
Home » Uncategorized (Page 44)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
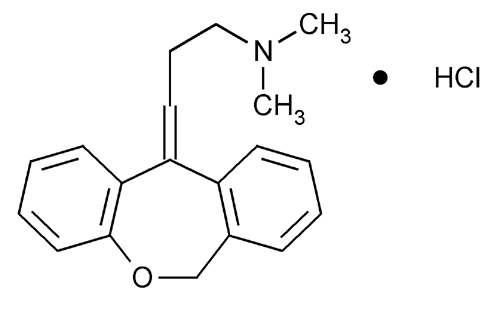
Doxepin, ドキセピン

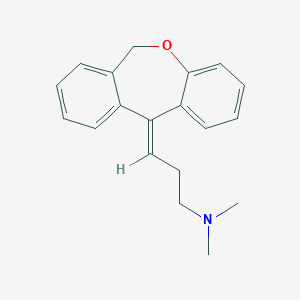
Doxepin
1668-19-5
1229-29-4 (hydrochloride), 4698-39-9 ((E)-isomer); 25127-31-5 ((Z)-isomer)
Launched – 1964
| Doxepin Hydrochloride | 3U9A0FE9N5 | 1229-29-4 |
NSC-108160
P-3693A
SO-101
Aponal
Quitaxon
Silenor
Sinequan
Sinquan
Xepin
Zonalon
USP
USP32/pub/data/v32270/usp32nf27s0_m28110
N,N-Dimethyldibenz[b,e]oxepin-D11(6H),![]() -propylamine hydrochloride
-propylamine hydrochloride ![]()
![]()
![]() [1229-29-4; 4698-39-9 ((E)-isomer); 25127-31-5 ((Z)-isomer)].
[1229-29-4; 4698-39-9 ((E)-isomer); 25127-31-5 ((Z)-isomer)].
DESCRIPTION
SINEQUAN® (doxepin hydrochloride) is one of a class of psychotherapeutic agents known as dibenzoxepin tricyclic compounds. The molecular formula of the compound is C19H21NO•HCl having a molecular weight of 316. It is a white crystalline solid readily soluble in water, lower alcohols and chloroform.
Inert ingredients for the capsule formulations are: hard gelatin capsules (which may contain Blue 1, Red 3, Red 40, Yellow 10, and other inert ingredients); magnesium stearate; sodium lauryl sulfate; starch.
Inert ingredients for the oral concentrate formulation are: glycerin; methylparaben; peppermint oil; propylparaben; water.
Chemistry
SINEQUAN (doxepin HCl) is a dibenzoxepin derivative and is the first of a family of tricyclic psychotherapeutic agents. Specifically, it is an isomeric mixture of: 1-Propanamine, 3-dibenz[b,e]oxepin-11(6H)ylidene-N,N-dimethyl-, hydrochloride.
 |
For Consumers
WHAT ARE THE POSSIBLE SIDE EFFECTS OF DOXEPIN (SINEQUAN) (SINEQUAN)?
Get emergency medical help if you have any of these signs of an allergic reaction: hives; difficulty breathing; swelling of your face, lips, tongue, or throat.
Report any new or worsening symptoms to your doctor, such as: mood or behavior changes, anxiety, panic attacks, trouble sleeping, or if you feel impulsive, irritable, agitated, hostile, aggressive, restless, hyperactive (mentally or physically), more depressed, or have thoughts about suicide or hurting yourself.
Synthesis Reference
Luigi Schioppi, Brian Talmadge Dorsey, Michael Skinner, John Carter, Robert Mansbach, Philip Jochelson, Roberta L. Rogowski, Cara Casseday, Meredith Perry, Bryan Knox, “LOW-DOSE DOXEPIN FORMULATIONS AND METHODS OF MAKING AND USING THE SAME.” U.S. Patent US20090074862, issued March 19, 2009.

DOI: 10.1007/BF00904459
DOI: 10.1007/BF00901313 US 3420851
DE 1232161
SYN 2
Synth Commun 1989, 19(19): 3349, US 3438981

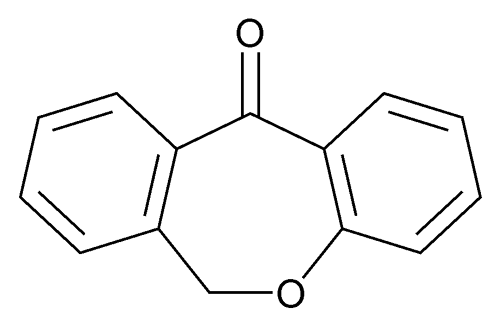
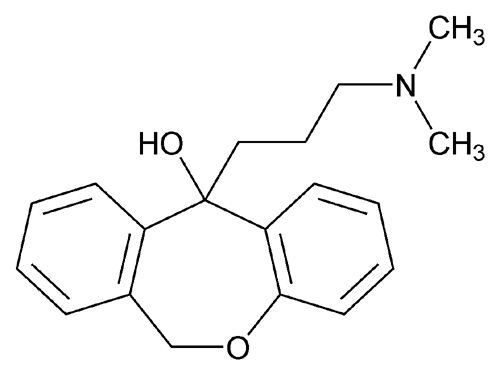
Condensation of dibenzo-oxepinone (I) with 3-(dimethylamino)propylmagnesium chloride (II), followed by a dehydration of the resultant tertiary alcohol with hot HCl gives the target 3-(dimethylamino)propylidene derivative.
SYN 3

Chlorination of 2-(phenoxymethyl)benzoic acid (I) with SOCl2 at 50 °C gives 2-(phenoxymethyl)benzoyl chloride (II), which undergoes cyclization in the presence of FeCl3 in toluene to furnish dibenzo[b,e]oxepin-11-one (III)
Grignard reaction of intermediate (III) with tert-butyl 3-chloropropyl ether (IV) using Mg in refluxing THF or Et2O provides 11-(3-tert-butoxypropyl)-6,11-dihydrodibenzo[b,e]oxepin-11-ol (V), which upon elimination by means of HCl in refluxing EtOH affords alkene (VI).
Treatment of tert-butyl ether (VI) with SOCl2 in refluxing toluene gives 11-(3-chloropropylidene)-6,11-dihydrodibenzo[b,e]oxepine (VII), which is then coupled with dimethylamine (VIII) in the presence of Ni(OAc)2, PPh3 and K2CO3 in DMF or in EtOH at 100 °C to furnish doxepin (VII) .
Finally, treatment of tertiary amine (VII) with HCl at 140 °C yields the target doxepin hydrochloride .
US 2014309437, CN 102924424
Doxepin is a tricyclic antidepressant (TCA) used as a pill to treat major depressive disorder, anxiety disorders, chronic hives, and for short-term help with trouble remaining asleep after going to bed (a form of insomnia).[8][7][9] As a cream it is used for short term treatment of itchiness due to atopic dermatitis or lichen simplex chronicus.[10]
At doses used to treat depression, doxepin appears to inhibit the reuptake of serotonin and norepinephrine and to have antihistamine, adrenergic and serotonin receptor antagonistic, and anticholinergic activities; at low doses used to treat insomnia it appears to be selective for the histamine H1 receptor.[11]
It was introduced under the brand names Quitaxon and Aponal by Boehringer, which discovered it, and as Sinequan by Pfizer,[12] and has subsequently been marketed under many other names worldwide.[2]
Medical uses
Doxepin is used as a pill to treat major depressive disorder, anxiety disorders, chronic hives, and for short-term help with trouble remaining asleep after going to bed (a form of insomnia).[8][7][9] As a cream it is used for short term treatment of itchiness to due atopic dermatitis or lichen simplex chronicus.[10]
In 2016 the American College of Physicians advised that insomnia be treated first by treating comorbid conditions, then with cognitive behavioral therapy and behavioral changes, and then with drugs; doxepin was among those recommended for short term help maintaining sleep, on the basis of weak evidence.[13][14] The 2017 American Academy of Sleep Medicine recommendations focused on treatment with drugs were similar.[13] A 2015 AHRQ review of treatments for insomnia had similar findings.[15]
A 2010 review found that topical doxepin is useful to treat itchiness.[16]
A 2010 review of treatments for chronic hives found that doxepin had been superseded by better drugs but was still sometimes useful as a second line treatment.[17]
Chemistry
Doxepin is a tricyclic compound, specifically a dibenzoxepin, and possesses three rings fused together with a side chain attached in its chemical structure.[38] It is the only TCA with a dibenzoxepin ring system to have been marketed.[64] Doxepin is a tertiary amine TCA, with its side chain–demethylated metabolite nordoxepin being a secondary amine.[40][41] Other tertiary amine TCAs include amitriptyline, imipramine, clomipramine, dosulepin (dothiepin), and trimipramine.[65][66] Doxepin is a mixture of (E) and (Z) stereoisomers (the latter being known as cidoxepin or cis-doxepin) and is used commercially in a ratio of approximately 85:15.[3][67] The chemical name of doxepin is (E/Z)-3-(dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine[38][68] and its free base form has a chemical formula of C19H21NO with a molecular weight of 279.376 g/mol.[68] The drug is used commercially almost exclusively as the hydrochloride salt; the free base has been used rarely.[3][69] The CAS Registry Number of the free base is 1668-19-5 and of the hydrochloride is 1229-29-4.[3][69]

