
Home » Posts tagged 'FDA 2022' (Page 3)
Tag Archives: FDA 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PF 04965842, Abrocitinib
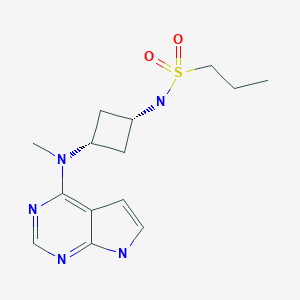

PF-04965842
PF 04965842, Abrocitinib
UNII: 73SM5SF3OR
CAS Number 1622902-68-4, Empirical Formula C14H21N5O2S, Molecular Weight 323.41
N-[cis-3-(Methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)cyclobutyl]-1-propanesulfonamide,
N-((1s,3s)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)cyclobutyl)propane-1-sulfonamide
1-Propanesulfonamide, N-(cis-3-(methyl-7H-pyrrolo(2,3-d)pyrimidin-4-ylamino)cyclobutyl)-
N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide
PHASE 3, for the potential oral treatment of moderate-to-severe atopic dermatitis (AD)
Jak1 tyrosine kinase inhibitor
UPDATE…… JAPAN APPROVED, 2021, 2021/9/27, CIBINQO
ALSO
fda 2022, APPROVALS 2022, 1/14/2022
THE US
In February 2018, the FDA granted Breakthrough Therapy designation for the treatment of patients with moderate-to-severe AD
PHASEIII
In December 2017, a randomized, double-blind, placebo-controlled, parallel-group, phase III trial (NCT03349060; JADE Mono-1; JADE; B7451012; 2017-003651-29) of PF-04965842 began in patients aged 12 years and older (expected n = 375) with moderate-to-severe AD
PRODUCT PATENT
| Pub. No.: | WO/2014/128591 | International Application No.: | PCT/IB2014/058889 | |||
| Publication Date: | 28.08.2014 | International Filing Date: | 11.02.2014 |
EXPIRY Roughly 2034
| form | powder |
| color | white to beige |
| solubility | DMSO: 10 mg/mL, clear |
| storage temp. | room temp |
- Biochem/physiol Actions
-
- PF-04965842 is a Janus Kinase (JAK) inhibitor selective for JAK1 with an IC50value of 29 nM for JAK1 compared to 803 nM for JAK2, >10000 nM for JAK3 and 1250 nM for Tyk2. JAKs mediate cytokine signaling, and are involved in cell proliferation and differentiation. PF-04965842 has been investigated as a possible treatment for psoriasis.
- Originator Pfizer
- Class Skin disorder therapies; Small molecules
- Mechanism of Action Janus kinase 1 inhibitors
Highest Development Phases
- Phase IIIAtopic dermatitis
- DiscontinuedLupus vulgaris; Plaque psoriasis
Most Recent Events
- 08 Mar 2018Phase-III clinical trials in Atopic dermatitis (In children, In adults, In adolescents) in USA (PO) (NCT03422822)
- 14 Feb 2018PF 4965842 receives Breakthrough Therapy status for Atopic dermatitis in USA
- 06 Feb 2018Pfizer plans the phase III JADE EXTEND trial for Atopic Dermatitis (In children, In adults, In adolescents) in March 2018 (PO) (NCT03422822)
This compound was developed by Pfizer for Kinase Phosphatase Biology research. To learn more about Sigma′s partnership with Pfizer and view other authentic, high-quality Pfizer compounds,
PF-04965842 is an oral Janus Kinase 1 inhibitor being investigated for treatment of plaque psoriasis.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified into tyrosine and serine/threonine kinases. Inappropriate kinase activity, arising from mutation, over-expression, or inappropriate regulation, dys-regulation or de-regulation, as well as over- or under-production of growth factors or cytokines has been i mplicated in many diseases, including but not limited to cancer, cardiovascular diseases, allergies, asthma and other respiratory diseases, autoimmune d iseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative disorders such as Alzheimer’s disease. Inappropriate kinase activity triggers a variety of biological cellular responses relating to cell growth, cell differentiation , survival, apoptosis, mitogenesis, cell cycle control, and cel l mobility implicated in the aforementioned and related diseases.
Thus, protein kinases have emerged as an important class of enzymes as targets for therapeutic intervention. In particular, the JAK family of cellular protein tyrosine kinases (JAK1, JAK2, JAK3, and Tyk2) play a central role in cytoki ne signaling (Kisseleva et al., Gene, 2002, 285 , 1; Yamaoka et al. Genome Biology 2004, 5, 253)). Upon binding to their receptors, cytokines activate JAK which then phosphorylate the cytokine receptor, thereby creating docking sites for signaling molecules, notably, members of the signal transducer and activator of transcription (STAT) family that ultimately lead to gene expression. Numerous cytokines are known to activate the JAK family. These cytokines include, the IFN family (IFN-alpha, IFN-beta, IFN-omega, Limitin, IFN-gamma, IL- 10, IL- 19, IL-20, IL-22), the gp 130 family (IL-6, IL- 11, OSM, LIF, CNTF, NNT- 1//SF-3, G-CSF, CT- 1, Leptin, IL- 12 , I L-23), gamma C family (IL-2 , I L-7, TSLP, IL-9, IL- 15 , IL-21, IL-4, I L- 13), IL-3 family (IL-3 , IL-5 , GM-CSF), single chain family (EPO, GH, PRL, TPO), receptor tyrosine kinases (EGF, PDGF, CSF- 1, HGF), and G-protein coupled receptors (ATI).
Abrocitinib, sold under the brand name Cibinqo, is a Janus kinase inhibitor medication used for the treatment of atopic dermatitis (eczema).[2] It was developed by Pfizer.[2]
Medical uses
Abrocitinib is indicated for the treatment of moderate-to-severe atopic dermatitis in adults who are candidates for systemic therapy.[2]
Side effects
The most common adverse effects in studies were upper respiratory tract infection, headache, nausea, and diarrhea.[3]
Pharmacology
Mechanism of action
It is a selective inhibitor of the enzyme janus kinase 1 (JAK1).[3]
Pharmacokinetics
Abrocitinib is quickly absorbed from the gut and generally reaches highest blood plasma concentrations within one hour. Only 1.0 to 4.4% of the dose are found unmetabolized in the urine.[4]
History
- April 2016: initiation of Phase 2b trial
- December 2017: initiation of JADE Mono-1 Phase 3 trial[5]
- May 2018: Results of Phase 2b trial posted
- October 2019: Results of Phase 3 trial presented[6]
- June 2020: Results of second Phase 3 trial published[7]
Society and culture
Legal status
In October 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Cibinqo, intended for the treatment of atopic dermatitis.[8] The applicant for this medicinal product is Pfizer Europe MA EEIG.[8] In December 2021, the European Commission approved abrocitinib for the treatment of atopic dermatitis.[2][9]
In January 2022, the United States Food and Drug Administration (FDA) approved abrocitinib for adults with moderate-to-severe atopic dermatitis.[10]
////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
EU
Click to access cibinqo-epar-public-assessment-report_en.pdf
Introduction
The finished product is presented as immediate release film-coated tablets containing 50 mg, 100 mg
or 200 mg of abrocitinib as active substance.
Other ingredients are:
Tablet core: microcrystalline cellulose (E460i), anhydrous dibasic calcium phosphate (E341ii), sodium
starch glycolate and magnesium stearate (E470b).
Film-coat: hypromellose (E464), titanium dioxide (E171), lactose monohydrate, macrogol (E1521),
triacetin (E1518) and red iron oxide (E172).
The product is available in high-density polyethylene (HDPE) bottles with polypropylene closure or
polyvinylidene chloride (PVDC) blisters with aluminium foil lidding film, as described in section 6.5 of
the SmPC.
The chemical name of abrocitinib is N-((1S,3S)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-
yl)amino)cyclobutyl)propane-1-sulfonamide corresponding to the molecular formula C14H21N5O2S. It
has a relative molecular mass of 323.42 Daltons and the following structure depicted in Figure 1:

The chemical structure of abrocitinib was elucidated by a combination of UV/VIS and IR spectroscopy,
mass spectrometry, NMR spectroscopy and X-ray diffraction.
The active substance is a white to pale-purple or pale pink crystalline powder. It is non-hygroscopic
and its solubility is pH dependent. Abrocitinib is classified as BCS Class II. The impact of particle size
on finished product uniformity of dosage units and dissolution has been studied (see finished product
section). Based on the abrocitinib finished product biopharmaceutics performance, stability, and
manufacturing experience, the active substance particle size specification was established.
Abrocitinib is an achiral molecule, but with 2 stereocentres.
Only one crystalline anhydrous form (Form 1) of abrocitinib has been identified. This form has been the
only form used in all toxicology and clinical studies. Extensive polymorph and hydrate screening have
been conducted to investigate if additional solid forms of abrocitinib could be discovered. Abrocitinib,
Form 1 was the only anhydrous crystalline form identified from these studies. No new anhydrous
polymorphs, hydrates or amorphous solids of abrocitinib were isolated from these screens.
Experiments with 1,4 dioxane and dimethyl sulfoxide yielded solvated forms of abrocitinib. When these
solvated structures were subjected to high temperature, these materials desolvated and converted to
Form 1, free base anhydrous form of abrocitinib. However, these are not relevant since the commercial
crystallisation step does not utilise either of these solvent systems.
It has been confirmed that the manufacturing process consistently yields polymorphic form I. This form
is physically and chemically stable under normal manufacturing and storage conditions as well as
under accelerated conditions. Hence the absence of control of form I is justified.
