
Home » Uncategorized (Page 151)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FONDAPARINUX

FONDAPARINUX
Fondaparinux is a drug belonging to the group of the antithrombotic agents and are used to prevent deep vein thrombosis in patients undergoing orthopedic surgery. It is also used for the treatment of severe venous thrombosis and pulmonary
114870-03-0 ………..10x SODIUM SALT
| CAS number | 114870-03-0 FREE FORM |
|---|
| MF | C31H43N3Na10O49S8 10X SODIUM |
|---|---|
| MW | 1726.77 g/mol 10X SODIUM |
GSK-576428 Org-31540 SR-90107SR-90107A
launched 2002
Arixtra, Quixidar, Fondaparinux sodium, Fondaparin sodium, Arixtra (TN), Fondaparinux, Org-31540, AC1LCS4W, SR-90107A
Fondaparinux (Arixtra) is a synthetic pentasaccharide anticoagulant. Apart from the O-methyl group at the reducing end of the molecule, the identity and sequence of the five monomeric sugar units contained in fondaparinux is identical to a sequence of five monomeric sugar units that can be isolated after either chemical or enzymatic cleavage of the polymeric glycosaminoglycan heparin and heparan sulfate (HS). This monomeric sequence in heparin and HS is thought to form the high affinity binding site for the natural anti-coagulant factor, antithrombin III (ATIII).
Binding of heparin/HS to ATIII has been shown to increase the anti-coagulant activity of antithrombin III 1000-fold. Fondaparinux potentiates the neutralizing action ofATIII on activated Factor X 300-fold. Fondaparinux may be used: to prevent venous thromboembolism in patients who have undergone orthopedic surgery of the lower limbs (e.g. hip fracture, hip replacement and knee surgery); to prevent VTE in patients undergoing abdominal surgery who are are at high risk of thromboembolic complications; in the treatment of deep vein thrombosis (DVT) and pumonary embolism (PE); in the management of unstable angina (UA) and non-ST segment elevation myocardial infarction (NSTEMI); and in the management of ST segment elevation myocardial infarction (STEMI).

FONDAPARINUX
Fondaparinux (trade name Arixtra) is an anticoagulant medication chemically related to low molecular weight heparins. It is marketed byGlaxoSmithKline. A generic version developed by Alchemia is marketed within the US by Dr. Reddy’s Laboratories.
Fondaparinux is a synthetic pentasaccharide Factor Xa inhibitor. Apart from the O-methyl group at the reducing end of the molecule, the identity and sequence of the five monomeric sugar units contained in fondaparinux is identical to a sequence of five monomeric sugar units that can be isolated after either chemical or enzymatic cleavage of the polymeric glycosaminoglycans heparin and heparan sulfate (HS). Within heparin and heparan sulfate this monomeric sequence is thought to form the high affinity binding site for the anti-coagulant factor antithrombin III (ATIII). Binding of heparin/HS to ATIII has been shown to increase the anti-coagulant activity of antithrombin III 1000 fold. In contrast to heparin, fondaparinux does not inhibit thrombin.
Fondaparinux is given subcutaneously daily. Clinically, it is used for the prevention of deep vein thrombosis in patients who have had orthopedic surgery as well as for the treatment of deep vein thrombosis and pulmonary embolism.
One potential advantage of fondaparinux over LMWH or unfractionated heparin is that the risk for heparin-induced thrombocytopenia (HIT) is substantially lower. Furthermore, there have been case reports of fondaparinux being used to anticoagulate patients with established HIT as it has no affinity to PF-4. However, its renal excretion precludes its use in patients with renal dysfunction.
Unlike direct factor Xa inhibitors, it mediates its effects indirectly through antithrombin III, but unlike heparin, it is selective for factor Xa.[1]
Fondaparinux is similar to enoxaparin in reducing the risk of ischemic events at nine days, but it substantially reduces major bleeding and improves long term mortality and morbidity.[2]
It has been investigated for use in conjunction with streptokinase.[3]
Fondaparinux sodium, a selective coagulation factor Xa inhibitor, was first launched in the U.S. in 2002 by GlaxoSmithKline in a subcutaneous injection formulation for the prophylaxis of deep venous thrombosis (DVT) which may lead to pulmonary embolism in patients at risk for thromboembolic complications who are undergoing hip replacement, knee replacement, hip fracture surgery or abdominal surgery. The product is available in Japan for the treatment of acute deep venous thrombosis and acute pulmonary thromboembolism. In 2004, GlaxoSmithKline launched fondaparinux as an injection to be used in conjunction with warfarin sodium for the subcutaneous treatment of acute pulmonary embolism and DVT.
In 2007, GlaxoSmithKline received approval in the E.U. for the treatment of acute coronary syndrome (ACS), specifically unstable angina or non-ST segment elevation myocardial infarction (UA/NSTEMI) and ST-segment elevation myocardial infarction (STEMI), while in the U.S. an approvable letter was received for this indication. Currently, the drug is in clinical development at GlaxoSmithKline for the treatment of venous limb superficial thrombosis.

Fondaparinux Molecule
GlaxoSmithKline had filed a regulatory application in the E.U. seeking approval of fondaparinux sodium for the prevention of venous thromboembolic events (VTE), however; in 2008, the application was withdrawn for commercial reasons. Commercial launch in Japan for the product for the prevention of venous thromboembolism in high risk patients undergoing surgery in the abdomen took place in 2008.
In 2010, the EMA approved a regulatory application filed by GlaxoSmithKline seeking approval of a once-daily formulation of fondaparinux sodium for the treatment of adults with acute symptomatic spontaneous superficial-vein thrombosis (SVT) of the lower limbs without concomitant DVT. Product launch took place in the U.K. for this indication the same year.
The antithrombotic activity of fondaparinux is the result of antithrombin III (ATIII)-mediated selective inhibition of Factor Xa. By selectively binding to ATIII, the drug potentiates (about 300 times) the innate neutralization of Factor Xa by ATIII. Neutralization of Factor Xa, in turn, interrupts the blood coagulation cascade and thus inhibits thrombin formation and thrombus development. Fondaparinux does not inactivate thrombin (activated Factor II) and has no known effect on platelet function. At the recommended dose, no effects have been demonstrated on fibrinolytic activity or bleeding time.
Originally developed by Organon and Sanofi (formerly known as sanofi-aventis), fondaparinux sodium is currently available in approximately 30 countries. In 2004, Organon transferred its rights to the drug to Sanofi in exchange for revenues based on future sales from jointly developed antithrombotic products and in early 2005, GlaxoSmithKline also acquired the antithrombotic.
At the beginning of 2005, GlaxoSmithKline signed a two-year agreement with Adolor (acquired by Cubist in 2011) for the copromotion of fondaparinux sodium in the U.S. In Sepetember 2013, Aspen Pharmacare acquired Arixtra global rights (excluding China, India and Pakistan) from GlaxoSmithKline for the treatment of thrombosis with GlaxoSmithKline commercializing the product in Indonesia under licence from Aspen.
Chemical structure
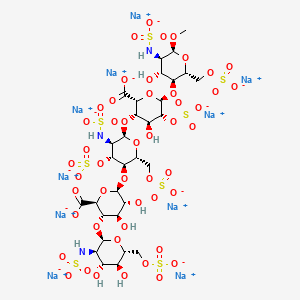
Abbreviations
- GlcNS6S = 2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside
- GlcA = β-D-glucopyranuronoside
- GlcNS3,6S = 2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl
- IdoA2S = 2-O-sulfo-α-L-idopyranuronoside
- GlcNS6SOMe = methyl-O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside
The sequence of monosaccharides is D-GlcNS6S-α-(1,4)-D-GlcA-β-(1,4)-D-GlcNS3,6S-α-(1,4)-L-IdoA2S-α-(1,4)-D-GlcNS6S-OMe, as shown in the following structure:

ARIXTRA (fondaparinux sodium) Injection is a sterile solution containing fondaparinux sodium. It is a synthetic and specific inhibitor of activatedFactor X (Xa). Fondaparinux sodium is methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-β-D-glucopyranuronosyl-( 1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-Osulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt.
The molecular formula of fondaparinux sodium is C31H43N3Na10O49S8 and its molecular weight is 1728. The structural formula is provided below:
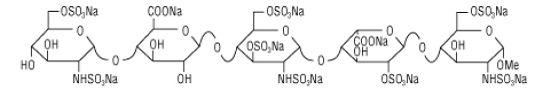
ARIXTRA is supplied as a sterile, preservative-free injectable solution for subcutaneous use.
Each single-dose, prefilled syringe of ARIXTRA, affixed with an automatic needle protection system, contains 2.5 mg of fondaparinux sodium in 0.5 mL, 5.0 mg of fondaparinux sodium in 0.4 mL, 7.5 mg of fondaparinux sodium in 0.6 mL, or 10.0 mg of fondaparinux sodium in 0.8 mL of an isotonic solutionof sodium chloride and water for injection. The final drug product is a clear and colorless to slightly yellow liquid with a pH between 5.0 and 8.0.
……………….
INTRODUCTION
In U.S. Patent No. 7,468,358, Fondaparinux sodium is described as the “only anticoagulant thought to be completely free of risk from HIT-2 induction.” The biochemical and pharmacologic rationale for the development of a heparin pentasaccharide in Thromb. Res., 86(1), 1-36, 1997 by Walenga et al. cited the recently approved synthetic pentasaccharide Factor Xa inhibitor Fondaparinux sodium. Fondaparinux has also been described in Walenga et al., Expert Opin. Investig. Drugs, Vol. 11, 397-407, 2002 and Bauer, Best Practice & Research Clinical Hematology, Vol. 17, No. 1, 89-104, 2004.
Fondaparinux sodium is a linear octasulfated pentasaccharide (oligosaccharide with five monosaccharide units ) molecule having five sulfate esters on oxygen (O-sulfated moieties) and three sulfates on a nitrogen (N- sulfated moieties). In addition, Fondaparinux contains five hydroxyl groups in the molecule that are not sulfated and two sodium carboxylates. Out of five saccharides, there are three glucosamine derivatives and one glucuronic and one L-iduronic acid. The five saccharides are connected to each other in alternate α and β glycosylated linkages (see Figure 1).
Figure 1 Fondaparinux Sodium

Monosaccharide E Monosaccharide D Monosaccharide C Monosaccharide B Monosaccharide A derived from derived from derived from derived from derived from
Monomer E Monomer D Monomer C Monomer B1 Monomer A2
Fondaparinux Sodium
Fondaparinux sodium is a chemically synthesized methoxy derivative of the natural pentasaccharide sequence, which is the active site of heparin that mediates the interaction with antithrombin (Casu et al., J. Biochem., 197, 59, 1981). It has a challenging pattern of O- and N- sulfates, specific glycosidic stereochemistry, and repeating units of glucosamines and uronic acids (Petitou et al, Progress in the Chemistry of Organic Natural Product, 60, 144-209, 1992).
The monosaccharide units comprising the Fondaparinux molecule are labeled as per the convention in Figure 1, with the glucosamine unit on the right referred to as monosaccharide A and the next, an uronic acid unit to its left as B and subsequent units, C, D and E respectively. The chemical synthesis of Fondaparinux starts with monosaccharides of defined structures that are themselves referred to as Monomers A2, Bl, C, D and E, for differentiation and convenience, and they become the corresponding monosaccharides in fondaparinux sodium.
Due to this complex mixture of free and sulfated hydroxyl groups, and the presence of N- sulfated moieties, the design of a synthetic route to Fondaparinux requires a careful strategy of protection and de-protection of reactive functional groups during synthesis of the molecule. Previously described syntheses of Fondaparinux all adopted a similar strategy to complete the synthesis of this molecule. This strategy can be envisioned as having four stages.
The strategy in the first stage requires selective de-protection of five out of ten hydroxyl groups. During the second stage these five hydroxyls are selectively sulfonated. The third stage of the process involves the de -protection of the remaining five hydroxyl groups. The fourth stage of the process is the selective sulfonation of the 3 amino groups, in the presence of five hydroxyl groups that are not sulfated in the final molecule. This strategy can be envisioned from the following fully protected pentasaccharide, also referred to as the late-stage intermediate.

In this strategy, all of the hydroxyl groups that are to be sulfated are protected with an acyl protective group, for example, as acetates (R = CH3) or benzoates (R = aryl) (Stages 1 and 2) All of the hydroxyl groups that are to remain as such are protected with benzyl group as benzyl ethers (Stage 3). The amino group, which is subsequently sulfonated, is masked as an azide (N3) moiety (Stage 4). R1 and R2 are typically sodium in the active pharmaceutical compound (e.g., Fondaparinux sodium).
This strategy allows the final product to be prepared by following the synthetic operations as outlined below: a) Treatment of the late- stage intermediate with base to hydrolyze (deprotect) the acyl ester groups to reveal the five hydroxyl groups. The two R1 and R2 ester groups are hydrolyzed in this step as well.

b) Sulfonation of the newly revealed hydroxyl groups.

c) Hydrogenation of the O-sulfated pentasaccharide to de-benzylate the five benzyl- protected hydroxyls, and at the same time, unmask the three azides to the corresponding amino groups.

d) On the last step of the operation, the amino groups are sulfated selectively at a high pH, in the presence of the five free hydroxyls to give Fondaparinux (Figure 1). While the above strategy has been shown to be viable, it is not without major drawbacks. One drawback lies in the procedure leading to the fully protected pentasaccharide (late stage intermediate), especially during the coupling of the D-glucuronic acid to the next adjacent glucose ring (the D-monomer to C-monomer in the EDCBA nomenclature shown in Figure 1). Sugar oligomers or oligosaccharides, such as Fondaparinux, are assembled using coupling reactions, also known as glycosylation reactions, to “link” sugar monomers together. The difficulty of this linking step arises because of the required stereochemical relationship between the D-sugar and the C-sugar, as shown below:

The stereochemical arrangement illustrated above in Figure 2 is described as having a β- configuration at the anomeric carbon of the D-sugar (denoted by the arrow). The linkage between the D and C units in Fondaparinux has this specific stereochemistry. There are, however, competing β- and α-glycosylation reactions.
The difficulties of the glycosylation reaction in the synthesis of Fondaparinux is well known. In 1991 Sanofi reported a preparation of a disaccharide intermediate in 51% yield having a 12/1 ratio of β/α stereochemistry at the anomeric position (Duchaussoy et al., Bioorg. & Med. Chem. Lett., 1(2), 99-102, 1991).
In another publication (Sinay et al, Carbohydrate Research, 132, C5-C9, 1984) yields on the order of 50% with coupling times on the order of 6- days are reported. U.S. Patent No. 4,818,816 {see e.g., column 31, lines 50-56) discloses a 50% yield for the β-glycosylation.
Alchemia’s U.S. Patent No. 7,541,445 is even less specific as to the details of the synthesis of this late-stage Fondaparinux synthetic intermediate. The ‘445 Patent discloses several strategies for the assembly of the pentasaccharide (1+4, 3+2 or 2+3) using a 2-acylated D-sugar (specifically 2-allyloxycarbonyl) for the glycosylation coupling reactions. However, Alchemia’s strategy involves late-stage pentasaccharides that all incorporate a 2-benzylated D- sugar.
The transformation of acyl to benzyl is performed either under acidic or basic conditions. Furthermore, these transformations, using benzyl bromide or benzyl trichloroacetimidate, typically result in extensive decomposition and the procedure suffers from poor yields. Thus, such transformations (at a disaccharide, trisaccharide, and pentasaccharide level) are typically not acceptable for industrial scale production.
Examples of fully protected pentasaccharides are described in Duchaussoy et al, Bioorg. Med. Chem. Lett., 1 (2), 99-102, 1991; Petitou et al, Carbohydr. Res., 167, 67-75, 1987; Sinay et al, Carbohydr. Res., 132, C5-C9, 1984; Petitou et al., Carbohydr. Res., 1147, 221-236, 1986; Lei et al., Bioorg. Med. Chem., 6, 1337-1346, 1998; Ichikawa et al., Tet. Lett., 27(5), 611-614, 1986; Kovensky et al, Bioorg. Med. Chem., 1999, 7, 1567-1580, 1999.
These fully protected pentasaccharides may be converted to the O- and N-sulfated pentasaccharides using the four steps (described earlier) of: a) saponification with LiOHZH2CVNaOH, b) O-sulfation by an Et3N- SO3 complex; c) de-benzylation and azide reduction via H2/Pd hydrogenation; and d) N-sulfation with a pyridine-SO3 complex.
Even though many diverse analogs of the fully protected pentasaccharide have been prepared, none use any protective group at the 2-position of the D unit other than a benzyl group. Furthermore, none of the fully protected pentasaccharide analogs offer a practical, scaleable and economical method for re-introduction of the benzyl moiety at the 2-position of the D unit after removal of any participating group that promotes β-glycosylation.
Furthermore, the coupling of benzyl protected sugars proves to be a sluggish, low yielding and problematic process, typically resulting in substantial decomposition of the pentasaccharide (prepared over 50 synthetic steps), thus making it unsuitable for a large [kilogram] scale production process.

Ref. 1. Sinay et al, Carbohydr. Res., 132, C5-C9, 1984.
Ref. 2. Petitou et al., Carbohydr. Res., 147, 221-236. 1986
It has been a general strategy for carbohydrate chemists to use base-labile ester-protecting group at 2-position of the D unit to build an efficient and stereoselective β-glycosidic linkage. To construct the β-linkage carbohydrate chemists have previously acetate and benzoate ester groups, as described, for example, in the review by Poletti et al., Eur. J. Chem., 2999-3024, 2003.
The ester group at the 2-position of D needs to be differentiated from the acetate and benzoates at other positions in the pentasaccharide. These ester groups are hydrolyzed and sulfated later in the process and, unlike these ester groups, the 2-hydroxyl group of the D unit needs to remain as the hydroxyl group in the final product, Fondaparinux sodium.
Some of the current ester choices for the synthetic chemists in the field include methyl chloro acetyl and chloro methyl acetate [MCA or CMA] . The mild procedures for the selective removal of theses groups in the presence of acetates and benzoates makes them ideal candidates. However, MCA/CMA groups have been shown to produce unwanted and serious side products during the glycosylation and therefore have not been favored in the synthesis of Fondaparinux sodium and its analogs. For by-product formation observed in acetate derivatives see Seeberger et al., J. Org. Chem., 2004, 69, 4081-93.
Similar by-product formation is also observed using chloroacetate derivatives. See Orgueira et al., Eur. J. Chem., 9(1), 140-169, 2003.
The levulinyl group can be rapidly and almost quantitatively removed by treatment with hydrazine hydrate as the deprotection reagent as illustrated in the example below. Under the same reaction conditions primary and secondary acetate and benzoate esters are hardly affected by hydrazine hydrate. See, e.g., Seeberger et al, J. Org. Chem., 69, 4081-4093, 2004.

The syntheses of Fondaparinux sodium described herein takes advantage of the levulinyl group in efficient construction of the trisaccharide EDC with improved β- selectivity for the coupling under milder conditions and increased yields.

