
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

ELLAGIC ACID A CANCER FIGHTING WONDER

ELLAGIC ACID
2,3,7,8-Tetrahydroxy-chromeno[5,4,3-cde]chromene-5,10-dione
as a very potent CK2 inhibitor

Ellagic acid is a natural phenol antioxidant found in numerous fruits and vegetables. The antiproliferative and antioxidant properties of ellagic acid have spurred preliminary research into the potential health benefits of ellagic acid consumption.
Ellagic acid is the dilactone of hexahydroxydiphenic acid.
Ellagic acid is an antioxidant and an anti-proliferative compound present in fruits, nuts and vegetables. In spite of evidences for anticancer activity in various cancer cell-lines, human cancer cells, the mechanistic role of ellagic acid is not conclusive enough to be recommended for a clinical use. The present review provides information about the chemopreventive role of ellagic acid in oral cancer and proposes molecular basis for ellagic acid’s inhibitory activity against oral cancer. We show that ellagic acid modulates growth of tumor cells through regulation of multiple cell signaling pathways including cell proliferation pathway (cyclin dependent kinase 2, cyclin A2, cyclin B1, cyclin D1, c-myc, PKCα), cell survival/apoptosis pathway (Bcl-XL, Bax, Caspase 9/3, Akt), tumor suppressor pathway (p53, p21), inflaming Metastasis pathways (IL-1 beta, TNF-α, matrix metalloproteinases 9/3, COX-2), angiogenesis pathways (VEGF), cell immortalization (TERT), NF-κβ.

Chinese medicine…Cordyceps ( dong chong xia cao ) 冬蟲草 དབྱར་རྩྭ་དགུན་འབུ་ ………..to treat many diseases related to lungs, kidney, and also used as a natural Viagra.

Ophiocordyceps sinensis (left) growing out of the head of a dead caterpillar
Ophiocordyceps sinensis is a fungus that parasitizes larvae of ghost moths and produces a fruiting body valued as an herbal remedy. The fungus germinates in the living larva, kills and mummifies it, and then the stalk-like fruiting body emerges from the corpse. It is known in English colloquially as caterpillar fungus, or by its more prominent foreign names (see below): yartsa gunbu or yatsa gunbu (Tibetan), or Dōng chóng xià cǎo (Chinese: 冬虫夏草; literally “winter worm, summer grass”). Of the various entomopathogenic fungi, Ophiocordyceps sinensis is one that has been used for at least 2000 years[2] to treat many diseases related to lungs, kidney, and also used as a natural Viagra. This fungus is not yet cultivated commercially,[3] despite the fact that several fermentable strains of Ophiocordyceps sinensis are isolated by Chinese Scientists.[4] Overharvesting and over exploitation have led to the classification of O. sinensis as an endangered species in China.[5] Additional research needs to be carried out in order to understand its morphology and growth habit for conservation and optimum utilization.

The moths in which O. sinensis grows are ambiguously referred to as “ghost moth”, which identifies either a single species or the genus Thitarodes, and the species parasitized by O. sinensis may be one of several Thitarodes that live on the Tibetan Plateau (Tibet, Qinghai, West-Sichuan, SW-Gansu & NW Yunnan), and the Himalayas (India, Nepal, Bhutan).
O. sinensis is known in the West as a medicinal mushroom, and its use has a long history in Traditional Chinese medicine as well as Traditional Tibetan medicine.[6] The hand-collected fungus-caterpillar combination is valued by herbalists and as a status symbol;[7] it is used as an aphrodisiac and treatment for ailments such as fatigue and cancer, although such use is mainly based on traditional Chinese medicine and anecdote. Recent research however seems to indicate a variety of beneficial effects in animal testing, including increased physical endurance through heightened ATP production in rats.[8]

| Cordyceps Sinensis |
| Cordyceps sinensis (Berk.) Sacc. and the usually the larvae are the remains of Hepialus varians |
| tonifies lung yin and kidney yang. For impotence, chronic lower back pain, afraid of cold, over abundance of mucus and tears, chronic cough and wheezing from deficiency, blood in phlegm from consumption due tokidney yang deficiency (shenyangxu). |
Cordyceps ( Dong Chong Cao ) 冬蟲草 Chinese Herbal Articles also known as chong cao, dong chong cao, yarsa gumba (Nepalese name of Tibetan origin), yartsa gunbu (dbyar rtswa dgun ‘bu) Tibetan name 蟲草, 冬蟲草. It belong to the “Ascomycetes or Clavicipitaceae” family.
Cordyceps ( Dong Chong Cao ) 冬蟲草 has a sweet, warm properties. It is use for treating the lung and kidney.

|
1. Improves auto-immune system.
2. Protects kidneys from toxins.
3. Protects kidneys from exhaustion.
4. Protects liver from toxins and treats and prevents cirrhosis of liver.
5. Protect the heart from the damaging effect of ouabain (C29H44O12.8H2O).
6. Anti-arrhythmia.
7. Anti-rejection effect in cornea transplant.
8. Antibiotic effect.
9. Inhibits contraction of smooth muscles.
|

Cordyceps ( Dong Chong Cao ) 冬蟲草 use in large dosages and/or long term usage can be toxic to kidneys.
According to the classics Medical Material, “Ben Cao Bei Yao” 本草備要, the best dong chong xia cao 冬蟲夏草, are produced in Sichuan. Today, most of them are produced in Xizang (Tibet) and Qinghai. Because the sizes the larvae are larger, they fetch higher prices.
According to the classics Medical Material, “Ben Cao Bei Yao” 本草備要, the best dong chong xia cao 冬蟲夏草, are produced in Sichuan. Today, most of them are produced in Xizang (Tibet) and Qinghai. Because the sizes the larvae are larger, they fetch higher prices.
Taxonomic History/ Systematics

Caterpillars with emergingOphiocordyceps sinensis
Morphological Features
Similar to other Cordyceps]] species, O. sinensis consists of two parts, a fungal endosclerotium (caterpillar) and stroma.[2] The stroma is the upper fungal part and is dark brown or black, but can be a yellow color when fresh and, longer than the caterpillar itself, usually 4–10 cm. It grows singly from the larval head, and is clavate, sublanceolate or fusiform and distinct from the stipe.[9] The stipe is slender, glabrous, and longitudinally furrowed or ridged. The fertile part of the stroma is the head. The head is granular due to the ostioles of the embedded perithecia.[2] The perithecia are ordinally arranged and ovoid [9] The asci are cylindrical or slightly tapering at both ends, and may be straight or curved, with a capitate and hemispheroid apex and may be two to four spored.[2] Similarly, ascospores are hyaline, filiform, multiseptate at a length of 5-12 um and subattenuated on both sides.[9] Perithecial, ascus and ascospore characters in the fruiting bodies are the key identification characteristics of O. sinensis. Ophiocordyceps (Petch) Kobayasi species produce whole ascospores and do not separate into part spores which is different from other Cordyceps species, which produce either immersed or superficial perithecia perpendicular to stromal surface and the ascospores at maturity are disarticulated into part spores.[10] Generally Cordyceps species possess brightly colored and fleshy stromata, but O. sinensis had dark pigments and tough to pliant stromata, a typical characteristic feature of most of the Ophiocordyceps species.[3]
Important developments in Classification
The species was first described scientifically by Miles Berkeley in 1843 as Sphaeria sinensis;[11] Pier Andrea Saccardo transferred the species to the genus Cordyceps in 1878.[12]The scientific name‘s etymology is from the Latin cord “club”, ceps “head”, and sinensis “from China“. The fungus was known as Cordyceps sinensis until 2007, when molecularanalysis was used to emend the classification of the Cordycipitaceae and the Clavicipitaceae, resulting in the naming of a new family Ophiocordycipitaceae and the transfer of several Cordyceps species to Ophiocordyceps.[10] Based on a molecular phylogenetic study, Sung et al. (2007) separated the megagenus Cordyceps into four genera as it was polyphyletic, viz. Cordyceps (40 spp.), Ophiocordyceps (146 spp.), Metacordyceps (6 spp.) and Elaphocordyceps (21 spp.), while the remaining 175 spp. were left in Cordyceps. As a result, C. sinensis was transferred to Ophiocordyceps, hence renamed as O. sinensis.[2]
Common Names[edit]
In Tibetan it is known as དབྱར་རྩྭ་དགུན་འབུ་ (ZYPY: yartsa gunbu, Wylie: dbyar rtswa dgun ‘bu, “summer grass winter bug”), which is the source of the Nepali यार्शागुम्बा, yarshagumba,yarchagumba or yarsagumba. The transliteration in Bhutan is Yartsa Guenboob. It is known as keera jhar, keeda jadi, keeda ghas or ‘ghaas fafoond in Hindi. Its name in Chinese Dōng chóng xià cǎo (冬蟲夏草) means “winter worm, summer grass” (i.e., “worm in the winter, [turns to] plant in the summer”). The Chinese name is a literal translation of the original Tibetan name, which was first recorded in the 15th Century by the Tibetan doctor Zurkhar Namnyi Dorje. In colloquial Tibetan Yartsa gunbu is often shortened to simply “bu” or “yartsa”.
In traditional Chinese medicine, its name is often abbreviated as chong cao (蟲草 “insect plant”), a name that also applies to other Cordyceps species, such as C. militaris. InJapanese, it is known by the Japanese reading of the characters for the Chinese name, tōchūkasō (冬虫夏草).
Strangely, sometimes in Chinese English language texts Cordyceps sinensis is referred to as aweto [Hill H. Art. XXXVI: The Vegetable Caterpillar (Cordiceps robertsii). Transactions and Proceedings of the Royal Society of New Zealand 1868-1961. Vol 34, 1901;396-401], which is the Māori name for Cordyceps robertsii, a species from New Zealand.
The English term “vegetable caterpillar” is a misnomer, as no plant is involved. “Caterpillar fungus” is a preferable term.
Nomenclature of the anamorph
Since the 1980s, 22 species in 13 genera have been attributed to the anamorph of O. sinensis. Of the 22 species, Cephalosporium acreomonium is the zygomycetous species ofUmbelopsis, Chrysosporium sinense has very low similarity in RAPD polymorphism, hence it is not the anamorph. Likewise, Cephalosporium dongchongxiacae, C. sp. sensu,Hirsutella sinensis and H. hepiali and Synnematium sinnense are synonymous and only H. sinensis is only validly published in articles. Cephalosporium sinensis possibly might be synonymous to H. sinensis but there is lack of valid information. Isaria farinose is combined to Paecilomyces farinosus and is not the anamorph. Several species like Isaria sp. Verticella sp. Scydalium sp. Stachybotrys sp. were identified only up to generic level, and thus it is dubious that they are anamorph. Mortierella hepiali is discarded as anamorph as it belongs to Zygomycota. Paecilomyces sinensis and Sporothrix insectorum are discarded based on the molecular evidence. P. lingi appeared only in one article and thus is discarded due to incomplete information. Tolypocladium sinense, P. hepiali, and Scydalium hepiali, have no valid information and thus are not considered as anamorph toOphiocordyceps sinensis. V. sinensis is not considered anamorph as there is no valid published information. Similarly, Metarhizium anisopliae is not considered anamorph as it has widely distributed host range, and is not restricted only in high altitude.[13] Thus Hirsutella sinensis is considered the validly published anamorph of O. sinensis. Cordyceps nepalensis and C. multiaxialis which had similar morphological characteristics to C. sinensis, also had almost identical or identical ITS sequences and its presumed anamorph, H. sinensis. This also confirms H. sinensis to be anamorph of O. sinensis and suggests C. nepalensis and C. multiaxialis are synonyms.[14] Evidence based on microcyclic conidiation from ascospores and molecular studies [2] support H. sinensis as the anamorph of the caterpillar fungus, O. sinensis.
Ecology
The caterpillars prone to infection by O. sinensis generally live 6 inches underground [4] in alpine grass and shrub-lands on the Tibetan Plateau and the Himalayas at an altitude between 3,000 and 5,000 m (9,800 and 16,400 ft). The fungus is reported from the northern range of Nepal, Bhutan, and also from the northern states of India, apart from northern Yunnan, eastern Qinghai, eastern Tibet, western Sichuan, southwestern Gansu provinces.[4] The fungus consumes its host from inside out as they hibernate in alpine meadows. Usually the larvae are more vulnerable after shedding their skin, during late summer. The fungal fruiting body disperses spores which infect the caterpillar. The infected larvae tend to remain vertical to the soil surface with their heads up. The fungus then germinates in the living larva, kills and mummifies it, and then the stalk-like fruiting body emerges from the head and the fungus finally emerges from the soil by early spring.[15] Fifty-seven taxa from seven genera (1 Bipectilus, 1 Endoclita, 1 Gazoryctra, 12 Hepialus, 2Magnificus, 3 Pharmacis, and 37 Thitarodes [3]) are recognized as potential hosts of O. sinensis.
Reproduction Biology
Ophiocordyceps sinensis has both teleomorphic and anamorphic phases. Spending up to five years underground before pupating, the Thitarodes caterpillar is attacked while feeding on roots. It is not certain how the fungus infects the caterpillar; possibly by ingestion of a fungal spore or by the fungus mycelium invading the insect through one of the insect’s breathing pores. The dark brown to black fruiting body (or mushroom) emerges from the ground in spring or early summer, the long, usually columnar fruiting body reaches 5–15 cm above the surface and releases spores.
In late autumn, chemicals on the skin of the caterpillar interact with the fungal spores and release the fungal mycelia, which then infects the caterpillar.[4] After invading a host larva, the fungus ramifies throughout the host and eventually kills it. Gradually the host larvae become rigid due to the production of fungal sclerotia. Fungal sclerotia are multihyphal structures that can remain dormant and then germinate to produce spores. After over-wintering, the fungus ruptures the host body, forming a sexual sporulating structure (a perithecial stroma) from the larval head in summer that is connected to the sclerotia (dead larva) below ground and grows upward to emerge from the soil.[16] The slow growing O. sinensis grows at a comparatively low temperature, i.e., below 21oC. Temperature requirements and growth rates are crucial factors that identify O. sinensis from other similar fungi.[3]
Use in medicine
It is used as a curative to many diseases, anti- aging,[17] hypoglycemic,[18] aphrodisiac and also treatment against cancer. Ophiocordyceps sinensis serves against kidney and lung problems and stimulates the immune system; it is used for treatment of fatigue, night sweating, respiratory disease, hyperglycemia, hyperlipidemia, asthenia after severe illness, arrhythmias and other heart diseases and liver disease.[4]
Traditional Asian medicines

Medicinal use of the caterpillar fungus apparently originated in Tibet and Nepal. So far the oldest known text documenting its use was written in the late fourteen hundreds by the Tibetan doctor Zurkhar Nyamnyi Dorje (Wylie: Zur mkhar mnyam nyid rdo rje)[1439-1475]) in his text: Man ngag bye ba ring bsrel (“Instructions on a Myriad of Medicines”). A translation is available at Winkler.[19]
The first mention of Ophiocordyceps sinensis in traditional Chinese Medicine was in Wang Ang’s 1694 compendium of materia medica, Ben Cao Bei Yao.[20] In the 18th Century it was listed in Wu Yiluo‘s Ben cao cong xin (“New compilation of materia medica”).[21] No sources have been published to uphold widespread claims of “thousands of years of use in Chinese medicine” or use of “chong cao since the 7th Century Tang Dynasty in China”. The ethno-mycological knowledge on caterpillar fungus among the Nepalese people is documented byDevkota(2006) The entire fungus-caterpillar combination is hand-collected for medicinal use.
The fungus is a medicinal mushroom which is highly prized by practitioners of Tibetan medicine, Chinese medicine and traditional Folk medicines, in which it is used as an aphrodisiac and as a treatment for a variety of ailments from fatigue to cancer. In Chinese medicine it is regarded as having an excellent balance of yin and yang as it is apparently both animal and vegetable. Assays have found thatOphiocordyceps species produce many pharmacologically active substances. They are now cultivated on an industrial scale for their medicinal value. However, no one has succeeded so far in growing the larva cum mushroom artificially. The biological process that forms the Ophiocordyceps is still unknown and true cultivation has yet to be realized.[3] All artificial products are derived from mycelia grown on grains or in liquids.
According to Bensky et al. (2004), laboratory-grown C. sinensis mycelia have similar clinical efficacy and less associated toxicity. He notes a toxicity case of constipation, abdominal distension, and decreased peristalsis, two cases of irregular menstruation, and one case report ofamenorrhea following ingestion of tablets or capsules containing C. sinensis. In Chinese medicine C. sinensis is considered sweet and warm, entering the lung and kidney channels; the typical dosage is 3–9 grams.[22]
Research

