

IUPAC Condensed
H-Ile-Asn-Leu-Lys-Ala-Leu-Ala-Ala-Leu-Ala-Lys-Lys-xiIle-Leu-NH2
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
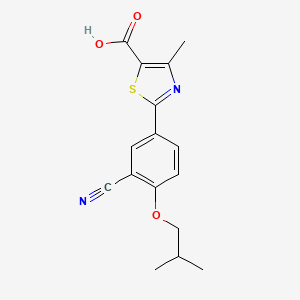
| Febuxostat; 144060-53-7; Uloric; Adenuric; Tei 6720; 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid; | |
| Molecular Formula: | C16H16N2O3S |
|---|---|
| Molecular Weight: | 316.37484 g/mol |
2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methyl-1,3-thiazole-5-carboxylic acid
Febuxostat is a thiazole derivative and inhibitor of XANTHINE OXIDASE that is used for the treatment of HYPERURICEMIA in patients with chronic GOUT.
CAS 144060-53-7
Febuxostat (INN; trade names Adenuric in Europe and New Zealand, Uloric in the US, Goturic in Latin America, Feburic in Japan) is a drug that inhibits xanthine oxidase, thus reducing production of uric acid in the body. It is used in the treatment of chronicgout and hyperuricemia.
Febuxostat was discovered by scientists at the Japanese pharmaceutical company Teijin in 1998. Teijin partnered the drug with TAP Pharmaceuticals in the US and Ipsen in Europe. Ipsen obtained marketing approval for febuxostat from the European Medicines Agency in April 2008, Takeda obtained FDA approval in February 2009, and Teijin obtained approval from the Japanese “Pharmaceuticals and Medical Devices Agency” in 2011.
Febuxostat is used to treat chronic gout and hyperuricemia.[2] National Institute for Health and Clinical Excellence concluded that febuxostat is more effective than standard doses of allopurinol, but not more effective than higher doses of allopurinol.[2]

Uloric 40 mg tablet
Febuxostat is in the US pregnancy category C; there are no adequate and well-controlled studies in pregnant women.[3]
The adverse effects associated with febuxostat therapy include nausea, diarrhea, arthralgia, headache, increased hepatic serum enzyme levels and rash.[3][4]
Febuxostat is contraindicated with concomitant use of theophylline and chemotherapeutic agents, namely azathioprine and 6-mercaptopurine, because it could increase blood plasma concentrations of these drugs, and therefore their toxicity.[3][5]
Febuxostat is a non-purine-selective inhibitor of xanthine oxidase.[3] It works by non-competitively blocking the molybdenum pterincenter which is the active site on xanthine oxidase. Xanthine oxidase is needed to successively oxidize both hypoxanthine andxanthine to uric acid. Hence, febuxostat inhibits xanthine oxidase, therefore reducing production of uric acid. Febuxostat inhibits both oxidized as well as reduced form of xanthine oxidase because of which febuxostat cannot be easily displaced from the molybdenum pterin site.[4]

Febuxostat was discovered by scientists at the Japanese pharmaceutical company Teijin in 1998.[6] Teijin partnered the drug withTAP Pharmaceuticals in the US and Ipsen in Europe.[7][8][9]
Ipsen obtained marketing approval for febuxostat from the European Medicines Agency in April 2008,[10] Takeda obtained FDA approval in February 2009,[11][12] and Teijin obtained approval from the Japanese authorities in 2011.[13] Ipsen exclusively licensed its European rights to Menarini in 2009.[14] Teijin partnered with Astellas for distribution in China and southeast Asia.[15][16]
In the UK, NICE has found that febuxostat has a higher cost/benefit ratio than allopurinol and on that basis recommended febuxostat as a second-line drug for people who cannot use allopurinol.[2]
Febuxostat is marketed as Adenuric in Europe and New Zealand, Uloric in the US, Goturic and Goutex in Latin America, Feburic in Japan, and is generic in several countries and is available by many names in those countries.[1]
Febuxostat (Formula I) is an inhibitor of xanthine oxidase, which was discovered by the Japanese company Teijin Pharma Ltd and it is indicated for use in the treatment of hyperuricemia and chronic gout. Its chemical name is 2-(3-cyano-4-isobutoxyphenyl)-4-methyl- l,3-thiazole-5-carboxylic acid. It is marketed under the brand names Adenuric in Europe, Feburic in Japan and Uloric in USA and Canada.

In EP0513379B1 Febuxostat is prepared from 4-hydroxy-3-nitrobenzaldehyde, according to the following scheme.

This particular process suffers from major drawbacks. Not only it is very long, including seven steps from the starting material to the final product, but, most importantly, it employs the use of cyanides, which are extremely toxic reagents. Cyanide salts are likely to generate hydrocyanide, which sets a high amount of risk in an industrial scale process.
In Japanese patent JP06345724A(JP2706037B) the intermediate ethyl ester of Febuxostat is prepared from p-cyano-nitrobenzene, in three steps. Febuxostat may, then, be prepared by alkaline hydrolysis, according to prior art.
MeCSNH,

The use of extremely toxic potassium cyanide makes this process unsuitable for manufacturing purposes.
Route A

In Japanese patent JP3202607B Febuxostat ethyl ester is prepared, according to the above scheme, through two similar routes. Route A uses flash column chromatography for the purification of the hydroxylamine reaction product, while Route B suffers from low yield and the use of chlorinated solvents for recrystallization. In addition, the reaction solvent is, in both cases, formic acid which causes severe skin burns and eye damage to humans. Formic acid is also corrosive towards metal-based materials of construction (MOC), like stainless steel and nickel alloys, limiting the options, essentially, to glass reactors or vessels. The drawbacks of using this solvent are also related to the high volumes of formic acid required per batch, which hinder the waste treatment.
In CN101723915B focus is made to the improvement of the hydroxylamine reaction. Formic acid is replaced with dimethylformamide (DMF) and other solvents. However, according to widely used organic chemistry textbooks, such as March’s Advanced Organic Chemistry, pi 287, 6th edition, M. B. Smith and J. March, ISBN 0-471-72091-7, the mechanism of the reaction involves the formation of an oxime, upon the action of hydroxylamine, which further dehydrates to form a nitrile, with the aid of a suitable reagent, for example formic acid, or acetic anhydride. In the absence of such a reagent, it is expected that the reaction will, at least, not lead to completion, thereby leading to low yields and undesired impurity levels, namely the intermediate oxime. Such impurities, arising from the reactions of the process and which exhibit similar structure of the desired product, are often difficult to remove with common industrial techniques, e.g. crystallization.
In WO2010142653A1 the intermediate Febuxostat ethyl ester is prepared from 4-cyanophenol, through a five-step process. Febuxostat can be prepared from its respective ethyl ester via alkaline hydrolysis, as in the previous case.
OH

1: patents US5614520 febuxostat synthetic process:

2: Patent JP1994329647 febuxostat synthesis


PATENT
https://www.google.com/patents/CN102936230A?cl=en
Gout occurs because the body produces too much uric acid and renal clearance capacity decreased, uric acid accumulation in the body, leading to urate crystals deposited in the joints and organs. Therefore, it means the treatment of gout usually taken to be: to promote uric acid excretion and suppression of uric acid, and the use of appropriate measures to improve symptoms. Uric acid formation and purine metabolism, the final step in the purine metabolism, hypoxanthine generation xanthine xanthine oxidoreductase (XOR) effect, further generate uric acid, inhibit the activity of the enzyme can effectively reduce uric acid production. Febuxostat is currently the world’s newly developed XOR inhibitors, which act by highly selective to the oxidase, reduce uric acid synthesis, reduce uric acid levels, so as to effectively treat the disease ventilation.
Compared with the traditional treatment of gout drug allopurinol, febuxostat has obvious advantages: (1) allopurinol reduced the XOR only inhibit rather than febuxostat of oxidized and reduced form are XOR significant inhibition, thus reducing the role of uric acid, which is more powerful and lasting; (2) Since allopurinol is a purine analogue, the inevitable result of the purine and other activity related to the impact of pyridine metabolism. So allopurinol treatment should be repeated large doses of the drug to maintain a high level. Which also brought serious or even fatal adverse reactions due to drug accumulation due.Instead of febuxostat non-purine XOR inhibitors, so it has better security.
Document TMX-67. Drugs Fut2001, 26, I, 32, and EP0513379, US5614520, W09209279, public
The detailed preparation febuxostat. Using 3-nitro-4-hydroxybenzaldehyde as the starting material is first reacted with hydroxylamine hydrochloride, to give 3-nitro-4-hydroxybenzonitrile. In effect then HCl, reaction with thioacetamide to give 3-nitro-4-hydroxy-thiobenzamide. Closed loop then reacted with 2-chloro ethyl acetoacetate to give 2- (3_ nitro-4-hydroxyphenyl) methyl-5-thiazolyl -4_ carboxylic acid ethyl ester. Followed by potassium carbonate effect, isobutane is reacted with bromo, to give 2- (3_ nitro-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester. Under the catalytic action of palladium on carbon, hydrogen reduction to give 2- (3-amino-4-isobutyloxyphenyl) -4-methyl-5-thiazole carboxylic acid ethyl ester. Followed by diazotization with sodium nitrite occur, was added cuprous cyanide and potassium cyanide, to give 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl-5-thiazolecarboxylic acid ethyl ester. Finally, under the effect of the hydrolysis of sodium hydroxide, to give the product 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl – thiazole-5-carboxylic acid, to obtain febuxostat.The process route is as follows:

This route in the preparation of febuxostat, there are many disadvantages: raw 3-nitro-4-hydroxybenzaldehyde in the country is difficult to buy; requires the use of palladium-carbon catalytic hydrogenation reaction under the factory equipment higher requirements, there is a certain danger; the cyano preparation, the need to use sodium nitrite diazotization, could easily lead to corrosion of equipment; the cyano preparation, the need to use toxic cyanide copper, potassium cyanide, pollution, higher risk.
Document JP1994329647, JP1998045733, US3518279 reported another synthesis of febuxostat
Methods. From 4-hydroxy-thiobenzamide as a starting material, and the cyclization reaction to give ethyl 2-bromo-acetyl occurred
2- (4_ hydroxyphenyl) -4_ methyl-5-carboxylic acid ethyl ester in polyphosphoric acid effect, HMTA (hexamethylene tetramine) reacts with 2- (3_ aldehyde – 4-hydroxyphenyl) methyl-5-thiazolyl -4_ carboxylic acid ethyl ester. Then two cases: the first case, the effect of potassium carbonate, is reacted with isobutane to give bromo-2- (4-isobutyloxyphenyl 3_ aldehyde) -4_-methyl-5- thiazole carboxylic acid ethyl ester, and then reacted with hydroxylamine hydrochloride to give 2- (3_-cyano-4-isobutyloxyphenyl) -4_-methyl-5-thiazole carboxylic acid ethyl ester; second case is the first with hydroxylamine hydrochloride to give 2- (3_ cyano-4-hydroxyphenyl) methyl-5-thiazolecarboxylic -4_ carboxylic acid ethyl ester, and then under the effect of potassium carbonate, and reacted with isobutane to give bromo-2- (3 _-cyano-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester.
Finally, under the effect of the hydrolysis of sodium hydroxide, to give the product 2- (3_-cyano-4-isobutyloxyphenyl) -4_ methyl – thiazole-5-carboxylic acid, i.e., to obtain febuxostat . The process route is as follows:

This synthesis route febuxostat process, since the introduction of aldehyde HMTA in PPA (polyphosphoric acid) effect. So there are a lot of phosphorus wastewater, serious environmental pollution, but also because PPA has great viscosity, and therefore difficult to stir the production, operation is extremely inconvenient.
Document Heterocyclesl998, 47,2,857 JP1994345724 also reported the synthesis method of febuxostat, using p-nitrophenyl-carbonitrile as a starting material in the reaction with potassium cyanide in DMSO solvent, and then the carbonate lower potassium catalyzed reaction of isobutane and brominated 1,3-cyano-4-diisobutoxybenzene ether. By reaction with thioacetamide to afford
3-cyano-4-isobutyloxyphenyl thiobenzamide. Under heating, and 2-chloro ethyl acetoacetate, ring closure reaction occurs to give 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl-5-carboxylic acid ethyl ester, and finally hydrolysis under the effect of sodium hydroxide, to give the product 2- (3-cyano-4-isobutyloxyphenyl) -4-methyl – thiazole-5-carboxylic acid, to obtain febuxostat.
The present invention febuxostat new technology system, comprising the steps of:
(1) 2-hydroxy-5-cyano – NaSH reacted with benzaldehyde to give 4-hydroxy-3- aldehyde thiobenzamide;

(2) the step (I) to give 4-hydroxy-3-aldehyde thiobenzamide reaction with ethyl 2-halo-acetyl, closed
Ring to give 2- (3-aldehyde-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;

X is a halogen, preferably Cl or Br;
(3) the step (2) to give 2- (3-aldehyde-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole with hydroxylamine in formic acid in the reaction solution to give 2- (3- cyano-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;

(4) The step (3) to give 2- (3-cyano-4-hydroxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole isobutane with halo effect in potassium carbonate, to give 2- (3-aldehyde-4-isobutyloxyphenyl) -4-methyl-5-ethoxycarbonyl thiazole;
(5) in step (4) to give 2- (3-aldehyde-4-isobutyloxyphenyl) -4-methyl-5-ethoxycarbonyl-thiazol-off hydrolyzable ester group, to obtain a non-Tendon Disposition Tanzania.
[0011] Scheme of the method is as follows:

X is halogen, may be Cl, Br;
Preparation 5 febuxostat Example
To a 500ml reaction flask was added 200ml of absolute ethanol, the product of Step 4 was added with stirring (60g, O. 174mol),
5% sodium hydroxide was added 100ml. Stirring heated to 40 degrees, until it is completely dissolved. 40 degrees heat, reaction 4h. The reaction by TLC tracking. After completion of the reaction, the reaction solution was added 10% hydrochloric acid to adjust the pH to 3, the precipitated solid was filtered. And dried to give a pale yellow solid. Dried over anhydrous recrystallized from methanol to give 31. 2g of white crystals, yield 56.7%.
TLC monitoring of the reaction. Eluent: petroleum ether / ethyl acetate = 3: 1 Melting point:. 201 · 7 ~202 30C (literature value 201 ~202 ° C)
1H-NMR δ:. 1 01 (m, 6H), 2.06 (m, lH), 2.57 (m, 3 H), 3.96 (d, 2H), 7.30 (d, lH), 8.13 (m, 1H), 8. 19 (d, 1H);
MS (m / z):. 316 O (M +)
Infrared detection: 3550-3400cm_1; 2961, 2933,2874; 2227cm_1; 1680U604U511cm_1; 1425cm_1; 1296U283CHT1;
Elemental analysis for C, Η, N, S purified product actual measurement of the content of C, H, N, S content: C:. 60 57%, H:. 5 32%, N:. 8 86%, S: 10. 16%; theoretical value: In C16H16N203S calculated C: 60 74%, H: 510%, N: 885%, S: 1014%..
CLIP
Facile OnePot Transformation of Arenes into Aromatic Nitriles …

Patent




Scheme-Ill:

Clip
synthesis describes synthesis of febuxostat (I) from 4-hydroxybenzonitrile (II) in six stages. The synthesis shown is a short, concise route and does not require use of poisonous reagents such as KCN (14). Compound II was converted to 4-hydroxybenzothioamide (III) with 85% yield using NaHS in the presence of hydrated magnesium chloride as Lewis acid. Intermediate III, on cyclization with ethyl-2-chloroacetoacetate, gave thiazole ester (IV) with quantitative yield. In these two stages, the source of potential impurities was identified as an ortho isomer (i.e., 2-hydroxybenzonitrile), which can lead to Impurity VIII and subsequently to Impurity IX . Impurities VIII and IX can be controlled in starting material II with appropriate specification.
|
The ortho formylation of hydroxyl compound IV by using Duff condition (hexamine/TFA) gave aldehyde V (15). The major impurity identified in this reaction was dialdehyde X. Although we have used only 1.0 equivalence of hexamine with respect to Compound IV, the dialdehyde X impurity was formed to a 5-10% ratio in only 2.5 h. It is, therefore, impossible to get rid of this impurity during the reaction, and only effective recrystallization will eliminate it. Impurity X was minimized (≤ 2%) by recrystallization using IPA/H2O (3:5) to get aldehyde V with 50% yield and & #8805; 97% HPLC purity.
Aldehyde V, on alkylation with isobutyl bromide in the presence of potassium carbonate base, gave compound VI with 90% yield. In this stage, Impurities XI and XII were alkylations of carryover Compound IV and dialdehyde, respectively. Two more isomeric impurities n-butyl-aldehyde XIII and 1-methyl propyl-aldehyde XIV were also identified in this stage. Both isomeric impurities can be controlled with appropriate specification for isobutyl bromide. The reaction of Compound VI with hydroxylamine hydrochloride and sodium formate in formic acid at reflux temperature gave Compound VII with 85% yield. Impurities XIII and XIV will also carry forward to impurities n-butyl-nitrile XV and 1-methyl propyl-nitrile XVI, respectively.
In the final step, Compound VII was hydrolyzed using sodium hydroxide in a MeOH:THF:H2O (1:1:1) solvent combination to yield febuxostat (85%). During saponification, methyl ester Impurity XVII was identified via trans-esterification. Its hydrolysis was comparatively slower than its ethyl isomer VII. One way to avoid Impurity XVII is to replace methanol with ethanol. Carryover impurities XI, XV, and XVI were also hydrolyzed to their respective acid derivatives impurities XVIII, XIX, and XX. However, the acid derivatives of impurities X and XII were unexpectedly absent as impurities. It is believed that, because they were present in low concentrations during workup, they were eliminated in the mother liquor. Two additional impurities, amide XXI and diacid XXII, formed by the side reaction of the febuxostat nitrile group with sodium hydroxide, were identified during saponification. The amide XXI and diacid XXII impurities can be controlled by using appropriate equivalence of sodium hydroxide and controlled reaction time. Febuxostat, on acetone recrystallization and seed Crystal A at 45°C, gave pure febuxostat with 75% yield.

http://www.pharmtech.com/investigation-various-impurities-febuxostat
Febuxostat is an inhibitor of xanthine oxidase, and was developed by Teijin pharma. This compound is known as a new drug that is effective against gout and hyperuricemia, and it has been 40 years since the last time a drug of this kind of drug was developed.
Febuxostat has therefore gained a lot of popularity and it has already been accepted as a drug in Europe, USA, Korea and Japan. The synthesis of this molecule have been reported in patents by Teijin pharma as shown below.[1,2]

Recently, Itami group was reported the rapoid synthesis of febxostat by using Ni-catalyzed direct coupling of azoles and arylhalides[3]
References
Sorbera, L.A.; Castaner, J.; Rabasseda, X.; Revel, L.; TMX-67. Drugs Fut 2001, 26, 1, 32
[1] Hasegawa, M.; A facile one-pot synthesis of 4-alkoxy-1,3-benzenedicarbonitrile. Heterocycles 1998, 47, 2, 857. [2] Hasegawa, M.; Hasegawa, M.; Komoriya, K. (Teijin Ltd.); Cyano cpds. and their preparation method. JP 1994345724 . [3] “Nickel-Catalyzed Biaryl Coupling of Heteroarenes and Aryl Halides/Triflates”
Canivet, J.; Yamaguchi, J.; Ban, I.; Itami, K. Org. Lett. 2009, 11, 1733-1736. DOI: 10.1021/ol9001587

Ni-based catalytic systems for the arylation of heteroarenes with aryl halides and triflates have been established. Ni(OAc)2/bipy is a general catalyst for aryl bromides/iodides, and Ni(OAc)2/dppf is effective for aryl chlorides/triflates. Thiazole, benzothiazole, oxazole, benzoxazole, and benzimidazole are applicable as heteroarene coupling partners. A rapid synthesis of febuxostat, a drug for gout and hyperuricemia, is also demonstrated.
A CLIP
A final example of a thiazole containing drug is given in the novel xanthine oxidase inhibitor febuxostat (359, Uloric) which was approved by the FDA in 2009. This inhibitor works by blocking xanthine oxidase in a non-competitive fashion. Consequently, the amount of the oxidation product uric acid is reduced. Thus it is an efficient treatment for hyperuricemia in gout. In order to prepare febuxostat first a synthesis of the noncommercial 4-isobutoxy-1,3-dicyanobenzene building block (363), has to be conducted. An elegant way of achieving this was shown through the reaction of 4-nitrocyanobenzene (360) with potassium cyanide in dry DMSO followed by quenching with isobutyl bromide under basic conditions (Scheme 70). It is suggested that a Meisenheimer-complex intermediate 361 is initially formed, which after rearomatisation, undergoes nucleophilic aromatic substitution of the nitro group by the DMSO solvent [107]. Upon hydrolysis and O-alkylation the desired 4-isobutoxy-1,3-dicyanobenzene (363) is obtained in good overall yield. Subsequently, the less hindered nitrile is converted to the corresponding thioamide 365 in an intriguing reaction using thioacetamide (364). The thiazole ring is then formed by condensation with chloroacetoacetate 366 followed by ester hydrolysis (Scheme 70).
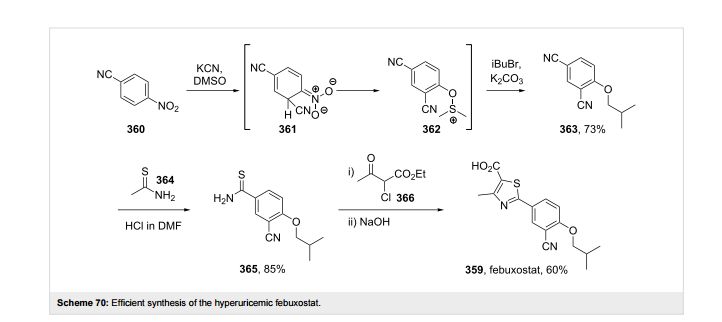
107 Hasegawa, M. Heterocycles 1998, 47, 857–864. doi:10.3987/COM-97-S(N)89
Paper | Special issue | Vol 47, No. 2, 1998, pp.857-864
Masaichi Hasegawa
*Teijin Institute, Bio-Medical Research, Asahigaoka 4-3-2, Hino, Tokyo 191, Japan
2-(3-Cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxlic acid (TEI-6720) was prepared. The introduction of cyano group to 4-nitrobenzonitrile with KCN in dry DMSO followed by quenching with alkyl halide afforded the key intermediates, 4-alkoky-1,3-benzenedicarbonitriles, in good yield. The reaction was completed in dry DMSO, while no reaction occurred in dry DMF. This observation can be suggested by the participation of DMSO in the reaction.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S70
A CLIP
Synthesis and characterization of process-related impurities of an anti-hyperuricemia drug-Febuxostat
Venkateswara Rao Vallu,$ Krunal Girishbhai Desai, Sandip Dhaya Patil, Rajendra Agarwal, Pratap Reddy Padi and Mahesh Reddy Ghanta
*Process Research Laboratory-I, Research & Development Centre, Macleods Pharmaceuticals Ltd, G-2, Mahakali Caves Road, Shantinagar, Andheri (East), Mumbai, Maharastra, India
$Department of Chemistry, Pacific University, Pacific Hills, Airport Road, Pratap Nagar Extension, Debari, Udaipur, Rajasthan, India _____________________________________________________________________
Der Pharma Chemica, 2014, 6(3):300-311 (http://derpharmachemica.com/archive.html)
http://derpharmachemica.com/vol6-iss3/DPC-2014-6-3-300-311.pdf
Synthesis of 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1) [10] A solution of 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1 tech grade, 5.0 g, 0.015 mol.) in methanol (50.0 mL) was heated the reaction mass at 60-65°C till clear solution was obtained. Water (50.0 mL) was added drop wise into reaction mass with in 30.0 min. at 60-65°C. Resultant white crystalline solid was filtrated, Mahesh Reddy Ghanta et al Der Pharma Chemica, 2014, 6 (3):300-311 _____________________________________________________________________________ 302 http://www.scholarsresearchlibrary.com washed with water (10.0 mL) and dried in vacuum tray drier at 50-55°C under vacuum to give
2-(3-cyano-4- isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (1). Yield: 95.0 % (4.75 g)
mp 239°C. Purity by HPLC: 99.74 % (10.2 min. retention time),
Anal. Calcd for C16H16N2O3S: C, 60.74; H, 5.10; N, 8.85. Found: C, 60.70; H, 5.11; N, 8.87 %;
IR (KBr) υmax (in cm−1): 3834.61, 3742.03, 3680.30, 3556.85, 3456.55, 2962.76, 2877.89, 2661.85, 2546.12, 2353.23, 2229.79, 2168.06, 2029.18, 1921.16, 1790.00, 1674.27, 1604.83, 1512.24, 1427.37, 1381.08, 1280.78, 1172.76, 1118.75, 1010.73, 918.15, 833.28, 771.55, 725.26, 648.10, 524.66, 462.93; 1H NMR (300 MHz, CDCl3 or DMSO-d6) δH (in ppm): 1.00-1.02 (d, 6H, (CH3)2-CH-), 2.49-2.50 (m, 1H, (CH3)2-CH-), 3.97-3.99 (d, 2H, -CH-CH2−), 7.33–8.25 (d, dd, 3H, Ar-H), 2.64 (s, 3H, -CH3), 13.39 (s, 1H, -COOH);
13C NMR (300 MHz, DMSO–d6) δC (in ppm) (Positiona ): 166.3 (l), 162.9 (p), 162.2 (n), 159.6 (e), 133.1 (g), 131.6 (i), 125.5 (m), 123.0 (h), 115.5 (k), 114.0 (f), 101.7 (j), 75.2 (d), 27.7 (b), 18.8 (a, c), 17.1 (o);
MS m/z (%) (70 eV): m/z =317.0 (100.0 %) [M+1], 318.0 (16.0 %) [M+2], 403.0 (63.0 %), 512.0 (47.0 %), 482.0 (46.0 %), 405.0 (27.0 %), 468.0 (25.0 %), 570.0 (24.0 %).
PATENT
| WO 2012066561 |
As per the present invention, hydroxylamine hydrochloride is added to compound of Formula-Ill in presence of a polar aprotic solvent like DMSO, DMA, ACN or DMF. To this reaction mixture acetyl halides or sulfonyl chlorides are added and temperature raised to 70 -80 °C. Acetyl halides are selected from acetyl bromide or acetyl chloride. Sulfonyl chlorides are selected from methane sulfonyl chloride or para toluene sulfonyl chloride. To this reaction mixture a base selected from alkali metal carbonates like potassium carbonate or sodium carbonate, preferably potassium carbonate and alkyl halide selected from isobutyl bromide is successively added. The reaction mass is washed with water and compound of Formula-II is isolated. In one embodiment the present invention provides, process for the preparation of Febuxostat comprising the steps of:
a) reacting the compound of Formula-III(a) with hydroxylamine hydrochloride in presence of organic solvent;

