WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Viltolarsen was approved for medical use in the United States in August 2020.[3][4] After golodirsen was approved in December 2019, viltolarsen is the second approved targeted treatment for people with this type of mutation in the United States.[3][5] Approximately 8% of people with DMD have a mutation that is amenable to exon 53 skipping.[3]
Medical uses
Viltolarsen is indicated for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping.[3][2]
DMD is a rare genetic disorder characterized by progressive muscle deterioration and weakness.[3] It is the most common type of muscular dystrophy.[3] DMD is caused by mutations in the DMD gene that results in an absence of dystrophin, a protein that helps keep muscle cells intact.[3] The first symptoms are usually seen between three and five years of age and worsen over time.[3] DMD occurs in approximately one out of every 3,600 male infants worldwide; in rare cases, it can affect females.[3]
Adverse effects
The most common side effects include upper respiratory tract infection, injection site reaction, cough, and pyrexia (fever).[3][4][2]
Although kidney toxicity was not observed in the clinical studies, the clinical experience is limited, and kidney toxicity, including potentially fatal glomerulonephritis, has been observed after administration of some antisense oligonucleotides.[3]
History
Viltolarsen was evaluated in two clinical studies with a total of 32 participants, all of whom were male and had genetically confirmed DMD.[3] The increase in dystrophin production was established in one of those two studies, a study that included sixteen DMD participants, with eight participants receiving viltolarsen at the recommended dose.[3] In the study, dystrophin levels increased, on average, from 0.6% of normal at baseline to 5.9% of normal at week 25.[3] Trial 1 provided data for evaluation of the benefits of viltolarsen.[4] The combined populations from both trials provided data for evaluation of the side effects of viltolarsen.[4] Trial 1 was conducted at six sites in the United States and Canada and Trial 2 was conducted at five sites in Japan.[4] All participants in both trials were on a stable dose of corticosteroids for at least three months before entering the trials.[4]
The U.S. Food and Drug Administration (FDA) concluded that the applicant’s data demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit in people with DMD who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping.[3] A clinical benefit of the drug has not been established.[3] In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease, and the lack of available therapies.[3]
The application for viltolarsen was granted priority review designation and the FDA granted the approval to NS Pharma, Inc.[3]
Hwang J, Yokota T (October 2019). “Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies”. Expert Rev Mol Med. 21: e5. doi:10.1017/erm.2019.5. PMID31576784.
Roshmi RR, Yokota T (October 2019). “Viltolarsen for the treatment of Duchenne muscular dystrophy”. Drugs Today. 55 (10): 627–639. doi:10.1358/dot.2019.55.10.3045038. PMID31720560.
Clinical trial number NCT02740972 for “Safety and Dose Finding Study of NS-065/NCNP-01 in Boys With Duchenne Muscular Dystrophy (DMD)” at ClinicalTrials.gov
ClassAntineoplastics; Ethers; Fluorobenzenes; Morpholines; Pyridines; Pyrimidinones; Pyrroles; Small molecules
Mechanism of Action Type 1 fibroblast growth factor receptor antagonists; Type 3 fibroblast growth factor receptor antagonists; Type 4 fibroblast growth factor receptor antagonists; Type-2 fibroblast growth factor receptor antagonists
Orphan Drug Status Yes – Myeloproliferative disorders; Lymphoma; Cholangiocarcinoma
MarketedCholangiocarcinoma
Phase IIBladder cancer; Lymphoma; Myeloproliferative disorders; Solid tumours; Urogenital cancer
Phase I/IICancer
05 Nov 2020Preregistration for Cholangiocarcinoma (Late-stage disease, Metastatic disease, First line therapy, Inoperable/Unresectable) in Japan (PO) in November 2020
05 Nov 2020Incyte Corporation stops enrolment in the FIGHT-205 trial for Bladder cancer due to regulatory feedback
26 Oct 2020Preregistration for Cholangiocarcinoma (Second-line therapy or greater, Inoperable/Unresectable, Late-stage disease, Metastatic disease) in Canada (PO)
Pemigatinib, also known as INCB054828, is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) types 1, 2, and 3 (FGFR1/2/3), with potential antineoplastic activity. FGFR inhibitor INCB054828 binds to and inhibits FGFR1/2/3, which may result in the inhibition of FGFR1/2/3-related signal transduction pathways. This inhibits proliferation in FGFR1/2/3-overexpressing tumor cells.
Pemigatinib (INN),[2] sold under the brand name Pemazyre, is a medication for the treatment of adults with previously treated, unresectable locally advanced or metastatic bile duct cancer (cholangiocarcinoma) with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement as detected by an FDA-approved test.[3][4] Pemigatinib works by blocking FGFR2 in tumor cells to prevent them from growing and spreading.[3]
Pemigatinib belongs to a group of medicines called protein kinase inhibitors.[5] It works by blocking enzymes known as protein kinases, particularly those that are part of receptors (targets) called fibroblast growth factor receptors (FGFRs).[5] FGFRs are found on the surface of cancer cells and are involved in the growth and spread of the cancer cells.[5] By blocking the tyrosine kinases in FGFRs, pemigatinib is expected to reduce the growth and spread of the cancer.[5]
The most common adverse reactions are hyperphosphatemia and hypophosphatemia (electrolyte disorders), alopecia (spot baldness), diarrhea, nail toxicity, fatigue, dysgeusia (taste distortion), nausea, constipation, stomatitis (sore or inflammation inside the mouth), dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain and dry skin.[3][4] Ocular (eye) toxicity is also a risk of pemigatinib.[3][4]
Medical uses
Cholangiocarcinoma is a rare form of cancer that forms in bile ducts, which are slender tubes that carry the digestive fluid bile from the liver to gallbladder and small intestine.[3] Pemigatinib is indicated for the treatment of adults with bile duct cancer (cholangiocarcinoma) that is locally advanced (when cancer has grown outside the organ it started in, but has not yet spread to distant parts of the body) or metastatic (when cancer cells spread to other parts of the body) and who have tumors that have a fusion or other rearrangement of a gene called fibroblast growth factor receptor 2 (FGFR2).[3] It should be used in patients who have been previously treated with chemotherapy and whose cancer has a certain type of abnormality in the FGFR2 gene.[6]
History
Pemigatinib was approved for use in the United States in April 2020 along with the FoundationOne CDX (Foundation Medicine, Inc.) as a companion diagnostic for patient selection.[3][4][7]
The approval of pemigatinib in the United States was based on the results the FIGHT-202 (NCT02924376) multicenter open-label single-arm trial that enrolled 107 participants with locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or rearrangement who had received prior treatment.[3][4][6] The trial was conducted at 67 sites in the United States, Europe, and Asia.[6] During the clinical trial, participants received pemigatinib once a day for 14 consecutive days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects.[3][4][6] To assess how well pemigatinib was working during the trial, participants were scanned every eight weeks.[3] The trial used established criteria to measure how many participants experienced a complete or partial shrinkage of their tumors during treatment (overall response rate).[3] The overall response rate was 36% (95% CI: 27%, 45%), with 2.8% of participants having a complete response and 33% having a partial response.[3] Among the 38 participants who had a response, 24 participants (63%) had a response lasting six months or longer and seven participants (18%) had a response lasting 12 months or longer.[3][4]
On 24 August 2018, orphan designation (EU/3/18/2066) was granted by the European Commission to Incyte Biosciences Distribution B.V., the Netherlands, for pemigatinib for the treatment of biliary tract cancer.[5] On 17 October 2019, orphan designation EU/3/19/2216 was granted by the European Commission to Incyte Biosciences Distribution B.V., the Netherlands, for pemigatinib for the treatment of myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB, or FGFR1, or with PCM1-JAK2.[10]
PATENT
US 20200281907
The present disclosure is directed to, inter alia, methods of treating cancer in a patient in need thereof, comprising administering pemigatinib, which is 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one, having the structure shown below:
Pemigatinib is described in U.S. Pat. No. 9,611,267, the entirety of which is incorporated herein by reference. Pemigatinib is further described in US Publication Nos.: 2019/0337948 and 2020/0002338, the entireties of which are incorporated herein by reference.
Provided herein is a method of treating cancer comprising administering a therapy to a patient in need thereof, wherein the therapy comprises administering a therapeutically effective amount of pemigatinib to the patient while avoiding the concomitant administration of a CYP3A4 perpetrator.
A mixture of 4-chloro-1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde (CAS #958230-19-8, Lakestar Tech, Lot: 124-132-29: 3.0 g, 17 mmol) and ethylamine (10M in water, 8.3 mL, 83 mmol) in 2-methoxyethanol (20 mL, 200 mmol) was heated to 130° C. and stirred overnight. The mixture was cooled to room temperature then concentrated under reduced pressure. The residue was treated with 1N HCl (30 mL) and stirred at room temperature for 1 h then neutralized with saturated NaHCO 3 aqueous solution. The precipitate was collected via filtration then washed with water and dried to provide the desired product (2.9 g, 92%). LC-MS calculated for C 10H 12N 3O [M+H] + m/z: 190.1; found: 190.1.
A mixture of 4-(ethylamino)-1H-pyrrolo[2,3-b]pyridine-5-carbaldehyde (7.0 g, 37 mmol), 2,6-difluoro-3,5-dimethoxyaniline (9.1 g, 48 mmol) and [(1S)-7,7-dimethyl-2-oxobicyclo[2.2.1]hept-1-yl]methanesulfonic acid (Aldrich, cat #21360: 2 g, 7 mmol) in xylenes (250 mL) was heated to reflux with azeotropic removal of water using Dean-Stark for 2 days at which time LC-MS showed the reaction was complete. The mixture was cooled to room temperature and the solvent was removed under reduced pressure. The residue was dissolved in tetrahydrofuran (500 mL) and then 2.0 M lithium tetrahydroaluminate in THF (37 mL, 74 mmol) was added slowly and the resulting mixture was stirred at 50° C. for 3 h then cooled to room temperature. The reaction was quenched by addition of water, 15% aqueous NaOH and water. The mixture was filtered and washed with THF. The filtrate was concentrated and the residue was washed with CH 2Cl 2 and then filtered to get the pure product (11 g, 82%). LC-MS calculated for C 18H 21F 2N 4O 2[M+H] + m/z: 363.2; found: 363.1.
A solution of triphosgene (5.5 g, 18 mmol) in tetrahydrofuran (30 mL) was added slowly to a mixture of 5-{[(2,6-difluoro-3,5-dimethoxyphenyl)amino]methyl}-N-ethyl-1H-pyrrolo[2,3-b]pyridin-4-amine (5.6 g, 15 mmol) in tetrahydrofuran (100 mL) at 0° C. and then the mixture was stirred at room temperature for 6 h. The mixture was cooled to 0° C. and then 1.0 M sodium hydroxide in water (100 mL, 100 mmol) was added slowly. The reaction mixture was stirred at room temperature overnight and the formed precipitate was collected via filtration, washed with water, and then dried to provide the first batch of the purified desired product. The organic layer in the filtrate was separated and the aqueous layer was extracted with methylene chloride. The combined organic layer was concentrated and the residue was triturated with methylene chloride then filtered and dried to provide another batch of the product (total 5.5 g, 92%). LC-MS calculated for C 19H 19F 2N 4O 3[M+H] + m/z: 389.1; found: 389.1.
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (900 mg, 2.32 mmol) in N,N-dimethylformamide (20 mL) cooled to 0° C. was added sodium hydride (185 mg, 4.63 mmol, 60 wt % in mineral oil). The resulting mixture was stirred at 0° C. for 30 min then benzenesulfonyl chloride (0.444 mL, 3.48 mmol) was added. The reaction mixture was stirred at 0° C. for 1.5 h at which time LC-MS showed the reaction completed to the desired product. The reaction was quenched with saturated NH 4Cl solution and diluted with water. The white precipitate was collected via filtration then washed with water and hexanes, dried to afford the desired product (1.2 g, 98%) as a white solid which was used in the next step without further purification. LC-MS calculated for C 25H 23F 2N 4O 5S [M+H] + m/z: 529.1; found: 529.1.
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one (1.75 g, 3.31 mmol) in tetrahydrofuran (80 mL) at −78° C. was added freshly prepared lithium diisopropylamide (1M in tetrahydrofuran (THF), 3.48 mL, 3.48 mmol). The resulting mixture was stirred at −78° C. for 30 min then N,N-dimethylformamide (1.4 mL, 18 mmol) was added slowly. The reaction mixture was stirred at −78° C. for 30 min then quenched with water and extracted with EtOAc. The organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 20% EtOAc in DCM to give the desired product as a white solid (1.68 g, 91%). LC-MS calculated for C 26H 23F 2N 4O 6S (M+H) + m/z: 557.1; found: 556.9.
To a solution 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde (1.73 g, 3.11 mmol) in dichloromethane (50 mL) was added morpholine (0.95 mL, 11 mmol), followed by acetic acid (2 mL, 30 mmol). The resulting yellow solution was stirred at room temperature overnight then sodium triacetoxyborohydride (2.3 g, 11 mmol) was added. The mixture was stirred at room temperature for 3 h at which time LC-MS showed the reaction went to completion to the desired product. The reaction was quenched with saturated NaHCO 3 then extracted with ethyl acetate (EtOAc). The organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 40% EtOAc in DCM to give the desired product as a yellow solid (1.85 g, 95%). LC-MS calculated for C 30H 32F 2N 5O 6S (M+H) + m/z: 628.2; found: 628.0.
To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′: 5,6]pyrido[4,3-d]pyrimidin-2-one (1.5 g, 2.4 mmol) in tetrahydrofuran (40 mL) was added tetra-n-butylammonium fluoride (1M in THF, 7.2 mL, 7.2 mmol). The resulting solution was stirred at 50° C. for 1.5 h then cooled to room temperature and quenched with water. The mixture was extracted with dichloromethane (DCM) and the organic extracts were combined then washed with water and brine. The organic layer was dried over Na 2SO 4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 10% MeOH in DCM to give the desired product as a white solid, which was further purified by prep HPLC (pH=2, acetonitrile/H 2O). LC-MS calculated for C 24H 28F 2N 5O 4 (M+H) + m/z: 488.2; found: 488.0. 1H NMR (500 MHz, DMSO) δ 12.09 (s, 1H), 8.06 (s, 1H), 7.05 (t, J=8.1 Hz, 1H), 6.87 (s, 1H), 4.78 (s, 2H), 4.50 (s, 2H), 4.17 (q, J=6.8 Hz, 2H), 3.97 (br, 2H), 3.89 (s, 6H), 3.65 (br, 2H), 3.37 (br, 2H), 3.15 (br, 2H), 1.37 (t, J=6.8 Hz, 3H).
The present disclosure is directed to, inter alia, solid forms, including crystalline forms and amorphous forms, of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)- 1 ,3,4,7 -tetrahydro-2H-pyrrolo [3 ‘,2’ : 5 ,6]pyrido [4,3 -d]pyrimidin-2-one
(Compound 1), and processes and intermediates for preparing the compound. The structure of Compound 1 is shown below.
Compound 1
Compound 1 is described in US Patent No. 9,611,267, the entirety of which is incorporated herein by reference.
Example 1
Synthesis of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-l^, 4,7-tetrahydro-2H-pyrrolo[3f,2f:5,6]pyrido[4r3-d]pyrimidin-2-one (Compound 1) Scheme 1.
