
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Moexipril


RS-10085-197
SPM-925
RS-10085 (free base)

Pharmacology
Moexipril is available as a prodrug moexipril hydrochloride, and is metabolized in the liver to form the pharmacologically active compound moexiprilat. Formation of moexiprilat is caused by hydrolysis of an ethyl ester group.[5] Moexipril is incompletely absorbed after oral administration, and its bioavailability is low.[6] The long pharmacokinetic half-life and persistent ACE inhibition of moexipril allows once-daily administration.[7]
Moexipril is highly lipophilic,[2] and is in the same hydrophobic range as quinapril, benazepril, and ramipril.[7] Lipophilic ACE inhibitors are able to penetrate membranes more readily, thus tissue ACE may be a target in addition to plasma ACE. A significant reduction in tissue ACE (lung, myocardium, aorta, and kidney) activity has been shown after moexipril use.[8]
It has additional PDE4-inhibiting effects.[9]
Side effects
Moexipril is generally well tolerated in elderly patients with hypertension.[10] Hypotension, dizziness, increased cough, diarrhea, flu syndrome, fatigue, and flushing have been found to affect less than 6% of patients who were prescribed moexipril.[3][10]
Mechanism of action
As an ACE inhibitor, moexipril causes a decrease in ACE. This blocks the conversion of angiotensin I to angiotensin II. Blockage of angiotensin II limits hypertension within the vasculature. Additionally, moexipril has been found to possess cardioprotective properties. Rats given moexipril one week prior to induction of myocardial infarction, displayed decreased infarct size.[11] The cardioprotective effects of ACE inhibitors are mediated through a combination of angiotensin II inhibition and bradykininproliferation.[8][12] Increased levels of bradykinin stimulate in the production of prostaglandin E2[13] and nitric oxide,[12] which cause vasodilation and continue to exert antiproliferative effects.[8] Inhibition of angiotensin II by moexipril decreases remodeling effects on the cardiovascular system. Indirectly, angiotensin II stimulates of the production of endothelin 1 and 3 (ET1, ET3)[14] and the transforming growth factor beta-1 (TGF-β1),[15] all of which have tissue proliferative effects that are blocked by the actions of moexipril. The antiproliferative effects of moexipril have also been demonstrated by in vitro studies where moexipril inhibits the estrogen-stimulated growth of neonatal cardiac fibroblasts in rats.[12] Other ACE inhibitors have also been found to produce these actions, as well.
WO 2014202659
http://www.google.com/patents/WO2014202659A1?cl=en
US4344949
http://www.google.co.in/patents/US4344949
References
- Hochadel, Maryanne, ed. (2006). The AARP Guide to Pills. Sterling Publishing Company. p. 640. ISBN 978-1-4027-1740-6. Retrieved2009-10-09.
- Belal, F.F, K.M. Metwaly, and S.M. Amer. “Development of Membrane Electrodes for the Specific Determination of Moexipril Hydrochloride in Dosage Forms and Biological Fluids.” Portugaliae Electrochimica Acta. 27.4 (2009): 463-475.
- Rodgers, Katie, Michael C Vinson, and Marvin W Davis. “Breakthroughs: New drug approvals of 1995 — part 1.” Advanstar Communications, Inc. 140.3 (1996): 84.
- Dart, Richard C. (2004). Medical toxicology. Lippincott Williams & Wilkins. p. 647. ISBN 978-0-7817-2845-4. Retrieved 2009-10-09.
- Kalasz, H, G. Petroianu, K. Tekes, I. Klebovich, K. Ludanyi, et al. “Metabolism of moexipril to moexiprilat: determination of in vitro metabolism using HPLC-ES-MS.” Medicinal Chemistry. 3 (2007): 101-106.
- Jump up^ Chrysant, George S, PK Nguyen. “Moexipril and left ventricular hypertrophy.” Vascular Health Risk Management. 3.1 (2007): 23-30.
- Cawello W, H. Boekens, J. Waitzinger, et al. “Moexipril shows a long duration of action related to an extended pharmacokinetic half-life and prolonged ACE-inhibition.” Int J Clin Pharmacol Ther. 40 (2002): 9-17.
- ^ Jump up to:a b c Chrysant, SG. “Vascular remodeling: the role of angiotensin-converting enzyme inhibitors.” American Heart Journal. 135.2 (1998): 21-30.
- Jump up^ Cameron, RT; Coleman, RG; Day, JP; Yalla, KC; Houslay, MD; Adams, DR; Shoichet, BK; Baillie, GS (May 2013). “Chemical informatics uncovers a new role for moexipril as a novel inhibitor of cAMP phosphodiesterase-4 (PDE4)”. Biochemical Pharmacology 85 (9): 1297–1305. doi:10.1016/j.bcp.2013.02.026. PMC 3625111. PMID 23473803.
- White, WB, and M Stimpel. “Long-term safety and efficacy of moexipril alone and in combination with hydrochlorothiazide in elderly patients with hypertension.” Journal of human hypertension. 9.11 (1995): 879-884.
- Rosendorff, C. “The Renin-Angiotensin System and Vascular Hypertrophy.” Journal of the American College of Cardiology. 28 (1996): 803-812.
- Hartman, J.C. “The role of bradykinin and nitric oxide in the cardioprotective action of ACE inhibitors.” The Annals of Thoracic Surgery. 60.3 (1995): 789-792.
- Jaiswal, N, DI Diz, MC Chappell, MC Khosia, CM Ferrario. “Stimulation of endothelial cell prostaglandin production by angiotensin peptides. Characterization of receptors.” Hypertension. 19.2 (1992): 49-55.
- Phillips, PA. “Interaction between endothelin and angiotensin II.” Clinical and Experimental Pharmacology and Physiology. 26.7. (1999): 517-518.
- Youn, TJ, HS Kim, BH Oh. “Ventricular remodeling and transforming growth factor-beta 1 mRNA expression after nontransmural myocardial infarction in rats: effects of angiotensin converting enzyme inhibition and angiotensin II type 1 receptor blockade.” Basic research in cardiology. 94.4 (1999): 246-253.
////////////
| Systematic (IUPAC) name | |
|---|---|
|
(3S)-2-[(2S)-2-{[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino}propanoyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
|
|
| Clinical data | |
| Trade names | Univasc |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a695018 |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 13-22% |
| Protein binding | 90% |
| Metabolism | Hepatic (active metabolite, moexiprilat) |
| Biological half-life | 1 hour; 2-9 hours (active metabolite) |
| Excretion | 50% (faeces), 13% (urine) |
| Identifiers | |
| CAS Registry Number | 103775-10-6 |
| ATC code | C09AA13 |
| PubChem | CID: 91270 |
| IUPHAR/BPS | 6571 |
| DrugBank | DB00691 |
| ChemSpider | 82418 |
| UNII | WT87C52TJZ |
| KEGG | D08225 |
| ChEMBL | CHEMBL1165 |
| Chemical data | |
| Formula | C27H34N2O7 |
| Molecular mass | 498.568 g/mol |
9-(5-oxotetrahydrofuran-2-yl)nonanoic acid methyl ester
9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester |
 |
| Name | 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester | ||
| Synonyms | |||
| Name in Chemical Abstracts | 2-Furannonanoic acid, tetrahydro-5-oxo-, methyl ester | ||
| CAS No | 22623-86-5 | ||
| Molecular formula | C14H24O4 | ||
| Molecular mass | 256.35 | ||
| SMILES code | O=C1OC(CC1)CCCCCCCCC(=O)OC | ||
1H NMR

| 1H-NMR: 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester | |||
| 500 MHz, CDCl3 | |||
| delta [ppm] | mult. | atoms | assignment |
| 1.24-1.45 | m | 10 H | 4-H, 5-H, 6-H, 7-H, 8-H |
| 1.57 | m | 2 H | 3-H |
| 1.70 | m | 1 H | 9-H |
| 1.82 | m | 1 H | 9-H |
| 2.27 | t | 2 H | 2-H |
| 2.30 | m | 2 H | 3-H (ring) |
| 2.50 | m | 2 H | 4-H (ring) |
| 3.67 | s | 3 H | O-CH3 |
| 4.48 | m | 1 H | 2-H (ring) |

13C NMR

| 13C-NMR: 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester | |||
| 125.7 MHz, CDCl3 | |||
| delta [ppm] | assignment | ||
| 24.9 | C3 | ||
| 25.2 | C9 | ||
| 28.0-29.2 | C4, C5, C6, C7, C8, C3 (ring) | ||
| 34.0 | C2 | ||
| 35.5 | C4 (ring) | ||
| 51.4 | O-CH3 | ||
| 81.0 | C2 (ring) | ||
| 174.2 | C1 (O-C(=O)-) | ||
| 177.2 | C5 (O-C(=O)-, ring) | ||
| 76.5-77.5 | CDCl3 | ||

