
Home » Posts tagged 'CHUGAI'
Tag Archives: CHUGAI
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
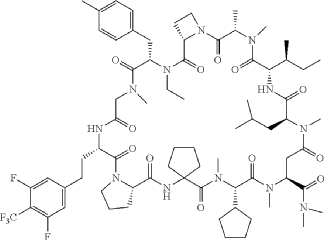
Paluratide
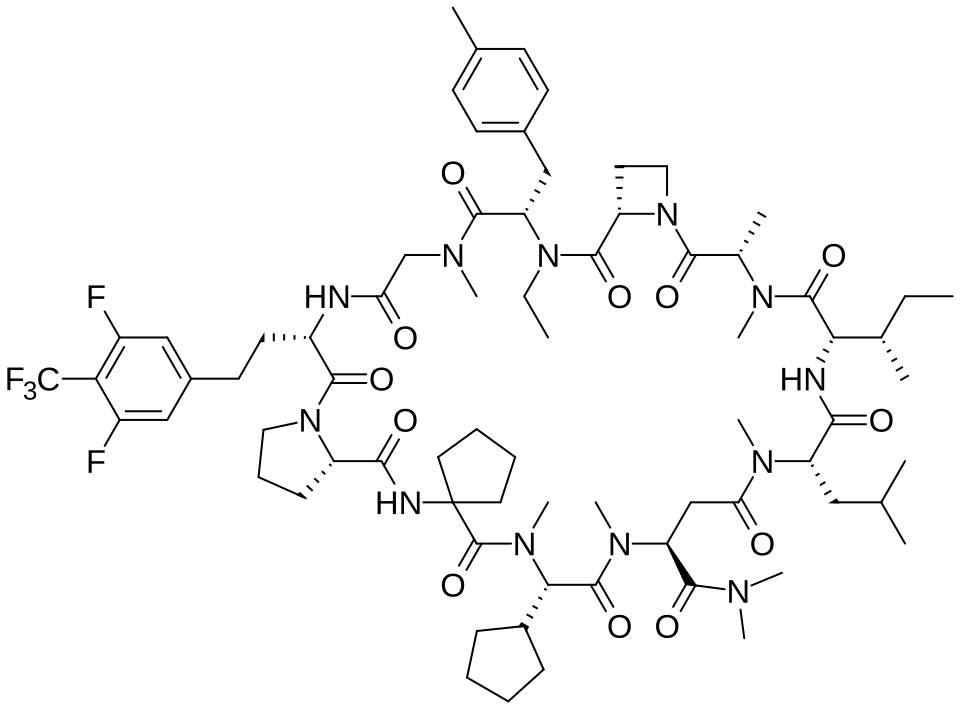