To a l-L flask was added 4-chloro-5-(l,3-dioxolan-2-yl)-l-(phenylsulfonyl)-lH-pyrrolo [2,3-b] pyridine (50.0 g, 137 mmol) (see, e.g., Example 2) and tetrahydrofuran (THF, 266 g, 300 mL) under N2. To this mixture at -70 °C was added 2.0 M lithium
diisopropylamide in THF/heptane/ethyl benzene (77.4 g, 95 mL, 190 mmol, 1.4 eq.). The mixture was stirred at -70 °C for 1 h. To the mixture was added /V- formyl morpholine (29.7 g, 258 mmol, 1.9 eq.) in THF (22. 2 g, 25 mL) dropwise. The reaction was done in 30 min after addition. LC/MS showed that the desired product, 4-chloro-5-(l, 3-dioxolan-2-yl)-l-(phenylsulfonyl)- 1 //-pyrrolo [2, 3-61 pyridine-2-carbaldehyde, was formed cleanly. The reaction was quenched with acetic acid (16.4 g, 15.6 mL, 274 mmol, 2.0 eq.) and the dry ice cooling was removed. To the mixture was added morpholine (33.7 g, 33.5 mL, 387 mmol, 2.83 eq.) followed by acetic acid (74.0 g, 70 mL, 1231 mmol, and 9.0 eq.) at 0 °C (internal temperature rose from 0 °C to 18 °C) and stirred overnight. Sodium triacetoxyborohydride (52.50 g, 247.7 mmol, 1.8 eq.) was added and the reaction mixture temperature rose from 20 °C to 32 °C. The mixture was stirred at room temperature for 30 min. HPLC & LC/MS indicated the reaction was complete. Water (100 g, 100 mL) was added followed by 2.0 M sodium carbonate (Na2C03) in water (236 g, 200 mL, 400 mmol, 2.9 eq.) slowly (off gas!). The mixture was stirred for about 30 min. The organic layer was separated and water (250 g, 250 mL) and heptane (308 g, 450 mL) were added. The resulting slurry was stirred for 1 h and the solid was collected by filtration. The wet cake was washed with heptane twice (75.00 mL x 2, 51.3 g x 2) before being dried in oven at 50 °C overnight to give the desired product, 4-((4-chloro-5-( 1 3-dioxolan-2-yl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridin-2-yl)methyl)morpholine as a light brown solid (52.00 g, 81.8 % yield): LCMS calculated for C21H23CIN2O5S [M+H]+: 464.00; Found: 464.0; ftf NMR ^OO MHz, DMSO-de) d 8.48 (s, 1 H), 8.38 (m, 2H), 7.72 (m, 1H), 7.64 (m, 2H), 6.83 (s, 1H), 6.13 (s, 1H), 4.12 (m, 2H), 4.00 (m, 2H), 3.92 (s, 2H), 3.55 (m, 4H), 2.47 (m, 4H).
Step 2: Synthesis of 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-lH-pyrrolo[2, 3-b] pyridine-5 -carbaldehyde
To a 2 L reactor with a thermocouple, an addition funnel, and a mechanical stirrer was charged 4-((4-chloro-5 -(1 ,3 -dioxolan-2-yl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo [2,3 -6]pyridin-2-yl)methyl)morpholine (20.00 g, 43.1 mmol) and dichloromethane (265 g, 200 mL) at room temperature. The resulting mixture was stirred at room temperature (internal temperature
was 19.5 °C) to achieve a solution. To the resulting solution was added an aqueous hydrochloric acid solution (0.5 M, 240 g, 200.0 ml, 100 mmol, 2.32 eq.) at room temperature in 7 min. After over 23 h agitations at room temperature, the bilayer reaction mixture turned into a thick colorless suspension. When HPLC showed the reaction was complete, the slurry was cooled to 0-5 °C and aqueous sodium hydroxide solution (1 N, 104 g, 100 mL, 100 mmol, and 2.32 eq.) was added in about 10 min to adjust the pH of the reaction mixture to 10-11. «-Heptane (164 g, 240 mL) was added and the reaction mixture and the mixture were stirred at room temperature for 1 h. The solid was collected by filtration and the wet cake was washed with water (2 x 40 mL), heptane (2 x 40 ml) before being dried in oven at 50 °C under vacuum to afford the desired product, 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-/i |pyridine-5-carbaldehyde as a light brown solid (16.9 g, 93% yield): LCMS calculated for C19H19CIN3O4S [M+H]+: 420.00; Found: 420.0; ¾ NMR (400 MHz, DMSO-de) d 10.33 (s, 1H), 8.76 (s, 1 H), 8.42 (m, 2H), 7.74 (m, 1H), 7.65 (m, 2H), 6.98 (s, 1H), 3.96 (m, 2H), 3.564 (m, 4H), 2.51 (m, 4H).
To a 2-L reactor equipped with a thermocouple, a nitrogen inlet and mechanical stirrer were charged AOV-dimethyl formamide (450 mL, 425 g), 4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridine-5-carbaldehyde (30.0 g, 71.45 mmol) and 2,6-difluoro-3,5-dimethoxyanihne (14.2 g, 75.0 mmol). To this suspension (internal temperature 20 °C) was added chlorotrimethylsilane (19.4 g, 22. 7 mL, 179 mmol) dropwise in 10 min at room temperature (internal temperature 20-23 °C). The suspension changed into a solution in 5 min after the chlorotrimethylsilane addition. The solution was stirred at room temperature for 1.5 h before cooled to 0-5 °C with ice-bath. Borane-THF complex in THF (1.0 M, 71.4 mL, 71.4 mmol, 64.2 g, 1.0 eq.) was added dropwise via additional funnel over 30 min while maintaining temperature at 0-5 °C. After addition, the mixture was stirred for 4 h. Water (150 g, 150 mL) was added under ice-bath cooling in 20 min, followed by slow addition of ammonium hydroxide solution (28% N¾, 15.3 g, 17 ml, 252 mmol, 3.53 eq.) to pH 9-10 while maintaining the temperature below 10 °C. More water (250 mL, 250 g) was added through the additional funnel. The slurry was stirred for 30 min and the solids were collected by filtration. The wet cake was washed with water (90 g x 2, 90 ml x 2) and heptane (61.6 g x2, 90 ml x 2). The product w as suction dried overnight to give the desired product LG-((4-chloro-2-(morphohnomethyl)-l-(phenylsulfonyl)-li/-pyrrolo[2,3-Z>]pyridin-5-yl)methyl)-2,6- difluoro-3,5-dimethoxyaniline (41.6 g, 96% yield): LCMS calculated for C27H28ClF2N405S[M+H]+: 593.10; Found: 593.1 ; ¾ NMR (400 MHz, DMSO-d6) 5 8.36 (m, 2H), 8.28 (s, 1H), 7.72 (m, 1H), 7.63 (m, 2H), 6.78 (s, 1H), 6.29 (m, 1H), 5.82 (m, 1H), 4.58 (m, 2H), 3.91 (s, 2H), 3.76 (s, 6H), 3.56 (m, 4H), 2.47 (m, 4H).
To a 2-L, 3-neck round bottom flask fitted with a thermocouple, a nitrogen bubbler inlet, and a magnetic stir were charged /V-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-li/-pyrrolo[2,3-b]pyridin-5-yl)methyl)-2,6-difluoro-3,5-dimethoxyaniline (67.0 g, 113 mmol) and acetonitrile (670 ml, 527 g). The suspension was cooled to 0-5 °C.
To the mixture was charged ethyl isocyanate (17.7 mL, 15.9 g, 224 mmol, 1.98 eq.) over 30 sec. The temperature stayed unchanged at 0.7 °C after the charge. Methanesulfonic acid (16.1 mL, 23.9 g, 248 mmol, 2.2 eq.) was charged dropwise over 35 min while maintaining the temperature below 2 °C. The mixture was warmed to room temperature and stirred overnight. At 24 h after addition showed that the product was 93.7%, unreacted SM was 0.73% and the major impurity (bis-isocyanate adduct) was 1.3%. The mixture was cooled with an ice-bath and quenched with sodium hydroxide (NaOH) solution (1.0M, 235 mL, 244 g, 235 mmol, 2.08 eq.) over 20 min and then saturated aqueous sodium bicarbonate
(NaHCCh) solution (1.07 M, 85 mL, 91 g, 0.091 mol, 0.80 eq.) over 10 min. Water (550 mL, 550 g) was added and the liquid became one phase. The mixture was stirred for 2 h and the solids were collected by filtration, washed with water (165 mL, 165 g) to give l-((4-chloro-2-(morpholinomethyl)- 1 -(phenylsulfonyl)- 1 //-pyrrolo| 2.3-6 |p\ ri din-5 -y l (methy l )- 1 -(2,6-difluoro-3,5-dimethoxyphenyl)-3-ethylurea ( 70.3 g, 93.7% yield).
The crude l-((4-chloro-2-(morpholinomethyl)-l -(phenylsulfonyl)- li/-pyrrolo [2, 3-61 pyridin-5-yl) methyl)- 1 -(2, 6-difluoro-3, 5-dimethoxyphenyl)-3-ethylurea (68.5 g, 103 mmol) was added in to acetonitrile (616 mL, 485 g). The mixture was heated 60-65 °C and an amber colored thin suspension was obtained. The solid was filtered off with celite and the celite was washed with acetonitrile (68.5 mL, 53.8 g). To the pale yellow filtrate was added water (685 g, 685 ml) to form a slurry. The slurry was stirred overnight at room temperature and filtered. The solid was added to water (685 mL, 685 g) and stirred at 60 °C for 2 h. The solid was filtered and re-slurred in heptane (685 mL, 469 g) overnight. The product was dried in an oven at 50 °C under vacuum for 48 h to afford l-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)- 1 //-pyrrolo|2.3-6 |pyridin-5-yl)methyl)- 1 -(2.6-difluoro-3.5-
dimethoxyphenyl)-3-ethylurea as a colorless solid (62.2 g, 90.8% yield, 99.9% purity by HPLC area%). KF was 0.028%. Acetonitrile (by ‘H NMR) was about 1.56%, DCM (by ‘H NMR) 2.0%: LCMS calculated for C30H33CIF2N5O6S [M+H]+: EM: 664.17; Found: 664.2; ¾ NMR (400 MHz, DMSO-de) d 8.33 (m, 2H), 8.31 (s, 1H), 7.72 (m, 1H), 7.64 (m, 1H), 6.96 (m, 2H), 6.73 (s, 1H), 6.43 (m, 1H), 4.87 (s, 2H), 3.90 (s, 2H), 3.77 (s, 6H), 3.54 (m, 4H),
To a 2000 mL flask equipped with a thermal couple, a nitrogen inlet, and a mechanical stirrer were charged dry l-((4-chloro-2-(morpholinomethyl)-l-(phenylsulfonyl)-1 //-pyrrolo| 2.3-6 |pyridin-5-yl)methyl)- 1 -(2.6-dinuoro-3.5-dimetho\yphenyl)-3-ethylurea (30.0 g, 45.2 mmol, KF=0. l l%) and tetrahydrofuran (1200 mL, 1063 g). To this suspension at room temperature was charged 1.0 M lithium hexamethyldisilazide in THF (62.3 mL, 55.5 g, 62.3 mmol, 1.38 eq). The mixture turned into a solution after the base addition. The reaction mixture was stirred for 2 h and HPLC shows the starting material was not detectable. To this mixture was added 1.0 M hydrochloric acid (18.1 mL, -18.1 g. 18.1 mmol, 0.4 eq.). The solution was concentrated to 600 mL and water (1200 mL, 1200 g) was added. Slurry was formed after water addition. The slurry was stirred for 30 min at room temperature and the solid was collected by filtration. The wet cake was washed with water twice (60 mLx2,
60 gx2) and dried at 50 °C overnight to give 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-l,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4, 3-d]pyrimidin-2-one as a light brown solid (26.58 g, as-is yield 93.7%): THF by ‘H NMR 0.32%, KF 5.26%, adjusted yield was 88.5%: LCMS calculated for C30H32F2N5O6S [M+H]+: EM: 628.20; Found: 628.2; ¾ NMR (400 MHz, DMSO-de) d 8.41 (m, 2H), 8.07 (s, 1H), 7.70 (m, 1H), 7.63 (m, 2H), 7.05 (m, 1H), 6.89 (s, 1H), 4.76 (s, 2H), 4.09 (m, 2H), 3.93 (s, 2H), 3.89 (s, 6H), 3.60 (m, 4H), 2.50 (m, 4H), 1.28 (m, 3H).
To a stirring suspension of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholinomethyl)-7-(phenylsulfonyl)-l,3,4,7-tetrahydro-2i/-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (10.0 g, 15.93 mmol) in l,4-dioxane (100 ml, 103 g) in a 500 mL flask equipped with a nitrogen inlet, a condenser, a thermocouple and a heating mantle was added 1 M aqueous sodium hydroxide (63.7 ml, 66.3 g, 63.7 mmol). The reaction mixture was heated at 75 °C for 18 h. LCMS showed the reaction was complete. Water (100 mL, 100 g) was added to give a thick suspension. This slurry was stirred at room temperature for 1 h and filtered. The cake was washed with water (3 x 10 mL, 3 x 10 g) and heptane (2 x 10 mL, 2 x 6.84 g). The cake was dried overnight by pulling a vacuum through the filter cake and then dried in an oven at 50 °C under vacuum overnight to give 3-(2,6-difluoro-3,5-dimethoxyphenyl)-l-ethyl-8-(morpholin-4-ylmethyl)-l,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5, 6]pyrido[4,3-d]pyrimidin-2-one (6.8 g, 87.6% yield): LCMS calculated for C24H28F2N5O4 [M+H]+: 488.20; Found: 488.2.
[0833] To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (Example 49, Step 3: 900 mg, 2.32 mmol) in N,N-dimethylformamide (20 mL) cooled to 0° C. was added sodium hydride (185 mg, 4.63 mmol, 60 wt % in mineral oil). The resulting mixture was stirred at 0° C. for 30 min then benzenesulfonyl chloride (0.444 mL, 3.48 mmol) was added. The reaction mixture was stirred at 0° C. for 1.5 h at which time LC-MS showed the reaction completed to the desired product. The reaction was quenched with saturated NH4Cl solution and diluted with water. The white precipitate was collected via filtration then washed with water and hexanes, dried to afford the desired product (1.2 g, 98%) as a white solid which was used in the next step without further purification. LC-MS calculated for C25H23F2N4O5S [M+H]+ m/z: 529.1; found: 529.1.
[0835] To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (1.75 g, 3.31 mmol) in tetrahydrofuran (80 mL) at −78° C. was added freshly prepared lithium diisopropylamide (1M in tetrahydrofuran (THF), 3.48 mL, 3.48 mmol). The resulting mixture was stirred at −78° C. for 30 min then N,N-dimethylformamide (1.4 mL, 18 mmol) was added slowly. The reaction mixture was stirred at −78° C. for 30 min then quenched with water and extracted with EtOAc. The organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 20% EtOAc in DCM to give the desired product as a white solid (1.68 g, 91%). LC-MS calculated for C26H23F2N4O6S (M+H)+ m/z: 557.1; found: 556.9.
