PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Naronapride (free base), also known as ATI-7505, is a highly selective, high-affinity 5-HT(4) receptor agonist for gastrointestinal motility disorders. ATI-7505 accelerates overall colonic transit and tends to accelerate GE and AC emptying and loosen stool consistency.
Investigated for use/treatment in gastroesophageal reflux disease (GERD) and gastroparesis.
Renexxion , presumed to have been spun-out from Armetheon , under license from ARYx Therapeutics is developing naronapride (ATI-7505; phase 2 clinical in February 2020), an analog of the gastroprokinetic 5-HT 4 agonist cisapride identified using ARYx’s RetroMetabolic platform technology (ARM), for the oral treatment of upper GI disorders. In September 2018, this was still the case . PATENT
Process for preparing trihydrate salt of naronapride hydrochloride as 5-HT 4 receptor agonist useful for treating gastrointestinal disorders such as dyspepsia, gastroparesis, constipation, post-operative ileus. Appears to be the first filing from the assignee and the inventors on this compound,
In some aspects, provided herein is a method of making a trihydrate form of (3S, 4R, 3’R)-6-[4-(4-amino-5-chloro-2-methoxy-benzoylamino)-3-methoxy-piperidin-l-yl]-hexanoic acid l-azabicyclo[2.2.2]oct-3’-yl ester di-hydrochloride salt, which has the following formula:
Example 5: NMR Characterization of the Trihydrate
[0282] ^-Nuclear Magnetic Resonance Spectroscopy (‘H-NMR) : Approximately 6 mg of the trihydrate was dissolved in in 1 g of deuterated solvent (dimethylsulfoxide (DMSO)-C45 99.9% d, with 0.05% v/v tetramethyl silane (TMS)). A Varian Gemini 300 MHz FT-NMR spectrometer was used to obtain the ¾-NMK spectrum. A list of the peaks is provided in Table 1 below. A representative ‘H-NMR spectrum is provided in FIG. 6.
Table 1. ‘H-NMR peak list for trihydrate
[0283] 13 C-Nuclear Magnetic Resonance Spectroscopy ( 13C-NMR ): Approximately 46 mg of the trihydrate was dissolved in 1 mL of deuterated solvent (deuterium oxide, Aldrich, 99.9% D, TPAS 0.75%). The 13C-NMR spectrum was obtained using a Varian Gemini 300 MHz FT-NMR spectrometer. A list of the peaks is provided in Table 2 below. A representative 13C-NMR spectrum is provided in FIG. 7.
Table 2. 13C-NMR peak list for trihydrate
PATENT
US10570127 claiming composition (eg tablet) comprising a trihydrate form of naronapride.
patent
ARYX THERAPEUTICS, WO2005/68461, A1, (2005)
Methods
titanium tetraethoxide; toluene;
Reactants can be synthesized in 1 step.
ARYX THERAPEUTICS, WO2005/68461, A1, (2005) The ester (1 part by weight) and (R)-3-Quinuclidinol (about 1.12 part by weight) were suspended in toluene before slowly adding titanium (IV) ethoxide (about 0.5 part by weight) to the stirred suspens ion. The mixture was heated to about 91 °C under a stream of nitrogen, and partial vacuum was applie d to the flask through a distillation apparatus in order to azeotropically remove the ethanol. Addit ional toluene was added as needed to maintain a minimum solvent volume in the flask. The reaction was considered complete after about 33 hours. The mixture was cooled to about room temperature and ext racted five times with water. The organic layer was concentrated under reduced pressure and the resulting residue was redissolved in EtOH/iPrOH (about 1: 1 v/v) and then filtered through a 0.45 micron membrane filter to remove any particulates. Concentrated hydrochloric acid was added slowly to the stirred filtrate to precipitate out the desired product as the dihydrochloride salt. The resulting s uspension was stirred for several hours at room temperature and collected under vacuum filtration and rinsed with EtOH/tPrOH (1: 1; v/v) to provide 0.53 part by weight of the crude product salt. Crude dihydrochloride salt was resuspended in ethanol and heated to reflux before cooling to room temperature over about 1 hour. The product was collected under vacuum filtration and rinsed with ethanol an d then air-dried. The solids were resuspended in ethanol and warmed to about 55 °C to give a clear s olution before adding warm isopropanol and the product was allowed to precipitate by slow cooling to room temperature. The resulting suspension was stirred for several hours before vacuum filtering and rinsing with, e. g., isopropanol. The product was vacuum dried, initially at room temperature for several hours and then at about 55 °C until a constant weight was achieved.
Reactants can be synthesized in 2 steps.
ARYX THERAPEUTICS, WO2007/28073, A2, (2007) Production of Compound IV and Compound VI[0394] A mixture of (+)-Comrhoound II (1 eq.), (R)-(-)-3-quinuclidinol HCl salt (1 eq.), EDAC (1 eq.) and DMAP (1 eq.) in DMF is heated at around 5OC overnight . After cooling and diluting with water, the mixture is purified by chromatography or by crystallization to provide Compound IV. Similarly, using (S)-(+)-quinuclidinol, Compound VI is obtained
REFERENCES
1: Jiang C, Xu Q, Wen X, Sun H. Current developments in pharmacological therapeutics for chronic constipation. Acta Pharm Sin B. 2015 Jul;5(4):300-9. doi: 10.1016/j.apsb.2015.05.006. Epub 2015 Jun 6. Review. PubMed PMID: 26579459; PubMed Central PMCID: PMC4629408.
2: Buchwald P, Bodor N. Recent advances in the design and development of soft drugs. Pharmazie. 2014 Jun;69(6):403-13. Review. PubMed PMID: 24974571.
3: Mozaffari S, Didari T, Nikfar S, Abdollahi M. Phase II drugs under clinical investigation for the treatment of chronic constipation. Expert Opin Investig Drugs. 2014 Nov;23(11):1485-97. doi: 10.1517/13543784.2014.932770. Epub 2014 Jun 24. Review. PubMed PMID: 24960333.
4: Shin A, Camilleri M, Kolar G, Erwin P, West CP, Murad MH. Systematic review with meta-analysis: highly selective 5-HT4 agonists (prucalopride, velusetrag or naronapride) in chronic constipation. Aliment Pharmacol Ther. 2014 Feb;39(3):239-53. doi: 10.1111/apt.12571. Epub 2013 Dec 5. Review. PubMed PMID: 24308797.
5: Stevens JE, Jones KL, Rayner CK, Horowitz M. Pathophysiology and pharmacotherapy of gastroparesis: current and future perspectives. Expert Opin Pharmacother. 2013 Jun;14(9):1171-86. doi: 10.1517/14656566.2013.795948. Epub 2013 May 11. Review. PubMed PMID: 23663133.
6: Tack J, Camilleri M, Chang L, Chey WD, Galligan JJ, Lacy BE, Müller-Lissner S, Quigley EM, Schuurkes J, De Maeyer JH, Stanghellini V. Systematic review: cardiovascular safety profile of 5-HT(4) agonists developed for gastrointestinal disorders. Aliment Pharmacol Ther. 2012 Apr;35(7):745-67. doi: 10.1111/j.1365-2036.2012.05011.x. Epub 2012 Feb 22. Review. PubMed PMID: 22356640; PubMed Central PMCID: PMC3491670.
7: Hoffman JM, Tyler K, MacEachern SJ, Balemba OB, Johnson AC, Brooks EM, Zhao H, Swain GM, Moses PL, Galligan JJ, Sharkey KA, Greenwood-Van Meerveld B, Mawe GM. Activation of colonic mucosal 5-HT(4) receptors accelerates propulsive motility and inhibits visceral hypersensitivity. Gastroenterology. 2012 Apr;142(4):844-854.e4. doi: 10.1053/j.gastro.2011.12.041. Epub 2012 Jan 4. PubMed PMID: 22226658; PubMed Central PMCID: PMC3477545.
8: Bowersox SS, Lightning LK, Rao S, Palme M, Ellis D, Coleman R, Davies AM, Kumaraswamy P, Druzgala P. Metabolism and pharmacokinetics of naronapride (ATI-7505), a serotonin 5-HT(4) receptor agonist for gastrointestinal motility disorders. Drug Metab Dispos. 2011 Jul;39(7):1170-80. doi: 10.1124/dmd.110.037564. Epub 2011 Mar 29. PubMed PMID: 21447732.
9: Tack J. Current and future therapies for chronic constipation. Best Pract Res Clin Gastroenterol. 2011 Feb;25(1):151-8. doi: 10.1016/j.bpg.2011.01.005. Review. PubMed PMID: 21382586.
10: Manabe N, Wong BS, Camilleri M. New-generation 5-HT4 receptor agonists: potential for treatment of gastrointestinal motility disorders. Expert Opin Investig Drugs. 2010 Jun;19(6):765-75. doi: 10.1517/13543784.2010.482927. Review. PubMed PMID: 20408739.
12: Vakil N. New pharmacological agents for the treatment of gastroesophageal reflux disease. Rev Gastroenterol Disord. 2008 Spring;8(2):117-22. Review. PubMed PMID: 18641594.
13: Bayés M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2007 Jun;29(5):359-73. PubMed PMID: 17805439.
14: Camilleri M, Vazquez-Roque MI, Burton D, Ford T, McKinzie S, Zinsmeister AR, Druzgala P. Pharmacodynamic effects of a novel prokinetic 5-HT receptor agonist, ATI-7505, in humans. Neurogastroenterol Motil. 2007 Jan;19(1):30-8. PubMed PMID: 17187586.
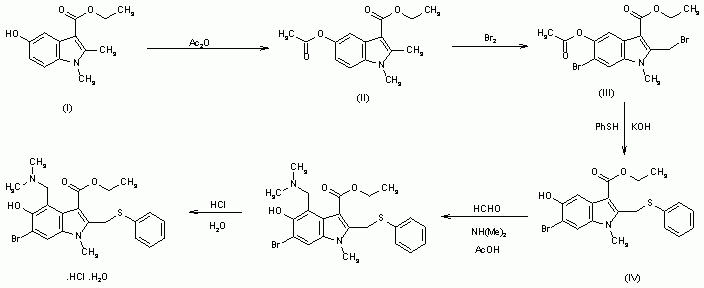
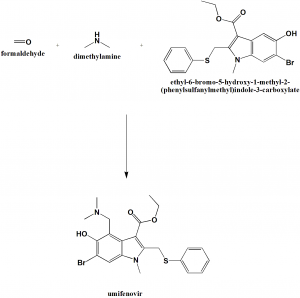
////////////NARONAPRIDE, ATI 7505, ATI 7505,PHASE 2
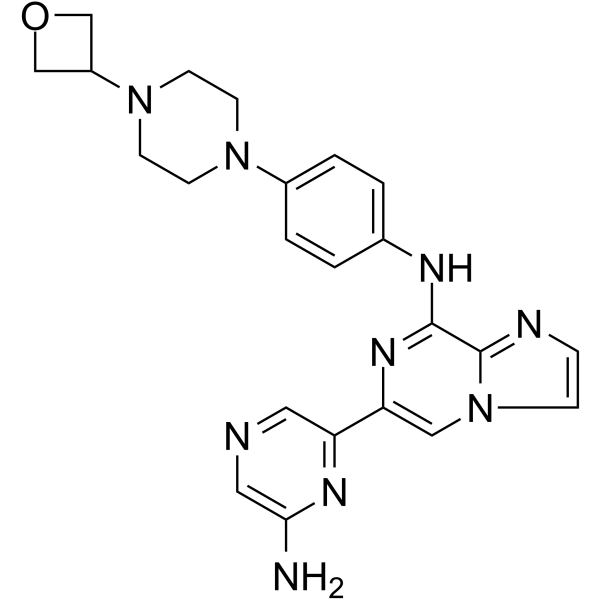
Aurinia Pharmaceuticals (following its merger with Isotechnika ), in collaboration with licensee Paladin Labs (a subsidiary of Endo International plc ), 3SBio ,and ILJIN , is developing a capsule formulation of the immunosuppressant calcineurin inhibitor peptide voclosporin for the treatment of psoriasis, the prevention of organ rejection after transplantation, autoimmune disease including systemic lupus erythematosus and lupus nephritis, and nephrotic syndrome including focal segmental glomerulosclerosis;
Voclosporin is an experimental immunosuppressant drug being developed by Aurinia Pharmaceuticals. It is being studied as a potential treatment for lupus nephritis (LN) and uveitis.[1] It is an analog of ciclosporin that has enhanced action against calcineurin and greater metabolic stability.[2] Voclosporin was discovered by Robert T. Foster and his team at Isotechnika in the mid 1990s.[3] Isotechnika was founded in 1993 and merged with Aurinia Pharmaceuticals in 2013.
Initially, voclosporin was a mixture of equal proporations of cis and trans geometric isomers of amino acid-1 modified cyclosporin. Later, in collaboration with Roche in Basel, Switzerland, voclosporin’s manufacturing was changed to yield the predominantly trans isomer which possesses most of the beneficial effect of the drug (immunosuppression) in the treatment of organ transplantation and autoimmune diseases.
Novel crystalline forms of voclosporin which is a structural analog of cyclosporine A as calcineurin signal-transduction pathway inhibitor useful for treating lupus nephritis.
Voclosporin is a structural analog of cyclosporine A, with an additional single carbon extension that has a double-bond on one side chain. Voclosporin has the chemical name (3S,6S,9S,l2R,l5S,l8S,2lS,24S,30S,33S)-30-Ethyl-33-[(lR,2R,4E)-l-hydroxy-2-methyl-4,6-heptadien-l-yl]-6,9,l8,24-tetraisobutyl-3,2l-diisopropyl-l,4,7,l0,l2,l5,l9,25,28-nonamethyl-l,4,7,l0,l3,l6,l9,22,25,28,3 l-undecaazacyclotritriacontane-2,5,8,l l,l4,l7,20,23,26,29,32-undecone and the following chemical structure:
Voclosporin is reported to be a semisynthetic structural analogue of cyclosporine that exerts its immunosuppressant effects by inhibition of the calcineurin signal-transduction pathway and is in Phase 3 Clinical Development for Lupus Nephritis.
[0003] Voclosporin and process for preparation thereof are known from International Patent Application No. WO 1999/18120.
[0004] Certain mixtures of cis and trans-isomers of cyclosporin A analogs referred to as
ISATX247 in different ratios are known from U.S. Patent No. 6,998,385, U.S. Patent No. 7,332,472 and U.S. Patent No. 9,765,119.
[0005] Polymorphism, the occurrence of different crystal forms, is a property of some molecules and molecular complexes. A single compound, like Voclosporin, may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – “TGA”, or differential scanning calorimetry – “DSC”), powder X-ray diffraction (PXRD) pattern, infrared absorption fingerprint, Raman absorption fingerprint, and solid state (13C-) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
[0006] Different salts and solid state forms (including solvated forms) of an active
pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, improving the dissolution profile, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also provide improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to use variations in the properties and characteristics of a solid active pharmaceutical ingredient for providing an improved product.
[0007] Discovering new salts, solid state forms and solvates of a pharmaceutical product can provide materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other salts or polymorphic forms. New salts, polymorphic forms and solvates of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product (dissolution profile, bioavailability, etc.). It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, e.g., a different crystal habit, higher crystallinity or polymorphic stability which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life.
[0008] For at least these reasons, there is a need for solid state forms (including solvated forms) of Voclosporin and salts thereof.
HPLC method:
Method description
Column: Zorbax SB C18, 1.8 pm, 100×2.1 mm
Mobile phase: A: 38 ACN : 7 TBME : 55 voda : 0.02 H3P04 (V/V/V/V)
B: 70 ACN : 7 TBME : 23 voda : 0.02 H P04 (V/V/V/V)
[0095] The starting material Voclosporin crude may be obtained according to ET.S. Patent No. 6,998,385 ETnless otherwise indicated, the purity is determined by HPLC (area percent). The crude product contained according to HPLC analysis 42.6 % trans-Voclosporin (further only Voclosporin), 40.2 % cis-Voclosporin and 2.9 % Cyclosporin A. The crude Voclosporin was purified by column chromatography on silica gel using a mixture of toluene and acetone 82 : 18 (v/v) as mobile phase. The fractions were monitored by HPLC. The appropriate fractions were joined and evaporated, obtaining purified Voclosporin as a white foam. According to HPLC analysis it contained 85.7 % Voclosporin, 3.6 % cis-Voclosporin and 2.6 % Cyclosporin A (further only purified Voclosporin).
[0096] The Voclosporin crude (containing about 42.6 % of Voclosporin) was used for further optimization of the chromatographic separation of cis-Voclosporin and Voclosporin and the effort resulted in improved process for chromatographic separation which includes purification by column chromatography on silica gel using a mixture of toluene and methylisobutylketone 38 : 62 as mobile phase. The fractions were monitored by HPLC. The appropriate fractions were joined and evaporated to a dry residue, weighing 31.0 grams. This residue was not analyzed. The material was dissolved in 25 ml of acetone and then 50 ml of water was added and the solution was let to crystallize for 2 hours in the refrigerator. Then the crystalline product was separated by filtration and dried in vacuum dryer (40 °C, 50 mbar, 12 hours), obtaining 29.6 g of dry product containing 90.6 % of Voclosporin, 0.4 % cis-Voclosporin and 3.7 % Cyclosporin A (further mentioned as final Voclosporin).
Example 1: Preparation of Voclosporin Form A
4.1 grams of Purified Voclosporin was dissolved in acetone and the solution was evaporated to 8.0 grams and the concentrate was diluted by 6 ml of water. The solution was let to crystallize in refrigerator at about 2 °C for 12 hours. The crystalline product was filtered off, washed by a mixture of acetone and water 1 : 1 (v/v) and dried on open air obtaining 2.6 grams of crystalline product Form A. Voclosporin form A was confirmed by PXRD as presented in Figure 1.
Example 2: Preparation of Voclosporin Form B
[0097] 1.0 gram of Purified Voclosporin was dissolved in a mixture of 1.5 ml acetone and 3.0 ml n-hexane. The solution was let to crystallize in refrigerator at about 2 °C for 12 hours. The crystalline product was filtered off, washed by a mixture of acetone and hexane 1 : 2 (v/v) and dried on open air obtaining 0.5 grams of crystalline product Form B. Voclosporin form B was confirmed by PXRD as presented in Figure 2.
Example 3: Preparation of Amorphous Voclosporin
[0098] 2.0 grams of Purified Voclosporin was dissolved in 40 ml of hot cyclohexane and the solution was stirred for 12 hours at room temperature. Then the crystalline product was filtered off and washed with 5 ml of cyclohexane and dried on open air, obtaining 1.3 grams of amorphous powder. Amorphous Voclosporin was confirmed by PXRD as presented in Figure 3
Example 4: Preparation of Voclosporin Form C
[0099] Final Voclosporin (2 grams) was dissolved in acetonitrile (20 ml) at 50 °C, water (6 ml) was added with stirring, and the clear solution was allowed to crystallize 5 days at 20 °C. Colorless needle crystals were directly mounted to the goniometer head in order to define the crystal structure. Voclosporin form C was confirmed by X-ray crystal structure determination.
References
^“Luveniq Approval Status”. Luveniq (voclosporin) is a next-generation calcineurin inhibitor intended for the treatment of noninfectious uveitis involving the intermediate or posterior segments of the eye.
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Synthesis
methanol; potassium carbonate;
Reactants can be synthesized in 7 steps.
Synthesis, vol. 44, 1, (2012), p. 63 – 68
Yield:60%
SYN 2
sulfuric acid; tetrahydrofuran;
ISOTECHNIKA INC., WO2004/89960, A2, (2004) 20 ml of THF were added and the reaction mixture was cooled to 0 °C. 2.7 ML (48.69 mmol, 3 equiv. ) of concentrated sulfuric acid were added. The temperature was raised to RT. After completion of the reaction (ca 1 hour), 100 ml of water were added. The organic phase was separated and washed 2 times with 50 ml water. The water phases were re-extracted sequentially with 50 ml dichloromethane. The c ombined organic phases were dried over NA2SO4, filtered and concentrated under reduced pressure at 3 0°C. The resulting white foam was re-dissolved in 250 ml MTBE and after a few minutes, the crystalli zation started. After stirring 15 min. at RT and 2 hours at 0-2 C, THE SUSPENSION WAS FILTERED. THE crystals were washed with 50 ml cold MTBE (-20 °C) and dried at 40-50 °C under reduced pressure to p rovide 19.2 g of (E) -acetyl-ISA247 as white powder in >98percent isomeric purity (400MHZ LH NMR). (E)-ACETYL-ISA247 can be RECRYSTALLIZED by dissolving the solid in dichloromethane at room temperatur e and exchanging the solvent to MTBE (by adding MTBE, concentrating the solution to half its volume under reduced pressure at 40°C and repeating these operation 2 to three times). The solution is cool ed to room temperature and the crystallization then starts within a few minutes. The suspension is s tirred at room temperature for 2h and 30min at 0°C. The crystals of (E) -acetyl-ISA247 are isolated after filtration, washing with MTBE and drying under reduced pressure at 40°C.iii) Peterson eliminat ion The CRUDE-TRIMETHYLSILYALCOHOL diastereomers mixture (11 g, maximum 4.056 mmol) was dissolved in 25 ml THF. 0.679 ml (12.16 mmol, 3 equiv.) concentrated sulfuric were added dropwise maintaining th e temperature between 20 °C and 25 °C. After 2 hours at RT, 50 ml half saturated aqueous NaCl soluti on were added. The resulting mixture was extracted twice with 50 ML MTBE. The organic phases were washed with 50ML of a half saturated aqueous NACL solution, combined, dried over NA2SO4 and concentrat ed under reduce pressure at 40°C. The resulting crude E-acetyl-ISA247 was re-dissolved in 20 ml dich loromethane and concentrated under reduced pressure. The crude product was dissolved in 60 ml MTBE. The crystallization started within 10 min. The suspension was stirred for an additional 15 min. at R T and 2 hours AT-10 °C. The crystals were isolated by filtration, washed with 20 ml cold MTBE (-20 ° C) and dried under reduced pressure to provide 3. 6 G of (E)-ACETYL-ISA247 in ca 98percent isomeric purity by NMR.iii) Peterson elimination After overnight reaction, the organic layer was separated an d the water phase was discarded. 50 ML THF were added to the organic phase. The solution was concent rated under reduced pressure at 30 °C to half its volume. 100 ML THP were added and the solution was concentrated to 80 ML. The volume was adjusted to 100 ml with THF and the solution was cooled to 0- 2 °C. 1. 812 ML (32. 46 MMOL, 2 equiv.) concentrated sulfuric acid were added dropwise over 5 min., maintaining the temperature below 5 °C. After addition, the reaction cooling bath was removed and th e temperature was raised to RT. After 4 hours reaction, 40 ML water were added followed by 20 ml MTB E. The aqueous layer was separated and discarded. The organic phase was washed with 40 ml NAHCO3 Q, 20 ML saturated NACLAQ, 40 ml saturated NaClaq, dried over Na2SO4, filtered and concentrated at 40 ° C under reduced pressure. The crude E-acetyl-ISA247 was RE-DISSOLVED in 200 ml MTBE and crystallizat ion started within a few minutes. After 15 min. at RT and 2.5 hours at 0 °C, the suspension was filt ered, the crystals were washed with 50 ML MTBE and dried at 50 °C under reduced pressure to give 18. 45 g of (E) -acetyl-ISA247 as a white powder (>98percent isomeric purity by NMR).iii) Peterson elim ination 5 ml THF were added to the organic phase and the solution was cooled to 0- 2 °C. 181 UL (3.2 46, 2 equiv. ) concentrated sulfuric acid were added. The reaction mixture was warmed up to RT. Afte r stirring overnight, 20 ml water were added. The aqueous layer was separated and discarded. The organic phase was washed with 20 ml of 5percent aqueous NAHCO3 solution, dried over MGS04, filtered and concentrated under reduced pressure at 40 °C to give 2 g of (E) -acetyl-ISA247 as a white foam in > 98percent double bond isomeric purity (by NMR).ii) Peterson elimination The crude product was dissol ved in 11.15 ML THF and 268 P1 concentrated sulfuric acid were added. The reaction mixture was heate d at 33 °C for 1.5 hour and then cooled to RT. 22 ml water were added and the reaction mixture was e xtracted with 22 ml MTBE. The aqueous phase was RE-EXTRACTED with 11 ml MTBE. The organic layer were washed with 11 ml water, combined, dried over NA2SO4, filtered and concentrated at 40 °C under redu ced pressure to give 1.89 g of crude (E) -acetyl-ISA247 as a beige powder. The crude product was re-dissolved in 20 ml MTBE at RT. The crystallization started within a few minutes. The suspension was stirred 30 min. at RT, 45 min. at-10 °C and was filtered. The solid was washed with cold MTBE and dr ied at 40 °C under reduced pressure to give 1.02 g of (E)-acetylISA247 as a white powder in ca 98per cent double bond isomeric purity (NMR). ii) Peterson elimination The crude product was dissolved in 8 ML THF at RT. The solution was cooled to 0-5 °C and 200 UL of concentrated sulfuric acid were adde d dropwise. The temperature was raised to RT and the reaction mixture was stirred 10 hours. 40 ml MTBE and 15 ml of water were added. The water phase was separated and discarded. The organic phase was washed 15 ml of a 5percent aqueous NAHCO3 solution, 15 ml of a half saturated aqueous NACL solution, dried over NA2SO4, filtered and concentrated under reduced pressure to give 1. 8 g of crude E-acet yl- ISA247. The crude diene was dissolved in 20 ml dichloromethane. 20 ML MTBE were added, and the s olution was concentrated at 40 °C under reduced pressure to half its volume. The last two operations was repeated three times to in order to exchange the solvent from dichloromethane to MTBE. The solution was cooled to RT and the crystallization started within a few minutes. The suspension was stirr ed 2 hours at RT and 30 min. at 0 °C. The suspension was filtered. The solid was washed with 15 ml M TBE and dried under reduced pressure at 40 °C to give 1.1 g OF E-ACETYL-ISA247 in >95percent double bond isomeric purity (NMR), as a white powder.ii) Peterson elimination The crude product was dissolv ed in 10 ml THF at RT. The solution was cooled to 0-5 °C and 200 UL of concentrated sulfuric acid we re added dropwise. The temperature was raised to RT and the reaction mixture was stirred overnight. 40 ml MTBE and 15 ML of water were added. The water phase was separated and discarded. The organic p hase was washed with 15 ml water, 15 ml of a 5percent aqueous NAHCO3 solution, 15 ml of a half saturated aqueous NaCl solution, filtered and concentrated under reduced pressure to give 1.8 g of crude E-ACETYL-ISA247. The crude diene was redissolved in 35 ml of MTBE. The crystallization started withi n a few minutes. The suspension was stirred 2 hours at RT and 30 min. at 0 °C. The suspension was fi ltered. The solid was washed with 15 ml MTBE and dried under reduced pressure at 40 °C to gi ve 1 g of E-acetyl-ISA247 in >95percent double bond isomeric purity (NMR), as a white powder.
REFERENCES
1: Mok CC. Calcineurin inhibitors in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2017 Jun;31(3):429-438. doi: 10.1016/j.berh.2017.09.010. Epub 2017 Oct 11. Review. PubMed PMID: 29224682.