Cordycepin, a compound isolated from the “Caterpillar fungus”.
Some work has been published in which Ophiocordyceps sinensis has been used to protect the bone marrow and digestive systems ofmice from whole body irradiation.[23] An experiment noted Ophiocordyceps sinensis may protect the liver from damage.[24] An experiment conducted with mice noted the mushroom may have an anti-depressant effect.[25] Researchers have noted that the caterpillar fungus has ahypoglycemic effect and may be beneficial for people with insulin resistance.[26][27][28][29][30] There is also experimental evidence of the supposed energizing effect of the fungus, as it has been shown to increase endurance through heightened ATP production in rats.[8]
A March 2013 study on Cordyceps Sinensis documented the medicinal fungus’ anti-inflammatory properties.[31] Scientists were able to show Cordyceps Sinensis’ ability to suppress interleukin-1b and interleukin-18 secretion by inhibiting both canonical and non-canonical inflammasomes. Inflammasomes have long been associated with auto-inflammatory diseases, such as gout. The study used a specific anamorphic mycelial form of Cordyceps Sinensis known as Hirsutella Sinensis.
Introduction to the Western world
| ) |

Ophiocordyceps sinensis
The Western world was largely unaware of Ophiocordyceps prior to 1993. The fungus dramatically caught the world’s eye due to the performance of three female Chinese athletes, Wang Junxia, Qu Yunxia, and Zhang Linli. These athletes broke five world records for 1,500, 3,000 and 10,000 meter dashes at the National Games in Beijing, China. The number of new world records set at a single track event attracted much attention and suspicion. Following the races, the women were expected by some to fail drug tests for anabolic steroids. However, the athletes’ tests revealed no illegal substances, and coach Ma Junren told the reporters that the runners were takingOphiocordyceps sinensis and turtle blood at his request. However for the 2000 Sydney Olympics, Ma Junren withdrew some of his athletes at the last minute. It was speculated that a new doping test would have revealed illegal substances, thus half a dozen Chinese field and track athletes were left at home.
Economics and impact

Many shops in downtown Lanzhouadvertise Dōng chóng xià cǎo (冬虫夏草) among other local specialties.
In rural Tibet, yartsa gunbu has become the most important source of cash income. The fungi contributed 40% of the annual cash income to local households and 8.5% to the GDP in 2004. Prices have increased continuously, especially since the late 1990s. In 2008, one kilogram traded for US$3,000 (lowest quality) to over US$18,000 (best quality, largest larvae). The annual production on the Tibetan Plateau was estimated in 2009 at 80–175 tons.[32] The Himalayan Ophiocordyceps production might not exceed a few tons.
In 2004 the value of a kilogram of caterpillars was estimated at about 30,000 to 60,000 Nepali rupees in Nepal, and about Rs 100,000 in India.[33] In 2011 the value of a kilogram of caterpillars was estimated at about 350,000 to 450,000 Nepali rupees in Nepal. A 2012 BBC article indicated that in north Indian villages a single fungus was worth Rs 150 (about £2 or $3), which is more than the daily wage of a manual laborer.[34]
According to Daniel Winkler, the price of Ophiocordyceps sinensis has risen dramatically on the Tibetan Plateau, basically 900% between 1998 and 2008, an annual average of over 20% (after inflation). However, the value of big sized caterpillar fungus has increased more dramatically than smaller size Cordyceps, regarded as lower quality.[20]
| Year | % Price Increase | Price/kg (Yuan) |
|---|---|---|
| 1980s | 1,800 | |
| 1997 | 467% (incl. inflation) | 8,400 |
| 2004 | 429% (incl. inflation) | 36,000 |
| 2005 | 10,000–60,000 | |
| 2013 | 125,000–500,000 |
Because of its high value, inter-village conflicts over access to its grassland habitats has become a headache for the local governing bodies and in several cases people were killed. In November 2011, a court in Nepal convicted 19 villagers over the murder of a group of farmers during a fight over the prized aphrodisiac fungus. Seven farmers were killed in the remote northern district of Manang in June 2009 after going to forage for Yarchagumba. [35]
Its value gave it a role in the Nepalese Civil War, as the Nepalese Maoists and government forces fought for control of the lucrative export trade during the June–July harvest season.[36] Collecting yarchagumba in Nepal had only been legalised in 2001, and now demand is highest in countries such as China, Thailand, Vietnam, Korea and Japan. By 2002, the herb was valued at R 105,000 ($1,435) per kilogram, allowing the government to charge a royalty of R 20,000 ($280) per kilogram.
The search for Ophiocordyceps sinensis is often perceived to pose a threat to the environment of the Tibetan Plateau where it grows. While it has been collected for centuries and is still common in such areas, current collection rates are much higher than in historical times.
Ophiocordyceps producers like to perpetuate the story that unscrupulous harvesters insert twigs into the ascocarps of wild C. sinensis to increase their weight and therefore the price paid. A tiny twig is only used when the ascocarp is broken from the caterpillar, and has nothing to do with artificially increasing weight. Supposedly, at some point in the past, someone inserted lead wires with which to increase weight; however, each year hundreds of millions of specimens are harvested and this appears to have been a one-time occurrence.
Cultivated C. sinensis mycelium is an alternative to wild-harvested C. sinensis, and producers claim it may offer improved consistency. Artificial culture of C. sinensis is typically by growth of pure mycelia in liquid culture (in China) or on grains (in the West). The first time in Vietnam, Prof. Aca. Dr. Dai Duy Ban together with scientists and DAIBIO Company and DAIBIO Great Traditional Medicine Family Clinic discovered the Cordyceps sinensis as Isaria cerambycidae N.SP. to develop Fermentation DAIBIO Cordyceps Sinensis.[37]Ascocarps are not produced through in vitro cultivation.
References
- “Ophiocordyceps sinensis (Berk.) G.H. Sung, J.M. Sung, Hywel-Jones & Spatafora 2007″. MycoBank. International Mycological Association. Retrieved 2011-07-19.
- Shrestha, B., Weimin, Z., Yongjie, Z., & Xingzhong, L. (2010). What is the Chinese caterpillar fungus Ophiocordyceps sinensis (Ophiocordycipitaceae)?. Mycology: An International Journal On Fungal Biology, 1(4), 228-236. doi:10.1080/21501203.2010.536791.
- Hsieh, C., et al., A Systematic Review of the Mysterious Caterpillar Fungus Ophiocordyceps sinensis in Dong-ChongXiaCao and Related Bioactive Ingredients. Vol. 3. 2013. 16-32.
- Zhu JS, Halpem GM, Jones K. 1998. The scientific rediscovery of an ancient Chinese herbal medicince: Cordyceps sinensis. I. J Alt Complem Med 4:289-303.
- Xiao-Liang, W., & Yi-Jian, Y. (2011). Host insect species of Ophiocordyceps sinensis: a review. Zookeys, 12743-59. doi:10.3897/zookeys.127.802
- Halpern, Miller (2002). Medicinal Mushrooms. New York, New York: M. Evans and Company, Inc. pp. 64–65. ISBN 0-87131-981-0
- http://www.npr.org/2011/10/09/141164173/caterpillar-fungus-the-viagra-of-the-himalayas
- ^ Jump up to:a b Rajesh Kumar, P.S. Negi, Bhagwat Singh, Govindasamy Ilavazhagan, Kalpana Bhargava, Niroj Kumar Sethy (2011). “Cordyceps sinensis promotes exercise endurance capacity of rats by activating skeletal muscle metabolic regulators”. Journal of Ethnopharmacology 136: 260–266.
- Sung, G. H., et al. (2007). “A multi-gene phylogeny of Clavicipitaceae (Ascomycota, Fungi): identification of localized incongruence using a combinational bootstrap approach.” Molecular Phylogenetics and Evolution 44(3): 1204-1223.
- Sung GH, Hywel-Jones NL, Sung JM, Luangsa-Ard JJ, Shrestha B, Spatafora JW. (2007). “Phylogenetic classification of Cordyceps and the clavicipitaceous fungi”.Studies in Mycology 57: 5–59. doi:10.3114/sim.2007.57.01. PMC 2104736.PMID 18490993.
- Berkeley MJ. (1843). “On some entomogenous Sphaeriae”. London Journal of Botany 2: 205–11.
- Saccardo PA. (1878). “Enumeratio Pyrenomycetum Hypocreaceorum hucusque cognitorum systemate carpologico dispositorum” (PDF). Michelia (in Latin) 1 (3): 277–325.
- Jiang, Y. Y., & Yao, Y. J. (n.d). Names related to Cordyceps sinensis anamorph. Mycotaxon, 84245-254.
- Liu, Z., Liang, Z., Liu, A., Yao, Y., Hyde, K. D., & Yu, Z. (n.d). Molecular evidence for teleomorph-anamorph connections in Cordyceps based on ITS-5.8S rDNA sequences. Mycological Research, 106(9), 1100-1108.
- Stone, R. (2008). Last Stand for the Body Snatcher of the Himalayas?. Science, (5905), 1182. doi:10.2307/20145300
- Xing, X. K., & Guo, S. X. (2008). The Structure and Histochemistry of Sclerotia of Ophiocordyceps sinensis. Mycologia, (4), 616. doi:10.2307/20444986.
- Ji DB, Ye J, Li CL, Wang YH, Zhao J, Cai SQ (2009) Antiaging effect of Cordyceps sinensis extract. Phytotherapy Research 23 (1): 116-122. Doi: 10.1002/ptr.2576
- Zhang GQ, Huang YD, Bian Y, Wong JH, Ng TB, Wang HX (2006) Hypoglycemic activity of the fungus Cordyceps militaris, Cordyceps sinensis, Tricholoma mongolicum and Omphalia lapidescens in streptozotocin-induced diabetic rats. Applied Microbiology and Biotechnology 72 (6): 1152-1156. Doi: 10.1007/s00253-006-0411-9.
- Winkler D. (2008). “The mushrooming fungi market in Tibet exemplified by Cordyceps sinensis and Tricholoma matsutake“. Journal of the International Association of Tibetan Studies. In: In the Shadow of the Leaping Dragon: Demography, Development, and the Environment in Tibetan Areas (4).
- Winkler D. (2008). “Yartsa Gunbu (Cordyceps sinensis) and the fungal commodification of the rural economy in Tibet AR”. Economic Botany 62 (3): 291–305.doi:10.1007/s12231-008-9038-3.
- Wu Y (1757). “Ben cao cong xin” – “New compilation of materia medica” (in Chinese).
- Jump up^ Bensky D, Gamble A, Clavey S, Stöger E, Bensky L. Lai (2004). Materia Medica: Chinese Herbal Medicine (3rd ed.). Seattle, Washington: Eastland Press. ISBN 978-0-939616-42-8.
- Liu W-C, Wang S-C, Tsai M-L, Chen, M-C, Wang Y-C, Hong J-H, McBride WH, Chiang C-S. (2006). “Protection against radiation-induced bone marrow and intestinal injuries byCordyceps sinensis, a Chinese herbal medicine”. Radiation Research 166 (6): 900–907.doi:10.1667/RR0670.1. PMID 17149981.
- WS, Hsu SL, Chyau CC, Chen KC, Peng RY. (July 2009). “Compound Cordyceps TCM-700C exhibits potent hepatoprotective capability in animal model”. Fitoterapia 81(1): 1–7. doi:10.1016/j.fitote.2009.06.018. PMID 19596425.
- Nishizawa K, Torii K, Kawasaki A, et al. (2007). “Antidepressant-like effect ofCordyceps sinensis in the mouse tail suspension test”. Biological and Pharmaceutical Bulletin 30 (9): 1758–62. doi:10.1248/bpb.30.1758. PMID 17827735.
- Kiho T, Hui J, Yamane A, Ukai S. (1993). “Polysaccharides in fungi. XXXII. Hypoglycemic activity and chemical properties of a polysaccharide from the cultural mycelium of Cordyceps sinensis“. Biological and Pharmaceutical Bulletin 16 (12): 1291–3. doi:10.1248/bpb.16.1291. PMID 8130781.
- Kiho T, Yamane A, Hui J, Usui S, Ukai S. (1996). “Polysaccharides in fungi. XXXVI. Hypoglycemic activity of a polysaccharide (CS-F30) from the cultural mycelium of Cordyceps sinensis and its effect on glucose metabolism in mouse liver”. Biological and Pharmaceutical Bulletin 19 (2): 294–6. doi:10.1248/bpb.19.294. PMID 8850325.
- Zhao CS, Yin WT, Wang JY, et al. (2002). “CordyMax Cs-4 improves glucose metabolism and increases insulin sensitivity in normal rats”. Journal of Alternative and Complementary Medicine 8 (3): 309–14. doi:10.1089/10755530260127998.PMID 12165188.
- Lo HC, Tu ST, Lin KC, Lin SC. (2004). “The anti-hyperglycemic activity of the fruiting body of Cordyceps in diabetic rats induced by nicotinamide and streptozotocin”. Life Sciences 74 (23): 2897–908. doi:10.1016/j.lfs.2003.11.003. PMID 15050427.
- Li SP, Zhang GH, Zeng Q, et al. (2006). “Hypoglycemic activity of polysaccharide, with antioxidation, isolated from cultured Cordyceps mycelia”. Phytomedicine 13 (6): 428–33.doi:10.1016/j.phymed.2005.02.002. PMID 16716913.
- Huang, T. et al. (March 2013). “Hirsutella sinensis mycelium suppresses interleukin-1b and interleukin-18 secretion by inhibiting both canonical and non-canonical inflammasomes.” (PDF). Scientific Report. 3, 1374;.
- Winkler, D. (2009). “Caterpillar Fungus (Ophiocordyceps sinensis) Production and Sustainability on the Tibetan Plateau and in the Himalayas”. Asian Medicine 5 (2): 291. doi:10.1163/157342109X568829.
- Sharma S. (2004). “Trade of Cordyceps sinensis from high altitudes of the Indian Himalaya: Conservation and biotechnological priorities” (PDF). Current Science 86(12): 1614–9.
- Jeffrey, Craig (2012-07-07). “The ‘Viagra’ transforming local economies in India”. BBC News. Retrieved July 9, 2012.
- Staff (14 November 2011) ‘Himalayan viagra’: Six men get life for Nepal murders BBC News Asia, Retrieved 9 July 2012
- Baral N, Heinen JT. (2005). “The Maoist people’s war and conservation in Nepal”.Politics and the Life Sciences 24 (1): 2–11. doi:10.2990/1471-5457(2005)24[2:TMPWAC]2.0.CO;2.
- DAIBIO Cordyceps Sinensis in Vietnam
- Winkler, D. 2005. Yartsa Gunbu – Cordyceps sinensis. Economy, Ecology & Ethno-mycology of a Fungus Endemic to the Tibetan Plateau. In: A.BOESI & F. CARDI (eds.). Wildlife and plants in traditional and modern Tibet: Conceptions, Exploitation and Conservation. Memorie della Società Italiana di Scienze Naturali e del Museo Civico di Storia Naturale di Milano, Vol. 33.1:69–85.
- Zhang Y., Zhang S., Wang M., Bai F. & Liu X. (2010). “High Diversity of the Fungal Community Structure in Naturally-Occurring Ophiocordyceps sinensis“. PLoS ONE 5(12): e15570. doi:10.1371/journal.pone.0015570.
External links
Yartsa Gunbu (Cordyceps sinensis) in Tibet
- Daniel Winkler’s Cordyceps blog
- Nepal’s Nature – The Himalayan Viagra
- Page at Everything2.com
- Image gallery of Cordyceps sinensis
- The first time in Vietnam, Prof.Aca.D.Sc Dai Duy Ban with his scientists discovered Cordyceps Sinensis as Isaria cerambycidae N.SP. and Fermentation Daibio Cordyceps Sinensis by Daibio Great Family Traditional Medicine Clinic Company
- Daibio Cordyceps Sinensis in Vietnam
- An Electronic Monograph of Cordyceps and Related Fungi
- Cordyceps information from Drugs.com
- Cordyceps sinensis (Berk.) Sacc. Medicinal Plant Images Database (School of Chinese Medicine, Hong Kong Baptist University) (English) (traditional Chinese)
- Chinese Caterpiller Fungus Chinese Medicine Specimen Database (School of Chinese Medicine, Hong Kong Baptist University) (English) (traditional Chinese)
- Tibet’s Golden “Worm” August 2012 National Geographic (magazine)
Dandelion, Burdock, and Cancer
 burdock
burdock dandelion
dandelionDandelion root and burdock root are my two most commonly prescribed herbs when chronic conditions require anti-inflammatory, blood purifying alteratives for gentle detoxification. This includes conditions such as arthritis and cancer. I’ve studied literally hundreds of herbs from around the world, and considering cost, availability, palatability (no small matter, as people with chronic disease like cancer need to be able to take their herbs at least three times a day for months) – there are probably no two more simple and powerful anticancer herbs on the planet than dandelion and burdock.*
After prescribing both of these in strong dose clinically for years with great results (patients feel better, or experience slowing or even complete remission of some cancers), I learned that many professional British medical herbalists also use the same two-herb combination for conditions requiring blood, lymphatic and liver detoxification.
http://www.planetherbs.com/michaels-blog/dandelion-burdock-and-cancer.html
Glenmark’s novel molecule ‘GRC 27864’ for chronic inflammatory diseases including pain entering human trials

