
Home » Posts tagged 'antiviral'
Tag Archives: antiviral
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Encofosbuvir
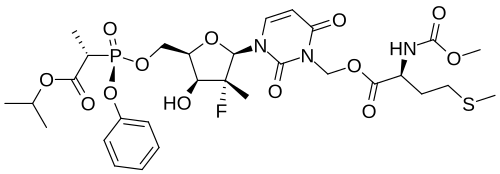
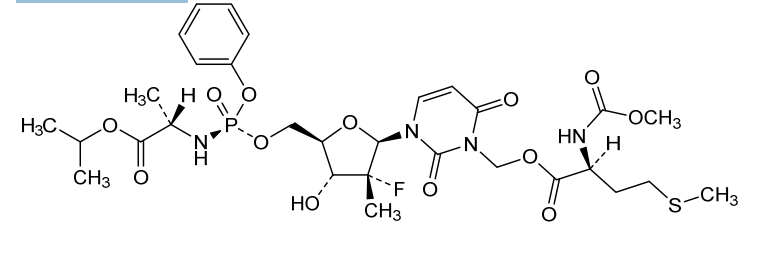
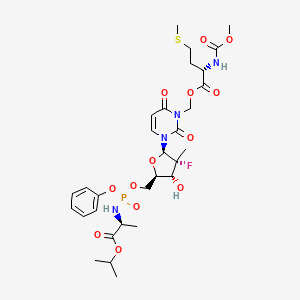
Encofosbuvir, Yiqibuvir
CAS 2232134-77-7
MF C30H42FN4O13PS MW 748.7 g/mol
- L-Alanine, O3-[N-(methoxycarbonyl)-L-methionyl]-[P(S),2’R]-2′-deoxy-2′-fluoro-3-(hydroxymethyl)-2′-methyl-P-phenyl-5′-uridylyl-, 1-methylethyl ester
- O3-[N-(Methoxycarbonyl)-L-methionyl]-[P(S),2’R]-2′-deoxy-2′-fluoro-3-(hydroxymethyl)-2′-methyl-P-phenyl-5′-uridylyl-L-alanine 1-methylethyl ester
[3-[(2R,3R,4R,5R)-3-fluoro-4-hydroxy-3-methyl-5-[[[[(2S)-1-oxo-1-propan-2-yloxypropan-2-yl]amino]-phenoxyphosphoryl]oxymethyl]oxolan-2-yl]-2,6-dioxopyrimidin-1-yl]methyl (2S)-2-(methoxycarbonylamino)-4-methylsulfanylbutanoate