FDA
U.S. FDA Approves Pfizer’s CIBINQO® (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis
CIBINQO is a once-daily oral treatment with proven efficacy to manage symptoms for adults who have not yet found relief with current options
NEW YORK–(BUSINESS WIRE)– Pfizer Inc. (NYSE: PFE) announced today that the United States (U.S.) Food and Drug Administration (FDA) approved CIBINQO® (abrocitinib), an oral, once-daily, Janus kinase 1 (JAK1) inhibitor, for the treatment of adults living with refractory, moderate-to-severe atopic dermatitis (AD) whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
CIBINQO is approved at the recommended doses of 100 mg and 200 mg, with the 200 mg dose being recommended for patients who are not responding to the 100 mg dose. Additionally, a 50 mg dose was approved to treat moderate-to-severe AD specifically in patients with moderate renal impairment (kidney failure), certain patients receiving treatment with inhibitors of cytochrome P450 (CYP) 2C19, or patients who are known or suspected to be poor metabolizers of CYP2C19. For patients with moderate renal impairment who are not responding to 50 mg once daily, 100 mg once daily may also be prescribed.
“The reality for patients living with chronic inflammatory skin disease such as moderate-to-severe atopic dermatitis is that many experience debilitating symptoms that are not managed by current treatment options. Today’s approval of CIBINQO will provide an important new oral option that could help those who have yet to find relief,” said Jonathan Silverberg, MD, PhD, MPH, Department of Dermatology, The George Washington University School of Medicine and Health Sciences. “In multiple large-scale clinical trials, CIBINQO demonstrated strong efficacy at clearing skin, improving itch, and managing the extent and severity of eczema, offering a benefit-risk profile that supports the use of this treatment in the FDA-approved patient population.”
The FDA approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The safety and efficacy of CIBINQO was evaluated in three randomized, placebo-controlled, Phase 3 trials. Additionally, safety was evaluated through a randomized, placebo-controlled, dose-ranging trial and an ongoing long-term open-label extension trial. Across the trials, CIBINQO demonstrated a consistent safety profile and profound improvements in skin clearance, extent of disease, and severity, as well as rapid improvement in itch after two weeks, for some people living with AD versus placebo. In addition, a higher proportion of subjects treated with CIBINQO in two monotherapy trials achieved improvement in itching at week 12 compared to placebo.
“The FDA’s approval offers hope to the millions of patients across the U.S. who are suffering daily with an immuno-inflammatory condition that can cause intense and persistent itching, pain, discomfort, and distress if left uncontrolled,” said Mike Gladstone, Global President of Pfizer Inflammation & Immunology. “CIBINQO, an efficacious once-daily pill, is a medical breakthrough made possible by Pfizer researchers and the people living with moderate-to-severe atopic dermatitis who participated in our clinical trials.”
“Atopic dermatitis is so much more than just a rash, and it goes beyond the surface of the skin. It’s a chronic condition that can both significantly disrupt patients’ daily lives and negatively impact their emotional well-being,” said Julie Block, President and CEO, National Eczema Association. “We appreciate Pfizer’s commitment to this resilient patient community and eagerly await the positive impact CIBINQO could have on the treatment landscape for moderate-to-severe atopic dermatitis.”
The most common adverse events reported in ≥5% of patients with CIBINQO included nasopharyngitis (12.4% with CIBINQO 100 mg, 8.7% with CIBINQO 200 mg, and 7.9%, with placebo), nausea (6%, 14.5%, and 2.1%, respectively), and headache (6%, 7.8%, and 3.5%, respectively).
The full prescribing information for CIBINQO can be found here. CIBINQO will be made available in the coming weeks.
Additional Details on the CIBINQO Clinical Trial Program
Five clinical trials in the CIBINQO JAK1 Atopic Dermatitis Efficacy and Safety (JADE) global development program were included in the New Drug Application (NDA) to support the FDA approval.
The safety and efficacy of CIBINQO was evaluated in three Phase 3, randomized, placebo-controlled clinical trials. The trials evaluated measures of improvements in skin clearance, itch, disease extent, and severity, including the Investigator Global Assessment (IGA), Eczema Area and Severity Index (EASI), and Peak Pruritus Numerical Ratings Scale (PP-NRS). In each of the trials, over 40% of patients had prior exposure to a systemic therapy:
- JADE MONO-1 and JADE MONO-2: A pair of randomized, double-blind, placebo-controlled trials designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO monotherapy in 778 patients 12 years of age and older with moderate-to-severe AD. The trials assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
- JADE COMPARE: A randomized, double-blind, placebo-controlled trial designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO in 837 adult patients with moderate-to-severe AD on background topical medicated therapy. The trial also included an active control arm with dupilumab, a biologic treatment administered by subcutaneous injection, compared with placebo. The trial assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
Select findings for CIBINQO 100 mg, 200 mg, and placebo follow (*p<0.01 or **p<0.001):
- JADE MONO-1:
- IGA Response Rate (Week 12): 24%*, 44%**, and 8%, respectively
- EASI-75 Response Rate (Week 12): 40%**, 62%**, and 12%, respectively
- JADE MONO-2
- IGA Response Rate (Week 12): 28%**, 38%**, and 9%, respectively
- EASI-75 Response Rate (Week 12): 44%**, 61%**, and 10%, respectively
- JADE COMPARE
- IGA Response Rate (Week 12): 36%**, 47%**, and 14%, respectively
- EASI-75 Response Rate (Week 12): 58%**, 68%**, and 27%, respectively
Safety was additionally evaluated through a randomized dose-ranging trial and a long-term, open-label, extension trial (JADE EXTEND).
U.S. IMPORTANT SAFETY INFORMATION
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, AND THROMBOSIS
Serious Infections
Patients treated with CIBINQO may be at increased risk for developing serious infections that may lead to hospitalization or death. The most frequent serious infections reported with CIBINQO were herpes simplex, herpes zoster, and pneumonia.
If a serious or opportunistic infection develops, discontinue CIBINQO and control the infection.
Reported infections from Janus kinase (JAK) inhibitors used to treat inflammatory conditions:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Test for latent TB before and during therapy; treat latent TB prior to use. Monitor all patients for active TB during treatment, even patients with initial negative, latent TB test.
- Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral (including herpes zoster), and other infections due to opportunistic pathogens.
Avoid use of CIBINQO in patients with an active, serious infection, including localized infections. The risks and benefits of treatment with CIBINQO should be carefully considered prior to initiating therapy in patients with chronic or recurrent infections or those who have resided or traveled in areas of endemic tuberculosis or endemic mycoses.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with CIBINQO, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
Consider yearly screening for patients in highly endemic areas for TB. CIBINQO is not recommended for use in patients with active TB. For patients with a new diagnosis of latent TB or prior untreated latent TB, or for patients with a negative test for latent TB but who are at high risk for TB infection, start preventive therapy for latent TB prior to initiation of CIBINQO.
Viral reactivation, including herpes virus reactivation (eg, herpes zoster, herpes simplex), was reported in clinical studies with CIBINQO. If a patient develops herpes zoster, consider interrupting CIBINQO until the episode resolves. Hepatitis B virus reactivation has been reported in patients receiving JAK inhibitors. Perform viral hepatitis screening and monitoring for reactivation in accordance with clinical guidelines before starting therapy and during therapy with CIBINQO. CIBINQO is not recommended for use in patients with active hepatitis B or hepatitis C.
Mortality
In a large, randomized postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another JAK inhibitor to TNF blocker treatment, a higher rate of all-cause mortality (including sudden cardiovascular death) was observed with the JAK inhibitor. CIBINQO is not approved for use in RA patients.
Malignancies
Malignancies, including non-melanoma skin cancer (NMSC), were reported in patients treated with CIBINQO. Lymphoma and other malignancies have been observed in patients receiving JAK inhibitors used to treat inflammatory conditions. Perform periodic skin examination for patients who are at increased risk for skin cancer. Exposure to sunlight and UV light should be limited by wearing protective clothing and using broad-spectrum sunscreen.
In a large, randomized postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. CIBINQO is not approved for use in RA patients. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Major Adverse Cardiovascular Events
Major adverse cardiovascular events were reported in patients treated with CIBINQO. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients. Patients who are current or past smokers are at additional increased risk. Discontinue CIBINQO in patients that have experienced a myocardial infarction or stroke.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur.
Thrombosis
Deep vein thrombosis (DVT) and pulmonary embolism (PE) have been reported in patients treated with CIBINQO. Thrombosis, including PE, DVT, and arterial thrombosis have been reported in patients receiving JAK inhibitors used to treat inflammatory conditions. Many of these adverse reactions were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of overall thrombosis, DVT, and PE were observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients.
Avoid CIBINQO in patients that may be at increased risk of thrombosis. If symptoms of thrombosis occur, discontinue CIBINQO and treat patients appropriately.
Contraindication
CIBINQO is contraindicated in patients taking antiplatelet therapies, except for low-dose aspirin (≤81 mg daily), during the first 3 months of treatment.
Laboratory Abnormalities
Hematologic Abnormalities: Treatment with CIBINQO was associated with an increased incidence of thrombocytopenia and lymphopenia. Prior to CIBINQO initiation, perform a complete blood count (CBC). CBC evaluations are recommended at 4 weeks after initiation and 4 weeks after dose increase of CIBINQO. Discontinuation of CIBINQO therapy is required for certain laboratory abnormalities.
Lipid Elevations: Dose-dependent increase in blood lipid parameters were reported in patients treated with CIBINQO. Lipid parameters should be assessed approximately 4 weeks following initiation of CIBINQO therapy, and thereafter patients should be managed according to clinical guidelines for hyperlipidemia. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Immunizations
Prior to initiating CIBINQO, complete all age-appropriate vaccinations as recommended by current immunization guidelines, including prophylactic herpes zoster vaccinations. Avoid vaccination with live vaccines immediately prior to, during, and immediately after CIBINQO therapy.
