
Home » Uncategorized (Page 81)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AZD 1080
 .
.
AZD 1080
2-Hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]-1H-indole-5-carbonitrile
2-hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]1H-indole-5-carbonitrile
AZD1080 is a selective, orally active, brain permeable GSK3 inhibitor, inhibits human GSK3α and GSK3β with Ki of 6.9 nM and 31 nM, respectively, shows >14-fold selectivity against CDK2, CDK5, CDK1 and Erk2.
Cas 612487-72-6, AZD1080,
AZD-1080, a glycogen synthase kinase 3 (GSK-3) inhibitor, had been in early clinical trials for the treatment of Alzheimer’s type dementia by AstraZeneca
| Astrazeneca Ab |
PATENTS
WO 2003082853
http://www.google.com/patents/WO2003082853A1?cl=en
PAPER
Organic Process Research & Development (2008), 12(3), 540-543.
http://pubs.acs.org/doi/abs/10.1021/op800020r

A mild and robust method for the large-scale palladium-catalysed cyanation of aryl bromides has been developed. The reaction is sensitive to cyanide poisoning of the catalyst, and it was found that the order of adding the reagents had a strong impact on the performance of the reaction. Addition of the cyanide source to a preheated mixture of the other reagents was critical for achieving a robust and scaleable process. This improved protocol allowed the reaction to be run to full conversion within 3 h at 50 °C on a 6.7 kg scale. Furthermore, it led to the identification of several new efficient catalysts for the reaction.
2-hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl]1H-indole-5-carbonitrile (2) (5.2 kg, 15.6 mol), 90% yield with a purity of >90% by HPLC. 1H NMR (d6-DMSO, 400 MHz) δ 14.79 (broad s, 1H), 10.86 (broad s, 1H), 8.08 (s, 1H), 7.95 (s, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.27 (dd,J = 8.0, 0.9 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 3.57 (t, J = 4.4 Hz, 4H), 3.36 (s, 2H), 2.36 (broad s, 4H); 13C NMR (d6-DMSO, 100 MHz) δ 168.8, 148.6, 141.8, 137.0, 136.1, 125.4, 123.9, 122.3, 121.1, 118.8, 118.3, 108.7, 101.3, 84.6, 66.1, 58.4, 52.8. MS (ES) m/z [M + 1] 335.
PAPER
Topics in Organometallic Chemistry (2012), 42(Organometallics as Catalysts in the Fine Chemical Industry), 125-134.
http://link.springer.com/chapter/10.1007%2F3418_2011_25
PATENT
https://www.google.co.in/patents/WO2007089193A1?cl=en

In the above scheme, preferably Rl is bromo and X is chloro.
Synthesis of 2-Hydroxy-3-[5-(morpholin-4-ylmethyl)pyridin-2-yl] lH-indole-5-carbonitrile citrate
Example 14
2-Hydroxy-3-r5-(moφholin-4-ylmethyl)pyridin-2-yl1 lH-indole-5-carbonitrile citrate salt 2-Hydroxy-3-[5-(moφholin-4-ylmethyl)pyridin-2-yl] lH-indole-5-carbonitrile (5.14 kg, 15.4 mol) was suspended in ethanol (54 L) at room temperature. The suspension was heated to an inner temperature of 700C and a solution of citric acid (3.424 kg, 17.82 mol, 1.300 eq)) in water (103 L) was added keeping the inner temperature above 650C. The mixture was heated to reflux. After this the resulting solution was mixed with activated charcoal (0.412 kg) and reflux continued for 3.5 h after which the reaction mixture was clear filtered at 830C followed by cooling to room temperature over 20 h. After filtration the precipitate was washed twice with a cold mixture of ethanol/water (6.9 L/13.7 L). Drying under vacuum at 5O0C gave 6.648 kg, 82.2% yield of 2-hydroxy-3-[5-(morpholin- 4-ylmethyl)pyridin-2-yl]lH-indole-5-carbonitrile citrate having a purity of at least 98%. The palladium content was less than 1 ppm and the zinc content was lower than 10 ppm. 1H NMR (Jd-DMSO3 400 MHz) δ 14.7 (br s, 1 H), 11.55 (s, 1 H), 10.98 (s, IH), 8.31 (s, 1 H), 8.08 (br d, J= 1.84Hz, IH), 8.02 (s, IH), 7.90 (br d, J = 8.92Hz, 1 H), 7.31 (d, J = 8.0 Hz, 1 H), 7.02 (d, J= 8.0Hz), 4.28 (s, 2 H), 3.97 (m, 2 H), 3.94 (m, 2H), 3.35 (m, 9H), 3.32 (m, 2H) ppm; 13C NMR (d6-DMSO, 400MHz) δ 168.9, 148.5, 142.7, 139.8, 137.5,126.4, 124.9, 124.8, 120.9, 119.4, 118.4, 113.3, 109.0, 101.6, 85.7, 63.1, 55.5, 50.3, 40.1, 39.9, 39.7, 39.2, 39.0, 38.8ppm; MS (ES) m/z [M++l] 335.
LIK 066, Licogliflozin diprolinate
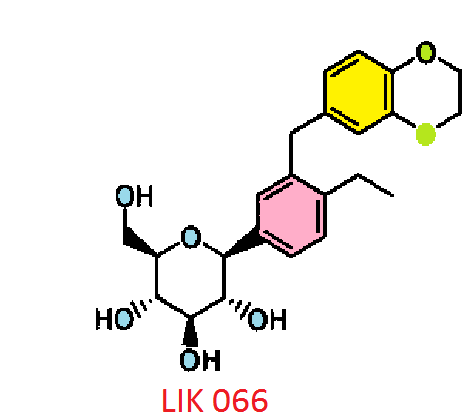
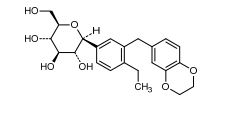
Licogliflozin
LIK 066
Licogliflozin diprolinate

LIK-066, a new flozin on the horizon
C23 H28 O7 . 2 C6 H11 N O, 642.7795, 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C…WO2011048112
CAS 1291095-45-8, (1S)-1,5-anhydro-1-C-[3-[(2,3-dihydro-1,4-benzodioxin-6-yl)methyl]-4-ethylphenyl]-D-glucitol (1:1) WITH L-Proline, compd., 1:1 Proline Co-crvstal , 1:1 Proline Co-crvstal …..…WO2011048112
CAS BASE 1291094-73-9, 416.46, C23 H28 O7
(1S)-1,5-Anhydro-1-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-D-glucitol bis[1-[(2S)-pyrrolidin-2-yl]ethanone]
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Sodium glucose transporter-2 inhibitor
SGLT 1/2 inhibitor
Novartis Ag innovator
Clinical trial……..https://clinicaltrials.gov/ct2/show/NCT01915849
https://clinicaltrials.gov/ct2/show/NCT02470403
- 10 Jun 2015 Novartis initiates enrolment in a phase II trial for Type 2 diabetes mellitus in USA (NCT02470403)
- 02 Apr 2014 Novartis terminates a phase II trial in Type-2 diabetes mellitus in USA, Poland, Argentina, Hungary, Puerto Rico and South Africa (NCT01824264)
- 01 Jan 2014 Novartis completes a phase II trial in Type 2 diabetes mellitus in USA (NCT01915849)
Licogliflozin, a SGLT-1/2 inhibitor, is in phase II clinical development at Novartis for the treatment of metabolic disorders, for the treatment of heart failure in patients with type 2 diabetes, for the treatment of obesity and for the treatment of polycystic ovary syndrome (PCOS) in overweight and obese women. Phase II trials for the treatment of type 2 diabetes had been discontinued.
EMA/415156/2014 European Medicines Agency decision P/0183/2014 of 24 July 2014 on the agreement of a paediatric investigation plan and on the granting of a deferral and on the granting of a waiver for (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3- dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran3,4,5-triol (2:1) (LIK066) (EMEA-001527-PIP01-13) in accordance with Regulation (EC) No 1901/2006 of the European Parliament and of the Council
1. Opinion of the Paediatric Committee on the agreement of a Paediatric Investigation Plan and a deferral and a waiver. 2014, EMEA-001527-PIP01-13 (here) [ Novartis revealed the IUPAC name here].
Where name is given
http://www.who.int/medicines/publications/druginformation/issues/DrugInformation2017_Vol31-4/en/


http://www.who.int/medicines/publications/druginformation/issues/PL_118.pdf?ua=1
SEE ALSO


LIK-066 is in phase II clinical studies at Novartis for the treatment of type 2 diabetes.
In June 2014, the EMA’s PDCO adopted a positive opinion on a pediatric investigation plan (PIP) for LIK-066 for type 2 diabetes
Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition. Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
Type 1 diabetes, also known as Insulin Dependent Diabetes Mellitus (IDDM), is characterized by loss of the insulin-producing β-cells of the islets of Langerhans of the pancreas leading to a deficiency of insulin. Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) – is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin). Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from β-cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
Chronic elevation of blood glucose level also leads to damage of blood vessels. In diabetes, the resultant problems are grouped under “microvascular disease” (due to damage of small blood vessels) and “macro vascular disease” (due to damage of the arteries). Examples of microvascular disease include diabetic retinopathy, neuropathy and nephropathy, while examples of macrovascular disease include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
Diabetic retinopathy, characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US. Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy. Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide. A high glycemic diet (i.e., a diet that consists of meals that give high postprandial blood sugar) is known to be one of the causative factors contributing to the development of obesity.
Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level. Therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
Glucose is unable to diffuse across the cell membrane and requires transport proteins. The transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase. Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
SGLT belongs to the sodium/glucose co-transporter family SLCA5. Two different SGLT isoforms, SGLT1 and SGLT2, have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different. SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
As a consequence, glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments. Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule. In healthy human, more than 99% of plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1 % of the total filtered glucose being excreted in urine. It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule. A drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient’s needs for both blood glucose control, while preserving insulin secretion. In addition, SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
Indeed small molecule SGLT2 inhibitors have been discovered and the anti-diabetic therapeutic potential of such molecules has been reported in literature [T-1095 (Diabetes, 1999, 48, 1794-1800, Dapagliflozin (Diabetes, 2008, 57, 1723-1729)].
SYNTHESIS
PATENT
WO 2011048112
https://www.google.com/patents/WO2011048112A1?cl=en
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
Example 61-62:

Ex. 61
Example 61 : Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
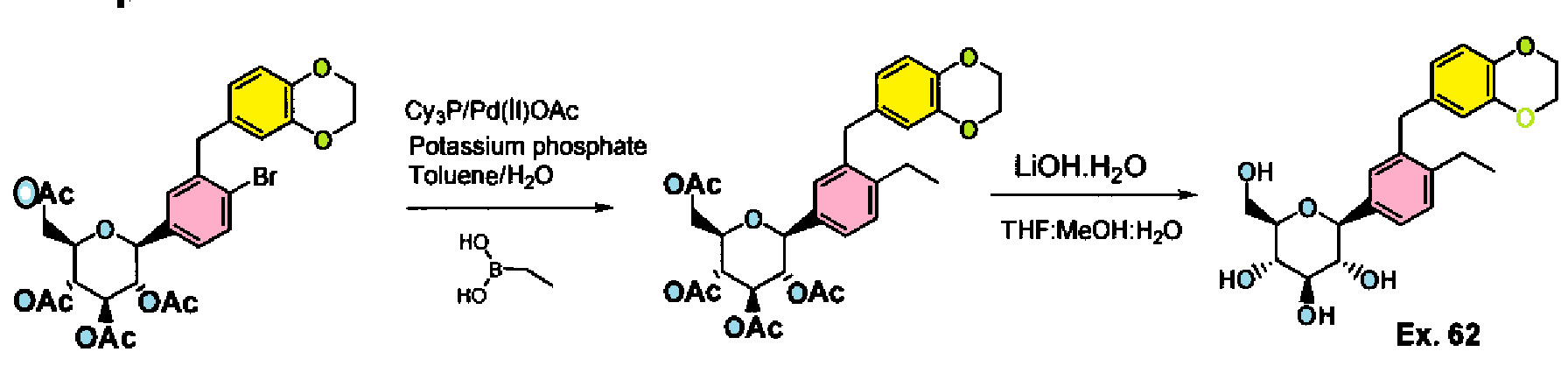
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 mL), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Example 62: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 mL) and washed with brine (75 mL), brine containing 5 mL of 5% aqueous KHS04 (75 mL), and brine (20 mL) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.59)
H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J – 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m z 434.2 (M+18).
PICK UP IDEAS FROM HERE
Examples 57-58:

Ex. 57 Ex. 58
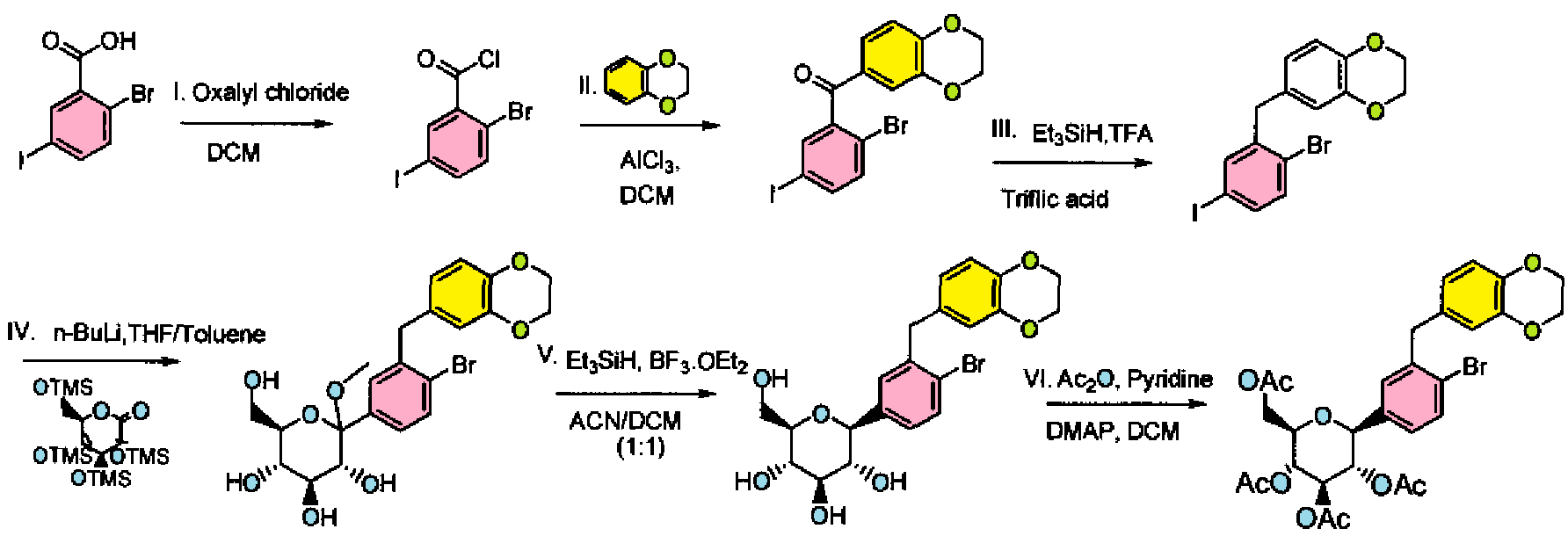
Step I: To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
Step II: To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2). The dichloromethane layers were combined and washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL X 2), and brine (200 mL), then dried over sodium sulfate and concentrated. The solid product was triturated with hexanes, and the triturated product was dried under vacuum to furnish (2-bromo-5-iodo-phenyl)-(2,3- dihydro-benzo[1 ,4]dioxin-6-yl)-methanone (30 g).
1H N R (400 MHz, DMSO-D6): δ 4.29-4.37 (m, 4H), 7.02 (d, J = 8.4 Hz, 1 H), 7.16 (d, J = 2.4 Hz, 1 H), 7.18-7.19 (m, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.77-7.81 (m, 1 H), 7.82 (d, J = 2.0 Hz, 1 H).
Step III: To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution (200 mL X 2), water (200 mL), and brine (200 mL), then dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish 6-(2-bromo-5-iodo-benzyl)-2,3- dihydro-benzo[1 ,4]dioxine (26.5 g). H NMR (400 MHz, DMSO-D6): δ 3.90 (s, 4H), 4.2 (s, 2H), 6.65 (dd, J = 8.4 Hz, J = 2.0 Hz, H), 6.68 (d, J = 2.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, H), 7.39 (d, J = 8.4 Hz, 1 H), 7.50 (dd, J = 8.4 Hz, J = 2.4 Hz 1 H), 7.67 (d, J = 2.8 Hz, 1 H).
Step IV: To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C. The reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C. After stirring for 1 h, 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq. NaHC03 solution (~75 mL) and extracted with ethyl acetate (250 mL X 3), dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish (3R,4S,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (28.4 g)
Example 57: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-1 ,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
Step V: To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C. After stirring for 4 h at 10°C, the reaction was quenched with saturated aqueous sodium bicarbonate (~ 100 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 X 150 mL). The organic layers were combined and dried over sodium sulfate, concentrated to furnish (3R,4R,5S,6R)-2- [4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol (28.4 g). Crude product was used for next reaction without purification. Example 58: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2!3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate Step V: To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature. After stirring for 2 h, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (500ml) and washed with 1 N HCI (200 mL X 2) followed by brine (200ml), then dried over sodium sulfate and
concentrated. The resulting crude compound was dissolved in ethanol (320 mL) at 65 °C and allowed to cool to room temperature while stirring. Light yellow solid formed was filtered and washed with cold ethanol (150 mL) followed by hexane (200 mL) to get acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin- 6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester powder (22.5 g, purity 98%).
COCRYSTAL
Example 75: 1:1 Proline Co-crvstal with f2S.3R.4R.5S.6R¾-2-r3-f2.3-Dihvdro- benzori.41dioxin-6-ylmethyl)-4-ethyl-phenvn-6-hvdroxymethyl-tetrahydro-pyran- 3.4.5-triol
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62) was completely amorphous initially but formed a crystalline complex with proline. This was confirmed by powder X-ray diffraction (PXRD) analysis. The stiochiometry of Example 62 and L- proline in the co-crystal prepared by method 1 was found to be 1 :1 by NMR
spectroscopy & HPLC. Characterization data for co-crystals of Example 62 and proline prepared by method 1 is shown in Table 3. Relative intensities of the most prominent powder x-ray diffraction peaks for co-crystals of Example 62 and proline are shown in Table 3A.
Table 3