Glenmark’s novel molecule ‘GRC 27864’ for chronic inflammatory diseases including pain entering human trials
- GRC 27864 is a potent, selective, orally bioavailable inhibitor of mPGES-1
- The molecule has successfully completed pre-clinical and Phase 1 enabling studies. Regulatory submission has been filed for Phase 1 trial (first-in-human)with MHRA, UK
- mPGES-1 inhibitors selectively block the production of PGE2 while sparing other prostanoids of physiological importance
- With this announcement, Glenmark has reaffirmed its position globally in the development of novel pain therapies
Mumbai, India: April 3, 2014: Glenmark Pharmaceuticals today announced that its Novel Chemical Entity (NCE) ‘GRC 27864’ is entering human trials. This NCE program targets Microsomal Prostaglandin E synthase-1 (mPGES-1) as a novel therapeutic target in pain management. Selective mPGES-1 inhibitors are expected to inhibit increased prostaglandin E2 (PGE2) production in the disease state without affecting other prostanoid metabolites and, consequently, may be devoid of the GI(gastrointestinal) and cardiovascular side effects seen with NSAIDs and COX-2 inhibitors, respectively.
Glenmark has completed preclinical studies and Phase 1 enabling GLP studies for its selected lead molecule, GRC 27864 and has filed a Phase 1 application forfirst-in-human trial with the MHRA, UK. The Phase 1 studies are to be initiated soon and are likely to get completed by January 2015. Following this, Glenmark will also be initiating a proof of concept study in patients with acute pain.
ANTHONY CRASTO’S NEW DRUG APPROVALS TOUCHES 2 LAKH VIEWS IN 179 COUNTRIES
ANTHONY CRASTO’S NEW DRUG APPROVALS TOUCHES 2 LAKH VIEWS IN 179 COUNTRIES

DR ANTHONY MELVIN CRASTO Ph.D
WORLDDRUGTRACKER,
OTHERS
- Eurekamoments in Organic Chemistry
- NEW DRUG APPROVALS
- WORLD DRUG TRACKER
- Green Chemistry International
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
- Drug Scaleup and Manufacturing International
SEE ALSO
- Organic Chemistry by Dr Anthony
- Technology Transfer
- Stereochemistry
- Spectroscopy
- Polymorphism
- Reactions in Org Chem
DR ANTHONY MELVIN CRASTO, Worlddrugtracker, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his PhD from ICT ,1991, Mumbai, India, in Organic chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with GLENMARK- GENERICS LTD, Research centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Prior to joining Glenmark, he worked with major multinationals like Hoechst Marion Roussel, now sSanofi, Searle India ltd, now Rpg lifesciences, etc. he is now helping millions, has million hits on google on all organic chemistry websites. His New Drug Approvals, Green Chemistry International, Eurekamoments in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules and implementation them on commercial scale over a 25 year tenure, good knowledge of IPM, GMP, Regulatory aspects, he has several international drug patents published worldwide . He gas good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, polymorphism etc He suffered a paralytic stroke in dec 2007 and is bound to a wheelchair, this seems to have injected feul in him to help chemists around the world, he is more active than before and is pushing boundaries, he has one lakh connections on all networking sites, He makes himself available to all, contact him on +91 9323115463, amcrasto@gmail.com
Personal Links
- my sites on the net
- DR ANTHONY MELVIN CRASTO
- GOOGLE GROUP ORGANIC PROCESS DEVELOPMENT
- mixxt
- epernicus
- scipeople
- jimdo
- yolasite
- my cv
- slidestaxx
- wordpress blog
- ABOUT ME
- BRANDSITE
- SKILLPAGES
- Academia.edu
- RESEARCHGATE
- DIIGO
- SLIDESHATE
- WIX
- WIX BLOG
- ISSUU
- SCRIBD
- BIZ
- GOOGLE BLOG
- APNACIRCLE
- Eurekamoments in Organic Chemistry
- Organic Chemistry by Dr Anthony
- Green Chemistry International
- Technology Transfer
- Stereochemistry
- Spectroscopy
- Polymorphism
- Reactions in Org Chem
- DR ANTHONY MELVIN CRASTO Ph.D
- Pharmaceuticals
- Medicinal chemistry
- Organic chemistry literature
- Patent related site
- Green chemistry
- Reagents
- R & D
- Molecules
- Heterocyclic chem
- Sourcing
- NEW DRUG APPROVALS
- WORLD DRUG TRACKER
- Green Chemistry International
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
- GOOGLE PLUS
- Drug Scaleup and Manufacturing International


amcrasto@gmail.com
email me if u like my posts
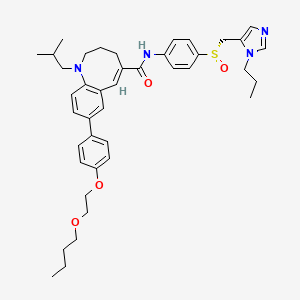
Cenicriviroc in Phase 2 for HIV by Takeda/Tobira

Cenicriviroc
TAK-652; TBR-652
(-)-(S)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-[4-(1-propyl-1H-imidazol-5-ylmethylsulfinyl)phenyl]-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide methanesulfonate
497223-25-3 , Molecular Formula: C41H52N4O4S Molecular Weight: 696.94098
497223-28-6 (mesylate) C41 H52 N4 O4 S . C H4 O3 S, 793.047
Cenicriviroc (TAK-652, TBR-652) is an experimental drug candidate for the treatment of HIV infection.[1] It is being developed by Takeda Pharmaceutical and Tobira Therapeutics.
TBR-652 (formerly TAK-652) is a highly potent and orally active CCR5 antagonist in phase II clinical trials at Takeda for the treatment of HIV infection. Tobira Therapeutics is evaluating the compound in preclinical studies for the treatment of rheumatoid arthritis.
TBR-652 binds CCR5 receptors to interfere with the entry of the HIV-1 virus into macrophages and activated T-cells by inhibiting fusion between viral and cellular membranes. This mechanism of action differs from currently used HIV treatments such as nucleoside reverse transcriptase inhibitors and protease inhibitors.
In 2007, Takeda entered into an agreement with Tobira pursuant to which Tobira obtained exclusive worldwide rights to develop, manufacture and commercialize TBR-652 for the treatment of HIV infection.
Cenicriviroc is an inhibitor of CCR2 and CCR5 receptors,[2] allowing it to function as an entry inhibitor which prevents the virus from entering into a human cell. Inhibition of CCR2 may have an anti-inflammatory effect.
A double-blind, randomized, placebo-controlled clinical study to assess the antiviral activity, safety, and tolerability of cenicriviroc was conducted in 2010. HIV-infected patients taking cenicriviroc had significant reductions in viral load, with the effect persisting up to two weeks after discontinuation of treatment.[3] Additional Phase II clinical trials are underway.[4]
Phase IIb data presented at the 20th Conference on Retroviruses and Opportunistic Infections (CROI) in March 2013 showed similar viral suppression rates of 76% for patients taking 100 mg cenicriviroc, 73% with 200 mg cenicriviroc, and 71% with efavirenz. Non-response rates were higher with cenicriviroc, however, largely due to greater drop-out of patients. A new tablet formulation with lower pill burden may improve adherence. Looking at immune and inflammatory biomarkers, levels of MCP-1 increased and soluble CD14 decreased in the cenicriviroc arms.[5]
Although HIV has been largely rendered a chronic infection, there remains a need for new drugs because of the virus’s propensity to develop resistance to the drugs used to keep it at bay.
Pfizer’s maraviroc was the first drug that acted on the cells to prevent viral entry by antagonising the CCR5 co-receptor. Several others have been investigated and have failed; another that is undergoing clinical trials is Takeda’s cenicriviroc, which has been licensed to Tobira Therapeutics. Unlike maraviroc, the new agent also acts at the CCR2 co-receptor, which is implicated in cardiovascular and metabolic diseases.
In a Phase I double blind, placebo controlled trial designed to study safety, efficacy and pharmacokinetics, treatment-experienced but CCR5 antagonist-naïve patients with HIV-1 were given doses of 25, 50, 75, 100 or 150mg of the drug, or placebo once a day for 10 days.2 The maximum median reductions in HIV-1 RNA values were 0.7, 1.6, 1.8 and 1.7 log10 copies/ml for the respective doses, with a median time to nadir of 10 to 11 days. The effect on CD-4 cell counts was negligible. There was also a significant reduction in levels of monocyte chemotactic protein 1, suggesting that CCR2 was also being blocked. The drug was both generally safe and well tolerated, and no patients withdrew from the trial due to adverse events.
In another Phase I trial, designed to look at pharmacokinetics and pharmacodynamics and carried out in a similar patient population, subjects were given the drug as oral monotherapy for 10 days, again in doses of 25, 50, 75, 100 and 150mg, or placebo.3 The drug was well absorbed into the systemic circulation, and the concentration levels declined slowly, with meant elimination half-lives of one to two days. Potent, dose-dependent reductions in viral load were seen, and again it was generally safe and well tolerated across all levels.
In June 2011, Tobira initiated a multi-centre, double blind, double dummy, 48-week comparative Phase IIb trial in 150 patients with HIV-1 infection. Subjects are being given 100 or 200mg once-daily doses of the drug to evaluate its efficacy, safety and tolerability.
PATENTS
WO 2003014105
WO 2003076411
WO 2005116013
WO 2007144720
WO 2011163389
US 20130079233
WO 2013167743
See also
ancriviroc (formerly known as SCH-C), vicroviroc which has the chemical name (4,6-dimethylprymidine-5-yl){4- [(3S)-4-{(1 R)-2-methoxy-1 -[4-(trifluoromethyl)phenyl]ethyl}-3-methylpiperazin-1 -yl]-4-methylpiperidin-1 – yljmethanone, PRO-140, apliviroc (formerly known as GW-873140, Ono-4128, AK-602), AMD-887, INC- B9471 , CMPD-167 which has the chemical name N-methyl-N-((1R,3S,4S)-3-[4-(3-benzyl-1-ethyl-1H- pyrazol-δ-yOpiperidin-i-ylmethylH-IS-fluorophenyllcyclopent-i-yll-D-valine), methyl1-endo-{8-[(3S)-3- (acetylamino)-3-(3-fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl}-2-methyl-4,5,6,7-tetrahydro-1 H- imidazo[4,5-c]pyridine-5-carboxylate, methyl 3-endo-{8-[(3S)-3-(acetamido)-3-(3-fluorophenyl)propyl]-8- azabicyclo[3.2.1]oct-3-yi}-2-methyl-4,5,6,7-tetrahydro-3H-imidazo[4,5-c]pyridine-5-carboxylate, ethyl 1- endo-{8-[(3S)-3-(acetylamino)-3-(3-fiuorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl}-2-methyl-4,5,6,7- tetrahydro-1 H-imidazo[4,5-c]pyridine-5-carboxylate and N-{(1S)-3-[3-endo-(5-lsobutyryl-2-methyl-4,5,6,7- tetrahydro-1H-imidazo[4,5-c]pyridin-1-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1-(3-fluorophenyl)propyl}acetamide) and pharmaceutically acceptable salts, solvates or derivatives of the above. The last four compounds are disclosed in WO 03/084954 and WO 05/033107.


