

| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9221751 | USE OF 1, 3-DIPHENYLPROP-2-EN-1-ONE DERIVATIVES FOR TREATING LIVER DISORDERS |
2014-10-24
|
2015-02-19
|
| US8058308 | SUBSTITUTED 1, 3-DIPHENYLPROP-2-EN-1-ONE DERIVATIVES, PREPARATION AND USES THEREOF |
2011-08-04
|
2011-11-15
|
| US8106097 | COMPOSITION BASED ON SUBSTITUTED 1, 3-DIPHENYLPROP-2-EN-1-ONE DERIVATIVES, PREPARATION AND USES THEREOF |
2010-05-13
|
2012-01-31
|
| US7566737 | Combinations of substituted 1, 3-diphenylprop-2-EN-1-one derivatives with other therapeutically active ingredients |
2007-02-08
|
2009-07-28
|
| US7943661 | Substituted 1, 3-diphenylprop-2-en-1-one derivatives and preparation and uses thereof |
2005-08-11
|
2011-05-17
|
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
Join me on Linkedin
Join me on Researchgate
Join me on Facebook
 FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
Googleplus
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
GoogleplusMYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64


GFT 505, Elafibranor, элафибранор , إيلافيبرانور , 依非兰诺

(E)-Elafibranor
- Molecular FormulaC22H24O4S
- Average mass384.489 Da
Elafibranor
CAS 824932-88-9 E Z MIXTURE USAN
CAS 923978-27-2 E ISOMER INN
2-(2,6-Dimethyl-4-{3-[4-(methylsulfanyl)phenyl]-3-oxo-1-propen-1-yl}phenoxy)-2-methylpropanoic acid
Elafibranor(GFT505)
GFT505;GFT-505;GFT 505
UNII:2J3H5C81A5
(E)-Elafibranor
2-(2,6-Dimethyl-4-{(1E)-3-[4-(methylsulfanyl)phenyl]-3-oxo-1-propen-1-yl}phenoxy)-2-methylpropanoic acid
2-(2,6-Dimethyl-4-{(1E)-3-[4-(methylsulfanyl)phenyl]-3-oxo-1-propen-1-yl}phenoxy)-2-methylpropansäure
2J3H5C81A5
CAS 923978-27-2 E ISOMER INN
Acide 2-(2,6-diméthyl-4-{(1E)-3-[4-(méthylsulfanyl)phényl]-3-oxo-1-propén-1-yl}phénoxy)-2-méthylpropanoïque[French] [ACD/IUPAC Name]
GFT505
Propanoic acid, 2-[2,6-dimethyl-4-[(1E)-3-[4-(methylthio)phenyl]-3-oxo-1-propen-1-yl]phenoxy]-2-methyl-
Treatment of Non-Alcoholic Steato-Hepatitis, Reducing Cardiometabolic Risk Factors in Patients with Diabetes and Pre-Diabetes
OTHERS
US7385082
US8058308
CN 106674069
WO 2016127019
WO 2018060373
WO 2018060372
INNOVATOR Genfit SA
FAST TRACK FDA
Fibrosis; Primary biliary cirrhosis; Cholangitis; Obesity; Non-alcoholic steatohepatitis; Lipid metabolism disorder; Cancer; Non-insulin dependent diabetes; Crohns disease
Genfit is developing elafibranor (GFT-505; structure shown), a PPAR alpha and delta agonist with antioxidant properties and an anti-inflammatory action, for the potential oral treatment of non-alcoholic steatohepatitis (NASH) dyslipidemia, type 2 diabetes, atherogenic dyslipidemia, abdominal obesity and primary biliary cholangitis (PBC)
REGULATORY
In November 2016, the EMA approved elafibranor’s Pediatric Investigation Plan (PIP) . In February 2017, the company expected to obtain conditional marketing authorization for elafibranor in NASH during the course of the second half of 2019 or first half of 2020 .
In February 2014, the FDA granted Fast Track designation for GFT-505 for the treatment of NASH
PHASE III
In March 2015, the company was planning to begin a late stage phase III trial in patients with seriously Ill NASH (expected n = 2,000)
EUROPE
| Active substance | Elafibranor |
|---|---|
| Decision number | P/0237/2016 |
| PIP number | EMEA-001857-PIP01-15 |
| Pharmaceutical form(s) | Capsule, hard; Coated tablet |
| Condition(s)/indication(s) | Treatment of non-alcoholic fatty liver disease (NAFLD) including non-alcoholic steatohepatitis (NASH) |
| Route(s) of administration | Oral use |
| PIP applicant | Genfit SA France Tel.+33 320164000 Fax +33 320164001 Email: contact@genfit.com |
| Decision type | P: decision agreeing on a investigation plan, with or without partial waiver(s) and or deferral(s) |
Doubts on drug substance
- Elafibranor
- GFT 505
- GFT-505
- UNII-2J3H5C81A5
scifinder refers to CAS Registry Number 923978-27-2 as E isomer
- 2-[2,6-Dimethyl-4-[(1E)-3-[4-(methylthio)phenyl]-3-oxo-1-propen-1-yl]phenoxy]-2-methylpropanoic acid
- GFT 505
SYNTHESIS
6 STEPS
WO 2005005369, WO 2004005233