Renal Impairment
Avoid use in patients with severe renal impairment or end stage renal disease, including those on renal replacement therapy.
Hepatic Impairment
Avoid use in patients with severe hepatic impairment.
Adverse Reactions
Most common adverse reactions (≥1%) in subjects receiving 100 mg and 200 mg include: nasopharyngitis, nausea, headache, herpes simplex, increased blood creatinine phosphokinase, dizziness, urinary tract infection, fatigue, acne, vomiting, oropharyngeal pain, influenza, gastroenteritis.
Most common adverse reactions (≥1%) in subjects receiving either 100 mg or 200 mg also include: impetigo, hypertension, contact dermatitis, upper abdominal pain, abdominal discomfort, herpes zoster, and thrombocytopenia.
Use in Pregnancy
Available data from pregnancies reported in clinical trials with CIBINQO are not sufficient to establish a drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Advise females of reproductive potential that CIBINQO may impair fertility.
There will be a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to CIBINQO during pregnancy. Pregnant women exposed to CIBINQO and health care providers are encouraged to call 1-877-311-3770.
Lactation
Advise women not to breastfeed during treatment with CIBINQO and for one day after the last dose.
Indication
CIBINQO is indicated for the treatment of adults with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
Limitations of Use: CIBINQO is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
About CIBINQO® (abrocitinib)
CIBINQO is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD, including interleukin IL-4, IL-13, IL-31, IL-22, and thymic stromal lymphopoietin (TSLP).
In addition to receiving regulatory approval in the U.S., CIBINQO has received marketing authorization in the European Union, Great Britain, Japan, Korea, the United Arab Emirates, Norway, Iceland, and Singapore.
About Atopic Dermatitis
AD is a chronic skin disease characterized by inflammation of the skin and skin barrier defects.i,ii Most people know AD is a skin condition. But many don’t realize it can be caused in part by an abnormal immune response beneath the skin. This dysregulated immune response is thought to contribute to inflammation within the skin and the signs of AD on the surface. Lesions of AD are characterized by erythema (red/pink or discolored skin patches, depending on normal skin color), itching, lichenification (thick/leathery skin), induration (hardening)/papulation (formulation of papules), and oozing/crusting.i,ii
AD is one of the most common inflammatory skin diseases, affecting approximately 5-10% of adults in the U.S.iii,iv Approximately 1 in 3 adults with AD have moderate-to-severe disease.v,vi
About Pfizer Inflammation & Immunology
At Pfizer Inflammation & Immunology, we strive to deliver breakthroughs that enable freedom from day-to-day suffering for people living with autoimmune and chronic inflammatory diseases, which can be debilitating, disfiguring and distressing, dramatically affecting what they can do. With a focus on immuno-inflammatory conditions in Rheumatology, Gastroenterology and Medical Dermatology, our current portfolio of approved medicines and investigational molecules spans multiple action and delivery mechanisms, from topicals to small molecules, biologics and biosimilars. The root cause of many immunological diseases is immuno-inflammation, which requires specifically designed agents. Our differentiated R&D approach resulted in one of the broadest pipelines in the industry, where we purposefully match molecules to diseases where we believe they can make the biggest difference. Building on our decades-long commitment and pioneering science, we continue to advance the standard of care for patients living with immuno-inflammatory diseases and are working hand-in-hand with patients, caregivers and the broader healthcare community on healthcare solutions for the many challenges of managing chronic inflammatory diseases, allowing patients to live their best lives.
Pfizer Inc.: Breakthroughs that Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety, and value in the discovery, development, and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments, and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments, and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
There remains a need for new compounds that effectively and selectively inhibit specific JAK enzymes, and JAK1 in particular, vs. JAK2. JAK1 is a member of the Janus family of protein kinases composed of JAK1, JAK2, JAK3 and TYK2. JAK1 is expressed to various levels in all tissues. Many cytokine receptors signal through pairs of JAK kinases in the following combinations: JAK1/JAK2, JAK1/JAK3, JAK1/TYK2 , JAK2/TYK2 or JAK2/JAK2. JAK1 is the most broadly
paired JAK kinase in this context and is required for signaling by γ-common (IL-2Rγ) cytokine receptors, IL—6 receptor family, Type I, II and III receptor families and IL- 10 receptor family. Animal studies have shown that JAK1 is required for the development, function and homeostasis of the immune system. Modulation of immune activity through inhibition of JAK1 kinase activity can prove useful in the treatment of various immune disorders (Murray, P.J.
J. Immunol., 178, 2623-2629 (2007); Kisseleva, T., et al., Gene, 285 , 1-24 (2002); O’Shea, J . J., et al., Ceil , 109, (suppl .) S121-S131 (2002)) while avoiding JAK2 dependent erythropoietin (EPO) and thrombopoietin (TPO) signaling (Neubauer H., et al., Cell, 93(3), 397-409 (1998);
Parganas E., et al., Cell, 93(3), 385-95 (1998)).

Tofacitinib (1), baricitinib (2), and ruxolitinib (3)
SYNTHESIS 5+1 =6 steps
Main synthesis
Journal of Medicinal Chemistry, 61(3), 1130-1152; 2018

INTERMEDIATE
CN 105732637
ONE STEP

CAS 479633-63-1, 7H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-7-[(4- methylphenyl)sulfonyl]-
![]()
Pfizer Receives Breakthrough Therapy Designation from FDA for PF-04965842, an oral JAK1 Inhibitor, for the Treatment of Patients with Moderate-to-Severe Atopic Dermatitis
Dateline:
Public Company Information:
NEW YORK–(BUSINESS WIRE)–Pfizer Inc. (NYSE:PFE) today announced its once-daily oral Janus kinase 1 (JAK1) inhibitor PF-04965842 received Breakthrough Therapy designation from the U.S. Food and Drug Administration (FDA) for the treatment of patients with moderate-to-severe atopic dermatitis (AD). The Phase 3 program for PF-04965842 initiated in December and is the first trial in the J AK1 A topic D ermatitis E fficacy and Safety (JADE) global development program.
“Achieving Breakthrough Therapy Designation is an important milestone not only for Pfizer but also for patients living with the often devastating impact of moderate-to-severe atopic dermatitis, their providers and caregivers,” said Michael Corbo, Chief Development Officer, Inflammation & Immunology, Pfizer Global Product Development. “We look forward to working closely with the FDA throughout our ongoing Phase 3 development program with the hope of ultimately bringing this important new treatment option to these patients.”
Breakthrough Therapy Designation was initiated as part of the Food and Drug Administration Safety and Innovation Act (FDASIA) signed in 2012. As defined by the FDA, a breakthrough therapy is a drug intended to be used alone or in combination with one or more other drugs to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If a drug is designated as a breakthrough therapy, the FDA will expedite the development and review of such drug.1
About PF-04965842 and Pfizer’s Kinase Inhibitor Leadership
PF-04965842 is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD including interleukin (IL)-4, IL-13, IL-31 and interferon gamma.
Pfizer has established a leading kinase research capability with multiple unique kinase inhibitor therapies in development. As a pioneer in JAK science, the Company is advancing several investigational programs with novel selectivity profiles, which, if successful, could potentially deliver transformative therapies for patients. Pfizer has three additional kinase inhibitors in Phase 2 development across multiple indications:
- PF-06651600: A JAK3 inhibitor under investigation for the treatment of rheumatoid arthritis, ulcerative colitis and alopecia areata
- PF-06700841: A tyrosine kinase 2 (TYK2)/JAK1 inhibitor under investigation for the treatment of psoriasis, ulcerative colitis and alopecia areata
- PF-06650833: An interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor under investigation for the treatment of rheumatoid arthritis
Working together for a healthier world®
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products. Our global portfolio includes medicines and vaccines as well as many of the world’s best-known consumer health care products. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 150 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
DISCLOSURE NOTICE: The information contained in this release is as of February 14, 2018. Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments.
This release contains forward-looking information about PF-04965842 and Pfizer’s ongoing investigational programs in kinase inhibitor therapies, including their potential benefits, that involves substantial risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Risks and uncertainties include, among other things, the uncertainties inherent in research and development, including the ability to meet anticipated clinical trial commencement and completion dates and regulatory submission dates, as well as the possibility of unfavorable clinical trial results, including unfavorable new clinical data and additional analyses of existing data; risks associated with preliminary data; the risk that clinical trial data are subject to differing interpretations, and, even when we view data as sufficient to support the safety and/or effectiveness of a product candidate, regulatory authorities may not share our views and may require additional data or may deny approval altogether; whether regulatory authorities will be satisfied with the design of and results from our clinical studies; whether and when drug applications may be filed in any jurisdictions for any potential indication for PF-04965842 or any other investigational kinase inhibitor therapies; whether and when any such applications may be approved by regulatory authorities, which will depend on the assessment by such regulatory authorities of the benefit-risk profile suggested by the totality of the efficacy and safety information submitted, and, if approved, whether PF-04965842 or any such other investigational kinase inhibitor therapies will be commercially successful; decisions by regulatory authorities regarding labeling, safety and other matters that could affect the availability or commercial potential of PF-04965842 or any other investigational kinase inhibitor therapies; and competitive developments.
A further description of risks and uncertainties can be found in Pfizer’s Annual Report on Form 10-K for the fiscal year ended December 31, 2016 and in its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results”, as well as in its subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov and www.pfizer.com .

# # # # #
1 Food and Drug Administration Fact Sheet Breakthrough Therapies at https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDASIA/ucm329491.htmaccessed on January 25, 2018
PATENT
CA 2899888
PATENT
WO 2014128591
PFIZER INC. [US/US]; 235 East 42nd Street New York, New York 10017 (US)
BROWN, Matthew Frank; (US).