http://pubs.acs.org/doi/full/10.1021/jm0509703

Compound (S)-(−)-5b (TAK-652) also inhibited the replication of six macrophage-tropic (CCR5-using or R5) HIV-1 clinical isolates in peripheral blood mononuclear cells (PBMCs) (mean IC90 = 0.25 nM).
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-propyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide ((S)–(−)-5a). The 1 N HCl (160 mL) was added to 1931 (35.68 g, 53.4 mmol), and the mixture was extracted with EtOAc. To the aqueous layer was added 25% aqueous K2CO3 (160 mL), and the mixture was extracted with a mixture of EtOAc and i-PrOH (4:1). The organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo to give (S)-18. To a solution of 16a (18.0 g, 41.1 mmol) and DMF (0.5 mL) in THF (180 mL) was added thionyl chloride (SOCl2) (4.50 mL, 61.7 mmol) at room temperature. After being stirred at room temperature for 1.5 h, the reaction mixture was concentrated in vacuo. A solution of the residue in THF (200 mL) was added dropwise to a mixture of (S)-18 and triethylamine (Et3N) (35.0 mL, 251 mmol) in THF (150 mL) under ice cooling. After being stirred at room temperature for 4 h, water was added to the reaction mixture. The mixture was washed with 10% aqueous AcOH, saturated aqueous NaHCO3, and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on a NH silica gel (hexane/EtOAc = 1:5 → 1:8 → 1:9) to give 21.14 g (75%) of (S)-(−)-5a as a yellow amorphous powder, [α]D = −132.5° (C = 0.507%, EtOH). 1H NMR (300 MHz, CDCl3) δ 0.87−1.03 (9H, m), 1.34−1.49 (2H, m), 1.50−1.85 (8H, m), 2.55−2.65 (2H, m), 3.15−3.25 (2H, m), 3.52−3.58 (4H, m), 3.75−3.83 (4H. m), 4.02 (1H, d, J = 13.8 Hz), 4.08−4.17 (3H, m), 6.56 (1H, d, J = 1.0 Hz), 6.80 (1H, d, J = 8.8 Hz), 6.96 (2H, d, J = 8.8 Hz), 7.31−7.46 (7H, m), 7.55 (1H, s), 7.76 (2H, d, J = 8.8 Hz), 7.98 (1H, s). Anal. (C40H50N4O4S·0.25H2O) C, H, N.
(S)–(−)-8-{4-[2-(Butoxy)ethoxy]phenyl}-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide methanesulfonate ((S)–(−)-5b). The free base of (S)-(−)-5b was prepared in 80% yield from 16band 19 by a method similar to that described for (S)-(−)-5a. To a solution of the free base of (S)-(−)-5b (64.91 g, 93.1 mmol) in EtOAc (600 mL) was added dropwise a solution of methanesulfonic acid (8.95 g, 93.1 mmol) in EtOAc (160 mL) at room temperature. After being stirred at room temperature for 4 h, the crystals were collected by filtration and washed with EtOAc to give 69.09 g (94%) of (S)-(−)-5b as yellow crystals. The crystals (68.0 g) were purified by recrystallization from 2-butanone to give 58.9 g (85%) of (S)-(−)-5b as yellow crystals, mp 145.5−147.5 °C, [α]D = −191.2° (c = 0.508%, EtOH). 1H NMR (300 MHz, DMSO-d6) δ 0.82−0.97 (12H, m), 1.29−1.39 (2H, m), 1.40−1.55 (4H, m), 1.65−1.85 (2H, m), 2.00−2.25 (1H, m), 2.29 (3H,s), 2.38−2.60 (2H, m), 3.10 (2H, d, J = 7.8 Hz), 3.30−3.60 (4H, m), 3.70 (2H, t, J = 4.8 Hz), 3.98 (2H, t,J = 6.6 Hz), 4.10 (2H, t, J = 4.8 Hz), 4.34 (1H, d, J = 15.0 Hz), 4.68 (1H, d, J = 15.0 Hz), 6.87 (1H, d, J = 8.7 Hz), 6.99 (2H, d, J = 8.7 Hz), 7.16 (1H, s), 7.42−7.60 (8H, m), 7.93 (2H, d, J = 8.7 Hz), 9.05 (1H, s), 10.18 (1H, s). Anal. (C42H56N4O7S2) C, H, N.
…………………
WO 2003014105 OR US20090030032
http://www.google.st/patents/US20090030032?hl=pt-PT&cl=un
EXAMPLE 7 Preparation of Compounds 9 and 10
8-[4-(2-Butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propyl-1H-imidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzazocin-5-carboxamide (317 mg) was resolved by using CHIRAKCEL OJ 50 mm ID×500 mL (hexane/ethanol) to give (−)-8-[4-(2-butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propylimidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzoazocine-5-carboxamide (142 mg) (Compound 9) and (+)-8-[4-(2-butoxyethoxy)phenyl]-1-propyl-N-[4-[[[1-propylimidazol-5-yl]methyl]sulfinyl]phenyl]-1,2,3,4-tetrahydro-1-benzoazocine-5-carboxamide (143 mg) (Compound 10).
Compound 9
[α]D=−127.4° (C=0.533% in ethanol).
Compound 10
[α]D=+121.0° (C=0.437% in ethanol).
………………………….
WO 2003076411
http://www.google.st/patents/WO2003076411A1?cl=en
http://www.google.st/patents/US20050107606?hl=pt-PT&cl=en

Example 21 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide
To a solution of 8-[4-(2-butoxyethoxy)phenyl]-1-isobutyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid (45 g) in tetrahydrofuran (135 ml) was added N,N-dimethylformamide (230 mg) and added dropwise thionyl chloride (12.45 g) at 10 to 15° C., and the resulting solution was stirred at the same temperature for 40 minutes to prepare an acid chloride.
Separately, to a solution of (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine in tetrahydrofuran (270 ml) was added pyridine (27.59 g), the resulting mixture was adjusted to 5° C. or lower, and then thereto was added dropwise the acid chloride solution at 5° C. or less, and the resulting mixture was stirred at the same temperature for 2 hours. To the mixture were added water (270 ml) and 20% aqueous citric acid solution (180 ml), tetrahydrofuran was distilled off under reduced pressure and the residue was extracted with ethyl acetate. The extract was sequentially washed with water, saturated sodium bicarbonate solution and water, and then the solvent was distilled off. To the residue was added ethyl acetate (360 ml), added heptane (360 ml) at 40° C. and added seed crystals of (−)-8-[4-(2-butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide (10 mg), and the mixture was stirred at 25° C. for 2 hours and stirred at 5° C. for 1 hour. The precipitated crystals were collected by filtration to obtain 63.97 g (yield: 92.1%) of the title compound. Melting point: 120-122° C.
Elemental analysis value: in terms of C41H52N4O4S
Calcd. value: C, 70.66; H, 7.52; N, 8.04.
Analytical value: C, 70.42; H, 7.52; N, 8.01
Industrial Applicability
According to the present invention, an optically active sulfoxide derivative having CCR5 antagonism or an intermediate compound thereof can be prepared without causing side reactions such as racemization and Pummerer rearrangement. In particular, Process 7 is industrially advantageous since it is possible to prepare an optically active Compound (II) by asymmetric oxidization in the presence of an optically active acid.
Example 20 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-propyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide.methanesulfonate
According to the same method as that described in Example 15, the title compound was produced from 8-[4-(2-butoxyethoxy)phenyl]-1-propyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid and (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine.
1H-NMR (CDCl3, δ, 300 MHz) 0.88-1.01 (9H, m), 1.37-1.42 (2H, m), 1.57-1.80 (8H, m), 2.63 (2H, br), 2.77 (3H, s), 3.27 (2H, br), 3.51-3.57 (4H, m), 3.77-3.86 (4H, m), 3.90-4.05 (1H, m), 4.14 (2H, t, J=4.6 Hz), 4.25 (1H, d, J=14.6 Hz), 6.73 (1H, s), 6.84 (1H, d, J=8.7 Hz), 6.93 (2H, d, J=8.8 Hz), 7.21 (2H, d, J=8.7 Hz), 7.40-7.48 (4H, m), 7.61 (1H, s), 7.89 (2H, d, J=8.7 Hz), 8.65 (1H, s), 9.27 (1H, br)
Elemental analysis value: in terms of C41H54N4O7S2
Calcd. value: C, 63.21; H, 6.99; N, 7.19; S, 8.23.
Analytical value: C, 63.00; H, 7.09; N, 7.41; S, 8.25
Example 15 (−)-8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-N-(4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenyl)-1,2,3,4-tetrahydro-1-benzazocine-5-carboxamide.methanesulfonate
8-[4-(2-Butoxyethoxy)phenyl]-1-isobutyl-1,2,3,4-tetrahydro-1-benzazocine-5-carboxylic acid (986 mg) was dissolved in tetrahydrofuran (3 ml) and thereto was added N,N-dimethylformamide (one drop). Subsequently, to the resulting solution was added dropwise oxalyl chloride (0.2 ml, 2.29 mmol) under ice-cooling and the mixture was stirred for 80 minutes under ice-cooling to prepare an acid chloride.
Separately, (−)-4-{[(1-propyl-1H-imidazol-5-yl)methyl]sulfinyl}phenylamine (689 mg) was added to tetrahydrofuran (7 ml) and the resulting solution was cooled to 5° C. To the solution was added dropwise pyridine (0.62 ml) and added dropwise the acid chloride solution at 3 to 5° C., and the mixture was stirred for 2 hours under ice-cooling. To the mixture was added water (20 ml) at 10° C. or lower and the mixture was extracted with ethyl acetate. The organic layer was sequentially washed with water, saturated sodium bicarbonate solution and water, and concentrated under reduced pressure. Thereto was added toluene and the mixture was concentrated under reduced pressure. Thereto was added acetonitrile and the mixture was concentrated under reduced pressure. The residue was dissolved in acetonitrile (7 ml) and acetone (7 ml), thereto was added dropwise methanesulfonic acid (209 mg), and added seed crystals and the mixture was stirred at room temperature for 100 minutes. Subsequently, to the mixture was added acetone-acetonitrile (1:1, 5 ml). After stirring at room temperature overnight, the mixture was stirred for 2.5 hours under ice-cooling. The precipitated crystals were collected by filtration and washed with the ice-cooled acetone (9 ml). The crystals were dried at 40° C. under reduced pressure to obtain 1.51 g (yield: 87%) of the title compound as yellow crystals.
1H-NMR (300 MHz, DMSO-d6, δ): 0.78-0.96 (12H, m), 1.25-1.40 (2H, m), 1.41-1.51 (4H, m), 1.65-1.85 (2H, m), 2.05-2.15 (1H, m), 2.30 (3H, s), 2.35-2.50 (2H, m), 3.05-3.15 (2H, m), 3.30-3.55 (4H, m), 3.65-3.70 (2H, m), 3.90-4.05 (2H, m), 4.05-4.10 (2H, m), 4.30 (1H, d, J=14.73 Hz), 4.65 (1H, d, J=14.73 Hz), 6.85 (1H, d, J=8.97 Hz), 6.97 (1H, d, J=8.79 Hz), 7.17 (1H, s), 7.35-7.75 (6H, m), 7.92 (2H, d, J=8.79 Hz), 9.08 (1H, s), 10.15 (1H, s).
Elemental analysis value: in terms of C41H52N4O4S.CH4SO3
Calcd. value: C, 63.61; H, 7.12; N, 7.06; S, 8.09.
Found value: C, 63.65; H, 7.23; N, 7.05; S, 8.08.
………………………….
References
- Klibanov, Olga M.; Williams, Shannon H.; Iler, Cameron A (2010). “Cenicriviroc, an orally active CCR5 antagonist for the potential treatment of HIV infection”. Current Opinion in Investigational Drugs 11 (8): 940–950. PMID 20721836.
- Baba, Masanori; Takashima, Katsunori; Miyake, Hiroshi; Kanzaki, Naoyuki; Teshima, Koichiro; Wang, Xin; Shiraishi, Mitsuru; Iizawa, Yuji (2005). “TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans”. Antimicrobial Agents and Chemotherapy 49 (11): 4584–4591. doi:10.1128/AAC.49.11.4584-4591.2005. PMC 1280155. PMID 16251299.
- C. Reviriego (2011). Drugs of the Future 36 (7): 511–517. doi:10.1358/dof.2011.36.7.1622066.
- “Tobira Therapeutics Initiates Phase 2b Trial of Cenicriviroc”. The Body. July 5, 2011.
- CROI 2013: CCR5/CCR2 Inhibitor Cenicriviroc Has Both Anti-HIV and Anti-inflammatory Effects. Highleyman, Liz. HIVandHepatitis.com. 7 March 2013.
|
11-26-2012
|
Chemokine receptor antagonists.
|
Journal of medicinal chemistry
|
|
|
6-1-2011
|
Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects.
|
Journal of acquired immune deficiency syndromes (1999)
|
|
|
8-1-2010
|
Cenicriviroc, an orally active CCR5 antagonist for the potential treatment of HIV infection.
|
Current opinion in investigational drugs (London, England : 2000)
|
|
|
3-1-2009
|
The relative activity of “function sparing” HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property?
|
Molecular pharmacology
|
|
|
2-1-2007
|
Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652.
|
Antimicrobial agents and chemotherapy
|
|
|
9-10-2006
|
[Progress in AIDS therapy].
|
Nihon Naika Gakkai zasshi. The Journal of the Japanese Society of Internal Medicine
|
|
|
3-23-2006
|
Highly potent and orally active CCR5 antagonists as anti-HIV-1 agents: synthesis and biological activities of 1-benzazocine derivatives containing a sulfoxide moiety.
|
Journal of medicinal chemistry
|
|
|
11-1-2005
|
TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans.
|
Antimicrobial agents and chemotherapy
|
|
|
1-27-2005
|
Stereoselective synthesis of [L-Arg-L/D-3-(2-naphthyl)alanine]-type (E)-alkene dipeptide isosteres and its application to the synthesis and biological evaluation of pseudopeptide analogues of the CXCR4 antagonist FC131.
|
Journal of medicinal chemistry
|
|
|
1-1-2005
|
TAK-652, a novel CCR5 inhibitor, has favourable drug interactions with other antiretrovirals in vitro.
|
Antiviral therapy
|
……………….
Chemical structures of selected small molecule CCR5 inhibitors. A. Maraviroc (MVC, Selzentry), B. Vicriviroc (VCV), C. Cenicriviroc (TBR-652), D. PF-232798.

ACH-702 the isothiazoloquinolone in preclinical from Achillion Pharmaceuticals (USA)

ACH-702
7-[3(R)-(2-Aminopropan-2-yl)pyrrolidin-1-yl]-9-cyclopropyl-6-fluoro-8-methoxy-2,3,4,9-tetrahydroisothiazolo[5,4-b]quinoline-3,4-dione
(7^-7-[3-(1-AMrNO-I-METHYLETHYL)PYRROLiDiN-I-YL]-P-CYCLOPROPYL-6-FLUORO-8-METHOXY-PH-ISOTHIAZOLO[5,4-B]QUINOLINE-3,4-DIONE
(R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-l-yl]-9-cyclopropyl-6-fluoro-8-methoxy- 9H-isothiazolo[5, 4-b] -quinoline-3, 4-dione
922491-46-1 free base
922491-09-6 (hydrochloride)468.973, C21 H25 F N4 O3 S . Cl H
ACH-0139586
ACH-702
Achillion Pharmaceuticals (USA)

pre clinical
Achillion Pharmaceuticals is working on the discovery of compounds in a new subclass of quinolones, the isothiazoloquinolones. The most advanced compound is ACH-702, which is at the pre-clinical stage of development [1-3].
 ACH 702
ACH 702
The utility of isothiazoloquinolines as pharmaceutical agents has been discussed in the literature. For example, Pinol, et al discussed the use of isothiazoloquinolines as medical bactericides in US Patent 5,087,621, including

The Proctor & Gamble Company discussed antimicrobial quinolones including the following compound:

in published application no. US 2003008894.
The use of isothiazoloquinoline compounds as TNF production inhibitors has also been discussed, for example by Sankyo Co., Ltd. in JPl 010149, which includes the following compound

Bayer Aktiengesellschaft has discussed bicycle[3.3.0]oct-7-yl containing compounds useful for treating H. pylori infections in WO 98/26768, including isothiazoloquinolines, having the general structure shown below in which Y may be sulfur joined to the carboxamide group to form a 5-membered ring

Otsuka Pharmaceutical Co., Ltd. has discussed the use of isothiazoloquinolines as antibacterial agents in JP 01193275, including the following carbamate-containing compound

Abbott Laboratories has discussed the use of isothiazoloquinolines as antineoplastic agents in US Patent No. 5,071,848 and has discussed the use of tricyclic quinolones as antibacterial agents in US 4,767,762. The Abbott compounds have hydrogen, halogen, or lower alkyl as substituents at the 6- and 8- positions of the isothiazoloquinoline core.
………………
Synthesis
WO2008021491A2
http://www.google.com/patents/WO2008021491A2?cl=en
EXAMPLE 1. SYNTHESIS OF (7^-7-[3-(1-AMrNO-I-METHYLETHYL)PYRROLiDiN-I-YL]-P-
CYCLOPROPYL-6-FLUORO-8-METHOXY-PH-ISOTHIAZOLO[5,4-B]QUINOLINE-3,4-DIONE (5). Step 1. Ethyl l-cyclopropyl-6, 7-difluoro-2-methanesulfonyl-8-methoxy-4-oxo-l,4-dihydro- quinoline-3-carboxylate (6)
Oxonβ®
MeOHZH2O


6
Water (180 mL), followed by Oxone® (Dupont Specialty Chemicals) (170 g, 277 mmol), is added to a suspension of 1 in MeOH (510 mL). The reaction mixture is heated with stirring at 55-60 0C for 3 h. The reaction mixture is cooled to room temperature, diluted with water (40 mL), and stirred at 5 0C (ice bath) for 30 min. The resulting crystals are collected by filtration, washed with water (2 x 100 mL), and dried to afford 6 (13.8 g). This material was used in the next step without further purification, mp 177-178 0C. 1H NMR (DMF-^7): J0.62 (m, IH), 1.11 (m, 2H), 1.29 (m, IH), 1.32 (t, JH-H = 7.0 Hz, 3H), 3.76 (s, 3H), 4.18 (m, IH), 4.21 (d, JH-F = 2.0 Hz, 3H), 4.33 (q, JH–H = 7.0 Hz, 2H), 7.64 (dd, JH-F = 10.0 Hz, 8.5 Hz, IH). Step 2 (R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-yl]-l-cyclopropyl-6-fluoro-2- methanesulfonyl-8-methoxy-4-oxo-l , 4-dihydro-quinoline-3-carboxylic acid ethyl ester (7)