SYN 2
CN106674069

Solubility (25°C)
| In vitro | DMSO | 76 mg/mL (197.66 mM) |
|---|---|---|
| Ethanol | 76 mg/mL (197.66 mM) | |
| Water | Insoluble |
Biological Activity
| Description | Elafibranor is an agonist of the peroxisome proliferator-activated receptor-α(PPAR-alpha) and peroxisome proliferator-activated receptor-δ(PPAR-δ). It improves insulin sensitivity, glucose homeostasis, and lipid metabolism and reduces inflammation. | ||
|---|---|---|---|
| Targets |
|
||
| In vitro | GFT505 is a novel PPAR modulator that shows a preferential activity on PPAR-α and concomitant activity on PPAR-δ[2]. | ||
| In vivo | Elafibranor (GFT505) is a dual PPARα/δ agonist that has demonstrated efficacy in disease models of nonalcoholic fatty liver disease (NAFLD)/NASH and liver fibrosis. In the rat, GFT505 concentrated in the liver with limited extrahepatic exposure and underwent extensive enterohepatic cycling. Elafibranor confers liver protection by acting on several pathways involved in NASH pathogenesis, reducing steatosis, inflammation, and fibrosis. GFT505 improved liver dysfunction markers, decreased hepatic lipid accumulation, and inhibited proinflammatory (interleukin-1 beta, tumor necrosis factor alpha, and F4/80) and profibrotic (transforming growth factor beta, tissue inhibitor of metalloproteinase 2, collagen type I, alpha 1, and collagen type I, alpha 2) gene expression[1]. |
* Please note that Selleck tests the solubility of all compounds in-house, and the actual solubility may differ slightly from published values. This is normal and is due to slight batch-to-batch variations.
Elafibranor (code name GFT505) is a multimodal and pluripotent medication for treatment of atherogenic dyslipidemia for an overweight patient with or without diabetes. It is an oral treatment that acts on the 3 sub-types of PPAR (PPARa, PPARg, PPARd) with a preferential action on PPARa. As of February 2016, elafibranor has completed 8 clinical trials and a phase III is in progress.
Elafibranor (INN,[2] code name GFT505) is an experimental medication that is being studied and developed by Genfit for the treatment of cardiometabolic diseases including diabetes, insulin resistance, dyslipidemia, and non-alcoholic fatty liver disease (NAFLD).[3][4][5]
Elafibranor is a dual PPARα/δ agonist.[6][7]
Elafibranor is an agonist of the peroxisome proliferator-activated receptor-α(PPAR-alpha) and peroxisome proliferator-activated receptor-δ(PPAR-δ). It improves insulin sensitivity, glucose homeostasis, and lipid metabolism and reduces inflammation
FT505 is an oral treatment that acts on the 3 sub-types of PPAR (PPARa, PPARg, PPARd) with a preferential action on PPARa. It has a sophisticated mechanism of action. It is able to differentially recruit cofactors to the nuclear receptor, which subsequently lead to differential regulation of genes and biological effect. Therefore, the ability to identify and profile the activity of selective nuclear receptor modulator (SNuRMs) is a powerful approach to select innovative drug candidates with improved efficacy and diminished side effects. These pluripotent and multimodal molecules have significant positive effects on obesity, insulin-resistance and diabetes, atherosclerosis, inflammation, and the lipid triad (increasing of HDL cholesterol, lowering of triglycerides and LDL cholesterol).
Clinical studies
Administered to over 800 patients and healthy volunteers to date, elafibranor has demonstrated:
- beneficial properties for non-alcoholic steatohepatitis (NASH)[8]
- improvement of insulin sensitivity and glucose homeostasis[9]
Phase 2b (GOLDEN) results were published online in Gastroenterology in February 2016[10] and will be fully available in the paper version in May 2016.
As of February 2016, elafibranor has completed 8 clinical trials and a phase III is in progress.[11]
Pre-clinical studies
Efficacy on histological NASH parameters (steatosis, inflammation, fibrosis) in animal disease models — anti-fibrotic activities.[12]
The absence of safety concern has been confirmed in a full toxicological package up to 2-year carcinogenicity studies and cardiac studies (in mice).[13]
PATENT
20060142611 or 20050176808
Patent
US20070032543
https://patents.google.com/patent/US20070032543A1/en
- Compound 29: 1-[4-methylthiophenyl]-3-[3,5-dimethyl-4-carboxydimethylmethyloxyphenyl]prop-2-en-1-one
-

-
This compound was synthesized from 1-[4-methylthiophenyl]-3-[3,5-dimethyl-4-isopropyloxycarbonyldimethylmethyloxyphenyl]prop-2-en-1-one (compound 28) according to general method 5 described earlier.
-
Purification was made by chromatography on silica gel (elution: dichloromethane/methanol 98:2).
-
1H NMR DMSO-d6 δppm: 1.39 (s, 6H), 2.22 (s, 6H), 2.57 (s, 3H), 7.40 (d, J=8.55 Hz, 2H), 7.57 (s, 2H), 7.62 (d, J=15.5 Hz, 1H), 7.83 (d, J=15.5 Hz, 1H), 8.1 (d, J=8.55 Hz, 2H), 12.97 (s, 1H).
-
MS (ES-MS): 383.3 (M−1).
PATENT
WO 2016127019
PATENT
CN 106674069
https://patents.google.com/patent/CN106674069A/enhttps://patents.google.com/patent/CN106674069A/en
The liver is one of the most important organs of the body, is one of the highest organ of risk. Many factors can lead to liver disease. For example, drinking too much can lead to cirrhosis, excessive medication can lead to liver damage and even obesity can lead to fatty liver. Thus, the pharmaceutical treatment of fatty liver diseases has become a hot spot of bio-pharmaceutical development.
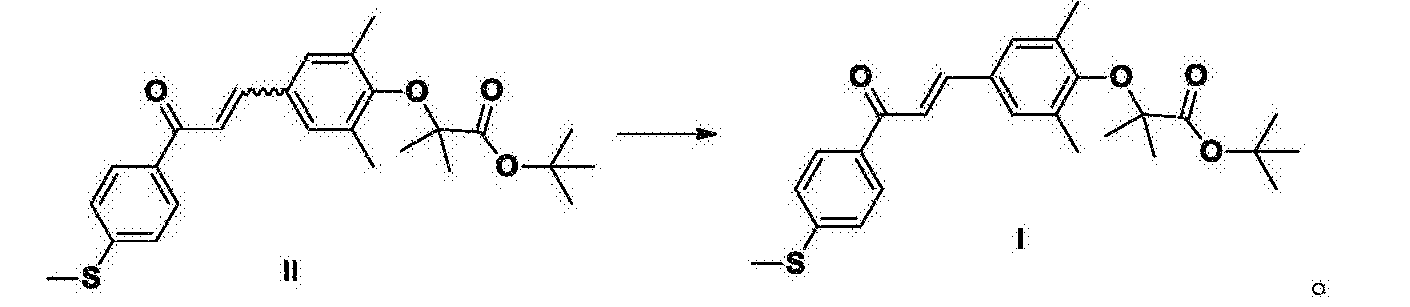
French Genf biopharmaceutical company said recently that the US Food and Drug Administration has agreed to continue the development of peroxisome proliferator-activated receptor α / δ dual agonist GFT505, and begin Phase IIb study in the United States. GFT 505 is expected to rule early diagnosis of fatty liver, heart disease and its complications, prevention and treatment of diabetes-related lipid hyperlipidemia. French Food and Drug Administration approval to a detailed in-depth far for preclinical and clinical data were analyzed based. Experts expressed the Authority, GFT505 to ensure safe operation and research and can lead to liver cancer or liver cirrhosis related biomarkers all favorable. GFT505 structure as shown in formula III.

