
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

WO 2015129603, NEW PATENT, Daiichi Sankyo Co Ltd, Edoxaban

HIGH-PURITY CRYSTALS OF ACTIVE BLOOD COAGULATION FACTOR X (FXA) INHIBITOR
DAIICHI SANKYO COMPANY,LIMITED [JP/JP]; 3-5-1,Nihonbashi Honcho,Chuo-ku, Tokyo 1038426 (JP)
Claims highly pure crystalline form of edoxaban p-toluenesulfonate monohydrate. Useful for treating thrombotic diseases. Daiichi Sankyo had developed and launched edoxaban for treating non-valvular atrial fibrillation, deep vein thrombosis and pulmonary embolism, the drug was recently launched in US (in February 2015) and approved in Europe (in June 2015).
The present invention addresses the problem of providing high-purity crystals of a compound which is represented by formula (1a) and is an active blood coagulation factor X (FXa) inhibitor. High-purity crystals of a compound represented by formula (1a) which: are characterised by being obtained by a step for dissolving crystals in a solvent and thereafter performing recrystallisation; have a 0.03% or less maximum content of one impurity as the impurity content by percentage; and have a 0.13% or less total impurity content.
In N represented 1 – (5-Chloro-2-yl) -N 2 – ((1S, 2R, 4S) -4 – [(dimethylamino) carbonyl] -2 – {[(5-methyl-4 , 5,6,7-tetrahydro thiazolone [5,4-c] pyridin-2-yl) carbonyl] amino} cyclohexyl) Etanjiamido p- toluenesulfonic acid monohydrate [hereinafter, may be referred to as compound (1a) is there
The obtained compound, in analysis using HPLC, as impurities, a peak of more impurities (both 0.03 wt%) is confirmed, the total of the impurities was 0.16 wt.% Since, its purity was 99.84% (Note that the content of% refers to% of the HPLC area value of the free form of formula (1a) compound).1 H-NMR (DMSO-d6) delta: 1.45-1.54 (1H, M), 1.66-1.78 (3H, M), 2.03-2.10 (2H, M), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3 .13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m) , 7.11 (2H, d, J = 7.8Hz), 7.46 (2H, d, J = 8.2Hz), 8.01 (2H, d, J = 1.8Hz), 8.46 ( 1H, t, J = 1.8Hz), 8.75 (1H, d, J = 6.9Hz), 9.10-9.28 (1H, br.s), 10.18 (1H, br.s ), 10.29 (1H, s).
Found: C; 50.25%, H; 5.36%, N; 13.32%

/////////////WO 2015129603, NEW PATENT, Daiichi Sankyo Co Ltd, Edoxaban
deleted
deleted
Chi-Med Says Fruquintinib Successful in Lung Cancer Trial

Fruquintinib
Phase 3…cancer
Hutchison Medipharma Enterprises Limited
Hutchison MediPharma for the treatment of locally advanced or metastatic colorectal cancer
C21H19N3O5
Exact Mass: 393.1325
cas 1194506-26-7, 6 ((6,7-dimethoxyquinazolin-4-yl) oxy) – N, 2-dimethylbenzofuran-3-carboxamide,
3-Benzofurancarboxamide, 6-[(6,7-dimethoxy-4-quinazolinyl)oxy]-N,2-dimethyl-
Synonym: Fruquintinib; HMPL-013; HMPL 013; HMPL013.
HPLC.http://www.medkoo.com/Product-Data/Fruquintinib/QC-Fruquintinib-CRB50706web.pdf
Fruquintinib, also known as HMPL-013, is an orally available, small molecule inhibitor of vascular endothelial growth factor receptors (VEGFRs), with potential anti-angiogenic and antineoplastic activities.
HMPL-013, a novel small molecule compound that selectively inhibits vascular endothelial growth factor receptor (VEGFR), is in phase III clinical studies at Hutchison MediPharma for the treatment of locally advanced or metastatic colorectal cancer. Phase II clinical trials are also ongoing for the treatment of non-squamous non-small cell lung cancer.
Early clinical development is under way at the company for the treatment of gastric cancer in combination with paclitaxel.
Fruquintinib’s mechanism of action is the inhibition of all three forms of VEGF receptors (VEGFR-1, 2, 3). Competitive advantages over currently marketed therapies are the compound’s unique kinase profile, a highly potent efficacy and excellent kinase selectivity, large safety margin, a broad spectrum antitumor activity and a low cost of goods.
Upon oral administration, fruquintinib inhibits VEGF-induced phosphorylation of VEGFRs 1, 2, and 3 which may result in the inhibition of migration, proliferation and survival of endothelial cells, microvessel formation, the inhibition of tumor cell proliferation, and tumor cell death. Expression of VEGFRs may be upregulated in a variety of tumor cell types.
In 2013, the company entered into a licensing, co-development, and commercialization agreement in China with Eli Lilly.
Angiogenesis is a physiological process of growing new blood vessels from pre-existing vessels. It takes place in a healthy subject to heal wounds, i.e., restoring blood flow to tissues after injury or insult.
Excessive angiogenesis may be triggered by certain pathological conditions such as cancer, age-related macular degeneration, and chronic inflammatory disease. As a result, new blood vessels feed diseased tissues and destroy normal tissues. In cancer, new blood vessels also allow tumor cells to escape into the circulation and lodge in other organs.
Vascular endothelial growth factor (VEGF), a homodimeric glycoprotein, and its receptors, e.g., kinase insert domain receptor (KDR), constitute an important angiogenic pathway. Studies have shown that inhibition of KDR resulted in endothelial cell apoptosis and, thus, suppression of angiogenesis. See Rubin M. Tuder, Chest, 2000; 117: 281. KDR inhibitors are therefore potential candidates for treating an angiogenesis-related disorder.
Chi-Med Says Fruquintinib Successful in Lung Cancer Trial
Written by Richard Daverman, PhD, Executive Editor, Greg B. Scott.
Hutchison MediPharma, a division of Chi-Med reported that fruquintinib met its primary endpoint in a second proof-of-concept China trial, this time as a treatment for advanced non-squamous non-small cell lung cancer. The company said fruquintinib “clearly” met its primary endpoint of progression-free survival, though specific data are being held for a scientific meeting. In 2013, Hutchison out-licensed China rights for the drug to Lilly. In May, the first proof-of-concept trial triggered two payments from Lilly to HMP totaling $18 million. More details…. http://www.chinabiotoday.com/articles/20150904
………….
Patent
US 20090281130
https://www.google.com.ar/patents/US20090281130
EXAMPLE 1 Synthesis of 6-(6,7-dimethoxyquinazolin-4-yloxy)-N,2-dimethylbenzofuran-3-carboxamide:

