
Home » Posts tagged 'BREAKTHROUGH THERAPY'
Tag Archives: BREAKTHROUGH THERAPY
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gildeuretinol
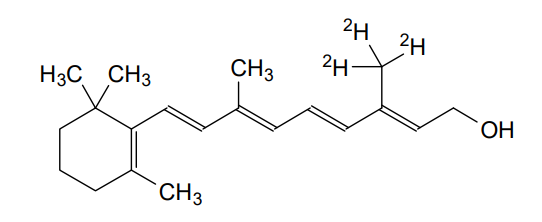
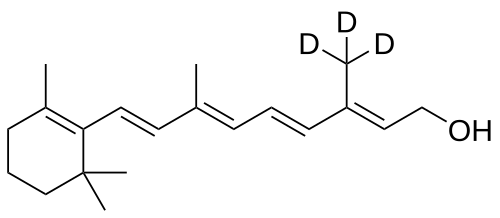
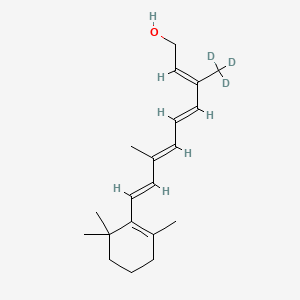
Gildeuretinol
CAS118139-35-8
MF C20H272H3O, MW 289.5 g/mol
(2E,4E,6E,8E)-3-(2H3)methyl-7-methyl-9-(2,6,6-trimethylcyclohex-1-en-1-yl)nona-2,4,6,8-tetraen-1-ol; (20,20,20-2H3)retinol
(2E,4E,6E,8E)-7-methyl-3-(trideuteriomethyl)-9-(2,6,6-trimethylcyclohexen-1-yl)nona-2,4,6,8-tetraen-1-ol
vitamin A analogue, Orphan Drug, Stargardt disease, breakthrough therapy, Pediatric Rare Disease designations, ALK-001, KL-49, ALK 001, KL 49
- OriginatorColumbia University
- DeveloperAlkeus Pharmaceuticals
- ClassEye disorder therapies; Retinoids; Vitamins
- Mechanism of ActionDimerisation inhibitors; Vitamin A replacements
- Orphan Drug StatusYes – Stargardt disease
- Phase II/IIIDry age-related macular degeneration
- Phase IIStargardt disease
- No development reportedRetinal dystrophies
- 08 Sep 2025Gildeuretinol – Alkeus Pharmaceuticals receives Orphan Drug status for Stargardt disease in European Union
- 09 Jan 2025Alkeus Pharmaceuticals announces intention to submit an NDA to US FDA for Stargardt disease in 2025
- 09 Jan 2025Efficacy and adverse event data from phase II trial for Stargardt disease released by Alkeus Pharmaceuticals
Gildeuretinol is an investigational new drug being developed by Alkeus Pharmaceuticals, Inc. for the treatment of retinal diseases, particularly Stargardt disease and geographic atrophy secondary to age-related macular degeneration (AMD). Stargardt disease is caused by a defect in the ABCA4 gene that clears toxic byproducts resulting from the dimerization of vitamin A. Gildeuretinol is new molecular entity designed to reduce the dimerization of vitamin A in the eye without affecting the visual cycle.[1]
Gildeuretinol has received breakthrough therapy, orphan drug and Pediatric Rare Disease designations from the U.S. Food and Drug Administration.[2]



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Zaydon YA, Tsang SH (July 2024). “The ABCs of Stargardt disease: the latest advances in precision medicine”. Cell & Bioscience. 14 (1) 98. doi:10.1186/s13578-024-01272-y. PMC 11282698. PMID 39060921.
- Fitch J (22 November 2024). “Gildeuretinol for Stargardt disease receives Rare Pediatric Disease, Fast Track Designations”. Contemporary Pediatrics.
| Clinical data | |
|---|---|
| Other names | ALK-001, KL-49 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 118139-35-8 |
| PubChem CID | 169490774 |
| UNII | PSZ7W5NR24 |
| KEGG | D12713 |
| ChEMBL | ChEMBL5314606 |
| Chemical and physical data | |
| Formula | C20H30D3O |
| Molar mass | 292.500 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Gildeuretinol, vitamin A analogue, Orphan Drug, Stargardt disease, breakthrough therapy, Pediatric Rare Disease designations, ALK-001, KL-49, ALK 001, KL 49, PSZ7W5NR24
Suzetrigine



Suzetrigine
CAS
2649467-58-1 |
Average: 473.4
Monoisotopic: 473.137396951
Chemical Formula
C21H20F5N3O4
FDA 1/30/2025, Journavx
To treat moderate to severe acute pain
Press Release
- 2-Pyridinecarboxamide, 4-[[[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)tetrahydro-4,5-dimethyl-5-(trifluoromethyl)-2-furanyl]carbonyl]amino]-
- 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)-4,5- dimethyl-5-(trifluoromethyl)oxolane-2- carboxamido]pyridine-2-carboxamide
- 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)-4,5-dimethyl-5-(trifluoromethyl)oxolane-2-amido]pyridine2-carboxamide
- 4-[[[(2R,3S,4S,5R)-3-(3,4-Difluoro-2-methoxyphenyl)tetrahydro-4,5-dimethyl-5-(trifluoromethyl)-2-furanyl]carbonyl]amino]-2-pyridinecarboxamide
- CS-0641183
- HY-148800
- VX 548
- VX-548
- VX548
- Management of
Acute, moderate pain
Suzetrigine, sold under the brand name Journavx, is a medication used for the management of pain.[1][2] It is a non-opioid, small-molecule analgesic that works as a selective inhibitor of Nav1.8-dependent pain-signaling pathways in the peripheral nervous system,[3][4] avoiding the addictive potential of opioids. Suzetrigine is taken by mouth.[1]
The most common adverse reactions include itching, muscle spasms, increased blood level of creatine kinase, and rash.[1][2]
It was developed by Vertex Pharmaceuticals,[5] and was approved for medical use in the United States in January 2025.[2][6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines.[2]
Medical uses
Suzetrigine is indicated for the treatment of moderate to severe acute pain in adults.[1][2]
FDA Approves Novel Non-Opioid Treatment for Moderate to Severe Acute Pain
First Drug Approved in New Class of Non-Opioid Pain Medicines; Agency Continues to Take Steps to Support New Approaches for Pain Management
For Immediate Release:January 30, 2025
Today, the U.S. Food and Drug Administration approved Journavx (suzetrigine) 50 milligram oral tablets, a first-in-class non-opioid analgesic, to treat moderate to severe acute pain in adults. Journavx reduces pain by targeting a pain-signaling pathway involving sodium channels in the peripheral nervous system, before pain signals reach the brain.
Journavx is the first drug to be approved in this new class of pain management medicines.
Pain is a common medical problem and relief of pain is an important therapeutic goal. Acute pain is short-term pain that is typically in response to some form of tissue injury, such as trauma or surgery. Acute pain is often treated with analgesics that may or may not contain opioids.
The FDA has long supported development of non-opioid pain treatment. As part of the FDA Overdose Prevention Framework, the agency has issued draft guidance aimed at encouraging development of non-opioid analgesics for acute pain and awarded cooperative grants to support the development and dissemination of clinical practice guidelines for the management of acute pain conditions.
“Today’s approval is an important public health milestone in acute pain management,” said Jacqueline Corrigan-Curay, J.D., M.D., acting director of the FDA’s Center for Drug Evaluation and Research. “A new non-opioid analgesic therapeutic class for acute pain offers an opportunity to mitigate certain risks associated with using an opioid for pain and provides patients with another treatment option. This action and the agency’s designations to expedite the drug’s development and review underscore FDA’s commitment to approving safe and effective alternatives to opioids for pain management.”
The efficacy of Journavx was evaluated in two randomized, double-blind, placebo- and active-controlled trials of acute surgical pain, one following abdominoplasty and the other following bunionectomy. In addition to receiving the randomized treatment, all participants in the trials with inadequate pain control were permitted to use ibuprofen as needed for “rescue” pain medication. Both trials demonstrated a statistically significant superior reduction in pain with Journavx compared to placebo.
The safety profile of Journavx is primarily based on data from the pooled, double-blind, placebo- and active-controlled trials in 874 participants with moderate to severe acute pain following abdominoplasty and bunionectomy, with supportive safety data from one single-arm, open-label study in 256 participants with moderate to severe acute pain in a range of acute pain conditions.
The most common adverse reactions in study participants who received Journavx were itching, muscle spasms, increased blood level of creatine phosphokinase, and rash. Journavx is contraindicated for concomitant use with strong CYP3A inhibitors. Additionally, patients should avoid food or drink containing grapefruit when taking Journavx.
The application received Breakthrough Therapy, Fast Track and Priority Review designations by the FDA.
The FDA granted approval of Journavx to Vertex Pharmaceuticals Incorporated.