GFT505 Intermediate I is a key intermediate GFT505III, the existing technology (e.g., Patent Document 1 ^ 1 ^ 20060142611 or 20050176808) are synthesized by the method of 4-methylthio-acetophenone and 3,5 dimethyl-4-hydroxybenzaldehyde GFT505 condensation of intermediate IV, with 2-bromo-iso-butyric acid tert-butyl ester obtained. Process GFT505 Intermediate I Z double bond configuration is a type, but the 4-methylthio-acetophenone and 3,5_-dimethyl-4-hydroxybenzaldehyde condensation process, the formation of a double bond, it is difficult GFT505 avoid intermediate IV of formula Z, E mixtures of formula, and then 2-bromo-iso-butyric acid tert-butyl ester to give GFT505 intermediate II, R is also of formula Z, E mixtures of formula. E-isomer and Z-type polarity very close to the crystallization purification difficult, very precise product by column chromatography is not suitable for industrial production.

Accordingly, a need to find an efficient synthesis, reducing the content of Z-isomer impurities to improve the purity and yield of the products, and to avoid use of column chromatography purification process difficult industrialization.
The present invention provides a method for the preparation of intermediate I GFT505, comprising the steps of: an organic solvent, a compound II with an alkali metal t-butoxide isomerization reaction to give intermediate I GFT505; the said compound II is a double bond in Z / E mixtures, according GFT505 intermediate I is a compound of formula E; the double bonds in Z / E mixtures of formula Z refers to the product from 0.1% to 99.0% of the total mass of the mixture (including 0.1%, comprising 99.0%); the compound of formula E E means that the content of the compound of formula more than 99.0% (including 99.0%);
In reaction I of the preparation of intermediates GFT505, the organic solvent is preferably a protic solvent, a polar aprotic organic solvent non-polar solvent, more preferably a non-polar solvent. The protic solvent is preferably & ~ (: 4 alcoholic solvent; the & ~ (: t-butanol 4 alcoholic solvent preferably the polar aprotic organic solvent is preferably C 1-C4 nitrile solvents, &. ~ C6 ketone solvents, C1-C4 one or more 4 sulfone amide solvents and C1-C solvent. C1-C4 of the nitrile solvents preferably acetonitrile. the C 1-C6 ketone solvent preferably acetone and / or methyl isobutyl ketone. C1-C4 of the amide-based solvent is preferably N, N- dimethylformamide. C 1-C4 of the sulfone solvent is preferably dimethylsulfoxide. the said nonpolar solvent is preferably aromatic hydrocarbon solvent; the aromatic hydrocarbon solvent preferably toluene.
Example 1: Preparation of intermediate IV GFT505 (refer to Patent W02011 / 144579)

A mixture of 4-mercapto-acetophenone (50g, 0.30 Imo 1), 3,5- dimethyl-4-hydroxybenzaldehyde (45g, 0.30 Imo 1) was added to a methanol solution of hydrogen chloride in 200ml (4moI / L) , 20 ~ 30 ° C for 3 hours, cooled to 0 ~ 10 ° C, stirred for 1 hour, filtered and dried to give 83g GFT505 intermediate (IV) as a yellow solid in 93% yield.
Example 2: Preparation of intermediate IV GFT505 (refer to Patent W02011 / 144579)
A mixture of 4-mercapto-acetophenone (I 9Kg, 114mo 1), 3,5- dimethyl-4-hydroxybenzaldehyde (I 7.1Kg, 114mo 1) was added to a methanol solution of hydrogen chloride in 76L (4mol / L ), 20 ~ 30 ° C for 3 hours, cooled to 0 ~ 10 ° C, stirred for 1 hour, centrifuged, 40 ° C and dried under vacuum for 12 hours to obtain 31.6Kg GFT505 intermediate (IV) as a yellow solid, yield 93% . LCMS: m / z = 299 (M + H) +.
Example 3: GFT505 intermediate II preparation (Ref US2006 / 142611)

The GFT505 Intermediate IV (78.8g, 0.263mol) was added to the reaction flask was added acetonitrile (480 ml of), potassium carbonate (54.5g, 0.395mol), tert-butyl 2-bromo-isobutyrate (39.3 g, 0.176mol), heated to 75 ~ 85 ° C for 10 hours, additional potassium carbonate (54.5g, 0.395mol), 2_ tert-butyl bromoisobutyrate (39.3g, 0.176mol) 10 hours, refed with potassium carbonate (54 · 5g, 0 · 395mol), 2- tert-butyl bromoisobutyrate (39 · 3g, 0 · 176mol) for 10 hours, until completion of the reaction compound, and concentrated under reduced pressure to dryness, was added 800g 400g of dichloromethane and water, layers were separated, washed with water, the organic phase dried over anhydrous sodium sulfate, filtered, the organic phase was concentrated to dryness, ethyl acetate and petroleum ether to give a solid compound II 81. Ig, yield 70% 〇
Example 4: GFT505 intermediate II preparation (Ref US2006 / 142611)
The GFT505 Intermediate IV (30Kg, 100mol) was added to acetonitrile (183L) was added potassium carbonate (21Kg, 152mol), 2- tert-butyl bromoisobutyrate (14 · 9Kg, 66 · 8mol), was heated to 75 ~ 85 ° C for 10 hours, additional potassium carbonate (21Kg, 152mol), 2- tert-butyl bromoisobutyrate (14.9Kg, 66.8mol) for 10 hours, refed with potassium carbonate (21Kg, 152mol), 2- tert-butyl bromoisobutyrate (14.9Kg, 66.8mol) for 10 hours, until the reaction was complete compound, 45 ~ 55 ° C was slowly concentrated under reduced pressure to distilled off, water was added and 300Kg 160Kg dichloromethane , the organic layer was separated out, IOOKg IOOKg water and washed with 10% concentration of aqueous sodium chloride solution (the mass concentration refers to the percentage by mass of the total mass of sodium chloride aqueous solution), 15 to 25 ° C was slowly distilled off under reduced pressure to concentrate. Ethyl acetate was added IOOKg was heated to 75 ~ 85 ° C a clear solution was added heptane 180Kg, cooled to stirred 15 ~ 25 ° C for 2-3 hours. Centrifugation, washed with n-heptane 40Kg, 40 ~ 50 ° C was dried in vacuo for 12 hours to obtain 31.6Kg GFT505 intermediate II, R a yield of 71.6%. LC-MS: m / z = 441 (M + H) + square
Example 5: Preparation of Intermediate I GFT505

