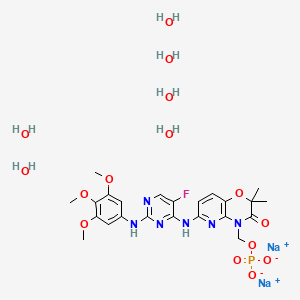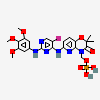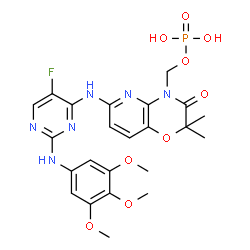
Fostamatinib
- Molecular FormulaC23H26FN6O9P
- Average mass580.459 Da
SQ8A3S5101
TAVALISSE [Trade name]
фостаматиниб [Russian] [INN]
فوستاماتينيب [Arabic] [INN]
[6-({5-Fluoro-2-[(3,4,5-trimethoxyphenyl)amino]-4-pyrimidinyl}amino)-2,2-dimethyl-3-oxo-2,3-dihydro-4H-pyrido[3,2-b][1,4]oxazin-4-yl]methyl dihydrogen phosphate[ACD/IUPAC Name]
2H-Pyrido[3,2-b]-1,4-oxazin-3(4H)-one, 6-[[5-fluoro-2-[(3,4,5-trimethoxyphenyl)amino]-4-pyrimidinyl]amino]-2,2-dimethyl-4-[(phosphonooxy)methyl]-[ACD/Index Name]
9022
Fostamatinib disodium hexahydrate
ホスタマチニブジナトリウム水和物
TAVALISSE™
(fostamatinib disodium hexahydrate) Tablets, for Oral Use
DESCRIPTION
Fostamatinib is a tyrosine kinase inhibitor. TAVALISSE is formulated with the disodium hexahydrate salt of fostamatinib, a phosphate prodrug that converts to its pharmacologically active metabolite, R406, in vivo.
The chemical name for fostamatinib disodium hexahydrate is disodium (6-[[5-fluoro-2-(3,4,5trimethoxyanilino) pyrimidin-4-yl]amino]-2,2-dimethyl-3-oxo-pyrido[3,2-b][1,4]oxazin-4-yl)methyl phosphate hexahydrate. The molecular formula is C23H24FN6Na2O9P·6H2O, and the molecular weight is 732.52. The structural formula is:
Fostamatinib disodium is a white to off-white powder that is practically insoluble in pH 1.2 aqueous buffer, slightly soluble in water, and soluble in methanol.
Each TAVALISSE oral tablet contains 100 mg or 150 mg fostamatinib, equivalent to 126.2 mg or 189.3 mg fostamatinib disodium hexahydrate, respectively.
The inactive ingredients in the tablet core are mannitol, sodium bicarbonate, sodium starch glycolate, povidone, and magnesium stearate. The inactive ingredients in the film coating are polyvinyl alcohol, titanium dioxide, polyethylene glycol 3350, talc, iron oxide yellow, and iron oxide red.

Fostamatinib, sold under the brand name Tavalisse, is a medication approved by the U.S. Food and Drug Administration since 2018 for the treatment of chronic immune thrombocytopenia (ITP). The drug is administered orally as a disodium hexahydrate salt, and is a prodrug of the active compound tamatinib (R-406),[1] which is an inhibitor of the enzyme spleen tyrosine kinase (Syk),[2] hence it is an syk inhibitor.
Fostamatinib has been investigated for the treatment and basic science of Rheumatoid Arthritis and Immune Thrombocytopenic Purpura (ITP). It was approved on April 17, 2018 under the trade name Tavalisse for use in ITP [8]. Fostamatinib has also been granted orphan drug status by the FDA [8].
Fostamatinib is indicated for use in the treatment of chronic immune thrombocytopenia (ITP) in patients who have had insufficient response to previous therapy [Label].