FENWICK, Ashley Edward; (US).
FLANAGAN, Mark Edward; (US).
GONZALES, Andrea; (US).
JOHNSON, Timothy Allan; (US).
KAILA, Neelu; (US).
MITTON-FRY, Mark J.; (US).
STROHBACH, Joseph Walter; (US).
TENBRINK, Ruth E.; (US).
TRZUPEK, John David; (US).
UNWALLA, Rayomand Jal; (US).
VAZQUEZ, Michael L.; (US).
PARIKH, Mihir, D.; (US)
COMPD 2

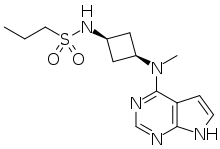
Example 2 : N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane- l -sulƒonamide
This compound was prepared using 1-propanesulfonyl chloride. The crude compound was purified by chromatography on silica gel eluting with a mixture of dichloromethane and methanol (93 : 7) to afford the title compound as a tan sol id (78% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.60 (br s, 1 H), 8.08 (s, 1 H), 7.46 (d, 1 H), 7.12 (d, 1 H), 6.61 (d, 1 H), 4.81-4.94 (m, 1 H), 3.47-3.62 (m, 1 H), 3.23 (s, 3 H), 2.87-2.96 (m, 2 H), 2.52-2.63 (m, 2 H), 2.14-2.27 (m, 2 H) 1.60- 1.73 (m, 2 H) 0.96 (t, 3 H). LC/MS (exact mass) calculated for C14H21N5O2S;
323.142, found (M + H+); 324.1.
PAPER
Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.

https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.7b01598
N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (25)
Schmieder, G.; Draelos, Z.; Pariser, D.; Banfield, C.; Cox, L.; Hodge, M.; Kieras, E.; Parsons-Rich, D.; Menon, S.; Salganik, M.; Page, K.; Peeva, E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study Br. J. Dermatol. 2017, DOI: 10.1111/bjd.16004
Compound 25, N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide is available through MilliporeSigma (cat. no. PZ0304).
CLIP
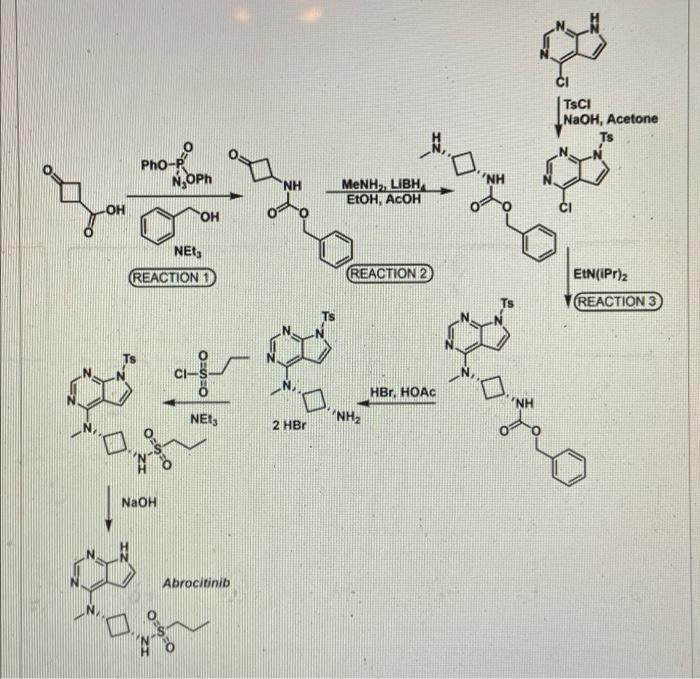
REFERENCES
1: Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, Kieras E, Parsons-Rich D, Menon S, Salganik M, Page K, Peeva E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study. Br J Dermatol. 2017 Sep 26. doi: 10.1111/bjd.16004. [Epub ahead of print] PubMed PMID: 28949012
2 Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.
- Originator Pfizer
- Class Anti-inflammatories; Antipsoriatics; Pyrimidines; Pyrroles; Skin disorder therapies; Small molecules; Sulfonamides
- Mechanism of Action Janus kinase 1 inhibitors
- Phase III Atopic dermatitis
- Discontinued Lupus vulgaris; Plaque psoriasis
- 21 May 2019Pfizer initiates enrolment in a phase I trial in Healthy volunteers in USA (PO) (NCT03937258)
- 09 May 2019 Pfizer plans a phase I pharmacokinetic and drug-drug interaction trial in healthy volunteers in May 2019 (NCT03937258)
- 30 Apr 2019 Pfizer completes a phase I trial (In volunteers) in USA (PO) (NCT03626415)
References[
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/213871s000lbl.pdf
- ^ Jump up to:a b c d e “Cibinqo EPAR”. European Medicines Agency (EMA). 11 October 2021. Retrieved 17 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. (October 2019). “Efficacy and Safety of Oral Janus Kinase 1 Inhibitor Abrocitinib for Patients With Atopic Dermatitis: A Phase 2 Randomized Clinical Trial”. JAMA Dermatology. 155 (12): 1371–1379. doi:10.1001/jamadermatol.2019.2855. PMC 6777226. PMID 31577341.
- ^ Peeva E, Hodge MR, Kieras E, Vazquez ML, Goteti K, Tarabar SG, et al. (August 2018). “Evaluation of a Janus kinase 1 inhibitor, PF-04965842, in healthy subjects: A phase 1, randomized, placebo-controlled, dose-escalation study”. British Journal of Clinical Pharmacology. 84 (8): 1776–1788. doi:10.1111/bcp.13612. PMC 6046510. PMID 29672897.
- ^ Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
- ^ “Pfizer Presents Positive Phase 3 Data at the 28th Congress of the European Academy of Dermatology and Venereology for Abrocitinib in Moderate to Severe Atopic Dermatitis”. Drugs.com. 12 October 2019.
- ^ Silverberg, J. I.; Simpson, E. L.; Thyssen, J. P.; Gooderham, M.; Chan, G.; Feeney, C.; Biswas, P.; Valdez, H.; Dibonaventura, M.; Nduaka, C.; Rojo, R. (3 June 2020). “Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial”. JAMA Dermatology. 156 (8): 863–873. doi:10.1001/jamadermatol.2020.1406. PMC 7271424. PMID 32492087.
- ^ Jump up to:a b “Cibinqo: Pending EC decision”. European Medicines Agency. 15 October 2021. Retrieved 15 October 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “European Commission Approves Pfizer’s Cibinqo (abrocitinib) for the Treatment of Adults with Moderate-to-Severe Atopic Dermatitis”. Pfizer Inc. (Press release). 10 December 2021. Retrieved 17 December 2021.
- ^ “U.S. FDA Approves Pfizer’s Cibinqo (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis”. Pfizer Inc. (Press release). 14 January 2022. Retrieved 16 January 2022.
External links
- “Abrocitinib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
- Clinical trial number NCT03575871 for “Study Evaluating Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-2)” at ClinicalTrials.gov
- {{ClinicalTrialsGov|NCT03720470|Study Evaluating Efficacy and Safety of PF-04965842 and Dupilumab in Adult Subjects With Moderate to Severe Atopic Dermatitis on Background Topical Therapy (JADE Compare)}
 |
|
| Clinical data | |
|---|---|
| Trade names | Cibinqo |
| Other names | PF-04965842 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Elimination half-life | 2.8–5.2 h |
| Excretion | 1.0–4.4% unchanged in urine |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.251.498 |
| Chemical and physical data | |
| Formula | C14H21N5O2S |
| Molar mass | 323.42 g·mol−1 |
| 3D model (JSmol) | |
/////////PF 04965842, Abrocitinib, Phase III, Atopic dermatitis, pfizer, fda 2022, APPROVALS 2022
CCCS(=O)(N[C@H]1C[C@@H](N(C)C2=C3C(NC=C3)=NC=N2)C1)=O
CCCS(=O)(=O)N[C@@H]1C[C@@H](C1)N(C)c2ncnc3[nH]ccc23
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
MITAPIVAT
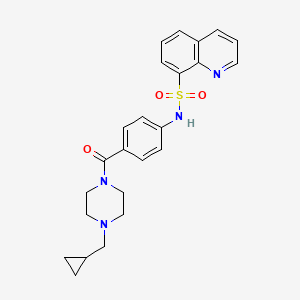

MITAPIVAT
CAS 1260075-17-9
MF C24H26N4O3S
MW 450.55
UPDATE…. FDA APPROVE 2/17/2022 To treat hemolytic anemia in pyruvate kinase deficiency, Pyrukynd
8-Quinolinesulfonamide, N-[4-[[4-(cyclopropylmethyl)-1-piperazinyl]carbonyl]phenyl]-
N-[4-[[4-(Cyclopropylmethyl)-1-piperazinyl]carbonyl]phenyl]-8-quinolinesulfonamide
- AG-348
- Originator Agios Pharmaceuticals
- Class Antianaemics; Piperazines; Quinolines; Small molecules; Sulfonamides
- Mechanism of Action Pyruvate kinase stimulants
- Orphan Drug Status Yes – Inborn error metabolic disorders
- New Molecular Entity Yes
- Phase III Inborn error metabolic disorders
- Phase II Thalassaemia
- 27 Feb 2019 Agios Pharmaceuticals plans a phase III trial for Inborn error metabolic disorders (Pyruvate kinase deficiency) (Treatment-experienced) in the US, Brazil, Canada, Czech Republic, Denmark, France, Germany, Ireland, Italy, Japan, South Korea, Netherlands, Portugal, Spain, Switzerland, Thailand, Turkey and United Kingdom in March 2019 (NCT03853798) (EudraCT2018-003459-39)
- 11 Dec 2018 Phase-II clinical trials in Thalassaemia in Canada (PO) (NCT03692052)
- 29 Aug 2018 Chemical structure information added
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Mitapivat sulfate | N4JTA67V3O | Not Available | Not applicable |
Mitapivat sulfate
CAS#: 2151847-10-6 (sulfate hydrate)
Chemical Formula: C48H60N8O13S3
Exact Mass: 1052.3442
Molecular Weight: 1053.23
Elemental Analysis: C, 54.74; H, 5.74; N, 10.64; O, 19.75; S, 9.13
N-(4-(4-(cyclopropylmethyl)piperazine-1-carbonyl)phenyl)quinoline-8-sulfonamide hemisulfate trihydrate
Related CAS #: 2151847-10-6 (sulfate hydrate) 1260075-17-9 (free) 2329710-91-8 (sulfate)
Mitapivat, sold under the brand name Pyrukynd, is a medication used to treat hemolytic anemia.[1] It is taken as the sulfate hydrate salt by mouth.[1]
Mitapivat is a pyruvate kinase activator.[1]
Mitapivat was approved for medical use in the United States in February 2022.[1][2][3]
Activator of pyruvate kinase isoenzyme M2 (PKM2), an enzyme involved in glycolysis. Since all tumor cells exclusively express the embryonic M2 isoform of PK, it is hypothesized that PKM2 is a potential target for cancer therapy. Modulation of PKM2 might also be effective in the treatment of obesity, diabetes, autoimmune conditions, and antiproliferation-dependent diseases.