2: Dang W, Yin Y, Wang Y, Wang W, Su J, Sprengers D, van der Laan LJW, Felczak K, Pankiewicz KW, Chang KO, Koopmans MPG, Metselaar HJ, Peppelenbosch MP, Pan Q. Inhibition of Calcineurin or IMP Dehydrogenase Exerts Moderate to Potent Antiviral Activity against Norovirus Replication. Antimicrob Agents Chemother. 2017 Oct 24;61(11). pii: e01095-17. doi: 10.1128/AAC.01095-17. Print 2017 Nov. PubMed PMID: 28807916; PubMed Central PMCID: PMC5655111.
3: Wong TC, Lo CM, Fung JY. Emerging drugs for prevention of T-cell mediated rejection in liver and kidney transplantation. Expert Opin Emerg Drugs. 2017 Jun;22(2):123-136. doi: 10.1080/14728214.2017.1330884. Epub 2017 May 22. Review. PubMed PMID: 28503959.
4: Chow C, Simpson MJ, Luger TA, Chubb H, Ellis CN. Comparison of three methods for measuring psoriasis severity in clinical studies (Part 1 of 2): change during therapy in Psoriasis Area and Severity Index, Static Physician’s Global Assessment and Lattice System Physician’s Global Assessment. J Eur Acad Dermatol Venereol. 2015 Jul;29(7):1406-14. doi: 10.1111/jdv.13132. Epub 2015 Apr 27. PubMed PMID: 25917315.
5: Simpson MJ, Chow C, Morgenstern H, Luger TA, Ellis CN. Comparison of three methods for measuring psoriasis severity in clinical studies (Part 2 of 2): use of quality of life to assess construct validity of the Lattice System Physician’s Global Assessment, Psoriasis Area and Severity Index and Static Physician’s Global Assessment. J Eur Acad Dermatol Venereol. 2015 Jul;29(7):1415-20. doi: 10.1111/jdv.12861. Epub 2015 Apr 27. PubMed PMID: 25917214.
6: Maya JR, Sadiq MA, Zapata LJ, Hanout M, Sarwar S, Rajagopalan N, Guinn KE, Sepah YJ, Nguyen QD. Emerging therapies for noninfectious uveitis: what may be coming to the clinics. J Ophthalmol. 2014;2014:310329. doi: 10.1155/2014/310329. Epub 2014 Apr 24. Review. PubMed PMID: 24868451; PubMed Central PMCID: PMC4020293.
7: Hardinger KL, Brennan DC. Novel immunosuppressive agents in kidney transplantation. World J Transplant. 2013 Dec 24;3(4):68-77. doi: 10.5500/wjt.v3.i4.68. Review. PubMed PMID: 24392311; PubMed Central PMCID: PMC3879526.
8: Ling SY, Huizinga RB, Mayo PR, Larouche R, Freitag DG, Aspeslet LJ, Foster RT. Cytochrome P450 3A and P-glycoprotein drug-drug interactions with voclosporin. Br J Clin Pharmacol. 2014 Jun;77(6):1039-50. doi: 10.1111/bcp.12309. PubMed PMID: 24330024; PubMed Central PMCID: PMC4093929.
9: Mayo PR, Ling SY, Huizinga RB, Freitag DG, Aspeslet LJ, Foster RT. Population PKPD of voclosporin in renal allograft patients. J Clin Pharmacol. 2014 May;54(5):537-45. doi: 10.1002/jcph.237. Epub 2013 Nov 30. PubMed PMID: 24243422.
10: Gubskaya AV, Khan IJ, Valenzuela LM, Lisnyak YV, Kohn J. Investigating the Release of a Hydrophobic Peptide from Matrices of Biodegradable Polymers: An Integrated Method Approach. Polymer (Guildf). 2013 Jul 8;54(15):3806-3820. PubMed PMID: 24039300; PubMed Central PMCID: PMC3770487.
11: Ling SY, Huizinga RB, Mayo PR, Freitag DG, Aspeslet LJ, Foster RT. Pharmacokinetics of voclosporin in renal impairment and hepatic impairment. J Clin Pharmacol. 2013 Dec;53(12):1303-12. doi: 10.1002/jcph.166. Epub 2013 Oct 8. PubMed PMID: 23996158.
12: Mayo PR, Huizinga RB, Ling SY, Freitag DG, Aspeslet LJ, Foster RT. Voclosporin food effect and single oral ascending dose pharmacokinetic and pharmacodynamic studies in healthy human subjects. J Clin Pharmacol. 2013 Aug;53(8):819-26. doi: 10.1002/jcph.114. Epub 2013 Jun 4. PubMed PMID: 23736966.
13: Schultz C. Voclosporin as a treatment for noninfectious uveitis. Ophthalmol Eye Dis. 2013 May 5;5:5-10. doi: 10.4137/OED.S7995. Print 2013. PubMed PMID: 23700374; PubMed Central PMCID: PMC3653814.
14: Gomes Bittencourt M, Sepah YJ, Do DV, Agbedia O, Akhtar A, Liu H, Akhlaq A, Annam R, Ibrahim M, Nguyen QD. New treatment options for noninfectious uveitis. Dev Ophthalmol. 2012;51:134-61. doi: 10.1159/000336338. Epub 2012 Apr 17. Review. PubMed PMID: 22517211.
15: Khan IJ, Murthy NS, Kohn J. Hydration-induced phase separation in amphiphilic polymer matrices and its influence on voclosporin release. J Funct Biomater. 2012 Oct 30;3(4):745-59. doi: 10.3390/jfb3040745. PubMed PMID: 24955746; PubMed Central PMCID: PMC4030927.
16: Roesel M, Tappeiner C, Heiligenhaus A, Heinz C. Oral voclosporin: novel calcineurin inhibitor for treatment of noninfectious uveitis. Clin Ophthalmol. 2011;5:1309-13. doi: 10.2147/OPTH.S11125. Epub 2011 Sep 13. PubMed PMID: 21966207; PubMed Central PMCID: PMC3180504.
17: Busque S, Cantarovich M, Mulgaonkar S, Gaston R, Gaber AO, Mayo PR, Ling S, Huizinga RB, Meier-Kriesche HU; PROMISE Investigators. The PROMISE study: a phase 2b multicenter study of voclosporin (ISA247) versus tacrolimus in de novo kidney transplantation. Am J Transplant. 2011 Dec;11(12):2675-84. doi: 10.1111/j.1600-6143.2011.03763.x. Epub 2011 Sep 22. PubMed PMID: 21943027.
18: Kuglstatter A, Mueller F, Kusznir E, Gsell B, Stihle M, Thoma R, Benz J, Aspeslet L, Freitag D, Hennig M. Structural basis for the cyclophilin A binding affinity and immunosuppressive potency of E-ISA247 (voclosporin). Acta Crystallogr D Biol Crystallogr. 2011 Feb;67(Pt 2):119-23. doi: 10.1107/S0907444910051905. Epub 2011 Jan 15. PubMed PMID: 21245533; PubMed Central PMCID: PMC3045272.
19: Kunynetz R, Carey W, Thomas R, Toth D, Trafford T, Vender R. Quality of life in plaque psoriasis patients treated with voclosporin: a Canadian phase III, randomized, multicenter, double-blind, placebo-controlled study. Eur J Dermatol. 2011 Jan-Feb;21(1):89-94. doi: 10.1684/ejd.2010.1185. PubMed PMID: 21227890.
20: Deuter CM. [Systemic voclosporin for uveitis treatment]. Ophthalmologe. 2010 Jul;107(7):672-5. doi: 10.1007/s00347-010-2217-5. German. PubMed PMID: 20571806.
CAS Name: (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-Dideoxy-3-C-methyl-3-O-methyl-a-L-ribo-hexopyranosyl)oxy]-2-ethyl-3,4,10-trihydroxy-3,5,6,8,10,12,14-heptamethyl-11-[[3,4,6-trideoxy-3-(dimethylamino)-b-D-xylo-hexopyranosyl]oxy]-1-oxa-6-azacyclopentadecan-15-one
Additional Names:N-methyl-11-aza-10-deoxo-10-dihydroerythromycin A; 9-deoxo-9a-methyl-9a-aza-9a-homoerythromycin A
Molecular Formula: C38H72N2O12
Molecular Weight: 748.98
Percent Composition: C 60.94%, H 9.69%, N 3.74%, O 25.63%
Literature References: Semi-synthetic macrolide antibiotic; related to erythromycin A, q.v. Prepn: BE892357; G. Kobrehel, S. Djokic, US4517359 (1982, 1985 both to Sour Pliva); of the crystalline dihydrate: D. J. M. Allen, K. M. Nepveux, EP298650; eidem, US6268489 (1989, 2001 both to Pfizer). Antibacterial spectrum: S. C. Aronoff et al.,J. Antimicrob. Chemother.19, 275 (1987); and mode of action: J. Retsema et al.,Antimicrob. Agents Chemother.31, 1939 (1987). Series of articles on pharmacology, pharmacokinetics, and clinical experience: J. Antimicrob. Chemother.31, Suppl. E, 1-198 (1993). Clinical trial in prevention of Pneumocystis carinii pneumonia in AIDS patients: M. W. Dunne et al.,Lancet354, 891 (1999). Review of pharmacology and clinical efficacy in pediatric infections: H. D. Langtry, J. A. Balfour, Drugs56, 273-297 (1998).
Azithromycin was discovered 1980 by Pliva, and approved for medical use in 1988.[5][6] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[7] The World Health Organization classifies it as critically important for human medicine.[8] It is available as a generic medication[9] and is sold under many trade names worldwide.[2] The wholesale cost in the developing world is about US$0.18 to US$2.98 per dose.[10] In the United States, it is about US$4 for a course of treatment as of 2018.[11] In 2016, it was the 49th most prescribed medication in the United States with more than 15 million prescriptions.[12]
Medical uses
Azithromycin is used to treat many different infections, including:
Prevention and treatment of acute bacterial exacerbations of chronic obstructive pulmonary disease due to H. influenzae, M. catarrhalis, or S. pneumoniae. The benefits of long-term prophylaxis must be weighed on a patient-by-patient basis against the risk of cardiovascular and other adverse effects.[13]
Community-acquired pneumonia due to C. pneumoniae, H. influenzae, M. pneumoniae, or S. pneumoniae[14]
Uncomplicated skin infections due to S. aureus, S. pyogenes, or S. agalactiae
Urethritis and cervicitis due to C. trachomatis or N. gonorrhoeae. In combination with ceftriaxone, azithromycin is part of the United States Centers for Disease Control-recommended regimen for the treatment of gonorrhea. Azithromycin is active as monotherapy in most cases, but the combination with ceftriaxone is recommended based on the relatively low barrier to resistance development in gonococci and due to frequent co-infection with C. trachomatis and N. gonorrhoeae.[15]
Genital ulcer disease (chancroid) in men due to H. ducrey
Acute bacterial sinusitis due to H. influenzae, M. catarrhalis, or S. pneumoniae. Other agents, such as amoxicillin/clavulanate are generally preferred, however.[17][18]
Acute otitis media caused by H. influenzae, M. catarrhalis or S. pneumoniae. Azithromycin is not, however, a first-line agent for this condition. Amoxicillin or another beta lactam antibiotic is generally preferred.[19]
Pharyngitis or tonsillitis caused by S. pyogenes as an alternative to first-line therapy in individuals who cannot use first-line therapy[20]
Bacterial susceptibility
Azithromycin has relatively broad but shallow antibacterial activity. It inhibits some Gram-positive bacteria, some Gram-negative bacteria, and many atypical bacteria.
A strain of gonorrhea reported to be highly resistant to azithromycin was found in the population in 2015. Neisseria gonorrhoeae is normally susceptible to azithromycin,[21] but the drug is not widely used as monotherapy due to a low barrier to resistance development.[15] Extensive use of azithromycin has resulted in growing Streptococcus pneumoniae resistance.[22]
Aerobic and facultative Gram-positive microorganisms
No harm has been found with use during pregnancy.[3] However, there are no adequate well-controlled studies in pregnant women.[23]
Safety of the medication during breastfeeding is unclear. It was reported that because only low levels are found in breast milk and the medication has also been used in young children, it is unlikely that breastfed infants would suffer adverse effects.[4] Nevertheless, it is recommended that the drug be used with caution during breastfeeding.[3]
Airway diseases
Azithromycin appears to be effective in the treatment of COPD through its suppression of inflammatory processes.[24] And potentially useful in asthma and sinusitis via this mechanism.[25] Azithromycin is believed to produce its effects through suppressing certain immune responses that may contribute to inflammation of the airways.[26][27]
Adverse effects
Most common adverse effects are diarrhea (5%), nausea (3%), abdominal pain (3%), and vomiting. Fewer than 1% of people stop taking the drug due to side effects. Nervousness, skin reactions, and anaphylaxis have been reported.[28]Clostridium difficile infection has been reported with use of azithromycin.[3] Azithromycin does not affect the efficacy of birth control unlike some other antibiotics such as rifampin. Hearing loss has been reported.[29]
Occasionally, people have developed cholestatic hepatitis or delirium. Accidental intravenous overdose in an infant caused severe heart block, resulting in residual encephalopathy.[30][31]
In 2013 the FDA issued a warning that azithromycin “can cause abnormal changes in the electrical activity of the heart that may lead to a potentially fatal irregular heart rhythm.” The FDA noted in the warning a 2012 study that found the drug may increase the risk of death, especially in those with heart problems, compared with those on other antibiotics such as amoxicillin or no antibiotic. The warning indicated people with preexisting conditions are at particular risk, such as those with QT interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or those who use certain drugs to treat abnormal heart rhythms.[32][33][34]
Pharmacology
Mechanism of action
Azithromycin prevents bacteria from growing by interfering with their protein synthesis. It binds to the 50S subunit of the bacterial ribosome, thus inhibiting translation of mRNA. Nucleic acid synthesis is not affected.[23]
Pharmacokinetics
Azithromycin is an acid-stable antibiotic, so it can be taken orally with no need of protection from gastric acids. It is readily absorbed, but absorption is greater on an empty stomach. Time to peak concentration (Tmax) in adults is 2.1 to 3.2 hours for oral dosage forms. Due to its high concentration in phagocytes, azithromycin is actively transported to the site of infection. During active phagocytosis, large concentrations are released. The concentration of azithromycin in the tissues can be over 50 times higher than in plasma due to ion trapping and its high lipid solubility.[citation needed] Azithromycin’s half-life allows a large single dose to be administered and yet maintain bacteriostatic levels in the infected tissue for several days.[35]
Following a single dose of 500 mg, the apparent terminal elimination half-life of azithromycin is 68 hours.[35]Biliary excretion of azithromycin, predominantly unchanged, is a major route of elimination. Over the course of a week, about 6% of the administered dose appears as unchanged drug in urine.
History
A team of researchers at the pharmaceutical company Pliva in Zagreb, SR Croatia, Yugoslavia, — Gabrijela Kobrehel, Gorjana Radobolja-Lazarevski, and Zrinka Tamburašev, led by Dr. Slobodan Đokić — discovered azithromycin in 1980.[6] It was patented in 1981. In 1986, Pliva and Pfizer signed a licensing agreement, which gave Pfizer exclusive rights for the sale of azithromycin in Western Europe and the United States. Pliva put its azithromycin on the market in Central and Eastern Europe under the brand name Sumamed in 1988. Pfizer launched azithromycin under Pliva’s license in other markets under the brand name Zithromax in 1991.[36] Patent protection ended in 2005.[37]
It is available as a generic medication.[9] The wholesale cost is about US$0.18 to US$2.98 per dose.[10] In the United States it is about US$4 for a course of treatment as of 2018.[11] In India, it is about US$1.70 for a course of treatment.[citation needed]
Available forms
Azithromycin is commonly administered in film-coated tablet, capsule, oral suspension, intravenousinjection, granules for suspension in sachet, and ophthalmic solution.[2]
Usage
In 2010, azithromycin was the most prescribed antibiotic for outpatients in the US,[38] whereas in Sweden, where outpatient antibiotic use is a third as prevalent, macrolides are only on 3% of prescriptions.[39]
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^Taylor SP, Sellers E, Taylor BT (2015). “Azithromycin for the Prevention of COPD Exacerbations: The Good, Bad, and Ugly”. Am. J. Med. 128 (12): 1362.e1–6. doi:10.1016/j.amjmed.2015.07.032. PMID26291905.
^Mandell LA, Wunderink RG, Anzueto A, Bartlett JG, Campbell GD, Dean NC, Dowell SF, File TM, Musher DM, Niederman MS, Torres A, Whitney CG (2007). “Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults”. Clin. Infect. Dis. 44 Suppl 2: S27–72. doi:10.1086/511159. PMID17278083.
^Rosenfeld RM, Piccirillo JF, Chandrasekhar SS, Brook I, Ashok Kumar K, Kramper M, Orlandi RR, Palmer JN, Patel ZM, Peters A, Walsh SA, Corrigan MD (2015). “Clinical practice guideline (update): adult sinusitis”. Otolaryngol Head Neck Surg. 152 (2 Suppl): S1–S39. doi:10.1177/0194599815572097. PMID25832968.
^Hauk L (2014). “AAP releases guideline on diagnosis and management of acute bacterial sinusitis in children one to 18 years of age”. Am Fam Physician. 89 (8): 676–81. PMID24784128.
^Neff MJ (2004). “AAP, AAFP release guideline on diagnosis and management of acute otitis media”. Am Fam Physician. 69 (11): 2713–5. PMID15202704.
^Randel A (2013). “IDSA Updates Guideline for Managing Group A Streptococcal Pharyngitis”. Am Fam Physician. 88 (5): 338–40. PMID24010402.
^Zarogoulidis, P.; Papanas, N.; Kioumis, I.; Chatzaki, E.; Maltezos, E.; Zarogoulidis, K. (May 2012). “Macrolides: from in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases”. European Journal of Clinical Pharmacology. 68 (5): 479–503. doi:10.1007/s00228-011-1161-x. ISSN1432-1041. PMID22105373.
^Mori F, Pecorari L, Pantano S, Rossi M, Pucci N, De Martino M, Novembre E (2014). “Azithromycin anaphylaxis in children”. Int J Immunopathol Pharmacol. 27 (1): 121–6. doi:10.1177/039463201402700116. PMID24674687.
^Dart, Richard C. (2004). Medical Toxology. Lippincott Williams & Wilkins. p. 23.
^Tilelli, John A.; Smith, Kathleen M.; Pettignano, Robert (2006). “Life-Threatening Bradyarrhythmia After Massive Azithromycin Overdose”. Pharmacotherapy. 26 (1): 147–50. doi:10.1592/phco.2006.26.1.147. PMID16506357.
^Baselt, R. (2008). Disposition of Toxic Drugs and Chemicals in Man (8th ed.). Foster City, CA: Biomedical Publications. pp. 132–133.
^Hicks, LA; Taylor TH, Jr; Hunkler, RJ (April 2013). “U.S. outpatient antibiotic prescribing, 2010”. The New England Journal of Medicine. 368 (15): 1461–1462. doi:10.1056/NEJMc1212055. PMID23574140.
^Hicks, LA; Taylor TH, Jr; Hunkler, RJ (September 2013). “More on U.S. outpatient antibiotic prescribing, 2010”. The New England Journal of Medicine. 369 (12): 1175–1176. doi:10.1056/NEJMc1306863. PMID24047077.
Substances Referenced in Synthesis Path CAS-RN Formula Chemical Name CAS Index Name 76801-85-9 C37H70N2O12 2-deoxo-9a-aza-9a-homoerythromycin A 1-Oxa-6-azacyclopentadecan-15-one, 13-[(2,6-dideoxy-3-C-methyl-3-O-methyl-α-L-ribo-hexopyranosyl)oxy]-2-eth- yl-3,4,10-trihydroxy-3,5,8,10,12,14-hexamethyl-11-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-, [2R-(2 R*,3S*,4R*,5R*,8R*,10R*,11R*,12S*,13S*,1 4R*)]-
Percent Composition: C 67.59%, H 8.19%, Cl 11.08%, N 13.14%
Literature References: Prepd by the condensation of 4,7-dichloroquinoline with 1-diethylamino-4-aminopentane: DE683692 (1939); H. Andersag et al.,US2233970 (1941 to Winthrop); Surrey, Hammer, J. Am. Chem. Soc.68, 113 (1946). Review: Hahn in Antibioticsvol. 3, J. W. Corcoran, F. E. Hahn, Eds. (Springer-Verlag, New York, 1975) pp 58-78. Comprehensive description: D. D. Hong, Anal. Profiles Drug Subs.5, 61-85 (1976). Comparative clinical trial with dapsone in rheumatoid arthritis: P. D. Fowler et al.,Ann. Rheum. Dis.43, 200 (1984); with penicillamine: T. Gibson et al.,Br. J. Rheumatol.26, 279 (1987).
Percent Composition: C 41.91%, H 6.25%, Cl 6.87%, N 8.15%, P 12.01%, O 24.81%
Properties: Bitter, colorless crystals. Dimorphic. One modification, mp 193-195°; the other, mp 215-218°. Freely sol in water; pH of 1% soln about 4.5; less sol at neutral and alkaline pH. Stable to heat in solns of pH 4.0 to 6.5. Practically insol in alcohol, benzene, chloroform, ether.
Chloroquine is a medication used primarily to prevent and to treat malaria in areas where that parasitic disease is known to remain sensitive to its effects.[1] A benefit of its use in therapy, when situations allow, is that it can be taken by mouth (versus by injection).[1] Controlled studies of cases involving human pregnancy are lacking, but the drug may be safe for use for such patients.[verification needed][1][2] However, the agent is not without the possibility of serious side effects at standard doses,[1][3] and complicated cases, including infections of certain types or caused by resistant strains, typically require different or additional medication.[1] Chloroquine is also used as a medication for rheumatoid arthritis, lupus erythematosus, and other parasitic infections (e.g., amebiasis occurring outside of the intestines).[1] Beginning in 2020, studies have proceeded on its use as a coronavirusantiviral, in possible treatment of COVID-19.[4]
Chloroquine, otherwise known as chloroquine phosphate, is in the 4-aminoquinoline class of drugs.[1] As an antimalarial, it works against the asexual form of the malaria parasite in the stage of its life cycle within the red blood cell.[1] In its use against rheumatoid arthritis and lupus erythematosus, its activity as a mild immunosuppressive underlies its mechanism.[1] Antiviral activities, established and putative, are attributed to chloroquines inhibition of glycosylation pathways (of host receptor sialylation or virus protein post-translational modification), or to inhibition of virus endocytosis (e.g., via alkalisation of endosomes), or other possible mechanisms.[5] Common side effects resulting from these therapeutic uses, at common doses, include muscle problems,[clarification needed] loss of appetite, diarrhea, and skin rash.[clarification needed][1] Serious side effects include problems with vision (retinopathy), muscle damage, seizures, and certain anemias.[1][6]
Distribution of malaria in the world:[11]
♦ Elevated occurrence of chloroquine- or multi-resistant malaria
♦ Occurrence of chloroquine-resistant malaria
♦ No Plasmodium falciparum or chloroquine-resistance
♦ No malaria
Chloroquine has been extensively used in mass drug administrations, which may have contributed to the emergence and spread of resistance. It is recommended to check if chloroquine is still effective in the region prior to using it.[14] In areas where resistance is present, other antimalarials, such as mefloquine or atovaquone, may be used instead. The Centers for Disease Control and Prevention recommend against treatment of malaria with chloroquine alone due to more effective combinations.[15]
Amebiasis
In treatment of amoebic liver abscess, chloroquine may be used instead of or in addition to other medications in the event of failure of improvement with metronidazole or another nitroimidazole within 5 days or intolerance to metronidazole or a nitroimidazole.[16]
Mental/mood changes (such as confusion, personality changes, unusual thoughts/behavior, depression, feeling being watched, hallucinating)[17][18]
Signs of serious infection (such as high fever, severe chills, persistent sore throat)[17]
Skin itchiness, skin color changes, hair loss, and skin rashes.[18][19]
Chloroquine-induced itching is very common among black Africans (70%), but much less common in other races. It increases with age, and is so severe as to stop compliance with drug therapy. It is increased during malaria fever; its severity is correlated to the malaria parasite load in blood. Some evidence indicates it has a genetic basis and is related to chloroquine action with opiate receptors centrally or peripherally.[20]
Unpleasant metallic taste
This could be avoided by “taste-masked and controlled release” formulations such as multiple emulsions.[21]
This manifests itself as either conduction disturbances (bundle-branch block, atrioventricular block) or Cardiomyopathy – often with hypertrophy, restrictive physiology, and congestive heart failure. The changes may be irreversible. Only two cases have been reported requiring heart transplantation, suggesting this particular risk is very low. Electron microscopy of cardiac biopsies show pathognomonic cytoplasmic inclusion bodies.
Chloroquine has not been shown to have any harmful effects on the fetus when used for malarial prophylaxis.[24] Small amounts of chloroquine are excreted in the breast milk of lactating women. However, this drug can be safely prescribed to infants, the effects are not harmful. Studies with mice show that radioactively tagged chloroquine passed through the placenta rapidly and accumulated in the fetal eyes which remained present five months after the drug was cleared from the rest of the body.[23][25] Women who are pregnant or planning on getting pregnant are still advised against traveling to malaria-risk regions.[24]
Elderly
There is not enough evidence to determine whether chloroquine is safe to be given to people aged 65 and older. Since it is cleared by the kidneys, toxicity should be monitored carefully in people with poor kidney functions.[23]
Chloroquine is very dangerous in overdose. It is rapidly absorbed from the gut. In 1961, a published compilation of case reports contained accounts of three children who took overdoses and died within 2.5 hours of taking the drug. While the amount of the overdose was not stated, the therapeutic index for chloroquine is known to be small.[26] One of the children died after taking 0.75 or 1 gram, or twice a single therapeutic amount for children. Symptoms of overdose include headache, drowsiness, visual disturbances, nausea and vomiting, cardiovascular collapse, seizures, and sudden respiratory and cardiac arrest.[23]
An analog of chloroquine – hydroxychloroquine – has a long half-life (32–56 days) in blood and a large volume of distribution (580–815 L/kg).[27] The therapeutic, toxic and lethal ranges are usually considered to be 0.03 to 15 mg/l, 3.0 to 26 mg/l and 20 to 104 mg/l, respectively. However, nontoxic cases have been reported up to 39 mg/l, suggesting individual tolerance to this agent may be more variable than previously recognised.[27]
Pharmacology
Chloroquine’s absorption of the drug is rapid. It is widely distributed in body tissues. It’s protein binding is 55%.[ It’s metabolism is partially hepatic, giving rise to its main metabolite, desethylchloroquine. It’s excretion os ≥50% as unchanged drug in urine, where acidification of urine increases its elimination It has a very high volume of distribution, as it diffuses into the body’s adipose tissue.
Accumulation of the drug may result in deposits that can lead to blurred vision and blindness. It and related quinines have been associated with cases of retinal toxicity, particularly when provided at higher doses for longer times. With long-term doses, routine visits to an ophthalmologist are recommended.