clip
https://www.sciencedirect.com/science/article/pii/S0040402007016079

History
Doxepin was discovered in Germany in 1963 and was introduced in the United States as an antidepressant in 1969.[38] It was subsequently approved at very low doses in the United States for the treatment of insomnia in 2010.[44][69]
Society and culture
Generic names
Doxepin is the generic name of the drug in English and German and its INN and BAN, while doxepin hydrochloride is its USAN, USP, BANM, and JAN.[3][69][70][2] Its generic name in Spanish and Italian and its DCIT are doxepina, in French and its DCF are doxépine, and in Latin is doxepinum.[2]
The cis or (Z) stereoisomer of doxepin is known as cidoxepin, and this is its INN while cidoxepin hydrochloride is its USAN.[3]
Brand names
It was introduced under the brand names Quitaxon and Aponal by Boehringer and as Sinequan by Pfizer.[12]
As of October 2017, doxepin is marketed under many brand names worldwide: Adnor, Anten, Antidoxe, Colian, Dofu, Doneurin, Dospin, Doxal, Doxepini, Doxesom, Doxiderm, Flake, Gilex, Ichderm, Li Ke Ning, Mareen, Noctaderm, Oxpin, Patoderm, Prudoxin, Qualiquan, Quitaxon, Sagalon, Silenor, Sinepin, Sinequan, Sinequan, Sinquan, and Zonalon.[2] It is also marketed as a combination drug with levomenthol under the brand name Doxure.[2]
Approvals
The oral formulations of doxepin are FDA-approved for the treatment of depression and sleep-maintenance insomnia and its topical formulations are FDA-approved the short-term management for some itchy skin conditions.[71] Whereas in Australia and the United Kingdom, the only licensed indication(s) is/are in the treatment of major depression and pruritus in eczema, respectively.[20][72]
Research
Antihistamine
As of 2017 there was no good evidence that topical doxepin was useful to treat localized neuropathic pain.[73] Cidoxepin is under development by Elorac, Inc. for the treatment of chronic urticaria (hives).[74] As of 2017, it is in phase II clinical trials for this indication.[74] The drug was also under investigation for the treatment of allergic rhinitis, atopic dermatitis, and contact dermatitis, but development for these indications was discontinued.[74]
Headache
Doxepin was under development by Winston Pharmaceuticals in an intranasal formulation for the treatment of headache.[75] As of August 2015, it was in phase II clinical trials for this indication.[75]
PATENT
https://patents.google.com/patent/US9486437B2/en
Doxepin:
Doxepin HCl is a tricyclic compound currently approved and available for treatment of depression and anxiety. Doxepin has the following structure:

For all compounds disclosed herein, unless otherwise indicated, where a carbon-carbon double bond is depicted, both the cis and trans stereoisomers, as well as mixtures thereof are encompassed.
Doxepin belongs to a class of psychotherapeutic agents known as dibenzoxepin tricyclic compounds, and is currently approved and prescribed for use as an antidepressant to treat depression and anxiety. Doxepin has a well-established safety profile, having been prescribed for over 35 years.
Doxepin, unlike most FDA approved products for the treatment of insomnia, is not a Schedule IV controlled substance. U.S. Pat. Nos. 5,502,047 and 6,211,229, the entire contents of which are incorporated herein by reference, describe the use of doxepin for the treatment chronic and non-chronic (e.g., transient/short term) insomnias at dosages far below those used to treat depression.
It is contemplated that doxepin for use in the methods described herein can be obtained from any suitable source or made by any suitable method. As mentioned, doxepin is approved and available in higher doses (75-300 milligrams) for the treatment of depression and anxiety. Doxepin HCl is available commercially and may be obtained in capsule form from a number of sources. Doxepin is marketed under the commercial name SINEQUAN® and in generic form, and can be obtained in the United States generally from pharmacies in capsule form in amounts of 10, 25, 50, 75, 100 and 150 mg dosage, and in liquid concentrate form at 10 mg/mL. Doxepin HCl can be obtained from Plantex Ltd. Chemical Industries (Hakadar Street, Industrial Zone, P.O. Box 160, Netanya 42101, Israel), Sifavitor S.p.A. (Via Livelli 1—Frazione, Mairano, Italy), or from Dipharma S.p.A. (20021 Baranzate di Bollate, Milano, Italy). Also, doxepin is commercially available from PharmacyRx (NZ) (2820 1st Avenue, Castlegar, B.C., Canada) in capsule form in amounts of 10, 25, 50, 75, 100 and 150 mg. Furthermore, Doxepin HCl is available in capsule form in amounts of 10, 25, 50, 75, 100 and 150 mg and in a 10 mg/ml liquid concentrate from CVS Online Pharmacy Store (CVS.com).
Also, doxepin can be prepared according to the method described in U.S. Pat. No. 3,438,981, which is incorporated herein by reference in its entirety. It should be noted and understood that although many of the embodiments described herein specifically refer to “doxepin,” other doxepin-related compounds can also be used, including, for example, pharmaceutically acceptable salts, prodrugs, metabolites, in-situ salts of doxepin formed after administration, and solid state forms, including polymorphs and hydrates.
Metabolites:
In addition, doxepin metabolites can be prepared and used. By way of illustration, some examples of metabolites of doxepin can include, but are not limited to, desmethyldoxepin, hydroxydoxepin, hydroxyl-N-desmethyldoxepin, doxepin N-oxide, N-acetyl-N-desmethyldoxepin, N-desmethyl-N-formyldoxepin, quaternary ammonium-linked glucuronide, 2-O-glucuronyldoxepin, didesmethyldoxepin, 3-O-glucuronyldoxepin, or N-acetyldidesmethyldoxepin. The metabolites of doxepin can be obtained or made by any suitable method, including the methods described above for doxepin.
Desmethyldoxepin has the following structure:

Desmethyldoxepin is commercially available as a forensic standard. For example, it can be obtained from Cambridge Isotope Laboratories, Inc. (50 Frontage Road, Andover, Mass.). Desmethyldoxepin for use in the methods discussed herein can be prepared by any suitable procedure. For example, desmethyldoxepin can be prepared from 3-methylaminopropyl triphenylphosphonium bromide hydrobromide and 6,11-dihydrodibenz(b,e)oxepin-11-one according to the method taught in U.S. Pat. No. 3,509,175, which is incorporated herein by reference in its entirety.
Hydroxydoxepin has the following structure:
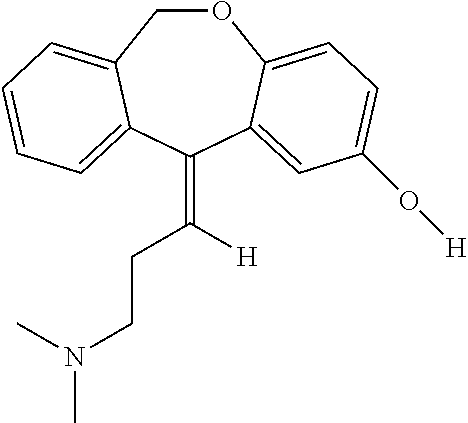
2-Hydroxydoxepin can be prepared by any suitable method, including as taught by Shu et al. (Drug Metabolism and Disposition (1990) 18:735-741), which is incorporated herein by reference in its entirety.
Hydroxyl-N-desmethyldoxepin has the following structure:

2-Hydroxy-N-desmethyldoxepin can be prepared any suitable method.
Doxepin N-oxide has the following structure:

Doxepin N-oxide can be prepared by any suitable method. For example, doxepin N-oxide can be prepared as taught by Hobbs (Biochem Pharmacol (1969) 18:1941-1954), which is hereby incorporated by reference in its entirety.
N-acetyl-N-desmethyldoxepin has the following structure:

N-acetyl-N-desmethyldoxepin can be prepared by any suitable means. For example, (E)-N-acetyl-N-desmethyldoxepin has been produced in filamentous fungus incubated with doxepin as taught by Moody et al. (Drug Metabolism and Disposition (1999) 27:1157-1164), hereby incorporated by reference in its entirety.
N-desmethyl-N-formyldoxepin has the following structure:

N-desmethyl-N-formyldoxepin can be prepared by any suitable means. For example, (E)-N-desmethyl-N-formyldoxepin has been produced in filamentous fungus incubated with doxepin as taught by Moody et al. (Drug Metabolism and Disposition (1999) 27:1157-1164), hereby incorporated by reference in its entirety.
N-acetyldidesmethyldoxepin has the following structure:

N-acetyldidesmethyldoxepin can be prepared by any suitable means. For example, (E)-N-acetyldidesmethyldoxepin has been produced in filamentous fungus incubated with doxepin as taught by Moody et al. (Drug Metabolism and Disposition (1999) 27:1157-1164), hereby incorporated by reference in its entirety.
Didesmethyldoxepin has the following structure:

Didesmethyldoxepin can be prepared by any suitable means. For example, (Z)- and (E)-didesmethyldoxepin have been isolated from plasma and cerebrospinal fluid of depressed patients taking doxepin, as taught by Deuschle et al. (Psychopharmacology (1997) 131:19-22), hereby incorporated by reference in its entirety.
3-O-glucuronyldoxepin has the following structure:

3-O-glucuronyldoxepin can be prepared by any suitable means. For example, (E)-3-O-glucuronyldoxepin has been isolated from the bile of rats given doxepin, as described by Shu et al. (Drug Metabolism and Disposition (1990) 18:1096-1099), hereby incorporated by reference in its entirety.
2-O-glucuronyldoxepin has the following structure:

2-O-glucuronyldoxepin can be prepared by any suitable means. For example, (E)-2-O-glucuronyldoxepin has been isolated from the bile of rats given doxepin, and also in the urine of humans given doxepin, as described by Shu et al. (Drug Metabolism and Disposition (1990) 18:1096-1099), hereby incorporated by reference in its entirety.
Quaternary ammonium-linked glucuronide of doxepin (doxepin N+-glucuronide) has the following structure:

N+-glucuronide can be obtained by any suitable means. For example, doxepin N+-glucuronide can be prepared as taught by Luo et al. (Drug Metabolism and Disposition, (1991) 19:722-724), hereby incorporated by reference in its entirety.
PATENT
https://patents.google.com/patent/CN105330638A/en
doxepin hydrochloride, the chemical name is N, N- dimethyl-3-dibenzo (b, e) _ oxepin -11 (6H) -1-propanamine salt subunit cistron iso the mixture body configuration. CAS Number 1229-29-4 thereof, of the formula
[0003]

[0004] Doxepin hydrochloride is a drug for the treatment of depression and anxiety neurosis that act to inhibit the central nervous system serotonin and norepinephrine reuptake, such that these two synaptic cleft neurotransmitter concentration increased and antidepressant effect, but also has anti-anxiety and sedative effects. Doxepin hydrochloride oral absorption, bioavailability of 13-45%, half-life (Shu 1/2) is 8-12 hours, to apparent volume of distribution (1) ^ 9-33171.Primarily metabolized in the liver to active metabolites thereof demethylation.Metabolite excretion from the kidney, elderly patients decline of metabolism and excretion ability of this product
[0005] Chinese Patent CN102924486A discloses a method for preparing a hydrochloride of doxepin. The method comprises the coupling reaction CN, i.e., the use of Ni (0Α〇) 2 / ΡΡ1 ^ φ to the amine-based compound. Although Ni catalyst the reaction step (OAc) 2 is more readily available and inexpensive, but the low yield of this step, and low product purity.
SUMMARY
[0006] Accordingly, the present invention provides a method of o-toluic acid synthesized multi doxepin hydrochloride, the higher the yield and purity of the obtained product was purified by this method.
[0007] – o-methylbenzoate method for the synthesis of doxepin hydrochloride, comprising the steps of:
[0008] (1) o-methylbenzoic acid with N- halosuccinimide benzylation halogenation reaction occurs in an acetonitrile solvent in the light conditions, to give o-halo-methylbenzoic acid (Compound J), the following reaction formula,
[0009]

[0010] (2) Compound J celite load cesium fluoride intramolecular substitution reaction, to give phthalide (Compound H) in an acetonitrile solvent and as a catalyst, the following reaction formula,
[0011]

[0012] (3) The phenol compound J with sodium methoxide in an alcohol solvent substitution reaction, to give a compound I, the following reaction formula,
[0013]

[0014] (4) The cyclization reaction of Compound I in a solvent in the catalytic DMS0 anhydrous aluminum chloride to give 6, 11-dihydro-dibenzo [b, e] oxepin -11- one (compound A), the following reaction formula,
[0015]

[0016] (5) 6, 11-dihydro-dibenzo [b, e] oxepin-11-one (Compound A) and 3-chloropropyl alkyl tert-butyl ether (compound B) is added magnesium powder and with THF and / or a nucleophilic addition of anhydrous diethyl ether under the conditions of the reaction solvent to give the hydroxy compound (compound C), the following reaction formula,
[0017]
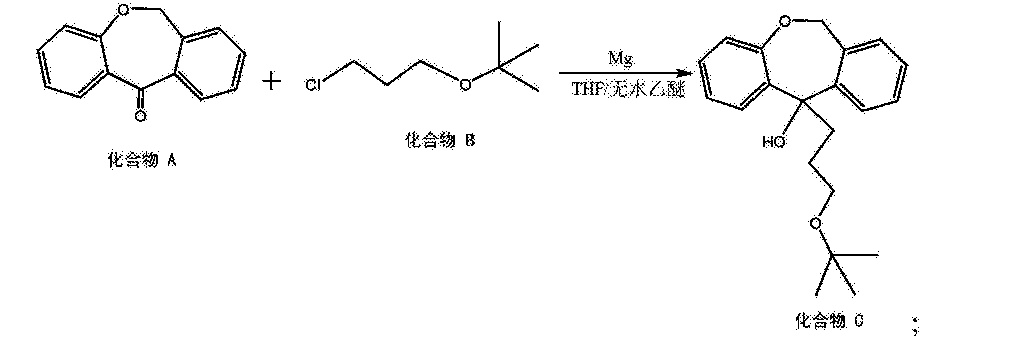
[0018] (6) heating elimination reaction to give an olefin compound (Compound D) in a strong base in an alcoholic solvent to the hydroxy compound, the following reaction formula,
[0019]

[0020] (7) to the olefinic compound in the nucleophilic substitution reaction of a hydrogen halide acid, to give halide (Compound E), the following reaction formula,

[0022] wherein the compound E X is a C1, Br, or a a I;
[0023] (8) the halide with dimethylamine in a solvent under an organic lithium compound is added in ether to nucleophilic substitution reaction to yield doxepin (Compound F.), The following reaction formula,
[0024]

[0025] (9) the doxepin neutralization reaction with hydrochloric acid to give sulfasalazine (Compound G), the following reaction formula,