[0837] To a solution 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-2-oxo-7-(phenylsulfonyl)-2,3,4,7-tetrahydro-1H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidine-8-carbaldehyde (1.73 g, 3.11 mmol) in dichloromethane (50 mL) was added morpholine (0.95 mL, 11 mmol), followed by acetic acid (2 mL, 30 mmol). The resulting yellow solution was stirred at room temperature overnight then sodium triacetoxyborohydride (2.3 g, 11 mmol) was added. The mixture was stirred at room temperature for 3 h at which time LC-MS showed the reaction went to completion to the desired product. The reaction was quenched with saturated NaHCO3 then extracted with ethyl acetate (EtOAc). The organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 40% EtOAc in DCM to give the desired product as a yellow solid (1.85 g, 95%). LC-MS calculated for C30H32F2N5O6S (M+H)+ m/z: 628.2; found: 628.0.
[0838] To a solution of 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-7-(phenylsulfonyl)-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidin-2-one (1.5 g, 2.4 mmol) in tetrahydrofuran (40 mL) was added tetra-n-butylammonium fluoride (1M in THF, 7.2 mL, 7.2 mmol). The resulting solution was stirred at 50° C. for 1.5 h then cooled to room temperature and quenched with water. The mixture was extracted with dichloromethane (DCM) and the organic extracts were combined then washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography eluted with 0 to 10% MeOH in DCM to give the desired product as a white solid, which was further purified by prep HPLC (pH=2, acetonitrile/H2O). LC-MS calculated for C24H28F2N5O4 (M+H)+ m/z: 488.2; found: 488.0. 1H NMR (500 MHz, DMSO) δ 12.09 (s, 1H), 8.06 (s, 1H), 7.05 (t, J=8.1 Hz, 1H), 6.87 (s, 1H), 4.78 (s, 2H), 4.50 (s, 2H), 4.17 (q, J=6.8 Hz, 2H), 3.97 (br, 2H), 3.89 (s, 6H), 3.65 (br, 2H), 3.37 (br, 2H), 3.15 (br, 2H), 1.37 (t, J=6.8 Hz, 3H).
^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3): 479. hdl:10665/330907.
Clinical trial number NCT02924376 for “Efficacy and Safety of Pemigatinib in Subjects With Advanced/Metastatic or Surgically Unresectable Cholangiocarcinoma Who Failed Previous Therapy – (FIGHT-202)” at ClinicalTrials.gov
Common side effects are diarrhea, palmar-plantar erythrodysesthesia (burning or tingling discomfort in the hands and feet), nausea, fatigue, hepatotoxicity (liver damage), vomiting, stomatitis (inflammation of the mouth and lips), decreased appetite, abdominal pain, headache, anemia and rash.[5][6] Pregnant or breastfeeding women should not take Tucatinib because it may cause harm to a developing fetus or newborn baby.[5]
Tucatinib was approved for medical use in Australia in August 2020.[7]
Medical uses
Tucatinib is a kinase inhibitor indicated in combination with trastuzumab and capecitabine for treatment of adults with advanced unresectable or metastatic HER2-positive breast cancer, including those with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting.[8]
Clinical trials
Two early stage clinical trials have reported encouraging results, both of which had options to enroll subjects with central nervous system (CNS) metastases.[2][9][10][11][12][10] HER2CLIMB is a Phase 2 randomized, double-blinded, placebo-controlled study of tucatinib in combination with trastuzumab and capecitabine in patients with pretreated, unresectable locally advanced or metastatic HER2-positive breast cancer.[13]
History
In April 2020, the U.S. Food and Drug Administration (FDA) approved tucatinib in combination with chemotherapy (trastuzumab and capecitabine) for the treatment of adults with advanced forms of HER2-positive breast cancer that can’t be removed with surgery, or has spread to other parts of the body, including the brain, and who have received one or more prior treatments.[5][6][14]
Tucatinib is a kinase inhibitor meaning it blocks a type of enzyme (kinase) and helps prevent the cancer cells from growing.[5] Tucatinib is approved for treatment after adults have taken one or more anti-HER2-based regimens in the metastatic setting.[5] The FDA approved tucatinib based on the results of the HER2CLIMB trial (NCT02614794) enrolling 612 subjects who had HER2-positive advanced unresectable or metastatic breast cancer and had prior treatment with trastuzumab, pertuzumab and ado-trastuzumab emtansine (T-DM1).[5][6] Subjects with previously treated and stable brain metastases, as well as those with previously treated and growing or untreated brain metastases, were eligible for the clinical trial, and 48% of enrolled subjects had brain metastases at the start of the trial.[5]
Subjects received either tucatinib 300 mg twice daily plus trastuzumab and capecitabine (tucatinib arm, n=410) or placebo plus trastuzumab and capecitabine (control arm, n=202).[6] The primary endpoint was progression-free survival (PFS), or the amount of time when there was no growth of the tumor, assessed by a blinded independent central review, evaluated in the initial 480 randomized patients.[5][6] The median PFS in subjects who received tucatinib, trastuzumab, and capecitabine was 7.8 months (95% CI: 7.5, 9.6) compared to 5.6 months (95% CI: 4.2, 7.1) in those subjects who received placebo, trastuzumab, and capecitabine (HR 0.54; 95% CI: 0.42, 0.71; p<0.00001).[5][6] Overall survival and PFS in subjects with brain metastases at baseline were key secondary endpoints.[5] The median overall survival in subjects who received tucatinib, trastuzumab, and capecitabine was 21.9 months (95% CI: 18.3, 31.0) compared to 17.4 months (95% CI: 13.6, 19.9) in subjects who received placebo, trastuzumab, and capecitabine (HR: 0.66; 95% CI: 0.50, 0.87; p=0.00480).[5][6] The median PFS in subjects with brain metastases at baseline who received tucatinib, trastuzumab and capecitabine was 7.6 months (95% CI: 6.2, 9.5) compared to 5.4 months (95% CI: 4.1, 5.7) in subjects who received placebo, trastuzumab and capecitabine (HR: 0.48; 0.34, 0.69; p<0.00001).[5][6]
Recently, the Mao team reported a new route for the efficient synthesis of Tucatinib.
The results were published on Synthesis (DOI: 10.1055/s-0037-1610706).
Previously, the synthesis report route of Tucatinib was published by Array BioPharma in a patent document (WO 2007059257, 2007). The synthetic route reported in the patent is shown in the figure below:
Using 4-nitro-2-cyanoaniline as the raw material, the first step is to condense with DMF-DMA to prepare imine 3 (yield 87%); subsequent catalytic hydrogenation of palladium on carbon to reduce the nitro group to obtain the amine 4 (90% yield); followed by 1,1&39;-thiocarbonyldiimidazole (TCDI) and The amino alcohol undergoes condensation to prepare the thiourea derivative 5 (yield is only 34%); further with the intermediate 6 to undergo ring-closure reaction to obtain the key intermediate 7 (yield 62%) ; Finally, under the action of p-toluenesulfonic acid, intramolecular dehydration and ring closure to form oxazoline, complete the synthesis of the target compound tucatinib.
Reverse synthesis analysis
The author broke the bond of Tucatinib from two points a and b and split them into three fragments. : Thioether oxazoline 17, nitrobenzene 3 and the key fragment of the original research route 6.
Preparation of key fragment 6
4-nitro-3-methylphenol 8 as a starting point The material, with pyridine derivative 9, undergoes aromatic affinity substitution reaction to prepare aryl ether 10 (yield 64%); then it is condensed with DMF-DMA, and then treated with hydroxylamine hydrochloride. The step yield was 81% to obtain the oxime derivative 12; subsequently, the ring was closed under the treatment of trifluoroacetic anhydride, the mostAfter palladium-catalyzed hydrogenation to reduce the nitro group, the key aniline triazole 6 was successfully prepared, with a total yield of 32.8%.
aromatic ring skeleton construction
fragment 3 was synthesized according to the method reported in the literature. The estimated aromatic ring fragment was then constructed with the aniline triazole 6 prepared above:
Compound 6 and fragment 3 were cyclized in acetic acid , 14 was successfully prepared, and finally the nitro group was reduced by palladium-catalyzed hydrogenation to obtain the key arylamine 15 with a two-step yield of 76.4%.
Fragment 17 and Tucatinib synthesis
amino alcohol and 1,1&39;-thiocarbonyl diimidazole (TCDI) The ring is closed to obtain 16, which is then treated with methyl trifluoromethanesulfonate to obtain oxazoline 17, with a total yield of 67.23% in the two steps.
oxazoline17 and arylamine 15 in the presence of cesium carbonate, heated in DMF for 20 hours, and finally completed the synthesis of Tucatinib with a yield of 76%.
Comparison of the new route and the patent route
The yield of the last step of the patent is unknown, starting with key intermediates 3 and 6, total income The rate is less than 19%.
The overview of the new route is as follows:
Correspondingly, starting from the intermediate 3 and 6, the total yield of the new route There is a significant improvement to 39%. Moreover, the purity of the product and other aspects also meet the requirements of API.
Comment
Tucatinib (Tukysa) Tucatinib/Tucatinib as a small-molecule oral tyrosine kinase (TKI) inhibitor for HER2 Positive breast cancer has highly specific targeting selectivity. The study of the new synthetic route
effectively improves the production efficiency in terms of ensuring the purity of the compound, and the raw materials used are relatively simple and easy to obtain.
Medicinal chemists have completed the research and development and synthesis of compounds (from 0 to 1), while process chemists have optimized the synthetic routes and processes, so that the compounds can be prepared more simply, efficiently, economically and environmentally.
SYN PATENT
CN 111825604
PAPER
Synthesis (2019), 51(13), 2660-2664
Abstract
A new and improved synthetic route to tucatinib is described that involves three key intermediates. The first of these, 4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylaniline, was prepared on a 100 g scale in 33% yield over five steps and 99% purity. Next, N 4-(4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)-3-methylphenyl)quinazoline-4,6-diamine was isolated in 67% yield over three steps and >99% purity. Then, 4,4-dimethyl-2-(methylthio)-4,5-dihydrooxazole trifluoromethanesulfonate was prepared under mild conditions in 67% yield over two steps. Finally, tucatinib was obtained in 17% yield over nine steps and in >99% purity (HPLC). Purification methods used to isolate the product and the intermediates involved in the route are also reported.
References
^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 161. hdl:10665/331046.
Clinical trial number NCT02614794 for “A Study of Tucatinib vs. Placebo in Combination With Capecitabine & Trastuzumab in Patients With Advanced HER2+ Breast Cancer (HER2CLIMB)” at ClinicalTrials.gov
Triheptanoin is a source of heptanoate fatty acids, which can be metabolized without the enzymes of long chain fatty acid oxidation.4 In clinical trials, patients with long chain fatty acid oxidation disorders (lc-FAODs) treated with triheptanoin are less likely to develop hypoglycemia, cardiomyopathy, rhabdomyolysis, and hepatomegaly.1,2 Complications in lc-FAOD patients are reduced from approximately 60% to approximately 10% with the addition of triheptanoin.2
Triheptanoin was granted FDA approval on 30 June 2020.4
Triheptanoin, sold under the brand name Dojolvi, is a medication for the treatment of children and adults with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD).[1][2][3]
The most common adverse reactions include abdominal pain, diarrhea, vomiting, and nausea.[1][2][3]
Triheptanoin was approved for medical use in the United States in June 2020.[4][2][3]
Since triheptanoin is composed of odd-carbon fatty acids, it can produce ketone bodies with five carbon atoms, as opposed to even-carbon fatty acids which are metabolized to ketone bodies with four carbon atoms. The five-carbon ketones produced from triheptanoin are beta-ketopentanoate and beta-hydroxypentanoate. Each of these ketone bodies easily crosses the blood–brain barrier and enters the brain.
Medical uses
Dojolvi is indicated as a source of calories and fatty acids for the treatment of children and adults with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD).[1][2]
Triheptanoin was approved for medical use in the United States in June 2020.[4][2]
The FDA approved triheptanoin based on evidence from three clinical trials (Trial 1/NCT018863, Trial 2/NCT022141 and Trial 3/NCT01379625).[3] The trials enrolled children and adults with LC-FAOD.[3] Trials 1 and 2 were conducted at 11 sites in the United States and the United Kingdom, and Trial 3 was conducted at two sites in the United States.[3]
Trial 1 and Trial 2 were used to evaluate the side effects of triheptanoin.[3] Both trials enrolled children and adults diagnosed with LC-FAOD.[3] In Trial 1, participants received triheptanoin for 78 weeks.[3] Trial 2 enrolled participants from other trials who were already treated with triheptanoin (including those from Trial 1) as well as participants who were never treated with triheptanoin before.[3] Trial 2 is still ongoing and is planned to last up to five years.[3]
The benefit of triheptanoin was evaluated in Trial 3 which enrolled enrolled children and adults with LC-FAOD.[3] Half of the participants received triheptanoin and half received trioctanoin for four months.[3] Neither the participants nor the investigators knew which treatment was given until the end of the trial.[3] The benefit of triheptanoin in comparison to trioctanoin was assessed by measuring the changes in heart and muscle function.[3]
Names
Triheptanoin is the international nonproprietary name.[17]
^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3): 694. hdl:10665/330879. License: CC BY-NC-SA 3.0 IGO.
Further reading
de Almeida Rabello Oliveira M, da Rocha Ataíde T, de Oliveira SL, de Melo Lucena AL, de Lira CE, Soares AA, et al. (March 2008). “Effects of short-term and long-term treatment with medium- and long-chain triglycerides ketogenic diet on cortical spreading depression in young rats”. Neurosci. Lett. 434 (1): 66–70. doi:10.1016/j.neulet.2008.01.032. PMID18281154. S2CID7754768.
“Triheptanoin”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT01379625 for “Study of Triheptanoin for Treatment of Long-Chain Fatty Acid Oxidation Disorder (Triheptanoin)” at ClinicalTrials.gov
ClassAntiparasitics; Heterocyclic compounds; Pyridines; Small molecules
Mechanism of ActionChelating agents; Metalloprotease inhibitors
Registered Pediculosis
27 Jul 2020Registered for Pediculosis (In adolescents, In children, In infants, In adults) in USA (Topical)
18 Jun 2020FDA assigns PDUFA action date of 12/08/2020 for Abametapir for Pediculosis (Dr Reddy’s Laboratories website, June 2020)
31 Mar 2019Abametapir is still in preregistration phase for Pediculosis in USA
Abametapir is a novel pediculicidal metalloproteinase inhibitor used to treat infestations of head lice.4 The life cycle of head lice (Pediculus capitis) is approximately 30 days, seven to twelve of which are spent as eggs laid on hair shafts near the scalp.2 Topical pediculicides generally lack adequate ovicidal activity,2 including standard-of-care treatments such as permethrin, and many require a second administration 7-10 days following the first to kill newly hatched lice that resisted the initial treatment. The necessity for follow-up treatment may lead to challenges with patient adherence, and resistance to agents like permethrin and pyrethrins/piperonyl butoxide may be significant in some areas.3
Investigations into novel ovicidal treatments revealed that several metalloproteinase enzymes were critical to the egg hatching and survival of head lice, and these enzymes were therefore identified as a potential therapeutic target.1 Abemetapir is an inhibitor of these metalloproteinase enzymes, and the first topical pediculicide to take advantage of this novel target. The improved ovicidal activity (90-100% in vitro) of abemetapir allows for a single administration, in contrast to many other topical treatments, and its novel and relatively non-specific mechanism may help to curb the development of resistance to this agent.1
Abametapir was first approved for use in the United States under the brand name Xeglyze on July 27, 2020.6
Abametapir, sold under the brand name Xeglyze, is a medication used for the treatment of head lice infestation in people six months of age and older.[1][2]
The most common side effects include skin redness, rash, skin burning sensation, skin inflammation, vomiting, eye irritation, skin itching, and hair color changes.[2]
Abametapir, a metalloproteinase inhibitor, demonstrated potent pediculicidal activity in preclinical studies. In vitro assays showed abametapir lotion (0.74–1% w/v) achieved >95% mortality of Pediculus humanus capitis adults and eggs within 10 minutes of exposure. Ex vivo human hair assays confirmed ovicidal efficacy, with >90% inhibition of egg hatching compared to vehicle controls. Mechanistic studies indicated abametapir disrupted metalloproteinase-dependent processes essential for louse development and egg viability. Toxicology studies in rodents and rabbits showed no significant systemic toxicity at topical doses up to 3% formulation, supporting its advancement into clinical evaluation as a single-application pediculicide.