10 6 7
A mixture containing compound (6) (3.88 g, 9.67 mmol), compound 10 (1.64 g, 12.8 mmol), anhydrous DIEA (5.05 g, 39.1 mmol, dried over 4A sieves), and anhydrous DMF (40 mL) is heated at 70 0C under an atmosphere of argon gas. After heating for 4.5 h (LC-MS analysis shows ~7% compound (6) remained), the reaction mixture is cooled to room temperature, diluted with EtOAc (200 mL), and washed with water (100 mL). The aqueous layer is extracted with EtOAc (100 mL), and the combined organic layers are washed with a saturated aqueous solution of sodium bicarbonate (100 mL). The organic layer is diluted with water (100 mL) and treated with an aqueous solution of HCl (4 N) until the aqueous layer is acidic (pH 2—3 after shaking the mixture vigorously). The organic layer is separated, and this process is repeated. The combined aqueous layers are diluted with EtOAc (100 mL) and treated with an aqueous solution of sodium hydroxide (6 N) until the aqueous layer is basic (pH ~8 after shaking the mixture vigorously). The aqueous layer is separated, and this process is repeated. The combined organic layers are dried over magnesium sulfate, filtered, and concentrated under reduced pressure giving an orange solid (3.27 g of an~80:20 mixture of compound (7) and impurity B). This solid is recrystallized from hot EtOAc (~60 mL) furnishing 2.18 g (44% yield) of pure compound 7 as a bright yellow solid. LC-MS mlz calcd for C24H32FN3O6S 509 ([M+]); found 510 ([M + H]+).
This reaction should not be allowed to proceed for more than a few hours (not overnight) as prolonged reaction time can lead to the formation of more side products. The product should be —95% pure (based on HPLC), with only a trace amount of impurity B. Step 3. (R)-7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-yl]-l-cyclopropyl-6-fluoro-2-mercapto-8- methoxy-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid ethyl ester (8)

7 8
Compound 7 (1.04 g, 2.04 mmol) is partially dissolved in DME (40 mL) under an atmosphere of argon. Sodium hydrosulfide hydrate (Aldrich, 72.6% by titration, 465 mg, 6.02 mmol) in water (3.0 mL) is added to this solution. The resulting mixture is sparged slowly with argon for 30 min.
The progress of the reaction is monitored by HPLC-MS, and judged to be complete (<2% of 7 remains) after 11.5 h. Excess sodium hydrosulfide is quenched upon addition of aq HCl (4.5 mL, 4 N).
The resulting orange solution (pH ~2) is sparged with argon (30 min) to remove the generated hydrogen sulfide. Step. 4 (R)- 7-[3-(l-amino-l-methyl-ethyl)-pyrrolidin-l-yl]-9-cyclopropyl-6-fluoro-8-methoxy- 9H-isothiazolo[5, 4-b] -quinoline-3, 4-dione (5)

A solution of potassium carbonate (4.26 g, 30.8 mmol) in water (25 mL) is next added to this solution to give a clear yellow solution (pH 9-10). The clear yellow solution is then sparged with argon for ~5 min. Finally, hydroxylamine-0-sulfonic acid (0.93 g, 8.2 mmol) is added portionwise as a solid, with immediate evolution of gas and formation of the product as a yellow precipitate. After stirring for 16 h, the reaction mixture (pH 10.2) is acidified with aq HCl to pH 8.3 (the approximate isoelectric point of 5) causing additional product to precipitate from solution. The reaction mixture is concentrated under reduced pressure (final volume -40 mL). The yellow precipitate is collected by centrifugation, washed with water (3 x 40 mL, with sonication), and lyophilized to give 0.80 g of 5.
……………………..
WO2007014308A1
http://www.google.com/patents/WO2007014308A1?cl=en
EXAMPLE 5. SYNTHESIS OF I-METHYL-I-PYRROLIDIN-3-YL ETHYLAMINE (5)
1 -Methyl- l-pyrrolidin-3-yl-ethylamine is prepared in accordance with the synthetic scheme below.
N O

5 P
Step 1. Synthesis of (S)-l-benzylpyrrolidin-3-yl methanesulfonate (N).
Methanesulfonyl chloride (15 mL, 0.19 mol) is added to a cooled (0 0C) solution of toluene (300 mL) containing (5)-l-benzylpyrrolidin-3-ol (24.5 g, 0.14 mol) and triethylamine (80 mL, 0.57 mol). The resulting mixture is stirred at 0 °C for 15 min, and allowed to warm to room temperature with stirring for 2h. The mixture is quenched with a 5% aqueous solution of sodium bicarbonate (250 mL). The organic layer is washed with a 5% aqueous solution of sodium bicarbonate (2 x 250 mL), washed with water (I x 250 mL), dried over magnesium sulfate, and concentrated under reduced pressure to give N (35.1 g, 99 %) as an orange oil. 1H NMR (300 MHz, CDCl3): £2.07 (m, IH), 2.30 (m, IH), 2.49 (m, IH), 2.75-2.90 (m, 3H), 2.98 (s, 3H), 3.61 (d, J= 13.0 Hz, IH), 3.68 (d, J= 13.0 Hz, IH), 5.18 (m, IH), 7.15-7.30 (m, 5H). LCMS mlz calcd for C12H17NO3S 255 ([M+]); found 256 ([M + H]+, 100%), 160 (40%). Steps 2 and 3. Syntheses of(R)-l-benzylpyrrolidine-3~carbonitrile (O) and 2-((R)-I- benzylpyrrolidin-3-yl)propan-2-amine (P).
The syntheses of O and P are described previously by Fedij et al. (Fedij, V.; Lenoir, E. A., Ill; Suto, M. J.; Zeller, J. R.; Wemple, J. Tetrahedron: Asymmetry 1994, J, 1131- 1134). Step 4. Synthesis ofl-((R)-Methyl~l-pyrrolidin-3-yl)-ethylamine (5).
A mixture containing P (7.4 g), 20% palladium hydroxide on carbon (7.5 g), and ethanol (75 niL) is stirred under an atmosphere of hydrogen gas (50 psi) at 45 °C for 24 h. The mixture is filtered and the filtrate is concentrated under reduced pressure to give 5 (4.1 g, 95 %) as a yellow oil. This material is stored under an atmosphere of argon gas. 1H NMR (300 MHz, CDCl3): J1.09 (s, 6H), 1.51 (m, IH), 1.64 (br s, 3H), 1.81 (m, IH), 2.06 (apparent pentet, J= 8.5 Hz, IH), 2.69 (dd, J= 11.0 Hz, J= 8.5 Hz, IH), 2.94 (m, 2H), 3.00 (dd, J= 11.0 Hz, J= 8.5 Hz, IH). LCMS mlz calcd for C7H16N2 128 ([M+]); found 129 ([M + H]+, 60%), 112 (100%).
EXAMPLE 6. GENERAL METHOD FOR THE FINAL AMINE-COUPLING STEP: SYNTHESIS OF 7-((R)-3-
(2-AMINOPROPAN-2-YL)PYRROLIDIN- 1 -YL)-9-CYCLOPROPYL-6-FLUORO-8- METHOXYISOTHIAZOLO[5,4-B]QUINOLINE-3 ,4(2H,9H)-DIONE HYDROCHLORIDE
[0261 ] 7-((R)-3-(2-Aminopropan-2-yl)pyrrolidin- 1 -yl)-9-cyclopropyl-6-fluoro-8- methoxyisothiazolo[5,4-b]quinoline-3,4(2H,9H)-dione hydrochloride is prepared in accordance with the synthetic scheme below.

Synthesis ofJ-ffRJS-^-aminopropan^-ylJpyrrolidin-l-ylJ-P-cyclopropyl-o-fluoro-S- methoxyisothiazolofS, 4-bJguinoline-3,4(2H, 9H)-dione hydrochloride (6).
Under an atmosphere of argon, a reaction vessel is charged with 5 (206.0 mg, 1.6 mmol), 3 (328.6 mg, 1.0 mmol), dimethyl sulfoxide (4.5 mL), and ΛζN-diisopropylethylamine (750 μL, 4.3 mmol). The resulting mixture is irradiated with microwaves (CEM Discover) at 125 0C for 1 h (conventional heating may also be used — 115 °C in an oil bath for 14 h), allowed to cool, and evaporated to dryness under reduced pressure (-70 °C/2-3 mm Hg). The oily residue is triturated with ethyl acetate (15 mL) and the resulting powder is collected by centrifugation. This solid is purified using preparative HPLC to give the desired product. Preparative HPLC is performed using a YMC Pack Pro C18 150 x 30.0 mm 5//m column coupled to a YMC Pack Pro 50 x 20 mm 5/an column with an isocratic elution of 0.37 min at 95:5 H2OiCHsCN containing 0.1% TFA followed by a 15.94 min linear gradient elution from 95:5 to 25:75, followed by a 0.69 min linear gradient from 25:75 to 5:95 at a flow rate of 30.0 mL/min with UV detection at 254. The crude material is loaded as a solution containing acetic acid (~2 mL), methanol (~1 mL), and water (~1 mL). The purified product is isolated as the TFA salt and is converted to the corresponding hydrochloride salt by addition of a solution of hydrogen chloride (~1.25 M in methanol) followed by evaporation; this process is repeated twice to give a yellow solid. Purity by HPLC: >99%; tR = 10.08 min. 1H NMR (300 MHz, TFA-d): δ 1.28 (m, 2H), 1.53 (m, 2H), 1.66 (s, 6H), 2.43 (m, IH), 2.57 (m, IH), 3.35 (m, IH), 3.97 (s, 3H), 4.01-4.38 (m, 5H), 8.17 (d, J= 12.0 Hz, IH, aromatic). 19F(1H) (282 MHz, TFA-J): δ-\ 18.0 (s). 13C(1H) (75 MHz, TFA-d): £13.5, 13.9, 25.0, 25.1, 29.1, 39.7, 49.6, 59.4 (br, W1/2 « 14 Hz), 59.8 (br, W1/2 « 14 Hz), 60.0, 66.8, 106.0, 112.1 (dJc_F = 23.0 Hz), 137.5 (br m, W1/2 « 24 Hz), 138.4, 144.8 (br, W1/2 » 10 Hz), 155.3 (dJc_F = 255.0 Hz), 169.8, 170.1, 171.5 (br, W1/2 « 9 Hz). LCMS mlz calcd for C21H25FN4O3S 432 ([M+]); found 433 ([M + H]+). Anal. Calcd for C21H25FN4O3S-l.5HCM.5H2O: C, 49.05; H, 5.78; N, 10.90; Cl, 10.34. Found: C, 49.30; H, 5.60; N, 10.83; Cl, 10.00.
EXAMPLE 3. SYNTHESIS OF 9-CYCLθPRθPYL-6,7-DiFLUθRθ-8-METHθχγ-9H-isoτHiAzθLθ[5,4- 5]QUlNOLlNE-3,4-DIONE (Compound 3).
9-Cyclopropyl-6,7-difluoro-8-methoxy-9H-isothiazolo[5,4-&]quinoline-3,4-dione (3) is prepared in accordance with the synthetic scheme below.