Paluratide
CAS 2676177-63-0
MFC73H105F5N12O12 MW 1437.7 g/mol
1,11-anhydro[N-methyl-L-alanyl-(2S)-azetidine-2-carbonyl-N-ethyl-4-methyl-L-phenylalanyl-N-methylglycyl-3-{[3,5-difluoro-4-(trifluoromethyl)phenyl]methyl}-L-alanyl-L-prolyl-2-
aminocyclopentane-1-carbonyl-(2S)-N-methyl-3-cyclopentylglycyl-1-
(dimethylamino)-N-methyl-L-aspart-4-yl-N-methyl-L-leucyl-Lisoleucine]
(3S,9S,12S,17S,20S,23S,27S,30S,36S)-20-[(2S)-butan-2-yl]-30-cyclopentyl-3-[2-[3,5-difluoro-4-(trifluoromethyl)phenyl]ethyl]-10-ethyl-N,N,7,17,18,24,28,31-octamethyl-9-[(4-methylphenyl)methyl]-23-(2-methylpropyl)-2,5,8,11,16,19,22,25,29,32,35-undecaoxospiro[1,4,7,10,15,18,21,24,28,31,34-undecazatricyclo[34.3.0.012,15]nonatriacontane-33,1′-cyclopentane]-27-carboxamide
G-protein Ras (rat sarcoma virus) inhibitor, antineoplastic, LUNA 18, CHUGAI, AW3YP3CD9X
Paluratide (development code LUNA18) was an investigational cyclic peptide KRAS inhibitor developed by Chugai Pharmaceutical, a member of the Roche Group, for the treatment of cancers with KRAS mutations.[1] The compound was notable as an orally bioavailable macrocyclic peptide that could target intracellular protein-protein interactions, a class of targets traditionally considered “undruggable.”[2]
Development was discontinued in July 2025 due to a narrow therapeutic window compared to competing KRAS inhibitors.[3]
Ras Inhibitor LUNA18 is an orally bioavailable cyclic peptide and Ras inhibitor, with potential antineoplastic activity. Upon oral administration, Ras inhibitor LUNA18 selectively targets, binds to and inhibits Ras, thereby inhibiting Ras-dependent signaling and inhibits proliferation of tumor cells in which Ras is overexpressed and/or mutated. Ras serves an important role in cell signaling, division and differentiation. Mutations of Ras may induce constitutive signal transduction leading to tumor cell growth, proliferation, invasion, and metastasis.
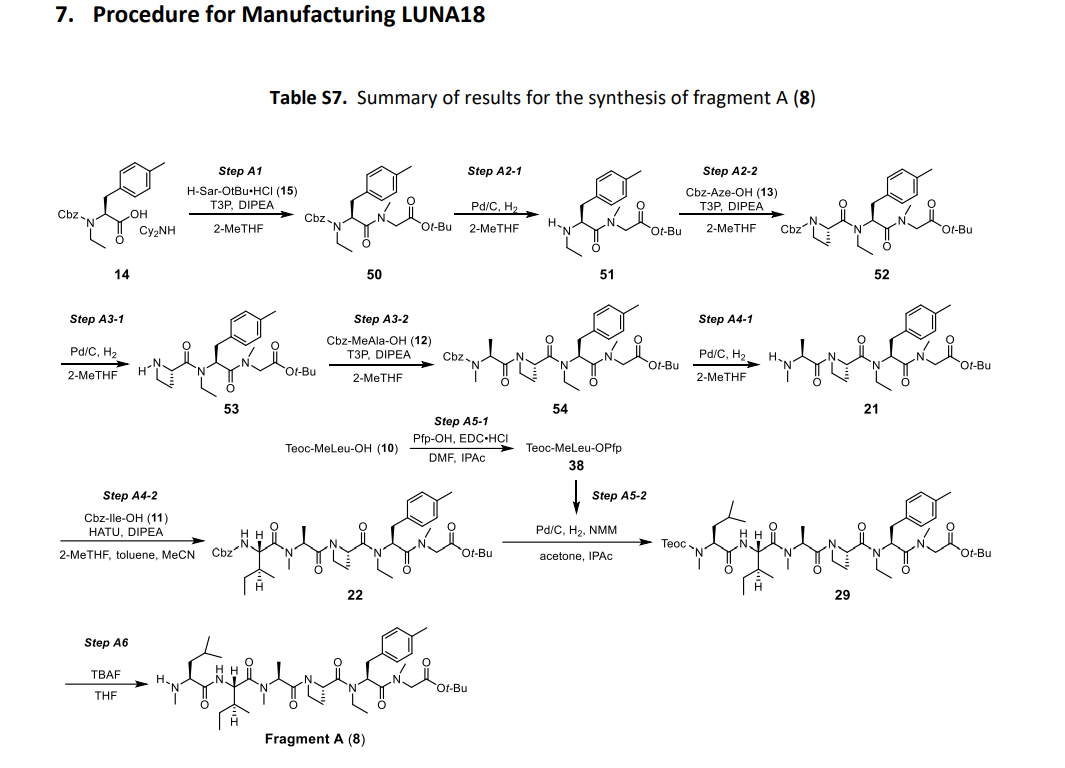
Paluratide (LUNA18 is synthesized using a novel liquid-phase peptide synthesis (LPPS) method, not traditional solid-phase methods, to overcome challenges with N-alkylated cyclic peptides. This process involves a convergent route of 24 telescoped chemical transformations, a final crystallization step, and a focus on specific strategies to manage side reactions like diketopiperazine formation and low reactivity of sterically hindered amino acids.