Substitution of the benzyl protecting group with a THP moiety provides its enhanced ability to be incorporated quantitatively in position-2 of the unit D of the pentasaccharide. Also, the THP group behaves in a similar manner to a benzyl ether in terms of function and stability. In the processes described herein, the THP group is incorporated at the 2-position of the D unit at this late stage of the synthesis (i.e., after the D and C units have been coupled through a 1,2-trans glycosidic (β-) linkage). The THP protective group typically does not promote an efficient β- glycosylation and therefore is preferably incorporated in the molecule after the construction of the β-linkage.
Fondaparinux and sodium salt thereof can be prepared from pure compound of Formula II by following the teachings from Bioorganic and Medicinal Chemistry Letters, 1(2), p. 95-98 (1991). A second aspect of the present invention provides a process for the preparation of 4-0- -D-glucopyranosyl-l,6-anhydro- -D-glucopyranose, represented by STR BELOW

……………………………..
SYNTHESIS
EP2464668A2 AND US8288515
The scheme below exemplifies some of the processes of the present invention disclosed herein.

The tetrahydropyranyl (THP) protective group and the benzyl ether protective group are suitable hydroxyl protective groups and can survive the last four synthetic steps (described above) in the synthesis of Fondaparinux sodium, even under harsh reaction conditions. Certain other protecting groups do not survive the last four synthetic steps in high yield.
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:


Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; ACN is acetonitrile; MS is molecular sieves; DMF is dimethyl formamide; PMB is p-methoxybenzyl; Bn is benzyl; DCM is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; CSA is camphor sulfonic acid; TEA is triethylamine; MeOH is methanol; DMAP is dimethylaminopyridine; RT is room temperature; CAN is ceric ammonium nitrate; Ac2O is acetic anhydride; HBr is hydrogen bromide; TEMPO is tetramethylpiperidine-N-oxide; TBACl is tetrabutyl ammonium chloride; EtOAc is ethyl acetate; HOBT is hydroxybenzotriazole; DCC is dicyclohexylcarbodiimide; Lev is levunlinyl; TBDPS is tertiary-butyl diphenylsilyl; TCA is trichloroacetonitrile; O-TCA is O-trichloroacetimidate; Lev2O is levulinic anhydride; DIPEA is diisopropylethylamine; Bz is benzoyl; TBAF is tetrabutylammonium fluoride; DBU is diazabicycloundecane; BF3.Et2O is boron trifluoride etherate; TMSI is trimethylsilyl iodide; TBAI is tetrabutylammonium iodide; TES-Tf is triethylsilyl trifluoromethanesulfonate (triethylsilyl triflate); DHP is dihydropyran; PTS is p-toluenesulfonic acid.
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:


The ester moieties in EDCBA Pentamer were hydrolyzed with sodium and lithium hydroxide in the presence of hydrogen peroxide in dioxane mixing at room temperature for 16 hours to give the pentasaccharide intermediate API1. The five hydroxyl moieties in API1 were sulfated using a pyridine-sulfur trioxide complex in dimethylformamide, mixing at 60° C. for 2 hours and then purified using column chromatography (CG-161), to give the pentasulfated pentasaccharide API2. The intermediate API2 was then hydrogenated to reduce the three azides on sugars E, C and A to amines and the reductive deprotection of the five benzyl ethers to their corresponding hydroxyl groups to form the intermediate API3. This transformation occurs by reacting API2 with 10% palladium/carbon catalyst with hydrogen gas for 72 hours. The three amines on API3 were then sulfated using the pyridine-sulfur trioxide complex in sodium hydroxide and ammonium acetate, allowing the reaction to proceed for 12 hours. The acidic work-up procedure of the reaction removes the THP group to provide crude fondaparinux which is purified and is subsequently converted to its salt form. The crude mixture was purified using an ion-exchange chromatographic column (HiQ resin) followed by desalting using a size exclusion resin or gel filtration (Biorad Sephadex G25) to give the final API, fondaparinux sodium
Experimental Procedures Preparation of IntD1 Bromination of Glucose Pentaacetate
To a 500 ml flask was added 50 g of glucose pentaacetate (C6H22O11) and 80 ml of methylene chloride. The mixture was stirred at ice-water bath for 20 min HBr in HOAc (33%, 50 ml) was added to the reaction mixture. After stirring for 2.5 hr another 5 ml of HBr was added to the mixture. After another 30 min, the mixture was added 600 ml of methylene chloride. The organic mixture was washed with cold water (200 ml×2), Saturated NaHCO3(200 ml×2), water (200 ml) and brine (200 ml×2). The organic layer was dried over Na2SO4 and the mixture was evaporated at RT to give white solid as final product, bromide derivative, IntD1 (˜95% yield). C14H19BrO9, TLC Rf=0.49, SiO2, 40% ethyl acetate/60% hexanes; Exact Mass 410.02.
Preparation of IntD2 by Reductive Cyclization
To a stirring mixture of bromide IntD1 (105 g), tetrabutylammonium iodide (60 g, 162 mmol) and activated 3 Å molecular sieves in anhydrous acetonitrile (2 L), solid NaBH4 (30 g, 793 mmol) was added. The reaction was heated at 40° C. overnight. The mixture was then diluted with dichloromethane (2 L) and filtered through Celite®. After evaporation, the residue was dissolved in 500 ml ethyl acetate. The white solid (Bu4NI or Bu4NBr) was filtered. The ethyl acetate solution was evaporated and purified by chromatography on silica gel using ethyl acetate and hexane as eluent to give the acetal-triacetate IntD2 (˜60-70% yield). TLC Rf=0.36, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD3 by De-Acetylation
To a 1000 ml flask was added triacetate IntD2 (55 g) and 500 ml of methanol. After stirring 30 min, the reagent NaOMe (2.7 g, 0.3 eq) was added and the reaction was stirred overnight. Additional NaOMe (0.9 g) was added to the reaction mixture and heated to 50° C. for 3 hr. The mixture was neutralized with Dowex 50Wx8 cation resin, filtered and evaporated. The residue was purified by silica gel column to give 24 g of trihydroxy-acetal IntD3. TLC Rf=0.36 in SiO2, 10% methanol/90% ethyl acetate.
Preparation of IntD4 by Benzylidene Formation
To a 1000 ml flask was added trihydroxy compound IntD3 (76 g) and benzaldehyde dimethyl acetate (73 g, 1.3 eq). The mixture was stirred for 10 min, after which D(+)-camphorsulfonic acid (8.5 g, CSA) was added. The mixture was heated at 50° C. for two hours. The reaction mixture was then transferred to separatory funnel containing ethyl acetate (1.8 L) and sodium bicarbonate solution (600 ml). After separation, the organic layer was washed with a second sodium bicarbonate solution (300 ml) and brine (800 ml). The two sodium carbonate solutions were combined and extracted with ethyl acetate (600 ml×2). The organic mixture was evaporated and purified by silica gel column to give the benzylidene product IntD4 (77 g, 71% yield). TLC Rf=0.47, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD5 by Benzylation
To a 500 ml flask was added benzylidene acetal compound IntD4 (21 g,) in 70 ml THF. To another flask (1000 ml) was added NaH (2 eq). The solution of IntD4 was then transferred to the NaH solution at 0° C. The reaction mixture was stirred for 30 min, then benzyl bromide (16.1 ml, 1.9 eq) in 30 ml THF was added. After stirring for 30 min, DMF (90 ml) was added to the reaction mixture. Excess NaH was neutralized by careful addition of acetic acid (8 ml). The mixture was evaporated and purified by silica gel column to give the benzyl derivative IntD5. (23 g) TLC Rf=0.69, SiO2 in 40% ethyl acetate/60% hexanes.
Preparation of IntD6 by Deprotection of Benzylidene
To a 500 ml flask was added the benzylidene-acetal compound IntD5 (20 g) and 250 ml of dichloromethane, the reaction mixture was cooled to 0° C. using an ice-water-salt bath. Aqueous TFA (80%, 34 ml) was added to the mixture and stirred over night. The mixture was evaporated and purified by silica gel column to give the dihydroxy derivative IntD6. (8 g, 52%). TLC Rf=0.79, SiO2 in 10% methanol/90% ethyl acetate.
Preparation of IntD7 by Oxidation of 6-Hydroxyl
To a 5 L flask was added dihydroxy compound IntD6 (60 g), TEMPO (1.08 g), sodium bromide (4.2 g), tetrabutylammonium chloride (5.35 g), saturated NaHCO3 (794 ml) and EtOAc (1338 ml). The mixture was stirred over an ice-water bath for 30 min To another flask was added a solution of NaOCl (677 ml), saturated NaHCO3 (485 ml) and brine (794 ml). The second mixture was added slowly to the first mixture (over about two hrs). The resulting mixture was then stirred overnight. The mixture was separated, and the inorganic layer was extracted with EtOAc (800 ml×2). The combined organic layers were washed with brine (800 ml). Evaporation of the organic layer gave 64 g crude carboxylic acid product IntD7 which was used in the next step use without purification. TLC Rf=0.04, SiO2 in 10% methanol/90% ethyl acetate.
Preparation of Monomer D by Benzylation of the Carboxylic Acid
To a solution of carboxylic acid derivative IntD7 (64 g) in 600 ml of dichloromethane, was added benzyl alcohol (30 g) and N-hydroxybenzotriazole (80 g, HOBt). After stirring for 10 min triethylamine (60.2 g) was added slowly. After stirring another 10 min, dicyclohexylcarbodiimide, (60.8 g, DCC) was added slowly and the mixture was stirred overnight. The reaction mixture was filtered and the solvent was removed under reduced pressure followed by chromatography on silica gel to provide 60.8 g (75%, over two steps) of product, Monomer D. TLC Rf=0.51, SiO2 in 40% ethyl acetate/60% hexanes.
Synthesis of the BA Dimer
Step 1. Preparation of BMod1, Levulination of Monomer B1
A 100 L reactor was charged with 7.207 Kg of Monomer B1 (21.3 moles, 1 equiv), 20 L of dry tetrahydrofuran (THF) and agitated to dissolve. When clear, it was purged with nitrogen and 260 g of dimethylamino pyridine (DMAP, 2.13 moles, 0.1 equiv) and 11.05 L of diisopropylethylamine (DIPEA, 8.275 kg, 63.9 moles, 3 equiv) was charged into the reactor. The reactor was chilled to 10-15° C. and 13.7 kg levulinic anhydride (63.9 mol, 3 equiv) was transferred into the reactor. When the addition was complete, the reaction was warmed to ambient temperature and stirred overnight or 12-16 hours. Completeness of the reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. When the reaction was complete, 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. The mixture was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L of water, 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight. The yield of the isolated syrup of BMod1 was 100%.
Synthesis of the BA Dimer
Step 2. Preparation of BMod2, TFA Hydrolysis of BMod1
A 100 L reactor was charged with 9296 Kg of 4-Lev Monomer B1 (BMod1) (21.3 mol, 1 equiv). The reactor chiller was turned to <5° C. and stirring was begun, after which 17.6 L of 90% TFA solution (TFA, 213 mole, 10 equiv) was transferred into the reactor. When the addition was complete, the reaction was monitored by TLC and HPLC. The reaction took approximately 2-3 hours to reach completion. When the reaction was complete, the reactor was chilled and 26.72 L of triethylamine (TEA, 19.4 Kg, 191.7 mole, 0.9 equiv) was transferred into the reactor. An additional 20 L of water and 20 L ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer was extracted (EtOAc layer) with 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 50:50, 80:20 (EtOAc/heptane), 100% EtOAc, 5:95, 10:90 (MeOH/EtOAc). The pure fractions were pooled and evaporated to a syrup. The yield of the isolated syrup, BMod2 was 90%.
Synthesis of the BA Dimer
Step 3. Preparation of BMod3, Silylation of BMod2
A 100 L reactor was charged with 6.755 Kg 4-Lev-1,2-DiOH Monomer B1 (BMod2) (17.04 mol, 1 equiv), 2328 g of imidazole (34.2 mol, 2 equiv) and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., then 5.22 L tert-butyldiphenylchloro-silane (TBDPS-Cl, 5.607 Kg, 20.4 mol, 1.2 equiv) was transferred into the reactor. When addition was complete, the chiller was turned off and the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (40% ethyl acetate/hexane) and HPLC. The reaction took approximately 3 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. Dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The yield of BMod3 was about 80%.
Synthesis of the BA Dimer
Step 4. Preparation of BMod4, Benzoylation
A 100 L reactor was charged with 8.113 Kg of 4-Lev-1-Si-2-OH Monomer B1 (BMod3) (12.78 mol, 1 equiv), 9 L of pyridine and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., after which 1.78 L of benzoyl chloride (2155 g, 15.34 mol, 1.2 equiv) was transferred into the reactor. When addition was complete, the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (40% ethyl acetate/heptane) and HPLC. The reaction took approximately 3 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The DCM solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). Isolated syrup BMod4 was obtained in 91% yield.
Synthesis of the BA Dimer
Step 5. Preparation of BMod5, Desilylation
A 100 L reactor was charged with 8.601 Kg of 4-Lev-1-Si-2-Bz Monomer B1 (BMod4) (11.64 mol, 1 equiv) in 30 L terahydrofuran. The reactor was purged with nitrogen and chilled to 0° C., after which 5.49 Kg of tetrabutylammonium fluoride (TBAF, 17.4 mol, 1.5 equiv) and 996 mL (1045 g, 17.4 mol, 1.5 equiv) of glacial acetic acid were transferred into the reactor. When the addition was complete, the reaction was stirred at ambient temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. The reaction took approximately 6 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min, after which the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20 (EtOAc/heptane) and 200 L 100% EtOAc. Pure fractions were pooled and evaporated to a syrup. The intermediate BMod5 was isolated as a syrup in 91% yield.
Synthesis of the BA Dimer
Step 6: Preparation of BMod6, TCA Formation
A 100 L reactor was charged with 5.238 Kg of 4-Lev-1-OH-2-Bz Monomer B1 (BMod5) (10.44 mol, 1 equiv) in 30 L of DCM. The reactor was purged with nitrogen and chilled to 10-15° C., after which 780 mL of diazabicyclo undecene (DBU, 795 g, 5.22 mol, 0.5 equiv) and 10.47 L of trichloroacetonitrile (TCA, 15.08 Kg, 104.4 mol, 10 equiv) were transferred into the reactor. Stirring was continued and the reaction was kept under a nitrogen atmosphere. After reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by HPLC and TLC (40:60 ethyl acetate/heptane). The reaction took approximately 2 hours to reach completion. When the reaction was complete, 20 L of water and 10 L of dichloromethane were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was separated with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/Heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of BMod6 was 73%.
Synthesis of the BA Dimer
Step 7. Preparation of AMod1, Acetylation of Monomer A2
A 100 L reactor was charged with 6.772 Kg of Monomer A2 (17.04 mole, 1 eq.), 32.2 L (34.8 Kg, 340.8 moles, 20 eq.) of acetic anhydride and 32 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C. When the temperature reached −20° C., 3.24 L (3.63 Kg, 25.68 mol, 1.5 equiv) of boron trifluoride etherate (BF3.Et2O) was transferred into the reactor. After complete addition of boron trifluoride etherate, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 3-5 hours for completion. When the reaction was complete, extraction was performed with 3×15 L of 10% sodium bicarbonate and 20 L of water. The organic phase (DCM) was evaporated to a syrup (bath temp. 40° C.) and allowed to dry overnight. The syrup was purified in a 200 L silica column using 140 L each of the following gradient profiles: 5:95, 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of AMod1 was 83%.
Synthesis of the BA Dimer
Step 8. Preparation of AMod3,1-Methylation of AMod1
A 100 L reactor was charged with 5891 g of acetyl Monomer A2 (AMod1) (13.98 mole, 1 eq.) in 32 L of dichloromethane. The reactor was purged with nitrogen and was chilled to 0° C., after which 2598 mL of trimethylsilyl iodide (TMSI, 3636 g, 18 mol, 1.3 equiv) was transferred into the reactor. When addition was complete, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 2-4 hours to reach completion. When the reaction was complete, the mixture was diluted with 20 L of toluene. The solution was evaporated to a syrup and was co-evaporated with 3×6 L of toluene. The reactor was charged with 36 L of dichloromethane (DCM), 3.2 Kg of dry 4 Å Molecular Sieves, 15505 g (42 mol, 3 equiv) of tetrabutyl ammonium iodide (TBAI) and 9 L of dry methanol. This was stirred until the TBAI was completely dissolved, after which 3630 mL of diisopropyl-ethylamine (DIPEA, 2712 g, 21 moles, 1.5 equiv) was transferred into the reactor in one portion. The completion of the reaction was monitored by HPLC and TLC (30:70 ethyl acetate/heptane). The reaction took approximately 16 hours for completion. When the reaction was complete, the molecular sieves were removed by filtration. Added were 20 L EtOAc and extracted with 4×20 L of 25% sodium thiosulfate and 20 L 10% NaCl solutions. The organic layer was separated and dried with 8-12 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 5:95, 10:90, 20:80, 30:70 and 40:60 (EtOAc/heptane). The pure fractions were pooled and evaporated to give intermediate AMod3 as a syrup. The isolated yield was 75%.
Synthesis of the BA Dimer
Step 9. Preparation of AMod4, DeAcetylation of AMod3
A 100 L reactor was charged with 4128 g of 1-Methyl 4,6-Diacetyl Monomer A2 (AMod3) (10.5 mol, 1 equiv) and 18 L of dry methanol and dissolved, after which 113.4 g (2.1 mol, 0.2 equiv) of sodium methoxide was transferred into the reactor. The reaction was stirred at room temperature and monitored by TLC (40% ethyl acetate/hexane) and HPLC. The reaction took approximately 2-4 hours for completion. When the reaction was complete, Dowex 50Wx8 cation resin was added in small portions until the pH reached 6-8. The Dowex 50Wx8 resin was filtered and the solution was evaporated to a syrup (bath temp. 40° C.). The syrup was diluted with 10 L of ethyl acetate and extracted with 20 L brine and 20 L water. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight at the same temperature. The isolated yield of the syrup AMod4 was about 88%.
Synthesis of the BA Dimer
Step 10. Preparation of AMod5,6-Benzoylation
A 100 L reactor was charged with 2858 g of Methyl 4,6-diOH Monomer A2 (AMod4) (9.24 mol, 1 equiv) and co-evaporated with 3×10 L of pyridine. When evaporation was complete, 15 L of dichloromethane, 6 L of pyridine were transferred into the reactor and dissolved. The reactor was purged with nitrogen and chilled to −40° C. The reactor was charged with 1044 mL (1299 g, 9.24 mol, 1 equiv) of benzoyl chloride. When the addition was complete, the reaction was allowed to warm to −10° C. over a period of 2 hours. The reaction was monitored by TLC (60% ethyl acetate/hexane). When the reaction was completed, the solution was evaporated to a syrup (bath temp. 40° C.). This was co-evaporated with 3×15 L of toluene. The syrup was diluted with 40 L ethyl acetate. Extraction was carried out with 20 L of water and 20 L of brine solution. The Ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 5:95, 10:90, 20:80, 25:70 and 30:60 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup. The isolated yield of the intermediate AMod5 was 84%.
Synthesis of the BA Dimer
Step 11. Preparation of BA1, Coupling of Amod5 with BMod6
A 100 L reactor was charged with 3054 g of methyl 4-Hydroxy-Monomer A2 (AMod5) from Step 10 (7.38 mol, 1 equiv) and 4764 g of 4-Lev-1-TCA-Monomer B1 (BMod6) from Step 6 (7.38 mol, 1 equiv). The combined monomers were dissolved in 20 L of toluene and co-evaporated at 40° C. Co evaporation was repeated with an additional 2×20 L of toluene, after which 30 L of dichloromethane (DCM) was transferred into the reactor and dissolved. The reactor was purged with nitrogen and was chilled to below −20° C. When the temperature was between −20° C. and −40° C., 1572 g (1404 mL, 11.12 moles, 1.5 equiv) of boron trifluoride etherate (BF3.Et2O) were transferred into the reactor. After complete addition of boron trifluoride etherate, the reaction was allowed to warm to 0° C. and stirring was continued. The completeness of the reaction was monitored by HPLC and TLC (40:70 ethyl acetate/heptane). The reaction required 3-4 hours to reach completion. When the reaction was complete, 926 mL (672 g, 6.64 mol, 0.9 equiv) of triethylamine (TEA) was transferred into the mixture and stirred for an additional 30 minutes, after which 20 L of water and 10 L of dichloromethane were transferred into the reactor. The solution was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was separated with 2×20 L water and 20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. The dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and used in the next step. The isolated yield of the disaccharide BA1 was about 72%.
Synthesis of the BA Dimer
Step 12, Removal of Levulinate Methyl [(methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate)-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl]-2-deoxy-α-D-glucopyranoside
A 100 L reactor was charged with 4.104 Kg of 4-Lev BA Dimer (BA1) (4.56 mol, 1 equiv) in 20 L of THF. The reactor was purged with nitrogen and chilled to −20 to −25° C., after which 896 mL of hydrazine hydrate (923 g, 18.24 mol, 4 equiv) was transferred into the reactor. Stirring was continued and the reaction was monitored by TLC (40% ethyl acetate/heptane) and HPLC. The reaction took approximately 2-3 hour for the completion, after which 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (ETOAc layer) was extracted with 20 L 25% brine solutions. The ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). The pure fractions were pooled and evaporated to dryness. The isolated yield of the BA Dimer was 82%. Formula: C42H43N3O13; Mol. Wt. 797.80.
Synthesis of the EDC Trimer
Step 1. Preparation of EMod1, Acetylation
A 100 L reactor was charged with 16533 g of Monomer E (45 mole, 1 eq.), 21.25 L acetic anhydride (225 mole, 5 eq.) and 60 L of dichloromethane. The reactor was purged with nitrogen and was chilled to −10° C. When the temperature was at −10° C., 1.14 L (1277 g) of boron trifluoride etherate (BF3.Et2O, 9.0 moles, 0.2 eq) were transferred into the reactor. After the complete addition of boron trifluoride etherate, the reaction was allowed to warm to room temperature. The completeness of the reaction was monitored by TLC (30:70 ethyl acetate/heptane) and HPLC. The reaction took approximately 3-6 hours to reach completion. When the reaction was completed, the mixture was extracted with 3×50 L of 10% sodium bicarbonate and SOL of water. The organic phase (DCM) was evaporated to a syrup (bath temp. 40° C.) and allowed to dry overnight. The isolated yield of EMod1 was 97%.
Synthesis of the EDC Trimer
Step 2. Preparation of EMod2, De-Acetylation of Azidoglucose
A 100 L reactor was charged with 21016 g of 1,6-Diacetyl Monomer E (EMod1) (45 mole, 1 eq.), 5434 g of hydrazine acetate (NH2NH2.HOAc, 24.75 mole, 0.55 eq.) and 50 L of DMF (dimethyl formamide). The solution was stirred at room temperature and the reaction was monitored by TLC (30% ethyl acetate/hexane) and HPLC. The reaction took approximately 2-4 hours for completion. When the reaction was completed, 50 L of dichloromethane and 40 L of water were transferred into the reactor. This was stirred for 30 minutes and the layers were separated. This was extracted with an additional 40 L of water and the organic phase was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.) and dried overnight at the same temperature. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of intermediate EMod2 was 100%.
Synthesis of the EDC Trimer
Step 3. Preparation of EMod3, Formation of 1-TCA
A 100 L reactor was charged with 12752 g of 1-Hydroxy Monomer E (EMod2) (30 mole, 1 eq.) in 40 L of dichloromethane. The reactor was purged with nitrogen and stirring was started, after which 2.25 L of DBU (15 moles, 0.5 eq.) and 15.13 L of trichloroacetonitrile (150.9 moles, 5.03 eq) were transferred into the reactor. Stirring was continued and the reaction was kept under nitrogen. After the reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (30:70 ethyl acetate/Heptane) and HPLC. The reaction took approximately 2-3 hours to reach completion. When the reaction was complete, 40 L of water and 20 L of DCM were charged into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 40 L water and the DCM solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90 (DCM/EtOAc/heptane), 20:5:75 (DCM/EtOAc/heptane) and 20:10:70 DCM/EtOAc/heptane). Pure fractions were pooled and evaporated to give Intermediate EMod3 as a syrup. Isolated yield was 53%.
Synthesis of the EDC Trimer
Step 4. Preparation of ED Dimer, Coupling of E-TCA with Monomer D
A 100 L reactor was charged with 10471 g of 6-Acetyl-1-TCA Monomer E (EMod3) (18.3 mole, 1 eq., FW: 571.8) and 6594 g of Monomer D (16.47 mole, 0.9 eq, FW: 400.4). The combined monomers were dissolved in 20 L toluene and co-evaporated at 40° C. This was repeated with co-evaporation with an additional 2×20 L of toluene, after which 60 L of dichloromethane (DCM) were transferred into the reactor and dissolved. The reactor was purged with nitrogen and was chilled to −40° C. When the temperature was between −30° C. and −40° C., 2423 g (2071 mL, 9.17 moles, 0.5 eq) of TES Triflate were transferred into the reactor. After complete addition of TES Triflate the reaction was allowed to warm and stirring was continued. The completeness of the reaction was monitored by HPLC and TLC (35:65 ethyl acetate/Heptane). The reaction required 2-3 hours to reach completion. When the reaction was completed, 2040 mL of triethylamine (TEA, 1481 g, 0.8 eq.) were transferred into the reactor and stirred for an additional 30 minutes. The organic layer (DCM layer) was extracted with 2×20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 15:85, 20:80, 25:75, 30:70 and 35:65 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The ED Dimer was obtained in 81% isolated yield.
Synthesis of the EDC Trimer
Step 5. Preparation of ED1 TFA, Hydrolysis of ED Dimer
A 100 L reactor was charged with 7.5 Kg of ED Dimer (9.26 mol, 1 equiv). The reactor was chilled to <5° C. and 30.66 L of 90% TFA solution (TFA, 370.4 mol, 40 equiv) were transferred into the reactor. When the addition was completed the reaction was allowed to warm to room temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexanes) and HPLC. The reaction took approximately 3-4 hours to reach completion. When the reaction was completed, was chilled and 51.6 L of triethylamine (TEA, 37.5 Kg, 370.4 mole) were transferred into the reactor, after which 20 L of water & 20 L ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. Ethyl acetate solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 20:80, 30:70, 40:60, 50:50, 60:40 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup. Isolated yield of ED1 was about 70%.
Synthesis of the EDC Trimer
Step 6. Preparation of ED2, Silylation of ED1
A 100 L reactor was charged with 11000 g of 1,2-diOH ED Dimer (ED1) (14.03 mol, 1 equiv), 1910.5 g of imidazole (28.06 mol, 2 equiv) and 30 L of dichloromethane. The reactor was purged with nitrogen and chilled to −20° C., after which 3.53 L butyldiphenylchloro-silane (TBDPS-Cl, 4.628 Kg, 16.835 mol, 1.2 equiv) was charged into the reactor. When the addition was complete, the chiller was turned off and the reaction was allowed to warm to ambient temperature. The reaction was monitored by TLC (50% ethyl acetate/hexane) and HPLC. The reaction required 4-6 hours to reach completion. When the reaction was completed, 20 L of water and 10 L of dichloromethane were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. Dichloromethane solution was dried in 4-6 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). Intermediate ED2 was obtained in 75% isolated yield.
Synthesis of the EDC Trimer
Step 7. Preparation of ED3, D-Levulination
A 100 L reactor was charged with 19800 g of 1-Silyl ED Dimer (ED2) (19.37 moles, 1 equiv) and 40 L of dry tetrahydrofuran (THF) and agitated to dissolve. The reactor was purged with nitrogen and 237 g of dimethylaminopyridine (DMAP, 1.937 moles, 0.1 equiv) and 10.05 L of diisopropylethylamine (DIPEA, 63.9 moles, 3 equiv) were transferred into the reactor. The reactor was chilled to 10-15° C. and kept under a nitrogen atmosphere, after which 12.46 Kg of levulinic anhydride (58.11 moles, 3 eq) was charged into the reactor. When the addition was complete, the reaction was warmed to ambient temperature and stirred overnight or 12-16 hours. The completeness of the reaction was monitored by TLC (40:60 ethyl acetate/hexane) and by HPLC. 20 L of 10% citric acid, 10 L of water and 25 L of ethyl acetate were transferred into the reactor. This was stirred for 30 min and the layers were separated. The organic layer (EtOAc layer) was extracted with 20 L of water, 20 L 5% sodium bicarbonate and 20 L 25% brine solutions. The ethyl acetate solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The ED3 yield was 95%.
Synthesis of the EDC Trimer
Step 8. Preparation of ED4, Desilylation of ED3
A 100 L reactor was charged with 19720 g of 1-Silyl-2-Lev ED Dimer (ED3) (17.6 mol, 1 equiv) in 40 L of THF. The reactor was chilled to 0° C., after which 6903 g of tetrabutylammonium fluoride trihydrate (TBAF, 26.4 mol, 1.5 equiv) and 1511 mL (26.4 mol, 1.5 equiv) of glacial acetic acid were transferred into the reactor. When the addition was complete, the reaction was stirred and allowed to warm to ambient temperature. The reaction was monitored by TLC (40:60 ethyl acetate/hexane) and HPLC. The reaction required 3 hours to reach completion. When the reaction was completed, 20 L of water and 10 L of dichloromethane were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified using a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20 (EtOAc/heptane) and 200 L 100% EtOAc. The pure fractions were pooled and evaporated to a syrup and used in the next step. The isolated yield of ED4 was about 92%.
Synthesis of the EDC Trimer
Step 9. Preparation of ED5, TCA Formation
A 100 L reactor was charged with 14420 g of 1-OH-2-Lev ED Dimer (ED4) (16.35 mol, 1 equiv) in 30 L of dichloromethane. The reactor was purged with nitrogen and stirring was begun, after which 1222 mL of diazabicycloundecene (DBU, 8.175 mol, 0.5 equiv) and 23.61 Kg of trichloroacetonitrile (TCA, 163.5 mol, 10 equiv) were transferred into the reactor. Stirring was continued and the reaction was kept under nitrogen. After reagent addition, the reaction was allowed to warm to ambient temperature. The reaction was monitored by HPLC and TLC (40:60 ethyl acetate/heptane). The reaction took approximately 2 hours for reaction completion. When the reaction was completed, 20 L of water and 10 L of DCM were transferred into the reactor and stirred for 30 min and the layers were separated. The organic layer (DCM layer) was extracted with 20 L water and 20 L 25% brine solutions. The dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The solution was evaporated to a syrup (bath temp. 40° C.). The crude product was purified using a 200 L silica column using 140-200 L each of the following gradient profiles: 10:90, 20:80, 30:70, 40:60 and 50:50 (EtOAc/heptane). The pure fractions were pooled and evaporated to a syrup and used in the next step. The isolated yield of intermediate ED5 was about 70%.
Synthesis of the EDC Trimer
Step 10.
Preparation of EDC Trimer, Coupling of ED5 with Monomer C 6-O-acetyl-2-azido-3,4-di-O-benzyl-2-deoxy-α-D-glucopyranosyl-(1→4)-benzyl (3-O-benzyl-2-O-levulinoyl)-β-D-glucopyranosyluronate-(1→4)-(3-O-acetyl-1,6-anhydro-2-azido)-2-deoxy-β-D-glucopyranose
A 100 L reactor was charged with 12780 g of 2-Lev 1-TCA ED Dimer (ED5) (7.38 mole, 1 eq., FW) and 4764 g of Monomer C (7.38 mole, 1 eq). The combined monomers were dissolved in 20 L toluene and co-evaporated at 40° C. Repeated was co-evaporation with an additional 2×20 L of toluene, after which 60 L of dichloromethane (DCM) was transferred into the reactor and dissolved. The reactor was purged with nitrogen and chilled to −20° C. When the temperature was between −20 and −10° C., 2962 g (11.2 moles, 0.9 eq) of TES Triflate were transferred into the reactor. After complete addition of TES Triflate the reaction was allowed to warm to 5° C. and stirring was continued. Completeness of the reaction was monitored by HPLC and TLC (35:65 ethyl acetate/Heptane). The reaction required 2-3 hours to reach completion. When the reaction was completed, 1133 g of triethylamine (TEA, 0.9 eq.) were transferred into the reactor and stirred for an additional 30 minutes. The organic layer (DCM layer) was extracted with 2×20 L 25% 4:1 sodium chloride/sodium bicarbonate solution. Dichloromethane solution was dried in 6-8 Kg of anhydrous sodium sulfate. The syrup was purified in a 200 L silica column using 140-200 L each of the following gradient profiles: 15:85, 20:80, 25:75, 30:70 and 35:65 (EtOAc/heptane). Pure fractions were pooled and evaporated to a syrup. The isolated yield of EDC Trimer was 48%. Formula: C55H60N6O18; Mol. Wt. 1093.09. The 1H NMR spectrum (d6-acetone) of the EDC trimer is shown in FIG. 3.