To a solution of 4-chloro-6,7-dimethoxyquinazoline (1 equiv.) in 2 ml CH3CN were added 6-hydroxy-N,2-dimethylbenzofuran-3-carboxamide (1 equiv.) and K2CO3 (1.5 equiv.). The mixture was refluxed under stirring for 10 hr. After the solvent was evaporated, the residue was washed with water, dried over MgSO4, filtered, concentrated, and purified by column chromatography to give the title compound in a yield of 85%.
1H NMR (DMSO-d6, 400 MHz) δ: 2.49 (s, 3H), 2.81 (d, J=8.4 Hz, 3H,10), 3.97 (s, 3H), 3.98 (s, 3H), 7.24 (dd, J=2.0, 8.4 Hz, 1H), 7.38 (s, 1H), 7.58 (s, 1H), 7.61 (d, J=2.0 Hz, 1H), 7.79 (d, J=8.4 Hz, 1H), 7.96 (m, 1H), 8.52 (s, 1H).
MS(m/e): 394.1 (M+1).
………………
WO 2009137797
https://www.google.com/patents/WO2009137797A2
……………….
CN 101575333
Example a: 6- (6,7-dimethoxy-quinazolin-4-oxo) -N, 2- dimethyl-benzofuran-3-carboxamide
[0048]

[0049] 4-Chloro-6,7-dimethoxy-quinazoline (1 mmol) was dissolved in 2 ml of acetonitrile, followed by addition of 6-hydroxy -N, 2- dimethyl-benzofuran-3- amide (1 mmol) and potassium carbonate (1.5 mmol). The reaction mixture was heated at reflux for 10 hours, concentrated to dryness, washed with water, and purified to give the desired product, yield 85%.
[0050] 1H NMR (DMS0-d6,400MHz) δ ppm:. 2 49 (s, 3H); 2.81 (d, J = 8. 4Hz; 3H, 10); 3.97 (s; 3H); 3.98 (s, 3H);. 7 24 (dd, J = 2. 0,8 4Hz;. 1H);. 7 38 (s, lH);. 7 58 (s, lH); 7.61 (d, J = 2. OHz; 1H);. 7 79 (d, J = 8. 4Hz; 1H);. 7 96 (m, 1H);. 8 52 (s, 1H).
[0051] MS (m / e)::. 394 1 (M + l).
………..
| EP1265874A2 * | Jan 23, 2001 | Dec 18, 2002 | Gödecke Gmbh | Method for the simplified production of (3-chloro-4-fluoro-phenyl)- 7-(3-morpholino-4-yl-propoxy)-6-nitro-quinazoline-4-yl]-amine or (3-chloro-4-fluoro-phenyl)- 7-(3-morpholino-4-yl-propoxy)-6-amino-quinazoline-4-yl]-amine |
| US20070208056 * | Jan 23, 2007 | Sep 6, 2007 | Bristol-Myers Squibb Company | Piperidinyl derivatives as modulators of chemokine receptor activity |
| US20080033000 * | May 15, 2007 | Feb 7, 2008 | Senex Biotechnology, Inc. | Identification of CDKI pathway inhibitors |
| 2 | See also references of EP2297115A2 | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8212033 * | Sep 29, 2010 | Jul 3, 2012 | Hutchison Medipharma Enterprises Limited | Use of substituted quinazoline compounds in treating angiogenesis-related diseases |
| US8497372 | Jun 4, 2012 | Jul 30, 2013 | Hutchison Medipharma Enterprises Limited | Use of substituted quinazoline compounds in treating age-related macular degeneration |
| US8575184 | Sep 1, 2010 | Nov 5, 2013 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
Hutchison Medipharma Enterprises Limited

Simon To, M.B.A.
Chairman

Mr To has been a Director since 2000 and an Executive Director and Chairman since 2006. He is also Chairman of the Remuneration Committee and a member of the Technical Committee of the Company. He is managing director of Hutchison Whampoa (China) Limited (“Hutchison China”) and has been with Hutchison China for over thirty years, building its business from a small trading company to a billion dollar investment group. He has negotiated major transactions with multinationals such as Procter & Gamble, Lockheed, Pirelli, Beiersdorf, United Airlines and British Airways.
Mr To’s career in China spans more than thirty years and he is well known to many of the top Government leaders in China. Mr To is the original founder of Hutchison Whampoa Limited’s healthcare business and has been instrumental in the acquisitions made to date. He received a First Class Honours Bachelor’s Degree in Mechanical Engineering from Imperial College, London and an MBA from Stanford University’s Graduate School of Business.
Christian Hogg, M.B.A.
Chief Executive Officer, Hutchison China MediTech Limited and Director, Hutchison MediPharma Holdings Limited

Mr Hogg has been an Executive Director and Chief Executive Officer since 2006. He is also a member of the Technical Committee of the Company. He joined Hutchison Whampoa (China) Limited in 2000 and has since led all aspects of the creation, implementation and management of the Company’s strategy, business and listing. This includes the creation of the Company’s start-up businesses and the acquisition and operational integration of assets that led to the formation of the Company’s China joint ventures.
Prior to joining Hutchison China, Mr Hogg spent ten years with Procter & Gamble starting in the US in Finance and then Brand Management in the Laundry and Cleaning Products Division. Mr Hogg then moved to China to manage P&G’s detergent business followed by a move to Brussels to run P&G’s global bleach business. Mr Hogg received a Bachelor’s degree in Civil Engineering from the University of Edinburgh and an MBA from the University of Tennessee.
Weiguo Su, Ph.D.
Executive Vice President and Chief Scientific Officer

Dr. Su has headed all drug discovery and research since he joined, including creating our R&D strategy, the formation and growth of research platform, and the research and discovery of each and every small molecule drug candidate in the Company’s portfolio.
Prior to joining in 2005, Dr. Su spent 15 years with Pfizer’s US R&D organization. Dr. Su delivered several high quality new drug candidates during his time with Pfizer, most recently as a director in the Medicinal Chemistry Department.
He received his Ph.D. and post-doctoral fellowship in Chemistry from Harvard University under the guidance of Nobel Laureate Professor E. J. Corey, and his Bachelor’s degree in Chemistry from Fudan University in Shanghai, China.
| R & D Center Address (A): Building 4, 720 Cailun Road Zhangjiang Hi-Tech Park Pudong, Shanghai, China Postal Code: 201203, China |
Head Office Address (B): Building 4, 917 Halei Road Zhangjiang Hi-Tech Park Pudong, Shanghai, China Postal Code: 201203, China |
Tel: +86 21 2067 3000 Email: BD@hmplglobal.com |
Addresses in Chinese:
R & D Center ( A): Chinese Cai Lun Road, Zhangjiang Hi-Tech Park in Pudong New Area, Shanghai, Lane 720 (intermediate哈雷路爱迪way out), Building 4
Head Office (B): Harley Road, Zhangjiang Hi-Tech Park, Pudong New Area, China, Shanghai, Lane 917, Building 4

![]()

///////
Indian pharma’s struggle to tighten standards paves way for M&A deals

MUMBAI – India’s smaller generic drugmakers, struggling to cope with a bruised reputation and tougher regulation in the United States, are under pressure to consider branching out to new, less-profitable markets or sell out to larger rivals.
Two years after its most high-profile regulatory setback to date in the United States – Ranbaxy’s $500 million U.S. fine for drug safety violations – India’s $15 billion a year generic drug industry is still rebuilding its image in its biggest market.
Many of its top firms are facing sanctions at some of their factories, as the U.S. Food and Drug Administration (FDA) tightens checks and its approvals process.
Combined with government-mandated price controls on drugs at home, that is piling pressure on smaller players.
“If they want to have a presence globally, they have to make investments. If they can’t, then they’ll have to focus on other markets or scale back their ambition outside of India, and that’s probably what will happen,” said Subhanu Saxena, CEO of Cipla , India’s fourth-largest drugmaker by revenue.
Ashok Anand, president of Hikal Ltd , a Mumbai-based drugmaker with a market value of $167 million, said some peers were putting themselves on the block.
“If they cannot deal with the stricter regulations, they might just prefer to sell out,” he said.
Pressure on U.S. sales has been felt across the Indian industry, with all drugmakers hit by delays in FDA approvals as the U.S. safety body overhauls its review process. Growth in U.S. revenue for drugmakers slowed to 14 percent in the year to March 2015, less than half what it was in the year to March 2012, according to brokerage Edelweiss.