Chloroquine is also a lysosomotropic agent, meaning it accumulates preferentially in the lysosomes of cells in the body. The pKa for the quinoline nitrogen of chloroquine is 8.5, meaning—in simplified terms, considering only this basic site—it is about 10% deprotonated at physiological pH (per the Henderson-Hasselbalch equation) This decreases to about 0.2% at a lysosomal pH of 4.6.Because the deprotonated form is more membrane-permeable than the protonated form, a quantitative “trapping” of the compound in lysosomes results.
Mechanism of action
Medical quinolines
Malaria
Hemozoin formation in P. falciparum: many antimalarials are strong inhibitors of hemozoin crystal growth.
The lysosomotropic character of chloroquine is believed to account for much of its antimalarial activity; the drug concentrates in the acidic food vacuole of the parasite and interferes with essential processes. Its lysosomotropic properties further allow for its use for in vitro experiments pertaining to intracellular lipid related diseases,[28][29] autophagy, and apoptosis.[30]
Inside red blood cells, the malarial parasite, which is then in its asexual lifecycle stage, must degrade hemoglobin to acquire essential amino acids, which the parasite requires to construct its own protein and for energy metabolism. Digestion is carried out in a vacuole of the parasitic cell.[citation needed]
Hemoglobin is composed of a protein unit (digested by the parasite) and a heme unit (not used by the parasite). During this process, the parasite releases the toxic and soluble molecule heme. The heme moiety consists of a porphyrin ring called Fe(II)-protoporphyrin IX (FP). To avoid destruction by this molecule, the parasite biocrystallizes heme to form hemozoin, a nontoxic molecule. Hemozoin collects in the digestive vacuole as insoluble crystals.[citation needed]
Chloroquine enters the red blood cell by simple diffusion, inhibiting the parasite cell and digestive vacuole. Chloroquine then becomes protonated (to CQ2+), as the digestive vacuole is known to be acidic (pH 4.7); chloroquine then cannot leave by diffusion. Chloroquine caps hemozoin molecules to prevent further biocrystallization of heme, thus leading to heme buildup. Chloroquine binds to heme (or FP) to form the FP-chloroquine complex; this complex is highly toxic to the cell and disrupts membrane function. Action of the toxic FP-chloroquine and FP results in cell lysis and ultimately parasite cell autodigestion. [31] Parasites that do not form hemozoin are therefore resistant to chloroquine.[32]
Since the first documentation of P. falciparum chloroquine resistance in the 1950s, resistant strains have appeared throughout East and West Africa, Southeast Asia, and South America. The effectiveness of chloroquine against P. falciparum has declined as resistant strains of the parasite evolved. They effectively neutralize the drug via a mechanism that drains chloroquine away from the digestive vacuole. Chloroquine-resistant cells efflux chloroquine at 40 times the rate of chloroquine-sensitive cells; the related mutations trace back to transmembrane proteins of the digestive vacuole, including sets of critical mutations in the P. falciparum chloroquine resistance transporter (PfCRT) gene. The mutated protein, but not the wild-type transporter, transports chloroquine when expressed in Xenopus oocytes (frog’s eggs) and is thought to mediate chloroquine leak from its site of action in the digestive vacuole.[33] Resistant parasites also frequently have mutated products of the ABC transporterP. falciparum multidrug resistance (PfMDR1) gene, although these mutations are thought to be of secondary importance compared to Pfcrt. Verapamil, a Ca2+ channel blocker, has been found to restore both the chloroquine concentration ability and sensitivity to this drug. Recently, an altered chloroquine-transporter protein CG2 of the parasite has been related to chloroquine resistance, but other mechanisms of resistance also appear to be involved.[34] Research on the mechanism of chloroquine and how the parasite has acquired chloroquine resistance is still ongoing, as other mechanisms of resistance are likely.[citation needed]
Chloroquine has antiviral effects.[36] It increases late endosomal or lysosomal pH, resulting in impaired release of the virus from the endosome or lysosome – release requires a low pH. The virus is therefore unable to release its genetic material into the cell and replicate.[37][38]
Chloroquine also seems to act as a zinc ionophore, that allows extracellular zinc to enter the cell and inhibit viral RNA-dependent RNA polymerase.[39][40]
Other
Chloroquine inhibits thiamine uptake.[41] It acts specifically on the transporter SLC19A3.
In Peru the indigenous people extracted the bark of the Cinchona plant[42] trees and used the extract (Chinchona officinalis) to fight chills and fever in the seventeenth century. In 1633 this herbal medicine was introduced in Europe, where it was given the same use and also began to be used against malaria.[43] The quinoline antimalarial drug quinine was isolated from the extract in 1820, and chloroquine is an analogue of this.
Chloroquine was discovered in 1934, by Hans Andersag and coworkers at the Bayer laboratories, who named it “Resochin”.[44] It was ignored for a decade, because it was considered too toxic for human use. During World War II, United States government-sponsored clinical trials for antimalarial drug development showed unequivocally that chloroquine has a significant therapeutic value as an antimalarial drug. It was introduced into clinical practice in 1947 for the prophylactic treatment of malaria.[45]
Society and culture
Resochin tablet package
Formulations
Chloroquine comes in tablet form as the phosphate, sulfate, and hydrochloride salts. Chloroquine is usually dispensed as the phosphate.[46]
Names
Brand names include Chloroquine FNA, Resochin, Dawaquin, and Lariago.[47]
Chloroquine has been recommended by Chinese, South Korean and Italian health authorities for the treatment of COVID-19.[52][53] These agencies noted contraindications for people with heart disease or diabetes.[54] Both chloroquine and hydroxychloroquine were shown to inhibit SARS-CoV-2 in vitro, but a further study concluded that hydroxychloroquine was more potent than chloroquine, with a more tolerable safety profile.[55] Preliminary results from a trial suggested that chloroquine is effective and safe in COVID-19 pneumonia, “improving lung imaging findings, promoting a virus-negative conversion, and shortening the disease course.”[56] Self-medication with chloroquine has caused one known fatality.[57]
On 24 March 2020, NBC News reported[58] a fatality due to misuse of a chloroquine product used to control fish parasites.[59]
Other viruses
In October 2004, a group of researchers at the Rega Institute for Medical Research published a report on chloroquine, stating that chloroquine acts as an effective inhibitor of the replication of the severe acute respiratory syndrome coronavirus (SARS-CoV) in vitro.[60]
Chloroquine was being considered in 2003, in pre-clinical models as a potential agent against chikungunya fever.[61]
^Mittra, Robert A.; Mieler, William F. (1 January 2013). Ryan, Stephen J.; Sadda, SriniVas R.; Hinton, David R.; Schachat, Andrew P.; Sadda, SriniVas R.; Wilkinson, C. P.; Wiedemann, Peter; Schachat, Andrew P. (eds.). Retina (Fifth Edition). W.B. Saunders. pp. 1532–1554 – via ScienceDirect.
^Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S (March 2020). “A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19”. Journal of Critical Care. doi:10.1016/j.jcrc.2020.03.005. PMID32173110.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^“Chloroquine (Base)”. International Drug Price Indicator Guide. Archived from the original on 27 August 2018. Retrieved 4 December 2015.
^CDC. Health information for international travel 2001–2002. Atlanta, Georgia: U.S. Department of Health and Human Services, Public Health Service, 2001.
^Ajayi AA (September 2000). “Mechanisms of chloroquine-induced pruritus”. Clinical Pharmacology and Therapeutics. 68 (3): 336. PMID11014416.
^Vaziri A, Warburton B (1994). “Slow release of chloroquine phosphate from multiple taste-masked W/O/W multiple emulsions”. Journal of Microencapsulation. 11 (6): 641–8. doi:10.3109/02652049409051114. PMID7884629.
^Tönnesmann E, Kandolf R, Lewalter T (June 2013). “Chloroquine cardiomyopathy – a review of the literature”. Immunopharmacology and Immunotoxicology. 35 (3): 434–42. doi:10.3109/08923973.2013.780078. PMID23635029.
^Hempelmann E (March 2007). “Hemozoin biocrystallization in Plasmodium falciparum and the antimalarial activity of crystallization inhibitors”. Parasitology Research. 100 (4): 671–6. doi:10.1007/s00436-006-0313-x. PMID17111179.
^Martin RE, Marchetti RV, Cowan AI, Howitt SM, Bröer S, Kirk K (September 2009). “Chloroquine transport via the malaria parasite’s chloroquine resistance transporter”. Science. 325 (5948): 1680–2. Bibcode:2009Sci…325.1680M. doi:10.1126/science.1175667. PMID19779197.
^Essentials of medical pharmacology fifth edition 2003, reprint 2004, published by-Jaypee Brothers Medical Publisher Ltd, 2003, KD Tripathi, pages 739,740.
^Alcantara LM, Kim J, Moraes CB, Franco CH, Franzoi KD, Lee S, et al. (June 2013). “Chemosensitization potential of P-glycoprotein inhibitors in malaria parasites”. Experimental Parasitology. 134 (2): 235–43. doi:10.1016/j.exppara.2013.03.022. PMID23541983.
^Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R (November 2003). “Effects of chloroquine on viral infections: an old drug against today’s diseases?”. The Lancet. Infectious Diseases. 3(11): 722–7. doi:10.1016/s1473-3099(03)00806-5. PMID14592603.
^Krafts K, Hempelmann E, Skórska-Stania A (July 2012). “From methylene blue to chloroquine: a brief review of the development of an antimalarial therapy”. Parasitology Research. 111 (1): 1–6. doi:10.1007/s00436-012-2886-x. PMID22411634.
^Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M (October 2004). “In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine”. Biochemical and Biophysical Research Communications. 323 (1): 264–8. doi:10.1016/j.bbrc.2004.08.085. PMID15351731.
^Devaux CA, Rolain JM, Colson P, Raoult D. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? Int J Antimicrob Agents. 2020 Mar 11:105938. doi:10.1016/j.ijantimicag.2020.105938PMID32171740
^Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. (March 2020). “In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)”. Clinical Infectious Diseases. doi:10.1093/cid/ciaa237. PMID32150618.
^Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M (October 2004). “In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine”. Biochemical and Biophysical Research Communications. 323 (1): 264–8. doi:10.1016/j.bbrc.2004.08.085. PMID15351731.
^Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R (November 2003). “Effects of chloroquine on viral infections: an old drug against today’s diseases?”. The Lancet. Infectious Diseases. 3(11): 722–7. doi:10.1016/S1473-3099(03)00806-5. PMID14592603.
^Savarino A, Lucia MB, Giordano F, Cauda R (October 2006). “Risks and benefits of chloroquine use in anticancer strategies”. The Lancet. Oncology. 7 (10): 792–3. doi:10.1016/S1470-2045(06)70875-0. PMID17012039.
^Sotelo J, Briceño E, López-González MA (March 2006). “Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial”. Annals of Internal Medicine. 144 (5): 337–43. doi:10.7326/0003-4819-144-5-200603070-00008. PMID16520474.
“Summaries for patients. Adding chloroquine to conventional chemotherapy and radiotherapy for glioblastoma multiforme”. Annals of Internal Medicine. 144 (5): I31. March 2006. doi:10.7326/0003-4819-144-5-200603070-00004. PMID16520470.
External links
“Chloroquine”. Drug Information Portal. U.S. National Library of Medicine.
Percent Composition: C 47.73%, H 2.47%, Cl 21.68%, N 8.56%, O 19.56%
Literature References: Prepn: GB824345 (1959 to Bayer), C.A.54, 15822b (1960). See also: E. Schraufstätter, R. Gönnert, US3079297; R. Strufe et al.,US3113067 (both 1963 to Bayer); Bekhli et al.,Med. Prom. SSSR1965, 25.
Properties: Pale yellow crystals, mp 225-230°. Practically insol in water. Sparingly sol in ethanol, chloroform, ether.
Melting point: mp 225-230°
Derivative Type: Ethanolamine salt
CAS Registry Number: 1420-04-8
Additional Names: Clonitrilide
Trademarks: Bayluscid (Bayer)
Molecular Formula: C13H8Cl2N2O4.C2H7NO
Molecular Weight: 388.20
Percent Composition: C 46.41%, H 3.89%, Cl 18.27%, N 10.82%, O 20.61%
Side effects include nausea, vomiting, abdominal pain, and itchiness.[2] It may be used during pregnancy and appears to be safe for the baby.[2] Niclosamide is in the anthelmintic family of medications.[3] It works by blocking the uptake of sugar by the worm.[4]
Side effects include nausea, vomiting, abdominal pain, constipation, and itchiness.[2] Rarely, dizziness, skin rash, drowsiness, perianal itching, or an unpleasant taste occur. For some of these reasons, praziquantel is a preferable and equally effective treatment for tapeworm infestation.[citation needed]
Mechanism of action
Niclosamide inhibits glucose uptake, oxidative phosphorylation, and anaerobic metabolism in the tapeworm.[8]
Other applications
Niclosamide’s metabolic effects are relevant to wide ranges of organisms, and accordingly it has been applied as a control measure to organisms other than tapeworms. For example, it is an active ingredient in some formulations such as Bayluscide for killing lamprey larvae,[9][10] as a molluscide,[11] and as a general purpose piscicide in aquaculture. Niclosamide has a short half-life in water in field conditions; this makes it valuable in ridding commercial fish ponds of unwanted fish; it loses its activity soon enough to permit re-stocking within a few days of eradicating the previous population.[11] Researchers have found that niclosamide is effective in killing invasive zebra mussels in cool waters.[12]
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^“Niclosamide”. International Drug Price Indicator Guide. Archived from the original on 10 May 2017. Retrieved 1 December 2016.
^Weinbach EC, Garbus J (1969). “Mechanism of action of reagents that uncouple oxidative phosphorylation”. Nature. 221 (5185): 1016–8. doi:10.1038/2211016a0. PMID4180173.
^Boogaard, Michael A. Delivery Systems of Piscicides “Request Rejected”(PDF). Archived(PDF) from the original on 2017-06-01. Retrieved 2017-05-30.
^Verdel K.Dawson (2003). “Environmental Fate and Effects of the Lampricide Bayluscide: a Review”. Journal of Great Lakes Research. 29 (Supplement 1): 475–492. doi:10.1016/S0380-1330(03)70509-7.
Percent Composition: C 46.90%, H 2.95%, N 13.67%, O 26.03%, S 10.44%
Literature References: Broad spectrum antiparasitic agent; inhibits pyruvate ferredoxin oxidoreductase. Prepn: J. F. Rossignol, R. Cavier, DE2438037; eidem,US3950351 (1975, 1976 both to S.P.R.L. Phavic); and antiparasitic activity: R. Cavier et al.,Eur. J. Med. Chem. – Chim. Ther.13, 539 (1978). Antibacterial spectrum in vitro: L Dubreuil et al.,Antimicrob. Agents Chemother.40, 2266 (1996). Toxicology: J. R. Murphy, J.-C. Friedmann, J. Appl. Toxicol.5, 49 (1985). Clinical pharmacokinetics: A. Stockis et al.,Int. J. Clin. Pharmacol. Ther.34, 349 (1996). Clinical trial in intestinal protozoan and helminthic infections: H. Abaza et al., Curr. Ther. Res.59, 116 (1998). Review of mechanism of action and clinical experience: H. M. Gilles, P. S. Hoffman, Trends Parasitol.18, 95-97 (2002).
Properties: Light yellow crystalline powder. Crystals from methanol, mp 202°. Poorly sol in ethanol. Practically insol in water. LD50 orally in male, female mice: 1350, 1380 mg/kg; in rats: >10 g/kg (Murphy, Friedmann).
Melting point: mp 202°
Toxicity data: LD50 orally in male, female mice: 1350, 1380 mg/kg; in rats: >10 g/kg (Murphy, Friedmann)
Chemically, nitazoxanide is the prototype member of the thiazolides, a class of drugs which are synthetic nitrothiazolyl-salicylamide derivatives with antiparasitic and antiviral activity.[4][6][8]Tizoxanide, an active metabolite of nitazoxanide in humans, is also an antiparasitic drug of the thiazolide class.[4][9]
Nitazoxanide alone has shown preliminary evidence of efficacy in the treatment of chronic hepatitis B over a one-year course of therapy.[17] Nitazoxanide 500 mg twice daily resulted in a decrease in serum HBV DNA in all of 4 HBeAg-positive patients, with undetectable HBV DNA in 2 of 4 patients, loss of HBeAg in 3 patients, and loss of HBsAg in one patient. Seven of 8 HBeAg-negative patients treated with nitazoxanide 500 mg twice daily had undetectable HBV DNA and 2 had loss of HBsAg. Additionally, nitazoxanide monotherapy in one case and nitazoxanide plus adefovir in another case resulted in undetectable HBV DNA, loss of HBeAg and loss of HBsAg.[18] These preliminary studies showed a higher rate of HBsAg loss than any currently licensed therapy for chronic hepatitis B. The similar mechanism of action of interferon and nitazoxanide suggest that stand-alone nitazoxanide therapy or nitazoxanide in concert with nucleos(t)ide analogs have the potential to increase loss of HBsAg, which is the ultimate end-point of therapy. A formal phase Ⅱ study is being planned for 2009.[19]
Chronic hepatitis C
Romark initially decided to focus on the possibility of treating chronic hepatitis C with nitazoxanide.[20] The drug garnered interest from the hepatology community after three phase II clinical trials involving the treatment of hepatitis C with nitazoxanide produced positive results for treatment efficacy and similar tolerability to placebo without any signs of toxicity.[20] A meta-analysis from 2014 concluded that the previous held trials were of low-quality and with held with a risk of bias. The authors concluded that more randomized trials with low risk of bias are needed to give any determine if Nitazoxanide can be used as an effective treatment for chronic hepatitis C patients.[21]
Clinical trials
Nitazoxanide has gone through Phase II clinical trials for the treatment of hepatitis C, in combination with peginterferon alfa-2a and ribavirin.[22][23]Romark Laboratories has announced encouraging results from international Phase I and II clinical trials evaluating a controlled release version of nitazoxanide in the treatment of chronic hepatitis C virus infection. The company used 675 mg and 1,350 mg twice daily doses of controlled release nitazoxanide showed favorable safety and tolerability throughout the course of the study, with mild to moderate adverse events. Primarily GI-related adverse events were reported.
A randomised double-blind placebo-controlled study published in 2006, with a group of 38 young children (Lancet, vol 368, page 124-129)[24] concluded that a 3-day course of nitazoxanide significantly reduced the duration of rotavirus disease in hospitalized pediatric patients. Dose given was “7.5 mg/kg twice daily” and the time of resolution was “31 hours for those given nitazoxanide compared with 75 hours for those in the placebo group.” Rotavirus is the most common infectious agent associated with diarrhea in the pediatric age group worldwide.
Teran et al.. conducted a study at the Pediatric Center Albina Patinö, a reference hospital in the city of Cochabamba, Bolivia, from August 2007 to February 2008. The study compared nitazoxanide and probiotics in the treatment of acute rotavirus diarrhea. They found Small differences in favor of nitazoxanide in comparison with probiotics and concluded that nitazoxanide is an important treatment option for rotavirus diarrhea.[17]
Lateef et al.. conducted a study in India that evaluated the effectiveness of nitazoxanide in the treatment of beef tapeworm (Taenia saginata) infection. They concluded that nitazoxanide is a safe, effective, inexpensive, and well-tolerated drug for the treatment of niclosamide- and praziquantel-resistant beef tapeworm (Taenia saginata) infection.[18]
A retrospective review of charts of patients treated with nitazoxanide for trichomoniasis by Michael Dan and Jack D. Sobel demonstrated negative result. They reported three case studies; two of which with metronidazole-resistant infections. In Case 3, they reported the patient to be cured with high divided dose tinidazole therapy. They used a high dosage of the drug (total dose, 14–56 g) than the recommended standard dosage (total dose, 3 g) and observed a significant adverse reaction (poorly tolerated nausea) only with the very high dose (total dose, 56 g). While confirming the safety of the drug, they showed nitazoxanide is ineffective for the treatment of trichomoniasis.[25]
The side effects of nitazoxanide do not significantly differ from a placebo treatment for giardiasis;[1] these symptoms include stomach pain, headache, upset stomach, vomiting, discolored urine, excessive urinating, skin rash, itching, fever, flu syndrome, and others.[1][26] Nitazoxanide does not appear to cause any significant adverse effects when taken by healthy adults.[1][2]
Overdose
Information on nitazoxanide overdose is limited. Oral doses of 4 grams in healthy adults do not appear to cause any significant adverse effects.[1][2] In various animals, the oral LD50 is higher than 10 g/kg.[1]
The anti-protozoal activity of nitazoxanide is believed to be due to interference with the pyruvate:ferredoxin oxidoreductase (PFOR) enzyme-dependent electron transfer reaction which is essential to anaerobic energy metabolism.[1][8] PFOR inhibition may also contribute to its activity against anaerobic bacteria.[27]
It has also been shown to have activity against influenza A virus in vitro.[28] The mechanism appears to be by selectively blocking the maturation of the viral hemagglutinin at a stage preceding resistance to endoglycosidase H digestion. This impairs hemagglutinin intracellular trafficking and insertion of the protein into the host plasma membrane.
Nitazoxanide modulates a variety of other pathways in vitro, including glutathione-S-transferase and glutamate-gated chloride ion channels in nematodes, respiration and other pathways in bacteria and cancer cells, and viral and host transcriptional factors.[27]
Pharmacokinetics
Following oral administration, nitazoxanide is rapidly hydrolyzed to the pharmacologically active metabolite, tizoxanide, which is 99% protein bound.[1][9] Tizoxanide is then glucuronide conjugated into the active metabolite, tizoxanide glucuronide.[1] Peak plasma concentrations of the metabolites tizoxanide and tizoxanide glucuronide are observed 1–4 hours after oral administration of nitazoxanide, whereas nitazoxanide itself is not detected in blood plasma.[1]
Roughly 2⁄3 of an oral dose of nitazoxanide is excreted as its metabolites in feces, while the remainder of the dose excreted in urine.[1] Tizoxanide is excreted in the urine, bile and feces.[1] Tizoxanide glucuronide is excreted in urine and bile.[1]
Chemistry
History
Nitazoxanide is the prototype member of the thiazolides, which is a drug class of structurally-related broad-spectrum antiparasitic compounds.[4] Nitazoxanide is a light yellow crystalline powder. It is poorly soluble in ethanol and practically insoluble in water.
Nitazoxanide was originally discovered in the 1980s by Jean-François Rossignol at the Pasteur Institute. Initial studies demonstrated activity versus tapeworms. In vitro studies demonstrated much broader activity. Dr. Rossignol co-founded Romark Laboratories, with the goal of bringing nitazoxanide to market as an anti-parasitic drug. Initial studies in the USA were conducted in collaboration with Unimed Pharmaceuticals, Inc. (Marietta, GA) and focused on development of the drug for treatment of cryptosporidiosis in AIDS. Controlled trials began shortly after the advent of effective anti-retroviral therapies. The trials were abandoned due to poor enrollment and the FDA rejected an application based on uncontrolled studies.
Subsequently, Romark launched a series of controlled trials. A placebo-controlled study of nitazoxanide in cryptosporidiosis demonstrated significant clinical improvement in adults and children with mild illness. Among malnourished children in Zambia with chronic cryptosporidiosis, a three-day course of therapy led to clinical and parasitologic improvement and improved survival. In Zambia and in a study conducted in Mexico, nitazoxanide was not successful in the treatment of cryptosporidiosis in advanced infection with human immunodeficiency virus at the doses used. However, it was effective in patients with higher CD4 counts. In treatment of giardiasis, nitazoxanide was superior to placebo and comparable to metronidazole. Nitazoxanide was successful in the treatment of metronidazole-resistant giardiasis. Studies have suggested efficacy in the treatment of cyclosporiasis, isosporiasis, and amebiasis.[29] Recent studies have also found it to be effective against beef tapeworm(Taenia saginata).[30]
Research
Nitazoxanide is also under investigation for the treatment of COVID-19.[31]
Pharmaceutical products
Dosage forms
Nitazoxanide is currently available in two oral dosage forms: a tablet (500 mg) and an oral suspension (100 mg per 5 ml when reconstituted).[1]
An extended release tablet (675 mg) has been used in clinical trials for chronic hepatitis C; however, this form is not currently marketed and available for prescription.[20]
Brand names
Nitazoxanide is sold under the brand names Adonid, Alinia, Allpar, Annita, Celectan, Colufase, Daxon, Dexidex, Diatazox, Kidonax, Mitafar, Nanazoxid, Parazoxanide, Netazox, Niazid, Nitamax, Nitax, Nitaxide, Nitaz, Nizonide, NT-TOX, Pacovanton, Paramix, Toza, and Zox.
^ Jump up to:abcdeStockis A, Allemon AM, De Bruyn S, Gengler C (May 2002). “Nitazoxanide pharmacokinetics and tolerability in man using single ascending oral doses”. Int J Clin Pharmacol Ther. 40 (5): 213–220. doi:10.5414/cpp40213. PMID12051573.
^“Nitazoxanide”. PubChem Compound. National Center for Biotechnology Information. Retrieved 3 January 2016.
^ Jump up to:abcdeDi Santo N, Ehrisman J (2013). “Research perspective: potential role of nitazoxanide in ovarian cancer treatment. Old drug, new purpose?”. Cancers (Basel). 5 (3): 1163–1176. doi:10.3390/cancers5031163. PMC3795384. PMID24202339. Nitazoxanide [NTZ: 2-acetyloxy-N-(5-nitro-2-thiazolyl)benzamide] is a thiazolide antiparasitic agent with excellent activity against a wide variety of protozoa and helminths. … Nitazoxanide (NTZ) is a main compound of a class of broad-spectrum anti-parasitic compounds named thiazolides. It is composed of a nitrothiazole-ring and a salicylic acid moiety which are linked together by an amide bond … NTZ is generally well tolerated, and no significant adverse events have been noted in human trials [13]. … In vitro, NTZ and tizoxanide function against a wide range of organisms, including the protozoal species Blastocystis hominis, C. parvum, Entamoeba histolytica, G. lamblia and Trichomonas vaginalis [13]
^White CA (2004). “Nitazoxanide: a new broad spectrum antiparasitic agent”. Expert Rev Anti Infect Ther. 2 (1): 43–9. doi:10.1586/14787210.2.1.43. PMID15482170.
^ Jump up to:abcdefRossignol JF (October 2014). “Nitazoxanide: a first-in-class broad-spectrum antiviral agent”. Antiviral Res. 110: 94–103. doi:10.1016/j.antiviral.2014.07.014. PMID25108173. Originally developed and commercialized as an antiprotozoal agent, nitazoxanide was later identified as a first-in-class broad-spectrum antiviral drug and has been repurposed for the treatment of influenza. … From a chemical perspective, nitazoxanide is the scaffold for a new class of drugs called thiazolides. These small-molecule drugs target host-regulated processes involved in viral replication. … A new dosage formulation of nitazoxanide is presently undergoing global Phase 3 clinical development for the treatment of influenza. Nitazoxanide inhibits a broad range of influenza A and B viruses including influenza A(pH1N1) and the avian A(H7N9) as well as viruses that are resistant to neuraminidase inhibitors. … Nitazoxanide also inhibits the replication of a broad range of other RNA and DNA viruses including respiratory syncytial virus, parainfluenza, coronavirus, rotavirus, norovirus, hepatitis B, hepatitis C, dengue, yellow fever, Japanese encephalitis virus and human immunodeficiency virus in cell culture assays. Clinical trials have indicated a potential role for thiazolides in treating rotavirus and norovirus gastroenteritis and chronic hepatitis B and chronic hepatitis C. Ongoing and future clinical development is focused on viral respiratory infections, viral gastroenteritis and emerging infections such as dengue fever.