PATENTS
https://patentimages.storage.googleapis.com/08/4f/6e/4f104b27a3772f/US11919887.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=US407339565&_cid=P22-M90R90-47554-1



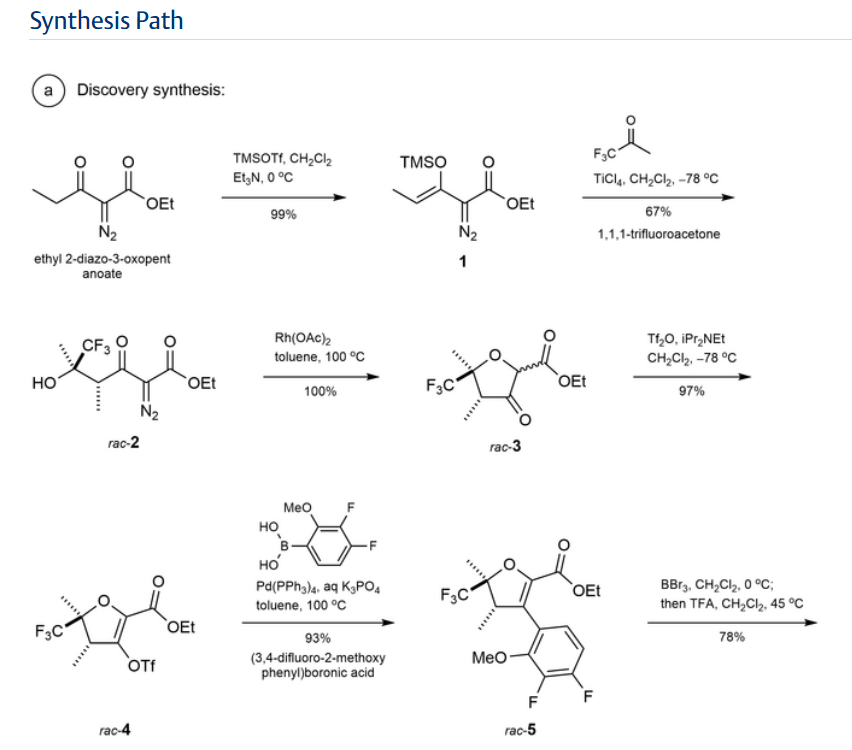
Step 1:
NEt₂ (7.7 mL, 55.2 mmol) was added to a solution of
ethyl 2-diazo-3-oxo-pentanoate (6.69 g, 39.3 mmol) in
DCM (80 mL) with stirring at 0° C. under nitrogen. Trimethylsilyl trifluoromethanesulfonate (8.5 mL, 47.0 mmol)
was added dropwise over 5 mins and the mixture was stirred
for a further 30 mins at 0° C. The reaction mixture was
diluted with pentane (100 mL), the layers separated and the
organic phase washed with dilute aqueous sodium bicarbonate (100 mL) and brine (100 mL). The organic layer was
dried (MgSO4), and concentrated in vacuo to give ethyl
(Z)-2-diazo-3-trimethylsilyloxy-pent-3-enoate (9.4 g, 99%)
as a red oil. H NMR (500 MHz, Chloroform-d) 8 5.33 (q,
J=7.0 Hz, 1H), 4.25 (q, J=7.1 Hz, 2H), 1.67 (d, J=7.0 Hz,
3H), 1.29 (t, J=7.1 Hz, 3H), 0.22 (s, 9H) ppm.
Step 2:
To a solution of 1,1,1-trifluoropropan-2-one (8 mL, 89.4
mmol) in DCM (80 mL) stirring at -78° C. was added TiCl
(70 mL of 1 M in DCM, 70.00 mmol) via cannula. To the
resulting solution, a solution of ethyl (Z)-2-diazo-3-trimethylsilyloxy-pent-3-enoate (36.1 g of 31.3% w/w, 46.6 mmol)
in 40 mL of DCM was added dropwise over 15 mins. After
100 mins the reaction was carefully quenched with water,
allowing the temperature to rise slowly, and then extracted
with DCM. The combined organic layers were dried
(MgSO), filtered, and concentrated in vacuo. Purification
by flash chromatography (330 g SiO₂, 0 to 20% EtOAc in
heptane) gave ethyl 2-diazo-6,6,6-trifluoro-5-hydroxy-4,5-
dimethyl-3-oxo-hexanoate (8.82 g, 67%), which was stored
as a solution in toluene. H NMR (500 MHz, Chloroform-d)
8 4.33 (q, J=7.1 Hz, 2H), 4.14 (q, J=7.0 Hz, 1H), 3.98 (s,
1H), 1.43 (q, J=1.2 Hz, 3H), 1.35 (t, J=7.1 Hz, 3H), 1.31 (dq.
J=7.0, 1.4 Hz, 3H) ppm. ESI-MS m/z calc. 282.08273, found
283.1 (M+1)*; 281.0 (M-1)-.
Step 3:
A solution of rhodium tetraacetate (245 mg, 0.55 mmol)
in benzene (32 mL) was heated at reflux for 10 min before
a solution of ethyl 2-diazo-6,6,6-trifluoro-5-hydroxy-4,5-
dimethyl-3-oxo-hexanoate (10 g, 35.4 mmol) in benzene (13
mL) was added slowly via addition funnel while refluxing
for 60 mins. The mixture was then concentrated in vacuo to
give ethyl rac-(4R, 5R)-4,5-dimethyl-3-oxo-5-(trifluoromethyl)tetrahydrofuran-2-carboxylate (9.0 g, 100%) as a
green coloured residue containing residual catalyst, and as a
mixture of epimers at the position next to the ester. This
material was used without further purification. H NMR
(500 MHz, Chloroform-d) 8 4.83-4.57 (m, 1H), 4.38-4.16
(m, 2H), 2.60 (dddd, J=9.3, 8.2, 5.6, 1.4 Hz, 1H), 1.73-1.63
(m, 3H), 1.30 (t, J=7.1 Hz, 3H), 1.24 (ddq, J=6.4, 4.1, 1.9
Hz, 3H) ppm.
Step 4:
To a stirred solution of ethyl rac-(4R,5R)-4,5-dimethyl- 5
3-oxo-5-(trifluoromethyl)tetrahydrofuran-2-carboxylate (48
g, 188.83 mmol) in DCM (400 mL) stirring at -78° C. was
added DIPEA (29.680 g, 40 mL, 229.64 mmol). A solution
of trifluoromethylsulfonyl trifluoromethanesulfonate
(53.440 g, 32 mL, 189.41 mmol) in DCM (200 mL) was 10
added to the reaction mixture at the same temperature over
1 h. The reaction mixture was stirred for 30 mins at 0° С.
before being quenched with 100 mL saturated aqueous
NaHCO3 solution. The organic layer was separated and
aqueous layer extracted with DCM (160 mL). The combined 15
organic layers were dried (MgSO) and concentrated in
vacuo to give ethyl rac-(4R,5R)-2,3-dimethyl-2-(trifluoromethyl)-4-(trifluoromethylsulfonyloxy)-3H-furan-5-carboxylate (71 g, 97%). H NMR (400 MHz, Chloroform-d) 8
4.38-4.32 (m, 2H), 3.29-3.23 (m, 1H), 1.64 (s, 3H), 1.37- 20
1.33 (m, 6H) ppm.
STEP 5
To stirred a solution of ethyl rac-(4R,5R)-2,3-dimethyl2-(trifluoromethyl)-4-(trifluoromethylsulfonyloxy)-3Hfuran-5-carboxylate (26 g, 67.311 mmol) in toluene (130.00
mL) was added (3,4-difluoro-2-methoxy-phenyl)boronic
acid (14 g, 74.5 mmol) followed by K3PO4 (100 mL of 2 M,
200.00 mmol) under an argon atmosphere. The reaction was
degassed before tetrakis(triphenylphosphine)palladium(0)
(4 g, 3.46 mmol) was added. After further degassing, the
reaction was heated at 100° C. for 2 hours. The reaction was
diluted in water and the aqueous layer extracted with EtOAc
(2×100 mL). The combined organic layers were concentrated in vacuo. Purification by flash chromatography (SiO.
0 to 10% EtOAc in heptane) gave ethyl 4-(3,4-difluoro-2- 35
methoxy-pheny1)-2,3-dimethyl-2-(trifluoromethyl)-3Hfuran-5-carboxylate (24.4 g, 93%) as a 6:1 diastereomeric
mixture, with the major isomer believed to be ethyl rac-(4R,
5R)-4-(3,4-difluoro-2-methoxy-phenyl)-2,3-dimethyl-2-
(trifluoromethyl)-3H-furan-5-carboxylate. Major isomer: H 40
NMR (400 MHz, Chloroform-d) 8 6.88-6.79 (m, 2H), 4.17-
4.09 (m, 2H), 3.90 (s, 3H), 3.46 (q, J=7.4 Hz, 1H), 1.67 (s,
3H), 1.12 (t, J=7.4 Hz, 3H), 1.06 (dd, J=5.4, 2.7 Hz, 3Н)
ppm. Minor isomer ¹H NMR (400 MHz, Chloroform-d) 8
6.88-6.79 (m, 2H), 4.17-4.09 (m, 2H), 3.88 (s, 3H), 3.76- 45
3.71 (m, 1H), 1.51 (s, 3H), 1.12 (t, J=7.4 Hz, 3H), 0.99 (dd,
J=5.4, 2.7 Hz, 3H) ppm. ESI-MS m/z calc. 380.1047, found
381.02 (M+1)+.
Step 6:
To an ice-cooled solution of ethyl 4-(3,4-difluoro-2- 50
methoxy-phenyl)-2,3-dimethyl-2-(trifluoromethyl)-3Hfuran-5-carboxylate (110 g, 243.0 mmol) in DCM (360 mL)
was added BBr, (370 mL of 1 M, 370.0 mmol) dropwise.