Compound II (81 · lg, 0.184mol) was added to 400g of toluene, cooled to 10 ~ 20 ° C, was added sodium tert-butoxide (26.8g, 0.279mol), heated to 50 ~ 60 ° C for 2 hours , 400g of water was added, layers were separated, washed with water, the organic phase concentrated to dryness under reduced pressure, methanol was added to 200ml, cooled to 0-10 ° C, stirred for 1 hour, filtered, 40 ~ 50 ° C (-0 · 08MPa ~ -0 · IMPa ) was dried in vacuo for 12 hours to give a yellow solid 78.8g GFT505 intermediate I, a yield of 97.0% APLC: 99.23% (in terms of E-form, Z configurational isomers accounted for 0.085%, largest other single impurity 0.41%).
Intermediate I the preparation of GFT505: 6 cases of Embodiment

Compound II (31Kg, 70.5mol) was added to 153Kg of toluene, cooled to 10 ~ 20 ° C, was added sodium tert-butoxide (10 · 3Kg, 107mol), warmed to 50 ~ 60 ° C for 2 hours, 160Kg of water, layered, and water IOOKg IOOKg mass concentration of the aqueous solution was washed with 10% sodium chloride (the concentration refers to the percentage by mass of the total mass of sodium chloride aqueous solution), 40 ~ 50 ° C Save concentrated under pressure to slowly distilled off, methanol was added to 60Kg, cooled to 0 ~ 10 ° C, stirred for 1 hour, centrifuged, washed with methanol 20Kg, 40 ~ 50 ° C (-0.08MPa ~ -0.1 MPa) was dried under vacuum for 12 hours to give 30.4 Kg GFT505 yellow solid intermediate I, 1.0 yield 98%. LC-MS: m / z = 441 (M + H) +; HPLC: 99 · 50% E configuration similar terms, Z configurational isomers accounted for 0.082%, largest other single impurity of 0.32%.
7 Example: Preparation of Intermediate I GFT505
The compound II (8.0g, 0.018mol) was added to 64g tert-butanol, cooled to 10 ~ 20 ° C, was added potassium tert-butoxide (6.05g, 0.054mol), heated to 70 ~ 80 ° C Reaction 4 to 5 hours, was added 200g of water, 60g extracted twice with isopropyl acetate, and the organic phase concentrated to dryness under reduced pressure, methanol was added 20ml, cooled to 0-10 ° C, stirred for 1 hour, filtered, 40 ~ 50 ° C (_ 0.08MPa ~ -0.1 MPa) was dried in vacuo for 12 hours to give 7.62g yellow solid GFT505 intermediate I, a yield of 95.2% dHPLC: 99.36% (in terms of E-form, Z configurational isomers accounted for 0.079%, single largest other 0.42% impurities).
Example 8: Preparation of Intermediate I GFT505
Compound II (8.Og, 0.018mo 1) was added to 16g N, N- dimethylformamide, cooled to 10 ~ 20 ° C, was added sodium tert-butoxide (2.17g, 0.023mol), heated to the reaction 90 ~ 100 ° C for 1-2 hours, was added 100g of water, 60g extracted twice with isopropyl acetate, the organic phase concentrated to dryness under reduced pressure, methanol was added 20ml, cooled to O-HTC, stirred for 1 hour, filtered, 40 ~ 50 ° C (-0.08MPa ~ -0 IMPa.) was dried in vacuo for 12 hours to give 7.34g yellow solid GFT505 intermediate I, a yield of 91.7% APLC: 99.21% E configuration similar terms, Z configurational isomers accounted 0.097%, the largest single other impurities 0.48%).
9 Example: Preparation of Intermediate I GFT505
The compound II (8.0g, 0.018mol) was added to 160g of acetonitrile, cooled to 10 ~ 20 ° C, was added lithium t (7.21g, 0.090mol) butanol, warmed to 40 ~ 50 ° C the reaction 9-10 hours, was added 160g of water, 90g extracted twice with isopropyl acetate, and the organic phase concentrated to dryness under reduced pressure, methanol was added 20ml, cooled to 0-10 ° C, stirred for 1 hour, filtered, 40 ~ 50 ° C (_ 0.08MPa ~ -0.1 MPa) was dried in vacuo for 12 hours to give 7.29g yellow solid GFT505 intermediate I, a yield of 91.1% dHPLC: 99.16% (in terms of E-form, Z configurational isomers accounted for 0.089%, largest other single impurity 0.49 %).
10 Example: Preparation of Intermediate I GFT505
The compound II (8.0g, 0.018mol) was added to 28g of dimethyl sulfoxide, cooled to 10 ~ 20 ° C, was added potassium t-butoxide (5.04g, 0.045mol), heated to 60 ~ 70 ° C the reaction 3 to 4 hours, was added 100g of water, 60g extracted twice with isopropyl acetate, and the organic phase concentrated to dryness under reduced pressure, methanol was added 20ml, cooled to O-UTC, stirred for 1 hour, filtered, 40 ~ 50 ° C (_ 0.08 MPa ~ -0.1 MPa) was dried in vacuo for 12 hours to give 7.33g yellow solid GFT505 intermediate I, a yield of 91.6% dHPLC: 99.46% (in terms of E-form, Z configurational isomers accounted for 0.077%, largest single impurity other 0.27%).
Preparation of GFT505III: 11 cases of Embodiment