^ Jump up to:abAnderson, V. R.; Curran, M. P. (2007). “Nitazoxanide: A review of its use in the treatment of gastrointestinal infections”. Drugs. 67(13): 1947–1967. doi:10.2165/00003495-200767130-00015. PMID17722965. Nitazoxanide is effective in the treatment of protozoal and helminthic infections … Nitazoxanide is a first-line choice for the treatment of illness caused by C. parvum or G. lamblia infection in immunocompetent adults and children, and is an option to be considered in the treatment of illnesses caused by other protozoa and/or helminths.
^ Jump up to:abKorba BE, Montero AB, Farrar K, et al. (January 2008). “Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication”. Antiviral Res. 77 (1): 56–63. doi:10.1016/j.antiviral.2007.08.005. PMID17888524.
^Shoff WH (5 October 2015). Chandrasekar PH, Talavera F, King JW (eds.). “Cyclospora Medication”. Medscape. WebMD. Retrieved 11 January 2016. Nitazoxanide, a 5-nitrothiazole derivative with broad-spectrum activity against helminths and protozoans, has been shown to be effective against C cayetanensis, with an efficacy 87% by the third dose (first, 71%; second 75%). Three percent of patients had minor side effects.
^Li TC, Chan MC, Lee N (September 2015). “Clinical Implications of Antiviral Resistance in Influenza”. Viruses. 7 (9): 4929–4944. doi:10.3390/v7092850. PMC4584294. PMID26389935. Oral nitazoxanide is an available, approved antiparasitic agent (e.g., against cryptosporidium, giardia) with established safety profiles. Recently, it has been shown (together with its active metabolite tizoxanide) to possess anti-influenza activity by blocking haemagglutinin maturation/trafficking, and acting as an interferon-inducer [97]. … A large, multicenter, Phase 3 randomized-controlled trial comparing nitazoxanide, oseltamivir, and their combination in uncomplicated influenza is currently underway (NCT01610245). Figure 1: Molecular targets and potential antiviral treatments against influenza virus infection
^ Jump up to:abTeran, C. G.; Teran-Escalera, C. N.; Villarroel, P. (2009). “Nitazoxanide vs. Probiotics for the treatment of acute rotavirus diarrhea in children: A randomized, single-blind, controlled trial in Bolivian children”. International Journal of Infectious Diseases. 13(4): 518–523. doi:10.1016/j.ijid.2008.09.014. PMID19070525.
^ Jump up to:abLateef, M.; Zargar, S. A.; Khan, A. R.; Nazir, M.; Shoukat, A. (2008). “Successful treatment of niclosamide- and praziquantel-resistant beef tapeworm infection with nitazoxanide”. International Journal of Infectious Diseases. 12 (1): 80–82. doi:10.1016/j.ijid.2007.04.017. PMID17962058.
^World Journal of Gastroenterology 2009 April 21, Emmet B Keeffe MD, Professor, Jean-François Rossignol The Romark Institute for Medical Research, Tampa
^Rossignol, Jean-François; Abu-Zekry, Mona; Hussein, Abeer; Santoro, M Gabriella (2006). “Effect of nitazoxanide for treatment of severe rotavirus diarrhoea: randomised double-blind placebo-controlled trial”. The Lancet. 368 (9530): 124–9. CiteSeerX10.1.1.458.1597. doi:10.1016/S0140-6736(06)68852-1. PMID16829296.
^Dan, M.; Sobel, J. D. (2007). “Failure of Nitazoxanide to Cure Trichomoniasis in Three Women”. Sexually Transmitted Diseases. 34 (10): 813–4. doi:10.1097/NMD.0b013e31802f5d9a. PMID17551415.
^White Jr, AC (2003). “Nitazoxanide: An important advance in anti-parasitic therapy”. Am. J. Trop. Med. Hyg. 68 (4): 382–383. doi:10.4269/ajtmh.2003.68.382. PMID12875283.
^Lateef, M.; Zargar, S. A.; Khan, A. R.; Nazir, M.; Shoukat, A. (2008). “Successful treatment of niclosamide- and praziquantel-resistant beef tapeworm infection with nitazoxanide”. International Journal of Infectious Diseases. 12 (1): 80–2. doi:10.1016/j.ijid.2007.04.017. PMID17962058.
^Cynthia Liu, Qiongqiong Zhou, Yingzhu Li, Linda V. Garner, Steve P. Watkins, Linda J. Carter, Jeffrey Smoot, Anne C. Gregg, Angela D. Daniels, Susan Jervey, Dana Albaiu. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Central Science 2020; doi:10.1021/acscentsci.0c00272
Hydroxychloroquine (HCQ), sold under the brand name Plaquenil among others, is a medication used for the prevention and treatment of certain types of malaria.[2] Specifically it is used for chloroquine-sensitive malaria.[3] Other uses include treatment of rheumatoid arthritis, lupus, and porphyria cutanea tarda.[2] It is taken by mouth.[2] It is also being used as an experimental treatment for coronavirus disease 2019 (COVID-19).[4]
Hydroxychloroquine was approved for medical use in the United States in 1955.[2] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[6] The wholesale cost in the developing world is about US$4.65 per month as of 2015, when used for rheumatoid arthritis or lupus.[7] In the United States the wholesale cost of a month of treatment is about US$25 as of 2020.[8] In the United Kingdom this dose costs the NHS about £ 5.15.[9] In 2017, it was the 128th most prescribed medication in the United States with more than five million prescriptions.[10]
In 2014, its efficacy to treat Sjögren syndrome was questioned in a double-blind study involving 120 patients over a 48-week period.[11]
Hydroxychloroquine is widely used in the treatment of post-Lymearthritis. It may have both an anti-spirochaete activity and an anti-inflammatory activity, similar to the treatment of rheumatoid arthritis.[12]
Contraindications
The drug label advises that hydroxychloroquine should not be prescribed to individuals with known hypersensitivity to 4-Aminoquinoline compounds.[13] There are a range of other contraindications[14][15] and caution is required if patients have certain heart conditions, diabetes, psoriasis etc.
Side effects[
The most common adverse effects are a mild nausea and occasional stomach cramps with mild diarrhea. The most serious adverse effects affect the eye, with dose-related retinopathy as a concern even after hydroxychloroquine use is discontinued.[2] For short-term treatment of acute malaria, adverse effects can include abdominal cramps, diarrhea, heart problems, reduced appetite, headache, nausea and vomiting.[2]
For prolonged treatment of lupus or rheumatoid arthritis, adverse effects include the acute symptoms, plus altered eye pigmentation, acne, anemia, bleaching of hair, blisters in mouth and eyes, blood disorders, convulsions, vision difficulties, diminished reflexes, emotional changes, excessive coloring of the skin, hearing loss, hives, itching, liver problems or liver failure, loss of hair, muscle paralysis, weakness or atrophy, nightmares, psoriasis, reading difficulties, tinnitus, skin inflammation and scaling, skin rash, vertigo, weight loss, and occasionally urinary incontinence.[2] Hydroxychloroquine can worsen existing cases of both psoriasis and porphyria.[2]
Children may be especially vulnerable to developing adverse effects from hydroxychloroquine.[2]
One of the most serious side effects is retinopathy (generally with chronic use).[2][16] People taking 400 mg of hydroxychloroquine or less per day generally have a negligible risk of macular toxicity, whereas the risk begins to go up when a person takes the medication over 5 years or has a cumulative dose of more than 1000 grams. The daily safe maximum dose for eye toxicity can be computed from one’s height and weight using this calculator. Cumulative doses can also be calculated from this calculator. Macular toxicity is related to the total cumulative dose rather than the daily dose. Regular eye screening, even in the absence of visual symptoms, is recommended to begin when either of these risk factors occurs.[17]
Toxicity from hydroxychloroquine may be seen in two distinct areas of the eye: the cornea and the macula. The cornea may become affected (relatively commonly) by an innocuous cornea verticillata or vortex keratopathy and is characterized by whorl-like corneal epithelial deposits. These changes bear no relationship to dosage and are usually reversible on cessation of hydroxychloroquine.
The macular changes are potentially serious. Advanced retinopathy is characterized by reduction of visual acuity and a “bull’s eye” macular lesion which is absent in early involvement.
Overdose
Due to rapid absorption, symptoms of overdose can occur within a half an hour after ingestion. Overdose symptoms include convulsions, drowsiness, headache, heart problems or heart failure, difficulty breathing and vision problems.
Hydroxychloroquine overdoses are rarely reported, with 7 previous cases found in the English medical literature. In one such case, a 16-year-old girl who had ingested a handful of hydroxychloroquine 200mg presented with tachycardia (heart rate 110 beats/min), hypotension (systolic blood pressure 63 mm Hg), central nervous system depression, conduction defects (ORS = 0.14 msec), and hypokalemia (K = 2.1 meq/L). Treatment consisted of fluid boluses and dopamine, oxygen, and potassium supplementation. The presence of hydroxychloroquine was confirmed through toxicologic tests. The patient’s hypotension resolved within 4.5 hours, serum potassium stabilized in 24 hours, and tachycardia gradually decreased over 3 days.[18]
Specifically, the FDA drug label for hydroxychloroquine lists the following drug interactions [13]:
Digoxin (wherein it may result in increased serum digoxin levels)
Insulin or antidiabetic drugs (wherein it may enhance the effects of a hypoglycemic treatment)
Drugs that prolong QT interval and other arrhythmogenic drugs (as Hydroxychloroquine prolongs the QT interval and may increase the risk of inducing ventricular arrhythmias if used concurrently)
Mefloquine and other drugs known to lower the convulsive threshold (co-administration with other antimalarials known to lower the convulsion threshold may increase risk of convulsions)
Antiepileptics (concurrent use may impair the antiepileptic activity)
Methotrexate (combined use is unstudied and may increase the frequency of side effects)
Cyclosporin (wherein an increased plasma cylcosporin level was reported when used together).
Pharmacology[
Pharmacokinetics
Hydroxychloroquine has similar pharmacokinetics to chloroquine, with rapid gastrointestinal absorption and elimination by the kidneys. Cytochrome P450 enzymes (CYP2D6, 2C8, 3A4 and 3A5) metabolize hydroxychloroquine to N-desethylhydroxychloroquine.[21]
Pharmacodynamics
Antimalarials are lipophilic weak bases and easily pass plasma membranes. The free base form accumulates in lysosomes (acidic cytoplasmic vesicles) and is then protonated,[22] resulting in concentrations within lysosomes up to 1000 times higher than in culture media. This increases the pH of the lysosome from 4 to 6.[23] Alteration in pH causes inhibition of lysosomal acidic proteases causing a diminished proteolysis effect.[24] Higher pH within lysosomes causes decreased intracellular processing, glycosylation and secretion of proteins with many immunologic and nonimmunologic consequences.[25] These effects are believed to be the cause of a decreased immune cell functioning such as chemotaxis, phagocytosis and superoxide production by neutrophils.[26] HCQ is a weak diprotic base that can pass through the lipid cell membrane and preferentially concentrate in acidic cytoplasmic vesicles. The higher pH of these vesicles in macrophages or other antigen-presenting cells limits the association of autoantigenic (any) peptides with class II MHC molecules in the compartment for peptide loading and/or the subsequent processing and transport of the peptide-MHC complex to the cell membrane.[27]
In 2003, a novel mechanism was described wherein hydroxychloroquine inhibits stimulation of the toll-like receptor (TLR) 9 family receptors. TLRs are cellular receptors for microbial products that induce inflammatory responses through activation of the innate immune system.[29]
As with other quinoline antimalarial drugs, the mechanism of action of quinine has not been fully resolved. The most accepted model is based on hydrochloroquinine and involves the inhibition of hemozoinbiocrystallization, which facilitates the aggregation of cytotoxic heme. Free cytotoxic heme accumulates in the parasites, causing their deaths.[citation needed]
Brand names
It is frequently sold as a sulfate salt known as hydroxychloroquine sulfate.[2] 200 mg of the sulfate salt is equal to 155 mg of the base.[2]
Brand names of hydroxychloroquine include Plaquenil, Hydroquin, Axemal (in India), Dolquine, Quensyl, Quinoric.[30]
Hydroxychloroquine and chloroquine have been recommended by Chinese and South Korean health authorities for the experimental treatment of COVID-19.[31][32]In vitro studies in cell cultures demonstrated that hydroxychloroquine was more potent than chloroquine against SARS-CoV-2.[33]
On 17 March 2020, the AIFA Scientific Technical Commission of the Italian Medicines Agency expressed a favorable opinion on including the off-label use of chloroquine and hydroxychloroquine for the treatment of SARS-CoV-2 infection.[34]
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^“NADAC as of 2019-08-07”. Centers for Medicare and Medicaid Services. Retrieved 19 March 2020. Typical dose is 600mg per day. Costs 0.28157 per dose. Month has about 30 days.
^British national formulary: BNF 69 (69 ed.). British Medical Association. 2015. p. 730. ISBN9780857111562.
^Effects of Hydroxychloroquine on Symptomatic Improvement in Primary Sjögren Syndrome, Gottenberg, et al. (2014) “Archived copy”. Archived from the original on 11 July 2015. Retrieved 10 July 2015.
^Steere, AC; Angelis, SM (October 2006). “Therapy for Lyme Arthritis: Strategies for the Treatment of Antibiotic-refractory Arthritis”. Arthritis and Rheumatism. 54 (10): 3079–86. doi:10.1002/art.22131. PMID17009226.
^Mohammad, Samya; Clowse, Megan E. B.; Eudy, Amanda M.; Criscione-Schreiber, Lisa G. (March 2018). “Examination of Hydroxychloroquine Use and Hemolytic Anemia in G6PDH-Deficient Patients”. Arthritis Care & Research. 70 (3): 481–485. doi:10.1002/acr.23296. ISSN2151-4658. PMID28556555.
^Hurst, NP; French, JK; Gorjatschko, L; Betts, WH (1988). “Chloroquine and Hydroxychloroquine Inhibit Multiple Sites in Metabolic Pathways Leading to Neutrophil Superoxide Release”. The Journal of Rheumatology. 15 (1): 23–27. PMID2832600.
^Fox, R (1996). “Anti-malarial Drugs: Possible Mechanisms of Action in Autoimmune Disease and Prospects for Drug Development”. Lupus. 5: S4–10. doi:10.1177/096120339600500103. PMID8803903.
^Waller; et al. Medical Pharmacology and Therapeutics (2nd ed.). p. 370.
Umifenovir[2] (trade names ArbidolRussian: Арбидол, Chinese: 阿比朵尔) is an antiviral treatment for influenza infection used in Russia[3] and China. The drug is manufactured by Pharmstandard (Russian: Фармстандарт). Although some Russian studies have shown it to be effective, it is not approved for use in other countries. It is not approved by FDA for the treatment or prevention of influenza.[4] Chemically, umifenovir features an indole core, functionalized at all but one positions with different substituents. The drug is claimed to inhibit viral entry into target cells and stimulate the immune response. Interest in the drug has been renewed as a result of the SARS-CoV-2 outbreak.
Umifenovir is manufactured and made available as tablets, capsules and syrup.
Testing of umifenovir’s efficacy has mainly occurred in China and Russia,[5][6] and it is well known in these two countries.[7] Some of the Russian tests showed the drug to be effective[5] and a direct comparison with Tamiflu showed similar efficiency in vitro and in a clinical setting.[8] In 2007, Arbidol (umifenovir) had the highest sales in Russia among all over-the-counter drugs.
Mode of action
Biochemistry
Umifenovir inhibits membrane fusion.[3] Umifenovir prevents contact between the virus and target host cells. Fusion between the viral envelope (surrounding the viral capsid) and the cell membrane of the target cell is inhibited. This prevents viral entry to the target cell, and therefore protects it from infection.[9]
Some evidence suggests that the drug’s actions are more effective at preventing infections from RNA viruses than infections from DNA viruses.[10]
More recent studies indicate that umifenovir also has in vitro effectiveness at preventing entry of Ebolavirus Zaïre Kikwit, Tacaribe arenavirus and human herpes virus 8 in mammalian cell cultures, while confirming umifenovir’s suppressive effect in vitro on Hepatitis B and poliovirus infection of mammalian cells when introduced either in advance of viral infection or during infection.[13][14]
Research
In February 2020, Li Lanjuan, an expert of the National Health Commission of China, proposed using Arbidol (umifenovir) together with darunavir as a potential treatment during the 2019–20 coronavirus pandemic.[15] Chinese experts claim that preliminary tests had shown that arbidol and darunavir could inhibit replication of the virus.[16][17] So far without additional effect if added on top of recombinant human interferon α2b spray.[18]
In 2007, the Russian Academy of Medical Sciences stated that the effects of Arbidol (umifenovir) are not scientifically proven.[20]
Russian media criticized lobbying attempts by Tatyana Golikova (then-Minister of Healthcare) to promote umifenovir,[21] and the unproven claim that Arbidol can speed up recovery from flu or cold by 1.3-2.3 days.[22] They also debunked claims that the efficacy of umifenovir is supported by peer-reviewed studies.[23][24]
Arbidol hydrochloride, chemical name: 6-bromo-4-(dimethylaminomethyl)-5-hydroxy-1-methyl-2-(phenylthiomethyl)-1H- Indole-3-carboxylic acid ethyl ester hydrochloride, the structural formula is as follows:
Arbidol hydrochloride is an antiviral drug developed by the Soviet Medicinal Chemistry Research Center. It was first listed in Russia in 1993. It is used as a monohydrate for medicinal purposes. This product not only has immunomodulatory and interferon-inducing effects, but also has good anti-influenza virus activity, and is clinically used for the prevention and treatment of influenza and other acute viral respiratory tract infections.
The preparation of Arbidol hydrochloride has multiple synthetic routes, Chinese patent CN1687033A and Wang Dun, Wu Xiujing, Gong Ping’s “Synthesis of Arbidol Hydrochloride” bibliographical report in Chinese Pharmaceutical Industry Magazine 2004,35(8) are Taking p-benzoquinone and 3-aminocrotonic acid ethyl ester as starting materials, through Neitzescu reaction, O-acylation, N-alkylation, bromination, thiophenol reaction, Mannich amine methylation reaction, hydrochloric acid acidification to obtain hydrochloric acid Arbidol, the total reaction yield was 22.9%.
The synthetic route is as follows:
The Nenitzescu reaction used in the synthesis of indole rings in this method, the reaction yield of this step is about 60%, resulting in a total yield of 22.9%.
U.S. Patent US5198552 and World Patent WO9008135 reported that 5-hydroxy-1,2-dimethylindole-3-ethyl carboxylate was used as raw material, and arbidol hydrochloride was prepared through bromination, condensation, Mannich reaction and salt-forming reaction you.
Although the synthesis steps of this method are short, the raw material 5-hydroxy-1,2-dimethylindole-3-carboxylic acid ethyl ester is not easy to obtain, and the large-scale application is difficult.
Song Yanling, Zhao Yanfang, Gong Pingren reported in the 3rd National Symposium on Pharmaceutical Engineering Technology and Education “Synthesis Research on Arbidol Hydrochloride” in the literature report using thiophenol as the starting material, and chloroacetoacetic acid. After the substitution reaction of the ethyl ester, the thiophenyl fragment in the molecule is introduced, which is then condensed with methylamine, followed by the Neitzescu reaction with p-benzoquinone, and the dimethylamine methyl group is introduced through the Mannich reaction. Reaction, then carry out deprotection reaction, and finally obtain the final product Arbidol hydrochloride through salification reaction
Its synthetic route is as follows:
Since the Nenitzescu reaction yield in this method is only 33.7%, the total yield is only 11.2%.
There are also bibliographical reports (Wen Yanzhen, Gao Zhiwei, Wei Wenlong, Zhi Cuimei, Wang Qi etc. in China Pharmaceutical Industry Journal 2006, “The Synthetic Route Diagram of Arbidol Hydrochloride” reported in 2006,37(12)) is based on ethyl acetoacetate. Ester and methylamine are used as starting materials, and Arbidol hydrochloride is obtained by Neitzescu reaction, acylation to protect hydroxyl group, bromination, thiophenol reaction, Mannich reaction, and acidification.
The synthetic route is as follows:
The method has relatively mild reaction conditions and relatively easy-to-obtain raw materials, but the total yield is still low, about 20%.
The above synthesis methods of Arbidol hydrochloride all use the Nenitzescu indole ring synthesis method to synthesize the indole ring of Arbidol hydrochloride, resulting in a low total reaction yield of about 10% to 20%.
SUMMARY OF THE INVENTION
In view of the above-mentioned problems, the object of the present invention is to provide a preparation method of Arbidol hydrochloride, the raw materials are easy to obtain, the reaction technical conditions are relatively simple, the reaction conditions are mild, and the total reaction yield is relatively high, reaching more than 30%. The cost is low, and it is suitable for industrial production. The method of the invention is based on the starting material of 3-iodo-4-nitrophenol, which is protected by a hydroxyl group, synthesized by indole ring, N-methylated, brominated, thiophenolated, and Mannich amine. Methylation reaction, acidification with hydrochloric acid, and purification to obtain Arbidol hydrochloride.
The reaction formula of the inventive method is as follows:
Fe stands for iron powder
CH 3 COOH stands for acetic acid
H 2 O is for water
(CH 3 ) 2 SO 4 Represents dimethyl sulfate
K 2 CO 3 stands for potassium carbonate
Example 1:
A preparation method of Arbidol hydrochloride, its steps are (preparation of compound 1):
A. Preparation of compound 1: 53 g of 3-iodo-4-nitrophenol was added to 160 g of acetone (drying over anhydrous potassium carbonate), 30.3 g of triethylamine was added, and 37.7 g of triethylamine was added dropwise at room temperature (20-25° C., the same below). g acetyl chloride, dripped in 1 hour, the reaction solution was automatically raised to reflux temperature T=56°C, reacted for 0.5h, cooled to room temperature T=25°C naturally, the reaction solution was poured into 1000g ice water, stirred, filtered, and the filter cake was washed with water , and vacuum-dried to obtain 57.4 g of compound 1 crude product with a yield of 93.6%. The next reaction was carried out directly without further purification.
B. Preparation of compound 2: 48.6 g of ethyl acetoacetate and 180 ml of freshly distilled tetrahydrofuran were added to a dry flask. Over 2 hours, 41.9 g of potassium tert-butoxide was added in portions with stirring. The temperature was raised to T=70°C (reflux), and the solution of 57.4 g of compound 1 obtained in the step and 75 mL of freshly distilled tetrahydrofuran was added dropwise, and the drop was completed in 2 hours. TLC plates monitor the reaction endpoint. After the reaction mixture was cooled to room temperature T=25°C, 93.5 ml of a 4 mol/L hydrochloric acid solution was added dropwise. The precipitated potassium chloride was removed by filtration, the solvent was evaporated under reduced pressure, and the obtained solid was washed with 45 mL of water and 60 mL of petroleum ether in turn to obtain 56.6 g of a crude product of compound 2 with a yield of 98%. The crude product can be recrystallized from the mixed solution of petroleum ether and ethyl acetate to obtain pure product.
C. Preparation of compound 3: add 56.6 g of compound 2, 160 mL of acetic acid and 160 mL of water to the flask, stir under nitrogen protection, add 30.8 g of iron powder in batches, stir vigorously, and heat the reaction mixture to T=80 °C for 4 h. End (TLC plate detection). Iron and its oxides were removed by filtration, water and acetic acid were distilled off under reduced pressure, neutralized with saturated sodium carbonate solution to weakly alkaline, extracted with ethyl acetate, dried over anhydrous magnesium sulfate, and concentrated to obtain 44.1 g of compound 3 crude product in a yield of 44.1 g. 92.3%.
D. Preparation of compound 3: add 10.0 g of compound 2, 28 mL of acetic acid and 28 mL of water to the flask, stir under nitrogen protection, add 7.2 g of iron powder in batches, stir vigorously, and simultaneously heat the reaction mixture to T=80 ° C, 4h reaction End (TLC plate detection). Iron and its oxides were removed by filtration, water and acetic acid were evaporated under reduced pressure, neutralized with saturated sodium carbonate solution to weakly alkaline, extracted with ethyl acetate, dried over anhydrous magnesium sulfate, and concentrated to obtain 7.5 g of compound 3 crude product, yield 89.4%.
E. Preparation of compound 4: 44.1 g of compound 3 prepared in step C was added to 230 ml of DMF, and after adding 35.0 g of anhydrous potassium carbonate, 31.9 g of dimethyl sulfate was slowly added dropwise at 100° C. under stirring, and the same temperature T= The reaction was carried out at 100 °C for 4 h. The reaction solution was cooled to room temperature of T=25°C, 280 ml of water was added under stirring, left to stand for crystallization, suction filtered, the filter cake was washed with water and dried to obtain 44.6 g of a crude compound 4, which was recrystallized with methanol to obtain 36.8 g of a refined compound of compound 4, Yield 79.3%
F. Preparation of compound 5: 36.8g of compound 4 was added to 200ml of carbon tetrachloride, 0.1g of benzoyl peroxide was added, heated to T=76°C and refluxed, 45.0g of bromine was added dropwise, and the reaction was completed within 2h. For 4 h, the reaction solution was cooled in an ice-water bath, filtered, and the filter cake was washed with a small amount of carbon tetrachloride, and dried to obtain 47.5 g of compound 5 crude product, with a yield of 82%.
G. Preparation of compound 6: dissolve 15.4 g of potassium hydroxide in 360 ml of methanol, stir, cool to 0-10° C. in an ice-water bath, add 12.7 g of thiophenol, react for 10 min, add 47.5 g of compound 5, and warm to room temperature, The reaction was carried out for 3 to 3.5 h, the reaction solution was poured into 1500 ml of ice water, adjusted to pH 2 with hydrochloric acid under stirring, filtered, the filter cake was washed with water, and dried in vacuo to obtain 42.9 g of crude compound 6 with a yield of 93.1%. The crude product was recrystallized with ethyl acetate, 10 g of activated carbon was decolorized, and 36.0 g of the dried compound 6 was purified, with a purification yield of 84%. The mother liquor of recrystallization is concentrated and recovered. Or to prepare compound 6, dissolve 3.3 g of potassium hydroxide in 75 ml of methanol, stir, cool to 0-10° C. in an ice-water bath, add 2.7 g of thiophenol, react for 10 min, add 10.0 g of compound 5, warm to room temperature, and react For 3-3.5 h, the reaction solution was poured into 300 ml of ice water, adjusted to pH 2 with hydrochloric acid under stirring, filtered, the filter cake was washed with water, and dried in vacuo to obtain 9.0 g of crude compound 6 with a yield of 93.1%. The crude product was recrystallized with isopropanol, 2 g of activated carbon was decolorized, and 6.3 g of the dried refined product of compound 6 was obtained, with a purification yield of 70%. The mother liquor of recrystallization is concentrated and recovered.