Example 1
[0043] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 10 ° C, under stirring for 4h. A known separation method, separation of o-bromomethyl-benzoic acid. This compound is named J.
[0044] placed in a 20L reaction container, Compound J, diatomaceous earth in an amount of 0.05 to load cesium fluoride (compound J as a mass basis), acetonitrile in an amount of 2.5 (in Compound J 1 is a mass basis), and the temperature was adjusted to 30 ° C, with stirring under reflux for 20h adjustment. Then, a known means for separating the reaction phthalide.
After [0045] placed in a 20L reaction vessel phthalide, 3 an amount of sodium methoxide in ethanol solvent (total mass of phenol phthalide and 1 meter), the reaction solution temperature adjusted to 50 ° C, was added dropwise start phenol was 1.05 mass (in mass was 1 meter phthalide), dropwise over lh. After the dropwise addition, the reaction temperature after 5h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0046] The above compound I, in an amount of 10% anhydrous aluminum chloride (mass of Compound I was 100% basis), the amount of DMS0 3 (mass basis Compound I 1) into a reaction vessel , the temperature was adjusted to 95 ° C. The reaction time is to be 12h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0047] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.1-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2 times the mass 6, 11-dihydro-dibenzo [b, e] oxepin-11-one magnesium in , taking all of fifths THF (5 to 6 times by mass, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) and heated to 35 ° C and allowed to react. After the reaction started, the remaining 3/5 of THF was added dropwise.Was added dropwise to the system to be completed into hydrogen, reflux. After a total reaction 5h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0048] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 1.5 times the mass of hydroxy compound class of sodium hydroxide (concentration l〇wt mass%), was heated to 65 ° C, 2h elimination reaction after the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0049] placed in a 20L reaction vessel of the olefin compound, in an aqueous solution plus 1 times the mass of the olefinic compound hydrochloride (concentration of 5wt%), and heated to 50 ° C, so that a nucleophilic substitution reaction . The reaction time is to be after 4h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0050] placed in a 20L reaction vessel above halide, 0.1 times the mass of methyl lithium halides to 2 times the mass of the halide in diethyl ether, heated to 40 ° C, so that the nucleophilic substitution reaction. The reaction time is to be after 5h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0051] 20L is placed in a pressure reactor above doxepin, 1.05 times the mass of material in the doxepin hydrochloride (concentration of 30wt%), the control pressure to 3 ~ 4MPa, and heated to 130 ° C , and among the responses. Time after to be reacted for 20 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 37.9%, measured by HPLC obtaining 99.2% purity.
[0052] Example 2
[0053] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 20 ° C, under stirring for 2h. A known separation method, separation of o-toluic acid halide.
[0054] placed in a 20L reaction container, Compound J, an amount of load of cesium fluoride Celite ~ 0.05 0.15 (in mass Compound J is 1 meter), in an amount of 2.5 to 8 acetonitrile (compound J as a mass basis), and the temperature was adjusted to 30 ~ 50 ° C, 12 ~ 20h at reflux with stirring under regulation. Then, a known means for separating the reaction phthalide.
After [0055] phthalide placed in 20L reaction vessel, an amount of sodium methoxide in 10 ethanol solvent (total mass of phenol phthalide and 1 meter), adjusting the temperature of the reaction solution was 60 ° C, was added dropwise start phenol was 1.15 mass (in mass was 1 meter phthalide), dropwise over lh.After the dropwise addition, the reaction temperature after 5h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0056] The above compound I, in an amount of 40% anhydrous aluminum chloride (mass of Compound I was 100% basis), in an amount of DMS0 8 (in compound I is a mass basis) into a reaction vessel , the temperature was adjusted to 105 ° C. The reaction time is to be for 6h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0057] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.5-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2.4 times the mass in 6, 11-dihydro-dibenzo [b, e] oxepin-11-one of magnesium, taking all fifths THF (5 to 7 times the mass in 6, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) is to make, and heated to 40 ° C reaction.After the reaction started, the remaining 3/5 of THF was added dropwise. Was added dropwise to the system to be completed into hydrogen, reflux. When the total reaction 2h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0058] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 5 times the mass of hydroxy compound class of sodium hydroxide (concentration of 70wt%), was heated to 80 ° C, the reaction was stopped after the elimination reaction LH, cooling, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0059] placed in a 20L reaction vessel of the olefin compound, in an aqueous solution of 2 times the mass of the olefinic compound added hydrobromic acid (concentration of 30wt%), and heated to 60 ° C, so that nucleophilic Substitution reaction. The reaction time is to be after the 1. 5h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0060] placed in a 20L reaction vessel above halide, 0.8 times the mass of phenyl lithium halide to 8 times the mass of the halide in diethyl ether, heated to 50 ° C, so that the nucleophilic substitution reaction. The reaction time is to be after 2h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0061] 20L is placed in a pressure reactor above doxepin, 1.2 times the mass of material in the doxepin hydrochloride (concentration of 38wt%), the control pressure to 3 ~ 4MPa, and heated to 150 ° C , and among the responses. Time after to be reacted for 16 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 39.7%, measured by HPLC obtaining 99.4% purity.
[0062] Example 3
[0063] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 15 ° C, under stirring for 3h. A known separation method, separation of o-bromomethyl-benzoic acid.
[0064] placed in a 20L reaction container, Compound J, an amount of load of cesium fluoride Celite ~ 0.05 0.15 (in mass Compound J is 1 meter), in an amount of 2.5 to 8 acetonitrile (compound J as a mass basis), and the temperature was adjusted to 30 ~ 50 ° C, 12 ~ 20h at reflux with stirring under regulation. Then, a known means for separating the reaction phthalide.
After [0065] phthalide placed in 20L reaction vessel, an amount of sodium methoxide in ethanol solvent 6 (total mass of phenol phthalide and 1 meter), adjusting the temperature of the reaction solution was 55 ° C, was added dropwise start phenol was 1.10 mass (in mass was 1 meter phthalide), dropwise over lh.After the dropwise addition, the reaction temperature after 3. 5h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0066] Anhydrous aluminum above compound I, in an amount of 25% of the chloride (compound I mass is 100% basis), in an amount of DMS0 6. 5 (in compound I is a mass basis) into the reaction vessel temperature is adjusted to 100 ° C. The reaction time is to be 9h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0067] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.3-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2.2 times the mass in 6, 11-dihydro-dibenzo [b, e] oxepin-11-one of magnesium, taking all fifths THF (5 to 7 times the mass in 6, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) is to make, and heated to 38 ° C reaction.After the reaction started, the remaining 3/5 of THF was added dropwise. Was added dropwise to the system to be completed into hydrogen, refluxed for 2h. After a total reaction 3. 5h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0068] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 3-hydroxysteroid times the mass of the compound of sodium hydroxide (concentration of 40wt%), and heated to 75 ° C, 1. 5h the reaction stopped after elimination the reaction was cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0069] placed in a 20L reaction vessel of the olefin compound, an aqueous solution of 1.5-fold increase in the mass of hydroiodic olefinic compounds (concentration of 18wt%), was heated to 55 ° C, so nucleophilic substitution reaction. The reaction time is to be after 2h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0070] placed in a 20L reaction vessel above halide, 0.4 times the mass of the halide in n-butyllithium, in diethyl ether five times the mass of halide and heated to 45 ° C, so that a nucleophilic substitution reaction . The reaction time is to be 3. After 5h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0071] 20L is placed in a pressure reactor above doxepin, 1.12 times the mass of material in the doxepin hydrochloride (concentration of 34wt%), the control pressure to 3 ~ 4MPa, and heated to 140 ° C , and among the responses. Time after to be reacted for 18 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 40.2%, measured by HPLC obtaining 99.5% purity.
[0072] Example 4
[0073] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 15 ° C, under stirring for 4h. A known separation method, separation of o-toluic acid halide.
[0074] placed in a 20L reaction container, Compound J, an amount of load of cesium fluoride Celite ~ 0.05 0.15 (in mass Compound J is 1 meter), in an amount of 2.5 to 8 acetonitrile (compound J as a mass basis), and the temperature was adjusted to 30 ~ 50 ° C, 12 ~ 20h at reflux with stirring under regulation. Then, a known means for separating the reaction phthalide.
After [0075] phthalide placed in 20L reaction vessel, 5 an amount of sodium methoxide in ethanol solvent (total mass of phenol phthalide and 1 meter), adjusting the temperature of the reaction solution was 55 ° C, was added dropwise start phenol was 1.15 mass (in mass was 1 meter phthalide), dropwise over lh.After the dropwise addition, the reaction temperature after 5h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0076] The above compound I, in an amount of 25% anhydrous aluminum chloride (mass of Compound I was 100% basis), in an amount of DMS0 8 (in compound I is a mass basis) into a reaction vessel , the temperature was adjusted to 100 ° C. The reaction time is to be 12h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0077] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.3-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2.4 times the mass in 6, 11-dihydro-dibenzo [b, e] oxepin-11-one of magnesium, taking all fifths THF (5 to 7 times the mass in 6, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) is to make, and heated to 40 ° C reaction.After the reaction started, the remaining 3/5 of THF was added dropwise. Was added dropwise to the system to be completed into hydrogen, reflux. When the total reaction 2h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0078] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 5 times the mass of hydroxy compound class of sodium hydroxide (concentration of 70wt%), was heated to 80 ° C, the reaction was stopped after the elimination reaction LH, cooling, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0079] placed in a 20L reaction vessel of the olefin compound, an aqueous solution of 1.5-fold increase in the mass of hydroiodic olefinic compounds (concentration of 30wt%), and heated to 60 ° C, so nucleophilic substitution reaction. The reaction time is to be after the 1. 5h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0080] placed in a 20L reaction vessel above halide, 0.8 times in mass n-butyl lithium halide, eight times the mass of the halide in diethyl ether, heated to 50 ° C, so that a nucleophilic substitution reaction . The reaction time is to be after 2h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0081] 20L is placed in a pressure reactor above doxepin, 1.2 times the mass of material in the doxepin hydrochloride (concentration of 38wt%), the control pressure to 3 ~ 4MPa, and heated to 150 ° C , and among the responses. Time after to be reacted for 16 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 41.6%, measured by HPLC obtaining 99.7% purity.
[0082] Example 5
[0083] placed in a 20L reaction vessel acetonitrile, o-methylbenzoic acid, N- bromosuccinimide, using a water bath temperature controlled at 15 ° C, the reaction 2. 5h under stirring. A known separation method, separation of o-bromomethyl-benzoic acid.
[0084] placed in a 20L reaction vessel o-bromomethyl benzoic acid, diatomaceous earth in an amount of load of cesium fluoride 0.05 ~ 0.15 (in mass Compound J is 1 meter), in an amount of 2. 5-8 acetonitrile (compound J as a mass basis), and the temperature was adjusted to 30 ~ 50 ° C, 12 ~ 20h at reflux with stirring under regulation. Then, a known means for separating the reaction phthalide.
After [0085] phthalide placed in 20L reaction vessel, 5 an amount of sodium methoxide in ethanol solvent (total mass of phenol phthalide and 1 meter), adjusting the temperature of the reaction solution was 55 ° C, was added dropwise start was 1.08 mass of phenol (mass was phthalide 1 meter), dropwise over lh.After the dropwise addition, the reaction temperature after 3h using known separation methods, to give o-methyl benzyl phenyl ether, this compound is named I.
[0086] Anhydrous aluminum above compound I, in an amount of 25% of the chloride (compound I mass is 100% basis), in an amount of DMS0 5 (in compound I is a mass basis) into a reaction vessel , the temperature was adjusted to 100 ° C.The reaction time is to be 8h. Using known separation means for separating the 6, 11-dihydro-dibenzo [b, e] oxepin-11-one.
[0087] placement 6, 11-dihydro-dibenzo in a reaction vessel and 20L [b, e] oxepin-11-one, 1.2-dihydro-fold of the mole of diphenyl at 6, 11 and [ b, e] oxepin-11-one 3-chloropropyl alkyl tert-butyl ether, 2.2 times the mass in 6, 11-dihydro-dibenzo [b, e] oxepin-11-one of magnesium, taking all fifths THF (5 to 7 times the mass in 6, 11-dihydro-dibenzo [b, e] THF oxepin-11-one) is to make, and heated to 38 ° C reaction.After the reaction started, the remaining 3/5 of THF was added dropwise. Was added dropwise to the system to be completed into hydrogen, reflux. When the total reaction 2h, the reaction was stopped. After the system was cooled and then poured into saturated ammonium chloride solution, extracted twice with ethyl acetate was added, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to give hydroxy compound.
[0088] placed in a 20L reaction vessel above hydroxy compound, an ethanol solution of 2 times the mass of hydroxy compound class of sodium hydroxide (concentration of 40wt%), was heated to 70 ° C, the reaction was stopped after the elimination reaction 2h, cooling, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the olefinic compounds.
[0089] placed in a 20L reaction vessel of the olefin compound, an aqueous solution of 1.5-fold increase in the mass of hydroiodic olefinic compounds (concentration of 15wt%), and heated to 50 ° C, so nucleophilic substitution reaction. The reaction time is to be after 4h, the reaction was stopped, cooled, the solvent was distilled off more of the obtained crude product was crystallized from acetonitrile to give the halides.
[0090] placed in a 20L reaction vessel above halide, 0.4 times the mass of the halide in n-butyl lithium, 2 to 8 times the mass of the halide in diethyl ether, heated to 45 ° C, so that nucleophilic Substitution reaction. The reaction time is to be after 3h, the reaction was stopped, reaction was complete and extracted with ethylacetate three times, dried over anhydrous sodium sulfate 5h, the resulting crude product was recrystallized from acetonitrile to obtain doxepin.
[0091] 20L is placed in a pressure reactor above doxepin, 1.12 times the mass of material in the doxepin hydrochloride (mass concentration 37. 6wt%), the control pressure to 3 ~ 4MPa, heated to 140 ° C, allowing the reaction among. Time after to be reacted for 20 h, cooled to room temperature and should be finished by filtration, and dried to give doxepin hydrochloride. In this embodiment overall yield 43.9%, measured by HPLC obtaining 99.9% purity.
PATENTS
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Sinequan, many others[2] |
| Synonyms | NSC-108160[3] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682390 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth, topical, intravenous, intramuscular injection[1] |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 13–45% (mean 29%)[5][6] |
| Protein binding | 76%[7] |
| Metabolism | Hepatic (CYP2D6, CYP2C19)[4][5] |
| Metabolites | Nordoxepin, glucuronide conjugates[4] |
| Elimination half-life | Doxepin: 8–24 hours (mean 17 hours)[7] Nordoxepin: 31 hours[7] |
| Excretion | Urine: ~50%[4][5] Feces: minor[5] |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C19H21NO |
| Molar mass | 279.376 g/mol |
| 3D model (JSmol) | |
- Virtanen R, Iisalo E, Irjala K: Protein binding of doxepin and desmethyldoxepin. Acta Pharmacol Toxicol (Copenh). 1982 Aug;51(2):159-64. [PubMed:7113722]
- Virtanen R, Scheinin M, Iisalo E: Single dose pharmacokinetics of doxepin in healthy volunteers. Acta Pharmacol Toxicol (Copenh). 1980 Nov;47(5):371-6. [PubMed:7293791]
- Negro-Alvarez JM, Carreno-Rojo A, Funes-Vera E, Garcia-Canovas A, Abellan-Aleman AF, Rubio del Barrio R: Pharmacologic therapy for urticaria. Allergol Immunopathol (Madr). 1997 Jan-Feb;25(1):36-51. [PubMed:9111875]
- Sansone RA, Sansone LA: Pain, pain, go away: antidepressants and pain management. Psychiatry (Edgmont). 2008 Dec;5(12):16-9. [PubMed:19724772]
- Kirchheiner J, Meineke I, Muller G, Roots I, Brockmoller J: Contributions of CYP2D6, CYP2C9 and CYP2C19 to the biotransformation of E- and Z-doxepin in healthy volunteers. Pharmacogenetics. 2002 Oct;12(7):571-80. [PubMed:12360109]
- ZONALON® (doxepin hydrochloride) CREAM, 5% [Link]
- FDA Label: SilenorTM (doxepin) tablets for oral administration [Link]
//////////////Doxepin, ドキセピン , NSC-108160 , P-3693A , SO-101
[H]C(CCN(C)C)=C1C2=CC=CC=C2COC2=CC=CC=C12
C19H21NO·HCl ![]()
![]()
![]()
![]()
![]() 315.84
315.84
N,N-Dimethyldibenz[b,e]oxepin-D11(6H),
USP Doxepin Hydrochloride RS.
USP Doxepin Related Compound A RS
 .
. USP Doxepin Related Compound B RS
 .
. USP Doxepin Related Compound C RS.
Identification—
Related compounds—
Chromatographic system (see Chromatography ![]() 621
621![]() )— The liquid chromatograph is equipped with a 215-nm detector and a 4.6-mm × 25-cm column that contains 5-µm packing L1. The flow rate is about 1 mL per minute. The column temperature is maintained at 30
)— The liquid chromatograph is equipped with a 215-nm detector and a 4.6-mm × 25-cm column that contains 5-µm packing L1. The flow rate is about 1 mL per minute. The column temperature is maintained at 30![]() . Chromatograph about 20 µL of the Standard solution, and record the peak areas as directed for Procedure: the resolution, R, between doxepin related compound A and doxepin related compound C is not less than 1.5; the resolution between doxepin related compound C and doxepin related compound B is not less than 1.5; and the signal-to-noise ratio for all the peaks is not less than 10. [NOTE—Use the approximate relative retention times given in Table 1 for the purpose of peak identification. The doxepin related compound C peak will be the largest peak in the Standard solution chromatogram.]
. Chromatograph about 20 µL of the Standard solution, and record the peak areas as directed for Procedure: the resolution, R, between doxepin related compound A and doxepin related compound C is not less than 1.5; the resolution between doxepin related compound C and doxepin related compound B is not less than 1.5; and the signal-to-noise ratio for all the peaks is not less than 10. [NOTE—Use the approximate relative retention times given in Table 1 for the purpose of peak identification. The doxepin related compound C peak will be the largest peak in the Standard solution chromatogram.]
| Name | Relative Retention Time (RRT) |
Limit (%) |
| Doxepin related compound A | 0.48 | 0.10 |
| Doxepin related compound C | 0.55 | 0.20 |
| Doxepin related compound B | 0.63 | 0.10 |
| Doxepin hydrochloride | 1.0 | — |
| Unknown impurity | — | 0.10 each |
Procedure— Inject a volume (about 20 µL) of the Test solution into the chromatograph, record the chromatogram for up to 2.2 times the retention time of doxepin, and measure the peak responses. Calculate the percentage of each individual doxepin related compound in the portion of Doxepin Hydrochloride taken by the formula:
in which rU is the individual peak response for each doxepin related compound obtained from the Test solution; rS is the response of the corresponding peak in theStandard solution; CS is the concentration, in mg per mL, of each doxepin related compound in the Standard solution; and CT is the concentration, in mg per mL, of Doxepin Hydrochloride in the Test solution. The related substance limits are listed in Table 1. [NOTE—Discard any peak with a relative retention time less than 0.25. This method is not intended to resolve the E- and Z-isomers of doxepin hydrochloride. Minor variations in the mobile phase composition could result in a shoulder in the trailing edge of doxepin. In cases where there may be separation, both the E- and Z-isomers should be used in the appropriate calculations.] Use the response of the doxepin peak obtained from the Standard solution and the concentration of doxepin hydrochloride in the Standard solution to calculate the percentage of unknown individual impurities.
Assay—
Procedure— Separately inject equal volumes (about 20 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the responses for the major peaks. Calculate the quantity, in mg, of C19H21NO·HCl in the portion of Doxepin Hydrochloride taken by the formula:
in which C is the concentration, in µg per mL, of USP Doxepin Hydrochloride RS in the Standard preparation, and rU(Z) and rU(E) are the respective peak responses of the (Z)- and (E)-isomers obtained from the Assay preparation, and rS(Z) and rS(E) are the respective peak responses of the (Z)- and (E)-isomers obtained from the Standard preparation. Calculate the percentage of the (Z)-isomer in the Assay preparation taken by the formula:
in which WS is the weight, in mg, of USP Doxepin Hydrochloride RS in the Standard preparation, WT is the weight, in mg, in the portion of Doxepin Hydrochloride taken, and PZ is the labeled percentage of (Z)-isomer in USP Doxepin Hydrochloride RS. Similarly calculate the percentage of (E)-isomer in the Assay preparationtaken by the formula:
in which PE is the labeled percentage of (E)-isomer in USP Doxepin Hydrochloride RS.
Auxiliary Information— Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
| Monograph | Ravi Ravichandran, Ph.D. Senior Scientist 1-301-816-8330 |
(MDPP05) Monograph Development-Psychiatrics and Psychoactives |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
Pharmacopeial Forum: Volume No. 32(2) Page 330
Chromatographic Column—
ONC201 disrupts mitochondrial function and kills breast cancer cells, reveals study — Med-Chemist