antiviral, HEC 110114; Yiqibuvir, CHINA 2025, APPROVALS 2025, 82E4Q8WQV7
Encofosbuvir is a novel, oral small-molecule direct-acting antiviral (DAA) drug used to treat the hepatitis C virus (HCV). Approved by China’s National Medical Products Administration (NMPA) in March 2025, it serves as a core component of a domestic, pan-genotypic treatment regimen.
🔬 Mechanism of Action
Encofosbuvir works by targeting the machinery the virus needs to replicate itself:
- Target Enzyme: It functions as an HCV NS5B RNA-dependent RNA polymerase inhibitor.
- Viral Suppression: By selectively binding to this polymerase, it blocks the synthesis of viral RNA, effectively halting the replication and spread of the hepatitis C virus in mammals
Clinical Indications and Usage
According to the official regulatory updates from the China NMPA, encofosbuvir is prescribed under specific clinical parameters:
- Combination Therapy: It must be used in combination with netanasvir (specifically netanasvir phosphate capsules).
- Target Genotypes: The regimen is highly effective across multiple viral strains, covering HCV genotypes 1, 2, 3, and 6.
- Patient Profile: It is indicated for adult patients who are treatment-naïve (never treated before) or who have been previously treated with interferon. It is safe for use in patients with or without compensated liver cirrhosis.
The drug is classified as a Class 1 innovative drug, representing a milestone in self-developed, domestic intellectual property:
- Developers: It was jointly developed and brought to market by Sunshine Lake Pharma (a subsidiary of HEC Pharm) and YiChang HEC ChangJiang Pharmaceutical Co., Ltd.
- Dosage Form: It is distributed commercially as 0.3g tablets.
- Therapeutic Context: This medication expands the developer’s innovative hepatitis C pipeline, building upon their previously approved portfolio like emitasvir phosphate
- OriginatorHEC Pharm; Sunshine Lake Pharma
- DeveloperSunshine Lake Pharma
- ClassAntivirals
- Mechanism of ActionHepatitis C virus NS 5 protein inhibitors
- RegisteredHepatitis C
- 27 Mar 2025Registered for Hepatitis C (Combination therapy, Treatment-naive) in China (PO)
- 08 Feb 2025Preregistered for Hepatitis C (Combination therapy, Treatment-experienced) in China (PO)
- 08 Feb 2025Registered for Hepatitis C (Combination therapy, Treatment-experienced) in China (PO)
Encofosbuvir is an antiviral drug used to treat hepatitis C virus (HCV). In China, encofosbuvir is approved for use in combination with netanasvir for the treatment of adult patients with chronic HCV genotypes 1, 2, 3, or 6, who are either treatment-naive or have been previously treated with interferon.[1]
SYN
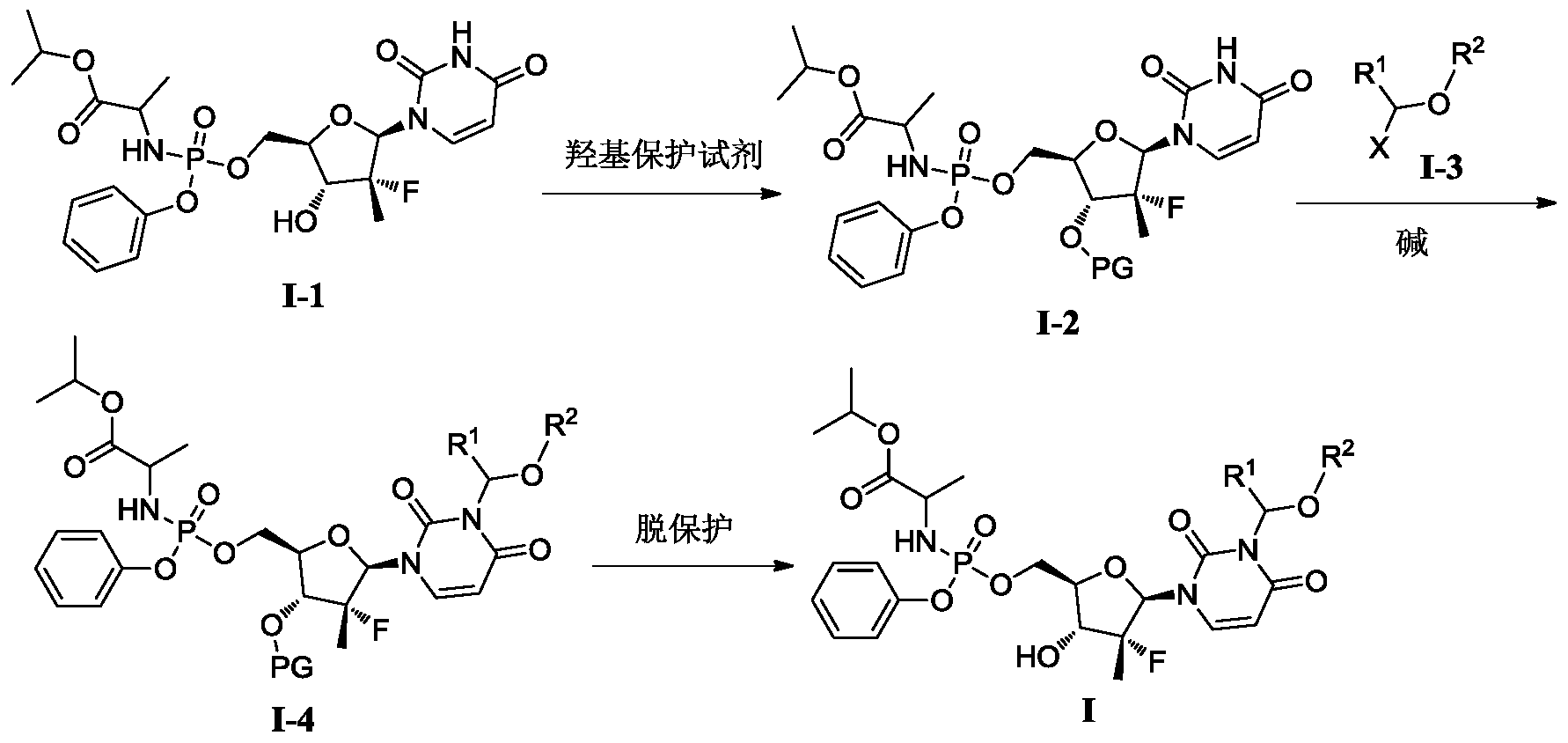
[0515](S)-(3-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-((((S)-(((S)-1-isopropoxy-1-oxopropyl-2-yl)amino)(phenoxy)phosphoryl)oxy)methyl)-3-methyltetrahydrofuran-2-yl)-2,6-dioxo-2,3-dihydropyrimidin-1(6H)-yl)methyl-2-((methoxycarbonyl)amino)-4-(methylthio)butyrate
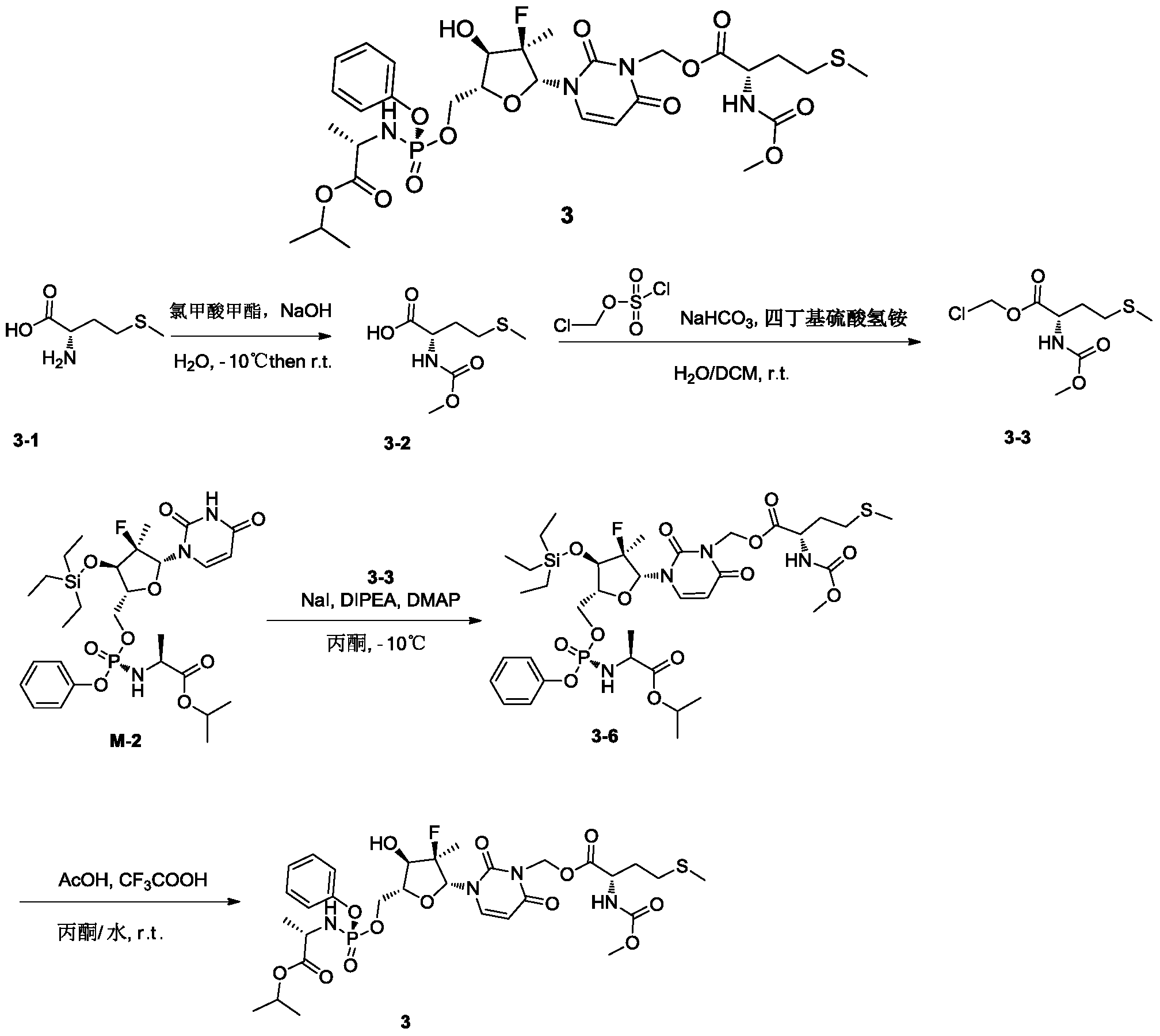
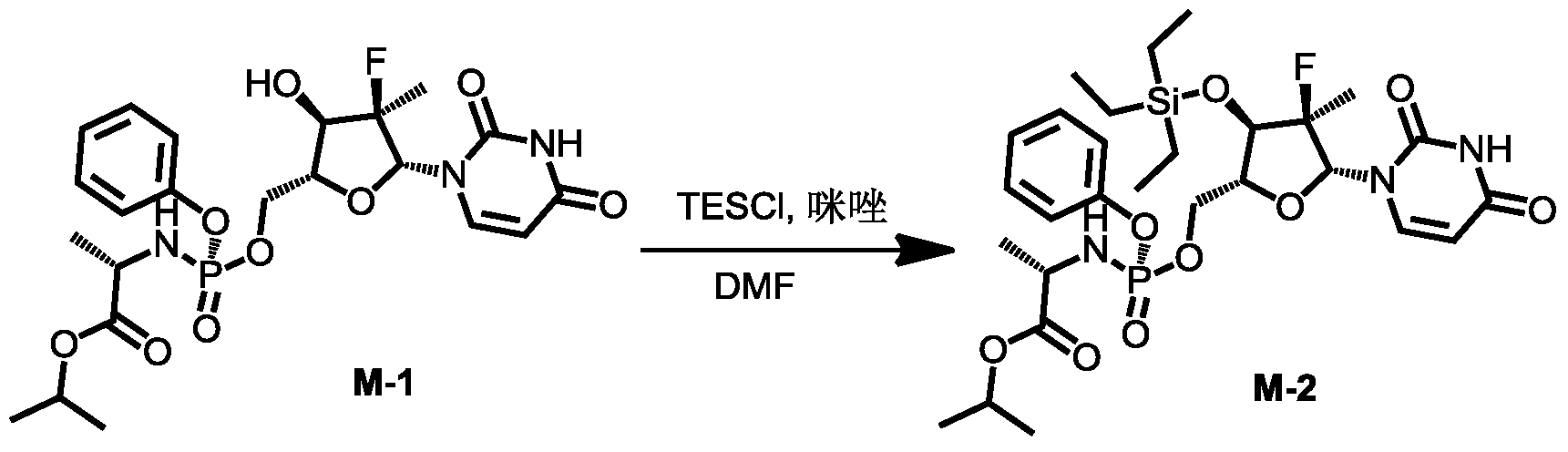
[0531]4) Synthesis of compound 3
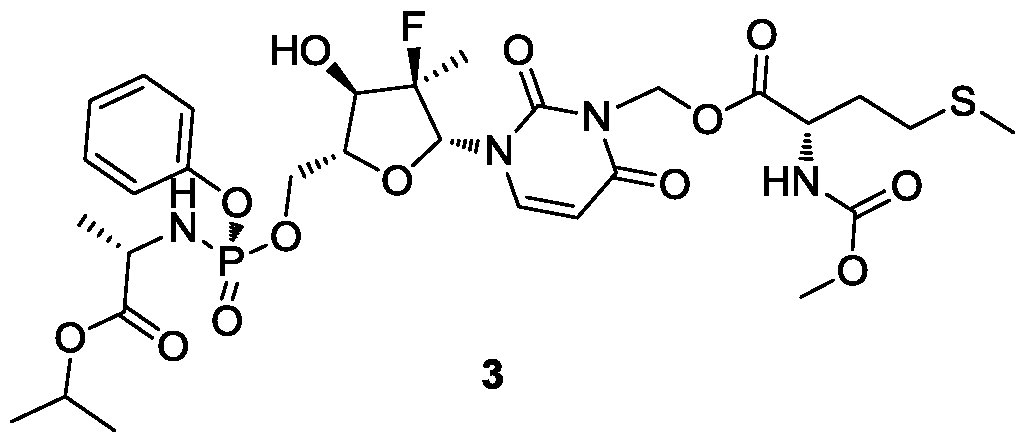
Compound 3-6 (3.63 g, 4.2 mmol, 1 eq) was dissolved in acetone (12 mL), and water (9 mL), trifluoroacetic acid (3 mL), and glacial acetic acid (12 mL) were added sequentially at room temperature, followed by a reaction time of 2 hours. After the reaction was monitored by TLC until complete, 30 mL of dichloromethane was added to the reaction solution, stirred thoroughly, and allowed to stand for phase separation. The organic phase was washed sequentially with water (10 mL × 3), saturated sodium chloride (10 mL), dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. The solution was purified by column chromatography using DCM:MeOH = 50:1 as the eluent, yielding 2.8 g of a white foamy solid.
[0534]MS-ESI: m/z 748.8[M+1] +;
[0535]
1H NMR(400MHz,CDCl 3)δ7.49(d,J=8.2Hz,1H),7.34(d,J=7.6Hz,2H),7.24–7.16(m,3H),6.19(d,J=17.3Hz,1H),6.07–5.94(m,2H),5.75(d,J=8.3Hz,1H),5.41(d,J=7.4Hz,1H),5.07–4.95(m,1H),4.58–4.39(m,3H),4.12(d,J=8.6Hz,1H),4.02–3.80(m,4H),3.67(s,3H),2.52(t,J=7.5Hz,2H),2.14–1.90(m,5H),1.37(dd,J=18.4,14.3Hz,6H),1.24(d,J=6.3Hz,6H)。
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
“Netanasvir Phosphate Capsules Approved for Marketing by China NMPA”. National Medical Products Administration. 2025-06-11.
| Clinical data | |
|---|---|
| Trade names | 英强布韦 |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2232134-77-7 |
| PubChem CID | 141522644 |
| UNII | 82E4Q8WQV7 |
| Chemical and physical data | |
| Formula | C30H42FN4O13PS |
| Molar mass | 748.71 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////encofosbuvir, anax labs, antiviral, HEC 110114; Yiqibuvir, CHINA 2025, APPROVALS 2025, 82E4Q8WQV7
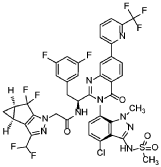
Suricapavir
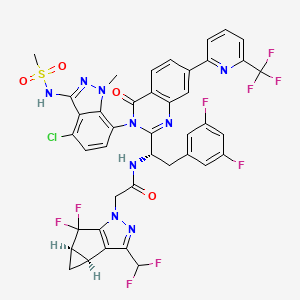
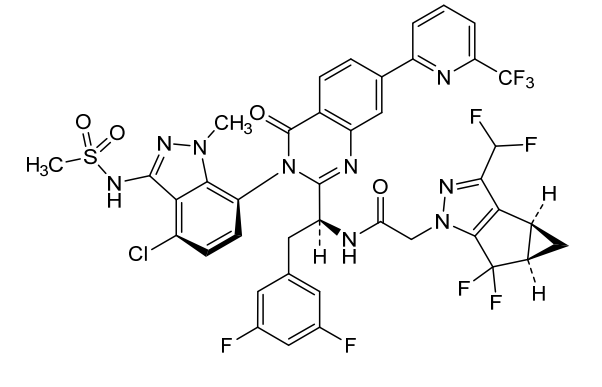
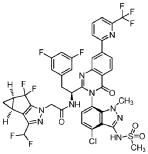
Suricapavir
CAS 2417270-21-2
MF C41H29ClF9N9O4S MF950.2 g/mol
N-[(1S)-1-[3-[4-chloro-3-(methanesulfonamido)-1-methylindazol-7-yl]-4-oxo-7-[6-(trifluoromethyl)-2-pyridinyl]quinazolin-2-yl]-2-(3,5-difluorophenyl)ethyl]-2-[(2S,4R)-9-(difluoromethyl)-5,5-difluoro-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetamide
N-[(1S)-1-[3-[4-chloro-3-(methanesulfonamido)-1-methyl-indazol-7-yl]-4-oxo-7-[6-(trifluoromethyl)-2-pyridyl]quinazolin-2-yl]-2-(3,5-difluorophenyl)ethyl]-2-[(2S,4R)-9-(difluoromethyl)-5,5-difluoro-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetamide
N-[(1S)-1-{(3P)-3-[4-chloro-3-(methanesulfonamido)-1-methyl-1Hindazol-7-yl]-4-oxo-7-[6-(trifluoromethyl)pyridin-2-yl]-3,4-
dihydroquinazolin-2-yl}-2-(3,5-difluorophenyl)ethyl]-2-[(3bS,4aR)-3-
(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-1Hcyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl]acetamide
inhibitor of viral replication, antiviral, ZZ799EX5KN
tructurally resembles:
- Lenacapavir-type macroheterocyclic capsid inhibitors (Gilead class)
PAT
Preparation of Example 59: N-((S)-1-((3P)-3-(4-chloro-1-methyl-3-(methylsulfonamido)-1H-indazol-7-yl)-4-oxo-7-(6-(trifluoromethyl)pyridin-2-yl)-3,4-dihydroquinazolin-2-yl)-2- (3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5- tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide.
The title compound was prepared according to General Procedure D using 2-chloro-6-(trifluoromethyl)pyridine as the coupling partner. The experiment afforded the title compound, N-((S)-1-((3P)-3-(4-chloro-1-methyl-3-(methylsulfonamido)-1H-indazol-7-yl)-4-oxo-7-(6-(trifluoromethyl)pyridin-2-yl)-3,4-dihydroquinazolin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide. The sample was analyzed using LCMS Method F: retention time = 1.51 min.; observed ion = 948.4 (M-H).1H NMR (METHANOL-d4, 500 MHz) Shift 8.66 (s, 1H), 8.4-8.4 (m, 3H), 8.22 (t, 1H, J=7.9 Hz), 7.88 (d, 1H, J=7.7 Hz), 7.28 (br d, 1H, J=8.0 Hz), 7.20 (d, 1H, J=7.7 Hz), 6.7-6.8 (m, 1H), 6.61 (dd, 2H, J=2.2, 8.2 Hz), 6.67 (br t, 2H, J=54.7 Hz), 4.5-4.6 (m, 2H), 3.61 (s, 3H), 3.4-3.5 (m, 1H), 3.2-3.2 (m, 3H), 3.1-3.2 (m, 1H), 2.41 (br dd, 2H, J=3.7, 7.3 Hz), 1.34 (br d, 1H, J=5.4 Hz), 0.99 (br dd, 1H, J=1.9, 3.7 Hz)
PAT
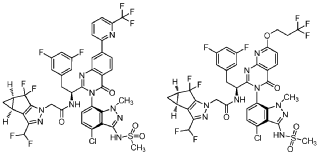
WO 2020/084492 and WO 2020/254985 disclose certain Capsid Inhibitor compounds including the two compounds shown below which will be referred to in this application as the compounds of Formula la and Formula lb.
PAT
- Inhibitors of human immunodeficiency virus replicationPublication Number: WO-2023062559-A1Priority Date: 2021-10-13
- Inhibitors of human immunodeficiency virus replicationPublication Number: US-2023149408-A1Priority Date: 2020-04-15
- Inhibitors of human immunodeficiency virus replicationPublication Number: WO-2021209900-A1Priority Date: 2020-04-15
- Pharmaceutical compositions comprising cabotegravirPublication Number: US-2023045509-A1Priority Date: 2019-12-09
- Inhibitors of human immunodeficiency virus replicationPublication Number: US-11541055-B2Priority Date: 2018-10-24Grant Date: 2023-01-03
- Inhibitors of human immunodeficiency virus replicationPublication Number: US-2020360384-A1Priority Date: 2018-10-24
- Inhibitors of human immunodeficiency virus replicationPublication Number: EP-3870577-B1Priority Date: 2018-10-24Grant Date: 2025-03-19
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////////////suricapavir, ANAX, inhibitor of viral replication, antiviral, ZZ799EX5KN
Pixavir marboxil
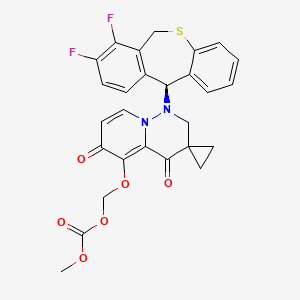
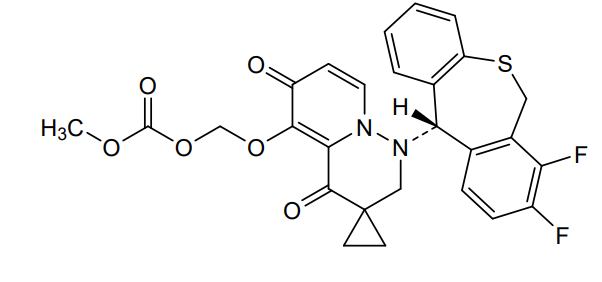
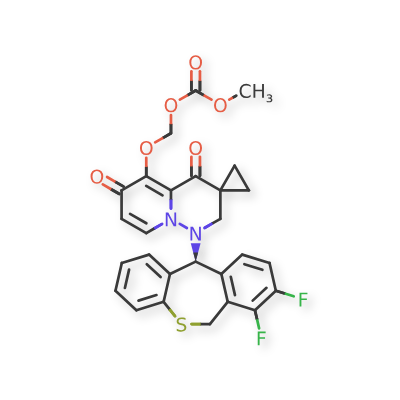
Pixavir marboxil
CAS 2365473-17-0
MF C27H22F2N2O6S MW540.535
(1-((11S)-7,8-DIFLUORO-6,11-DIHYDROBENZO(C)(1)BENZOTHIEPIN-11-YL)-4,6-DIOXOSPIRO(2H-PYRIDO(1,2-B)PYRIDAZINE-3,1′-CYCLOPROPANE)-5-YL)OXYMETHYL METHYL CARBONATE
({1′-[(11S)-7,8-difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-4′,6′-dioxo1′,2′,4′,6′-tetrahydrospiro[cyclopropane-1,3′-pyrido[1,2-b]pyridazin]-5′-yl}oxy)methyl methyl carbonate
antiviral, TG 1000, Yi Li Kang, SV42843XSX, Cap-dependent endonuclease-IN-1, Influenza virus infections, TaiGen Biotechnology
- OriginatorTaiGen Biotechnology
- Class3-ring heterocyclic compounds; Antivirals; Benzene derivatives; Carbonates; Cyclopropanes; Dibenzothiepins; Esters; Ethers; Fluorobenzenes; Organic sulfur compounds; Pyridazines; Pyridones; Small molecules; Spiro compounds
- Mechanism of ActionEndonuclease inhibitors; Virus replication inhibitors
- MarketedInfluenza virus infections
- 27 Feb 2026Launched for Influenza virus infections (In adults, In adolescents) in China (PO), prior to February 2026 (TaiGen Biotechnology pipeline, February 2026)
- 26 Jan 2026Pixavir marboxil licensed to Boryung Biopharma for commercialization in South Korea
- 16 Dec 2025Chemical structure information added.
Pixavir marboxil (also known as TG-1000) is an investigational antiviral drug designed to treat and inhibit influenza virus infections. It belongs to a class of compounds known as cap-dependent endonuclease (CEN) inhibitors, which target a key viral enzyme necessary for influenza virus replication.
Mechanism of Action
- Blocks viral replication: Pixavir marboxil works by inhibiting the influenza virus’s cap-dependent endonuclease, a part of the viral RNA polymerase complex the virus needs to “snatch” capped RNA fragments from host cell mRNA. Without this process, the virus cannot efficiently produce its own viral proteins or replicate.
What Viruses It Targets
Pixavir marboxil has shown activity against:
- Influenza A viruses
- Influenza B viruses
- Certain drug-resistant influenza strains
This broad spectrum makes it useful for seasonal flu and potentially strains less responsive to older antiviral drugs
Clinical Development & Approval Status
Phase Trials & Results
- Completed Phase III: Clinical trials in adults and adolescents (age ≥12) showed that a single dose shortened time to symptom relief compared to placebo (e.g., median ~60.9 h vs ~87.9 h).
- Symptom relief benefits: The data indicated statistically significant improvement in flu symptoms and faster viral inactivation in treated patients versus placebo.
- Pediatric Formulation: China’s health authority approved pediatric Phase III studies for Pixavir (children <12), indicating further development for younger patients.
Regulatory Filings
- NDA (New Drug Application): Pixavir marboxil has been submitted for approval to the National Medical Products Administration (NMPA) in mainland China based on Phase III results.
- Generic Name Approved: The drug has been officially recognized with the generic name “Pixavir marboxil,” moving it closer to commercialization.
Pixavir marboxil is a small molecule drug. The usage of the INN stem ‘-xavir’ in the name indicates that Pixavir marboxil is a influenza CAP-dependent endonuclease inhibitor. Pixavir marboxil has a monoisotopic molecular weight of 540.12 Da.
SYN
PAT
- Cap-dependent endonuclease inhibitorsPublication Number: US-10596171-B2Patent Family: AU-2019209426-A1; AU-2019209426-B2; BR-112020014810-A2; CA-3078391-A1; CA-3078391-C; CL-2020001919-A1; CN-110300753-A; CN-110300753-B; CO-2020006411-A2; EA-202090658-A1; EP-3743424-A1; EP-3743424-A4; IL-274199-A; IL-274199-B; JO-P20200159-A1; JP-2021512146-A; JP-6994121-B2; KR-102432975-B1; KR-20200086385-A; MX-2020007722-A; MY-197875-A; NZ-763248-A; PE-20211240-A1; PH-12020550921-A1; SG-11202003014V-A; TW-201938166-A; TW-I714951-B; US-10596171-B2; US-2019224198-A1; WO-2019144089-A1; ZA-202002037-BPriority Date: 2018-01-22Grant Date: 2020-03-24Inventor(s): LIN CHU-CHUNG; CHEN HUNG-CHUAN; CHIANG CHIAYN; YEN CHI-FENG; HSU MING-CHUAssignee(s): TAIGEN BIOTECHNOLOGY CO LTD; HSU MING CHUClassification: A61K31/5025; A61P31/16Abstract: Provided is a compound of Formula (I) below, or a pharmaceutically acceptable salt, metabolite, or prodrug thereof: n nwherein: A 1 is CR 4 or N; A 2 is CR 5 R 6 or NR 7 ; A 3 is CR 5 ′R 6 ′ or NR 7 ′; each of R 1 , R 2 , R 2 ′, R 3 , R 3 ′, R 4 , R 5 , R 5 ′, R 6 , R 6 ′, R 7 , and R 7 ′, independently, is hydrogen, deuterium, halogen, cyano, hydroxyl, carboxyl, amino, formyl, nitro, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, C 2-6 alkenyloxy, C 1-6 alkylcarbonyl, C 1-6 alkyloxycarbonyl, C 1-6 alkylamine, C 3-20 carbocyclyl, or C 3-20 heterocyclyl; or R 5 and R 6 , R 5 ′ and R 6 ′, or R 5 and R 5 ′, together with the adjacent atom to which they are each attached, form C 3-10 carbocyclyl or C 3-10 heterocyclyl. Further provided are a method of using the above-described compound, or the pharmaceutically acceptable salt, metabolite, or prodrug thereof for treating influenza and a pharmaceutical composition containing same.Linked Compounds: 274Linked Substances: 411
- Cap-dependent endonuclease inhibitorsPublication Number: US-2019224198-A1Priority Date: 2018-01-22Linked Compounds: 275Linked Substances: 411
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////pixavir marboxil, antiviral, TG 1000, Yi Li Kang, SV42843XSX, Cap-dependent endonuclease-IN-1, Influenza virus infections, TaiGen Biotechnology
Dezecapavir