H. Preparation of compound 7:
In 320ml of ethanol, add, (33%) dimethylamine aqueous solution 29.2g, (37-40%) formaldehyde aqueous solution 23.8g, stir for 10min, add 36.0g compound 6, react at 60°C for 5h, the reaction is completed, 5.0g activated carbon decolorization, Filtration while hot, tetrahydrofuran was distilled off from the filtrate under reduced pressure, and dried to obtain 40.4 g of compound 7 crude product, with a yield of 99.0%. Or to prepare compound 7, under stirring and cooling conditions, 8.1 g of (33%) dimethylamine aqueous solution, 6.6 g of (37-40%) formaldehyde solution and 10 g of compound 6 were sequentially added to 100 ml of glacial acetic acid, and placed in 70 The reaction was carried out at °C for 6 hours. After the completion of the reaction, the reaction solution was concentrated under reduced pressure, 100 ml of water was added, and the pH was adjusted to 12 with trimethylamine solution. The aqueous phase was extracted three times with dichloromethane (20 ml×3), and the organic phase was dried over anhydrous sodium sulfate. Concentrate under reduced pressure and dry to obtain 9.6 g of crude compound 7, with a yield of 85.0%.
J. Preparation of compound 8:
40.4 g of the crude product of compound 7 obtained in the above step H was heated and dissolved in 150 ml of acetone, adjusted to pH=2 with hydrochloric acid while hot, a solid was precipitated, cooled to about 0°C in an ice-water bath, filtered, and the filter cake was washed with frozen acetone and dried in vacuo to obtain compound 8 Crude product 40.5g, yield 89.8%.
The above crude compound J was recrystallized from acetone-ethanol-water (3:1:1). 36.5 g of product were obtained, and the yield was 90.0%.
Research and develop a kind of method for efficient green synthesis of arbidol hydrochloride intermediate, the structural formula of arbidol hydrochloride is as follows:
Example 1: Ethyl 5-acetoxy-1,2-dimethylindole-3-carboxylate
After the device was installed, 150 mL of acetic anhydride solvent was added to the three-necked flask, and then solid ethyl 5-hydroxy-1,2-dimethylindole-3-carboxylate (23.3 g, 0.1 mol) was added while stirring. After all dissolved, heated to reflux for 4 h, after the reaction was completed, the reaction solution was cooled, and the solid was obtained by suction filtration. Wash the solid with water for 4 times (150 mL-200 mL of water each time), and slowly add 0.15 mol/L ammonia water to the solution in the third time to control the pH of the mixed system by adding water to the solid to be 8 to 9. Finally, suction filtration A solid was obtained, which was dried in an oven at 70° C. for 5 h to obtain a crude product. Recrystallization from methanol gave 18.8 g of ethyl 5-acetoxy-1,2-dimethylindole-3-carboxylate as brown crystals. Yield 65%.
Example 2: Ethyl 5-acetoxy-6-bromo-2-bromomethyl-1-methylindole-3-carboxylate
After installing the device, in a three-necked flask, ethyl 5-acetoxy-1,2-dimethylindole-3-carboxylate (17.9g, 0.065mol), catalyst (p-cymene)- Ruthenium dichloride dimer (4.0g, 0.0065moL), N-bromosuccinimide NBS (46.28g, 0.26moL) and 200mL dimethylacetamide DMA, slowly warmed to 90°C in oil bath under nitrogen protection , maintain the reaction temperature for 24h, after the reaction is over; cool the reaction solution to room temperature, add an appropriate amount of water to the reaction solution, extract 5 times with ethyl acetate, combine the organic phases, dry, spin dry the solvent to obtain a solid, use acetone After recrystallization, a white powdery solid was precipitated, which was dried in vacuo to obtain 23.3 g of ethyl 5-acetoxy-6-bromo-2-bromomethyl-1-methylindole-3-carboxylate. Yield 80%.
Example 3: Ethyl 6-bromo-5-hydroxy-1-methyl-2-phenylthiomethylindole-3-carboxylate
Install the device, add 150 mL of methanol solvent to the three-necked flask, slowly add 8.6 g of solid potassium hydroxide under stirring, cool to room temperature after all dissolved, then add thiophenol (6.2 g, 0.05 mL) under stirring, After about 15 min, ethyl 5-acetoxy-6-bromo-2-bromomethyl-1-methylindole-3-carboxylate (23.3 g, 0.05 moL) was finally added, and the reaction was stirred at room temperature for 4 h. After the reaction is completed. 10% acetic acid was added dropwise to the reaction solution until the pH of the reaction solution was 3-4. After a large amount of yellow solid was precipitated, the solid was obtained by suction filtration, washed once with water, filtered with suction, and dried at 70 °C for 5 h in a drying box. get crude products. Recrystallization from ethyl acetate gave 12.6 g of ethyl 6-bromo-5-hydroxy-1-methyl-2-phenylthiomethylindole-3-carboxylate as yellow-white crystals. Yield 60%.
Example 4: Arbidol
After installing the device, add 100 mL of glacial acetic acid solution to the three-necked flask, cool it to 0 °C, slowly add 40 mL of 40% methylamine aqueous solution, and then add 10 mL of 37% formaldehyde aqueous solution, and after the reaction is stirred for 15 min, add 6- Ethyl bromo-5-hydroxy-1-methyl-2-phenylthiomethylindole-3-carboxylate (12.6g, 0.03moL), stirred uniformly for 10 min, then began to heat up to 80°C, maintaining the reaction temperature , and react for 4 h after complete dissolution. After the reaction is over, pour the reaction solution into water, add an appropriate amount of 20% potassium hydroxide solution to neutralize it with stirring, adjust the pH of the solution to 7.0, precipitate solids, filter with suction, and wash with water once. The solid was obtained by suction filtration, and dried in an oven at 70 °C for 5 h to obtain a crude product. Recrystallize with acetonitrile, after complete dissolution, add 1 g of activated carbon to reflux for 30 min, filter hot, cool, and precipitate 8.5 g of brown solid Arbidol. Yield 60%.
Example 5: Arbidol hydrochloride
Install the device, add an appropriate amount of acetone solvent to the three-necked flask, add Arbidol (8.5g, 0.018moL) under stirring, heat to reflux, add 10mL of concentrated hydrochloric acid dropwise, reflux for 30 min, and after the reaction is over, cool the reaction The liquid was brought to room temperature, and filtered with suction to obtain crude Arbidol hydrochloride, which was dried in an oven at 50 °C for 3 h. Recrystallize with acetone:ethanol (3:2) solvent, cool at room temperature for 10 h, freeze in refrigerator for 10 h, suction filtration, wash the solid with a small amount of acetone, and obtain 7.0 g of refined Arbidol hydrochloride in a yield of 75%. MS (EI): m/z: 513.8754 ([M]+).
1,2-Dimethyl-5-hydroxyindole-3-acetic acid ethyl ester (I) is acetylated with acetic anhydride affording the O-acyl derivative (II) , which is brominated to the corresponding dibromide compound (III) . The reaction of (III) with thiophenol in KOH yields (IV) , which is then submitted to a conventional Mannich condensation with formaldehyde and dimethylamine in acetic acid, giving the free base of arbidol (V), which is treated with aqueous hydrochloric acid .
Umifenovir (Arbidol®) is an indole derivative first marketed in 1993 for the prophylactic treatment of infections caused by influenza A and B viruses [74]. Produced by Pharmstandard, it is still currently used in Russia and China to treat influenza infections [75]. Umifenovir is marketed in 50 and 100 mg capsules, being administered orally. The pharmacokinetics is limited, presenting rapid absorption and reaching the maximum concentration in 1.6–1.8 h. It is a slow elimination drug, with a half-life of 16 to 21 h, and may be administered twice a day [76].
The drug’s anti-influenza mechanism of action is related to arbidol’s ability to bind to the haemagglutinin (HA) protein [77]. The haemagglutinin (HA) protein is a homotrimeric glycoprotein found on the surface of the influenza virus, and it is essential for its infectivity. This protein is responsible for allowing the influenza virus binding to the sialic acid present on the surface of the target cells (respiratory tract cells or erythrocytes). As a result of this interaction, the virus is internalized in the host cell. Once umifenovir binds to the HA protein, this glycoprotein is prevented from binding to sialic acid, so the virus is no longer able to penetrate the host cell [78].
The structural similarity between the SARS-CoV-2 peak and the influenza virus (H3N2) HA glycoproteins justifies the fact that drugs that are capable of binding to HA can also do so to the SARS-CoV-2 spike protein. This fact was evidenced by molecular modeling studies, wherein was demonstrated that umifenovir is able to bind to the protein peak, preventing its trimerization, which would be a determining factor for the mechanism of cell adhesion (Fig. 8) [78].
Fig. 8. Umifenovir (in orange) binding region in SARS-CoV-2 spike glycoprotein. Reprinted from International Journal of Antimicrobial Agents, 56, N. Vankadari, “Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein”, Page 2, with permission of Elsevier. Copyright 2020. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Recently in 2020, in vitro studies performed with Vero cells confirmed that arbidol efficiently inhibits SARS-CoV-2 infection with an EC50 of 4.11 μM. The author also determined that arbidol was able to efficiently block both viral entry and post-entry stages, and also concluded that the drug prevented the viral attachment and release of SARS-CoV-2 from the intracellular vesicles. Importantly, the EC50 of arbidol against SARS-CoV-2 led the authors to suggest that the dose of arbidol currently recommended by the Chinese Guidelines (200 mg, 3 times/day) should be elevated in order to achieve ideal therapeutic efficacy to inhibit the SARS-CoV-2 infection [79].
A clinical trial was conducted at Wuhan Jinyintan Hospital, in 2020, from February 2 to March 20 conducted to evaluate the effectiveness and safety of umifenovir in the treatment of COVID-19 patients. In this study, 81 patients were evaluated: 45 received 200 mg of umifenovir three times a day, and 36 were in the control group. The authors concluded that baseline clinical and laboratory characteristics were similar in the two groups, and patients in the umifenovir group had a longer hospital stay than those in the control [80]. Although such results may seem discouraging, further clinical trials with higher doses of umifenovir may be required in order to verify its clinical efficiency against the SARS-CoV-2 infection.
The synthesis of umifenovir was described in 2006 starting from the reaction between ethyl acetoacetate 63 and methylamine, giving enaminone 64, which next undergoes a Nentizescu condensation reaction with 1,4-benzoquinone to produce indole derivative 65 (Scheme 9). Then, an acetylation reaction is carried out to protect the hydroxyl group in 65, producing 66, which is converted to 67 after a bromination step. The reaction of intermediate 67 with thiophenol in basic medium leads to the formation of 68, which finally affords umifenovir after a Mannich reaction [81].
Drug-repurposing studies are testing a range of compounds to treat COVID-19, but manufacturers may struggle to meet demand if any of these candidates prove effective against SARS-CoV-2. The pandemic has already strained global supply chains and limited the availability of a number of products, including hand sanitizer and diagnostic test reagents. The raw materials needed to make a new antiviral drug would most likely face similar pressures. But a team led by Tim Cernak of the University of Michigan has used an AI-based retrosynthesis program called Synthia to devise alternative routes to 12 leading drug candidates under investigation. The work appears on a preprint server and has not been peer reviewed (ChemRxiv 2020, DOI: 10.26434/chemrxiv.12765410.v1). “If the world runs out of one of the drugs currently in the clinic, we are providing a backup recipe,” Cernak says. Using alternative starting materials that are readily available, the researchers aimed to find routes of similar length and cost to those of existing syntheses. For each compound, the researchers whittled down a long list of options offered by Synthia to identify the most suitable synthetic strategies. Then the team tested some of these syntheses in the lab, including four new routes to the antiviral umifenovir, currently being investigated in eight clinical trials against COVID-19. Cernak says this approach could be used more generally to rapidly identify alternative synthetic routes whenever crises cause supply chain disruptions in drug manufacturing.
Artificial intelligence finds alternative routes to COVID-19 drug candidates
If drug-repurposing studies hit pay dirt, backup recipes could help antiviral manufacturers avoid supply chain problems
The research on the disease COVID-19 is an ongoing process since its outbreak as a pandemic. The repurposing of existing approved drugs has received priority attention due to some promising results obtained regarding COVID-19. In this article, some of the important chemical methodologies adopted for the synthesis of umifenovir, (s)-cidofovir, ribavirin, and ruxolitinib have been discussed. The repurposing of these approved drugs has received priority attention due to some promising results obtained regarding COVID-19 and some drugs are under more therapeutic trials. This manuscript has highlighted the synthetic strategies of four heterocyclic-based approved drugs, umifenovir, (s)-cidofovir, ribavirin, and ruxolitinib, repurposed for the treatment of COVID-19.
Ethyl 5-acetoxy-2-methyl-1H-indole-3-carboxylate 1a: Acetic anhydride (25.9 ml, 274 mmol 20 eq.) was added to a stirred solution of ethyl 5-hydroxy-2-methyl-1H-indole- 3-carboxylate 1 (3.00 g, 13.6 mmol, 1.0 eq.) in pyridine (3.32 mL, 41.1 mmol, 3.0 eq.) and the reaction heated to reflux. After 1 h, the reaction was allowed to cool back to rt before pouring the mixture into a solution of aqueous saturated sodium bicarbonate (40 mL). The product was extracted with ethyl acetate (3 x 40 ml) and the combined organic layers were washed with water (40 mL), dried (Na 2 SO 4 ) and concentrated In vacua to yield the product as a white solid which was used without further purification (3.4g, 96%). NMR: δH ( 400 MHz, CDCl 3) 8.34 (1H, s; NH), 7.75 (1H, s, H 4 ), 7.21 (1H, d, J 8.5, H 6 ), 6.89 (1H, d, J 8.5, H 7 ), 4.38 (2H , q, J 7.1 , CO 2 CH 2 CH 3 ), 2.71 (3H, s, C 1 CH 3 ), 2.34 (3H, s, CO 2 CH 3 ), 1.43 (3H, t, J 7.1, CO 2 CH 2 CH 3 ). δ c (100 MHz, CDCl 3 ) 170.8<a name=”
0.31 (40% ethyl acetate in hexane), HRMS. (ESI-TOF): C 14 H 15 O 4 N ([M+H] + ) requires 262.1074, found 262.1074.
Ethyl 5-acetoxy-1,2-dimethyl-1H-indole-3-carboxylate 1b: Protected indole 1b (1.35 g, 5.17 mmol, 1 eq.) was dissolved in DMF (15 mL). To this solution, methyl iodide (0.965 ml, 15.5 mmol, 3.0 eq.) was added and the resulting mixture was cooled on ice. Sodium hydride (0.186 g, 7.75 mmol, 1.5 eq.) was added and the reaction was left to stir on ice for 1.5 h. After this time, a small amount of water (5.0 mL) was added to the reaction and the solvents removed in vacuo. The resultant brown oil was then purified directly by column chromatography (30% ethyl acetate in petrol) to yield the title compound as a pale yellow solid (1.50 g, 95%). NMR: δ Η (500 MHz, CDCl 3 ) 7.79 (s, 1H, H 4 ), 7.26 (m, 1H, H 6 ), 6.96 (ddd, J = 8.8, 2.4, 0.8 Hz, 1H, H7 ), 4.38 (f, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.69 (s, 3H, NCH 3 ), 2.77 (d, J = 1.3 Hz, 3H, ΑrCΗ 3 ), 2.33 (s , 3H, CO 2 CH 3 ), 1.43 (t, J= 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ C (150 MHz, CDCl 3 ) 170.5 (CO 2 CH 3 ), 166.0 (CO 2 Et), 146.5 (C 5 ), 146.0 (C 2 ), 134.5 (C 8 ), 127.2 (C 3 ), 116.2 ( C6 ), 114.0 (C4 ) , 109.6 (C7 ), 104.4 (C 1 ), 59.6 (CO 2 CH 2 CH 3 ), 29.9 (NCH 3 ), 21.3 (C 1 CH 3 ), 14.8 (CO 2 CH 2 CH 3 ), 12.1 (CO 2 CH 3 ) . R f : 0.4 (30% ethyl acetate in hexane). HRMS (ESI-TOF): C 15 H 17 O 4 N ,([M+H] + ) requires 276.1230, found 276.1229.
Ethyl 6-bromo-2-(bromomethyl)-5-hydroxy-1-methyl-1H-indole-3-carboxylate 2: Bromine (558 μL, 10.9 mmol, 3.0 eq.) was added to a stirred solution of protected indole ( 1b, 1.00 g, 3.63 mmol, 1.0 eq.) in carbon tetrachloride (100 mL). After refluxing for 16 h, the reaction was cooled and aqueous sodium thlosulphate (10%. w/v, 100 mL) was added and left to stir for 20 min until the orange color disappeared. After this time, the organic layer was separated, washed with water (2 x 100 mL), dried (Na 2 SO 4 and concentrated in vacuo to yield a pale yellow solid, which was used without further purification (1.40 g, 99%) NMR: δ Η (400 MHz, PDCl 3 ) 7.86 (1H, s, H 4 ), 7.54 (1H, s, H 7), 5.05 (2H, s, CH 2 Br), 4.41 (2H, q, J 7.1, CO 2 CH 2 CH 3 ), 3.69 (3H, s, NCH 3 ), 2.39 (3H, s, CO 2 CH 3 ), 1.45 (3H, t, J 7.1, CO 2 CH 2 CH 3 ), δ C (100 MHz, CDCl 3 )
1.4.5 (CO 2 CH 2 CH 3 ), R f : 0.75 (CH 2 Cl 2 ). HRMS (ESI-TOF): C 15 Η 15 O 3 ΝΒr ([M+H] + ) requires 431.9441, found 431.9441.<a name=”
Ethyl 6-bromo-5-hydroxy-1-methyl-2-((phenylthio)methyl)-1H-indole-3-carboxylate 3: Thiophenol (99.8 μL, 0.972 mmol, 1.0 eq.) was added to a solution of potassium hydroxide (164 mg, 2.92 mmol, 3.0 eq.) in methanol (2 ml) and left to stir at room temperature for 15 min. After this time, the solution was cooled on ice and bromo indole 2 (880 mg, 0.972 mmol, 10 eq.) in CH 2 Cl 2 (5 mL) was added. The reaction was left to stir for 3 h before neutralization with acetic acid. The solvent was removed in vacuo and columned directly (20% EtOAc in petrol) to yield the title product as a pale yellow solid (362 mg, 86%). NMR: δ Η (600 MHz, CDCl 3 ) 7.74 (s, 1 H, Hr), 7.43 (s, 1H, H 4 ), 7.36 (dq, J = 5.2, 3.4, 2.4 Hz, 2H, H10 ), 7.25 (dd, J = 5.2, 1.9 Hz, 3H, H 11 and 5.33 (s, 2H, SCH 2 ), 4.29 (q, J = 7.3 Hz, 2H, CO 2 CH 2 CH 3 ), 3.60 (d, J = 18.1 Hz, 3H, NCH 3 ), 1.38 (t, J = 7.3 Hz, 3H, CO 2 CH 2 CH 3 ), δ c (150 MHz, CDCl 3 ) 165.1, 147.7, 144.2, 134.1 R f : 0.35 (20% EtOAc in petrol) HRMS (ESI-TOF)-: C 19 H 18 BrNO 3 S ([M+H] + ) requires 420.0263, found 420.0260.
Arbidol [Ethyl 6-bromo-4((dimethylamino)methyl)-5-hydroxy-1-methyl-2-((phenylthio)methyl)-1H-indole-3-carboxylate] 4: 1 Indole 3 (200 mg, 0.476 mmol, 1.0 eq.) and N, N, N’, N’-tetramethylaminomethane (1-95 μL, 1.43 mmol, 3.0 eq.) were dissolved in 1,4-dioxane (2 mL). The reaction was. heated to reflux for 3.5 h before removing the solvent in vacuo. The reaction was then re-dissolved in ethyl acetate and 1 M HCl was added to the solution causing the title product to crash out as a pale yellow solid (117 mg, 51%). NMR-: δ B (500 MHz, MeOD) 7.87 (s, 1 H, H 7 ), 7.39 (dd, J = 7.4, 2.2 Hz, 2H, H 10 ), 7.35 – 7.31 (m, 3H, H 11 and H12 ) . 4.87 (s, 2H, SCH 2), 4.71 (s, 2H, CH 2 NMe 2 ), 4.33 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CB 3 ), 3.63 (s, 3H, NCH 3 ), 2.97 (s, 6H, N (CH 3 ) 2 ), 1.39 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, MeOD) 169.7 (CO 2 Et), 152.7 (C s ), 149.0 (C), 137.7 (C 10 ), 136.4 (C 3 ), 136.0 (C 8 ), 132.3 (C 11 ), 131.3 (CH), 129.3 (C12 ) , 119.8 (C7 ) , 113.1 (C2), 111.3 ( C6), 108.3 (C4 ) , 64.2. (CO 2 CH 2 CH 3 ), 57.4 (CH 2 NMe 2 ), 45.4 (CH 2 N(CR 3 ) 2 ); 33.7 (CH 2 SPh), 32.9 (NCH 3 ), 16.6 (CO 2 CH 2 CH 3 ). R f : 0.25 (EtOAc). HRMS (ESI-TOF): G 22 H 25 BrN 2 O 3 S ([M+H] + ) requires 477.0842, found 477.0844.
Synthesis of Arbidol Analogues
Ethyl 5-acetoxy-6-bromo-2-(((3-hydroxyhpenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 8a: 3-hydroxythiophenol. (117 μL, 1.15 mmol, 1.0 eq.) was added to a solution of sodium carbonate (367 mg, 3.46 mmol, 3.0: eq.) and bromo indole 2 (500 mg, 1.15 mmol, 1.0 eq.) in dry ethyl acetate (10mL). The reaction was heated to 100°C and stirred for 5 h before<a name=”cooling, filtering and removing the solvent in vacuo. The compound was purified by column chromatography (40% EtOAc in Hexanes) to produce the title product as a pale yellow solid (240 mg, 44%). NMR: δ H (500 MHz,. CDCl 3 ) 7.85 (s, 1H, H 7 ), 7.56 (s, 1 H, H 4 ), 7.12 (t, J = 7.9 Ηz, 1Η, H 13 ), 6.95 – 6.90.(m, 1H, Η 14 ), 6.78 (s, 1H, H 10 ), 6.75-6.71 (m, 1H,.H 12 ), 4.69 (s, 2H, SCH 2 ), 4.30 (q, J = 7.4 Hz, 3H, CO 2 CH 2 CH 3 ), -3.66 (s, 3H, NGH 3 ), 2.4Q (s, 3H, COCH 3), 1.38 (t, J = 7.4 Hz, 3H, CO 2 CH 2 CH 3 ). Δ C (150 MHz, CDCL 3 ) 169.8, 165.0, 156.1, 144.6, 143.3, 135.6, 135.1, 130.1, 126.1, 124.8, 119.3, 113.9, 111.1, 105.8, 60.1, 30.4, 29.9, 21.0, 14. . R f : OAS (30% EtOAc in Hexane). HRMS (ESS-TOF): C 21 H 20 SrNO 5 S ([M+H] + ) requires 473.0318, found 478.0317.
Ethyl 6-bromo-5-hydroxy-24(((3-hydroxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 8: Sodium carbonate (106 mg. 1.00 mmol, 2.0 eq.) was added to a stirred solution of meta-hydroxy indole 8a (240 mg, 0.502 mmol, 1.0 eq.) in methanol (40 ml) and left to stir for 2h, The solution was then filtered and the solvent removed in vacuo, The product was re -dissolved in ethyl acetate (10 mL) and washed once with water (40 mL) before drying (Na 2 SO 4 and concentrating in vacuo to give the title product as a white solid, which could be used without further purification (160 mg, 67%), NMR-: δ H (600 MHz, MeOD) 7.60 (s, 1H, H 7 ), 7.58 (s, 1 H, H 4 ), 7.07 (dd, J = 8.2, 7.7 Hz, 1H, H 13), 6.83 – 6.81 (m, 1Η, Η 14 ), 6.79 (ddd, J = 7.7, 1.8, 0.9, 1H, H 10 ), 6.7.0 (dd, J = 8.2, 1.8, 0.9 Hz, 1H, H 12 ), 4.70 (s, 2H, SCH 2 ), 4.26 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.64 (s, 3H, NCH 3 ), 1.39 (t, J = 7.1 Hz , 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, MeOD) 166.9, 158.9, 150.6, 145.5, 136.4, 133.6, 131.0,. 130.7, 128.0, 1.24.9, 120.5, 116.0, 114.8, 107.9, 104.8, 60.8, 30.5, 30.4, 14.8. R f : 0.45 (1% MeOH in CΗ 2 Cl 2 ). HRMS (ESI-TOF): C 7 PM18 BrNO 4 S ([M+H] + ) requires 436.0213, found 436.0215.
Ethyl 2-(((3-aminophenyl)thio)methyl)-6-bromo-5-hydroxy-1-methyl-1H-indole-3-carboxyiate 9: 3-aminothiophenol (54.3 μL, 0.511 mmol, 1.0 eq.) was added to a solution of potassium hydroxide (86 mg, 1.53 mmol, 3.0 eq.) in methanol (2 ml) and left to stir at room temperature for 15 min. After this time, the solution was cooled on ice and bromo indole 2 (200 mg, 0.511 mmol, 1.0 eq.) in CH 2 Cl 2 (5 ml) was added. The reaction was left to stir for 3 h before neutralization with acetic-acid. The solvent was removed in vacuo and purified directly by preparative TLC (1% MeOH in CH 2 Cl 2 ) to yield the title product as a pale yellow solid (138 mg, 62%), NMR: δ H (500 MHz, CDCl 3) 7.74 (d, J = 1.8 Hz, 1H, H 7 ), .7.42 (d, J = 1.8 Hz., 1H, H 4 ), 7.03 (t, J = 8.1 Hz, 1 H, H 13 ), 6.75 (d, J= 7.7 Hz, 1H, H 14 ), 6.68 (s, 1H, H 10 ), 6.55 (d, J = 6.1 Hz, 1H, H 12 ), 4.68 (d, J = 1.9 Hz, 2H, SCH 2 ), 4.35 ™ 4.30 (m, 2H, COCH 2 GH 3 ),. 3.60 (d, J = 1.9 Hz, 3H, NCH 3 ), 1.40 (td, J = 7.1, 1.8 Hz, 3H, COCH 2 CH 3 ). δ c (150 MHz, CDCl 3 ) 1-66.9, 150.6, 149.6,<a name=”
146.0, 136.0, 133.5, 1 . 30.5, 128.1, 123.1, 120.2, 117.6, 115.8, 114.8, 107.9, 104.6, 68.1, 60.8, 30.4, 14.8. R f : 0.85 (1% MeOH in CH 2 Cl 2 ), HRMS (ESI-TOF): C 19 H 19 BrN 2 O 3 S ([M+H] + ) requires 435.0372, found 435.0370.