TRAIL, a member of the TNF family of ligands, causes caspase-dependent apoptosis through activation of its receptors, death receptor 4 and DR5.ONC201 was originally identified as a small molecule that inhibits both Akt and ERK, resulting in dephosphorylation of Foxo3a and thereby induces TRAIL transcription.Recently, two independent groups, Wafik El Deiry at Fox Chase and…
via ONC201 disrupts mitochondrial function and kills breast cancer cells, reveals study — Med-Chemist
FDA approves new treatment Xeljanz (tofacitinib) for moderately to severely active ulcerative colitis
| The U.S. Food and Drug Administration today expanded the approval of Xeljanz (tofacitinib) to include adults with moderately to severely active ulcerative colitis. Xeljanz is the first oral medication approved for chronic use in this indication. Other FDA-approved treatments for the chronic treatment of moderately to severely active ulcerative colitis must be administered through an intravenous infusion or subcutaneous injection. |
May 30, 2018
Release
The U.S. Food and Drug Administration today expanded the approval of Xeljanz (tofacitinib) to include adults with moderately to severely active ulcerative colitis. Xeljanz is the first oral medication approved for chronic use in this indication. Other FDA-approved treatments for the chronic treatment of moderately to severely active ulcerative colitis must be administered through an intravenous infusion or subcutaneous injection.
“New treatments are needed for patients with moderately to severely active ulcerative colitis,” said Julie Beitz, M.D., director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research. “Today’s approval provides an alternative therapy for a debilitating disease with limited treatment options.”
Ulcerative colitis is a chronic, inflammatory bowel disease affecting the colon. Patients experience recurrent flares of abdominal pain and bloody diarrhea. Other symptoms include fatigue, weight loss and fever. More than 900,000 patients are affected in the U.S., many of them experiencing moderately to severely active ulcerative colitis, and there is currently no cure.
The efficacy of Xeljanz for the treatment of moderately to severely active ulcerative colitis was demonstrated in three controlled clinical trials. This included two 8-week placebo-controlled trials that demonstrated that 10 mg of Xeljanz given twice daily induces remission in 17 to 18 percent of patients by week eight. In a placebo-controlled trial among patients who achieved a clinical response by week eight, Xeljanz, at a 5 mg or 10 mg dose given twice daily, was effective in inducing remission by week 52 in 34 percent and 41 percent of patients, respectively. Among patients who achieved remission after 8 weeks of treatment, 35 percent and 47 percent achieved sustained corticosteroid-free remission when treated with 5 mg and 10 mg, respectively.
The safety of chronic use of Xeljanz for ulcerative colitis was studied in the 52-week placebo- controlled trial. Additional supportive safety information was collected from patients who received treatment in an open-label long-term study.
The most common adverse events associated with Xeljanz treatment for ulcerative colitis were diarrhea, elevated cholesterol levels, headache, herpes zoster (shingles), increased blood creatine phosphokinase, nasopharyngitis (common cold), rash and upper respiratory tract infection.
Less common serious adverse events included malignancy and serious infections such as opportunistic infections. Xeljanz has a boxed warning for serious infections and malignancy. Patients treated with Xeljanz are at increased risk for developing serious infections that may lead to hospitalization or death. Lymphoma and other malignancies have been observed in patients treated with Xeljanz.
Use of Xeljanz in combination with biological therapies for ulcerative colitis or with potent immunosuppressants, such as azathioprine and cyclosporine, is not recommended.
Xeljanz, made by Pfizer Labs, was previously approved in 2012 for rheumatoid arthritis and in 2017 for psoriatic arthritis.
/////////////Xeljanz, tofacitinib, pfizer, fda 2017, psoriatic arthritis, ulcerative colitis
Specific Stereoisomeric Conformations Determine the Drug Potency of Cladosporin Scaffold against Malarial Parasite
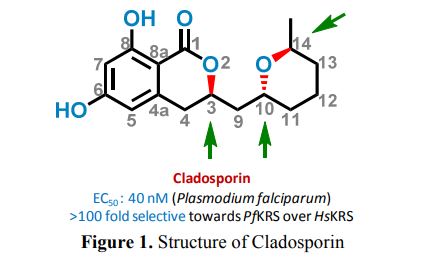
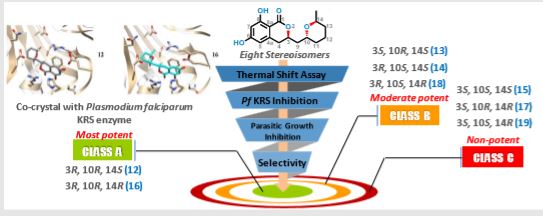