Formula-III(a)
b) adding acyl halides or sulfonyl chlorides to the reaction mixture;
c) optionally isolating compound of Formula- IV (a)

Formula-IV(a)
d) reacting with isobutyl bromide in presence of base;
e) isolating the compound of Formula-II(a); and

FormuIa-II(a)
f) hydrolyzing the compound of Formula-II(a) to get Febuxostat.
The following examples are provided to illustrate the process of the present invention. They, are however, not intended to limiting the scope of the present invention in any way and several variants of these examples would be evident to person ordinarily skilled in the art. Experimental procedure:
Example – 1: Preparation of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyI thiozole -5-carboxylate
A mixture of 10. Og of Ethyl -2-(3-formyl-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate and 2.85 g of hydroxylamine hydrochloride were stirred for 30 minutes in 40 g of Dimethyl sulfoxide. To this reaction mixture 3.3 grams of acetyl chloride was added and stirred at 70 -80°C for 2-3 hours. Reaction mass was cooled to room temperature and to this 19 g of potassium carbonate and 19 g of isobutyl bromide was added successively. The reaction mass was stirred for 5 hours at 70-80°C. Reaction mass was diluted with 200 ml of purified water. The reaction mass was filtered and washed with purified water to give 10.0 g of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxyltae (yield 84.0%)
Example – 2: Preparation of Ethyl-2-(3-cyano-4-hydroxyphenyl)-4-methyl thiozole – 5-carboxylate
A mixture of 10. Og of Ethyl-2-(3-formyl-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate and 2.85 g of hydroxylamine hydrochloride were stirred for 30 minutes in 30 g of Dimethylformamide. To this reaction mixture 3.3 grams of acetyl chloride was added and stirred at 90°C for 2-3 hours. Reaction mass was cooled to room temperature and diluted with 100 ml of water and stir for 2 hours. The reaction mass was filtered and washed with purified water to give 10.0 g of Ethyl-2-(3-cyano-4-hydroxy phenyl)-4- methyl thiozole -5-carboxyltae (yield 99.0%).
Example – 3: Preparation of Ethyl 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylate
A mixture of 10. Og of Ethyl-2-(3-cyano-4-hydroxy phenyl)-4-methyl thiozole -5- carboxylate, 30 g of NMP, 9.6 g of potassium carbonate and 7.2 g of isobutyl bromide were stirred for 3 hours at 90°C. Reaction mass was diluted with 100 ml of purified water. The reaction mass was filtered and washed with purified water and ethanol to give 10.5 g of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxyltae (yield 88.0%). Example – 4: Preparation of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5- carboxylic acid
A mixture of 10. Og of Ethyl-2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5- carboxyltae, 2.0g of sodium hydroxide was heated at 45-60°C in 75 ml of aqueous methanol for 1 hour. Reaction mass was cooled to ambient temperature and pH adjusted to 2.0 to 2.5 with dilute hydrochloric acid and precipitated crystal was collected by filtration to give 8.8g of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid (yield 95.8%).
Example – 5-13: Preparation of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole – 5-carboxylic acid
The above compound was prepared by following the procedure as disclosed in Example- 4, using the below listed solvents instead of aqueous methanol.

Example – 14: Preparation of pure 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid
10.0 g of 2-(3-cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid was dissolved in 100 ml of ethanol at reflux temperature. After dissolution reaction mass was cooled and precipitated crystal was collected by filtration to give 9.6 g of pure 2-(3- cyano-4-isobutoxy phenyl)-4-methyl thiozole -5-carboxylic acid (yield 96%).
PATENT
KR 201603732
PATENT
WO 2015018507
https://www.google.com/patents/WO2015018507A2?cl=en



EXPERIMENTAL
of compound of formula lib

Dissolve 14.14g of ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula III) in 55 ml dimethylformamide, at ambient temperature. Add 40g of potassium carbonate, along with 15.9 ml isobutyl bromide. Heat the reaction to 75-80 °C and stir for 4 hours. Cool to 25-30 °C, while 165 ml process water is added. Further cool to 0-5 °C and stir for 30 minutes at this temperature. Filter off the precipitated solid and wash the filter cake with 55 ml process water. The wet cake is dried under vacuum at 40 °C for 7 hours, to furnish 16.43 g of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula lib).
of compound of formula Illb

In a 25 mL round-bottomed flask charge under stirring at 25-30 °C, 1.0 g (2.88 mmol) of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate in 3.0 mL dimethylformamide. Add 34 mg (0.19 mmol) copper acetate under stirring at 25-30 °C. Flush with oxygen (02) and add 0.66 ml (34.92 mmol) 25% aqueous ammonia. Flush again with 02. Heat the reaction mixture to 80-82 °C overnight. Check the progress of the reaction by TLC (cyclohexanerethyl acetate 3:1). Cool reaction mass to 25-30 °C. Add 25mL ethyl acetate and 25mL brine at the reaction mass, separate organic layer and extract aqueous layer twice with 25mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate till dry. The residue is purified with column chromatography (cyclohexane:ethyl acetate 9:1). afforded 0.754g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb) Yield: 75.4%.
EXAMPLE 3: Preparation of compound of formula Illb

In a 25 mL round-bottomed flask charge under stirring at 25-30 °C, 0.17 g (0.49 mmol) of ethyl 2-(3-formyl-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate in 2.5 mL tetrahydrofuran. Add 2.9 mL (153.43 mmol) 25% aqueous ammonia, under stirring at 25-30 °C. Add 137 mg (0.54 mmol) iodine (I2) to the reaction mass, stir the reaction mixture at 25-30 °C for 15-30 min. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 3:1). Starting material is consumed. Add 2.5 mL 5% w/v aqueous sodium thiosulfate Na2S203 and 15mL ethyl acetate at the reaction mass, separate organic layer and extract twice aqueous layer with 15mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate till dry. 0.158g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb) are collected.
EXAMPLE 4: Preparation of Febuxostat

In a 100 ml 2-neck round-bottomed flask charge 2.407g of ethyl-2-(3-cyano-4-isobutoxyphenyl)-4-methylhiazole-carboxylate in 20ml tetrahydrofuran under stirring, at 25-35 °C, 0.748g of sodium hydroxide and heat reaction mass to 60-65 °C for approximately 8 hrs. Check the progress of the reaction by TLC (cyclohexane:ethyl acetate 3:1). Cool reaction mass to 0-5 °C and add 50 ml process water keeping temperature within 0-5 °C. Adjust pH to 1-2 with 4.5 ml 6 N hydrochloric acid, keeping temperature within 0-5 °C. Warm up reaction mass to 25-30 °C and stir reaction mass at the above temperature for 15 min. Filter off the precipitated solid through Buchner funnel under reduced pressure, spray wash with 2 ml process water and suck dry for 20-30 min. Transfer the crude solid in a 50 ml round-bottomed flask, charge 12 ml process water and 12 ml acetone at 25-30°C. Heat the reaction mass to 50-60 °C for 60 min. Cool down reaction mass to 0-5 °C and stir for 60 min at the above temperature. Filter off the precipitated solid though Buchner funnel under reduced pressure, spray wash with 2 ml of a 1 : 1 mixture of acetone and process water and suck dry for 30-45 min. Dry under vacuum at 60 °C. 1.821g of (compound I) Febuxostat are collected, Purity: 82.6%, Yield: 0.62w/w.
on of compound of formula Ilia

In a 50 mL round-bottomed flask charge under stirring 0.5g (1.72 mmol) of ethyl 2-(3-formyl-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate in 8.6 mL THF, at 25-30 °C. Add 10.3 mL (544.94 mmol) 25% aqueous ammonia, under stirring at 25-30 °C. Add 480 mg (1.89 mmol) iodine (I2) to the reaction mass, stir the reaction mixture at 25-30 °C for 15-30 min. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 1 :1). Starting material is consumed. Add 8.6 mL 5% w/v aqueous thiosulfate and 40 mL ethyl acetate at the reaction mass, separate organic layer and extract aqueous layer twice with 40 mL ethyl acetate. Combine organic layers, dry over anhydrous sodium sulfate, filter off and concentrate to dryness. Purification of the residue with column chromatography (cyclohexane: ethyl acetate 3: 1) afforded 0.213 g of ethyl 2-(3-cyano-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula Ilia). Yield : 42.6%.
EXAMPLE 6: Preparation of compound Illb
Dissolve 2.2 g of ethyl 2-(3-cyano-4-hydroxyphenyl)-4-methylthiazole-5-carboxylate (Formula VI) in 7 ml dimethylformamide and to this mixture add 6.6 g of potassium carbonate and 3.14 g of isobutyl bromide. Stir the reaction at 75 °C for 15 hours and then cool to 40 °C. Add 15 ml process water and cool to 0-5 °C. Filter the precipitated solid off and wash with 15 ml process water, which, after drying, affords 2.28 g of ethyl 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylate (Formula Illb).
EXAMPLE 7: Preparation of compound I (Febuxostat)
In a 100 ml 2-neck round-bottomed flask charge 2.131 g of ethyl-2(3-cyano-4-isobutoxyphenyl)-4-methylhiazole-carboxylate, 64 ml methanol and 2.5 ml process water are added under stirring at 25-35 °C. Add 1.718 g potassium carbonate and heat reaction mass to reflux for approximately 2-3 hrs. Check the progress of the reaction by TLC (cyclohexane: ethyl acetate 3:1). Cool reaction mass to 20-25 °C. Concentrate solvent at below 40 °C. To the residue add 43 ml process water, 21 ml ethyl acetate and stir for 30 min at 25-35 °C. Separate layers and transfer aqueous layer in a 100 ml round-bottomed flask. Adjust pH to 2.3-2.7 with 25 ml 1 N hydrochloric acid, at 25-35 °C. Warm up reaction mass to 40 °C and stir reaction mass at this temperature for 60-90 min. Cool down reaction mass to 25-35 °C. Filter off the precipitated solid through Buchner funnel under reduced pressure, spray wash with 5 ml process water and suck dry for 30-45 min. Dry under vacuum at 60 °C. 1.708g of (compound I) Febuxostat are collected, Purity: 86.7%, Yield: 0.69w/w.
EXAMPLE 8: Preparation of Febuxostat crystalline form III
In a 250 mL round-bottomedflask charge under stirring at 25-30 °C 10 g of crude 2-(3-cyano-4-isobutoxyphenyl)-4-methylthiazole-5-carboxylic acid (Febuxostat) in 200 mL ethyl acetate. Heat reaction mass to reflux and stir for 30 min. Cool reaction mass to 25-30°C. Warm again reaction mass and partially distill off solvent from the reaction mass at temperature below 40 °C under reduced pressure. Cool reaction mass to 25-30°C. Filter off the precipitated solid through Buchner funnel under reduced pressure and spray wash with 10 mL ethyl acetate. Dry under vacuum at 60°C. 8.5 g of Febuxostat are collected. Yield: 85 % w/w. XRPD of crystalline compound is in accordance with the one reported in Chinese patent CN101412700B.
PATENT
CN 104418823
https://www.google.com/patents/CN104418823A?cl=zh

PATENT
CN 103588723
https://www.google.com/patents/CN103588723A?cl=zh
Chinese patent CN1275126 described by the Japanese company Teijin invention relates febuxostat Form A, B, C, D, G, and six kinds of amorphous and crystalline preparation method, reported in the literature Form A relatively stable . The method used is a solvent of methanol and water, patent phase diagram (Figure 7 Zone I) can be obtained in anhydrous crystalline Form A (hereinafter referred to as “Form A”), the mixing process by a temperature and the formation of methanol and water to determine the composition of the solvent, and the need to add a certain amount of Form a as a seed crystal to induce precipitation of crystals to control crystallization conditions are very harsh, operable range is very small, easy to form methanol solvate, hydrate or stable crystalline type C, to obtain reproducible single crystal type a low, it is difficult to achieve industrial production, and no mention of the preparation of Form a yield and purity in this patent.
[0011]
[0012] Chinese patent CN102267957A invention discloses a method for preparing febuxostat Form A, the solvent is preferably acetone, dissolved into 25 ~ 40 ° C was allowed to stand, when there began to crystallize when stirred for 20 to 40 minutes, then placed in -15 ~ 0 ° C to continue the crystallization of 8 to 10 hours. The crystallization process need to well below zero, when industrial mass production, resulting in high production costs, is not conducive to industrial production, the process yield up to 95.4%.
[0013] Chinese patent CN101139325 of Example 7 discloses the preparation of Form A with acetone method, although the process is simple, but the yield is low, only 50%.
[0014] Although the Chinese patent CN101684108A isopropyl alcohol as a solvent is disclosed a method for preparing crystalline form, the crystalline form of preparation is used to cool and heat a phased manner was allowed to stand, the crystallization temperature, long crystallization time, about 30 hours, the yield is low, and its products are not crystalline Form A.
[0015] In addition, Chinese patent CN101525319A, CN101805310, CN101926795A, CN101926794, W02012020272A2 are disclosed ethanol as a solvent or aqueous ethanol as a solvent preparation methods, and its products are crystalline ethanol solvate.
[0016] World Patent W02011139886A2 discloses the use of a mixed solvent of alcohol, and its products are not obtained polymorph A0
PAPER
Letters in Organic Chemistry (2015), 12(3), 217-221
Author(s): Xiao Long Li, Rui Qiu, Wei Li Wan, Xu Cheng, Li Hai and Yong Wu
Affiliation: Key Laboratory of Drug Targeting of Education Ministry, West China School of Pharmacy, Sichuan University, Chengdu 610041, China.
Graphical Abstract:

Total synthesis of three Febuxostat metabolites, named 67M-1, 67M-2, and 67M-4,is described in this article. Through condensation of the key intermediate compound A with different side chains, and then oxidation and hydrolysis, we obtained three target compounds with an overall yield of 19.5%-28.0%.
VOLUME: 12
ISSUE: 3
Page: [217 – 221]
Pages: 5
DOI: 10.2174/1570178612666150108000805, http://www.eurekaselect.com/127479/article

ULORIC (febuxostat) is a xanthine oxidase inhibitor. The active ingredient in ULORIC is 2-[3-cyano-4-(2-methylpropoxy) phenyl]-4-methylthiazole-5-carboxylic acid, with a molecular weight of 316.38. The empirical formula is C16H16N2O3S.
The chemical structure is:
 |
Febuxostat is a non-hygroscopic, white crystalline powder that is freely soluble in dimethylformamide; soluble in dimethylsulfoxide; sparingly soluble in ethanol; slightly soluble in methanol and acetonitrile; and practically insoluble in water. The melting range is 205°C to 208°C.
LORIC tablets for oral use contain the active ingredient, febuxostat, and are available in two dosage strengths, 40 mg and 80 mg. Inactive ingredients include lactose monohydrate, microcrystalline cellulose, hydroxypropyl cellulose, sodium croscarmellose, silicon dioxide and magnesium stearate. ULORIC tablets are coated with Opadry II, green.
| CN1642546A * | Mar 28, 2003 | Jul 20, 2005 | Teijin Ltd. | Containing a single crystalline solid preparation |
| CN102471295A * | Jul 14, 2010 | May 23, 2012 | Teijin Pharma Ltd. | The method of manufacturing the poor solvent additive method of 2- (3-cyano-4-isobutyl-phenyl) -4-methyl-5-carboxylic acid crystalline polymorph of |
| EP2502920A1 * | Mar 25, 2011 | Sep 26, 2012 | Sandoz Ag | Crystallization process of Febuxostat from A |
| JP2011020950A* | Title not available |
| WO2015018507A3 * | Jul 30, 2014 | Oct 22, 2015 | Pharmathen S.A. | A novel process for the preparation of febuxostat |
| CN103304512A * | Jun 4, 2013 | Sep 18, 2013 | 华南理工大学 | Preparation method for febuxostat |
| WO2011031409A1 * | Aug 12, 2010 | Mar 17, 2011 | Teva Pharmaceutical Industries Ltd. | Processes for preparing febuxostat |
| JP2834971B2 | Title not available | |||
| JP3202607B2 | Title not available | |||
| JPH1045733A * | Title not available | |||
| US5614520 | Jan 30, 1995 | Mar 25, 1997 | Teijin Limited | 2-arylthiazole derivatives and pharmaceutical composition thereof |
| CN102229581A * | Nov 15, 2010 | Nov 2, 2011 | 邹巧根 | Preparation method for febuxostat intermediate |
| JPH1045733A * | Title not available |
| CN101497589A * | Feb 26, 2009 | Aug 5, 2009 | 沈阳药科大学 | Method for synthesizing 2-(3-cyano-4- isobutoxy phenyl)-4-methyl-carboxylate |
| CN101863854A * | Jun 29, 2010 | Oct 20, 2010 | 沈阳药科大学 | Synthesis method of 2-(3-cyan-4-isobutoxy) phenyl-4-methyl-5-thiazole formic acid |
| JP2706037B2 * | Title not available |
| Reference | ||
|---|---|---|
| 1 | * | HASEGAWA, M. ET AL.: ‘A facile one-pot synthesis of 4-alkoxy-1,3-benzenedicarbonitrile‘ HETEROCYCLES vol. 47, no. 2, 1998, pages 857 – 864 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2012131590A1 * | Mar 28, 2012 | Oct 4, 2012 | Sandoz Ag | An improved process for preparation of febuxostat and its polymorphic crystalline form c thereof |
| WO2014009817A1 * | Mar 19, 2013 | Jan 16, 2014 | Alembic Pharmaceuticals Limited | Pharmaceutical composition of febuxostat |
| WO2014057461A1 | Oct 10, 2013 | Apr 17, 2014 | Ranbaxy Laboratories Limited | Process for the preparation of crystalline form g of febuxostat |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(3-cyano-4-isobutoxyphenyl)-4-methyl-
1,3-thiazole-5-carboxylic acid |
|
| Clinical data | |
| Trade names | Uloric, Adenuric, Atenurix, Feburic, Goturic, Goutex. Generic in several countries.[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609020 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | ~49% absorbed |
| Protein binding | ~99% to albumin |
| Metabolism | via CYP1A2, 2C8, 2C9,UGT1A1, 1A3, 1A9, 2B7 |
| Biological half-life | ~5-8 hours |
| Excretion | Urine (~49% mostly as metabolites, 3% as unchanged drug); feces (~45% mostly as metabolites, 12% as unchanged drug) |
| Identifiers | |
| CAS Number | 144060-53-7 |
| ATC code | M04AA03 (WHO) |
| PubChem | CID 134018 |
| IUPHAR/BPS | 6817 |
| DrugBank | DB04854 |
| ChemSpider | 118173 |
| UNII | 101V0R1N2E |
| KEGG | D01206 |
| ChEMBL | CHEMBL1164729 |
| Chemical data | |
| Formula | C16H16N2O3S |
| Molar mass | 316.374 g/mol |
/////////Febuxostat, 144060-53-7, Uloric, Adenuric, Tei 6720, thiazole derivative, inhibitor of XANTHINE OXIDASE, treatment of HYPERURICEMIA, chronic GOUT, FBX, Febugood, Feburic, Febutaz, TMX 67, Zurig
CC1=C(SC(=N1)C2=CC(=C(C=C2)OCC(C)C)C#N)C(=O)O

The heterogeneous palladium-catalyzed Suzuki reactions between model aryl bromides (4-bromoanisole, 4-bromoaniline, 4-amino-2-bromopyridine, and 2-bromopyridine) and phenylboronic acid have been successfully conducted in water with no added ligand at the 100 mL scale using 20–40 mmol of aryl bromide. The product yields associated with these substrates were optimized, and key reaction parameters affecting the yields were identified. The results clearly indicate that the reaction parameters necessary to achieve high yields are substrate-dependent. In addition, it is demonstrated that aqueous Suzuki reactions of substrates containing basic nitrogen centers can produce quantitative yields of desired products in the absence of added ligand.