Step 1. Synthesis of 2,4, 5-trifluoro-3-methoxybenzoyl chloride (A)
A mixture of 2,4,5-trifluoro-3-methoxybenzoic acid (154 mg, 0.75 mmol) and thionyl chloride (8 mL) is refluxed for 4 h. Excess thionyl chloride is removed in vacuo, and the remaining residue is used directly in the next synthetic step. Step 2. Synthesis of (Z)-ethyl 3-hydroxy-3-(2,4,5-trifluoro-3-methoxyphenyl)aaγlate (B).
Compound B is prepared using the general method of Wierenga and Skulnick (Wierenga, W.; Skulnick, H. I. J. Org. Chein. (1979) 44: 310-311). H-Butyllithium (1.6 M in hexanes) is added to a cooled (-78 °C) solution of tetrahydrofliran (10 mL) containing ethyl hydrogen malonate (180 juL, 1.50 mmol) and 2,2′-bipyridyl (~1 mg as indicator). The temperature of the reaction mixture is allowed to rise to ca. -5 0C during the addition of n- butyllithium. Sufficient n-butyllithium (2.8 mL, 4.48 mmol) is added until a pink color persists at -5 0C for 5-10 min. A solution of 2,4,5-trifluoro-3-methoxybenzoyl chloride (0.75 mmol, vide supra) in tetrahydrofuran (~3 mL) is added in one portion to the reaction mixture that had been recooled to -78 0C. The resulting mixture is allowed to warm to room temperature, diluted with ethyl acetate (50 mL), and quenched with a 1 M aqueous solution of hydrochloric acid. The organic layer is washed with a 5% aqueous solution of sodium bicarbonate (2 x 30 mL), followed by brine (2 x 50 mL), dried over sodium sulfate, and evaporated under reduced pressure to give the crude product. This material is purified by flash column chromatography (eluting with 20% v/v ethyl acetate in hexanes) to give pure B as a white solid. 1H NMR (300 MHz, CDCl3): (enol, predominant tautomer, >90%) δ 1.32 (t, JH–H = 7.0 Hz, 3H, CO2CH2CH3), 4.02 (apparent t, JH–F = 1.0 Hz, 3H, OCH3), 4.25 (q, JH–H = 7.0 Hz, 2H, CO2CH2CH3), 5.79 (s, IH, CH3C(OH)=CH- CO2CH2CH3), 7.39 (ddd, JH_F= 11.0 Hz, 8.5 Hz, 6.5 Hz, IH, aromatic), 12.68 (s, IH, OH). 19F(1H) NMR (282 MHz, CDCl3): <5-146.8 (dd, JF_F = 21.5 Hz, 10.5 Hz, IF), -140.2 (dd, JF_F = 21.5 Hz, 13.5 Hz, IF), -131.3 (dd, JF_F = 13.5 Hz, 10.5 Hz, IF).
Step 3. Synthesis ofζEyethy^-^ZJ-N-cyclopropy^methylthioJcarbonoimidoylJS-hydroxyS- (2, 4, 5-trifluoro-3-methoxyphenyl)acrylate (C)
Sodium hydride (60% in mineral oil, 31 mg, 0.78 mmol) is added portionwise to a cooled (0 °C) solution containing B (200 mg, 0.73 mmol), cyclopropyl isothiocyanate (120 /JL, 1.2 mmol), and dimethylformamide (2 mL). The resulting mixture is allowed to warm to room temperature with stirring overnight (18 h). Methyl iodide (80 juL, 1.2 mmol) is added to the resulting solution and stirred for an additional 4 h (until TLC indicated the complete consumption of B). The reaction mixture is diluted with ethyl acetate (100 mL) and quenched by addition of a saturated aqueous solution of ammonium chloride (30 mL). The organic layer is washed with brine (4 x 30 mL), dried over sodium sulfate, and evaporated under reduced pressure to give the crude product. This material is purified by flash column chromatography (eluting with 40% v/v ethyl acetate in hexanes) to give C as a yellow oil. 1H NMR (300 MHz, CDCl3): (50.86 (m, 2H, cyclopropyl CH2), 0.97 (m, 5H), 2.52 (s, 3H, SCH3), 3.00 (m, IH, cyclopropyl CH), 3.96 (q, JH–H = 7.0 Hz, 2H, CO2CH2CH3), 4.02 (apparent t, JH–F = 1.0 Hz, 3H, OCH3), 6.96 (m, IH, aromatic), 11.71 (s, IH). 19F(1H) NMR (282 MHz, CDCl3): £-149.9 (br, IF), -141.4 (br, IF), -135.7 (br, IF).
Step 4. Synthesis of ethyl l-cyclopropyl-6,7-difluoro-8-methoxy-2-(methylthio)-4-oxo-l,4- dihydroquinoline-3-carboxylate (D)
Sodium hydride (60% in mineral oil, 82 mg, 2.1 mmol) is added portionwise to a solution of C (760 mg, 1.95 mmol) in dimethylformamide (15 mL) at room temperature. The reaction mixture is heated at 80 0C for 3 d (until TLC indicates the complete consumption of B), cooled to room temperature, and quenched by addition of a saturated aqueous solution of ammonium chloride (10 mL). The mixture is extracted with ethyl acetate (3 x 50 mL). The combined organic extracts are washed with brine (4 x 30 mL), dried over sodium sulfate, and evaporated under reduced pressure to give crude D. This material is purified by flash column chromatography (eluting with 30% v/v ethyl acetate in hexanes) to D as a pale yellow oil.1H NMR (300 MHz, CDCl3): £0.73 (m, 2H, cyclopropyl CH2), 1.19 (m, 2H, cyclopropyl CH2), 1.38 (t, JH–H = 7.0 Hz, 3H, CO2CH2CH3), 2.66 (s, 3Η, SCH3), 3.74 (m, IH, cyclopropyl CH), 4.08 (d, JH–F = 2.5 Hz 3H, OCH3), 4.40 (q, JH_H = 7.0 Hz, 2H, CO2CH2CH3), 7.76 (dd, JH_F = 10.5 Hz, 8.5 Hz IH, aromatic). 19F(1H) NMR (282 MHz, CDCl3): £-146.8 (d, JF_F = 21.0 Hz, IF), – 137.7 (d, JF–F = 21.0 Hz, IF). LCMS mlz calcd for C17H17F2NO4S 369 ([M+]); found 370 ([M + H]+).
Step 5. Synthesis of ethyl l-cyclopropyl-6,7-difluoro-8-methoxy-2-(methylsulfinyl)-4-oxo-l,4- dihydroquinoline-3-carboxylate (E)
m-Chloroperoxybenzoic acid (<77%, 34 mg, 0.15 mmol) is added in one portion to a solution of D (50 mg, 0.14 mmol) in methylene chloride (3 mL) at room temperature. The reaction mixture is stirred for 1 h, diluted with ethyl acetate (20 mL), and washed with a 5% aqueous solution of sodium bicarbonate (2 x 10 mL). The organic layer is dried over sodium sulfate and evaporated under reduced pressure to give the crude product. This material is purified by preparative thin-layer chromatography (eluting with 10% v/v hexanes in ethyl acetate) to give pure E as a white solid. 1H NMR (300 MHz, CDCl3): £0.62 (m, IH, cyclopropyl CH2), 1.00 (m, IH, cyclopropyl CH2), 1.13 (m, IH, cyclopropyl CH2), 1.29 (m, IH, cyclopropyl CH2), 1.36 (t, JH_H = 7.5 Hz, 3H, CO2CH2CH3), 3.22 (s, 3Η, S(O)CH3), 3.85 (m, IH, cyclopropyl CH), 4.09 (d, JH-F = 2.5 Hz, 3H, OCH3), 4.37 (q, JH–H = 7.5 Hz, 2H, CO2CH2CH3), 7.75 (dd, JH–F = 10.0, 8.0 Hz, IH, aromatic). 19F(1H) NMR (282 MHz, CDCl3): £-145.2 (d, JF_F = 21.0 Hz, IF), -136.2 (d, JF_F = 21.0 Hz, IF). LCMS mlz calcd for C17H17F2NO5S 385 ([M+]); found 386 ([M + H]+).
Step 6. Synthesis of ethyl l-cyclopropyl-βJ-difluoro-l-mercaptoS-methoxy-^oxo-lA- dihydroquinoline-3-carboxylate (F).
Anhydrous sodium hydrogen sulfide (Alfa Aesar, 20 mg, 0.36 mmol) is added in one portion to a solution of DMF (6 mL) containing E (93 mg, 0.24 mmol) at room temperature. The resulting solution is heated at 40 0C for 2-3 h (until TLC indicated complete consumption of E) and allowed to cool to room temperature. The reaction mixture is quenched by addition of a 5% aqueous solution of hydrochloric acid (20 mL) and extracted with ethyl acetate (2 x 25 mL). The combined organic extracts are washed with brine (4 x 25 mL), dried over sodium sulfate, and evaporated to dryness under reduced pressure to give crude F in quantitative yield. This material is used directly in the next synthetic step to prevent its oxidative degradation. LCMS mlz calcd for C16H15F2NO4S 355 ([M+]); found 356 ([M + H]+) Step 7. Synthesis of9-cyclopropyl-6,7-difluoro-8-methoxyisothiazolo[5,4-b]quinoline- 3,4(2H,9H)-dione (3).
A solution of sodium bicarbonate (820 mg, 9.8 mmol) in water (14 mL) is added to a solution of F (348 mg, 0.98 mmol) in tetrahydrofuran (10 mL) at room temperature. Hydroxylamine-O-sulfonic acid (465 mg, 4.1 mmol) is added in one portion to this mixture. The reaction mixture is stirred at room temperature for ~3 h and quenched by addition of an aqueous solution of 5% hydrochloric acid (100 mL). The precipitate that formed is collected by filtration, washed with water (3 x 5 mL), and dried in vacuo to give 3 as a white solid. This product is of sufficient purity (>95% by 1H NMR spectroscopy) to use directly in the final amine-coupling step. 1HNMR (300 MHz, DMSO-J6): Jl.12 (m, 4H, cyclopropyl CH2), 3.85 (m, IH, cyclopropyl CH), 4.01 (d, JH–F= 1.5 Hz, 3H, OCH3), 7.85 (dd, JH_F = 11.0 Hz, 9.0 Hz, IH, aromatic). 19F(1H) NMR (282 MHz, DMSO-J6): £-146.4 (d, JF_F = 23.0 Hz, IF), -140.2 (d, JF_ F = 23.0 Hz, IF). LCMS mlz calcd for C14H10F2N2O3S 324 ([M*]); found 325 ([M + H]+).
REFERENCES
- Achillion Pharmaceuticals. About ACH-702. Available online: http://www.achillion.com/PL/pdf/04_ach_702_bg.pdf (accessed on 2 May 2013).
- Pucci, M.J.; Podos, S.D.; Thanassi, J.A.; Leggio, M.J.; Bradbury, B.J.; Deshpande, M. In vitro and in vivoprofiles of ACH-702, an isothiazoloquinolone, against bacterial pathogens. Antimicrob. Agents Chemother. 2011, 55, 2860–2871, doi:10.1128/AAC.01666-10.
- Achillion Pharmaceuticals, Inc. SEC filling form 10-Q quarterly report filed August 7, 2013. Available online: http://ir.achillion.com/secfiling.cfm?filingID=1193125–13–324297 (accessed on 28 September 2013).
- An efficient method for the synthesis of (R)-3-(1-amino-1-methylethyl)pyrrolidines for the antiinfective agent, PD 138312
Tetrahedron Asymmetry 1994, 5(7): 1131 - WO 2007014308
- WO 2008021491
-
WO2011031745A1 Sep 8, 2010 Mar 17, 2011 Achaogen, Inc. Antibacterial fluoroquinolone analogs -
HASHIMOTO, A. ET AL.: “Practical synthesis and molecular structure of a potent broad-spectrum antibacterial isothiazoloquinolone” ORG. PROCESS RESEARCH & DEVELOPMENT, vol. 11, 16 March 2007 (2007-03-16), pages 389-398, XP002465315 2 * WANG, Q. ET AL.: “Isothiazoloquinolones with Enhanced Antistaphylococcal Activities against Multidrug-Resistant Strains: Effects of Structural Modifications at the 6-, 7-, and 8-Positions” J. MED. CHEM., vol. 50, 2007, pages 199-210, XP002465316 -
WO2005019228A1 * Aug 4, 2004 Mar 3, 2005 Achillion Pharmaceuticals Inc Isothiazoloquinolones and related compounds as anti-infective agents WO2006118605A2 * Nov 10, 2005 Nov 9, 2006 Achillion Pharmaceuticals Inc 8a, 9-dihydro-4a-h-isothiazolo[5,4-b] quinoline-3, 4-diones and related compounds as anti-infective agents WO2007014308A1 * Jul 27, 2006 Feb 1, 2007 Achillion Pharmaceuticals Inc 8-methoxy-9h-isothiazolo[5,4-b]quinoline-3,4-diones and related compounds as anti-infective agents -
Citing Patent Filing date Publication date Applicant Title WO2008021491A2 * Aug 16, 2007 Feb 21, 2008 Achillion Pharmaceuticals Inc Method for synthesis of 8-alkoxy-9h-isothiazolo[5,4-b]quinoline-3,4-diones WO2011031745A1 Sep 8, 2010 Mar 17, 2011 Achaogen, Inc. Antibacterial fluoroquinolone analogs EP2488532A2 * Oct 15, 2010 Aug 22, 2012 Rib-X Pharmaceuticals, Inc. Antimicrobial compounds and methods of making and using the same US7902365 Aug 16, 2007 Mar 8, 2011 Achillion Pharmaceuticals, Inc. Method for synthesis of 8-alkoxy-9H-isothiazolo[5,4-B]quinoline-3,4-diones US8138346 Mar 4, 2011 Mar 20, 2012 Achillion Pharmaceuticals, Inc. Method for synthesis of 8-alkoxy-9H-isothiazolo[5,4-B]quinoline-3,4-diones
MG 96077 in Pre-Clinical for Gram-negative bacteria

MG 96077
poster
MG96077 – MethylGene
………..http://methylgene.solocom.biz/files/2011/10/poster102.pdf ……………..lot of data presented
Mirati Therapeutics (USA)
Pre-Clinical for Gram-negative bacteria
Beta-Lactamase Inhibitors—Non-beta-Lactam Phosphonate-Based
Mirati Therapeutics is seeking partners to continue the development of the compound MG96077, a non-beta-lactam phosphonate-based beta-lactamase inhibitor that has shown an inhibitory profile for both class A and class C beta-lactamase enzymes [1,2].
Potent, irreversible inhibitor of serine β-lactamases that efficiently protects βlactams from hydrolysis in a variety of class
A- and class C-producing organisms-
 |
September 14, 2009 13:23 ET
MethylGene Presents Preclinical Data for Its Beta-Lactamase Inhibitor, MG96077, at the 49th Annual ICAAC Meeting
MONTREAL, QUEBEC–(Marketwire – Sept. 14, 2009) – MethylGene Inc. (TSX:MYG) today disclosed preclinical data for MG96077, a novel, broad spectrum, non-beta-lactam beta-lactamase inhibitor (BLI). MG96077 possesses a broad-spectrum inhibitory profile for both class A and class C beta-lactamase enzymes, including extended spectrum beta-lactamases (ESBLs). In addition, the compound overcomes resistance in beta-lactam-resistant organisms such as Pseudomonas aeruginosa. The data were presented in a poster session at the 49th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) Annual Meeting in San Francisco, California.
Poster C1-1373: Novel Beta-Lactamase Inhibitor Potentiates and Extends the Antibacterial Activity of Imipenem against Beta-Lactam-Resistant P. aeruginosa and K. pneumoniae
MG96077 was tested in combination with imipenem, a commonly-used antibiotic agent for a variety of serious infections.
A series of in vitro and in vivo preclinical studies focused on comparing the combination of MG96077 and imipenem to imipenem alone, or imipenem plus currently approved BLIs, were performed. Greater than 90 percent of imipenem-resistant clinical isolates of Pseudomonas aeruginosa and Klebsiella pneumoniae were rendered susceptible with the addition of MG96077 to imipenem. The combination of imipenem and any of the three currently approved BLIs did not achieve greater than 61 percent coverage.
Furthermore, the combination of imipenem and MG96077 in vivo demonstrated 3-6 log reduction in colony forming units (CFU) and a 100 percent survival rate in combating imipenem-resistant P. aeruginosa infections of mouse spleen and lung. The pharmacokinetic properties of MG96077 were similar to imipenem in preclinical studies with no observable drug-drug interactions.
Thus, MG96077 is a novel beta-lactamase inhibitor that restores efficacy to imipenem against a high percentage of imipenem-resistant Pseudomonas and Klebsiella strains and, therefore, may address the clinical need for antibacterial therapies with more potent coverage of resistant gram-negative organisms.
MethylGene retains exclusive rights to MG96077 and a series of related molecules. Additional data has been developed regarding MG96077 compared to other beta-lactam antibiotics, as well as other compounds in the series paired with various beta-lactam antibiotics.
“Antibiotic resistance rates are increasing among several problematic gram-negative pathogens, including P. aeruginosa, K. pneumoniae, Acinetobacter spp. and Enterobacteriaceae that are often responsible for serious hospital-acquired infections. In these studies, MG96077 appears to demonstrate activity in a variety of organisms and we look forward to further evaluation of this compound in what is a growing antibiotic market in need of novel treatments,” said Donald F. Corcoran, President and Chief Executive Officer of MethylGene.
About MethylGene
MethylGene Inc. (TSX:MYG) is a publicly-traded, clinical stage, biopharmaceutical company focused on the discovery, development and commercialization of novel therapeutics with a focus on cancer. The Company’s product candidates include: MGCD265, an oral, multi-targeted kinase inhibitor targeting the c-Met, VEGF, Ron and Tie-2 receptor tyrosine kinases that is in Phase I and Phase II clinical trials for cancers; MGCD290, a fungal Hos2 inhibitor being developed for use in combination with fluconazole for serious fungal infections that is in Phase I clinical studies; and MGCD0103, an oral, isoform-selective HDAC inhibitor which has been in multiple clinical trials for solid tumors and hematological malignancies and is licensed to Taiho Pharmaceutical Co. Ltd. A fourth compound discovered using MethylGene’s HDAC platform, EVP-0334 – a potential cognition enhancing agent, is in a Phase I study sponsored by EnVivo Pharmaceuticals Inc. MethylGene also has a funded collaboration with Otsuka Pharmaceutical Co. Ltd. for applications in ocular diseases using the Company’s proprietary kinase inhibitor chemistry. Please visit our website at www.methylgene.com.
- Martell, L.A.; Rahil, G.; Vaisburg, A.; Young, K.; Hickey, E.; Hermes, J.; Dininno, F.; Besterman, J.M. A Novel Beta-Lactamase Inhibitor Potentiates and Extends the Antibacterial Activity of Imipenem against β-Lactam-Resistant P. aeruginosa and K. pneumoniae. In Proceedings of 49th ICAAC Annual Meeting, San Francisco, CA, USA, 14 September 2009.
- Mirati Therapeutics. MG96077. Available online: http://mirati.com/other-pipeline-assets/mg96077(accessed on 9 July 2013).
- 49th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) Annual Meeting in San Francisco, California.
Poster C1-1373: Novel Beta-Lactamase Inhibitor Potentiates and Extends the Antibacterial Activity of Imipenem against Beta-Lactam-Resistant P. aeruginosa and K. pneumoniae
ATL1102 for MS – Toxicology Study Main Findings
| Sequence Type: DNA fragment | |
| CTGAGTCTGTTTTCCATTCT |
ATL 1102
The antisense oligonucleotide is complementary to a region in the 3’UTR of human ITGA4 (integrin alpha 4) cDNA whose sequence is 5′-CTGAGTCTGTTTTCCATTCT-3′
Phosphorothioate antisense oligonucleotide consisting of a 9-nucleotide central region of deoxynucleotides flanked by 3 2′-O-methoxyethyl (2′-MOE) nucleotides on the 5′ end and 8 2′-MOE nucleotides on the 3′ end.
TOORAK, Australia, April 1, 2014 /PRNewswire/ — Antisense Therapeutics Limited (“ANP” or the “Company”) is pleased to advise that results from a chronic toxicity study in monkeys indicate that ATL1102, an antisense oligonucleotide currently under development for the treatment of multiple sclerosis (MS), was well-tolerated when given subcutaneously for a 6-month dosing period at the 2 dose levels tested (1.5 and 3mg/kg/dose). The Company believes that the preclinical and clinical experience to date with ATL1102 should allow dosing in future trials at or above the 1.5 mg/kg/dose level.
read at
http://www.sys-con.com/node/3037721
ATL-1102
ISIS-107248
TV-1102
ITGA4 Expression Inhibitors
Signal Transduction Modulators
PHASE 2
Antisense Therapeutics
Isis Pharmaceuticals
Antisense Therapeutics Limited (ASX: ANP) is an Australian publicly listed biopharmaceutical drug discovery and development company. Its mission is to create, develop and commercialise second generation antisense pharmaceuticals for large unmet markets. ANP has 4 products in its development pipeline that it has in-licensed from Isis Pharmaceuticals Inc., world leaders in antisense drug development and commercialisation – ATL1102 (injection) which has successfully completed a Phase II efficacy and safety trial, significantly reducing the number of brain lesions in patients with multiple sclerosis, ATL1103 a second-generation antisense drug designed to block GHr production and thereby lower blood IGF-I levels and is in clinical development as a potential treatment for growth and other GH-IGF-I disorders, ATL1102 (inhaled) which is at the pre-clinical research stage as a potential treatment for asthma and ATL1101 a second-generation antisense drug at the pre-clinical stage being investigated as a potential treatment for cancer.
ATL1102 is a second generation antisense inhibitor of CD49d, a subunit of VLA-4 (Very Late Antigen-4). In inflammation, white blood cells (leukocytes) move out of the bloodstream into the inflamed tissue, for example, the Central Nervous System (CNS) in MS, and the lung airways in asthma. The inhibition of VLA-4 may prevent white blood cells from entering sites of inflammation, thereby slowing progression of the disease. VLA-4 is a clinically validated target in the treatment of MS. Antisense inhibition of VLA-4 has demonstrated positive effects in a number of animal models of inflammatory disease including MS with the MS animal data having been published in a peer reviewed scientific journal. ATL1102 was previously shown by Antisense Therapeutics to be highly effective in reducing MS lesions in a Phase IIa clinical trial in MS patients.
ATL-1102 is an antisense oligonucleotide in phase II clinical trials at Isis Pharmaceuticals and Antisense Therapeutics for the treatment of relapsing-remitting multiple sclerosis (MS) in a subcutaneous injection formulation. Phase I clinical trials in a subcutaneous injections for stem cell mobilization and preclinical studies of an inhalation formulation of the drug candidate for the treatment of asthma are also being conducted at Antisense Therapeutics.
ATL-1102 is complementary to nt 4288-4207 (3’UTR) of human integrin alpha 4 (ITGA4) cDNA, and thus inhibits ITGA4 expression, blocking the synthesis of CD49d, a subunit of very late antigen-4 (VLA-4). VLA-4 is known to play a part in both the onset and progression of MS, and its inhibition may prevent white blood cells from entering the central nervous system.
ATL-1102 was originally developed at Isis Pharmaceuticals. In December 2001, Isis and Circadian Technologies formed Antisense Therapeutics, established to focus on the discovery and development of antisense therapeutics. As part of the company’s formation, Antisense Therapeutics received a license to ATL-1102 and entered into a five-year antisense drug discovery and development program with Isis. In 2008, Antisense licensed ATL-1102 to Teva. In 2010, Teva terminated its licensee agreement with Antisense for the development of ATL-1102 for the treatment of relapsing-remitting multiple sclerosis. The company stated that the compound was not on line with its preferred product pipeline. In 2001, ATL-1102 was licensed to Antisense Therapeutics by Isis Pharmaceuticals. In 2012, development and commercialization rights to the product were licensed to Tianjin International Joint Academy of Biotechnology and
Contact Information:
Website: www.antisense.com.au
Managing Director: Mark Diamond +61 (3) 9827 8999
USA Investor/Media: Joshua Drumm +(1) 212 375 2664;jdrumm@tiberend.com
Australia Investor/Media: Simon Watkin +61 (0)413 153 272;simon@marketconnect.com.au
SOURCE Antisense Therapeutics Limited
MK 2048 an HIV integrase inhibitor from Merck