Medical uses
Abametapir is indicated for the topical treatment of head lice infestation in people six months of age and older.[1][2]
History
The U.S. Food and Drug Administration (FDA) approved abametapir based on evidence from two identical clinical trials of 699 participants with head lice.[2] The trials were conducted at fourteen sites in the United States.[2]
The benefit and side effects of abametapir were evaluated in two clinical trials that enrolled participants with head lice who were at least six months old.[2]
About half of all enrolled participants was randomly assigned to abametapir and the other half to placebo.[2] Abametapir lotion or placebo lotion were applied once as a ten-minute treatment to infested hair.[2] The benefit of abametapir in comparison to placebo was assessed after 1, 7 and 14 days by comparing the counts of participants in each group who were free of live lice.[2]
SYN
Ronald Harding, Lewis David Schulz, Vernon Morrison Bowles, “Pediculicidal composition.” WIPO Patent WO2015107384A2, published July, 2015.
Percent Composition: C 41.81%, H 4.56%, N 14.63%, O 27.85%, S 11.16%
Literature References: Prepn from 5-nitrofurfural and 4-amino-3-methyltetrahydro-1,4-thiazine 1,1-dioxide: Herlinger et al.,DE1170957 corresp to US3262930 (1964 and 1966 to Bayer). Series of articles on pharmacology and clinical findings: Arzneim.-Forsch.22, 1563-1642 (1972). Toxicity data: K. Hoffmann, ibid. 1590.
Properties: Orange-red crystals from dil acetic acid, mp 180-182°. LD50 in mice, rats (mg/kg): 3720, 4050 by gavage (Hoffmann).
Melting point: mp 180-182°
Toxicity data: LD50 in mice, rats (mg/kg): 3720, 4050 by gavage (Hoffmann)
Common side effects include abdominal pain, headache, nausea, and weight loss.[1] There are concerns from animal studies that it may increase the risk of cancer but these concerns have not be found in human trials.[5] Nifurtimox is not recommended in pregnancy or in those with significant kidney or liver problems.[5] It is a type of nitrofuran.[5]
Chagas disease, caused by a parasite known as Trypanosoma cruzi (T.cruzi), is a vector-transmitted disease affecting animals and humans in the Americas. It is commonly known as American Trypanosomiasis.11
The CDC estimates that approximately 8 million people in Central America, South America, and Mexico are infected with T. cruzi, without symptoms. If Chagas disease is left untreated, life-threatening sequelae may result.11
Nifurtimox, developed by Bayer, is a nitrofuran antiprotozoal drug used in the treatment of Chagas disease. On August 6 2020, accelerated FDA approval was granted for its use in pediatric patients in response to promising results from phase III clinical trials. Continued approval will be contingent upon confirmatory data.10 A convenient feature of Bayer’s formulation is the ability to divide the scored tablets manually without the need for pill-cutting devices.10
Medical uses
Nifurtimox has been used to treat Chagas disease, when it is given for 30 to 60 days.[7][8] However, long-term use of nifurtimox does increase chances of adverse events like gastrointestinal and neurological side effects.[8][9] Due to the low tolerance and completion rate of nifurtimox, benznidazole is now being more considered for those who have Chagas disease and require long-term treatment.[5][9]
In the United States nifurtimox is indicated in children and adolescents (birth to less than 18 years of age and weighing at least 2.5 kilograms (5.5 lb) for the treatment of Chagas disease (American Trypanosomiasis), caused by Trypanosoma cruzi.[2]
Nifurtimox has also been used to treat African trypanosomiasis (sleeping sickness), and is active in the second stage of the disease (central nervous system involvement). When nifurtimox is given on its own, about half of all patients will relapse,[10] but the combination of melarsoprol with nifurtimox appears to be efficacious.[11] Trials are awaited comparing melarsoprol/nifurtimox against melarsoprol alone for African sleeping sickness.[12]
Combination therapy with eflornithine and nifurtimox is safer and easier than treatment with eflornithine alone, and appears to be equally or more effective. It has been recommended as first-line treatment for second-stage African trypanosomiasis.[13]
Pregnancy and breastfeeding
Use of nifurtimox should be avoided in pregnant women due to limited use.[5][8][14] There is limited data shown that nifurtimox doses up to 15 mg/kg daily can cause adverse effects in breastfed infants.[15] Other authors do not consider breastfeeding a contraindication during nifurtimox use.[15]
Side effects
Side effects occur following chronic administration, particularly in elderly people. Major toxicities include immediate hypersensitivity such as anaphylaxis and delayed hypersensitivity reaction involving icterus and dermatitis. Central nervous system disturbances and peripheral neuropathy may also occur.[8]
Contraindications
Nifurtimox is contraindicated in people with severe liver or kidney disease, as well as people with a background of neurological or psychiatric disorders.[5][16][20]
Mechanism of action
Nifurtimox forms a nitro-anion radical metabolite that reacts with nucleic acids of the parasite causing significant breakdown of DNA.[8] Its mechanism is similar to that proposed for the antibacterial action of metronidazole. Nifurtimox undergoes reduction and creates oxygen radicals such as superoxide. These radicals are toxic to T. cruzi. Mammalian cells are protected by presence of catalase, glutathione, peroxidases, and superoxide dismutase. Accumulation of hydrogen peroxide to cytotoxic levels results in parasite death.[8]
Manufacturing and availability
A bottle of nifurtimox
Nifurtimox is sold under the brand name Lampit by Bayer.[3] It was previously known as Bayer 2502.
Nifurtimox is only licensed for use in Argentina and Germany,[citation needed] where it is sold as 120-mg tablets. It was approved for medical use in the United States in August 2020.[3]
Nifurtimox, 1,1-dioxide 4-[(5-nitrofuryliden)amino]-3-methylthiomorpholine (37.4.7), is made by the following scheme. Interaction of 2-mercaptoethanol with propylene oxide in the presence of potassiumhydroxide gives (2-hydroxyethyl)-(2-hydroxypropylsul-fide) (37.4.3), which undergoes intramolecular dehydration using potassium bisulfate to make 2-methyl-1,4-oxithiane (37.4.4). Oxidation of this using hydrogen peroxide gives 2-methyl-1,4-oxithian-4,4-dioxide (37.4.5), which when reacted with hydrazine transforms to 4-amino-3-methyltetrahydro-1,4-thiazin-1,1-dioxide (37.4.6). Reacting this with 5-nitrofurfurol gives the corresponding hydrazone—the desired nifurtimox [58,59].
58. H. Herlinger, K.H. Heinz, S. Petersen, M.Bock, Ger. Pat. 1.170.957 (1964).
59. H. Herlinger, K.H. Heinz, S. Petersen, M. Bock, U.S. Pat. 3.262.930 (1966)
^ Jump up to:abcWorld Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^Pepin J, Milord F, Mpia B, et al. (1989). “An open clinical trial of nifurtimox for arseno-resistant T. b. gambiense sleeping sickness in central Zaire”. Trans R Soc Trop Med Hyg. 83(4): 514–7. doi:10.1016/0035-9203(89)90270-8. PMID2694491.
^Castro, José A.; de Mecca, Maria Montalto; Bartel, Laura C. (2006-08-01). “Toxic side effects of drugs used to treat Chagas’ disease (American trypanosomiasis)”. Human & Experimental Toxicology. 25 (8): 471–479. doi:10.1191/0960327106het653oa. ISSN0960-3271. PMID16937919.
^“Chagas disease”. World Health Organization. Archived from the original on 2014-02-27. Retrieved 2016-11-08.
^Clinical trial number NCT00601003 for “Study of Nifurtimox to Treat Refractory or Relapsed Neuroblastoma or Medulloblastoma” at ClinicalTrials.gov. Retrieved on July 10, 2009.
External links
“Nifurtimox”. Drug Information Portal. U.S. National Library of Medicine.
Pralsetinib, sold under the brand name Gavreto, is a medication for the treatment of metastatic RET fusion-positive non-small cell lung cancer (NSCLC).[1] Pralsetinib is a tyrosine kinase inhibitor. It is taken by mouth.[1]
The most common adverse reactions include increased aspartate aminotransferase (AST), decreased hemoglobin, decreased lymphocytes, decreased neutrophils, increased alanine aminotransferase (ALT), increased creatinine, increased alkaline phosphatase, fatigue, constipation, musculoskeletal pain, decreased calcium, hypertension, decreased sodium, decreased phosphate, and decreased platelets.[1]
Pralsetinib was approved for medical use in the United States in September 2020.[1][2][3][4]
Medical uses
Pralsetinib is indicated for the treatment of adults with metastatic RET fusion-positive non-small cell lung cancer (NSCLC) as detected by an FDA approved test.[1][4]
History
Efficacy was investigated in a multicenter, open-label, multi-cohort clinical trial (ARROW, NCT03037385) with 220 participants aged 26-87 whose tumors had RET alterations.[1][4] Identification of RET gene alterations was prospectively determined in local laboratories using either next generation sequencing, fluorescence in situ hybridization, or other tests.[1] The main efficacy outcome measures were overall response rate (ORR) and response duration determined by a blinded independent review committee using RECIST 1.1.[1] The trial was conducted at sites in the United States, Europe and Asia.[4]
Efficacy for RET fusion-positive NSCLC was evaluated in 87 participants previously treated with platinum chemotherapy.[1] The ORR was 57% (95% CI: 46%, 68%); 80% of responding participants had responses lasting 6 months or longer.[1] Efficacy was also evaluated in 27 participants who never received systemic treatment.[1] The ORR for these participants was 70% (95% CI: 50%, 86%); 58% of responding participants had responses lasting 6 months or longer.[1]
Step 7: Synthesis of (1R,4S)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexane-carboxamide (Compound 129) and (1S,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxamide (Compound 130)
[0194]
[0195]
The title compounds were prepared from methyl 1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxylate (192 mg, 0.53 mmol) using the same two-step procedure (hydrolysis and amide coupling) outlined in Synthetic Protocols 1 and 2, with PyBOP as the amide coupling reagent instead of HATU. The products were initially isolated as a mixture of diastereomers (190 mg), which was then dissolved in 6 mL methanol and purified by SFC (ChiralPak AD-H 21×250 mm, 40% MeOH containing 0.25% DEA in CO2, 2.5 mL injections, 70 mL/min). Peak 1 was concentrated to give (1R,4S)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxamide (29 mg, 10%) as a white solid. Peak 2 was concentrated to give (1s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexane-carboxamide (130 mg, 46%) as a white solid.
Example 6. Synthesis of Compound 149Step 1: Synthesis of Methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate
[0196]
[0197]
Methyl 4-iodo-1-methoxycyclohexanecarboxylate (3.37 g, 11.3 mmol) was dissolved in dimethylacetamide (38 mL) in a pressure vessel under a stream of N2. Rieke Zinc (17.7 mL of a 50 mg/mL suspension in THF, 13.6 mmol) was added quickly via syringe, and the vessel was capped and stirred at ambient temperature for 15 minutes. The vessel was opened under a stream of N2 and 2,4-dichloro-6-methylpyrimidine (1.84 g, 11.3 mmol) was added followed by PdCl2dppf (826 mg, 1.13 mmol). The vessel was capped and heated to 80° C. for one hour, then cooled to room temperature. The reaction mixture was diluted with EtOAc, filtered through celite, and the filtrate was washed with H2O (3×), brine, dried over sodium sulfate, filtered, and concentrated. The resulting residue was purified by flash-column chromatography on silica gel (gradient elution, 0 to 50% EtOAc-hexanes) to give methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate (74 mg, 2.2%) as a colorless oil. MS (ES+) C14H19ClN2O3 requires: 298, found: 299 [M+H]+.
Step 2: Synthesis of tert-Butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate
[0198]
[0199]
Methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate (70.5 mg, 0.236 mmol), tert-butyl 3-amino-5-methyl-1H-pyrazole-1-carboxylate (69.8 mg, 0.354 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphine (20.0 mg, 0.2 equiv.), Pd2(dba)3 (21.6 mg, 0.1 equiv.), and potassium acetate (70 mg, 0.71 mmol) were combined in a vial under nitrogen and 0.98 mL dioxane was added. The reaction mixture was heated to 115° C. for 2 h, then cooled to ambient temperature. The reaction mixture was diluted with EtOAc, filtered through celite, concentrated onto silica gel, and the resulting residue was purified by flash-column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-hexanes) to give tert-butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate (48 mg, 44%) as a yellow oil. MS (ES+) C23H33N5O5 requires: 459, found: 460 [M+H]+.
Step 3: Synthesis of 1-Methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid
[0200]
[0201]
Lithium hydroxide monohydrate (13 mg, 0.31 mmol) was added to a solution of tert-butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate (47.7 mg, 0.104 mmol) in THF/MeOH/H2O (17:1:1, 1.8 mL). The reaction mixture was heated to 60° C. and stirred for 16 h. The reaction mixture was then cooled to ambient temperature and concentrated to give crude 1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid (57 mg, crude) which was used in the subsequent amide coupling without any further purification. MS (ES+) C17H23N5O3 requires: 345, found: 346 [M+H]+.
Step 4: Synthesis of (1s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxamide (Compound 149)
[0202]
[0203]
The title compound was prepared from 1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid (57 mg, 0.104 mmol) using the same procedured (amide coupling) outlined in Synthetic Protocols 1 and 2, with PyBOP as the amide coupling reagent instead of HATU. The products were initially isolated as a mixture of diastereomers (36 mg), which was then dissolved in 6 mL methanol-DCM (1:1) and purified by SFC (ChiralPak IC-H 21×250 mm, 40% MeOH containing 0.25% DEA in CO2, 1.0 mL injections, 70 mL/min). Peak 1 was an undesired isomer, and Peak 2 was concentrated to give (1 s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxamide (13.4 mg, 24%) as a white solid.