Agios Pharmaceuticals is developing AG-348 (in phase 3 , in June 2019), an oral small-molecule allosteric activator of the red blood cell-specific form of pyruvate kinase (PK-R), for treating PK deficiency and non-transfusion-dependent thalassemia.
Mitapivat is a novel, first-in-class pyruvate kinase activator. It works to increase the activity of erythrocyte pyruvate kinase, a key enzyme involved in the survival of red blood cells. Defects in the pyruvate kinase enzyme in various red blood cells disorders lead to the lack of energy production for red blood cells, leading to lifelong premature destruction of red blood cells or chronic hemolytic anemia.1
On February 17, 2022, the FDA approved mitapivat as the first disease-modifying treatment for hemolytic anemia in adults with pyruvate kinase (PK) deficiency, a rare, inherited disorder leading to lifelong hemolytic anemia.6 Mitapivat has also been investigated in other hereditary red blood cell disorders associated with hemolytic anemia, such as sickle cell disease and alpha- and beta-thalassemia.1
SYN
WO 20100331307

CAS 59878-57-8 TO CAS 57184-25-5
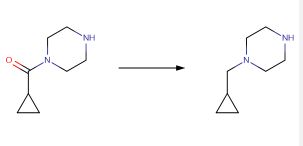
Eisai Co., Ltd., EP1508570, Lithium aluminium hydride (770 mg, 20.3 mmol) was suspended in tetrahydrofuran (150 mL), 1-(cyclopropylcarbonyl)piperazine (1.56 g, 10.1 mmol) was gradually added thereto, and the reaction mixture was heated under reflux for 30 minutes. The reaction mixture was cooled to room temperature, and 0.8 mL of water, 0.8 mL of a 15percent aqueous solution of sodium hydroxide and 2.3 mL of water were seque ntially gradually added thereto. The precipitated insoluble matter was removed by filtration through Celite, and the filtrate was evaporated to give the title compound (1.40g) as a colorless oil. The product was used for the synthesis of (8E,12E,14E)-7-((4-cyclopropylmethylpiperazin-1-yl)carbonyl)oxy-3,6,16,21-tetrahydroxy-6,10,12,16,20-pentamethyl-18,19-epoxytricosa-8,12,14-trien-11-olide (the co mpound of Example 27) without further purification.1H-NMR Spectrum (CDCl3,400MHz) delta(ppm): 0.09-0.15(2H,m), 0.48-0.56(2H,m),0.82-0.93(1H,m),2.25(2H,d,J=7.2Hz) 2.48-2.65(4H,m),2.90-2.99(4H,m).
CAS 91-22-5 TO CAD 18704-37-5
chlorosulfonic acid;
Russian Journal of Organic Chemistry, vol. 36, 6, (2000), p. 851 – 853
NMR
US2010/331307
dimethylsulfoxide-d6, 1H
1H NMR (400 MHz, DMSO-d6) δ: 1.2 (t, 2H), 1.3 (t, 2H), 1.31-1.35 (m, 1H), 2.40 (s, 2H), 3.68 (br s, 4H), 3.4-3.6 (m, 4H), 7.06 (m, 6H), 7.25-7.42 (m, 3H), 9.18 (s, 1H) 10.4 (s, 1H)
1H NMR (400 MHz, DMSO-d6) δ: 0.04-0.45 (m, 2H), 0.61-0.66 (m, 2H), 1.4-1.6 (m, 1H), 2.21-2.38 (m, 4H), 2.61 (d, 2H), 3.31-3.61 (br s, 4H), 6.94-7.06 (m, 4H), 7.40 (d, 2H), 7.56-7.63 (m, 2H), 8.28 (d, 1H), 9.18 (s, 1H), 10.4 (s, 1H)
SYN
SYN
J. Med. Chem. 2024, 67, 4376−4418
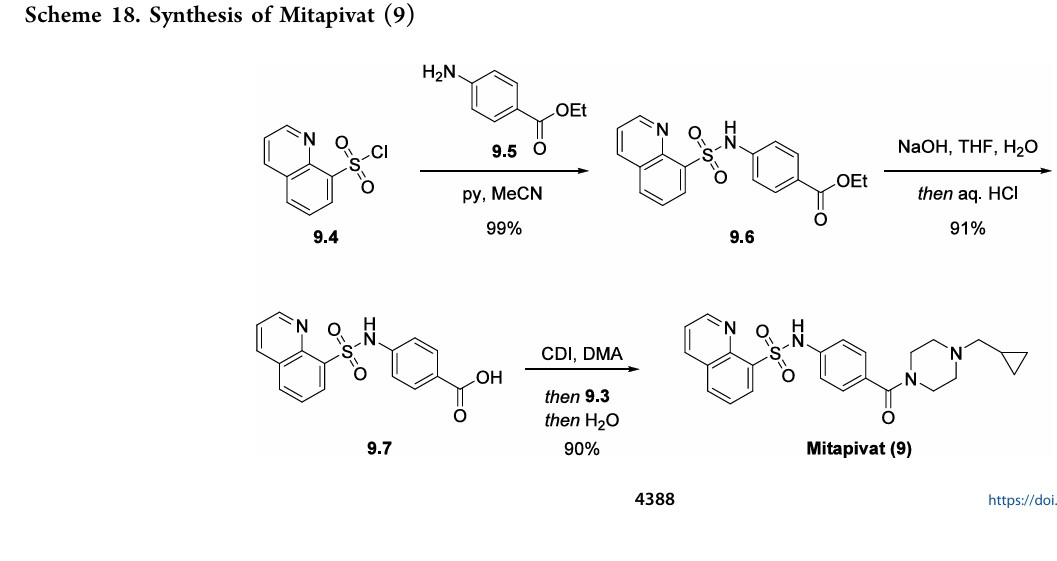
Mitapivat (Pyrukynd). Mitapivat (9), developed by Agios Pharmaceuticals, Inc., was approved in the EU and US as an oral treatment of hemolytic anemia and pyruvate kinase deficiency (PKD), a rare, inherited condition associated with chronic hemolytic anemia. 66 Previous clinical management of PKD has been limited to only supportive therapies associated with short- and long-term risks, including red cell transfusions and splenectomy. 67 Mitapivat is a first-in-class activator of erythrocyte pyruvate kinase and has been shown to increase hemoglobin levels in patients who were not receiving regular red cell transfusions. In addition to its approved treatment of PKD, it is also currently in clinical studies for the treatment of sickle cell disease and thalassemia. 66 68
As compared to the linear medicinal chemistry route utilized for analog synthesis, Agios Pharmaceuticals, Inc. developed a convergent, high-yielding route to mitapivat depicted in Scheme 17 and Scheme 18.
69 Piperazine 9.3 was synthesized in two steps from tert-butyl piperazine-1-carboxylate (9.1) in
92% yield (Scheme 17) beginning with a reductive amination in the presence of excess cyclopropanecarbaldehyde (9.2) to give the desired tertiary amine. This was carried forward as a
solution into the N-Boc deprotection, precipitating the piperazine as bis-HCl salt 9.3. The second module of mitapivat, sulfonamide 9.7, was prepared in two steps beginning with the reaction of sulfonyl chloride 9.4 with aniline 9.5 to afford ester 9.6 in 99% yield (Scheme 18). Saponification of ester 9.6 under basic
conditions provided carboxylic acid 9.7 in 91% yield. Efficient coupling was demonstrated on the kilogram scale by activation of carboxylic acid 9.7 with CDI, followed by addition of piperazine 9.3. Finally, addition of water to the reaction mixture induced crystallization of the product, providing mitapivat in 90% isolated yield.
(66) Pilo, F.; Angelucci, E. Mitapivat for sickle cell disease and
thalassemia. Drugs Today (Barc) 2023, 59, 125−134.
(67) Al-Samkari, H.; Galacteros, F.; Glenthoej, A.; Rothman, J. A.;
Andres, O.; Grace, R. F.; Morado-Arias, M.; Layton, D. M.; Onodera,
K.; Verhovsek, M.; et al. Mitapivat versus placebo for pyruvate kinase
deficiency. N. Engl. J. Med. 2022, 386, 1432−1442.