Upon completion the mixture was quenched by addition of
water and aqueous sodium bicarbonate solution, the aqueous 55
layer extracted with DCM and the combined organic layers
dried (MgSO) and concentrated in vacuo. The residue was
dissolved in DCM (430 mL) at ambient temperature and
TFA (40 mL, 519.2 mmol) was added, then the reaction was
heated to 45° C. Upon completion, the mixture was
quenched by addition of aqueous sodium bicarbonate solution and the aqueous layer extracted with DCM, dried
(MgSO) and concentrated in vacuo to give the desired
product in a 5:1 mixture of diastereomers. Recrystallization
was carried out by solubilizing the crude in the smallest
possible amount of DCM and adding a layer of heptane on
top of this solution (liquid-liquid diffusion). After approx. 1



https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021113627&_cid=P22-M90RUB-70989-1

Example 6
rel-(2S,3R,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (20), (2S,3R,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)- 5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (21), rel- (2R,3S,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2- carbonyl]amino]pyridine-2-carboxamide (22), and (2R,3S,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy- phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (23)
[00676] Step 7:
[00677] (4-[[3-(3-Chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (420 mg, 0.8827 mmol) was separated by chiral SFC [(R,R)-Whelk-O1 column, 5 µm particle size, 25 cm x 21.2 mm from Regis Technologies, MeOH, 20 mM NH3], followed by further purification of one or more of the fractions by chiral SFC using a Chiralpak IC column, 5 µm particle size, 25 cm x 20 mm from Daicel or a Chiralpak ID column, 5 µum particle size, 25 cm x 20 mm from Daicel to give:
[00678] First Eluting Isomer: rel-(2S,3R,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (20, 30 mg, 7.1%) (further purified by chiral SFC using Chiralpak IC column). 1H NMR (500 MHz, Chloroform-d) δ 8.92 (s, 1H), 8.47 (d, J = 5.5 Hz, 1H), 8.21 (dd, J = 5.6, 2.1 Hz, 1H), 8.09 (d, J = 2.2 Hz, 1H), 7.87 (d, J = 4.1 Hz, 1H), 7.26 (dd, J = 8.8, 5.8 Hz, 1H), 7.03 (t, J = 8.4 Hz, 1H), 5.87 – 5.82 (m, 1H), 4.77 (d, J = 10.6 Hz, 1H), 3.98 (td, J = 11.2, 8.3 Hz, 1H), 3.88 (s, 3H), 2.51 (dd, J = 13.2, 11.7 Hz, 1H), 2.42 (dd, J = 13.2, 8.3 Hz, 1H), 1.69 (s, 3H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00679] Second Eluting Isomer: (2S,3R,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (21, 29 mg, 6.7%) (further purified by chiral SFC using Chiralpak ID column). 1H NMR (500 MHz, Chloroform-d) δ 8.56 (s, 1H), 8.48 (d, J = 5.5 Hz, 1H), 8.08 (dd, J = 5.5, 2.2 Hz, 1H), 7.98 (d, J = 2.1 Hz, 1H), 7.86 (d, J = 4.4 Hz, 1H), 7.23 (dd, J = 8.8, 5.8 Hz, 1H), 7.01 (t, J = 8.4 Hz, 1H), 5.86 (d, J = 4.2 Hz, 1H), 4.80 (d, J = 9.7 Hz, 1H), 4.10 – 4.00 (m, 1H), 3.93 (s, 3H), 3.52 – 3.48 (m, 1H), 2.86 (dd, J = 13.9, 8.4 Hz, 1H), 2.16 -2.07 (m, 1H), 1.64 (s, 2H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00680] Third Eluting Isomer: rel-(2R,3S,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (22, 42 mg, 9.5%).
1H NMR (500 MHz, Chloroform-d) δ 8.87 (s, 1H), 8.33 (d, J = 5.6 Hz, 1H), 8.08 (dd, J = 5.6, 2.2 Hz, 1H), 7.98 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 4.5 Hz, 1H), 7.12 (dd, J = 8.8, 5.8 Hz, 1H), 6.89 (t, J = 8.4 Hz, 1H), 5.79 (d, J = 4.5 Hz, 1H), 4.63 (d, J = 10.7 Hz, 1H), 3.85 (td, J = 11.2, 8.4 Hz, 1H), 3.74 (s, 3H), 2.37 (dd, J = 13.2, 11.7 Hz, 1H), 2.28 (dd, J = 13.1, 8.4 Hz, 1H), 1.55 (s, 3H) ppm. ESI-MS m/z calc.
475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00681] Fourth Eluting Isomer: (2R,3S,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (23, 40 mg, 8.8%).
1H NMR (500 MHz, Chloroform-d) δ 8.43 (s, 1H), 8.35 (d, J = 5.5 Hz, 1H), 7.95 (dd, J = 5.5, 2.2 Hz, 1H), 7.85 (d, J = 2.2 Hz, 1H), 7.73 (d, J = 4.3 Hz, 1H), 7.10 (dd, J = 8.8, 5.9 Hz, 1H), 6.87 (t, J = 8.4 Hz, 1H), 5.76 – 5.71 (m, 1H), 4.67 (d, J = 9.7 Hz, 1H), 3.97 – 3.87 (m, 1H), 3.80 (s, 3H), 2.73 (dd, J = 13.9, 8.4 Hz, 1H), 1.98 (dd, J = 13.9, 11.6 Hz, 1H), 1.51 (s, 3H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00682] Compound 22 – Solid Form A
Efficacy
When people used suzetrigine in clinical studies conducted through 2024, there was a reduction in pain typically from seven to four on the standard numerical scale used to rate pain.[7][8] Suzetrigine provided pain relief equal to a combination of hydrocodone and paracetamol (acetaminophen) (5 mg of hydrocodone bitartrate and 325 mg of acetaminophen).[8][9]
Suzetrigine suppresses pain at the same level as an opioid, but without the risks of addiction, sedation, or overdose.[10] An alternative to opioids, it is the first pain medication to be approved by the Food and Drug Administration in two decades.[10]
The efficacy of suzetrigine was evaluated in two randomized, double-blind, placebo- and active-controlled trials of acute surgical pain, one following abdominoplasty and the other following bunionectomy.[2] Both trials found that suzetrigine reduced pain more effectively than a placebo.[2]
Contraindications
Concomitant use of suzetrigine with strong CYP3A inhibitors is contraindicated.[1][2]
Adverse effects
Common adverse effects of suzetrigine may include itching, rash, muscle spasms, and increased levels of creatine kinase.[2] Mild side effects may include nausea, constipation, headache, and dizziness.[7][8] As of 2024, long-term safety and side effects remain undetermined.[8]
In preliminary research, suzetrigine had no serious neurological, behavioral, or cardiovascular effects.[3]
Interactions
Consuming grapefruit while using suzetrigine may cause an adverse grapefruit–drug interaction.[1][2]
Mechanism of action
Suzetrigine operates on peripheral nerves, avoiding the addictive potential of opioids which affect the central nervous system.[3][4][7] Unlike opioid medications, which reduce pain signals in the brain, suzetrigine works by closing sodium channels in peripheral nerves, inhibiting pain-signaling nerves from transmitting painful sensations to the brain.[3][4][7]
In pharmacological studies, suzetrigine selectively inhibited Nav1.8 channels, but not other voltage-gated sodium channels, and bound to a unique site on these sodium channels with a novel allosteric mechanism, by binding to the channel’s second voltage sensing domain, thereby stabilizing the closed state, causing tonic inhibition. It exerts its action on dorsal root ganglion.[3]
History
Vertex Pharmaceuticals announced in January 2024 that suzetrigine had successfully met several endpoints in its Phase III clinical trials.[5] The company announced in July 2024 that the FDA had accepted a new drug application for suzetrigine.[11] The FDA granted the application for suzetrigine priority review, fast track, and breakthrough therapy designations.[2][11] In January 2025, the FDA granted approval of Journavx to Vertex Pharmaceuticals.[2]
Society and culture
Legal status
Suzetrigine was approved for medical use in the United States in January 2025.[2]
Names
Suzetrigine is the international nonproprietary name.[12]
Suzetrigine is sold under the brand name Journavx.[1][2]





References
a) WO2021113627A1 (Vertex, 10.06.2021; USA-prior. 06.12.2019).
US11834441B2 (Vertex, 05.12.2023; USA-prior. 06.12.2019).
b) WO2022256660A1 (Vertex, 08.12.2022; USA-prior. 04.06.2021).
WO2024123815A1 (Vertex, 13.06.2024; USA-prior. 06.12.2022).
WO2022256708A1 (Vertex, 08.12.2022; USA-prior. 04.06.2021, 02.12.2021).
Source:
Suzetrigine, in Kleemann A., Kutscher B., Reichert D., Bossart M., Pharmaceutical Substances, Thieme. https://pharmaceutical-substances.thieme.com/lexicon/KD-19-0151, accessed: 05-29-2025
| Clinical data | |
|---|---|
| Pronunciation | /suˈzɛtrɪdʒiːn/ soo-ZE-tri-jeen |
| Trade names | Journavx |
| Other names | VX-548 |
| AHFS/Drugs.com | Journavx |
| License data | US DailyMed: Suzetrigine |
| Routes of administration | By mouth |
| Drug class | Nav1.8 sodium channel blocker; Analgesic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2649467-58-1 |
| PubChem CID | 156445116 |
| DrugBank | DB18927 |
| ChemSpider | 128942439 |
| UNII | LOG73M21H5 |
| KEGG | D12860 |
| ChEMBL | ChEMBL5314487 |
| Chemical and physical data | |
| Formula | C21H20F5N3O4 |
| Molar mass | 473.400 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f g h “Journavx- suzetrigine tablet, film coated”. DailyMed. 6 February 2025. Retrieved 2 April 2025.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves Novel Non-Opioid Treatment for Moderate to Severe Acute Pain” (Press release). U.S. Food and Drug Administration (FDA). 30 January 2025. Archived from the original on 7 February 2025. Retrieved 30 January 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e Osteen, Jeremiah D.; Immani, Swapna; Tapley, Tim L.; Indersmitten, Tim; Hurst, Nicole W.; Healey, Tiffany; et al. (January 2025). “Pharmacology and Mechanism of Action of Suzetrigine, a Potent and Selective NaV1.8 Pain Signal Inhibitor for the Treatment of Moderate to Severe Pain”. Pain and Therapy. doi:10.1007/s40122-024-00697-0. PMID 39775738.