Table 3A

3.70 15.78 18.36 25.18
9.68 10.68 18.88 36.33
11.07 21.21 20.42 69.29
14.26 14.81 21.18 27.94
14.80 22.97 22.50 12.25
15.40 4 98 23.78 33.08
16.12 8.45 24.56 6.92
16.59 18.78 25.79 21.69
17.31 100.0 27.46 8.90
17.60 20.35 31.97 7.65
17.98 47.20 32.46 5.98
1:1 Proline Co-crvstal
Example 77: 1:1 Proline Co-crvstal with (2S.3R.4R.5S.6Ri-2-f3-(2.3-Dihvdro- benzoh .41dioxin-6-ylmethvh-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 2:
1 :1 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1500mg,3.6mmol), L- proline (415mg, 3.6mmol) and ethanol (23 ml_) were added to a 50 mL 3-neck round bottom flask equipped with nitrogen purging, magnetic stirring bar,
thermometer pocket & calcium chloride guard tube and the mixture was stirred at 25-30°C for 30 min., then heat to reflux. A clear solution was observed which was refluxed for 30 min., then slowly cool to 25-30°C causing percipitation. Di- isopropyl ether (DIPE, 23 mL) was added while maintaining the mixture at 25-30°C and stirring continuously for additional one to two hours at the same temperature. The precipitate was collected by filtration using vacuum (Nitrogen atmosphere), and the filter cake was washed with ethanol-DIPE mixture (1 :1 v/v, 10ml) followed by DIPE (23 mL). The product was vacuum dried at 65-70°C for 5-6 hrs.
1:1 Proline Co-crvstal (ΔΗ 53 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig. 1. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 2.
2:1 Proline Co-crvstal
Example 78: 2:1 Proline Co-crvstal with f2S.3R.4R.5S.6R>-2-r3-f2.3-Pihvdro-benzof1.41dioxin-6-ylmethvH-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 3: 1 :2 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1 kg) was added to 15 L of ethanol with agitation while maintaining the mixture at 20-25 °C. The mixture was stirred for 10 min at 20-25 °C, then L-proline (537 gm) was added while maintaining the mixture at 20-25 °C. The mixture was stirred at this temperature for 30 min., then heated to reflux and refluxed for 30 min. The mixture was slowly cooled to 25-30°C then stired for 1 hr. DIPE (15 L) was added while maintaining the temperature at 25-30 °C and the mixture was stirred at this temperature for 1 hr. The precipitated product was collected by filtration and the product was washed with DIPE (5 L). The product was air dried at 65-70 °C to yield 1.22 kg
(79%) of a 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C (ΔΗ 85 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig.
3. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 4. Relative
intensities of the most prominent powder x-ray diffraction peaks for the 1 :2 co- crystals of Example 62 and proline are shown in Table 5.
Table 5

PATENT
WO 2012140597
http://www.google.co.in/patents/WO2012140597A1?cl=en
. TABLE 2:

Intermediate 2: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-

Intermediate 2
Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- 4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for 1 h at same temperature, reaction was quenched with the addition of 1 N HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic layers were washed with brine (10 ml), dried over sodium sulfate, concentrated and purified by preparative HPLC to give 220 mg of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J = 6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H), 3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H), 4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57 (dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m, 3H). MS (ES) m/z 553.3 (M+1 ).
Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2 mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it was stirred at room temperature for 20 min. Reaction mixture was cooled to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5 ml). The reaction mixture was stirred at same temperature for 5 min and then at room temperature for 2 h. Reaction mixture was cooled to 0°C and quenched by the addition of 10% sodium bisulfite solution (5 ml). This was extracted with ether (3 X 10 ml). Combined organic layer was washed with brine (5 ml), dried over sodium sulfate and concentrated to give 700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6- [3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro- pyran-2-ylmethyl ester.
To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.
![]()
PATENT
SEE INDIAN PATENT
IN 2009DE02173
Glycoside derivatives and uses thereof
REFERENCES
Pediatric investigation plan (PIP) decision: (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2:1) ( LIK066) (EMEA-001527-PIP01-13)
European Medicines Agency (EMA) Web Site 2014, July 24
Safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) assessment of LIK066 in healthy subjects and in patients with type 2 diabetes mellitus (T2DM) (NCT01407003)
ClinicalTrials.gov Web Site 2011, August 07
IN 2009DE02173
| WO2001016147A1 | 24 Aug 2000 | 8 Mar 2001 | Kissei Pharmaceutical | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 | 2 Oct 2000 | 19 Apr 2001 | Bruce Ellsworth | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 | 15 Mar 2001 | 20 Sep 2001 | Hideki Fujikura | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 | 29 Mar 2001 | 11 Oct 2001 | Squibb Bristol Myers Co | O-aryl glucoside sglt2 inhibitors and method |
| WO2003020737A1 | 5 Sep 2002 | 13 Mar 2003 | Squibb Bristol Myers Co | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003043985A1 | 20 Nov 2002 | 30 May 2003 | Andrew Thomas Bach | Heterocyclic compounds and methods of use |
| WO2004018491A1 | 21 Aug 2003 | 4 Mar 2004 | Nobuhiko Fushimi | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004078163A2 | 26 Feb 2004 | 16 Sep 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004080990A1 | 12 Mar 2004 | 23 Sep 2004 | Kazuhiro Ikegai | C-glycoside derivatives and salts thereof |
| WO2004099230A1 | 30 Apr 2004 | 18 Nov 2004 | Eikyu Yoshiteru | Monosaccharide compounds |
| WO2004103995A1 | 19 May 2004 | 2 Dec 2004 | Gary Michael Ksander | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2005011592A2 | 29 Jul 2004 | 10 Feb 2005 | Janssen Pharmaceutica Nv | Substituted indazole-o-glucosides |
| WO2005021566A2 | 20 Aug 2004 | 10 Mar 2005 | Barsoumian Edward Leon | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085237A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085265A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006011502A1 | 27 Jul 2005 | 2 Feb 2006 | Chugai Pharmaceutical Co Ltd | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006054629A1 | 17 Nov 2005 | 26 May 2006 | Kissei Pharmaceutical | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2008016132A1 | 3 Aug 2007 | 7 Feb 2008 | Daiichi Sankyo Co Ltd | Benzyl phenyl glucopyranoside derivative |
| WO2011048112A1 * | 19 Oct 2010 | 28 Apr 2011 | Novartis Ag | Glycoside derivatives and uses thereof |
| US20030114390 * | 4 Oct 2002 | 19 Jun 2003 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| US20040018998 | 21 Sep 2001 | 29 Jan 2004 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US20060009400 | 28 Jun 2005 | 12 Jan 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948 | 15 Jul 2005 | 26 Jan 2006 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349 | 27 Jul 2005 | 2 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060035841 | 9 Aug 2005 | 16 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031 | 30 Sep 2005 | 6 Apr 2006 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060293252 | 14 Aug 2006 | 28 Dec 2006 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014 | 26 Jul 2007 | 31 Jan 2008 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015032272A1 * | 19 Aug 2014 | 12 Mar 2015 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9034921 | 1 Jun 2012 | 19 May 2015 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
INVENTORS OF LIK 066
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
| BEBERNITZ, Gregory, Raymond; (US). BOCK, Mark, G.; (US). REDDY, Dumbala Srinivas; (IN). HAJARE, Atul Kashinath; (IN). VYAVAHARE, Vinod; (IN). BHOSALE, Sandeep Bhausaheb; (IN). KURHADE, Suresh Eknath; (IN). SALUNKHE, Videsh; (IN). SHAIKH, Nadim, S.; (IN). BHUNIYA, Debnath; (IN). PALLE, P., Venkata; (IN). FENG, Lili; (US). LIANG, Jessica; (US) |
 Mark G Bock
Mark G Bock
BEBERNITZ, Gregory, Raymond….PIC NOT AVAILABLE

Dr. Srinivasa Reddy
 NADEEM SHAIKH
NADEEM SHAIKH
 Venkata Palle
Venkata Palle
ONLY FEW…………………….
//////Licogliflozin diprolinate
see……..http://medcheminternational.blogspot.in/2015/11/lik-066-novartis-for-treatment-of-type.html
Zydus Cadila’s new 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds in pipeline for diabetes type 2
List of compounds as DPP-IV inhibitors


Watch out on this post as I get to correct structure………..
![]()

2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds

One Example of 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds
CAS 1601479-87-1
(2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[5-(methylsulfonyl)-3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]tetrahydro-2H-pyran-3-amine
MW 399.45, C18 H23 F2 N3 O3 S
INTRODUCTION
Dipeptidyl peptidase IV , CD26; DPP-IV; DP-IV inhibitors acting as glucose lowering agents reported to be useful for the treatment of type 2 diabetes. compound inhibited human DPP-IV enzyme activity (IC50 < 10 nM) in fluorescence based assays.
It lowered glucose levels (with -49.10% glucose change) when administered to C57BL/6J mice at 0.3 mg/kg p.o. in oral glucose tolerance test (OGTT).
Compound displayed the following pharmacokinetic parameters in Wistar rats at 2 mg/kg p.o.: Cmax = 459.04 ng/ml, t1/2 = 59.48 h and AUC = 4751.59 h·ng/ml.
Dipeptidyl peptidase 4 (DPP-IV) inhibitor that inhibited human DPP-IV enzyme activity with an IC50 of < 10 nM in a fluorescence based assay.
Watch out on this post as I get to correct structure………..![]()

PATENT
http://www.google.com/patents/WO2014061031A1?cl=en
Compound 8: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz): 7.32-7.28 (m, IH), 7.26-7.23 (m, 2H), 4.77 (d, IH, J= 10Hz), 4.32(dd, IH, J,= 2.0Hz, J2= 10.8Hz), 4.19 (s, 4H), 3.89-3.83 (m, 4H), 3.70- 3.65 (m, IH), 3.61 (t, IH, J= 11.6Hz), 3.53-3.46 (m, IH), 3.04 (s, 3H), 2.65-2.62 (dd, IH, Ji= 1.2Hz, J2= 12Hz), 1.84 (q, IH, J = 12 Hz); ESI-MS: (+ve mode) 400.0 (M+H)+ (100 %); HPLC: 99.4 %.
Compound 4: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(hexahydropyrrolo[3, 4-c Jpyrrol- 2(lH)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz):

.23-7.20 (m, 2H), 4.64 (d, IH, J= 10.4 Hz), 4.38-4.35 (dd, IH, J,= 2.4Hz, J2= 10.4Hz), 3.69 (t, IH, J= 11Hz), 3.57-3.53 (m, 4H), 3.34-3.30 (m, 8H), 2.68-2.65 (m, IH), 2.04 (q, IH, J = 1 1.6 Hz); ESI-MS: (+ve mode) 323.9 (M+H)+ (100 %), 345.9 (M+Na)+ (20%); HPLC: 98.6 %

PATENT
IN 2012MU03030
“NOVEL DPP-IV INHIBITORS”
3030/MUM/2012
Abstract:
The present invention relates to novel compounds of the general formula (I) their tautomeric forms, their enantiomers, their diastereoisomers, their pharmaceutically accepted salts, or pro-drugs thereof, which are useful for the treatment or prevention of diabetes mellitus (DM), obesity and other metabolic disorders. The invention also relates to process for the manufacture of said compounds, and pharmaceutical compositions containing them and their use.

Pankaj R. Patel (right), Chairman and Managing Director,
////////////2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds, DPP-IV inhibitors
DC_AC50, selective way of blocking copper transport in cancer cells

DC_AC50
3-amino-N-(2-bromo-4,6-difluorophenyl)-6,7-dihydro-5H- cyclopenta [b] thieno [3,2-e] pyridine-2-carboxamide
licensed DC_AC50 to Suring Therapeutics, in Suzhou, China
INNOVATORS Jing Chen of Emory University School of Medicine, Hualiang Jiang of the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences, Chuan He of the University of Chicago, and coworkers

Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation
- Nature Chemistry, (2015)
- doi:10.1038/nchem.2381
Jing Chen of Emory University School of Medicine, Hualiang Jiang of the Shanghai Institute of Materia Medica of the Chinese Academy of Sciences, Chuan He of the University of Chicago, and coworkers have now developed a selective way of blocking copper transport in cancer cells (Nat. Chem. 2015, DOI: 10.1038/nchem.2381). By screening a database of 200,000 druglike small molecules, the researchers discovered a promising compound, DC_AC50, for cancer treatment. They zeroed in on the compound by testing how well database hits inhibited a protein-protein interaction leading to copper transport and reduced proliferation of cancer cells.

Scientists had already found a molecule, tetrathiomolybdate, that interferes with copper trafficking and have tested it in clinical trials against cancer. But tetrathiomolybdate is a copper chelator: It inhibits copper transport in cells by nonselectively sequestering copper ions. Sometimes, the chelator snags too much copper, inhibiting essential copper-based processes in normal cells and causing side effects.
In contrast, DC_AC50 works by inhibiting interactions between proteins in the copper-trafficking pathway: It prevents chaperone proteins, called Atox1 and CCS, from passing copper ions to enzymes that use them to run vital cellular processes. Cancer cells are heavy users of Atox1 and CCS, so DC_AC50 affects cancer cells selectively.
The team has licensed DC_AC50 to Suring Therapeutics, in Suzhou, China, for developing anticancer therapies. The group also plans to further tweak DC_AC50 to develop more-potent versions.
Thomas O’Halloran of Northwestern University, who has studied tetrathiomolybdate, comments that “the challenge in drug design is hitting one of these copper-dependent processes without messing with housekeeping functions that normal cells depend upon. DC_AC50 appears to block the function of copper metallochaperone proteins without interacting directly with their cargo, copper ions. As the first member of a new class of inhibitors, it provides a new way to interrogate the physiology of copper trafficking disorders and possibly intervene.”
PATENT
http://www.google.com/patents/WO2014116859A1?cl=en

COMPD IS LC-1 COMPD 50
Scheme 1 (Compounds LCI -LCI 9):

Experimental procedure for Scheme 1 :
Step a: To 1 equivalent of sodium metal in anhydrous diethyl ether is added 1-2 equivalents of ethyl formate and 1-2 equivalents of cyclopentanone. The resulting mixture is stirred overnight. The mother liquor is filtered by suction filtration to obtain crude intermediate 2.
Step b: To a solution of intermediate 2 in an organic solvent, is added 0.1 to 1 equivalent of glacial acetic acid. The reaction is stirred at 50-100 °C, then 2′ and 0.1 to 1 equivalent of glacial acetic acid are added. The resulting reaction mixture is refluxed for 1-5 hours, filtered and recrystallized to produce product 3; the said organic solvent may optionally be tetrahydrofuran, ether, dimethylformamide, ethyleneglycol dimethyl ether, ethylene glycol diethyl ether, dioxane, ethanol, methanol, ethyl acetate, or dichloromethane. Step c: To a solution of compound 3 in an organic solvent, is added 1 equivalent of methyl bromoacetate and an appropriate amount of base. The reaction mixture is stirted at room temperature to produce intermediate 4. The said organic solvent may optionally be tetrahydrofuran, aether, dimethylformamide, ethylene glycol dimethyl ether, ethylene glycol diethyl ether, dioxane, ethanol, methanol, ethyl acetate, or dichloromethane. The said base may optionally be potassium hydroxide, sodium hydroxide, sodium carbonate, potassium carbonate, cesium carbonate, and their aqueous solution in various concentrations.
Step d: The base described in Step c is added to a solution of compound 4 in an organic solvent. The reaction mixture is stirred and heated to produce intermediate 5. Step e: An appropriate amount of di-tert-butyl dicarbonate and alkali are added to a solution of compound 5 in an organic solvent. The reaction is stirred to produce intermediate 6.
Step f: An appropriate amount of base is added to a solution of compound 6 in an organic solvent, which is then hydro lyzed to produce intermediate 7.
Step g: 3′ and a stoichiometric amount of condensing agent are added to a solution of compound 7 in an organic solvent. The reaction mixture is stirred until 3′ reacts completely to produce the final product. The said organic so ί vers t may optional iy be tetrahydrofuran, aether, dimethyl formamide, ethylene glycol dimethyl ether, ethylene glycol diethyl ether, dioxane, ethanol, methanol, ethyl acetate, or dichloromethane. The said condensing agent may optionally be DCC, EDO, HOBt, and GDI. Step h: To a solution of compound 7 in an organic solvent is added aqueous hydrochloric acid or trifluoroacetic acid. The reaction mixture is stirred vigorously to yield the BOC- deprotected final product.
Scheme 2 (Compounds LCI -LCI 9)

LCI ~LC39
Experimental procedure for Scheme 2(Compounds LC1-LC19):
Step a: Dissolve 1 equivalent of sodium in anhydrous ether, which shall be added slowly under an ice bath and rapid stirring condition. Add 1 equivalent of ethyl formate and 1 equivalent of cyclopentanone in a constant pressure dropping funnel, add 0.5 ml ethanol as an initiator, after 1 hour of stirring in ice bath, and stir overnight at room temperature until the reaction of sodium is finished. Perform suction filtration, wash with absolute ether to produce crude product for the following steps of reaction.
Step b: Dissolve the product in above steps directly in ethanol and control its amount, add an appropriate amount of glacial acetic acid, and stir and reflux under 70°C. Add cyano- sulfamide into the reaction solution, and add an appropriate amount of glacial acetic acid, react and reflux for about 3 hours. Recrystallize with ethanol to produce crude product.
Step c: Add 1 equivalent of the appropriate aniline or phenol and 2 equivalents of potassium carbonate solid in a round-bottomed flask that is placed in ice bath, add anhydrous THF to fully dissolve the solid, add 1.5 equivalents of bromoacetyl bromide into a constant pressure dropping funnel and dilute with THF, which is slowly dropped into the former said round- bottomed flask that is moved to room temperature in 10 min late and react for 1 hour; extract and dry with anhydrous sodium sulfate, filtrate by suction, and perform rotary evaporation to remove the solvent, and the crude product is obtained, which is to be used directly in the next step of reaction.
Step d: Dissolve the product from Step 2 into DMF under normal temperature by mixing, add 3 equivalents of 10% KOH solution, which is then transferred to an oil bath of 70°C and react, and add I equivalent of the product from step 3. Stir for about 3 hours and then extract directly with ethyl acetate, and recrystallize the crude product with ethanol to produce pure end product.
Steps a and b: Intermediate 3 is prepared in accordance with the method outlined in Scheme 1. Step c: 3′ and bromoacetyl bromide are condensed in the presence of a suitable base to produce intermediate 9. The said base may optionally be potassium hydroxide, sodium hydroxide, sodiumcarbonate, potassium carbonate, cesium carbonate, and their aqueous solution in various concentrations.
Step d: An appropriate amount of base is added to a solution of compound 3 in an organic solvent, and the reaction mixture is heated to 40-100 °C. Intermediate 9 is added, and the heated solution is stirred for 1-10 hours to yield the final product. The said organic solvent may optionally be tetrahydrofuran, aether, dimethylformamide, ethylene glycol dimethyl ether, ethylene glycol diethyl ether, dioxane, ethanol, methanol, ethyl acetate, or dichloromethane. The said base may optionally be potassium hydroxide, sodium hydroxide, sodium carbonate, potassium carbonate, cesium carbonate, and their aqueous solution in various concentrations.
NMR and mass spectral data: LC-1 (Compound 50)- 3-amino-N-(2-bromo-4,6-difluorophenyl)-6,7-dihydro-5H- cyclopenta [b] thieno [3,2-e] pyridine-2-carboxamide

1H NMR (CDCI3, 400 MHz) δ 9.15 (s, 1H), 7.61 (s, 1H), 7.13(m, 1H), 6.60 (m, 1H), 6.27 (s, 2H), 3.20 (t, 2H), 2.98 (t, 2H), 2.39 (m, 2H); ESI-MS (EI) m/z 422 (M+)
/////
ZYD 1/ZYDPLA 1 From Zydus Cadila, a New NCE in Gliptin class of Antidiabetic agents.