Key aspects of the synthesis
- Liquid-phase synthesis: A novel, high-yielding LPPS process was developed to enable the large-scale production of paluratide. This is a departure from traditional solid-phase methods, which have limitations with solubility and waste.
- Convergent synthetic route: The synthesis uses a convergent approach, meaning smaller fragments of the peptide are synthesized separately and then joined together. The overall process includes 24 telescoped chemical transformations followed by a final crystallization step.
- Addressing synthesis challenges: Specific strategies were employed to overcome key difficulties:
- Low reactivity: Amino acids with N-alkylation are sterically hindered, so more reactive and stable protecting groups were used to ensure efficient coupling.
- Side reactions: The method was designed to prevent side reactions like diketopiperazine formation in intermediates and incomplete hydrolysis of active esters.
- Instability: The peptide backbone is sensitive to acidic conditions, so a mildly acidic aqueous medium was chosen for workup and purification to maintain stability.
- Protecting group selection: Cbz-protected amino acid active esters were preferred over Boc-protected ones because they are less prone to forming N-carboxyanhydrides (NCA) under activating conditions, which can reduce yield and purity.
- Purification: A final crystallization step is used for purification.
PAT
- Method for producing eutectic of cyclic peptidePublication Number: WO-2024195801-A1Priority Date: 2023-03-20
- Method for producing cyclic peptide crystalsPublication Number: WO-2024085235-A1Priority Date: 2022-10-20
- Composition containing peptide, surfactant, and polymerPublication Number: WO-2024080308-A1Priority Date: 2022-10-12
- Methods for producing cyclic compounds comprising n-substituted amino acid residuesPublication Number: EP-4086272-A1Priority Date: 2021-05-07
- Methods for producing cyclic compounds comprising n-substituted amino acid residuesPublication Number: US-2022411462-A1Priority Date: 2021-05-07
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.5c00260?ref=PDF



PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US383248369&_cid=P20-MI3YXS-80609-1



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Mechanism of action
Paluratide functions as a pan-RAS inhibitor, targeting multiple RAS isoforms including KRAS, NRAS, and HRAS.[1] The compound binds with high affinity to KRASG12D, with a dissociation constant (Kd) of 0.043 nM, and blocks the interaction between KRASG12D and the guanine nucleotide exchange factor SOS1 with an IC50 of less than 2.2 nM.[4]
Unlike covalent KRAS inhibitors that target specific mutations (such as sotorasib for KRASG12C), paluratide was designed to inhibit RAS proteins through disruption of protein-protein interactions with guanine nucleotide exchange factors (GEFs).[1] This mechanism allows the drug to affect RAS signalling regardless of the specific mutation, theoretically providing broader applicability across different KRAS-mutant cancers. The compound also demonstrates activity against downstream signalling pathways, affecting ERK and AKT phosphorylation.[4]
Medical uses
Paluratide was being developed for the treatment of locally advanced or metastatic solid tumors harbouring RAS gene alterations.[5] The drug demonstrated significant cellular activity against multiple cancer types with KRAS mutations in preclinical studies, including colorectal cancer, gastric cancer, non-small cell lung cancer, and pancreatic cancer.[1]
Chemistry
Paluratide is an 11-member (11-mer) cyclic peptide with a molecular weight in the range of 1000–2000 g/mol, classified as a “middle-size” cyclic peptide.[1] The compound features extensive N-alkylation, a modification that reduces hydrogen bond donors and improves oral absorption while maintaining cellular permeability.[2] Its structure allows it to navigate the challenging boundary between small molecules and biologics, achieving properties of both classes. The compound demonstrated oral bioavailability ranging from 21% to 47% in preclinical animal studies without requiring special formulations.[1]
Discovery
Paluratide was discovered through Chugai Pharmaceutical’s cyclic peptide platform using an mRNA display library screening approach.[1] The initial hit compound, designated AP8747, was identified from the mRNA display library and subsequently underwent extensive chemical optimization without scaffold hopping (maintaining the basic cyclic peptide structure).[1] The optimization focused on increasing plasma stability, improving absorption, reducing clearance, and reducing hydrogen bond donors to achieve oral bioavailability.
The final clinical compound, LUNA18, emerged after modifications to four amino acid positions (positions 5, 7, 10, and 11) from an intermediate compound (compound 40). Key structure-activity relationship findings included: the side chain at position 5 preferring aromatic over aliphatic groups; physicochemical properties being adjustable at position 11; and biological activity enhancement through modifications at positions 7 and 10.[1]
Chugai also developed a novel synthetic methodology that enabled the broadly applicable synthesis of highly N-alkylated cyclic peptide-like drugs.[6] This method overcame three major technical challenges: formation of diketopiperazine, insufficient reactivity of amidation due to steric hindrance, and instability of cyclic peptides under acidic conditions. Using this approach, more than 4,000 cyclic peptides were synthesized with a process yield of 31% and final product purity of 97%.[6]
Clinical trials
A Phase 1 dose-escalation and cohort expansion study (NCT05012618) was initiated in August 2021 to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary activity of paluratide administered as a single agent or in combination with other anti-cancer drugs.[5] The study, in the United States and Japan, was designed to enrol approximately 195 patients with locally advanced or metastatic solid tumors positive for documented RAS alterations.[5]
Paluratide was administered orally as capsules.[5] The study also evaluated combination therapy with cetuximab, an EGFR inhibitor.[5]
References
- Tanada M, Tamiya M, Matsuo A, Chiyoda A, Takano K, Ito T, et al. (August 2023). “Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor”. Journal of the American Chemical Society. 145 (30): 16610–16620. Bibcode:2023JAChS.14516610T. doi:10.1021/jacs.3c03886. PMID 37463267.