The GFT505 Intermediate I (77.9g, 0.177mol, may be prepared as described in Example 10) was added to the reaction flask was added 790g of dichloromethane was added trifluoroacetic acid (209.7g, 1.84mol), 20 ~ 30 ° C the reaction for 5-6 hours, concentrated to dryness, was added 600ml ethyl acetate and 600ml of water, layers were separated, washed with water, dried over anhydrous sodium sulfate, filtered, concentrated to a small volume the organic phase, 10-20 ° C for 2 hours crystallization, filtration, under -0.08MPa ~ -0.1 MPa, 40 ° C ~ 50 ° C was dried in vacuo 12 hours to give 60.1 g as a yellow solid. 25〇1 yellow solid was recrystallized from ethyl acetate to give 52.98 ^ as a yellow solid 6? 505 (111), a yield of 77.8%.
LC-MS: m / z = 385 (M + H) +; HPLC: 99 · 86%, largest single impurity 0.5 06%.
GFT505III prepared: Example 12 Embodiment
The GFT505 Intermediate I (30Kg, 68.2mol, may be prepared as described in Example 9) was added to 307Kg dichloromethane was added trifluoroacetic acid (80.8Kg, 709mol), 20-30 ° C the reaction 5-6 h, concentrated to dryness, ethyl acetate and water 197Kg 231Kg, layered, and water IOOKg IOOKg concentration of 10 mass% aqueous sodium chloride concentration (which refers to the quality of the aqueous solution of sodium chloride percentage of total mass) washing, 40 ~ 50 ° C to about 80Kg concentrated under reduced pressure, cooled to IO ~ 20 ° C for 2 hours crystallization, centrifugation was washed with ethyl acetate 20Kg, at -0.08MPa ~ -O.IMPa, 40 ~ 50 ° C was dried in vacuo for 12 hours to give a yellow solid was 23.2Kg. As a yellow solid was obtained as a yellow solid GFT505III 20.9Kg 82Kg recrystallized from ethyl acetate, 5.8 79% yield. LCMS: m / z = 385 (M + H) +; HPLC: 99 · 95%, largest single impurity 0.5 03%.
References
- Jump up^ Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. (2011). “Effects of the New Dual PPAR / Agonist GFT505 on Lipid and Glucose Homeostasis in Abdominally Obese Patients with Combined Dyslipidemia or Impaired Glucose Metabolism”. Diabetes Care. 34 (9): 2008–2014. doi:10.2337/dc11-0093. PMC 3161281
 . PMID 21816979.
. PMID 21816979. - Jump up^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 74” (PDF). World Health Organization. p. 10. Retrieved 9 November 2016.
- Jump up^ “Advanced Compound Status” (Press release). Genfit.
- Jump up^ “GFT505 Broadens Its Therapeutic Potential” (PDF) (Press release). Retrieved 31 Mar 2013.
- Jump up^ Cariou, Bertrand; Staels, Bart (2014-10-01). “GFT505 for the treatment of nonalcoholic steatohepatitis and type 2 diabetes”. Expert Opinion on Investigational Drugs. 23 (10): 1441–1448. doi:10.1517/13543784.2014.954034. ISSN 1744-7658. PMID 25164277.
- Jump up^ US Patent No. 7655641 “96 dpi image of original patent USPTO 7655641” (PDF). Retrieved 31 Mar 2013.
- Jump up^ “GFT-505” (PDF). Drugs of the Future. 37 (8): 555–559. 2012.[permanent dead link]
- Jump up^ Staels, Bart; Rubenstrunk, Anne; Noel, Benoit; Rigou, Géraldine; Delataille, Philippe; Millatt, Lesley J.; Baron, Morgane; Lucas, Anthony; Tailleux, Anne (2013-12-01). “Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis”. Hepatology. 58 (6): 1941–1952. doi:10.1002/hep.26461. ISSN 1527-3350.
- Jump up^ Cariou, Bertrand; Hanf, Rémy; Lambert-Porcheron, Stéphanie; Zaïr, Yassine; Sauvinet, Valérie; Noël, Benoit; Flet, Laurent; Vidal, Hubert; Staels, Bart (2013-05-28). “Dual Peroxisome Proliferator–Activated Receptor α/δ Agonist GFT505 Improves Hepatic and Peripheral Insulin Sensitivity in Abdominally Obese Subjects”. Diabetes Care. 36: DC_122012. doi:10.2337/dc12-2012. ISSN 0149-5992. PMC 3781493 . PMID 23715754.
- Jump up^ “Elafibranor, an Agonist of the Peroxisome Proliferator-activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening – Gastroenterology”. http://www.gastrojournal.org. Retrieved 2016-03-08.
- Jump up^ clinical trials involving GFT505
- Jump up^ Quintero, Pablo; Arrese, Marco (2013-12-01). “Nuclear control of inflammation and fibrosis in nonalcoholic steatohepatitis: therapeutic potential of dual peroxisome proliferator-activated receptor alpha/delta agonism”. Hepatology. 58 (6): 1881–1884. doi:10.1002/hep.26582. ISSN 1527-3350. PMID 23787705.
- Jump up^ Hanf, Rémy; Millatt, Lesley J.; Cariou, Bertrand; Noel, Benoit; Rigou, Géraldine; Delataille, Philippe; Daix, Valérie; Hum, Dean W.; Staels, Bart (2014-11-01). “The dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 exerts anti-diabetic effects in db/db mice without peroxisome proliferator-activated receptor gamma-associated adverse cardiac effects”. Diabetes & Vascular Disease Research. 11 (6): 440–447. doi:10.1177/1479164114548027. ISSN 1752-8984. PMID 25212694.
External links
- Genfit Pharmaceutical
- NashBiotechs Several articles on drug candidates in NASH
 |
|
| Clinical data | |
|---|---|
| Synonyms | GFT505, SureCN815512 |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| Chemical and physical data | |
| Formula | C22H24O4S |
| Molar mass | 384.489 g/mol |
| 3D model (JSmol) | |
/////////////////Elafibranor, E Elafibranor, 923978-27-2, GFT-505, UNII-2J3H5C81A5, GFT505, GFT 505, элафибранор , إيلافيبرانور , 依非兰诺 , PHASE 3, FAST TRACK
CC1=CC(=CC(=C1OC(C)(C)C(=O)O)C)C=CC(=O)C2=CC=C(C=C2)SC
Specific Stereoisomeric Conformations Determine the Drug Potency of Cladosporin Scaffold against Malarial Parasite