http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00180
//////////
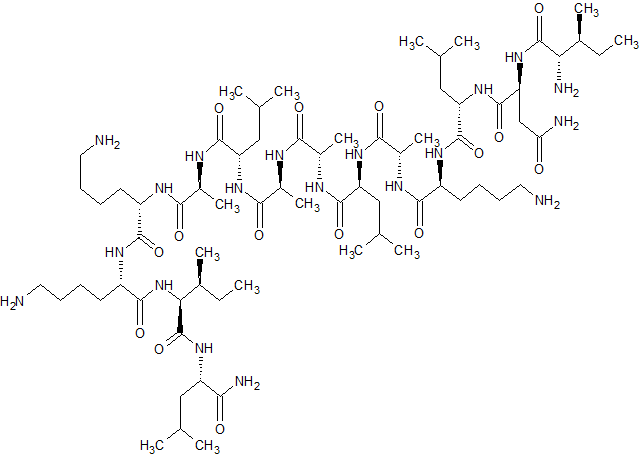
Mastoparan, Peptide (H-INLKALAALAKKIL-NH2)
H-Ile-Asn-Leu-Lys-Ala-Leu-Ala-Ala-Leu-Ala-Lys-Lys-xiIle-Leu-NH2
PEPTIDE1{I.N.L.K.A.L.A.A.L.A.K.K.[*N[C@H](C(=O)*)C(C)CC |$_R1;;;;;_R2;;;;$|].L.[am]}$$$$

| Ile – Asn – Leu – Lys – Ala – Leu – Ala – Ala – Leu – Ala – Lys – Lys – Ile – Leu -NH2 |
| Mastoparan; Mast cell degranulating peptide (Vespula lewisii); NSC351907; CAS 72093-21-1; | |
| Molecular Formula: | C70H131N19O15 |
|---|---|
| Molecular Weight: | 1478.90744 g/mol |
Description
| Physical State: | Solid |
| Derived from: | Synthetic. Originally isolated from wasp venom (Vespula lewisii) |
| Solubility: | Soluble in water (2.6 mg/ml), and 100% ethanol. |
| Storage: | Store at -20° C |
| Refractive Index: | n20D 1.53 |
| IC50: | Na+,K+-ATPase: IC50 = 7.5 µM |
Mastoparan is a peptide toxin from wasp venom. It has the chemical structure Ile-Asn-Leu-Lys-Ala-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2.[2]
The net effect of mastoparan’s mode of action depends on cell type, but seemingly always involves exocytosis. In mast cells, this takes the form of histamine secretion, while in platelets and chromaffin cells release serotonin and catecholamines are found, respectively. Mastoparan activity in the anterior pituitary gland leads to prolactin release.
In the case of histamine secretion, the effect of mastoparan takes place via its interference with G protein activity. By stimulating theGTPase activity of certain subunits, mastoparan shortens the lifespan of active G protein. At the same time, it promotes dissociation of any bound GDP from the protein, enhancing GTP binding. In effect, the GTP turnover of G proteins is greatly increased by mastoparan. These properties of the toxin follow from the fact that it structurally resembles activated G protein receptors when placed in a phospholipid environment. The resultant G protein-mediated signaling cascade leads to intracellular IP3 release and the resultant influx of Ca2+.
In an experimental study conducted by Tsutomu Higashijima and his counterparts, mastoparan was compared to melittin, which is found in bee venom.[2] Mainly, the structure and reaction to phosphate was studied in each toxin. Using Circular Dichroism (CD), it was found that when mastoparan was exposed to methanol, an alpha helical form existed. It was concluded that strong intramolecular hydrogen bonding occurred. Also, two negative bands were present on the CD spectrum. In an aqueous environment, mastoparan took on a nonhelical, unordered form. In this case, only one negative band was observed on the CD spectrum. Adding phosphate buffer to mastoparan resulted in no effect.
Melittin produced a different conformational change than mastoparan. In an aqueous solution, melittin went from a nonhelical form to an alpha helix when phosphate was added to the solution. The binding of melittin to the membrane was believed to result fromelectrostatic interactions, not hydrophobic interactions.
Infections caused by multidrug resistant bacteria are currently an important problem worldwide. Taking into account data recently published by the WHO, lower respiratory infections are the third cause of death in the world with around 3.2 million deaths per year, this number being higher compared to that related to AIDS or diabetes mellitus [1]. It is therefore important to solve this issue, although the perspectives for the future are not very optimistic. During the last 30 years an enormous increase has been observed of superbugs isolated in the clinical setting, especially from the group called ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter spp.) which show high resistance to all the antibacterial agents available [2]. We will focus on Acinetobacter baumannii, the pathogen colloquially called “iraquibacter” for its emergence in the Iraq war. It is a Gram-negative cocobacillus and normally affects people with a compromised immune system, such as patients in the intensive care unit (ICU) [3] and [4]. Together with Escherichia coliand P. aeruginosa, A. baumannii are the most common cause of nosocomial infections among Gram-negative bacilli. The options to treat infections caused by this pathogen are diminishing since pan-drug resistant strains (strains resistant to all the antibacterial agents) have been isolated in several hospitals [5]. The last option to treat these infections is colistin, which has been used in spite of its nephrotoxic effects [6]. The evolution of the resistance of A. baumannii clinical isolates has been established by comparing studies performed over different years, with the percentage of resistance to imipenem being 3% in 1993 increasing up to 70% in 2007. The same effect was observed with quinolones, with an increase from 30 to 97% over the same period of time[7]. In Spain the same evolution has been observed with carbapenems; in 2001 the percentage of resistance was around 45%, rising to more than 80% 10 years later [8]. Taking this scenario into account, there is an urgent need for new options to fight against this pathogen. One possible option is the use of antimicrobial peptides (AMPs) [9],[10] and [11], and especially peptides isolated from a natural source [12]. One of the main drawbacks of using peptides as antimicrobial agents is the low stability or half-life in human serum due to the action of peptidases and proteases present in the human body[13], however there are several ways to increase their stability, such as using fluorinated peptides [14] and [15]. One way to circumvent this effect is to study the susceptible points of the peptide and try to enhance the stability by protecting the most protease labile amide bonds, while at the same time maintaining the activity of the original compound. Another point regarding the use of antimicrobial peptides is the mechanism of action. There are several mechanisms of action for the antimicrobial peptides, although the global positive charge of most of the peptides leads to a mechanism of action involving the membrane of the bacteria [16]. AMPs has the ability to defeat bacteria creating pores into the membrane [17], also acting as detergents [18], or by the carpet mechanism [19]. We have previously reported the activity of different peptides against colistin-susceptible and colistin-resistant A. baumannii clinical isolates, showing that mastoparan, a wasp generated peptide (H-INLKALAALAKKIL-NH2), has good in vitro activity against both colistin-susceptible and colistin-resistant A. baumannii [20]. Therefore, the aim of this manuscript was to study the stability of mastoparan and some of its analogues as well as elucidate the mechanism of action of these peptides.
Paper
Volume 101, 28 August 2015, Pages 34–40

http://www.sciencedirect.com/science/article/pii/S0223523415300933
doi:10.1016/j.ejmech.2015.06.016
The treatment of some infectious diseases can currently be very challenging since the spread of multi-, extended- or pan-resistant bacteria has considerably increased over time. On the other hand, the number of new antibiotics approved by the FDA has decreased drastically over the last 30 years. The main objective of this study was to investigate the activity of wasp peptides, specifically mastoparan and some of its derivatives against extended-resistant Acinetobacter baumannii. We optimized the stability of mastoparan in human serum since the specie obtained after the action of the enzymes present in human serum is not active. Thus, 10 derivatives of mastoparan were synthetized. Mastoparan analogues (guanidilated at the N-terminal, enantiomeric version and mastoparan with an extra positive charge at the C-terminal) showed the same activity against Acinetobacter baumannii as the original peptide (2.7 μM) and maintained their stability to more than 24 h in the presence of human serum compared to the original compound. The mechanism of action of all the peptides was carried out using a leakage assay. It was shown that mastoparan and the abovementioned analogues were those that released more carboxyfluorescein. In addition, the effect of mastoparan and its enantiomer against A. baumannii was studied using transmission electron microscopy (TEM). These results suggested that several analogues of mastoparan could be good candidates in the battle against highly resistant A. baumannii infections since they showed good activity and high stability.

Patent IDDatePatent TitleUS20160672612016-03-10SERCA INHIBITOR AND CALMODULIN ANTAGONIST COMBINATION
| Mastoparan | |
|---|---|

Solution structure of mastoparan from Vespa simillima xanthoptera.[1]
|
|
| Identifiers | |
| Symbol | Mastoparan_2 |
| Pfam | PF08251 |
| InterPro | IPR013214 |
| TCDB | 1.C.32 |
| OPM superfamily | 160 |
| OPM protein | 2czp |
///////Peptide, Antimicrobial peptide, Mastoparan, Acinetobacter baumannii, NSC351907, 72093-21-1, NSC 351907
CCC(C)C(C(=O)NC(CC(=O)N)C(=O)NC(CC(C)C)C(=O)NC(CCCCN)C(=O)NC(C)C(=O)NC(CC(C)C)C(=O)NC(C)C(=O)NC(C)C(=O)NC(CC(C)C)C(=O)NC(C)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(C(C)CC)C(=O)NC(CC(C)C)C(=O)N)N
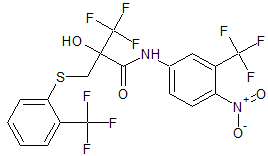
Cas 1929605-82-2

SYNTHESIS

Scheme .
Synthetic strategy used in the synthesis of 52. Reagents and conditions: (a) NaH (1 equiv.), THF, 0 °C to RT, 3 h; (b) KCN (1.2 equiv.), 25% H2SO4, 0 °C to RT, 20 h; c) HCl, AcOH, reflux, 24 h; (d) 8, SOCl2(1.3 equiv.), DMA, RT, 72 h.
3-Bromo-1,1,1-trifluoroacetone (48) was coupled with thiophenol 47 to afford 49, which was then converted into cyano derivative 50 using potassium cyanide and 25% sulfuric acid [16]. Intermediate 51 was obtained after refluxing 50 in concentrated HCl and glacial acetic acid. Coupling of 51 with commercially available 4-nitro-3-(trifluoromethyl)aniline 8yielded the desired amide 52.
To a mixture of NaH (10.47 mmol) in 10 mL anhydrous THF was added a solution of 2-(trifluoromethyl)benzenethiol (10.47 mmol) in 2mL anhydrous THF at 0 °C. This mixture was stirred for 20 min. 3-Bromo-1,1,1-trifluoropropan-2-one was then added dropwise to the mixture at 0 °C, the reaction was warmed to r.t. and stirred for 12 h. The mixture was filtered trough celite, the filtered pad was washed with THF, and the filtrate was evaporated to dryness. The residue was purified by flash column chromatography eluting with n-hexane/EtOAc 100:0 v/v increasing to n-hexane/EtOAc 85:15 v/v to give a pale yellow oil in 93% yield. 1H-NMR (CDCl3): d 7.76-7.69 (m, 2H), 7.60-7.53 (m, 1H), 7.42-7.38 (m, 1H), 3.44 (s, 2H). 19F-NMR (CDCl3): d -59.91 (s, 3F), -85.26 (s, 3F). 13C-NMR (CDCl3): d 189.6, 137.7, 135.9, 134.5, 133.2, 130.6, 129.6 (q, J= 26.3 Hz), 127.0 (q, J= 3.8 Hz), 124.3 (q, J= 4.1 Hz), 124.0 (q, J= 3.7 Hz), 94.4 (q, J= 30.4 Hz), 40.4.
A 20% aqueous solution of H2SO4 (3.4 mL) was added dropwise to a mixture of 49 (11.03 mmol) and KCN (13.24 mmol) in 5 mL H2O at 0 °C. The reaction mixture was warmed to r.t. and stirred for 20 h. The mixture was then diluted with water (50 mL) and extracted with Et2O (3 x 150 mL). The organic extracts were washed with sat. aq. NaHCO3 and brine, dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash column chromatography eluting with n-hexane/EtOAc 100:0 v/v increasing to n-hexane/EtOAc 95:5 v/v to give a pale yellow oil in 86% yield. 1H-NMR (CDCl3): d 7.80 (d, J= 7.8 Hz, 1H), 7.77-7.76 (m, 1H), 7.72-7.59 (m, 1H), 7.52-7.49 (m, 1H), 4.36 (bs, 1H), 3.58 (d, J= 14.6 Hz, 1H), 3.44 (d, J= 14.6 Hz, 1H). 19F-NMR (CDCl3): d -57.08 (s, 3F), -79.51 (s, 3F). 13C-NMR (CDCl3): d 135.4, 132.8, 132.5 (q, J= 30.1 Hz), 129.1, 128.7 (q, J= 5.5 Hz), 126.7, 124.9, 124.6, 122.6, 122.4, 120.4, 114.0, 71.4 (q, J= 32.9), 40.75.
A mixture of 51 (6.89 mmol), concentrated HCl (23.4 mL) and AcOH (4.1 mL) was refluxed o.n. with vigorous stirring. The mixture was then diluted with water (100 mL) and extracted with Et2O (4 x 100 mL), which was in turn washed with sat. aq. NaHCO3 (4 x 100 mL). The water solution was acidified with concentrated HCl to pH 1 and extracted with Et2O (4x 150 mL). The Et2O extracts were dried over Na2SO4, filtered and concentrated to dryness to give a pale yellow waxy solid in 41% yield. 1H-NMR (CDCl3): d 9.57 (bs, 1H), 7.70 (d, J= 7.7 Hz, 1H), 7.67 (d, J= 7.7 Hz, 1H), 7.54-7.51 (m, 1H), 7.39-7.36 (m, 1H), 3.60 (s, 2H). 19F-NMR (CDCl3): d -60.10 (s, 3F), -77.7 (s, 3F). 13C-NMR (CDCl3): d 172.0, 134.1, 134.0, 131.2 (q, J= 30.1 Hz), 127.5, 126.7 (q, J= 5.6 Hz), 124.2 (q, J= 121.9 Hz), 121.9 (q, J= 126.7 Hz), 78.2 (q, J= 28.7 Hz), 37.7.
Thionyl chloride (1.16 mmol) was added dropwise to a stirring solution of 51 in anhydrous DMA at -10 °C under Ar atmosphere. The reaction mixture was stirred for 1 h, then a solution of 8 in 2 mL anhydrous DMA was added dropwise. The reaction mixture was warmed to r.t. and stirred for 72 h. The mixture was then diluted with sat. aq. NaHCO3 (40 mL) and extracted with Et2O (3 x 40 mL). The organic extracts were filtered trough celite, dried over Na2SO4 and evaporated to dryness. The residue was purified by flash column chromatography eluting with n-hexane/EtOAc 100:0 v/v increasing to n-hexane/EtOAc 80:20 v/v to give a pale yellow solid in 13% yield.
1H-NMR (CDCl3): d 8.93 (bs, 1H), 7.94 (d, J= 8.8 Hz, 1H), 7.87 (d, J= 2.2 Hz, 1H), 7.72 (d, J= 8.1 Hz, 1H), 7.69 (dd, J= 8.8 Hz, 2.2 Hz, 1H), 7.50-7.47 (m, 2H), 7.26-7.23 (m, 1H), 4.41 (s, 1H), 4.19 (d, 14.7 Hz, 1H), 3.45 (d, J= 14.7 Hz, 1H).
19F-NMR (CDCl3): d -59.7 (s, 3F), -60.12 (s, 3F), -77.4 (s, 3F).
13C-NMR (CDCl3): d 164.6, 143.8, 140.0, 134.7, 132.6, 131.1 (q, J= 29.8 Hz), 130.5, 128.3, 126.8 (q, J= 5.5 Hz), 126.7, 125.2 (q, J= 36.3 Hz), 124.5, 123.9, 122.6, 122.4, 122.2, 121.7, 120.4, 118.2 (q, J= 5.8 Hz), 76.3 (q, J= 27.8 Hz), 38.5.
MS [ESI, m/z]: 523.0 [M+H]+.
EI-HMRS (M-H)– found 521.0215, calculated for C18H0N2O4F9S 521.0218.
HPLC (method 1): retention time = 23.84 min.
clips
Prostate cancer (PC) is a leading cause of male death worldwide and it is the most frequently diagnosed cancer among men aged 65–74 [1]. The prognosis varies greatly, being highly dependent on a number of factors such as stage of diagnosis, race and age. Currently, PC treatment includes androgen deprivation, surgery, radiation, endocrine therapy and radical prostatectomy.
PC cell growth is strongly dependent on androgens, therefore blocking their effect can be beneficial to the patient’s health. Such outcomes can be achieved by antagonism of the androgen receptor (AR) using anti-androgen drugs, which have been extensively explored either alone or in combination with castration [2]. Flutamide (Eulexin®) (1) (in its active form as hydroxyflutamide (2)), bicalutamide (Casodex®) (3), nilutamide (Niladron®) (4) and enzalutamide (previously called MDV3100) (Xtandi®) (5) are all non-steroidal androgen receptor antagonists approved for the treatment of PC (Fig. 1). In many cases, after extended treatment over several years, these anti-androgens become ineffective and the disease may progress to a more aggressive and lethal form, known as castration resistant prostate cancer (CRPC). The major cause of this progressive disease is the emergence of different mutations on the AR, which cause the anti-androgen compounds to function as agonists, making them tumour-stimulating agents[3].

Fig. 1.
Structure of anti-androgen small molecules approved by FDA or in clinical development for the treatment of PC.
Among the drugs used for the treatment of PC, bicalutamide and enzalutamide selectively block the action of androgens while presenting fewer side effects in comparison with other AR antagonists [4], [5] and [6]. The structure of these molecules is characterised by the presence of a trifluoromethyl substituted anilide, which appears to be critical for biological activity (Fig. 1). As a means to improve the anti-proliferative activity of these compounds, and in order to exploit the well established potential of the fluorine atom in enhancing the pharmacological properties and drug-like physicochemical characteristics of candidate compounds [7], [8] and [9], a wide array of diverse new structures has been rationally designed and synthesised, through the introduction of fluoro-, trifluoromethyl- and trifluoromethoxy groups in diverse positions of both aromatic rings of the parent scaffolds. Our modifications resulted in a marked improvement of in vitro anti-proliferative activities on a range of human PC cell lines (VCap, LNCaP, DU-145 and 22RV1). In addition, we probed full versus partial AR antagonism for our new compounds.
Paper

Volume 118, 8 August 2016, Pages 230–243

School of Pharmacy and Pharmaceutical Sciences, Redwood Building, King Edward VII Avenue, CF10 3NB, Cardiff, Wales, UK
Prostate cancer (PC) is one of the major causes of male death worldwide and the development of new and more potent anti-PC compounds is a constant requirement. Among the current treatments, (R)-bicalutamide and enzalutamide are non-steroidal androgen receptor antagonist drugs approved also in the case of castration-resistant forms. Both these drugs present a moderate antiproliferative activity and their use is limited due to the development of resistant mutants of their biological target.
Insertion of fluorinated and perfluorinated groups in biologically active compounds is a current trend in medicinal chemistry, applied to improve their efficacy and stability profiles. As a means to obtain such effects, different modifications with perfluoro groups were rationally designed on the bicalutamide and enzalutamide structures, leading to the synthesis of a series of new antiproliferative compounds. Several new analogues displayed improved in vitro activity towards four different prostate cancer cell lines, while maintaining full AR antagonism and therefore representing promising leads for further development.
Furthermore, a series of molecular modelling studies were performed on the AR antagonist conformation, providing useful insights on potential protein-ligand interactions.
http://www.sciencedirect.com/science/article/pii/S0223523416303452

Professor Chris McGuigan, 57, was trying to invent new drugs to use in the fight against the disease.
But the tragic scientist, who was head of medicinal chemistry at Cardiff University’s School of Pharmacy and Pharmaceutical Sciences, died after his own fight with cancer.
A spokesman for Cardiff University said: “Professor McGuigan had been at the heart of scientific research for more than 30 years. He was an exceptionally gifted inventor and chemist.
“His loss will be felt cross the university and the wider scientific community.

“He had a strong drive to use his scientific ideas for social good, working tirelessly to address medical needs where they were unmet.
“Our thoughts are with his family, friends and close colleagues at this very sad time.”
Prof McGuigan’s research led him to try and develop new drugs for cancer, HIV, hepatitis B and C, shingles, measles, influenza and central nervous system (CNS) disease.
He also invented four new experimental drugs that were used in human clinical trials.
Prof McGuigan, who lived in Cardiff, is survived by wife Maria, 50, and his two young daughters Phoebe and Grace.
///////////1929605-82-2, bicalutamide and enzalutamide derivatives, antiproliferative agents, treatment of prostate cancer, School of Pharmacy and Pharmaceutical Sciences, Redwood Building, King Edward VII Avenue, CF10 3NB, Cardiff, Wales, UK
FC(F)(F)c1cc(ccc1[N+]([O-])=O)NC(=O)C(O)(CSc2ccccc2C(F)(F)F)C(F)(F)F
 .CF3COOH
.CF3COOH
PHASE 1 CHINA



The main purpose: to determine HAO472 treatment of relapsed / refractory C the maximum tolerated dose (MTD). Secondary objectives: 1) evaluation of drug safety and tolerability; 2) study HAO472 in pharmacokinetic characteristics of the human body; 3) the effectiveness of HAO472 treatment of relapsed / refractory M2b type of AML.
Acute myelogenous leukemia
HAO472
Phase I
Test Number: CTR20150246
Sponsor Name:
Jiangsu Hengrui Medicine Co., Ltd. 1/
2 Ruijin Hospital, Shanghai Jiaotong University School of Medicine /
3 Jiangsu Hengrui Medicine Co., Ltd. /
4 Shanghai Hengrui Medicine Co., Ltd. /

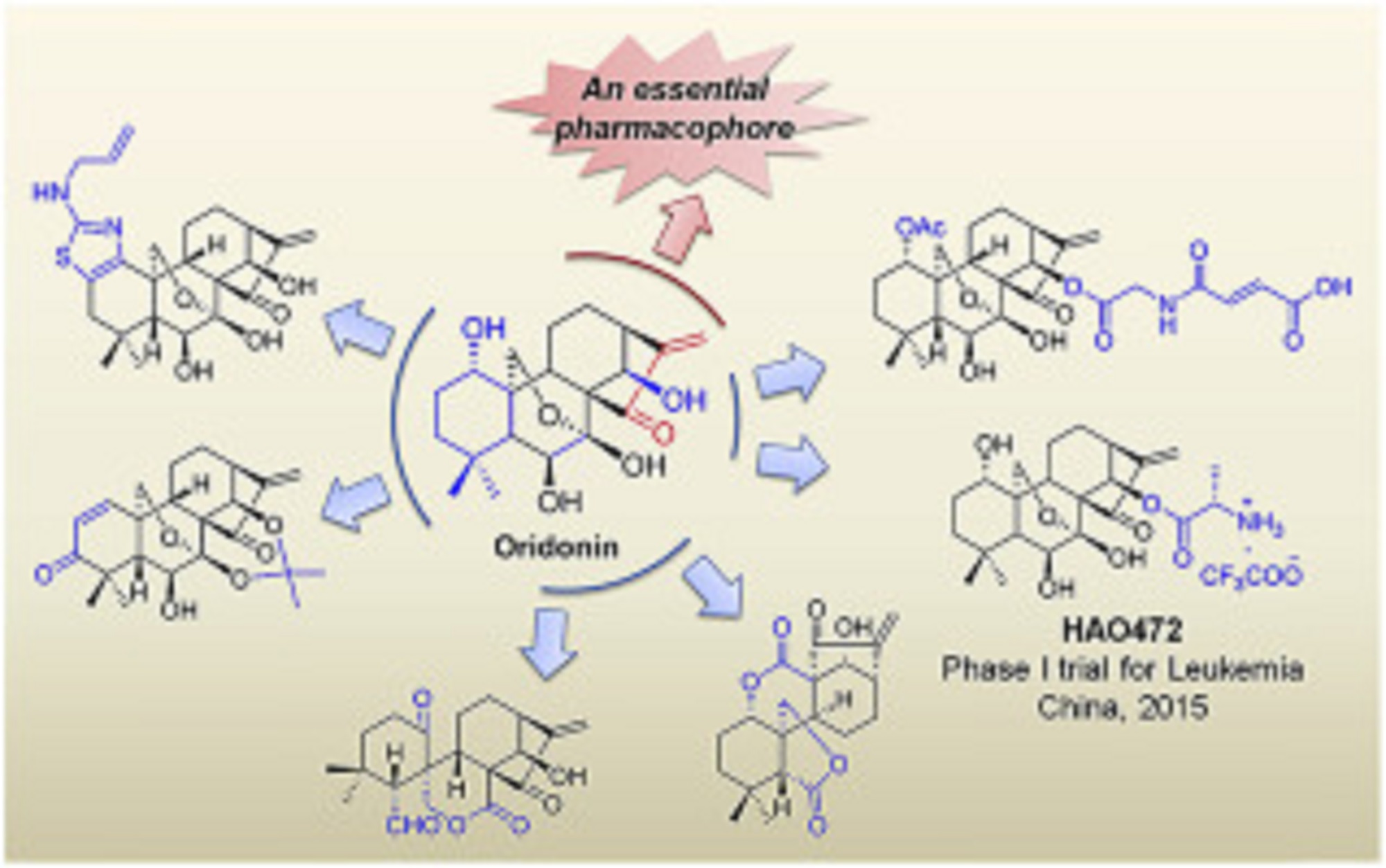
Natural products have historically been, and continue to be, an invaluable source for the discovery of various therapeutic agents. Oridonin, a natural diterpenoid widely applied in traditional Chinese medicines, exhibits a broad range of biological effects including anticancer and anti-inflammatory activities. To further improve its potency, aqueous solubility and bioavailability, the oridonin template serves as an exciting platform for drug discovery to yield better candidates with unique targets and enhanced drug properties. A number of oridonin derivatives (e.g. HAO472) have been designed and synthesized, and have contributed to substantial progress in the identification of new agents and relevant molecular mechanistic studies toward the treatment of human cancers and other diseases. This review summarizes the recent advances in medicinal chemistry on the explorations of novel oridonin analogues as potential anticancer therapeutics, and provides a detailed discussion of future directions for the development and progression of this class of molecules into the clinic.
Volume 122, 21 October 2016, Pages 102–117