Dezecapavir
CAS 2570323-59-8
MF C37H29ClF9N9O5S MW918.19
1H-Cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-1-acetamide, N-[(1S)-1-[(3S)-3-[4-chloro-1-methyl-3-[(methylsulfonyl)amino]-1H-indazol-7-yl]-3,4-dihydro-4-oxo-7-(3,3,3-trifluoropropoxy)pyrido[2,3-d]pyrimidin-2-yl]-2-(3,5-difluorophenyl)ethyl]-3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-, (3bS,4aR)-
N-[(1S)-1-[(3P)-3-[4-chloro-3-(methanesulfonamido)-1-methyl-1H-indazol-7-yl]-4-oxo-7-(3,3,3-
trifluoropropoxy)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl]-2-(3,5-difluorophenyl)ethyl]-2-[(3bS,4aR)-3-
(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-1Hcyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-
yl]acetamide
inhibitor of viral replication, antiviral, SEK9LN2LSM, VH 4011499, VH4011499, VH-4011499, VH 499, GSK2838232, GSK 2838232
Dezecapavir is a potent experimental antiviral compound, specifically a novel HIV-1 capsid inhibitor, developed to block HIV replication by targeting the virus’s capsid protein, showing high effectiveness in lab settings (low nM range EC50) and representing a new class of drugs for HIV treatment, potentially for long-acting injectable therapies. It’s a complex molecule with a unique structure designed to disrupt the HIV capsid assembly, halting the virus’s life cycle early on.
Key Characteristics:
- Mechanism: Inhibits HIV-1 capsid assembly, a crucial step in the viral lifecycle.
- Potency: Very effective in cell cultures, with a low nanomolar EC50 (effective concentration).
- Class: Belongs to a new class of antivirals, distinct from integrase or reverse transcriptase inhibitors, offering a novel approach to HIV treatment.
- Development: Under investigation, often mentioned as a potential candidate for long-acting injectable (LAI) treatments due to its potency.
What it does:
Dezecapavir binds to the HIV capsid, preventing the virus from uncoating and maturing, thereby stopping new infections from forming.
Significance:
It represents a promising new option for HIV treatment, especially in the context of growing resistance to existing drugs, and could be part of future long-acting regimens.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020254985&_cid=P20-MKHOOT-76990-1

Preparation of Example 1: N-((S)-l-((3P)-3-(4-chloro-l-methyl-3-(methylsulfonamido)-lH- indazol-7-yl)-4-oxo-7-(3,3,3-trifluoropropoxy)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)-2-(3,5- difluorophenyl)ethyl)-2-((3bSr4aR)-3-(difluoromethyl)-5r5-difluoro-3br4r4ar5-tetrahydro-lH- cyciopropa[3, 4]cydopenta[ l,2-c]pyrazoi-l-yi)acetamide

A solution of diisopropyl (E)-diazene-l,2-dicarboxylate (“DIAD”, 0.125 ml, 0.637 mmol) in THF (0.2 mL) was added dropwise to a mixture of N-(l-((3P)-3-(4-chloro-3-(N-(4-methoxybenzyl)methylsulfonamido)-l-methyl-lH-indazol-7-yl)-7-hydroxy-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-lH-cyclopropa[3,4]cyclopenta[l,2-c]pyrazol-l-yl)acetamide (0.2 g, 0.212 mmol)), 3,3,3-trifluoropropan-l-ol (0.073 g, 0.637 mmol) and triphenylphosphine (0.178 g, 0.679 mmol) in Tetrahydrofuran (2.1 mL) at rt. The reaction mixture was stirred for 18 h at rt and then was concentrated in vacuo. The residue was purified on silica gel (24 g RediSep Gold column) using a gradient of 0-60 % ethyl acetate in hexanes over 15 CV, and then holding at 60 % ethyl acetate in hexanes for 5 CV. Fractions containing the pure product were pooled and then concentrated to give a yellow solid. This solid was taken up in DCM (1 mL):TFA (0.5 mL); the solution was cooled to 0 °C; and to the solution was added triflic acid (0.057 mL, 0.637 mmol). The mixture was stirred for 1 h and then concentrated in vacuo. The residue was taken up in ethyl acetate; washed with 1 N
NaOH; washed with 0.5M citric acid; dried over Na2SC>4; filtered; and then was concentrated in vacuo. The residue was subjected to silica gel chromatography (24 g RediSep Gold column) using 0-60 % ethyl acetate in hexanes over 20 CV, then at 60 % ethyl acetate for 10 CV. Fractions containing the pure product were pooled and then concentrated in vacuoto give N-(l-((6P)-3-(4-chloro-l-methyl-3-(methylsulfonamido)-lH-indazol-7-yl)-4-oxo-7-(3,3,3-trifluoropropoxy)-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-3-(difluoromethyl)-5,5-difluoro-3b,4,4a,5-tetrahydro-lH-cyclopropa[3,4]cyclopenta[l,2-c]pyrazol-l-yl)acetamide (0.078 g, 0.081 mmol, 38.0 % yield) as a brown solid. *H NMR (500 MHz, METHANOL-d^ d ppm 8.46 – 8.53 (m, 1 H) 7.28 – 7.34 (m, 1 H) 7.19 – 7.24 (m, 1 H) 7.03 – 7.09 (m, 1 H) 6.53 – 6.81 (m, 4 H) 4.80 (dd, J=5.96, 2.98Hz, 3 H) 4.49 – 4.62 (m, 2 H) 3.58 – 3.62 (m, 3 H) 3.40 – 3.49 (m, 1 H) 3.22 – 3.24 (m, 3 H) 3.06 – 3.14 (m, 1 H) 2.80 – 2.89 (m, 2 H) 2.37 – 2.44 (m, 2 H) 1.32 – 1.37 (m, 1 H) 0.96 – 1.01 (m, 1 H). LCMS Analysis Method: Column = Acquity UPLC BEH C18, 2.1 x 100 mm, 1.7 pm particles; Injection Volume = 5.00 pL; Flowrate = 0.80 mL/min; Solvent A = 95:5
WatenMeCN w/ 0.1% v/v formic acid; Solvent B = 5:95 WatenMeCN w/ 0.1% v/v formic acid;
Elution profile = Start %B: 0, End %B: 100, Gradient Time: 3.5 min. then hold at 100% B for 1 min.; Detection wavelength 1 = 220 nm, wavelength 2 = 254 nm. LCMS retention time = 3.097 min; m/z = 918.05 [M+l]+.