GENERAL STRUCTURE

3-[4-(5-methyl-1,3,4-oxadiazol-2-yl)phenoxy]-5-[[(3R)-1-methyl-2-oxo-3-pyrrolidinyl]oxy]-N-2-thiazolyl- Benzamide
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide……S CONF…..WO2011013141A2
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide…..R CONF…..WO2011013141A2
CAS 1263402-84-1 R CONF
CAS 1263402-76-1 S CONF
ZYD 1/ZYDPLA 1……….Probable Representative structure only, I will modify it as per available info
Watch out on this post as I get to correct structure………..![]()

ZYDPLA1 is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre, the NCE research wing of Zydus. ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents. It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1.
By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
In October 2013, Zydus received IND approval from the US FDA to initiate a phase I trial in type II diabetes
Clinical trials..Type 2 Diabetes Mellitus
NCT01972893; ZYD1/1001;
CTRI/2011/04/001684;
ZYD1
ZYD1/1001
ZYD1 is a novel GLP-1 receptor agonist. The ZYD1 exhibits increased stability to proteolytic cleavage, especially against dipeptidyl peptidase-4 (DPP-IV).ZYD1 is a potent antidiabetic agent without gastrointestinal side-effects. A first in human (FIH) Phase I study intends to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of ZYD1 in normal healthy adult volunteers……..https://clinicaltrials.gov/show/NCT01972893
A randomized, double blind, placebo controlled Phase I clinical study to evaluate the safety, tolerability and pharmacokinetics of ZYD1, a selective GLP-1 agonist, following the subcutaneous administrations in healthy volunteers …………http://www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=2263&EncHid=&modid=&compid=%27,%272263det%27
Some clippings I found

ONE MORE……………

Zydus announces data presentations on ZYDPLA1 “A once-weekly small molecule DPP-IV inhibitor for treating diabetes”, at the ENDO conference in Chicago, Illinois, USA. Ahmedabad, India June 9, 2014 The Zydus group will be presenting data on its molecule ZYDPLA1 a novel compound in the Gliptin class of anti-diabetic agents during the joint meeting of the International Society of Endocrinology and the Endocrine Society: ICE/ENDO 2014 to be held from June 21-24, 2014 in Chicago, Illinois.
ZYDPLA1, currently in Phase I clinical evaluation in USA, is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre. ZYDPLA1 works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP- 1 levels, ZYDPLA1 glucose-dependently increases insulin secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
The Chairman & Managing Director of Zydus, Mr. Pankaj R. Patel said, “Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.”
The abstract of Poster Number: LB-PP02-4 can also be viewed on the ENDO web program at https://endo.confex.com/endo/2014endo/webprogram/authora.html. The Poster Preview is scheduled on Sunday, June 22, 2014 at McCormick Place West.
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPP IV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
About Zydus
Headquartered in Ahmedabad, India, Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 16,000 people worldwide including over 1100 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global researchbased pharmaceutical company by 2020. The group has a strong research pipeline of NCEs, biologics and vaccines which are in various stages of clinical trials including late stage.
About Zydus Research Centre
The Zydus Research Centre has over 20 discovery programmes in the areas of cardio-metabolic disorders, pain, inflammation and oncology. Zydus has in-house capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. The Zydus Research group had identified and developed Lipaglyn™ (Saroglitazar) which has now become India’s first NCE to reach the market. Lipaglyn™ is a breakthrough therapy in the treatment of diabetic dyslipidemia and Hypertriglyceridemia. The company recently announced the commencement of Phase III trials of LipaglynTM (Saroglitazar) in patients suffering from Lipodystrophy.
PATENT
http://www.google.com/patents/WO2011013141A2?cl=en
Rajendra Kharul, Mukul R. Jain, Pankaj R. Patel
Substituted benzamide derivatives as glucokinase (gk) activators

Scheme 2:

Scheme 3:

Scheme 4A:


Scheme 4B.
] Scheme 5 A:

Scheme 5B:

Scheme 6:

Example 1
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
4-(Dimethylamino)pyridine (DMAP) (0.149 g), N-(3-Dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDCI.HC1) (0.524 g) were added to a solution of 3-
( 1 -Methoxypropan-2-yloxy)-5-(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl) phenoxy) benzoic acid (0.5 g) (Intermediate 1) in dry DCM under nitrogen at 0-5 0C. 2-Aminothiazole (0.134 g) was added and the mixture was stirred for 16 h at room temperature. It was diluted with commercially available DCM. Organic phase was washed with dil HCl, saturated solution of NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude residue. The residue was chromatographed using silica gel as stationary phase and MeOH: CHCl3 gradient as mobile phase up to yield the product (0.3 g) as a white solid.
1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.92-2.01 (m, 1 H), 2.59 (s, 3 H), 2.60-2.65 (m,
I H), 2.79 (s, 3 H), 3.31-3.34 (m, 1 H), 3.36-3.44 (m ,1 H), 5.15 (t, J = 7.6 Hz, 1 H),
7.08 (s, 1 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.27-7.29 (m, 1 H), 7.40 (s, 1 H), 7.54 (s, 1 H),
7.62 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H), 12.60 (bs, 1 H); ESI-MS mix (relative intensities): 492.03 (M+H)+ (100 %), 514.02 (M+Na)+(15 %); UPLC Purity: 93.59 %, Rettime: 3.59 min.
Intermediate 1: 3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxo pyrrolidin -3-yloxy)benzoic acid
A solution of Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl- 2-oxopyrrolidin-3-yloxy)benzoate (7 g) (Intermediate 2) in a mixture of THF and methanol (1 :1 ratio) was treated with a solution of sodium hydroxide (2 g) in water and the reaction mixture was stirred for 1 h at room temperature. The resulting solution was concentrated under vacuum to remove THF and methanol, diluted with water, and washed with EtOAc. The aqueous phase was cooled and acidified with 0.1 N HCl and extracted with DCM, combined organic extracts washed with brine, dried over Na2SO4 and concentrated in vacuo to give the product (3.5 g) as white solid.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.20-2.27 (m, 1 H), 2.59-2.67 (m, 1 H), 2.77 (s, 3 H), 2.95 (s, 3 H), 3.38-3.44 (m, 1 H), 3.49-3.54 (m, 1 H), 4.96 (t, J = 7.2 Hz, 1 H), 6.93-6.95 (m, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.32-7.34 (m, 1 H), 7.52 (d, J= 8.8 Hz, 2 H), 9.96-9.98 (m, 2 H); ESI-MS (relative intensities): 431.9 (M+ Na)+ (70%).
Intermediate 2: Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2- oxo- pyrrolidin-3-yloxy)benzoate
To a stirred mixture of Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy) benzoate (15 g) (Intermediate 3), N,N-dimethylglycine hydrochloride (2.3 g), copper (II) iodide (1 g) in dry 1,4-dioxane was added 2-(4-iodophenyl)-5 -methyl- 1,3,4- oxadiazole (15.4 g) (Intermediate 4) under nitrogen. The reaction mixture was refluxed for 24 h. The reaction mixture was cooled, quenched with water and extracted with DCM. Combined organic washings were washed with water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude product. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether (9:1) as mobile phase to give the product (7 g) as thick liquid. 1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.91-1.98 (m, 1 H), 2.49-2.54 (m, 1 H), 2.56 (s, 3 H), 2.77 (s, 3 H), 3.34-3.41 (m, 2 H), 3.81 (s, 3 H), 5.12 (t, J= 7.6 Hz, 1 H), 7.13- 7.15 (m, 2 H), 7.22 (d, J = 8.8 Hz, 2 H), 7.42 (s, 1 H), 7.97 (d, J = 8.8 Hz, 2 H); ESI- MS (relative intensities): 423.9 (M+H)+ (100%), 446.2 (M+ Na)+ (30%).
Intermediate 3: Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy)benzoate
To a stirred solution of Methyl 3, 5-dihydroxybenzoate (20 g) [CAS No. 2150- 44-9] in dry DMF was added potassium carbonate (48 g) and the suspension stirred at ambient temperature under nitrogen. To this 3-Bromo-l-methyl-pyrrolidin-2-one (4Og) (Intermediate 5) [J. Med. Chem., 1987, 30, 1995-98] was added in three equal portions in 4 h intervals at room temperature and stirred overnight at ambient temperature. It was then quenched with water. The aqueous suspension was extracted with DCM. The combined extracts were washed with water, brine, dried over Na2SO4, and filtered, concentrated under reduced pressure to get the thick liquid residue. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether as a mobile phase to yield the product as white solid (15 g).1H NMR (CDCl3, 400 MHz) δ ppm: 2.08-2.10 (m, 1 H), 2.60-2.67 (m, 1 H), 3.04 (s, 3 H), 3.40-
3.43 (m, 1 H), 3.48-3.51 (m, 1 H), 3.87 (s, 3 H), 4.91 (t, J = 7.2 Hz, 1 H), 6.59- 6.61 (m, 1 H), 7.07-7.09 (m, 1 H), 7.09-7.13 (m, 1 H), 8.02 (s, 1 H); ESI-MS (relative intensities): 287.9 (M+ Na)+ (30%).
Example 68…. S CONFIGURATION
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of S-(-)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5- [(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (3.5 g) (Intermediate 13) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, 4- (dimethylamino)pyridine (2.24 g) followed by N-(3-Dimethy lam inopropy I)-N5– ethylcarbodiimide hydrochloride (EDCI. HCl) (3.3 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.94 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (200 mL), washed with dil HCl (20 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (3.5 g). The crude brown solid was purified by solvent trituration.
1H ΝMR (CDCl3, 400 MHz) δ ppm: 2.13-2.22 (m, 1 H), 2.62 (s, 3 H), 2.56-2.64 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.92 (t, J= 7.2 Hz, 1 H), 7.01 (s, 1 H), 7.04 (t, J= 2 Hz, 1 H), 7.21 (d, J = 8.8 Hz, 2 H), 7.26 (s, 1 H), 7.36 (s, 1 H), 7.44 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 513.8 (M+Νa)+ (10 %); UPLC Purity: 98.13 %, Ret. time: 3.577 min. Chiral Purity by HPLC: 97.31 %, Ret. time: 22.93 min. % ee: 94.62 %
Intermediate 13: S-(-)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2- oxo-pyrro- lidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 1.5 g) was added to a stirring mixture of (.S)-(-)-Methyl 3- [4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy] benzoate (5.3g) (Intermediate 14) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (3.5 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.17-2.22 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.99 (t, J= 7.2 Hz, 1 H), 6.89 (t, J = 2.4 Hz, 1 H), 7.07 (d, J = 8.8 Hz, 2 H), 7.28 (s, 1 H), 7.53 (s, 1 H), 7.95 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410 (M+H)+ (100 %); UPLC Purity: 97.85 %, Ret. time: 3.136 min. Chiral Purity by HPLC: 99.59 %, Ret. Time: 57.46 min. % ee: 99.18 %
Intermediate 14: (S) -(-) -Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate
Sodium hydride suspension (0.71 g, 50 %) was added to a stirring solution of (£)-(-)- methyl 3 -(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (5.5 g) (Intermediate 15) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.91 mL) was added and stirred till the reaction completion. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vaccuo to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product (5.3 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.14-2.21 (m, 1 H), 2.58-2.63 (m, 1 H), 2.64 (s, 3 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m , 1 H), 3.89 (s, 3 H), 4.99 (t, J = 7.2 Hz, 1 H), 6.99 (t, J = 2 Hz, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.35 (s, 1 H), 7.53 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 424.1 (M+H)+ (100 %); UPLC Purity: 96.1 1 %, Ret. time: 3.68 min. Chiral Purity by HPLC: 92.05 %, Ret. Time: 39.33 min.
Intermediate 15: (S) -(-) -Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxo pyrrolidin-3-yl)oxy) benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (7 g) (Intermediate 7) and (/?)-(+)-3-hydroxy-2-pyrrolidinone (Intermediate 16) (2.4g) in dry THF (200 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (1 1.3 g) was added. Diisopropyl azodicarboxylate (DIAD) (6.2 mL) in dry THF (10 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (6 g). 1H NMR (CDCl3, 400 MHz) δ ppm: 2.26-2.33 (m, 1 H), 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.47 (m, 1 H), 3.51-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.07 (bs, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (100 %); UPLC Purity: 98.35 %, Ret. time: 3.47 min. Chiral Purity by HPLC: 95.31 %, Ret. Time: 47.97 min. ee: 90.62 %.
Intermediate 16: (R)-(+)-3-Hydroxy-2-pyrrolidinone
To a stirring mixture of 4-Nitrobenzoic acid (21.5 g) and (5)-(-)-3-hydroxy-2- pyrrolidinone (11.8 g) (Intermediate 17) in dry THF (360 mL) taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (61.2 g) was added. To this reaction mixture, diisopropyl diazodicarboxylate (DIAD) (34 mL) was added drop wise in three portions at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was concentrated under vacuum to obtain residue. Methanol (360 mL) was added to the residue followed by potassium carbonate (10 g) at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was diluted with CHCl3 and filtered through celite. Celite bed was successively washed with 1 % MeOH:CHCl3. The filtrates were combined and concentrated to dryness to remove solvents. The residues were partitioned between EtOAc: dil. HCl (200 mL, 9:1) and stirred for 15 min. Layers were separated, aq. layer was washed with EtOAc thrice until all organic impurities were washed out. The aq. Layer was concentrated to dryness to remove the water and solid residues were obtained. The residues obtained were washed with 1-2 % MeOH: CHCl3 (3 x 100 mL), dried over sodium sulfate, filtered trough cotton, concentrated to get brown thick liquid product.
1U NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.46-2.54 (m, 1 H), 3.28-3.35 (m, IH), 3.38-3.48 (m, 1 H), 4.50 (t, J = 8.4 Hz, 1 H), 4.55 (bs, 1 H), 7.02 (bs, 1 H); [α]D25: + 68, c = l, CHCl3
Intermediate 17: (S)-(-)-3-hydroxy-2-pyrrolidinone
Cone. H2SO4 (14.8 g, 8 mL) was added drop wise over 5 min to the stirring solution of (5)-(-)-4-Amino-2-hydroxybutyric acid (15 g) [CAS No. 40371-51-5] in MeOH (95 rnL) under dry conditions using anhydrous CaCl2 guard tube. After refluxing for 4 h, the reaction mixture was allowed to cool to room temperature and diluted with water (15 mL). Potassium carbonate (24 g) was added in portions to the reaction mixture and stirred overnight (20 h). Reaction mixture was diluted with CHCl3, filtered through celite. Celite bed was thoroughly washed with 1 % MeOHiCHCl3. The filtrates were combined and evaporated to dryness to obtain thick liquid residue. The residue was subjected to aging using 1-2 % MeOHiCHCl3 and then filtered. Organic layers were combined, dried over anhydrous sodium sulphate, filtered and concentrated to obtain the white solid. (1 1.8 g)
1H NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.48-2.55 (m, 1 H), 3.30-3.35
(m, IH), 3.36-3.50 (m, 1 H), 4.34 (t, J = 8.4 Hz, 1 H), 6.51 (bs, 1 H); [α]D25: + 98, c =
1, CHCl3
Following examples (Example 70-76) were prepared by using similar procedure as that of example lwith suitable modifications as are well within the scope of a skilled person
Example 77 R CONFIGURATION
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide
CORRECTED AS (R)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of (/?j-(+)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-
[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (0.2 g) (Intermediate 18) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, N.ΛP-dimethylamino pyridine (0.060 g) followed by EDCI. HCl (0.23 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.054 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (20 mL), washed with dil HCl (5 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (0.080 g). The crude brown solid was purified by solvent trituration.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.15-2.20 (m, 1 H), 2.55-2.60 (m, 1 H), 2.62 (s, 3 H), 2.93 (s, 3 H), 3.38-3.43 (m, 1 H), 3.47-3.53 (m, 1 H), 4.91 (t, J= 6.8 Hz, 1 H), 6.99 (d, J= 8.8 Hz, 2 H), 7.10-7.14 (m, 2 H), 7.23-7.26 (m, 1 H), 7.36 (s, 1 H), 7.43 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H), 10.75 (bs, 1 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 514.0 (M+Na)+ (20 %); UPLC Purity: 95.25 %, Ret.time: 3.578 min. Chiral Purity by HPLC: 95.93 %, Ret.time: 14.17min. % ee: 91.86 %
Intermediate 18: (R)-(+)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl- 2-oxo- pyrrolidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 0.35 g) was added To a stirring mixture of (/?)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate (1.1 g) (Intermediate 19) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (0.76 g).
1H NMR (DMSO-J6, 400 MHz) δ ppm: 1.92-1.99 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 3.31 (s, 3 H), 3.32-3.40 (m, 2 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.08 (s, 1 H), 7.14 (s, 1 H), 7.23 (d, J= 8.8 Hz, 2 H), 7.40 (s, 1 H), 7.99 (d, J= 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (65 %), 410.1 (M+H)+ (100 %); UPLC Purity: 96.95 %, Ret. time: 3.12 min. Chiral Purity by HPLC: 89.04 %, Ret. Time: 48.15 min. Intermediate 19: (R)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate:
Sodium hydride suspension (0.16 g, 50 %) was added to a stirring solution of (R)- (+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (1.5 g) (Intermediate 20) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.20 mL) was added and stirred till the reaction completed. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vacuum to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product
(1.2 g).
1U NMR (DMSO-J6, 400 MHz) δ ppm: 1.95-1.98 (m, 1 H), 2.51-2.55 (m, 1 H), 2.56 (s, 3 H), 2.88 (s, 3 H), 3.29-3.34 (m, 1 H), 3.37-3.40 (m ,1 H), 3.81 (s, 3 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.13-7.17 (m, 2 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.41 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 423.9 (M+H)+ (100 %); UPLC Purity: 90.38 %, Ret. time: 3.68 min.
Intermediate 20: (R)-(+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxopyrrolidin -3-yl)oxy)benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (2.5 g) (Intermediate 7) and (5)-(-)-3-hydroxy-2-pyrrolidinone (Intermediate 17) (0.8 g) in dry THF (70 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (3.77 g) was added. Diisopropyl azodicarboxylate (DIAD) (2.1 mL) in dry THF (2 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (2 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.23-2.30 (m, 1 H); 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.46 (m, 1 H), 3.50-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (45 %); UPLC Purity: 96.40 %, Ret. time: 3.48 min. Chiral Purity by HPLC: 90.92 %, Ret. Time: 48.36 min.


http://zyduscadila.com/wp-content/uploads/2015/09/ZYDPLA1-a-Novel-LongActing-DPP-4-Inhibitor.pdf
http://zyduscadila.com/wp-content/uploads/2015/05/PressNote23-10-13.pdf
http://zyduscadila.com/wp-content/uploads/2015/07/annual_report_14-15.pdf
http://pharmaxchange.info/press/2012/08/glucokinase-activators-gkas-in-diabetes-management/
LB-PP02-4 ZYDPLA1, a novel long-acting DPP-4 inhibitor
Jt Int Congr Endocrinol Annu Meet Endocr Soc (ICE/ENDO) (June 21-24, Chicago) 2014, Abst LBSU-1075
LB-PP02-4 ZYDPLA1, a Novel Long-Acting DPP-4 Inhibitor
Session: LBSU 1074-1087-Diabetes & Obesity
Translational
Disclosure: MRJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. AAJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. RB: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. HP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. SK: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. KP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VKR: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PRP: Chairman, Cadila Healthcare Limited, Ahmedabad, India. RD: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India.