////////Natural product, Oridonin, Diterpenoids, Anticancer agents, Drug discovery, Chemical biology, AML, HAO 472, relapsed / refractory AML. Jiangsu Hengrui Medicine Co., Ltd, PHASE1, LEUKEMIA
C[C@H](N)C(=O)O[C@]15OC[C@@]2([C@H](O)CCC(C)(C)[C@@H]2[C@H]1O)[C@H]3CC[C@@H]4C(=C)C(=O)[C@@]35C4O
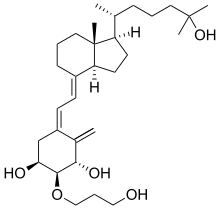
Eldecalcitol
(1S,2S,3S,5Z)-5-[(2E)-2-[(1R,3aS,7aR)-1-[(2R)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1H-inden-4-ylidene]ethylidene]-2-(3-hydroxypropoxy)-4-methylidenecyclohexane-1,3-diol
(1R,2R,3R,5Z,7E)-2-(3-Hydroxypropyloxy)-9,10-secocholesta-5,7,10(19)-triene-1,3,25-triol
| AC1O5QQ2; CAS 104121-92-8; AN-3697; ED 71, Edirol® | |
| Molecular Formula: | C30H50O5 |
|---|---|
| Molecular Weight: | 490.715 g/mol |
APPROVED JAPAN , 2011-01-21, Chugai (Originator) , Roche,Taisho Toyama
Eldecalcitol was approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on January 21, 2011. It was developed by Chugai Pharmaceutical (a member of Roche) and marketed as Edirol® by Chugai Pharmaceutical and Taisho.
Eldecalcitol is an orally active vitamin D analogue leading to greater absorption of bind calcium. It is usually used to treat osteoporosis.
Edirol® is available as capsule for oral use, containing 0.5 μg or 0.75 μg of free Eldecalcitol, and the recommended dose is 0.75 μg once daily.
ED-71, a vitamin D analog, is a more potent inhibitor of bone resorption than alfacalcidol in an estrogen-deficient rat model of osteoporosis. ED-71, effectively and safely increased lumbar and hip bone mineral density (BMD) in osteoporotic patients who also received vitamin D3 supplementation.
Eldecalcitol is a drug used in Japan for the treatment of osteoporosis.[1] It is an analog of vitamin D.[2] Osteoporosis is a common bone disease among the older generation, with an estimated prevalence of over 200 million people.[1] This condition often results in bone fractures due to abnormally low bone mass density, and is a leading cause of disability, especially among developed countries with longer average life spans. Osteoporosis is more common in women than with men.
Chugai Pharmaceutical/Roche are the originators of the medicinal drug eldecalcitol through Taisho Pharmaceutical Holdings and Chugai Pharmaceutical. The trade name of eldecalcitol is Edirol, and its Chemical Abstracts Service (CAS) registry number is 104121-92-8. Eldecalcitol was approved for use in Japan on January 2011. The approval came from the Japanese Ministry of Health, Labor, and Welfare for the objective of a treatment for osteoporosis.[3]
Clinical trials have suggested that eldecalcitol, a vitamin D analog, has strong effects to reduce calcium reabsorption into the body from bones, therefore increasing bone mineral density, and to increase calcium absorption in intestines.[4] In animals, eldecalcitol inhibits the activity of osteoclasts for the function to reduce bone degradation for calcium, while still able to maintain osteoblast function so as to not hinder bone formation.[5] Unlike other vitamin D analogs, eldecalcitol does not significantly suppress parathyroid hormone levels, promising a better treatment for osteoporosis in comparison to other medications.[6] Bone mineral density increases with eldecalcitol use, in addition to strengthening bone structure. This occurs due to the function of the eldecalcitol drug, which decreases bone reabsorption as observed through a bone reabsorption marker. Bone geometry assessments show that eldecalcitol increases cortical bone area in patients with osteoporosis more so than other vitamin D analogs, such as alfacalcidol. There was also the maintenance of thickness of cortical bone mass, strongly indicating that eldecalcitol improves the strength and mass of bone, specifically cortical bone structure.[7] Adverse effects of eldecalcitol include an increase in blood and urinary calcium levels. Abnormally high levels of calcium can lead to problems associated with hypercalcemia.
Eldecalcitol can be used for the treatment of hypocalcaemia or osteoporosis. Calcium absorption increases with the presence of eldecalcitol by the body, occurring in the intestines, which is useful for those who have low calcium levels. Eldecalcitol is more often used due to its effects to treat osteoporosis. In the aging population, the bone matrix becomes weakened through untreated osteoporosis. This leads to an increased risk of severe fractures that include spinal and hip fractures in addition to vertebral and wrist fractures. This creates a burden on the health care system due to a decline in the quality of life for the individuals that suffer from this condition. Some risk factors leading to the predisposition of developing osteoporosis are previous incidents of bone fractures and a reduction in bone mineral density.[1] These factors expectantly increase as age increases. Bone health is reliant on maintaining physiologically needed levels of calcium, where the body constantly maintains this calcium homeostasis through osteoblast and osteoclast activity. Osteoblast activity serves this function of maintaining appropriate calcium levels by depositing calcium in bones when blood calcium levels are above normal. In contrast, osteoclasts break down bone tissue to increase blood calcium levels if they are low.[8] This activity is performed after absorption of calcium by the body, which requires the actions of vitamin D. The active metabolite of vitamin D, calcitriol, performs its function through interactions with the calcitriol receptor. This nuclear hormone receptor is responsible for calcium absorption which, in turn, is involving in bone depletion and formation. The new analogs of vitamin D, such as eldecalcitol, are observed to have stronger effects in preventing bone loss, fractures, and falls in comparison to calcitriol.[9] Eldecalcitol is even more effective than its counterpart alfacalcidol, another vitamin D analog. Studies have shown eldecalcitol is more effective than alfacalcidol in preventing vertebral and wrist fractures, and even falls, with osteoporotic patients with vitamin D insufficiencies.[10] Eldecalcitol is also more effective at preventing fractures than vitamin D and calcium supplements.[1] Eldecalcitol increases calcium absorption for vitamin D deficient patients, and therefore could be used for osteoporosis treatment for all age groups.
Analogs of vitamin D are being explored intensely for their regulatory effects on calcium metabolism with the purpose of treating osteoporosis, a skeletal disease associated with low bone mass and deterioration of bone tissue. Vitamin D is imperative for absorption of calcium to maintain bone strength.
Eldecalcitol is an orally administered drug to patients, which binds to vitamin D receptors and binding protein for the goal of achieving greater specificity to bind calcium for its absorption. This greater affinity is 2.7-fold that of the active vitamin D form of calcitriol. Eldecalcitol is readily absorbed into the body, with a long elimination half-life of over eight hours, reaching maximum absorption in 3.4 hours.[1]
Eldecalcitol is present in the form of pills for oral administration. In preclinical models with healthy male volunteers, oral doses of eldecalcitol ranged from 0.1 to 1.0 micrograms once daily to show an increase in bone mineral density.[11] Preclinical trials show improvements for doses at 0.5 and 0.75 micrograms, which are the recommended dosage amounts for the Edirol product as approved by the Japanese Ministry of Health, Labor, and Welfare for treating osteoporosis.[3]
The class of eldecalcitol is a vitamin D3 derivative. This molecule has a molecular weight of 490.71 grams per mole. The eldecalcitol analog of calcitriol, contains a hydroxypropyl group in the lower cyclohexane ring. The synthesis of eldecalcitol incorporates two units assembled together. The IUPAC names include (3S, 4S, 5R)-oct-1-en-7-yne-3,4,5-triol that is fused to a bicyclic system, (R)-6-((1R, 3aR, 7aR, E)-4-(bromomethylene)-7a-methyloctahydro-1H-inden-1-yl)-2-methylheptan-2-ol. The assembly process includes a Diels-Alder reaction to give the fully protected eldecalcitol. In order to get the parent molecule, the hydroxyl groups have to be deprotected. The chemistry of eldecalcitol allows for its binding 2.7-fold more potently than calcitriol. In addition, some vitamin D derivatives have been known to inhibit the serum parathyroid hormone. Eldecalcitol only weakly inhibits the serum parathyroid hormone, making it an even more appealing medicinal drug for its physiological uses in the treatment of osteoporosis.[3] Animal studies of eldecalcitol, in ovariectomized rats, show improvements in bone mass while lowering bone reabsorption to demonstrate its effectiveness in osteoporosis treatment.[5]
PAPER
Heterocycles, Vol 92, No. 6, 2016, pp.1013-1029
Published online, 22nd March, 2016
Noboru Kubodera*
*International Institute of Active Vitamin D Analogs, 35-6, Sankeidai, Mishima, Shizuoka 411-0017, Japan
Presented herein are diverse and important contributions by medicinal chemists to different stages of pharmaceutical development. The conceptual elements reviewed, which are intended for young chemists who engage in drug discovery research, draw upon the author’s experience in developing eldecalcitol, an active vitamin D3 analog used to treat osteoporosis. The review covers exploratory research for a lead candidate compound; process development for practical manufacturing; and synthesis of other compounds relevant to the program, such as tritiated compounds, postulated metabolites, and miscellaneous analogs for mode of action studies.
PAPER
Eldecalcitol [1α,25-dihydroxy-2β-(3-hydroxypropoxy)vitamin D3], an analog of calcitriol (1α,25-dihydroxyvitamin D3), possesses a hydroxypropoxy substituent at the 2β-position of calcitriol. Eldecalcitol has potent biological effects on bone disease such as osteoporosis. The marketing of eldecalcitol has very recently started in Japan. In consideration of this, we have been investigating practical synthesis of eldecalcitol for industrial-scale production. Eldecalcitol was initially synthesized in a linear manner. The 27-step linear sequence was, however, suboptimal due to its lengthiness and low overall yield (ca. 0.03%). Next, we developed a convergent approach based on the Trost coupling reaction, in which the A-ring fragment (ene-yne part obtained in 10.4% overall yield) and the C/D-ring fragment (bromomethylene part obtained in 27.1% overall yield) are coupled to produce the triene system of eldecalcitol (15.6%). Although the overall yield of the convergent synthesis was better than that of the linear synthesis, significant improvements were still necessary. Therefore, additional biomimetic studies were investigated. Process development for the practical production of eldecalcitol is described herein.
http://ar.iiarjournals.org/content/32/1/303/F3.expansion.html
Convergent synthesis of eldecalcitol (5) by coupling A-ring fragment 37 with C/D-ring fragment 40. Reagents and conditions: a: HO(CH2)3OH/t-BuOK, 120°C. b: t-BuCOCl/pyridine/CH2Cl2, rt. c: H2/Pd(OH)2/MeOH, rt. d: Me2C(OMe)2/TsOH/acetone, rt. e: DMSO/(COCl)2/CH2Cl2, −60°C. f: CH2=CHMgBr/THF, −60°C. g: t-BuCOCl/Et3N/DMAP/CH2Cl2, rt. h: 1 M HCl/MeOH, rt. i: Ph3P/DEAD/benzene, reflux. j: LiC ≡ CTMS/BF3-OEt2, −78°C. k: 10 N NaOH/MeOH, rt. l: TBSOTf/Et3N/CH2Cl2, 0°C. m: TESOTf/Et3N/CH2Cl2, 0°C. n: O3/CH2Cl2/MeOH, −78°C then NaBH4/MeOH, −78°C. o: NMO/TPAP/4Ams/CH2Cl2, rt. p: Ph3P+CH2BrBr−/NaHMDS/ THF, −60°C to rt. q: (dba)3Pd2-CHCl3/PPh3/Et3N/toluene, reflux. r: TBAF/THF/toluene, reflux.

Industrial synthesis of alfacalcidol (4) and biomimetic synthesis of eldecalcitol (5) from cholesterol (42). Reagents and conditions: a: [Al(Oi-Pr)3]/cyclohexanone. b: DDQ/AcOEt. c: NaOEt/EtOH. d: NaBH4/MeOH/THF. e: Ac2O/DMPA/pyridine, rt. f: NBS/AIBN/n-hexane, reflux. g: γ-collidine/toluene, reflux. h: KOH/MeOH, rt. i: PTAD/CH2Cl2, rt. j: TBSCl/imidazole. k: MCPBA/CH2Cl2. l: DMI, 140°C. m: TBAF/THF. n: NaBH4/EtOH. o: 400 W high pressure mercury lamp/THF, 0°C then reflux without mercury lamp. p: HO(CH2)3OH/t-BuOK, 110°C. q: Microbial 25-hydroxylation.

Reference:1. US4666634A / EP184206A2.

Reference:1. Anticancer. Res. 2012, 32, 303-310.
2. Drugs. Fut. 2005, 30, 450-461.

Reference:1. J. Am. Chem. Soc. 2001, 123, 3716-3722.

Reference:1. Bioorg. Med. Chem. Lett. 1997, 7, 2871-2874.
2. Anticance. Res. 2009, 29, 3571-3578.

Reference:1. EP0503630A1.
2. Drugs Fut. 2005, 30, 450-461.

Reference:1. Bioorg. Med. Chem. 1998, 6, 2517-2523.
| No. | Major Technical Classification | Publication No. | Patent No. | Legal Status | Filling Date | Estimated Expiry Date |
| 1 | Preparation | CN85108857A | CN1008368B | Granted/expired | 1985/12/4 | 2005/12/4 |
| 2 | Crystal | CN1223639A | CN1216861C | Granted | 1997/6/16 | 2017/6/16 |
| 3 | Preparation | CN1637017A | CN1276927C |
| Patent ID | Date | Patent Title |
|---|---|---|
| US7927613 | 2011-04-19 | Pharmaceutical co-crystal compositions |
| US7323580 | 2008-01-29 | CRYSTALS OF A VITAMIN D DERIVATIVE AND A METHOD FOR THE PREPARATION THEREOF |
| US7235679 | 2007-06-26 | Crystals of a vitamin D derivative and a method for the preparation thereof |
| EP0924199 | 2006-05-10 | CRYSTALS OF VITAMIN D DERIVATIVES AND PROCESS FOR THE PREPARATION THEREOF |
| US2005009794 | 2005-01-13 | Crystals of a vitamin D derivative and a method for the preparation thereof |
| US6831183 | 2004-12-14 | Crystals of a vitamin D derivative and a method for the preparation thereof |
| US6448421 | 2002-09-10 | CRYSTALS OF VITAMIN D DERIVATIVES AND PROCESS FOR THE PREPARATION THEREOF |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(1S,2S,3S,5Z,7E)-2-(3-Hydroxypropoxy)-9,10-secocholesta-5,7,10-triene-1,3,25-triol
|
|
| Clinical data | |
| Trade names | Edirol |
| Identifiers | |
| CAS Number | 104121-92-8 |
| ATC code | None |
| PubChem | CID 6438982 |
| ChemSpider | 4943418 |
| Chemical data | |
| Formula | C30H50O5 |
| Molar mass | 490.715 g/mol |
///////////eldecalcitol, active vitamin D3 analog, treat osteoporosis, AC1O5QQ2, 104121-92-8, AN-3697, ED 71, ED-71, Edirol®, PMDA, JAPAN
O[C@H]1CC(\C(=C)[C@H](O)[C@H]1OCCCO)=C\C=C2/CCC[C@]3([C@H]2CC[C@@H]3[C@H](C)CCCC(O)(C)C)C
OR
CC(CCCC(C)(C)O)C1CCC2C1(CCCC2=CC=C3CC(C(C(C3=C)O)OCCCO)O)C

Rifaximin;
Rifaxidin; Rifacol; Xifaxan; Normix; Rifamycin L 105;L 105 (ansamacrolide antibiotic), L 105SV
(2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro[4,5-e]pyrido[1,2-á]-benzimidazole-1,15(2H)-dione,25-acetate
CAS 80621-81-4, 4-Deoxy-4-methylpyrido[1,2-1,2]imidazo[5,4-c]rifamycin SV,
4-Deoxy-4′-methylpyrido[1′,2′-1,2]imidazo[5,4-c]rifamycin SV, Rifacol
| C43H51N3O11 | |
| Molecular Weight: | 785.87854 g/mol |
|---|

XIFAXAN tablets for oral administration are film-coated and contain 200 mg or 550 mg of rifaximin.
Rifaximin is an orally administered, semi-synthetic, nonsystemic antibiotic derived from rifamycin SV with antibacterial activity. Rifaximin binds to the beta-subunit of bacterial DNA-dependent RNA polymerase, inhibiting bacterial RNA synthesis and bacterial cell growth. As rifaximin is not well absorbed, its antibacterial activity is largely localized to the gastrointestinal tract.
Rifaximin (trade names:RCIFAX, Rifagut, Xifaxan, Zaxine) is a semisynthetic antibiotic based on rifamycin. It has poor oral bioavailability, meaning that very little of the drug will be absorbed into the blood stream when it is taken orally. Rifaximin is used in the treatment of traveler’s diarrhea, irritable bowel syndrome, and hepatic encephalopathy, for which it receivedorphan drug status from the U.S. Food and Drug Administration in 1998.
Rifaximin is licensed by the U.S. Food and Drug Administration to treat traveler’s diarrhea caused by E. coli.[1] Clinical trials have shown that rifaximin is highly effective at preventing and treating traveler’s diarrhea among travelers to Mexico, with fewside effects and low risk of developing antibiotic resistance.[2][3][4] It is not effective against Campylobacter jejuni, and there is no evidence of efficacy against Shigella or Salmonella species.
| Launched – 1988 | Alfa Wassermann | Infection, bacterial |
| Launched – 2004 | Salix | Traveler’s diarrhea |
| Launched – 2010 | Salix | Encephalopathy, hepatic |
| Launched – 2015 | Salix | Irritable bowel syndrome (Diarrhea predominant) |
| Launched | Alfa Wassermann Merck & Co. |
Hyperammonemia |
The drug is also at Salix in phase II trials for the treatment of Crohn’s disease. Alfa Wasserman is also conducting phase II trials for Crohn’s disease. The product was approved and launched in the U.S. for the maintenance of remission of hepatic encephalopathy in 2010. Mayo Clinic is conducting phase II clinical trials in the U.S. for the treatment of primary sclerosing cholangitis and the University of Hong Kong is also conducting Phase II trials for the treatment of functional dyspepsia.
It may be efficacious in relieving chronic functional symptoms of bloating and flatulence that are common in irritable bowel syndrome (IBS),[5][6] especially IBS-D.
In February 1998, Salix was granted orphan drug designation by the FDA for the use of rifaximin to treat hepatic encephalopathy. In 2009, a codevelopment agreement was established between Lupin and Salix in the U.S. for the development of a new formulation using Lupin’s bioadhesive drug delivery technology.
There was recentlya pilot-study done on the efficacy of rifaximin as a means of treatment for rosacea, according to the study, induced by the co-presence of small intestinal bacterial overgrowth.[7]
In the United States, rifaximin has orphan drug status for the treatment of hepatic encephalopathy.[8] Although high-quality evidence is still lacking, rifaximin appears to be as effective as or more effective than other available treatments for hepatic encephalopathy (such as lactulose), is better tolerated, and may work faster.[9] Hepatic encephalopathy is a debilitating condition for those with liver disease. Rifaximin is an oral medication taken twice daily that helps patients to avoid reoccurring hepatic encephalopathy. It has minimal side effects, prevents reoccurring encephalopathy and high patient satisfaction. Patients are more compliant and satisfied to take this medication than any other due to minimal side effects, prolong remission, and overall cost.[10] Rifaximin helps patients avoid multiple readmissions from hospitals along with less time missed from work as well. Rifaximin should be considered a standard prescribed medication for those whom have episodes of hepatic encephalopathy.
The drawbacks to rifaximin are increased cost and lack of robust clinical trials for HE without combination lactulose therapy.
Also treats hyperammonemia by eradicating ammoniagenic bacteria.
Rifaximin interferes with transcription by binding to the β-subunit of bacterial RNA polymerase.[11] This results in the blockage of the translocation step that normally follows the formation of the first phosphodiester bond, which occurs in the transcription process.[12]
Efficacy
A 2011 study in patients with IBS (sans constipation) indicated 11% showed benefits over a placebo.[13] The study was supported by Salix Pharmaceuticals, the patent holder.[13] A 2010 study in patients treated for Hepatic Cirrhosis with hospitalization involving Hepatic encephalopathy resulted in 22% of the rifaxmin treated group experiencing a breakthrough episode of Hepatic encephalopathy as compared to 46% of the placebo group. The majority patients were also receivingLactulose therapy for prevention of hepatic encephalopathy in addition to Rifaximin.[14] Rifaximin shows promising results, causing remission in up to 59% of people with Crohn’s disease and up to 76% of people with Ulcerative Colitis.[15]
In the United States, Salix Pharmaceuticals holds a US Patent for rifaximin and markets the drug under the name Xifaxan, available in tablets of 200 mg and 550 mg.[16][17] In addition to receiving FDA approval for traveler’s diarrhea and (marketing approved for)[17] hepatic encephalopathy, Xifaxan received FDA approval for IBS in May 2015.[18] No generic formulation is available in the US and none has appeared due to the fact that the FDA approval process was ongoing. If Xifaxan receives full FDA approval for hepatic encephalopathy it is likely that Salix will maintain marketing exclusivity and be protected from generic formulations until March 24, 2017.[17] Price quotes received on February 21, 2013 for Xifaxan 550 mg in the Denver Metro area were between $23.57 and $26.72 per tablet. A price quote received on June 24, 2016 for Xifaxan 550 mg was $31.37 per tablet.
Rifaximin is approved in 33 countries for GI disorders.[19][20] On August 13, 2013, Health Canada issued a Notice of Compliance to Salix Pharmaceuticals Inc. for the drug product Zaxine.[21] In India it is available under the brand names Ciboz and Xifapill.[
SPECTRA
LINK IS CLICK
APT 13C NMR RIFAXIMIN
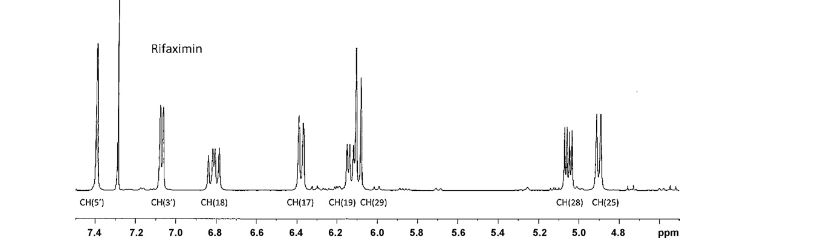
1H NMR PARTIAL
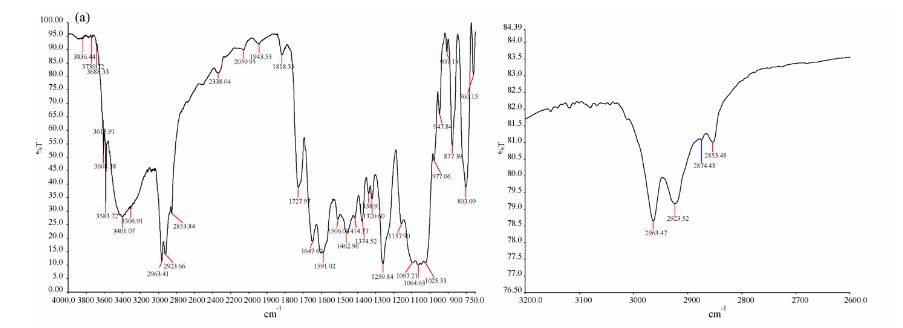
IR
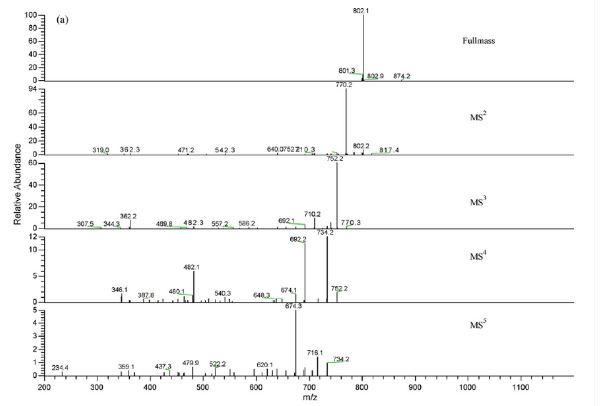
Direct infusion mass analysis ESI (+)
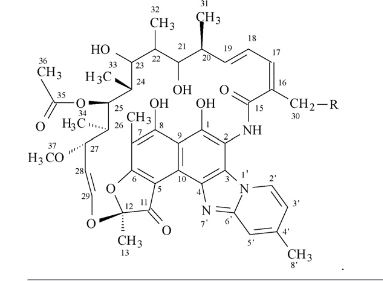
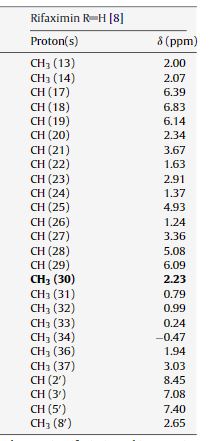
IH NMR
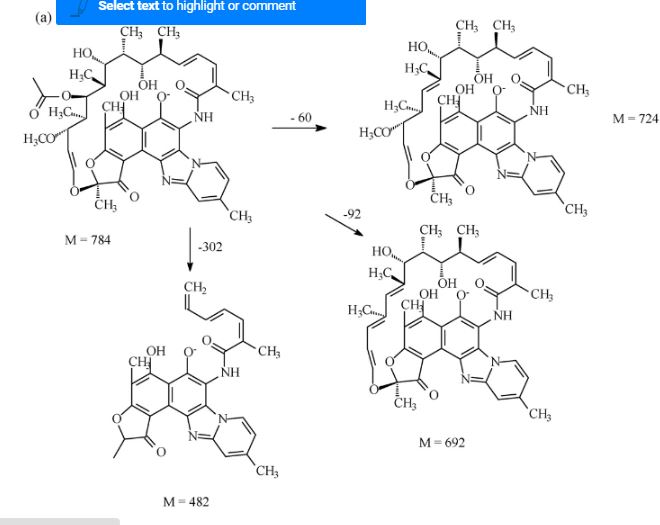
Synthesis
Rifaximin is a broad-spectrum antibiotic belonging to the family of Rifamycins and shows its antibacterial activity, in the gastrointestinal tract against localized bacteria that cause infectious diarrhoea, irritable bowel syndrome, small intestinal bacterial overgrowth, Crohn’s disease, and/or pancreatic insufficiency.
Rifaximin is sold under the brand name Xifaxan® in US for the treatment of Travellers’ diarrhoea and Hepatic Encephalopathy. The chemical name of Rifaximin is (2S , 16Z, 18E,20S ,21 S ,22R,23R,24R,25S ,26S ,27S ,28E)-5,6,21 ,23 ,25-pentahydroxy-27-methoxy-2,4,1 l,16,20,22,24,26-octamethyl-2,7(epoxypentadeca-[l,l l,13]trienimino) benzofuro[4,5-e]pyrido[l,2-a]-benzimidazole-l,15(2H)-dione,25-acetate and the molecular formula is G^HsiNsOn with a molecular weight of 785.9. The structural formula of Rifaximin is:

Formula I
Rifaximin was first described and claimed in Italian patent IT 1154655 and U.S. Pat. No.4,341,785. These patents disclose a process for the preparation of Rifaximin and a method for the crystallisation thereof. The process for the preparation of Rifaximin is as depicted in scheme I given below:

Scheme -I
U.S. Pat. No. 4,179,438 discloses a process for the preparation of 3-bromorifamycin S which comprises reaction of rifamycin S with at least two equivalents of bromine, per one mole of rifamycin S in the presence of at least one mole of pyridine per each equivalent of bromine and in the presence of ethanol, methanol or mixtures thereof with water at a
temperature not above the room temperature. The process is shown in the scheme given below:

Rifamycin S 3-Bromo-Rifamycin-S
U.S. Patent No.4,557, 866 discloses a process for one step synthesis of Rifaximin from Rifamycin O, which is shown in scheme II given below:

Rifamycin O Rifaximin
Scheme -II
US ‘866 patent also discloses purification of Rifaximin by performing crystallization of crude Rifaximin from a 7:3 mixture of ethyl alcohol/water followed by drying both under atmospheric pressure and under vacuum. The crystalline form which is obtained has not been characterized.
U.S. Patent No. 7,045,620 describes three polymorphic forms α, β and γ of Rifaximin. Form a and β show pure crystalline characteristics while the γ form is poorly crystalline. These polymorphic forms are differentiated on the basis of water content and PXRD. This patent also discloses processes for preparation of these polymorphs which involve use of specific reaction conditions during crystallization like dissolving Rifaximin in ethyl alcohol at 45-65°C, precipitation by adding water to form a suspension, filtering suspension and washing the resulted solid with demineralized water, followed by drying at room temperature under vacuum for a period of time between 2 and 72 hours. Crystalline forms a and β are obtained by immediate filtration of suspension when temperature of reaction mixture is brought to 0°C and poorly crystalline form γ is obtained when the reaction mixture is stirred for 5-6 hours at 0°C and then filtered the suspension. In addition to above these forms are also characterized by specific water content. For a form water content should be lower than 4.5%, for β form it should be higher than 4.5% and to obtain γ form, water content should be below 2%.
U.S. Patent No. 7,709,634 describes an amorphous form of Rifaximin which is prepared by dissolving Rifaximin in solvents such as alkyl esters, alkanols and ketones and precipitating by addition of anti-solvents selected from hydrocarbons, ethers or mixtures thereof.
U.S. Patent No. 8,193,196 describes two polymorphic forms of Rifaximin, designated δ and ε respectively. Form δ has water content within the range from 2.5 to 6% by weight (preferably from 3 to 4.5%).
U.S. Patent No 8,067,429 describes a-dry, β-1, β-2, ε-dry and amorphous forms of Rifaximin.
U.S. Patent No. 8,227,482 describes polymorphs Form μ, Form π, Form Omicron, Form Zeta, Form Eta, Form Iota and Form Xi of Rifaximin.
International application publications WO 2008/035109, WO 2008/155728, WO 2012/035544, WO 2012/060675, and WO 2012/156533 describes various amorphous or poorly crystalline forms of Rifaximin.
These polymorphic forms are obtained under different experimental conditions and are characterized by XRPD pattern.
The polymorphic forms of Rifaximin obtained from the prior art methods have specific water content. Transition between different polymorphic forms of Rifaximin occurs by drying or wetting of the synthesized Rifaximin. Hence, it is evident from above that Rifaximin can exist in number of polymorphic forms, formation of these polymorphic forms depends upon specific reaction conditions applied during crystallization and drying.
Rifaximin is a semi-synthetic, rifamycin-based non-systematic antibiotic. It is chemically termed as (2S,16Z,18E,20S,21S,22R,23R,24R,25S,26 S,27S, 28E)-5,6,21,23,255-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro[4,5-e]pyrido[1,2-a]-benzimida-zole-1,15(2H)-dione,25-acetate (I).

Rifaximin is used for treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli.
Rifaximin was first disclosed in US4341785 which also discloses a process for its preparation and a method for crystallization of rifaximin using suitable solvents or mixture of solvents. However, this patent does not mention the polymorphism of rifaximin.
Canadian patent CA1215976 discloses a process for the synthesis of imidazo rifamycins which comprises reacting rifamycin S with 2-amino-4-methyl pyridine.
US4557866 discloses a process for preparation of rifaximin, but does not mention the polymorphs of rifaximin.
US7045620 discloses crystalline polymorphic forms of rifaximin which are termed as rifaximin α, rifaximin β and rifaximin γ. These polymorphic forms are characterized using X-ray powder diffraction. Further this patent mentions that γ form is poorly crystalline with a high content of amorphous component. This patent also discloses processes for preparation of these polymorphs which involve use of processes of crystallization and drying as disclosed in US4557866along with control of temperature at which the product is crystallized, drying process, water content thereof. Further, according to this patent, crystal formation depends upon the presence of water within the crystallization solvent.
The above patent discloses rifaximin α which is characterized by water content lower than 4.5% & powder X-ray diffractogram having significant peaks are at values of diffraction angles 2θ of 6.6°; 7.4°; 7.9°, 8.8°, 10.5°, 11.1 °, 11.8°, 12.9°, 17.6°, 18.5°, 19.7°, 21.0°, 21.4°, 22.1°; rifaximin β which is characterized by water content higher than 4.5% & powder X-ray diffractogram having significant peaks are at values of diffraction angles 2θ of 5.4°; 6.4°; 7.0°, 7.8°, 9.0°, 10.4°, 13.1°, 14.4°, 17.1°, 17.9°, 18.3°, 20.9° and rifaximin γ which is characterized by poorer powder X-ray diffractogram because of poor crystallinity. The significant peaks are at values of diffraction angles 2θ of 5.0°; 7.1°; 8.4°.
US2005/0272754 also discloses polymorphs of rifaximin namely rifaximin α form, rifaximin β form & rifaximin γ form characterized by powder X-ray diffractogram, intrinsic dissolution rates and processes of preparation of polymorphic forms of rifaximin. However, none of the above patents disclose a wholly amorphous form of rifaximin.
It is a well known fact that different polymorphic forms of the same drug may have substantial differences in certain pharmaceutically important properties. The amorphous form of a drug may exhibit different dissolution characteristics and in some case different bioavailability patterns compared to crystalline forms.
Further, amorphous and crystalline forms of a drug may have different handling properties, dissolution rates, solubility, and stability.
Furthermore, different physical forms may have different particle size, hardness and glass transition temperatures. Amorphous materials do not exhibit the three-dimensional long-range orders found in crystalline materials, but are structurally more similar to liquids where the arrangement of molecules is random.
Amorphous solids do not give a definitive x-ray diffraction pattern (XRD). In addition, amorphous solids do not give rise to a specific melting point and tend to liquefy at some point beyond the glass transition temperature. Because amorphous solids do not have lattice energy, they usually dissolve in a solvent more rapidly and consequently may provide enhanced bioavailability characteristics such as a higher rate and extent of absorption of the compound from the gastrointestinal tract. Also, amorphous forms of a drug may offer significant advantages over crystalline forms of the same drug in the manufacturing process of solid dosage form such as compressibility.
PATENT
https://www.google.com/patents/EP2069363B1?cl=e
The schematic representation for preparation of amorphous rifaximin is as follows :

Amorphous rifaximin according to the present invention can be characterized by various parameters like solubility, intrinsic dissolution, bulk density, tapped density.
Rifaximin is known to exist in 3 polymorphic Forms namely α Form, β Form & γ Form of which the α Form is thermodynamically the most stable. Hence, the amorphous form of rifaximin was studied in comparison with α Form.
Further, when intrinsic dissolution of amorphous rifaximin is carried out against the α Form, it is observed that the amorphous rifaximin has better dissolution profile than α Form which is shown in table below (this data is also shown graphically in Figure 3):
Dissolution medium : 1000 ml of 0.1M Sodium dihydrogen phosphate monohydrate + 4.5g of sodium lauryl sulphate
Temperature : 37±0.5°C
Rotation speed : 100 rpm
Particle size : Amorphous rifaximin – 11 microns
α Form of rifaximin – 13 microns
| Time in minutes | % Release of Amorphous Rifaximin | % Release of α Form of Rifaximin |
| 15 | 1.1 | 0.8 |
| 30 | 1.9 | 1.8 |
| 45 | 2.9 | 3.0 |
| 60 | 3.7 | 4.4 |
| 120 | 8.1 | 11.0 |
| 180 | 12.6 | 18.0 |
| 240 | 16.6 | 24.6 |
| 360 | 24.7 | 38.7 |
| 480 | 32.0 | 47.5 |
| 600 | 39.5 | 52.7 |
| 720 | 46.4 | 56.4 |
| 960 | 60.4 | 62.9 |
| 1200 | 72.9 | 67.8 |
| 1400 | 83.0 | 72.7 |
CLIP
Rifaximin (CAS NO.: 80621-81-4), with other name of 4-Deoxy-4-methylpyrido[1,2-1,2]imidazo[5,4-c]rifamycin SV, could be produced through many synthetic methods.
Following is one of the reaction routes:

The reaction of rifamycin S (I) with pyridine perbromide (II) in 2-propanol/chloroform (70/30) mixture at 0 C gives 3-bromorifamicin S (III), which is then condensed with 2-amino-4-methyl-pyridine (IV) at 10 C. The o-quinoniminic compound (V) is then obtained. This compound is finally reduced with ascorbic acid.
POLYMORPHISM
Rifaximin (INN; see The Merck Index, XIII Ed., 8304) is an antibiotic belonging to the rifamycin class, exactly it is a pyrido-imidazo rifamycin described and claimed in Italian Patent IT 1154655, while European Patent EP 0161534 describes and claims a process for its production starting from rifamycin O (The Merck Index, XIII Ed., 8301).
Both these patents describe the purification of rifaximin in a generic way stating that crystallization can be carried out in suitable solvents or solvent systems and summarily showing in some examples that the reaction product can be crystallized from the 7:3 mixture of ethyl alcohol/water and can be dried both under atmospheric pressure and under vacuum without specifying in any way either the experimental conditions of crystallization and drying, or any distinctive crystallographic characteristic of the obtained product.
The presence of different polymorphs had just not been noticed and therefore the experimental conditions described in both patents had been developed with the goal to get a homogeneous product having a suitable purity from the chemical point of view, independent from the crystallographic aspects of the product itself.
It has now been found, unexpectedly, that there are several polymorphous forms whose formation, besides the solvent, depends on time and temperature conditions under which both crystallization and drying are carried out.
In the present application, these orderly polymorphous forms will be, later on, conventionally identified as rifaximin α (FIG. 1) and rifaximin β (FIG. 2) on the basis of their respective specific diffractograms, while the poorly crystalline form with a high content of amorphous component will be identified as rifaximin γ (FIG. 3).
Rifaximin polymorphous forms have been characterized through the technique of the powder X-ray diffraction.
The identification and characterization of these polymorphous forms and, simultaneously, the definition of the experimental conditions for obtaining them is very important for a compound endowed with pharmacological activity which, like rifaximin, is marketed as medicinal preparation, both for human and veterinary use. In fact it is known that the polymorphism of a compound that can be used as active ingredient contained in a medicinal preparation can influence the pharmaco-toxicologic properties of the drug. Different polymorphous forms of an active ingredient administered as drug under oral or topical form can modify many properties thereof like bioavailability, solubility, stability, colour, compressibility, flowability and workability with consequent modification of the profiles of toxicological safety, clinical effectiveness and productive efficiency.
What mentioned above is confirmed by the fact that the authorities that regulate the grant of marketing authorization of the drugs market require that the manufacturing methods of the active ingredients are standardized and controlled in such a way that they give homogeneous and sound results in terms of polymorphism of production batches (CPMP/QWP/96, 2003—Note for Guidance on Chemistry of new Active Substance; CPMP/ICH/367/96—Note for guidance specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances; Date for coming into operation: May 2000).
The need for the above-mentioned standardization has further been strengthened in the field of the rifamycin antibiotics by Henwood S. Q., de Villiers M. M., Liebenberg W. and Lotter A. P., Drug Development and Industrial Pharmacy, 26 (4), 403-408, (2000), who have ascertained that different production batches of the rifampicin (INN) made from different manufacturers differ from each other in that they show different polymorphous characteristics, and as a consequence they show different dissolution profiles, along with a consequent alteration of the respective pharmacological properties.
By applying the crystallization and drying processes generically disclosed in the previous patents IT 1154655 and EP 0161534 it has been found that under some experimental conditions a poorly crystalline form of rifaximin is obtained, while under other experimental conditions other polymorphic crystalline forms of Rifaximin are obtained. Moreover it has been found that some parameters, absolutely not disclosed in the above-mentioned patents, like for instance preservation conditions and the relative ambient humidity, have the surprising effect to determine the polymorph form.
The polymorphous forms of rifaximin object of the present patent application were never seen or hypothesized, while thinking that, whichever method was used within the range of the described condition, a sole homogeneous product would always have been obtained, irrespective of crystallizing, drying and preserving conditions. It has now been found that the formation of α, β and γ forms depends both on the presence of water within the crystallization solvent, on the temperature at which the product is crystallized and on the amount of water present in the product at the end of the drying phase. Form α, form β and form γ of rifaximin have then been synthesized and they are the object of the invention.
Moreover it has been found that the presence of water in rifaximin in the solid state is reversible, so that water absorption and/or release can take place in time in presence of suitable ambient conditions; consequently rifaximin is susceptible of transition from one form to another, also remaining in the solid state, without need to be again dissolved and crystallized. For instance polymorph α, getting water by hydration up to a content higher than 4.5%, turns into polymorph β, which in its turn, losing water by drying up to a content lower than 4.5%, turns into polymorph α.
These results have a remarkable importance as they determine the conditions of industrial manufacturing of some steps of working which could not be considered critical for the determination of the polymorphism of a product, like for instance the washing of a crystallized product, or the preservation conditions of the end product, or the characteristics of the container in which the product is preserved.
The above-mentioned α, β and γ forms can be advantageously used as pure and homogeneous products in the manufacture of medicinal preparations containing rifaximin.
As already said, the process for manufacturing rifaximin from rifamycin O disclosed and claimed in EP 0161534 is deficient from the point of view of the purification and identification of the product obtained; it shows some limits also from the synthetic point of view as regards, for instance, the very long reaction times, from 16 to 72 hours, not very suitable to an industrial use and moreover because it does not provide for the in situ reduction of rifaximin oxidized that may be formed within the reaction mixture.
Therefore, a further object of the present invention is an improved process for the industrial manufacturing of the α, β and γ forms of rifaximin, herein claimed as products and usable as defined and homogeneous active ingredients in the manufacture of the medicinal preparations containing such active ingredient.
PATENT
https://www.google.com/patents/US20090234114
FIG. 1 is a powder X-ray diffractogram of rifaximin polymorphic form α.
FIG. 2 is a powder X-ray diffractogram of rifaximin polymorphic form β.
FIG. 3 is a powder X-ray diffractogram of rifaximin polymorphic form γ.

Rifaximin (INN; see The Merck Index, XIII Ed., 8304, CAS no. 80621-81-4), IUPAC nomenclature (2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25 pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-(1,11,13)trienimino)benzofuro(4,5-e)pyrido(1,2,-a)benzimidazole-1,15(2H)-dione,25-acetate) is a semi-synthetic antibiotic belonging to the rifamycin class of antibiotics. More precisely rifaximin is a pyrido-imidazo rifamycin described in the Italian patent IT 1154655, whereas the European patent EP 0161534 discloses a process for rifaximin production using rifamycin O as starting material (The Merck Index, XIII Ed., 8301).
U.S. Pat. No. 7,045,620, US 2008/0262220, US 7,612,199, US 2009/0130201 and Cryst. Eng. Comm., 2008, 10 1074-1081 (2008) disclose new forms of rifaximin.
WO 2008/035109 A1 discloses a process to prepare amorphous rifaximin, which comprises reaction of rifamycin S with 2-amino-4 picoline in presence of organic solvent like dichloromethane, ethylacetate, dichloroethylene, chloroform, in an inert atmosphere. When water is added to the reaction mixture, a solid precipitate corresponding to amorphous rifaximin is obtained.
The process described in this document can be assimilated to a crash precipitation, wherein the use of an anti-solvent causes the precipitation of rifaximin without giving any information about the chemical physical and biological characteristics of the rifaximin obtained.
WO 2009/108730 A2 describes different polymorphous forms of rifaximin and also amorphous forms of rifaximin. Amorphous forms are prepared by milling and crash precipitation and with these two different methods the amorphous rifaximin obtained from these two different processes has the same properties.

FIG. 5: FT-IR spectrum of rifaximin obtained by spray drying process.

Patent
Rifaximin, lUPAC name:
(2S,16Z,18E,20S,21 S,22H,23H,24H,25S,26S,27S,28£)-5,6,21 ,23,25-pentahydroxy- 27-methoxy-2,4,1 1 ,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1 ,1 1 ,13]-trienimmino)-benzofuro-[4,5-e]-pirido-[1 ,2-oc]-benzimidazol-1 , 15(2 -/)-dione,25-acetate, is the compound of formula (I):

Rifaximin is a broad-spectrum antibiotic belonging to the family of rifamycins, devoid of systemic activity. In view of its physicochemical properties, it is not adsorbed in the gastrointestinal tract and therefore exerts its antimicrobial action inside the gastrointestinal tract. Rifaximin therefore has applications in the treatment of diarrhoea and of microbial infections of the gastrointestinal tract typically caused by E. coli, a microorganism which, being incapable of passing through the mucosa of the gastrointestinal tract, remains in contact with the gastrointestinal fluids. Rifaximin also has applications for the treatment of irritable bowel syndrome, Crohn’s disease, diverticulitis and for antibiotic prophylaxis preceding surgical operations on the intestines.
Rifaximin was obtained and described for the first time in the EP161534 starting from rifamycin O and 2-amino-4-picoline in the presence of ethanol/water and
ascorbic acid/HCI to obtain raw rifaximin which is then treated with Ethanol/water to obtain crystallized rifaximin.
Polymorphic forms of rifaximin, and processes for their synthesis and purification, are described in various documents of the known art.
Rifaximin K was firstly described in WO2012/156951 . Such a crystalline form resulted to be more stable in the presence of humidity than the other known crystalline forms of rifaximin, thus enabling the storage, even for prolonged periods. Such a polymorph was obtained by a process starting from rifaximin comprising the following steps: -suspending or dissolving rifaximin in a 1 ,2-dimethoxyethane based solvent, recovering the product and drying to remove said 1 ,2-dimethoxyethane based solvent. In one of the embodiments of the invention 1 ,2-dimethoxyethane is used as the unique solvent of rifaximin, in other 1 ,2-dimethoxyethane is described as used in combination of n-heptane, methanol, acetonitrile, R-COO-R1 esters wherein R and R1 are independently C3-C6 alkyl radicals, and C3-C7 alkyl ketones, ethanol, isopropanol and water.
The synthesis of 4-deoxypyrido(1′,2′-1,2)imidazo(5,4-c)rifamycin SV derivatives
J Antibiot 1984, 37(12): 1611
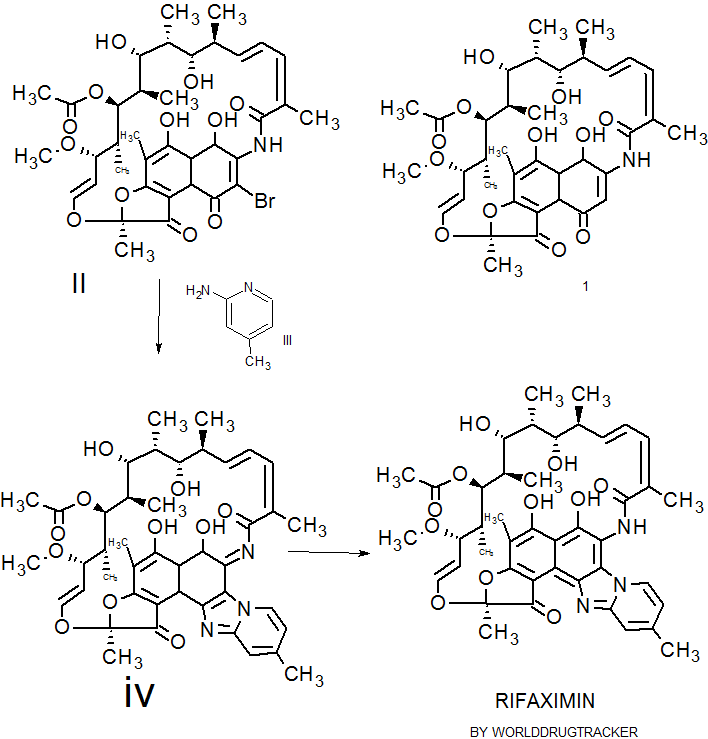
LAST STEP DEPICTED AGAIN
Treatment of rifamycin S (I) with Pyr·Br2 in 2-PrOH/CHCl3 gives 3-bromorifamycin S (II) (1), which upon cyclocondensation with 2-amino-4-methyl-pyridine (III) (1,2,3) in CHCl3 (2) or EtOH (3) yields imine derivative (IV). Finally, reduction of (IV) with L-(+)- ascorbic acid (1,2,3) in MeOH (2) or EtOH (3) provides the target rifaximin (1,2,3).
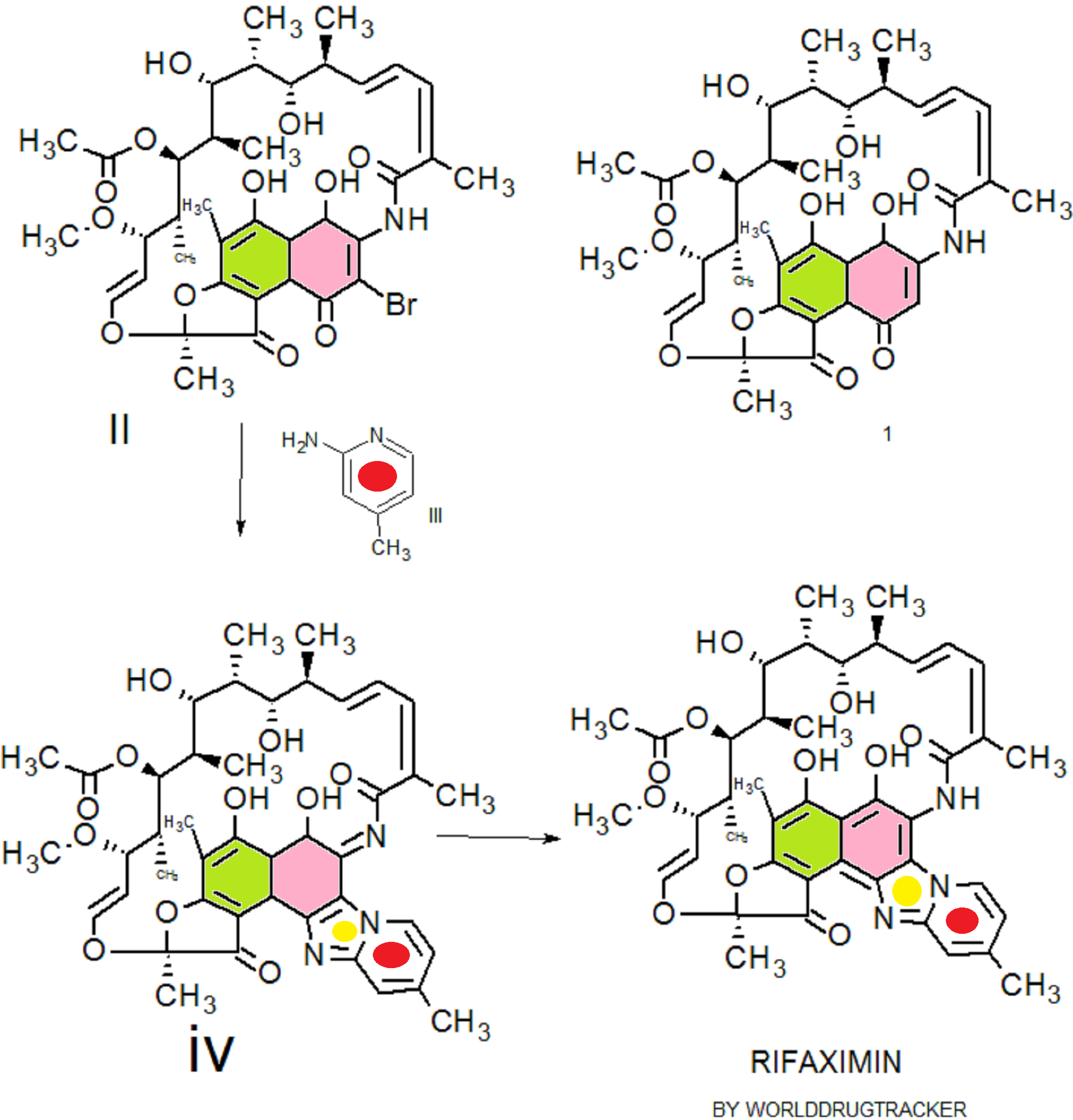
PATENT
WO 2005044823, WO 2012035544, WO 2015014984
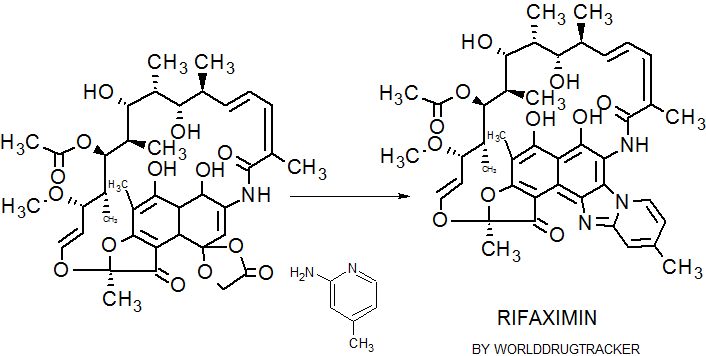
Rifaximin is prepared by the cyclocondensation of rifamycin-O with 2-amino-4-picoline in a solvent mixture such as acetone, acetonitrile, EtOH, MIBK, propylene glycol, i-PrOH or t-BuOH and H2O at 50 °C or EtOH/aceone/H2O or optionally in the presence of I2 in CH2Cl2
PATENT
The process is shown in the scheme given below:

Rifamycin-S
3-halo-Rifamycin-S

Examples
Example 1;
5g of Rifamycin S, 3.1 gms of 2-amino-4-methyl pyridine, 0.45 g of iodine, 1.65 ml of acetic acid and 20ml of acetonitrile were charged in a clean and dry round bottom flask followed by stirring the resultant reaction mixture at about 30°C for about 30 hours. The reaction progress was monitored by TLC, after completion of reaction, the reaction mass was quenched by adding a mixture of 4.0g of ascorbic acid dissolved in 20 ml of water. The resultant reaction suspension was stirred at about 25°C for about 15mins. 25 ml of dichloromethane was charged and stirred for about 15mins. followed by separation of organic and aqueous phases. The aqueous phase was extracted with 25 ml of dichloromethane followed by separation of organic and aqueous phases. The organic phases were combined and distilled at below about 50°C to yield Rifaximin as residue. 11.25ml of purified water and 11.25ml of ethanol were charged to the residue and stirred at about 30°C for about 15 mins. The resultant reaction
suspension was heated to about 75°C and stirred for about 30mins. The resultant reaction solution further cooled to about 25 °C and stirred for about 2 hours followed by further cooling to about 5°C for about 3 hours. The solid precipitated was filtered and the solid was washed with a mixture of 2.5ml of ethanol and 2.5 ml of purified water. The solid obtained was dried at about 50°C for about 10 hours to afford 3 g. of Rifaximin as crystalline form. Purity by HPLC: 99.85 area %.
PAPER
European journal of medicinal chemistry (2015), 103, 551-62
Patent
https://www.google.com/patents/WO2013027227A1?cl=en
Examples
Example 1 : Purification of Rifamycin S
Rifamycin S (500g) and Ethanol (1.5L) were stirred and refluxed for 1 hour. The reaction mixture was then cooled slowly to ambience, stirred at this temperature for 2 hour and filtered. The product dried in vacuum oven at 40 °C to obtain 475g of pure Rifamycin S showing the des acetyl impurity below to 0.6%.
Example 2: Preparation of rifaximin
Rifamycin S (300 g) was stirred in dichloromethane (900 ml) at room temperature for 15 minutes to get a clear solution and then 2-Amino-4-methyl pyridine (139.2g) was added at room temperature under nitrogen atmosphere. Iodine (57. Og) dissolved in dichloromethane (2100ml), was added drop wise in 30-45 minutes at room temperature. The reaction mass was stirred for 22-24 hours at 25-30 °C. After completion of the reaction, a 20% solution of L(-) ascorbic acid in water (300 ml) was added. The reaction mixture was stirred for 45-60 minutes at room temperature and then cooled to 10-15 °C. The pH of the resulting solution was adjusted to 1.5-2.0 with slow addition of dilute hydrochloric acid under stirring. The reaction mass was stirred for 15-20 minutes and layers were separated. The organic layer was washed with demineralized water (1500 ml), 10% sodium thiosulfate solution (1500 ml) and with demineralized water till pH was neutral. The solvent was distilled off under vacuum at 40-45 °C to get a residue which was taken in cyclohexane (1500 ml) and stirred for 1 hour. The resulting solid was filtered, washed with cyclohexane (300 ml) crystallized from a mixture of ethyl alcohol and water (600ml; 420ml ethyl alcohol and 180 ml water) to get 240g of crude rifaximin having purity 99.3% by HPLC.
Example 3: Preparation of rifaximin
Step-1: Preparation of crude rifaximin
Rifamycin S (300 g) was stirred in dichloromethane (900 ml) at room temperature for 15 minutes to get a clear solution and then 2-amino-4-methyl pyridine (139.2g) was added at room temperature under nitrogen atmosphere. Iodine (57. Og) dissolved in dichloromethane (2100ml), was added drop wise in 30-45 minutes at room temperature and was stirred for 22-24 hours. After completion of the reaction, a 20% solution of L (-) ascorbic acid in water (300 ml) was added and stirred for 45-60 minutes. The reaction mass was cooled to 10-15 °C and pH of the resulting solution was adjusted to 1.5-2.0 with slow addition of dilute hydrochloric acid under stirring. The reaction mass was stirred for 15-20 minutes and layers were separated and the organic layer was washed with demineralized water (1500 ml), with 10% sodium thiosulfate solution (1500 ml) and demineralized water till pH was neutral. The solvent was distilled off under vacuum at 40-45 °C to obtain a residue which was crystallized from a mixture of ethyl alcohol and water (378ml ethyl alcohol and 162 ml water) and dried at 35-40 °C to obtain 240g crude rifaximin having purity 98.8% by HPLC. Step-2: Purification of crude rifaximin
Crude rifaximin (240g) was stirred in dichloromethane (2400ml) at room temperature, a neutral alumina (240g) was added, stirred for 1 hour and filtered. The solvent was then distilled off and residue was treated with ethyl acetate (2400ml) and stirred to dissolution. The resulting residue was crystallized from a mixture of ethyl alcohol and water (302ml ethyl alcohol and 130ml water) and dried at 35-40 “C to obtain 192g of rifaximin having purity 99.8% by HPLC.
PATENT
https://www.google.com/patents/US9018225
PAPER

PATENTS
| US4341785 | May 11, 1981 | Jul 27, 1982 | Alfa Farmaceutici S.P.A. | Imidazo-rifamycin derivatives with antibacterial utility |
| US4557866 | Apr 26, 1985 | Dec 10, 1985 | Alfa Farmaceutici S.P.A. | Process for the synthesis of pyrido-imidazo rifamycins |
| US7045620 | Dec 5, 2003 | May 16, 2006 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in medicinal preparations |
| US7612199 | Jun 4, 2009 | Nov 3, 2009 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β, and γ of rifaximin |
| US7902206 | Mar 8, 2011 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β and γ of rifaximin | |
| US7906542 | May 13, 2008 | Mar 15, 2011 | Alfa Wassermann, S.P.A. | Pharmaceutical compositions comprising polymorphic forms α, β, and γ of rifaximin |
| US7915275 | Mar 29, 2011 | Alfa Wassermann, S.P.A. | Use of polymorphic forms of rifaximin for medical preparations | |
| US7923553 | Apr 12, 2011 | Alfa Wassermann, S.P.A. | Processes for the production of polymorphic forms of rifaximin | |
| US7928115 | Apr 19, 2011 | Salix Pharmaceuticals, Ltd. | Methods of treating travelers diarrhea and hepatic encephalopathy | |
| US8158644 | Apr 17, 2012 | Alfa Wassermann, S.P.A. | Pharmaceutical compositions comprising polymorphic forms α, β, and γ of rifaximin | |
| US8158781 | Mar 4, 2011 | Apr 17, 2012 | Alfa Wassermann, S.P.A. | Polymorphic forms α, β and γ of rifaximin |
| US8193196 | Feb 27, 2006 | Jun 5, 2012 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20050272754 * | May 24, 2005 | Dec 8, 2005 | Alfa Wassermann S.P.A. | Polymorphic forms of rifaximin, processes for their production and uses thereof |
| Reference | ||
|---|---|---|
| 1 | Viscomi, G. C., et al., “Crystal forms of rifaximin and their effect on pharmaceutical properties“, Cryst Eng Comm, 2008, 10, 1074-1081, (May 28, 2008), 1074-1081. | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US9186355 | Mar 30, 2015 | Nov 17, 2015 | Novel Laboratories | Rifaximin crystalline forms and methods of preparation thereof |
| WO2008035109A1 * | Sep 24, 2007 | Mar 27, 2008 | Cipla Limited | Rifaximin |
| WO2009108730A2 * | Feb 25, 2009 | Sep 3, 2009 | Salix Pharmaceuticals, Ltd. | Forms of rifaximin and uses thereof |
| WO2011080691A1 * | Dec 27, 2010 | Jul 7, 2011 | Silvio Massimo Lavagna | Method for the production of amorphous rifaximin |
| EP1698630A1 * | Mar 3, 2005 | Sep 6, 2006 | ALFA WASSERMANN S.p.A. | New polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20080262220 * | May 13, 2008 | Oct 23, 2008 | Giuseppe Claudio Viscomi | Polymorphic forms alpha, beta and gamma of rifaximin |
| US20090082558 * | Sep 20, 2007 | Mar 26, 2009 | Apotex Pharmachem Inc. | Amorphous form of rifaximin and processes for its preparation |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015014984A1 * | Aug 1, 2014 | Feb 5, 2015 | Clarochem Ireland Ltd. | A process for preparing rifaximin k |
| CN103360357A * | Aug 7, 2013 | Oct 23, 2013 | 中国药科大学 | A simvastatin-gliclazide co-amorphous compound |
| US9359374 | Jun 13, 2013 | Jun 7, 2016 | Apotex Pharmachem Inc. | Polymorphic forms of rifaximin |
| US4341785 * | May 11, 1981 | Jul 27, 1982 | Alfa Farmaceutici S.P.A. | Imidazo-rifamycin derivatives with antibacterial utility |
| US4557866 * | Apr 26, 1985 | Dec 10, 1985 | Alfa Farmaceutici S.P.A. | Process for the synthesis of pyrido-imidazo rifamycins |
| US7045620 * | Dec 5, 2003 | May 16, 2006 | Alfa Wassermann, S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in medicinal preparations |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8518949 | Jun 4, 2012 | Aug 27, 2013 | Alfa Wassermann S.P.A. | Polymorphous forms of rifaximin, processes for their production and use thereof in the medicinal preparations |
| US20140079783 * | Jul 3, 2013 | Mar 20, 2014 | Alfa Wassermann Spa | Pharmaceutical Compositions Comprising Rifaximin and Amino acids, Preparation Methods and Use Thereof |
| CN101836959A * | May 20, 2010 | Sep 22, 2010 | 山东达因海洋生物制药股份有限公司 | Method for preparing almost bitterless rifaximin dry suspension |
| CN103269587A * | Jun 3, 2011 | Aug 28, 2013 | 萨利克斯药品有限公司 | New forms of rifaximin and uses thereof |
| WO2011153444A1 * | Jun 3, 2011 | Dec 8, 2011 | Salix Pharmaceuticals, Ltd | New forms of rifaximin and uses thereof |
| Patents |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2S,16Z,18E,20S,21S,22R,23R,24R,25S,26S,27S,28E)-5,6,21,23,25-pentahydroxy-27-methoxy-2,4,11,16,20,22,24,26-octamethyl-2,7-(epoxypentadeca-[1,11,13]trienimino)benzofuro
[4,5-e]pyrido[1,2-a]-benzimida-zole-1,15(2H)-dione,25-acetate |
|
| Clinical data | |
| Trade names | Xifaxan, Xifaxanta, Normix, Rifagut |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a604027 |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | < 0.4% |
| Metabolism | Hepatic |
| Biological half-life | 6 hours |
| Excretion | Fecal (97%) |
| Identifiers | |
| CAS Number | 80621-81-4 |
| ATC code | A07AA11 (WHO) D06AX11(WHO) QG51AA06 (WHO)QJ51XX01 (WHO) |
| PubChem | CID 6436173 |
| DrugBank | DB01220 |
| ChemSpider | 10482302 |
| UNII | L36O5T016N |
| KEGG | D02554 |
| ChEBI | CHEBI:75246 |
| ChEMBL | CHEMBL1617 |
| Chemical data | |
| Formula | C43H51N3O11 |
| Molar mass | 785.879 g/mol |
Giuseppe Viscomi, Manuela Campana, Dario Braga, Donatella Confortini, Vincenzo Cannata, Paolo Righi, Goffredo Rosini, “Polymorphic forms of rifaximin, processes for their production and uses thereof.” U.S. Patent US20050272754, issued December 08, 2005.
///////Rifaximin, Rifaxidin, Rifacol, Xifaxan, Normix, Rifamycin L 105, 80621-81-4
CC1C=CC=C(C(=O)NC2=C(C3=C(C4=C(C(=C3O)C)OC(C4=O)(OC=CC(C(C(C(C(C(C1O)C)O)C)OC(=O)C)C)OC)C)C5=C2N6C=CC(=CC6=N5)C)O)C

TAK-243, AOB 87172, MLN-7243
CAS 1450833-55-2
Chemical Formula: C19H20F3N5O5S2
Molecular Weight: 519.5142
Sulfamic acid, [(1R,2R,3S,4R)-2,3-dihydroxy-4-[[2-[3-[(trifluoromethyl)thio]phenyl]pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]methyl ester
((lR,2R,3S,4R)-2,3-dihydroxy-4-(2-(3-(trifluoromethylthio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamate
methyl ((1S,2R,3S,4R)-2,3-dihydroxy-4-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)cyclopentyl)sulfamate
Phase I
Millennium Pharmaceuticals, Inc. INNOVATOR
Roushan AFROZE, Indu T. Bharathan,Jeffrey P. CIAVARRI, Paul E. Fleming,Jeffrey L. Gaulin, Mario Girard, Steven P. Langston, Francois R. SOUCY, Tzu-Tshin WONG, Yingchun Ye,
A UAE inhibitor potentially for the treatment of solid tumors.
TAK-243, also known as MLN7243 and AOB87172, is a small molecule inhibitor of ubiquitin-activating enzyme (UAE), with potential antineoplastic activity. UAE inhibitor MLN7243 binds to and inhibits UAE, which prevents both protein ubiquitination and subsequent protein degradation by the proteasome. This results in an excess of proteins in the cells and may lead to endoplasmic reticulum (ER) stress-mediated apoptosis. This inhibits tumor cell proliferation and survival. UAE, also called ubiquitin E1 enzyme (UBA1; E1), is more active in cancer cells than in normal, healthy cells.
![]()
Research Code TAK-243; MLN-7243, TAK-243; TAK 243; TAK243; MLN7243; MLN-7243; MLN 7243; AOB87172; AOB-87172; AOB 87172.
CAS No. 1450833-55-2(MLN 7243)
Cancer is the second most common cause of death in the U.S. and accounts for one of every eight deaths globally (American Cancer Society, Cancer Facts and Figures, 2014). The American Cancer Society expects that in 2014 at least 1,665,540 new cancer cases will be diagnosed in the US and 585,720 Americans are expected to die of cancer, almost 1 ,600 people per day. Currently available paradigms for treating solid tumors may include systemic treatment such as chemotherapy, hormonal therapy, use of targeted agents and biological agents, either as single agents or in combination. These treatments can be delivered in combination with localized treatments such as surgery or radiotherapy. These anti-cancer paradigms can be use in the curative setting as adjuvant or neo-adjuvant treatments or in the metastatic setting as palliative case for prolonged survival and to help manage symptoms and side-effects. In hematological cancers, stem cell transplants may also be an option in certain cancers as well as chemotherapy and/or radiation. Although medical advances have improved cancer survival rates, there remains a continuing need for new and more effective treatments.
Ubiquitin is a small 76-amino acid protein that is the founding member of a family of posttranslational modifiers known as the ubiquitin-like proteins (Ubls). Ubls play key roles in controlling many biological processes including cell division, cell signaling and the immune response. There are 8 known human Ubl activating enzymes (known as Els) (Schulman, B.A., and J.W. Harper, 2009, Ubiquitin-like protein activation by El enzymes: the apex for downstream signalling pathways, Nat Rev Mol Cell Biol 10:319-331). Ubiquitin and other Ubls are activated by a specific El enzyme which catalyzes the formation of an acyl-adenylate intermediate with the C-terminal glycine of the Ubl. The activated Ubl molecule is then transferred to the catalytic cysteine residue within the El enzyme through formation of a thioester bond intermediate. The El -Ubl intermediate and an E2 enzyme interact, resulting in a thioester exchange wherein the Ubl is transferred from the El to active site cysteine on the E2. The Ubl is then conjugated, i.e. transferred, to the target protein, either directly or in conjunction with an E3 ligase enzyme, through isopeptide bond formation with the amino group of a lysine side chain in the target protein. Eukaryotic cells possess ~35 ubiquitin E2 enzymes and >500 ubiquitin E3 enzymes. The E3 enzymes are the specificity factors of the ubiquitin pathway which mediate the selective targeting of specific cellular substrate proteins (Deshaies, R.J., and C.A. Joazeiro, 2009, RING domain E3 ubiquitin ligases, Annu Rev Biochem 78:399-434; Lipkowitz, S., and A.M. Weissman, 2011, RTNGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis, Nat Rev Cancer 11 :629-643; Rotin, D., and S. Kumar, 2009, Physiological functions of the HECT family of ubiquitin ligases, Nat Rev Mol Cell Biol 10:398-409).
Two El enzymes have been identified for ubiquitin, UAE (ubiquitin-activating enzyme) and UBA6 (Jin, J., et al., 2007, Dual El activation systems for ubiquitin differentially regulate E2 enzyme charging, Nature 447: 1135-1138). UAE is the El responsible for the majority (>99%) of ubiquitin flux within the cell. UAE is capable of charging each of the approximately -35 E2 enzymes with the exception of Usel, which is the only E2 known to exclusively work with UBA6 (Jin et al., 2007). Inhibition of UAE is sufficient to dramatically impair the great majority of ubiquitin-dependent cellular processes (Ciechanover, A., et al., 1984, Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85, Cell 37:57-66; Finley, D., A. et al., 1984, Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85, Cell 37:43-55).
The cellular signals generated by ubiquitin are diverse. Ubiquitin can be attached to substrates as a single entity or as polyubiquitin polymers generated through isopeptide linkages between the C-terminus of one ubiquitin and one of the many lysines on a second ubiquitin. These varied modifications are translated into a variety of cellular signals. For example, conjugation of a lysine 48 -linked polyubiquitin chain to a substrate protein is predominantly associated with targeting the protein for removal by the 26S proteasome. A single ubiquitin modification, or monoubiquination, typically affects protein localization and/or function. For example, monoubiquitination modulates the following: 1) the function of Histones 2a and 2b (Chandrasekharan, M.B., et al., 2010, Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation, Epigenetics 5:460-468), 2) controls the nucleo-cytoplasmic shuttling of PTEN (Trotman, L,C, et al., 2007, 3) ubiquitination regulates PTEN nuclear import and tumor suppression, Cell 128: 141-156), 4) drives localization of the FANCD2 protein to sites of DNA damage (Gregory, R.C., et al., 2003, Regulation of the Fanconi anemia pathway by monoubiquitination, Semin Cancer Biol 13:77-82) and 5) promotes the internalization and endosomal/lysosomal turnover of some cell surface receptors, like EGFR (Mosesson, Y., and Y. Yarden, 2006, Monoubiquitylation: a recurrent theme in membrane proteintransport. Isr Med Assoc J 8:233-237). Other forms of polyubiquitination chains occur on lysine positions 11, 29 and 63, impacting various cellular roles including cell cycle, DNA repair and autophagy (Behrends, C, and J.W. Harper, 2011, Constructing and decoding unconventional ubiquitin chains, Nat Struct Mol Biol 18:520-528; Bennett, E.J., and J.W. Harper, 2008, DNA damage: ubiquitin marks the spot, Nat Struct Mol Biol 15:20-22; Komander, D., 2009, The emerging complexity of protein ubiquitination, Biochem Soc Trans 37:937-953).
UAE-initiated ubiquitin conjugation plays an important role in protein homeostasis, cell surface receptor trafficking, transcription factor turnover and cell cycle progression. Many of these processes are important for cancer cell survival and it is believed that tumor cells may have increased sensitivity to UAE inhibition as a result of their rapid growth rate, increased metabolic demands and oncogene fueled protein stress. Preclinical studies with PYZD-4409, a UAE inhibitor, demonstrated this compound induced cell death in both leukemia and myeloma cell lines and induced anti-tumor activity in a mouse acute myeloid leukemia (AML model). (Xu, W.G., et al., 2010, The ubiquitin-activating enzyme El as a therapeutic target for the treatment of leukemia and multipie myeloma, Blood, 115:2251-59). Thus, UAE represents a protein homeostasis target opportunity for the treatment of cancer.
Clinical results of VELCADE® (bortezomib) For Injection have prompted evaluation of other enzymes within the ubiquitin proteasome system (UPS) as druggable targets for human cancer. We have identified a first in class investigational drug, TAK-243 (MLN7243), which targets the ubiquitin activating enzyme, UAE (UBA1), an essential cellular enzyme responsible for activating > 99% of all cellular ubiquitin. Ubiquitin is involved in multiple cellular processes including ubiquitin-dependent protein turnover, cell cycle progression, regulation of apoptosis, protein localization and response to DNA damage. Experiments combining targeted siRNA knockdown with TAK-243 identified DNA damage repair genes necessary for UAE inhibitor-induced cell death. A more focused approach revealed TAK-243 treatment blocked essential monoubiquitination events within the Translesion synthesis (TLS), Fanconi Anemia (FA) and Homologous recombination (HR) pathways. Inhibition of UAE prevented mono-ubiquitin signaling of key mediators within these pathways, including PCNA and FANCD2, by blocking formation of their specific E2-ubiquitin thioesters. In vitro cell-based assays combining TAK-243 with ultraviolet (UV) and radiation, both known to induce DNA damage, yielded inhibition of cell growth and enhanced DNA damage as observed through colony formation assays and Comet assay detection, respectively. Xenograft tumor bearing mice were treated with carboplatin or docetaxel, combined with TAK-243, to evaluate combination benefits in vivo. Synergistic and additive anti-tumor combination benefits were observed in animals treated with TAK-243 + carboplatin and TAK-243 + docetaxel. These important mechanistic in vitro and in vivo studies indicate the dependency of ubiquitination signaling in DNA damage repair and provide a mechanistic rationale for combining radiation, carboplatin or docetaxel with TAK-243 in the clinical setting. Currently, TAK-243 is being evaluated in a solid tumor phase I clinical trial evaluating safety, tolerability, pharmacokinetics, pharmacodynamics and anti-tumor activity (ClinicalTrials.gov identifier: NCT02045095).
Citation Format: Michael A. Milhollen, Judi Shi, Tary Traore, Jessica Huck, Darshan Sappal, Jennifer Duffy, Eric Lightcap, Yuko Ishii, Jeff Ciavarri, Paul Fleming, Neil Bence, Marc L. Hyer. The small molecule UAE inhibitor TAK-243 (MLN7243) prevents DNA damage repair and reduces cell viability/tumor growth when combined with radiation, carboplatin and docetaxel. [abstract]. In: Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2015 Nov 5-9; Boston, MA. Philadelphia (PA): AACR; Mol Cancer Ther 2015;14(12 Suppl 2):Abstract nr A164.
PATENT
WO 2013123169
https://www.google.com/patents/WO2013123169A1?cl=en
Scheme 1 : General route for 2-substituted ((1R,2R,3S,4R)-2,3-dihydroxy-4- (pyrazolo[1,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates

A genera! route for the synthesis of compounds represented by structure iv wherein Z is an optionally substituted fused or non-fused aryl or heteroaryl ring is outlined above in Scheme 1. Compound i (obtained by coupling an appropriately protected cyclopentylamine or salt thereof with 2-bromo-7-chloropyrazolo[1 ,5-a]pyrimidine in the presence of a suitable base as described below in the procedure of Examples 1a and 1b) is transformed to a compound of formula iii by coupling with a metal substituted compound Z-M via a palladium catalyzed reaction. A compound of formula iii can also be obtained by first transforming i to a metal substituted compound of formula ii using suitable boron or tin containing reagents, and then coupling with a halogen substituted compound Z-X via a palladium catalyzed reaction. Compounds of formula iv are then obtained by reaction with an appropriate sulfamating reagent (for example chlorosulfonamide or see Armitage, I. et. al. U.S. Patent Application US2009/0036678, and Armitage, I. et. al. Org. Lett., 2012, 14 (10), 2626-2629) followed by appropriate deprotection conditions.
Scheme 2: General route for 5-halogen substituted, 2 -substituted ((1R,2R,3S,4R)- 2,3-dihydroxy-4-(pyrazolo[1,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates


A general route for the synthesis of compounds represented by structure ix wherein Z is an optionally substituted fused or non-fused aryl or heteroaryl ring and X is a halogen is outlined above in Scheme 2. Cyclization of amino-pyrazole v with a suitable diester and an appropriate base at an elevated temperature is followed by reaction with an appropriate halogenating reagent such as POCI3 at an elevated temperature to give compounds of formula vii. Compounds of formula viii are then obtained by reaction with an appropriately protected cyc!opentylamine or a salt thereof in the presence of a suitable base. Sulfamation and deprotection following Method 1 as described previously provides compounds of formula ix.
Scheme 3: General route for 5-alkyl substituted, 2-substituted ((1R,2R,3S,4R)-2,3- dihydroxy-4-(pyrazolo[1 ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamates

SIMILAR COMPD
Example 17. Synthesis of (s.e.)-{(1 ,2R,3S,4R)-4-[(3,6-dichloro-2-{3- [(trifluoromethyl)sulfanyl]phenyl}pyrazolo[1,5-a]pyrimidin-7-yl)amino]-2,3- dihydroxycyclopentyl}methyl sulfamate (1-124) and (s.e.)-{(1 ,2R,3S,4R)-4-[(6-chloro-2-{3- [(trifluoromethyl)sulfanyl]phenyl}pyrazolo[1,5^]pyrimidin-7-y[)arnino]-2,3- dihydroxycyclopentyl}methyl sulfamate 0-125).