IR |

| IR: 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester | |||
| [Film, T%, cm-1] | |||
| [cm-1] | assignment | ||
| 2931, 2856 | aliph. C-H valence | ||
| 1776 | C=O valence, lactone | ||
| 1737 | C=O valence, ester | ||
| Cu |
  nonanoic acid methyl ester")  |
|
Synthesis of 9-(5-oxotetrahydrofuran-2-yl)nonanoic acid methyl ester |
| Reaction type: | addition to alkenes, radical reaction, ring closure reaction |
| Substance classes: | alkene, halogencarboxylic acid ester, lactone |
| Techniques: | working with cover gas, stirring with magnetic stir bar, heating under reflux, evaporating with rotary evaporator, filtering, recrystallizing, heating with oil bath |
| Degree of difficulty: | Easy |
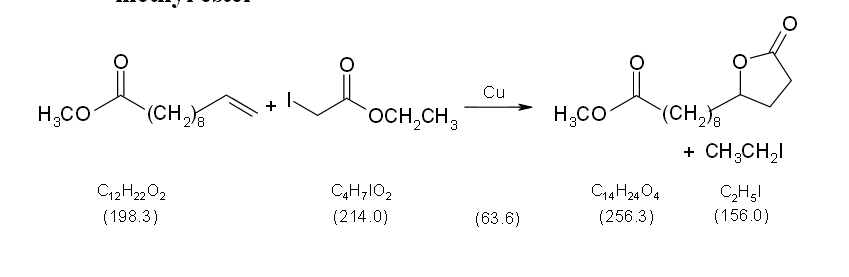
Operating scheme
 Instructions
Instructions
http://www.oc-praktikum.de/nop/en/instructions/pdf/4005_en.pdf
Instruction (batch scale 100 mmol)
Equipment 250 mL two-neck flask, protective gas supply, reflux condenser, heatable magnetic stirrer, magnetic stir bar, rotary evaporator, Buechner funnel, suction flask, desiccator, oil bath Substances undecenoic acid methyl ester (bp 248 °C) 19.8 g (22.3 mL, 100 mmol) iodoacetic acid ethyl ester (bp 73-74 °C/ 21 hPa) 27.8 g (15.4 mL, 130 mmol) copper powder (finely powdered, >230 mesh ASTM) 30.5 g (480 mmol) tert-butyl methyl ether (bp 55 °C) 130 mL petroleum ether (bp 60-80 °C) 300 mL Reaction In a 250 mL two-neck flask with magnetic stir bar and a reflux condenser connected with a protective gas piping 19.8 g (22.3 mL, 100 mmol) undecenoic acid methyl ester and 27.8 g (15.4 mL, 130 mmol) iodoacetic acid ethyl ester are mixed with 30.5 g (480 mmol) copper powder under a protective gas atmosphere. Afterwards the reaction mixture is stirred at 130 °C oil bath temperature under protective gas for 4 hours. (Reaction monitoring see Analytics.)
Work up The reaction mixture is cooled down to room temperature, 30 mL tert-butyl methyl ether are added, the mixture is stirred for 5 minutes and filtered off. The copper powder on the filter is washed four times with 25 mL tert-butyl methyl ether each. Filtrates and wash solutions are combined, the solvent is evaporated at the rotary evaporator. A yellow oil remains as crude product. Crude yield: 25.4 g.
The crude product is dissolved in 300 mL petroleum ether under reflux. The solution is allowed to cool down to room temperature, then it is stored in the refrigerator over night for complete crystallization. The crystalline product is sucked off over a Buechner funnel and dried in the vacuum desiccator. The mother liquor is stored again in the refrigerator for a check of complete crystallization. Yield: 19.5 g (76.1 mmol, 76%); white solid, mp 34 °C Comments In order to achieve a quantitative reaction within 4 hours, a fivefold excess of copper is used.
Waste management Recycling The copper powder can be used three times.
Waste disposal Waste Disposal evaporated tert-butyl methyl ether (might contain iodoethane) organic solvents, containing halogen mother liquor from recrystallization organic solvents, containing halogen copper powder solid waste, free from mercury, containing heavy metals
Time 6-7 hours
Break After heating and before recrystallizing
Degree of difficulty Easy
Analytics Reaction monitoring with TLC Sample preparation: Using a Pasteur pipette, two drops of the reaction mixture are taken and diluted with 0.5 mL diethyl ether. TLC-conditions: adsorbant: TLC-aluminium foil (silica gel 60) eluent: petroleum ether (60/80) : acetic acid ethyl ester = 7 : 3 visualisation: The TLC-aluminium foil is dipped in 2 N H2SO4 and then dried with a hot air dryer. Reaction monitoring with GC Sample preparation: Using a Pasteur pipette, one drop of the reaction mixture is taken and diluted with 10 mL dichloromethane. From this solution, 0.2 µL are injected. 10 mg from the solid product are dissolved in 10 mL dichloromethane. From this solution, 0.2 µL are injected. GC-conditions: column: DB-1, 28 m, internal diameter 0.32 mm, film 0.25 µm inlet: on-column-injection carrier gas: hydrogen (40 cm/s) oven: 90 °C (5 min), 10 °C/min to 240 °C (40 min) detector: FID, 270 °C Percent concentration was calculated from peak areas.
Chromatogram |

| GC: crude product | |
| column | DB-1, L=28 m, d=0.32 mm, film=0.25 µm |
| inlet | on column injection, 0.2 µL |
| carrier gas | H2, 40 cm/s |
| oven | 90°C (5 min), 10°C/min –> 240°C (40 min) |
| detector | FID, 270°C |
| integration | percent concentration calculated from relative peak area |

| GC: pure product | |
| column | DB-1, L=28 m, d=0.32 mm, film=0.25 µm |
| inlet | on column injection, 0.2 µL |
| carrier gas | H2, 40 cm/s |
| oven | 90°C (5 min), 10°C/min –> 240°C (40 min) |
| detector | FID, 270°C |
| integration | percent concentration calculated from relative peak area |
Substances required |
| Batch scale: | 0.01 mol | 0.1 mol | 10-Undecenoic acid methyl ester |
| Educts | Amount | Risk | Safety | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 10-Undecenoic acid methyl ester |
|
19.8 g | H- EUH- | P- | |||||
| Iodoacetic acid ethyl ester |
|
27.8 g | H300 H314 EUH- | P264 P280 P305 + 351 + 338 P310 | |||||
| Reagents | Amount | Risk | Safety | ||||||
| Copper powder |
|
30.5 g | H400 EUH- | P273 | |||||
| Solvents | Amount | Risk | Safety | ||||||
| tert-Butyl methyl ether |
|
130 mL | H225 H315 | P210 | |||||
| Petroleum ether (60-80) |
|
300 mL | H225 H304 H315 H336 H411 EUH- | P210 P261 P273 P301 + 310 P331 | |||||
| Others | Amount | Risk | Safety | ||||||
| Sulfuric acid 2N |
|
H314 H290 EUH- | P280 P301 + 330 + 331 P305 + 351 + 338 P309 + 310 | ||||||
| Solvents for analysis | Amount | Risk | Safety | ||||||
| Petroleum ether (60-80) |
|
H225 H304 H315 H336 H411 EUH- | P210 P261 P273 P301 + 310 P331 | ||||||
| Acetic acid ethyl ester |
|
H225 H319 H336 EUH066 | P210 P261 P305 + 351 + 338 | ||||||
| Dichloromethane |
|
H351 H315 H319 H335 H336 H373 | P261 P281 P305 + 351 + 338 |
Substances produced |
| Batch scale: | 0.01 mol | 0.1 mol | 10-Undecenoic acid methyl ester |
| Products | Amount | Risk | Safety | |
|---|---|---|---|---|
| 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester |
Equipment |
| Batch scale: | 0.01 mol | 0.1 mol | 10-Undecenoic acid methyl ester |
 |
two-necked flask 250 mL |  |
protective gas piping | |
 |
reflux condenser |  |
heatable magnetic stirrer with magnetic stir bar | |
 |
rotary evaporator |  |
suction filter | |
 |
suction flask |  |
exsiccator with drying agent | |
 |
oil bath |
Simple evaluation indices |
| Batch scale: | 0.01 mol | 0.1 mol | 10-Undecenoic acid methyl ester |
| Atom economy | 53.9 | % | |
| Yield | 76 | % | |
| Target product mass | 19.5 | g | |
| Sum of input masses | 370 | g | |
| Mass efficiency | 53 | mg/g | |
| Mass index | 19 | g input / g product | |
| E factor | 18 | g waste / g product |
………………
………
Aseptic Manufacturing Operation: Chinese Company Zhuhai United Laboratories does not comply with EU GMP
DRUG REGULATORY AFFAIRS INTERNATIONAL


While the focus of attention has been on Indian manufacturers during the last 2 years now also Chinese manufacturers are in the spot light. On 15 June 2015 the National Agency for Medicines and Medical Devices of Romania entered a GMP Non-Compliance Report for Zhuhai United Laboratories into EudraGMDP. Read more about the GMP deviations observed at Zhuhai United.