PAT
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: US-2021323967-A1Priority Date: 2019-06-19
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: US-2025019383-A1Priority Date: 2019-06-19
- Pyrido[2,3-D]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: KR-20220024608-APriority Date: 2019-06-19
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: EP-3986561-A1Priority Date: 2019-06-19
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: EP-3986561-B1Priority Date: 2019-06-19Grant Date: 2024-02-14
- Pyrido [2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: US-12129255-B2Priority Date: 2019-06-19Grant Date: 2024-10-29
- Pyrido[2,3-d]pyrimidine derivatives as inhibitors of human immunodeficiency virus replicationPublication Number: WO-2020254985-A1Priority Date: 2019-06-19
- Inhibitors of human immunodeficiency virus replicationPublication Number: EP-4415685-A1Priority Date: 2021-10-13
- Inhibitors of human immunodeficiency virus replicationPublication Number: WO-2023062559-A1Priority Date: 2021-10-13
- Inhibitors of human immunodeficiency virus replicationPublication Number: US-2024423985-A1Priority Date: 2021-10-13
- Inhibitors of human immunodeficiency virus replicationPublication Number: US-2023149408-A1Priority Date: 2020-04-15
- Pharmaceutical compositions comprising cabotegravirPublication Number: US-2023045509-A1Priority Date: 2019-12-09
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.

Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
NEXT
ADVERTISEMENT
Advect Process Systems Ltd.
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1
Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.

Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com
//////////dezecapavir, inhibitor of viral replication, antiviral, SEK9LN2LSM, VH 4011499, VH4011499, VH-4011499, VH 499, GSK2838232, GSK 2838232
Limnetrelvir



Limnetrelvir
CAS 2923500-04-1
MF C27H23F4N5O4 MW 557.50
N-[(3R)-1-[4-cyano-2-(morpholine-4-carbonyl)-6-(trifluoromethyl)phenyl]pyrrolidin-3-yl]-8-fluoro-2-oxo-1H-quinoline-4-carboxamide
N-{(3R)-1-[4-cyano-2-(morpholine-4-carbonyl)-6-
(trifluoromethyl)phenyl]pyrrolidin-3-yl}-8-fluoro-2-oxo1,2-dihydroquinoline-4-carboxamide
antiviral, ABBV-903, ABBV 903, 4TPS988XGG
Limnetrelvir (ABBV-903) is a MPro inhibitor. Limnetrelvir could be used in antiviral research.
SYN

Example 1 – Synthesis of Compound (2) (R)-N-(1-(4-cyano-2-(morpholine-4-carbonyl)-6-(trifluoromethyl)phenyl)pyrrolidin-3-yl)-8-fluoro-2-oxo-1,2-dihydroquinoline-4-carboxamide

Compound 2F – Synthesis of 8-fluoro-2-oxo-1,2-dihydroquinoline-4-carboxylic acid

[00035] A suspension of 7-fluoroindoline-2,3-dione (55 g, 333 mmol), malonic acid (41.6 g, 400 mmol) and sodium acetate (68.3 g, 833 mmol) in acetic acid (500 mL) was heated at 112 °C overnight. The reaction mixture was cooled to room temperature and poured into cold 0.4 M aqueous HCl (2200 mL). The precipitate was collected by filtration and rinsed thoroughly with ice-cold water (~250 mL) followed by methyl tert-butyl ether (~100 mL) and then concentrated twice from acetonitrile with high vacuum. The materials were largely dissolved into 1 M aqueous NaOH (370 mL) and filtered through diatomaceous earth with a 0.1 M aqueous NaOH (50 mL) rinse. Then the filtrate was washed thrice with dichloromethane (3 x 200 mL) which removed the color. After this aqueous layer was filtered again through diatomaceous earth, it was acidified by the dropwise addition of concentrated aqueous HCl (33 mL, ~0.4 moles). The material was collected by filtration. After prolonged drying under heat and vacuum, the material was treated with water (1 L) and the mixture was made acidic by the addition of a small amount of 1 M aqueous HCl. The suspension was heated to 80 °C and then allowed to slowly cool to room temperature. The resulting material was collected by filtration, washed with 0.01 M aqueous HCl (150 mL) and dried under vacuum at 80 °C to provide the title compound (2F).1H NMR (500 MHz, DMSO-d6) δ ppm 14.00 (bs, 1H), 12.07 (bs, 1H), 8.00 (dd, J = 8.2, 1.2 Hz, 1H), 7.49 (ddd, J = 11.0, 8.1, 1.2 Hz, 1H), 7.23 (ddd, J = 8.2, 8.1, 5.2 Hz, 1H), 6.95 (s, 1H); 13C NMR (101 MHz, DMSO-d6, 90 °C) δ ppm 165.75 – 165.73 (m), 160.30, 148.75 (d, J = 246.0 Hz), 140.85 – 140.80 (m), 128.11 (d, J = 13.7 Hz), 123.85, 121.60 – 121.53 (m), 121.53 – 121.43 (m), 117.70 – 117.65 (m), 115.33 (d, J = 17.2 Hz); 19F NMR (376 MHz, DMSO-d6, 90 °C) δ ppm -130.47 (dd, J = 10.9, 5.3 Hz); MS (APCI, M+H+) m/z 208.
Compound 2G – Synthesis of (R)-N-(1-(4-cyano-2-(morpholine-4-carbonyl)-6-(trifluoromethyl)phenyl)pyrrolidin-3-yl)-8-fluoro-2-oxo-1,2-dihydroquinoline-4-carboxamide (2)

00036] To a mixture of Compound 2F (29.84 g, 144 mmol) in anhydrous N,N-dimethylformamide (360 mL) was added DMTMM (4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-
methylmorpholinium chloride) (43.17 g, 156 mmol) over twelve minutes at room temperature. After the suspension had been stirred forty minutes, it was added over eight minutes to a suspension of Compound 2E (≤120 mmol) and N-methylmorpholine (16 mL, 146 mmol) in N,N-dimethylformamide (120 mL) with a N,N-dimethylformamide (20 mL) rinse. After forty minutes, the reaction mixture was added to rapidly stirred 0.1 M aqueous K2HPO4 (2.5 L) and extracted four times with 4:1 isopropyl acetate / heptanes then once with isopropyl acetate alone. The product, which had begun to precipitate out from the combined extracts, was separated by decantation and filtration, then washed with dichloromethane. The remaining aqueous phase was extracted twice more with isopropyl acetate and all the organic extracts were combined, then washed with additional 0.1 M aqueous K2HPO4 followed by water, dried (Na2SO4), and filtered. The filtrate was concentrated with the dichloromethane wash of the material collected above. The residue was concentrated, dissolved in acetonitrile / CH2Cl2, filtered, and purified by chromatography on silica (20 to 100% acetonitrile / CH2Cl2). The collected fractions were concentrated to a small volume, and stirred in ethyl acetate overnight.
[00037] The suspension was heated at 70 °C for twenty minutes, then allowed to slowly cool to room temperature. Methyl tert-butyl ether was stirred in, the suspension was cooled to 0 °C, and the purified product was collected by filtration with rinses of 1:1 ethyl acetate / methyl tert-butyl ether followed by methyl tert-butyl ether before being dried under vacuum with heat. The material obtained previously from the early extracts were also stirred in ethyl acetate, heated at 70 °C, then allowed to slowly cool to room temperature. Methyl tert-butyl ether was stirred in, the suspension was cooled to 0 °C, and the purified product was collected by filtration with rinses of 1:1 ethyl acetate / methyl tert-butyl ether rinse followed by methyl tert-butyl ether. The material was dried overnight under vacuum to provide the title compound (2).1H NMR (500 MHz, DMSO-d6) δ ppm 11.96 (s, 1H), 9.03 – 8.84 (m, 1H), 8.21 – 8.18 (m, 1H), 7.98 – 7.93 (m, 1H), 7.56 – 7.50 (m, 1H), 7.49 – 7.43 (m, 1H), 7.21 – 7.15 (m, 1H), 6.66 – 6.59 (m, 1H), 4.51 – 4.40 (m, 1H), 3.73 – 3.55 (m, 6H), 3.54 – 3.22 (m, 6H), 2.29 – 2.18 (m, 1H), 2.07 – 1.95 (m, 1H); 1H NMR (400 MHz, DMSO-d6, 90 °C) δ ppm 11.47 (bs, 1H), 8.77 – 8.47 (m, 1H), 8.09 (d, J = 2.1 Hz, 1H), 7.88 (d, J = 2.1 Hz, 1H), 7.54 (dd, J = 8.1, 1.2 Hz, 1H), 7.39 (ddd, J = 11.0, 8.1, 1.2 Hz, 1H), 7.15 (ddd, J = 8.1, 8.1, 5.1 Hz, 1H), 6.60 (s, 1H), 4.54 – 4.43 (m, 1H), 3.74 – 3.20 (m, 12H), 2.31 – 2.21 (m, 1H), 2.06 – 1.96 (m, 1H); 13C NMR (101 MHz, DMSO-d6, 90 °C) δ
ppm 166.61, 165.97, 161.31, 149.59 (d, J = 246.3 Hz), 148.95, 146.10 – 146.03 (m), 136.00, 135.65, 133.13 (q, J = 6.1 Hz), 128.84 – 128.63 (m), 123.76 (q, J = 273.7 Hz), 122.19, 122.16, 122.12, 121.60, 118.99 – 118.91 (m), 117.75, 116.17 (d, J = 17.3 Hz), 105.57, 66.04, 57.95, 51.06, 50.35, 47.74, 42.35, 31.54; 19F NMR (376 MHz, DMSO-d6) δ ppm -57.54 – -58.10 (m), -130.02 – -130.15 (m); 19F NMR (376 MHz, DMSO-d6, 90 °C) δ ppm -58.37 – -58.97 (m), -130.96 (dd, J = 11.0, 5.1 Hz). MS (APCI, M+H+) m/z 558.






PAT
- Pyrrolidine Main Protease Inhibitors as Antiviral AgentsPublication Number: US-2024158368-A1Priority Date: 2022-10-14
- Pyrrolidine main protease inhibitors as antiviral agentsPublication Number: WO-2024081351-A1Priority Date: 2022-10-14
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////limnetrelvir, antiviral, ABBV-903, ABBV 903, 4TPS988XGG
Iscartrelvir



Iscartrelvir
CAS 2921711-74-0
MF 2921711-74-0, 526.4 g/mol
N-{(1S,2R)-2-[4-bromo-2-(methylcarbamoyl)-6-nitroanilino]cyclohexyl}isoquinoline-4-carboxamide
antiviral, WU-04, WU 04, W2LTV65R5E
Iscartrelvir is an investigational new drug developed by the Westlake University for the treatment of COVID-19. It targets the SARS-CoV-2 3CL protease, which is crucial for the replication of the virus responsible for COVID-19.[1][2]
Iscartrelvir is a small molecule drug. The usage of the INN stem ‘-trelvir’ in the name indicates that Iscartrelvir is a antiviral 3CL protease inhibitor. Iscartrelvir has a monoisotopic molecular weight of 525.1 Da.
PAT
WO2022150962A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022150962&_cid=P11-MJKTXT-76321-1

SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN331401594&_cid=P11-MJKTO7-65334-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024243841&_cid=P11-MJKTO7-65334-1
N-((1S,2R)-2-((4-bromo-2-(methylcarbamoyl)-6-nitrophenyl)amino)cyclohexyl)isoquinoline-4-carboxamide, and its structure is as follows:

Example 1: Preparation of Compound 1
[0189]A free, amorphous compound 1, a yellow solid, was prepared according to the method disclosed in paragraphs [00121]-[00128] of WO2022150962A1, and was used in the following examples. The specific synthetic steps are shown in steps a to d:

The reagents and conditions for steps a to d are further described below: (a) 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), CH₂Cl₂
or
dichloromethane (DCM), 0°C, 2 h; (b) DIPEA, dimethylformamide (DMF), 80°C, 16 h; (c) 3M ethyl hydrochloride (HCl·EA), CH₂Cl₂ , 1
h ; (d ) HATU, DIPEA, DMF, room temperature, 12 h.
[0191]Step a: Synthesis of N-methyl-5-bromo-2-fluoro-3-nitrobenzamide (I-1)
[0192]A solution of 5-bromo-2-fluoro-3-nitrobenzoic acid (0.8 g, 3.80 mmol) in dichloromethane (20 mL) was stirred at 0 °C. Then, HATU (2.0 g, 5.25 mmol), DIPEA (1.88 mL, 11.4 mmol), and methylamine hydrochloride (0.31 g, 4.5 mmol) were added to the reaction mixture. The mixture was stirred at 0 °C for 2 hours until it became clear. The mixture was extracted three times with dichloromethane, and the combined organic layers were washed with a saturated brine solution. The organic phase was then dried over anhydrous Na₂SO₄ and concentrated
under vacuum. Finally, the mixture was purified by chromatography to give compound I-1 (0.8 g, 76% yield) as a yellow solid.
[0193]Step b: Synthesis of tert-butyl 2-((4-bromo-2-(methylcarbamoyl)-6-nitrophenyl)amino)cyclohexyl)carbamate (I-2)
[0194]A solution of compound I-1 (0.8 g, 2.9 mmol) in 15 mL of DMF was stirred at room temperature. Then, tert-butyl ((1S,2R)-2-aminocyclohexyl)carbamate (0.75 g, 3.5 mmol) (the corresponding stereoisomer of this reagent can be used to synthesize the stereoisomer of compound I-2) and DIPEA (1.44 mL, 8.7 mmol) were added to the reaction mixture. The mixture was heated to 80 °C and stirred for 16 hours. The mixture was extracted three times with ethyl acetate, and the combined organic layers were washed with saturated salt solution. The organic phase was then dried over anhydrous Na₂SO₄ and concentrated under vacuum to give compound
I -2 as a yellow solid, requiring no further purification.