Syk is a protein tyrosine kinase associated with various inflammatory cells, including macrophages, which are presumed to be the cells responsible for ITP platelet clearance.[3] When FcγRs I, IIA, and IIIA bind to their ligands, the receptor complex becomes activated and triggers the phosphorylation of the immunoreceptor-activating motifs (ITAMs). This leads to various genes becoming activated, which causes a cytoskeletal rearrangement that mediates phagocytosis in cells of the monocyte/macrophage lineage. Because Syk plays an important role in FcγR-mediated signal transduction and inflammatory propagation, it is considered a good target for the inhibition of various autoimmune conditions, including rheumatoid arthritis and lymphoma.
Clinical trials
Fostamatinib has been in clinical trials for rheumatoid arthritis, autoimmune thrombocytopenia, autoimmune hemolytic anemia, IgA nephropathy, and lymphoma.[1][4]
The investigation of fostamatinib began with studies involving the treatment of mouse models with cytopenia. Mice were used to measure the effectiveness of R788, a small molecule prodrug of the biologically active R406, a Syk inhibitor. In animal models, treatment with R406/R788 was shown to be safe and effective in reducing inflammation and joint damage in immune-mediated rheumatoid arthritis. The models responded favorably to treatment so the study progressed to Phase 2 trials involving humans. Human studies have shown that R788 has good oral bioavailability, biologic activity, is well tolerated, and does not exhibit collagen or ADP-induced platelet aggregation. In NCT00706342, 16 adults with chronic ITP were entered into an open-label, single-arm cohort dose-escalation trials beginning with 75 mg and rising to 175 mg twice a day. The dose was increased until a persistent response was evident, toxicity was reached, or 175 mg twice a day was met. 8 patients achieved persistent responses with platelet counts greater than 50,000 mm3/L on more than 67% of their visits. 3 of these patients had not persistently responded to thrombopoietic agents. 4 others had nonsustained responses. Mean peak platelet count exceeded 100,000 mm3/L in these 12 patients. Toxicity was evidenced primarily in GI-related side effects, notable diarrhea, urgency, and vomiting. 2 patients developed transaminitis.[5]
Rheumatoid arthritis
A phase II study of rheumatoid arthritis patients failing to respond to a biologic agent showed little efficacy as compared to placebo, but the drug was well tolerated. In patients with high inflammatory burden, measured by levels of C-reactive protein, ACR20 was achieved by a significantly higher portion of those in the fostamatinib group (42%) versus the placebo group (26%).[6]
Autoimmune thrombocytopenia
Immune thrombocytopenic purpura (ITP) is an autoimmune disease where the immune system attacks and destroys platelets in the blood, causing abnormally low platelet counts. It is characterized by the antibody-mediated destruction of platelets. Patients with ITP have accelerated clearance of circulating IgG-coated platelets via Fcγ receptor-bearing macrophages in the spleen and liver, leading to different levels of thrombocytopenia and variable degrees of mucocutaneous bleeding.[7] Recent studies of ITP pathophysiology suggest decreased platelet production may also be an important component of the thrombocytopenia. Many patients exhibit responses to established therapies, including corticosteroids, IV immunoglobulin, anti-D, splenectomy, and rituximab. However, there are a significant minority of patients who retain persistently low platelet counts despite treatment. These patients are consistently at risk of intracranial hemorrhage and other bleeding complications. Several thrombopoiesis-stimulating therapies including eltrombopag and AMG 531 are being investigated to help combat low platelet counts in ITP patients. Rigel reported results from two Phase III clinical trials for fostamatinib as an ITP treatment in August and October 2016. The study is the second Phase 3, multi-center, randomized, double-blind, placebo controlled, study of fostamatinib disodium in the treatment of persistent/chronic immune thrombocytopenic purpura that Rigel has conducted. Primary outcome measures are defined as a stable platelet response by the end of the study (week 24) of at least 50,000/µL on at least 4 of the 6 visits between weeks 14-24. Participants received either a placebo, 100 mg, or 150 mg of the drug in the morning and evening for 24 full weeks. The first study, FIT 1 (047) met the primary endpoint in a statistically significant manner, with 18% of patients hitting the 50,000 platelets/µL of blood and no patients receiving the placebo meeting that criteria. As of June 2016, the open-label, long term extension study (049) is currently tracking 118 patients who opted to receive fostamatinib after completing either study 047 or 048.[8]