////////Dipeptidyl Peptidase IV, CD26, DPP-IV, DP-IV, Inhibitors
GKM 001 in pipeline for Diabetes by Advinus




GKM 001……Several probables
Watch out on this post as I get to correct structure………..![]()
Advinus Therapeutics Private L,
![]()
A glucokinase activator for treatment of type II diabetes
In October 2012, Takeda and Advinus have entered into an agreement to initiate a three-year discovery collaboration program focused on novel targets for inflammation, CNS, and metabolic diseases.
| Company | Advinus Therapeutics Ltd. |
| Description | Activator of glucokinase (GCK; GK) |
| Molecular Target | Glucokinase (GCK) (GK) |
| Mechanism of Action | Glucokinase activator |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase I/II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |

Advinus chief executive officer/MD Dr. Rashmi Barbhaiya.

PATENT
https://www.google.co.in/patents/WO2009047798A2?cl=en
Example Cl : (-)-{5-ChIoro-2-[2-(4-cyclopropanesulfonylphenyI)-2-(2,4- difluorophenoxy)acetylamino]thiazol-4-yl}-acetic acid, ethyl ester


Step I: Preparation of (-)-(4-Cyclopropanesulfonylphenyl)-(2,4- difluorophenoxy)acetic acid (Cl-I):
To a solution of (4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (obtained in example Al -step III) in ethyl acetate was added (S)-(-)-l-phenylethylamine drop wise at -15 °C. After completion of addition the reaction was stirred for 4-6 hours. Solid was filtered and washed with ethyl acetate. The solid was then taken in IN HCl and extracted with ethyl acetate, ethyl acetate layer was washed with brine, dried over anhydrous sodium sulfate. Solvent was removed under reduced pressure to obtain (-)-(4- cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid. Enantiomeric enrichment was done by repeating the diasteriomeric crystallization. [α]23 589 = – 107.1 ° (c = 2%Chloroform) Enantiomeric purity > 99. % (chiral HPLC)
Step II: (-)-{5-Chloro-2-[2-(4-cyclopropanesulfonylphenyl)-2-(2,4- difluorophenoxy)acetyIamino]thiazol-4-yl}-acetic acid ethyl ester : To a solution of (-)-4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (Cl-I) in DCM, was added DMF and cooled to 0 °C, followed by the addition of oxalyl chloride under stirring. Stirring was continued for 1 hour at the same temperature. The resulting mixture was further cooled to -35 °C, and to that, a solution of excess (2- amino-5-chlorothiazol-4-yl)acetic acid ethyl ester in DCM was added drop wise. After completion of reaction, the reaction mixture was poured into IN aqueous HCl under stirring, organic layer was washed with IN HCl, followed by 5% brine, dried over anhydrous sodium sulfate, solvent was removed under reduced pressure to get the crude compound which was purified by preparative TLC to get the title compound. [α]23 589 = – ve (c = 2%Chloroform)
1H NMR(400 MHz, CDCl3): δ 1.06-1.08 (m, 2H), 1.30 (t, J=7.2 Hz, 3H), 1.33-1.38 (m, 2H), 2.42-2.50 (m, IH), 3.73 (d, J=2 Hz, 2H), 4.22 (q, J=7.2 Hz ,2H), 5.75 (s, IH), 6.76- 6.77 (m, IH), 6.83-6.86 (m, IH), 6.90-6.98 (m, IH), 7.73 (d, J=8.4 Hz, 2H), 7.96 (d, J=8.4 Hz, 2H), 9.96 (bs, IH). MS (EI) m/z: 571.1 and 573.1 (M+ 1; for 35Cl and 37Cl respectively).
Examples C2 and C3 were prepared in analogues manner of example (Cl) from the appropriate chiral intermediate:

Example Dl : (+)-{5-Chloro-2-[2-(4-cyclopropanesulfonylphenyl)-2-(2,4- difluorophenoxy)acetylamino]thiazol-4-yl}acetic acid, ethyl ester

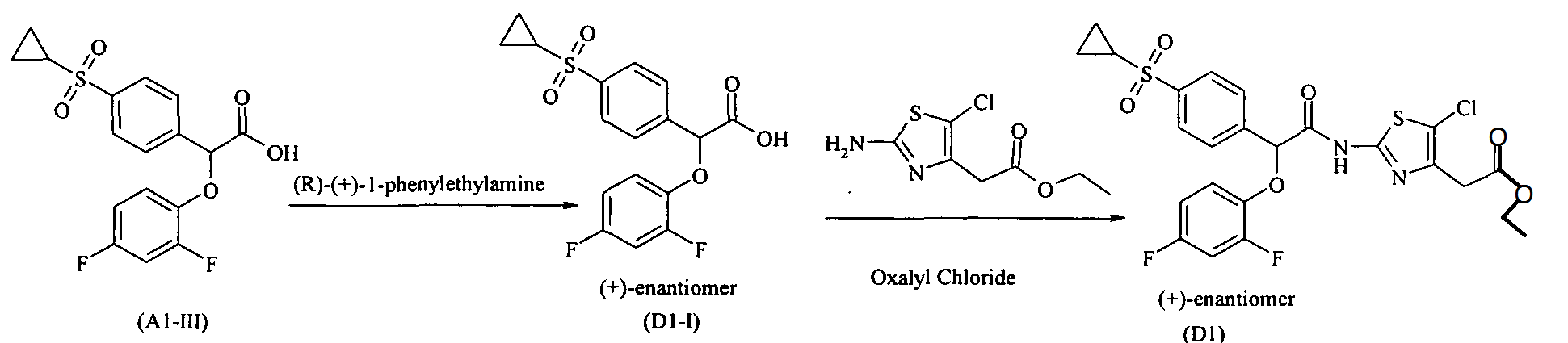
Preparation of (+)-(4-Cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (Dl-I):
To a solution of (4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (obtained in example Al -step III) in ethyl acetate, was added (R) (+)-l- phenylethylamine drop wise at -15 °C. After completion of addition the reaction was stirred for 4-6 hours. Solid was filtered and washed with ethyl acetate. The solid was then taken in IN HCl and extracted with ethyl acetate, ethyl acetate layer was washed with brine, dried over anhydrous sodium sulfate. Solvent was removed under reduced pressure to obtain (+)-(4-Cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid. Enantiomeric enrichment was done by repeating the diasteriomeric crystallization. [α]23 589 = +93.07° (c = 2%Chloroform) Enantiomeric purity > 99. % (by chiral HPLC)
(+)-(4-CyclopropanesuIfonylphenyI)-(2,4-difluorophenoxy)acetic acid ethyl ester (Dl)
The example Dl was prepared using (+)-4-cyclopropanesulfonylphenyl)-(2,4- difluorophenoxy)acetic acid (Dl-I), and following the same reaction condition for amide coupling as described in example Cl, [ot]23 589 = + ve (c = 2%Chloroform)
PATENT
https://www.google.co.in/patents/WO2008104994A2?cl=en
Synthesis Type-P
Example Pl : {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)- propionylamino]-thiazol-4-yI}-acetic acid
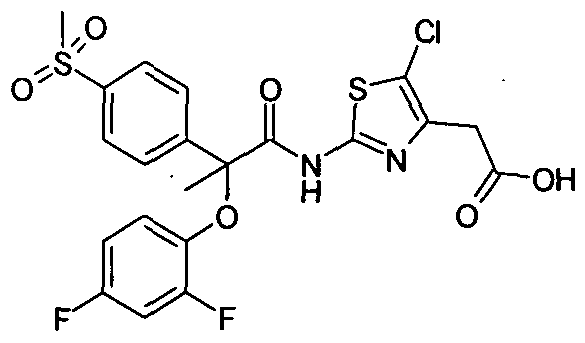
To a solution of {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl- phenyl)-propionylamino]-thiazol-4-yl}-acetic acid methyl ester (0.03 g, 0.05 mmol) in THF: Ethanol: water ( ImI + 0.3ml + 0.3 ml) was added lithium hydroxide (0.0046 g, 0.11 mmol). The resulting mixture was stirred for 5 hours at room temperature followed by removal of solvent under reduced pressure. The residue was suspended in water (15 ml), extracted with ethyl acetate to remove impurities. The aqueous layer was acidified with IN HCl (0.5 ml) and extracted with ethyl acetate (2×10 ml), This ethyl acetate layer was washed with water (15 ml), brine (20 ml), dried over anhydrous sodium sulfate and solvent was removed under reduced pressure to give solid product {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4- methanesulfonyl-phenyl)-propionylamino]-thiazol-4-yl} -acetic acid (9 mg). 1H NMR (400 MHz, CDCl3): δ 1.85 (s, 3H) , 3.07 (s, 3H) , 3.72 ( s, 2H), 6.64-6.69 ( m, 2H ) , 6.89-6.91 (m, IH ), 7.84 ( d, J – 8.4 Hz, 2H), 8.00 ( d, J = 8.8 Hz, 2H). MS (EI) mlz: 530.70 (M + 1), mp: 109-111 0C.
Preparation of {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)- propionylamino)-thiazol-4-yl}-acetic acid methyl ester used in Example Pl:

To a mixture of 2-(2, 4-Difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)-propionic acid (0.110 g, 0.22 mmol), (2-Amino-5-chloro-thiazol-4-yl)-acetic acid methyl ester (0.071 g, 0.32 mmol), HOBt (0.052g, 0.38 mmol), and EDCI (0.074 g, 0.38 mmol) in methylene dichloride (10 ml) was added N-methylmorpholine (0.039 g, 0.38 mmol). The resulting mixture was stirred at room temperature for overnight followed by dilution with 10 ml methylene dichloride. The reaction mixture was poured onto water (20 ml), and organic layer separated, washed with water (2x 20 ml), brine (20 ml), dried over sodium sulfate and solvent evaporated to get residue which was purified by preparative TLC using 50% ethyl acetate in hexane as mobile. To give desired compound (0.30 g). 1H NMR (400 MHz, CDCl3): δ 1.45 (t, J = 7.2 Hz, 3H), 1.93 (s, 3H), 3.14 (s, 3H), 3.77 (d, J = 2.8 Hz, IH), 4.26 (q, J = 7.2 Hz, IH), 6.69-6.77(m, 2H), 6.96-7.02 (m, IH), 7.89 (d, J = 8.4 Hz, 2H), 8.07 (d, J= 8.4Hz, IH).; MS (EI) m/z: 559 .00 (M + 1).
PATENT
http://www.google.com/patents/WO2012020357A1?cl=en
4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]- thiazol-5-yloxy}-benzoic acid, cas 1359151-08-8

Step I: (4-Cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester:
A1C13 (7.98 g, 48.42 mmole) was suspended in DCM (50 mL) and cooled to 0 C under argon atmosphere. To this suspension was added chlorooxo ethylacetate (4.5 mL, 39.98 mmol) at 0 °C and stirred for 45 min. followed by addition of a solution of cyclopropylsulfanyl-benzene (5 g, 33.28 mmol) in DCM (10 mL) and stirred at 25 °C for 2 hr. Reaction mixture was slowly poured over crushed ice, organic layer was separated and aqueous layer was extracted with DCM (3 X 50 mL), combined organic layer was washed with brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain (4- cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester (3.1 g) as an oily product.
*H NMR (400 MHz, CDC13): δ 0.72-0.73 (m, 2H), 1.15-1.17 (m, 2H), 1.40 (t, J = 6.6 Hz, 3H), 2.18-2.21 (m, 1H), 4.41 (q, J = 6.8 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H); MS (EI) m/z: 250.9 (M+l).
Step II: (4-Cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester:
(4-Cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester (3.1 g, 12.53 mmole) in DCM (50 mL) was cooled to 0-5 °C followed by addition of mCPBA (9.8 g , 31.33 mmol) in portion wise at 0 °C. After stirring at 25 °C for 4 hr, the reaction mixture was filtered; filtrate was washed with saturated aq. Na2S203 and satd. aq. sodium bicarbonate solution followed by brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give (4-cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester (3 g).
*H NMR (400 MHz, CDC13): δ 1.05-1.10 (m, 2H), 1.36-1.39 (m, 2H), 1.40 (t, J = 6.8 Hz, 3H), 2.45-2.50 (m, 1H), 4.42 (q, J = 7.2 Hz, 2H), 8.01 (d, J = 8.4 Hz, 2H), 8.20 (d, J = 8.4 Hz, 2H); MS (EI) m/z: 297.1 (M+NH4).
Step III: p-Toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester:
A mixture of (4-cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester (0.5 g, 1.77 mmole) and p-toluene sulfonyl hydrazide (0.48 g , 2.3 mmol) in toluene (15 mL) was refluxed for 16 hr using a Dean-Stark apparatus. Reaction mixture was concentrated to give the crude product which was purified by column chromatography over silica gel using 20-25% ethyl acetate in hexane as eluent to provide p-toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester (0.5 g).
MS (EI) m/z 451.0 (M+l).
Step IV: (4-Cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester:
To a solution of p-toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester (0.5 g, 1.23 mmol) in dry DCM (6 mL), was added triethylamine (0.17 mL, 1.35 mmol) and stirred at 25 °C for 1 hr. Reaction mixture was concentrated to provide (4- cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester (0.5 g) which was used in next reaction without any purification.
MS (EI) m/z: 295.1 (M+l).
Step V: Cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester:
(4-Cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester (1 g, 3.37 mmol) was dissolved in DCM (16 mL) under argon atmosphere. To this solution, cyclopentanol (0.77 mL, 8.44 mmol) was added followed by rhodium(II)acetate dimer (0.062 g, 0.14 mmol). Mixture was stirred at 25 C for 12 hr. Reaction mixture was diluted with DCM (25 mL), organic layer was washed with water followed by brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a crude product which was purified by column chromatography using 25-35% ethyl acetate in hexane as eluent to provide cyclopentyloxy-(4- cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester (0.35 g).
*H NMR (400 MHz, CDC13): δ 1.02-1.05 (m, 2H), 1.24 (t, J = 6.8 Hz, 3H), 1.35-1.37 (m, 2H), 1.53-1.82 (m, 8H), 2.42-2.50 (m, 1H), 4.02-4.04 (m, 1H), 4.15-4.22 (m, 2H), 5.00 (s, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H); MS (EI) m/z: 370.0 (M+18).
Step VI: Cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid:
To cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester (0.35 g, 0.99 mmol) was added a solution of lithium hydroxide (0.208 g, 4.97 mmol) in water (4 mL) followed by THF (2 mL) and methanol (1 drop) and stirred for 12 hours at 25 0 C. Organic solvents were evaporated from the reaction mixture and aqueous layer was acidified IN HCl, extracted with ethyl acetate (3 X 10 mL), organic layer was washed with brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide cyclopentyloxy-(4- cyclopropanesulfonyl-phenyl)-acetic acid (0.210 g).
*H NMR (400 MHz, CDC13): δ 1.02-1.07 (m, 2H), 1.34-1.38 (m, 2H), 1.55-1.62 (m, 2H), 1.69- 1.82 (m, 6H), 2.43-2.47 (m, 1H), 4.08-4.10 (m, 1H), 5.02 (s, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H); MS (EI) m/z: 342.0 (M+18)
Example Al: 4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]-

To a mixture of cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid (Preparation 1) (0.1 g, 0.30 mmol), 4-(2-Amino-thiazol-5-yloxy)-benzoic acid methyl ester (0.085 g, 0.33 mmol), HOBt (0.045 g, 0.33 mmol), and EDCI (0.063 g, 0.33 mmol) in DCM (5 mL), was added N-methyl morpholine (0.033 g, 0.30 mmol). The resulting mixture was stirred at room temperature overnight followed by dilution with methylene chloride (20 mL). The reaction mixture was poured into water; organic layer was washed with water, brine, dried over sodium sulfate, and the organic solvent evaporated to get a residue which was purified by preparative TLC to provide the title compound (0.145 g).
*H NMR (400 MHz, CDC13): δ 1.03-1.05 (m, 2H), 1.34-1.38 (m, 2H), 1.58- 1.65 (m, 2H), 1.76- 1.81 (m, 6H), 2.42-2.45 (m, 1H), 3.89 (s, 3H), 4.05-4.15 (m, 1H), 5.08 (s, 1H), 7.07 (d, J = 8.8 Hz, 2H), 7.15 (s, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.8 Hz, 2H), 9.72 (s, 1H); MS (EI) m/z: 556.9 (M + 1).
Example Bl: 4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]- thiazol-5-yloxy}-benzoic acid:
4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]-thiazol-5-yloxy}- benzoic acid methyl ester (0.145 g, 0.26 mmol, obtained in example Al) was taken in H20: THF (1 :2, 6 mL) to it was added MeOH (1 drop) followed by LiOH (0.054 g, 1.30 mmol) and stirred for 12 hr. After completion of the reaction, organic solvent was removed under reduced pressure. The aqueous layer was washed with diisopropyl ether then acidified with 1 N HC1 to pH 4. The solid formed was filtered, washed with water, diisopropyl ether & dried under vacuum to get the title_compound (0.12 g).
IH NMR- (400 MHz DMSO-ifc):- δ 1.01-1.05 (m, 2H), 1.09-1.13 (m, 2H), 1.22-1.49 (m, 2H), 1.59-1.73 (m, 6H), 2.82-2.86 (m, IH), 3.99-4.01 (m, IH), 5.31 (s, IH), 7.16 (d, J = 8.4 Hz, 2H), 7.37 (s, IH), 7.74 (d, J = 8.4 Hz, 2H), 7.91 (m, 4H), 12.55 (br. s, IH), 12.90 (br.s, IH); MS (EI) m/z: 542.9 (M+l)
CLIPPINGS
Advinus’ GK-activator Achieves Early POC for Diabetes
November 29 2011
Partnership Dialog Actively Underway
Advinus Therapeutics, a research-based pharmaceutical company founded by globally experienced industry executives and promoted by the TATA Group, announced that it has successfully completed a 14-day POC study in 60 Type II diabetic patients on its lead molecule, GKM-001, a glucokinase activator. The results of the trial show effective glucose lowering across all doses tested without any incidence of hypoglycemia or any other clinically relevant adverse events.
The clinical trials on GKM-001 validate the company’s pre-clinical hypothesis that a liver selective Glucokinase activator would not cause hypoglycemia (very low blood sugar), while showing robust efficacy.
“GKM-001 is differentiated from most other GK molecules that are in development, or have been discontinued, due to its novel liver selective mechanism of action. GKM-001 has a prolonged pharmacological effect and a half-life that should support a once a day dosing as both mono and combination therapy.” said Dr. Rashmi Barbhaiya, MD & CEO, Advinus Therapeutics. He added that Advinus is actively exploring partnership options to expedite further development and global marketing of GKM-001.
GKM-001 belongs to a novel class of molecules for treatment of type II diabetes. It is an activator of Glucokinase (GK), a glucose-sensing enzyme found mainly in the liver and pancreas. Being liver selective, GKM-001 mostly activates GK in the liver and not in pancreas, which is its key differentiation from most competitor molecules that activate GK in pancreas as well. The resulting increase in insulin secretion creates a potential for hypoglycemia-a risk GKM-001 is designed to avoid. Advinus has the composition of matter patent on GKM-001 for all major markets globally. Both the Single Ascending Dose data, in healthy and type II diabetics, and the Multiple Ascending Dose Study in Type II diabetics has shown that the molecule shows effective glucose lowering in a dose dependent manner and has excellent safety and tolerability profile over a 40-fold dose range. The pharmacokinetic properties of the molecule support once a day dosing. GKM-001 has the potential to be “First-in-Class” drug to address this large, growing and yet poorly addressed market.
Advinus also has identified a clinical candidate as a back-up to GKM-001, which is structurally different. In its portfolio, the company has a growing pipeline for COPD, sickle cell disease, inflammatory bowel disease, type 2 diabetes, acute and chronic pain and rheumatoid arthritis in various stages of late discovery and pre-clinical development.
About the Diabetes Market:
The present 300 million diabetics population is estimated to jump to 450 million by 2030 worldwide. A large proportion of these patients are poorly controlled despite multiple therapies. Total sales of diabetic prescription products were $32 billion in 2010.
Advinus Therapeutics team discovers novel molecule for treatment of diabetes
- The first glucokinase modulator discovered and developed in India
- A new concept for the management of diabetes for patients, globally
- 100 per cent ‘made in India’ molecule for the treatment of diabetes
- IND approved by DGCI, Phase I clinical trial shows excellent safety and tolerance profiles with efficacy
Bangalore: Advinus Therapeutics (Advinus), the research-based pharmaceutical company founded by leading global pharmaceutical executives and promoted by the Tata group, today, announced the discovery of a novel molecule for the treatment of type II diabetes — GKM-001.The molecule is an activator of glucokinase; an enzyme that regulates glucose balance and insulin secretion in the body.
GKM-001 is a completely indigenously developed molecule and the initial clinical trials have shown excellent results for both safety and efficacy.
“Considering past failures of other companies on this target, our discovery programme primarily focused on identifying a molecule that would be efficacious without causing hypoglycaemia; a side effect associated with most compounds developed for this target.
“Recently completed Phase I data indicate that Advinus’ GKM–001 is a liver selective molecule that has overcome the biggest clinical challenge of hypoglycaemia. GKM-001 is differentiated from most other GK molecules in development due to this novel mechanism of action,” said Dr Rashmi Barbhaiya, MD and CEO, Advinus Therapeutics.
He further added, “We are very proud that GKM-001 is 100 per cent Indian. Advinus’s discovery team in Pune discovered the molecule and entire preclinical development was carried out at our centre in Bangalore. The Investigational New Drug (IND) application was filed with the DGCI for approval to initiate clinical trials in India within 34 months of initiation of the discovery programme. Subsequent to the approval of the IND, we have completed the Phase I Single Ascending Dose study in India within two months.”
GKM-001 is a novel molecule for the treatment of type II diabetes. It is the first glucokinase modulator discovered and developed in India and has potential to be both first or best in class. The success in discovering GKM-001 is attributed to the science-driven efforts in Advinus laboratories and ‘breaking the conventional mold’ for selection of a drug candidate. Advinus has ‘composition of matter’ patent on the molecule for all major markets globally. Glucokinase as a class of target is considered to be novel as currently there is no product in the market or in late clinical trials. The strategy for early clinical development revolved around assessing safety (particularly hypoglycaemia) and early assessment of therapeutic activity (glucose lowering and other biomarkers) in type II diabetics. The Phase I data, in both healthy and type II diabetics, shows excellent safety and tolerability over a 40-fold dose range and desirable pharmacokinetic properties consistent with ‘once a day’ dosing. The next wave of clinical studies planned continues on this strategy of early testing in type II diabetics.
Right behind the lead candidate GKM-001, Advinus has a rich pipeline of back up compounds on the same target. These include several structurally different compounds with diverse potency, unique pharmacology and tissue selectivity. Having discovered the molecule with early indication of wide safety margins, desired efficacy and pharmacokinetic profiles, the company now seeks to out-licence GKM-001 and its discovery portfolio.
Patent
wo 2008104994
| WO2008104994A2 * | 25 Feb 2008 | 4 Sep 2008 | Advinus Therapeutics Private L | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2008104994A2 * | Feb 25, 2008 | Sep 4, 2008 | Advinus Therapeutics Private L | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2009047798A2 * | Oct 7, 2008 | Apr 16, 2009 | Advinus Therapeutics Private L | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
///////GKM 001, pipeline, Diabetes, Advinus, type II diabetes, glucokinase modulator, Rashmi Barbhaiya
Some pics