While the focus of attention has been on Indian manufacturers during the last 2 years now also Chinese manufacturers are again in the spot light. Just recently the EU found serious GMP deviations at an API manufacturer (Huzhou Sunflower Pharmaceuticals) and on 15 June 2015 the National Agency for Medicines and Medical Devices of Romania entered a GMP Non-Compliance Report for Zhuhai United Laboratories Co., LTD located at Sanzao Science &Technology Park, National Hi-Tech Zone, Zhuhai, Guangdong, 519040, China into EudraGMDP.
According to the report issued by the…
View original post 253 more words
Inna Ben-Anat, Global QbD Director of Teva Pharmaceuticals
DRUG REGULATORY AFFAIRS INTERNATIONAL

Meet Inna Ben-Anat, Global QbD Director of Teva Pharmaceuticals. Inna is a key thought leader in Quality by Design for generics.
https://www.linkedin.com/pub/inna-ben-anat/6/47a/670
![]() Ben-Anat, InnaASSOCIATE DIRECTOR, HEAD OF QDD STRATEGY | TEVA PHARMACEUTICALSAssociate Director, Head of QbD Strategy Chemical Engineer with a degree in Quality Assurance and Reliability (Technion-Israel Institute of Technology). QbD Strategy Leader at Teva (USA). Headed the implementation of a global QbD training programme. More than 12 years of pharmaceutical development experience.
Ben-Anat, InnaASSOCIATE DIRECTOR, HEAD OF QDD STRATEGY | TEVA PHARMACEUTICALSAssociate Director, Head of QbD Strategy Chemical Engineer with a degree in Quality Assurance and Reliability (Technion-Israel Institute of Technology). QbD Strategy Leader at Teva (USA). Headed the implementation of a global QbD training programme. More than 12 years of pharmaceutical development experience.

Inna Ben-Anat is a Quality by Design (QbD) Strategy Leader in Teva Pharmaceuticals USA. In this role, Inna has implemented global QbD training program, and is supporting R&D teams in developing Quality by Design strategies, optimizing formulations and processes and assisting develop product specifications. Additionally, Inna supports Process Engineering group with process optimization during scale-up and supports Operations in identification and resolution of any technical issues. Inna has extensive expertise in process development, design…
View original post 1,144 more words
Determining Criticality-Process Parameters and Quality Attributes
DRUG REGULATORY AFFAIRS INTERNATIONAL
Determining Criticality-Process Parameters and Quality Attributes Part I: Criticality as a Continuum
As the pharmaceutical industry tries to embrace the methodologies of quality by design (QbD) provided by the FDA’s process validation (PV) guidance (1) and International Conference on Harmonization (ICH) Q8/Q9/Q10 (2-4), many companies are challenged by the evolving concept of criticality as applied to quality attributes and process parameters. Historically, in biopharmaceutical development, criticality has been a frequently arbitrary categorization between important high-risk attributes or parameters and those that carry little or no risk. This binary designation was usually determined during early development for the purposes of regulatory filings, relying heavily on scientific judgment and limited laboratory studies.
|

With the most recent ICH and FDA guidances…
View original post 8,374 more words
Alternative solvents can make preparative liquid chromatography greener

DOI: 10.1039/C5GC00887E, Paper
Alternative solvents can make preparative liquid chromatography greener
E-mail: lvy33@163.com
Alternative solvents can make preparative liquid chromatography greener
To make preparative Reversed-Phase High Performance Liquid Chromatography (RP-pHPLC) greener, alternative solvents were considered among others in terms of toxicity, cost, safety, workability, chromatographic selectivity and elution strength. The less toxic solvents ethanol, acetone and ethyl acetate were proposed as possible greener replacements for methanol, acetonitrile and tetrahydrofuran (THF).
For testing their feasibility, five ginkgo terpene trilactones were used as model analytes. The best “traditional” eluent, i.e., methanol–THF–water (2 :1:7) was used as the benchmark. A generic two-step chromatographic optimization procedure by UHPLC consisting of (1) a simplex design using the Snyder solvent triangle and (2) HPLC modelling software was used.
:1:7) was used as the benchmark. A generic two-step chromatographic optimization procedure by UHPLC consisting of (1) a simplex design using the Snyder solvent triangle and (2) HPLC modelling software was used.
In the first step, two ternary mixtures were found (acetone–ethyl acetate–water (20.25:3.75:76) and ethanol–ethyl acetate–water (9.5:7.5:83)), which already gave better results than the benchmark. The second step in which the influence of the gradient time, temperature and ratio of the two best ternary isocratic solvents was studied, led to an optimal 10.5 min gradient and a minimum resolution of 5.76.
In the final step, scale-up from 2.1 to 22 mm i.d. pHPLC columns proceeded successfully. When 0.5 g of the sample was injected, baseline separation was maintained. Chromatographic and absolute purities for products exceeded 99.5% and 95% respectively. This example shows that using less toxic and cheaper solvents for pHPLC can go hand in hand with higher productivity and less waste.
SEE
http://www.rsc.org/suppdata/c5/gc/c5gc00887e/c5gc00887e1.pdf
ML-236B, Mevastatin (compactin)
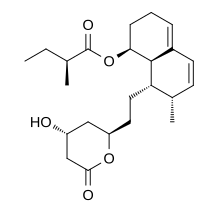
Mevastatin (compactin, ML-236B) is a hypolipidemic agent that belongs to the statins class.
It was isolated from the mold Penicillium citrinum by Akira Endo in the 1970s, and he identified it as a HMG-CoA reductase inhibitor,[1] i.e., a statin. Mevastatin might be considered the first statin drug;[2] clinical trials on mevastatin were performed in the late 1970s in Japan, but it was never marketed.[3] The first statin drug available to the general public was lovastatin.
In vitro, it has antiproliferative properties.[4]
A British group isolated the same compound from Penicillium brevicompactum, named it compactin, and published their results in 1976.[5] The British group mentions antifungal properties with no mention of HMG-CoA reductase inhibition.
High doses inhibit growth and proliferation of melanoma cells.[6]
| Systematic (IUPAC) name | |
|---|---|
| (1S,7R,8S,8aR)-8-{2-[(2R,4R)-4-Hydroxy-6-oxotetrahydro-2H-pyran-2-yl]ethyl}-7-methyl-1,2,3,7,8,8a-hexahydronaphthalen-1-yl (2S)-2-methylbutanoate | |
| Clinical data | |
| Identifiers | |
| 73573-88-3 |
|
| None | |
| PubChem | CID: 64715 |
| IUPHAR/BPS | 3031 |
| DrugBank | DB06693 |
| ChemSpider | 58262 |
| UNII | 1UQM1K0W9X |
| KEGG | C13963 |
| ChEBI | CHEBI:34848 |
| ChEMBL | CHEMBL54440 |
| Chemical data | |
| Formula | C23H34O5 |
| 390.513 g/mol | |
 |
|
Title: Mevastatin
CAS Registry Number: 73573-88-3
CAS Name: (2S)-2-Methylbutanoic acid (1S,7S,8S,8aR)-1,2,3,7,8,8a-hexahydro-7-methyl-8-[2-[(2R,4R)-tetrahydro-4-hydroxy-6-oxo-2H-pyran-2-yl]ethyl]-1-naphthalenyl ester
Additional Names: 7-[1,2,6,7,8,8a-hexahydro-2-methyl-8-(methylbutyryloxy)naphthyl]-3-hydroxyheptan-5-olide; 2b-methyl-8a-(2-methyl-1-oxobutoxy)mevinic acid lactone; compactin; 6-demethylmevinolin
Manufacturers’ Codes: CS-500; ML-236 B
Molecular Formula: C23H34O5
Molecular Weight: 390.51
Percent Composition: C 70.74%, H 8.78%, O 20.49%
Literature References:
Fungal metabolite which is a potent inhibitor of HMG-CoA reductase, the rate controlling enzyme in cholesterol biosynthesis. Isoln from Penicillium citrinum: A. Endo et al., DE 2524355 corresp to US 3983140 (1975, 1976 to Sankyo).
Isoln from P. brevicompactum, crystal and molecular structure: A. G. Brown et al., J. Chem. Soc. Perkin Trans. 1 1976,1165.
Inhibition of HMG-CoA reductase activity: A. Endo et al., FEBS Lett. 72, 323 (1976); M. S. Brown et al., J. Biol. Chem. 253,1121 (1978).
Therapeutic effects in primary hypercholesterolemia: A. Yamamoto et al., Atherosclerosis 35, 259 (1980).
Total synthesis: N. Y. Wang et al., J. Am. Chem. Soc. 103, 6538 (1981); M. Hirama, M. Uei, ibid. 104, 4251 (1982); N. N. Girotra, N. L. Wendler, Tetrahedron Lett. 23, 5501 (1982); C.-T. Hsu et al., J. Am. Chem. Soc. 105, 593 (1983); P. A. Grieco et al., ibid. 1403; D. L. J. Clive et al., J. Am. Chem. Soc. 110, 6914 (1988). Review of syntheses: T. Rosen, C. H. Heathcock, Tetrahedron 42,4909-4951 (1986).
Review of mevastatin and related compounds: A. Endo, J. Med. Chem. 28, 401-405 (1985).
Properties: Crystals from aq ethanol, mp 152°. [a]D22 +283° (c = 0.48 in acetone). uv max: 230, 237, 246 nm (log e 4.28, 4.30, 4.11).
Melting point: mp 152°
Optical Rotation: [a]D22 +283° (c = 0.48 in acetone)
Absorption maximum: uv max: 230, 237, 246 nm (log e 4.28, 4.30, 4.11)
|
References
- Endo, Akira; Kuroda M.; Tsujita Y. (December 1976). “ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium”. Journal of Antibiotics (Tokyo) 29 (12): 1346–8. doi:10.7164/antibiotics.29.1346. PMID 1010803.
- “The story of statins”.
- Endo, Akira (Oct 2004). “The origin of the statins”. Atheroscler Suppl. 5 (3): 125–30. doi:10.1016/j.atherosclerosissup.2004.08.033.PMID 15531285.
- Wachtershauser, A.; Akoglu, B; Stein, J (2001). “HMG-CoA reductase inhibitor mevastatin enhances the growth inhibitory effect of butyrate in the colorectal carcinoma cell line Caco-2”. Carcinogenesis 22 (7): 1061–7. doi:10.1093/carcin/22.7.1061. PMID 11408350.
- Brown, Allan G.; Smale, Terry C.; King, Trevor J.; Hasenkamp, Rainer; Thompson, Ronald H. (1976). “Crystal and molecular structure of compactin, a new antifungal metabolite from Penicillium brevicompactum.”. J. Chem. Soc., Perkin Trans. 1 (11): 1165–1170.doi:10.1039/P19760001165. PMID 945291.
- ^ Glynn, Sharon A; O’Sullivan, Dermot; Eustace, Alex J; Clynes, Martin; O’Donovan, Norma (2008). “The 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors, simvastatin, lovastatin and mevastatin inhibit proliferation and invasion of melanoma cells”. BMC Cancer8: 9. doi:10.1186/1471-2407-8-9. PMC 2253545. PMID 18199328.
The present invention
http://www.google.com/patents/US6204032
is related to a new method for producing ML-236B, a precursor of pravastatin sodium, in particular to a method for producing ML-236B lactone form(I), free acid form (II), and sodium salt(III) shown in the following formulae by using a new microorganism isolated from soil. ML-236B is obtained from the culture broth of this microorganism and it is used as a substrate of pravastatin sodium which is a potent cholesterol-lowering agent used in treatment for hypercholesterolemia.