Ethyl 2-(((3-aminophenyl)thio)methyl)-6-bromo-5-hydroxy-1-methyl-1H-indole-3-carboxylate 10: 2-napthalenethiol (82.0 mg, 0.511 mmot, 1.0 eq.) was added to a solution of potassium hydroxide (86 mg, 1.53 mmol, 3.0 eq.) in methanol (2 mL) and left to stir at room temperature for 15 min. After this time, the solution was cooled on ice and bromo indole 2 (200 mg, 0.511 mmol, 1.0 eq.) in CH 2 Cl 2 (5 mL) was added. The reaction was left to stir for 3 h before neutralization with acetic acid. The solvent was removed in vacuo and purified directly by preparative TLC (1% MeOH in CH 2 Cl 2 ) to yield the title product as a pale yellow solid (118 mg, 50%). NMR: δR(600 MHz, DMSO) 9.77 (s, 1H, OH), 7.83 (d, J = 1.8 Hz, 1H, Ar), 7.81 -7.79 (m, 1H, Ar), 7.75 (d, J = 8.6 Hz, 1H , Ar), 7.72 – 7.70 (m, 1H, Ar), 7.66 (s, 1H, Hz), 7.46 (s, 1H, H 4 ), 7.45 – 7.40 (m, 2H, Ar), 7.34 (dd, J = 8.5, 1.9 Hz, 1H, Ar), 4.82 (s, 2H, CH 2 SPh), 4.04 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.63 (s, 3H, NC. %), 1.14 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, DMSO) 164.3, 149.3, 143.4, 133.2, 132.0, 131.7, 131.6, 128.9, 128.4, 128.4, 127.7, 127.2, 126.8, 126.3, 1:26.0, 116.3, 116.3, 116.3 06.3, 103.3, 59.2, 30.3, 28.1, 14.3. R f : 0.75 (1% MeOH in CH2 Cl 2 ). HRMS (ESI-TOF): C 23 H 20 BrNO 3 S (fM+Hf) requires 470.0420, found 470.0420.
Ethyl 6-bromo-4-((dimethylammino)methyl)-5-hydroxy-2-(((3-hydroxyohenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 11; Meta-hydroxy indole 8 (30.0 mg, 0.069 mmol, 1.0 eq,) and N, N,N’,N’-tetramethyldiaminomethane (47.0 μL, 0.344 mmol, 5.0 eq.) were dissolved in CH 2 Cl 2 (30 mL) . The reaction was heated to reflux for 3.5 h before removing the solvent in vacuo to, yield the title product as a pale yellow solid (34 mg. 99%). NMR; δH (500 MHz, CDCl 3 ) 7.47 (s, 1H, H 7 ), 7.12 (t, J = 7.9 Hz, 1H, H 13 ), 6.90 (d, J = 7.9 Hz, 1H, H 14 ), 6.90 ( d, J = 7.9 Hz, 1H, H 12 ), 6.66 (s, 1H, H 10 ), 4.41 (s, 2H, CH 2 NMe2 ), 4.34 (s, 2H, CH 2 SPh), 4.15 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.60 (s, 3H, NCH 3 ), 2.55 (s, 6H, CH 2N (CH 3 ) 2 ), . 1.33 – 1.21 (m, 3H, CO 2 CH 2 CH 3 ). δ c ( . 150 MHz, CDCl 3 ) 165.9, 156.7, 150.9, 142.6, 135.1, 132.2, 131.0, 130.0, 128.9, 124.6, 124.3, 119.3, 115.5, 113.4, 106.8, 106.8, 106.8, 106.8, 165.5 58.7, 44.0, 30.4, 29.9, 14.3, R f : 0.15 (10% MeOH in CH 2 Cl 2 ). HRMS (ESS-TOF): C22 H 25 BrN 2 O 4 S ([M+H] + ) requires 493.0791, found 493.0792,
0.0419 mmol, 1.0 eq.) and 1-(2-((trimethylsilyl)oxy)ethyl)piperazine (25 mg, 0.126 mmol, 3.0 eq.) were dissolved in 1,4-dioxane (2 mL) and refluxed overnight. The solvent was then removed in vacuo and the reaction columned directly to yield the title product as a yellow solid (12 mg, 51%). NMR: δ Η (400 MHz, MeOD) 7.56 (s, 1.H, H 7 ), 7.22 – 7.30 (m, 5H, SPh), 4.57 (s, 2H, CH 2 SPh), 4.14 – 4.19 (m, 4H, CH 2 NR 2 and CΟ 2 CH 2 CΗ 3 ), 3.68 (t, J = 5.9 Hz, 2H, . CH 2 OH), 3.60 (s, 3H, NCH 3 ), 2.53 – 2.70 (m, 10H, piperazine ring and CH 2 CH2 CH 2 OH), . 134 – 1.30 (rn, 3H, CO 2 CH 2 CH 3 ), δ c (150 MHz, MeOD) 167.2, 151.7, 143.9, 135.4, 134.2, 133.4, 130.1, 129.0, 125.5, 114.1, 113.6, 107.0, 108.9, 108.9, 108.9 61.5, 61.1, 59.8, 59.1, 54.3, 52.9, 30.9, 30.5, 14.7. R f : 0.15 (5% MeOH in CH 2 Cl 2 ), HRMS (ESI-TOF): C 26 H 32 BrN 3 O 4 S ([M+H] + ) requires 562.1370, found 562.1368,
Ethyl 6-bromo-5-hydroxy-2-(((2-hydroxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 16: 2-hydroxythiophenol (26.0 μL, 0.256 mmol, 1.0 eq.) was added to a solution of sodium carbonate (81.0 mg, 0.767 mmol, 3.0 eq.) and promo indole 2 (100 mg, 0.256 mmol, 1.0 eq.) in ethyl acetate (2 mL). The reaction was heated to 50°C and stirred for 2 h before cooling and removing the solvent in vacuo. The product was then re-dissolved in methanol (2 mL) and potassium hydroxide (21.5 mg, 0.384 mmol, 1.5 eq.) was added. The reaction was stirred at room temperature for 3 h before direct purification by preparative TLC (2% MeOH in CH 2 Cl 2 ) to yield the title product as. a white solid (20.5 mg, 1.8%), NMR: δ H (500 MHz, MeOD) 7.59 (s, 1H, H 7), 7.54 (s, 1H, Η 4 ), 7.17 (t, J = 7.7 Hz, 1H, H 12 ), 7.08 (d, J = 7.7 Hz, 1H, H 14 ), 6.85 (d, J =7.7 Hz , 1H, H 13 ), 6.66 (t, J = 7.7 Hz, 1H, H 15 ), 4.58 (s, 2H, SCH 2 ), 4.24 (q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ) , 3.59 (s, 3H, NCH 3 ), 1.40 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, MeOD) 161.3, 146.4, 143.3, 134.8, 132.0, 130.4, 129.4, 123.7, 123.0, 121.8, 120.5, 114.4, 113.6, 110.0, 102.7, 60.04, 390.2, 390.2, 390.04 R f : 0.6 (2% MeOH in CH 2 Cl2 ). HRMS (ESI-TOF): C 19 H 18 BrNO 4 S ([M+H] + ) requires 436.0213, found 436.0212.
Ethyl 6-bromo-5-hydroxy-2-(((4-hydroxyphenyl)thmetihoyl)-)1-methyl-1H-indole-3-carboxylate 17: 4-hydroxythiophenol (26.0 μL, 0.256 mmol, 1.0 eq.) was added to a solution of sodium carbonate (81.0 mg, 0.767 mmol, 3.0 eq.) and bromo indole 2 (100 mg, 0.256 mmol, 1.0 eq.) in ethyl acetate (2 mL). The reaction was heated to 50°C. and stirred for 2 hours before cooling<a name=”and removing the solvent in vacuo. The product was then re-dissolved in methanol (2 mL) and potassium hydroxide (21.5 mg, 0.384 mmol, 1.5 eq.) was added. The reaction was stirred at room temperature for 3 h before direct purification by preparative TLC (2% MeOH in CH 2 Cl 2 ) to yield the title product as a white solid (2.5 mg, 2%). NMR: δ Η (600 MHz, DMSO) 7.65 (s, 1H, H 7 ), 7.49 (s, 1 H, H 4 ), 7.03 (d, J = 8.7 Hz, 2H , H 12 ), 6.58 (d, J = 8.7 Hz, 2. HH 13 ) , 4.52 (s, 2H, SCH 2 ), 4.08 (q, J = 7.2 Hz, 2H, CO 2 CH 2 CH 3 ), 3.51 (s, 3H, NCH3 ), 1.23 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, DMSO) 164.2, 157.9, 149.2, 144.3, 135.7, 131.4, 126.1, 1214, 115.9, 114.1, 106.3, 102.9, 79.2, 59.0, 55.4, 30.0, R 14.3, f ; 0.5 (2% MeOH in CH 2 Cl 2 ). HRMS (ESI-TOF): C 19 H 18 BrNO 4 S ([Μ+H] + ) requires 436.0213, found 436.0213.
Ethyl 5-acetoxy-6-bromo-2-(((3-methoxyphenyl) thio)methyl)-1-methyl-1H-indole-3-carboxylate 18a: 3-methpxythiophenol (14.6 μL, 0.118 mmol, 1.0 eq.) was added to a solution of sodium carbonate (37.4 mg, 0.353 mmol, 3.0 eq.) and bromo indoles 2 (46.0 mg, 0.118 mmol, 1.0 eq.) in dry ethyl acetate (20 ml). The reaction was heated to 50°C and stirred for 2 h before addition of water. The organic layer was separated, dried (Na 2 SO 4 ) and concentrated in vacuo. The compound was purified by column chromatography (20% EtOAc in Hexanes) to produce the title product as a white solid (34 mg, 59%). UMR; δ Η (600 MHz, DMSO) 7.92 (s, 1 H, H 7 ), 7.6.6 (s, 1 H, H 4 ), 7.13 (t, J = 7.9 Hz, 1H, H 13), 6.8.7 – 6.84 (m, 1 H, H 14 , 6.79 – 6.74 (m, 2H, H 10 and H 1 2 ), 4.77 (s, 2H, SCH 2 ), 4.13 (q, J = 7.1 Hz , 2H, CO 2 CH 2 CH 3 ), 3.70 (s, 3H, NCH 3 ), 3.58 (s, 3H, SPhOCH 3 ), 2.27 (s, 3H, COCH 3 ), 1.20 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CW 3 ).δ c (150 MHz, DMSO) 169.1, 163.9, 159.4, 144.9, 142.6, 135.1, 135.0, 129.9, 125.2, 123.3, 116.2, 115.1, 114.73, 110.73, 110.2 104.3, 59.5, 55.1, 30.6, 28.1, 20.7, 14.3 R f : 0.4 (20% EtOAc in Hexane) HRMS (ESI-TOF): C 22H 22 BrNO s S ([M+H] + ) requires 492.0475, found 492.0472.
Ethyl 6-bromo-5-hydroxy-2-((3(-methoxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 18: Sodium carbonate (41.3 mg, 0.390 mmol, 2.0 eq.) was added to a stirred solution of meta-methoxy indole 18a (96.0 mg, 0.195 mmot, 1.0 eq.) in methanol (10 mL) and left to stir for 2h, the solution was then filtered and the solvent removed in vacuo. The product-was re-dissolved in ethyl acetate (10 mL) and washed once with water (10 mL) before drying (Na 2 SO 4 ) and concentrating in vacuo to give the title product as a white solid, which could he used without further purification (80 mg, 91%), NMR: δ Η (600 MHz, CDCl 3 ) 7.73 (s, 1H, Η 7 ), 7.41 (s, 1H, H 4 ), 7.17 – 7.13 (m, H 13), 7.07 (m, 1H, H 14 ), 6.96 (dt, J = 7.7, 1.3, 1H, H 10 ), 6.85 (m, 1H, H 12 ),4.71 (s, 2H, SCH 2 ), 4.30 ( q, J = 7.1 Hz, 2H, CO 2 CH 2 CH 3 ), 3.63 (s, 3H, NCH 3 ), 1.41 (t, J = 7.1 Hz, 3H, CO 2 CH 2 CH 3 ). Δ C (150 MHz, CDCL 3 ) 165.2, 159.8, 147.7, 135.4, 132.6, 129.8, 12.7.2, 124.6, 119.7, 117.3, 114.1, 112.5, 107.5, 107.9, 55.4, 29.9, 29.6, 14.7 Rf :. _<a name=”0.55 (1% MeOH in CH 2 Cl 2 ). HRMS (ESl-TOF): C 20 H 20 BrNO 4 S ([M+H] + ) requires 450.0369, found 450.0367.
Ethyl 6-bromo-4-((dimethylamino)methy-5-hydroxy-2-(((2-hydroxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylatete 19: Ortho-hydroxy indole: 16 (13.5 mg, 0.0309 mmol, 1.0 eq.) and N, N, N’,N’-tetramethyldiaminomethane (12.7 μL, 0.0928 mmol, 3.0 eq.) were dissolved in 1,4-dioxane (2.0 mL). reaction was heated io reflux for 3.5 h before removing the solvent in vacuo to yield the title product as a white solid (13 mg, 85 %).HMR: δ H (500 MHz, MeOD) 7.53 (s, 1H, H 7 ) , 7.19 – 7.11 (m, 1H, H 4 ), 7.03 (dd, J = 7.6, 1.7 Hz, 1 H, H 12 ), 6.86 – 6.78 (m, TH, H 14 ), 6.63 (dt, J = 13.7 , 7.6 Hz, 1H, H 15 ), 4.48 (s, 2H, CH 2 Sph), 4.34 (s, 2H, CH 2NMe 2 ), 4.22 (dq, J = 10.8, 7.1, 6.3 Hz, 2H, CO 2 CH 2 CH 3 ), 3.56 (s, 3H, NCH 3 ), 2.49 (d, J = 11.4 Hz, 6H, CH 2 N(CH 3 ) 2 ), 1.42 – 1.37 (m, 3H, CO 2 CH 2 CH 3 ). Δ C (150 MHz, MEOD) 167.5, 160.4, 137.0, 136.6, 132.5, 131.6, 130.6, 126.1, 114.5, 112.6, 110.6, 106.0, 68.1, 61.4, 60.1, 43.5, 29.9, 29.9 14.6. R f : 0.4 (5% MeOH in CH 2 Cl 2 ), HRMS (ESI-TOF): C 22 H 25 BrN 2 O4 S ([M+H] + ) requires 493.0791, found 493.0793.
Ethyl 6-bromo-4-((dimethylamino)methyl-5-hydroxy-2-(((3-methoxyphenyl)thio)methyl)-1-methyl-1H-indole-3-carboxylate 21: Sodium carbonate (17.5 mg, 0165 mmol, 3.0 eq.) was added to a stirred solution of meta-methoxy indole 18 (27.0 mg, 0.055 mmol, 1 .0 eq.) in ethyl acetate (8 mL) and methanol (1 mL). to stir for 3h before filtering and removing the solvent in vacuo.The compound was then re-dissolved in 1,4-dioxane (5 mL) and N, N, N’,N’-tetramethyldiaminomethane (5.5 μL, 0.04 mmol, 3.0 eq.) as added. The reaction was heated to reflux overnight before removing the solvent in vacuo. Purification by preparative TLC (5% MeOH In CH 2 Cl 2 ) yielded the title product as a pale yellow solid (7 mg, 24%) .NMR: δ Μ (600 MHz, CDCl 3) 7.44 (s, 1H, H 7 ), 7.19 (t, J = 7.9 Hz, 1 H, H 15 ), 6.96 (rn, 1H, H 14 ), 6.82 (m, 1H, W 10 ), 6.77 (m , 1H, H 12 ), 4.52 (s, 2H, CH 2 SPh), 4.21 (qd, J = 7.2, 0.8 Hz, 2H, CO 2 CH 2 CH 3 ), 4.17 (s, 2H, CH 2 NMe 2 ), 3.66 (s, 3H, NCH 3 ), 3.58 (s, 3H, OCH 3 ), 2.38 (s, 6H, CH 2 H(CH 3 ) 2 ), 1.34 (m, 3H, CO 2 CH 2 CH 3 ). δ c (150 MHz, CDCl 3) 165.6, 159.9, 151.7, 141.7, 135.5, 131.9, 129.9, 124.7, 117.5, 114.2, 113.1, 112.6, 108.6. 106.2, 60.5, 59.9, 55.3, 44.2, 30.5, 29.9, 14.5. R f : 0.35 (5% MeOH in CH 2 Cl 2 ). HUMS (ESI-TOF): C 23 H 27 BrN 2 O 5 S ((M+H] + ) requires 523.0897, found
^ Jump up to:abLeneva IA, Russell RJ, Boriskin YS, Hay AJ (February 2009). “Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol”. Antiviral Research. 81 (2): 132–40. doi:10.1016/j.antiviral.2008.10.009. PMID19028526.
^Wang MZ, Cai BQ, Li LY, Lin JT, Su N, Yu HX, Gao H, Zhao JZ, Liu L (June 2004). “[Efficacy and safety of arbidol in treatment of naturally acquired influenza]”. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. Acta Academiae Medicinae Sinicae. 26 (3): 289–93. PMID15266832.
^Boriskin YS, Leneva IA, Pécheur EI, Polyak SJ (2008). “Arbidol: a broad-spectrum antiviral compound that blocks viral fusion”. Current Medicinal Chemistry. 15 (10): 997–1005. doi:10.2174/092986708784049658. PMID18393857.
^Leneva IA, Burtseva EI, Yatsyshina SB, Fedyakina IT, Kirillova ES, Selkova EP, Osipova E, Maleev VV (February 2016). “Virus susceptibility and clinical effectiveness of anti-influenza drugs during the 2010-2011 influenza season in Russia”. International Journal of Infectious Diseases. 43: 77–84. doi:10.1016/j.ijid.2016.01.001. PMID26775570.
^Shi L, Xiong H, He J, Deng H, Li Q, Zhong Q, Hou W, Cheng L, Xiao H, Yang Z (2007). “Antiviral activity of arbidol against influenza A virus, respiratory syncytial virus, rhinovirus, coxsackie virus and adenovirus in vitro and in vivo”. Archives of Virology. 152 (8): 1447–55. doi:10.1007/s00705-007-0974-5. PMID17497238.
^Glushkov RG, Gus’kova TA, Krylova LIu, Nikolaeva IS (1999). “[Mechanisms of arbidole’s immunomodulating action]”. Vestnik Rossiiskoi Akademii Meditsinskikh Nauk (in Russian) (3): 36–40. PMID10222830.
^Hulseberg CE, Fénéant L, Szymańska-de Wijs KM, Kessler NP, Nelson EA, Shoemaker CJ, Schmaljohn CS, Polyak SJ, White JM. Arbidol and Other Low-Molecular-Weight Drugs That Inhibit Lassa and Ebola Viruses. J Virol. 2019 Apr 3;93(8). pii: e02185-18. doi:10.1128/JVI.02185-18PMID30700611
Umifenovir is an indole-based, hydrophobic, dual-acting direct antiviral/host-targeting agent used for the treatment and prophylaxis of influenza and other respiratory infections.13 It has been in use in Russia for approximately 25 years and in China since 2006. Its invention is credited to a collaboration between Russian scientists from several research institutes 40-50 years ago, and reports of its chemical synthesis date back to 1993.13 Umifenovir’s ability to exert antiviral effects through multiple pathways has resulted in considerable investigation into its use for a variety of enveloped and non-enveloped RNA and DNA viruses, including Flavivirus,2 Zika virus,3 foot-and-mouth disease,4 Lassa virus,6 Ebola virus,6 herpes simplex,8, hepatitis B and C viruses, chikungunya virus, reovirus, Hantaan virus, and coxsackie virus B5.13,9 This dual activity may also confer additional protection against viral resistance, as the development of resistance to umifenovir does not appear to be significant.13
Umifenovir is currently being investigated as a potential treatment and prophylactic agent for COVID-19 caused by SARS-CoV2 infections in combination with both currently available and investigational HIV therapies.1,16,17
Indication
Umifenovir is currently licensed in China and Russia for the prophylaxis and treatment of influenza and other respiratory viral infections.13 It has demonstrated activity against a number of viruses and has been investigated in the treatment of Flavivirus,2 Zika virus,3 foot-and-mouth disease,4 Lassa virus,6 Ebola virus,6 and herpes simplex.8 In addition, it has shown in vitro activity against hepatitis B and C viruses, chikungunya virus, reovirus, Hantaan virus, and coxsackie virus B5.13,9
Umifenovir is currently being investigated as a potential treatment and prophylactic agent for the prevention of COVID-19 caused by SARS-CoV-2 infections.1,16
Pharmacodynamics
Umifenovir exerts its antiviral effects via both direct-acting virucidal activity and by inhibiting one (or several) stage(s) of the viral life cycle.13 Its broad-spectrum of activity covers both enveloped and non-enveloped RNA and DNA viruses. It is relatively well-tolerated and possesses a large therapeutic window – weight-based doses up to 100-fold greater than those used in humans failed to produce any pathological changes in test animals.13
Umifenovir does not appear to result in significant viral resistance. Instances of umifenovir-resistant influenza virus demonstrated a single mutation in the HA2 subunit of influenza hemagglutinin, suggesting resistance is conferred by prevention of umifenovir’s activity related to membrane fusion. The mechanism through which other viruses may become resistant to umifenovir requires further study.13
Mechanism of action
Umifenovir is considered both a direct-acting antiviral (DAA) due to direct virucidal effects and a host-targeting agent (HTA) due to effects on one or multiple stages of viral life cycle (e.g. attachment, internalization), and its broad-spectrum antiviral activity is thought to be due to this dual activity.13 It is a hydrophobic molecule capable of forming aromatic stacking interactions with certain amino acid residues (e.g. tyrosine, tryptophan), which contributes to its ability to directly act against viruses. Antiviral activity may also be due to interactions with aromatic residues within the viral glycoproteins involved in fusion and cellular recognition,5,7 with the plasma membrane to interfere with clathrin-mediated exocytosis and intracellular trafficking,10 or directly with the viral lipid envelope itself (in enveloped viruses).13,12 Interactions at the plasma membrane may also serve to stabilize it and prevent viral entry (e.g. stabilizing influenza hemagglutinin inhibits the fusion step necessary for viral entry).13
Due to umifenovir’s ability to interact with both viral proteins and lipids, it may also interfere with later stages of the viral life cycle. Some virus families, such as Flaviviridae, replicate in a subcellular compartment called the membranous web – this web requires lipid-protein interactions that may be hindered by umifenovir. Similarly, viral assembly of hepatitis C viruses is contingent upon the assembly of lipoproteins, presenting another potential target.13
Absorption
Umifenovir is rapidly absorbed following oral administration, with an estimated Tmax between 0.65-1.8 hours.14,15,13 The Cmax has been estimated as 415 – 467 ng/mL and appears to increase linearly with dose,14,15 and the AUC0-inf following oral administration has been estimated to be approximately 2200 ng/mL/h.14,15
Volume of distribution
Data regarding the volume of distribution of umifenovir are currently unavailable.
Protein binding
Data regarding protein-binding of umifenovir are currently unavailable.
Metabolism
Umifenovir is highly metabolized in the body, primarily in hepatic and intestinal microsomess, with approximately 33 metabolites having been observed in human plasma, urine, and feces.14 The principal phase I metabolic pathways include sulfoxidation, N-demethylation, and hydroxylation, followed by phase II sulfate and glucuronide conjugation. In the urine, the major metabolites were sulfate and glucuronide conjugates, while the major species in the feces was unchanged parent drug (~40%) and the M10 metabolite (~3.0%). In the plasma, the principal metabolites are M6-1, M5, and M8 – of these, M6-1 appears of most importance given its high plasma exposure and long elimination half-life (~25h), making it a potentially important player in the safety and efficacy of umifenovir.14
Enzymes involved in the metabolism of umifenovir include members of the cytochrome P450 family (primarily CYP3A4), flavin-containing monooxygenase (FMO) family, and UDP-glucuronosyltransferase (UGT) family (specifically UGT1A9 and UGT2B7).14,11
Lu H: Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci Trends. 2020 Jan 28. doi: 10.5582/bst.2020.01020. [PubMed:31996494]
Haviernik J, Stefanik M, Fojtikova M, Kali S, Tordo N, Rudolf I, Hubalek Z, Eyer L, Ruzek D: Arbidol (Umifenovir): A Broad-Spectrum Antiviral Drug That Inhibits Medically Important Arthropod-Borne Flaviviruses. Viruses. 2018 Apr 10;10(4). pii: v10040184. doi: 10.3390/v10040184. [PubMed:29642580]
Fink SL, Vojtech L, Wagoner J, Slivinski NSJ, Jackson KJ, Wang R, Khadka S, Luthra P, Basler CF, Polyak SJ: The Antiviral Drug Arbidol Inhibits Zika Virus. Sci Rep. 2018 Jun 12;8(1):8989. doi: 10.1038/s41598-018-27224-4. [PubMed:29895962]
Herod MR, Adeyemi OO, Ward J, Bentley K, Harris M, Stonehouse NJ, Polyak SJ: The broad-spectrum antiviral drug arbidol inhibits foot-and-mouth disease virus genome replication. J Gen Virol. 2019 Sep;100(9):1293-1302. doi: 10.1099/jgv.0.001283. Epub 2019 Jun 4. [PubMed:31162013]
Kadam RU, Wilson IA: Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc Natl Acad Sci U S A. 2017 Jan 10;114(2):206-214. doi: 10.1073/pnas.1617020114. Epub 2016 Dec 21. [PubMed:28003465]
Hulseberg CE, Feneant L, Szymanska-de Wijs KM, Kessler NP, Nelson EA, Shoemaker CJ, Schmaljohn CS, Polyak SJ, White JM: Arbidol and Other Low-Molecular-Weight Drugs That Inhibit Lassa and Ebola Viruses. J Virol. 2019 Apr 3;93(8). pii: JVI.02185-18. doi: 10.1128/JVI.02185-18. Print 2019 Apr 15. [PubMed:30700611]
Zeng LY, Yang J, Liu S: Investigational hemagglutinin-targeted influenza virus inhibitors. Expert Opin Investig Drugs. 2017 Jan;26(1):63-73. doi: 10.1080/13543784.2017.1269170. Epub 2016 Dec 14. [PubMed:27918208]
Li MK, Liu YY, Wei F, Shen MX, Zhong Y, Li S, Chen LJ, Ma N, Liu BY, Mao YD, Li N, Hou W, Xiong HR, Yang ZQ: Antiviral activity of arbidol hydrochloride against herpes simplex virus I in vitro and in vivo. Int J Antimicrob Agents. 2018 Jan;51(1):98-106. doi: 10.1016/j.ijantimicag.2017.09.001. Epub 2017 Sep 7. [PubMed:28890393]
Pecheur EI, Borisevich V, Halfmann P, Morrey JD, Smee DF, Prichard M, Mire CE, Kawaoka Y, Geisbert TW, Polyak SJ: The Synthetic Antiviral Drug Arbidol Inhibits Globally Prevalent Pathogenic Viruses. J Virol. 2016 Jan 6;90(6):3086-92. doi: 10.1128/JVI.02077-15. [PubMed:26739045]
Blaising J, Levy PL, Polyak SJ, Stanifer M, Boulant S, Pecheur EI: Arbidol inhibits viral entry by interfering with clathrin-dependent trafficking. Antiviral Res. 2013 Oct;100(1):215-9. doi: 10.1016/j.antiviral.2013.08.008. Epub 2013 Aug 25. [PubMed:23981392]
Song JH, Fang ZZ, Zhu LL, Cao YF, Hu CM, Ge GB, Zhao DW: Glucuronidation of the broad-spectrum antiviral drug arbidol by UGT isoforms. J Pharm Pharmacol. 2013 Apr;65(4):521-7. doi: 10.1111/jphp.12014. Epub 2012 Dec 24. [PubMed:23488780]
Teissier E, Zandomeneghi G, Loquet A, Lavillette D, Lavergne JP, Montserret R, Cosset FL, Bockmann A, Meier BH, Penin F, Pecheur EI: Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PLoS One. 2011 Jan 25;6(1):e15874. doi: 10.1371/journal.pone.0015874. [PubMed:21283579]
Blaising J, Polyak SJ, Pecheur EI: Arbidol as a broad-spectrum antiviral: an update. Antiviral Res. 2014 Jul;107:84-94. doi: 10.1016/j.antiviral.2014.04.006. Epub 2014 Apr 24. [PubMed:24769245]
Deng P, Zhong D, Yu K, Zhang Y, Wang T, Chen X: Pharmacokinetics, metabolism, and excretion of the antiviral drug arbidol in humans. Antimicrob Agents Chemother. 2013 Apr;57(4):1743-55. doi: 10.1128/AAC.02282-12. Epub 2013 Jan 28. [PubMed:23357765]
Liu MY, Wang S, Yao WF, Wu HZ, Meng SN, Wei MJ: Pharmacokinetic properties and bioequivalence of two formulations of arbidol: an open-label, single-dose, randomized-sequence, two-period crossover study in healthy Chinese male volunteers. Clin Ther. 2009 Apr;31(4):784-92. doi: 10.1016/j.clinthera.2009.04.016. [PubMed:19446151]
Wang Z, Chen X, Lu Y, Chen F, Zhang W: Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Biosci Trends. 2020 Feb 9. doi: 10.5582/bst.2020.01030. [PubMed:32037389]
Nature Biotechnology: Coronavirus puts drug repurposing on the fast track [Link]
Lanraplenib (GS-9876) is a highly selective and orally active SYK inhibitor (IC50=9.5 nM) in development for the treatment of inflammatory diseases. Lanraplenib (GS-9876) inhibits SYK activity in platelets via the glycoprotein VI (GPVI) receptor without prolonging bleeding time (BT) in monkeys or humans.