Autoimmune hemolytic anemia
Approval for treatment of autoimmune hemolytic anemia (AIHA) is in Stage 1 of Phase II trials. This study is a Phase 2, multi-center, open label, Simon two-stage study to evaluate the safety and efficacy of fostamatinib disodium in the treatment of warm antibody autoimmune hemolytic anemia. Primary outcome measures examined include a hemoglobin response measured by levels higher than 10 g/dL and 2 g/dL higher than the baseline hemoglobin. Responses were studied for a period of 12 weeks and for a dose of 150 mg in the morning and evening. The study began in April 2016 and is estimated to conclude in September 2017. The study is currently recruiting participants from U.S. states including Arizona, California, D.C., Massachusetts, New York, North Carolina, and Texas. Subjects must have had a diagnosis of primary or secondary warm antibody AIHA, and must have failed at least 1 prior treatment regimen for AIHA. Subjects cannot have a platelet count less than 30,000/µL, have AIHA secondary to autoimmune disease, have uncontrolled or poorly controlled hypertension, or have cold antibody AIHA, cold agglutinin syndrome, mixed type AIHA, or paroxysmal cold hemoglobinuria.[9]
Immunoglobulin A nephropathy
Fostamatinib as a treatment for IgA nephropathy (IgAN) is in Phase II trials, which will conclude at the end of 2016. IgAN is a chronic autoimmune disease associated with inflammation in the kidneys that reduces their ability to successfully filter blood. There are currently no disease-targeted therapies for IgAN. Participants are currently being recruited from the US, Austria, Germany, Hong Kong, Taiwan, and the UK. Patients must be between 18 and 70 years old, have renal biopsy findings consistent with IgA nephropathy, have been treated with an Angiotensin Converting Enzyme inhibitor (ACEi) and/or an Angiotensin II Receptor Blocker (ARB) for at least 90 days at the maximum approved dose, have a proteinuria > 1 gm/day at diagnosis of IgA nephropathy and a level > 0.5 gm/day at the second screening visit, and a blood pressure controlled to ≤ 1302/80 with angiotensin blockade. Eligible candidates cannot have recently used cyclophosphamide, mycophenolate mofetil, azathioprine, Rituximab, or > 15 mg/day of prednisone or any other corticosteroid equivalent. The study investigates whether fostamatinib is a safe and effective treatment for IgAN. It is a Phase 2, multi-center, randomized, double-blind, ascending-dose, placebo-controlled clinical study. Primary outcome measures include the mean change in proteinuria as measured by spot urine protein/creatinine ratio (sPCR). Effects were evaluated for 100 mg, 150 mg, and placebo formulations taken twice daily by mouth for 24 weeks. The study began in October 2014 and is expected to complete by June 2017.[10]
Synthesis

PATENTS
https://patents.google.com/patent/WO2008064274A1/en14
Suitable active 2,4-pyrimidinediamine compounds are described, for example, in U.S. application Serial No. 10/355,543 filed January 31 , 2003 (US2004/0029902A1), international application Serial No. PCT/US03/03022 filed January 31, 2003 (WO 03/063794), U.S. application Serial No. 10/631,029 filed July 29, 2003 (US 2005/0028212), international application Serial No. PCT/US03/24087 (WO2004/014382), U.S. application Serial No. 10/903,263 filed July 30, 2004 (US2005/0234049), and international application Serial No.
PCT/US2004/24716 (WO 2005/016893), the disclosures of which are incorporated herein by reference. In such 2,4-pyrimidinediamine compounds, the progroup(s) Rp can be attached to any available primary or secondary amine, including, for example, the N2 nitrogen atom of the 2,4-pyrimidinediamine moiety, the N4 nitrogen atom of the 2,4-pyrimidinediamine moiety, and/or a primary or secondary nitrogen atom included in a substituent on the 2,4-pyrimidinediamine compound. The use of phosphate-containing progroups Rp is especially useful for 2,4-pyrimidinediamine compounds that exhibit poor water solubility under physiological conditions (for example, solubilities of less than about 10 μg/ml). While not intending to be bound by any theory of operation, it is believed that the phosphate-containing progroups aid the solubility of the underlying active 2,4-pyrimidinediamine compound, which in turn increases its bioavailability when administered orally. It is believed that the phosphate progroups Rp are metabolized by phosphatase enzymes found in the digestive tract, permitting uptake of the underlying active drug.