Annual day party at Advinus !!!with Rashmi Barbhaiya
Dr. Rashmi Barbhaiya, MD & CEO, Advinus Therapeutics Pvt.
 .
.
with Kaushal Joshi, Vishal Pathade, Ramanareddy Jinugu, Mohammed Kakajiwala, Vishal Baxi and Dilip Reddy.
///////
New route for Expensive drug Ivacaftor synthesis from CSIR-NCL, Pune, India



IVACAFTOR
Breaking and Making of Rings: A Method for the Preparation of 4-Quinolone-3-carboxylic Acid Amides and the Expensive Drug Ivacaftor
Article first published online: 3 NOV 2015
DOI: 10.1002/ejoc.201501048
http://onlinelibrary.wiley.com/doi/10.1002/ejoc.201501048/abstract
SUPPORTING INFO……….http://onlinelibrary.wiley.com/store/10.1002/ejoc.201501048/asset/supinfo/ejoc_201501048_sm_miscellaneous_information.pdf?v=1&s=2b5b6ac6456ec88f478c07a692e49254e7239f80
Abstract
A simple and convenient method to access 4-quinolone-3-carboxylic acid amides from indole-3-acetic acid amides through one-pot oxidative cleavage of the indole ring followed by condensation (Witkop–Winterfeldt type oxidation) was explored. The scope of the method was confirmed with more than 20 examples and was successfully applied to the synthesis of the drug Ivacaftor, the most expensive drug on the market.


REFERENCES
N. Vasudevan, Gorakhnath R. Jachak And D. Srinivasa Reddy, Breaking and Making of Rings: A Method for the Preparation of 4-Quinolone-3-carboxylic Acid Amides and the Expensive Drug Ivacaftor, Eur. J. Org. Chem., , 0000 (2015), DOI:10.1002/ejoc.201501048. 
http://academic.ncl.res.in/publications/index/select-faculty/2015/ocd
Breaking and Making of Rings: A Method for the Preparation …
6 days ago – European Journal of Organic Chemistry … 20 examples and was successfully applied to the synthesis of the drug Ivacaftor, the most expensive …
European Journal of Organic Chemistry – Wiley Online Library
European Journal of Organic Chemistry ….. examples and is successfully applied to the synthesis of the drug Ivacaftor, the most expensive drug on the market.
Breaking and making – Wiley Online Library
6 days ago – … for the Preparation of 4-Quinolone-3-carboxylic Acid Amides and the Expensive Drug Ivacaftor … European Journal of Organic Chemistry.
READ ABOUT DR SRINIVASA REDDY at…………
ONE ORGANIC CHEMIST ONE DAY BLOG……..LINK
Dr. Srinivasa Reddy of CSIR-NCL bags the
prestigious Shanti Swarup Bhatnagar Prize

AN INTRODUCTION
Ph.D., University of Hyderabad, 2000 (Advisor: Professor Goverdhan Mehta).
Post-doctoral with Profs. Sergey A. Kozmin(University of Chicago, USA) and Prof.
Jeffrey Aubé (University of Kansas, USA)
Experienced in leading drug discovery programs (Dr. Reddy’s & TATA Advinus – 7
years of pharma experience)
Acquired skills in designing novel small molecules and lead optimization
Experienced in planning and execution of total synthesis of biologically active
molecules with moderate complexity
One of the molecules is currently in human clinical trials.

SILICO LINEZOLID, SILINEZOLID, NDS 10024

Therapeutic options for brain infections caused by pathogens with a reduced sensitivity to drugs are limited. Recent reports on the potential use of linezolid in treating brain infections prompted us to design novel compounds around this scaffold. Herein, we describe the design and synthesis of various oxazolidinone antibiotics with the incorporation of silicon.
Our findings in preclinical species suggest that silicon incorporation is highly useful in improving brain exposures. Interestingly, three compounds from this series demonstrated up to a 30-fold higher brain/plasma ratio when compared to linezolid thereby indicating their therapeutic potential in brain associated disorders
Design, Synthesis, and Identification of Silicon Incorporated Oxazolidinone Antibiotics with Improved Brain Exposure
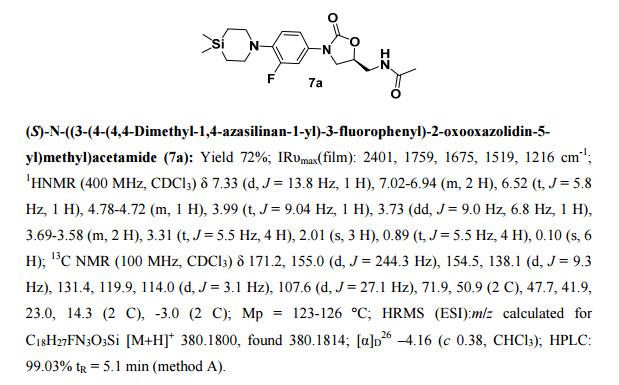


Examples from patent

- (S)—N((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2 oxooxazolidin-5-yl)methyl)acetamide
- NDS 10024
- Preparation of (S)—N((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2 oxooxazolidin-5-yl)methyl)acetamide (12)
-
-
To a solution of 8 (50 mg, 0.135 mmol) in dimethylformamide (DMF), lithium-t-butoxide (LiOtBu) (32.3 mg, 0.4 mmol) is added. The mixture is stirred at 25° C. for 15 min, followed by the addition of MeOH (0.01 mL, 0.27 mmol). 6 (52 mg, 0.27 mmol) is then added and the reaction mixture is allowed to stir at 25° C. for 24 h. Glacial acetic acid is then added and the organic phase is extracted with EtOAc and washed with brine solution. The crude material is purified by column chromatography on silica gel using hexane-EtOAC mixtures to furnish the pure product 12. The analogous procedure for the corresponding morpholine analogue was adapted from Lu, C. V.; Chen, J. J.; Perrault, W. R.; Conway, B. G.; Maloney, M. T.; Wang, Y. Org. Pro. Res. and Development. 2006, 10, 272-277.
-
1H NMR (200 MHz, CDCl3): δ 7.33 (d, J=13.8 Hz, 1H), 7.02-6.94 (m, 2H), 6.52 (t, J=5.8 Hz, 1H), 4.77-4.73 (m, 1H), 3.99 (t, J=9.04 Hz, 1H), 3.72 (dd, J=9.0 Hz, 6.8 Hz, 1H), 3.69-3.58 (m, 2H), 3.31 (t, J=5.5 Hz, 4H), 2.01 (s, 3H), 0.89 (t, J=5.5 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ171.2, 155.0 (d, J=244.3 Hz), 154.5, 138.2 (d, J=9.3 Hz), 131.5, 119.9, 114.0 (d, J=3.4 Hz), 107.6 (d, J=27.1 Hz), 71.9, 50.9, 47.7, 41.9, 23.0, 14.3, −2.9.
- Preparation of Bis(bromomethyl)dimethylsilane (2) (as per scheme 2)
-

-
HBr gas is bubbled to a solution of dimethyl divinylsilane 1 (10.0 g, 89.28 mmols), and dibenzoylperoxide (DBP, 100 mg), in heptane (100 mL) at 0° C. for 30 min. The Reaction mixture (RM) is allowed to stir at room temperature (25° C.) for 18 h, water (200 mL) is added to the reaction mixture and the organic layer is separated. The heptane layer is washed with 2N NaOH (2 100 mL), dried and concentrated to obtain the product 2 as a colourless liquid (24.5 g) in 100% yield.
-
1H NMR (200 MHz, CDCl3): δ 3.58-3.49 (m, 4H), 1.45-1.40 (m, 4H), 0.09 (s, 6H).
-

-
Benzylamine (20 mL, 182 mmol) and Et3N (15.2 mL, 109 mmol) are added to a solution of bis-(bromomethyl) dimethylsilane 2 (10 g, 36.5 mmol) in chloroform (100 mL). The mixture is then refluxed for 16 h. 5% sodiumhydroxide solution (150 mL) is then added and the aqueous layer is extracted with dichloromethane (DCM, 2×100 mL). It is then washed with brine (200 mL), dried and concentrated. The product is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product 3 as a light yellow liquid (4.3 g) in 54% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.23-7.35 (m, 5H), 3.66 (s, 2H), 2.68 (t, J=6.3 Hz, 4H), 0.75 (t, J=6.3 Hz, 4H), 0.04 (s, 6H).
- Preparation of 1-benzyl-4,4-dimethyl-1,4-azasilinane (3)
Preparation of 4,4-dimethyl-1,4-azasilinane hydrochloride (4)
-

-
To a solution of 4,4-dimethyl-1,4-azasilinane 3 (2.3 g, 10.5 mmol) in EtOH (20 mL), 6N hydrochloricacid (1.75 mL, 10.5 mmol) is added and the solvent is removed under reduced pressure. The reaction mixture is co-evaporated with EtOH (2×10 mL) and recrystallized from EtOH-diethyl ether. To a slurry of Pd/C (50 mg) in EtOH (15 mL) an ethanolic solution of above prepared HCl salt is added drop wise and stirred at 25° C. under hydrogen atmosphere for 20 h. The reaction mixture is filtered through celite and washed with 2×20 mL of MeOH. The filtrate is then concentrated under reduced pressure to give viscous oil which was triturated with diethyl ether to obtain the product 4 as a white solid (950 mg) in 70% yield.
Preparation of 1-(2-fluoro-4-nitrophenyl)-4,4-dimethyl-1,4-azasilinane (9)
-

-
To a solution of 4,4-dimethyl-1,4-azasilinane hydrochloride 4 (500 mg, 3.85 mmol) in EtOAc (15 mL), triethylamine (1.3 mL, 9.63 mmol) is added and stirred at 25° C. for 10 min. The reaction mixture is cooled to 0° C. and 3,4-difluoronitrobenzene (612 mg, 3.85 mmol) is added drop wise and allowed to stir at 25° C. for 6 h. Water is then added and the organic layer is separated. The aqueous layer is extracted with EtOAc (2×10 mL) and the solvent is removed under reduced pressure. The product is purified by column chromatography using hexane-EtOAc mixtures and a crystalline yellow solid 9 (721 mg) is obtained in 70% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.93-7.84 (m, 2H), 6.86 (t, J=4 Hz, 1H), 3.70-3.67 (m, 4H), 0.91-0.85 (m, 4H), 0.12 (s, 6H). 13C NMR (50 MHz, CDCl3): δ 151.1 (d, J=246.71 Hz), 144.4 (d, J=7.13 Hz), 137.8 (d, J=8.59 Hz), 121.4, 115.9 (d, J=4.61 Hz), 113.2 (J=27.78 Hz), 49.4, 13.8, −2.8. IR (CHCl3): ν 2948, 2894, 1603, 1523, 1492, 1400, 1342, 1223, 983, 832, 742 cm−1′. M.P: 70-72° C.
Preparation of benzyl 4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenylcarbamate (10)
-

-
To a solution of compound 9 (610 mg, 2.28 mmol) in THF (25 mL), Pd/C (30 mg) is added and hydrogenated under a pressure of 35 psi in a par hydrogenator for 8 h. The reaction mixture is filtered through celite. Celite pad is washed with THF (2×20 mL). To the filtrate, saturated NaHCO3 (420 mg, 5.01 mmol) and CBzCl (427 mg, 2.5 mmol) are added at 0° C. and stirred at 25° C. for 5 h. 10 mL water is added to reaction mixture and the aqueous layer is extracted with EtOAc (2×20 mL). The crude mixture is then subjected to column chromatography on silica gel using hexane-EtOAc mixtures to afford the product as a viscous liquid 10 (690 mg) in 82% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.41-7.37 (m, 5H), 6.94-6.93 (m, 2H), 6.68 (s, 1H), 5.21 (s, 1H), 3.3 (t, J=6.38 Hz, 4H), 0.93 (t, J=6.08 Hz, 4H), −0.13 (s, 6H). 13C NMR (50 MHz, CDCl3): 155.4 (d, 244.4 Hz), 153.6, 136.1, 135.9, 128.6, 128.5, 128.3, 120.4, 117.2 (d, 18.7 Hz), 114.7, 108.3 (20.5 Hz), 67.1, 51.4, 14.4, −3.0. IR (CHCl3): ν 3317, 2953, 2803, 1706, 1594, 1521, 1271, 1221, 1058, 869, 759 cm−1. M.P: 80-82° C.
Preparation of (S)-5-(aminomethyl)-3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)oxazolidin-2-one (11) (NDS-10057)
-

-
To a solution of 10 (1.20 g, 3.23 mmol) and (S)-tert-butyl 3-chloro-2-hydroxypropylcarbamate (1.35 g, 6.47 mmol) in DMF (10 mL), LiOtBu (1.03 g, 12.94 mmol) is added at 0° C. The mixture is stirred at 25° C. for 45 h. The starting material 10 is not consumed completely. Saturated NH4Cl is then added; the organic phase is extracted with EtOAc (2×20 mL), washed with brine solution, dried and concentrated. The crude residue is dissolved in 20 mL of DCM-TFA mixture (8:2) and stirred at 25° C. for 3 h. RM is concentrated and dissolved in water (10 mL), the aqueous layer is washed with diethyl ether (2×50 mL), basified with saturated NaHCO3 and extracted with DCM (2×50 mL). The DCM layer is dried and concentrated. The crude is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product as an off-white solid (500 mg) in 45% (based on recovery of starting material) over 2 steps.
-
1H NMR (400 MHz, CDCl3): δ 7.36 (dd, J=14.2 Hz, 2.3 Hz, 1H), 7.09 (dd, J=8.8 Hz, 1.7 Hz, 1H), 6.96 (t, J=9.5 Hz, 1H), 4.72-4.59 (m, 1H), 4.00 (t, J=8.3 Hz, 1H), 3.79 (dd, J=8.7 Hz, 6.8 Hz, 1H), 3.30 (t, J=6.2 Hz, 4H), 3.03 (dq, J=13.6 Hz, 4.2 Hz, 2H), 0.90 (t, J=6.2 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 155.1 (d, J=244.3 Hz), 154.7, 137.9 (d, J=9.0 Hz), 132.1 (d, J=10.3 Hz), 112.0 (d, J=4.3 Hz), 113.8 (d, J=3.2 Hz), 107.4 (d, J=26.9 Hz), 73.8, 51.0, 47.8, 45.01, 14.4, −2.9. IR (CHCl3): ν 3685, 3021, 2955, 2809, 2401, 1747, 1515, 1416, 1219, 1029, 991, 870, 771, 667 cm−1. M.P: 94-96° C. ESI-MS: 360.11 (M+Na).
Preparation of (S)—N-((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2-oxooxazolidin-5-yl)methy)acetamide (12) (NDS 10024)
-

-
To solution of amine 11 (300 mg, 0.9 mmol) and DIPEA (0.3 mL, 1.78 mmol) in dry THF (4.0 mL), acetylchloride (0.08 mL, 1.07 mmol) is added at 0° C., and stirred at 25° C. for 3 h. Further, saturated NaHCO3 (5.0 mL) is added to the reaction mixture and extracted with EtOAc (2×5 mL). The organic layer is washed with brine, dried and concentrated. The product is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product as an off-white solid (170 mg) in 50% yield.
-
1HNMR (400 MHz, CDCl3): δ 7.33 (d, J=13.8 Hz, 1H), 7.02-6.94 (m, 2H), 6.52 (t, J=5.8 Hz, 1H), 4.77-4.73 (m, 1H), 3.99 (t, J=9.04 Hz, 1H), 3.72 (dd, J=9.0 Hz, 6.8 Hz, 1H), 3.69-3.58 (m, 2H), 3.31 (t, J=5.5 Hz, 4H), 2.01 (s, 3H), 0.89 (t, J=5.5 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ171.2, 155.0 (d, J=244.3 Hz), 154.5, 138.2 (d, J=9.3 Hz), 131.5, 119.9, 114.0 (d, J=3.4 Hz), 107.6 (d, J=27.1 Hz), 71.9, 50.9, 47.7, 41.9, 23.0, 14.3, −2.9. IR (CHCl3): ν 2401, 1759, 1675, 1519, 1216, 759, 669 cm−1 M.P: 123-126° C. ESI-MS: 380.10 (M+H).