But for larger players who want to plug gaps or, for the likes of Glenmark and Aurobindo who aim to grow in the United States, this pressure has lowered prices and could pave the way for attractive deals, bankers said.
“Now that some of the smaller companies are reeling under intensive regulatory scrutiny and want to cash out on their investments, valuations would be much more realistic,” said the head of India M&A at a large European bank in Mumbai.
SPENDING SPREE
Indian manufacturers say they have spent millions in high-end testing equipment, improved training and have hired larger teams in quality control since Ranbaxy was fined for manipulating clinical data.
Some consultants estimate spending on compliance has more than doubled to reach about 6 to 7 percent of sales for the larger companies.
But while the number of U.S. export bans issued to Indian companies fell to eight in 2014 from 21 in 2013, according to FDA data, the agency continues to find manufacturing violations at the plants of some of the biggest drugmakers in the country, an indication of the pervasiveness of the problem.
Sun Pharmaceutical Industries , Wockhardt , Dr Reddy’s Laboratories and Cadila Healthcarehave all faced FDA rebukes over the past year.
Smaller firms Ipca and Aarti Drugs faced FDA bans on their plants this year.
These failures – which executives blame on India’s “quick fix” culture and consultants blame on a failure to prioritize compliance – have clouded short-term growth prospects and added to pressure on smaller players, pushing some to look elsewhere.
“They can choose to be in lesser-regulated markets, such as Latin America, where there is a lot of demand. But they will have to live with much thinner margins,” said the finance director of a small Indian drugmaker, who did not want to be named. “It’s survival of the fittest.” REUTERS
http://m.todayonline.com/business/indian-pharmas-struggle-tighten-standards-paves-way-ma-deals
///////
China Generic Drugmakers Poaching Indian Execs

China Generic Drugmakers Poaching Indian Execs
Written by Richard Daverman, PhD, Executive Editor, Greg B. Scott.
In the competition between China and India pharmas, China’s generic drug industry leads in the supply of APIs to global drugmakers, but India supplies more finished generic drugs to the world’s marketplace. That may be changing. According to press reports,
China drugmakers have begun hiring experienced Indian pharma execs, offering them two to three times their present salaries.
The China companies are willing to pay at these levels because the Indian professionals have two skills the Chinese want: drug formulation experience and English.
China’s drugmakers want help as they target the western world’s lucrative generic drug market.
More details…. http://www.chinabiotoday.com/articles/20150903

///////////
Tocagen’s Double Action Glioblastoma Treatment Receives FDA Orphan Drug Designation

Toca 511 and Toca FC, developed by Tocagen, is a combination treatment currently being investigated in phase I/II trials for recurrent high grade glioma including the notoriously difficult to treat glioblastoma multiforme. Toca 511 (vocimagene amiretrorepvec) is a nonlytic retroviral replicating vector (RRV) that encodes the transgene cytosine deaminase (CD). This enzyme is used to catalyze the conversion of Toca FC, a novel oral extended-release prodrug 5-fluorocytosine (5-FC) to the active 5-fluorouracil (5-FU). Intravenous or intracranial injection of Toca 511 takes place during initial treatment and 3-7 weeks later the patient starts cyclic administration of Toca FC.1,2,3 The phase I/II trials in humans have shown similar results of patients exceeding the average life expectancy of high grade gliomas.4
Clinical stage immuno-oncology company, Tocagen, Inc., announced the US Food and Drug Administration has granted its primary immuno-oncology candidate orphan drug designation as a promising and much-needed treatment of glioblastoma, the most common form of primary brain cancer. Every year, over 10,000 people are diagnosed with glioblastoma in the United States. The new designation brings the company’s Toca 511 & Toca FC closer to helping patients suffering with this type of tumor. Tocagen is preparing to proceed with a pivotal clinical trials later this year.

ANNA TAN
Glioblastoma is known to be extremely aggressive, with newly diagnosed patients expecting a mere five-year survival rate of less than 5 percent, along with a high likelihood of tumor recurrence despite completion of standard treatment. Once the tumor recurs, the average survival is only 8 months.

Toca 511 is a retroviral replicating vector (RRV) that selectively delivers a gene for the enzyme cytosine deaminase into the tumor. Patients then take oral cycles of Toca FC, a novel formulation of an antifungal drug, which is converted within infected cancer cells into the FDA-approved anticancer drug, 5-fluorouracil (5 FU). Toca 511 & Toca FC work by programming cancer cells to convert the prodrug 5-FC into the anticancer drug 5-FU, effectively causing tumor cell death and stimulating the immune system through a combination of mechanisms.