Synthesis of IntermediatesExample 7. Synthesis of Ketone and Boronate IntermediatesA. Methyl 1-methoxy-4-oxocyclohexane-1-carboxylate
[0204]
[0205]
The title compound was prepared as described in WO 2014/130810 A1 page 86.
B. Ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate
[0206]
Step 1: Synthesis of ethyl 8-ethoxy-1,4-dioxaspiro[4.5]decane-8-carboxylate
[0207]
A solution of 1,4-dioxaspiro[4.5]decan-8-one (20.0 g, 128 mmol) in CHBr3 (3234 g, 1280 mmol) was cooled to 0° C. and potassium hydroxide (57.5 g, 1024 mmol) in EtOH (300 mL) was added dropwise over 2.5 hrs. After stirring the mixture for 23 h, the mixture was concentrated, and the residue was partitioned between EtOAc and H2O. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give crude product, which was purified by flash column chromatography on silica gel (gradient elution, PE:EA=15:1 to 10:1) to obtain the title compound (18.0 g).
Step 2: Synthesis of ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate
[0208]
To a solution of ethyl 8-ethoxy-1,4-dioxaspiro[4.5]decane-8-carboxylate (10 g, 43 mmol) in 1,4-dioxane (250 mL) was added aqueous HCl (6 M, 92.5 mL), and the mixture was stirred for 23 h at ambient temperature. The mixture was then diluted with H2O and extracted with EtOAc.
[0209]
The organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a crude residue, which was purified by flash column chromatography on silica gel (PE:EA=15:1) to obtain the product (8.0 g). 1H NMR (400 MHz, DMSO) δ 4.20-4.13 (m, 2H), 3.43 (q, J=6.9 Hz, 1H), 2.48-2.39 (m, 1H), 2.24-2.12 (m, 2H), 2.10-2.01 (m, 1H), 1.22 (t, J=7.1 Hz, 2H), 1.17 (t, J=7.0 Hz, 2H).
C. Ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
[0210]
Step 1: Synthesis of ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate
[0211]
A solution of methylmagnesium bromide (3M, 109.8 mL, 329.4 mmol) was added dropwise to a suspension of CuCN (14.75 g, 164.7 mmol) in diethyl ether (50 mL) at 0° C. The mixture was stirred for 30 min at 0° C. and then cooled to −78° C. The solution of ethyl 2-methyl-4-oxocyclohex-2-ene-1-carboxylate (10 g, 54.9 mmol) in diethyl ether (10 mL) was then added dropwise. The mixture was stirred between −40° C. to −20° C. for 2 h, then was warmed to ambient temperature for 16 h. The reaction mixture was carefully added to a saturated solution of ammonium chloride. The aqueous layer was extracted twice with diethyl ether, and the organic layers were combined. The combined organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography on silica gel (PE:EA=10:1) to give ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate (1.16 g).
Step 2: Synthesis of ethyl 6,6-dimethyl-4-(((trifluoromethyl)sulfonyl)oxy)cyclohex-3-ene-1-carboxylate
[0212]
Ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate (1.16 g, 5.85 mmol) and DIPEA (3.03 g, 23.4 mmol) were dissolved in dry toluene (2 mL) and heated at 45° C. for 10 minutes. Trifluoromethanesulfonic anhydride (6.61 g, 23.4 mmol) in DCM (20 mL) was added dropwise over 10 min and the mixture was heated at 45° C. for 2 h. The mixture was allowed to cool to room temperature, concentrated, diluted with water (60 mL) and extracted with DCM (2×40 mL). The organic layer was washed with saturated sodium bicarbonate solution (20 mL) and brine (20 mL), dried over sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-petroleum ether) to afford ethyl 6,6-dimethyl-4-(((trifluoromethyl)sulfonyl)oxy)cyclohex-3-ene-1-carboxylate (1 g).
Step 3: Synthesis of ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
[0213]
Ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate (1 g, 3.03 mmol), 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1.15 g, 4.54 mmol), Pd(dppf)Cl2 (73.5 mg, 0.09 mmol) and potassium acetate (891 mg, 9.08 mmol) were suspended in 1,4-dioxane (20 mL). The reaction mixture was flushed with nitrogen, then heated to 100° C. for 2 h. The mixture was cooled to room temperature, filtered, and concentrated, and the resulting brown oil was purified by flash column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-petroleum ether) to afford ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate (618 mg).
D. Ethyl 6-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
[0214]
[0215]
Ethyl 6-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate was prepared using the same synthetic protocol as described above using ethyl 2-methyl-4-oxocyclohexane-1-carboxylate as the starting material.
E. Methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate
[0216]
Step 1: Synthesis of methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate
[0217]
A mixture of acrylaldehyde (120 g, 2.14 mol), methyl methacrylate (200 g, 2.00 mol) and hydroquinone (2.2 g, 20 mmol) were heated in a sealed steel vessel at 180° C. for one h. The mixture was then cooled to ambient temperature and concentrated. The residue was purified by silica gel column chromatography (gradient elution, petroleum ether:ethyl acetate=100:1 to 80:1) to give methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate (70 g, 22% yield) as a pale yellow oil. 1H-NMR (400 MHz, CDCl3): δ 6.38 (d, J=6.4 Hz, 1H), 4.73-4.70 (m, 1H), 3.76 (s, 3H), 2.25-2.22 (m, 1H), 1.99-1.96 (m, 2H), 1.79-1.77 (m, 1H), 1.49 (s, 3H).
Step 2: Synthesis of methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate
[0218]
To a solution of methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate (20.0 g, 128 mmol) in anhydrous tetrahydrofuran (200 mL) was added borane (67 mL, 1 M in tetrahydrofuran) dropwise at −5° C. The reaction mixture was stirred at 0° C. for 3 hours. This reaction was monitored by TLC. The mixture was quenched by a solution of sodium acetate (10.5 g, 128 mmol) in water (15 mL). Then the mixture was treated with 30% hydrogen peroxide solution (23.6 g, 208.2 mmol) slowly at 0° C. and stirred at 30° C. for 3 h. The mixture was then partitioned between saturated sodium sulfite solution and tetrahydrofuran. The aqueous layer was further extracted with tetrahydrofuran (2×). The combined organic layers were washed with saturated brine, dried over sodium sulfate and concentrated in vacuo. The residue was purified by a silica gel column chromatography (gradient elution, petroleum ether:ethyl acetate=10:1 to 1:1) to give crude methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate (18 g, crude) as a pale yellow oil, which used directly for next step.
Step 3: Synthesis of methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate
[0219]
To a solution of methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate (18.0 g, 103 mmol) in anhydrous dichloromethane (200 mL) was added PCC (45.0 g, 209 mmol) in portions. The reaction mixture was stirred at ambient temperature until TLC indicated the reaction was completed. Petroleum ether (500 mL) was then added and the mixture was filtered. The filter cake was washed with petroleum ether (100 mL), and the filtrate was concentrated under vacuum to give methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate (15 g, 84% yield) as a pale yellow oil. 1H-NMR (400 MHz, CDCl3): δ 4.25 (d, J=17.6 Hz, 1H), 4.07 (d, J=17.6 Hz, 1H), 3.81 (s, 3H), 2.52-2.44 (m, 3H), 2.11-2.04 (m, 1H), 1.53 (s, 3H).
Example 8. Synthesis of Iodide IntermediatesA. Methyl 1-methoxy-4-iodocyclohexane-1-carboxylate
[0220]
Step 1: Synthesis of methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate
[0221]
Methyl 1-methoxy-4-oxocyclohexanecarboxylate (4.00 g, 21.5 mmol) was dissolved in methanol (100 mL) and the solution was cooled to 0° C. Sodium borohydride (2.03 g, 53.7 mmol) was added in portions over 20 min. The reaction mixture was stirred for 30 min, then was quenched by addition of aqueous saturated NH4Cl solution. The quenched reaction mixture was evaporated to remove the MeOH, then the aqueous suspension was extracted with DCM (3×). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to yield a residue that was purified by flash-column chromatography on silica gel (gradient elution, 5% to 100% ethyl acetate-hexanes) to afford methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate (2.00 g, 49.5%) as a colorless oil. MS (ES+) C9H16O4 requires: 188, found: 211 [M+Na]+.
Step 2: Synthesis of methyl 1-methoxy-4-iodocyclohexane-1-carboxylate
[0222]
Methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate (2.00 g, 10.6 mmol) was dissolved in THF (20 mL) and imidazole (723 mg, 10.6 mmol) and triphenylphosphine (3.34 g, 12.8 mmol) were added. The mixture was cooled to 0° C., and then a solution of iodine (3.24 g, 12.8 mmol) in THF (10 mL) was added dropwise over 15 min. The reaction mixture was allowed to warm to ambient temperature and was then stirred for 2 days, after which it was poured over saturated sodium thiosulfate solution and extracted with EtOAc. The organic layer was dried over sodium sulfate, filtered, concentrated, and the residue was triturated with hexane (40 mL, stir for 20 min). The mixture was filtered, and the filtrate was evaporated to provide a residue that was purified by flash-column chromatography on silica gel (gradient elution, 0 to 30% ethyl acetate-hexanes) to give the title compound (2.37 g, 75%) as a pale yellow oil. MS (ES+) C9H15IO3 requires: 298, found: 299 [M+H]+.
B. Ethyl 1-ethoxy-4-iodocyclohexane-1-carboxylate
[0223]
[0224]
The title compound was prepared as described above using ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate as a starting material. C11H19IO3 requires: 326, found: 327 [M+H]−.
Example 9. Synthesis of Amine IntermediatesA. (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine
[0225]
Step 1: Synthesis of 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one
[0226]
4-Fluoro-1H-pyrazole (4.73 g, 55 mmol) and potassium carbonate (17.27 g, 125 mmol) were combined and stirred in N,N-dimethylformamide (41.7 mL) for 10 minutes in an open sealed tube before addition of 2-bromo-5-acetylpyridine (10 g, 50 mmol). The reaction tube was sealed and stirred for 20 hours at 100° C. The reaction mixture was then cooled to room temperature and poured into water (˜700 mL). The mixture was sonicated and stirred for 20 minutes, after which a beige solid was isolated by filtration, washed with small amounts of water, and dried to yield 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one (9.81 g, 96% yield). MS: M+1=206.0.
Step 2: Synthesis of (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide
[0227]
To a stirred room temperature solution of 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one (9.806 g, 47.8 mmol) in THF (96 mL) was added (R)-(+)-t-Butylsulfinamide (5.79 g, 47.8 mmol) followed by titanium (IV) ethoxide (21.8 g, 96 mmol). The solution was stirred at 75° C. on an oil bath for 15 hours. The reaction solution was cooled to room temperature and then to −78° C. (external temperature) before the next step. To the −78° C. solution was added dropwise over nearly 55 minutes L-Selectride (143 mL of 1N in THF, 143 mmol). During addition, some bubbling was observed. The reaction was then stirred after the addition was completed for 15 minutes at −78° C. before warming to room temperature. LC-MS of sample taken during removal from cold bath showed reaction was completed. The reaction was cooled to −50° C. and quenched slowly with methanol (˜10 mL), then poured into water (600 mL) and stirred. An off-white precipitate was removed by filtration, with ethyl acetate used for washes. The filtrate was diluted with ethyl acetate (800 mL), the layers were separated, and the organic layer was dried over sodium sulfate, filtered, and concentrated down. The crude was purified by silica gel chromatography to yield (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide (10.5 g, 99% purity, 70.3% yield) as a light yellow solid. MS: M+1=311.1.
Step 3: Synthesis of (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine
[0228]
A solution of (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide (10.53 g, 33.9 mmol)) in methanol (79 mmol) and 4N HCl/dioxane (85 mL, 339 mmol) was stirred for 2.5 hours, at which point LC-MS showed reaction was complete. The reaction solution was poured into diethyl ether (300 mL) and a sticky solid was formed. The mixture was treated with ethyl acetate (200 mL) and sonicated. The solvents were decanted, and the sticky solid was treated with more ethyl acetate (˜200 mL), sonicated and stirred. The bulk of the sticky solid was converted to a suspension. A light yellow solid was isolated by filtration, washed with smaller amounts of ethyl acetate, and dried to yield (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine (7.419 g, 78% yield). LC-MS confirmed desired product in high purity. MS: M+1=207.1.
“Pralsetinib”. Drug Information Portal. U.S. National Library of Medicine.
“Pralsetinib”. NCI Drug Dictionary. National Cancer Institute.
Clinical trial number NCT03037385 for “Phase 1/2 Study of the Highly-selective RET Inhibitor, Pralsetinib (BLU-667), in Patients With Thyroid Cancer, Non-Small Cell Lung Cancer, and Other Advanced Solid Tumors (ARROW)” at ClinicalTrials.gov
Roche is investing $775 million in cash and equity for access to Blueprint Medicines’ oncology drug candidate pralsetinib, which is under review by the US Food and Drug Administration.
Pralsetinib is a small-molecule inhibitor of RET alterations—rare genetic fusions or mutations that occur at low levels across lung, thyroid, and many other cancers.
The drug will go up against Eli Lilly and Company’s Retevmo, an RET inhibitor that received FDA approval in May for certain lung and thyroid cancers. Lilly acquired Retevmo in its $8 billion purchase of Loxo Oncology in 2019, a deal to obtain Loxo’s pipeline of small molecules for genetically defined tumors.
But SVB Leerink analyst Andrew Berens points out that Retevmo has side effects: it can cause an irregular heart rhythm called QT prolongation and hemorrhagic events. That leaves room for pralsetinib, which Roche will be better able to get in front of oncologists, Berens argues. In addition to a vast commercial network, Roche brings diagnostic tools to help identify cancer patients whose tumors feature RET alterations.
The FDA has a deadline of Nov. 23 to decide on approving the drug for lung cancer.
Roche’s move lowers the likelihood of a takeover of Blueprint, which had appeared on many investors’ short lists of acquisition targets. “We were surprised by the profuse language framing this deal as ensuring Blueprint’s independence,” Piper Sandler stock analyst Christopher J. Raymond told investors in a note.