Preparation of EDC1
Step 1:
Anhydro Ring Opening & Acetylation 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-1,3,6-tri-O-acetyl-β-D-glucopyranose
7.0 Kg (6.44 mol) of EDC Trimer was dissolved in 18 L anhydrous Dichloromethane. 6.57 Kg (64.4 mol, 10 eq) of Acetic anhydride was added. The solution was cooled to −45 to −35° C. and 1.82 Kg (12.9 mol, 2 eq) of Boron Trifluoride etherate was added slowly. Upon completion of addition, the mixture was warmed to 0-10° C. and kept at this temperature for 3 hours until reaction was complete by TLC and HPLC. The reaction was cooled to −20° C. and cautiously quenched and extracted with saturated solution of sodium bicarbonate (3×20 L) while maintaining the mixture temperature below 5° C. The organic layer was extracted with brine (1×20 L), dried over anhydrous sodium sulfate, and concentrated under vacuum to a syrup. The resulting syrup of EDC1 (6.74 Kg) was used for step 2 without further purification. The 1H NMR spectrum (d6-acetone) of the EDC-1 trimer is shown in FIG. 4.

Preparation of EDC2
Step 2:
Deacetylation 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-β-D-glucopyranose
The crude EDC1 product obtained from step 1 was dissolved in 27 L of Tetrahydrofuran and chilled to 15-20° C., after which 6 Kg (55.8 mol) of benzylamine was added slowly while maintaining the reaction temperature below 25° C. The reaction mixture was stirred for 5-6 hours at 10-15° C. Upon completion, the mixture was diluted with ethyl acetate and extracted and quenched with 10% citric acid solution (2×20 L) while maintaining the temperature below 25° C. The organic layer was extracted with 10% NaCl/1% sodium bicarbonate (1×20 L). The extraction was repeated with water (1×10 L), dried over anhydrous sodium sulfate and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel yielded 4.21 Kg (57% yield over 2 steps) of EDC2[ also referred to as 1-Hydroxy-6-Acetyl EDC Trimer]. The 1H NMR spectrum (d6-acetone) of the EDC-2 trimer is shown in FIG. 5.

Preparation of EDC3
Step 3:
Formation of TCA Derivative 6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-1-O-trichloroacetimidoyl-β-D-glucopyranose
4.54 Kg (3.94 mol) of EDC2 was dissolved in 20 L of Dichloromethane. 11.4 Kg (78.8 mol, 20 eq) of Trichloroacetonitrile was added. The solution was cooled to −15 to −20° C. and 300 g (1.97 mol, 0.5 eq) of Diazabicycloundecene was added. The reaction was allowed to warm to 0-10° C. and stirred for 2 hours or until reaction was complete. Upon completion, water (20 L) was added and the reaction was extracted with an additional 10 L of DCM. The organic layer was extracted with brine (1×20 L), dried over anhydrous sodium sulfate, and concentrated under vacuum to a syrup. Column chromatographic separation using silica gel and 20-60% ethyl acetate/heptane gradient yielded 3.67 Kg (72% yield) of 1-TCA derivative, EDC3. The 1H NMR spectrum (d6-acetone) of the EDC-3 trimer is shown in FIG. 6.

Preparation of EDCBA1
Step 4:
Coupling of EDC3 with BA Dimer Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl)-(1→4)-O-[benzyl 3-O-benzyl-2-O-levulinoyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
3.67 Kg (2.83 mol) of EDC3 and 3.16 Kg (3.96 mol, 1.4 eq) of BA Dimer was dissolved in 7-10 L of Toluene and evaporated to dryness. The resulting syrup was coevaporated with Toluene (2×15 L) to remove water. The dried syrup was dissolved in 20 L of anhydrous Dichloromethane, transferred to the reaction flask, and cooled to −15 to −20° C. 898 g (3.4 mol, 1.2 eq) of triethylsilyl triflate was added while maintaining the temperature below −5° C. When the addition was complete, the reaction was immediately warmed to −5 to 0° C. and stirred for 3 hours. The reaction was quenched by slowly adding 344 g (3.4 mol, 1.2 eq) of Triethylamine. Water (15 L) was added and the reaction was extracted with an additional 10 L of DCM. The organic layer was extracted with a 25% 4:1 Sodium Chloride/Sodium Bicarbonate solution (2×20 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. The resulting syrup of the pentasaccharide, EDCBA1 was used for step 5 without further purification. The 1H NMR spectrum (d6-acetone) of the EDCBA-1 pentamer is shown in FIG. 7.

Preparation of EDCBA2
Step 5:
Hydrolysis of Levulinyl moiety Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl)-(1→4)—O-[benzyl 3-O-benzyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl)-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
The crude EDCBA1 from step 4 was dissolved in 15 L of Tetrahydrofuran and chilled to −20 to −25° C. A solution containing 679 g (13.6 mol) of Hydrazine monohydrate and 171 g (1.94 mol) of Hydrazine Acetate in 7 L of Methanol was added slowly while maintaining the temperature below −20° C. When the addition was complete, the reaction mixture was allowed to warm to 0-10° C. and stirred for several hours until the reaction is complete, after which 20 L of Ethyl acetate was added and the reaction was extracted with 10% citric acid (2×12 L). The organic layer was washed with water (1×12 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel and 10-45% ethyl acetate/heptane gradient yielded 2.47 Kg (47.5% yield over 2 steps) of EDCBA2. The 1H NMR spectrum (d6-acetone) of the EDCBA-2 pentamer is shown in FIG. 8.