2. Description of the Prior Art
It has been known that heart disease such as myocardial infarction, arteriosclerosis have been caused mainly by hyperlipidemia, especially hypercholesterolemia. It was reported by U.S. Pat. No. 3,983,140 and UK. Patent No. 1,453,425 that a cholesterol-lowering compound called ML-236B produced by a fungus Penicillium sp. had been discovered. ML-236B is produced by soil microorganisms or chemical conversion. It was reported that Penicillium brevicompactin, Penicilmyces sp., Trichoderma longibraiatum, Trichoderma pseudokoningi, Hyphomyces chrisopomus and Penicillium citrium produced ML-236B(David et al., “Biotechnology of filamentous fungi”, p241; JP Publication No. Pyung 4-349034).
Particularly, Sankyo Pharmaceutical Company, Japan, had developed Penicillium citrium SANK 18767 by mutation of a strain Penicillium citrium NRRL-8082 which was reported in 1971. By continuing strain development for 14 years, they had obtained Penicillium citrium Thom SANK 13380. ML-236B productivity had risen from 1.75 mg/l to 42.5 mg/l.
However, the method above described required so much time about 14 years to develop a strain with high ML-236B productivity. It also needed a little long cultivation time, 14 days, and showed relatively low ML-236B productivity.



The invention will be described in more detail in the drawings.
FIG. 1 is the IR spectrum of ML-236B obtained from this invention;

 and
and
FIG. 2 is the 13C-NMR spectrum of ML-236B obtained from this invention.

The physical properties such as appearance, melting point. molecular weight, elemental analysis, formular, UV spectrum, IR spectrum, solubility and specific rotation of ML-236B obtained from Example 2, 3 and Comparative Example are described in Table 1.
| TABLE 1 | ||
| COMPARATIVE | ||
| Article | EXAMPLE 2, 3 | EXAMPLE |
| Appearance | white crystal | white crystal |
| Melting point (° C.) | 150˜152 | 150˜152 |
| Molecular weight | calculated 390.2635 | experimental 390.2392 |
| experimental 390.2392 | ||
| Elemental | C 70.74, H 8.77, O 20.49 | C 70.74, O 20.49, H 8.77 |
| Analysis (%) | C 70.55 , H 8.69 | |
| calculated | C 70.85 , H 8.02 | |
| experimental | ||
| Formula | C23H34O5 | C23H34O5 |
| UV spectrum | 230, 237, 246 | 230, 237, 246 |
| (nm, MeOH) | ||
| IR spectrum | 3509, 2964, 2938, 2884, | 3509, 2964, 2938, 2884, |
| (cm−1, KBr ) | 1744, 1698, 1445, 1385, | 1744, 1699, 1445, 1385, |
| 1236, 1206, 1182, 1151, | 1236, 1206, 1182, 1150, | |
| 1077, 1056 | 1076, 1056 | |
| Solubility | methanol, chloroform, | methanol, chloroform, |
| soluble | ethanol, ethyl acetate | ethanol, ethyl acetate |
| insoluble | water | water |
| Specific rotation | +283n | +283n |
| [α]D | ||
13C NMR data of ML-236B are shown in Table 2 and FIG. 2.
| TABLE 2 | |||||
| The | δ c(ppm) | The | δ c(ppm) | ||
| number | EX- | COMPAR- | number | EX- | COMPAR- |
| of | AMPLE | ATIVE | of | AMPLE | ATIVE |
| carbon | 2,3 | EXAMPLE | carbon | 2,3 | EXAMPLE |
| C-1 | 171.50 | 170.67 | C-13 | 124.48 | 123.33 |
| C-2 | 39.31 | 38.44 | C-14 | 134.35 | 133.38 |
| C-3 | 63.18 | 62.12 | C-15 | 128.96 | 127.96 |
| C-4 | 36.88 | 35.84 | C-16 | 133.49 | 132.37 |
| C-5 | 77.22 | 76.26 | C-17 | 31.66 | 30.70 |
| C-6 | 33.75 | 32.82 | C-18 | 14.66 | 13.64 |
| C-7 | 24.83 | 23.78 | C-19 | — | — |
| C-8 | 37.66 | 36.67 | C-20 | 177.79 | 176.55 |
| C-9 | 38.31 | 37.40 | C-21 | 42.56 | 41.50 |
| C-10 | 68.45 | 67.51 | C-22 | 27.55 | 26.48 |
| C-11 | 27.06 | 26.30 | C-23 | 12.59 | 11.49 |
| C-12 | 21.74 | 20.74 | C-24 | 17.74 | 16.64 |
By using a new microorganism which was obtained from this invention, the productivity of pravastatin precursor was elevated highly and the pravastatin precursor could be prepared in a simple way in short time.
Therefore, the present invention could be used effectively in production of pravastatin precursor.
DR ANTHONY MELVIN CRASTO

MY BLOGS ON MED CHEM

FLAGS AND HITS
…………..today on this blog
Pravastatin


Pravastatin (marketed as Pravachol or Selektine) is a member of the drug class of statins, used in combination with diet, exercise, and weight-loss for lowering cholesterol and preventing cardiovascular disease.
Medical uses
Pravastatin is primarily used for the treatment of dyslipidemia and the prevention of cardiovascular disease.[1] It is recommended to be used only after other measures such as diet, exercise, and weight reduction have not improved cholesterol levels.[1]
The evidence for the use of pravastatin is generally weaker than for other statins. The antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT), failed to demonstrate a difference in all-cause mortality or nonfatal myocardial infarction/fatal coronary heart disease rates between patients receiving pravastatin 40mg daily (a common starting dose) and those receiving usual care.[2]
Mechanism of action


Pravastatin acts as a lipoprotein-lowering drug through two pathways. In the major pathway, pravastatin inhibits the function of hydroxymethylglutaryl-CoA (HMG-CoA) reductase. As a reversiblecompetitive inhibitor, pravastatin sterically hinders the action of HMG-CoA reductase by occupying the active site of the enzyme. Taking place primarily in the liver, this enzyme is responsible for the conversion of HMG-CoA to mevalonate in the rate-limiting step of the biosynthetic pathway for cholesterol. Pravastatin also inhibits the synthesis of very-low-density lipoproteins, which are the precursor to low-density lipoproteins (LDL). These reductions increase the number of cellular LDL receptors and, thus, LDL uptake increases, removing it from the bloodstream.[6] Overall, the result is a reduction in circulating cholesterol and LDL. A minor reduction in triglycerides and an increase in high-density lipoproteins (HDL) are common.
History
Initially known as CS-514, it was originally identified in a bacterium called Nocardia autotrophica by researchers of the Sankyo Pharma Inc..[7] It is presently being marketed outside Japan by thepharmaceutical companyBristol-Myers Squibb. In 2005, Pravachol was the 22nd highest-selling brand-name drug in the United States, with sales totaling $1.3 billion.[8]
The U.S. Food and Drug Administration approved generic pravastatin for sale in the United States for the first time on April 24, 2006. Generic pravastatin sodium tablets are manufactured byBiocon Ltd, India and TEVA Pharmaceuticals in Kfar Sava, Israel.[8]