Description
Lanraplenib (GS-9876) is a highly selective and orally active SYK inhibitor (IC50=9.5 nM) in development for the treatment of inflammatory diseases. Lanraplenib (GS-9876) inhibits SYK activity in platelets via the glycoprotein VI (GPVI) receptor without prolonging bleeding time (BT) in monkeys or humans[1][2][3].
Lanraplenib (GS-9876) inhibits anti-IgM stimulated phosphorylation of AKT, BLNK, BTK, ERK, MEK, and PKCδ in human B cells with EC50 values of 24-51 nM. Lanraplenib (GS-9876) inhibits anti-IgM mediated CD69 and CD86 expression on B-cells (EC50=112±10 nM and 164±15 nM, respectively) and anti-IgM /anti-CD40 co-stimulated B cell proliferation (EC50=108±55 nM). In human macrophages, Lanraplenib (GS-9876) inhibits IC-stimulated TNFα and IL-1β release (EC50=121±77 nM and 9±17 nM, respectively)[1].
Lanraplenib (GS-9876) inhibits glycoprotein VI (GPVI)-induced phosphorylation of linker for activation of T cells and phospholipase Cγ2, platelet activation and aggregation in human whole blood, and platelet binding to collagen under arterial flow[2].
Spleen tyrosine kinase (SYK) is a critical regulator of signaling in a variety of immune cell types such as B-cells, monocytes, and macrophages. Accordingly, there have been numerous efforts to identify compounds that selectively inhibit SYK as a means to treat autoimmune and inflammatory diseases. We previously disclosed GS-9973 (entospletinib) as a selective SYK inhibitor that is under clinical evaluation in hematological malignancies. However, a BID dosing regimen and drug interaction with proton pump inhibitors (PPI) prevented development of entospletinib in inflammatory diseases. Herein, we report the discovery of a second-generation SYK inhibitor, GS–9876 (lanraplenib), which has human pharmacokinetic properties suitable for once-daily administration and is devoid of any interactions with PPI. Lanraplenib is currently under clinical evaluation in multiple autoimmune indications.
Step 6. 6-(6-Aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)imidazo|1,2-a]pyrazin-8-amine (39). To a solution of tert-butyl(6-(6-(bis(tert-butoxycarbonyl)amino)pyrazm-2-yl)imidazo[1,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin1yl)phenyl)carbamate 45 (200 mg, 0.269 mmol) in DCM (2 ml) was added TFA (0.5 ml, 6.578 mmol). The reaction was stirred at room temperature for 16h, treated with saturated sodium bicarbonate, extracted with EtOAc, and purified on silica gel, eluting with 5%MeOH / EtOAc to 20%MeOH / EtOAc. The desired fractions were combined and concentrated to provide 100 mg (83% yield) of the title compound 39. m/z calcd for C23H25N9O [M+H] + 444.23, found LCMS-ESI+ (m/z): [M+H] + 444.20. 1H NMR (300 MHz d6-DMSO) δ: 9.5 (s,lH), 8.588 (s, 1H), 8.47 (s, 1H), 8.12 (d, 1H), 7.95-7.92 (d5 2H), 7.88 (s, 1H), 7.62 (s, 1H), 6.99-6.96 (d, 2H), 6.46 (s, 2H), 4.57- 4.53 (m, 2H), 4.48-4.44 (m, 2H), 3.43 (m, 1H), 3.15-3.12 (m, 4H), 2.41- 2.38 (m, 4H).
Protein kinases, the largest family of human enzymes, encompass well over 500 human proteins. Spleen Tyrosine Kinase (Syk) is a member of the Syk family of tyrosine kinases, and is a regulator of early B-cell development as well as mature B-cell activation, signaling, and survival.
Acute Graft Versus Host Disease (aGVHD), also known as fulminant Graft Versus Host Disease, generally presents symptoms within the first 100 days following allogenic hematopoietic stem cell transplantation and is generally characterized by selective damage to the skin, liver, mucosa, and gastrointestinal tract. Chronic Graft Versus Host Disease (cGVHD) occurs in recipients of allogeneic hematopoietic stem cell transplant (HSCT). GVHD is considered chronic when it occurs >100 days post-transplant, though aspects of cGVHD may manifest themselves prior to the 100 day point and overlap with elements of aGVHD. The disease has a cumulative incidence of 35-70% of transplanted patients, and has an annual incidence of approximately 3,000-5,000 and a prevalence of approximately 10,000 in the US. cGVHD is difficult to treat and is associated with worse outcomes compared to those without cGVHD. Current standard of care includes a variety of approaches including systemic corticosteroids often combined with calcineurin inhibitors, mTOR inhibitors, mycophenylate mofetil, or rituximab. Despite treatment, response rates are poor (40-50%) and cGVHD is associated with significant morbidity such as serious infection and impaired quality of life; the 5-year mortality is 30-50% (Blazar et al., Nature Reviews Immunology 12, 443-458, June 2012).
Human and animal models have demonstrated that aberrant B-lymphocyte signaling and survival is important in the pathogenesis of cGVHD. B-cell targeted drugs, including SYK inhibitors (fostamatinib – Sarantopoulos et al, Biology of Blood and Marrow Transplantation, 21(2015) S 11 -S 18) and BTK inhibitors (ibrutinib – Nakasone et al, Int. J. HematoL- 27 March 2015), have been shown to selectively reduce the function and frequency of aberrant GVHD B-cell populations ex vivo.
There remains a need for new methods, pharmaceutical compositions, and regimens for the treatment of GVHD, including aGVHD and cGVHD.
Example 2. Preparation of 6-(6-aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-l- yl)phenyl)imid azo [ 1,2-a] pyrazin-8-amine (2)
2-Bis(tert-butoxycarbonyl)amino-6-bromopyrazine XIV: To a mixture of 6-bromopyrazin-2-amine (5 g, 28.7 mmol) and di-tert-butyl dicarbonate (25.09 g, 1 14.94 mmol) was added DCM (10 ml) followed by DMAP (0.351 g, 29 mmol). The reaction was heated to 55 °C for lh, cooled to RT, the reaction was partitioned between water and DCM, purified on silica gel and concentrated to provide of 2-bis(tert-butoxycarbonyl)amino-6-bromopyrazine XIV. LCMS-ESI+ (m/z): [M+H]+: 374.14. XH NMR (DMSO) δ: 8.84(d, 2H), 1.39 (s, 18H).
tert-Butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)carbamate XVI – CHEMISTRY A route: tert-Butyl 4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl(6-(tributylstannyl)imidazo[l,2-a]pyrazin-8-yl)carbamate V (215 mg, 0.291 mmol), was combined with 2-bis(tert-butoxycarbonyl)amino-6-bromopyrazine XIV (217.58 mg, 0.581 mmol),
bis(triphenylphosphine)palladium(II) dichloride(30.61 mg, 0.044 mmol) and 1,4-dioxane (5ml). The reaction mixture was stirred in a microwave reactor at 120 °C for 30 min. The reaction mixture was quenched with saturated KF, extracted with EtOAc, purified on silica gel, eluted with EtOAc. The desired fractions were combined and concentrated to provide 100 mg (46% yield) of tert-butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)carbamate XVI. LCMS-ESI+ (m/z): [M+H]+: 744.4. lH NMR (300 MHz d6-DMSO) δ: 9.37 (s, 1H), 9.18 (s, 1H), 8.77 (s, 1H), 8.33 (d, 1H), 7.87 (d, 1H), 7.28-7.25 (d, 2H), 6.92-6.89 (d, 2H), 4.55-4.41 (m, 4H), 3.4 (m, lH), 3.14-3. 11 (m,4H), 2,37-2.34 (m, 4H), 1.37 (s, 18H), 1.3 (s, 9H).
tert-Butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)carbamate XVI – CHEMISTRY B route: Step 1 : To a dry 250 mL round-bottomed flask was added 2-bis(tert-butoxycarbonyl)amino-6-bromopyrazine XIV (l .Og, l .Oequiv, 2.67mmol), KOAc (790mg, 8.02mmol, 3.0equiv), 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(l ,3,2-dioxaborolane) (750mg, 2.94mmol, l . l equiv), Pd(dba) (171mg, 0.187mmol, 0.07equiv) and X-phos (128mg, 0.267mmol, O. lequiv) followed by 1,4-dioxane (25mL) and the solution was sonicated for 5 min and then purged with N2 gas for 5 min. The flask with contents was then placed under N2 atmosphere and heated at 1 10 °C for 90 min. Once full conversion to the pinacolboronate was achieved by LCMS, the reaction was removed from heat and allowed to cool to RT. Once cool, the reaction contents were filtered through Celite and the filter cake was washed 3 x 20 mL EtOAc. The resultant solution was then concentrated down to a deep red-orange
syrup providing N, N-BisBoc 6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyrazin-2-amine XV, which was used directly in the next step.
Step 2: The freshly formed N, N-BisBoc 6-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyrazin-2-amine XV (2.67 mmol based on 100% conversion, 2.0 equiv based on bromide) was dissolved in 20 Ml of 1,2-dimethoxy ethane and to that solution was added tert-butyl (6-bromoimidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)carbamate IV (707mg, 1.34mmol, l .Oequiv), Na2CC>3 (283mg, 2.67mmol, 2.0equiv), Pd(PPh3)4 (155mg, 0.134mmol, 0.1 equiv) and water (l OmL) and the solution was degassed for 5 min using N2 gas. The reaction was then placed under N2 atmosphere and heated at 110 °C for 90 min. LCMS showed complete consumption of the bromide starting material and the reaction was removed from heat and allowed to cool to RT. The reaction was diluted with 100 mL water and 100 mL 20% MeOH/DCM and the organic layer was recovered, extracted 1 x sat. NaHCCb, 1 x sat brine and then dried over Na2SC>4. The solution was then filtered and concentrated down to an orange-red solid. The sample was then slurried in warm MeOH, sonicated then filtered, washing 2 x 20 mL with cold MeOH and then the cream-colored solid was dried on hi-vacuum overnight to yield 905 mg of tert-butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin- 1 -yl)phenyl)carbamate XVI.
6-(6-Aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)imidazo[l,2-a]pyrazin-8-amine (2): To a solution of tert-butyl (6-(6-(bis(tert-butoxycarbonyl)amino)pyrazin-2-yl)imidazo[l,2-a]pyrazin-8-yl)(4-(4-(oxetan-3-yl)piperazin-l -yl)phenyl)carbamate XVI (200 mg, 0.269 mmol) in DCM (2 ml) was added TFA (0.5 ml, 6.578 mmol). The reaction was stirred at rt for 16h, saturated sodium bicarbonate was added, extracted with EtOAC and purified on silica gel, eluted with 5%MeOH / EtOAc, 20%MeOH / EtOAc. The desired fractions were combined and concentrated to provide the title compound 2. LCMS-ESI+(m/z): [M+H]+: 444.2. lH NMR (300 MHz d6-DMSO) δ: 9.5 (s, lH), 8.588 (s, IH), 8.47 (s, IH), 8. 12 (d, IH), 7.95-7.92 (d, 2H), 7.88 (s, IH), 7.62 (s, IH), 6.99-6.96 (d, 2H), 6.46 (s, 2H), 4.57-4.53 (m, 2H), 4.48-4.44 (m, 2H), 3.43 (m, IH), 3.15-3.12 (m, 4H), 2.41 -2.38 (m, 4H).
To a 720 L reactor, was added di-fer/-butyl (6-bromopyrazin-2-yl)imidodicarbonate (18.5 kg, 1.41 equiv, 49 mol), bis(pinacolato)diboron (13.8 kg, 1.56 equiv, 54 mol), potassium propionate (11.9 kg, 3.02 equiv, 106 mol), and bis(di-fer/-butyl(4-dimethylaminophenyl) phosphine)dichloropalladium (1.07 kg, 0.0043 equiv, 1.5 mol), followed by degassed toluene (173 L). The mixture was degassed then heated at 65 °C until the reaction was deemed complete (0% tert-butyl 2-((6-bromopyrazin-2-yl)(tert-butoxycarbonyl)amino)-2-oxoacetate) by UPLC. Upon completion, the reaction was cooled to 23 °C. Once cooled, 6-bromo-N-(4-(4-(oxetan-3-yl)piperazin-l -yl)phenyl)imidazo[l ,2-a]pyrazin-8-amine (15.0 kg, 1.00 equiv, 35 mol) was added and the mixture was degassed. A degassed aqueous potassium carbonate solution prepared using water (54 L) and potassium carbonate (20.6 g, 4.26 equiv, 149 mol) was then added to the reaction mixture and the reactor contents was degassed. The reactor contents was heated at 65 °C until reaction was deemed complete (1% 6-bromo-N-(4-(4-(oxetan-3-yl)piperazin-l-yl)phenyl)imidazo[l,2-a]pyrazin-8-amine) by UPLC. Upon completion, the reaction was cooled to 24 °C.
The cooled mixture was concentrated and then diluted with dichloromethane (300 L), transferred to a 1900 L reactor and rinsed forward with dichloromethane (57 L). N-acetyl-L-cysteine (3.8 kg) was charged and the mixture was agitated for 15 h. Water (135 L) was then added and the mixture was filtered and rinsed forward with dichloromethane (68 L). The organic layer was recovered and washed with a brine solution prepared using water (68 L) and sodium chloride (7.5 kg).
The resultant organic layer was polish filtered then concentrated and fert-butyl methyl ether (89.9 kg) was slowly charged keeping the temperature at 31 °C. The contents was cooled to 0 °C and aged, then filtered and rinsed with tert-butyl methyl ether (32.7 kg) and dried at 40 °C to give 17.2 kg of di-tert-butyl {6-[8-({4-[4-(oxetan-3-yl)piperazin-l-yl]phenyl} amino)imidazo[l,2-a]pyrazin-6-yl]pyrazin-2-yl}imidodicarbonate.
To a slurry of di-tert-butyl {6-[8-({4-[4-(oxetan-3-yl)piperazin-l -yl]phenyl} amino)imidazo[l,2-a]pyrazin-6-yl]pyrazin-2-yl}imidodicarbonate (225 g, 0.35 mol, 1 mol eq.) in water (12 parts) was added a solution of sulfuric acid (3.1 parts, 6.99 mol, 20 mol eq.) in water (5 parts). The reaction was heated to ca. 40 °C and stirred at this temperature for ca. 4 h at which point the reaction is deemed complete. The reaction mixture was cooled to ca. 22 °C, acetone (3 parts) was charged and a solution of sodium carbonate (4.1 parts, 8.75 mol, 25.0 mol eq.) in water (15 parts) was added. The resulting slurry was filtered and the wet cake was washed with water in portions (4 x 1 parts), then with fert-butyl methyl ether (4 parts). The wet cake (Example 2 free base) was dried at ca. 60 °C. To the slurry of dry Example 2 free base in 2-propanol (2.3 parts) was added a solution of succinic acid (Based on the isolated Example 2 free base: 0.43 parts, 1.6 mol eq.) in 2-propanol (15 parts). The resulting slurry was heated to ca. 40 °C and stirred at this temperature for ca. 2 h and then cooled to ca. 22 °C, followed by a stir period of ca. 16 h. The slurry was filtered at ca. 22 °C and the wet cake was washed with 2-propanol (5 parts) and dried at ca. 60 °C to afford the product.
Tranilast (INN, brand name Rizaben) is an antiallergic drug. It was developed by Kissei Pharmaceuticals and was approved in 1982 for use in Japan and South Korea for bronchial asthma. Indications for keloid and hypertrophic scar were added in the 1980s.
Kissei has developed and launched tranilast in Japan and South Korea for the treatment of allergic rhinitis, asthma and atopic dermatitis. Kissei, in collaboration with GlaxoSmithKline was additionally developing tranilast for the prevention of restenosis following percutaneous transluminal coronary angioplasty.
It should not be taken women who are or might become pregnant, and it is secreted in breast milk.[1]
Interactions
People who are taking warfarin should not also take tranilast, as they interact.[1] It appears to inhibit UGT1A1 so will interfere with metabolism of drugs that are affected by that enzyme.[1]
Adverse effects
When given systemically, tranilast appears to cause liver damage; in a large well-conducted clinical trial it caused elevated transaminases three times the upper limit of normal in 11 percent of patients, as well as anemia, kidney failure, rash, and problems urinating.[1]
As of March 2018 it was marketed in Japan, China, and South Korea under the brand names Ao Te Min, Arenist, Brecrus, Garesirol, Hustigen, Krix, Lumios, Rizaben, Tramelas, Tranilast and it was marketed as a combination drug with salbutamol under the brand name Shun Qi.[2]
In 2016 the FDA proposed that tranilast be excluded from the list of active pharmaceutical ingredients that compounding pharmacies in the US could formulate with a prescription.[1]
Pharmacology
It appears to work by inhibiting the release of histamine from mast cells; it has been found to inhibit proliferation of fibroblasts but its biological target is not known.[3] It has been shown to inhibit the release of many cytokines in various cell types, in in vitro studies.[3] It has also been shown to inhibit NALP3 inflammasome activation and is being studied as a treatment for NALP3-driven inflammatory diseases.[4]
Chemistry
Tranilast is an analog of a metabolite of tryptophan, and its chemical name is 3′,4′-dimethoxycinnamoyl) anthranilic acid (N-5′).[3]
It is almost insoluble in water, easily soluble in dimethylsulfoxide, soluble in dioxane, and very slightly soluble in ether. It is photochemically unstable in solution.[3]
Orally active anti-allergic agent. Prepn: K. Harita et al., DE 2402398; idem, US 3940422 (1974, 1976 both to Kissei).
Y. Kamijo, M. Kobayashi, and A. Ajisawa, Jpn. Kokai, 77/83,428 (1977) via Chem. Abstr.,
As of 2016, Altacor was developing a formulation of tranilast to prevent of scarring following glaucoma surgery and had obtained an orphan designation from the EMA for this use.[7][8]
History
It was developed by Kissei and first approved in Japan and South Korea for asthma in 1982, and approved uses for keloid and hypertrophic scars were added later in the 1980s.[3]
PATENT
tranilast product case US03940422 , expired in all the regional territories.
PATENT
WO2013144916 claiming tranilast complexes and cocrystals with nicotinamide, saccharin, gentisic acid, salicylic acid, urea, 4-aminobenzoic acid and 2,4-dihydroxybenzoic acid
Novel crystalline forms of tranilast or its salts as histamine H1 receptor antagonist useful for treating allergy, allergic rhinitis and atopic dermatitis.
Tranilast, (2-[[3-(3,4-dimethoxyphenyl)-l-oxo-2-propenyl]amino] benzoic acid, shown below), was originally developed as an anti-allergy drug due to its ability to inhibit the release of inflammatory mediators, such as histamine, from mast cells and basophils (P. Zampini. IntJ Immunopharmacol. 1983;
Tranilast
Tranilast has been marketed in Japan, China and South Korea by Kissei Pharmaceutical Co. Ltd, for allergic conditions such as allergic conjunctivitis, bronchial asthma, allergic rhinitis and atopic dermatitis, under the Rizaben® brand name for more than thirty years. More recently tranilast has also been shown to have anti-proliferative properties. Tranilast was shown to inhibit the proliferation of fibroblasts and suppress collagen synthesis (M. Isaji. Biochem Pharmacol. 1987; 36: 469-474) and also to inhibit the transformation of fibroblasts to myofibroblasts and their subsequent contraction (M. Isaji. Life Sci. 1994; 55: 287-292). This additional behaviour led to tranilast gaining additional approval for the treatment of keloids and hypertrophic scars.
[004] Over recent years many researchers have explored the anti-proliferative effects of tranilast to assess its potential in fibrotic and cancerous conditions. Its anti-proliferative action is believed to be due to its ability to inhibit transforming growth factor beta (TGF-b) (H. Suzawa. Jpn J Pharmacol. 1992 Oct; 60(2): 91-96). Fibrosis is a condition that can affect most organs of the body and fibroblast proliferation, differentiation and collagen synthesis are known to be key factors in the progression of most types of fibrosis. Tranilast has been shown in-vivo to have potential beneficial effects in
numerous fibrotic conditions. Tranilast has been shown in-vivo to have potential in lung fibrosis (M. Kato. Eur RespirJ. 2013; 42(57): 2330), kidney fibrosis (DJ Kelly, J Am Soc Nephrol. 2004; 15(10): 2619-29), cardiac fibrosis (J Martin, Cardiovasc Res. 2005; 65(3): 694-701), ocular fibrosis (M J Moon, BMC Opthalmol. 2016; 16: 166) and liver fibrosis (M Uno, Hepatology. 2008; 48(1): 109-18.
[005] Tranilast’s anti-tumor action has also recently been demonstrated, in-vitro and in-vivo. Tranilast has been shown to inhibit the proliferation, apoptosis and migration of several cell lines including breast cancer (R. Chakrabarti. Anticancer Drugs. 2009 Jun; 20(5): 334-45) and prostate cancer (S. Sato. Prostate. 2010 Feb; 70(3): 229-38) cell lines. In a study of mammary carcinoma in mice tranilast was found to produce a significant reduction in metastasis (R. Chakrabarti. Anticancer Drugs. 2009 Jun; 20(5): 334-45). In a pilot study in humans, tranilast was shown to have the potential to improve the prognosis of patients with advanced castration-resistant prostate cancer (K. Izumi. Anticancer Research. 2010 Jul; 30: 73077-81). In-vitro studies also showed the therapeutic potential of tranilast in glioma (M Platten. IntJ Cancer. 2001; 93:53-61), pancreatic cancer (M Hiroi, J Nippon Med Sch. 2002; 69: 224-234) and gastric carcinoma (M Yashiro, Anticancer Res. 2003; 23: 3899-3904).
[006] Given the wide range of fibrotic conditions and cancers for which tranilast could have a potential therapeutic benefit, as well as the different patient types and specific areas of the body requiring treatment, it is anticipated that patients would benefit from having multiple delivery methods for the administration of tranilast so as to best suit the patient’s needs. The pharmaceutical compositions could include, for example, a solid oral dosage, a liquid oral dosage, an injectable composition, an inhalable composition, a topical composition or a transdermal composition.
[007] Kissei Pharmaceutical Co. Ltd explored the anti-proliferative effect of tranilast in the prevention of restenosis associated with coronary intervention. In a Phase II clinical study Kissei found that the current approved dose of tranilast (300 mg/day) was insufficient to prevent restenosis and that a higher dose of 600 mg/day was needed to achieve a decrease in restenosis rates (H. Tamai, Am Heart J.1999; 138(5): 968-75). However, it was found that a 600 mg daily dosage can result in a ten-fold inter-patient variation in plasma concentrations of the drug (30-300 pmol/L) (H Kusa ma. Atherosclerosis. 1999; 143: 307-313) and in the Phase III study of tranilast for the prevention of restenosis the dose was further increased to 900mg daily (D Holmes, Circulation. 2002; 106(10): 1243-1250).
[008] The marketed oral form of tranilast (Rizaben®) contains tranilast in its pure crystalline form. Crystalline tranilast has extremely low aqueous solubility (solubility of 14.5 pg/ml in water and 0.7 pg/ml in pH 1.2 buffer solution (Society of Japanese Pharmacopoeia. 2002)). Whilst, high energy amorphous forms are often used as a means of improving the solubility of poorly soluble drug
compounds, literature shows that an amorphous form of tranilast is not completely photostable in the solid state and that it undergoes photodegradation on storage when exposed to light (S. Onoue. EurJ Pharm Sci. 2010; 39: 256-262).
[009] It is expected that the very low solubility of tranilast is a limiting factor in the oral bioavailability of the drug. Given the limited time any drug has to firstly dissolve in the
gastrointestinal tract and then be absorbed into the bloodstream, this issue will become even more limiting as the oral dose of tranilast is increased. The poor solubility of tranilast is also possibly a key factor in the high inter-patient variability reported for higher dose tranilast pharmacokinetics. As a BCS class II drug (low solubility/high permeability) it is expected that absorption from the gastrointestinal tract is hampered by the dissolution rate of the drug in gastrointestinal media as well as its overall solubility. For treatment of chronic proliferative diseases such as fibrosis and cancer it is vital for the delivery method of a drug to produce consistent, predictable plasma levels that are maintained above the minimum effective concentration. To achieve efficacious oral delivery of tranilast at higher doses there is a need for new solid forms of the drug with both high solubility and rapid dissolution rates.
[010] Given the severity of conditions involving cancer or fibrosis there is also a need for systemic treatment options by which tranilast can be delivered by healthcare specialists that do not require the patient to swallow solid oral dosage forms. Alternative dosage forms suitable for these needs could include, for example, injectable compositions, liquid oral formulations or nebulized inhaled formulations. These would require a liquid formulation of tranilast suitable for systemic delivery. [Oil] Given the potential of tranilast to treat ocular diseases, such as allergic conjunctivitis, Kissei Pharmaceutical Co. Ltd recognised the need to develop an eye drop formulation of tranilast for localised treatment. However, as well as having very low aqueous solubility, tranilast is also photochemically unstable when stored in solution, resulting in significant degradation (N Hori, Chem. Pharm. Bull. 1999; 47(12): 1713-1716). Therefore, the only way Kissei were able to achieve an eye drop liquid composition of tranilast was to use both solubilising and stabilising agents in the formulation (US Patent 5356620). The resulting 0.5% (w/v) eye drop formulation is currently also marketed under the Rizaben® brand name. However, the focus of this formulation and of the subsequent research that has attempted to produce alternative solution formulations of tranilast has always been solely on external delivery of tranilast using compositions such as eye drops and skin ointments etc. None of the liquid formulations of tranilast previously described have been produced for systemic delivery such as for oral or IV delivery. Excipients used in the previously reported external preparations are not suitable for systemic delivery. Also, despite the successful
development of an eye drop formulation of tranilast, the package insert of the marketed Rizaben® eye drops states that the product should not be stored in a refrigerator as crystals may precipitate.