Step c: Synthesis of 2-(2-aminocyclohexyl)amino)-5-bromo-N-methyl-3-nitrobenzamide hydrochloride (I-3)
[0196]A solution of compound I-2 (90 mg, 0.19 mmol) (or the corresponding stereoisomer) in anhydrous dichloromethane (6 mL) was stirred at room temperature. Then, HCl (4 mL, 3 M in ethyl acetate) was added. The mixture was stirred at room temperature for 2 hours. The mixture was concentrated under vacuum to give compound I-3 as a yellow solid, requiring no further purification.
[0197]Step d: Synthesis of N-((1S,2R)-2-((4-bromo-2-(methylcarbamoyl)-6-nitrophenyl)amino)cyclohexyl)isoquinoline-4-carboxamide
[0198]At room temperature, a solution of the corresponding isoquinoline-4-carboxylic acid (1 equivalent) and HATU (1.5 equivalent) in anhydrous DMF (6 mL) was stirred. Then, compound I-3 and DIPEA (5.0 equivalent) were added. The mixture was stirred overnight at room temperature. The mixture was extracted three times with ethyl acetate, and the combined organic layers were washed with saturated brine. The organic phase was then dried over anhydrous Na₂SO₄ and
concentrated under vacuum. Finally, the mixture was purified by chromatography to give compound 1 as a free amorphous solid in yellow form.
PAT
- Aromatic ring-containing pyridone amide compoundsPublication Number: CN-119100980-APriority Date: 2023-06-07
- Crystal of viral protease inhibitor and usePublication Number: WO-2024243841-A1Priority Date: 2023-05-31
- Protease inhibitors, their preparation and usePublication Number: CN-113072497-BPriority Date: 2021-01-12Grant Date: 2023-04-14
- Protease inhibitors, their preparation and usePublication Number: CN-113072497-APriority Date: 2021-01-12
- Protease inhibitors, their preparation and usePublication Number: CN-116751164-APriority Date: 2021-01-12
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | WPV01; WU-04 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2921711-74-0 |
| PubChem CID | 156774920 |
| ChemSpider | 129307041 |
| UNII | W2LTV65R5E |
| PDB ligand | J7R (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C24H24BrN5O4 |
| Molar mass | 526.391 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Yang L, Wang Z (September 2023). “Bench-to-bedside: Innovation of small molecule anti-SARS-CoV-2 drugs in China”. European Journal of Medicinal Chemistry. 257 115503. doi:10.1016/j.ejmech.2023.115503. PMC 10193775. PMID 37229831.
- Hou N, Shuai L, Zhang L, Xie X, Tang K, Zhu Y, et al. (February 2023). “Development of Highly Potent Noncovalent Inhibitors of SARS-CoV-2 3CLpro”. ACS Central Science. 9 (2): 217–227. doi:10.1021/acscentsci.2c01359. PMC 9885526. PMID 36844503.
- Resistance mechanisms of SARS-CoV-2 3CLpro to the non-covalent inhibitor WU-04Publication Name: Cell DiscoveryPublication Date: 2024-04-09PMCID: PMC11003996PMID: 38594245DOI: 10.1038/s41421-024-00673-0
- Identification of Ebselen derivatives as novel SARS-CoV-2 main protease inhibitors: Design, synthesis, biological evaluation, and structure-activity relationships explorationPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2023-12-15PMID: 37972434DOI: 10.1016/j.bmc.2023.117531
- The molecular mechanism of non-covalent inhibitor WU-04 targeting SARS-CoV-2 3CLpro and computational evaluation of its effectiveness against mainstream coronavirusesPublication Name: Physical chemistry chemical physics : PCCPPublication Date: 2023-09-13PMID: 37655706DOI: 10.1039/d3cp03828a
- Bench-to-bedside: Innovation of small molecule anti-SARS-CoV-2 drugs in ChinaPublication Name: European Journal of Medicinal ChemistryPublication Date: 2023-09-05PMCID: PMC10193775PMID: 37229831DOI: 10.1016/j.ejmech.2023.115503
- Development of Highly Potent Noncovalent Inhibitors of SARS-CoV-2 3CLproPublication Name: ACS Central SciencePublication Date: 2023-01-25PMCID: PMC9885526PMID: 36844503DOI: 10.1021/acscentsci.2c01359
////////iscartrelvir, antiviral, WU-04, WU 04, W2LTV65R5E
Odentegravir
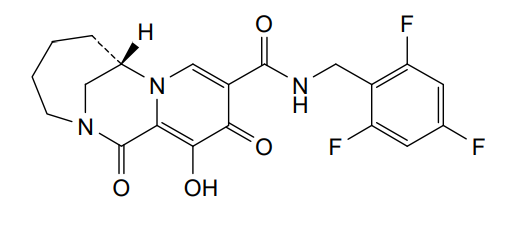

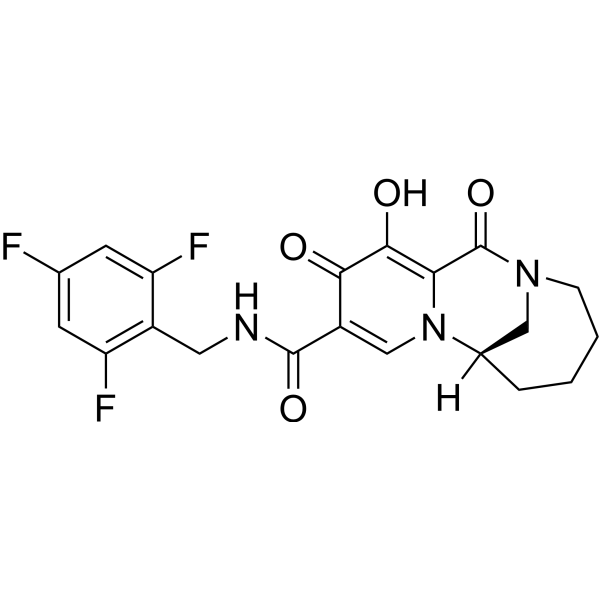
Odentegravir
CAS 2495436-99-0
MF C20H18F3N3O4 MW421.4 g/mol
(7S)-12-hydroxy-1,11-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-1,4,5,6,7,11-hexahydro-3H-2,7-
methanopyrido [1,2-a][1,4]diazonine-10-carboxamide
(7S)-1,4,5,6,7,11-HEXAHYDRO-12-HYDROXY-1,11-DIOXO-N-((2,4,6-TRIFLUOROPHENYL)METHYL)-3H-2,7-METHANOPYRIDO(1,2-A)(1,4)DIAZONINE-10-CARBOXAMIDE
(7S)-12-HYDROXY-1,11-DIOXO-N-((2,4,6-TRIFLUOROPHENYL)METHYL)-1,4,5,6,7,11-HEXAHYDRO-3H-2,7-METHANOPYRIDO(1,2-A)(1,4)DIAZONINE-10-CARBOXAMIDE
3H-2,7-METHANOPYRIDO(1,2-A)(1,4)DIAZONINE-10-CARBOXAMIDE, 1,4,5,6,7,11-HEXAHYDRO-12-HYDROXY-1,11-DIOXO-N-((2,4,6-TRIFLUOROPHENYL)METHYL)-, (7S)-
antiviral, H8B26JZ4A4, orb2664247
Odentegravir is a small molecule drug classified as a
HIV integrase inhibitor, indicated by the “-tegravir” stem in its name. It is a chemical compound with the molecular formula
has been used in research for its antiviral properties.
- Drug Class: HIV integrase inhibitor
- Chemical Formula:
C20H18F3N3O4cap C sub 20 cap H sub 18 cap F sub 3 cap N sub 3 cap O sub 4𝐶20𝐻18𝐹3𝑁3𝑂4
- Molecular Weight:
421.12421.12421.12 Da (monoisotopic)
- Classification: Small molecule drug
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020197991&_cid=P12-MHY8KB-06018-1
Example 23: Preparation of racemic-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26), (7R)-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-
methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26-1) and (7S)-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26-2):