- ^ Jump up to:a b c Jones, Jim; Correll, Darin J.; Lechner, Sandra M; Jazic, Ina; Miao, Xiaopeng; Shaw, David; et al. (August 2023). “Selective Inhibition of NaV1.8 with VX-548 for Acute Pain”. The New England Journal of Medicine. 389 (5): 393–405. doi:10.1056/NEJMoa2209870. PMID 37530822. S2CID 260377748.
- ^ Jump up to:a b “Vertex Announces Positive Results From the VX-548 Phase 3 Program for the Treatment of Moderate-to-Severe Acute Pain” (Press release). Vertex. 30 January 2024. Archived from the original on 25 December 2024. Retrieved 31 January 2025 – via Business Wire.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ Jump up to:a b c d Broadfoot, Marla (20 August 2024). “New Painkiller Could Bring Relief to Millions — without Addiction Risk”. Scientific American. Archived from the original on 30 December 2024. Retrieved 31 January 2025.
- ^ Jump up to:a b c d Hang Kong, Aaron Yik; Tan, Hon Sen; Habib, Ashraf S. (September 2024). “VX-548 in the Treatment of Acute Pain”. Pain Management. 14 (9): 477–486. doi:10.1080/17581869.2024.2421749. PMC 11721852. PMID 39552600.
- ^ Kingwell, Katie (December 2024). “NaV1.8 inhibitor poised to provide opioid-free pain relief”. Nature Reviews. Drug Discovery. 24 (1): 3–5. doi:10.1038/d41573-024-00203-3. PMID 39668193.
- ^ Jump up to:a b Dolgin, Elie (January 2025). “US drug agency approves potent painkiller – the first non-opioid in decades”. Nature. 638 (8050): 304–305. doi:10.1038/d41586-025-00274-1. PMID 39885357.
- ^ Jump up to:a b “Vertex Announces FDA Acceptance of New Drug Application for Suzetrigine for the Treatment of Moderate-to-Severe Acute Pain” (Press release). Vertex. 30 July 2024. Retrieved 31 January 2025 – via Business Wire.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 90”. WHO Drug Information. 37 (3). hdl:10665/373341.
Further reading
- Oliver, Brian; Devitt, Catherine; Park, Grace; Razak, Alina; Liu, Sun Mei; Bergese, Sergio D. (2025). “Drugs in Development to Manage Acute Pain”. Drugs. 85 (1): 11–19. doi:10.1007/s40265-024-02118-0. PMID 39560856.
External links
- “Suzetrigine (Code C199115)”. NCI Thesaurus.
- Clinical trial number NCT05661734 for “A Single-arm Study to Evaluate Safety and Effectiveness of VX-548 for Acute Pain” at ClinicalTrials.gov
- Clinical trial number NCT05558410 for “Evaluation of Efficacy and Safety of VX-548 for Acute Pain After an Abdominoplasty” at ClinicalTrials.gov
//////////Suzetrigine, Journavx, FDA 2025, APPROVALS 2025, CS-0641183, HY-148800, VX 548, VX-548, VX548, Breakthrough Therapy, Fast Track, Priority Review
Avalglucosidase alfa
QQGASRPGPR DAQAHPGRPR AVPTQCDVPP NSRFDCAPDK AITQEQCEAR GCCYIPAKQG
LQGAQMGQPW CFFPPSYPSY KLENLSSSEM GYTATLTRTT PTFFPKDILT LRLDVMMETE
NRLHFTIKDP ANRRYEVPLE TPRVHSRAPS PLYSVEFSEE PFGVIVHRQL DGRVLLNTTV
APLFFADQFL QLSTSLPSQY ITGLAEHLSP LMLSTSWTRI TLWNRDLAPT PGANLYGSHP
FYLALEDGGS AHGVFLLNSN AMDVVLQPSP ALSWRSTGGI LDVYIFLGPE PKSVVQQYLD
VVGYPFMPPY WGLGFHLCRW GYSSTAITRQ VVENMTRAHF PLDVQWNDLD YMDSRRDFTF
NKDGFRDFPA MVQELHQGGR RYMMIVDPAI SSSGPAGSYR PYDEGLRRGV FITNETGQPL
IGKVWPGSTA FPDFTNPTAL AWWEDMVAEF HDQVPFDGMW IDMNEPSNFI RGSEDGCPNN
ELENPPYVPG VVGGTLQAAT ICASSHQFLS THYNLHNLYG LTEAIASHRA LVKARGTRPF
VISRSTFAGH GRYAGHWTGD VWSSWEQLAS SVPEILQFNL LGVPLVGADV CGFLGNTSEE
LCVRWTQLGA FYPFMRNHNS LLSLPQEPYS FSEPAQQAMR KALTLRYALL PHLYTLFHQA
HVAGETVARP LFLEFPKDSS TWTVDHQLLW GEALLITPVL QAGKAEVTGY FPLGTWYDLQ
TVPIEALGSL PPPPAAPREP AIHSEGQWVT LPAPLDTINV HLRAGYIIPL QGPGLTTTES
RQQPMALAVA LTKGGEARGE LFWDDGESLE VLERGAYTQV IFLARNNTIV NELVRVTSEG
AGLQLQKVTV LGVATAPQQV LSNGVPVSNF TYSPDTKVLD ICVSLLMGEQ FLVSWC
(Disulfide bridge:26-53, 36-52, 47-71, 477-502, 591-602, 882-896)
Avalglucosidase alfa
アバルグルコシダーゼアルファ (遺伝子組換え)
Avalglucosidase alfa (USAN/INN);
Avalglucosidase alfa (genetical recombination) (JAN);
Avalglucosidase alfa-ngpt
To treat late-onset Pompe disease
| Formula | C4490H6818N1197O1299S32 |
|---|---|
| CAS | 1802558-87-7 |
| Mol weight | 99375.4984 |
FDA APPROVED Nexviazyme, 2021/8/6, Enzyme replacement therapy product
Treatment of Pompe disease
Biologic License Application (BLA): 761194
Company: GENZYME CORP
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-pompe-diseaseFor Immediate Release:August 06, 2021
Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
Patients with Pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure. Normally, glycogen—the stored form of glucose—breaks down to release glucose into the bloodstream to be used as fuel for the cells.
“Pompe disease is a rare genetic disease that causes premature death and has a debilitating effect on people’s lives,” said Janet Maynard, M.D., deputy director of the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine in the FDA’s Center for Drug Evaluation and Research. “Today’s approval brings patients with Pompe disease another enzyme replacement therapy option for this rare disease. The FDA will continue to work with stakeholders to advance the development of additional new, effective and safe therapies for rare diseases, including Pompe disease.”
Nexviazyme, an enzyme replacement therapy, is an intravenous medication that helps reduce glycogen accumulation. The effectiveness of Nexviazyme for the treatment of Pompe disease was demonstrated in a study of 100 patients who were randomized to take Nexviazyme or another FDA-approved enzyme replacement therapy for Pompe disease. Treatment with Nexviazyme improved lung function similar to the improvement seen with the other therapy.
The most common side effects included headache, fatigue, diarrhea, nausea, joint pain (arthralgia), dizziness, muscle pain (myalgia), itching (pruritus), vomiting, difficulty breathing (dyspnea), skin redness (erythema), feeling of “pins and needles” (paresthesia) and skin welts (urticaria). Serious reactions included hypersensitivity reactions like anaphylaxis and infusion-associated reactions, including respiratory distress, chills and raised body temperature (pyrexia). Patients susceptible to fluid volume overload or with compromised cardiac or respiratory function may be at risk for serious acute cardiorespiratory failure.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Nexviazyme also received an orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Nexviazyme to Genzyme Corporation.
###
NEW DRUG APPROVALS
one time
$10.00
FDA grants priority review for avalglucosidase alfa, a potential new therapy for Pompe disease
- The FDA decision date for avalglucosidase alfa, an investigational enzyme replacement therapy, is set for May 18, 2021
- Regulatory submission based on positive data from two trials in patients with late-onset and infantile-onset Pompe disease, respectively
- Avalglucosidase alfa received FDA Breakthrough Therapy and Fast Track designations for the treatment of people with Pompe Disease
- Pompe disease, a rare degenerative muscle disorder, affects approximately 3,500 people in the U.S.
- Milestone reinforces 20+year commitment to Pompe disease community
PARIS – November 18, 2020 – The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application (BLA) for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease (acid α-glucosidase deficiency). The target action date for the FDA decision is May 18, 2021.
Avalglucosidase alfa is an investigational enzyme replacement therapy designed to improve the delivery of acid alpha-glucosidase (GAA) enzyme to muscle cells, and if approved, would offer a potential new standard of care for patients with Pompe disease.
In October, the European Medicines Agency accepted for review the Marketing Authorization Application for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease. The Medicines and Healthcare Products Regulatory Agency in the UK has granted Promising Innovative Medicine designation for avalglucosidase alfa.
“The hallmarks of Pompe disease are the relentless and debilitating deterioration of the muscles, which causes decreased respiratory function and mobility,” said Karin Knobe, Head of Development for Rare Diseases and Rare Blood Disorders at Sanofi. “Avalglucosidase alfa is specifically designed to deliver more GAA enzyme into the lysosomes of the muscle cells. We have been greatly encouraged by positive clinical trial results in patients with late-onset and infantile-onset Pompe disease.”