Preparation of EDCBA Pentamer
Step 6:
THP Formation Methyl O-6-O-acetyl-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-[benzyl 3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronate]-(1→4)-O-2-azido-2-deoxy-3,6-di-O-acetyl-α-D-glucopyranosyl-(1→4)-O-[methyl 2-O-benzoyl-3-O-benzyl-α-L-Idopyranosyluronate]-(1→4)-2-azido-6-O-benzoyl-3-O-benzyl-2-deoxy-α-D-glucopyranoside
2.47 Kg (1.35 mol) of EDCBA2 was dissolved in 23 L Dichloroethane and chilled to 10-15° C., after which 1.13 Kg (13.5 mol, 10 eq) of Dihydropyran and 31.3 g (0.135 mol, 0.1 eq) of Camphorsulfonic acid were added. The reaction was allowed warm to 20-25° C. and stirred for 4-6 hours until reaction was complete. Water (15 L) was added and the reaction was extracted with an additional 10 L of DCE. The organic layer was extracted with a 25% 4:1 Sodium Chloride/Sodium Bicarbonate solution (2×20 L), dried over anhydrous sodium sulfate, and evaporated under vacuum to a syrup. Column chromatographic separation using silica gel and 10-35% ethyl acetate/heptane gradient yielded 2.28 Kg (88.5% yield) of fully protected EDCBA Pentamer. The 1H NMR spectrum (d6-acetone) of the EDCBA pentamer is shown in FIG. 9.

Preparation of API1
Step 1:
Saponification Methyl O-2-azido-2-deoxy-3,4-di-O-benzyl-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-azido-2-deoxy-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-α-L-Idopyranosyluronosyl-(1→4)-2-azido-3-O-benzyl-2-deoxy-α-D-glucopyranoside disodium salt
To a solution of 2.28 Kg (1.19 mol) of EDCBA Pentamer in 27 L of Dioxane and 41 L of Tetrahydrofuran was added 45.5 L of 0.7 M (31.88 mol, 27 eq) Lithium hydroxide solution followed by 5.33 L of 30% Hydrogen peroxide. The reaction mixture was stirred for 10-20 hours to remove the acetyl groups. Then, 10 L of 4 N (40 mol, 34 eq) sodium hydroxide solution was added. The reaction was allowed to stir for an additional 24-48 hours to hydrolyze the benzyl and methyl esters completely. The reaction was then extracted with ethyl acetate. The organic layer was extracted with brine solution and dried with anhydrous sodium sulfate. Evaporation of the solvent under vacuum gave a syrup of API1 [also referred to as EDCBA(OH)5] which was used for the next step without further purification.
Preparation of API2
Step 2:
O-Sulfonation Methyl O-2-azido-2-deoxy-3,4-di-O-benzyl-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-azido-2-deoxy-3,6-di-O-sulfo-α-D-glucopyranosyl-(1→4)-O-3-O-benzyl-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-azido-2-deoxy-6-O-sulfo-α-D-glucopyranoside, heptasodium salt
The crude product of API1 [aka EDCBA(OH)5] obtained in step 1 was dissolved in 10 L Dimethylformamide. To this was added a previously prepared solution containing 10.5 Kg (66 moles) of sulfur trioxide-pyridine complex in 10 L of Pyridine and 25 L of Dimethylformamide. The reaction mixture was heated to 50° C. over a period of 45 min. After stiffing at 1.5 hours at 50° C., the reaction was cooled to 20° C. and was quenched into 60 L of 8% sodium bicarbonate solution that was kept at 10° C. The pH of the quench mixture was maintained at pH 7-9 by addition of sodium bicarbonate solution. When all the reaction mixture has been transferred, the quench mixture was stirred for an additional 2 hours and pH was maintained at pH 7 or greater. When the pH of quench has stabilized, it was diluted with water and the resulting mixture was purified using a preparative HPLC column packed with Amberchrom CG161-M and eluted with 90%-10% Sodium Bicarbonate (5%) solution/Methanol over 180 min. The pure fractions were concentrated under vacuum and was then desalted using a size exclusion resin or gel filtration (Biorad) G25 to give 1581 g (65.5% yield over 2 steps) of API2 [also referred to as EDCBA(OSO3)5]. The 1H NMR spectrum (d6-acetone) of API-2 pentamer is shown in FIG. 10.

Preparation of API3
Step 3:
Hydrogenation Methyl O-2-amino-2-deoxy-6-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2-O-tetrahydropyranyl-β-D-glucopyranosyluronosyl-(1→4)-O-2-amino-2-deoxy-3,6-di-O-sulfo-α-D-glucopyranosyl-(1→4)-O-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-amino-2-deoxy-6-O-sulfo-α-D-glucopyranoside, heptasodium salt
A solution of 1581 g (0.78 mol) of O-Sulfated pentasaccharide API2 in 38 L of Methanol and 32 L of water was treated with 30 wt % of Palladium in Activated carbon under 100 psi of Hydrogen pressure at 60-65° C. for 60 hours or until completion of reaction. The mixture was then filtered through 1.0μ and 0.2μ filter cartridges and the solvent evaporated under vacuum to give 942 g (80% yield) of API3 [also referred to as EDCBA(OSO3)5(NH2)3]. The 1H NMR spectrum (d6-acetone) of API-3 pentamer is shown in FIG. 11.

Preparation of Fondaparinux Sodium
Step 4:
N-Sulfation & Removal of THP Methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)—O-β-D-glucopyranuronosyl-(1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-O-sulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt
To a solution of 942 g (0.63 mol) of API3 in 46 L of water was slowly added 3.25 Kg (20.4 mol, 32 eq) of Sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2 N sodium hydroxide solution. The reaction was allowed to stir for 4-6 hours at pH 9.0-9.5. When reaction was complete, the pH was adjusted to pH 7.0 using 50 mM solution of Ammonium acetate at pH 3.5. The resulting N-sulfated EDCBA(OSO3)5(NHSO3)3 mixture was purified using Ion-Exchange Chromatographic Column (Varian Preparative 15 cm HiQ Column) followed by desalting using a size exclusion resin or gel filtration (Biorad G25). The resulting mixture was then treated with activated charcoal and the purification by ion-exchange and desalting were repeated to give 516 g (47.6% yield) of the purified Fondaparinux Sodium form.
Analysis of the Fondaparinux sodium identified the presence of P1, P2, P3, and P4 in the fondaparinux. P1, P2, P3, and P4 were identified by standard analytical methods.
INTERMEDIATES
The monomers used in the processes described herein may be prepared as described in the art, or can be prepared using the methods described herein.

The synthesis of Monomer A-2 (CAS Registry Number 134221-42-4) has been described in the following references: Arndt et al., Organic Letters, 5(22), 4179-4182, 2003; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; and Sakairi et al., Journal of the Chemical Society, Chemical Communications, (5), 289-90, 1991, and the references cited therein, which are hereby incorporated by reference in their entireties.

Monomer C(CAS Registry Number 87326-68-9) can be synthesized using the methods described in the following references: Ganguli et al., Tetrahedron: Asymmetry, 16(2), 411-424, 2005; Izumi et al., Journal of Organic Chemistry, 62(4), 992-998, 1997; Van Boeckel et al., Recueil: Journal of the Royal Netherlands Chemical Society, 102(9), 415-16, 1983; Wessel et al.,Helvetica Chimica Acta, 72(6), 1268-77, 1989; Petitou et al., U.S. Pat. No. 4,818,816 and references cited therein, which are hereby incorporated by reference in their entireties.

Monomer E (CAS Registry Number 55682-48-9) can be synthesized using the methods described in the following literature references: Hawley et al., European Journal of Organic Chemistry, (12), 1925-1936, 2002; Dondoni et al., Journal of Organic Chemistry, 67(13), 4475-4486, 2002; Van der Klein et al., Tetrahedron, 48(22), 4649-58, 1992; Hori et al., Journal of Organic Chemistry, 54(6), 1346-53, 1989; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; Tailler et al.,Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio–Organic Chemistry, (23), 3163-4, (1972-1999) (1992); Paulsen et al., Chemische Berichte, 111(6), 2334-47, 1978; Dasgupta et al., Synthesis, (8), 626-8, 1988; Paulsen et al., Angewandte Chemie, 87(15), 547-8, 1975; and references cited therein, which are hereby incorporated by reference in their entireties.

Monomer B-1 (CAS Registry Number 444118-44-9) can be synthesized using the methods described in the following literature references: Lohman et al., Journal of Organic Chemistry, 68(19), 7559-7561, 2003; Orgueira et al., Chemistry—A European Journal, 9(1), 140-169, 2003; Manabe et al., Journal of the American Chemical Society, 128(33), 10666-10667, 2006; Orgueira et al., Angewandte Chemie, International Edition, 41(12), 2128-2131, 2002; and references cited therein, which are hereby incorporated by reference in their entireties.
Synthesis of Monomer D
Monomer D was prepared in 8 synthetic steps from glucose pentaacetate using the following procedure:

Pentaacetate SM-B was brominated at the anomeric carbon using HBr in acetic acid to give bromide derivative IntD1. This step was carried out using the reactants SM-B, 33% hydrogen bromide, acetic acid and dichloromethane, stirring in an ice water bath for about 3 hours and evaporating at room temperature. IntD1 was reductively cyclized with sodium borohydride and tetrabutylammonium iodide in acetonitrile using 3 Å molecular sieves as dehydrating agent and stirring at 40° C. for 16 hours to give the acetal derivative, IntD2. The three acetyl groups in IntD2 were hydrolyzed by heating with sodium methoxide in methanol at 50° C. for 3 hours and the reaction mixture was neutralized using Dowex 50WX8-100 resin (Aldrich) in the acid form to give the trihydroxy acetal derivative IntD3.
The C4 and C6 hydroxyls of IntD3 were protected by mixing with benzaldehyde dimethyl acetate and camphor sulphonic acid at 50° C. for 2 hours to give the benzylidene-acetal derivative IntD4. The free hydroxyl at the C3 position of IntD4 was deprotonated with sodium hydride in THF as solvent at 0° C. and alkylated with benzyl bromide in THF, and allowing the reaction mixture to warm to room temperature with stirring to give the benzyl ether IntD5. The benzylidene moiety of IntD5 was deprotected by adding trifluoroacetic acid in dichloromethane at 0° C. and allowing it to warm to room temperature for 16 hours to give IntD6 with a primary hydroxyl group. IntD6 was then oxidized with TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxide) in the presence of tetrabutylammonium chloride, sodium bromide, ethyl acetate, sodium chlorate and sodium bicarbonate, with stirring at room temperature for 16 hours to form the carboxylic acid derivative IntD7. The acid IntD7 was esterified with benzyl alcohol and dicyclohexylcarbodiimide (other reactants being hydroxybenzotriazole and triethylamine) with stirring at room temperature for 16 hours to give Monomer D.
Synthesis of the BA Dimer
The BA Dimer was prepared in 12 synthetic steps from Monomer B1 and Monomer A2 using the following procedure:


The C4-hydroxyl of Monomer B-1 was levulinated using levulinic anhydride and diisopropylethylamine (DIPEA) with mixing at room temperature for 16 hours to give the levulinate ester BMod1, which was followed by hydrolysis of the acetonide with 90% trifluoroacetic acid and mixing at room temperature for 4 hours to give the diol BMod2. The C1 hydroxyl of the diol BMod2 was silylated with tert-butyldiphenylsilylchloride by mixing at room temperature for 3 hours to give silyl derivative BMod3. The C2-hydroxyl was then benzoylated with benzoyl chloride in pyridine, and mixed at room temperature for 3 hours to give compound BMod4. The silyl group on BMod4 was then deprotected with tert-butyl ammonium fluoride and mixing at room temperature for 3 hours to give the C1-hydroyl BMod5. The C1-hydroxyl is then allowed to react with trichloroacetonitrile in the presence of diazobicycloundecane (DBU) and mixing at room temperature for 2 hours to give the trichloroacetamidate (TCA) derivative BMod6, which suitable for coupling, for example with Monomer A-2.
Monomer A-2 was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1.
Monomer A2 was prepared for the coupling reaction by opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1. This transformation occurs using boron trifluoride etherate, acetic anhydride and dichloromethane, between −20° C. and room temperature for 3 hours. The C1-Acetate of AMod1 was then hydrolyzed and methylated in two steps to give the diacetate AMod3. That is, first AMod1 was reacted with trimethylsilyl iodide and mixed at room temperature for 2 hours, then reacted with and tetrabutyl ammonium iodide. This mixture was reacted with diisoproylethylamine and methanol and stirred for 16 hours at room temperature, thus forming AMod3. The C4 and C6 acetates of AMod3 are hydrolyzed with sodium methoxide to give the diol Amod4. The AMod3 mixture was also subjected to mixing at room temperature for 3 hours with Dowex 50 Wx4x8-100 resin in the acid form for neutralization. This formed Amod4. The C6-hydroxyl of AMod4 is then benzoylated by treating with benzoyl chloride in pyridine at −40° C. and then allowing it to warm up to −10° C. over 2 hours to give AMod5.
Coupling of monomer AMod5 with the free C4-hydroxyl group of BMod6 was performed in the presence of BF3.Et2O and dichloromethane with mixing between −20° C. and room temperature for 3 hours to provide disaccharide BA1. The C4-levulinyl moiety of the disaccharide was then hydrolyzed with hydrazine to give the BA Dimer, which is suitable for subsequent coupling reactions.
Synthesis of EDC Trimer
The EDC Trimer was prepared in 10 synthetic steps from Monomer E, Monomer D and Monomer C using the following procedure:


Monomer E was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give diacetate EMod1. This occurs by the addition of Monomer E with boron trifluoride etherate, acetic anhydride and dichloromethane at −10° C., and allowing the reaction to warm to room temperature with stirring for 3 hours. The C1-Acetate of EMod1 is then hydrolyzed to give the alcohol, EMod2. This occurs by reacting Emod1 with hydrazine acetate and dimethylformamide and mixing at room temperature for 3 hours. The C1-hydroxyl of Emod2 is then reacted with trichloroacetonitrile to give the trichloro acetamidate (TCA) derivative EMod3 suitable for coupling, which reaction also employs diazabicycloundecane and dichloromethane and mixing at room temperature for 2 hours.
Monomer D, having a free C4-hydroxyl group, was coupled with monomer EMod3 in the presence of triethylsilyl triflate with mixing at −40° C. for 2 hours to give the disaccharide ED Dimer. The acetal on ring sugar D of the ED Dimer is hydrolyzed to give the C1,C2-diol ED1. This occurs by reacting the ED Dimer with 90% trifluoro acetic acid and mixing at room temperature for 4 hours. The C1-hydroxyl moiety of ED1 was then silylated with tert-butyldiphenylsilyl chloride to give the silyl derivative ED2. The C2-hydroxyl of ED2 was then allowed to react with levulinic anhydride in the presence of dimethylaminopyridine (DMAP) and diethylisopropylamine for approximately 16 hours to give the levulinate ester ED3. The TBDPS moiety is then deprotected by removal with tert-butylammonium fluoride in acetic acid with mixing at room temperature for 3 hours to give ED4 having a C1-hydroxyl. The C1-hydroxyl moiety of ED4 was then allowed to react with trichloroacetonitrile to give the TCA derivative ED5, which is suitable for coupling.
The C1-hydroxyl moiety of ED4 is then allowed to react with trichloroacetonitrile to give the TCA derivative ED5 suitable for coupling using diazabicycloundecane and dichloromethane, and mixing at room temperature for 2 hours. Monomer C, havinga free C4-hydroxyl group, was then coupled with the disaccharide ED5 in the presence of triethylsilyl triflate and mixed at −20° C. for 2 hours to give the trisaccharide EDC Trimer.
Synthesis of the EDCBA Pentamer
The EDCBA Pentamer was prepared using the following procedure:

The preparation of EDCBA Pentamer is accomplished in two parts as follows. In part 1, the EDC Trimer, a diacetate intermediate, is prepared for the coupling reaction with Dimer BA by initially opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the tetraacetate derivative EDC1. This occurs by reacting the EDC Trimer with boron trifluoride etherate, acetic anhydride and dichlormethane and stirring between −10° C. and room temperature for 3 hours. The C1-Acetate of EDC1 is then hydrolyzed to give the alcohol, EDC2, by reacting EDC1 with benzylamine [BnNH2] and tetrahydrofuran and mixing at −10° C. for 3 hours. The C1-hydroxyl of EDC2 is then reacted with trichloroacetonitrile and diazabicycloundecane, with mixing at room temperature for 2 hours, to give the trichloro acetamidate (TCA) derivative EDC3 suitable for coupling.


In Part 2 of the EDCBA Pentameter synthesis, the Dimer BA, having a free C4-hydroxyl group, is coupled with trisaccharide EDC3 in the presence of triethylsilyltriflate at −30° C. mixing for 2 hours to give the pentasaccharide EDCBA1. The levulinyl ester on C2 of sugar D in EDCBA1 is hydrolyzed with a mixture of deprotecting agents, hydrazine hydrate and hydrazine acetate and stiffing at room temperature for 3 hours to give the C2-hydroxyl containing intermediate EDCBA2. The C2-hydroxyl moiety on sugar D of EDCBA2 is then alkylated with dihydropyran (DHP) in the presence of camphor sulfonic acid (CSA) and tetrahydrofuran with mixing at room temperature for 3 hours to give the tetrahydropyranyl ether (THP) derivative, EDCBA Pentamer.
…………………………
A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodium, Org Process Res Dev 2013, 17: 869, http://pubs.acs.org/doi/full/10.1021/op300367c

An improved method for the simultaneous removal of O-benzyl and N-carboxybenzyl groups as well as reducing azide groups to amines in protected heparin-like pentasaccharides, a key process in fondaparinux sodium synthesis, is reported. Under catalytic transfer hydrogenation conditions, using readily available and inexpensive ammonium formate, the hydrogenolysis is done in less than an hour in good yield and purity. This procedure represents a major advantage over the previously published procedures, the latter of which involve several hours/days of hydrogenation reaction under catalytic reduction using gaseous hydrogen.