[0024] It has been discovered that the water solubility and oral bioavailability of a particular biologically active 2,4-pyrimidinediamine compound, illustrated below (Compound 1), increased dramatically when formulated to include a progroup Rp of the formula -CH2-O-P(O)(OH)2 at the ring nitrogen atom highlighted with the asterisk (Compound 4):

Compound 4
EXAMPLES
1. Synthesis of Prodrug Compound 4
1.1 N4-(2,2-dimethyl-4-[(di-tert-butyl phosphonoxy)methyl]-3- oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5- trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3)

4 days

[0260] N4-(2,2-dimethyl-3-oxo-4H-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (1, 1.0 g, 2.12 mmol), Cs2CO3 (1.0 g, 3.07 mmol) and di-tert-butyl chloromethyl phosphate (2, 0.67 g, 2.59 mmol) in acetone (20 mL) was stirred at room temperature under nitrogen atmosphere. Progress of the reaction was monitored by LC/MS. Crude reaction mixture displayed three product peaks with close retention times with M++H 693 (minor-1), 693 (major; 3) and 477 (minor-2) besides starting material (Compound 1). Upon stirring the contents for 4 days (70% consumption), the reaction mixture was concentrated and diluted with water. The resultant pale yellow precipitate formed was collected by filtration and dried. The crude solid was purified by silica gel (pretreated with 10%NEt3/CH2Cl2 followed by eluting with hexanes) column chromatography by gradient elution with 70% EtOAc / hexanes-100% EtOAc). The fractions containing Compound 1 and M++H 693 were collected and concentrated. The resulting crude white solid was subjected to repurifϊcation in the similar manner as described previously but by eluting with 30%-50%-75%-100% EtOAc/hexanes. The major product peak with M++H 693 was collected as a white solid (270 mg, 18%) and was characterized as N4-(2,2-dimethyl-4-[(di-tert-butyl phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3). 1H NMR (DMSO-d6): δ 9.21 (s, IH), 9.17 (s, IH), 8.16 (d, IH, J = 2.6 Hz), 7.76 (d, IH, J = 8.5 Hz), 7.44 (d, IH, J = 8.5 Hz), 7.02 (s, 2H), 5.78 (d, IH, J3PH = 6.1 Hz), 3.64 (s, 6H), 3.58 (s, 3H), 1.45 (s, 6H), 1.33 (s, 9H). LCMS: ret. time: 14.70 min.; purity: 95%; MS (m/e): 693 (MH+). 31P NMR (DMSO-d6): -11.36.
1.2. N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3- oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5- trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 4)
[0261] Trifluoroacetic acid (1.5 mL) was added dropwise as a neat for 5 min to N4-(2,2-dimethyl-4-[(di-tert-butyl phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3, 120 mg, 0.173 mmol ) dissolved in CH2Cl2 (10 mL) at 00C under nitrogen atmosphere. The contents were allowed to stir for 1.5 h. Progress of the reaction mixture was monitored by LC/MS. After complete consumption of the starting material, reaction mixture was concentrated, dried and triturated with ether. The ethereal layer was decanted and dried to provide the crude solid. LC/MS analysis of the crude displayed three peaks with M++H 581, 471 and 501. The peak corresponding to M++H 581 was collected by preparative HPLC chromatographic purification. The fractions were lyophilised and dried to provide 53 mg (52%) of off white fluffy solid and characterized as N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[ 1 ,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 4). 1H NMR (DMSO-d6): δ 9.21 (br s, 2H), 8.16 (d, IH, J = 2.6 Hz), 7.93 (d, IH, J = 8.5 Hz), 7.39 (d, IH, J = 8.5 Hz), 7.05 (s, 2H), 5.79 (d, IH, J3PH = 6.6 Hz), 3.67 (s, 6H), 3.59 (s, 3H), 1.44 (s, 6H). LCMS: ret. time: 8.52 min.; purity: 95%; MS (m/e): 581 (MH+). 31P NMR (DMSO-d6): -2.17.