“There’s an extraordinary need for new treatment options for patients with this devastating disease,” said Harry Gruber, M.D., chief executive officer of Tocagen. “We believe FDA’s granting of both orphan drug and Fast Track designations to Toca 511 & Toca FC will enable us to more efficiently advance our program, which we hope will ultimately offer physicians and patients a new option in the fight against brain cancer.”
ICT-107 is a dendritic cell-based immunotherapy targeting multiple tumor-associated antigens on glioblastoma stem cells. The trial will be a randomized, double-blind, placebo-controlled, and will aim to enroll around 400 HLA-A2 positive patients. The study will be conducted across 120 sites in the US, Canada, and the European Union.
Mechanism of action
Retroviruses, once inside the target cell, use reverse transcriptase to produce DNA from the RNA present in the virus. Toca 511 is based on the gamma retrovirus, murine leukemia (MLV).5 The virus has many innate properties that are suitable for targeted cancer treatment. One of the most important properties is the reproduction mechanism that occurs without cytolysis of the host cell. In non-lytic reproduction, the infected cell continuously forms small buds that are pinched off containing the virus to allow rapid infection. Another property is the requirement for cell division. Infection is limited to mitotically active cells. These two properties present an ideal candidate vector for modification. The lack of cytolysis in the host cell prevents an immune response and the necessity for the cell to be dividing allows localization to cancerous tumors. As an oncolytic agent, the mechanism uses the rapid mitotic activity of the cancerous tumor cells to spread the therapeutic gene in an effective and controlled manner.5 In Toca 511, the insertion of the CD transgene into the active tumor catalyzes the treatment. The expression of CD by the tumor allows intratumoral conversion of 5-FC to 5-FU.6 This allows the cytotoxic 5-FU to be maintained within the tumor cell. A second mechanism of action is proposed based upon recent data. Post-treatment, a systemic anticancer immune response is present that selectively acts against the cancerous cells.4,7
Design
The design of the Toca 511 RRV is based upon the vector design by Logg et al.5 Multiple changes facilitated selection of a clinically efficacious RRV. The original ecotropic envelope was changed to an amphotropic sequence. In the IRES-CD cassette, multiple small repeats were removed to allow for decreased instability during homologous recombination. A restriction site Psi I was placed at the 3′ of IRES for the insertion of the CD transgene. The resulting vector consists of the following, 5′ to 3′: CMV-R-U5, PBS, 5′ SS, gag, pol (with a 3′ SS), 4070A env, IRES, Psi I, yCD2, Not I, PPT, and the U3-R-U5.8
Clinical trials
Toca 511 and Toca FC combination therapy is currently being investigated for recurrent and progressive Grade III or IV glioma.1,2,3 The initial clinical study is the first to use a RRV to facilitate gene transfer into gliomas. In a recent presentation by Tocagen, researchers expressed the safety and efficacy of the therapy in the first two trials. Minimal treatment toxicity was reported. The landmark six and twelve month survival rates were higher than previously published data in both studies.4 Following positive results with the initial two trials, investigation into the intravenous efficacy is currently being determined.7
Preclinical investigations
Two important discoveries that led to the creation of Toca 511/FC treatment are the optimization of yeast CD and modifications to the vector backbone for genomic replication stability. The optimization of the yeast CD involved the modification of the codon sequence at three amino acids to a known preferred human codon sequence. This did not change the amino acid sequence. This resulted in stability at 37°C compared to the previous 26°C. The vector backbone modification at the env-3′ untranslated boundary created a vector with higher fidelity than the wild type.8 In studies of mice with implanted gliomas, Toca 511 and Toca FC therapy resulted in an unprecedented survival rate.6,8 Furthermore, when the mice were re-implanted with the same glioma post-treatment, memory T lymphocytes remained active and the growth was inhibited.6 The combination of these findings led to the clinical candidate that is currently undergoing trials.
References
1. Tocagen Inc. A Phase 1 Ascending Dose Trial of the Safety and Tolerability of Toca 511 in Patients With Recurrent High Grade Glioma. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2014 June 12]. Available from: http://clinicaltrials.gov/show/NCT01156584 NLM Identifier: NCT01156584.
2. Tocagen Inc. A P1 Ascending Dose Trial of Safety and Tolerability of Toca 511, a Retroviral Replicating Vector, Administered to Subjects at the Time of Resection for Recurrent High Grade Glioma & Followed by Treatment With Toca FC, Extended-Release 5-FC. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2014 June 12]. Available from: http://clinicaltrials.gov/show/NCT01470794 NLM Identifier: NCT01470794.
3. Tocagen Inc. A P1 Ascending Dose Trial of the Safety and Tolerability of Toca 511, a Retroviral Replicating Vector, Administered Intravenously Prior to, and Intracranially at the Time of, Subsequent Resection for Recurrent HGG & Followed by Treatment With Extended-Release 5-FC. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2014 June 12]. Available from: http://clinicaltrials.gov/show/NCT01985256 NLM Identifier: NCT01985256.
4. Interim Clinical Data for Tocagen’s Toca 511 & Toca FC in Patients with High Grade Glioma Presented at American Association of Neurological Surgeons Annual Meeting. Tocagen Inc., 10 April 2014. Web. 10 June 2014. .
5. Logg, C. R.; Robbins, M. J. Retroviral Replicating Vectors in Cancer. Methods in Enzymology 2012, 507, 199-228.
6. Ostertag, D.; Amundson, K. K.; Espinoza, F. L.; Martin, B. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-flurouracil using a nonlytic retroviral replicating vector. Neuro-Oncology 2012, 14(2), 145-159.
7. Tocagen Doses First Patient Intravenously in Clinical Trial of
Selective Cancer Therapy, Toca 511 & Toca FC. Tocagen Inc., 11 March 2014. Web. 10 June 2014. http://www.tocagen.com/press/tocagen-doses-first-patient-intravenously-in-clinical-trial-of-selective-cancer-therapy-toca-511-toca-fc/
8. Perez, O. D.; Logg, C. R.; Hiraoka, K.; Diago, O. Design and Selection of Toca 511 for Clinical Use: Modified Retroviral Replicating Vector With Improved Stability and Gene Expression. Molecular Therapy 2012, 20(9), 1689-1698.
Anna Tan, RN

Anna Tan, R.N. – Managing Editor | BioNews Services
///
Christopher Conway has been appointed to lead AMRI ’s discovery business strategy as SrVP of Discovery and Development and Global Commercial Sales.

Christopher Conway, Senior Vice President,
Global Sales and Marketing
Christopher Conway has been appointed to lead the company’s discovery business strategy as Senior Vice President of Discovery and Development and Global Commercial Sales.

September 3, 2015
SEE AN UPDATEFRO AMRI
QUOTE
Dear industry colleague,
As we continue to support the research and development that leads to the commercialization of pharmaceutical products, it is critical that we align our focus on R&D with the commercial demands of the market. Today, AMRI has announced an organizational change in our Discovery and Development Services (DDS) business. These changes are expected to drive top and bottom line growth for the Discovery and Development Solutions (DDS) business through strong commercial leadership; strengthen the DDS strategy and aggressively pursue the most valuable growth opportunities, externally and internally; and ensure that our service offerings are well aligned with your needs and the needs of the market.
Effective immediately, Christopher Conway has been appointed to lead the company’s discovery business strategy as Senior Vice President of Discovery and Development and Global Commercial Sales. In this role, Chris will head up the global Discovery and Development Solutions (DDS) business leading these businesses in the United States, Europe and Asia. Sales and marketing will also continue to report to him. He succeeds Michael A. Luther, Ph.D., MBA, who will be leaving AMRI to pursue other opportunities. We thank Dr. Luther for his efforts in moving the DDS business along over the last year and wish him the best in his future endeavors.
Related to this announcement, we would like to take the opportunity to announce the hiring of Rory Curtis, Ph.D., who has joined AMRI as Vice President of Discovery Biology and Pharmacology. Rory will also serve as site head with responsibility for scientific operations at AMRI’s Buffalo, N.Y. location. Rory was most recently Senior Director of Human Diseases in Discovery at Cubist Pharmaceuticals, where he developed Cubist’s antibacterial drug discovery into new disease areas such as pain, inflammation and gastro-intestinal disease. Before this, he held positions of increasing responsibility at Elixir Pharmaceuticals, Millennium Pharmaceuticals and Regeneron Pharmaceuticals.

Rory Curtis
Vice President of Discovery Biology and Pharmacology, Site Head AMRI Buffalo
In addition to Chris’ current direct reports and Rory, he will have a scientific leadership team reporting into him, which includes Michael P. Trova, Ph.D., Senior Vice President of Chemistry; Raj Shenoy, Senior Director of Global Chemical Development; and Pete Michels, Ph.D., Senior Director of Metabolism and Biotransformations.
Michael P. Trova, Ph.D.

Pete C. Michels, Ph.D., Senior Director, Chemical Development, Fermentation and Biocatalysis, AMRI
We are very excited about the future of AMRI Drug Discovery and Development and are pleased to welcome Rory to the AMRI discovery team. Market demand for our DDS services continues to grow and these changes will help us increase our market share and strengthen AMRI’s global position in Discovery and Development.
As we approach the second half of 2015, we are looking forward to working with you on a great number of new opportunities in 2016 and beyond. We appreciate your loyalty and support, and continue to remain dedicated to enhancing your pharmaceutical services experience from early discovery through to the commercialization and delivery of drug product. If you have any questions, please feel free to read today’s related press release at www.amriglobal.com, or contact us here.
Sincerely,

William S. Marth
President and CEO
Albany Molecular Research Inc. (AMRI)
www.amriglobal.com

William S. Marth. President and Chief Executive Officer Albany Molecular Research, Inc.