Synthesis of Compound 1 (FONDAPARINUX)
………………
SYNTHESIS
In the synthesis of Fondaparinux sodium, the monomers XII, XVIII, XXVII, XXXVIII, XXXXI and dimers XIX, XX, XL described herein may be made either by processes described in the art or, by a process as described herein. The XII and XVIII monomers may then linked to form a disaccharide XX, XXXIX and XXVII monomers may then linked to form a disaccharide XL, XLIII and XX dimers may then linked to form a tetrasaccharide, XLVII tetramer and XLV monomer may be linked to form a pentasaccharide (XLVIII) pentamer. The XLVIII pentamer is an intermediate that may be converted through a series of reactions to fondaparinux sodium. This strategy described herein provides an efficient method for multi-kilogram preparation of fondaparinux in high yields and highly stereoselective purity.
Fondaparinux sodium (LIII) was prepared in 3 synthetic steps from O – S pentasaccharide (L) using the following procedure:

Fondaparinux Sodium (LIII)
Preparation of Fondaparinux sodium (LIII)—
N- sulfonation of Deprotected Pentasaccharide (LI) methyl 0-2-deoxy-3,6-di-0- sulfo-2-(sulfoamino)-a-D-glucopyranosyl-(l— >4)-0-2-0-sulfo-a-L- idopyranurosyl-( 1— >4)-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D-glucopyranoside,decasodium salt
A solution of deprotected pentasaccharide (LI) (145 gm) in water (2.54 V) was adjusted to a pH of 9.5 – 10.5 with 1 N NaOH solution. S03-pyridine complex (156 gm) was added into 3 lots every 15 min, the pH being maintained at 9.5-10.5 by automatic addition of 1 N NaOH. The mixture was stirred for 2 hrs at RT, during this aqueous NaOH (IN solution) was added to maintain pH at 9.5 – 10.5. After neutralization to pH 7 – 7.5 by addition of HC1 solution, the mixture was evaporated using vacuum. The residue was dissolved in water (1.6 L) at RT, to this solution was added acetone (1.6 L) at RT. The reaction mass was cooled to 5°C – 1 0 °C and stirred for 1 hr. The solid was filtered and washed with cold acetone: water (1 :1). The clear filtrate was distilled off completely under vacuum below 55°C. The residue was dissolved in water (1.6 L) at RT, and to this solution was added acetone(1.6 L) at RT. The mixture was cooled to 5 to 10°C and stirred for 1 hr. The solid was filtered and washed with cold acetone/water (1 :1). The clear filtrate was distilled off completely under vacuum below 55°C. The residue was dissolved in water (0.7 L) and charcoal (40 gm) was added at RT. The mixture was stirred for 30 min at RT then filtered. To the filtrate was added charcoal (40 gm) at RT. The mixture was stirred for 30 min at RT then filtered. To the filtrate was added charcoal (40 gm) at RT. The mixture was stirred for 30 min at RT then filtered. The pH of the clear filtrate was adjusted to 8.0 – 8.5 with IN NaOH solution and distilled off completely under vacuum below 55 °C. The residue was dissolved in 0.5 M NaCl solution and layered onto a column of Dowex® 1×2 -400 resins using a gradient of NaCl solution (0.5 to 10M). The product fractions were combined and distilled off under vacuum below 55 °C up to 1 – 2 L volume. The solid was filtered off and the clear filtrate was distilled off under vacuum below 55 °C up to slurry stage and subjected to azeotropic distillation with methanol two times. The solid residue was stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The wet solid was again stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The wet solid was again stirred with methanol (2.13 L) at RT for 1 hr and the solid was filtered off and washed with methanol. The above solid was dissolved in water and the pH adjusted to 4 – 4.5 with IN HC1 solution and charcoalized three times with 26 gm of charcoal at RT for 15-30 minutes and filtered off. To the clear filtrate was added 0.39 kg of NaCl, then methanol was added (35 volume) at RT and the mixture was stirred for 15-30 minutes. The solution was decanted and the sticky mass was stirred with methanol (0.65 L) at RT for 15-30 minutes. The solid was filtered off and dissolved in water, and the pH adjusted to 8 – 8.5 with IN NaOH solution. The solution was filtered through 0.45 micron paper & distilled off completely under vacuum below 55°C. The solution was subjected to azeotropic distillation with methanol to give highly pure fondaparinux sodium (97.17 gm) (HPLC purity 99.7%).
SOR Results
Three batches of product made in accordance with the present processes provided the following stereoisomeric optical rotation results:
Specification: Between +50.0° and +60.0°.
Batch- 1 = +55.1°
Batch-2 = +55.7° Batch-3 = +55.4°.
INTERMEDIATES
Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; MS is molecular sieve; DMF is dimethyl formamide; Bn is benzyl; MDC is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; MeOH is methanol; RT is room temperature; Ac2O is acetic anhydride; HBr is hydrogen bromide; EtOAc is ethyl acetate; Cbz is benzyloxycarbonyl; CADS is chloro acetyl disaccharide; HDS is hydroxy disaccharide; NMP is N-methylpyrrolidone.
Methyl 3-O-benzyl-4-O-monochloro acetyl-β-L-idopyranuronate

Route of Synthesis for α-Methyl-6-o-acetyl-3-o-benzyl-2-(benzyloxy carbonyl)amino-2-deoxy-α-D-glucopyranoside

Methyl 6-O-acetyl-3-O-benzyl-2-(benzyloxy carbonyl)amino-2-deoxy-4-O-(methyl-2-O acetyl-3-O-benzyl-α-L-idopyranosyluronate)-glucopyranoside

Route of Synthesis for 1,6-Anhydro-2-azido-3-O-acetyl-2-deoxy-beta-D-glucopyranose

Route of synthesis for Methyl 2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranuronate

Route of synthesis for 3-O-Acetyl-1,6-anhydro-2-azido-4-O-2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranosyl methyluronate-beta-D-glucopyranose
(or)
3-O-Acetyl-1,6-anhydro-2-azido-2-deoxy-4-O-(methyl 2,3-di-O-benzyl-4-O-chloroacetyl-beta-D-glucopyranosyluronate)-beta-D-glucopyranose

Route of Synthesis for 1,6-Anhydro-2-azido-3,4-di-O-benzyl-2-deoxy-beta-D-glucopyranose

Synthesis of Disaccharide XLIII
Disaccharide XLIII was prepared in 2 synthetic steps from CADS sugar (XL) using the following procedure:

CADS sugar XL was acetylated at the anomeric carbon using AC2O and TFA to give acetyl derivative XLII. This step was carried out using the reactants CADS, AC2O and TFA, stirring in an ice water bath for about 5-24 hours, preferably 20 hours, and evaporating to residue under vacuum. Residue was recrystallized in ether. Acetyl CADS (XLII) was brominated at the anomeric carbon using titanium tetra bromide in MDC andethylacetate and stirring at 20° C.-50° C. for 6-16 hours, preferably 6 hours, to give the bromo derivative, (XLIII) after work-up and recrystallization from solvent/alcohol.
Synthesis of the Monosaccharide (XLV)
The monosaccharide (XLV) was prepared in 2 synthetic steps from monomer (XLI) using the following procedure:

Mono sugar (XLI) was acetylated at the anomeric carbon using AC2O and TFA to give acetyl derivative (XLIV). This step was carried out using the reactants Mono sugar (XLI), AC2O and TFA, stirring in an ice water bath for about 5-24 hours, preferably 24 hours, and evaporating to residue under vacuum. Residue was recrystallized in ether. Acetyl Mono sugar (XLIV) was brominated at the anomeric carbon using titanium tetra bromide in MDC and ethyl acetate and stirring at 20° C.-50° C. for 6-20 hours, preferably 16 hours, to give the bromo derivative, (XLV) after work-up and recrystallization from ether.
Synthesis of the Hydroxy Tetrasaccharide (XLVII)
The hydroxy tetrasaccharide (XLVII) was prepared in 2 synthetic steps from disaccharide (XLIII) and HDS (XX) using the following procedure:

Disaccharide (XLIII), was coupled with disaccharide (XX) in the presence of silver carbonate, silver per chlorate and 4 A° MS in MDC and stirred at ambient temperature for 5-12 hrs, preferably 4-6 hours, in the dark followed by work-up and purification in water/methanol to give the tetrasaccharide (XLVI). The d echloroacetylation of tetrasaccharide (XLVI) was carried out in THF, ethanol and pyridine in the presence of thiourea at reflux for 6 to 20 hrs, preferably 12 hours, to give the hydroxy tetrasaccharide (XLVIII).
Synthesis of the Pentasaccharide (XLVIII)
The pentasaccharide (XLVIII) was prepared in 2 synthetic steps from monosaccharide (XLV) and tetrasaccharide (XLVII) using the following procedure:

Monosaccharide (XLV), was coupled with tetrasaccharide (XLVII) in the presence of 2,4,6-collidine, silver triflate and 4 A° MS in MDC and stirred at −10° C. to −20° C. for 1 hr in the dark followed by work-up and purification by column chromatography to give the pentasaccharide (XLVIII).
Synthesis of OS Pentasaccharide (L)
The OS pentasaccharide (L) was prepared in 2 synthetic steps from pentasaccharide (XLVIII) using the following procedure:

Pentasaccharide (XLVIII) was deacetylated in the presence of NaOH in mixture of solvents of MDC, methanol and water at 0° C. to 35° C., for 1-2 hrs followed by work-up and distillation to obtain deacetylated pentasaccharide (XLIX) which was subjected to O-sulfonation in DMF in the presence of SO3-trimethylamine (TMA) at 50° C. to 100° C., preferably 50° C.-55° C., for 6-24 hrs, preferably 12 hours, followed by salt removal through Sephadex® resin and column chromatography purification, then pH adjustment by dilute NaOH to give OS pentasaccharide (L).
…………………………
INTERMEDIATE
highly pure 4-Ο-β-ϋ- glucopyranosyl- 1 ,6-anhydro- -D-glucopyranose

Example 1 : Preparation and purification of 4-0- -D-grucopyranosyl-L6-anhvdro- -D- glucopyranose
A solution of pentachlorophenyl 2,3,6,2′,3′,4′,6′-hepta-(9-acetyl- -D-ceilobioside represented by Formula I;

(400 g) in isopropyl alcohol (4 L) at ambient temperature was cooled to 2°C to 5°C and pulverized potassium hydroxide (355 g) was added to it. This reaction mixture was stirred and the temperature was allowed to rise to ambient temperature. At ambient temperature, the mixture was stirred until the reaction was complete (about 2 hours). The mixture was then heated to 50°C to 55°C and stirred for 30 minutes. The solid obtained was filtered and washed with isopropyl alcohol (400 mL). The solid was stirred with isopropyl alcohol (2.8 L) at 50°C for 30 minutes followed by filtering and washing with isopropyl alcohol (400 mL). The resultant solid was suspended into methanol (800 mL to 1600 mL) followed by cooling to 2°C to 5°C. The pH of the suspension was adjusted to 2 to 3 using 15% methanolic hydrochloride. The solid so obtained was filtered and washed with methanol (400 mL). Solvent was recovered from the filtrate to dryness under vacuum to obtain the pure compound of Formula II as foamy solid.
Yield: 142 g
Example 2: Preparation and purification of 4-Q- -D-grucopyranosyl-l,6-anhvdro- -D- glucopyranose
A solution of pentachlorophenyl 2,3,6,2 ,3 ^ ^‘-hepta-O-acetyl- -D-cellobioside of Formula I (100 g) in methanol (300 mL) at ambient temperature was cooled to 2°C to 5°C and pulverized potassium hydroxide (88.6 g) was added to it. This reaction mixture was stirred and the temperature was allowed to rise to ambient temperature. At ambient temperature, the mixture was stirred until the reaction was complete (about 2 hours). The mixture was cooled to 2°C to 5°C and 15% methanolic hydrogen chloride was added to it until the pH of the mixture reached 2 to 3. At this pH, the reaction mixture was filtered and the residual solid was washed with methanol (100 mL). The solvent was recovered from the filtrate under vacuum. The solid material so obtained was stirred with dichloromethane (500 mL) followed by removal of solvent through decantation/filtration. The resultant solid was stirred with isopropyl alcohol (500 mL), filtered and dried to obtain the pure compound of Formula II.
Yield: 29 g
………………………
SYNTHESIS
Synthesis of Fondaparinux
Fondaparinux was prepared using the following procedure:
Conversion of FPP (also referred to a Fully Protected Pentamer) to FondaparinuxSodium:

Reagents: 1. NaOH, H202, LiOH, Dioxane, RT, 24-48 h; 2. Py.S03, DMF, 60°C, 2h, CG-161 purification; 3. 10% Pd/C, H2, 72h; 4. (a) Py.S03, NaOH, NH4OAc, 12h, (b) HiQ NH4OAc/ NaCl ion-exchange, Sephadex Desalt and (c) HiQ NaCl ion-exchange, Sephadex Desalt. The ester moieties in EDCBA Pentamer-CB were hydrolyzed with sodium and lithium hydroxide in the presence of hydrogen peroxide in dioxane mixing at room temperature for 24- 48 hours to give the pentasaccharide intermediate API1-CB. The five hydroxyl moieties in API1-CB were sulfated using a pyridine-sulfur trioxide complex in dimethylformamide, mixing at 60°C for 2 hours and then purified using column chromatography (CG-161), to give the pentasulfated pentasaccharide API2-CB. The intermediate API2-CB was then hydrogenated to reduce the three azides on sugars E, C and A to amines and the reductive deprotection of the six benzyl ethers to their corresponding hydroxyl groups to form the intermediate API3-CB. This transformation occurs by reacting API2-CB with 10% palladium/carbon catalyst with hydrogen gas for 72 hours. The three amines on API3-CB were then sulfated using the pyridine-sulfur trioxide complex in sodium hydroxide and ammonium acetate, allowing the reaction to proceed for 12 hours . The crude fondaparinux is purified and is subsequently converted to its salt form. The crude mixture was purified using an ion-exchange chromatographic column (HiQ resin) followed by desalting using a size exclusion resin or gel filtration (Biorad Sephadex G25) to give the final product, fondaparinux sodium.
Preparation of Fondaparinux Sodium – Step 4: N-Sulfation of API-3-CB:
Methyl 0-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D-glucopyranosyl-(l→4)-0^-D- glucopyranuronosyl-(l→4)-0-2-deoxy-3,6-di-0-sulfo-2-(sulfoamino)-a-D-glucopyranosyl- (l→4)-0-2-0-sulfo-a-L-idopyranuronosyl-(l→4)-2-deoxy-6-0-sulfo-2-(sulfoamino)-a-D- glucopyranoside, decasodium salt
To a solution of 25.4 gram (16.80 mmol, leq) of API-3-CB in 847 mL of water was slowly added 66.85 gram (446.88 mmol, 25eq) of sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2N sodium hydroxide solution. The reaction was allowed to stir for 4 hours at pH 9.0 – 9.5. When reaction was completed, the pH was adjusted 7.0 by using 70 mL of 50 mmol Ammonium acetate solution pH -3.5. The resulting N-Sulfated Cellobiose mixture was purified using Ion-Exchange
Chromatographic Column followed by desalting using size exclusion resin to gave gram ( %) of the purified Fondaparinux Sodium form.
To a solution of 942 g (0.63 mol) of API3 in 46 L of water was slowly added 3.25 Kg (20.4 mol, 32 eq) of Sulfur trioxide-pyridine complex, maintaining the pH of the reaction mixture at pH 9-9.5 during the addition using 2 N sodium hydroxide solution. The reaction was allowed to stir for 4-6 hours at pH 9.0-9.5. When reaction was complete, the pH was adjusted to pH 7.0 using 50 mM solution of Ammonium acetate at pH 3.5. The resulting N- sulfated EDCBA(OS03)5(NHS03)3 mixture was purified using Ion-Exchange Chromatographic Column (Varian Preparative 15 cm HiQ Column) followed by desalting using a size exclusion resin or gel filtration (Biorad G25). The resulting mixture was then treated with activated charcoal and the purification by ion-exchange and desalting were repeated to give 516 g (47.6% yield) of the purified Fondaparinux sodium form.
INT
SCHEME 1 – Synthesis of Monomer A-2 & AMod5 fBuildinq Block Al

Reagents: 1. NaOMe, MeOH, RT, 2hr, 50wx resin; 2. (Bu3Sn)20 (0.8equiv), ACN, MS, reflux, 3h; 3.l2 (1.5 equiv), 5°C to RT, 2h; 4. NaH (2 equiv), DMF, p-MeOC6H4CH2Br (PMB-Br, 2.5 equiv), -20°C to RT, 2h; 5. NaN3, DMF, 120°C, 12h; 6. NaH, DMF, BnBr, 0°C to RT, 3h.; 7. BF3.Et20, Ac20, DCM, -20°C to RT, 3h; 8. (a) TMS-I, TBAI, RT, 2h; (b) DIPEA, MeOH, 16h, RT; 9. NaOMe, Dowex 50WX8-100 resin H+ form, RT, 3h; 10. Pyridine, Bz-CI, -40°C to -10°C, 2h;
Scheme 2 – Synthesis of Monomer B-1 and BMod6 fBuildinq Block B1

Reagents: 1. NaH, BnBr, THF, DMF, 0° to 65°C, 3h; 2. 66% Acetic Acid/H20, 40 °C, 16h; 3. Nal04, (Bu)4NBr, DCM, H20, Dark, 3h; 4. (PhS)3CH, n-BuLi, THF, -78 °C, 3h; 5. CuCI2/CuO, MeOH, H20, 3h; 6. 90% TFA/H20, DCM, RT, 2h; 7. DMF, CSA 2-methoxypropene, 0° to RT, 16hrs; MeOH, TEA. 8. Lev20, DIPEA, RT, 16h; 9. 90% TFA, RT, 4h; 10. Imidazole, TBDPSi-CI, RT, 3h; 11. Pyridine, BzCI, RT, 3h; 12. TBAF, RT, 3h; 13. TCA, DBU, RT, 2h; Also see, e.g., Bonnaffe et al., Tetrahedron Lett., 41, 307-311, 2000; Bonnaffe et al., Carbohydr. Res., 2003, 338, 681-686, 2003; and Seeberger et al., J. Org. Chem., 2003, 68, 7559- 7561, 2003.
……………………..
Carbohydrate Research, 2012 , vol. 361, p. 155 – 161
1H NMR (D2O) δ: 5.68 (d, J = 3.8 Hz, 1H, H-1D), 5.56 (d, J = 3.4 Hz, 1H, H-1F), 5.24 (d, J = 3.8 Hz, 1H, H-1G), 5.07 (d, J = 3.5 Hz, 1H, H-1H), 4.68 (d, J = 7.9 Hz, 1H, H-5G), 4.54 (dd, J = 11.4, 2.2 Hz, 1H, H-1E), 4.48-4.34 (m, 6H, H-6F, 6H, 6’H, 6D, 3E, 2G), 4.33-4.30 (m, 1H, H-6’F), 4.25-4.17 (m, 4H, H-4G, 3G, 6’D, 5F), 4.06-3.98 (m, 2H, H-4F, 5H), 3.94 (dd, J = 9.7, 2.2 Hz, 1H, H-5D), 3.92-3.86 (m, 2H,H-3E, H-4E), 3.85-3.80 (m, 2H, H-5E, 4H), 3.73-3.60 (m, 3H, H-3H, 3D, 4D), 3.53-3.44 (m, 2H, H-2F, H-2E), 3.47 (s, 3H, OMe), 3.34 (dd, J = 10.2, 3.7 Hz, 1H, H-2H), 3.31(dd, J = 10.2, 3.7 Hz, 1H, H-2D)