…………………………..
BIOCON LIMITED Patent: WO2005/19155 A1, 2005 ; Location in patent: Page/Page column 8 ;http://google.com/patents/WO2005019155A1?cl=en
The present invention relates to a novel process for the preparation of substantially pure l^^^^^δa-hexahydro-beta,delta/6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-/ (beta R, delta R, lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid, sodium salt.
BACKGROUND OF THE INVENTION
US 4,346,227 discloses l,2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid, sodium salt. The compound is also known by the synonyms 3-beta-Hydroxycompactin; Eptastatin and Pravastatin. The compound is used as cholestrerol lowering agent which inhibit the enzyme H G CoA reductase.
The step of conversion of l,2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid to its sodium salt is crilcial. The prior art methods convert the acid form into sodium salt form as final step to afford the sodium salt. The prior art methods for the preparation of sodium salt from the l,2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-t(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)-1-Naphthaleneheptanoic acid are disclosed herein as reference.
WO 98/45410 discloses preparation of 1,2,6,7,8,8a-hexahydro-beta,delta, 6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid sodium by feeding compactin sodium to the microorganism Streptomyces exfoliatus and recovering the hydroxylated compactin sodium (l,2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid sodium salt) by extraction, purification by semi preparative HPLC and crystallization.
The process involves use of HPLC, which is a tedious and expensive technique and cannot be scaled up beyond a limit.
WO 00/46175 discloses a process for preparation of
l/2,6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-1-oxobutoxy]-, (beta R,delta R,lS,2S,6S,8S,8aR)- 1-Naphthaleneheptanoic acid sodium salt from lactone by hydrolyzing with sodium hydroxide.
Also amine salts can be transformed to sodium salt by treating with sodium hydroxide and/or sodium alkoxide.
When amine salts are employed, it involves an extra step i.e., the preparation of the amine salt.
US 2003/0050502 discloses a process for preparation of sodium salt of a statin by contacting a solution of hydroxy acid of the statin with sodium-2-ethylhexanoate and recovering the corresponding sodium salt.
The process involves use of expensive reagent sodium-2-ethyl hexanoate.
The prior art methods suffer from one or more disadvantages like use of expensive reagents, need of special equipment to carry out the operation or increased number of steps for the preparation of sodium salt of l,2,6,7,8,8a-hexahydro-beta,delta/6-trihydroxy-2-methyl-8-[(2S)-2-me hyl-l-oxobutoxy]-, (beta R,delta
R/lS^δS/δS/δaR)- 1-Naphthaleneheptanoic acid.
The present invention relates to a process, which overcomes all the disadvantages of the prior art and results in substantially pure product in high yields.
Example 1
To a solution of 3,5-Dihydroxy-7-[6-hydroxy-2-methyl-δ-(2-methyl-butyryloxy)-l,2,6,7,δ,δa-hexahydro-naphthalen-l-yl]-heptanoic acid ( 70 g, 0.165 mol) in ethyl acetate (500 ml), solid sodium carbonate (δ.76 g, 0.0δ25 mol) was added and stirred for 2 hours. l,2/6,7,8,8a-hexahydro-beta,delta,6-trihydroxy-2-methyl-8-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)-1-Naphthaleneheptanoic acid sodium salt was precipitated. The reaction mixture was filtered and cake was washed with ethyl acetate to get free flowing crystals of l,2,6,7,δ,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)- 1-Naphthaleneheptanoic acid sodium (FORMULA I). Yield: 65 g, δδ% Example 2
To a solution of 3,5~Dihydroxy-7-[6-hydroxy-2-methyl-δ-(2-methyl-butyryloxy)-l,2,6,7,δ,δa-hexahydro-naphthalen-l-yl]-heptanoic acid (10 Kg, 23.6 mol) in isobutyl acetate (60 L), solid sodium carbonate (1.25 Kg, 11.8 moi) was added and stirred for 3 hoursl,2,6,7,8,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)-1-Naphthaleneheptanoic acid sodium salt was precipitated. The reaction mixture was filtered and cake was washed with isobutyl acetate to get free flowing crystals of l,2,6,7,δ,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)- 1-Naphthaleneheptanoic acid sodium (FORMULA I). Yield: 9 Kg, δ5%
Example 3
To a solution of 3,5-Dihydroxy-7-[6-hydroxy-2-methyl-δ-(2-methyl-butyryloxy)-l,2,6,7,δ,δa-hexahydro-naphthalen-l-yl]-heptanoic acid (100. Kg, 236 mol) in butyl acetate (600 L), solid sodium carbonate (12.5 Kg, 118 mol) was added and stirred for 3 hours. l,2,6,7,8,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)-1-Naphthaleneheptanoic acid sodium salt was precipitated. The reaction mixture was filtered and cake was washed with butyl acetate to get free flowing crystals of l,2,6,7,δ,δa-hexahydro-beta,delta,6-trihydroxy-2-methyl-δ-[(2S)-2-methyl-l-oxobutoxy]-, (beta R,delta R,lS,2S,6S,δS,δaR)- 1-Naphthaleneheptanoic acid sodium (FORMULA I). Yield: 95 Kg, 90%

FORMULA I
…………………………
US2005/113446 A1, ; Page/Page column 6 ;
……………………………
http://www.google.com/patents/EP0975738A1?cl=en
Description of the Drawings
The invention will be described in more detail in the drawings. Fig. 1 is the IR spectrum of pravastatin sodium Fig. 2 is the13C-NMR spectrum of pravastatin sodium Fig. 3 is the H-NMR spectrum of pravastatin sodium



EXPERIMENTAL EXAMPLE
The physical properties of pravastatin sodium obtained from Example 1 and Comparative Example are described in Table 3.
Table 3

IR spectrum, “C-NMR spectrum, H-NMR spectrum of pravastatin sodium obtained from this invention are represented in Fig. 1, Fig. 2 and Fig. 3, respectively. By using a new microorganism Streptomyces exfoliatus YJ-118 isolated from this invention, ML-236B concentration in culture broth could be raised to 0.5% (w/v) and pravastatin sodium productivity was increased up to 600—1,340 mg/ / much higher than that of other microorganisms (60 mg/ / ) .
EXAMPLE 1
To 125 ml Erlenmeyer flask containing 20 ml seed culture medium(I) that comprises glucose 1%, yeast extract 0.2%, skim milk 0.2%, casein hydrolyte (N-Z amine) 0.5%, pH 7.0. 0.02% (w/v) ML-236B was added and Streptomyces exfoliatus YJ-118 isolated from manufacturing Example was inoculated. The cultivation was done at 27° C., 200 rpm, for 2 days on a rotary shaker. 20 ml of seed culture above was inoculated in 2 l Erlenmeyer flask containing 400 ml production medium(II) that comprises glucose 1.0%, yeast extract 1.0%, polypeptone 0.5%. K2HPO4 0.1%, MgSO4.7H2O 0.05%, NaCl 0.01˜0.1%, pH 7.2 and the flask was cultured at 27° C., 150 rpm. One day after cultivation, 0.05% (w/v) ML-236B (formula II-a) was added every day till the final concentration of ML-236B in culture broth became 0.2% (w/v). The cultivation was continued at 27° C., 150 rpm for 6 days and 0.3% glucose was fed once every two days 2 times in total. After then, the culture broth was adjusted to pH 9.0 and stirred for 3 hr. After centrifugation cell mass was removed and the supernatant was applied to a column of HP-20 500 ml. After washed with water, pravastatin sodium was eluted with 25% acetone solution. Pravastatin sodium fraction was concentrated in vacuo and the residue was applied to semi preparative HPLC(Kromasil C18 resin). Pravastatin sodium was eluted with 35% acetonitrile solution and was obtained as white crystal 1,254 mg (627 mg/l),
References
- “Prevachol”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- No Authors Listed (2002). “Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT)”. JAMA288 (23): 2998–3007. doi:10.1001/jama.288.23.2998. PMID12479764.
- Pfeffer MA, Keech A, Sacks FM, et al. “Safety and tolerability of pravastatin in long-term clinical trials: prospective Pravastatin Pooling (PPP) Project.” Circulation 2002;105:2341-2346
- Williams, Eni. “Pravachol Side Effects Center”. RxList. Retrieved 1 December 2012.
- “Pravastatin”. LactMed. U.S. National Library of Medicine. Retrieved 1 December 2012.
- Vaughan, C. J., and A. M. Gotto, Jr. 2004. Update on statins: 2003. Circulation 110: 886–892.
- Yoshino G, Kazumi T, Kasama T, et al. (1986). “Effect of CS-514, an inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase, on lipoprotein and apolipoprotein in plasma of hypercholesterolemic diabetics”. Diabetes Res. Clin. Pract.2 (3): 179–81. doi:10.1016/S0168-8227(86)80020-1. PMID3091343.
- “FDA Approves First Generic Pravastatin”. Retrieved 2008-01-20.
| WO2001044144A2 * | Dec 14, 2000 | Jun 21, 2001 | M Lakshmi Kumar | Process for the preparation of sodium salts of statins |
| US20020082295 * | Oct 5, 2001 | Jun 27, 2002 | Vilmos Keri | Pravastatin sodium substantially free of pravastatin lactone and epi-pravastatin, and compositions containing same |