SCHEME2


SCHEME 3

SCHEME 4



Dr. D. Srinivasa Reddy of NCL winner Shanti Swarup Bhatnagar Award 2015
see
http://oneorganichemistoneday.blogspot.in/2015/02/dr-d-srinivasa-reddy.html
Dr. Srinivasa Reddy of CSIR-NCL bags the
prestigious Shanti Swarup Bhatnagar Prize
AN INTRODUCTION
Ph.D., University of Hyderabad, 2000 (Advisor: Professor Goverdhan Mehta).
Post-doctoral with Profs. Sergey A. Kozmin(University of Chicago, USA) and Prof.
Jeffrey Aubé (University of Kansas, USA)
Experienced in leading drug discovery programs (Dr. Reddy’s & TATA Advinus – 7
years of pharma experience)
Acquired skills in designing novel small molecules and lead optimization
Experienced in planning and execution of total synthesis of biologically active
molecules with moderate complexity
One of the molecules is currently in human clinical trials.
MYSELF WITH HIM
OTHER AUTHORS




////////
C[Si]1(C)CCN(CC1)c2ccc(cc2F)N3C[C@H](CNC(C)=O)OC3=O
CEP 18770, Delanzomib

CEP-18770, Delanzomib
cas 847499-27-8
Chemical Formula: C21H28BN3O5
Exact Mass: 413.21220, UNII-6IF28942WO;
CT-47098
NPH 007098
NPH007098
[(1R)-1-[[(2S,3R)-3-Hydroxy-2-[[(6-phenylpyridin-2-yl)carbonyl]amino]-1-oxobutyl]amino]-3-methylbutyl]boronic acid
[(lR)-l-[[(2S,3R)-3-hydroxy-2- [6-phenyl-pyridine-2-carbonyl)amino]-l-oxobutyl]amino]-3-methylbutylboronic acid,
Boronic acid, ((1R)-1-(((2S,3R)-3-hydroxy-1-oxo-2-(((6-phenyl-2-pyridinyl)carbonyl)amino)butyl)amino)-3-methylbutyl)-
In phase 2, multiple mylenoma, Ethical Oncology Science (EOS), licensee
CEP-18770 was discovered through collaboration between Cephalon and Novuspharma/CTI.
Cephalon, Inc., 145 Brandywine Parkway, West Chester, Pennsylvania 19380, and Cell Therapeutics Europe S.r.l., Via L. Ariosto, 23, I-20091 Bresso, Italy
Cephalon was acquired by Teva in October 2011. In 2013, EOS was acquired by Clovis Oncology.
Chemical Process Research and Development, Teva Branded Pharmaceutical Products R&D Inc., 383 Phoenixville Pike, Malvern, Pennsylvania 19355, United States

CEP-18770 is a reversible P2 threonine boronic acid inhibitor of the chymotrypsin-like activity of the proteasome. Displays anti-multimyeloma (MM) effect.

HPLC………http://www.apexbt.com/downloader/document/A4009/HPLC.pdf
NMR………http://www.apexbt.com/downloader/document/A4009/NMR.pdf


CLICK ON IMAGE FOR CLEAR VIEW
Delanzomib, also known as CEP-18770, is An orally bioavailable synthetic P2 threonine boronic acid inhibitor of the chymotrypsin-like activity of the proteasome, with potential antineoplastic activity. Proteasome inhibitor CEP 18770 represses the proteasomal degradation of a variety of proteins, including inhibitory kappaBalpha (IkappaBalpha), resulting in the cytoplasmic sequestration of the transcription factor NF-kappaB; inhibition of NF-kappaB nuclear translocation and transcriptional up-regulation of a variety of cell growth-promoting factors; and apoptotic cell death in susceptible tumor cell populations. In vitro studies indicate that this agent exhibits a favorable cytotoxicity profile toward normal human epithelial cells, bone marrow progenitors, and bone marrow-derived stromal cells relative to the proteasome inhibitor bortezomib. The intracellular protein IkappaBalpha functions as a primary inhibitor of the proinflammatory transcription factor NF-kappaB
New series of dipeptidyl boronate inhibitors of 20S proteasome were identified to be highly potent drug-like candidates with IC50 values of 1.2 and 1.6 nM, respectively, which showed better activities than the drug bortezomib on the market
ref
The potent, selective, and orally bioavailable threonine-derived 20S human proteasome inhibitor that has been advanced to preclinical development, [(1R)-1-[ [ (2S,3R)- 3-hydroxy-2-[ (6-phenylpyridine- 2-carbonyl) amino]-1 -oxobutyl] amino]- 3-methylbutyl] boronic acid (CEP-18770, has been reported
ref .
Dorsey BD, Iqbal M, Chatterjee S, Menta E, Bernardini R, Bernareggi A, et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J Med Chem. 2008;51:1068–1072. [PubMed]
Further, the anti-multiple myeloma protea-some inhibitor CEP-18770 enhanced the anti-myeloma activity of bortezomib and melphalan. The combination of anti-multiple myeloma proteasome inhibitor CEP-18770 intravenously and bortezomib exhibited complete regression of bortezomib-sensitive tumours. Moreover, this combination markedly delayed progression of bortezomib-resistant tumours compared to treatment with either agent alone
Paper
Development and scale-up of an optimized route to the peptide boronic acid, CEP-18770
Org Process Res Dev 2013, 17(3): 422
http://pubs.acs.org/doi/abs/10.1021/op400010u
 USED AS PRODRUG
USED AS PRODRUGCEP-18770 is an unstable peptide boronic acid and an amorphous solid, making it a challenging synthetic target. Process R&D led to a new process that avoided chromatography through crystalline intermediates, increased atom and volume efficiency, provided a chromophore, and gave higher yields and purity. A stable, crystalline diethanolamine adduct was discovered that has the potential to be used as a prodrug.

Compound 8 proved to be a direct substitute for delanzomib in the formulation process. In the first step of the IV formulation process, delanzomib is dissolved in water along with several excipients. Predictably, the delanzomib degrades during this process. It was found that upon dissolution in the lyophilization medium, 8 hydrolyzes to delanzomib,
N-[(1S,2R)-1-[[[(1R)-1–1[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-methano-1,3,2-benzodioxaborol-2-yl]-3-methylbutyl]amino]carbonyl]-2-hydroxypropyl]-6-phenyl-2-pyridinecarboxamide (5)
PAPER
Discovery of a Potent, Selective, and Orally Active Proteasome Inhibitor for the Treatment of Cancer
http://pubs.acs.org/doi/abs/10.1021/jm7010589

The ubiquitin−proteasome pathway plays a central role in regulation of the production and destruction of cellular proteins. These pathways mediate proliferation and cell survival, particularly in malignant cells. The successful development of the 20S human proteasome inhibitor bortezomib for the treatment of relapsed and refractory multiple myeloma has established this targeted intervention as an effective therapeutic strategy. Herein, the potent, selective, and orally bioavailable threonine-derived 20S human proteasome inhibitor that has been advanced to preclinical development, [(1R)-1-[[(2S,3R)-3-hydroxy-2-[(6-phenylpyridine-2-carbonyl)amino]-1-oxobutyl]amino]-3-methylbutyl]boronic acid 20 (CEP-18770), is disclosed.
[(1R)-1-[[(2S,3R)-3-Hydroxy-2-[(6-phenylpyridine-2-carbonyl)amino]-1-oxobutyl]amino]-3-methylbutyl]boronic Acid (20)
Patent
http://www.google.com/patents/WO2010056733A1?cl=en
Preferred among these compounds is [(lR)-l-[[(2S,3R)-3-hydroxy-2- [6-phenyl-pyridine-2-carbonyl)amino]-l-oxobutyl]amino]-3-methylbutylboronic acid, also known as CEP- 18770, which has the following structure:

PATENT
http://www.google.co.in/patents/WO2005021558A2
NOT SAME BUT SIMILAR
Example E.4 Boronic acid, [(lR)-l-[[(2S,3R)-3-hydroxy-2-[[4-(3-pyridyl)benzoyl]amino]-l- oxobutyI]amino]-3-methyIbutyl].

[00275] A mixture of 4-(pyridin-3-yl)benzamide, N-[(1S,2R)-1-[[[(1R)-1-
[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-methano-l,3,2-benzodioxaborol-2- yl]-3-methylbutyl]amino]carbonyl]-2-hydroxypropyl]- of Example D.8.3 (155 mg, 0.283 mmol), 2-methylpropylboronic acid (81 mg, 0.793 mmol) and 2N aqueous hydrochloric acid (0.3 ml) in a heterogeneous mixture of methanol (3 ml) and hexane (3 ml) was stirred at room temperature for 24 hours. The hexane layer was removed and the methanolic layer was washed with fresh hexane (about 5 ml). Ethyl acetate (10 ml) was added to the methanol layer which was then concentrated. The residue was taken up with ethyl acetate and the mixture was concentrated. This step was repeated (2-3 times) until an amorphous white solid was obtained. The solid was then triturated with diethyl ether (5 ml) and the surnatant was removed by decantation. This step was repeated. The residue (126 mg) was combined with the product of a similar preparation (140 mg) and dissolved in ethyl acetate (about 40 ml) and a small amount of methanol (2-3 ml). The solution was washed with a mixture of NaCl saturated solution (7 ml) and 10% NaHCO3 (2 ml). The layers were separated and the aqueous phase was further washed with ethyl acetate (2 x 20 ml). The combined organic phases were dried over sodium sulfate and concentrated. The residue was taken up with ethyl acetate (about 20 ml) and the minimum amount of methanol, and then concentrated to small volume (about 5 ml). The resulting white was collected by filtration and dried under vacuum at 50°C (160 mg, 65% overall yield).
1H NMR (MeOH-d4): 8.90 (IH, s); 8.49 (IH, d, J=4.0); 8.20 (IH, d, J=8.1); 8.06 (2H, d, J=8.1); 7.85 (2H, d, J=8.1); 7.58 (IH, t br., J=6.0); 4.80 (IH, d, J=3.9); 4.40-4.29 (IH, m); 2.78 (IH, t, J=7.5); 1.73-1.61 (IH, m); 1.38 (2H, t, J=6.9); 1.31 (3H, d, J=6.3); 0.94 (6H, d, J=6.31). [00276] Further compounds prepared according to the above procedure for
Example E.4 are reported in Table E-4. Table E-4

E.4.3 IS THE COMPD
D.8.12 Chemical Name: 6-Phenyl-2-pyridinecarboxamide,N-[(lS,2R)-l-[[[(lR)- l-[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-
 THIS IS PRECURSOR OF FINAL PDT
THIS IS PRECURSOR OF FINAL PDTmethano-l,3,2-benzodioxaborol-2-yl]-3- methylbutyl]amino]carbonyl]-2-hydroxypropyl]. Analytical Data: Η -NMR (DMSO-d6): 9.20-8.95 (IH, m); 8.76 (IH, d, J=8.55 Hz); 8.26-8.16 (4H, m); 8.12 (IH, t, J= 7.77 Hz); 8.02 (IH, d, J= 7.56 Hz); 7.60-7.47 (4H, m); 5.27 (IH, d, J= 4.97 Hz); 4.50 (IH, dd, J= 4.22 Hz, J= 8.50 Hz); 4.16-4.07 (2H, m); 2.65-2.56 (IH, m); 2.25-2.15 (IH, m); 2.09-1.98 (IH, m); 1.84 (IH, t, J= 5.62 Hz); 1.79- 1.73 (IH, m); 1.73-1.66 (IH, m); 1.66-1.59 (IH, m); 1.40-1.26 (4H, m); 1.23 (7H, d, J= 10.89 Hz); 1.15-1.10 (4H, m); 0.85 (7H, d, J= 6.56 Hz); 0.79 (IH, bs).
|
References |
1. Fuchs, Ota. Proteasome inhibition as a therapeutic strategy in patients with multiple myeloma. Multiple Myeloma (2009), 101-125. CODEN: 69MVM2 AN 2010:737549
2. Genin, E.; Reboud-Ravaux, M.; Vidal, J. Proteasome inhibitors: recent advances and new perspectives in medicinal chemistry. Current Topics in Medicinal Chemistry (Sharjah, United Arab Emirates) (2010), 10(3), 232-256. CODEN: CTMCCL ISSN:1568-0266. CAN 152:516315 AN 2010:423458
3. Sanchez, Eric; Li, Mingjie; Steinberg, Jeffrey A.; Wang, Cathy; Shen, Jing; Bonavida, Benjamin; Li, Zhi-Wei; Chen, Haiming; Berenson, James R. The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib and melphalan. British Journal of Haematology (2010), 148(4), 569-581. CODEN: BJHEAL ISSN:0007-1048. AN 2010:353952
4. Dick, Lawrence R.; Fleming, Paul E. \Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discovery Today (2010), 15(5/6), 243-249. CODEN: DDTOFS ISSN:1359-6446. AN 2010:318415
5. Ruggeri, Bruce; Miknyoczki, Sheila; Dorsey, Bruce; Hui, Ai-Min. The development and pharmacology of proteasome inhibitors for the management and treatment of cancer. Advances in Pharmacology (San Diego, CA, United States) (2009), 57(Contemporary Aspects of Biomedical Research: Drug Discovery), 91-135. CODEN: ADPHEL ISSN:1054-3589. AN 2010:62762
6. Chen-Kiang, Selina; Di Liberto, Maurizio; Huang, Xiangao. Targeting CDK4 and CDK6 kinases or genes thereof in cancer therapy for sensitizing drug-resistant tumors. PCT Int. Appl. (2009), 149pp. CODEN: PIXXD2 WO 2009061345 A2 20090514 CAN 150:531264 AN 2009:586623
7. Rickles, Richard; Lee, Margaret S. Use of adenosine A2A receptor agonists and phosphodiesterase (PDE) inhibitors for the treatment of B-cell proliferative disorders, and combinations with other agents. PCT Int. Appl. (2009), 70 pp. CODEN: PIXXD2 WO 2009011893 A2 20090122 CAN 150:160095 AN 2009:86451
8. Rickles, Richard; Pierce, Laura; Lee, Margaret S. Combinations for the treatment of B-cell proliferative disorders. PCT Int. Appl. (2009), 79pp. CODEN: PIXXD2 WO 2009011897 A1 20090122 CAN 150:160094 AN 2009:83374
9. Hoveyda, Hamid; Fraser, Graeme L.; Benakli, Kamel; Beauchemin, Sophie; Brassard, Martin; Drutz, David; Marsault, Eric; Ouellet, Luc; Peterson, Mark L.; Wang, Zhigang. Preparation and methods of using macrocyclic modulators of the ghrelin receptor. U.S. Pat. Appl. Publ. (2008), 178pp. CODEN: USXXCO US 2008194672 A1 20080814 CAN 149:288945 AN 2008:975261
10. Piva, Roberto; Ruggeri, Bruce; Williams, Michael; Costa, Giulia; Tamagno, Ilaria; Ferrero, Dario; Giai, Valentina; Coscia, Marta; Peola, Silvia; Massaia, Massimo; Pezzoni, Gabriella; Allievi, Cecilia; Pescalli, Nicoletta; Cassin, Mara; di Giovine, Stefano; Nicoli, Paola; de Feudis, Paola; Strepponi, Ivan; Roato, Ilaria; Ferracini, Riccardo; Bussolati, Benedetta; Camussi, Giovanni; Jones-Bolin, Susan; Hunter, Kathryn; Zhao, Hugh; Neri, Antonino; Palumbo, Antonio; Berkers, Celia; Ovaa, Huib; Bernareggi, Alberto; Inghirami, Giorgio. CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood (2008), 111(5), 2765-2775. CODEN: BLOOAW ISSN:0006-4971. CAN 149:486154 AN 2008:292777
11. Dorsey, Bruce D.; Iqbal, Mohamed; Chatterjee, Sankar; Menta, Ernesto; Bernardini, Raffaella; Bernareggi, Alberto; Cassara, Paolo G.; D’Arasmo, Germano; Ferretti, Edmondo; De Munari, Sergio; Oliva, Ambrogio; Pezzoni, Gabriella; Allievi, Cecilia; Strepponi, Ivan; Ruggeri, Bruce; Ator, Mark A.; Williams, Michael; Mallamo, John P. Discovery of a Potent, Selective, and Orally Active Proteasome Inhibitor for the Treatment of Cancer. Journal of Medicinal Chemistry (2008), 51(4), 1068-1072. CODEN: JMCMAR ISSN:0022-2623. CAN 148:345774 AN 2008:146611
12. Dorsey, Bruce D.; Menta, Ernesto; Bernardini, Raffaella; Bernareggi, Alberto; Casara, Paolo G.; D’Arasmo, Germano; Ferretti, Edmondo; De Munari, Sergi; Oliva, Ambrogio; Iqbal, Mohamed; Chatterjee, Sankar; Ruggeri, Bruce; Ator, Mark A.; Williams, Michael; Mallamo, John P. CEP-18770: Discovery of a Potent, Selective and Orally Active Proteasome Inhibitor for the Treatment of Cancer. Frontiers in CNS and Oncology Medicinal Chemistry, ACS-EFMC, Siena, Italy, October 7-9 (2007), COMC-027. CODEN: 69KAR2 AN 2007:1171000
13. Marblestone Jeffrey G Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs : the investigational drugs journal (2009), 12(12), 750-3.
| Patent | Submitted | Granted |
|---|---|---|
| Proteasome inhibitors and methods of using the same [US7576206] | 2005-05-19 | 2009-08-18 |
| PROTEASOME INHIBITORS AND METHODS OF USING THE SAME [US7915236] | 2009-11-26 | 2011-03-29 |
| BORONATE ESTER COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF [US2009325903] | 2009-12-31 |
| US7442830 * | 6 Aug 2007 | 28 Oct 2008 | Millenium Pharmaceuticals, Inc. | Proteasome inhibitors |
| US7687662 * | 2 Jul 2008 | 30 Mar 2010 | Millennium Pharmaceuticals, Inc. | Proteasome inhibitors |
| US8003819 * | 12 Feb 2010 | 23 Aug 2011 | Millennium Pharmaceuticals, Inc. | Proteasome inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8962572 | 4 Oct 2011 | 24 Feb 2015 | Fresenius Kabi Usa, Llc | Bortezomib formulations |
| WO2012177835A1 | 21 Jun 2012 | 27 Dec 2012 | Cephalon, Inc. | Proteasome inhibitors and processes for their preparation, purification and use |
/////CEP-18770, delanzomib
B(C(CC(C)C)NC(=O)C(C(C)O)NC(=O)C1=CC=CC(=N1)C2=CC=CC=C2)(O)O
SCYX 7158
SCYX-7158
[4-fluoro-N-(1-hydroxy-3,3-dimethyl-1,3-dihydro-benzo[c]oxaborol-6-yl-2-trifluoromethyl benzamide]
- C17H14BF4NO3
- Average mass 367.103 Da
Human African trypanosomiasis (HAT) is an important public health problem in sub-Saharan Africa, affecting hundreds of thousands of individuals. An urgent need exists for the discovery and development of new, safe, and effective drugs to treat HAT, as existing therapies suffer from poor safety profiles, difficult treatment regimens, limited effectiveness, and a high cost of goods. We have discovered and optimized a novel class of small-molecule boron-containing compounds, benzoxaboroles, to identify SCYX-7158 as an effective, safe and orally active treatment for HAT.
The presence of a boron atom in the heterocyclic core structure has been found essential for trypanocidal activity of orally active series of benzoxaborole-6-carboxamides in murine models of human African trypanosomiasis. SCYX-7158 has been identified as an effective, safe and orally active treatment for human African trypanoso-miasis to enter preclinical studies, with expected progression to phase 1 clinical trials in 2011 ………http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3764666/
A drug discovery project employing integrated biological screening, medicinal chemistry and pharmacokinetic characterization identified SCYX-7158 as an optimized analog, as it is active in vitro against relevant strains of Trypanosoma brucei, including T. b. rhodesiense and T. b. gambiense, is efficacious in both stage 1 and stage 2 murine HAT models and has physicochemical and in vitro absorption, distribution, metabolism, elimination and toxicology (ADMET) properties consistent with the compound being orally available, metabolically stable and CNS permeable.
In a murine stage 2 study,SCYX-7158 is effective orally at doses as low as 12.5 mg/kg (QD×7 days). In vivo pharmacokinetic characterization of SCYX-7158 demonstrates that the compound is highly bioavailable in rodents and non-human primates, has low intravenous plasma clearance and has a 24-h elimination half-life and a volume of distribution that indicate good tissue distribution.
Most importantly, in rodents brain exposure of SCYX-7158 is high, with Cmax >10 µg/mL and AUC0–24 hr >100 µg*h/mL following a 25 mg/kg oral dose. Furthermore, SCYX-7158 readily distributes into cerebrospinal fluid to achieve therapeutically relevant concentrations in this compartment.