Synthesis of 12-Hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26):
[0335] 12-(Benzyloxy)-1,11-dioxo-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxylic acid (57 mg, 0.155 mmol) was dissolved in DCM (2 mL) with (2,4,6-trifluorophenyl)methanamine (27 mg, 0.17 mmol) and triethylamine (60 mg, 0.464 mmol). HATU (60 mg, 0.186 mmol) was added and the mixture was stirred at room
temperature. After overnight reaction, the reaction was concentrated to dryness, purified by silicon gel chromatography to obtain compound 12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a) MS (m/z) 512.06 [M+H]+.
[0336] Compound 12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a) (7 mg, 0.014 mmol) was dissloved in Tolune (1 mL), then followed by the addition of TFA (1 mL). The resulting mixture was stirred at rt for overnight. The solvent was removed under vacuo an the residue was purifed by HPLC to obtain the title compound (26). MS (m/z) 422.091 [M+H]+.1H NMR (400 MHz, DMSO-d6) d 10.39 (t, J = 5.8 Hz, 1H), 8.45 (s, 1H), 7.24 – 7.11 (m, 2H), 4.72 (dd, J = 5.9, 2.9 Hz, 1H), 4.54 (dd, J = 6.0, 2.4 Hz, 2H), 4.11 (d, J = 13.3 Hz, 1H), 3.88 – 3.79 (m, 1H), 3.64 (dd, J = 14.7, 1.9 Hz, 1H), 3.05 (dq, J = 9.5, 3.4 Hz, 1H), 2.06 – 1.91 (m, 1H), 1.89 – 1.74 (m, 3H), 1.61 (d, J = 7.7 Hz, 1H), 1.11 (d, J = 12.7 Hz, 1H).
Synthesis of (7S)-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26-2) and (7R)-12-hydroxy-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26-1):
[0337] Racemic 12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a) was separated by chiral HPLC separation (SFC chromatography on an IB 4.6X100mm 5mic column using MeOH(20) as co-solvent) to obtain compounds (7R)-12-(Benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a-1) and (7S)-12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a-2)
[0338] Compound (7S)-12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a-2) (20 mg, 0.039 mmol) was dissloved in Tolune (1 mL), then followed by the addition of TFA (1 mL). The resulting mixture was stireed at rt for overnight. The solvent was removed under vacuo an the residue was purifed by HPLC to obtain the title compound (26-2). (MS (m/z) 422.123 [M+H]+. 1H NMR (400 MHz, DMSO-d6) d 10.59 (s, 1H), 10.39 (d, J = 5.9 Hz, 1H), 8.45 (s, 1H), 7.18 (t, J = 8.6 Hz, 2H), 4.72 (s, 1H), 4.59 – 4.48 (m, 2H), 4.11 (d, J = 13.2 Hz, 1H), 3.85 (d, J = 14.6 Hz, 1H), 3.69 – 3.59 (m, 1H), 3.05 (ddd, J = 11.3, 6.7, 3.6 Hz, 1H), 1.97 (m, 1H), 1.87 – 1.71 (m, 3H), 1.67 – 1.55 (m, 1H), 1.10 (m, 1H).
[0339] Compound (7R)-12-(benzyloxy)-1,11-dioxo-N-(2,4,6-trifluorobenzyl)-1,4,5,6,7,11-hexahydro-3H-2,7-methanopyrido[1,2-a][1,4]diazonine-10-carboxamide (26a-1) ((20 mg, 0.039 mmol) was dissloved in Tolune (1 mL), then followed by the addition of TFA (1 mL). The resulting mixture was stireed at rt for overnight. The solvent was removed under vacuo an the residue was purifed by HPLC to obtain the title compound (26-1). MS (m/z) 422.116 [M+H]+. 1H NMR (400 MHz, DMSO-d6) d 10.58 (s, 1H), 10.39 (t, J = 5.8 Hz, 1H), 8.45 (s, 1H), 7.18 (dd, J = 9.2, 8.0 Hz, 2H), 4.73 (s, 1H), 4.58 – 4.49 (m, 2H), 4.11 (d, J = 13.3 Hz, 1H), 3.85 (d, J = 14.6 Hz, 1H), 3.65 (d, J = 14.2 Hz, 1H), 3.10 – 3.00 (m, 1H), 1.96 (m, 1H), 1.82 (d, J = 12.2 Hz, 3H), 1.61 (d, J = 7.4 Hz, 1H), 1.18 – 1.05 (m, 1H).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023196875&_cid=P12-MHY8FJ-02517-1
PAT
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usesPublication Number: JP-2025013503-APriority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usesPublication Number: KR-102714084-B1Priority Date: 2019-03-22Grant Date: 2024-10-08
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: KR-20240151256-APriority Date: 2019-03-22
- Bridged Tricyclic Carbamoylpyridone Compounds and Their Pharmaceutical UsePublication Number: ES-2927041-T3Priority Date: 2019-03-22Grant Date: 2022-11-03
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-11548902-B1Priority Date: 2019-03-22Grant Date: 2023-01-10
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-2023027019-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: AU-2020245350-B2Priority Date: 2019-03-22Grant Date: 2023-04-20
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-2023203061-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-11084832-B2Priority Date: 2019-03-22Grant Date: 2021-08-10
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: AU-2020245350-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: EP-3938047-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: EP-3938047-B1Priority Date: 2019-03-22Grant Date: 2022-06-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: EP-4122537-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and uses thereofPublication Number: US-2023339971-A1Priority Date: 2022-04-06
- Bridged tricyclic carbamoylpyridone compounds and uses thereofPublication Number: US-2023339972-A1Priority Date: 2022-04-06
- Bridged tricyclic carbamoylpyridone compounds and uses thereofPublication Number: WO-2023196875-A1Priority Date: 2022-04-06
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: US-2020317689-A1Priority Date: 2019-03-22
- Bridged tricyclic carbamoylpyridone compounds and their pharmaceutical usePublication Number: WO-2020197991-A1Priority Date: 2019-03-22
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////Odentegravir, antiviral, H8B26JZ4A4, orb2664247
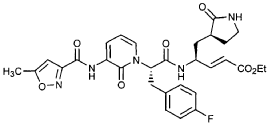
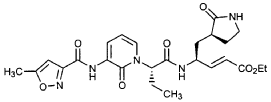
Imocitrelvir
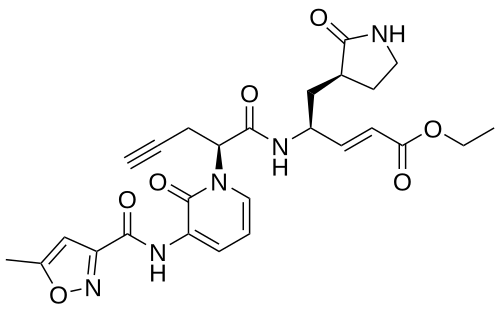
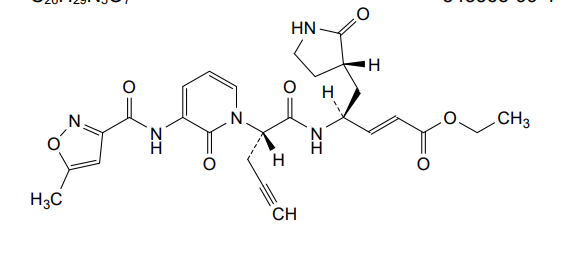
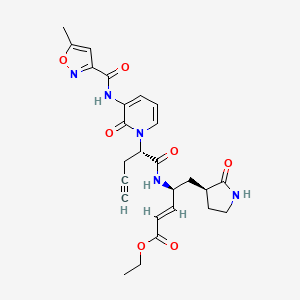
Imocitrelvir
CAS 343565-99-1
MFC26H29N5O7 MW523.5 g/mol
ethyl (2E,4S)-4-{(2S)-2-[3-(5-methyl-1,2-oxazole-3-carboxamido)-2-oxopyridin-1(2H)-yl]pent-4-ynamido}-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
ethyl (E,4S)-4-[[(2S)-2-[3-[(5-methyl-1,2-oxazole-3-carbonyl)amino]-2-oxo-1-pyridinyl]pent-4-ynoyl]amino]-5-[(3S)-2-oxopyrrolidin-3-yl]pent-2-enoate
protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
Imocitrelvir is an investigational new drug that is being evaluated for the treatment of viral infections. It is a 3C protease inhibitor in picornaviruses. Originally developed by Pfizer for treating human rhinovirus infections,[1] this small molecule has shown promise against a broader range of viruses, including polioviruses.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2003-09-17
PMID: 14521419
DOI: 10.1021/jm030166l
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016044656&_cid=P21-MHBDH2-20719-1
PAT
PAT
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001040189&_cid=P21-MHBDI9-21481-1
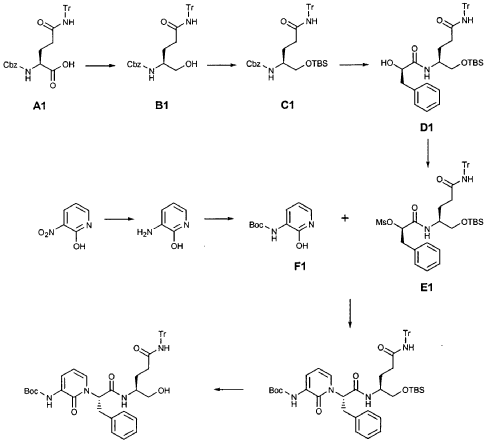
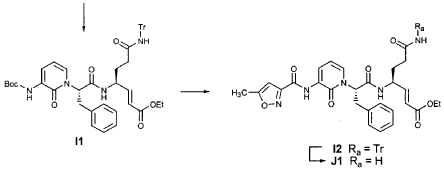
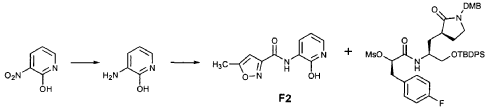
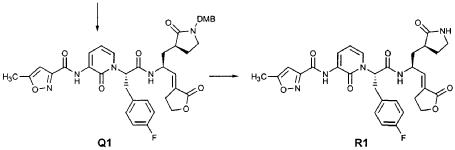
EXAMPLE 21
Preparation of Compound 22: tra«5-(4S,3″”S)-4-(2′-{3″-[(5′”-Methylisoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin- 1 “-yl} acetylamino)-5-(2″”-oxopyrrilidin-3″”-yl)pent-2-enoic Acid Ethyl Ester
Preparation of Intermediate {3-[(5′-Methylisoxazole-3′-carbonyl)amino]-2-oxo-2H-pyridin-l-yl} acetic Acid tert-Butyl Ester
To a solution of 5-methylisoxazole-3-carboxylic acid (2′-hydroxy-4′-methylpyridin-3′-yl)amide (F2, Example 19) (0.520 g, 2.37 mmol, 1 equiv) in TΗF (20 mL) at 0 °C was added NaΗ (0.095 g, 2.37 mmol, 1.0 equiv). The resulting mixture was stirred at 0 °C for 20 min, and then t-butyl bromoacetate (0.385 mL, 2.61 mmol, 1.1 equiv) was added. The reaction mixture was stirred and warmed to room temperature for 30 min, then was partitioned between 0.5 N ΗC1 (100 mL) and EtOAc (2 x 100 mL). The combined organic layers were dried over Na2SO and were concentrated. Purification of the residue by flash column chromatography (30% EtOAc in hexanes) provided the title intermediate (0.628 g, 79%) as a white solid: IR (cm-1) 3343, 1743, 1651, 1581, 1156; Η NMR (CDC13) δ 1.52 (s, 9H), 2.53 (s, 3H), 4.65 (s, 2H), 6.32 (t, 1H, 7= 7.2), 6.51 (s, IH), 7.01 (dd, 1H, 7= 6.9, 1.8), 8.50 (dd, 1H, 7= 7.5, 1.8), 9.63 (s, br. IH); Anal. C16H19N3O5: C, H, N.
Preparation of Compound 22
The preceding intermediate was transformed into Compound 22 by a process that was analogous to that described in Example 25 for the transformation of V3 to product R3: mp = 102-106 °C; IR (cm”1) 3336, 1684, 1534, 1457; JH NMR (CDCI3) δ 1.27 (t, 3H, 7= 7.2), 1.67-1.75 (m, IH), 1.98-2.09 (m, IH), 2.37-2.49 (m, IH), 2.53 (s, 3H), 2.55-2.61 (m, IH), 3.34-3.46 (m, 2H), 3.51-3.52 (m, IH), 4.17 (q, 2H, 7= 7.2), 4.61-4.78 (m, 3H), 5.98 (dd, IH, 7 = 15.6, 1.5), 6.20 (s, br. IH), 6.35 (t, 1H, 7= 7.8), 6.51 (s, IH), 6.85 (dd, IH, 7= 15.6, 5.1), 7.17 (d, IH, 7= 7.2), 8.33 (d, IH, 7= 7.2), 8.49 (d, IH, 7= 7.5), 9.57 (s, br. IH); Anal.
C23H27N5O7: C, H, N.
EXAMPLE 24
Preparation of Compound 25: trans-(2’S,3″”‘S,4S)-4-(3,-(4″-Fluorophenyl)-2′-{3″‘-[(5″”-methylisoxazole-3″”-carbonyl)amino]-2′”-oxo-2′”H-pyridin- “-yl}propionylamino)-5-(2″ oxopyrrolidin-3′””-yl)pent-2-enoic Acid Ethyl Ester
The title compound was prepared from F2 (Example 19) in a manner analogous to that described for the conversion of U2 to 13 in Example 23 utilizing intermediate Y2 (Example 25) where appropriate: IR (cm-1) 3331, 1690, 1590, 1531, 1455; !H NMR (CDCI3) δ 1.30 (t, 3H, 7= 7.0), 1.45-1.55 (m, IH), 1.64-1.75 (m, IH), 2.03-2.31 (m, 3H), 2.49 (s, 3H), 3.10 (dd, IH, 7= 13.7, 7.9), 3.20-3.46 (m, 3H), 4.20 (q, 2H, 7= 7.0), 4.36-4.47 (m, IH), 5.67 (dd, IH, 7 = 15.7, 1.4), 5.85-5.92 (m, IH), 6.29 (t, 1H, 7= 7.2), 6.45 (s, IH), 6.70 (dd, IH, 7= 15.7, 5.7), 6.86 (s, IH), 6.90-6.97 (m, 2H), 7.10-7.16 (m, 2H), 7.60 (dd, IH, 7= 7.2, 1.6), 8.37 (dd, IH, 7 = 7.2, 1.6), 8.51 (d, IH, 7= 6.6), 9.47 (s, IH).
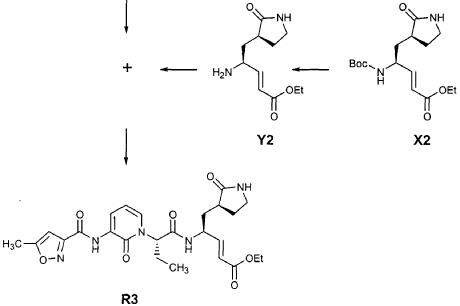
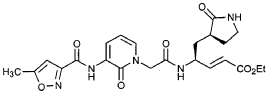
EXAMPLE 25
Preparation of Compound 26: tr_.«5-(2’S,3″”S,4S)-4-(2′-{3″-[(5″‘-Methyl-isoxazole-3′”-carbonyl)amino]-2″-oxo-2″H-pyridin-l”-yl}butyrylamino)-5-(2″”-oxopyrrolidin-3″”-yl)pent-2-enoic Acid Ethyl Ester (R3)
Preparation of Intermediate (2R)-2-Trifluoromethanesulfonyl-oxybutyric acid tert-butyl ester (U3)
Commercially available T3 (0.575 g, 3.59 mmol, 1 equiv) was dissolved in CH2CI2 (25 mL) and cooled in an ice bath. 2,6-Lutidine (0.836 mL, 7.18 mmol, 2 equiv) and trifluoromethanesulfonic anhydride (1.15 mL, 6.84 mmol, 1.9 equiv) were added and the reaction mixture was stirred 30 min. It was then diluted with MTBE (400 mL), washed with a mixture of brine and 1 N HCl (2:1, 100 mL) and brine (100 mL), dried over Na2SO4 and evaporated to provide the title intermediate which was used without further purification.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyri din- l’-yl} butyric Acid tert-Butyl Ester (V3)
Intermediate F2 from above (0.200 g, 0.912 mmol, 1.1 equiv) was suspended in TΗF (6 mL). Sodium hydride (60% dispersion in mineral oil, 0.0332 g, 0.830 mmol, 1 equiv) was added in one portion. After stirring 30 min, a solution of intermediate U3 (0.830 mmol, 1 equiv, based on T3) in TΗF (7 mL) was added dropwise. The resulting mixture was stirred 2 hours, then diluted with EtOAc (200 mL) and washed with brine (2 x 50 mL). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (25% EtOAc in hexanes) to provide the title intermediate (0.178 g, 59%) as an oil: R/= 0.30 (25% EtOAc in hexanes); IR (cm”1) 3331, 1731, 1690, 1649, 1602, 1531 ; *Η NMR (CDCI3) δ 0.93 (t, 3H, 7= 7.3), 1.45 (s, 9H), 1.83-2.01 (m, IH), 2.17-2.31 (m, IH), 2.50 (s, 3H), 5.44-5.51 (m, IH), 6.32 (t, IH, 7= 7.2), 6.48 (s, IH), 7.10 (dd, IH, 7= 7.2, 1.8), 8.45 (dd, 1H, 7= 7.2, 1.8), 9.64 (s, IH); Anal. C18H23N3O5: C, H, N.
Preparation of Intermediate (2S)-2- {3′-[(5″-Methylisoxazole-3″-carbonyl)amino]-2′-oxo-2’H-pyridin-l’-yl}butyric Acid (W3)
Intermediate V3 from above (0.143 g, 0.397 mmol, 1 equiv) was stirred for 1 h in a solution of TFA (2 mL) in CΗ2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Intermediate trα«5-(3’S,4S)-4-Amino-5-(2′-oxopyrrolidin-3′-yl)pent-2-enoic Acid Ethyl Ester (Y2)
Intermediate X2, prepared according to the method disclosed in the co-pending application, U.S. Provisional Patent Application No. 60/150,358, filed August 24, 1999(0.130 g, 0.398 mmol, 1 equiv), was stirred for 30 min in a solution of TFA (2 mL) in CH2CI2 (3 mL). The volatiles were evaporated. The residue was suspended in toluene (10 mL) and concentrated to dryness, providing the title intermediate which was used without further purification.
Preparation of Product R3 (Compound 26)
Intermediates W3 and Y2 (as prepared above) were combined in CH2CI2 (7 mL) and cooled in an ice bath. HOBt (0.064 g, 0.47 mmol, 1.2 equiv), iP^NEt (0.484 mL, 2.78 mmol, 7 equiv) and EDC (0.084 g, 0.44 mmol, 1.1 equiv) were added sequentially. The reaction mixture was allowed to warm to 23 °C overnight, then diluted with EtOAc (500 mL) and washed with 5% KHSO4 , half saturated NaHCO3, and brine (100 mL each). The organic phase was dried over MgSO4 and evaporated. The residue was purified by flash column chromatography (gradient elution, 2→3% CH3OH in CH2CI2) to provide the title intermediate (0.119 g, 58%) as a white foam: IR (cm”1) 3331, 1684, 1649, 1590, 1531; JH NMR (CDCI3) δ 0.92 (t, 3H, J = 7.3), 1.29 (t, 3H, J = 7.1), 1.47-1.58 (m, IH), 1.62-1.77 (m, IH), 1.85-2.00 (m, IH), 2.08-2.33 (m, 4H), 2.49 (s, 3H), 3.25-3.42 (m, 2H), 4.19 (q, 2H, J = 7.1), 4.39-4.50 (m, IH), 5.73 (dd, IH, J = 8.8, 6.8), 5.97 (dd, IH, J = 15.7, 1.4), 6.34 (t, IH, J = 7.2), 6.46 (s, IH), 6.86 (dd, IH, J = 15.7, 5.9), 7.18 (s, IH), 7.59 (dd, IH, J = 7.2, 1.8), 8.42 (dd, IH, J = 7.2, 1.8), 8.58-8.62 (m, IH), 9.56 (s, 1); Anal. C25H31N5O7O.5OH2O: C, H, N.
PAT
- Treatment of infection by human enterovirus d68Publication Number: US-2020016243-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: WO-2016044656-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus d68Publication Number: US-2021052708-A1Priority Date: 2014-09-17
- Treatment of infection by human enterovirus D68Publication Number: US-11191817-B2Priority Date: 2014-09-17Grant Date: 2021-12-07
- Therapeutic compounds and methodsPublication Number: US-2025051283-A1
- Protease Inhibitors for Treatment or Prevention of Coronavirus DiseasePublication Number: US-2023192660-A1Priority Date: 2020-05-08
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-2019030027-A1Priority Date: 2016-01-29
- Composition and combined medication method for treating enterovirus infectionPublication Number: US-10864210-B2Priority Date: 2016-01-29Grant Date: 2020-12-15
- Treatment of infection by human enterovirus D68Publication Number: US-10328128-B2Priority Date: 2014-09-17Grant Date: 2019-06-25
- Treatment of infection by human enterovirus d68Publication Number: US-2017290893-A1Priority Date: 2014-09-17
- Nucleotide and nucleoside therapeutic compositions, combinations and related uses thereofPublication Number: CN-117881402-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: EP-4333859-A1Priority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations, and related usesPublication Number: JP-2024517807-APriority Date: 2021-05-05
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: WO-2022235874-A1Priority Date: 2021-05-05
- Protease inhibitors for treatment or prevention of coronavirus diseasePublication Number: EP-4146267-A1Priority Date: 2020-05-08
- 4′-substituted nucleosides and nucleotides as antiviral agentsPublication Number: WO-2024227159-A2Priority Date: 2023-04-28
- Therapeutic compoundsPublication Number: WO-2024206284-A2Priority Date: 2023-03-27
- Antibody molecules binding to sars-cov-2Publication Number: WO-2024168061-A2Priority Date: 2023-02-07
- Predictive model for variants associated with drug resistance and theranostic applications thereofPublication Number: WO-2023172635-A1Priority Date: 2022-03-08
- Nucleotide and nucleoside therapeutic compositions, combinations and uses related theretoPublication Number: CA-3216679-A1Priority Date: 2021-05-05
LIT
- Structure and inhibition of SARS-CoV-1 and SARS-CoV-2 main proteases by oral antiviral compound AG7404Publication Name: Antiviral ResearchPublication Date: 2022-12PMCID: PMC9632241PMID: 36336176DOI: 10.1016/j.antiviral.2022.105458
- Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug DesignPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-09-30PMID: 34591488DOI: 10.1021/acs.jmedchem.1c01215
- In Vitro Antiviral Activity of New Oxazoline Derivatives as Potent Poliovirus InhibitorsPublication Name: Journal of Medicinal ChemistryPublication Date: 2018-12-04PMCID: PMC9169555PMID: 30512950DOI: 10.1021/acs.jmedchem.8b01482
- A Novel Series of Highly Potent Small Molecule Inhibitors of Rhinovirus ReplicationPublication Name: Journal of Medicinal ChemistryPublication Date: 2017-06-15PMID: 28581749DOI: 10.1021/acs.jmedchem.7b00175
- Anti-poliovirus activity of protease inhibitor AG-7404, and assessment of in vitro activity in combination with antiviral capsid inhibitor compoundsPublication Name: Antiviral ResearchPublication Date: 2013-05PMID: 23499651DOI: 10.1016/j.antiviral.2013.03.003