Specific Stereoisomeric Conformations Determine the Drug Potency of Cladosporin Scaffold against Malarial Parasite
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b00565
Pronay Das†ab, Palak Babbar†c, Nipun Malhotra†c, Manmohan Sharmac , Goraknath R. Jachakab , Rajesh G. Gonnadebd, Dhanasekaran Shanmugambe, Karl Harlosf , Manickam Yogavelc , Amit Sharmac *, and D. Srinivasa Reddyab* †All three have contributed equally to this work.
aOrganic Chemistry Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune 411008, India
b Academy of Scientific and Innovative Research (AcSIR), New Delhi 110025, India
cMolecular Medicine Group, International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi 110067, India dCenter for Material Characterization, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune 411008, India
e Biochemical Sciences Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune 411008, India
fDivision of Structural Biology, Welcome Trust Centre for Human Genetics, The Nuffield Department of Medicine, University of Oxford, Oxford OX3 7BN, UK
J. Med. Chem., Just Accepted Manuscript
DOI: 10.1021/acs.jmedchem.8b00565
Publication Date (Web): May 21, 2018
Copyright © 2018 American Chemical Society
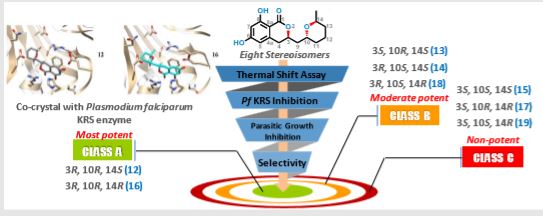
The dependence of drug potency on diastereomeric configurations is a key facet. Using a novel general divergent synthetic route for a three-chiral centre anti-malarial natural product cladosporin, we built its complete library of stereoisomers (cladologs) and assessed their inhibitory potential using parasite-, enzyme- and structure-based assays.
We show that potency is manifest via tetrahyropyran ring conformations that are housed in the ribose binding pocket of parasite lysyl tRNA synthetase (KRS). Strikingly, drug potency between top and worst enantiomers varied 500-fold, and structures of KRS-cladolog complexes reveal that alterations at C3 and C10 are detrimental to drug potency where changes at C3 are sensed by rotameric flipping of Glutamate332.
Given that scores of anti-malarial and anti-infective drugs contain chiral centers, this work provides a new foundation for focusing on inhibitor stereochemistry as a facet of anti-microbial drug development.
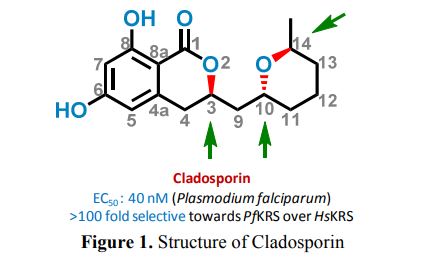
Cladosporin (12) displays exquisite selectivity for the parasite lysyl-tRNA synthetase over human enzyme. This species specific selectivity of cladosporin has been previously described through comprehensive sequence alignment, where the residues val329 and ser346 seem to be sterically crucial for accommodating the methyl moiety of THP ring10. The structural features of compound 12 clearly indicate the presence of three stereocenters, and therefore 2n (n=3) i.e., eight stereoisomers are possible (Fig.1). Till date, only one asymmetric total synthesis of cladosporin13 has been achieved which was followed by another report of formal syntheses14. Here, we have developed a general chemical synthesis route to synthetically access all the eight possible stereoisomers of compound 12.
cladosporin (compound 12) (0.052 g) as a white solid with a yield of 54 %. Melting point: 171-173 °C; [α]25 D = -15.75 (c = 0.6, EtOH); IR υmax(film): cm-1 3416, 3022, 1656, 1218; 1H NMR (400 MHz, CDCl3): δ 11.06 (s, 1H), 7.47 (br. s., 1H), 6.29 (s, 1H), 6.16 (s, 1H), 4.68 (t, J = 9.8 Hz, 1H), 4.12 (s, 1H), 4.01 (s, 1H), 2.89 – 2.75 (m, 2H), 2.00 – 1.94 (m, 1H), 1.87 – 1.81 (m, 1H), 1.70 – 1.63 (m, 4H), 1.35 (d, J = 6.1 Hz, 2H), 1.23 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 169.9, 164.3, 163.1, 141.8, 106.7, 102.0, 101.5, 76.3, 68.0, 66.6, 39.3, 33.6, 30.9, 18.9, 18.1; HRMS calculated for C16H21O5 [M + H]+ 293.1384, observed 293.1379.

Dr. D. Srinivasa Reddy has been appointed as an editor of Bioorganic & Medicinl Chemistry Letters, Elsevier Publications. Congratulation Sir !
Click here for details. https://www.journals.elsevier.com/bioorganic-and-medicinal-chemistry-letters
The research interests of his group lie in issues related to application of oriented organic synthesis, in particular total synthesis of biologically active natural products, medicinal chemistry and crop protection. This team has been credited with having accomplished total synthesis of more than 25 natural products with impressive biological activities. “Some of our recent achievements include identification of potential leads, like antibiotic compound based on hunanamycin natural product for treating food infections, anti-diabetic molecule in collaboration with an industry partner and anti-TB compound using a strategy called ‘re-purposing of a drug scaffold’,” said Reddy.
A total of two awardees out of four were from CSIR institutes. In addition to Reddy, Rajan Shankarnarayanan, CSIR – CCMB, Hyderabad (basic sciences), also was conferred with the award. Vikram Mathews, CMC, Vellore (medical research) and Prof Ashish Suri, AIIMS, New Delhi (clinical research), were the others to receive the awards.
With more than 80 scientific publications and 35 patents, Reddy is one of the most prominent scientists in the city and has already been honoured with the Shanti Swarup Bhatnagar prize in chemical sciences. Reddy is also a nominated member of the scientific body of Indian Pharmacopoeia, government of India and was elected as a fellow of the Telangana and Maharashtra Academies of Sciences in addition to the National Academy of Sciences, India (NASI).
//////////CLADOSPORIN, NCL, CSIR, SRINIVASA REDDY, PUNE, MALARIA
PF 06650833


PF-06650833
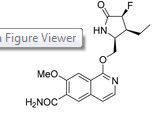
1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide
CAS 1817626-54-2
Chemical Formula: C18H20FN3O4
Molecular Weight: 361.3734
- Originator Pfizer
- Class Anti-inflammatories; Antirheumatics
- Mechanism of Action Interleukin-1 receptor-associated kinase inhibitors
- Phase II Rheumatoid arthritis
- Phase I Lupus vulgaris
- 01 Aug 2018 Pfizer completes a phase II trial in Rheumatoid arthritis (Treatment-experienced) in USA, Ukraine, Taiwan, Serbia, Russia, Romania, Poland, Mexico, South Korea, Georgia, Bosnia-Herzegovina, Australia, Croatia, Spain, Slovakia, Czech Republic, Hungary, Germany, Bulgaria (PO) (NCT02996500)
- 28 Jul 2018 No recent reports of development identified for phase-I development in Lupus(In volunteers) in USA (PO, Controlled release)
- 28 Jul 2018 No recent reports of development identified for phase-I development in Lupus(In volunteers) in USA (PO, Immediate release)
- PF-06650833 is an inhibitor of Interleukin-1 receptor associated kinase 4 (IRAK4). RAK4 is located proximal to TLR/IL-1 receptors, and in preclinical studies, inhibits downstream signaling from these receptors. The development of novel small molecule inhibitors of this kinase has the potential to lead to new therapeutics to treat diseases such as rheumatoid arthritis, lupus, and lymphomas.
Interleukin-1 receptor associated kinase 4 (IRAK-4) is a serine threonine kinases that plays a key role in innate immune signaling. IRAK-4 is activated by the interleukin (IL-1) family receptors (IL-1R, IL-18R, and IL-33R), as well as the Toll-like receptors (TLRs). Inhibition of IRAK-4 blocks the production of inflammatory cytokines such as type I interferons, tumor necrosis factor (TNF), IL-1, IL-6, and IL-12 that are key drivers of autoimmune and inflammatory diseases. IRAK-4 is an attractive therapeutic target for diseases associated with dysregulated inflammation, such as systemic lupus erythematosus and rheumatoid arthritis.