Pompe disease is a rare, degenerative muscle disorder that can impact an individual’s ability to move and breathe. It affects an estimated 3,500 people in the U.S. and can manifest at any age from infancy to late adulthood.i
The BLA is based on positive data from two trials:
- Pivotal Phase 3, double-blind, global comparator-controlled trial (COMET), which evaluated the safety and efficacy of avalglucosidase alfa compared to alglucosidase alfa (standard of care) in patients with late-onset Pompe disease. Results from this trial were presented during a Sanofi-hosted virtual scientific session in June 2020 and in October 2020 at World Muscle Society and the American Association of Neuromuscular and Electrodiagnostic Medicine.
- The Phase 2 (mini-COMET) trial evaluated the safety and exploratory efficacy of avalglucosidase alfa in patients with infantile-onset Pompe disease previously treated with alglucosidase alfa. Results from this trial were presented at the WORLDSymposium, in February 2020.
Delivery of GAA to Clear Glycogen
Pompe disease is caused by a genetic deficiency or dysfunction of the lysosomal enzyme GAA, which results in build-up of complex sugars (glycogen) in muscle cells throughout the body. The accumulation of glycogen leads to irreversible damage to the muscles, including respiratory muscles and the diaphragm muscle supporting lung function, and other skeletal muscles that affect mobility.
To reduce the glycogen accumulation caused by Pompe disease, the GAA enzyme must be delivered into the lysosomes within muscle cells. Research led by Sanofi has focused on ways to enhance the delivery of GAA into the lysosomes of muscle cells by targeting the mannose-6-phosphate (M6P) receptor that plays a key role in the transport of GAA.
Avalglucosidase alfa is designed with approximately 15-fold increase in M6P content, compared to standard of care alglucosidase alfa, and aims to help improve cellular enzyme uptake and enhance glycogen clearance in target tissues.ii The clinical relevance of this difference has not been confirmed.
Avalglucosidase alfa is currently under clinical investigation and its safety and efficacy have not been evaluated by any regulatory authority worldwide.
| About Sanofi Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, Empowering Life |
/////////Avalglucosidase alfa, FDA 2021, Nexviazyme, APPROVALS 2021, PEPTIDE, Enzyme replacement therapy , Pompe disease, アバルグルコシダーゼアルファ (遺伝子組換え), Fast Track, Priority Review, Breakthrough Therapy, orphan drug designation, genzyme, sanofi
BELUMOSUDIL

BELUMOSUDIL
MW 452.5
911417-87-3, SLx-2119, KD-025, KD 025, WHO 11343
2-[3-[4-(1H-indazol-5-ylamino)quinazolin-2-yl]phenoxy]-N-propan-2-ylacetamide
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
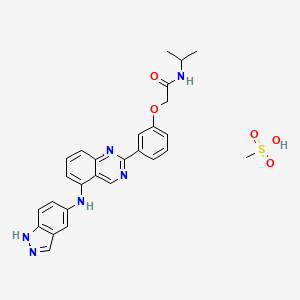
Belumosudil mesylate
KD025 mesylate
2109704-99-4
UPDATE FDA APPROVED 7/16/2021 To treat chronic graft-versus-host disease after failure of at least two prior lines of systemic therapy, Rezurock
New Drug Application (NDA): 214783
Company: KADMON PHARMA LLC
200 MG TABLET
FDA approves belumosudil for chronic graft-versus-host disease
On July 16, 2021, the Food and Drug Administration approved belumosudil (Rezurock, Kadmon Pharmaceuticals, LLC), a kinase inhibitor, for adult and pediatric patients 12 years and older with chronic graft-versus-host disease (chronic GVHD) after failure of at least two prior lines of systemic therapy.
Efficacy was evaluated in KD025-213 (NCT03640481), a randomized, open-label, multicenter dose-ranging trial that included 65 patients with chronic GVHD who were treated with belumosudil 200 mg taken orally once daily.
The main efficacy outcome measure was overall response rate (ORR) through Cycle 7 Day 1 where overall response included complete response (CR) or partial response (PR) according to the 2014 criteria of the NIH Consensus Development Project on Clinical Trials in Chronic Graft-versus-Host Disease. The ORR was 75% (95% CI: 63, 85); 6% of patients achieved a CR, and 69% achieved a PR. The median time to first response was 1.8 months (95% CI: 1.0, 1.9). The median duration of response, calculated from first response to progression, death, or new systemic therapies for chronic GVHD, was 1.9 months (95% CI: 1.2, 2.9). In patients who achieved response, no death or new systemic therapy initiation occurred in 62% (95% CI: 46, 74) of patients for at least 12 months since response.
The most common adverse reactions (≥ 20%), including laboratory abnormalities, were infections, asthenia, nausea, diarrhea, dyspnea, cough, edema, hemorrhage, abdominal pain, musculoskeletal pain, headache, phosphate decreased, gamma glutamyl transferase increased, lymphocytes decreased, and hypertension.
The recommended dosage of belumosudil is 200 mg taken orally once daily with food.
View full prescribing information for Rezurock.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Australia’s Therapeutic Goods Administration, Health Canada, Switzerland’s Swissmedic, and the United Kingdom’s Medicines and Healthcare products Regulatory Agency.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application 6 weeks ahead of the FDA goal date.
This application was granted priority review and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Belumosudil mesylate is an orally available rho kinase 2 (ROCK 2) inhibitor being developed at Kadmon. In 2020, the drug candidate was submitted for a new drug application (NDA) in the U.S., under a real-time oncology review pilot program, for the treatment of chronic graft-versus-host disease (cGVHD). The compound is also in phase II clinical development for the treatment of idiopathic pulmonary fibrosis and diffuse cutaneous systemic sclerosis. Formerly, the company had also been conducting clinical research for the treatment of psoriasis and non-alcoholic steatohepatitis (NASH); however, no further development has been reported for these indications. Originally developed by Nano Terra, the product was licensed to Kadmon on an exclusive global basis in 2011. In 2019, Kadmon entered into a strategic partnership with BioNova Pharmaceuticals and established a joint venture, BK Pharmaceuticals, to exclusively develop and commercialize KD-025 for the treatment of graft-versus-host disease in China. The compound has been granted breakthrough therapy designation in the U.S. for the treatment of cGVHD and orphan drug designations for cGVHD and systemic sclerosis. In the E.U. belumosudil was also granted orphan drug status in the E.U. for the treatment of cGVHD.
Kadmon , under license from NT Life Sciences , is developing belumosudil as mesylate salt, a ROCK-2 inhibitor, for treating IPF, chronic graft-versus-host disease, hepatic impairment and scleroderma. In July 2021, belumosudil was reported to be in pre-registration phase.
Belumosudil (formerly KD025 and SLx-2119) is an experimental drug being explored for the treatment of chronic graft versus host disease (cGvHD), idiopathic pulmonary fibrosis (IPF), and moderate to severe psoriasis. It is an inhibitor of Rho-associated coiled-coil kinase 2 (ROCK2; ROCK-II).[1] Belumosudil binds to and inhibits the serine/threonine kinase activity of ROCK2. This inhibits ROCK2-mediated signaling pathways which play major roles in pro- and anti-inflammatory immune cell responses. A genomic study in human primary cells demonstrated that the drug also has effects on oxidative phosphorylation, WNT signaling, angiogenesis, and KRAS signaling.[2] Originally developed by Surface Logix, Inc,[1] Belumosudil was later acquired by Kadmon Corporation. As of July 2020 the drug was in completed or ongoing Phase II clinical studies for cGvHD, IPF and psoriasis.[3]
cGvHD is a complication that can follow stem cell or hematopoietic stem cell transplantation where the transplanted cells (graft) attack healthy cells (host). This causes inflammation and fibrosis in multiple tissues. Two cytokines controlled by the ROCK2 signaling pathway, IL-17 and IL-21, have a major role in the cGvHD response. In a 2016 report using both mouse models and a limited human clinical trial ROCK2 inhibition with belumosudil targeted both the immunologic and fibrotic components of cGvHD and reversed the symptoms of the disease.[4] In October 2017 KD025 was granted orphan drug status in the United States for treatment of patients with cGvHD.[5]
IPF is a progressive fibrotic disease where the lining of the lungs become thickened and scarred.[6] Increased ROCK activity has been found in the lungs of humans and animals with IPF. Treatment with belumosudil reduced lung fibrosis in a bleomycin mouse model study.[7] Belumosudil may have a therapeutic benefit in IPF by targeting the fibrotic processes mediated by the ROCK signaling pathway.
Psoriasis is an inflammatory skin condition where patients experiences eruptions and remissions of thickened, erythematous, and scaly patches of skin. Down-regulation of pro-inflammatory responses was observed with KD025 treatment in Phase 2 clinical studies in patients with moderate to severe psoriasis.[8]
“Substance Name:Substance Name: Belumosudil [USAN]”.
PATENT
| WO2012040499 |
https://patents.google.com/patent/WO2012040499A2/en
PATENT
| CN106916145 |
https://patents.google.com/patent/CN106916145A/en

WO 2014055996, WO 2015157556



Patent
WO-2021129589
Novel crystalline polymorphic forms (N1, N2 and N15) of KD-025 (also known as belumosudil ), useful as a Rho A kinase 2 (ROCK-2) inhibitor for treating multiple sclerosis, psoriasis, rheumatoid arthritis, idiopathic pulmonary fibrosis (IPF), atherosclerosis, non-alcoholic fatty liver and systemic sclerosis. Represents the first filing from Sunshine Lake Pharma or its parent HEC Pharm that focuses on belumosudil.KD-025 is a selective ROCK2 (Rho-associated protein kinase 2, Rho-related protein kinase 2) inhibitor. It has multiple clinical indications such as the treatment of multiple sclerosis, psoriasis, rheumatoid arthritis, and Primary pulmonary fibrosis, atherosclerosis, non-alcoholic fatty liver, etc., among which many indications are in clinical phase I, and psoriasis and systemic sclerosis are in clinical phase II.