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | AG-7404, V-7404 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 343565-99-1 |
| PubChem CID | 5280053 |
| IUPHAR/BPS | 13223 |
| UNII | VQ1AN3OO42 |
| ChEMBL | ChEMBL141157 |
| Chemical and physical data | |
| Formula | C26H29N5O7 |
| Molar mass | 523.546 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “Imocitrelvir”. PatSnap.
- Xie H, Rhoden EE, Liu HM, Ogunsemowo F, Mainou BA, Burke RM, et al. (November 2024). “Antiviral Development for the Polio Endgame: Current Progress and Future Directions”. Pathogens. 13 (11). Basel, Switzerland: 969. doi:10.3390/pathogens13110969. PMC 11597170. PMID 39599522.
- Bandyopadhyay AS, Burke RM, Hawes KM (June 2024). “Polio Eradication: Status, Struggles and Strategies”. The Pediatric Infectious Disease Journal. 43 (6): e207-211. doi:10.1097/INF.0000000000004330. PMID 38564755.
////////Imocitrelvir, protease inhibitor, antiviral, AG-7404, V-7404, AG 7404, V 7404, VQ1AN3OO42
Nirsevimab
(Heavy chain)
QVQLVQSGAE VKKPGSSVMV SCQASGGLLE DYIINWVRQA PGQGPEWMGG IIPVLGTVHY
GPKFQGRVTI TADESTDTAY MELSSLRSED TAMYYCATET ALVVSETYLP HYFDNWGQGT
LVTVSSASTK GPSVFPLAPS SKSTSGGTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP
AVLQSSGLYS LSSVVTVPSS SLGTQTYICN VNHKPSNTKV DKRVEPKSCD KTHTCPPCPA
PELLGGPSVF LFPPKPKDTL YITREPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP
REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL
PPSREEMTKN QVSLTCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT
VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK
(Light chain)
DIQMTQSPSS LSAAVGDRVT ITCQASQDIV NYLNWYQQKP GKAPKLLIYV ASNLETGVPS
RFSGSGSGTD FSLTISSLQP EDVATYYCQQ YDNLPLTFGG GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H153-H209, H229-L214, H235-H’235, H238-H’238, H270-H330, H376-H434, H’22-H’96, H’153-H’209, H’229-L’214, H’270-H’330, H’376-H’434, L23-L88, L’23-L’88, L134-L194, L’134-L’194)
>Heavy_chain QVQLVQSGAEVKKPGSSVMVSCQASGGLLEDYIINWVRQAPGQGPEWMGGIIPVLGTVHY GPKFQGRVTITADESTDTAYMELSSLRSEDTAMYYCATETALVVSETYLPHYFDNWGQGT LVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFP AVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPA PELLGGPSVFLFPPKPKDTLYITREPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKP REEQYNSTYRVVSVLTVLHQDWLEGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTL PPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLT VDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
>Light_chain DIQMTQSPSSLSAAVGDRVTITCQASQDIVNYLNWYQQKPGKAPKLLIYVASNLETGVPS RFSGSGSGTDFSLTISSLQPEDVATYYCQQYDNLPLTFGGGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
Nirsevimab
EMS APPROVED 2022/10/31, Beyfortus, AstraZeneca AB
| Formula | C6494H10060N1708O2050S46 |
|---|---|
| CAS | 1989556-22-0 |
| Mol weight | 146334.5658 |
Monoclonal antibody
Prevention of respiratory syncytial virus infection
- Immunoglobulin g1-kappa, anti-(human respiratory syncytial virus fusion glycoprotein f0 (protein f))human monoclonal antibody.gamma.1 heavy chain (1-456) (human vh (homo sapiens ighv1-69*01(ighd)-ighj4*01 (90.1%)) (8.8.19) (1-126) -homo sapiens ighg1*03
- Immunoglobulin g1, anti-(human respiratory syncytial virus fusion protein)(human monoclonal med18897 .gamma.1-chain), disulfide with monoclonal med18897 .kappa.-chain, dimer
Synthesis Reference
Khan, AA et al. (2020) Dosage regimens for and compositions including anti-rsv antibodies. (U.S. Patent No. 2020/0347120 A1). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/6b/d2/10/a841b66e0c90cf/US20200347120A1.pdf
Nirsevimab, sold under the brand name Beyfortus, is a human recombinant monoclonal antibody with activity against respiratory syncytial virus, or RSV for infants.[2][3] It is under development by AstraZeneca and Sanofi.[2][3] Nirsevimab is designed to bind to the fusion protein on the surface of the RSV virus.[4][5]
The most common side effects reported for nirsevimab are rash, pyrexia (fever) and injection site reactions (such as redness, swelling and pain where the injection is given).[6]
Nirsevimab was approved for medical use in the European Union in November 2022.[1][7]
Nirsevimab (MEDI8897) is a recombinant human immunoglobulin G1 kappa (IgG1ĸ) monoclonal antibody used to prevent respiratory syncytial virus (RSV) lower respiratory tract disease in neonates and infants.6 It binds to the prefusion conformation of the RSV F protein, a glycoprotein involved in the membrane fusion step of the viral entry process, and neutralizes several RSV A and B strains.6,1 Compared to palivizumab, another anti-RSV antibody, nirsevimab shows greater potency at reducing pulmonary viral loads in animal models. In addition, nirsevimab was developed as a single-dose treatment for all infants experiencing their first RSV season, whereas palivizumab requires five monthly doses to cover an RSV season.5 This is due to a modification in the Fc region of nirsevimab that grants it a longer half-time compared to typical monoclonal antibodies.1,6
On November 2022, nirsevimab was approved by the EMA for the prevention of RSV lower respiratory tract disease in newborns and infants during their first RSV season.6
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | F protein of RSV |
| Clinical data | |
| Trade names | Beyfortus |
| Other names | MED-18897, MEDI8897 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1989556-22-0 |
| PubChem SID | 384585358 |
| DrugBank | DB16258 |
| UNII | VRN8S9CW5V |
| KEGG | D11380 |
| ChEMBL | ChEMBL4297575 |
| Chemical and physical data | |
| Formula | C6494H10060N1708O2050S46 |
| Molar mass | 146336.58 g·mol−1 |
Adverse effects
No major hypersensitivity reactions have been reported, and adverse events of grade 3 or higher were only reported in 8% (77 of 968) of participants in clinical trial NCT02878330.[8][4]
Pharmacology
Mechanism of action
Nirsevimab binds to the prefusion conformation of the RSV fusion protein, i.e. it binds to the site at which the virus would attach to a cell; effectively rendering it useless. It has a modified Fc region, extending the half-life of the drug in order for it to last the whole RSV season.[4]
History
The opinion by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) is based on data from two randomized, double-blind, placebo-controlled multicenter clinical trials that investigated the efficacy and safety of nirsevimab in healthy preterm (premature) and full-term infants entering their first respiratory syncytial virus (RSV) season.[6] These studies demonstrated that nirsevimab prevents lower respiratory tract infection caused by RSV requiring medical attention (such as bronchiolitis and pneumonia) in term and preterm infants during their first RSV season.[6]
The safety of nirsevimab was also evaluated in a phase II/III, randomized, double‑blind, multicenter trial in infants who were born five or more weeks prematurely (less than 35 weeks gestation) at higher risk for severe RSV disease and infants with chronic lung disease of prematurity (i.e. long-term respiratory problems faced by babies born prematurely) or congenital heart disease.[6] The results of this study showed that nirsevimab had a similar safety profile compared to palivizumab (Synagis).[6]
Society and culture
Legal status
On 15 September 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Beyfortus, intended for the prevention of respiratory syncytial virus (RSV) lower respiratory tract disease in newborns and infants.[9][6] Beyfortus was reviewed under EMA’s accelerated assessment program.[9] The applicant for this medicinal product is AstraZeneca AB.[9] Nirsevimab was approved for medical use in the European Union in November 2022.[1][7]
Research
Nirsevimab is being investigated as an experimental vaccine against respiratory syncytial virus, RSV, in the general infant population.[2][3] The MELODY study is an ongoing, randomized, double-blind, placebo-controlled to evaluate the safety and efficacy of nirsevimab in late preterm and term infants. Initial results have been promising, with nirsevimab reducing LRTI (lower respiratory tract infections) by 74.5% compared to placebo in infants born at term or late preterm.[5][10][11]
Ongoing trials for nirsevimab are:
- “Evaluate the Safety and Efficacy of Nirsevimab in Healthy Preterm and Term Infants in China (CHIMES)”.
- “A Study to Evaluate the Safety and Efficacy of MEDI8897 for the Prevention of Medically Attended Lower Respiratory Tract Infection Due to Respiratory Syncytial Virus in Healthy Late Preterm and Term Infants (MELODY)”.
- “Evaluate the Safety and Tolerability, for Nirsevimab in Immunocompromised Children (MUSIC)”.
References
- ^ Jump up to:a b c “Beyfortus”. Union Register of medicinal products. 3 November 2022. Retrieved 6 November 2022.
- ^ Jump up to:a b c “Nirsevimab demonstrated protection against respiratory syncytial virus disease in healthy infants in Phase 3 trial” (Press release). Sanofi. 26 April 2021. Archived from the original on 27 December 2021. Retrieved 27 December 2021.
- ^ Jump up to:a b c “Nirsevimab MELODY Phase III trial met primary endpoint of reducing RSV lower respiratory tract infections in healthy infants” (Press release). AstraZeneca. 26 April 2021. Archived from the original on 26 December 2021. Retrieved 27 December 2021.
- ^ Jump up to:a b c Griffin MP, Yuan Y, Takas T, Domachowske JB, Madhi SA, Manzoni P, et al. (Nirsevimab Study Group) (July 2020). “Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants”. The New England Journal of Medicine. 383 (5): 415–425. doi:10.1056/NEJMoa1913556. PMID 32726528. S2CID 220876651.
- ^ Jump up to:a b Hammitt LL, Dagan R, Yuan Y, Baca Cots M, Bosheva M, Madhi SA, et al. (March 2022). “Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants”. The New England Journal of Medicine. 386 (9): 837–846. doi:10.1056/NEJMoa2110275. PMID 35235726. S2CID 247220023.
- ^ Jump up to:a b c d e f “New medicine to protect babies and infants from respiratory syncytial virus (RSV) infection”. European Medicines Agency (EMA) (Press release). 16 September 2022. Archived from the original on 19 September 2022. Retrieved 18 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Beyfortus approved in the EU for the prevention of RSV lower respiratory tract disease in infants”. AstraZeneca (Press release). 4 November 2022. Retrieved 6 November 2022.
- ^ Clinical trial number NCT02878330 at ClinicalTrials.gov
- ^ Jump up to:a b c “Beyfortus: Pending EC decision”. European Medicines Agency (EMA). 15 September 2022. Archived from the original on 19 September 2022. Retrieved 18 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Zacks Equity Research (25 March 2022). “Pfizer’s (PFE) RSV Jab Gets Another Breakthrough Therapy Tag”. Nasdaq. Archived from the original on 8 April 2022. Retrieved 8 April 2022.
- ^ “Nirsevimab significantly protected infants against RSV disease in Phase III MELODY trial”. AstraZeneca (Press release). 3 March 2022. Retrieved 6 November 2022.
////////////Nirsevimab, EU 2022, APPROVALS 2022, PEPTIDE, Monoclonal antibody, respiratory syncytial virus infection, ANTIVIRAL, 1989556-22-0, MED-18897, MEDI8897, AstraZeneca AB