Albany Molecular Research Inc. (AMRI)
26 Corporate Circle
Albany, NY 12203
Albany Molecular Research Inc. provides global contract research and manufacturing services to the pharmaceutical and biotechnology industries. Our services include Drug Discovery, such as medicinal chemistry, discovery biology and in vitro ADME; Development, such as pre-formulation, formulation and validation; and Manufacturing, such as cGMP API manufacturing.
SINGAPORE RESEARCH CENTRE
 AMRI’s Singapore Research Centre, Pte. Ltd. provides chemistry and biology services to support drug discovery and development programs. AMRI is one of the first drug discovery R&D companies to establish operations in Singapore. Fully integrated with AMRI’s locations in the United States, Asia, and Europe, the Singapore centre offers medicinal chemistry services such as hit-to-lead andlead optimization as well as focused library synthesis / custom synthesis. In the area of biology / in vitro pharmacology, the Singapore Research Centre provides target validation; assay development; HTS; rapid production of SAR quality data; and in vitro ADMET support, including CYP inhibition, metabolic stability (liver microsome assays), and aqueous solubility. As a signatory to the World Patent Treaty, Singapore provides an environment that protects intellectual property, enabling our scientists to conduct proprietary and cutting-edge research on behalf of our customers.
AMRI’s Singapore Research Centre, Pte. Ltd. provides chemistry and biology services to support drug discovery and development programs. AMRI is one of the first drug discovery R&D companies to establish operations in Singapore. Fully integrated with AMRI’s locations in the United States, Asia, and Europe, the Singapore centre offers medicinal chemistry services such as hit-to-lead andlead optimization as well as focused library synthesis / custom synthesis. In the area of biology / in vitro pharmacology, the Singapore Research Centre provides target validation; assay development; HTS; rapid production of SAR quality data; and in vitro ADMET support, including CYP inhibition, metabolic stability (liver microsome assays), and aqueous solubility. As a signatory to the World Patent Treaty, Singapore provides an environment that protects intellectual property, enabling our scientists to conduct proprietary and cutting-edge research on behalf of our customers.
Areas of Expertise: Discovery Services
Contact Information:
61 Science Park Road
#05-01 The Galen
Singapore Science Park II
Singapore 117525
Phone: +65-6398-5500
Fax: +65-6398-5511
HYDERABAD RESEARCH CENTRE
 The Hyderabad Research Centre, Pvt. Ltd. (AMRHRC) is located in Hyderabad, India, an emerging technology metropolis located in South Central India.
The Hyderabad Research Centre, Pvt. Ltd. (AMRHRC) is located in Hyderabad, India, an emerging technology metropolis located in South Central India.
All fully integrated with AMRI’s U.S.-based resources, this Centre’s core area of expertise is in the areas of medicinal chemistrysupport, chemical development, custom synthesis of scaffolds and building blocks, process development, GMP analytical services,scale-up and preparation of reference standards.
Areas of Expertise: Chemical Development and Small Scale Manufacturing
Contact Information:
Hyderabad Research Centre Pvt. Ltd.
Plot # 9, Alexandria Knowledge Park, Turkapally, Shameerpet
Genome Valley, RR District, Hyderabad – 500 078, India
Tel : +91 – 40 – 6687 6666 (Board)
Fax : +91 – 40 – 6687 6600

GRANT CARR, PH.D. 
SENIOR DIRECTOR, LEAD DISCOVERY, AMRI

ROBERT B. KARGBO, PH.D.
SENIOR RESEARCH SCIENTIST I, AMRI
Novartis launches first US ‘biosimilar’ drug at 15 percent discount

Zarxio, filgrastim-sndz
Novartis launches first US ‘biosimilar’ drug at 15 percent discount
LONDON/ZURICH: Novartis kicked off a new era in U.S. medicine on Thursday with the launch of the first “biosimilar” copy of a biotechnology drug approved in the United States, at a discount of 15 percent to the original.
The Swiss drugmaker’s generics unit Sandoz said Zarxio, its form of Amgen’s white blood cell-boosting product Neupogen, would increase access to an important treatment by offering a “high-quality, more affordable version”.
U.S. biotech group Amgen had tried to stop the sale of Zarxio, also known as filgrastim-sndz, but the Washington-based appeals court rejected its attempt to block the launch…..http://www.channelnewsasia.com/news/health/novartis-makes-history-wi/2097550.html