//////////Pralsetinib, GAVRETO, 2020 APPROVALS, FDA 2020
The medication, developed by Cassiopea and Intrepid Therapeutics,[2] was approved by the US Food and Drug Administration (FDA) for acne in August 2020.[6][7]
Medical uses
Clascoterone is indicated for the topical treatment of acne vulgaris in females and males age 12 years and older.[1][8] It is applied to the affected skin area in a dose of 1 mg cream (or 10 mg clascoterone) twice per day, once in the morning and once in the evening.[1] The medication should not be used ophthalmically, orally, or vaginally.[1]
Available forms
Clascoterone is available in the form of a 1% (10 mg/g) cream for topical use.[1]
The incidences of local skin reactions with clascoterone were similar to placebo in two large phase 3 randomized controlled trials.[1][9] Suppression of the hypothalamic–pituitary–adrenal axis (HPA axis) may occur during clascoterone therapy in some individuals due to its cortexolonemetabolite.[1][8] HPA axis suppression as measured by the cosyntropin stimulation test was observed to occur in 3 of 42 (7%) of adolescents and adults using clascoterone for acne.[1][8] HPA axis function returned to normal within 4 weeks following discontinuation of clascoterone.[1][8]Hyperkalemia (elevated potassium levels) occurred in 5% of clascoterone-treated individuals and 4% of placebo-treated individuals.[1]
Steady-state levels of clascoterone occur within 5 days of twice daily administration.[1] At a dosage of 6 g clascoterone cream applied twice daily, maximal circulating levels of clascoterone were 4.5 ± 2.9 ng/mL, area-under-the-curve levels over the dosing interval were 37.1 ± 22.3 h*ng/mL, and average circulating levels of clascoterone were 3.1 ± 1.9 ng/mL.[1] In rodents, clascoterone has been found to possess strong local antiandrogenic activity, but negligible systemic antiandrogenic activity when administered via subcutaneous injection.[10] Along these lines, the medication is not progonadotropic in animals.[10]
Clascoterone is rapidly hydrolyzed into cortexolone (11-deoxycortisol) and this compound is a possible primary metabolite of clascoterone based on in-vitro studies in human liver cells.[1][8] During treatment with clascoterone, cortexolone levels were detectable and generally below or near the low limit of quantification (0.5 ng/mL).[1] Clascoterone may also produce other metabolites, including conjugates.[1]
The elimination of clascoterone has not been fully characterized in humans.[1]
Clascoterone, also known as cortexolone 17α-propionate or 11-deoxycortisol 17α-propionate, as well as 17α,21-dihydroxyprogesterone 17α-propionate or 17α,21-dihydroxypregn-4-en-3,20-dione 17α-propionate, is a syntheticpregnanesteroid and a derivative of progesterone and 11-deoxycortisol (cortexolone).[11] It is specifically the C17α propionateester of 11-deoxycortisol.[10]
C17α esters of 11-deoxycortisol were unexpectedly found to possess antiandrogenic activity.[10] Clascoterone, also known as cortexolone 17α-propionate, was selected for development based on its optimal drug profile.[10] The medication was approved by the US Food and Drug Administration (FDA) for the treatment of acne in August 2020.[6]
Two large phase 3randomized controlled trials evaluated the effectiveness of clascoterone for the treatment of acne over a period of 12 weeks.[1][8][9] Clascoterone decreased acne symptoms by about 8 to 18% more than placebo.[1][9] The defined treatment success endpoint was achieved in about 18 to 20% of individuals with clascoterone relative to about 7 to 9% of individuals with placebo.[1][8][9] The comparative effectiveness of clascoterone between males and females was not described.[1][9]
A small pilot randomized controlled trial in 2011, found that clascoterone cream decreased acne symptoms to a similar or significantly greater extent than tretinoin 0.05% cream.[8][13] No active comparator was used in the phase III clinical trials of clascoterone for acne.[8] Hence, it’s unclear how clascoterone compares to other therapies used in the treatment of acne.[8]
The FDA approved clascoterone based on evidence from two clinical trials (Trial 1/NCT02608450 and Trial 2/NCT02608476) of 1440 participants 9 to 58 years of age with acne vulgaris.[14] The trials were conducted at 99 sites in the United States, Poland, Romania, Bulgaria, Ukraine, Georgia, and Serbia.[14]
Participants applied clascoterone or vehicle (placebo) cream twice daily for 12 weeks.[14] Neither the participants nor the health care providers knew which treatment was being given until after the trial was completed.[14] The benefit of clascoterone in comparison to placebo was assessed after 12 weeks of treatment using the Investigator’s Global Assessment (IGA) score that measures the severity of disease (on a scale from 0 to 4) and a decrease in the number of acne lesions.[14]
Dissolving the compound 11-deoxycortisol (1.04g, 3.0mmol, 1eq.) in 10mL of anhydrous pyridine, dissolving dried DMTrCl (1.2-1.5eq) in 5mL of anhydrous dichloromethane, dropwise adding a dichloromethane solution of DMTrCl into the reactant solution at room temperature, and reacting for 4 hours at room temperature; the reaction was quenched with methanol and the solvent was evaporated to dryness with an oil pump to give intermediate I in 85% yield (the next reaction was carried out without work-up, the solvent environment and catalyst were similar to the reaction of this step).
MS + 303(DMTr protecting group fragment), 649[M + H] +
Melting point: 95-97 deg.C
Example 2:
preparation of intermediate II
Wherein R is DMTr
Under the protection of nitrogen, dissolving the intermediate product I (1eq.) in 5mL of anhydrous dichloromethane, adding DMAP (0.1eq.) into the solution, dropwise adding triethylamine (1.2eq.) and propionic anhydride or propionyl chloride (1.2eq. ), reacting at 40 ℃ for 12 hours after dropwise adding, and evaporating the solvent to obtain an intermediate product II.
Or under the protection of nitrogen, dissolving the intermediate product I (1eq.) in 5mL of anhydrous pyridine, adding DMAP (0.1eq.) into the solution, dropwise adding triethylamine (1.2eq.) and propionic anhydride or propionyl chloride (1.2eq .), reacting at 80 ℃ for 4 hours after dropwise adding, and evaporating the solvent to obtain an intermediate product II. (the reaction in the step can be directly carried out for the next step of removing DMTr protecting group to obtain the reaction after solvent evaporation without strict purification post-treatment)
MS + :303(DMTr protecting group fragment), 727[ M + Na [)] + ,768[M+Na+CH 3 CN] + .
Example 3:
preparation of target Compound 1 (21-hydroxy-17- (1-oxopropoxy) pregn-4-ene-3, 20-dione)
Dissolving the concentrated intermediate product II in an ethyl acetate solution, slowly dropwise adding 0.5M hydrochloric acid solution or 2% trifluoroacetic acid-ethyl acetate solution at 0 ℃, reacting for 5 minutes at 0 ℃, removing DMTr protective groups, adding 5% sodium bicarbonate aqueous solution at 0 ℃, stirring, neutralizing acid in a reaction system, washing an ethyl acetate organic layer twice by using 5% sodium bicarbonate aqueous solution, removing acid and other water-soluble impurities in the ethyl acetate organic layer, drying the ethyl acetate organic layer by anhydrous sodium sulfate, evaporating to remove part of ethyl acetate solvent, adding petroleum ether into the remaining small amount of ethyl acetate solution, and recrystallizing in a system with 10 times of solvent amount of ethyl acetate-petroleum ether (5:1) to obtain a target product with high purity of 90%. The total yield from 11-deoxycortisol is up to 70%. The final product was free of isomerized by-products by HPLC and was not found by LCMS.
Alcoho lysis with CCL of cortexolone 17α, 21-dipropionate
Add butanol (0.4g, 5.45 mmoles) and CCL (17.4g, 3.86 U/mg, FLUKA) to a solution of cortexolone- 17α,21-dipropionate (0.5g, 1.09 mmoles) in toluene (50ml). Maintain the mixture under stirring, at 30 0C, following the progress of the reaction in TLC (Toluene/ethyl acetate 6/4) until the initial material is dissolved (24h). Remove the enzyme by means of filtration using a Celite layer. Recover the cortexolone 17α-propionate (0.437, 99%) after evaporation under low pressure. Through crystallisation, from diisopropyl ether you obtain a product with a purity >99% in HPLC.
(s, 3H, CH3– 18). P.f. 135-136 0C (acetone/hexane).
Example 5
Alcoho lysis with CALB of cortexolone- 17α, 21-dipropionate
Dissolve cortexolone, 17α, 2-dipropionate (0.5g, 1 .09 mmoles) in acetonitrile
(40ml), add CALB (2.3g, 2.5 U/mg Fluka) and octanol (0.875ml). Leave the mixture under stirring, at 30 0C, for 76 hrs. Remove the enzyme by means of filtration using a paper filter. Once the solvents evaporate, recover a solid
(0.4758) which upon analysis 1H-NMR shall appear made up of cortexolone- 17α- propionate at 91%.
Example 6
Crystallisation
Add the solvent (t-butylmethylether or diisopropylether) to the sample according to the ratios indicated in Table 3. Heat the mixture to the boiling temperature of the solvent, under stirring, until the sample dissolves completely. Cool to room temperature and leave it at this temperature, under stirring, for 6 hours. Filter using a buchner funnel and maintain the solid obtained, under low pressure, at a room temperature for 15 hours and then, at 400C, for 5 hours.
Example 7
Precipitation Disslove the sample in the suitable solvent (dichloromethane, acetone, ethyl acetate or ethanol) according to the ratios indicated in table 3 and then add the solvent, hexane or water, according to the ratios indicated in table 3, maintaining the mixture, under stirring, at room temperature. Recover the precipitate by filtration using a buchner funnel and desiccate as in example 6. Example 8.
Obtaining a pharmaceutical form containing the medication in a defined crystalline form.
Prepare a fluid cream containing 2 % cetylic alcohol, 16% glyceryl monostearate, 10% vaseline oil, 13 % propylene glycol, 10% poly ethylengly col with low polymerization 1.5% polysorbate 80 and 47.5 % purified water. Add 1 g of cortexolone 17α-propionate of crystalline form III to 100 g of this cream and subject the mixture to homogenisation by means of a turbine agitator until you obtain homogeneity. You obtain a cream containing a fraction of an active ingredient dissolved in the formulation vehicle and a non-dissolved fraction of an active ingredient, present as a crystal of crystalline form III. This preparation is suitable for use as a formulation vehicle for skin penetration tests on Franz cells, where a coefficient of penetration in the range of 0.04 to 0.03 cm/h is observed on the preparation. Example 9.
Obtaining the pharmaceutical form containing the medication in solvate form IV for replacing the solvent during the galenic formulation procedure Dissolve lOOg of cortexolone 17α-propionate of crystalline form III in 2500 g of propylene glycol under stirring at room temperature. Separately prepare, by using a turbo emulsifϊer raising the temperature up to about 700C, an emulsion with 250 g of Cetylic alcohol, 1500 g of glyceryl monostearate, 1000 g of liquid paraffin, 5 g of mixed tocopherols, 100 g of polysorbate 80 and 4650 g of water. After cooling the emulsion up to about 300C, add – under stirring and under negative pressure – the cortexolone 17α-propionate solution in propylene glycol. Maintain the emulsioned cream under stirring until you obtain homogeneity, making sure the temperature remains low by means the circulation of a coolant. The cream contains a dispersed crystalline fraction, made up of an active ingredient in solvate crystalline form IV, formed due to the precipitation of the active ingredient itself from the glycolic solution which contained it when the latter was added to the predominantly aqueous formulation. The DRX spectra of the crystalline form present in the cream are indicated in Fig. 30.
Several 17α-monoesters of cortexolone and its Δ9-derivative are endowed with antiandrogenic activity. Their synthesis can be accomplished by means of a lipase-catalyzed chemoselective alcoholysis of the corresponding 17α,21-diesters.
Cortexolone derivatives in which the hydroxyl group at position C-17α is esterified with short chain aliphatic or aromatic acids and the derivatives of the corresponding 9,11-dehydro derivative, are known to have an antiandrogenic effect.
[0002]
EP 1421099 describes cortexolone 17α-propionate and 9,11-dehydro-cortexolone-17-α-butanoate regarding a high antiandrogenic biological activity demonstrated both “in vitro” and “in vivo” on the animal.
[0003]
US3530038 discloses the preparation of a crystalline form of cortexolone-17α-propionate having a melting point of 126-129 °C and an IR spectrum with bands at (cm-1): 3500, 1732, 1713, 1655 and 1617.
[0004]
A method for obtaining the above mentioned derivatives is described by Gardi et al. (Gazz. Chim. It. 63, 43 1,1963) and in the United States patent US3152154 providing for the transformation of cortexolone, or transformation of 9,11-dehydrocortexolone, in the intermediate orthoester using orthoesters available in the market as a mixture of aprotic solvents such as cyclohexane and DMF, in presence of acid catalysis (ex. PTSA.H20). The intermediate orthoester thus obtained can be used as is or upon purification by suspension in a solvent capable of solubilising impurities, preferably in alcohols. The subsequent hydrolysis in a hydroalcoholic solution, buffered to pH 4-5 preferably in acetate buffer, provides the desired monoester.
[0005]
Such synthesis is indicated in the diagram 1 below
[0006]
However, the monoesters thus obtained were, in the reaction conditions, unstable and, consequently hard to manipulate and isolate (R. Gardi et al Tetrahedron Letters, 448, 1961). The instability is above all due to the secondary reaction of migration of the esterifying acyl group from position 17 to position 21.
[0007]
It is thus known that in order to obtain the above mentioned monoesters with a chemical purity in such a manner to be able to proceed to the biological tests, it is necessary to use, at the end of the synthesis, a purification process which is generally performed by means of column chromatography.
[0008]
Furthermore, US3152154 describes how the hydrolysis of the diester in a basic environment is not convenient due to the formation of a mixture of 17α,21-diol, of 17- and 21 -monoesters, alongside the initial non-reacted product.
[0009]
Now, it has been surprisingly discovered that an alcoholysis reaction using a lipase from Candida as a biocatalyst can be usefully applied during the preparation of 17α monoesters of cortexolone, or its 9,11-dehydroderivatives.
[0010]
As a matter of fact, it has been discovered that such enzymatic alcoholysis of the 17,21-diester of the cortexolone, or of its derivative 9,11-dehydro, selectively occurs in position 21 moving to the corresponding monoester in position 17, as shown in diagram 2 below:
[0011]
The chemoselectivity of the special enzymatic reaction in alcoholysis conditions, according to the present invention, opens new perspectives for preparation, at industrial level with higher yields, of 17α-monoesters with respect to the methods already indicated in literature.
[0012]
The diesters serving as a substrate for the reaction of the invention can be prepared according to the prior art, for example following the one described in B.Turner, (Journal of American Chemical Society, 75, 3489, 1953) which provides for the esterification of corticosteroids with a linear carboxylic acid in presence of its anhydride and PTSA monohydrate.
EXAMPLES
Example 1
Alcoholysis with CCL of cortexolone 17α, 21-dipropionate
[0055]
Add butanol (0.4g, 5.45 mmoles) and CCL (17.4g, 3.86 U/mg, FLUKA) to a solution of cortexolone-17α,21-dipropionate (0.5g, 1.09 mmoles) in toluene (50ml). Maintain the mixture under stirring, at 30 °C, following the progress of the reaction in TLC (Toluene/ethyl acetate 6/4) until the initial material is dissolved (24h). Remove the enzyme by means of filtration using a Celite layer. Recover the cortexolone 17α-propionate (0.437, 99%) after evaporation under low pressure. Through crystallisation, from diisopropyl ether you obtain a product with a purity >99% in HPLC.
Alcoholysis with CALB of cartexolone-17α, 21-dipropionate
[0061]
Dissolve cortexolone, 17α, 2-dipropionate (0.5g, 1.09 mmoles) in acetonitrile (40ml), add CALB (2.3g, 2.5 U/mg Fluka) and octanol (0.875ml). Leave the mixture under stirring, at 30 °C, for 76 hrs. Remove the enzyme by means of filtration using a paper filter. Once the solvents evaporate, recover a solid (0.4758) which upon analysis 1H-NMR shall appear made up of cortexolone-17α-propionate at 91%.
Example 6
Crystallisation
[0062]
Add the solvent (t-butylmethylether or diisopropylether) to the sample according to the ratios indicated in Table 3. Heat the mixture to the boiling temperature of the solvent, under stirring, until the sample dissolves completely. Cool to room temperature and leave it at this temperature, under stirring, for 6 hours. Filter using a buchner funnel and maintain the solid obtained, under low pressure, at a room temperature for 15 hours and then, at 40°C, for 5 hours.