2. Alternative Synthesis of Prodrug Compound 4
[0262] An alternative method of synthesizing prodrug Compound 4 which alleviates the need for column chromatography and HPLC purification is provided below.
2.1 Synthesis of N4-(2,2-dimethyl-4- [(di-tert-butyl
phosphonoxy)methyl] -3-oxo-5-pyrido [ 1 ,4] oxazin-6-yl)-5- fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine
(Compound 3)

rt
92% conversion
majoπminor 6.5:1

[0263] N4-(2,2-dimethyl-3-oxo-4H-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 1, 19.73 g, 41.97 mmol),
Cs2CO3 (15.04 g, 46.16 mmol) and di-tert-butyl chloromethyl phosphate (13.0 g, 50.38 mmol) in DMF (100 mL) was stirred at room temperature under nitrogen atmosphere. Progress of the reaction was monitored by in process LC/MS. Crude reaction mixture displayed two product peaks (ratio 1 :6.5) with close retention times displaying M++H 693 (minor) and 693 (major) besides starting material (Compound 1). Initial yellow reaction mixture turned to olive green as the reaction progressed. Workup was carried out as follows 1). Upon stirring the contents for 30 h (92% consumption), reaction mixture was poured onto ice-water (400 mL) and stirred the contents by adding brine solution (200 mL). Fine yellow tan solid formed was filtered, washed with water and dried overnight.
2). The solid (35 g) was dissolved in MTBE (500 mL) and washed with water (40OmL). Aqueous layer was extracted with MTBE (2 X 350 mL) till the absence of UV on TLC. Combined organic layers were dried over anhydrous Na2SO4 and decanted.
Note: step 2 can be done directly, however, DMF extraction back into solution leads to difficulty in the crystallization step.
3). The dark red clear solution was subjected to 10 g of activated charcoal treatment, heated to boil and filtered.
4). The dark red clear solution was concentrated by normal heating to 400 mL of its volume and left for crystallization. The solid crystallized as granules was filtered, crushed the granules to powder, washed with MTBE (400 mL) and dried under high vacuum. See step 7 for the workup of mother liquor. Weight of the solid: 17 g; purity: 90% (Compound 3), 6.26% (Compound 1), 1.8% (minor M+ 693).
5). At this stage solid was taken in 500 ml of ethyl ether and heated to boil. Cooled and filtered to remove undissolved material. Filtrate was concentrated.
6). Above concentrate was subjected to crystallization in MTBE (300 mL).
The white solid formed was filtered, washed with MTBE (100 mL) and dried under high vacuum to provide the desired N4-(2,2-dimethyl-4-[(di-tert-butyl
phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3) in 97% purity. 1H NMR (DMSO-d6): δ 9.21 (s, IH), 9.17 (s, IH), 8.16 (d, IH, J = 2.6 Hz), 7.76 (d, IH, J = 8.5 Hz), 7.44 (d, IH, J = 8.5 Hz), 7.02 (s, 2H), 5.78 (d, IH, J3PH = 6.1 Hz), 3.64 (s, 6H), 3.58 (s, 3H), 1.45 (s, 6H), 1.33 (s, 9H). LCMS: ret. time: 14.70 min.; purity: 95%; MS (m/e): 693 (MH+). 31P NMR (DMSO-d6): -11.36. Weight of the solid: 15.64 g (yield: 55%); purity: 97% (Compound 3), 3% (Compound 1).
7). The mother liquor was concentrated and steps 5 and 6 were repeated to provide Compound 3.
2.2. Synthesis of N4-(2,2-dimethyl-4-[(dihydrogen
phosphonoxy)methyl] -3-oxo-5-pyrido [ 1 ,4] oxazin-6-yl)-5- fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine
(Compound 4)
[0264] N4-(2,2-dimethyl-4-[(di-tert-butyl phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 3); (15.0 g, 21.67 mmol) dissolved in AcOH:H20 (225 niL, 4:1) was heated at 65 0C (oil bath temp). The progress of the reaction was monitored by in process LC/MS. The reaction mixture transformed to faint tan white solid after Ih of heating. At this point most of Compound 3 converted to mono des t-butyl product. After 3h of heating, consumption of SM and complete conversion of intermediate (mono des t-butylated) to product was observed.