On March 6, 2015, FDA approved the first biosimilar under the Biologics Price Competition and Innovation Act (BPCIA), Sandoz’s Zarxio®. Sandoz submitted Zarxio®as a highly similar, not interchangeable biosimilar, for the same indications as the referenced product. The BPCIA was signed into law in March 2010.
FDA designated “filgrastim-sndz” as the placeholder nonproprietary name rather than the innovator’s name, filgrastim. FDA said that this nonproprietary name “should not be viewed as reflective of the agency’s decision on a comprehensive naming policy for biosimilar and other biological products. While the FDA has not yet issued draft guidance on how current and future biological [biosimilar?] products marketed in the United States should be named, the agency intends to do so in the near future.”
Accompanying the news release was a document “Biosimilars: More Treatment Options Are on the Way”. The document includes various quotes and paraphrased statements by Leah Christl, Ph.D., Associate Director for Therapeutic Biologics, to help describe to consumers what biosimilar medications are. Below are some quotes and information from that document:
Biologics are medicines that generally come from living organisms, which can include humans, animals and microorganisms such as yeast and bacteria.
. . .
“Biologics are different from conventional medications. Conventional medications—drugs—are generally made from chemicals, or chemically synthesized, and therefore their structure can be relatively easily defined,” explains Christl.
Unlike conventional medications, biologics can’t be made by following a chemical “recipe.” “Biologics come from living organisms which are variable in nature. In addition, they are generally more complex and not as easy to define and characterize,” Christl explains. For that reason, manufacturing biologics is a far more complex process than manufacturing drugs.
Just as it does for drugs, FDA rigorously and thoroughly evaluates a biologic’s safety and effectiveness before granting it licensure (approval). Currently, biologics are among the fastest growing segments of the prescription product market.
. . .
Christl explains that a biosimilar is a type of biologic that is highly similar to another, already FDA-approved biologic (known as the reference product).
“It is important to note that a biosimilar is not just like a generic drug,” she adds. “Because of the differences in complexity of the structure of the biologic and the process used to make a biologic, biosimilars are not as easy to produce as generics, which are copies of brand name drugs.” A biosimilar is not an exact duplicate of another biologic; rather, a biosimilar is highly similar to the reference product.
Before approving a biosimilar, FDA experts must also first verify that there are no clinically meaningful differences between the biosimilar and its reference product. In other words, it will work the same way as the reference product for its approved indications.
Also, the biosimilar must have the same strength and dosage form (injectable, for example) and route of administration as the reference product. The biosimilar must be manufactured followingCurrent Good Manufacturing Practices.
“Patients can rest assured that they’ll be able to rely upon the safety and effectiveness of an FDA-approved biosimilar, just as they can rely on the reference product that the biosimilar was compared to,” Christl says. Like other biologics, biosimilars generally must be prescribed by a physician.
. . .
“Biosimilars are likely to create greater competition in the medical marketplace,” saysChristl. This could not only increase treatment options for patients, but also lead to less expensive alternatives to comparable products. With an increasing number of biosimilars on the market, consumers may expect to get equally safe and effective treatment, but at lower costs, she says.
Despite the significant achievement for FDA to approve the first biosimilar under the BPCIA, significant questions other than nonproprietary naming remain. First, Sandoz chose not to take advantage of the pre-approval patent exchange mechanism of the BPCIA, which could have addressed possible patent challenges that may prevent Sandoz from marketing Zarxio®until certain patents are invalidated, are found unenforceable, or have expired. Second, because this and other non-interchangeable versions of biosimilars are not expected to have automatic substitution based on the BPCIA, it remains unclear how ready physicians or patients will be to try a biosimilar version over its referenced product. Third, company representatives from Sandoz and other biosimilar manufacturers have not indicated at what price their biosimilar products will be sold, at times suggesting “at parity,” which may cause reimbursement issues. Fourth, many states have enacted rules that include special physician notification provisions, even when interchangeable biosimilars are dispensed to patients. And there are still issues surrounding pharmacovigilance and risk management when there are innovator and corresponding biosimilar versions marketed. Nevertheless, FDA proclaims that more biosimilars are on the way, as additional companies have indicated that they have submitted or FDA has filed their biosimilar applications. Sandoz’s Zarxio® then is just the tip of the iceberg of what is coming with more issues to be resolved along the way.
ZARXIO (filgrastim-sndz) is a 175 amino acid human granulocyte colony-stimulating factor (G-CSF) manufactured by recombinant DNA technology.
ZARXIO is produced by Escherichia coli (E coli) bacteria into which has been inserted the human granulocyte colony-stimulating factor gene. ZARXIO has a molecular weight of 18,800 daltons. The protein has an amino acid sequence that is identical to the natural sequence predicted from humanDNA sequence analysis, except for the addition of an N-terminal methioninenecessary for expression in E coli. Because ZARXIO is produced in E coli, the product is non-glycosylated and thus differs from G-CSF isolated from a human cell.
ZARXIO injection is a sterile, clear, colorless to slightly yellowish , preservative-free liquid containing filgrastimsndz at a specific activity of 1.0 x 108 U/mg (as measured by a cell mitogenesis assay). The product is available in single-use prefilled syringes. The single-use prefilled syringes contain either 300 mcg/0.5 mL or 480 mcg/0.8 mL of filgrastim-sndz. See table below for product composition of each single-use prefilled syringe.
| 300 MCG/0.5 ML SYRINGE | 480 MCG/0.8 ML SYRINGE | |
| Filgrastim-sndz | 300 mcg | 480 mcg |
| Glutamic Acid | 0.736 mg | 1.178 mg |
| Polysorbate 80 | 0.02 mg | 0.032 mg |
| Sorbitol | 25 mg | 40 mg |
| Sodium hydroxide | q.s. | qs. |
| Water for Injection | ||
| USP q.s. ad* | ad 0.5 mL | ad 0.8 mL |
Vandetanib
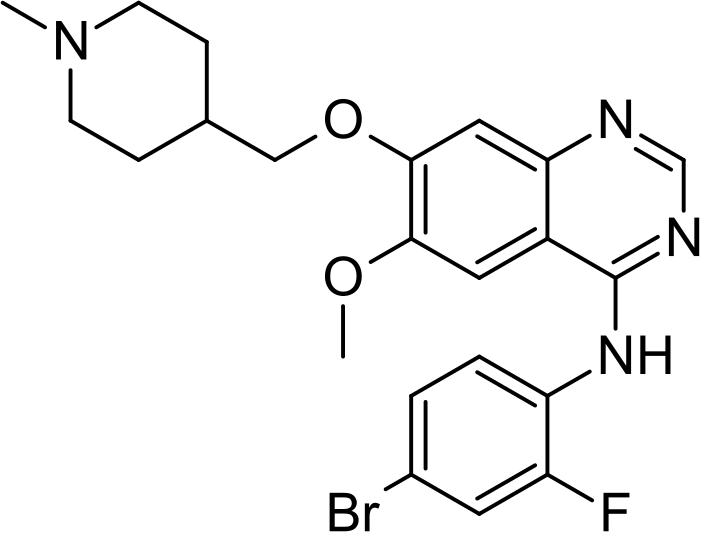
Vandetanib; 443913-73-3; Zactima; ZD6474; Caprelsa; ZD 6474; ch 331, azd 6474
cas 338992-00-0 free form
338992-48-6 HCl
338992-53-3 monotrifluoroacetate
N-(4-Bromo-2-fluorophenyl)-6-methoxy-7-(1-methylpiperidin-4-ylmethoxy)quinazolin-4-amine
Vandetanib (INN, trade name Caprelsa) is an anti-cancer drug that is used for the treatment of certain tumours of the thyroid gland. It acts as a kinase inhibitor of a number of cell receptors, mainly the vascular endothelial growth factor receptor (VEGFR), theepidermal growth factor receptor (EGFR), and the RET-tyrosine kinase.[1][2] The drug was developed by AstraZeneca.
Orphan drug designation has been assigned in the E.U. for the treatment of medullary thyroid carcinoma. In 2005, orphan drug designation was also assigned in the U.S. for several indications, including treatment of patients with follicular thyroid carcinoma, medullary thyroid carcinoma, anaplastic thyroid carcinoma, and locally advanced and metastatic papillary thyroid carcinoma. In 2013, orphan drug designation has been assigned in Japan as well for the treatment of thyroid cancer.
Approvals and indications
Vandetanib was the first drug to be approved by FDA (April 2011) for treatment of late-stage (metastatic) medullary thyroid cancer in adult patients who are ineligible for surgery.[3] Vandetanib was first initially marketed without a trade name,[4] and is being marketed under the trade name Caprelsa since August 2011.[5]
Vandetanib is an orally active vascular endothelial growth factor receptor-2 (VEGFR-2/KDR) tyrosine kinase inhibitor, originally developed by AstraZeneca, which was filed for approval in the U.S. and the E.U. for the treatment of non-small cell lung cancer (NSCLC) in combination with chemotherapy, in patients previously treated with one prior anticancer therapy.
However, in late 2009 the company withdrew both the U.S and the EU applications. In 2010, AstraZeneca discontinued development of this compound for the treatment of NSCLC. In 2011, the FDA approved vandetanib for the treatment of medullary thyroid cancer. Also in 2011, a positive opinion was assigned to the regulatory application filed in the E.U. for this indication and in Japan was filed for approval.
Final EMA approval was granted in February 2012 and first E.U. launch took place in the U.K. in 2012.
2011 年 4 月 6 by the FDA-approved surgical resection can not be used for locally advanced or metastatic medullary thyroid cancer (medullary thyroid cancer, MTC) of the drug. Vandetanib is vascular endothelial growth factor receptors (vascular endothelial growth factor receptor, VEGFR) and epidermal growth factor receptor (epidermal growth factor receptor, EGFR) antagonists, tyrosine kinase inhibitors (tyrosine kinase inhibitor). Produced by AstraZeneca.
The synthetic route is as follows:
………………

………………………..