Example 7 (comparative)
Precipitation
[0063]
Disslove the sample in the suitable solvent (dichloromethane, acetone, ethyl acetate or ethanol) according to the ratios indicated in table 3 and then add the solvent, hexane or water, according to the ratios indicated in table 3, maintaining the mixture, under stirring, at room temperature. Recover the precipitate by filtration using a buchner funnel and desiccate as in example 6.
Example 8.
Obtaining a pharmaceutical form containing the medication in a defined crystalline form.
[0064]
Prepare a fluid cream containing 2 % cetylic alcohol, 16% glyceryl monostearate, 10% vaseline oil, 13 % propylene glycol, 10% polyethylenglycol with low polymerization 1.5% polysorbate 80 and 47.5 % purified water. Add 1 g of cortexolone 17α-propionate of crystalline form III to 100 g of this cream and subject the mixture to homogenisation by means of a turbine agitator until you obtain homogeneity. You obtain a cream containing a fraction of an active ingredient dissolved in the formulation vehicle and a non-dissolved fraction of an active ingredient, present as a crystal of crystalline form III. This preparation is suitable for use as a formulation vehicle for skin penetration tests on Franz cells, where a coefficient of penetration in the range of 0.04 to 0.03 cm/h is observed on the preparation.
^ Jump up to:abRosette C, Rosette N, Mazzetti A, Moro L, Gerloni M (February 2019). “Cortexolone 17α-Propionate (Clascoterone) is an Androgen Receptor Antagonist in Dermal Papilla Cells In Vitro”. J Drugs Dermatol. 18 (2): 197–201. PMID30811143.
^ Jump up to:abcRosette C, Agan FJ, Mazzetti A, Moro L, Gerloni M (May 2019). “Cortexolone 17α-propionate (Clascoterone) Is a Novel Androgen Receptor Antagonist that Inhibits Production of Lipids and Inflammatory Cytokines from Sebocytes In Vitro”. J Drugs Dermatol. 18 (5): 412–418. PMID31141847.
^Celasco G, Moroa L, Bozzella R, Ferraboschi P, Bartorelli L, Di Marco R, Quattrocchi C, Nicoletti F (2005). “Pharmacological profile of 9,11-dehydrocortexolone 17alpha-butyrate (CB-03-04), a new androgen antagonist with antigonadotropic activity”. Arzneimittelforschung. 55 (10): 581–7. doi:10.1055/s-0031-1296908. PMID16294504.
^Trifu V, Tiplica GS, Naumescu E, Zalupca L, Moro L, Celasco G (2011). “Cortexolone 17α-propionate 1% cream, a new potent antiandrogen for topical treatment of acne vulgaris. A pilot randomized, double-blind comparative study vs. placebo and tretinoin 0·05% cream”. Br. J. Dermatol. 165 (1): 177–83. doi:10.1111/j.1365-2133.2011.10332.x. PMID21428978. S2CID38404925.
^World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3): 106. hdl:10665/330879.
^Der Sarkissian SA, Sun HY, Sebaratnam DF (August 2020). “Cortexolone 17 α-proprionate for hidradenitis suppurativa”. Dermatol Ther: e14142. doi:10.1111/dth.14142. PMID32761708.
External links
“Clascoterone”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02608450 for “A Study to Evaluate the Safety and Efficacy of CB-03-01 Cream, 1% in Subjects With Facial Acne Vulgaris (25)” at ClinicalTrials.gov
Clinical trial number NCT02608476 for “A Study to Evaluate the Safety and Efficacy of CB-03-01 Cream, 1% in Subjects With Facial Acne Vulgaris (26)” at ClinicalTrials.gov
Somapacitan, also known as NNC0195-0092,3 is a growth hormone analog indicated to treat adults with growth hormone deficiency.2,6 This human growth hormone analog differs by the creation of an albumin binding site, and prolonging the effect so that it requires weekly dosing rather than daily.5
Somapacitan was granted FDA approval on 28 August 2020.7
The most common side effects include: back pain, joint paint, indigestion, a sleep disorder, dizziness, tonsillitis, swelling in the arms or lower legs, vomiting, adrenal insufficiency, hypertension, increase in blood creatine phosphokinase (a type of enzyme), weight increase, and anemia.[2]
It was approved for medical use in the United States in August 2020.[2][3][4]
Somapacitan (Sogroya) is the first human growth hormone (hGH) therapy that adults only take once a week by injection under the skin; other FDA-approved hGH formulations for adults with growth hormone deficiency must be administered daily.[2]
Medical uses
Somapacitan is indicated for replacement of endogenous growth hormone in adults with growth hormone deficiency.[2]
Contraindications
Somapacitan should not be used in people with active malignancy, any stage of diabetic eye disease in which high blood sugar levels cause damage to blood vessels in the retina, acute critical illness, or those with acute respiratory failure, because of the increased risk of mortality with use of pharmacologic doses of somapacitan in critically ill individuals without growth hormone deficiency.[2]
History
Somapacitan was evaluated in a randomized, double-blind, placebo-controlled trial in 300 particpants with growth hormone deficiency who had never received growth hormone treatment or had stopped treatment with other growth hormone formulations at least three months before the study.[2] Particpants were randomly assigned to receive injections of weekly somapacitan, weekly placebo (inactive treatment), or daily somatropin, an FDA-approved growth hormone.[2] The effectiveness of somapacitan was determined by the percentage change of truncal fat, the fat that is accumulated in the trunk or central area of the body that is regulated by growth hormone and can be associated with serious medical issues.[2]
At the end of the 34-week treatment period, truncal fat decreased by 1.06%, on average, among particpants taking weekly somapacitan while it increased among particpants taking the placebo by 0.47%.[2] In the daily somatropin group, truncal fat decreased by 2.23%.[2] Particpants in the weekly somapacitan and daily somatropin groups had similar improvements in other clinical endpoints.[2]
It was approved for medical use in the United States in August 2020.[2][4] The U.S. Food and Drug Administration (FDA) granted the approval of Sogroya to Novo Nordisk, Inc.[2][4]
A Trial Investigating the Safety, Tolerability, Availability and Distribution in the Body of Once-weekly Long-acting Growth Hormone (Somapacitan) Compared to Once Daily Norditropin NordiFlex® in Adults With Growth Hormone Deficiency
A Trial Investigating Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of a Single Dose of Long-acting Growth Hormone (Somapacitan) Compared to Daily Dosing of Norditropin® SimpleXx® in Children With Growth Hormone Deficiency
Investigating Efficacy and Safety of Once-weekly NNC0195-0092 (Somapacitan) Treatment Compared to Daily Growth Hormone Treatment (Norditropin® FlexPro®) in Growth Hormone Treatment naïve Pre-pubertal Children With Growth Hormone Deficiency
A Trial to Evaluate the Safety of Once Weekly Dosing of Somapacitan (NNC0195-0092) and Daily Norditropin® FlexPro® for 52 Weeks in Previously Human Growth Hormone Treated Japanese Adults With Growth Hormone Deficiency
Investigation of Pharmacokinetics, Pharmacodynamics, Safety and Tolerability of Multiple Doses of Somapacitan in Subjects With Mild and Moderate Degrees of Hepatic Impairment Compared to Subjects With Normal Hepatic Function.
A Trial to Compare the Safety of Once Weekly Dosing of Somapacitan With Daily Norditropin® FlexPro® for 26 Weeks in Previously Human Growth Hormone Treated Adults With Growth Hormone Deficiency
Trial to Compare the Efficacy and Safety of NNC0195-0092 (Somapacitan) With Placebo and Norditropin® FlexPro® (Somatropin) in Adults With Growth Hormone Deficiency.
Investigation of Pharmacokinetics, Pharmacodynamics, Safety and Tolerability of Multiple Doses of Somapacitan in Subjects With Various Degrees of Impaired Renal Function Compared to Subjects With Normal Renal Function
A dose-finding trial evaluating the effect and safety of once-weekly treatment of somapacitan compared to daily Norditropin® in children with short stature born small for gestational age with no catch-up growth by 2 years of age or older
A randomised, multinational, active-controlled,(open-labelled), dose finding, (double-blinded), parallel group trial investigating efficacy and safety of once-weekly NNC0195-0092 treatment compared to daily growth hormone treatment (Norditropin® FlexPro®) in growth hormone treatment naïve pre-pubertal children with growth hormone deficiency
A multicentre, multinational, randomised, open-labelled, parallel-group, active-controlled trial to compare the safety of once weekly dosing of NNC0195-0092 with daily Norditropin® FlexPro® for 26 weeks in previously human growth hormone treated adults with growth hormone deficiency
A multicentre, multinational, randomised, parallel-group, placebo-controlled (double blind) and active-controlled (open) trial to compare the efficacy and safety of once weekly dosing of NNC0195-0092 with once weekly dosing of placebo and daily Norditropin® FlexPro® in adults with growth hormone deficiency for 35 weeks, followed by a 53-week open-label extension period
A randomised, open-labelled, active-controlled, multinational, dose-escalation trial investigating safety, tolerability, pharmacokinetics and pharmacodynamics of a single dose of long-acting growth hormone (NNC0195-0092) compared to daily dosing of Norditropin® SimpleXx® in children with growth hormone deficiency
Phase 1
Ongoing, Completed
2013-12-09
///////////Somapacitan, PEPTIDE.2020 APPROVALS, FDA 2020, ソマパシタン, NN8640
Copper Cu 64 dotatate, sold under the brand name Detectnet, is a radioactive diagnostic agent indicated for use with positron emission tomography (PET) for localization of somatostatin receptor positiveneuroendocrine tumors (NETs) in adults.[1]
Common side effects include nausea, vomiting and flushing.[2]
It was approved for medical use in the United States in September 2020.[1][2]
History
The U.S. Food and Drug Administration (FDA) approved copper Cu 64 dotatate based on data from two trials that evaluated 175 adults.[3]
Trial 1 evaluated adults, some of whom had known or suspected NETs and some of whom were healthy volunteers.[3] The trial was conducted at one site in the United States (Houston, TX).[3] Both groups received copper Cu 64 dotatate and underwent PET scan imaging.[3] Trial 2 data came from the literature-reported trial of 112 adults, all of whom had history of NETs and underwent PET scan imaging with copper Cu 64 dotatate.[3] The trial was conducted at one site in Denmark.[3] In both trials, copper Cu 64 dotatate images were compared to either biopsy results or other images taken by different techniques to detect the sites of a tumor.[3] The images were read as either positive or negative for presence of NETs by three independent image readers who did not know participant clinical information.[3]
Known imaging techniques with tremendous importance in medical diagnostics are positron emission tomography (PET), computed tomography (CT), magnetic resonance imaging (MRI), single photon computed tomography (SPECT) and ultrasound (US). Although today’s imaging technologies are well developed they rely mostly on non-specific, macroscopic, physical, physiological, or metabolic changes that differentiate pathological from normal tissue.
[0003]
Targeting molecular imaging (MI) has the potential to reach a new dimension in medical diagnostics. The term “targeting” is related to the selective and highly specific binding of a natural or synthetic ligand (binder) to a molecule of interest (molecular target) in vitro or in vivo.
[0004]
MI is a rapidly emerging biomedical research discipline that may be defined as the visual representation, characterization and quantification of biological processes at the cellular and sub-cellular levels within intact living organisms. It is a novel multidisciplinary field, in which the images produced reflect cellular and molecular pathways and in vivo mechanism of disease present within the context of physiologically authentic environments rather than identify molecular events responsible for disease.
[0005]
Several different contrast-enhancing agents are known today and their unspecific or non-targeting forms are already in clinical routine. Some examples listed below are reported in literature.
[0006]
For example, Gd-complexes could be used as contrast agents for MRI according to “Contrast Agents I” by W. Krause (Springer Verlag 2002, page one and following pages). Furthermore, superparamagnetic particles are another example of contrast-enhancing units, which could also be used as contrast agents for MRI (Textbook of Contrast Media, Superparamagnetic Oxides, Dawson, Cosgrove and Grainger Isis Medical Media Ltd, 1999, page 373 and following pages). As described in Contrast Agent II by W. Krause (Springer Verlag 2002, page 73 and following pages), gas-filled microbubbles could be used in a similar way as contrast agents for ultrasound. Moreover “Contrast Agents II” by W. Krause (Springer Verlag, 2002, page 151 and following pages) reports the use of iodinated liposomes or fatty acids as contrast agents for X-Ray imaging.
[0007]
Contrast-enhancing agents that can be used in functional imaging are mainly developed for PET and SPECT.
[0008]
The application of radiolabelled bioactive peptides for diagnostic imaging is gaining importance in nuclear medicine. Biologically active molecules which selectively interact with specific cell types are useful for the delivery of radioactivity to target tissues. For example, radiolabelled peptides have significant potential for the delivery of radionuclides to tumours, infarcts, and infected tissues for diagnostic imaging and radiotherapy.
[0009]
DOTA (1,4,7,10-tetrakis(carboxymethyl)-1,4,7,10tetraazacyclododecane) and its derivatives constitute an important class of chelators for biomedical applications as they accommodate very stably a variety of di- and trivalent metal ions. An emerging area is the use of chelator conjugated bioactive peptides for labeling with radiometals in different fields of diagnostic and therapeutic nuclear oncology.
[0010]
There have been several reports in recent years on targeted radiotherapy with radiolabeled somatostatin analogs.
[0011]
US2007/0025910A1 discloses radiolabled somatostatin analogs primarily based on the ligand DOTA-TOC. The radionucleotide can be (64)Copper and the somatostatin analog may be octreotide, lanreotide, depreotide, vapreotide or derivatives thereof. The compounds of US2007/0025910A1 are useful in radionucleotide therapy of tumours.
[0012]
US2007/0025910A1 does not disclose (64)Cu-DOTA-TATE. DOTA-TATE and DOTA-TOC differ clearly in affinity for the 5 known somatostatin receptors (SST1-SST2). Accordingly, the DOTA-TATE has a 10-fold higher affinity for the SST2 receptor, the receptor expressed to the highest degree on neuroendocrine tumors. Also the relative affinity for the other receptor subtypes are different. Furthermore, since 177Lu-DOTATATE is used for radionuclide therapy, only 64Cu-DOTATATE and not 64Cu-DOTATOC can be used to predict effect of such treatment by a prior PET scan.
[0013]
There exists a need for further peptide-based compounds having utility for diagnostic imaging techniques, such as PET.