Medicinal Chemistry Synthesis of SCYX-7158 SCHEME1
While the original route was eff ective for producing multi-gram quantities of the API, it was not amenable to scale-up. The route started with 2, a relatively expensive aryl boronic acid. This was protected as borocan 3 and halogen-lithium exchange followed by reaction with acetone and subsequent deprotection provided the oxaborole 4. This protection/alkylation/deprotection sequence added two steps to the overall synthesis and the metalation was not reliable. However, the biggest concern in the sequence was nitration of 4 to give 5. This was accomplished by adding a concentrated solution of 4 to cold fuming nitric acid. Besides the signifi cant safety considerations, the reaction did not scale well. Reduction of the nitro group to give aniline 6 was followed by amide formation to provide 1. While this end game was effi cient, the material produced was dark in color. The colored impurities were not removed by crystallization of 1 and furthermore a mixture of two polymorphs was formed under the original conditions.

The process chemistry route to SCYX-7158 is shown in Scheme 2. When considering alternative routes to 1, the readily available and inexpensive methyl 2-bromobenzoate (8) was identifi ed as an attractive starting point. Gratifyingly, treatment of 8 with methylmagnesium bromide aff orded 2-bromocumyl alcohol (9) in high yield using simple operating conditions. Lithiumhalogen exchange followed by reaction with triisopropyl borate and acidic work-up provided benzoxaborole 4, along with cumyl alcohol (10). While this conversion was not completely atom-effi cient, it was easily scalable and several strategies are available to suppress the by-product in the future.
With benzoxaborole 4 in hand, attention turned to the introduction of a nitrogen-linked amide at the C(6) position. This was accomplished using the same nitration/reduction/acylation strategy used in Scheme 1. Yet signifi cant changes to the chemistry were required for safety and reliability reasons. The fi rst task was introduction of the nitrogen. Nitration was demonstrated using acetic anhydride/nitric acid. However, due to slow rates of nitration and potential for accumulation of a reactive intermediate, alternative conditions had to be identifi ed. These limitations were overcome by use of trifl uoroacetic anhydride/nitric acid, which provided a more reactive nitrating intermediate, thus improving the rate of nitration and aff ording a process in which nitric acid was slowly added until 4 was consumed. Full safety assessment of the nitration reaction, including extensive calorimetry studies, demonstrated the safety of this reaction. This process was used to prepare kilogram quantities of 5.
Following reduction of nitrobenzoxaborole 5 to aniline 6 under standard catalytic hydrogenation conditions, acylation with 7 provided the fi nal drug candidate in high chemical yield. Two challenges remained which needed to be addressed through further optimization of the process. The fi rst challenge was color and purity of the API, which derived from a highly colored impurity generated in the nitration reaction which carried through to fi nal product and was not removed by crystallization. The second challenge was to consistently obtain a single polymorph of the API. Both challenges were addressed by isolation of crystalline isopropyl boronate 11 which rejected colored impurities, followed by regeneration of 1 through addition of water and azeotropic removal of isopropanol. This crystallization provided the API as a single polymorph. The API was isolated in good yield, very high purity and was white in color.
PATENT
https://www.google.co.in/patents/WO2011019616A1?cl=en
N-(3,3-Dimethyl-l-phenyl-2,3-dihvdro-lH-benzotblborol-6-yl)-4-fluoro-2- trifluoromethylbenzatnide
HNO3

 This is not the compd, see precursor
This is not the compd, see precursorTo a suspension of 2-bromophenylboronic acid (75.Og, 373.4 mmol) in toluene (525 niL) was added JV-butyldiethanolamine (64.ImL, 392.1 mmol, 1.05 equiv.) via a syringe. The mixture was heated at 50 0C for two hours. After cooling to room temperature, the toluene was evaporated under reduced pressure and the remaining clear colorless oil was treated with heptanes (500 mL). The heptanes mixture was then sonicated for 5 min and the resulting suspension was allowed to stand at room temperature overnight. The solid that precipitated was collected by filtration, washed with heptanes, and dried in a vacuum oven overnight to yield 2-(2′- bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan as a white solid. Data: 1H NMR (400 MHz, CHLOROFORM-^) δ ppm 0.86 (t, J=7.4 Hz, 3 H) 1.14 – 1.25 (m, 2 H) 1.51 – 1.62 (m, 2 H) 2.61 – 2.70 (m, 2 H) 3.01 – 3.11 (m, 2 H) 3.26 – 3.37 (m, 2 H) 4.09 – 4.26 (m, 4 H) 7.10 (td, J=7.6, 2.0 Hz, 1 H) 7.24 (td, J=7.3, 1.1 Hz, 1 H) 7.51 (d, J=7.9 Hz, 1 H) 7.81 (dd, J=IA, 1.9 Hz, 1 H). Amount obtained, 123.7 g (98.6% yield).
To a solution of 2-(2′-bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan (30.0g, 89.2 mmol) in THF (740 mL) at -78 0C was added /?-BuLi (42.8 mL, 2.5M in hexane, 107.0 mmol, 1.2 equiv.) dropwise via a syringe over a period of 10 min while maintaining reaction temperature at -78 0C. After the addition the reaction solution was stirred for 20 min at -78 0C before acetone (7.5 mL, 124.8 mmol, 1.4 equiv.) was added dropwise via a syringe over a period of 10 min while maintaining the reaction temperature at -78 0C. The resulting mixture was allowed to stir for 20 min at -78 0C then warm to room temperature gradually. Once the reaction vessel reached room temperature, 6N HCl solution (150 mL) was added and the mixture was stirred for an additional 30 min. The mixture was extracted with EtOAc (3X). The EtOAc extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The light yellow oil was then subjected to flash chromatography (Isco Companion, 8Og SiO2 cartridge, solid loaded SiO2, neat heptanes to 20:80 EtOAc gradient at 60 ml/min for 90 min). 3,3-Dimethyl-3H-benzo[c][l,2]oxaborol-l-ol was recovered as clear colorless oil. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.44 (s, 6 H) 7.31 (d, J=Ll Hz, 1 H) 7.38 – 7.47 (m, 2 H) 7.66 (d, J=7.2 Hz, 1 H) 8.99 (s, 1 H). Amount obtained: 9.4O g (65.2 % yield).
To 60 mL fuming HNO3 at -45 0C was slowly added a solution of 3,3- dimethyl-3H-benzo[c][l,2]oxaborol-l-ol (9.4 g, 58.0 mmol) in 11.9 mL nitrobenzene via a syringe while maintaining the reaction temperature between -40 to -45 0C. Once the addition was complete the resulting solution was allowed to stir at -45 ° C for an additional 45 min before poured into crushed ice. The ice mixture was allowed to melt and the aqueous solution was extracted with DCM (3X). The combined DCM extracts were dried over Na2SO4 then evaporated. The crude oil remaining was mixed with one liter 1 : 1 DCM/heptanes. The volume of the solution was reduced under reduced pressure by half and the resulting solution was allowed to stand overnight in a -20 0C freezer. The precipitate formed was filtered out, washed with heptanes and vacuum dried to give 3,3-dimethyl-6-nitro-3H-benzo[c][1.2]oxaborol-l-ol as a white solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.46 (s, 6 H) 7.69 (d, J=8.4 Hz, 1 H) 8.28 (dd, J=8.4, 2.3 Hz, 1 H) 8.48 (d, J=2.2 Hz, 1 H) 9.41 (br. s., 1 H). Amount obtained: 7.31 g (60.4 % yield).
To a solution of 3,3-dimethyl-6-nitro-3H-benzo[c][l .2]oxaborol-l-ol (6.98 g, 33.3 mol) in THF ( 277 mL) was added 6N HC1( 16.6 mL, 100.2 mmol, 3.0 equiv.). The vessel was vacuum/N2 purged three times and 5% Pd/C (3.5 g) was added. The mixture was again vacuum/N2 purged three times then vacuum purged again. H2 was then introduced from a balloon and the reaction was allowed to stir at room
temperature over night. The reaction solution was filtered through a short pad of celite and the filtrate was evaporated to yield 6-amino-3, 3 -dimethyl -3H- benzo[c][l,2]oxaborol-l-ol HCl salt as a dark brown foamy solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). Amount obtained: 8.29 g (100% yield).
To a solution of 6-amino-3, 3 -dimethyl -3H-benzo[c][l,2]oxaborol-l-ol HCl salt (8.29 g, 33.3 mmol) in DCM (170 mL) was added Et3N (11.6 mL, 83.2 mmol, 2.5 equiv.). The mixture was cooled to 0 0C and 2-trifluoromethyl-4- fluorobenzoyl chloride (6.1 mL, 39.9 mmol, 1.2 equiv.) was added slowly via a syringe. The resulting solution was allowed to warm to room temperature gradually and stir for 2 hours. The reaction solution was diluted with DCM, washed with IN HCl, H2O, brine and then dried over Na2SO4, filtered and the filtrate was concentrated under reduced pressure to give an off- white solid. The solid was recrystallized from DCM/heptanes to give 4-fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihydro- benzo[c][l,2]oxaborol-6-yl-2-trifluoromethyl benzamide as a white solid. LCMS (M/Z) : 368 (M+H); 1H NMR (DMSO-d6) δ: 10.58 (s, IH), 9.11 (s, IH), 8.02 (d, J = 1.7 Hz, IH), 7.75 – 7.83 (m, 2H), 7.60 – 7.71 (m, 2H), 7.38 (d, J = 8.2 Hz, IH), 1.44 (s, 6H). Amount obtained: 11.7 g (96% yield)………IS SCYX 7158
BELOW NOT SCYX 7158
The title compound was prepared using a similar procedure to that of N-(I- phenyl- 1 ,3 -dihydrobenzo [c] [ 1 ,2]oxaborol-6-yl)-2-trifluoromethylbenzamide with phenyl magnesium bromide replacing p-to IyI magnesium bromide and 4-fluoro-iV-(l- hydroxy-3,3-dimethyl-2,3-dihydro-lH-benzo[b]borol-6-yl)-2-trifluoromethyl benzamide replacing N-(I -hydroxy-1 ,3-dihydrobenzo[c] [ 1 ,2]oxaborol-6-yl)-2- trifiuoromethylbenzamide. Data: LCMS m/e: 428 (M+H); 1H NMR (400 MHz, DMSO-J6) δ ppm 1.59 (s, 6 H) 7.46 – 7.62 (m, 4 H) 7.71 (td, J=8.5, 2.7 Hz,l H) 7.77 – 7.90 (m, 3 H) 8.00 – 8.09 (m, 2 H) 8.39 (d, J=2.0 Hz, 1 H) 10.66 (s, 1 H). 10 N-fl-p-Tolyl-lJ-dihydro-benzofcIflJIoxaborol-ό-vD-benzatnide
PATENT
82 4-Fluow-N-(l-hydwxy-3,3-dimethyl-l,3-dihydw-benzofcIfl,2Ioxabowl-6- yl-2-trifluoromethyl benzamide

To a suspension of 2-bromophenylboronic acid (10. Og, 49.7 mmol) in toluene (70 niL) was added N-butyldiethanolamine (8.5 mL, 52.2 mmol, 1.05 equiv.) via a syringe. The mixture was heated at 50 0C for two hours. After cooling to room temperature, the toluene was evaporated under reduced pressure and the remaining clear colorless crude oil was treated with heptanes (~ 500 mL). The heptanes mixture was then sonicated ~ 5 min and the resulting suspension was allowed to stand at room temperature overnight. The solid that precipitated was collected by filtration, washed with heptanes, and dried in a vacuum oven overnight to yield a white solid as the titled compound. 1U NMR (400 MHz, CHLOROFORM-J) δ ppm 0.86 (t, J=7.4 Hz, 3 H) 1.14 – 1.25 (m, 2 H) 1.51 – 1.62 (m, 2 H) 2.61 – 2.70 (m, 2 H) 3.01 – 3.11 (m, 2 H) 3.26 – 3.37 (m, 2 H) 4.09 – 4.26 (m, 4 H) 7.10 (td, J=7.6, 2.0 Hz, 1 H) 7.24 (td, J=7.3, 1.1 Hz, 1 H) 7.51 (d, J=7.9 Hz, 1 H) 7.81 (dd, J=IA, 1.9 Hz, 1 H). Amount obtained, 16.0 g, (98 % yield).
To a solution of 2-(2′-bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan (3.0g, 9.2 mmol) in THF (76 mL) at -78 0C was added /?-BuLi (4.4 mL, 2.5M in hexane, 11.0 mmol, 1.2 equiv.) dropwise via a syringe over a period of 10 min while maintaining reaction temperature at -78 0C. After the addition the reaction solution was stirred 20 min at -78 0C before acetone (946 μL, 12.8 mmol, 1.4 equiv.) was added dropwise via a syringe over a period of 10 min while maintaining the reaction temperature at -78 0C. The resulting mixture was allowed to stir for 20 min at -78 0C then warm to room temperature gradually. Once the reaction vessel reached room temperature, 6M HCl solution (30 mL) was added and the mixture was stirred for 30 min. The mixture was extracted with EtOAc (3X). The EtOAc extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The crude slightly yellow in color residual oil remaining was then subjected to flash chromatography (Isco Companion, 8Og SiO2 cartridge, solid loaded SiO2, neat heptane to 20:80 EtOAc gradient at 60 ml/min for 90 min). The product was recovered as clear colorless oil. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.44 (s, 6 H) 7.31 (d, J=Ll Hz, 1 H) 7.38 – 7.47 (m, 2 H) 7.66 (d, J=7.2 Hz, 1 H) 8.99 (s, 1 H). Amount obtained: 1.76 g (61%).
To 14.2 ml fuming HNO3 at -45 0C was added a solution of 3,3-dimethyl- 3H-benzo[c][l,2]oxaborol-l-ol (2.28 g, 14.1 mmol) in 3.0 ml nitrobenzene slowly via a syringe while maintaining the reaction temperature between -40 to -45 0C. Once the addition was complete the resulting solution was allowed to stir at -45 ° C for an additional 45 min before poured into crushed ice (600 g). The ice mixture was allowed to melt and the aqueous solution was extracted with dichloromethane. The combined dichloromethane extracts were dried over Na2SO4 then evaporated. The crude oil remaining was mixed with one liter 1 : 1 DCM:heptane. The volume of the solution was reduced on a rotovap by half and the resulting solution was allowed to stand overnight in a -20 0C freezer overnight. The precipitate formed was filtered out, washed with heptanes and vacuum dried to give the titled compound as a white solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.46 (s, 6 H) 7.69 (d, J=8.4 Hz, 1 H) 8.28 (dd, J=8.4, 2.3 Hz, 1 H) 8.48 (d, J=2.2 Hz, 1 H) 9.41 (br. s., 1 H). Amount obtained: 2.01 g (68%).
To a solution of 3,3-dimethyl-6-nitro-3H-benzo[c][1.2]oxaborol-l-ol (790 mg, 3.8 mmol) in THF ( 20 mL) was added HOAc (1.7 mL, 30 mmol). The vessel was vacuum/N2 purged three times and 5% Pd/C (200 mg) was added. The mixture was again vacuum/N2 purged three times then vacuum purged again. H2 was then introduced from a balloon and the reaction was allowed to stir for 2.5 hours. The reaction solution was filtered through a short pad of celite and the filtrate was evaporated to yield the title compound as a dark brown foamy solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). Amount obtained: 670 mg (89%). [0382] To a solution of 6-amino-3, 3 -dimethyl -3H-benzo[c][l,2]oxaborol-l-ol acetate salt (100 mg, 0.42 mmol) in DCM (2 niL) was added Et3N ( 117.3 μL, 0.84 mmol). The mixture was cooled to 0 0C and the 2-trifluoromethyl-4-fluorobenzoyl chloride (70.0 μL, 0.46 mmol) was added slowly via a syringe. The resulting solution was allowed to warm to room temperature gradually and stir for 2 hours. The reaction solution was diluted with DCM, washed with IN HCl, H2O and then dried over Na2SO4, filtered and the filtrate was concentrated under reduced pressure and the crude material was subjected to flash chromatography (Isco Companion, 4 g SiO2 cartridge, SiO2 solid load, neat heptanes to neat EtOAc gradient over 45 min, flow rate = 18 ml/min). The title compound was recovered as a white foam. LCMS (M/Z) : 368 (M+H); 1H NMR (DMSO-d6) δ: 10.58 (s, IH), 9.11 (s, IH), 8.02 (d, J = 1.7 Hz, IH), 7.75 – 7.83 (m, 2H), 7.60 – 7.71 (m, 2H), 7.38 (d, J = 8.2 Hz, IH), 1.44 (s, 6H). Amount obtained: 144.6 mg (93% yield).
Alternate Synthesis