[012] Thus, there remains a need for aqueous pharmaceutical compositions of tranilast suitable for systemic delivery. Given the potential photochemical degradation issue of long term storage of tranilast in solution and also the disadvantage of the larger storage facilities needed to store bulkier solution based formulations it would also be advantageous to develop a stable highly soluble solid form of tranilast that can be quickly dissolved at the time of treatment by the patient or healthcare provider to produce the required liquid formulation.
[013] Following efforts to make a liquid formulation of tranilast, Kissei made the statement that tranilast and pharmaceutically acceptable salts thereof are too insoluble in water to prepare an aqueous solution (US Patent 5356620). Since that US patent the only crystalline pharmaceutically acceptable salt to have been published is the sodium salt (N Geng, Cryst. Growth Des. 2013; 13: 3546-3553). In line with the findings of Kissei the authors of this paper stated that the apparent solubility of the crystalline tranilast sodium salt is even less than that of pure tranilast. Also, when they performed a dissolution study of tranilast in a sodium containing media they found that as the tranilast dissolved it gradually precipitated out of solution as its sodium salt indicating that the sodium salt has a lower thermodynamic solubility than the pure drug. The authors of this paper also successfully prepared the non-pharmaceutically acceptable crystalline cytosine salt of tranilast. Despite this crystalline cytosine salt showing approximately a two-fold solubility improvement over pure crystalline tranilast, not only would this crystalline cytosine salt not be suitable for systemic delivery to a patient due to cytosine not having FDA acceptability but this improvement in solubility would not be great enough to produce high dose tranilast liquid formulations such as an injectable formulation.
[014] Patent application EP1946753 discloses an attempt to prepare an external preparation of tranilast and claims the preparation of ionic liquid salts of tranilast with organic amines. The inventors claim that blending tranilast with the organic amine results in a liquid form. This application does not disclose the formation of any solid state, crystalline tranilast salts with organic amines. They demonstrate that these ionic liquid forms of tranilast have higher solubility in solvents suitable for external application to the skin and that these preparations have higher photostability than pure tranilast in the same formulation. However, this improved photostability still results in a significant proportion of the tranilast being photo-degraded and would not be suitable for long term storage. Also, the solvents used for preparation of these ionic liquid salt formulations are not suitable for internal delivery of tranilast. Moreover, there is no mention in EP1946753 of improved solubility in aqueous or bio-relevant media.
Tranilast, (2-[[3-(3,4-dimethoxyphenyl)-1-oxo-2-propenyl]amino]benzoic acid), shown below, is a therapeutic agent that exhibits an anti-allergic effect. It has been shown to inhibit the release of inflammatory mediators, such as histamine, from mast cells and basophils (P. Zampini. Int J Immunopharmacol. 1983; 5(5): 431-5). Tranilast has been used as an anti-allergic treatment, for several years in Japan and South Korea, for conditions such as allergic conjunctivitis, bronchial asthma, allergic rhinitis and atopic dermatitis.
[0004]
Tranilast is currently marketed in Japan and South Korea by Kissei Pharmaceutical Co. Ltd under the Rizaben® brand name. As well as displaying an anti-allergic effect tranilast has been shown to possess anti-proliferative properties. Tranilast was found to inhibit the proliferation of fibroblasts and suppress collagen synthesis (M. Isaji. Biochem Pharmacol. 1987; 36: 469-474) and also to inhibit the transformation of fibroblasts to myofibroblasts and their subsequent contraction (M. Isaji. Life Sci. 1994; 55: 287-292). On the basis of these effects tranilast is now also indicated for the treatment of keloids and hypertrophic scars. Its anti-fibrotic action is believed to be due to its ability to inhibit transforming growth factor beta (TGF-β) (H. Suzawa. Jpn J Pharmacol. 1992 October; 60(2): 91-96). TGF-β induced fibroblast proliferation, differentiation and collagen synthesis are known to be key factors in the progression of idiopathic pulmonary fibrosis and tranilast has been shown in-viva to have potential in the treatment of this chronic lung disease (T. Jiang. Afr J Pharm Pharmaco. 2011; 5(10): 1315-1320). Tranilast has also been shown in-vivo to be have potential beneficial effects in the treatment of airway remodelling associated with chronic asthma (S. C. Kim. J Asthma 2009; 46(9): 884-894.
[0005]
It has been reported that tranilast also has activity as an angiogenesis inhibitor (M. Isaji. Br. J Pharmacol. 1997; 122(6): 1061-1066). The results of this study suggested that tranilast may be beneficial for the treatment of angiogenic diseases such as diabetic retinopathy and age related macular degeneration. As well as showing inhibitory effects on mast cells and fibroblasts, tranilast has also demonstrated an ability to diminish tumor necrosis factor-alpha (TNF-α) from cultured macrophages (H. O. Pae. Biochem Biophys Res Commun. 371: 361-365) and T-cells (M. Platten. Science. 310: 850-855), and inhibited NF-kB-dependent transcriptional activation in endothelial cells (M. Spieker. Mol Pharmacol. 62: 856-863). Recent studies have revealed that tranilast attenuates inflammation and inhibits bone destruction in collagen induced arthritis in mice suggesting the possible usefulness of tranilast in the treatment of inflammatory conditions such as arthritis (N. Shiota. Br. Pharmacol. 2010; 159 (3): 626-635).
[0006]
As has recently been demonstrated, in-vitro and in-vivo, tranilast also possesses an anti-tumor action. Tranilast has been shown to inhibit the proliferation, apoptosis and migration of several cell lines including breast cancer (R. Chakrabarti. Anticancer Drugs. 2009 June; 20(5): 334-45) and prostate cancer (S. Sato. Prostate. 2010 February; 70(3): 229-38) cell lines. In a study of mammary carcinoma in mice tranilast was found to produce a significant reduction in metastasis (R. Chakrabarti. Anticancer Drugs. 2009 June; 20(5): 334-45). In a pilot study in humans, tranilast was shown to have the potential to improve the prognosis of patients with advanced castration-resistant prostate cancer (K. Izurni. Anticancer Research. 2010 July; 30: 73077-81).
[0007]
It has been reported that tranilast has the ability to induce or enhance neurogenesis and, therefore, could be used as an agent to treat neuronal conditions such as cerebral ischernia, glaucoma, multiple sclerosis, amyotrophic lateral sclerosis, Alzheimer’s disease, neurodegenerative trinucleotide repeat disorders, neurodegenerative lyosomal storage diseases, spinal cord injury and trauma, dementia, schizophrenia and peripheral neuropathy (A. Schneider. EP2030617).
[0008]
Tranilast’s beneficial properties have been reported to have utility in several ocular conditions. Tranilast is currently approved in Japan and Korea far the treatment of allergic conjunctivitis. WO2010137681 claims the use of tranilast as a prophylactic or therapeutic agent for the treatment of retinal diseases. The anti-fibrotic properties of tranilast have been reported to be of benefit in maintaining the filtering blob during glaucoma surgery and this has been demonstrated in a pilot study in humans (E. Chihara.J Glaucoma. 1999; 11(2): 127-133). There have also been several reported cases of the beneficial use of tranilast in the prevention of postoperative recurrence of pterygium (C. Fukui. Jap J Opthalmol. 1999; 12: 547-549). Tsuji recently reported that tranilast may be beneficial not only in the prevention of ptergium recurrence, but also for the inhibition of symblepharon and granuloma formation (A. Tsuji. Tokai J Exp Clin Med. 2011; 36(4): 120-123). Collectively it has been demonstrated that tranilast possesses anti-allergic, anti-fibrotic, anti-inflammatory, anti-tumor, neurogenesis enhancing end angiogenesis inhibitory properties and as such may be useful for the treatment of diseases associated with such properties.
[0009]
Tranilast occurs as a yellow crystalline powder that is identified by CAS Registry Number: 53902-12-8. As is typical of cinnamic acid derivatives (G. M. J. Schmidt J Chem. Soc. 1964: 2000) tranilast is photochemically unstable when in solution, tranforming into cis-isomer and dimer forms on exposure to light (N. Hori. Cehm Pharm Bull. 1999; 47: 1713-1716). Although pure crystalline tranilast is photochemically stable in the solid state it is practically insoluble in water (14.5 μg/ml) and acidic media (0.7 μg/ml in pH 1.2 buffer solution) (Society of Japanese Pharmacopoeia. 2002). Although tranilast has shown activity in various indications, it is possible that the therapeutic potential of the drug is currently limited by its poor solubility and photostability. High energy amorphous forms are often used as a means of improving the solubility of poorly soluble APIs, however, literature shows that amorphous solid dispersions of tranilast are not completely photostable in the solid state and that they undergo photodegradation on storage when exposed to light (S. Onoue. Eur J Pharm Sci. 2010; 39: 256-262). US20110136835 describes a combination of tranilast and allopurinol and its use in the treatment of hyperuricemia associated with gout and has one mention of a “co-crystal form”, but lacks any further description or characterization.
JP2011225626A *2001-02-012011-11-10Rohto Pharmaceutical Co LtdEye lotion
US6585997B22001-08-162003-07-01Access Pharmaceuticals, Inc.Mucoadhesive erodible drug delivery device for controlled administration of pharmaceuticals and other active compounds
CA2548281C2003-12-092013-11-12Medcrystalforms, LlcMethod of preparation of mixed phase co-crystals with active agents
JP2005314229A *2004-03-312005-11-10Rohto Pharmaceut Co LtdTranilast-containing medicine composition
JP2007051089A *2005-08-182007-03-01Medorekkusu:KkPreparation for external use
WO2007046544A1 *2005-10-212007-04-26Medrx Co., Ltd.Preparation for external application comprising salt of mast cell degranulation inhibitor having carboxyl group with organic amine
WO2008078730A1 *2006-12-262008-07-03Translational Research, Ltd.Preparation for transnasal application
EP2030617A12007-08-172009-03-04Sygnis Bioscience GmbH & Co. KGUse of tranilast and derivatives thereof for the therapy of neurological conditions
CN101683330A *2008-09-232010-03-31沈阳三川医药科技有限公司Oral compound pharmaceutic preparation containing tranilast and salbutamol
US20110136835A1 *2009-09-142011-06-09Nuon Therapeutics, Inc.Combination formulations of tranilast and allopurinol and methods related thereto
WO2010137681A12009-05-292010-12-02参天製薬株式会社Prophylactic or therapeutic agent for retinal diseases comprising tranilast, method for prevention or treatment of retinal diseases, and tranilast or pharmaceutically acceptable salt thereof and use thereof
JP2011093849A *2009-10-302011-05-12Kissei Pharmaceutical Co LtdEasily dissolvable powder inhalant composed of tranilast
^Holmes, D. R; Savage, M; Lablanche, J. M; Grip, L; Serruys, P. W; Fitzgerald, P; Fischman, D; Goldberg, S; Brinker, J. A; Zeiher, A. M; Shapiro, L. M; Willerson, J; Davis, B. R; Ferguson, J. J; Popma, J; King Sb, 3rd; Lincoff, A. M; Tcheng, J. E; Chan, R; Granett, J. R; Poland, M (2002). “Results of Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) Trial”. Circulation. 106 (10): 1243–50. doi:10.1161/01.CIR.0000028335.31300.DA. PMID12208800.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....














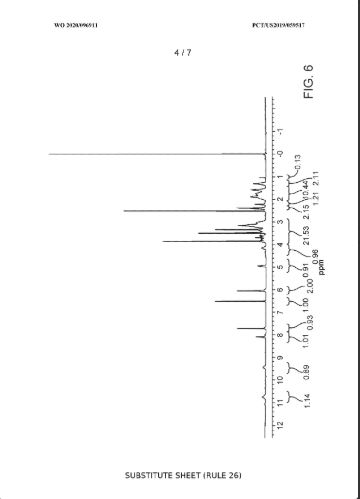

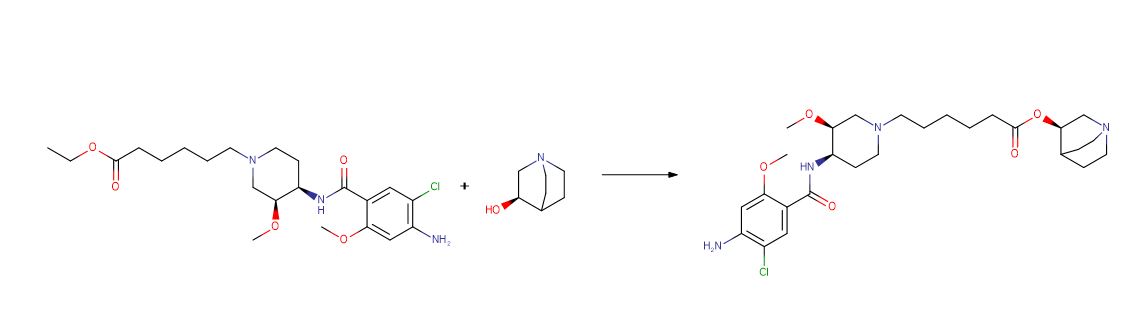
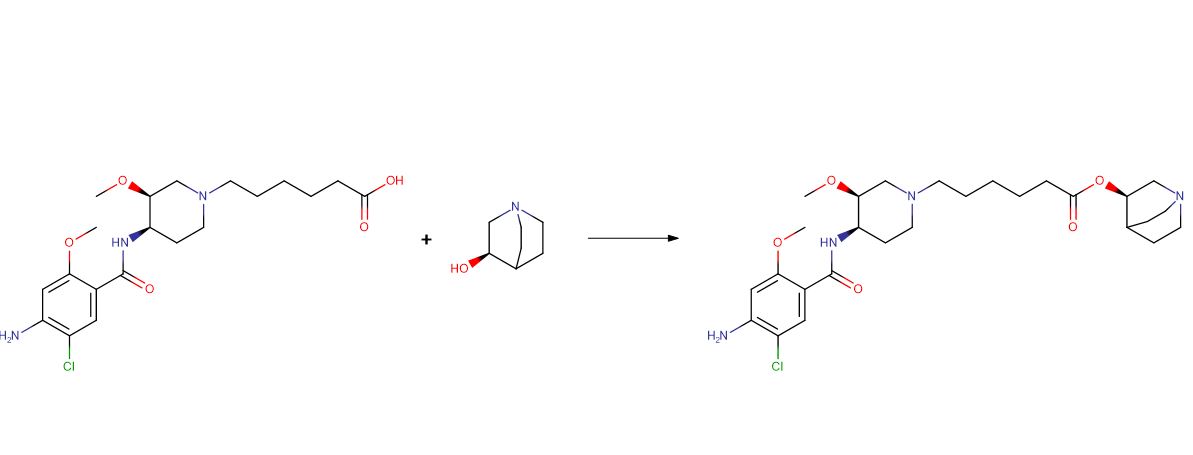






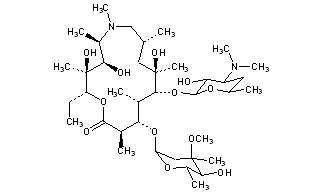
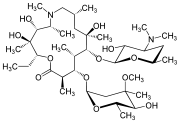
_tablets.jpg)
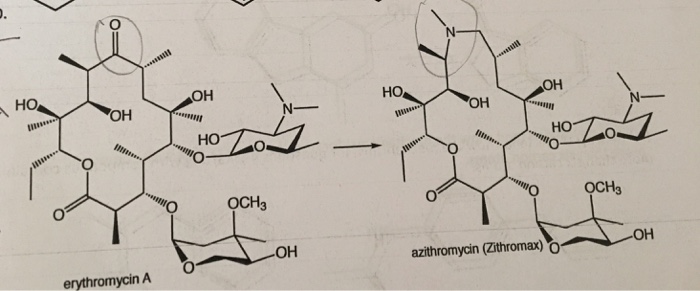
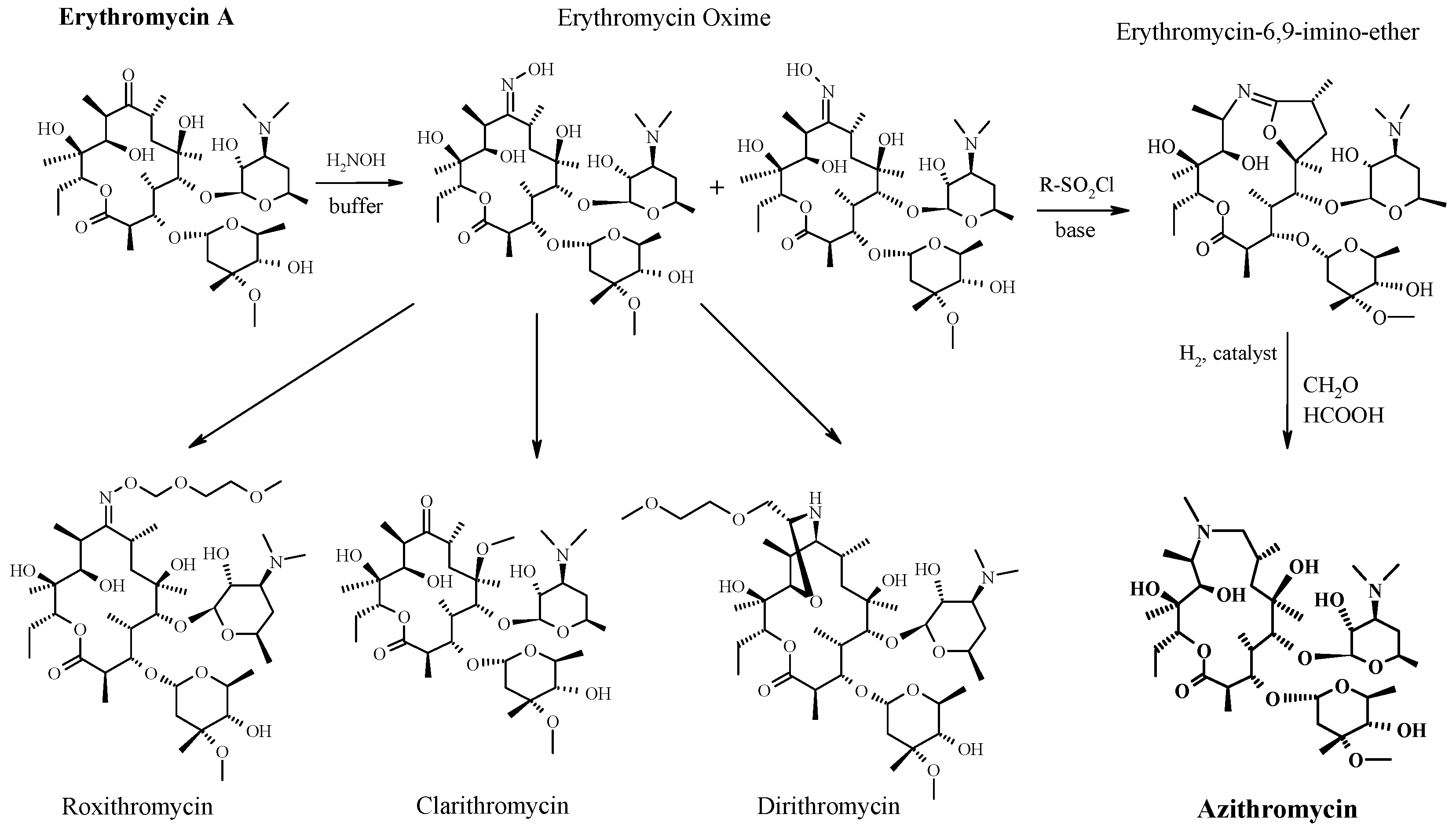


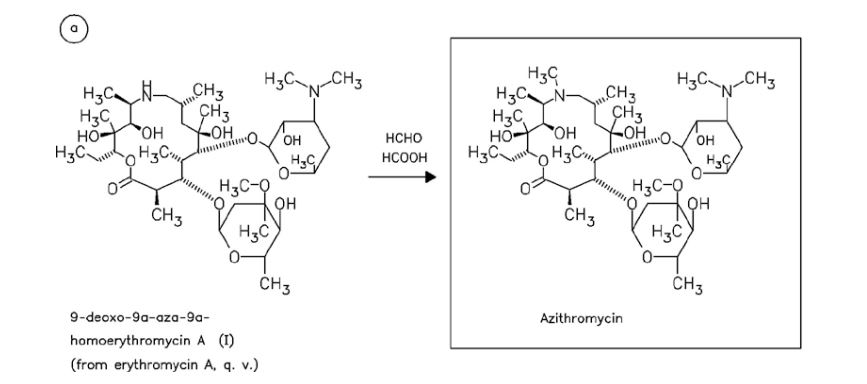
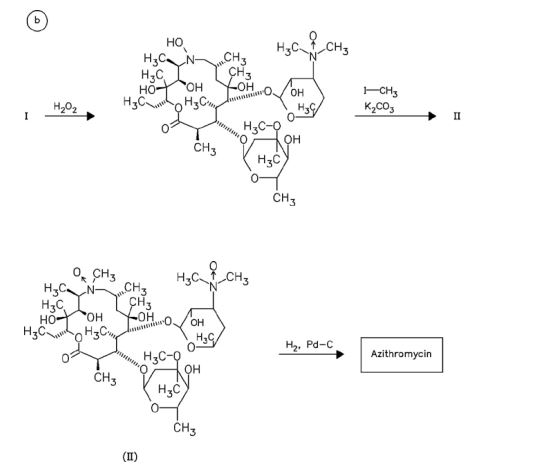
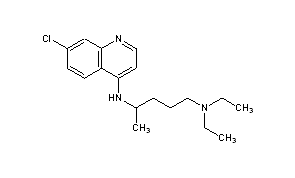
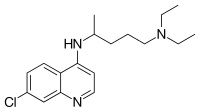



.jpg)


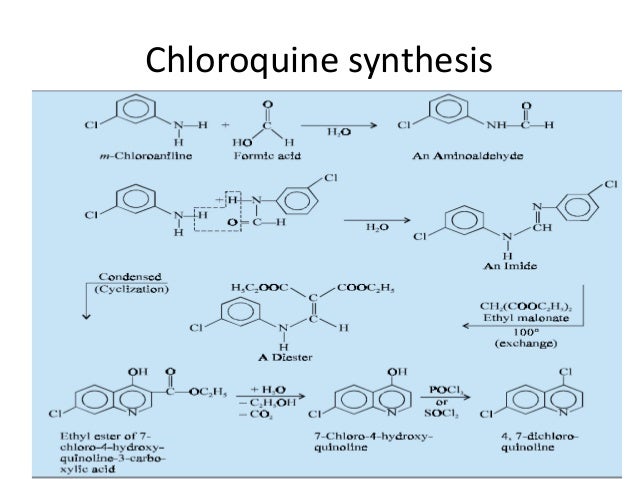
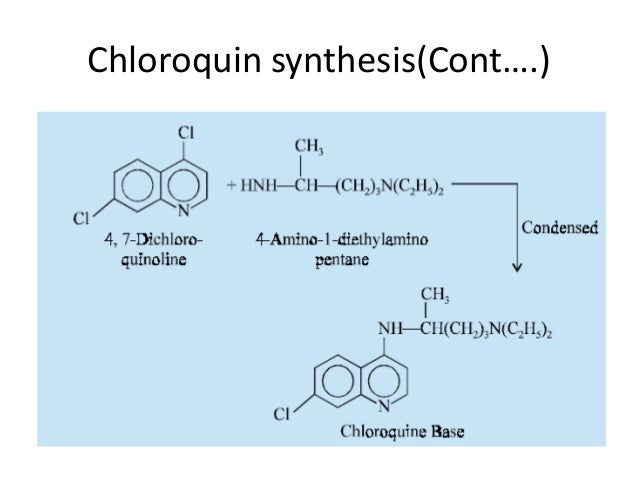

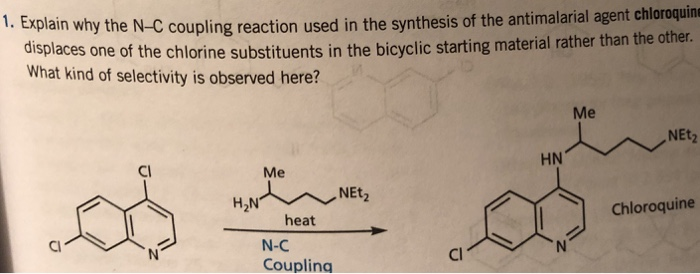















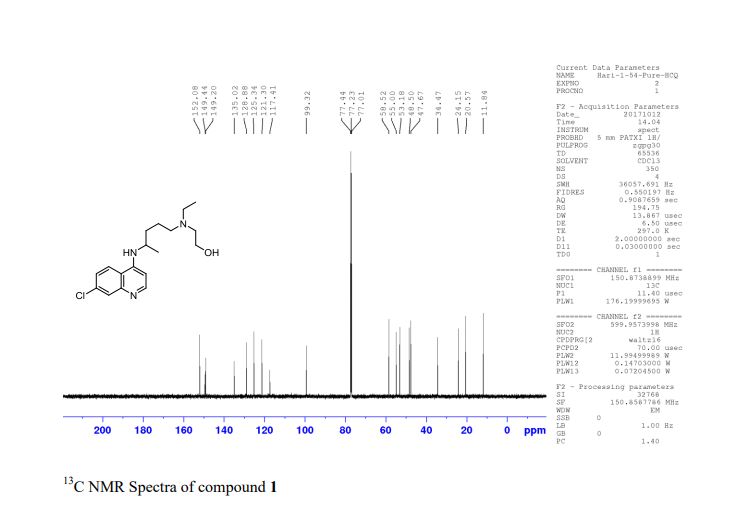
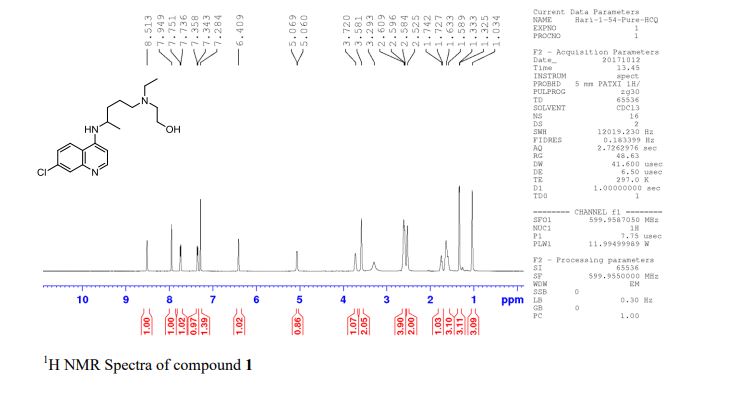


.jpg)