The structure of KD-025 is shown in the following formula (1).
Example 1 Preparation method of crystal form N1 of KD-025[0222]300mg of KD-025 solid was suspended and stirred in 10mL methanol at room temperature. After 22h, it was filtered, suction filtered and placed in a drying oven at 50°C under vacuum overnight to obtain 262mg of powder. The obtained crystal was detected by XPRD and confirmed to be KD-025 crystal form N1; its X-ray powder diffraction pattern was basically the same as that of Fig. 1, its DSC pattern was basically the same as that of Fig. 2, and the TGA pattern was basically the same as that of Fig. 3.
PATENT
WO2006105081 ,
Belumosudil product pat,
protection in the EU states until March 2026, expires in the US in May 2029 with US154 extension.
Example 82
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
[0257] A suspension of 2-(3-(4-(lH-indazol-5-ylamino)qumazolin-2-yl)ρhenoxy)acetic acid (70 mg, 0.14 mmol), PyBOP® (40 mg, 0.077 mmol), DlEA (24 μL, 0.14 mmol) in dry CH2Cl2 : DMF (2 : 0.1 mL) was stirred at RT for 15 minutes. To this solution of activated acid was added propan-2-amine (5.4 mg, 0.091 mmol). After 30 minutes, 1.0 equivalent of DIEA and 0.55 equivalents of PyBOP® were added. After stirring the solution for 15 minutes, 0.65 equivalents of propan-2-aminewere added and the mixture was stirred for an additional 30 minutes. The solvent was removed in vacuo and the crude product was purified using prep HPLC (25-50 90 rnins) to afford 2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide. (40 mg, 0.086 mmol, 61 %).
References
- ^ Jump up to:a b Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, et al. (October 2008). “Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin”. Blood Coagulation & Fibrinolysis. 19 (7): 709–18. doi:10.1097/MBC.0b013e32830b2891. PMC 2713681. PMID 18832915.
- ^ Park J, Chun KH (5 May 2020). “Identification of novel functions of the ROCK2-specific inhibitor KD025 by bioinformatics analysis”. Gene. 737: 144474. doi:10.1016/j.gene.2020.144474. PMID 32057928.
- ^ “KD025 – Clinical Trials”. ClinicalTrials.gov. Retrieved 25 July 2020.
- ^ Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. (April 2016). “Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism”. Blood. 127 (17): 2144–54. doi:10.1182/blood-2015-10-678706. PMC 4850869. PMID 26983850.
- ^ Shanley M (October 6, 2017). “Therapy to Treat Transplant Complications Gets Orphan Drug Designation”. RareDiseaseReport. Retrieved 25 July 2018.
- ^ “Pulmonary Fibrosis”. The Mayo Clinic. Retrieved July 25, 2018.
- ^ Semedo D (June 5, 2016). “Phase 2 Study of Molecule Inhibitor for Idiopathic Pulmonary Fibrosis Begins”. Lung Disease News. BioNews Services, LLC. Retrieved 25 July 2018.
- ^ Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, et al. (May 2017). “Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10”. Journal of Immunology. 198 (10): 3809–3814. doi:10.4049/jimmunol.1602142. PMC 5421306. PMID 28389592.
| Clinical data | |
|---|---|
| Routes of administration |
Oral administration (tablets or capsules) |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 911417-87-3 |
| PubChem CID | 11950170 |
| UNII | 834YJF89WO |
| CompTox Dashboard (EPA) | DTXSID80238425 |
| Chemical and physical data | |
| Formula | C26H24N6O2 |
| Molar mass | 452.518 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////BELUMOSUDIL, SLx-2119, KD-025, KD 025, WHO 11343, PHASE 2, cGvHD, IPF, psoriasis, Breakthrough Therapy, Orphan Drug Designation
CC(C)NC(=O)COC1=CC=CC(=C1)C2=NC3=CC=CC=C3C(=N2)NC4=CC5=C(C=C4)NN=C5
NEW DRUG APPROVALS
ONE TIME
$10.00
Naxitamab

(Heavy chain)
QVQLVESGPG VVQPGRSLRI SCAVSGFSVT NYGVHWVRQP PGKGLEWLGV IWAGGITNYN
SAFMSRLTIS KDNSKNTVYL QMNSLRAEDT AMYYCASRGG HYGYALDYWG QGTLVTVSSA
STKGPSVFPL APSSKSTSGG TAALGCLVKD YFPEPVTVSW NSGALTSGVH TFPAVLQSSG
LYSLSSVVTV PSSSLGTQTY ICNVNHKPSN TKVDKRVEPK SCDKTHTCPP CPAPELLGGP
SVFLFPPKPK DTLMISRTPE VTCVVVDVSH EDPEVKFNWY VDGVEVHNAK TKPREEQYNS
TYRVVSVLTV LHQDWLNGKE YKCKVSNKAL PAPIEKTISK AKGQPREPQV YTLPPSRDEL
TKNQVSLTCL VKGFYPSDIA VEWESNGQPE NNYKTTPPVL DSDGSFFLYS KLTVDKSRWQ
QGNVFSCSVM HEALHNHYTQ KSLSLSPGK
(Light chain)
EIVMTQTPAT LSVSAGERVT ITCKASQSVS NDVTWYQQKP GQAPRLLIYS ASNRYSGVPA
RFSGSGYGTE FTFTISSVQS EDFAVYFCQQ DYSSFGQGTK LEIKRTVAAP SVFIFPPSDE
QLKSGTASVV CLLNNFYPRE AKVQWKVDNA LQSGNSQESV TEQDSKDSTY SLSSTLTLSK
ADYEKHKVYA CEVTHQGLSS PVTKSFNRGE C
(Disulfide bridge: H22-H95, H146-H202, H222-L211, H228-H’228, H231-H’231, H263-H323, H369-H427, H’22-H’95, H’146-H’202, H’222-L’211, H’263-H’323, H’369-H’427, L23-L88, L131-L191, L’23-L’88, L’131-L’191)
Naxitamab
ナキシタマブ;
Antineoplastic, Anti-GD2 antibody
| Formula | C6414H9910N1718O1996S44 |
|---|---|
| CAS | 1879925-92-4 |
| Mol weight | 144434.4882 |
FDA APPROVED 2020/11/25, Danyelza
FDA grants accelerated approval to naxitamab for high-risk neuroblastoma in bone or bone marrow
On November 25, 2020, the Food and Drug Administration granted accelerated approval to naxitamab (DANYELZA, Y-mAbs Therapeutics, Inc.) in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) for pediatric patients one year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow demonstrating a partial response, minor response, or stable disease to prior therapy.
Efficacy was evaluated in patients with relapsed or refractory neuroblastoma in the bone or bone marrow enrolled in two single-arm, open-label trials: Study 201 (NCT 03363373) and Study 12-230 (NCT 01757626). Patients with progressive disease following their most recent therapy were excluded. Patients received 3 mg/kg naxitamab administered as an intravenous infusion on days 1, 3, and 5 of each 4-week cycle in combination with GM-CSF subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. At the investigator’s discretion, patients were permitted to receive pre-planned radiation to the primary disease site in Study 201 and radiation therapy to non-target bony lesions or soft tissue disease in Study 12-230.
The main efficacy outcome measures were confirmed overall response rate (ORR) per the revised International Neuroblastoma Response Criteria (INRC) and duration of response (DOR). Among 22 patients treated in the multicenter Study 201, the ORR was 45% (95% CI: 24%, 68%) and 30% of responders had a DOR greater or equal to 6 months. Among 38 patients treated in the single-center Study 12-230, the ORR was 34% (95% CI: 20%, 51%) with 23% of patients having a DOR greater or equal to 6 months. For both trials, responses were observed in either the bone, bone marrow or both.
The prescribing information contains a Boxed Warning stating that naxitamab can cause serious infusion-related reactions and neurotoxicity, including severe neuropathic pain, transverse myelitis and reversible posterior leukoencephalopathy syndrome (RPLS). To mitigate these risks, patients should receive premedication prior to each naxitamab infusion and be closely monitored during and for at least two hours following completion of each infusion.
The most common adverse reactions (incidence ≥25% in either trial) in patients receiving naxitamab were infusion-related reactions, pain, tachycardia, vomiting, cough, nausea, diarrhea, decreased appetite, hypertension, fatigue, erythema multiforme, peripheral neuropathy, urticaria, pyrexia, headache, injection site reaction, edema, anxiety, localized edema, and irritability. The most common Grade 3 or 4 laboratory abnormalities (≥5% in either trial) were decreased lymphocytes, decreased neutrophils, decreased hemoglobin, decreased platelet count, decreased potassium, increased alanine aminotransferase, decreased glucose, decreased calcium, decreased albumin, decreased sodium and decreased phosphate.
The recommended naxitamab dose is 3 mg/kg/day (up to 150 mg/day) on days 1, 3, and 5 of each treatment cycle, administered after dilution as an intravenous infusion in combination with GM-CSF, subcutaneously at 250 µg/m2/day on days -4 to 0 and at 500 µg/m2/day on days 1 to 5. Treatment cycles are repeated every 4 to 8 weeks.
View full prescribing information for DANYELZA. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761171lbl.pdf
This review used the Real-Time Oncology Review (RTOR) pilot program and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted accelerated approval based on overall response rate and duration of response. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials.