FONDAPARINUX
……………………………………..
Synthesis of intermediates
Synthetic Procedures
The following abbreviations are used herein: Ac is acetyl; ACN is acetonitrile; MS is molecular sieves; DMF is dimethyl formamide; PMB is p-methoxybenzyl; Bn is benzyl; DCM is dichloromethane; THF is tetrahydrofuran; TFA is trifluoro acetic acid; CSA is camphor sulfonic acid; TEA is triethylamine; MeOH is methanol; DMAP is dimethylaminopyridine; RT is room temperature; CAN is ceric ammonium nitrate; Ac2O is acetic anhydride; HBr is hydrogen bromide; TEMPO is tetramethylpiperidine-N-oxide; TBACl is tetrabutyl ammonium chloride; EtOAc is ethyl acetate; HOBT is hydroxybenzotriazole; DCC is dicyclohexylcarbodiimide; Lev is levunlinyl; TBDPS is tertiary-butyl diphenylsilyl; TCA is trichloroacetonitrile; O-TCA is O-trichloroacetimidate; Lev2O is levulinic anhydride; DIPEA is diisopropylethylamine; Bz is benzoyl; TBAF is tetrabutylammonium fluoride; DBU is diazabicycloundecane; BF3.Et2O is boron trifluoride etherate; TMSI is trimethylsilyl iodide; TBAI is tetrabutylammonium iodide; TES-Tf is triethylsilyl trifluoromethanesulfonate (triethylsilyl triflate); DHP is dihydropyran; PTS is p-toluenesulfonic acid.
The monomers used in the processes described herein may be prepared as described in the art, or can be prepared using the methods described herein.
The synthesis of Monomer A-2 (CAS Registry Number 134221-42-4) has been described in the following references: Arndt et al., Organic Letters, 5(22), 4179-4182, 2003; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; and Sakairi et al., Journal of the Chemical Society, Chemical Communications, (5), 289-90, 1991, and the references cited therein, which are hereby incorporated by reference in their entireties.
Monomer C(CAS Registry Number 87326-68-9) can be synthesized using the methods described in the following references: Ganguli et al., Tetrahedron: Asymmetry, 16(2), 411-424, 2005; Izumi et al., Journal of Organic Chemistry, 62(4), 992-998, 1997; Van Boeckel et al., Recueil: Journal of the Royal Netherlands Chemical Society, 102(9), 415-16, 1983; Wessel et al.,Helvetica Chimica Acta, 72(6), 1268-77, 1989; Petitou et al., U.S. Pat. No. 4,818,816 and references cited therein, which are hereby incorporated by reference in their entireties.
Monomer E (CAS Registry Number 55682-48-9) can be synthesized using the methods described in the following literature references: Hawley et al., European Journal of Organic Chemistry, (12), 1925-1936, 2002; Dondoni et al., Journal of Organic Chemistry, 67(13), 4475-4486, 2002; Van der Klein et al., Tetrahedron, 48(22), 4649-58, 1992; Hori et al., Journal of Organic Chemistry, 54(6), 1346-53, 1989; Sakairi et al., Bulletin of the Chemical Society of Japan, 67(6), 1756-8, 1994; Tailler et al.,Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio–Organic Chemistry, (23), 3163-4, (1972-1999) (1992); Paulsen et al., Chemische Berichte, 111(6), 2334-47, 1978; Dasgupta et al., Synthesis, (8), 626-8, 1988; Paulsen et al., Angewandte Chemie, 87(15), 547-8, 1975; and references cited therein, which are hereby incorporated by reference in their entireties.
Monomer B-1 (CAS Registry Number 444118-44-9) can be synthesized using the methods described in the following literature references: Lohman et al., Journal of Organic Chemistry, 68(19), 7559-7561, 2003; Orgueira et al., Chemistry—A European Journal, 9(1), 140-169, 2003; Manabe et al., Journal of the American Chemical Society, 128(33), 10666-10667, 2006; Orgueira et al., Angewandte Chemie, International Edition, 41(12), 2128-2131, 2002; and references cited therein, which are hereby incorporated by reference in their entireties.
Synthesis of Monomer D
Monomer D was prepared in 8 synthetic steps from glucose pentaacetate using the following procedure:
Pentaacetate SM-B was brominated at the anomeric carbon using HBr in acetic acid to give bromide derivative IntD1. This step was carried out using the reactants SM-B, 33% hydrogen bromide, acetic acid and dichloromethane, stirring in an ice water bath for about 3 hours and evaporating at room temperature. IntD1 was reductively cyclized with sodium borohydride and tetrabutylammonium iodide in acetonitrile using 3 Å molecular sieves as dehydrating agent and stirring at 40° C. for 16 hours to give the acetal derivative, IntD2. The three acetyl groups in IntD2 were hydrolyzed by heating with sodium methoxide in methanol at 50° C. for 3 hours and the reaction mixture was neutralized using Dowex 50WX8-100 resin (Aldrich) in the acid form to give the trihydroxy acetal derivative IntD3.
The C4 and C6 hydroxyls of IntD3 were protected by mixing with benzaldehyde dimethyl acetate and camphor sulphonic acid at 50° C. for 2 hours to give the benzylidene-acetal derivative IntD4. The free hydroxyl at the C3 position of IntD4 was deprotonated with sodium hydride in THF as solvent at 0° C. and alkylated with benzyl bromide in THF, and allowing the reaction mixture to warm to room temperature with stirring to give the benzyl ether IntD5. The benzylidene moiety of IntD5 was deprotected by adding trifluoroacetic acid in dichloromethane at 0° C. and allowing it to warm to room temperature for 16 hours to give IntD6 with a primary hydroxyl group. IntD6 was then oxidized with TEMPO (2,2,6,6-tetramethyl-1-piperidine-N-oxide) in the presence of tetrabutylammonium chloride, sodium bromide, ethyl acetate, sodium chlorate and sodium bicarbonate, with stirring at room temperature for 16 hours to form the carboxylic acid derivative IntD7. The acid IntD7 was esterified with benzyl alcohol and dicyclohexylcarbodiimide (other reactants being hydroxybenzotriazole and triethylamine) with stirring at room temperature for 16 hours to give Monomer D.
Synthesis of the BA Dimer
The BA Dimer was prepared in 12 synthetic steps from Monomer B1 and Monomer A2 using the following procedure:
The C4-hydroxyl of Monomer B-1 was levulinated using levulinic anhydride and diisopropylethylamine (DIPEA) with mixing at room temperature for 16 hours to give the levulinate ester BMod1, which was followed by hydrolysis of the acetonide with 90% trifluoroacetic acid and mixing at room temperature for 4 hours to give the diol BMod2. The C1 hydroxyl of the diol BMod2 was silylated with tert-butyldiphenylsilylchloride by mixing at room temperature for 3 hours to give silyl derivative BMod3. The C2-hydroxyl was then benzoylated with benzoyl chloride in pyridine, and mixed at room temperature for 3 hours to give compound BMod4. The silyl group on BMod4 was then deprotected with tert-butyl ammonium fluoride and mixing at room temperature for 3 hours to give the C1-hydroyl BMod5. The C1-hydroxyl is then allowed to react with trichloroacetonitrile in the presence of diazobicycloundecane (DBU) and mixing at room temperature for 2 hours to give the trichloroacetamidate (TCA) derivative BMod6, which suitable for coupling, for example with Monomer A-2.
Monomer A-2 was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1.
Monomer A2 was prepared for the coupling reaction by opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the triacetate derivative AMod1. This transformation occurs using boron trifluoride etherate, acetic anhydride and dichloromethane, between −20° C. and room temperature for 3 hours. The C1-Acetate of AMod1 was then hydrolyzed and methylated in two steps to give the diacetate AMod3. That is, first AMod1 was reacted with trimethylsilyl iodide and mixed at room temperature for 2 hours, then reacted with and tetrabutyl ammonium iodide. This mixture was reacted with diisoproylethylamine and methanol and stirred for 16 hours at room temperature, thus forming AMod3. The C4 and C6 acetates of AMod3 are hydrolyzed with sodium methoxide to give the diol Amod4. The AMod3 mixture was also subjected to mixing at room temperature for 3 hours with Dowex 50 Wx4x8-100 resin in the acid form for neutralization. This formed Amod4. The C6-hydroxyl of AMod4 is then benzoylated by treating with benzoyl chloride in pyridine at −40° C. and then allowing it to warm up to −10° C. over 2 hours to give AMod5.
Coupling of monomer AMod5 with the free C4-hydroxyl group of BMod6 was performed in the presence of BF3.Et2O and dichloromethane with mixing between −20° C. and room temperature for 3 hours to provide disaccharide BA1. The C4-levulinyl moiety of the disaccharide was then hydrolyzed with hydrazine to give the BA Dimer, which is suitable for subsequent coupling reactions.
Synthesis of EDC Trimer
The EDC Trimer was prepared in 10 synthetic steps from Monomer E, Monomer D and Monomer C using the following procedure:
Monomer E was prepared for coupling by opening the anhydro moiety with BF3.Et2O followed by acetylation of the resulting hydroxyl groups to give diacetate EMod1. This occurs by the addition of Monomer E with boron trifluoride etherate, acetic anhydride and dichloromethane at −10° C., and allowing the reaction to warm to room temperature with stirring for 3 hours. The C1-Acetate of EMod1 is then hydrolyzed to give the alcohol, EMod2. This occurs by reacting Emod1 with hydrazine acetate and dimethylformamide and mixing at room temperature for 3 hours. The C1-hydroxyl of Emod2 is then reacted with trichloroacetonitrile to give the trichloro acetamidate (TCA) derivative EMod3 suitable for coupling, which reaction also employs diazabicycloundecane and dichloromethane and mixing at room temperature for 2 hours.
Monomer D, having a free C4-hydroxyl group, was coupled with monomer EMod3 in the presence of triethylsilyl triflate with mixing at −40° C. for 2 hours to give the disaccharide ED Dimer. The acetal on ring sugar D of the ED Dimer is hydrolyzed to give the C1,C2-diol ED1. This occurs by reacting the ED Dimer with 90% trifluoro acetic acid and mixing at room temperature for 4 hours. The C1-hydroxyl moiety of ED1 was then silylated with tert-butyldiphenylsilyl chloride to give the silyl derivative ED2. The C2-hydroxyl of ED2 was then allowed to react with levulinic anhydride in the presence of dimethylaminopyridine (DMAP) and diethylisopropylamine for approximately 16 hours to give the levulinate ester ED3. The TBDPS moiety is then deprotected by removal with tert-butylammonium fluoride in acetic acid with mixing at room temperature for 3 hours to give ED4 having a C1-hydroxyl. The C1-hydroxyl moiety of ED4 was then allowed to react with trichloroacetonitrile to give the TCA derivative ED5, which is suitable for coupling.
The C1-hydroxyl moiety of ED4 is then allowed to react with trichloroacetonitrile to give the TCA derivative ED5 suitable for coupling using diazabicycloundecane and dichloromethane, and mixing at room temperature for 2 hours. Monomer C, having a free C4-hydroxyl group, was then coupled with the disaccharide ED5 in the presence of triethylsilyl triflate and mixed at −20° C. for 2 hours to give the trisaccharide EDC Trimer.
Synthesis of the EDCBA Pentamer
The EDCBA Pentamer was prepared using the following procedure:
The preparation of EDCBA Pentamer is accomplished in two parts as follows. In part 1, the EDC Trimer, a diacetate intermediate, is prepared for the coupling reaction with Dimer BA by initially opening the anhydro moiety and acetylation of the resulting hydroxyl groups to give the tetraacetate derivative EDC1. This occurs by reacting the EDC Trimer with boron trifluoride etherate, acetic anhydride and dichlormethane and stirring between −10° C. and room temperature for 3 hours. The C1-Acetate of EDC1 is then hydrolyzed to give the alcohol, EDC2, by reacting EDC1 with benzylamine [BnNH2] and tetrahydrofuran and mixing at −10° C. for 3 hours. The C1-hydroxyl of EDC2 is then reacted with trichloroacetonitrile and diazabicycloundecane, with mixing at room temperature for 2 hours, to give the trichloro acetamidate (TCA) derivative EDC3 suitable for coupling.
In Part 2 of the EDCBA Pentameter synthesis, the Dimer BA, having a free C4-hydroxyl group, is coupled with trisaccharide EDC3 in the presence of triethylsilyltriflate at −30° C. mixing for 2 hours to give the pentasaccharide EDCBA1. The levulinyl ester on C2 of sugar D in EDCBA1 is hydrolyzed with a mixture of deprotecting agents, hydrazine hydrate and hydrazine acetate and stiffing at room temperature for 3 hours to give the C2-hydroxyl containing intermediate EDCBA2. The C2-hydroxyl moiety on sugar D of EDCBA2 is then alkylated with dihydropyran (DHP) in the presence of camphor sulfonic acid (CSA) and tetrahydrofuran with mixing at room temperature for 3 hours to give the tetrahydropyranyl ether (THP) derivative, EDCBA Pentamer.
………………………………
Intermediates listed on the internet
Fondaparinux sodium Intermediates
Fondaparinux sodium N-4

……………………………….
Fondaparinux sodium N-3
114903-05-8

a-D-Glucopyranoside, Methyl O-2-azido-2-deoxy-3,4-bis-O-(phenylMethyl)-a-D-glucopyranosyl-(14) -O-2,3-bis-O-(phenylMethyl)-b-D-glucopyranuronosyl-(14)-O-2-azido- 2-deoxy-a-D-glucopyranosyl-(14)-O-3-O-(phenylMethyl)-a-L-idopyranu ronosyl-(14)-2-deoxy-2
FSC

114903-05-8

|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||
|
|||||||
| Description | |||||||
|
|||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
 |
|||||||||
|
|||||||||
| Description | |||||||||
|
|||||||||
References
- “Medscape.com”. Retrieved 2009-01-23.
- “NEJM — Comparison of Fondaparinux and Enoxaparin in Acute Coronary Syndromes”. Retrieved 2009-01-23.
- Peters RJ, Joyner C, Bassand JP, et al. (February 2008). “The role of fondaparinux as an adjunct to thrombolytic therapy in acute myocardial infarction: a subgroup analysis of the OASIS-6 trial”.Eur. Heart J. 29 (3): 324–31. doi:10.1093/eurheartj/ehm616. PMID 18245119.
- WO 2013003001
- Synthesis of heparin fragments: A methyl alpha-pentaoside with high affinity for antithrombin III
Carbohydr Res 1987, 167: 67 - A fast and effective hydrogenation process of protected pentasaccharide: A key step in the synthesis of fondaparinux sodiumOrg Process Res Dev 2013, 17: 869, http://pubs.acs.org/doi/full/10.1021/op300367c
- WO 2012047174
- US 2012116066
- WO 2013011460 RANBAXY
- WO 2013115817
- The unique antithrombin III binding domain of heparin: A lead to new synthetic antithrombotics
Angew Chem Int Ed Engl 1993, 32(12): 1671 - Bioorganic and Medicinal Chemistry Letters, 1(2), p. 95-98 (1991).
- Carbohydrate Research, 101, p. 148-151 (1982),
- Chemistry – A European Journal, 2012 , vol. 18, 34 pg. 10643 – 10652
- Carbohydrate Research, 2012 , vol. 361, p. 155 – 161
- Analytical Chemistry, 2006 , vol. 78, 6 pg. 1774 – 1779
PATENTS
| US4818816 * | Oct 26, 1987 | Apr 4, 1989 | Choay, S.A. | Process for the organic synthesis of oligosaccharides and derivatives thereof |
| US6376663 * | Nov 29, 1996 | Apr 23, 2002 | Macquarie Research Ltd. | Desalting and purification of oligosaccharides and their derivatives |
| US7541445 * | Sep 6, 2002 | Jun 2, 2009 | Alchemia Limited | Synthetic heparin pentasaccharides |
| US20040048785 * | Jun 18, 2003 | Mar 11, 2004 | Societe L’oreal S.A. | C-glycoside compounds for stimulating the synthesis of glycosaminoglycans |
| US20040149200 * | Jun 11, 2002 | Aug 5, 2004 | Tsuyoshi Shimose | Crystals of an oligosaccharides and process for preparation thereof |
| US20110105418 * | Jul 30, 2010 | May 5, 2011 | Reliable Biopharmaceutical Corporation | Process for preparing fondaparinux sodium and intermediates useful in the synthesis thereof |
| WO2011014793A2 * | Jul 30, 2010 | Feb 3, 2011 | Reliable Biopharmaceutical Corporation | Process for preparing fondaparinux sodium and intermediates useful in the synthesis thereof | |
| AU2008200616A1 | Title not available | ||||
| JPS63218691A * | Title not available | ||||
| US4818816 | Oct 26, 1987 | Apr 4, 1989 | Choay, S.A. | Process for the organic synthesis of oligosaccharides and derivatives thereof | |
| US7468358 | Oct 27, 2004 | Dec 23, 2008 | Paringenix, Inc. | Method and medicament for sulfated polysaccharide treatment of heparin-induced thrombocytopenia (HIT) syndrome | |
| US84771910 | Title not available | ||||
| USPP23055709 | Title not available |
FONDAPARINUX



The three specialties available in the United States – dalteparin (Fragmin, Pfizer), enoxaparin (Lovenox, Sanofi-Aventis) and tinzaparin (Innohep, Bristol-Myers Squibb) – the first two are found in Brazil, enoxaparin under the names Lovenox, Cutenox and Dripanina.

Lysosomal Storage Disorders: Advocacy Group Receives FDA Orphan Designations
This is the second Blog Post in a series over the next week that will examine Lysosomal Storage Disorders (LSDs) in the rare disease and orphan drug space. This Blog Post presents an advocacy group that receives two FDA Orphan Drug Designations (ODDs) in 2013 for treatment of rare diseases. The chart below identifies the gene therapy that receives FDA ODD where the sponsor is a rare disease advocacy group.
Row Num | Generic Name | Designation Date | Orphan Designation |
1 | recombinant adeno- associated virus vector AAV2/rh8 expressing human B-hexosaminidase A and B subunits | 03-25-2013 | Sandhoff Disease |
2 | recombinant adenovirus vector AAV2/rh8 expressing human B-hexosaminidase A & B subunits | 03-25-2013 | Tay-Sachs Disease |
.
** “Generic Name” Column Link = Is the FDA Orphan Drug Product Designation Database Record.
The National Tay-Sachs and Allied Diseases Association (NTSAD) announces in June 2013 that the FDA…
View original post 211 more words
FDA Breakthrough Therapy Designation: 32 And Counting
On February 3rd, GlaxoSmithKline (GSK) announces that Promacta (US)/Revolade (Europe) (Eltrombopag) receives the coveted FDA Breakthrough Therapy Designation (BTD) for cytopenias in patients with Severe Aplastic Anemia (SAA), who have had insufficient response to Immunosuppressive Therapy (IST). The drug is not approved or licensed anywhere in the world for this indication.
SAA is a rare disorder where the bone marrow fails to make enough new blood cells. There are currently no therapies approved for this indication. About forty percent (40%) of patients who do not respond to initial IST die within 5 years of diagnosis.
Regulatory Actions
• Receives FDA ODD in November 2013 for Aplastic Anemia
• Receives FDA BTD in February 2014 for Aplastic Anemia
• Receives FDA ODD in May 2008 & FDA approval in November 2008 for Idiopathic Thrombocytopenia Purpura.
This is the 32nd BTD that is announced by a sponsor company since…
View original post 150 more words
Topiroxostat 托匹司他 for gout and hyperuricemia


Topiroxostat
托匹司他
FUJI YAKUHIN ……..INNOVATOR
Approved in japan PMDA JUNE 28 2013
Xanthine oxidase inhibitor
FOR GOUT AND HYPERURICEMIA
Launched – 2013, Fuji YakuhinSanwa, Topiloric Uriadec
IUPAC Name: 4-(5-pyridin-4-yl-1H-1,2,4-triazol-3-yl)pyridine-2-carbonitrile
CAS Registry Number: 577778-58-6
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1)
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
3-(3-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole
Synonyms: 4-(5-PYRIDIN-4-YL-1H-1,2,4-TRIAZOL-3-YL)PYRIDINE-2-CARBONITRILE,
AC1NRB9T, Topiroxostat (JAN/INN), DB01685, D09786, FYX-051
SK-0910
4-[5-PYRIDIN-4-YL-1H-[1,2,4]TRIAZOL-3-YL]-PYRIDINE-2-CARBONITRILE,
C13H8N6 MF,248.2482 MW
 TOPIROXOSTAT
TOPIROXOSTAT
托匹司他
A xanthine oxidase inhibitor used to treat gout and hyperuricemia.