- Ohta A, Tanada M, Shinohara S, Morita Y, Nakano K, Yamagishi Y, et al. (November 2023). “Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets”. Journal of the American Chemical Society. 145 (44): 24035–24051. Bibcode:2023JAChS.14524035O. doi:10.1021/jacs.3c07145. PMID 37874670.
- Taylor NP (24 October 2025). “Roche axes 4 Chugai solid tumor assets in early-phase clear-out”. Fierce Biotech.
- “LUNA18 (Paluratide) – KRAS Inhibitor, ERK Inhibitor, RAS Inhibitor”. MedChemExpress.
- “A Dose-escalation Study of LUNA18 in Patients With Locally Advanced or Metastatic Solid Tumors (With Expansion)”. ClinicalTrials.gov. 29 July 2025. NCT05012618.
- Nomura K, Hashimoto S, Takeyama R, Tamiya M, Kato T, Muraoka T, et al. (October 2022). “Broadly Applicable and Comprehensive Synthetic Method for N-Alkyl-Rich Drug-like Cyclic Peptides”. Journal of Medicinal Chemistry. 65 (19): 13401–13412. doi:10.1021/acs.jmedchem.2c01296. PMID 36109865.
- “Chugai Announces 2025 2nd Quarter Results” (Press release). Chugai Pharmaceutical. 24 July 2025.
External links
- Phase 1 Clinical Trial Information at ClinicalTrials.gov
- Development of LUNA18 at Journal of the American Chemical Society
| Clinical data | |
|---|---|
| Other names | LUNA18 |
| Routes of administration | Oral administration |
| Legal status | |
| Legal status | Development discontinued |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2676177-63-0 |
| PubChem CID | 166509683 |
| ChemSpider | 129321315 |
| UNII | AW3YP3CD9X |
| Chemical and physical data | |
| Formula | C73H105F5N12O12 |
| Molar mass | 1437.707 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////Paluratide, antineoplastic, LUNA 18, CHUGAI, AW3YP3CD9X
RG-1577, EVT 302, Sembragiline, RO-4602522
RG-1577, EVT 302, Sembragiline, RO-4602522
CAS 676479-06-4, MW 342.36
- C19 H19 F N2 O3
- Acetamide, N-[(3S)-1-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-3-pyrrolidinyl]-
UNII-K3W9672PNJ
RG-1577, a selective and reversible monoamine oxidase B inhibitor, for treating AD (phase 2 clinical, as of May 2015).
Family members of the product case for RG-1577 (WO2004026825) hold protection in EU until 2023 and expire in US in 2024 with US154 extension. Follows on from WO2006097197, claiming a process for preparing RG-1577.
Alzheimer‘s Disease is a brain disease that slowly destroys memory and thinking skills, up to loss of the ability to carry out the simplest tasks. It is the most common cause of dementia among older people. Mild Alzheimer‘s Disease manifests itself in memory loss and small changes in other cognitive abilities, e.g getting lost, trouble handling money and managing daily tasks, having some mood and personality changes, etc.
In the stage of Moderate Alzheimer‘s Disease, the control of language, reasoning, sensory processing, and conscious thought are impacted. Memory loss and con usion grow worse, e.g patients have problems recognizing family and friends and become unable to learn new things, etc. hallucinations, delusions, and paranoia may occur. .Severe Alzheimer‘s Disease is the final stage. Patients cannot communicate anymore and are completely dependent.
N-[(3S)-l-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide has previously been described in the art. 1 WO 2006/097197 2 and WO 2006/0972703 relate to methods for preparing enantiomerically pure 4-pyrrolidinophenylbenzyl ether derivatives.
![]()
The processes of the prior art hamper from several drawbacks (e.g. long reaction sequence, low overall yield also due to loss of half of the product in the classical resolution step, the need for a chromatographic purification to remove by-products formed in the Mitsunobu reaction) and are therefore less suitable for the preparation of N-[(3S)-l-[4-[(3-fluorophenyl) methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide on large scale.
Most Recent Events
- 01 Aug 2014Roche completes a phase I trial in volunteers in USA (NCT02104648)
- 14 May 2014Roche completes enrolment in the MAyflOwer RoAD trial for Alzheimer’s disease (combination therapy, adjunctive treatment) in Australia, Canada, Czech Republic, France, Germany, Italy, Poland, South Korea, Spain, Sweden the United Kingdom and the USA (NCT01677754)
- 01 Apr 2014Roche initiates enrolment in a phase I trial in healthy volunteers in USA (NCT02104648)
http://www.evotec.com/uploads/media_library/10/2012-09_Evotec_Company_presentation_September_e.pdf