This application was granted priority review, breakthrough therapy, and orphan drug designation. A priority review voucher was issued for this rare pediatric disease product application. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
////////////Naxitamab, priority review, breakthrough therapy, orphan drug, FDA 2020, 2020 APPROVALS, Danyelza, MONOCLONAL ANTIBODY, PEPTIDE, ナキシタマブ,
FDA approves third oncology drug Rozlytrek (entrectinib) that targets a key genetic driver of cancer, rather than a specific type of tumor
FDA also approves drug for second indication in a type of lung cancer
The U.S. Food and Drug Administration today granted accelerated approval to Rozlytrek (entrectinib), a treatment for adult and adolescent patients whose cancers have the specific genetic defect, NTRK (neurotrophic tyrosine receptor kinase) gene fusion and for whom there are no effective treatments.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” said FDA Acting Commissioner Ned Sharpless, M.D. “Using the FDA’s expedited review pathways, including breakthrough therapy designation and accelerated approval process, we’re supporting this innovation in precision oncology drug development and the evolution of more targeted and effective treatments for cancer patients. We remain committed to encouraging the advancement of more targeted innovations in oncology treatment and across disease types based on our growing understanding of the underlying biology of diseases.”
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than the location in the body where the tumor originated. The approval marks a new paradigm in the development of cancer drugs that are “tissue agnostic.” It follows the policies that the FDA developed in a guidance document released in 2018. The previous tissue agnostic indications approved by the FDA were pembrolizumab for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib for NTRK gene fusion tumors in 2018.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK-fusion-positive tumors by relying on efficacy information obtained primarily in adults. The FDA continues to encourage the inclusion of adolescents in clinical trials. Traditionally, clinical development of new cancer drugs in pediatric populations is not started until development is well underway in adults, and often not until after approval of an adult indication,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
The ability of Rozlytrek to shrink tumors was evaluated in four clinical trials studying 54 adults with NTRK fusion-positive tumors. The proportion of patients with substantial tumor shrinkage (overall response rate) was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for nine months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid and colon/rectum.
Rozlytrek was also approved today for the treatment of adults with non-small cell lung cancer whose tumors are ROS1-positive (mutation of the ROS1 gene) and has spread to other parts of the body (metastatic). Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
Rozlytrek’s common side effects are fatigue, constipation, dysgeusia (distorted sense of taste), edema (swelling), dizziness, diarrhea, nausea, dysesthesia (distorted sense of touch), dyspnea (shortness of breath), myalgia (painful or aching muscles), cognitive impairment (confusion, problems with memory or attention, difficulty speaking, or hallucinations), weight gain, cough, vomiting, fever, arthralgia and vision disorders (blurred vision, sensitivity to light, double vision, worsening of vision, cataracts, or floaters). The most serious side effects of Rozlytrek are congestive heart failure (weakening or damage to the heart muscle), central nervous system effects (cognitive impairment, anxiety, depression including suicidal thinking, dizziness or loss of balance, and change in sleep pattern, including insomnia and excessive sleepiness), skeletal fractures, hepatotoxicity (damage to the liver), hyperuricemia (elevated uric acid), QT prolongation (abnormal heart rhythm) and vision disorders. Health care professionals should inform females of reproductive age and males with a female partner of reproductive potential to use effective contraception during treatment with Rozlytrek. Women who are pregnant or breastfeeding should not take Rozlytrek because it may cause harm to a developing fetus or newborn baby.
Rozlytrek was granted accelerated approval. This approval commits the sponsor to provide additional data to the FDA. Rozlytrek also received Priority Review, Breakthrough Therapy and Orphan Drug designation. The approval of Rozlytrek was granted to Genentech, Inc.
///////////////Rozlytrek, entrectinib, accelerated approval, priority Review, Breakthrough Therapy, Orphan Drug designation, fda 2019, Genentech, cancer
Tagraxofusp タグラクソフスプ
MGADDVVDSS KSFVMENFSS YHGTKPGYVD SIQKGIQKPK SGTQGNYDDD WKGFYSTDNK
YDAAGYSVDN ENPLSGKAGG VVKVTYPGLT KVLALKVDNA ETIKKELGLS LTEPLMEQVG
TEEFIKRFGD GASRVVLSLP FAEGSSSVEY INNWEQAKAL SVELEINFET RGKRGQDAMY
EYMAQACAGN RVRRSVGSSL SCINLDWDVI RDKTKTKIES LKEHGPIKNK MSESPNKTVS
EEKAKQYLEE FHQTALEHPE LSELKTVTGT NPVFAGANYA AWAVNVAQVI DSETADNLEK
TTAALSILPG IGSVMGIADG AVHHNTEEIV AQSIALSSLM VAQAIPLVGE LVDIGFAAYN
FVESIINLFQ VVHNSYNRPA YSPGHKTRPH MAPMTQTTSL KTSWVNCSNM IDEIITHLKQ
PPLPLLDFNN LNGEDQDILM ENNLRRPNLE AFNRAVKSLQ NASAIESILK NLLPCLPLAT
AAPTRHPIHI KDGDWNEFRR KLTFYLKTLE NAQAQQTTLS LAIF
(disulfide bridge: 187-202, 407-475)

methionyl (1)-Corynebacterium diphtheriae toxin fragment (catalytic and transmembrane domains) (2-389, Q388R variant)-His390-Met391-human interleukin 3 (392-524, natural P399S variant) fusion protein, produced in Escherichia coli antineoplastic,https://www.who.int/medicines/publications/druginformation/issues/PL_118.pdf

Tagraxofusp
タグラクソフスプ
| CAS: | 2055491-00-2 |
|
C2553H4026N692O798S16, 57694.4811
|
FDA 2018/12/21, Elzonris APPROVED
Antineoplastic, Immunotoxin, Peptide
DT-3881L3 / DT388IL3 / Molecule 129 / Molecule-129 / SL-401
UNII8ZHS5657EH
Diphteria toxin fusion protein with peptide and interleukin 3 Treatment of blastic plasmacytoid dendritic cell neoplasm (CD123-directed)
FDA approves first treatment for rare blood disease
>>tagraxofusp<<< MGADDVVDSSKSFVMENFSSYHGTKPGYVDSIQKGIQKPKSGTQGNYDDDWKGFYSTDNK YDAAGYSVDNENPLSGKAGGVVKVTYPGLTKVLALKVDNAETIKKELGLSLTEPLMEQVG TEEFIKRFGDGASRVVLSLPFAEGSSSVEYINNWEQAKALSVELEINFETRGKRGQDAMY EYMAQACAGNRVRRSVGSSLSCINLDWDVIRDKTKTKIESLKEHGPIKNKMSESPNKTVS EEKAKQYLEEFHQTALEHPELSELKTVTGTNPVFAGANYAAWAVNVAQVIDSETADNLEK TTAALSILPGIGSVMGIADGAVHHNTEEIVAQSIALSSLMVAQAIPLVGELVDIGFAAYN FVESIINLFQVVHNSYNRPAYSPGHKTRPHMAPMTQTTSLKTSWVNCSNMIDEIITHLKQ PPLPLLDFNNLNGEDQDILMENNLRRPNLEAFNRAVKSLQNASAIESILKNLLPCLPLAT AAPTRHPIHIKDGDWNEFRRKLTFYLKTLENAQAQQTTLSLAIF
December 21, 2018
Release
The U.S. Food and Drug Administration today approved Elzonris (tagraxofusp-erzs) infusion for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN) in adults and in pediatric patients, two years of age and older.
“Prior to today’s approval, there had been no FDA approved therapies for BPDCN. The standard of care has been intensive chemotherapy followed by bone marrow transplantation. Many patients with BPDCN are unable to tolerate this intensive therapy, so there is an urgent need for alternative treatment options,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
BPDCN is an aggressive and rare disease of the bone marrow and blood that can affect multiple organs, including the lymph nodes and the skin. It often presents as leukemia or evolves into acute leukemia. The disease is more common in men than women and in patients 60 years and older.
The efficacy of Elzonris was studied in two cohorts of patients in a single-arm clinical trial. The first trial cohort enrolled 13 patients with untreated BPDCN, and seven patients (54%) achieved complete remission (CR) or CR with a skin abnormality not indicative of active disease (CRc). The second cohort included 15 patients with relapsed or refractory BPDCN. One patient achieved CR and one patient achieved CRc.
Common side effects reported by patients in clinical trials were capillary leak syndrome (fluid and proteins leaking out of tiny blood vessels into surrounding tissues), nausea, fatigue, swelling of legs and hands (peripheral edema), fever (pyrexia), chills and weight increase. Most common laboratory abnormalities were decreases in lymphocytes, albumin, platelets, hemoglobin and calcium, and increases in glucose and liver enzymes (ALT and AST). Health care providers are advised to monitor liver enzyme levels and for signs of intolerance to the infusion. Women who are pregnant or breastfeeding should not take Elzonris because it may cause harm to a developing fetus or newborn baby.
The labeling for Elzonris contains a Boxed Warning to alert health care professionals and patients about the increased risk of capillary leak syndrome which may be life-threatening or fatal to patients in treatment.
The FDA granted this application Breakthrough Therapy and Priority Reviewdesignation. Elzonris also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Elzonris to Stemline Therapeutics.