NEW DRUG APPROVALS
ONE TIME
$10.00
Ropeginterferon alfa-2b
PCDLPQTHSL GSRRTLMLLA QMRRISLFSC LKDRHDFGFP QEEFGNQFQK AETIPVLHEM
IQQIFNLFST KDSSAAWDET LLDKFYTELY QQLNDLEACV IQGVGVTETP LMKEDSILAV
RKYFQRITLY LKEKKYSPCA WEVVRAEIMR SFSLSTNLQE SLRSKE
(Disulfide bridge: 2-99, 30-139)
Ropeginterferon alfa-2b
- AOP2014
CAS 1335098-50-4
UNII981TME683S
FDA APPROVED, 2021/11/12, BESREMI
PEPTIDE, Antineoplastic, Antiviral
Polycythemia vera (PV) is the most common Philadelphia chromosome-negative myeloproliferative neoplasm (MPN), characterized by increased hematocrit and platelet/leukocyte counts, an increased risk for hemorrhage and thromboembolic events, and a long-term propensity for myelofibrosis and leukemia.1,2 Interferon alfa-2b has been used for decades to treat PV but requires frequent dosing and is not tolerated by all patients.2 Ropeginterferon alfa-2b is a next-generation mono-pegylated type I interferon produced from proline-IFN-α-2b in Escherichia coli that has high tolerability and a long half-life.4,6 Ropeginterferon alfa-2b has shown efficacy in PV in in vitro and in vivo models and clinical trials.3,4
Ropeginterferon alfa-2b was approved by the FDA on November 12, 2021, and is currently marketed under the trademark BESREMi by PharmaEssentia Corporation.6
Ropeginterferon alfa-2b, sold under the brand name Besremi, is a medication used to treat polycythemia vera.[1][2][3][4] It is an interferon.[1][3] It is given by injection.[1][3]
The most common side effects include low levels of white blood cells and platelets (blood components that help the blood to clot), muscle and joint pain, tiredness, flu-like symptoms and increased blood levels of gamma-glutamyl transferase (a sign of liver problems).[3] Ropeginterferon alfa-2b can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness.[2] Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).[2]
It was approved for medical use in the European Union in February 2019,[3] and in the United States in November 2021.[2][5] Ropeginterferon alfa-2b is the first medication approved by the U.S. Food and Drug Administration (FDA) to treat polycythemia vera that people can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.[2]
https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-rare-blood-disease#:~:text=FDA%20NEWS%20RELEASE-,FDA%20Approves%20Treatment%20for%20Rare%20Blood%20Disease,FDA%2DApproved%20Option%20Patients%20Can%20Take%20Regardless%20of%20Previous%20Therapies,-ShareFor Immediate Release:November 12, 2021
Today, the U.S. Food and Drug Administration approved Besremi (ropeginterferon alfa-2b-njft) injection to treat adults with polycythemia vera, a blood disease that causes the overproduction of red blood cells. The excess cells thicken the blood, slowing blood flow and increasing the chance of blood clots.
“Over 7,000 rare diseases affect more than 30 million people in the United States. Polycythemia vera affects approximately 6,200 Americans each year,” said Ann Farrell, M.D., director of the Division of Non-Malignant Hematology in the FDA’s Center for Drug Evaluation and Research. “This action highlights the FDA’s commitment to helping make new treatments available to patients with rare diseases.”
Besremi is the first FDA-approved medication for polycythemia vera that patients can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.
Treatment for polycythemia vera includes phlebotomies (a procedure that removes excess blood cells though a needle in a vein) as well as medicines to reduce the number of blood cells; Besremi is one of these medicines. Besremi is believed to work by attaching to certain receptors in the body, setting off a chain reaction that makes the bone marrow reduce blood cell production. Besremi is a long-acting drug that patients take by injection under the skin once every two weeks. If Besremi can reduce excess blood cells and maintain normal levels for at least one year, then dosing frequency may be reduced to once every four weeks.
The effectiveness and safety of Besremi were evaluated in a multicenter, single-arm trial that lasted 7.5 years. In this trial, 51 adults with polycythemia vera received Besremi for an average of about five years. Besremi’s effectiveness was assessed by looking at how many patients achieved complete hematological response, which meant that patients had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots. Overall, 61% of patients had a complete hematological response.
Besremi can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness. Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).
Interferon alfa products like Besremi may cause or worsen neuropsychiatric, autoimmune, ischemic (not enough blood flow to a part of the body) and infectious diseases, which could lead to life-threatening or fatal complications. Patients who must not take Besremi include those who are allergic to the drug, those with a severe psychiatric disorder or a history of a severe psychiatric disorder, immunosuppressed transplant recipients, certain patients with autoimmune disease or a history of autoimmune disease, and patients with liver disease.
People who could be pregnant should be tested for pregnancy before using Besremi due to the risk of fetal harm.
Besremi received orphan drug designation for this indication. Orphan drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted the approval of Besremi to PharmaEssentia Corporation.
Medical uses
In the European Union, ropeginterferon alfa-2b is indicated as monotherapy in adults for the treatment of polycythemia vera without symptomatic splenomegaly.[3] In the United States it is indicated for the treatment of polycythemia vera.[1][2][5]
History
The effectiveness and safety of ropeginterferon alfa-2b were evaluated in a multicenter, single-arm trial that lasted 7.5 years.[2] In this trial, 51 adults with polycythemia vera received ropeginterferon alfa-2b for an average of about five years.[2] The effectiveness of ropeginterferon alfa-2b was assessed by looking at how many participants achieved complete hematological response, which meant that participants had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots.[2] Overall, 61% of participants had a complete hematological response.[2] The U.S. Food and Drug Administration (FDA) granted the application for Ropeginterferon_alfa-2b orphan drug designation and granted the approval of Besremi to PharmaEssentia Corporation[2]
REF
- Bartalucci N, Guglielmelli P, Vannucchi AM: Polycythemia vera: the current status of preclinical models and therapeutic targets. Expert Opin Ther Targets. 2020 Jul;24(7):615-628. doi: 10.1080/14728222.2020.1762176. Epub 2020 May 18. [Article]
- How J, Hobbs G: Use of Interferon Alfa in the Treatment of Myeloproliferative Neoplasms: Perspectives and Review of the Literature. Cancers (Basel). 2020 Jul 18;12(7). pii: cancers12071954. doi: 10.3390/cancers12071954. [Article]
- Verger E, Soret-Dulphy J, Maslah N, Roy L, Rey J, Ghrieb Z, Kralovics R, Gisslinger H, Grohmann-Izay B, Klade C, Chomienne C, Giraudier S, Cassinat B, Kiladjian JJ: Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J. 2018 Oct 4;8(10):94. doi: 10.1038/s41408-018-0133-0. [Article]
- Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, Wolf D, Kralovics R, Gisslinger B, Strecker K, Egle A, Melchardt T, Burgstaller S, Willenbacher E, Schalling M, Them NC, Kadlecova P, Klade C, Greil R: Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015 Oct 8;126(15):1762-9. doi: 10.1182/blood-2015-04-637280. Epub 2015 Aug 10. [Article]
- EMA Approved Products: Besremi (ropeginterferon alfa-2b ) solution for injection [Link]
- FDA Approved Drug Products: BESREMi (ropeginterferon alfa-2b-njft) injection [Link]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d e https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761166s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA Approves Treatment for Rare Blood Disease”. U.S. Food and Drug Administration (FDA) (Press release). 12 November 2021. Retrieved 12 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g “Besremi EPAR”. European Medicines Agency (EMA). Retrieved 14 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Wagner SM, Melchardt T, Greil R (March 2020). “Ropeginterferon alfa-2b for the treatment of patients with polycythemia vera”. Drugs of Today. Barcelona, Spain. 56 (3): 195–202. doi:10.1358/dot.2020.56.3.3107706. PMID 32282866. S2CID 215758794.
- ^ Jump up to:a b “U.S. FDA Approves Besremi (ropeginterferon alfa-2b-njft) as the Only Interferon for Adults With Polycythemia Vera” (Press release). PharmaEssentia. 12 November 2021. Retrieved 14 November 2021 – via Business Wire.
External links
- “Ropeginterferon alfa-2b”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01193699 for “Safety Study of Pegylated Interferon Alpha 2b to Treat Polycythemia Vera (PEGINVERA)” at ClinicalTrials.gov
- Clinical trial number NCT02218047 for “AOP2014 vs. BAT in Patients With Polycythemia Vera Who Previously Participated in the PROUD-PV Study. (CONTI-PV)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Besremi |
| Other names | AOP2014, ropeginterferon alfa-2b-njft |
| License data | EU EMA: by INNUS DailyMed: Ropeginterferon_alfa |
| Pregnancy category | Contraindicated |
| Routes of administration | Subcutaneous |
| Drug class | Interferon |
| ATC code | L03AB15 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2]EU: Rx-only [3] |
| Identifiers | |
| CAS Number | 1335098-50-4 |
| DrugBank | DB15119 |
| UNII | 981TME683S |
| KEGG | D11027 |
/////////Ropeginterferon alfa-2b, FDA 2021, APPROVALS 2021, BESREMI, PEPTIDE, Antineoplastic, Antiviral, AOP 2014, PharmaEssentia

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}