……………………..
WO2004026825
http://www.google.com/patents/WO2004026825A1?cl=en
………………….
WO2006097197
http://www.google.com/patents/WO2006097197A1?cl=en
……………………………………………..
PATENT
WO 2015063001
Novel, crystalline polymorphic forms A and B of a pyrrolidone derivative ie RG-1577, useful for treating Alzheimer’s disease (AD). Roche and its Japanese subsidiary Chugai, under license from Evotec, which previously licensed the drug from Roche, are developing RG 1577
formula 1 via the following routes

In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route A

1
In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route B

In a certain embodiment, present invention relates to a crystalline polymorph of a compound of formula 1.

synthesize a compound of formula 1 from a compound of formula 7

compound of formula 6 to a compound of formula 7

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 6 via the intermediate 6a to a compound of formula 7

further comprising reacting a compound of formula 3 with a compound of formula 5 to a compound of formula 6

comprising reacting a compound of formula 2 to a compound of formula 3

2 3
In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 10 to a compound of formula 6

eacting a compound of formula 9 with a compound of formula 5 to a compound of formula 10

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 8 to a compound of formula 9

(lS’)-N-[l-[4-(3-fluoro-benzyloxy)-phenyl]-5-oxo-pyrrolidin-3-yl-]acetamide (1)
To a suspension of chloride (7) (37.9 g, 100 mmol) in 2-methyltetrahydrofurane (600 ml) was added under vigorous stirring at 0°C 1.65 M potassium ie/t-butoxide in THF (75.5 ml, 125 mmol, ACROS) over 2.5 h. After additional stirring at 0°C for 1 h, the cold suspension was hydrolyzed with 0.1 M HCl (600 ml) and the reaction mixture was stirred at 30°C for 0.5 h. The organic layer was washed with water (300 ml), dried (Na2S04) and filtered. Removal of the solvent by rotary evaporation (50°C/>10 mbar) afforded 32.1 g crystalline residue, which was dissolved in 2-butanone (400 ml) at ca. 95°C and hot filtered. Crystallization, which was induced by seeding and cooling to room temperature and 0°C (4 h) afforded 25.4 g (74.2%) of the titled compound (1) as an off-white, crystalline powder,
Mp. 162-164°C (polymorph B).
Ee >99.8%, [cc]D20 = – 17.8 (DMF; c = 1).
1H NMR (400 MHz, DMSO- 6) δ ppm 1.82 (s, 3H), 2.34 (dd, J1=n. l, J2=3.9, 1H), 2.84 (dd, J/=17.1, J2=8.2, 1H), 3.55 (dd, J/=10.2, J2=3.2, 1H), 4.07 (dd, J/=10.2, J2=6.7, 1H), 4.32-4.41 (m, 1H), 5.13 (s, 2H), 7.02 & 7.55 (d, J=9.1, each 1H), 7.11-7.19 (m, 1H), 7.24-7.31 (m, 1H), 7.40-7.47 (m, 1H), 8.40 (d, J=6.4, 1H).
ESI-MS (m/z) 343 [M+H]+, 365 [M+Na]\. Anal.Calcd for Ci9H19FN203 (342.37): Calcd. C, 66.66; H, 5.59; N, 8.18; F, 5.02; O, 14.02. Found C, 66.76; H, 5.48; N, 8.13; F, 5.03; O, 13.99.
Crystallized (1) form previous step (9.5 g, 0.028 mol) was dissolved in 2-butanone (290 mL) upon heating. The hot solution was filtered over charcoal. The solution was concentrated by removal of 2-butanone (200 mL) by distillation prior to seeded cooling crystallization. Filtration, washing with chilled 2-butanone and drying at 50°C/25 mbar/16h afforded 9.18 g (93.9% corrected yield) of the title compound (1) as a crystalline powder of polymorphic form B with an assay of 100.4 %(w/w) and a purity of 99.97 %(area) (by HPLC).
Alternatively, to a stirred suspension of hydroxyamide (6) (30.0 g, 0.083 mol) in toluene (500 ml) was added at 50°C within 45 minutes thionyl chloride (10.40 g, 0.087 mol) and the resulting mixture was stirred for 3h at 50°C. The mixture was then heated up to 92°C and subsequently stirred at this temperature for 15 h. The Suspension was then cooled to 50°C and toluene was removed by distillation under reduced pressure. The distillation residue was cooled to ambient temperature and treated with N-methylpyrrolidone (210 ml) to obtain an almost clear solution. This solution was then cooled to -10°C and subsequently treated at this temperature within 2h with a solution of potassium iert-butoxide (12.40 g, 0.111 mol) in THF (60 g). The resulting mixture was stirred for another 60 minutes at -10°C, then warmed up to room temperature within 60 minutes and subsequently stirred at room temperature for 6 h. The reaction mixture was quenched with water (150 g) and the pH was adjusted with acetic acid (approx. 1.8 g) to pH 7-8. The mixture was then heated to 30-45°C and THF and toluene were distilled off under reduced pressure (<200 mbar) to obtain a clear NMP/water mixture (400 ml). This mixture was heated to 45°C and 260 mg of seed crystals were added. Water (320 ml) was then added within 3 h whereby the product crystallized. The resulting suspension was cooled to room temperature within 3 h and subsequently stirred at this temperature for 2 h. Filtration and washing of the filter cake with a mixture of water (100 ml) and N-methylpyrrolidone (20 ml) and subsequently only with water (150 ml) afforded after drying (70°C/10 mbar/20 h) 26.2 g (92%) of the title compound (1) as a crystalline powder with an assay of 99.6 %(w/w) and a purity of 99.7 %(area) (by HPLC).