(68) Salituro, F. G.; Saunders, J. O.; Yan, S. Preparation of
aroylpiperazines and related compounds as pyruvate kinase M2
modulators useful in treatment of cancer. U.S. Patent US
20100331307, 2010.
(69) Sizemore, J. P.; Guo, L.; Mirmehrabi, M.; Su, Y. Preparation of
amorphous and crystalline forms of N-(4-(4-(cyclopropylmethyl)
piperazine-1-carbonyl)phenyl)quinoline-8-sulfonamide useful for
treatment of pyruvate kinase associated disorders. WO 2019104134,
2019.

Development Overview
Introduction
Mitapivat (designated AG 348), an orally available, first-in-class, small molecule stimulator of pyruvate kinase (PK), is being developed by Agios Pharmaceuticals for the treatment of pyruvate kinase deficiency (Inborn error metabolic disorders in development table) and thalassemia. Mitapivat is designed to activate the wild-type (normal) and mutated PK-R (the isoform of pyruvate kinase that is present in erythrocytes), in order to correct the defects in red cell glycolysis found within mutant cells. Clinical development is underway for inborn error metabolic disorders in the US, Spain and Denmark and for Thalassaemia in Canada.
Mitapivat emerged from Agios’ research programme focussed on the discovery of small molecule therapeutics for inborn metabolic disorders [see Adis Insight Drug Profile 800036791].
Key Development Milestones
In June 2018, Agios Pharmaceuticals initiated the phase III ACTIVATE trial to evaluate the efficacy and safety of orally administered mitapivat as compared with placebo in participants with pyruvate kinase deficiency (PKD), who are not regularly receiving blood transfusions (NCT03548220; AG348-C-006). The randomised, double-blind, placebo-controlled global trial intends to enrol 80 patients in the US, Canada, Denmark, France, Germany, Italy, Japan, South Korea, Netherlands, Poland, Portugal, Spain, Switzerland, Thailand and United Kingdom. The study design has two parts. Part 1 is a dose optimisation period where patients start at 5mg of mitapivat or placebo twice daily, with the flexibility to titrate up to 20mg or 50mg twice daily over a three month period to establish their individual optimal dose, as measured by maximum increase in hemoglobin levels. After the dose optimisation period, patients will receive their optimal dose for an additional three months in part 2. The primary endpoint of the study is the proportion of patients who achieve at least a 1.5 g/dL increase in haemoglobin sustained over multiple visits in part 2 of the trial
In February 2018, Agios Pharmaceuticals initiated the phase III ACTIVATE-T trial to assess the efficacy and safety of mitapivat in regularly transfused adult subjects with pyruvate kinase deficiency (Inborn error metabolism disorders in development table) (EudraCT2017-003803-22; AG348-C-007). The open label trial will enrol approximately 20 patients in Denmark and Spain and will expand to Canada, France, Italy, Japan, the Netherlands, the UK and the US
In December 2018, Agios Pharmaceuticals initiated a phase II study to assess the safety, efficacy, pharmacokinetics and pharmacodynamics of mitapivat (50mg and 100mg) for the treatment of patients with non-transfusion-dependent thalassemia (AG348-C-010; EudraCT2018-002217-35; NCT03692052). This study will include a 24-week core period followed by a 2-year extension period for eligible participants. The open-label trial intends to enrol approximately 17 patients. Enrolment has been initiated in Canada and may expand to the US and the UK
Agios Pharmaceuticals, in June 2015 initiated the phase II DRIVE PK trial to evaluate the safety, efficacy, pharmacokinetics and pharmacodynamics of mitapivat in adult transfusion-independent patients with pyruvate kinase deficiency (Inborn error metabolism disorders in development table) (AG348-C-003; NCT02476916). The trial will include two arms with 25 patients each. The patients in the first arm will receive 50mg twice daily, and the patients in the second arm will receive 300mg twice daily. The study will include a six-month dosing period with the opportunity for continued treatment beyond six months based on safety and clinical activity. The open-label, randomised trial completed enrolment of targeted 52 patients in the US, in November 2016. Preliminary data from the trial was presented at the 21st Congress of the European Haematology Association (EHA-2016). Updated results were presented by Agios at the 58th Annual Meeting and Exposition of the American Society of Haematology in December 2016. Based on results of the DRIVE PK trial, Agios plans to develop a registration path for mitapivat. Updated data from the trial was presented at the 22nd Congress of the European Haematology Association (EHA-2017)
In June 2018, Agios Pharmaceuticals completed a phase I trial in healthy male volunteers to assess the absorption, distribution, metabolism, excretion and absolute bioavailability of AG 348 (AG348-C-009; NCT03703505). Radiolabelled analytes of AG 348 ([14C]AG 348 and [13C6]AG 348) were administered in a single oral and intravenous dose on day 1. The open label trial was initiated in May 2018 and enrolled 8 volunteers in the US
In November 2017, Agios Pharmaceuticals completed a phase I trial that evaluated the relative bioavailability and safety of the mitapivat tablet and capsule formulations after single-dose administration in healthy adults (AG348-C-005; NCT03397329). The open-label trial enrolled 26 subjects in the US and was initiated in October 2017
In October 2017, Agios Pharmaceuticals completed a phase I trial that evaluated the pharmacokinetics, safety and effect on QTc interval of mitapivat in healthy volunteers (AG348-C-004; NCT03250598). This single-dose, open-label trial was initiated in August 2017 and enrolled 60 volunteers in the US
In November 2014, Agios completed a randomised, double-blind, placebo-controlled phase I trial that assessed the safety, pharmacokinetics and pharmacodynamics of multiple escalating doses of mitapivat in healthy volunteers (MAD; AG-348MAD; AG348-C-002; NCT02149966). Mitapivat was dosed daily for 14 days. The trial recruited 48 subjects in the US. In June 2015, positive results from the trial were presented at the 20th congress of the European Haematology Association (EHA-2015). Mitapivat showed a favourable pharmacokinetic profile with rapid absorption, low to moderate variability and a dose-proportional increase in exposure following multiple doses and serum hormone changes consistent with reversible aromatase inhibition were also observed
Agios Pharmaceuticals completed a randomised, double-blind, placebo-controlled phase I clinical trial of mitapivat in August 2014 (AG-348 SAD; AG348-C-001; NCT02108106). The study evaluated the safety, pharmacokinetics and pharmacodynamics of single escalating doses of the agent in healthy volunteers. Potential metabolic biomarkers were also explored. The trial enrolled 48 participants in the US
Patent Information
As of January 2018, Agios Pharmaceuticals owned approximately six issued US patents, 65 issued foreign patents, five pending US patent applications and 55 pending foreign patent applications in a number of jurisdictions directed to PK deficiency programme, including mitapivat (AG 348). The patents are valid till at least 2030
- Route of administrationPO
- FormulationTablet, unspecified
- ClassAntianaemics, Piperazines, Quinolines, Small molecules, Sulfonamides
- Mechanism of ActionPyruvate kinase stimulants
- WHO ATC codeA16A-X (Various alimentary tract and metabolism products)B03 (Antianemic Preparations)B06A (Other Hematological Agents)
- EPhMRA codeA16A (Other Alimentary Tract and Metabolism Products)B3 (Anti-Anaemic Preparations)B6 (All Other Haematological Agents)
- Chemical nameN-[4-[4-(cyclopropylmethyl)piperazine-1-carbonyl]phenyl]quinoline-8-sulfonamide
- Molecular formulaC24 H26 N4 O3 S
References
-
Agios Reports First Quarter 2017 Financial Results.
Media Release -
Agios Announces Initiation of Global Phase 3 Trial (ACTIVATE) of AG-348 in Adults with Pyruvate Kinase Deficiency Who Are Not Regularly Transfused.
Media Release -
A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of AG-348 in Not Regularly Transfused Adult Subjects With Pyruvate Kinase Deficiency
ctiprofile -
Agios Provides Business Update on Discovery Research Strategy and Pipeline, Progress on Clinical Programs, Commercial Launch Preparations and Reports First Quarter 2018 Financial Results at Investor Day.
Media Release -
An Open-Label Study To Evaluate the Efficacy and Safety of AG-348 in Regularly Transfused Adult Subjects With Pyruvate Kinase (PK) Deficiency
ctiprofile -
A Phase 2, Open-label, Multicenter Study to Determine the Efficacy, Safety, Pharmacokinetics, and Pharmacodynamics of AG-348 in Adult Subjects With Non-transfusion-dependent Thalassemia
ctiprofile -
Agios Announces Key Upcoming Milestones to Support Evolution to a Commercial Stage Biopharmaceutical Company in 2017.
Media Release -
Agios to Present Clinical and Preclinical Data at the 20th Congress of the European Hematology Association.
Media Release -
Agios Announces Updated Data from Fully Enrolled DRIVE PK Study Demonstrating AG-348s Potential as the First Disease-modifying Treatment for Patients with Pyruvate Kinase Deficiency.
Media Release -
Agios Announces New Data from AG-348 and AG-519 Demonstrating Potential for First Disease-modifying Treatment for Patients with PK Deficiency.
Media Release -
Agios Provides Update on PKR Program.
Media Release -
AG-348 Achieves Proof-of-Concept in Ongoing Phase 2 DRIVE-PK Study and Demonstrates Rapid and Sustained Hemoglobin Increases in Adults with Pyruvate Kinase Deficiency.
Media Release -
Agios Reports New, Final Data from Phase 1 Multiple Ascending Dose (MAD) Study in Healthy Volunteers for AG-348, an Investigational Medicine for Pyruvate Kinase (PK) Deficiency.
