol. Exp. Ther. 295, 492 (2000).
- Mechanism of action study: A. Ishii-Watabe et al., Biochem. Pharmacol. 59, 1345 (2000).
- Clinical safety assessment and evaluation in acute coronary syndromes: R. F. Storey et al., Thromb. Haemostasis 85, 401 (2001); in angina pectoris and non-Q-wave myocardial infarction: F. Jacobsson et al., Clin. Ther. 24, 752 (2002).
- Clinical pharmacodynamics compared with clopidogrel: R. F. Storey et al., Platelets 13, 407 (2002).
- Review of clinical development: S. C. Chattaraj, Curr. Opin. Invest. Drugs2, 250-255 (2001).
- WO2013/103567 A2,
- Journal of Medicinal Chemistry, 1999 , vol. 42, 2 p. 213 – 220
Phase 2, Sarepta Therapeutics, Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy
Eteplirsen, also called AVI-4658, is an experimental drug, currently in clinical trials. It is designed for treatment of some mutations which cause Duchenne muscular dystrophy (DMD), a genetic degenerative muscle disease. Eteplirsen is a product of Sarepta Therapeutics Inc.
s excision of exon 51 during pre-mRNA splicing of the dystrophin RNA transcript. Skipping exon 51 changes the downstream reading frame of dystrophin;[1] giving eteplirsen to a healthy person would result in production of dystrophin mRNA which would not code for functional dystrophin protein but, for DMD patients with particular frameshifting mutations, giving eteplirsen can restore the reading frame of the dystrophin mRNA and result in production of functional (though internally-truncated) dystrophin.[2] Eteplirsen is given by intravenous infusion for systemic treatment of DMD.
Clinical studies
Several clinical trials have been conducted to test eteplirsen, one in the UK involving local injection to the foot,[3][4] one in the UK involving systemic injection at low doses[5][6] and one in the USA at higher systemic doses[7] that progressed to a rollover extension study.[8]
References
- “Exon Skipping Quantification by qRT-PCR in Duchenne Muscular Dystrophy Patients Treated with the Antisense Oligomer Eteplirsen”. Hum Gene Ther Methods.. 17 Oct 2012.
- “Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy.”. Biochim Biophys Acta. 1798 (12): 2296–303.. 17 Feb 2010.
- Gary Roper/Manager Clinical Research Governance Organisation, Imperial College London. “Safety and Efficacy Study of Antisense Oligonucleotides in Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Lancet Neurol. 8 (10): 918–28. 25 Aug 2009.
- Professor Francesco Muntoni, University College of London Institute of Child Health. “Dose-Ranging Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy (DMD) Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- “Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study.”. Lancet. 378 (9791): 595–605. 23 Jul 2011.
- Sarepta Therapeutics. “Efficacy Study of AVI-4658 to Induce Dystrophin Expression in Selected Duchenne Muscular Dystrophy Patients”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
- Sarepta Therapeutics. “Efficacy, Safety, and Tolerability Rollover Study of Eteplirsen in Subjects With Duchenne Muscular Dystrophy”. ClinicalTrials.gov. US Government, NIH. Retrieved 30 October 2012.