EXAMPLE
[0033]
Preparation of “Cu-Dotatate-DOTA-TATE
[0034]
64Cu was produced using a GE PETtrace cyclotron equipped with a beamline. The 64Cu was produced via the 64Ni (p,n) 64Cu reaction using a solid target system consisting of a water cooled target mounted on the beamline. The target consisted of 64Ni metal (enriched to >99%) electroplated on a silver disc backing. For this specific type of production a proton beam with the energy of 16 MeV and a beam current of 20 uA was used. After irradiation the target was transferred to the laboratory for further chemical processing in which the 64Cu was isolated using ion exchange chromatography. Final evaporation from aq. HCl yielded 2-6 GBq of 64Cu as 64CuCl2 (specific activity 300-3000 TBq/mmol; RNP >99%). The labeling of 64Cu to DOTA-TATE was performed by adding a sterile solution of DOTA-TATE (0.3 mg) and Gentisic acid (25 mg) in aq Sodium acetate (1 ml; 0.4M, pH 5.0) to a dry vial containing 64CuCl2 (˜1 GBq). Gentisic acid was added as a scavenger to reduce the effect of radiolysis. The mixture was left at ambient temperature for 10 minutes and then diluted with sterile water (1 ml). Finally, the mixture was passed through a 0.22 μm sterile filter (Millex GP, Millipore). Radiochemical purity was determined by RP-HPLC and the amount of unlabeled 64Cu2+ was determined by thin-layer chromatography. All chemicals were purchased from Sigma-Aldrich unless specified otherwise. DOTA-Tyr3-Octreotate (DOTA-TATE) was purchased from Bachem (Torrance, Calif.). Nickel-64 was purchased in +99% purity from Campro Scientific Gmbh. All solutions were made using Ultra pure water (<0.07 μSimens/cm). Reversed-phase high pressure liquid chromatography was performed on a Waters Alliance 2795 Separations module equipped with at Waters 2489 UV/Visible detector and a Caroll Ramsey model 105 S-1 radioactivity detector—RP-HPLC column was Luna C18, HST, 50×2 mm, 2.5 μm, Phenomenex. The mobile phase was 5% aq. acetonitrile (0.1% TFA) and 95% aq. acetonitrile (0.1% TFA).
[0035]
Thin layer chromatography was performed with a Raytest MiniGita Star TLC-scanner equipped with a Beta-detector. The eluent was 50% aq methanol and the TLC-plate was a Silica60 on Al foil (Fluka). Ion exchange chromatography was performed on a Dowex 1×8 resin (Chloride-form, 200-400 mesh).
The FDA has approved copper Cu 64 dotatate injection (Detectnet) for the localization of somatostatin receptor–positive neuroendocrine tumors (NETs), according to an announcement from RadioMedix Inc. and Curium Pharma.1
The positron emission tomography (PET) diagnostic agent is anticipated to launch immediately, according to Curium. Doses will be accessible through several nuclear pharmacies or through the nuclear medicine company.
“Detectnet brings an exciting advancement in the diagnosis of NETs for healthcare providers, patients, and their caregivers,” Ebrahim Delpassand MD, CEO of RadioMedix, stated in a press release. “The phase 3 results demonstrate the clinical sensitivity and specificity of Detectnet which will provide a great aid to clinicians in developing an accurate treatment approach for their [patients with] NETs.”
Copper Cu 64 dotatate adheres to somatostatin receptors with highest affinity for subtype 2 receptors (SSTR2). Specifically, the agent binds to somatostatin receptor–expressing cells, including malignant neuroendocrine cells; these cells overexpress SSTR2. The agent is a positron-producing radionuclide that possesses an emission yield that permits PET imaging.
“Perhaps most exciting is that the 12.7-hour half-life allows Detectnet to be produced centrally and shipped to sites throughout the United States,” added Delpassand. “This will help alleviate shortages or delays that have been experienced with other somatostatin analogue PET agents.”
Two single-center, open-label studies confirmed the efficacy of the diagnostic agent, according to Curium.2 In Study 1, investigators conducted a prospective analysis of 63 patients, which included 42 patients with known or suspected NETs according to histology, conventional imaging, or clinical evaluations, and 21 healthy volunteers. The majority of the participants, or 88% (n = 37) had a history of NETs at the time that they underwent imaging. Just under half of patients (44%; n = 28) were men and the majority were white (86%). Moreover, patients had a mean age of 54 years.
Images produced by the PET agent were interpreted to be either positive or negative for NET via 3 independent readers who had been blinded to the clinical data and other imaging information. Moreover, the results from the diagnostic agent were compared with a composite reference standard that was comprised of 1 oncologist’s blinded evaluation of patient diagnosis based on available histopathology results, reports of conventional imaging that had been done within 8 weeks before the PET imaging, as well as clinical and laboratory findings, which involved chromogranin A and serotonin levels.
Additionally, the percentage of patients who tested positive for disease via composite reference as well as through PET imaging was used to quantify positive percent agreement. Conversely, the percentage of participants who did not have disease per composite reference and who were determined to be negative for disease per PET imaging was used to quantify negative percent agreement.
Results showed that the percent reader agreement for positive detection in 62 scans was 91% (95% CI, 75-98) and negative detection was 97% (95% CI, 80-99). For reader 2, these percentages were 91% (95% CI, 75-98) and 80% (95% CI, 61-92), respectively, for 63 scans. Lastly, the percent reader agreement for reader 3 in 63 scans was 91% (95% CI, 75-98) positive and 90% (95% CI, 72-97) negative.
Study 2 was a retrospective analysis in which investigators examined published findings collected from 112 patients; 63 patients were male, while 43 were female. The mean age of patients included in the analysis was 62 years. All patients had a known history of NETs. Results demonstrated similar performance with the PET imaging agent.
In both safety and efficacy trials, a total of 71 patients were given a single dose of the diagnostic agent; the majority of these patients had known or suspected NETs and 21 were healthy volunteers. Adverse reactions such as nausea, vomiting, and flushing were reported at a rate of less than 2%. In all clinical experience that has been published, a total of 126 patients with a known history of NETs were given a single dose of the PET diagnostic agent. A total of 4 patients experienced nausea immediately after administration.
“Curium is excited to bring the first commercially available Cu 64 diagnostic agent to the US market,” Dan Brague, CEO of Curium, North America, added in the release. “Our unique production capabilities and distribution network allow us to deliver to any nuclear pharmacy, hospital, or imaging center its full dosing requirements first thing in the morning, to provide scheduling flexibility to the institution and its patients. We look forward to joining with healthcare providers and our nuclear pharmacy partners to bring this highly efficacious agent to the market.”
References
1. RadioMedix and Curium announce FDA approval of Detectnet (copper Cu 64 dotatate injection) in the US. News release. RadioMedix Inc and Curium. September 8, 2020. Accessed September 9, 2020. https://bit.ly/3m6iC0q.
2. Detectnet. Prescribing information. Curium Pharma; 2020. Accessed September 9, 2020. https://bit.ly/32eZxS3.
///////////////Copper Cu 64 dotatate, 銅(Cu64)ドータテート , FDA 2020, 2020 APPROVALS, Diagnostic, neuroendocrine tumors, Radioactive agent,

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











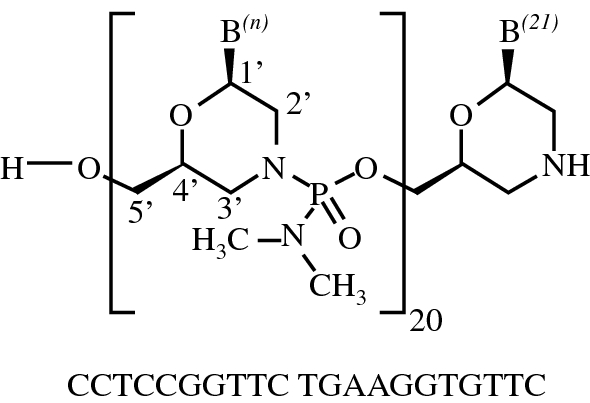

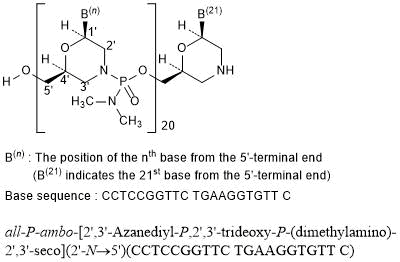















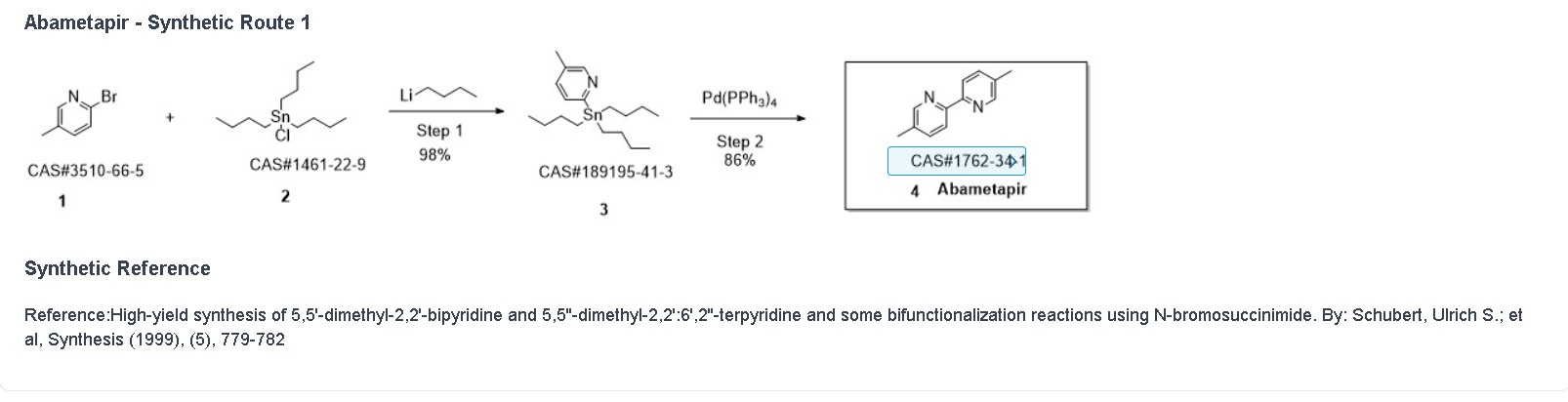

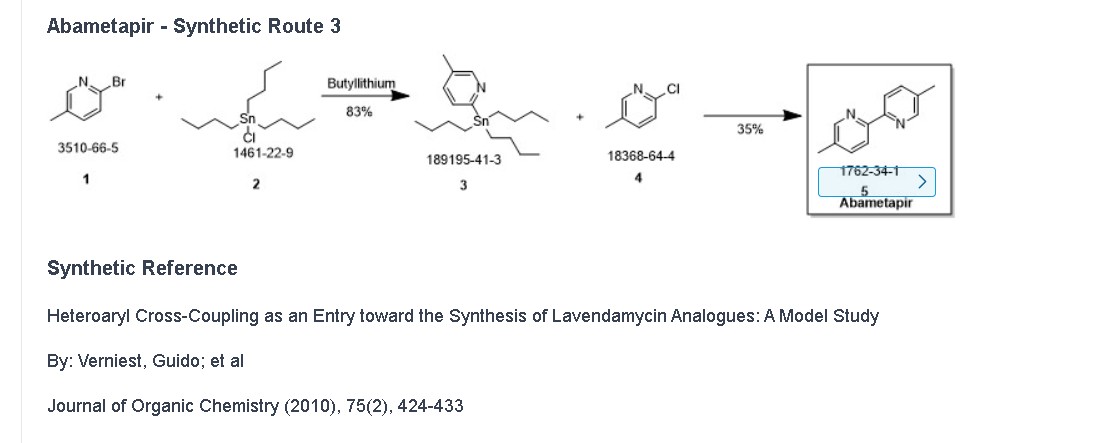

.jpg)



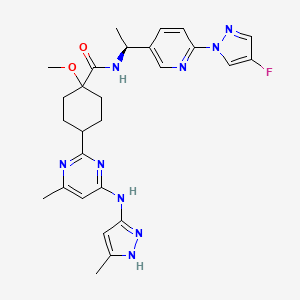




















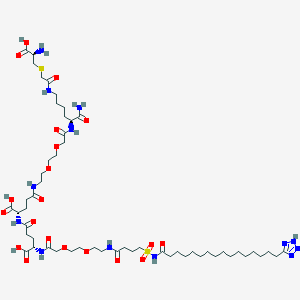

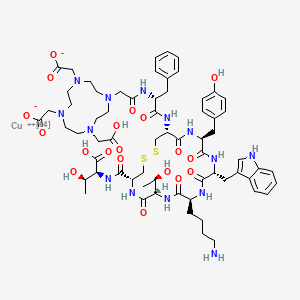

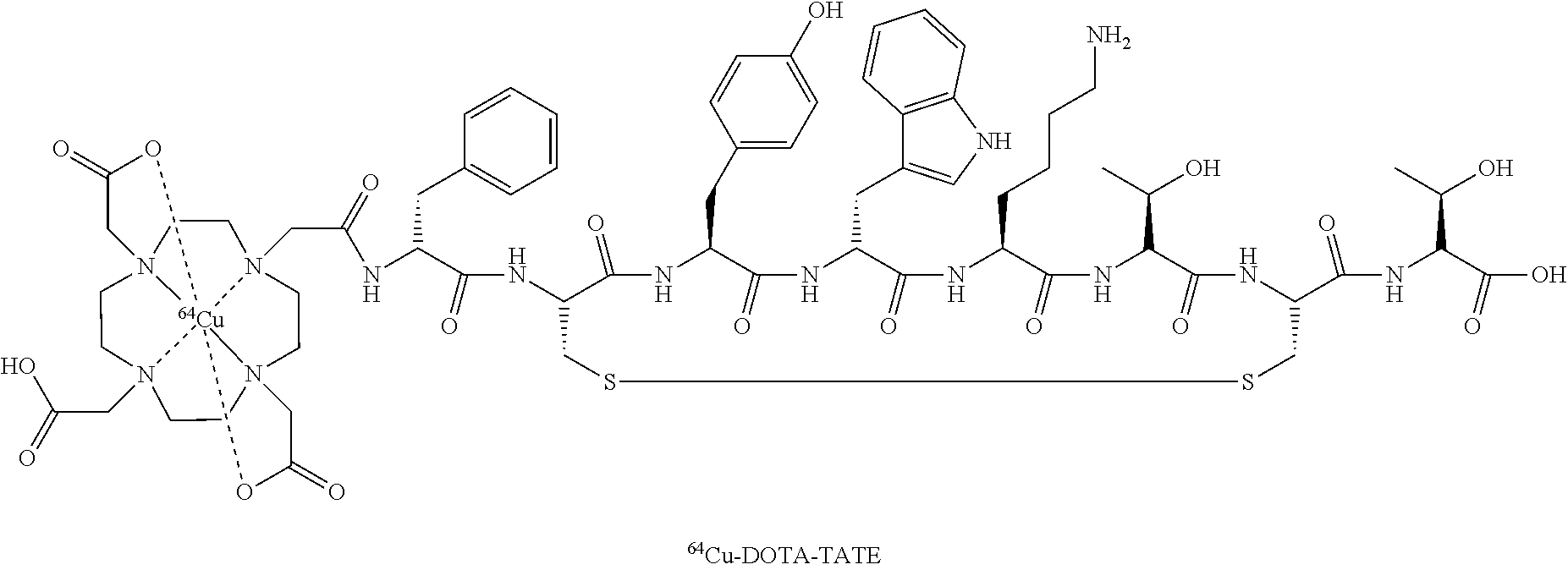




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}