Gatifloxacin



- 1.
Mather, R.; Karenchak, L. M.; Romanowski, E. G.; Kowalski, R. P. Am. J. Ophthalmol.2002, 133 ( 4) 463
- 2.
Corey, E. J.; Czakó, B.; Kürti, L. Molecules and Medicine; Wiley: NJ, 2007; p 135.
- 3.
Masuzawa, K.; Suzue, S.; Hirai, K.; Ishizaki, T. 8-Alkoxyquinolonecarboxylic acid and salts thereof excellent in the selective toxicity and process of preparing the same EP 0 230 295 A3, 1987.
- 4.
Niddam-Hildesheim, V.; Dolitzky, B.-Z.; Pilarsky, G.; Steribaum, G. Synthesis of Gatifloxacin WO 2004/069825 A1, 2004.
- 5.
Ruzic, M; Relic, M; Tomsic, Z; Mirtek, M. Process for the preparation of Gatifloxacin and regeneration of degradation products WO 2006/004561 A1, 2006.
- 6.
Iwata, M.; Kimura, T.; Fujiwara, Y.; Katsube, T. Quinoline-3-carboxylic acid derivatives, their preparation and use EP 0 241 206 A2, 1987.
- 7.
Sanchez, J. P.; Gogliotti, R. D.; Domagala, J. M.; Garcheck, S. J.; Huband, M. D.; Sesnie,J. A.; Cohen, M. A.; Shapiro, M. A. J. Med. Chem. 1995, 38, 4478
- 8.
Satyanarayana, C.; Ramanjaneyulu, G. S.; Kumar, I. V. S. Novel crystalline forms of Gatifloxacin WO 2005/009970 A1 2005.
- 9.
Takagi, N.; Fubasami, H.; Matsukobo, H.; (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture EP 0 464 823 A1, 1991.
………………………….
WO 2005009970
http://www.google.com/patents/WO2005009970A1?cl=en
preparation of Gatifloxacin hemihydrate from Ethyl-1- Cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate through boron difluoride chelate. Ethyl-1-cyclopropyl- 6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylate is reacted with aqueous hydrofluoroboric acid followed by condensation with 2-methyl piperazine in polar organic solvent resulting in an intermediate l-Cyclopropyl-7- (3-methyl piperazin-1- yl). -6-fluoro-8-methoxy-4-oxo-l, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. This intermediate may be further hydrolyzed to yield Gatifloxacin. Gatifloxacin so obtained may needs purification to yield high purity product. However to obtain directly high purity Gatifloxacin it is desirable to isolate the intermediate by cooling to low temperatures . Treating with an alcohol or mixture of alcohols purifies this intermediate. The purified condensed chelate in aqueous ethanol on hydrolysis with triethylamine followed by crystallization in ethanol gives Gatifloxacin hemihydrate with high purity.
STAGE – I:

Ethyl l-cyclopropyl-6,7-difluoro-8-met oxy l-Cycloproρyl-6, 7-difluoro-8-methoxy -4-oxo-l, -dihydro-3-quinoline -4-oxo-l, 4-dihydro-3-quinoline carboxylate carboxylic acid boron difluoride chelate
STAGE – II :

l-Cycloprop l-7- ( 3-methylpiperazin-l-yl.
6-fluoro~8-methoxy-4-oxo-l , 4-dihydro-3- carboxylicacid borondifluoride chelate quinoline carboxylicacid borondifluoride chelate
STAGE -III :

l-Cyclopropyl-7- (3- ethylpiperaz.in-l-yl . GATIFLOXACIN
-6-fluoro-8-methoxy-4-oxo-l , 4-dihydro-3- quinoline carboxylicacid borondifluoride chelate
Example-I: Preparation of Gatifloxacin • with isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-di luoro-8-methoxy-4-oxo- 1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate. Ethyl-l-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, -dihydro-3- quinόline carboxylate (100g)is suspended in ,40%aq..hydrofluoroboric acid -(1000 ml). Temperature of • the reaction mass is raised and maintained at 95°C to 100°C for 5hrs followed by cooling to 30°C – 35°C. Water (400 ml) is added and maintained at 25°C – 30°C for 2hrs . Product is filtered, washed with water (500 ml) and dried at 40°C – 45°C to constant weight. Dry weight of the product: 101.6 g (Yield: 95.8 %)
Stage-2: Preparation of 1- Cyclopropyl-7- (3-methylpiperazin-l-yl) – 6-fluoro-8-methoxy-4-oxo-l, -dihydro-3-quinoline carboxylic acid boron difluoride chelate
100 g of Boron difluoride chelate derivative prepared as above in stage-1 is suspended in acetonitrile (800 ml) , to that 2-methyl piperazine (44.0 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs followed by cooling to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr. The product is filtered and dried at 45°C – 50°C to constant weight. Dry weight of the product: 116.0 g (Yield: 93.9 %) .
The condensed chelate (100 g) prepared as above is suspended in methanol (1500 ml), maintained at 40°C – 45°C for 30 min. The reaction mass is gradually cooled, maintained for 1 hr at -5°C to 0°C. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. Dry weight of the product: 80.0 g (Yield: 80.0 %)
Stage -3: Preparation of Gatifloxacin (Crude)
The pure condensed chelate (100.0 g) prepared as above in stage-2 is suspended in 20% aq. ethanol (1000 ml) , the temperature is raised and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles, distilled under vacuum to remove solvent. Fresh ethanol (200 ml) is added and solvent is removed under vacuum at temperature below 50°C. Ethanol (200 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is mixed at -10°C to -5°C for 1 hr and then filtered. The wet cake is washed with ethanol (25 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 83.3 g (Yield: 91.7 %)
Stage- 4: Purification of crude Gatifloxacin
Crude Gatifloxacin (100.0 g) prepared as above in stage-3 is suspended in methanol (4000 ml), the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. Activated carbon (5 g) is added, maintained for 30 min and the solution is filtered. The filtrate is concentrated to one third of its original volume under vacuum at temperature below 40°C. The reaction mass is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight. The dry weight of the pure Gatifloxacin is 76.0 g (Yield: 76.0 %)
Example-II: Preparation of Gatifloxacin without isolation of intermediate (boron difluoride chelate derivative)
Stage-1: Preparation of l-cyclopropyl-6, 7-difluoro-8-methoxy-4- oxo-1, 4-dihydro-3-quinoline carboxylic acid boron difluoride chelate.
Ethyll-cyclopropyl-6, 7-difluoro-8-methoxy-4-oxo-l, 4-dihydro-3- quinoline carboxylate (lOOg) is suspended in 40% aq. hydrofluoroboric acid (1000 ml) . Temperature of the reaction mass is raised and maintained at 95°C to 100°C for 5 hrs followed by cooling to 30°C – 35°C. 400 ml DM water is added, maintained at 25°C – 30°C for 2hrs . The product is filtered, washed with DM water (500 ml) and dried at 40°C – 45°C to constant weight. The dry wt is 102.5 g (Yield: 96.6 %)
Stage – 2: Preparation of Gatifloxacin (Crude)
The boron difluoride chelate derivative (100 g) prepared as above in stage-1 is suspended in acetonitrile (800 ml) , 2-methyl piperazine (44 g, 1.5 mole equiv.) is added and mixed for 15 min to obtain a clear solution. The reaction mass is maintained at 30°C – 35°C for 12 hrs. Removed the solvent by vacuum distillation. 20% Aq. ethanol (1000 ml) is added, raised the temperature and maintained at 75°C to 80°C for 2 hrs. The reaction mass is cooled, filtered to remove insolubles. The filtrate is distilled under vacuum to remove solvent completely. Fresh ethanol (250 ml) is added and distilled under vacuum at temperature below 50°C. Fresh Ethanol (250 ml) is added to the residue and gradually cooled to -10°C to -5°C. The reaction mass is maintained at -10°C to -5°C for 1 hr and filtered. The wet cake is washed with ethanol (30 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the Gatifloxacin is 73.5 g (Yield: 65.4 %)
Stage -3: Purification of crude Gatifloxacin
Crude Gatifloxacin (80.0 g) prepared as above in stage-2 is suspended in methanol (2000 ml) , the temperature is raised and maintained at 60°C to 65°C for 20 min. to get a clear solution. The reaction mixture is filtered. The filtrate is gradually cooled and maintained at -10°C to -5°C for 2 hrs. The product is filtered, washed with methanol (50 ml) and dried at 45°C – 50°C to constant weight.
The dry weight of the pure Gatifloxacin is 56.0 g (Yield: 70.0 %)
……………………….
WO 2005047260
http://www.google.co.in/patents/WO2005047260A1?cl=en
Gatifloxacin is the international common name of l-cyclopropyl-6-fluoro-l, 4-dihydro-8-methoxy- 1- (3-methyl-l-piperazinyl) -4-oxo-3-guinolin-carboxylic acid of formula (I) , with application in medicine and known for its antibiotic activity:

European patent application EP-A-230295 discloses a process for obtaining gatifloxacin that consists on the reaction of compound (II) with 2-

In this process the gatifloxacin is isolated in the form of a hemihydrate after a laborious process of column chromatography and recrystallisation in methanol, which contributes towards making the final yield lower than 20% by weight. Moreover, in said process an undesired by-product is formed, resulting from demethylation at position 8 of the ring. European patent application EP-A-241206 discloses a process for preparing gatifloxacin, whose final steps are as follows:

(III) H ft N Me H DMSO
Gatifloxacin (I)

(IV) This process uses the intermediate compound (III) , which has been prepared and isolated in a separate operation, while the intermediate compound (IV) is also isolated before proceeding to its conversion into gatifloxacin by treatment with ethanol in the presence of triethylamine. The overall yield from these three steps is lower than 40%. These disadvantages — a synthesis involving several steps, low yields, and the need to isolate the intermediate products — hinder the production of gatifloxacin on an industrial scale. There is therefore a need to provide a process for preparing gatifloxacin with a good chemical yield, without the need to isolate the intermediate compounds and that substantially avoids demethylation in position 8 of the ring. The processes termed in English “one pot” are characterised in that the synthesis is carried out in the same reaction vessel, without isolating the intermediate compounds, and by means of successive addition of the reacting compounds. The authors of the present invention have discovered a simplified process for preparing gatifloxacin which does not require isolation of the intermediate compounds .
Example 1: Preparing gatifloxacin from compound (II) 10 g (0.0339 moles, 1 equivalent) of compound
(II) is placed in a flask, 30 ml of acetonitryl (3 volumes) is added and this is heated to a temperature of 76-80° C.

Once reflux has been attained, and being the temperature maintained, 3.28 g (0.0203 moles, 0.6 equivalents) of hexamethyldisilazane (HMDS) is added with a compensated adding funnel. Once addition is completed, the reaction is maintained with stirring for 1 hour at a temperature of 76-80° C. Once this period has elapsed, the reaction mixture is cooled to a temperature ranging between 0 and 15° C, and 5.78 g (0.0407 moles, 1.2 equivalents) of boron trifluoride ethyletherate is added while keeping the temperature below 15° C. Once addition is completed, the temperature is allowed to rise to 15- 25° C and it is kept under these conditions for approximately 2 hours. The pH of the mixture is then adjusted to an approximate value of 9 with triethylamine (approximately 2 ml) . To the resulting suspension is added a solution of 10.19 g (0.1017 moles, 3 equivalents) of 2-methylpiperazine in 28 ml of acetonitryl, while maintaining the temperature between 15 and 25° C. The resulting amber solution is kept with stirring under these conditions for approximately 3 hours . Once the reaction has been completed, the solution is distilled at low pressure until a stirrable paste is obtained. At this point 50 ml of methanol is added, the resulting suspension is raised to a temperature of 63-67° C and is kept under these conditions for approximately 5 hours . Once the reaction has been completed, the mixture is cooled to a temperature of 25-35° C in a water bath, and then at a temperature of 0-5° C in a water/ice bath for a further 1 hour. The resulting precipitate is filtered, washed with cold methanol (2 x 10 ml) and dried at 40° C in a vacuum oven to constant weight. 10.70 g of crude gatifloxacin is obtained, having a water content of 2.95% by weight. The yield of the process is 81.8%.
The crude product is crystallised in methanol by dissolving 20 g of crude gatifloxacin in 1 1 of methanol (50 volumes) at a temperature of 63-67° C. Once all the product has been dissolved, the solution is left to cool to a temperature of 30-40° C, and then to a temperature of 0-5° C in a water/ice bath, maintaining it under these conditions for 1 hour. The resulting suspension is filtered and the solid retained is washed with 20 ml (1 volume) of cold methanol. The solid obtained is dried at 40° C in a vacuum oven to provide 18.65 g of gatifloxacin with a water content of 2.36% by weight.
The overall yield from the compound (II) is 77.7%, with a purity exceeding 99.8% as determined by HPLC chromatography. The content of by-product resulting from demethylation in position 8 of the ring is lower than 0.1% as determined by HPLC chromatography.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 1-cyclopropyl-6-fluoro- 8-methoxy-7-(3-methylpiperazin-1-yl)- 4-oxo-quinoline-3-carboxylic acid | |
| Clinical data | |
| Trade names | Zymar |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a605012 |
|
|
| Oral (discontinued), Intravenous(discontinued) ophthalmic |
|
| Pharmacokinetic data | |
| Protein binding | 20% |
| Half-life | 7 to 14 hours |
| Identifiers | |
| 112811-59-3 |
|
| J01MA16 S01AE06 | |
| PubChem | CID: 5379 |
| DrugBank | DB01044 |
| ChemSpider | 5186 |
| UNII | 81485Y3A9A |
| KEGG | D08011 |
| ChEBI | CHEBI:5280 |
| ChEMBL | CHEMBL31 |
| NIAID ChemDB | 044913 |
| Chemical data | |
| Formula | C19H22FN3O4 |
| 375.394 g/mol | |
PAPER

An improved process to obtain gatifloxacin (1) through use of boron chelate intermediates has been developed. The methodology involves an initial activation step which accelerates the formation of the first chelate under low-temperature conditions and prevents demethylation of the starting material. To increase the overall yield and to avoid the isolation and manipulation of the resulting intermediates, the process has been designed to be carried out in one pot. As a result, we present here an easy, scaleable and substantially impurity-free process to obtain gatifloxacin (1) in high yield.
A High-Throughput Impurity-Free Process for Gatifloxacin
DSC analysis showed two endothermic peaks at 166.2 °C (T onset = 164.3 °C) and 190.0 °C (T onset = 188.2 °C) and an exothermic one at 168.1 °C. The shape of this DSC curve is characteristic of a monotropic transition between crystalline forms
Although there are several hydrates described for gatifloxacin such as, among others, the hemimydrate, sesquihydrate, and pentahydrate(Raghavan, K. S.; Ranadive, S. A.;Gougoutas, J. Z.; Dimarco, J. D.; Parker, W. L.; Dovich, M.; Neuman, A.Gatifloxacin pentahydrate. WO 2002/22126 A1, 2002) , the Gatifloxacin obtained by the present procedure does not seem to form a stoichometric hydrate, but instead it retains moisture.
Thus, the product is usually obtained with a Karl-Fischer value below 1% after drying, but it can absorb moisture until a final content of about 3%. This water content can vary between 2.0% and 3.5%, depending on the relative humidity of the environment. DSC analysis revealed a broad endothermic signal with minimum at 76 °C, while TGA analysis showed that the product loses all the water below 80 °C.
No loss of weight is registered when the product melts, and the weight is constant until the decomposition of the material at about 200 °C. On the basis of these results, it can be said that the water content of the gatifloxacin obtained by the present process is retained moisture instead of water belonging to the lattice. The shape of the derivative of the weight curve at the beginning of the analysis shows that the sample has already lost part of the moisture when the register starts. This is probably due to the sample starting to lose weight when makes contact with the dry atmosphere of the TGA oven that could explain the different values obtained for water content of the analyzed sample by TGA (1.90%) and Karl-Fischer (2.64%) methods.
Side-effects and removal from the market
A Canadian study published in the New England Journal of Medicine in March 2006 claims Tequin can have significant side effectsincluding dysglycemia.[2] An editorial by Dr. Jerry Gurwitz in the same issue called for the Food and Drug Administration (FDA) to consider giving Tequin a black box warning.[3] This editorial followed distribution of a letter dated February 15 by Bristol-Myers Squibb to health care providers indicating action taken with the FDA to strengthen warnings for the medication.[4] Subsequently it was reported on May 1, 2006 that Bristol-Myers Squibb would stop manufacture of Tequin, end sales of the drug after existing stockpiles were exhausted, and return all rights to Kyorin.[5]
Union Health and Family Welfare Ministry of India on 18 March 2011 banned the manufacture, sale and distribution of Gatifloxacin as it caused certain adverse side effects[6]
Contraindications
Availability
Gatifloxacin is currently available only in the US and Canada as an ophthalmic solution.
In China it is sold in tablet as well as in eye drop formulations.
Ophthalmic anti-infectives are generally well tolerated. The concentration of the drug observed following oral administration of 400 mg gatifloxacin systemically is approximately 800 times higher than that of the 0.5% Gatifloxacin eye drop. Given as an eye drop, Gatifloxacin Ophthalmic Solution 0.3% & 0.5% cause very low systemic exposures. Therefore, the systemic exposures resulting from the gatifloxacin ophthalmic solution are not likely to pose any risk for systemic toxicities.