[0265] Reaction mixture was cooled, poured onto ice-water (200 mL), stirred for 20 min and filtered. The clear white filter cake was washed with water (600 ml) and acetone (200 mL) successively, dried for 2h followed by drying under high vacuum over P2O5 in a desiccator. Weight of the solid: 12.70 g; purity: 97% (Compound 3) and 3% (Compound 1) 1H NMR indicated acetic acid presence (1 :1)
[0266] To remove acetic acid, the solid was taken in acetonitrile (300 mL) and concentrated by rotovap vacuum. This process was repeated 2 times with acetonitrile and toluene (3 X 300 mL). The solid obtained was dried under high vacuum at 50 OC. [0267] Finally, the solid was taken in acetone (400 mL), filtered and dried to provide solid N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 4). 1H NMR (DMSO-d6): δ 9.21 (br s, 2H), 8.16 (d, IH, J = 2.6 Hz), 7.93 (d, IH, J = 8.5 Hz), 7.39 (d, IH, J = 8.5 Hz), 7.05 (s, 2H), 5.79 (d, IH, J3PH = 6.6 Hz), 3.67 (s, 6H), 3.59 (s, 3H), 1.44 (s, 6H). LCMS: ret. time: 8.52 min.; purity: 95%; MS (m/e): 581 (MH+). 31P NMR (DMSO-d6): -2.17.
3. Synthesis of N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo- 5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4- pyrimidinediamine mono calcium salt (Prodrug Salt 6)

[0268] Aqueous (10 niL) NaHCO3 (0.17 g, 2.02 mmol) solution was added dropwise to a suspension of N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (0.5 g, 0.86 mmol) in water (5 mL) at room temperature while stirring the contents. The clear solution formed was treated with aqueous (10 mL) CaCl2 (0.11 g in 10 mL water, 0.99 mmol) n a dropwise manner at room temperature. The addition resulted in the precipitation of a white solid from reaction mixture. Upon completion of addition, the contents were stirred for a period of 30 min, filtered, washed with water (40 mL) and dried. The clear white solid was taken in water (30 mL) and heated on a stir plate to boil. The solution was cooled, filtered and dried. The white solid collected and further dried under high vacuo at 80 0C for 32 h to provide 0.41 g (83%) of solid N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[ 1 ,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine mono calcium salt (Prodrug Salt 6).
[0269] Ca(OAc)2 may also used in place Of CaCl2 in this preparation.
4. Synthesis of N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo- 5-pyrido[l,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4- pyrimidinediamine disodium salt hexahydrate and monosodium salt
hydrate

[0270] A round-bottomed flask was charged with 10.00 g N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[ 1 ,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (Compound 4) and 140 mL water into a round bottom flask to form a slurry having a pH between 3.6 and 3.7. The pH was adjusted to in the range of 9.3 to 10.3 by addition of 1 M aqueous NaOH, initially forming a turbid solution, which returned to a suspension upon prolonged stirring. The mixture was heated at reflux, then the turbid solution was hot filtered through filter paper. The solid collected in the filter paper was rinsed with 10 mL hot water.
Isopropanol (75 mL) was added to the filtrate, yielding a clear solution, which was allowed to cool to room temperature over about 1.5 hours with stirring, during which time a solid precipitated. The precipitate was collected by filtration, rinsed with 47 mL isopropanol, and taken up in 73 mL acetone to form a slurry, which was stirred for 1.5 hours at room temperature. The solid was again collected by filtration and rinsed with 18 mL acetone, then dried at about 40 0C under vacuum until substantially all isopropanol and acetone was removed (i.e., below 0.5 wt% each). The product was exposed to air at about 40% relative humidity and room temperature until the water content stabilized at about 15% by Karl Fisher titration, yielding 8.18 g of the title compound. 1H NMR (D2O): δ 7.67 (d, IH, J = 3.8 Hz), 7.49 (d, IH, J = 8.8 Hz), 6.87 (d, IH, J = 8.8 Hz), 6.50 (s, 2H), 5.52 (d, IH, J3PH = 2.0 Hz), 3.53 (s, 3H), 3.47 (s, 6H), 1.32 (s, 6H). 31P NMR (D2O): 2.75. The prodrug salt hydrate was obtained as a pure-white, highly crystalline material. Microscopic investigation indicated that the crystallites are plate-like with a particle size of less than 10 μm. Polarized light microscopy revealed birefringence corroborating the crystalline nature of the hydrate. [0271] The monosodium salt can be prepared from N4-(2,2-dimethyl-4-[(dihydrogen phosphonoxy)methyl]-3-oxo-5-pyrido[l,4]oxazin-6-yl)-5-fiuoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine and sodium hydroxide by a proper pH control; pH of 5-5.5 results in predominantly the formation of monosodium salt.