82e
82b
A 500 mL round-bottomed-flask equipped with a magnetic stir bar and ice- H2O bath was charged with 82a (18.4g, 85.5 mmol) and anhydrous THF (200 mL). MeMgCl (68 mL, 3.0M in 2-methylTHF) was added dropwise through an additional funnel. The mixture was allowed to warm to rt. gradually and stirred overnight. After cooling back to 0 0C, the white milky suspension was carefully treated with HCl (3M) until the upper layer turned clear with white precipitate at the bottom of the flask (pH = 6). The upper clear solution was decanted into a separatory funnel. The precipitate was rinsed with methyl tert-butyl ether (MTBE) (100 mL) 3 times. Combined MTBE with the clear solution and the mixture was washed with H2O (100 mL) 3 times, brine (100 niL), dried over MgSO4, filtered and concentrated under reduced pressure to give 82b as a light yellow oil (20.2g, 100%).
82c
A 50 mL round-bottomed-flask equipped with a magnetic stir bar and ice- H2O bath was charged with 82b (860 mg, 4.0 mmol) and anhydrous THF (20 mL). MeMgBr (1.3 mL, 2.0 M in THF) was slowly added via a syringe. The mixture was stirred at 0 0C for 10 minute and the ice bath was replaced with a dry ice-acetone bath at -40 0C. BuLi (1.9 mL, 2.5 M in hexanes) was added dropwise via a syringe. The resulting mixture was stirred at -40 0C for another 2h before B(O-ipr)3 (1.4 mL, 4.8 mmol) was added dropwise. The mixture was allowed to warm up to rt gradually and stirred overnight. After carefully quenched the reaction with H2O (1 mL), HCl (3M, 10 mL) was added and the mixture was stirred at rt for Ih. The mixture was extracted with EtOAc (20 mL) 3 times. Combined extracts was washed with H2O (20 mL), brine (20 mL), dried over MgSO4, filtered and concentrated under reduced pressure to give a clear oil. The oil solidified overnight to give 82c as a pale yellow waxy solid (544mg, 82.4%).
82d
A 3 L round-bottomed-flask equipped with a mechanical stirrer, thermocouple and ice bath was charged with 82c (86.2 g of 58 wt%, 309 mmol) and trifluoroacetic acid (259 mL). Trifluoroacetic anhydride (129 mL, 926 mmol) was added in one portion. An exotherm of 18 0C was observed. The solution was again cooled to 0 0C and 90% nitric acid (18.0 mL, 386 mmol) was added via syringe pump over 2 h. After the addition was complete, the solution was aged for 1 h. Water (1.75 L) was added. Note: Initially the quench is quite exothermic. Add the water in 5 mL aliquots until the exotherm subsides. The resulting suspension was stirred for 16 h while warming to rt. The solids were collected on a frit, rinsed with water (2 x 500 mL), and air dried to constant weight to provide 50.3 g of crude 82d as a free-flowing orange solid. Note: the crude 82d can be carried forward without recrystallization. The solid was charged to a IL three-necked round-bottomed-flask equipped with a nitrogen inlet adapter, thermocouple, heating mantle and mechanical stirrer.
Isopropylacetate (IPAc, 75 mL) was added and the resulting slurry was warmed to 75 0C and heptanes (250 mL) was added over 15 min while maintaining an internal temp of > 65 0C. The slurry was allowed to cool to rt over night. The solids were collected on a frit and rinsed with 10% IP Ac/heptanes (100 mL) and then heptanes (100 rnL). The product was air dried to constant weight to provide a tan solid (31.7 g, 58%).
82e
A 500 mL round-bottomed-flask equipped with a magnetic stir bar, thermocouple and septum was charged with 82d (29.7 g, 192 mmol) and THF (150 mL, anhydrous stabilizer free). The vessel was inerted by cycling vacuum the nitrogen three times and 5% Pd/C (6.0 g, 50% wet, Degussa type NO/W) was added. The vessel was again inerted by cycling vacuum then nitrogen three times. A hydrogen filled balloon was attached via needle and the atmosphere was changed by cycling vacuum the hydrogen three times. The slurry was stirred vigorously for 16 h. The atmosphere was changed again to nitrogen by cycling vacuum then nitrogen three times. The mixture was filtered through a 1″ pad of celite and the cake was rinsed with THF (50 mL). Concentration in vacuo provided a light tan powder (26.82 g). In a 500 mL round bottomed-flask, the solids were slurried in IPAc (50 mL) and warmed in an 80 0C water bath. Heptanes (150 mL) were added over 10 min. The resulting slurry was allowed to cool to rt and stir for 16 h. The solids were collected on a frit, rinsed with heptanes (50 mL) and air dried to provide an off- white solid (24.39 g, 96%).
4-Fluoro-N-(l-hvdroxy-3,3-dimethyl-l,3-dihvdro-benzofcJfl,2Joxaborol-6-yl-2- triβuoromethyl benzatnide
A lL three-necked round-bottomed-flask equipped with a nitrogen inlet adapter, mechanical stirrer and thermocouple was charged with 82e (15.7g, 88.4 mmol), THF (160 mL, anhydrous, stabilizer free) and K2CO3 (14.7g, 106 mmol). The suspension was stirred at rt and 4-fluoro-2-(trifluoromethyl)benzoyl chloride (22.Og, 97.3 mmol) was added over 10 min. The resulting suspension was aged for 24 h at rt. Water (80 mL) and isopropyl acetate (160 mL) were added and the phases were partitioned. The organic phase was further extracted with water (80 mL) and then brine (50 mL). The organic phase was dried over MgSO4 (20 g) and concentrated in vacuo to provide a tan solid (34.26 g). The solid was dissolved with acetone (195 mL) and transferred to a mechanically stirred IL round-bottomed-flask. Distilled water (113 mL) was added in one portion and the mixture was stirred for 30 min to produce a seed bed and then additional distilled water (60 mL) was added over 30 min. The suspension was stirred at rt overnight and the solids were collected on a frit. The cake was rinsed with 1 : 1 acetone/water (100 rnL) and air dried to constant weight to provide an off-white solid (30.5 g, 94%).
Alternate Synthesis
HNO3 CF3CO2H (CF3CO)2O

to RT


1-1
A 72 L round-bottomed-flask was equipped with a cold bath, mechanical stirrer, nitrogen inlet adaptor, oxygen sensor, thermowell and 2 L dropping funnel. The flask was charged with methyl 2-bromobenzoate (2513 g, 11.7 mol) and the system was flushed with nitrogen to <0.1% O2. THF (18L, anhydrous, inhibitor free) was added and the cold bath was charged with ice and acetone. When the internal temp reached -4 0C, MeMgBr (11.6 L of a 3M solution in ether, 34.8 mol) was added via dropping funnel over 3 h. The internal temp was maintained below 15 0C throughout. At the end of addition, the cold bath was drained and the reaction was aged overnight at ambient temperature. The bath was again charged with ice and acetone and the suspension cooled to below 15 0C. HPLC indicated incomplete conversion (92:8 product, starting ester), so additional MeMgBr (2.3L of a 3M solution in ether) was added. After Ih, HPLC showed the conversion to be >99: 1. The reaction was quenched by slow addition of IN HCl (42 L) keeping the internal temp below 15 0C throughout. At the end of the quench, the pH was adjusted to 6 with IN HCl. The mixture was extracted with MTBE (10 L then 2x5L). The combined organic phases were dried over MgSO4, filtered and concentrated via rotary evaporation to provide 2482 g of 2-(2-bromophenyl)-propan-2-ol as a pale yellow oil. 1H NMR (CHLOPvOFORM-d) δ: 7.62 – 7.67 (m, IH), 7.53 – 7.58 (m, IH), 7.24 – 7.30 (m, IH), 7.03 – 7.10 (m, IH), 1.70 – 1.75 (m, 6H). 1-2
A 72L round-bottomed-flask was equipped with a mechanical stirrer, O2 sensor, thermowell, 2L dropping funnel, N2inlet adaptor, and cold bath. The vessel was inerted to 0.01% O2 and charged with THF (27L, anhydrous, inhibitor free). The resulting solution was cooled to -70 0C using dry ice and acetone and n-BuLi (8.2 L of a 2.5M solution in heptane, 20.5 mol) was added over Ih. 2-(2-Bromophenyl)- propan-2-ol (1994 g, 9.27 mol) was dissolved in THF (9L) and the solution was added to the BuLi via dropping funnel over 2h, keeping the internal temp below -70 0C. The resulting thin yellow suspension was aged for 30 min then B(OiPr)3 (244 Ig, 13.0 mol) was added rapidly via addition funnel. The cold bath was drained and the misture was allowed to warm to room temperature while aging over night. HPLC analysis shows an 81 :19 ratio of desired product: 2-phenyl-2-propanol. The mixture was cooled to -10 0C and 2N HCl (9.3 L) was added via dropping funnel over 30 min, keeping the reaction mixture below 10 0C. After 3 h, the pH was adjusted to 4 with additional HCl. The reaction mixture was extracted with MTBE (2 x 4L). The combined organic phases were concentrated to provide 2028 g of a heavy oil. The oil was dissolved in MTBE (14L) and extracted with IN NaOH (4.6, then 5, then 4L). The aqueous phases were combined and acidified with 2N HCl (6.8 L) to a pH of 4-5. The mixture was extracted with MTBE (5L). The organic phase was dried over MgSO4 (282 g) and concentrated to provide 1450 g (ca 60 wt%) of 3,3-dimethyl-3H- benzo[c][l,2]oxaborol-l-ol as a waxy white solid. LC/MS: m/z 163 (M+H)+; 1H NMR (DMSO-de) δ: 8.96 (br. s., IH), 7.62 (d, J = 7.2 Hz, IH), 7.33 – 7.45 (m, 2H), 7.25 – 7.30 (m, IH), 1.40 (s, 6H).
1-3
A 22 L round-bottomed-flask equipped with a mechanical stirrer, thermocouple, 2 L dropping funnel and cold bath was charged with 3,3-dimethyl-3H- benzo[c][l,2]oxaborol-l-ol (508 g, 300 g contained, 1.85 mol) and trifluoroacetic acid (1.54 L). The solution was cooled to 5 0C. Trifluoroacetic anhydride (722 mL, 5.56 mol, 3.00 eq) was added via dropping funnel over 15 min. After aging at 0 – 3 0C for 30 min, nitric acid (90% fuming, 108 mL, 2.31 mol, 1.5 eq) was added dropwise over 2h 50 min keeping the internal temp below 5 0C. After aging for 1 h, icewater (10.4L) was added over 50 min maintaining the reaction temp below 15 0C to provide a slurry. The slurry was aged at 0 0C overnight to provide an orange suspension. The solids were collected on a frit, rinsed with cold water (5L) and air dried under a stream of air to constant weight (ca 24h) to provide 364 g of 3,3- dimethyl-6-nitro-3H-benzo[c][l,2]oxaborol-l-ol as a 92.4 wt% pure solid (88%). LC/MS : m/z 208 (M+H)+; 1H NMR (DMSO-d6) δ: 8.52 (d, J = 2.2 Hz, IH), 8.32 (dd, J = 8.4, 2.2 Hz, IH), 7.74 (d, J = 8.4 Hz, IH), 1.50 (s, 6H)
1-4
A 2 gallon stirred pressure vessel was charged with 3,3-dimethyl-6-nitro- 3H-benzo[c][l,2]oxaborol-l-ol (966 g, 812 g corrected, 3.92 mol), 5% Pd/C (193 g, 50% wet, Degussa type 101 NO/W) and THF (4.83 L, inhibitor free). The vessel was sealed, the atmosphere was changed to H2 (5 psi) and the reaction was fun for 16 h. An exotherm to 30 0C was observed over about 30 min. The vessel was purged with N2, and completion of reaction was determined by HPLC. The reaction was vacuum filtered through a pad of celite (very slow filtration) and the filter cake was rinsed with THF (2L). The filtrate was concentrated via rotary evaporation to provide 982 g of a dark brown solid. This was transferred to a 22L round-bottomed-flask and warmed to 80 0C in iPAc (1.83 L) to provide a dark brown slurry. The slurry was cooled to 60 0C and heptanes (5.49L) were added over 2 h. The slurry was allowed to age with stirring over night while cooling to room temperature. The solids were collected on a frit, rinsed with heptanes (4L) and air dried to provide a dark brown solid (747 g).
The solids (747 g) were transferred to a 22L rbf and slurried in iPAc (3 L) at 70 0C. The batch was allowed to cool to 40 0C and heptanes (3L) were added over 5 h. The slurry was aged at room temperature over night and the solids were collected on a frit, rinsed with 1 : 1 iP Ac/heptanes (2L) then heptanes (IL) and air dried to provide 554 g of 6-amino-3,3-dimethyl-3H-benzo[c][l,2]oxaborol-l-ol as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). 4-Fluow-N-(l-hvdwxy-3,3-dimethyl-l,3-dihvdw-benzofcJfl,2Joxabowl-6-yl)-2- triβuoromethyl benzatnide
A 22L four-necked round-bottomed-flask equipped with a nitrogen inlet adapter, mechanical stirrer and thermocouple was charged with 6-amino-3,3- dimethyl-3H-benzo[c][l,2]oxaborol-l-ol (554g, 3.13 mol), THF (5.5 L, anhydrous, stabilizer free) and K2CO3 (865 g, 6.26 mol). The suspension was stirred at room temperature for 30 min and 4-fluoro-2-(trifluoromethyl)benzoyl chloride (780 g, 3.44 mol) was added over 30 min. The resulting suspension was aged for 24 h at room temperature. HPLC showed unreacted 6-amino-3,3-dimethyl-3H-benzo[c][l,2] oxaborol-1-ol so an additional 42 niL of the acid chloride was added. After 30 min, water (2.8 L) and isopropyl acetate (5.5 L) were added and the phases were partitioned. The organic phase was further extracted with water (2.8 L) and then brine (2.8 L). The organic phase was dried over MgSO4 and concentrated in vacuo to provide a tan solid. The solid was dissolved with acetone (3.0 L) and transferred to a mechanically stirred 5OL round-bottomed-flask. Distilled water (2.0 L) was added in one portion and the mixture was stirred for 30 min to produce a seed bed and then additional water (1.0 L) was added over 30 min. The suspension was stirred at room temperature overnight and the solids were collected on a frit. The cake was rinsed with 1 : 1 acetone/water (1.0 L) and air dried to constant weight to provide 4-fluoro-N- ( 1 -hydroxy-3 ,3 -dimethyl- 1 ,3 -dihydro-benzo [c] [ 1 ,2]oxaborol-6-yl)-2-trifluoromethyl benzamide as a dark tan solid (1.3 kg).
Recrystallization of4-Fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihvdro- benzotcl t 1,21 oxaborol-6-yl)-2-trifluoromethyl benzamide
A 22 L round-bottomed-flask was charged with the dark tan crude 4- fluoro-N-(l -hydroxy-3, 3 -dimethyl- 1 ,3-dihydro-benzo[c] [ 1 ,2]oxaborol-6-yl)-2- trifluoromethyl benzamide (1.3 kg), acetone (8L) and Darco G-60 (55 g, 400 mesh) and water (5.3L). The resulting suspension was stirred for 15 min, filtered through a pad of celite (ca 500 g) to provide a brown solution. The celite pad was washed with 60% acetone/water (8L). The combined filtrate and rinse were transferred to a 50 L round-bottomed-flask and water (2L) was added. The solution was seeded (5 g) to initiate crystallization and additional water (2.2 L) was added slowly via addition funnel. After aging at room temperature overnight, the solids were collected and the filter cake was rinsed with 30% acetone/water (4L). The solids were air dried for 24 h then dried in a room temperature vacuum oven for 5 days to constant weight to provide 969 g (72% recovery) of 4-fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihydro- benzo[c][l,2]oxaborol-6-yl)-2-trifluoromethyl benzamide as a light tan solid.
LC/MS: m/z 368 (M+H)+;
1H NMR (400 MHz, DMSO-d6) δ ppm 1.44 (s, 5 H) 1.49 (s, 2 H) 7.39 (d, J=8.2 Hz, 1 H) 7.61 – 7.76 (m, 2 H) 7.77 – 7.84 (m, 2 H) 7.86 – 7.90 (m, 0 H) 8.03 (d, J=I.7 Hz, 1 H) 9.09 (s, 1 H) 10.58 (s, 1 H).
POTASSIUM SALT
Formation of potassium salt

To a 50OmL three-neck flask fitted with a mechanical stirrer was charged KOH (1.51 g, 26.9 mmol, 1.0 eq.). Under a nitrogen atmosphere, anhydrous acetone (140 mL) and H2O (2.5 mL, 5 eq.) were added via syringe. A solution of 4-fluoro-N- (l-hydroxy-3,3-dimethyl-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl-2-trifluoromethyl benzamide (10.0 g, 27.2 mmol, 1.0 eq.) in anhydrous acetone (60 mL) was added to the flask with vigorous stirring. The resulting clear solution was stirred at room temperature. The potassium salt precipitated from the solution over ca. 4 hours to afford a thick suspension. The precipitate was collected by filtration, washed with acetone (200 mL) and dried in a vacuum oven overnight to afford a white solid (10.6g, 91.9% yield). 1H NMR (methanol-d4) δ: 7.70 – 7.76 (m, IH), 7.53 – 7.60 (m, 2H), 7.47 – 7.53 (m, IH), 7.33 – 7.36 (m, IH), 7.01 – 7.06 (m, IH), 1.46 (s, 6H); M.P. (range) 197 – 200 0C; Elemental analysis: Theory: C 48.25%, H 3.57%, N 3.31%, K 9.24%; Found: C 48.70%, H 3.41%, N 3.25%, K 9.19%.
REFERENCES
http://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0001151
- touguia J, Costa J (1999) Therapy of human African trypanosomiasis: current situation. Mem Inst Oswaldo Cruz 94: 221–224
- Barrett MP, Boykin DW, Brun R, Tidwell RR (2007) Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br J Pharmacol 152: 1155–1171.
- 1986. Epidemiology and control of African trypanosomiasis. Report of a WHO expert committee. World Health Organization. Geneva, Switzerland. Technical Report Series, No. 739. 126 pp.
- Benzoxaboroles: a new class of potential drugs for human African trypanosomiasis. Robert T Jacobs, Jacob J Plattner, Bakela Nare, Stephen A Wring, Daitao Chen, Yvonne Freund, Eric G Gaukel, Matthew D Orr, Joe B Perales, Matthew Jenks, Robert A Noe, Jessica M Sligar, Yong-Kang Zhang, Cyrus J Bacchi, Nigel Yarlett, and Robert Don. Future Medicinal Chemistry. August 2011. Vol. 3, No. 10. Pages 1259-1278.
http://www.swisstph.ch/fileadmin/user_upload/Pdfs/Events/2010_09_Jacobs.pdf ……….POWERPOINT
Lead optimization investigation of oxaboroles for the treatment of human African trypanosomiasis
238th Am Chem Soc (ACS) Natl Meet (August 16-20, Washington) 2009, Abst MEDI 345
LINK
https://www.acsmedchem.org/ama/orig/abstracts/mediabstractf2009.pdf
Robert Jacobs, bob.jacobs@scynexis.com
Daitao Chen1 , Matt Orr1 , Jessica Sligar1 , Matt. Jenks1 , Andy Noe1 , Bakela Nare2 , Luke T. Mercer2 , Tana S. Bowling2 , Cindy Rewerts1 , Stephen Wring1 , Cyrus Bacchi3 , Nigel Yarllet3 , Charles Ding4 , Yvonne Freund5 , Kurt Jarnagin5 , Jacobs Plattner5 , and Robert Don6 . (1) Scynexis Inc, Duhram, NC 27713, (2) SCYNEXIS, Inc, Research Triangle Park, NC 27709-2878, (3) Pace University, New York, NY, (4) Anacor Pharmaceuticals, Inc, Palo Alto, CA, (5) Anacor Pharmaceuticals, Inc, (6) Drugs for Neglected Diseases initiative, Geneva, Switzerland

///////////SCYX-7158