MK 2048
Molecular Formula: C21H21ClFN5O4 Molecular Weight: 461.873943
869901-69-9, 3oyl, 3oyn
| (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide |
6(S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide
|
5-27-2009
|
Hiv Integrase Inhibitors
|
MK-2048 is a second generation integrase inhibitor, intended to be used against HIV infection. It is superior to the first available integrase inhibitor,raltegravir, in that it inhibits the HIV enzyme integrase 4 times longer. It is being investigated for use as part of pre-exposure prophylaxis (PrEP). [1]
It is being developed by Merck & Co.[2]
![]()
MK-2048 is a second generation integrase inhibitor for HIV-1 integrase. MK-2048 inhibits subtype B and subtype C integrase activities. MK-2048 inhibits R263K mutants slightly more effectively than G118R mutants.
MK-2048 inhibits S217H intasome and, by contrast, MK2048 remains fully active against the N224H intasome. MK2048 displays substantially lower dissociation rates compared with raltegravir, another integrase inhibitor.
MK-2048 is active against viruses resistant to RAL and EVG. MK-2048 exposure leads to the selection of G118R as a possible novel resistance mutation after 19 weeks. MK-2048, with continued pressure, subsequently leads to an additional substitution, at position E138K, after 29 weeks, within the IN gene.
Although the G118R mutation alone confers only slight resistance to MK-2048 but not to RAL or EVG, its presence arouses a dramatic reduction in viral replication capacity compared to wild-type NL4-3. E138K both partially restores viral replication capacity and also contributes to increased levels of resistance against MK-2048.

Structure of MK-2048 with important pharmacophore highlighted

…………………..
Synthesis
WO2005110415A1
http://www.google.as/patents/WO2005110415A1?cl=en
EXAMPLE 62 6(S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide

Step 1: te rt-Butyl[( 1 S)-2-(ethylamino)- 1 -methylethyl] carbamate To a cold (0 °C) solution of N-(tø/ -butoxycarbonyl)-L-alanine N’-methoxy-N’- methylamide (15.6 g, 67.2 mmol) in anhydrous THF (150 mL) and diethyl ether (400 mL), solid lithium aluminum hydride (5.1 g, 134.3 mmol) was added portionwise over a period of 30 minutes. The mixture was stirred at room temperature for 3 hours and cooled back to 0 °C. The reaction was treated carefully with an aqueous solution of potassium hydrogen sulfate (250 mL, 1M). The resultant mixture was diluted with diethyl ether.
The organic extract was washed successively with dilute hydrochloric acid, and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to provide the corresponding aldehyde as colorless solid. Without further purification, a cold (0 °C), stirred solution of the intermediate aldehyde (10.7 g, 61.8 mmol) and ethylamine hydrogen chloride (10.1 g, 123.5 mmol) in methanol (72 mL) was treated with sodium triacetoxyborohydride (17.2 g, 80.9 mmol) in one portion. The mixture was allowed to warm up to room temperature.
After stirring at room temperature overnight, the solution was concentrated under vacuum. The residue was partitioned between diethyl ether and cold aqueous sodium hydroxide (1.5 M). The ethereal extract was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to provide the titled compound. lH NMR (400 MHz, CDCI3) δ 4.68 (br s, IH), 3.75 (br t, IH), 2.62 (m, 5 H), 1.13 (d, J = 6.7 Hz, 3H),
1.09 (t, J = 7.0 Hz, 3H). ES MS M+l = 203
Step 2: ført-Butyl { ( 1 S)-2-[(bromoacetyl)ethylamino] – 1 -methylethyl } carbamate To a cold (0 °C) stirred solution of ?ert-butyl[(lS)-2-(ethylamino)-l- methylethyl]carbamate (11.0 g, 54.6 mmol) in a mixture of ethyl acetate (107 mL) and saturated aqueous sodium bicarbonate (65 mL), bromoacetyl bromide (12.1 g, 60.0 mmol) was added portionwise under an atmosphere of nitrogen. The mixture was allowed to warm up to room temperature over a period of 3.5 hours. The organic phase was separated, washed successively with saturated aqueous sodium bicarbonate, and brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was concentrated as a solution in toluene under vacuum to afford the title compound. ES MS M+l = 323, 325.
Step 3: fe7 -Butyl (2S)-4-ethyl-2-methyl-5-oxopiperazine-l-carboxylate To a stirred slurry of sodium hydride (1.7 g, 69.8 mmol) in anhydrous THF (800 mL), a solution of tert-butyl{(lS)-2-[(bromoacetyl)ethylamino]-l-methylethyl}carbamate (17.4 g, 53.7 mmol) in anhydrous THF (100 mL) was added dropwise over a period of 1 hour under an atmosphere of nitrogen. The reaction mixture was stirred at room temperature for two hours, cooled in an ice-water bath, and quenched with dropwise addition of aqueous citric acid (80 mL, 1M). The mixture was concentrated under vacuum. The residue was partitioned between chloroform and saturated aqueous sodium bicarbonate. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a gradient of 0-15% acetonitrile in chloroform. Collection and concentration of appropriate fractions provided the title compound. lH NMR (400 MHz, CDCI3) δ 4.46 (br s, IH), 4.24 (d, J = 18.4 Hz, 1 H), 3.78 (d, J = 18.4 Hz, 1 H),
3.64 (dd, J = 12.3, 4.2 Hz, 1 H), 3.54 (heptet, J = 7.1 Hz, 1 H), 3.38 (heptet, J = 7.1 Hz, 1 H), 2.99 (dd, J = 12.3, 1.8 Hz, 1 H), 1.47 (s, 9H), 1.21 (d, J = 6.8 Hz, 3H), 1.14 (t, J = 7.1 Hz, 3H). ES MS M+l = 243.
Step 4: (5S)-l-Ethyl-5-methylpiperazin-2-one hydrochloride Anhydrous hydrogen chloride gas was bubbled into a cold (-20 °C) solution of tert-butyl (2S)-4-ethyl-2-methyl-5-oxopiperazine-l-carboxylate (10.5 g, 43.4 mmol) in ethyl acetate (250 mL) under nitrogen. After the solution was saturated with hydrogen chloride, the reaction mixture was stirred in an ice-water bath for 30 minutes. The product mixture was purged with nitrogen, concentrated under vacuum to provide the title hydrogen chloride salt as pale yellow solid. lH NMR (400 MHz, DMSO-d6) δ 10.00 (br d, 2H), 3.72 (d, J = 16.6 Hz, 1 H), 3.62(d, J = 16.6 Hz, 1 H),
3.49-3.35 (m, 5 H), 3.29 (heptet, /= 7.3 Hz, 1 H), 1.31 (d, / = 6.6 Hz, 3H), 1.05 (t, J = 7.1 Hz, 3H).
Step 5: Ethyl (4S)-2-ethyl-8-hydroxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7- carboxy late Anhydrous ammonia gas was bubbled into a cold (0 °C) solution of (5S)-l-Ethyl-5- methylpiperazin-2-one hydrochloride (5.8 g, 32.3 mmol) in chloroform for 30 minutes. The resultant slurry was filtered and concentrated under vacuum. The residual oil was concentrated as a solution in toluene under vacuum, redissolved in toluene (120 mL) and treated with diethyl ethoxymethylenemalonate (7.0 g, 32.3 mmol) and heated in a sealed flask in an oil bath at 100 °C overnight. The resultant solution was concentrated under vacuum. The residual oil was concentrated as a solution in toluene under vacuum to provide the corresponding diethyl { [(2S)-4-ethyl-2-methyl-5- oxopiperazin-l-yl]methylene}malonate. Without further purification, to a solution of the malonate (10.5 g, 33.5 mmol) in anhydrous THF (330 mL) warmed with an external oil bath at 65 °C under an atmosphere of nitrogen, a solution of lithium bis(trimethylsilyl)amide (35.1 mL, 1 M, 35.1 mmol) was added. The solution was heated at the same temperature for one hour and concentrated under vacuum. The residue was partitioned between dichloromethane and hydrochloric acid (1M). The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum. The residue was triturated with diethyl ether. The solid precipitated was filtered, washed with diethyl ether to provide the title compound as pale brown solid. lH NMR (400 MHz, CDCI3) δ 8.43 (s, IH), 7.11 (s, IH), 4.32 (q, J = 7.1 Hz, 2H), 4.24 (m, IH), 3.65-
3.35 (m, 4H), 1.51 (d, J = 6.4 Hz, 3H), 1.36 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.0 Hz, 3H). ES MS M+l = 267
Step 6: Ethyl (4S)-2-ethyl-8-methoxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7- carboxylate A mixture of ethyl (4S)-2-ethyl-8-hydroxy-4-methyl-l -oxo- 1,2,3, 4-tetrahydropyrrolo[ 1,2- a]pyrazin-7-carboxylate (6.6 g, 24.8 mmol), anhydrous potassium carbonate (13.7 g, 99.1 mmol, 325 mesh), and iodomethane (4.2 g, 29.7 mmol) in anhydrous DMF (123 mL) was stirred at room temperature overnight. The mixture was filtered and concentrated under vacuum. The residue was partitioned between chloroform and dilute hydrochloric acid. The organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a gradient of 0-3% methanol in chloroform. Collection and concentration of appropriate fractions provided the title compound. Residual methanol was removed by concentrating from its solution in toluene under vacuum. lH NMR (400 MHz, CDCI3) δ 7.19 (s, IH), 4.29 (q, J = 7.1 Hz, 2 H), 4.24 (m, IH), 4.03 (s, 3H), 3.70-
3.32 (m, 4 H), 1.52 (d, J = 6.6 Hz, 3H), 1.35 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.2 Hz, 3H). ES MS M+l = 281
Step 7: Ethyl (4S)-6-bromo-2-ethyl-8-methoxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2- a]pyrazin-7-carboxylate To a mixture of ethyl (4S)-2-ethyl-8-(methoxy)-4-methyl-l-oxo-l,2,3,4- tetrahydropyrrolo[l,2- ]pyrazine-7-carboxylate (6.2 g, 22.1 mmol) and sodium bicarbonate (20.0 g, 238.0 mmol) in dichloromethane (500 mL) at 0 °C, a solution of bromine in dichloromethane (24.2 mmol, 0.5 M) was added over a period of 60 minutes. The reaction mixture was stirred at room temperature for 2 h, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluted with ethyl acetate. Collection and concentration of appropriate fractions provided the corresponding bromide. Residual ethyl acetate was removed by concentrating from its solution in benzene under vacuum. lH NMR (400 MHz, CDCI3) δ 4.58 (br m, IH), 4.34 (m, IH), 3.99 (s, 3H), 3.92 (dd, J = 13.0, 4.0 Hz,
IH), 3.67 (heptet, J = 7.1 Hz, 1 H), 3.49 (heptet, J = 7.1 Hz, 1 H), 3.23 (d, J = 13.0 Hz, IH), 1.40 (d, J = 7.1 Hz, 3H), 1.38 (t, 7 = 7.0 Hz, 3H), 1.20 (t, J = 7.0 Hz, 3H). ES MS M+l = 359, 361.
Step 8: Ethyl (4S)-2-ethyl-8-(methoxy)-6-[methoxy(oxo)acetyl]-4-methyl-l-oxo-l,2,3,4- tetrahydropyrrolo[ 1 ,2- ]pyrazine-7-carboxylate To a cold (-78 °C) solution of ethyl (4S)-6-bromo-2-ethyl-8-methoxy-4-methyl-l-oxo- l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7-carboxylate (8.51 g, 23.7 mmol) in anhydrous THF (800 mL) under an atmosphere of dry nitrogen, a solution of n-BuLi in hexane (10.5 mL, 26.3 mmol, 2.5 M) was added. The resultant mixture was stirred at -78 °C for 20 minutes. A solution of dimethyl oxalate (6.4 g, 53.8 mmol; dried from concentration from benzene under vac) in anhydrous THF (30 mL) was added. The reaction mixture was stirred at -78 °C for 1 hour and cannulated into a mixture of aqueous sulfuric acid (240 mL, 2M) and THF (200 mL) maintained between at -5 to -35 °C. The mixture was extracted with ethyl acetate (3 times). The organic extracts were combined, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluted with 40 to 100% ethyl acetate- hexane gradient. Collection and concentration of appropriate fractions provided the titled compound. lH NMR (400 MHz, CDCI3) δ 5.07 (m, IH), 4.29 (q, J = 7.2 Hz, 2H), 4.00 (s, 3H), 3.99-3.93 (m, IH), 3.89 (s, 3H), 3.74-3.66 (m, IH), 3.53-3.48 (m, IH), 3.23 (dd, J = 1.3, 13.2 Hz, IH), 1.46 (d, J = 6.6 Hz, 3H), 1.36 (t, J = 7.2 Hz, 3H), 1.22 (t, 7= 7.1 Hz, 3H). ES MS M+l = 367
Step 9: (6S)-8-Ethyl-10-methoxy-6-methyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carbohydrazide A mixture of ethyl (4S)-2-ethyl-8-(methoxy)-6-[methoxy(oxo)acetyl]-4-methyl-l-oxo- l,2,3,4-tetrahydropyrrolo[l,2-α]pyrazine-7-carboxylate (3.3 g, 8.9 mmol) and anhydrous hydrazine (1.7 mL, 53.7 mmol) in methanol (400 mL) was stirred at room temperature for one hour. The reaction mixture was concentrated under vacuum. The residue was concentrated from toluene. The resultant gummy solid was treated with methanol (20 mL). Diethyl ether was added to the resultant slurry which was filtered to provide the title compound as white solid. lH NMR (400 MHz, CDCI3) δ 8.99 (br s, 2H), 5.54 (br m, IH), 4.12 (m, IH), 4.10 (s, 3H), 3.81 (m, IH),
3.39 (m, IH), 3.21 (d, 7 = 12.6 Hz, IH), 1.44 (d, 7 = 6.4 Hz, 3H), 1.23 (t, 7 = 7.3 Hz, 3H). ES MS M+l =
335
Step 10: (6S)-8-Ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide To a solution of (6S)-8-ethyl-10-methoxy-6-methyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carbohydrazide (0.39 g, 1.2 mmol) and methylamine (5.9 mL, 11.8 mmol; 2 M in THF) in anhydrous dichloromethane (25 mL) in a water bath at room temperature, a solution of iodine (0.60 g, 2.4 mmol) in dichloromethane was added dropwise.
After the addition was completed, an aqueous solution of sodium sulfite was added and the mixture was stirred vigorously for 10 minutes. The organic phase was separated, diluted with chloroform, and washed with brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was triturated with a mixture of ethanol (7 mL) and diethyl ether (25 mL). The white solid precipitated was obtained by filtration and dried from its solution in toluene under vacuum. 1H NMR (400 MHz, CDCI3) δ 11.57 (s, IH), 7.38 (m, IH), 5.95 (br m, IH), 4.17 (s, 3H), 4.03 (dd, 7 =
13.4, 3.8 Hz, 1 H), 3.76 (heptet, 7 = 7.1 Hz, 1 H), 3.50 (heptet, 7 = 7.1 Hz, 1 H), 2.99 (dd, 7 = 12.9, 1.0 Hz, 1 H), 3.03 (d, 7 = 5.0 Hz, 3H), 1.44 (d, 7 = 6.6 Hz, 3H), 1.23 (t, 7 = 7.2 Hz, 3H). ES MS M+l = 334 Step 11: (6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[r,2′: l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide To a cold (0 °C) solution of (6S)-8-ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[l’,2′: l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.58 g, 4.73 mmol) in anhydrous DMF (50 mL), a solution of lithium bis(trimethylsilyl)amide (4.97 mL, 4.97 mmol, 1 M in THF) was added. After stirring at the same temperature for 25 minutes, 3-chloro-4-fluorobenzyl bromide (1.27 g, 5.68 mmol) was added. The reaction mixture was stirred at room temperature for 10 minutes and concentrated under vacuum. The residue was partitioned between chloroform and brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a 1-5% methanol in ethyl acetate gradient. Collection and concentration of appropriate fractions provided the title compound. lH NMR (400 MHz, CDCI3) δ 7.46 (dd, 7 = 6.9, 2.2 Hz, IH), 7.32 (m, IH), 7.09 (t, 7 = 7.6 Hz, IH), 7.03
(br signal, IH), 5.92 (m, IH), 5.32 (d, 7 = 14.1 Hz, IH), 5.26 (d, 7= 14.1 Hz, IH), 4.14 (s, 3H), 3.97 (dd, 7 = 13.2, 3.7 Hz, IH), 3.73 (heptet, 7 = 7.2 Hz, 1 H), 3.51 (heptet, 7 = 7.1 Hz, IH), 3.21 (dd, 7= 13.2, 1.7 Hz, IH), 3.03 (d, 7 = 5.0 Hz, 3H), 1.42 (d, 7 = 6.6 Hz, 3H), 1.23 (t, 7 = 7.1 Hz, 3H). ES MS M+l = 476
Step 12:
(6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d3pyridazine-4-carboxamide
To a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-l,9- dioxo-l,2,6,7,8,9-hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.15 g, 2.41 mmol) in anhydrous dichloromethane (800 mL), a solution of boron tribromide in dichloromethane (3.14 mL, 3.14 mmol; 1 M) was added. After stirring at room temperature for 5 minutes, the reaction mixture was treated with anhydrous methanol, stirred for 30 minutes, and concentrated under vacuum. The procedure was repeated twice. The residue was dissolved in a mixture of methanol and acetonitrile and treated with aqueous sodium hydroxide. The mixture was subjected to purification on preparative reverse phase high pressure column chromatography. Collection and lyophilization of appropriate fractions provided the title compound as white amorphous solid.
MK 2048
lH NMR (400 MHz, CDCI3) δ 7.48 (dd, 7 = 7.0, 2.2 Hz, IH), 7.33 (m, IH), 7.09 (t, 7 = 8.7 Hz, IH), 6.01 (m, IH), 5.33 (d, 7= 14.1 Hz, IH), 5.27 (d, 7 = 14.1 Hz, IH), 3.99 (dd, 7= 12.8, 4.0 Hz, 1 H), 3.71(heptet, 7 = 7.1 Hz, 1 H), 3.49 (heptet, 7 = 7.1 Hz, 1 H), 3.24 (dd, 7 = 13.2, 1.5 Hz, 1 H), 3.03 (d, 7 = 5.1 Hz, 3H), 1.42 (d, 7 = 6.6 Hz, 3H), 1.24 (t, 7 = 7.3 Hz, 3H). ES MS M+l = 462
The amorphous product was dissolved in boiling methanol (1.4 g/200 mL). Upon cooling in an ice-water bath, a precipitate formed which was separated by obtained by filtration to afford a white crystalline solid.
MK 2048sodium salt
The corresponding sodium salt was prepared by treatment of a solution of (6S)-2-(3- chloro-4-fluorobenzyl)-8-ethyl- 10-hydroxy-N,6-dimethyl-l ,9-dioxo- 1 ,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (920 mg, 1.99 mmol) in aqueous acetonitrile with aqueous sodium hydroxide (1.03 equivalent), followed by lyophilization of the resultant solution.
(5S)-1-ethyl-5-methylpiperazin-2-one
 1,5-Cyclooctadiene-iridium(I) chloride dimer, Chloro(1,5-cyclooctadiene)iridium(I) dimer, Di-μ-chlorobis[(1,2,5,6-η)-1,5-cyclooctadiene]diiridium, Iridium(I) chloride 1,5-cyclooctadiene complex dimer, [Ir(1,5-cod)Cl]2, [Ir(1,5-cod)Cl]2, [Ir(cod)Cl]2
1,5-Cyclooctadiene-iridium(I) chloride dimer, Chloro(1,5-cyclooctadiene)iridium(I) dimer, Di-μ-chlorobis[(1,2,5,6-η)-1,5-cyclooctadiene]diiridium, Iridium(I) chloride 1,5-cyclooctadiene complex dimer, [Ir(1,5-cod)Cl]2, [Ir(1,5-cod)Cl]2, [Ir(cod)Cl]2