- The reaction of 1-bromo-2,4,5-trifluoro-3-methoxybenzene (I) with CuCN and N-methyl-2-pyrrolidone at 150 C gives 2,4,5-trifluoro-3-methoxybenzonitrile (II), which by treatment with concentrated H2SO4 yields the benzamide (III) The hydrolysis of (III) with H2SO4 -. water at 110 C affords 2,4,5-trifluoro-2-methoxybenzoic acid (IV), which by reaction with SOCl2 is converted into the acyl chloride (V). The condensation of (V) with diethyl malonate by means of magnesium ethoxide in toluene affords diethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) malonate (VI), which by treatment with p-toluenesulfonic acid in refluxing water gives ethyl 2- (2,4,5-trifluoro-3-methoxybenzoyl) acetate (VII). The condensation of (VII) with triethyl orthoformate in refluxing acetic anhydride yields 3-ethoxy -2- (2,4,5-trifluoro-3-methoxybenzoyl) acrylic acid ethyl ester (VIII), which is treated with cyclopropylamine (IX) to afford the corresponding cyclopropylamino derivative (X). The cyclization of (X) by means of NaF in refluxing DMF gives 1-cyclopropyl-6,7-difluoro-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid ethyl ester (XI), which is hydrolyzed with H2SO4 in acetic acid to yield the corresponding free acid (XII). Finally, this compound is condensed with 2-methylpiperazine (XIII) in hot DMSO.
 |
|
Title: Gatifloxacin
CAS Registry Number: 112811-59-3
CAS Name: 1-Cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid
Trademarks: Tequin (BMS); Zymar (Allergan)
Molecular Formula: C19H22FN3O4
Molecular Weight: 375.39
Percent Composition: C 60.79%, H 5.91%, F 5.06%, N 11.19%, O 17.05%
Literature References: Fluorinated quinolone antibacterial. Prepn: K. Masuzawa et al., EP 230295; eidem, US 4980470 (1987, 1990 both to Kyorin); J. P. Sanchez et al., J. Med. Chem. 38, 4478 (1995); of the sesquihydrate: T. Matsumoto et al., US5880283 (1999 to Kyorin). In vitro antibacterial activity: A. Bauernfeind, J. Antimicrob. Chemother. 40, 639 (1997); H. Fukuda et al., Antimicrob. Agents Chemother. 42, 1917 (1998). Clinical pharmacokinetics: M. Nakashima et al., ibid. 39, 2635 (1995). Clinical study in urinary tract infection: H. Nito, 10th Mediterranean Congr. Chemother. 1996, 327; in respiratory tract infection: S. Sethi, Expert Opin. Pharmacother. 4, 1847 (2003).
Properties: Pale yellow prisms from methanol as hemihydrate, mp 162°.
Melting point: mp 162°
Derivative Type: Sesquihydrate
CAS Registry Number: 180200-66-2
Manufacturers’ Codes: AM-1155
Molecular Formula: C19H22FN3O4.1½H2O
Molecular Weight: 384.40
Percent Composition: C 59.37%, H 6.03%, F 4.94%, N 10.93%, O 18.73%
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Synthetic); Quinolones and Analogs
|
References
- Burka JM, Bower KS, Vanroekel RC, Stutzman RD, Kuzmowych CP, Howard RS (July 2005). “The effect of fourth-generation fluoroquinolones gatifloxacin and moxifloxacin on epithelial healing following photorefractive keratectomy”. Am. J. Ophthalmol. 140 (1): 83–7. doi:10.1016/j.ajo.2005.02.037.PMID 15953577.
- Park-Wyllie, Laura Y.; David N. Juurlink; Alexander Kopp; Baiju R. Shah; Therese A. Stukel; Carmine Stumpo; Linda Dresser; Donald E. Low; Muhammad M. Mamdani (March 2006).“Outpatient Gatifloxacin Therapy and Dysglycemia in Older Adults”. The New England Journal of Medicine 354 (13): 1352–1361. doi:10.1056/NEJMoa055191. PMID 16510739. Retrieved 2006-05-01. Note: publication date 30 March; available on-line 1 March
- Gurwitz, Jerry H. (March 2006). “Serious Adverse Drug Effects — Seeing the Trees through the Forest”. The New England Journal of Medicine 354 (13): 1413–1415.doi:10.1056/NEJMe068051. PMID 16510740. Retrieved2006-05-01.
- Lewis-Hall, Freda (February 15, 2006). “Dear Healthcare Provider:” (PDF). Bristol-Myers Squibb. Retrieved May 1, 2006.
- Schmid, Randolph E. (May 1, 2006). “Drug Company Taking Tequin Off Market”. Associated Press. Archived from the original on November 25, 2007. Retrieved 2006-05-01.[dead link]
- “Two drugs banned”. The Hindu (Chennai, India). 19 March 2011.
- Peggy Peck (2 May 2006). “Bristol-Myers Squibb Hangs No Sale Sign on Tequin”. Med Page Today. Retrieved 24 February2009.
| EP0610958A2 * | 20 Jul 1989 | 17 Aug 1994 | Ube Industries, Ltd. | Intermediates in the preparation of 4-oxoquinoline-3-carboxylic acid derivatives |
| ES2077490A1 * | Title not available |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008126384A1 | 31 Mar 2008 | 23 Oct 2008 | Daiichi Sankyo Co Ltd | Method for producing quinolone carboxylic acid derivative |
| CN101659654B | 28 Aug 2008 | 6 Nov 2013 | 四川科伦药物研究有限公司 | 2-Methylpiperazine fluoroquinolone compound and preparation method and application thereof |
| CN102351843A * | 18 Aug 2011 | 15 Feb 2012 | 张家口市格瑞高新技术有限公司 | Synthesis method of 2-methyl piperazine lomefloxacin |
| EP1832587A1 * | 2 Mar 2007 | 12 Sep 2007 | Quimica Sintetica, S.A. | Method for preparing moxifloxacin and moxifloxacin hydrochloride |
| US7365201 | 2 Mar 2006 | 29 Apr 2008 | Apotex Pharmachem Inc. | Process for the preparation of the boron difluoride chelate of quinolone-3-carboxylic acid |
| US7875722 | 30 Sep 2009 | 25 Jan 2011 | Daiichi Sankyo Company, Limited | Method for producing quinolone carboxylic acid derivative |
| EP0464823A1 * | Jul 4, 1991 | Jan 8, 1992 | Kyorin Pharmaceutical Co., Ltd. | (6,7-Substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid-O3,O4)bis(acyloxy-O)borates and the salts thereof, and methods for their manufacture |
| US4997943 * | Mar 31, 1987 | Mar 5, 1991 | Sankyo Company Limited | Quinoline-3-carboxylic acid derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101659654B | Aug 28, 2008 | Nov 6, 2013 | 四川科伦药物研究有限公司 | 2-Methylpiperazine fluoroquinolone compound and preparation method and application thereof |
| CN102351843A * | Aug 18, 2011 | Feb 15, 2012 | 张家口市格瑞高新技术有限公司 | Synthesis method of 2-methyl piperazine lomefloxacin |
TAKE A TOUR
TAKE A TOUR
Amritsar, punjab, India
-
Amritsar – Wikipedia, the free encyclopedia
https://en.wikipedia.org/?title=AmritsarAmritsar is one of the largest cities of the Punjab state in India. The city origin lies in the village of Tung, and was named after the lake founded by the fourth Sikh …
 GOLDEN TEMPLE
GOLDEN TEMPLE

 .
.






 Tandoori chicken at Surjit Food Plaza. amritsar
Tandoori chicken at Surjit Food Plaza. amritsar
Bullet marks on the walls of the park premises







The Jallianwalla Bagh in 1919, months after the massacre
Mealtime at the Golden Temple Amritsar
 Golden Temple – Harmandir Sahib: Free food for everyone
Golden Temple – Harmandir Sahib: Free food for everyone
Sri Guru Ram Dass Jee International Airport in Amritsar
Amritsar – Wagah Border – Street food stall | Explore bernic… |

-

Golden Temple
-

Maharaja Ranjit Singh’s Ram Bagh Gardens
-

Golden Temple
-

Durgiana Temple
-

The holy water
-

Jallianwala Bagh
-

Jallianwala Bagh
-

The holy water
-

Golden Temple
-

Golden Temple
-

Sikh Gurdwara
-

The holy water
 Night view of the Harmandir Sahib///////////
Night view of the Harmandir Sahib/////////// -



%27,_oil_on_canvas_painting_by_Charles_W._Bartlett,_1920,_Honolulu_Academy_of_Arts.jpg)
 tandoori chicken
tandoori chicken













WHO publishes final Guideline for Hold-Time Studies
DRUG REGULATORY AFFAIRS INTERNATIONAL
After the WHO had released the second draft of the guideline for the design of hold time studies in March already, it now released the final version as part of the Technical Report Series 992. Find out more about the Guideline for Hold Time Studies.
read
After the World Health Organisation (WHO) had released the second draft of the guideline for the design of hold-time studies in March already, it now released the final version as part of the Technical Report Series 992 (TRS 992, Annex 4).
The GMP regulations require that raw materials, packaging materials, intermediate, bulk and finished products need to be stored under suitable conditions. This also includes the definition of maximum hold-times for intermediate and bulk products prior to their further processing. The definition of these times should be justified on the basis of scientific data. This guideline aims at reflecting aspects that…
View original post 137 more words
DR ANTHONY MELVIN CRASTO
ORGANIC SPECTROSCOPY