PATENT EXP 3/12/22, US /EU/CN

FYX-051, TOPIROXOSTAT is a xanthine oxidase inhibitor. This agent was approved in Japan by Fuji Yakuhin and Sanwa for the treatment of gout and hyperuricemia in 2013 and launched at the same year. In 2009, the compound was licensed to Sanwa by Fuji Yakuhin in Japan for the codevelopment and commercialization of gout.
The number of patients with hyperuricemia in Japan is reported to be 1.25 million and the number suffering from asymptomatic hyperuricemia is estimated to reach several millions. Hyperuricemia is becoming a popular disease.
Presently, hyperuricemia and gout due to hyperuricemia are treated by improving the living environment and administering various drug therapies for each period when an attack of gout is predicted to occur (presymptomatic period), when an attack of gout occurs, or when an attack of gout subsides. That is, preventive therapy is conducted in the presymptomatic period by administering colchicines as well as controlling the daily living environment. When an attack occurs, drug therapy using non-steroidal or steroidal anti-inflammatory agents is mainly conducted. After the attack subsides, patients are given guidance to improve their lifestyle. When improvement is judged insufficient, an assessment is made as to whether hyperuricemia is caused by reduced excretion of uric acid or by increased production of uric acid followed by treatment with drugs, which exhibit a uricosuric effect, such as probenecid and benzbromarone, those which inhibit resorption of uric acid, such as sulfinpyrazone, those which improve acidurea conditions, such as citrates, and xanthine oxidase inhibitors which inhibit production of uric acid, such as allopurinol. Colchicine is said to be able to prevent about 90% of attacks through inhibiting chemotaxis and phagocytosis of leukocytes, such as neutrophils, if administration thereof has been completed within a few hours before the attack. Since colchicine has various adverse effects, however, the use thereof is limited to the minimum and it is therefore difficult to timely administer it.
Accordingly, drug therapies are mainly adopted, but only allopurinol is available for the treatment of a disease caused by increased production of uric acid. However, a metabolite of allopurinol, oxypurinol, tends to accumulate and may cause calculi formation. Furthermore, this drug has been reported to induce adverse events such as rash, a decreased renal function and hepatitis, and it is not easy to administer.
Examples of compounds having xanthine oxidase inhibiting activity that can be used for treating gout caused by increased production of uric acid and that are effective for hyperuricemia and gout due to hyperuricemia have been described in J. Medicinal Chemistry, 1975, Vol. 18, No. 9, pp. 895–900, Japanese Patent Publication No. 49-46622 and Japanese Patent Publication No. 50-24315, which disclose some 1,3,5-substituted or 3,5-substituted 1,2,4-triazole compounds.
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1) has a xanthine oxidase inhibitory activity and serum uric acid level known as the agent that reduces (Patent Document 1).

The method for producing the compound (1), for example, 2 by Reissert Henze reaction isonicotinic acid methyl N-oxide – is a cyano isonicotinate, and the hydrazide which is then, 4 – this condensed cyanopyridine After obtaining a hydrazide of isonicotinic acid N-oxide (Patent Document 1, Example 12) and method, a cyano group after introduction, 4 by Reissert Henze reaction – method of condensing a cyano pyridine is known (Patent Document 1, Example 39).Further, 4 – as a starting material cyano-N-oxide, a triazole ring after construction (Patent Document 3), Reissert Henze unprotected or (Patent Document 2) to protect the ring condensed with isonicotinic acid hydrazide method of obtaining the compound (1) by introducing a cyano group by the reaction have also been reported.
The crystalline polymorph, yet the same molecule with the same chemical composition, the molecular arrangement in the crystal are different, and are different crystalline states. The pharmaceutical compounds having crystal polymorphism such the differences in physicochemical properties, affect pharmacological activity, solubility, bioavailability, stability and the like are known.Therefore, when the crystal polymorphism is present in a pharmaceutically useful compound, producing compounds of the crystalline form highly useful from polymorphs thereof is desirable.
WO 2003/064410 discloses WO 2005/009991 discloses Japanese Patent Publication No. 2005-41802
However, 4 of the above Patent Document – no description about the presence of crystalline polymorph on carbonitrile – pyridine-2-[yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol] It has not been, to these manufacturing methods, it is disclosed a method for the purpose of improving the chemical purity and yield, there is no description of the crystallographic plane.
Method of producing topiroxostat, useful for preventing or treating gout; and its intermediates. Picks up from WO2012060308, claiming the use of this topiroxostat for treating renal dysfunction. Along with the concurrently published WO2014017515, claiming crystalline Forms I and II of this compound, which, Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017516
Crystalline Forms I and II of topiroxostat, useful for preventing or treating gout. Along with the concurrently published WO2014017516, claiming a method of producing this compound. Picks up from WO2012060308, claiming a method of treating renal dysfunction using topiroxostat, which Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017515
novel 1,2,4-triazole compounds having an optionally substituted 2-cyanopyridin-4-yl group at 3-position and an optionally substituted aromatic group at 5-position inhibit a xanthine oxidase and are useful for treatment of gout and hyperuricemia, and have previously filed a patent application (Patent Document 1). The compounds can be prepared according to a method shown by the following reaction scheme:
-
 wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
Patent Document 1: JP-A-2002-017825 -
-
A compound represented by formula (1) which is a starting material may be prepared by a method described in, for example, JP-A-47-7120, JP-A-61-152661A, JP-A-62-149673, JP-A-2002-528447, or European Patent Application No. 559363 specification. However, it is preferable to prepare compound (1) according to the following reaction scheme:
-

-


SYNTHESIS



PATENT
- Example 2
-
To the toluene solution obtained in Example 1 (2) was added 2-propanol (700 mL), and the mixture was stirred. To the resulting solution was added p-toluenesulfonic acid monohydrate (151.16 g) and the resulting mixture was stirred for 8 hours at an internal temperature of 80°C. The mixture was brought to room temperature, and the precipitated crystals were taken out and washed with 2-propanol (210 mL×2). The white crystals were dried under reduced pressure at 60°C for 15 hours to give 106.0 g of the captioned compound as white crystals. Subsequently, 90.0 g of the crystals was suspended in a mixture of 2-butanol (49 mL) and water (491 mL) and heated to an internal temperature of 80°C for 1 hour. The internal temperature was brought to room temperature, and the crystals were filtered and washed with a mixture of 2-butanol and water (1:10) (270 mL×3). The resulting crystals were dried under reduced pressure at 60°C for 15 hours to give 75.7 g of the captioned compound in a high purity.
-
1H―NMR(DMSO-d6)δppm:2.29(s,3H), 7.11 (m,2H), 7.48 (dd, 2H, J=6.48, 1.62Hz) , 8.32-8.35(m, 3H) , 8.57(dd, 1H, J=1.62, 0.81Hz) , 8.94-8.98(m, 3H)
- Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonate
Example 3
Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
-
To the white crystals (50.5g) obtained in Example 2 was added 2-propanol (937.5 mL) and water (312.5 mL), and the resulting mixture was heated and dissolved at an internal temperature of 80°C. Immediately thereafter, the solution was filtered and the filtrate was cooled to an internal temperature of 20°C. To the resulting suspension was added dropwise 0.52 mol/l of an aqueous sodium hydrogen carbonate solution (250 mL), and the mixture was stirred at room temperature for 2 hours. Then the crystals were filtered and washed with water (150 mL×3) and 2-butanol (150 mL×2). The crystals were dried under reduced pressure at 80°C for 15 hours to give 29.4 g of the captioned compound as pale yellow crystals.
-
1H―NMR(DMSO-d6)δppm:8.02(dd, 2H, J=4.59, 1.62Hz),8.32(dd, 1H, J=5.13, 1.62Hz), 8.55(dd, 1H, J=1.62, 1.08Hz), 8.80(dd, 2H, J=4.59, 1.62Hz), 8.93 (dd, 1H, J=5.13, 1.08Hz)
SYNTHESIS
Example 12
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of methyl isonicotinate N-oxide
13.9 g of isonicotinic acid N-oxide was added to 209 ml of methylene chloride, 29.7 g of 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline was further added thereto, and the mixture was stirred under argon atmosphere at room temperature for one hour. 32.1 g of methanol was added to this mixture, which was stirred at room temperature for 17 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (3:1) was used as an eluent to yield 11.1 g of a white powder.
1H-NMR (CDCl3) δppm: 3.95 (3H, s), 7.88 (2H, d, J=7.25 Hz), 8.22 (2H, J=7.25 Hz)
2) Production of Methyl 2-cyanoisonicotinate
11.1 g of the crystal obtained in 1) was dissolved in 170 ml of acetonitrile, 14.6 g of triethylamine and 21.5 g of trimethylsilylnitrile were added thereto, and the mixture was refluxed under argon atmosphere for 16 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (95:5) was used as an eluent to yield 8.44 g of a pale yellow powder.
1H-NMR (CDCl3) δppm: 4.01 (3H, s), 8.08 (1H, d, J=5.45 Hz), 8.24 (1H, s), 8.90 (1H, d, J=5.45 Hz)
3) Production of 2-cyanoisonicotinic acid hydrazide
8.44 g of the crystal obtained in 2) was added to 85 ml of methanol, 1.84 g of hydrazine was further added thereto, and the mixture was stirred under argon temperature for 2 hours. After the solvent was evaporated under reduced pressure, chloroform was added to the residue, which was stirred at room temperature for one hour. The precipitated crystal was filtered, washed with chloroform and dried with a vacuum pump to yield 4.15 g of a pale yellow powder.
1H-NMR (DMSO-d6) δppm: 4.72 (2H, s), 8.05 (1H, d, J=5.12 Hz), 8.31 (1H, s),8.90 (1H, d, J=5.12 Hz), 10.23 (1H, s)
4) Production of the Object Compound
2.67 g of 4-cyanopyridine was dissolved in 40 ml of methanol, 0.83 g of sodium methoxide was added thereto, and the mixture was stirred at room temperature for one hour. Then 4.15 g of the crystal obtained in 3) was added and the mixture was refluxed for 37 hours. After the reaction completed, the precipitated solid was filtered, washed with methanol and dried with a vacuum pump to yield 3.66 g of the object compound as a yellow powder.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54, 1.57 Hz), 8.31 (1H, dd, J=5.11, 1.65 Hz), 8.53 (1H, dd, J=1.65, 0.50 Hz), 8.80 (2H, dd, J=4.54, 1.57 Hz), 8.93 (1H, dd, J=5.11, 0.50 Hz)
Example 39
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of isonicotinic acid (N-2-tert-butoxycarbonyl)hydrazide-1-oxide
585 ml of methylene chloride was added to 39.0 g of isonicotinic acid N-oxide, and after 34.0 g of triethylamine was further added thereto, the mixture was cooled under argon atmosphere to −15° C. 33.5 g of ethyl chlorocarbonate in 117 ml of methylene chloride was added dropwise to this mixture, which was stirred at a temperature from −5 to −10° C. for one hour. Then 44.4 g of tert-butyl ester of carbamic acid in 117 ml of methylene chloride was added dropwise to this mixture and it was allowed to slowly rise to room temperature while it was stirred. The precipitated solid was filtered after 15 hours, washed with methylene chloride, and dried with a vacuum pump to yield 49.7 g of white crystal.
1H-NMR (DMSO-d6) δppm: 1.42 (9H, s), 7.82 (2H, d, J=7.09 Hz), 8.33 (2H, d, J=7.09 Hz), 9.02 (1H, s), 10.44 (1H, s)
Production of 2-cyanoisonicotinic acid hydrazine 1½ P-Toluenesulfonic acid salt
228 ml of dioxane was added to 30.4 g of the crystal obtained in 1), and after 13.1 g of trimethylsilyl cyanide and 38.8 g of N,N-dimethylcarbamoyl chloride were further added thereto, the mixture was stirred under argon atmosphere at 60° C. for 5 hours. After the solvent was evaporated under reduced pressure, the residue was dissolved in ethyl acetate and subsequently washed with 1.5 M sodium carbonate aqueous solution and a saturated saline solution and dried over magnesium sulfate. After the magnesium sulfate was filtered off, the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue, 68.5 g of p-toluenesulfonic acid monohydrate was added thereto, and the mixture was stirred at room temperature for 22 hours. The precipitated crystal was filtered, washed with ethyl acetate, and dried with a vacuum pump to yield 40.3 g of white crystal 2).
1H-NMR (DMSO-d6) δppm: 2.28 (4.5H, s), 7.12 (3H, dd, J=7.92 & 0.66 Hz), 7.48 (3H, dd, J=7.92 & 0.66 Hz), 8.10 (1H, dd, J=5.11 & 1.81 Hz), 8.39 (1H, dd, J=1.81 & 0.33 Hz), 8.99 (1H, dd, J=5.11 & 0.33 Hz)
3) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
9.98 g of 4-cyanopyridine was dissolved in 250 ml of methanol, and after 7.77 g of sodium methoxide was added thereto, the mixture was stirred at room temperature for one hour. Then 40.3 g of the crystal obtained in 2) was added and the mixture was refluxed for 24 hours. After the reaction completed, the precipitated crystal was filtered, washed with methanol, and dried with a vacuum pump to yield 16.3 g of yellow crystal.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54 & 1.57 Hz), 8.31 (1H, dd, J=5.11 & 1.65 Hz), 8.53 (1H, dd, J=1.65 & 0.50 Hz), 8.80 (2H, dd, J=4.54 & 1.57 Hz), 8.93 (1H, dd, J=5.11 & 0.50 Hz)
4) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
45 ml of ethanol and 15 ml of 1-methyl-2-pyrrolidone were added to 3.0 g of the crystal obtained in 3), and the mixture was heated and stirred at 80° C. for 19 hours. The crystal was filtered, subsequently washed with a mixture of ethanol and 1-methyl-2-pyrrolidone (3:1) and ethanol, and dried with a vacuum pump to yield 2.71 g of yellow crystal.
5) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonic acid salt
5 ml of ethanol and 30 ml of water were added to 2.48 g of the crystal obtained in 4), and after 3.8 g of p-toluenesulfonic acid monohydrate was further added thereto, the mixture was stirred at room temperature for 5 hours. The precipitated crystal was filtered, subsequently washed with a mixture of ethanol and water (1:6), water and then ethanol, and dried with a vacuum pump to yield 3.5 g of white crystal.
1H-NMR (DMSO-d6) δppm: 2.28 (3H, s), 7.12 (2H, dd, J=7.75 & 0.50 Hz), 7.48 (2H, dd, J=7.75 & 0.50 Hz), 8.33 (1H, dd, J=5.12 & 1.65 Hz), 8.45 (2H, d, J=6.11 Hz), 8.57 (1H, dd, J=1.65 & 0.66 Hz), 8.96˜9.02 (3H, m)
6) Production of the object compound
17 ml of ethanol and 17 ml of water were added to 3.36 g of the crystal obtained in 5), and the mixture was stirred at room temperature for 30 minutes. A solution of sodium carbonate (0.74 g of sodium carbonate in 17 ml of water) was further added, and the mixture was stirred at room temperature for 2 hours. The precipitated crystal was filtered, subsequently washed with water and ethanol, and dried with a vacuum pump to yield 1.89 g of the object compound as a pale yellow crystal.
TOPIROXOSTAT
SYNTHESIS

(First step)
The first step, 4 – is a step of obtaining a compound (3) is reacted in the presence of an alkali metal alkoxide, cyano-N-oxide and (2), and isonicotinic acid hydrazide.
4 used in this reaction – isonicotinic acid hydrazide and (2) a cyano-N-oxide is a known compound both, I can be prepared by known means.
The alkali metal alkoxide is used, 6 alkoxide alkali metal C 1-C are preferred, sodium methylate, sodium ethylate and the like can be given as specific examples. The reaction is preferably carried out in a solvent, as the solvent, alcohol solvents such as methanol, ethanol and the like are preferable.
The reaction is preferably first in a solvent, is treated with an alkali metal alkoxide compound (2) and then to react the isonicotinic acid hydrazide. First, heated to reflux under cooling, at 80 ℃ from 15 ℃ preferably, 30 minutes and 12 hours in general, the reaction temperature in the reaction with an alkali metal alkoxide (2) with the compound is reacted 1-4 hours, preferably about. Under the temperature conditions, using an excess amount or one equivalent of 30 minutes to 12 hours usually, reaction with isonicotinic acid hydrazide Subsequent to reaction for 1 to 5 hours, preferably.
Example 1:
Synthesis 4 oxide (3) – – – (4 – pyridin-carbonyl) -4 – N “pyridine hydrazide imide -1 was suspended in 40mL of methanol cyanopyridine-N-oxide and (2) 5.00g, sodium was added to methylate 22.4mg, and the mixture was stirred for 2 hours under 40 ℃ nitrogen atmosphere. was cooled to room temperature. reaction solution was stirred for 4 hours at 40 ℃ was added isonicotinic acid hydrazide 5.71g at the same temperature, precipitated The filtrated crystals were, washed with methanol 15mL, and dried 15 hours at 80 ℃, N “- to give (3) 9.60g oxide – (4 – pyridin) -4 – pyridine-hydrazide imide -1.
1 H-NMR (DMSO-d 6) δ (ppm): 6.98 (br, 2H), 7.81 (d, 2H, J = 5.77Hz), 7.85 (d, 2H, J = 7 .09 Hz), 8.29 (d, 2H, J = 7.09Hz), 8.73 (d, 2H, J = 5.77Hz), 10.37 (br, 1H)
MS m / z: 256 [M-H] –
(Second step)
The second step is a step of obtaining compound (4) by cyanation agent cyano compound (3).
As the cyanation agent used, trialkyl cyanide alkali metal cyanide, sodium cyanide, potassium cyanide and the like, zinc cyanide, trimethylsilyl cyanide and the like.
The cyanation reaction is preferably, for example, be carried out (Heterocycles, Vol.22, No.5, 1994) by Reissert Henze reaction. This reaction, for example, to give compound (4) by an organic solvent in the compound (3), and after activation with carbamoyl halide, and reacting the cyano agent. The alkylcarbamoyl halide used in the carbamoylation is a first step in Reissert Henze reaction, 6 alkylcarbamoyl halide di C 1-C dimethylcarbamoyl chloride, and di-propyl carbamoyl chloride can be used, preferably, dimethylcarbamoyl is chloride. The solvent used in this reaction, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, tetrahydrofuran and acetonitrile can be used, however, N, N-dimethylformamide is preferred. Further, 15 ~ 60 ℃, more preferably 30 ~ 50 ℃ reaction temperature. The reaction time is preferably 1 to 24 hours, more preferably 1 to 3 hours. As the cyanation agent used in the cyanation reaction followed, cyano agents above can be used, sodium cyanide, potassium cyanide, zinc cyanide, and trimethylsilyl cyanide, and more preferably, it is sodium cyanide . -20 ~ 60 ℃ is preferred, more preferably -10 ~ 40 ℃, reaction temperature is 1-4 hours.
Is a novel compound (4) The compound obtained in this second step, it is useful as an intermediate for the production of compound (1). If through Compound (4) can be synthesized in good yield and easily without the need for purification in the second step is also possible, and can be produced (1) Compound industrially efficiently compound (4).
Synthetic N “hydrazide (4) – (4 – pyridine carbonyl) -4 – pyridine carboxylic acid N’-(carboxylic imidoyloxy – 2 – – cyano-4)
Example 2
4 pyridine hydrazide imide -1 – oxide ( was suspended in N, N-dimethylformamide 48mL and 3) 10.0g, under nitrogen atmosphere, followed by stirring for 1 hour was added dimethylcarbamoyl chloride 9.20g at 40 ℃. was added sodium cyanide 2.48g at the same temperature, After cooling to 5 ℃ below. reaction mixture was stirred for 1 hour, the crystals were collected by filtration. precipitate was successively added dropwise a 5% aqueous sodium bicarbonate solution 100mL, and 100mL water, and washed with water 100mL, at 80 ℃ for 15 h and dried under reduced pressure to give 4 – hydrazide (4) 9.28g of pyridine-carboxylic acid N’-(carboxylic imide yl – 2 – cyano-4).
1 H-NMR (DMSO-d 6) δ (ppm): 7.15 (br, 2H), 7.82 (d, 2H, J = 5.61Hz), 8.14 (d, 1H, J = 5 .11 Hz), 8.37 (s, 1H), 8.75 (d, 2H, J = 5.61Hz), 8.86 (d, 1H, J = 5.11Hz), 10.47 (br, 1H )
MS m / z: 265 [M-H] –