Step 1. To a vial containing s.e {(1 ,2 ,3S,4 )-2,3-dihydroxy-4-t(2-{3- [(t rif I u orometh y l)sulf a nyl] phen l}p^
sulfamate (0.82 g, 0.0015 mol) and cooled to 0 °C is added N-chlorosuccinimide (126 mg, 0.000943 mol) as a solution in 12 mL of N,N-dimethy)formamide. The reaction mixture is stirred overnight with warming to rt. Saturated sodium bicarbonate solution is added and the reaction mixture is extracted with ethyl acetate, washed with brine, dried over sodium sulfate and concentrated in vacuo. The crude material is first purified by column chromatography (eluent: methanol/methylene chloride) and then purified by HPLC to afford both the dichloro (LCMS: (FA) +1 588) and mono chloro (LCMS: (FA) M+1 554) titlecompounds.
PATENT
UAE inhibitors are disclosed in patent application publications WO2013/123169 and US 2014/0088096. In one embodiment, the UAE inhibitor is a compound having the following structure (Compound 1):

(Compound 1);
or a pharmaceutically acceptable salt thereof. The Compound 1 is named ((lR,2R,3S,4R)-2,3-dihydroxy-4-(2-(3-(trifluoromethylthio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-ylamino)cyclopentyl)methyl sulfamate.
process for making Compound 1 :

Compound 1;
or pharmaceutically acceptable salt thereof, comprising the steps of:
a) contacting Compound 9 or a salt, solvate or hydrate thereof with 2,2-dimethyl-l,3-dioxane-4,6-dione (Meldrum’s acid):

Compound 9
under coupling conditions to provide compound 8 or a salt, solvate or hydrate thereof:

Compound 8
b) subjecting compound 8 or a salt, solvate or hydrate thereof to cyclization conditions to provide compound 7 or a salt, solvate or hydrate thereof

Compound 7
c) contacting Compound 7 or a salt, solvate or hydrate thereof with benzotriazole under chlorination displacement conditions to provide Compound 5 or a salt, complex, solvate or hydratei thereof

; Compound 5
d) contacting Compound 5 or a salt, complex, solvate or hydrate thereof with Compound 6 or a solvate or hydrate thereof:

; Compound 6
under displacement reaction conditions to provide Compound 3 or a salt, solvate or hydrate thereof
solvate or hydrate thereof with Compound

Cl ; Compound 4
under sulfamoylating reaction conditions to provide Compound 2 or a salt, solvate or hydrate thereof

; Compound 2
f) contacting Compound 2 or a salt, solvate or hydrate thereof with an acid under sulfamoylation conditions to provide Compound 1 or a pharmaceutically acceptable salt thereof
 COMPD1
COMPD1
Example 1: Synthesis of S-iB-Ktrifluoromethyltsulfanyllphenyll-lH-pyrazol-S-amine
Step A: 3-((trifluoromethyl)thio)benzoate
[0148] To dimethylcarbonate (68 mL) was added 3-((trifluoromethyl)thio)benzoic acid (100 g, Beta Pharma Scientific) and a catalytic amount of sulfuric acid (2.4 mL). The mixture was then heated to 90°C for 5h. The reaction was then cooled to room temperature and quenched with sodium bicarbonate (1.0 L). To the aqueous layer was with ethyl acetate (1.0 L). The phases were separated and this process was repeated with ethyl acetate (1.0 L). The organic layers were combined and concentrated with a rotovap to give a light orange oil. The methyl 3-((trifluoromethyl)thio)benzoate (105g, 99%) was taken on crude to the next reaction. Ή NMR (300 MHz, CHLOROFORM-^ δ ppm 3.99 (s, 3 H) 7.49 – 7.58 (m, 1 H) 7.85 (d, J=l.62 Hz, 1 H) 8.17 (dt, J=7.69, 1.43 Hz, 1 H) 8.32 – 8.44 (m, 1 H).
[0149] Step B: 3-oxo-3-(3-((trifluoromcthvnthio)phcnyl>proDaneiiitrilc
[0150] Methyl 3-((trifluoromethyl)thio)benzoate (100.0 g) in tetrahydrofuran (1.0 L) was added acetonitrile (44.2 mL, 847 rnmol) and 1M (in THF) potassium tert-butoxide (95.01 g). The reaction was complete in 10 min by HPLC analysis. The reaction was quenched with 1M HC1 (1.0 L) and then extracted with three times with (1.0 L) of ethyl acetate. The organic layers with 3-oxo-3-(3-((trifluoromethyl)thio)phenyl)propanenitrile were then concentrated to dryness. This material (lOO.Og, 96.3%) was taken on crude with further purification. Ή NMR (300 MHz, CHLOROFORM-rf) δ ppm 4.12 (s, 2 H) 7.51 – 7.75 (m, 1 H) 7.89 – 8.01 (m, 1 H) 8.01 – 8.10 (m, 1 H) 8.20 (s, 1 H)
[0151] Step C: 3-}3-htrifliioromethv sulfan llphenyl}-lH-pyrazol-5-amine
[0152] To 3-oxo-3-{3-[(trifluoromethyl)sulfanyl]phenyl}propanenitrile (100.0 g,) in ethanol (1000.0 mL) was added hydrazine hydrate (59.52 mL). The reaction was heated to 100°C for lh at which point HPLC analysis showed the reaction was complete. The reaction was concentrated to dryness on a rotovap to give a brown oil. The oil was taken up in ethyl acetate (1.0 L) and extracted with water (1.0 L). The phases were separated and the organic phase was concentrated. Upon concentration 3-{3-[(trifluoromethyl)sulfanyl]phenyl}-lH-pyrazol-5-amine was obtained (80.8 g; Yield = 76.4%) . !H NMR (300 MHz, CHLOROFORM-^ δ ppm 5.95 (s, 1 H) 6.73 (br s, 1 H) 7.13 – 7.34 (m, 2 H) 7.42 – 7.74 (m, 3 H) 7.85 (s, 1 H).
[0153] Example 2: f R.2R.3St4RV2.3-dihvdroxy-4-ff2-r3- ((trifluoromethylHhio)phenvnpyrazolo[l,5-alpyrimidin-7-yl¼mino)cvclopentyl)metliyl sulfamate
[0154] Step 1: f2.2-dimethyl-5-ffl3-(3-((triiluoromethvnthio phenvn-lH-pyrazol-5- amino methyleBC>-1.3-dioxane-4,6-dione)
[0155] To trimethoxy orthoformate (2.0 L), at 20°C and under a blanket of nitrogen, was added 2,2-dimethyl-l,3-dioxane-4,6-dione (361.35 g). The resulting white suspension went clear within minutes and was heated to 85°C over 15 minutes. The reaction was held at 85°C for 120 minutes. While the reaction was heated and stirred another solution of 3-(3-((trifluoromethyl)thio)pheny])-lH-pyrazol-5-amine (500.0 g) was made. To a 4L RBF was added 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (500.0 g) and then trimethoxy orthoformate (1.4 L) added into this solid. This solution was mixed to dissolve the solids and resulted a dark brown solution. The solution of 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (-1.8L in trimethoxy orthoformate) was added to the reactor over 30 minutes while maintaining the reaction temperature at 85°C. The reaction was then stirred for 20 minutes with white solids forming in the solution. After 20 minutes the reaction was sampled and the UPLC showed the complete conversion of 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5 -amine to 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l ,3-dioxane-4,6-dione. The reaction was cooled to 20 °C over 20 minutes and maintained at that temperature for 20 additional minutes. At this point, a thick white slurry had formed and the reaction was filtered using a Nutche Filter over 15 minutes. The reactor was washed with 1L of ethyl acetate and this solution was then mixed with the filter cake and removed by filtration. The cake was dried for -40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16 hours). The reaction was then analyzed by FfPLC and NMR to give 2,2-dimethyl-5-(((3-(3 -((trifluoromethyl)thio)phenyi lH-pyrazol-5-yl)amino)methylene)- 1 ,3-dioxane-4,6-dione (635.3 g, 79%) XH NMR (300 MHz, DMSO-cfe) δ ppm 1.68 (s, 6 H) 7.05 (d, J=2.05 Hz, 1 H) 7.64 -7.77 (m, 2 H) 7.77 – 8.03 (m, 1 H) 8.12 (s, 1 H) 8.72 (d, J=14.36 Hz, 1 H) 1 1.35 (d, J=14.66 Hz, 1 H) 13.47 (s, 1 H).
[0156] Step 2: 2-( 3-f(trifluoromethyl)thio phenyl)pyrazoIo [1,5-al pyrimidin-7-ol
[0157] A solution of 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l,3-dioxane-4,6-dione (615.00 g) in 1,2-dichIorobenzene (6.3 L) was stirred at ambient temperature for 10 minutes. The solution was then heated to 150°C over 75 minutes. The reaction was maintained at this temperature for 16 hours. An sample was taken after 16 hours and the UPLC analysis showed the complete conversion of 2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yI)amino)methylene)-l,3-dioxane-4,6-dione to 2-(3- ((trifluoromethyl)tmo)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol. The reaction was cooled to 20°C over 130 minutes. At this point, a thick white slurry had formed and the reaction was filtered using a Nutche Filter over 15 minutes. The reactor was washed with 1.8 L of acetonitrile and this solution was then mixed with the filter cake and then the solvent was removed by filtration. The cake was dried for ~40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16 hours). The reaction was then analyzed by HPLC and NMR to give 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (331.2 g, 72%) Ή NMR (300 MHz, METHANOL-^) δ ppm 6.55 (d, J=7.33 Hz, 1 H) 7.59 (s, 1 H) 8.40 – 8.52 (m, 1 H) 8.53 – 8.64 (m, 1 H) 8.69 (d, J=7.62 Hz, 1 H) 9.01 (dt, J=7.77, 1.39 Hz, 1 H) 9.12 (s, 1 H) 13.34 (s, 1 H).
[0158] Step 3: l-(2-(3-(f trffluoromethvmhiotohenvnpyrazolo n.5-al pyrimidin-7-vn-lH-benzofdiri.2.31triazole: triethylamine: hydrochloride complex (1:1.25:1.25 molesimolestmolest
[0159] To a solution of 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (30.00 g), benzotriazole (287.02 g) in acetonitrile (3000 mL) and triethylamine (403.00 mL) at 0°C, was added phosphoryl chloride (108 mL) slowly under a blanket of nitrogen, maintaining < 10°C. The reaction was then warmed to 80°C over 45 minutes and stirred for 240 minutes. HPLC indicated complete
consumption of starting material. To the reaction mixture was added acetonitrile (3000 mL) while maintaining the temperature at 80°C. The reaction was then cooled to 20°C over 80 minutes. The reaction was then stirred at ambient temperature for 14 hours. At this point, a thick slurry had formed and the reaction was filtered using a Nutche filter over 15 minutes. The reactor was washed twice with 900 mL of acetonitrile and this solution was then mixed with the filter cake and then the solvent was removed by filtration. The cake was dried for -40 minutes on the filter and then transferred to a vacuum oven and heated at 40°C under full vacuum overnight (16h). The reaction was then analyzed by HPLC and NMR to give l-(2-(3-((trifluorometJiyl)thio)phe
triethylamine: hydrochloride complex (1:1.25:1.25 moles:moles:moles) (438.1 g, 83%). ¾ NMR (300 MHz, DMSO-</6) δ ppm 1.19 (t, J=7.33 Hz, 12 H) 3.07 (qd, J=7.28, 4.84 Hz, 8 H) 7.60 – 7.78 (m, 6 H) 7.80 – 7.87 (m, 1 H) 8.15 (dt, J=7.99, 1.28 Hz, 1 H) 8.24 (s, 1 H) 8.33 (dt, J=8.14, 0.92 Hz, 1 H) 8.85 (d, J=4.69 Hz, 1 H).
[0160] Step 4: ff3aR4R.6R.6aS 2.2-dimethyl-6-ff2-f3~mrifluoromethyl)thio)phenvnpyrazoloil.5-alD\timidin-7-yl¼mino)tctralivdro-3aH-cvcLoDentaldlll,31dioxol-4-vnincthanol
[0161] To the reactor was added l-(2 3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)-lH-benzo[d][l,2,3]triazole: triethylamine: hydrochloride complex (1 :1.25: 1.25 moles :moles:moles) (430.0 g) and ((3aR,4R,6R,6aS)-6-amino-2,2-dimethyltetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol hydrochloride (209.0 g) and then triethylamine (2103 mL) was added. The reaction was then heated to 80°C, under a blanket of nitrogen. After 360 minutes, HPLC analysis indicated that the reaction mixture contained <1% starting material and the reaction was cooled to 20°C over 60 minutes. To the reaction was added ethyl acetate (3.5 L) and water (3.5 L). After stirring for 10 minutes the phases were separated and the aqueous layer was back extracted with ethyl acetate (3.5 L). The organic layers were combined and concentrated to form a dark, brown oil. Acetonitrile (4.5 L) was added and the solution was concentrated to dryness to give an orange solid. The solids was transferred back to the reaction with water (4.3 L), heated to 50°C, and stirred for 20 minutes. White solids formed in this hot solution and were isolated by filtration using a Nutche Filter over 15 minutes. The solids were dried under vacuum for 15 minutes on the filter and then dissolved in acetonitrile (4.0 L) at 0°C. The solution was stirred for 1 minutes. The solution was then filtered through a fritted funnel to remove the hydrolysis solid by product and the solution was concentrated to dryness. The solids were dried in a vacuum oven at full vacuum overnight (40°C, 16 hours). The reaction was then analyzed by HPLC and NMR to give ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5 -a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanoI (349.2 g, 88%). Ή NMR (300 MHz, DMSO-<¾) δ ppm 1.25 (s, 3 H) 1.47 (s, 3 H) 1.76 – 1.90 (m, 1 H) 2.25 (br d, J-3.22 Hz, 1 H) 2.33 – 2.47 (m, 1 H) 3.46 – 3.67 (m, 2 H) 4.08 (br d, J=5.57 Hz, 1 H) 4.48 – 4.64 (m, 2 H) 5.19 (t, J=4.40 Hz, 1 H) 6.28 (d, J=5.28 Hz, 1 H) 7.06 (s, 1 H) 7.58 – 7.71 (m, 1 H) 7.72 – 7.80 (m, 1 H) 8.12 – 8.24 (m, 2 H) 8.31 (d, J=7.62 Hz, 1 H) 8.42 (s, 1 H).
[0162] Step 5: ((3aR.4R.6R.6aS 2.2-dimethyl-6-ff2-f3-fftrifluoroinethYmhio)phenvnpyrazolo[1.5-al Dyrimidin-7-vnan] iiio>tetrahvdro-3aH-cvclonen ta [dl [1,31 dioTOl-4-yl )meth yl tert-bntoxycarbonylsulfamate
[0163] ((3aR,4R,6R,6aS)-2,2-dime l-6-((2-(3-((trifluorome
7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol (6.0 g) was dissolved in 2-methyltetrahedrafuran (60.0 mL) and to this solution was added pyridinium p-toluenesulfonate (5.9 g). This formed a precipitated and to this white slurry was added (4-aza-l-azoniabicyclo[2.2.2]oct-l-ylsulfonyl)(tert-butoxycarbonyl)azanide-l,4-diazabicyclo[2.2.2]octane (1 :1) hydrochloride1 (17.0 g). The mixture was stirred at ambient temperature until the HPLC showed <1% ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol remaining starting material (-300 minutes). The reaction was quenched with water (60 mL) and the phases were separated. To the organic layer was added acetonitrile (60 mL) and the mixture was concentrated using a rotovap at 50°C to ~60 mL. The mixture was allowed to cool to room temperature and stirred overnight. During this time a white slurry formed. White solids were filtered using a medium fritted filter. The solid was dried in a vacuum oven at full vacuum overnight (40 °C). The reaction was then analyzed by HPLC and NMR to give ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyI)tM^
cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (5.03 g, 68%). [H NMR (300 MHz, DMSO- 6) δ ppm 1.26 (s, 3 H) 1.42 (s, 9 H) 1.51 (s, 3 H) 2.33 – 2.48 (m, 2 H) 3.30 (br s, 1 H) 4.06 – 4.21 (m, 1 H) 4.29 (d, J=5.28 Hz, 2 H) 4.52 (dd, J=7.18, 5.13 Hz, 1 H) 4.76 (dd, J=7.18, 4.54 Hz, 1 H) 6.35 (d, J=5.57 Hz, 1 H) 7.08 (s, 1 H) 7.63 – 7.72 (m, 1 H) 7.74 – 7.82 (m, 1 H) 8.01 (d, ^=7. 2 Hz, 1 H) 8.21 (d, J=5.28 Hz, 1 H) 8.31 (dt, J=7.84, 1.36 Hz, 1 H) 8.48 (s, 1 H) 1 1.92 (br s, 1 H)
[0164] Step 6: f R,2R3S.4R)-2J-dihvdroxy-4-((2-(3-fftrifluoromethvDthio^phenvnpyrazolori.5-a]pyrimidin-7-yl)aminokvcl nent\l)methyl sulfamate
[0165] To a solution of ((3aR,4R,6R!6aS)-2,2-dimethyl-6-((2-(3- ((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (2.0 g) in acetonitrile (11 mL) at 0°C was added phosphoric acid (1 1 mL) while maintaining the temperature below 10°C. This mixture was warmed to ambient temperature and stirred for 4 hours. At this time HPLC analysis showed that <1% ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate starting material or reaction intermediates remained. To the reaction was added ethyl acetate (1 1 mL) and water (11 mL) and saturated Na2C03 (10 mL) dropwise. After this addition was complete saturated Na2C03 was added until the pH was between 6-7. The phases were separated and to the organic layer was added acetonitrile (30 mL) and the mixture was concentrated on a rotovap to ~16 mL. The mixture was stirred overnight. During this time a white slurry formed. The white solids were filtered using a medium filtted filter. The solid was dried in a vacuum oven at full vacuum overnight (40°C). The reaction was then analyzed by HPLC and NMR to give ((lR,2R,3S,4R)-2,3-dihydroxy-4-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)cyclopentyl)methyl sulfamate (1.5g ,84%). lH NMR (300 MHz, DMSO-c¾) δ ppm 1.44 – 1.61 (m, 1 H) 2.20 – 2.42 (m, 2 H) 3.78 (q, J-4.50 Hz, 1 H) 3.90 – 4.09 (m, 3 H) 4.09 – 4.22 (m, 1 H) 4.80 (d, ^5.28 Hz, 1 H) 5.03 (d, J=5.28 Hz, 1 H) 6.31 (d, J=5.57 Hz, 1 H) 7.05 (s, 1 H) 7.48 (s, 2 H) 7.62 – 7.72 (m, 1 H) 7.77 (d, J=7.92 Hz, 2 H) 8.17 (d, J=5.28 Hz, 1 H) 8.31 (dt, ^7.70, 1.43 Hz, 1 H) 8.47 (s, 1 H).
[0166] Example 3: fflR.2R.3S.4RV2.3-dihvdrosy-4-ff2-f3- ( ( trifluoroniethyl )thio)ph en vDpyrazolo 11,5-a I pyi Lmidin-7-Yl)amino)cvclopcntyl>m ethyl sulfama te
[0167] Step 1: .2-dimethyl-5-ff -(3-frtrifluoromethvnthio)phenvn-lH-pyrazol-5-yl)ainino)methylene -l,3-dioxane-4,6-dione)
[0168] Under a blanket of nitrogen at 20°C, Meldrum’s acid (18.6 Kg) and isopropanol (33 L) were placed in a 100 L glass-lined reactor. Trimethyl orthoformate (15.5 Kg (16.0L)) and isopropanol (11 L) were added and the mixture was heated to 80 °C for 40 min, whereby a small amount of methanol distilled off (<0.5 L). The mixture was stirred for 2 h at 80 °C. in a separate 160 L glass-lined reactor under nitrogen at 20 °C, 3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-amine (prepared in the manner described above) was mixed with isopropanol ( 10.9 kg, 42.0 mmol) and heated up to 80 °C within 60 min. The content of the 100 L reactor was transferred into the reaction mixture in the 160 L reactor at 80 °C, which was completed after 3 min. The reaction mixture was stirred for 30 min at 78 °C, the reaction was then cooled to 60 °C. HPLC analysis showed the reaction was 99.56% complete (product%/(product%+starting material0/.). The reaction mixture was cooled to 20 °C within 100 min, then the mixture was stirred for further 100 min at 20 °C. The suspension was then transferred onto a pressure filter. At 1.2 bar nitrogen, the solids were collected on the filter. The filter cake was washed 4 x with ethyl acetate (18 L each time). The wet cake was dried on the filter for 17 h at 20°C using a slight stream of nitrogen/vacuum (200-100 mbar). The wet product (14.7 kg) was further dried at the rotavap for approx. 24 h at 40-50 °C. 11,75 kg of the crude title compound was obtained (68% yield). NMRspectrum was consistent with that described above in Example 2.
[0169] Step 2: 2-(3-fftrifluoromethvnthio)phenYnpyrazolori.S-a1pyrimidin-7-ol
[0170] Under nitrogen at 20 °C, (2,2-dimethyl-5-(((3-(3-((trifluoromethyl)thio)phenyl)-lH-pyrazol-5-yl)amino)methylene)-l ,3-dioxane-4,6-dione) was placed in the reactor. 1 ,2-Dichlorobenzene (117 L) was added. The suspension was heated to 147°C for 90 min to give a solution, then it was stirred at 147°C for 18 h. Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 92.28% completion (product%/(product%+starting material%). The mixture was heated up again to 147°C and stirred for further 5 h at this temperature. HPLC analysis showed the reaction was 96.51% complete (product%/(product%+starting material%). The mixture was then stirred for 48 hours at 20°C, then it was heated again to 147°C und stirred at this temperature for 5 h. Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 98.47% completion (product%/(product%+starting material%). The mixture was heated up again to 146°C and stirred for further 5 h at this temperature.
Before sampling, the reaction was cooled to 60°C. HPLC analysis showed the reaction was 99.35% complete (product%/(product%+starting material%). The reaction was cooled to 20°C and the suspension was transferred in a pressure filter. The solids were collected on the filter at 1.8-3 bar N2 over a greater than 10 hour period. The filter cake was washed 4 x with acetonitrile (17 L), then it was dried on the filter for 18 h at 20°C/200-100 mbar, using a slight stream of N2. The material was transferred to a 50 L flask and dried on a rotavap at 50-60°C / 24-14 mbar for 2 d. 6.118 kg of the crude title compound was obtained (70% yield). NMR spectrum was consistent with that described above in Example 2.
[0171] Step 3: l-f2-f3- trifluoromethYnthio^phenvnpyrazoIo[1.5-alpyriinidiii-7-vn-lH-benzofdl [1.2.31 triazolc: triethylamine: hydrochloride complex ( 1 :0.21:0.21 moles:moles:moles)
[0172] Under N2 at 20°C, acetonitrile (30 L) was placed in the reactor, 2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-ol (6.00 kg) and lH-benzotriazol (5.83 kg) was added. A further portion of acetonitrile (30 L) was added, then the mixture was stirred at 20°C. Stirring proceeded over night. Triethylamine (8.16 L) was added at 20°C over 6 min. The yellow suspension was heated up to 45°C for 40 min. While stirring at 150 rpm, phosphoryl chloride (4.562 kg) was slowly added for 45 min. By controlling the addition, the reagent was dropped directly into the mixture to avoid the formation of lumps. The addition was exothermic, a maximum temperature of 53°C was observed. The brown suspension was heated up to 80°C over 1 h, then the reaction mixture was stirred for 5 h at this temperature. Acetonitrile (30 L) was added over 20 min keeping the internal temperature between 75-80°C. HPLC analysis showed the reaction was 98.31% completion (product%/(product%+starting material%).The mixture (brown suspension) was further stirred at 80°C for 70 min. HPLC analysis showed the reaction was 99.48% completion (product%/(product%+starting material%). Acetonitrile (61 L) was added over 30 min maintaining the temperature between 75-80°C. The pale brown suspension was stirred at 80°C for 90 min, then it was cooled to 20°C over 2.5 h. The mixture was stirred for 12 h at 20°C. The mixture was transferred in a pressure filter. The filter cake was washed twice with acetonitrile ( 18 L). Both wash steps were done at 3.5-4 bar N2. Each of these filtrations took overnight to go to completion. The filter cake was dried on the filter for 7.5 h. The material was transferred in a 50 L flask and dried at the rotavap at Ta 40-50°C / 50-11 mbar for 3 d to get a dry mass of 99.88% . The yield of l -(2-(3-((trifluoromethyl)t]iio)phenyl)pyrazolo[l ,5-a]pyrimidin-7-yl)-lH-benzo[d][l,2,3]triazole: triethylamine: hydrochloride complex (1 :0.21 :0.21 moles:moles:moles) was 7.948 kg (75%). NMR spectrum was consistent with that described above in Example 2.
[0173] Step 4: 3aR4R.6R,6aS)-2,2-dimethYl-6-f(2-f3-ffMfluoromethvnthio phenvnpyrazolori.5-alDyrimidin-7-yl)amino)tetrahvdro-3aH- vclopenta Idl [1.31 dioxol-4-vDmethanol
[0174] Under N2 in a 160 L glasslined reactor, triethylamine (21%) compound with l -(2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5 -a] pyrimidin-7-yI) – 1 H-benzo [d] [ 1 ,2,3 Jtriazole (21 %) hydrochloride (7.86 kg) was dissolved in triethylamine (23.3 L) at 20°C. ((3aR,4R,6R,6aS)-6-amino-2,2-dimethyltetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol hydrochloride (4.49 kg) was added, followed by triethylamine (23 L). The reaction mixture was heated up to 80°C over 1 h, and then the mixture was stirred for 8 h at 80°C. The mixture was then cooled to 20°C. HPLC analysis showed the reaction was 99.97% complete (product%/(product%+starting material%). Water (66 L) was then added over 30 min at 20-25°C (exotherm), whereby a brown suspension was obtained. The mixture was concentrated at 60°C, 150-95 mbar, until 42 L solvent was distilled off. The suspension was heated to 50°C, and the solids were collected on a 90 L pressure filter (1.2 bar N2), which took 40 min. During this process, the material on the filter was not actively heated. The remaining solids in the reactor were rinsed with 15 L of the mother liquor. The wet filter cake was transferred back in the reactor. Water (64 L) was added. The mixture was heated up to 50°C over 30 min. The washed solids were collected on the 90 L pressure filter. Remaining mother liquor in the filter cake was pressed off at 1.2 bar N2 for 50 min (50 L mother liquor was used to rinse the reactor). The filter cake was dried on the pressure filter for 13.5 h, applying a slight stream of N2 / vac at 20°C to afford 10.247 kg of crude ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)tWo)phenyl)pyrazolo[l ,5-a]pyrimidin-7-yl)ammo)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methanol. The wet filter cake was isolated. The wet filter cake was loaded into the reactor. Acetonitrile (65 L) was added, followed by activated charcoal (6.59 kg). The mixture was heated to 50°C for 30 min and stirred for 2 h at 50°C. Meanwhile a bed of celite (4.25 kg) had been prepared in the 90 L pressure filter, using acetonitrile (20 L) for conditioning. The bed was heated at 50°C. The black suspension was transferred on the filter and pushed through the Celite plug at 2 bar. The filtrate was transferred to a 200 L stirring tank via a heat resistant tube and a 0.45 μιη inline filter. The operation needed 18 min for completion. For washing, acetonitrile (50 L) which had been warmed up in the reactor to 50°C and transferred over the warmed filter cake and pushed through at 2 bar. Again, the filtrate was transferred in the 200 L stirring tank via a heat resistant tube and a 0.45 μιη inline filter. The operation needed 10 min for completion. The reactor was cleaned to remove attached charcoal (abrasive cleaning, using NaCl /acetone). The filtrate in the stirring tank was transferred in the reactor and concentrated at 50°C / 120 mbar until 63 L were distilled off. While well stirring (300 rpm) and 50°C, Water (1 10 L) was slowly added over 2 h. A pale yellow suspension was formed. The concentrate was cooled to 20°C for 3 h, then stirred at this temperature for 13 h. The solids were collected on a 50 L filter, using 1.2 bar N2 to push the filtrate through. The filter cake was washed twice with water (18 L), then dried on the filter for 24 h at 200-100 mbar, using a slight stream of N2. 4.563 kg of the title compound was obtained 55% yield. NMR spectrum was consistent with that described above in Example 2.
[0175] Step 5: (f3aR,4R,6R,6aS)-2^-dimethyl-6-(f2-f3-fftrifluorQmethvnthio phenvnpyrazolo[1.5- |pyrimidm-7-vnamino)teti ahYclro-3aH-cvclopenta|d||1.3ldioxol-4-yl mcthyl tert-butoxycarbonylsutfamatc
[0176] Under N2 at 20°C, ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3- ((trifluoromethyl)thio)phenyl)pyrazolo[ 1 , 5 -a]pyrimidin-7-yl)amino)tetrahydro-3 aH-cyclopenta[d][l,3]dioxol-4-yl)methanol (4.019 kg) was placed in a 160 L glasslined reactor, then 2-methyl-tetrahydrofuran (40 L) was added. The mixture was stirred at 150 rpm for 30 min at 20°C, whereby a clear solution was formed. A KF measurement was taken and showed the water content to be 0.036% H20. The solution was stirred over night at 20 °C. The next morning, PPTS (2.2 kg) was loaded into the reactor. At 20°C, (4-aza-l-azoniabicyclo[2.2.2]oct-l-yIsulfonyl)(tert-butoxyc£u-bonyl)azanide-l,4-diazabicyclo[2.2.2]octane (1:1) hydrochloride (10.2 kg) was added. Stirring of the heterogeneous mixture was started at 130 rpm. The reaction was stirred with 200 rpm for 1 h at 20°C, then with increased speed of 250 rpm for an additional hour. HPLC analysis showed the conversion to be 87.3%. The reaction mass was stirred with 300 rpm for 2 h at 20°C. HPLC analysis showed the conversion to be 95.6%. The reaction mass was stirred with 300 rpm for 2 h at 20°C. HPLC analysis showed the conversion to be 97.7%. NaHC03 3.7% (40 L) was added to the mixture at 20°C and the reaction was stirred at 300 rpm for 10 min. Most of the solids from the reaction mixture went into solution. To dissolve remaining material which was attached at the top of the reactor, the bilayered mixture was stir up shortly by a N2 stream from the bottom. The layers were separated, which was completed after 13 min. The aqueous layer was discharged, the organic layer remained in the reactor. The org. layer was a brown solution, the aqueous layer was colorless and turbid. The pH of aqueous layer was approx. 8 (pH stick). NaHC03 3.7% (40 L) was added to the mixture at 20°C and it was stirred at 300 rpm for 10 min. The layers were separated, which was completed after 27 min. The aqueous layer was discharged, the organic layer remained in the reactor. The organic layer was a brown solution, the aqueous layer was colorless and turbid. The pH of aqueous layer was approx. 8-9 (pH stick) and the pH of organic layer was approx. 8 (pH stick, wet). The product in organic layer was transferred in the feeding tank and stored temporarily (approx. 30 min) at 20°C. The reactor was optically cleaned using a mixture of 2-methyltetrahydrofuran (30 L) and H20 (20 L). The org. layer was placed in the reactor and stored at -20°C for 14.5 h . While stirring at 150 rpm, the org. layer (suspension) was diluted with acetonitrile (16 L) and water (15 L) and warmed up to 5°C. At 5°C, acetic acid (0.172 kg) was added over 5 min. to a pH of 6; resulting in a mixture that was a pale brown solution. ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate (2.0 g; prepared in a similar manner to that described above Example 2, Step 5) was added as seed. At 5°C, acetic acid (0.515 kg) was added over 15 min. to pH 4-5; a suspension formed. The feeding tank was rinsed with water (1.6 L). The mixture was stirred at 5°C with 90 rpm for 1.5 h, then it was transferred in a 50 L filter and filtered at 1.2 bar N2, in only 4 min. The filter cake was washed 4 x with cold acetonitrile (8 L, 0-5°C), then it was dried on the filter at 20°C for 8 h at 200 mbar, using a slight stream of N2. The yield of the title compound was 3.594 kg (62%). MR spectrum was consistent with that described above in Example 2.
[0177] Step 6: friR.2R.3S.4R 2.3-dihvdroxY-4-ff2-f3-fftrifluoromethvntliio phenvnDyrazolori.5-alpyrimidin-7-yl)aminokvciopent>T)mcthyl sulfamate Compound 1
[0178] 3.538 kg of ((3aR,4R,6R,6aS)-2,2-dimethyl-6-((2-(3-((trifluoromethyl)thio)phenyl)pyrazolo[l,5-a]pyrimidin-7-yl)amino)tetrahydro-3aH-cyclopenta[d][l,3]dioxol-4-yl)methyl tert-butoxycarbonylsulfamate was suspended in 13.5 kg of acetonitrile and cooled to 5°C. To this mixture was added 27.3 kg of H3PO4 over 1 hour and 50 minutes. The reaction was warmed to 20°C over 50 minutes and then stirred for 8h at 22°C. HPLC analysis showed the reaction was 99.69% complete. To the first portion (50% of the reaction mixture) was added 8.9 kg of water and 7.95 kg of ethyl acetate. The pH was then adjusted to 6.5 with 48 L of saturated sodium carbonate. 7.7 kg of ethyl acetate was added and the phases were separated. To the second portion (50% of the reaction mixture) was added 8.9 kg of water and 7.95 kg of ethyl acetate. The pH was then adjusted to 6.15 with 48 L of saturated sodium carbonate. 7.7 kg of ethyl acetate was added and the phases were separated. The organic phases were combined in a vessel (rinsed with 1.8 kg of ethyl acetate) and washed with 17.8 kg of water. The phases were separated and 17.8 kg of water and 0.237 kg of NaCl were added and the phases were separated. A repeat of wash with 17.8 kg of water and 0.237 kg of NaCl was added and the phases were separated. The organic layers were then combined and the temperature of the mixture was raised to 40°C and the pressure was reduced to 300-142 mbar. 27 L of liquid was distilled off over 4h. 31.7 kg of acetonitrile were then added to the solution and the temperature of the mixture was raised to 38°C and the pressure was reduced to 320-153 mbar. 26 L of liquid was distilled over 3h. 31.7 kg of acetonitrile were then added to the solution and the temperature of the mixture was raised to 37°C and the pressure was reduced to 320-153 mbar. 34 L of liquid was distilled over 2h. The suspension was stirred for lh at 50°C and then cooled to 20-25°C over 3h. The reaction was stirred overnight and the product was filtered and washed with 8.9 kg of acetonitrile twice. The cake was dried for 2h at 20°C (33 mbar) then at 40-45°C (1 mbar) to afford 2.08 kg (75.8%) of the title compound. 2.066 kg of ((lR,2R,3S,4R)-2,3-dihydroxy-4-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 , 5 -a]pyrimidin-7-yl)amino)cyclopenty l)methy 1 sulfamate was loaded into a reactor with 9.76 kg of acetronitrile and 4.12 kg of water and heated at a temperature of 56 °C for 1 hour and 10 minutes until dissolved. The solution was polished filtered and the filter was
rinsed with 3.16 kg acetonitrile and 1.37 kg of water. To the resulting solution was added with 11.0 kg of water over 45 minutes while maintaining the reaction temperature between 52-55°C. 0.009 kg of (( 1 R,2R,3S,4R)-2,3 -dihydroxy-4-((2-(3 -((trifluoromethyl)thio)phenyl)pyrazolo[ 1 ,5-a]pyrimidin-7-yl)amino)cyclopentyl)methyl sulfamate was added as seed (prepared in a similar manner to that described above Example 2, Step 5). A suspension was visible after 10 minutes of stirring. To the solution was added 9.62 kg of water over 3h while maintaining the reaction temperature between 50-55°C. The suspension was then cooled over 3h to 20°C and stirred for 12h at 22-23°C. The suspension was then filtered and washed twice with 13.7 kg of water. The product was dried at 40°C. 1.605 kg of the title compound was obtained in 78% yield. NMR spectrum was consistent with that described above in Example 2.
PATENT
WO2016069392
SYNTHESIS
![]()
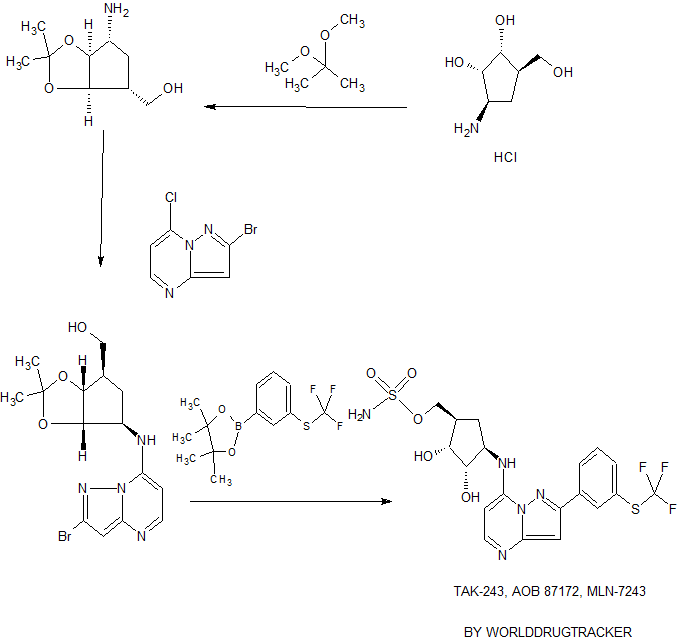
![]()
PF-05387252
CAS 1604034-71-0
| C25H27N5O2 | |
| MW | 429.51418 g/mol |
|---|
2-methoxy-3-[3-(4-methylpiperazin-1-yl)propoxy]-11H-indolo[3,2-c]quinoline-9-carbonitrile
IRAK4 inhibitor
Rheumatoid arthritis;
SLE
Preclinical
In the past decade there has been considerable interest in targeting the innate immune system in the treatment of autoimmune diseases and sterile inflammation. Receptors of the innate immune system provide the first line of defense against bacterial and viral insults. These receptors recognize bacterial and viral products as well as pro-inflammatory cytokines and thereby initiate a signaling cascade that ultimately results in the up-regulation of inflammatory cytokines such as TNFα, IL6, and interferons. Recently it has become apparent that self-generated ligands such as nucleic acids and products of inflammation such as HMGB1 and Advanced Glycated End-products (AGE) are ligands for Toll-like receptors (TLRs) which are key receptors of the innate immune system.
This demonstrates the role of TLRs in the initiation and perpetuation of inflammation due to autoimmunity.
Interleukin-1 receptor associated kinase (IRAK4) is a ubiquitously expressed serine/threonine kinase involved in the regulation of innate immunity. IRAK4 is responsible for initiating signaling from TLRs and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice lead to reductions in TLR and IL-1 induced pro-inflammatory cytokines. and 7 IRAK-4 kinase-dead knock-in mice have been shown to be resistant to induced joint inflammation in the antigen-induced-arthritis (AIA) and serum transfer-induced (K/BxN) arthritis models. Likewise, humans deficient in IRAK4 also display the inability to respond to challenge by TLR ligands and IL-1
However, the immunodeficient phenotype of IRAK4-null individuals is narrowly restricted to challenge by gram positive bacteria, but not gram negative bacteria, viruses or fungi. This gram positive sensitivity also lessens with age implying redundant or compensatory mechanisms for innate immunity in the absence of IRAK4.These data suggest that inhibitors of IRAK4 kinase activity will have therapeutic value in treating cytokine driven autoimmune diseases while having minimal immunosuppressive side effects. Additional recent studies suggest that targeting IRAK4 may be a viable strategy for the treatment of other inflammatory pathologies such as atherosclerosis.
Indeed, the therapeutic potential of IRAK4 inhibitors has been recognized by others within the drug-discovery community as evidenced by the variety of IRAK4 inhibitors have been reported to-date.12, 13, 14, 15 and 16 However, limited data has been published about these compounds and they appear to suffer from a variety of issues such as poor kinase selectivity and poor whole-blood potency that preclude their advancement into the pre-clinical models. To the best of our knowledge, no in vivo studies of IRAK4 inhibitors have been reported to-date in the literature. Herein we report a new class of IRAK4 inhibitors that are shown to recapitulate the phenotype observed in IRAK4 knockout and kinase-dead mice.
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(9), 2066-2072.
doi:10.1016/j.bmcl.2014.03.056
http://www.sciencedirect.com/science/article/pii/S0960894X14002832
![]()