……….
Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors
J Med Chem 1999, 42(26): 5369
http://pubs.acs.org/doi/abs/10.1021/jm990345w
………………………
Radiosynthesis of [(11)C]Vandetanib and [(11)C]chloro-Vandetanib as new potential PET agents for imaging of VEGFR in cancer
Bioorg Med Chem Lett 2011, 21(11): 3222
Novel 4-anilinoquinazolines with C-7 basic side chains: Design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors
J Med Chem 2002, 45(6): 1300
A novel approach to quinazolin-4(3H)-one via quinazoline oxidation: An improved synthesis of 4-anilinoquinazolines
Tetrahedron 2010, 66(4): 962
………………………………
CN 104098544
http://www.google.com/patents/CN104098544A?cl=en
Vandetanib is a synthetic Anilinoquinazoline, advanced medullary thyroid cancer can not be used for the treatment of surgical treatment (medullary thyroid cancer), chemical name: 4- (4-bromo-2- fluoroanilino) _6_ methoxy -7 – [(l- methylpiperidin-4-yl) methoxy] quinazoline, having the following structural formula I:

[0004] The present method of synthesizing the compound are as follows:
[0005] US Patent US7173038 AstraZeneca announced the following methods:
[0006] Method One:
[0007]

Method two:

A structure in which the synthesis of compounds of formula as follows:

the process is cumbersome, long synthetic route, therefore a need to provide a new synthetic way to overcome these problems.
An aspect provides a compound having the structure of formula II:

Another aspect provides a process for preparing a compound of the structural formula II, a compound of formula III with a compound of formula IV in the presence of a base to give a compound of the structural formula II,

where Μ for methylphenylsulfonyl, methylsulfonyl.
Example: 4- (4-bromo-2-fluoroanilino) -6_ methoxy-7 – [(1-formyl-4-yl) methoxy] quinazoline preparation
[0026] in 50mL two-neck flask was added 4- (4-bromo-2-fluoroanilino) -6-methoxy-7-hydroxy-quinazoline (3. 64g, 0 · Olmol), 1- formyl- 4-p methylsulfonyloxy- methylpiperazine steep (3. 56g, 0 · 012mol) and potassium carbonate (4. 14g, 0.03mol), yellow turbid solution was stirred and heated to 100 ° C, TLC detection to feed completion of the reaction. Down to room temperature, the reaction mixture was slowly poured into l〇〇mL water, stirred, filtered, then the filter cake was washed with 50mL water, 15mL of ethyl acetate and then slurried, filtered and dried to give a pale green solid 4- (4- bromo-2-fluoroanilino) -6-methoxy -7 – [(l- carboxylic acid piperidin-4-yl) methoxy] quinazoline 3. 9g, 80% yield.
[0027] ^ NMR (400Mz, DMS0): δ = 1 1〇-1 29 (m, 2H), δ = 1 40-1 43 (m, 2H), δ = 2 15 (s,….. 1H), δ = 2. 64-2. 73 (m, 1H), δ = 3. 06-3. 12 (m, 1H), δ = 3. 71-3. 74 (d, 1H), δ = 3. 95 (s, 3H), δ = 4 • 03-4. 05 (d, 2H), δ = 4. 20-4. 23 (d, 1H), δ = 7. 20 (s, 1H), δ = 7. 46-7. 48 (m, 1H), δ = 7. 51-7 • 53 (m, 1H), δ = 7. 65-7. 67 (d, 1H), δ = 7. 80 (s, 1H), δ = 8. 01 (s, 1H), δ = 8. 35 (s, 1H), δ = 9. 54 (s, 1H).
[0028] Example 2: Preparation of 4- (4-bromo-2-fluoroanilino) -6-methoxy-7 – [(1-methyl-piperidin-4-yl) methoxy] quinazoline preparation
[0029] 4- (4-bromo-2-fluoroanilino) in 100mL three-necked flask, 6-methoxy-7 – [(1-formyl-4-yl) methoxy] quinoline oxazoline (0 · 98g, 2. Ommol), zinc (0 · 6g, 4. 4mmol) and tetrahydrofuran (20mL), stirred pale yellow turbid liquid. At room temperature was added portionwise sodium borohydride (0. 15g, 4. OmmoL), little change in the temperature. Heating
……………………………….
CN 104211649
http://www.google.com/patents/CN104211649A?cl=en
Pharmacokinetics
Vandetanib is well absorbed from the gut, reaches peak blood plasma concentrations 4 to 10 hours after application, and has a half-life of 120 hours days on average, per Phase I pharmacokinetic studies. It has to be taken for about three months to achieve a steady-state concentration. In the blood, it is almost completely (90–96%) bound to plasma proteins such as albumin. It is metabolised to N-desmethylvandetanib via CYP3A4 and to vandetanib-N-oxide via FMO1 and 3. Both of these are active metabolites. Vandetanib is excreted via the faeces (44%) and the urine (25%) in form of the unchanged drug and the metabolites.[2][9][10]

Metabolites of vandetanib (top left): N-desmethylvandetanib (bottom left, via CYP3A4), vandetanib-N-oxide (bottom right, via FMO1 andFMO3), both pharmacologically active, and a minor amount of aglucuronide.[10]
Clinical trials
Non-small cell lung cancer
The drug underwent clinical trials as a potential targeted treatment for non-small-cell lung cancer. There have been some promising results from a phase III trial withdocetaxel.[11] There have also been ambivalent results when used with pemetrexed.[12] Another trial with docetaxel was recruiting in July 2009.[13]
AstraZeneca withdrew EU regulatory submissions for vandetanib (under the proposed trade name Zactima) in October 2009 after trials showed no benefit when the drug was administered alongside chemotherapy.[14]
References
- “Definition of vandetanib”. NCI Drug Dictionary. National Cancer Institute.
- “Vandetanib Monograph”. Drugs.com. Retrieved 29 August 2012.
- “FDA approves new treatment for rare form of thyroid cancer”. Retrieved 7 April 2011.
- “FDA approves orphan drug vandetanib for advanced medullary thyroid cancer” (Press release). AstraZeneca. Retrieved 2011-08-17.
- “AstraZeneca announces trade name CAPRELSA® for vandetanib” (Press release). AstraZeneca. Retrieved 2011-08-17.
- Khurana V, Minocha M, Pal D, Mitra AK (March 2014). “Role of OATP-1B1 and/or OATP-1B3 in hepatic disposition of tyrosine kinase inhibitors.”. Drug Metabol Drug Interact.0 (0): 1–11. doi:10.1515/dmdi-2013-0062. PMID 24643910.
- Haberfeld, H, ed. (2012). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag.
- Khurana V, Minocha M, Pal D, Mitra AK (May 2014). “Inhibition of OATP-1B1 and OATP-1B3 by tyrosine kinase inhibitors.”. Drug Metabol Drug Interact. 0 (0): 1–11.doi:10.1515/dmdi-2014-0014. PMID 24807167.
- Martin, P.; Oliver, S.; Kennedy, S. J.; Partridge, E.; Hutchison, M.; Clarke, D.; Giles, P. (2012). “Pharmacokinetics of Vandetanib: Three Phase I Studies in Healthy Subjects”.Clinical Therapeutics 34 (1): 221–237. doi:10.1016/j.clinthera.2011.11.011.PMID 22206795.
- “Clinical Pharmacology Review: Vandetanib” (PDF). US Food and Drug Administration, Center for Drug Evaluation and Research. 20 August 2010. Retrieved29 August 2012.
- “Vandetanib Shows Clinical Benefit When Combined With Docetaxel For Lung Cancer”. ScienceDaily. 3 June 2009.
- “IASLC: Vandetanib Fails to Improve NSCLC Outcomes with Pemetrexed”. Medpage today. 5 Aug 2009.
- Clinical trial number NCT00687297 for “Study of Vandetanib Combined With Chemotherapy to Treat Advanced Non-small Cell Lung Cancer” at ClinicalTrials.gov
- “Zactima”. European Medicines Agency.
External links
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-(4-bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy]quinazolin-4-amine
|
|
| Clinical data | |
| Trade names | Caprelsa |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a611037 |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Protein binding | 90–96% |
| Metabolism | CYP3A4, FMO1, FMO3 |
| Biological half-life | 120 hours (mean) |
| Excretion | 44% faeces, 25% urine |
| Identifiers | |
| CAS Registry Number | 443913-73-3 |
| ATC code | L01XE12 |
| PubChem | CID: 3081361 |
| IUPHAR/BPS | 5717 |
| DrugBank | DB08764 |
| ChemSpider | 2338979 |
| UNII | YO460OQ37K |
| ChEBI | CHEBI:49960 |
| ChEMBL | CHEMBL24828 |
| Synonyms | ZD6474 |
| Chemical data | |
| Formula | C22H24BrFN4O2 |
| Molecular mass | 475.354 g/mol |
//////
Bococizumab