Specific Stereoisomeric Conformations Determine the Drug Potency of Cladosporin Scaffold against Malarial Parasite
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b00565

Dr. D. Srinivasa Reddy has been appointed as an editor of Bioorganic & Medicinl Chemistry Letters, Elsevier Publications. Congratulation Sir !
Click here for details. https://www.journals.elsevier.com/bioorganic-and-medicinal-chemistry-letters
The research interests of his group lie in issues related to application of oriented organic synthesis, in particular total synthesis of biologically active natural products, medicinal chemistry and crop protection. This team has been credited with having accomplished total synthesis of more than 25 natural products with impressive biological activities. “Some of our recent achievements include identification of potential leads, like antibiotic compound based on hunanamycin natural product for treating food infections, anti-diabetic molecule in collaboration with an industry partner and anti-TB compound using a strategy called ‘re-purposing of a drug scaffold’,” said Reddy.
A total of two awardees out of four were from CSIR institutes. In addition to Reddy, Rajan Shankarnarayanan, CSIR – CCMB, Hyderabad (basic sciences), also was conferred with the award. Vikram Mathews, CMC, Vellore (medical research) and Prof Ashish Suri, AIIMS, New Delhi (clinical research), were the others to receive the awards.
With more than 80 scientific publications and 35 patents, Reddy is one of the most prominent scientists in the city and has already been honoured with the Shanti Swarup Bhatnagar prize in chemical sciences. Reddy is also a nominated member of the scientific body of Indian Pharmacopoeia, government of India and was elected as a fellow of the Telangana and Maharashtra Academies of Sciences in addition to the National Academy of Sciences, India (NASI).
FDA Approves Tavalisse (fostamatinib disodium hexahydrate) for Chronic Immune Thrombocytopenia — Med-Chemist

Rigel Pharmaceuticals, Inc. announced that the U.S. Food and Drug Administration (FDA) approved Tavalisse (fostamatinib disodium hexahydrate) for the treatment of thrombocytopenia in adult patients with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment. Tavalisse is an oral spleen tyrosine kinase (SYK) inhibitor that targets the underlying autoimmune cause of the…
Mibefradil, a new class of compound to study TRPM7 channel function — Sussex Drug Discovery Centre

Transient receptor potential (TRPM) is a family of non-selective cation channels that are widely expressed in mammalian cells. TRP channels are composed of six transmembrane domains and the family consists of eight different channels, TRPM1–TRPM8. TRPM7 is compromised of an ion channel moiety essential for the ion channel function, which serves to increase intracellular calcium […]
via Mibefradil, a new class of compound to study TRPM7 channel function — Sussex Drug Discovery Centre
Dark Chocolate improves vision with 2 hours — ClinicalNews.Org

Dark Chocolate improves vision with 2 hours Contrast sensitivity and visual acuity were significantly higher 2 hours after consumption of a dark chocolate bar compared with a milk chocolate bar, but the duration of these effects and their influence in real-world performance await further testing. Rabin JC, Karunathilake N, Patrizi K. Effects of Milk vs […]
via Dark Chocolate improves vision with 2 hours — ClinicalNews.Org
What are the drugs of the future? — All About Drugs
A cartoon representing how, in history, we are continuously faced with new scientific advancements that make us question what the future holds and whether what we currently have is still useful or should be replaced. What are the drugs of the future? Med. Chem. Commun., 2018, Advance ArticleDOI: 10.1039/C8MD90019A, Opinion Huy X. Ngo, Sylvie Garneau-Tsodikova…
Roquinimex
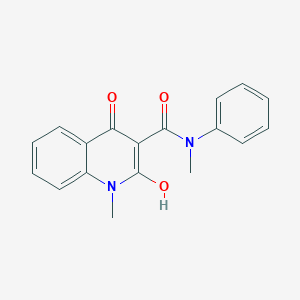

Roquinimex
- Molecular FormulaC18H16N2O3
- Average mass308.331 Da
LS-2616
PNU-212616
Roquinimex (Linomide) is a quinoline derivative immunostimulant which increases NK cell activity and macrophage cytotoxicity. It also inhibits angiogenesis and reduces the secretion of TNF alpha.
Investigated as a treatment for some cancers (including as adjuvant therapy after bone marrow transplantation in acute leukemia) and autoimmune diseases, such as multiple sclerosis and recent-onset type I diabetes.
Roquinimex has been investigated as a treatment for some cancers (including as adjuvant therapy after bone marrow transplantation in acute leukemia) and autoimmune diseases, such as multiple sclerosis and recent-onset type I diabetes. Several trials have been terminated due to serious cardiovascular toxicity.
Synthesis

Roquinimex synthesis:[1]
Ethyl 2-(methylamino)benzoate is condensed with ethyl malonate. Amine-ester ineterchange of that compound with N-methylanilineresults in formation of the amide roquinimex.
PAPER
Using DOE to Achieve Reliable Drug Administration: A Case Study

Design of experiments (DOE), a statistical tool, and mathematical modeling techniques are established and proven methodologies for process and product improvements in the pharmaceutical industry. This contribution presents a case study where an unsatisfactory dissolution capacity for the drug Roquinimex was overcome by investigating the process parameters with the help of an experimental design. By elucidating the detailed effects of temperature, dosing time, and dilution, conformity in the particle size distribution of the active pharmaceutical ingredient (API) from batch to batch in full-scale manufacturing could be ensured. As a direct result the manufactured drug met its specified dissolution capacity, which was a prerequisite for obtaining the desired bioavailability of the pharmaceutical oral formulation. This work demonstrates how the use of DOE in chemical process development adds value by allowing efficient and reliable improvements of a given synthetic step.
1 H NMR (4): δ 12.4 (broad s, 1H, OH), 8.1 (m, 1H, Ar), 7.5 (m, 1H, Ar), 7.1 (m, 7H, Ar), 3.5 (s, 3H, NCH3 ), 3.3 5 (s, 3H, NCH3 ).
SYN
BE 0904431; DE 3609052; GB 2172594; JP 1986221194; US 4672057
 By condensation of 4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid ethyl ester (I) with N-methylaniline (II) by heating at 125 C and distillation of the ethanol formed.
By condensation of 4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid ethyl ester (I) with N-methylaniline (II) by heating at 125 C and distillation of the ethanol formed.
CLIP
https://www.sciencedirect.com/science/article/abs/pii/S0731708597001076
 1H-NMR spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
1H-NMR spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
 13C-DEPT experiment of linomide in DMSO at 298 K recorded on a Bruker AC
13C-DEPT experiment of linomide in DMSO at 298 K recorded on a Bruker AC