First Discovery Synthesis of 1
Conditions: (a) LDA (1.2 equiv), TMSCl (1.3 equiv), THF, −60 °C, 30 min; (b) allyl methyl carbonate (1.1 equiv), Pd(OAc)2 (0.05 equiv), THF, 65 °C, 2 h, 73% (2 steps); (c) LiThCN (1.5 equiv), EtMgCl (1.5 equiv), TMSCl (2.0 equiv), THF, −78 °C, 6 h, 90%; (d) LDA (1.8 equiv), NFSI (1.25 equiv), THF, −78 °C, 1 h, 23% (8), 45% (9); (e) pTsOH (0.05 equiv), MeCN, H2O, 90 °C, 2 h, 97%; (f) 3 (0.9 equiv), KHMDS (2.0 equiv), DMF, THF, −10 °C, 30 min, 84%; (g) H2O2 (10 equiv), K2CO3 (4.0 equiv), DMSO, 20 °C, 2 h, 97%.
CLIP
Target: Interleukin-1 receptor associated kinase 4 (IRAK4): This kinase is important in innate immunity, and its inhibition is predicted to be beneficial in treating inflammatory diseases.
Disease: Rheumatoid arthritis, inflammatory bowel disorder
Notes: PF06650833 came from a screening assay that used nuclear magnetic resonance spectroscopy to determine binding between molecular fragments and IRAK4. The initial hit, which bound weakly to IRAK4, was optimized with structure- and property-based medicinal chemistry to generate a series of potent inhibitors, said Katherine Lee, an associate research fellow at Pfizer.
Paper
Improvements to Enable the Large Scale Synthesis of 1-{[(2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide (PF-06650833)
Stephen W. Wright*§  , Bryan Li‡ , Zhihui Peng‡, Lulin Wei‡, Emma McInturff‡, David Place‡, David B. Damon‡, and Robert A. Singer‡
, Bryan Li‡ , Zhihui Peng‡, Lulin Wei‡, Emma McInturff‡, David Place‡, David B. Damon‡, and Robert A. Singer‡
, Bryan Li‡ , Zhihui Peng‡, Lulin Wei‡, Emma McInturff‡, David Place‡, David B. Damon‡, and Robert A. Singer‡ § Medicine Design, Pfizer Worldwide Research and Development, 445 Eastern Point Road, Groton, Connecticut 06340, United States
‡ Chemical Research and Development, Pfizer Worldwide Research and Development, 445 Eastern Point Road, Groton, Connecticut 06340, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.8b00386
*E-mail: stephen.w.wright@pfizer.com.

Senior Principal Scientist at Pfizer Inc.
New London/Norwich, Connecticut Area

Robert Singer
Process Chemist -Assoc Research Fellow at Pfizer
New London/Norwich, Connecticut Area
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00386/suppl_file/op8b00386_si_001.pdf

An improved process for the large scale synthesis of 1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide (1), a candidate currently in clinical development, was developed. Key objectives were to eliminate chromatographic purifications, to maximize the reproducibility of each step, and to improve the yield and efficiency of each step relative to the previous discovery syntheses of 1. This work was focused on improvements to the synthesis of the stereochemically complex lactam 2. Steps of particular concern were the preparation of the unsaturated lactam 6, the cuprate conjugate addition reaction to produce 7, and the conversion of 7 to 8 with a high degree of diastereoselection. The solutions to these challenges have permitted the synthesis of 2 in excess of 100 kg, which in turn has permitted 1 to be prepared in sufficient amounts to support further development.
1 (31.3 kg, 91%, 82% overall) as a white, free-flowing powder.
1H NMR (500 MHz, DMSO): δ 8.86 (s, 1H), 8.16 (s, 1H), 7.90 (d, J = 5.9 Hz, 1H), 7.84 (br. s., 1H), 7.74 (s, 1H), 7.70 (br. s., 1H), 7.42 (d, J = 5.9 Hz, 1H), 4.90 (dd, J = 5.9, 53.8 Hz, 1H), 4.54 (dd, J = 3.5, 11.1 Hz, 1H), 4.26 (dd, J = 6.4, 11.0 Hz, 1H), 4.13–4.05 (m, 1H), 3.97 (s, 3H), 2.69–2.54 (m, 1H), 1.68–1.53 (m, 2H), 1.02 (t, J = 7.3 Hz, 3H).
13C NMR{1H} (126 MHz, DMSO): δ 171.0 (d, J = 19.4 Hz), 166.4, 158.4, 155.1, 137.7, 131.8, 130.3, 128.4, 120.3, 115.2, 103.2 (d, J = 4.2 Hz), 90.0 (d, J = 179.2 Hz), 66.3, 56.0, 54.1, 42.2 (d, J = 19.4 Hz), 16.4 (d, J = 8.4 Hz), 12.1.
19F NMR (H decoupled, 376 MHz, DMSO-d6): δ −199.26.
LCMS: 362 (MH+).


//////////////PF-06650833, PF 06650833, PF06650833, PF-6650833, PF 6650833, PF6650833.
O=C(C1=CC2=C(C(OC[C@H]([C@H](CC)[C@@H]3F)NC3=O)=NC=C2)C=C1OC)N
/////////////////PF-06650833, PF 06650833, Phase 3, Atopic dermatitis, PFIZER, Breakthrough Therapy Designation
LRH-1 agonism favours an immune-islet dialogue which protects against diabetes mellitus
Sreeni Labs Private Limited