(S)-1-[(R)-2-Di-(4-methoxy-3,5-dimethylphenyl-phosphino)ferrocenyl]-ethyl-dicyclohexylphosphine
SL-J006-2
(5S)-l-Ethyl-5-methylpiperazin-2-one was alternatively prepared as follows:
Step 1: N^rf-Butoxycarbonyl-N^ethylglycinamide Ethylamine (37 g, 0.82 mol) was condensed into a pressure vessel at 0 °C. N-(tert- butoxycarbonyl)glycine methyl ester (50 mL, 0.34 mol) was added. The vessel was sealed and the mixture was stirred at room temperature overnight. The product mixture was concentrated under vacuum and the residue was passed through a pad of silica gel eluting with ethyl acetate. The solution was concentrated under vacuum to provide the title compound as a clear oil. lH NMR (400 MHz, CDCI3) δ 6.11 (br s, IH), 5.18 (br s, IH), 3.77 (d, 7 = 5.7 Hz, 2H), 3.31 (q, 7 = 7.1
Hz, 2H), 1.15 (t, 7 = 7.1 Hz, 3H).
Step 2: l-Ethyl-5-methylpyrazin-2(lH)-one A cold (0 °C) solution of N^tø^butoxycarbonyl-N^ethylglycinamide (68.0 g, 0.33 mol) in anhydrous dichloromethane (500 mL) was saturated with anhydrous hydrogen chloride gas. After stirring at the same temperature for 1.5 hours, the solution was recharged with more hydrogen chloride gas and stirred for additional 15 minutes. The reaction mixture was concentrated under vacuum. The residue was dissolved in methanol, diluted with toluene, and concentrated under vacuum to afford the intermediate N-ethylglycinamide HCI salt.
This was stored under vacuum overnight and used without further purification. A solution of N-ethylglycinamide HCI salt (44.2 g, 0.32 mol), aqueous sodium hydroxide (640 mL, 1M), water (350 mL), pyruvic aldehyde (20.9 mL, 40% solution in water) was heated in an oil bath at 120 °C for one hour. The reaction mixture was cooled and saturated with solid sodium chloride. The mixture was extracted with chloroform (4×250 mL).
The combined organic extract was dried over anhydrous sodium sulfate, filtered, and passed through a plug of silica gel. The silica gel was rinsed successively with ethyl acetate and then 2% methanol in ethyl acetate. The eluted fractions were combined and concentrated under vacuum. The residual solid was recrystallized from diethyl ether to afford the title compound as pale yellow solid. lH NMR (400 MHz, CDCI3) δ 8.11 (s, IH), 6.92 (s, IH), 3.92 (q, 7 = 7.2 Hz, 2H), 2.28 (s, 3H), 1.37 (t, 7 = 7.2 Hz, 3H).
Step 3: (5 S)- 1 -Ethyl-5-methylpiperazin-2-one
A mixture of chloro-l,5-cyclooctadiene iridium (I) dimer (34 mg, 51 μmol) and (S)-l-[(R)-2-di-(3,5-bis(trifluoromethyl)phenyl)phosphino)ferrocenyl]ethyldicyclohexylphosphine (44 mg, 51 μmol; Solvias AG, SL-J006-2) in a mixture of 1:2 toluene and methanol (100 mL; purged with nitrogen for 15 minutes) was sonicated under an atmosphere of nitrogen for 15 minutes. To the resultant mixture, iodine (0.39 g, 1.52 mmol) and l-ethyl-5-methylpyrazin-2(lH)-one (7.0 g, 50.66 mmol) was added. The resultant mixture was heated in an oil bath at 50 °C under an atmosphere of hydrogen gas at 800 psi for 48 hours. The product mixture was filtered through a pad of Celite. The filtrate was concentrated under vacuum. The residue was treated with chloroform saturated with ammonia gas (100 mL). The resultant suspension was filtered through a pad of Celite, which was the rinsed with chloroform saturated with ammonia gas. The combined filtrate was concentrated under vacuum. The residue was concentrated as a solution in toluene for subsequent reaction. lH NMR (400 MHz, CDCI3) δ 3.58 (d, 7 = 17.2 Hz, IH), 3.53(d, 7 = 17.2 Hz, IH), 3.49-3.35 (m, 2H),
1.19 (d, 7 = 5.9 Hz, 3H), 1.14 (t, 7 = 7.2 Hz, 3H).
……………..
US 7538112
http://www.google.com/patents/US7538112
Step 12: (6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide
To a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.15 g, 2.41 mmol) in anhydrous dichloromethane (800 mL), a solution of boron tribromide in dichloromethane (3.14 mL, 3.14 mmol; 1 M) was added. After stirring at room temperature for 5 minutes, the reaction mixture was treated with anhydrous methanol, stirred for 30 minutes, and concentrated under vacuum. The procedure was repeated twice. The residue was dissolved in a mixture of methanol and acetonitrile and treated with aqueous sodium hydroxide. The mixture was subjected to purification on preparative reverse phase high pressure column chromatography. Collection and lyophilization of appropriate fractions provided the title compound as white amorphous solid.
1H NMR (400 MHz, CDCl3) δ 7.48 (dd, J=7.0, 2.2 Hz, 1H), 7.33 (m, 1H), 7.09 (t, J=8.7 Hz, 1H), 6.01 (m, 1H), 5.33 (d, J=14.1 Hz, 1H), 5.27 (d, J=14.1 Hz, 1H), 3.99 (dd, J=12.8, 4.0 Hz, 1 H), 3.71 (heptet, J=7.1 Hz, 1 H), 3.49 (heptet, J=7.1 Hz, 1 H), 3.24 (dd, J=13.2, 1.5 Hz, 1 H), 3.03 (d, J=5.1 Hz, 3H), 1.42 (d, J=6.6 Hz, 3H), 1.24 (t, J=7.3 Hz, 3H). ES MS M+1=462
The amorphous product was dissolved in boiling methanol (1.4 g/200 mL). Upon cooling in an ice-water bath, a precipitate formed which was separated by obtained by filtration to afford a white crystalline solid.
The corresponding sodium salt was prepared by treatment of a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (920 mg, 1.99 mmol) in aqueous acetonitrile with aqueous sodium hydroxide (1.03 equivalent), followed by lyophilization of the resultant solution.
References
- Keith Alcorn. Ralvetgravir shows potential for use as PrEP drug AIDSmap.com. 28 April 2009. Accessed 8 Nov 2009.
- Mark Mascolini. Merck Offers Unique Perspective on Second-Generation Integrase Inhibitor. 10th International Workshop on Clinical Pharmacology of HIV Therapy, April 15–17, 2009, Amsterdam. Accessed 8 Nov 2009.
| WO2011121105A1 | 1 Apr 2011 | 6 Oct 2011 | Tibotec Pharmaceuticals | Macrocyclic integrase inhibitors |
| EP1756114A2 * | 3 May 2005 | 28 Feb 2007 | Merck and Co., Inc. | Hiv integrase inhibitors |
| US7517929 | 3 Dec 2004 | 14 Apr 2009 | Momentive Performance Materials Inc. | Star-branched silicone polymers as anti-mist additives for coating applications |
| US5693640 * | 6 Jun 1995 | 2 Dec 1997 | Merck, Sharp & Dohme, Ltd. | Pyridazino-indole derivatives |
| US5756501 * | 3 Dec 1996 | 26 May 1998 | American Home Products Corporation | Saturated and unsaturated pyridazino 4,5-B! indolizines useful as antidementia agents |
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