A 2D 13C–1H COLOC experiment of linomide in DMSO at 298 K recorded on

 COSY 45° spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
COSY 45° spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
PATENT
https://patents.google.com/patent/US5912349
U.S. Pat. No. 4,738,971 discloses roquinimex and a method to produce it. The disclosed method starts with N-methylisatoic anhydride (I) and requires three steps. The improved process of the present invention starts with the same N-methylisatoic anhydride (I) and requires fewer steps.
The process of the present invention is practiced according to EXAMPLE 2. It is preferred to perform the claimed process in an aprotic solvent. Suitable aprotic solvents include DMF, THF, glyme, dioxane and ether and mixtures thereof.
The roquinimex produced by the process of the invention (EXAMPLE 2) can be upgraded or purified by the process of EXAMPLE 3.
Roquinimex is known to be useful as a pharmaceutical agent, see U.S. Pat. No. 4,738,971. It is preferably used in treating multiple sclerosis, in particular the treatment of relapsing remitting and secondary progressive multiple sclerosis. In treating multiple sclerosis roquinimex is administered in an oral dose of from about 2.0 to about 5.0 mg/day.
Example 1
N-Methyl-N-Phenyl-α-Carbomethoxyacetamide (V)
Mono-methyl malonate potassium salt also known as potassium methyl malonate (73.32 g, 0.47 mol) and water (50 ml) are cooled to 5° with an ice bath, and concentrated hydrochloric acid (40 ml) is added over a 30 minute period while the temperature is maintained below 10°. The mixture is filtered with suction to remove potassium chloride, and the precipitate washed with methyl t-butylether (75 ml). The aqueous layer of the filtrate is separated and washed with methyl t-butyl ether (3×50 ml). The combined methyl t-butyl ether extracts are dried over anhydrous sodium sulfate; then the solvent was removed under reduced pressure at 45-50° to give carbomethoxy acetic acid. This product was checked by NMR for complete removal of the methyl t-butyl ether solvent.
Carbomethoxy acetic acid (100 g, 0.84 mol) is dissolved in methylene chloride (400 ml). Thionyl chloride (100 g, 0.84 mol) is added via a dropping funnel. It can be added rapidly as there is little, if any, exotherm produced during the addition. After addition, the reaction is refluxed at 40-45° for 1 hr. At the end of the reflux period, 50% of the methylene chloride is removed (200 ml) by distillation at atmospheric pressure and 40-45°. Fresh methylene chloride is added (200 ml) followed by distillation to again remove 50% of the total volume. This add-distillation procedure is repeated two times to give the carbomethoxy acetyl chloride.
The carbomethoxy acetyl chloride mixture is cooled in an ice-salt bath to -5 to 0° and N-methyl aniline (55.64 g, 0.52 mol) in methylene chloride (200 ml) is added at a rate so as to maintain the temperature of the reaction mixture between -5 to 0°. The addition is performed using an addition funnel and can normally be carried out over a 3-5 min time period to control the slight exotherm. Pyridine (66.36 g, 0.84 mol) in methylene chloride (200 ml) is then added to the above mixture. The addition rate is adjusted so as to keep the temperature of the reaction between -5 to 0° during the addition. The addition is performed using an addition funnel and can normally be carried out over a 3-5 min time period to control the slight exotherm. After the addition is complete (as measured by HPLC) the reaction is quenched by pouring the reaction mixture into water (500 ml) and stirring continued for 30 min. The reaction is equilibrated and the methylene chloride layer separated. Additional methylene chloride (400-500 ml) is added and the methylene chloride mixture is washed successively with hydrochloric acid (1N, 2×300 ml), saturated sodium bicarbonate solution (2×300 ml), saline (1×600 ml) and the methylene chloride mixture dried through anhydrous sodium sulfate. Concentration of the mixture under reduced pressure at 40-45° gives the title compound, HPLC (Nucleosil column; acetonitrile/water, 45/55, 1 ml/min, UV=229 nm; Retention times for N-methyl-N-phenyl-α-carbomethoxyacetamide˜6.0 min; N-methyl aniline˜11.0-12.0 min.
Example 2
Preparation of Roquinimex (IV) from N-Methylisotoic anhydride (I) and N-Methyl-α-carbomethoxyacetamide (V)
N-Methyl-N-phenyl-α-carbomethoxyacetamide (V, EXAMPLE 1, 139 g, 0.671 mole) and DMF (695 mL). The mixture is subject to reduced pressure and purged with nitrogen three times. While at room temperature (20-25°), potassium t-butoxide solution (1.714 M in THF, 367 mL, 0.630 mole) is added in one portion. A small exotherm and slight darkening of the mixture followed this addition. The mixture is heated to 80-90° and kept at this temperature for 1.5 hr.
A -78° cooling bath is placed on the receiving flask of the distillation assembly, the nitrogen flow is shut off and the mixture is subject to reduced pressure over 0.5 hr to remove the THF solvent. The pot temperature at the end of the distillation is 72-76°. The amount of distillate collected should be nearly identical to the amount of potassium tert-butoxide reagent used, (367 ml). The mixture is then heated to 80-85° and N-methylisotoic anhydride (I, 70.72 g, 0.400 mole) is added in one portion followed by a 5-10 mL DMF wash. Gas evolution with foaming followed the addition and subsequent wash. The equipment is modified at this point to include a reflux condenser with a vacuum port. With the temperature still at 80-85°, the mixture is placed under reduced pressure and the mixture refluxed for 30 min. After refluxing the temperature is 79°. The reduced pressure and heat source are removed, the system is repressurized with nitrogen and the temperature is allowed to drop to 30° (±2°). Hydrochloric acid (0.6 N, 2.295 L) is added slowly via an addition funnel attached to the claisen head over 2.5 hr, to pH=1.0-1.5, making sure the temperature does not exceed 32°. The temperature control is especially critical at the beginning of the addition when a mild exotherm occurs. The temperature at the end of the addition is nearly room temperature (24-25°). When the acid addition is complete, the resulting slurry is stirred for 30 min and then let stand overnight before filtration. The solids are washed with water (2 ×330 mL) and dried on a nitrogen press to give the title compound, HPLC (Nucleosil column; acetonitrile/water, 45/55, 1 ml/min, UV=229 nm; Retention times 2.29 min.
Example 3
Purification of Roquinimex (IV)
Roquinimex crude is taken up in water (1.5 L) and the slurry is stirred vigorously at 20-25°. The pH is adjusted to 7.5-7.7 with sodium hydroxyde (7%, about 170 mL). (The base can be added as fast as possible but requires longer pH equilibration near the end of the addition (about 1-2 hr total addition time). It is recommended that 85% of the base is initially added to a stable pH and the rest is added dropwise until the pH has stabilized and falls into the desired range of 7.5-7.7.) Nearly all solids should be dissolved (some may remain however). After the base is added and the pH is stabilized for more than 30 min, Darco (charcoal, 15.00 g) is added and the mixture is stirred for 30 min. The mixture is filtered through a 0.45 micron Millipore filter and the filter cake is washed with water (2×175 mL). The filtrate is transferred to a flask.
The mixture is stirred vigorously, heated to 28-32° and hydrochloric acid (6 N, about 120 mL) is added over 30 to 45 min to a pH of 0.5 to 1.0. After the addition is over, the mixture is stirred for 15 min then allowed to stand, without stirring, at the above temperature for 2 hr before filtration. The filter cake is washed with water (2×180 mL) and dried on a nitrogen press to give essentially pure title compound.
PATENT
https://patents.google.com/patent/US6605616
The present invention relates to novel substituted quinoline-3-carboxamide derivatives, to methods for their preparation, to compositions containing them, and to methods and use for clinical treatment of diseases resulting from autoimmunity, such as multiple sclerosis, insulin-dependent diabetes mellitus, systemic lupus erythematosus, rheumatoid arthritis, inflammatory bowel disease and psoriasis and, furthermore, diseases where pathologic inflammation plays a major role, such as asthma, atherosclerosis, stroke and Alzheimer’s disease. More particularly, the present invention relates to novel quinoline derivatives suitable for the treatment of, for example, multiple sclerosis and its manifestations.
BACKGROUND OF THE INVENTION
Autoimmune diseases, e.g., multiple sclerosis (MS), insulin-dependent diabetes mellitus (IDDM), systemic lupuis erythematosus (SLE), rheumatoid arthritis (RA), inflammatory bowel disease (IBD) and psoriasis represent assaults by the body’s immune system which may be systemic in nature, or else directed at individual organs in the body. They appear to be diseases in which the immune system makes mistakes and, instead of mediating protective functions, becomes the aggressor (1).
MS is the most common acquired neurologic disease of young adults in western Europe and North America. It accounts for more disability and financial loss, both in lost income and in medical care, than any other neurologic disease of this age group. There are approximately 250.000 cases of MS in the United States. Although the cause of MS is unknown, advances in brain imaging, immunology, and molecular biology have increased researchers’ understanding of this disease. Several therapies are currently being used to treat MS, but no single treatment has demonstrated dramatic treatment efficacy. Current treatment of MS falls into three categories: treatment of acute exacerbations, modulation of progressive disease, and therapy for specific symptoms.
MS affects the central nervous system and involves a demyelination process, i.e., the myelin sheaths are lost whereas the axons are preserved. Myelin provides the isolating material that enables rapid nerve impulse conduction. Evidently, in demyelination, this property is lost. Although the pathogenic mechanisms responsible for MS are not understood, several lines of evidence indicate that demyelination has an immunopathologic basis. The pathologic lesions, the plaques, are characterized by infiltration of immunologically active cells such as macrophages and activated T cells (2).
In U.S. Pat. No. 4,547,511 and in U.S. Pat. No. 4,738,971 and in EP 59,698 some derivatives of N-aryl-1,2-dihydro-4-substituted-1-alkyl-2-oxo-quinoline-3-carboxamide are claimed as enhancers of cell-mediated immunity. The compound

known as roquinimex (Merck Index 12th Ed., No. 8418; Linomide®, LS2616, N-phenyl-N-methyl-1,2-dihydro-4-hydroxy-1-methyl-2-oxo-quinoline-3-carboxamide) belongs to this series of compounds. Roquinimex has been reported to have multiple immunomodulatory activities not accompanied with general immunosuppression (3-12). Furthermore, in U.S. Pat. No. 5,580,882 quinoline-3-carboxarnide derivatives are claimed to be useful in the treatment of conditions associated with MS. The particular preferred compound is roquinimex. In U.S. Pat. No. 5,594,005 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of type I diabetes. The particular preferred compound is roquinimex. In WO 95/24195 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of inflammatory bowel disease. Particularly preferred compounds are roquinimex or a salt thereof. In WO95/24196 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of psoriasis. Particularly preferred compounds are roquinimex or a salt thereof.
In clinical trials comparing roquinimex to placebo, roquinimex was reported to hold promise in the treatment of conditions associated with MS (13, 14). There are, however, some serious drawbacks connected to roquinimex. For example, it has been found to be teratogenic in the rat, and to induce dose-limiting side effects in man, e.g., a flu-like syndrome, which prevents from using the full clinical potential of the compound.
Further, in WO 92/18483 quinoline derivatives substituted in the 6-position with a RAS (O)n-group (RA=lower alkyl or aryl; n=0−2) are claimed, which possess an immunomodulating, anti-inflammatory and anti-cancer effect.
PAPER
Modified synthesis and antiangiogenic activity of linomide
https://www.sciencedirect.com/science/article/pii/S0960894X00006995?via%3Dihub