References
- ^ Jump up to:a b S.P. McAdoo; F.W.K. Tam (2011). “Fostamatinib Disodium”. Drugs of the Future. 36 (4): 273–280.
- Jump up^ Braselmann, S; Taylor, V; Zhao, H; Wang, S; Sylvain, C; Baluom, M; Qu, K; Herlaar, E; et al. (2006). “R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation”. The Journal of Pharmacology and Experimental Therapeutics. 319 (3): 998–1008. doi:10.1124/jpet.106.109058. PMID 16946104.
- Jump up^ Linger, Rachel M. A.; Keating, Amy K.; Earp, H. Shelton; Graham, Douglas K. (2008-01-01). “TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer”. Advances in cancer research. 100: 35–83. doi:10.1016/S0065-230X(08)00002-X. ISSN 0065-230X. PMC 3133732
 . PMID 18620092.
. PMID 18620092.
- Jump up^ Morales-Torres, Jorge (2010). “R788 (fostamatinib disodium): a novel approach for the treatment of rheumatoid arthritis”. International Journal of Clinical Rheumatology. 5 (1): 9–15. doi:10.2217/ijr.09.69.
- Jump up^ “Pilot Study of Fostamatinib Disodium/R935788 for the Treatment of Adult Refractory Immune Thrombocytopenic Purpura (ITP) – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2016-11-19.
- Jump up^ “Fostamatinib Shown to Be Safe but Not Effective in Rheumatoid Arthritis Patients Unresponsive to Biologic Agents”. Science Daily. Jan 27, 2011. Retrieved Dec 13, 2012.
- Jump up^ Podolanczuk, Anna; Lazarus, Alan H.; Crow, Andrew R.; Grossbard, Elliot; Bussel, James B. (2009-04-02). “Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk”. Blood. 113 (14): 3154–3160. doi:10.1182/blood-2008-07-166439. ISSN 0006-4971. PMID 19096013.
- Jump up^ “A Efficacy and Safety Study of Fostamatinib in the Treatment of Persistent/Chronic Immune Thrombocytopenic Purpura (ITP) – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2016-11-19.
- Jump up^ “A Safety and Efficacy Study of R935788 in the Treatment of Warm Antibody Autoimmune Hemolytic Anemia (AIHA) – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2016-11-19.
- Jump up^ “Safety and Efficacy Study of Fostamatinib to Treat Immunoglobin A (IgA) Nephropathy – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2016-11-19.
-
-
FDA Orange Book Patents
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
from FDA Orange Book
///////////SQ8A3S5101, TAVALISSE , фостаматиниб , وستاماتينيب , 福他替尼 , FDA 2018, fostamatinib disodium hexahydrate, fostamatinib , ホスタマチニブジナトリウム水和物
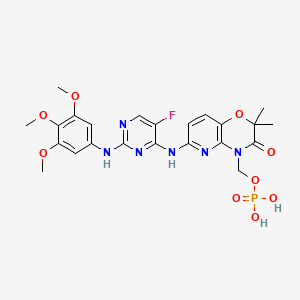
COC1=CC(NC2=NC=C(F)C(NC3=NC4=C(OC(C)(C)C(=O)N4COP(O)(O)=O)C=C3)=N2)=CC(OC)=C1OC

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....




















 ONPATTRO™ (patisiran) packaging and product vial (Photo: Business Wire)
ONPATTRO™ (patisiran) packaging and product vial (Photo: Business Wire)