IRAK4 is responsible for initiating signaling from Toll-like receptors (TLRs) and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice cause reductions in TLR induced pro-inflammatory cytokines and these mice are resistant to various models of arthritis. Herein we report the identification and optimization of a series of potent IRAK4 inhibitors. Representative examples from this series showed excellent selectivity over a panel of kinases, including the kinases known to play a role in TLR-mediated signaling. The compounds exhibited low nM potency in LPS- and R848-induced cytokine assays indicating that they are blocking the TLR signaling pathway. A key compound (26) from this series was profiled in more detail and found to have an excellent pharmaceutical profile as measured by predictive assays such as microsomal stability, TPSA, solubility, and c log P. However, this compound was found to afford poor exposure in mouse upon IP or IV administration. We found that removal of the ionizable solubilizing group (32) led to increased exposure, presumably due to increased permeability. Compounds 26 and 32, when dosed to plasma levels corresponding to ex vivo whole blood potency, were shown to inhibit LPS-induced TNFα in an in vivo murine model. To our knowledge, this is the first published in vivo demonstration that inhibition of the IRAK4 pathway by a small molecule can recapitulate the phenotype of IRAK4 knockout mice.


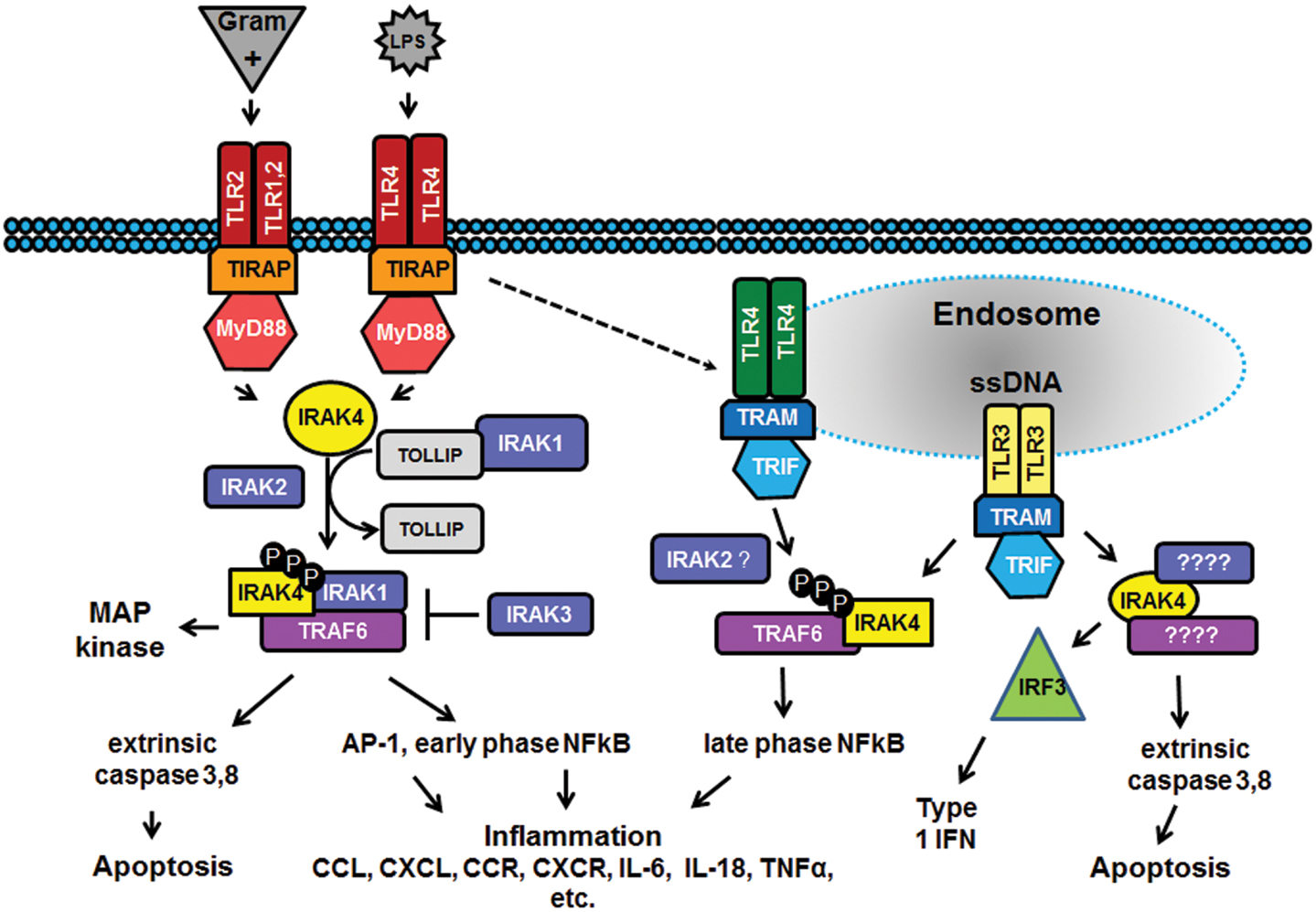
SYNTHESIS

////////PF-05387252, 1604034-71-0, PF 05387252, TLR signaling, Indoloquinoline, IRAK4, Kinase inhibitor, Inflammation, PRECLINICAL
N1(CCN(CC1)CCCOc3c(cc2c4nc5cc(ccc5c4cnc2c3)C#N)OC)C
OR
CN1CCN(CC1)CCCOC2=C(C=C3C(=C2)N=CC4=C3NC5=C4C=CC(=C5)C#N)OC
PF-05388169
CAS 1604034-78-7, MF C22 H21 N3 O4
MW 391.42
Rheumatoid arthritis;
SLE
Preclinical
![]()
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(9), 2066-2072.
http://www.sciencedirect.com/science/article/pii/S0960894X14002832
IRAK4 is responsible for initiating signaling from Toll-like receptors (TLRs) and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice cause reductions in TLR induced pro-inflammatory cytokines and these mice are resistant to various models of arthritis. Herein we report the identification and optimization of a series of potent IRAK4 inhibitors. Representative examples from this series showed excellent selectivity over a panel of kinases, including the kinases known to play a role in TLR-mediated signaling. The compounds exhibited low nM potency in LPS- and R848-induced cytokine assays indicating that they are blocking the TLR signaling pathway. A key compound (26) from this series was profiled in more detail and found to have an excellent pharmaceutical profile as measured by predictive assays such as microsomal stability, TPSA, solubility, and c log P. However, this compound was found to afford poor exposure in mouse upon IP or IV administration. We found that removal of the ionizable solubilizing group (32) led to increased exposure, presumably due to increased permeability. Compounds 26 and 32, when dosed to plasma levels corresponding to ex vivo whole blood potency, were shown to inhibit LPS-induced TNFα in an in vivo murine model. To our knowledge, this is the first published in vivo demonstration that inhibition of the IRAK4 pathway by a small molecule can recapitulate the phenotype of IRAK4 knockout mice.
SYNTHESIS
//////////PF-05388169, TLR signaling, Indoloquinoline, IRAK4, Kinase inhibitor, Inflammation, PRECLINICAL, 1604034-78-7
C(COC)OCCOc4c(cc3\C2=N\c1cc(ccc1/C2=C/Nc3c4)C#N)OC