Media Release -
Grace RF, Layton DM, Galacteros F, Rose C, Barcellini W, Morton DH, et al. Results Update from the DRIVE PK Study: Effects of AG-348, a Pyruvate Kinase Activator, in Patients with Pyruvate Kinase Deficiency. ASH-Hem-2017 2017; abstr. 2194.
Available from: URL: https://ash.confex.com/ash/2017/webprogram/Paper102236.html -
A Phase 2, Open Label, Randomized, Dose Ranging, Safety, Efficacy, Pharmacokinetic and Pharmacodynamic Study of AG-348 in Adult Patients With Pyruvate Kinase Deficiency
ctiprofile -
A Phase I, Open-label Study to Evaluate the Absorption, Distribution, Metabolism, and Excretion and to Assess the Absolute Bioavailability of AG-348 in Healthy Male Subjects Following Administration of a Single Oral Dose of [14C]AG-348 and Concomitant Single Intravenous Microdose of [13C6]AG-348
ctiprofile -
A Phase 1, Randomized, Open-Label, Two-Period Crossover Study Evaluating the Relative Bioavailability and Safety of the AG-348 Tablet and Capsule Formulations After Single-Dose Administration in Healthy Adults
ctiprofile -
A Phase 1, Single-Dose, Open-Label Study to Characterize and Compare the Pharmacokinetics, Safety, and Effect on QTc Interval of AG-348 in Healthy Subjects of Japanese Origin and Healthy Subjects of Non-Asian Origin
ctiprofile -
Agios Pharmaceuticals Initiates Multiple Ascending Dose Trial in Healthy Volunteers of AG-348 for the Potential Treatment of PK Deficiency, a Rare, Hemolytic Anemia.
Media Release -
A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose, Safety, Pharmacokinetic, and Pharmacodynamic Study of Orally Administered AG-348 in Healthy Volunteers
ctiprofile -
Agios Initiates Phase 1 Study of AG-348, a First-in-class PKR Activator, for Pyruvate Kinase Deficiency.
Media Release -
A Phase I, Randomized, Double-Blind, Placebo-Controlled, Single Ascending Dose, Safety, Pharmacokinetic and Pharmacodynamic Study of Orally Administered AG-348 in Healthy Volunteers
ctiprofile -
Agios Pharmaceuticals Reports First Quarter 2014 Financial Results.
Media Release -
Agios Pharmaceuticals Reports Third Quarter 2013 Financial Results.
Media Release -
Agios Pharmaceuticals to Present Preclinical Research at the 2013 American Society of Hematology Annual Meeting.
Media Release -
Agios Presents Preclinical Data from Lead Programs at American Society of Hematology Annual Meeting.
Media Release -
Agios Pharmaceuticals Form 10-K, February 2018. Internet-Doc 2018;.
Available from: URL: https://www.sec.gov/Archives/edgar/data/1439222/000143922218000004/agio-123117x10k.htm -
Agios Outlines Key 2018 Priorities Expanding Clinical and Research Programs to Drive Long Term Value.
Media Release -
Grace RF, Layton DM, Galacteros F, Rose C, Barcellini W, Morton DH, et al. Effects of Ag-348, a Pyruvate Kinase Activator, in Patients with Pyruvate Kinase Deficiency: Updated Results from the Drive Pk Study. EHA-2017 2017; abstr. S451.
Available from: URL: https://learningcenter.ehaweb.org/eha/2017/22nd/181738/rachael.f.grace.effects.of.ag-348.a.pyruvate.kinase.activator.in.patients.with.html?f=m3e1181l15534 -
Agios Presents Updated Data from DRIVE PK Study Demonstrating AG-348 is Well-Tolerated and Results in Clinically Relevant, Rapid and Sustained Hemoglobin Increases in Patients with Pyruvate Kinase Deficiency.
Media Release
Medical uses
Mitapivat is indicated for the treatment of hemolytic anemia in adults with pyruvate kinase deficiency.[1][3]Pharmacology
Mechanism of action
Mitapivat binds to and activates pyruvate kinase, thereby enhancing glycolytic pathway activity, improving adenosine triphosphate (ATP) levels and reducing 2,3-diphosphoglycerate (2,3-DPG) levels.[4] Mutations in pyruvate kinase cause deficiency in pyruvate kinase which prevents adequate red blood cell (RBC) glycolysis, leading to a buildup of the upstream glycolytic intermediate 2,3-DPG and deficiency in the pyruvate kinase product ATP.[4][5]Society and culture
Names
Mitapivat is the international nonproprietary name (INN).[6]References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216196s000lbl.pdf
- ^ “Agios Announces FDA Approval of Pyrukynd (mitapivat) as First Disease-Modifying Therapy for Hemolytic Anemia in Adults with Pyruvate Kinase Deficiency” (Press release). Agios Pharmaceuticals. 17 February 2022. Retrieved 19 February 2022 – via GlobeNewswire.
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/216196Orig1s000ltr.pdf
- ^ Jump up to:a b “Mitapivat (Code C157039)”. NCI Thesaurus. 31 January 2022. Retrieved 19 February 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “PK-R allosteric activator AG-348”. NCI Drug Dictionary. National Cancer Institute. Retrieved 19 February 2022. This article incorporates text from this source, which is in the public domain.
- ^ World Health Organization (2017). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 78”. WHO Drug Information. 31 (3): 539. hdl:10665/330961.
Further reading
- Kung C, Hixon J, Kosinski PA, Cianchetta G, Histen G, Chen Y, et al. (September 2017). “AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency”. Blood. 130 (11): 1347–1356. doi:10.1182/blood-2016-11-753525. PMC 5609468. PMID 28760888.
- Rab MA, Van Oirschot BA, Kosinski PA, Hixon J, Johnson K, Chubukov V, et al. (January 2021). “AG-348 (Mitapivat), an allosteric activator of red blood cell pyruvate kinase, increases enzymatic activity, protein stability, and ATP levels over a broad range of PKLR genotypes”. Haematologica. 106 (1): 238–249. doi:10.3324/haematol.2019.238865. PMC 7776327. PMID 31974203.
External links
- “Mitapivat sulfate”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03548220 for “A Study to Evaluate Efficacy and Safety of AG-348 in Not Regularly Transfused Adult Participants With Pyruvate Kinase Deficiency (PKD)” at ClinicalTrials.gov
- Clinical trial number NCT03559699 for “A Study Evaluating the Efficacy and Safety of AG-348 in Regularly Transfused Adult Participants With Pyruvate Kinase Deficiency (PKD)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Pyrukynd |
| Other names | AG-348, Mitapivat sulfate (USAN US) |
| License data | |
| Routes of administration | By mouth |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| DrugBank |
|
| ChemSpider | |
| UNII |
|
| KEGG |
|
| ChEMBL |
|
| Chemical and physical data | |
| Formula | C24H26N4O3S |
| Molar mass | 450.56 g·mol−1 |
| 3D model (JSmol) | |
Mavacamten
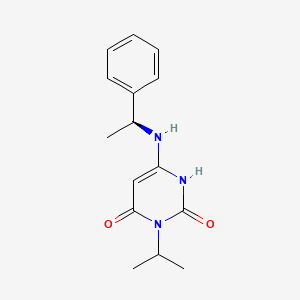 Mavacamten
SAR-439152; SAR 439152; SAR439152; MYK-461; MYK 461; MYK461; Mavacamten
(S)-3-isopropyl-6-((1-phenylethyl)amino)pyrimidine-2,4(1H,3H)-dione
cas 1642288-47-8
Chemical Formula: C15H19N3O2
Molecular Weight: 273.336
マバカムテン;
Mavacamten
SAR-439152; SAR 439152; SAR439152; MYK-461; MYK 461; MYK461; Mavacamten
(S)-3-isopropyl-6-((1-phenylethyl)amino)pyrimidine-2,4(1H,3H)-dione
cas 1642288-47-8
Chemical Formula: C15H19N3O2
Molecular Weight: 273.336
マバカムテン;
- UNII-QX45B99R3J
- QX45B99R3J
- HCM 1; MYK-461; SAR-439152
- Originator MyoKardia
- Class Cardiovascular therapies; Small molecules
- Mechanism of Action Myosin inhibitors
- Orphan Drug Status Yes – Hypertrophic cardiomyopathy
Highest Development Phases
- Phase III Hypertrophic cardiomyopathy
Most Recent Events
- 30 May 2018 Phase-III clinical trials in Hypertrophic cardiomyopathy in USA (PO) (NCT03470545)
- 08 May 2018 MyoKardia plans a long-term extension (LTE) trial of patients who complete the phase III EXPLORER-HCM or the phase II MAVERICK-HCM trial for Hypertrophic cardiomyopathy by the end of 2018
- 26 Apr 2018 MyoKardia initiates the PIONEER-OLE trial (an extension trial of phase II PIONEER trial) for Hypertrophic cardiomyopathy in USA (PO) (NCT03496168)




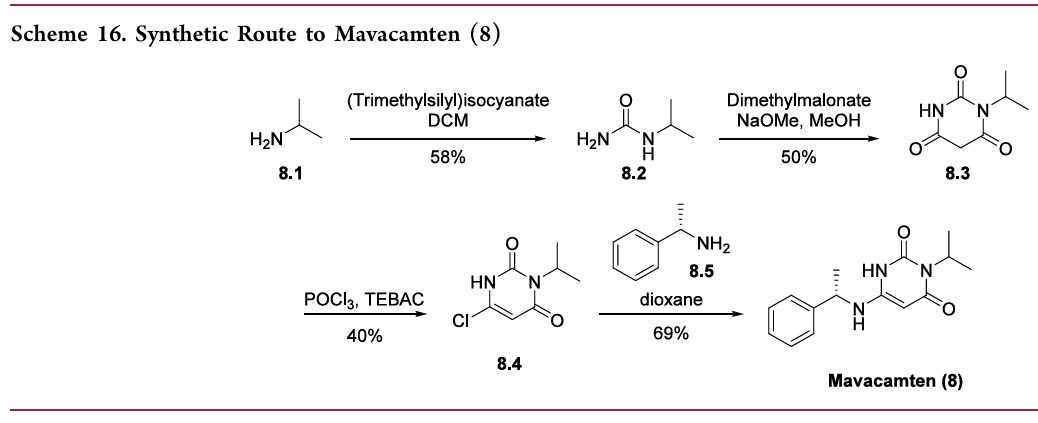
Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0° C. was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0° C., CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1:20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65° C. After cooling to ambient temperature and then to 0° C., the pH was carefully adjusted to 3 using aqueous concentrated HCl. The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20:1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. LC/MS: m/z (ES+) 171 (M+H)+. 1 1H-NMR (300 MHz, d6-DMSO): δ 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J=6.0 Hz, 6H).
Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50° C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtOAc/petroleum ether (1:1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J=6.0 Hz, 6H).
Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl)amino)pyrimidine-2, 4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3, 1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-α-methylbenzylamine (Sigma-Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80° C. for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous 1N HCl (2×50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SO4 and then concentrated under reduced pressure to half the original volume to yield a precipitate. Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, d6-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J=6.8 Hz, 1H), 4.87 (m, 1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J=6.7 Hz, 3H), 1.36 (m, 6H).
PATENT
https://patents.google.com/patent/US9181200/zh-CN Genetic (heritable) hypertrophic cardiomyopathy (HCM) comprises a group of highly penetrant, monogenic, autosomal dominant myocardial diseases. HCM is caused by one or more of over 1,000 known point mutations in any one of the structural protein genes contributing to the functional unit of myocardium, the sarcomere. About 1 in 500 individuals in the general population are found to have left ventricular hypertrophy unexplained by other known causes (e.g., hypertension or valvular disease), and many of these can be shown to have HCM, once other heritable (e.g., lysosomal storage diseases), metabolic, or infiltrative causes have been excluded. [0004] Sarcomere gene mutations that cause HCM are highly penetrant, but there is wide variability in clinical severity and clinical course. Some genotypes are associated with a more malignant course, but there is considerable variability between and even within families carrying the same mutation. Sex differences have also been noted, with male patients generally more severely affected than female patients. While many patients with HCM report minimal or no symptoms for extended periods of time, HCM is a progressive disease with a significant cumulative burden of morbidity. Symptoms of effort intolerance predominate, and can be exacerbated by exercise and other maneuvers that increase heart rate and/or decrease preload. As with many other disorders, symptoms tend to worsen with age. By far the most prevalent clinical burden for patients with HCM is exertional dyspnea, which limits their activities of daily living and can be debilitating. [0005] Patients with HCM are often symptomatic in the absence of documented hemodynamic abnormalities like left ventricular outflow tract obstruction (with or without mitral regurgitation). Patients’ symptoms of exertional dyspnea can rapidly worsen with the onset of atrial fibrillation, a common complication of HCM that can precipitate acute pulmonary edema that increases the risk of systemic arterial thromboembolic disease, including stroke. Other adverse events associated with HCM include intolerance of hypovolemia or hypervolemia, and syncope. Concomitant coronary artery disease may confer a higher risk of acute coronary syndromes than in patients without HCM. Sudden cardiac death (SCD) in patients with HCM is both uncommon and difficult to predict but is a leading cause of non-traumatic death in young adults. For survivors of SCD, ICD placement is standard practice, and in other HCM patients risk profiling, while imprecise, is used to identify those for whom ICD placement for primary prevention is deemed prudent. [0006] Medical therapy for HCM is limited to the treatment of symptoms and does not address the fundamental, underlying cause of disease – disruptions in normal sarcomere function. Currently available therapies are variably effective in alleviating symptoms but typically show decreased efficacy with increasing disease duration. Patients are thus empirically managed with beta-blockers, non-dihydropyridine calcium channel blockers, and/or disopyramide. None of these agents carry labeled indications for treating HCM, and essentially no rigorous clinical trial evidence is available to guide their use. Compounding this unfortunate situation is the fact that no new medical therapies for HCM have been identified for many years. For patients with hemodynamically significant outflow tract obstruction (resting gradient >30mmHg), in appropriately selected patients surgical myectomy or alcohol septal ablation is usually required to alleviate the hemodynamic obstruction. Provided are new therapeutic agents and methods that remedy the long-felt need for improved treatment of HCM and related cardiac disorders. [0072] Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0 °C was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0 °C, CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1 :20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
[0072] Compound 1.1. Isopropylurea. To a stirred solution of isopropylamine (15.3 g, 0.258 mol, 1.0 equiv) in CH2Cl2 (200 mL) under argon at 0 °C was added dropwise trimethylsilyl isocyanate (30 g, 0.26 mol, 1.0 equiv). The resulting mixture was allowed to reach ambient temperature and stirred overnight. After cooling to 0 °C, CH3OH (100 mL) was added dropwise. The resulting solution was stirred for 2 hours (h) at room temperature and then concentrated under reduced pressure. The crude residue was recrystallized from CH3OH:Et2O (1 :20) to yield 15.4 g (58%) the title compound as a white solid. LC/MS: m/z (ES+) 103 (M+H)+.
 [0073] Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65 °C. After cooling to ambient temperature and then to 0 °C, the pH was carefully adjusted to 3 using aqueous concentrated HCl . The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20: 1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. . LC/MS: m/z (ES+) 171 (M+H)+.1 1H-NMR (300 MHz, de-DMSO): 5 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J = 6.0 Hz, 6H).
[0073] Compound 1.2. 1-Isopropyl barbituric acid. To a stirred solution of 1.1 (14.4 g, 0.14 mol, 1.00 equiv) in CH3OH (500 mL) were added dimethyl malonate (19.55 g, 0.148 mol, 1.05 equiv) and sodium methoxide (18.9 g, 0.35 mol, 2.50 equiv). The resulting mixture was stirred overnight at 65 °C. After cooling to ambient temperature and then to 0 °C, the pH was carefully adjusted to 3 using aqueous concentrated HCl . The resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (200 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using CH2Cl2/CH3OH (20: 1) as eluent to yield 16.8 g (50%) of the title compound as a white solid. . LC/MS: m/z (ES+) 171 (M+H)+.1 1H-NMR (300 MHz, de-DMSO): 5 11.19 (s, 1H), 4.83 (m, 1H), 3.58 (s, 2H), 1.32 (d, J = 6.0 Hz, 6H).
 [0074] Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50 °C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4 , and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtO Ac/petroleum ether (1 : 1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J = 6.0 Hz, 6H).
[0074] Compound 1.3. 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione. To a 100-mL round-bottom flask containing compound 1.2 (11.4 g, 66.99 mmol, 1.00 equiv) under argon were added triethylbenzylammonium chloride (21.3 g, 93.51 mmol, 1.40 equiv) and POCl3 (30 mL). The resulting mixture was stirred overnight at 50 °C. After cooling to room temperature, the mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (150 mL) followed by slow addition of H2O (100 mL). The phases were separated and the organic layer was washed with H2O (100 mL), dried with anhydrous Na2SO4 , and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography using EtO Ac/petroleum ether (1 : 1) as eluent to yield 5.12 g (40%) of the title compound as a light yellow solid. 1H-NMR (300 MHz, d6-DMSO): δ 12.22 (s, 1H), 5.88 (s, 1H), 4.95 (m, 1H), 1.34 (d, J = 6.0 Hz, 6H).
 [0075] Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl) amino) pyrimidine-2,
4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3,
1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-a-methylbenzylamine (Sigma- Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80 °C for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous IN C1 (2 x 50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SC”4 and then
concentrated under reduced pressure to half the original volume to yield a precipitate.
Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, de-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J= 6.8 Hz, 1H), 4.87 (m,
1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H ), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J = 6.7 Hz, 3H), 1.36 (m, 6H).
[0075] Compound 1. (S)-3-Isopropyl-6-((1-phenylethyl) amino) pyrimidine-2,
4(1H,3H)-dione. To a solution of 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione (1.3,
1.0 g, 5.31 mmol) in 1,4-dioxane (20 mL) was added (S)-a-methylbenzylamine (Sigma- Aldrich, 1.43 g, 11.7 mmol, 2.2 equiv). The reaction mixture was stirred at 80 °C for 24 h. After cooling to ambient temperature, the mixture was concentrated under reduced pressure. The residual was taken up in EtOAc (70 mL) and washed with aqueous IN C1 (2 x 50 mL) and brine (40 mL). The organic layer was dried with anhydrous Na2SC”4 and then
concentrated under reduced pressure to half the original volume to yield a precipitate.
Hexane (20 mL) was added and the mixture was stirred at room temperature. The resulting solid was collected by filtration, washed with hexane (20 mL), and dried to yield 1.0 g (69%) of the title compound as a white solid. LC/MS: m/z (ES+) 274 (M+H)+. 1H-NMR (400 MHz, de-DMSO): δ 9.77 (s, 1H), 7.32 (m, 4H), 7.24 (m, 1H), 6.50 (d, J= 6.8 Hz, 1H), 4.87 (m,
1H), 4.52 (m, 1H), 4.31 (d, J=6.8 Hz, 1H), 1.37 (m, 3H ), 1.24 (m, 6H). 1H NMR (400 MHz, CD3OD) δ ppm 7.39-7.20 (m, 5H), 5.01 (m, 1H), 4.48 (m, 1H), 1.49 (d, J = 6.7 Hz, 3H), 1.36 (m, 6H).