HPLC
Purity (HPLC): Column: XSelect Phenyl Hexyl x2, 150 x 4.6mm, 3.5um. Starting
Pressure: 226 bar; temp.: 50°C. Inj. vol.: 2.0 μΐ^ + wash. Flow: 1.0 ml/min. Det: 204 nm. A: Water + 5% ACN, 77-2% in 7 min., hold for 1 min.; B: 0.1% HCOOH, 18% isocratic; C: MeOH, 5-80% in 7 min., hold for 1 min. Sample prep.: 2 mg/ml ACN. Retention times: β-acid 5.93 min., diacid 6.18 min., cc-acid 6.89 min., diester 6.96 min.
ee determination(HPLC): Column: Chiralpak IA-3 100 x 4.6mm, 3um; 91 bar, 2ml/min; temp.: 30°C. Inj. vol.: 10.0 μL· Det.: 206 nm. A: n-heptane, 80%; B: EtOH, 20%. Sample prep.: 4 mg/ml EtOH. Retention times: D-enantiomer 2.21 min., L-enantiomer 2.71 min
………………….
US 20050065204
EXAMPLE 11
Preparation of (S)-1-(4-Hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic Acid
8.00 g Polyethyleneglycol 6000 was dissolved in 150 mL (100 mM) magnesium acetate buffer pH 6.0 under stirring, and the solution added to a stirred suspension of 10.00 g (42.51 mmol) (RS)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid methyl ester (99.7%) in 40 mL methylcyclohexane. The mixture was heated to 28° C. and the pH readjusted to 6.0 with 2 M NaOH. The reaction was started by adding 33.2 mg Candida cylindraceae cholesterase (16.88 kU/g), and the pH was maintained at 6.0 by the controlled addition of 1.0 M NaOH solution under stirring. After a total consumption of 20.35 mL (20.35 mmol) 1.0 M sodium hydroxide solution (after 17.1 h; 47.9% conversion) the reaction mixture was passed through a sintered glass filter. The filtrate spontaneously separated into an aqueous and an organic phase.The aqueous phase was washed with 2×200 mL ethyl acetate to remove uncleaved ester. The aqueous phase was set to pH 4.0 with 25% sulfuric acid and concentrated in vacuo to a volume of ca. 80 mL (bath 60° C.). The solution was cooled to 1° C. (formation of white precipitate/crystals) and the pH set to 1.5 with 25% sulfuric acid. The precipitate/crystals were stirred overnight at 1° C., filtered off on a sintered glass filter (washed with a minimum amount of water) and dried overnight on high vacuum (RT, 6×10−2 mbar) to give 4.32 g (19.53 mmol; 45.9%) (S)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid. Analysis: HPLC (area A226nm): 99.3%, 0.7% ester. 98.9%ee. The product contains 5.3% water (according to Karl Fischer determination) and 2.1% (w/w) PEG (according to NMR).
| Company | Evotec AG |
| Description | Small molecule monoamine oxidase B (MAO-B) inhibitor |
| Molecular Target | Monoamine oxidase B (MAO-B) |
| Mechanism of Action | Monoamine oxidase B (MAO-B) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Alzheimer’s disease (AD) |
| Indication Details | Treat Alzheimer’s disease (AD) |
| Regulatory Designation | |
| Partner |
//////////
Chūō, japan




A Chūō Line (Rapid) E233 series (right) and A Chūō-Sōbu Line E231 series (June 2007)
 Chuo Dori street on a weekend afternoon
Chuo Dori street on a weekend afternoon


{kind=link}