Dr.Sreenivasa Mundla Reddy, Email Sreeni@sreenilabs.com
Phone :+91-9866092626, +91-40-27173353, Telangana, INDIA Web Sreeni Labs
SREENI LABS CONTRIBUTION
Customer requested Sreeni Labs to make BL001 first on few mg scale. Sreeni labs synthesized and supplied in a short time with full characterization data. Later, customer requested us to make it on several gram scale and we synthesized and delivered as custom synthesis project.
LRH-1 agonism favours an immune-islet dialogue which protects against diabetes mellitus
NATURE COMMUNICATIONS | (2018) 9:1488 |DOI: 10.1038/s41467-018-03943-0 | http://www.nature.com/naturecommunications
Type 1 diabetes mellitus (T1DM) is due to the selective destruction of islet beta cells by
immune cells. Current therapies focused on repressing the immune attack or stimulating beta
cell regeneration still have limited clinical efficacy. Therefore, it is timely to identify innovative
targets to dampen the immune process, while promoting beta cell survival and function. Liver
receptor homologue-1 (LRH-1) is a nuclear receptor that represses inflammation in digestive
organs, and protects pancreatic islets against apoptosis. Here, we show that BL001, a small
LRH-1 agonist, impedes hyperglycemia progression and the immune-dependent inflammation
of pancreas in murine models of T1DM, and beta cell apoptosis in islets of type 2 diabetic
patients, while increasing beta cell mass and insulin secretion. Thus, we suggest that LRH-1
agonism favors a dialogue between immune and islet cells, which could be druggable to
protect against diabetes mellitus.
//////////////SREENI LABS
FDA approves new drug Doptelet (avatrombopag) for patients with chronic liver disease who have low blood platelets and are undergoing a medical procedure
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
FDA Approves Tavalisse (fostamatinib disodium hexahydrate) for Chronic Immune Thrombocytopenia — Med-Chemist

Rigel Pharmaceuticals, Inc. announced that the U.S. Food and Drug Administration (FDA) approved Tavalisse (fostamatinib disodium hexahydrate) for the treatment of thrombocytopenia in adult patients with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment. Tavalisse is an oral spleen tyrosine kinase (SYK) inhibitor that targets the underlying autoimmune cause of the…
Mibefradil, a new class of compound to study TRPM7 channel function — Sussex Drug Discovery Centre

Transient receptor potential (TRPM) is a family of non-selective cation channels that are widely expressed in mammalian cells. TRP channels are composed of six transmembrane domains and the family consists of eight different channels, TRPM1–TRPM8. TRPM7 is compromised of an ion channel moiety essential for the ion channel function, which serves to increase intracellular calcium […]
via Mibefradil, a new class of compound to study TRPM7 channel function — Sussex Drug Discovery Centre
Dark Chocolate improves vision with 2 hours — ClinicalNews.Org

Dark Chocolate improves vision with 2 hours Contrast sensitivity and visual acuity were significantly higher 2 hours after consumption of a dark chocolate bar compared with a milk chocolate bar, but the duration of these effects and their influence in real-world performance await further testing. Rabin JC, Karunathilake N, Patrizi K. Effects of Milk vs […]
via Dark Chocolate improves vision with 2 hours — ClinicalNews.Org
FDA approves new uses for two drugs Tafinlar (dabrafenib) and Mekinist (trametinib) administered together for the treatment of BRAF-positive anaplastic thyroid cancer


FDA approves new uses for two drugs Tafinlar (dabrafenib) and Mekinist (trametinib) administered together for the treatment of BRAF-positive anaplastic thyroid cancer
The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive). Continue reading.
May 4, 2018
Release
The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive).
“This is the first FDA-approved treatment for patients with this aggressive form of thyroid cancer, and the third cancer with this specific gene mutation that this drug combination has been approved to treat,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval demonstrates that targeting the same molecular pathway in diverse diseases is an effective way to expedite the development of treatments that may help more patients.”
Thyroid cancer is a disease in which cancer cells form in the tissues of the thyroid gland. Anaplastic thyroid cancer is a rare, aggressive type of thyroid cancer. The National Institutes of Health estimates there will be 53,990 new cases of thyroid cancer and an estimated 2,060 deaths from the disease in the United States in 2018. Anaplastic thyroid cancer accounts for about 1 to 2 percent of all thyroid cancers.
Both Tafinlar and Mekinist are also approved for use, alone or in combination, to treat BRAF V600 mutation-positive metastatic melanoma. Additionally, Tafinlar and Mekinist are approved for use, in combination, to treat BRAF V600E mutation-positive, metastatic non-small cell lung cancer.
The efficacy of Tafinlar and Mekinist in treating ATC was shown in an open-label clinical trial of patients with rare cancers with the BRAF V600E mutation. Data from trials in BRAF V600E mutation-positive, metastatic melanoma or lung cancer and results in other BRAF V600E mutation-positive rare cancers provided confidence in the results seen in patients with ATC. The trial measured the percent of patients with a complete or partial reduction in tumor size (overall response rate). Of 23 evaluable patients, 57 percent experienced a partial response and 4 percent experienced a complete response; in nine (64 percent) of the 14 patients with responses, there were no significant tumor growths for six months or longer.
The side effects of Tafinlar and Mekinist in patients with ATC are consistent with those seen in other cancers when the two drugs are used together. Common side effects include fever (pyrexia), rash, chills, headache, joint pain (arthralgia), cough, fatigue, nausea, vomiting, diarrhea, myalgia (muscle pain), dry skin, decreased appetite, edema, hemorrhage, high blood pressure (hypertension) and difficulty breathing (dyspnea).
Severe side effects of Tafinlar include the development of new cancers, growth of tumors in patients with BRAF wild-type tumors, serious bleeding problems, heart problems, severe eye problems, fever that may be severe, serious skin reactions, high blood sugar or worsening diabetes, and serious anemia.
Severe side effects of Mekinist include the development of new cancers; serious bleeding problems; inflammation of intestines and perforation of the intestines; blood clots in the arms, legs or lungs; heart problems; severe eye problems; lung or breathing problems; fever that may be severe; serious skin reactions; and high blood sugar or worsening diabetes.
Both Tafinlar and Mekinist can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
The FDA granted Priority Review and Breakthrough Therapy designation for this indication. Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases, was also granted for this indication.
The FDA granted this approval to Novartis Pharmaceuticals Corporation.
///////////////Tafinlar, dabrafenib, Mekinist, trametinib, fda 2018, Priority Review, Breakthrough Therapy designation, Orphan Drug designation, Novartis Pharmaceuticals Corporation,
What are the drugs of the future? — All About Drugs
A cartoon representing how, in history, we are continuously faced with new scientific advancements that make us question what the future holds and whether what we currently have is still useful or should be replaced. What are the drugs of the future? Med. Chem. Commun., 2018, Advance ArticleDOI: 10.1039/C8MD90019A, Opinion Huy X. Ngo, Sylvie Garneau-Tsodikova…
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