PAPER
https://pubs.acs.org/doi/full/10.1021/jm031044w
1H NMR (CDCl3) δ 3.28 (s, br, 3H, 1-NCH3), 3.50 (s, 3H, 12-NCH3), 7.1−7.3 (m, 7H, 6,8,2‘,3‘,4‘,5‘,6‘-aromatic CH), 7.56 (dt, JHCCH = 7.5 and 8.5 Hz, JHCCCH = 1.5 Hz, 1H, 7-aromatic CH), 8.09 (dd, JHCCH = 8.0 Hz, JHCCCH = 1.5 Hz, 1H, 5-aromatic CH), 12.3 (s, br, 1H, 4-OH). 13C NMR (CDCl3) δ 28.7 (1C, 1-NCH3), 38.3 (1C, br, 12-NCH3), 104.6 (1C, 3-C), 113.5 (1C, 8-CH), 115.3 (1C, 10-C), 121.4 (1C, 6-CH), 124.6 (1C, 5-CH), 125.5 (2C, 2‘,6‘-CH), 126.7 (1C, 4‘-CH), 128.5 (2C, 3‘,5‘-CH), 132.3 (1C, 7-CH), 140.1 (1C, 9-C), 143.8 (1C, 1‘-C), 158.8 (1C, 2-CO), 164.3 (1C, 4-C), 169.4 (1C, 11-CO). MS-ESI: m/z 309 [MH]+. Anal. (C18H16N2O3) C, H, N.
PATENT
1,2-dihydro-4-hydroxy-2-oxo-quinoline-3-carboxanilides have been described in the literature since the 1970s (refs 1-4). The most well-known compound in this class, roquinimex (Linomide), was first described by AB Leo as an immuno-stimulating agent (ref 4) but was later also found to have immuno-modulating effects, as well as anti-angiogenetic effects (refs 5a, b). Roquinimex has been claimed beneficial for the treatment of autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, systemic lupus erythematosus, inflammatory bowel disease, diabetes type 1, and psoriasis, as well as for the treatment of cancer (refs 6a-d, 9d and refs therein).


The compound laquinimod (a 5-Cl, N-Et carboxanilide derivative) has been reported by Active Biotech AB to convey a better therapeutic index compared with roquinimex (refs 7a, b) and is currently in phase III clinical studies for the treatment of multiple sclerosis. Laquinimod has also entered clical trials in Crohn’s disease and
SLE. Two other compounds in the same class under clinical evaluation are tasquinimod (prostate cancer) and paquinimod (systemic sclerosis). Recently, a molecular target for laquinimod was identified as S100A9 (ref 8).
Fujisawa has reported on similar compounds with inhibitory activity on nephritis and on B16 melanoma metastases (refs 9a-d). Also the closely related thieno-pyridone analogs have been described as immunomodulating compounds with anti-inflammatory properties (ref
10).
Another closely related compound class are the corresponding N-pyridyl-carboxamide derivatives, which have been reported to have antitubercular activity as well as anti-inflammatory properties (ref
11). However, according to litterature (ref 10) these derivatives are less active as immunomodulating agents.
The N-hydrogen 3-carboxanilides (“N-H derivatives”) and the N-alkyl 3-carboxanilides (“N-alkyl derivatives”), respectively, are described in the prior art documents relating to inflammation, immunomodulation, and cancer as a homogenous group of compounds in terms of biological effects. Prior art also teaches that the N-alkyl derivatives are the preferred compound derivatives.

In fact, very few studies (refs 4, 9d) of N-hydrogen derivatives, especially in vivo studies, have been reported. Furthermore, no fundamental biological differences between the N-alkyl derivatives and the N-hydrogen derivatives, respectively, have been described.
However, some chemical properties of the N-hydrogen and the N-alkyl derivatives are different (ref 12). N-Alkyl derivatives adopt a twisted 3D-structure, whereas the N-H derivatives are stabilized by intramolecular hydrogen bonds in a planar structure. The N-alkyl derivatives are more soluble in aqueous media, but also inherently unstable towards nucleophiles, such as amines and alcohols (refs 12, 13).
The N-alkyl derivatives roquinimex (N-Me) and laquinimod (N-Et) have been reported to be metabolized in human microsomes to give the corresponding N-hydrogen derivatives, via N-dealkylation catalyzed mainly by CYP3A4 (refs 14a, b).
bHLH-PAS (basic helix-loop-helix Per-Arnt-Sim) proteins constitute a recently descovered protein family functioning as transcripon factors as homo or hetero protein dimers (refs 15a, b). The N-terminal bHLH domain is responsible for DNA binding and contributes to dimerization with other family members. The PAS region (PAS-A and PAS-B) is also involved in protein-protein interactions determining the choice of dimerization partner and the PAS-B domain harbors a potential ligand binding pocket.
The aryl hydrocarbon receptor (AhR or dioxin receptor) and its dimerization partner ARNT (AhR nuclear translocator) were the first mammalian protein members to be identified. AhR is a cytosolic protein in its non-activated form, associated in a protein complex with Hsp90, p23, and XAP2. Upon ligand activation, typically by chlorinated aromatic hydrocarbons like TCDD, the Ahr enters the nucleus and dimerizes with ARNT. The AhR/ARNT dimer recognizes specific xenobiotic response elements (XREs) to regulate TCDD-responsive genes. The ligand binding domain of AhR (AhR-LBD) resides in the PAS-B domain.
Recently, it has been demonstrated that AhR is involved in Thl7 and Treg cell development and AhR has been proposed as a unique target for therapeutic immuno-modulation (refs 16a-c). The AhR ligand TCDD was shown to induce development of Treg (FoxP3+) cells, essential for controlling auto-immunity, and to suppress symptoms in the EAE model. In addition, activation of AhR has been shown essential for the generation of IL-10 producing regulatory Trl cells (ref 16d), and Ahr ligands have also been proven efficacious in other models of auto-immunity, e.g. diabetes type 1, IBD, and uveitis (refs 16e-h). Apart from controlling autoimmune disorders, AhR activation and Treg cell development have been implicated as a therapeutic strategy for other conditions with an immunological component, such as allergic lung inflammation, food allergy, transplant rejection, bone loss, and type 2 diabetes and other metabolic disorders (refs 17a-e).
Apart from its role as a transcription factor, AhR has been reported to function as a ligand-dependent E3 ubiguitin ligase (ref 18), and ligand-induced degradation of β-catenin has been demonstrated to suppress intestinal cancer in mice (ref 19). In addition, activation of AhR has been implicated to play a protective role in prostate cancer (ref 20).
Other members of the bHLH-PAS family are the HIF-α (hypoxia inducible factor alpha) proteins, which also hetero-dimerize with ARNT. In conditions with normal oxygen levels (normoxia), HIF-α proteins are rapidly degraded by the ubiquitin-proteasome system and they are also inactivated by asparagine hydroxylation. Under hypoxic conditions, however, the proteins are active and upregulate genes as a response to the hypoxic state, e.g. genes for erythropoietin and vascular endothelial growth factor (VEGF). VEGF is essential for blood vessel growth (angiogenesis) and is together with HIF-1α considered as interesting targets for anti-angiogenetic tumour theraphy (ref 21). HIF-α proteins can be negatively and indirectly regulated by AhR ligands, which upon binding with AhR reduce the level of the common dimerization partner ARNT. Anti-angiogenetic effects can possibly also be achieved directly by AhR activity via upregulation of thrombospondin-1 (ref 22).
 |
|
Title: Roquinimex
CAS Registry Number: 84088-42-6
CAS Name: 1,2-Dihydro-4-hydroxy-N,1-dimethyl-2-oxo-N-phenyl-3-quinolinecarboxamide
Additional Names: N-phenyl-N-methyl-1,2-dihydro-4-hydroxy-1-methyl-2-oxoquinoline-3-carboxamide; 1,2-dihydro-4-hydroxy-N,1-dimethyl-2-oxo-3-quinolinecarboxanilide
Manufacturers’ Codes: LS-2616
Trademarks: Linomide (Pfizer)
Molecular Formula: C18H16N2O3
Molecular Weight: 308.33
Percent Composition: C 70.12%, H 5.23%, N 9.09%, O 15.57%
Literature References: Biological response modifier. Prepn: E. Eriksoo et al., EP 59698; eidem, US 4738971 (1982, 1988 both to AB Leo). Immunopharmacology: A. Tarkowski et al., Immunology 59, 589 (1986). Mechanism of action study: E.-L. Larsson et al.,Int. J. Immunopharmacol. 9, 425 (1987). Clinical evaluation in cancer patients: J. C. S. Bergh et al., Cancer Invest. 15, 204 (1997).
Properties: Crystals from pyridine, mp 200-204°.
Melting point: mp 200-204°
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Immunomodulators.
|
 |
|
| Clinical data | |
|---|---|
| ATC code | |
| Pharmacokinetic data | |
| Biological half-life | 26-42 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ECHA InfoCard | 100.163.758 |
| Chemical and physical data | |
| Formula | C18H16N2O3 |
| Molar mass | 308.331 g/mol |
| 3D model (JSmol) | |
| |
|
Roquinimex (Linomide) is a quinoline derivative immunostimulant which increases NK cell activity and macrophage cytotoxicity. It also inhibits angiogenesis and reduces the secretion of TNF alpha.
/////////////////Roquinimex, Linomide, FCF-89, LS-2616, PNU-212616
CN1C2=CC=CC=C2C(=O)C(=C1O)C(=O)N(C)C3=CC=CC=C3
The Green ChemisTREE: 20 years after taking root with the 12 principles

The Green ChemisTREE: 20 years after taking root with the 12 principles
View original post 314 more words