Tagraxofusp is an IL-3 conjugated truncated diphtheria toxin.[4] It is composed by the catalytic and translocation domains of diphtheria toxin fused via Met-His linker to a full-length human IL-3.[6, 7] Tagraxofusp was developed by Stemline Therapeutics Inc and FDA approved on December 21, 2018, as the first therapy for blastic plasmacytoid dendritic cell neoplasm.[3] This drug achieved approval after being designed with the title of breakthrough therapy, priority review, and orphan drug status.[2] Tagraxofusp has been designed as an orphan drug in EU since November 2015.[7]
Tagraxofusp is indicated for the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN) in adults and pediatric patients over 2 years old. This treatment allows an alternative for the previous intense treatment which consisted of intensive chemotherapy followed by bone marrow transplantation.[2]
BPDCN is a rare hematologic malignancy derived from plasmacytoid dendritic cells. It is characterized by the significantly increased expression of cells expressing CD4/CD56/CD123 and other markers restricted to plasmacytoid dendritic cells and a lack of expression of lymphoid, natural killer or myeloid lineage-associated antigens.[1] A key feature of the malignant cells is the overexpression of CD123, also known as interleukin-3 receptor, and the constant requirement of IL-3 for survival.[6]
Associated Conditions
PharmacodynamicsIn vitro studies showed that BPDCN blasts are ultrasensitive to tagraxofusp by presenting IC50 values in the femtomolar scale.[6] One of the main physiological changes of BPDCN is the presence of elevated interferon alpha and to produce an inflammatory response. In trials with tagraxofusp and following cell depletion, there was observed a significant reduction in the levels of interferon alpha and interleukin 6.[5]
In clinical trials, tagraxofusp reported complete remission and complete remission with a skin abnormality not indicative of active disease in 54% of the treated patients.[2]
Mechanism of actionTagraxofusp binds to cells expressing the IL-3 receptor and delivers in them the diphtheria toxin after binding. This is very useful as the malignant cells in BPDCN present a particularly high expression of IL-3 receptor (CD123+ pDC).[5] To be more specific, tagraxofusp gets internalized to the IL-3 receptor-expressing cell allowing for diphtheria toxin translocation to the cytosol and followed by the binding to ADP-ribosylation elongation factor 2 which is a key factor for protein translation. Once the protein synthesis is inhibited, the cell goes under a process of apoptosis.[4,6]
As the apoptosis induction requires an active state of protein synthesis, tagraxofusp is not able to perform its apoptotic function in dormant cells.[6]
Absorption
The reported Cmax in clinical trials was of around 23 ng/ml.[6] After a 15 min infusion of a dose of 12 mcg/kg the registered AUC and Cmax was 231 mcg.h/L and 162 mcg/L respectively.[Label]
Volume of distributionIn BPDCN patients, the reported volume of distribution is of 5.1 L.[Label]
Protein bindingTagraxofusp is not a substrate of p-glycoprotein and other efflux pump proteins associated with multidrug resistance.[6]
MetabolismFor the metabolism, as tagraxofusp is a fusion protein, it is expected to get processed until small peptides and amino acids by the actions of proteases.
Route of eliminationTagraxofusp is eliminated as small peptides and amino acids. More studies need to be performed to confirm the main elimination route.
Half lifeThe reported half-life of tagraxofusp is of around 51 minutes.[6]
ClearanceThe clearance of tagraxofusp was reported to fit a mono-exponential model.[6] The reported clearance rate is reported to be of 7.1 L/h.[Label]
ToxicityThere haven’t been analysis observing the carcinogenic, mutagenic potential nor the effect on fertility. However, in studies performed in cynomolgus monkeys at an overdose rate of 1.6 times the recommended dose, it was observed severe kidney tubular degeneration. Similar studies at the recommended dose reported the presence of degeneration and necrosis of choroid plexus in the brain were. This effect seems to be progressive even 3 weeks after therapy withdrawal.[Label]
- Kharfan-Dabaja MA, Lazarus HM, Nishihori T, Mahfouz RA, Hamadani M: Diagnostic and therapeutic advances in blastic plasmacytoid dendritic cell neoplasm: a focus on hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2013 Jul;19(7):1006-12. doi: 10.1016/j.bbmt.2013.01.027. Epub 2013 Feb 5. [PubMed:23396213]
- FDA news [Link]
- FDA approvals [Link]
- Oncology nursing news [Link]
- Stemline therapeutics news [Link]
- Blood journal [Link]
- NHS reports [Link]
FDA label, Download (455 KB)
/////////Antineoplastic, Immunotoxin, Peptide, Tagraxofusp, Elzonris, タグラクソフスプ , Stemline Therapeutics, Breakthrough Therapy, Priority Review designation, Orphan Drug designation, fda 2018, DT-3881L3 , DT388IL3 , Molecule 129 , Molecule-129 , SL-401,
FDA approves first treatment Firdapse (amifampridine) for Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder
FDA approves first treatment Firdapse (amifampridine) for Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder
The U.S. Food and Drug Administration today approved Firdapse (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in adults. LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. This is the first FDA approval of a treatment for LEMS.
November 28, 2018
Release
The U.S. Food and Drug Administration today approved Firdapse (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in adults. LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. This is the first FDA approval of a treatment for LEMS.
“There has been a long-standing need for a treatment for this rare disorder,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “Patients with LEMS have significant weakness and fatigue that can often cause great difficulties with daily activities.”
In people with LEMS, the body’s own immune system attacks the neuromuscular junction (the connection between nerves and muscles) and disrupts the ability of nerve cells to send signals to muscle cells. LEMS may be associated with other autoimmune diseases, but more commonly occurs in patients with cancer such as small cell lung cancer, where its onset precedes or coincides with the diagnosis of cancer. The prevalence of LEMS is estimated to be three per million individuals worldwide.
The efficacy of Firdapse was studied in two clinical trials that together included 64 adult patients who received Firdapse or placebo. The studies measured the Quantitative Myasthenia Gravis score (a 13-item physician-rated categorical scale assessing muscle weakness) and the Subject Global Impression (a seven-point scale on which patients rated their overall impression of the effects of the study treatment on their physical well-being). For both measures, the patients receiving Firdapse experienced a greater benefit than those on placebo.
The most common side effects experienced by patients in the clinical trials were burning or prickling sensation (paresthesia), upper respiratory tract infection, abdominal pain, nausea, diarrhea, headache, elevated liver enzymes, back pain, hypertension and muscle spasms. Seizures have been observed in patients without a history of seizures. Patients should inform their health care provider immediately if they have signs of hypersensitivity reactions such as rash, hives, itching, fever, swelling or trouble breathing.
The FDA granted this application Priority Review and Breakthrough Therapydesignations. Firdapse also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Firdapse to Catalyst Pharmaceuticals, Inc.
///////////Priority Review, Breakthrough Therapy, Firdapse, Orphan Drug designation, fda 2018, amifampridine
FDA approves first treatment Libtayo (cemiplimab-rwlc) for advanced form of the second most common skin cancer
FDA approves first treatment for advanced form of the second most common skin cancer
New drug targets PD-1 pathway
September 28, 2018
Release
The U.S. Food and Drug Administration today approved Libtayo (cemiplimab-rwlc) injection for intravenous use for the treatment of patients with metastatic cutaneous squamous cell carcinoma (CSCC) or locally advanced CSCC who are not candidates for curative surgery or curative radiation. This is the first FDA approval of a drug specifically for advanced CSCC.
Libtayo works by targeting the cellular pathway known as PD-1 (protein found on the body’s immune cells and some cancer cells). By blocking this pathway, the drug may help the body’s immune system fight the cancer cells.
“We’re continuing to see a shift in oncology toward identifying and developing drugs aimed at a specific molecular target. With the Libtayo approval, the FDA has approved six immune checkpoint inhibitors targeting the the PD-1 / PD-L1 pathway for treating a variety of tumors, from bladder to head and neck cancer, and now advanced CSCC,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This type of cancer can be difficult to treat effectively when it is advanced and it is important that we continue to bring new treatment options to patients.”
CSCC is the second most common human cancer in the United States with an estimated annual incidence of approximately 700,000 cases. The most common form of skin cancer is basal cell cancer. Squamous cells are thin, flat cells that look like fish scales and are found in the tissue that forms the surface of the skin. CSCC usually develops in skin areas that have been regularly exposed to the sun or other forms of ultraviolet radiation. While the majority of patients with CSCC are cured with surgical resection, a small percentage of patients will develop advanced disease that no longer responds to local treatments including surgery and radiation. Advanced CSCC may cause disfigurement at the site of the tumor and local complications such as bleeding or infection, or it may spread (metastasize) to local lymph nodes, distant tissues and organs and become life-threatening.
The safety and efficacy of Libtayo was studied in two open label clinical trials. A total of 108 patients (75 with metastatic disease and 33 with locally-advanced disease) were included in the efficacy evaluation. The study’s primary endpoint was objective response rate, or the percentage of patients who experienced partial shrinkage or complete disappearance of their tumor(s) after treatment. Results showed that 47.2 percent of all patients treated with Libtayo had their tumors shrink or disappear. The majority of these patients had ongoing responses at the time of data analysis.
Common side effects of Libtayo include fatigue, rash and diarrhea. Libtayo must be dispensed with a patient Medication Guide that describes uses of the drug and its serious warnings. Libtayo can cause the immune system to attack normal organs and tissues in any area of the body and can affect the way they work. These reactions can sometimes become severe or life-threatening and can lead to death. These reactions include the risk of immune-mediated adverse reactions including lung problems (pneumonitis), intestinal problems (colitis), liver problems (hepatitis), hormone gland problems (endocrinopathies), skin (dermatologic) problems and kidney problems. Patients should also be monitored for infusion-related reactions.
Libtayo can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
The FDA granted this application Breakthrough Therapy and Priority Reviewdesignations.
The FDA granted the approval of Libtayo to Regeneron Pharmaceuticals, Inc.

{kind=link}
{kind=link}
{kind=link}