Bococizumab
PF-04950615, RN-316, RN316
PCSK9 (proprotein convertase subtilisin/kexin type 9, neural apoptosis-regulated convertase 1, NARC1, NARC-1, proproteine convertase 9, PC9) [Homo sapiens]
IgG2 – kappa
Hypercholesterolemia
Cardiovascular diseases
STRUCTURAL FORMULA
Heavy chain
QVQLVQSGAE VKKPGASVKV SCKASGYTFT SYYMHWVRQA PGQGLEWMGE 50
ISPFGGRTNY NEKFKSRVTM TRDTSTSTVY MELSSLRSED TAVYYCARER 100
PLYASDLWGQ GTTVTVSSAS TKGPSVFPLA PCSRSTSEST AALGCLVKDY 150
FPEPVTVSWN SGALTSGVHT FPAVLQSSGL YSLSSVVTVP SSNFGTQTYT 200
CNVDHKPSNT KVDKTVERKC CVECPPCPAP PVAGPSVFLF PPKPKDTLMI 250
SRTPEVTCVV VDVSHEDPEV QFNWYVDGVE VHNAKTKPRE EQFNSTFRVV 300
SVLTVVHQDW LNGKEYKCKV SNKGLPSSIE KTISKTKGQP REPQVYTLPP 350
SREEMTKNQV SLTCLVKGFY PSDIAVEWES NGQPENNYKT TPPMLDSDGS 400
FFLYSKLTVD KSRWQQGNVF SCSVMHEALH NHYTQKSLSL SPGK 444
Light chain
DIQMTQSPSS LSASVGDRVT ITCRASQGIS SALAWYQQKP GKAPKLLIYS 50′
ASYRYTGVPS RFSGSGSGTD FTFTISSLQP EDIATYYCQQ RYSLWRTFGQ 100′
GTKLEIKRTV AAPSVFIFPP SDEQLKSGTA SVVCLLNNFY PREAKVQWKV 150′
DNALQSGNSQ ESVTEQDSKD STYSLSSTLT LSKADYEKHK VYACEVTHQG 200′
LSSPVTKSFN RGEC 214′
Disulfide bridges location
22-96 22”-96” 23′-88′ 23”’-88”’ 132-214′ 132”-214”’
134′-194′ 134”’-194”’ 145-201 145”-201” 220-220” 221-221”
224-224” 227-227” 258-318 258”-318” 364-422 364”-422”
Bococizumab nonproprietary drug name
RN-316, PF-04950615
target-PC9
USAN (AB-55) BOCOCIZUMAB
PRONUNCIATION boe” koe siz’ ue mab
THERAPEUTIC CLAIM Treatment of dyslipidemia
CHEMICAL NAME
1. Immunoglobulin G2, anti-(human neural apoptosis-regulated proteinase
1)(human-Mus musculus monoclonal PF-04950615 heavy chain), disulfide
with human-Mus musculus monoclonal PF-04950615 light chain, dimer
2. Immunoglobulin G2-kappa, anti-[human proprotein convertase subtilisin/hexin type 9 (neural apoptosis-regulated convertase 1, PC9)], humanized mouse monoclonal antibody; gamma 2 heavy chain (1-444) [humanized VH (Homo sapiens IGHV1-46-1*03 (90.8%) -(IGHD)-IGHJ6*01) [8.8.11] (1-118)-Homo sapiens IGHG2*01 CH2A100>S(327),CH2P101>S(328) (119-444)] (132-214′)-
disulfide with kappa light chain (1′-214′) [humanized V-KAPPA (Homo sapiensIGKV1-39*01 (88.2%)-IGKJ2*01 [6.3.9] (1′-107′)-IGKC*01 (108′-214′)]; dimer
(220-220”:221-221”:224-224”:227-227”)-tetrakisdisulfide
MOLECULAR FORMULA C6414H9918N1722O2012S54
MOLECULAR WEIGHT 145.1 kDa
TRADEMARK None as yet
SPONSOR Pfizer, Inc.
CODE DESIGNATIONS RN316, PF-04950615
CAS REGISTRY NUMBER 1407495-02-6
WHO NUMBER 9840
Bococizumab[1] (RN316)[2] is a drug in development by Pfizer targeting PCSK9 to reduce LDL cholesterol.[3]
![]()
Description
Bococizumab is a monoclonal antibody that inhibits PCSK9, a protein that interferes with the removal of LDL. LDL levels are a major risk factor for cardiovascular disease.
Clinical trials
A phase 2b study of statin patients was presented at the 2014 American College of Cardiology. Monthly or bimonthly injections resulted in significantly reduced LDL-C at week 12.
The Phase 3 SPIRE trials plan to enroll 17,000 patients to measure cardiovascular risk. High risk and statin intolerant subjects will be included.

References
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Bococizumab” (PDF). American Medical Association.
- World Health Organization (2013). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 110”(PDF). WHO Drug Information 27 (4).
- “Bococizumab (RN316) Significantly Reduced LDL Cholesterol In Statin-Treated Adults With High Cholesterol In A Phase 2b Study”. Retrieved 29 December 2014.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized (from mouse) |
| Target | Proprotein convertase subtilisin/kexin type 9 (PCSK9) |
| Clinical data | |
| Legal status |
|
| Routes of administration |
Subcutaneous injection |
| Identifiers | |
| CAS Registry Number | 1407495-02-6 |
| ATC code | None |
| PubChem | SID: 194168554 |
| IUPHAR/BPS | 7730 |
| ChEMBL | CHEMBL3137349 |
| Chemical data | |
| Formula | C6414H9918N1722O2012S54 |
| Molecular mass | 145.1 kDa |
//////
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}