(Third step)
The third step is a step of obtaining a compound (1) by the presence of an acid catalyst, the cyclization reaction of the compound (4).
As the acid, organic phosphoric acid, p-toluenesulfonic acid, such as hydrochloric acid, inorganic acids can be used, inorganic acids, phosphoric acid is particularly preferable. As the reaction solvent, water, 2 – butanol, 2 – mixed solvent of alcohol and water or alcohol, propanol, ethanol and the like can be used, but water and 2 – I was mixed 5:1 to 10:1 butanol solvent. The reaction temperature and time, 60 ~ 100 ℃, preferably 2 to 12 hours at 70 ~ 90 ℃, I want to 8-10 hours, preferably.
Intermediates and compounds of the present invention the method (1) can be isolated and purified from the washed reaction mixture, recrystallization, by means of various conventional chromatography.
Example 3:
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile 4 Synthesis of (1) – pyridine-carboxylic acid N’- (2 – cyano-4 – carboxylic imide yl) water 82mL, 2 hydrazide (4) 9.25g – butanol was added 8.2mL, phosphate 4.00g, was stirred for 8 h at 80 ℃. After cooling to room temperature, the reaction mixture was precipitated crystals were collected by filtration, water: 2 – were washed with a mixed solution of 92.5mL butanol = 10:1. The 13 h and dried under reduced pressure at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl) – 1 H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1 I got a) 7.89g.
Topiroxostat
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
MS m / z: 247 [M-H] –
PATENT
Synthetic water-carbonitrile p-toluenesulfonate – pyridine Example 1: 4 – [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol]: 2 – butanol = was added monohydrate 6.62g p-toluenesulfonic acid in a mixed solution of 55mL of 10:1, 4 at 80 ℃ – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl] pyridine-2 – – triazol-3 was added carbonitrile 7.85g, and the mixture was stirred at the same temperature for 1 hour. After cooling to room temperature, the reaction mixture, and the precipitated crystals were collected by filtration, and water: 2 – were washed with a mixed solution of 40mL of butanol = 10:1. The dried under reduced pressure for 10 hours at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile p-toluene I got a sulfonate 12.6g.
1 H-NMR (DMSO-d 6) δ (ppm): 2.29 (s, 3H), 7.11 (m, 2H), 7.48 (dd, 2H, J = 6.48,1.62 Hz ) ,8.32-8 .35 (m, 3H), 8.57 (dd, 1H, J = 1.62,0.81 Hz) ,8.94-8 .98 (m, 3H)
– [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazole and potassium carbonate 8.22g, 4 in a mixed solution of 80mL of ethanol = 9:1: preparation water of crystal form I: Example 2 I was dissolved carbonitrile p-toluenesulfonate 10.0g – -3 – yl] pyridine-2. After stirring for 5 hours plus 15mL 6M hydrochloric acid at 20 ℃, was the precipitated crystals were collected by filtration, and washed with water 100mL. The 23 h and dried under reduced pressure at 80 ℃, 4 – to obtain carbonitrile 5.78g – pyridin-2 [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazole. Having a DSC as shown in FIG 4 and the powder X-ray diffraction pattern shown in FIG 1, the resulting crystals were type-I crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
N, N carbonitrile 40.0g – preparation of 4 Form II – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl – triazol-3]-2: Example 3 – dimethylformamide was added 300mL, and stirred for 25 min at 150 ℃. After cooling to room temperature the solution, and the precipitated crystals were collected by filtration, and washed twice with water 200mL, 4 and dried under reduced pressure overnight at 80 ℃ the crystal – [5 – (pyridin-4 – yl)-1H-1 , 2,4 – I got carbonitrile 30.4g – yl] pyridine-2 – triazole-3. Having a DSC as shown in FIG 5 and powder X-ray diffraction pattern shown in FIG 2, the resulting crystals were type II crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
The 25 ℃, about 2g carbonitrile, – preparation of the hydrate 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2: Example 4 I was stored for 14 days under conditions of relative humidity 97%. Having a DSC as shown in FIG 7 and the powder X-ray diffraction pattern shown in FIG 3, the obtained crystal was a hydrate.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
Test Example: solubility test Type I crystal by crystal form, II-type crystal, and water solubility of the hydrate was calculated by absorbance measurement method, a saturated solution concentration of each sample. I Figure 8 shows the results.Whereas the 6.2μg/mL water solubility of crystalline Form I, II type crystal 4.2μg/mL, hydrate was 1.9μg/mL.
From Figure 8, the water solubility of Form II and Form I crystals is good, water-soluble type I crystal is particularly good.
NMR
BMCL Volume 19, Issue 21, 1 November 2009, Pages 6225–6229
http://www.sciencedirect.com/science/article/pii/S0960894X09012372?np=y
view compd 39 and ignore rest
 TOPIROXOSTAT, FYX O51
TOPIROXOSTAT, FYX O51
view compd 39 and ignore rest
| 1 | * | Baldwin, J.J., J. Med. Chem.; 1975; 18(9); 895-900, especially p. 898, lines 3-5. |
| 2 | * | Geldard, J.F. et al., J. Org. Chem.; 1965; 30(1); 318-319, especially p. 319, starting line 33. |
| 3 | * | Lever, A.B.P., Inorg. Chem; 1990; 29; 1271-1285, especially p. 1275, line 18 and 19. |
Nucleosides, Nucleotides and Nucleic Acids, 2008 , vol. 27, 6-7 pg. 888 – 893
Inoue, Tsutomu; Sato, Takahiro; Ashizawa, Naoki; Iwanaga, Takashi; Matsumoto, Koji; Nagata, Osamu; Nakamura, Hiroshi
Bioorganic and Medicinal Chemistry Letters, 2009 , vol. 19, 21 pg. 6225 – 6229
WO 2012060308
WO 2007148835
WO 2005009991
| WO2003064410A1 * | Dec 3, 2002 | Aug 7, 2003 | Naoki Ashizawa | Novel 1,2,4-triazole compound |
| US3882134 * | May 21, 1973 | May 6, 1975 | Merck & Co Inc | 1-Substituted-3,5-dipyridyl-1,2,4-triazoles |
| US3947577 * | Jan 8, 1975 | Mar 30, 1976 | Merck & Co., Inc. | Anti-hyperuricemia composition |
| US3984558 * | Nov 29, 1974 | Oct 5, 1976 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds used as bronchodilators |
| US4011218 * | Dec 3, 1974 | Mar 8, 1977 | Merck & Co., Inc. | 1,2,4-triazoles |
| US4104393 * | Sep 2, 1977 | Aug 1, 1978 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds |
| US5571897 * | Dec 5, 1991 | Nov 5, 1996 | Wallac Oy | Luminescent lanthanide chelates |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
US-9199970-B2 |
2015-12-01 |
|
|||
|
2.
US-20150322006-A1 |
2015-11-12 |
|
|||
|
3.
US-20150309021-A1 |
2015-10-29 |
|
|||
|
4.
US-20150291543-A1 |
2015-10-15 |
|
|||
|
5.
EP-2927219-A1 |
2015-10-07 |
EN
|
|
||
|
6.
US-20150274680-A1 |
2015-10-01 |
|
|||
|
7.
EP-2913053-A1 |
2015-09-02 |
EN
|
|
||
|
8.
EP-2511844-B1 |
2015-08-12 |
EN
|
|
||
|
9.
EP-2712861-B1 |
2015-07-29 |
EN
|
|
||
|
10.
US-20150203490-A1 |
2015-07-23 |
|
|||
|
11.
US-20150191463-A1 |
2015-07-09 |
|
|||
|
12.
US-20150166510-A1 |
2015-06-18 |
|
|||
|
13.
EP-2878594-A1 |
2015-06-03 |
EN
|
|
||
|
14.
EP-2878598-A1 |
2015-06-03 |
E
|
|
||
|
15.
EP-2878595-A1 |
2015-06-03 |
EN
|
|
||
|
16.
US-20150126558-A1 |
2015-05-07 |
|
|||
|
17.
US-8987473-B2 |
2015-03-24 |
|
|||
|
18.
EP-2842948-A1 |
2015-03-04 |
EN
|
|
||
|
19.
EP-2776028-A1 |
2014-09-17 |
EN
|
|
||
|
20.
US-20140256748-A1 |
2014-09-11 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-[5-(4-Pyridinyl)-1H-1,2,4-triazol-3-yl]-2-pyridinecarbonitrile
|
|
| Clinical data | |
| Trade names | Topiloric, Uriadec |
| Legal status |
|
| Identifiers | |
| CAS Number | 577778-58-6 |
| ATC code | None |
| PubChem | CID: 5288320 |
| ChemSpider | 4450517 |
| Chemical data | |
| Formula | C13H8N6 |
| Molecular mass | 248.24 g/mol |
/////////////
C1=CN=CC=C1C2=NC(=NN2)C3=CC(=NC=C3)C#N
Pamicogrel KB 3022 for Coagulation Disorders Therapy


 PAMICOGREL
PAMICOGRELPamicogrel (CAS NO.: 101001-34-7), with its systematic name of 1H-Pyrrole-1-acetic acid, 2-(4,5-bis(4-methoxyphenyl)-2-thiazolyl)-, ethyl ester, could be produced through many synthetic methods.
Following is one of the synthesis routes:
alpha-Bromo-4,4-dimethoxidesoxybenzoin (I) is cyclized with pyrrole-2-carbothioamide (II) in hot acetonitrile to produce 4,5-bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole (III), which is then condensed with ethyl bromoacetate (IV) in the prsence of NaOH and tetrabutylammonium bromide in refluxing dichloromethane – water.

- Reaction Scheme-I:
-


wherein R is as defined above, and Y is a halogen such as chlorine, bromine or iodine, or p-toluenesulfonyloxy group.
-
The process of the above reaction scheme-I can be carried out by reacting a compound (II) and an equimolar or excess amount of a compound (III) in the presence of a base or a phase transfer catalyst. In case of using a base such as metallic potassium, metallic sodium, potassium tert-butoxide etc.; the reaction is carried out in a solvent of tetrahydrofuran or dimethoxyethane at a temperature of from room temperature to a boiling point of the solvent for 1 to 24 hours. In case of using a phase transfer catalyst such as a quaternary ammonium salt (e.g. tetra-n-butylammonium bromide, methyltrioctylammonium chloride, etc.), the reaction is carried out in two phases of benzene or dichloromethane and 50 % aqueous sodium hydroxide or 60 % aqeuous potassium hydroxide at a temperature of from 0°C to a boiling point of the solvent for one minute to 24 hours.


- Reaction Scheme-II:
-

wherein X is a halogen such as bromine or chlorine.
-
The above process can be carried out by reacting a compound (IV) and an equimolar amount of a compound (V) in a solvent such as acetonitrile, dimethylformamide (DMF), dimethyl sulfoxide (DMSO), or an alcohol (e.g. ethanol) at a temperature of from 50°C to a boling point of the solvent for 10 minutes to 4 hours.

- Reaction Scheme-III:
-




- Reaction Scheme-IV:
-

wherein R1 is as defined above.
-
The process can be carried out by converting a compound (VII) into an oxime (VIII) by a conventional oxime forming reaction, heating the oxime (VIII) in acetic anhydride to obtain a compound (IX), and treating the compound (IX) with hydrogen sulfide, that is, by blowing hydrogen sulfide gas into a reaction system containing the compound (IX) in a solvent such as DMF, DMSO or pyridine in the presence of 0.5 to 5 equimolar amount of a base such as a tertiary amine (e.g. triethylamine) at a temperature of from 0° to 40°C for 3 to 24 hours

- Reaction Scheme-V:
-


- Reference Example 6
- 4,5-Bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole [compound of the formula (II)]:
-
Pyrrole-2-carbothioamide (cf. J. Org. Chem., 38, 667, 1973) (1.51 g, 12 mmole) and α-bromo-4,4′-dimethoxy- deoxybenzoin (cf. Aust. J. Chem., 8, 385. 1955) (4.02 g, 12 mmole) are dissolved in acetonitrile (120 ml). The mixture is stirred at 60°C for 50 minutes. After the reaction, the reaction mixture is distilled under reduced pressure to remove the solvent. To the resulting residue are added chloroform and aqueous solution of sodium carbonate, and the mixture is shaken. The chloroform layer is taken, and the aqueous layer is further extracted with chloroform. The chloroform layers are combined, dried over anhydrous magnesium sulfate, and distilled under reduced pressure to remove the solvent. The residue is recrystallized from ligroin to give 4,5-bis(4-methoxyphenyl)-2-(pyrrol-2-yl)-thiazole (3.74 g, yield: 86 %).
-
M.p. 131.5 – 134.0°C
-
NMR (CDCl3, δ ppm): 3.7 (6H) , 6.1 (1H, dd) , 6.5-6.9 (6H), 7.1-7.5 (4H), 9.4-9.8 (lH).
-
Elementary analysis for C21H18N2O2S:


- Example 14
-
Ethyl 2-[4,5-bis(4-methoxyphenyl)thiazol-2-yl]-pyrrole-1-acetate (compound of the formula (I) wherein R1 = -CH2-COOC2H5):
- 4,5-Bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole obtained in the same manner as described in Reference Example 6 (3.62 g, 10 mmole), ethyl bromoacetate (1.67 g, 10 mmole), and tetra-n-butylammonium bromide (0.32 g, 1 mmole) are refluxed with vigorous stirring in two phases of dichloromethane (40 ml) and 50 % aqueous sodium hydroxide (40 ml) at room temperature for 2 minutes. To the mixture are added water and dichloromethane under ice-cooling, and the mixture is shaken. The dichloromethane layer is taken, and the aqueous layer is further extracted with dichloromethane. The dichloromethane layers are combined, dried over anhydrous magnesium sulfate, and distilled under reduced pressure to remove the solvent. The residue is recrystallized from ligroin to give ethyl 2-[4,5-bis(4-methoxyphenyl)thiazol-2-yl]pyrrole-1-acetate (3.64 g, yield: 81 %).
-
M.p. 132.5 – 135.5°C
-
NMR (CDCl3, δ ppm): 1.2 (3H, t), 3.8 (6H), 4.15 (2H, q), 5.25 (2H, s), 6.25 (1H, dd), 6.7-6.95 (6H), 7.2-7.55 (4H).
-
Elementary analysis for C25H24N2O4S:


Determination of the antiplatelet agent. KB-3022, and its metabolite by high-performance liquid chromatography.Nakada Y, Ikuta Y, Kawashima T, Awata N.Chem Pharm Bull (Tokyo). 1990 Apr;38(4):1093-5.
 pamicogrel
pamicogrel| EP0037274A1 * | 30 Mar 1981 | 7 Oct 1981 | Eli Lilly And Company | Substituted triaryl thiazole compounds |
| EP0077024A2 * | 7 Oct 1982 | 20 Apr 1983 | Schering Aktiengesellschaft | Imidazole derivatives, process for their preparation and pharmaceutical products containing them |
| US4168315 * | 28 Sep 1977 | 18 Sep 1979 | The Upjohn Company | Dianisyl thiazole compound, compositions and method of antithrombotic treatment |
TAKEDA PHARMACEUTICALS 武田薬品工業株式会社 ON THE RISE
Tadataka Yamada, M.D., Chief Medical & Scientific Officer of Takeda

TAKEDA US CHICAGO OFFICE
TAKEDA PIPELINE SEE LINKS BELOW
1 https://www.takeda.com/investor-information/annual/files/ar2013_10_en.pdf
2. http://www.takeda.com/research/files/pipeline_20131031_en.pdf
3 http://www.takeda.com/research/pipeline/
- 2012 Download Entire File
 PDF 0.4MB 34P
PDF 0.4MB 34P
Takeda’s top executives had frequently pointed to TAK-875 as one of their best shots at coming up with an important new approach to treating diabetes. The drug is designed to spur insulin secretion in the pancreas and Takeda had confidently projected an approval in Japan in 2015 with a follow-up approval in the big U.S. market a year or two later.
The termination of the high-profile program caused some anxiety among investors. Takeda’s shares plunged 8% on the loss as analysts wondered how the pharma company could counter the loss of Actos, a $3.7 billion drug that accounted for about a quarter of its revenue in 2011.
Takeda won an approval on a trio of DPP-4 diabetes drugs–Nesina (alogliptin) and two combos with alogliptin, dubbed Oseni and Kazano–at the beginning of the year. But Takeda suffered some big delays in gaining acceptance, a common fate in this field, where regulators are particularly cautious about new drugs. And Merck had already solidified its lead in the DPP-4 market with Januvia whileOnglyza trailed closely behind it. Takeda had hoped that a combination of TAK-875 and Januvia could help regain some lost market territory–but that dream has clearly vanished as well.
| January 27, 2014 | |
| January 22, 2014 | |
| January 17, 2014 | |
| January 14, 2014 | |
| January 10, 2014 |
2013
| December 27, 2013 | ||
| December 25, 2013 | ||
| December 25, 2013 | ||
| December 24, 2013 | ||
| December 20, 2013 | ||
| December 20, 2013 | ||
| December 19, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 9, 2013 | ||
| December 5, 2013 | ||
| December 4, 2013 | ||
| November 30, 2013 | ||
| November 21, 2013 | ||
| November 19, 2013 | ||
| November 14, 2013 | ||
| November 12, 2013 | ||
| November 12, 2013 | ||
| October 21, 2013 | ||
| October 7, 2013 | ||
| October 2, 2013 | ||
| October 1, 2013 |
| September 26, 2013 | ||
| September 26, 2013 | ||
| September 24, 2013 | ||
| September 20, 2013 | ||
| September 20, 2013 | ||
| September 13, 2013 | ||
| September 13, 2013 | ||
| September 5, 2013 | ||
| September 2, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 22, 2013 | ||
| August 13, 2013 | ||
| August 1, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 30, 2013 | ||
| July 29, 2013 | ||
| July 26, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 18, 2013 | ||
| July 10, 2013 | ||
| July 1, 2013 |
CLIPPED
Takeda isn’t quite in the top 10 among global drugmakers, but the company boasts the 7th-largest pipeline in the industry, according to its presentation at the conference. Yamada noted that 31% of the pipeline assets are in late-stage trials. Millennium is leading development of three late-stage contenders, TAK-700 for prostate cancer, MLN9708 for multiple myeloma and MLN0002 for ulcerative colitis andCrohn’s disease.
In an effort to revive its diabetes franchise, Takeda is in the final stage of development for a first-of-a-kind GPR40 agonist called TAK-875, designed to provide glucose-dependent insulin secretion.
With a rich late-stage pipeline at Takeda, Yamada wants the company to focus on growing its ranks of earlier-stage drug candidates. To do this the company has landed a variety of deals, including the purchase of Intellikine for $310 million to acquire anti-cancer drugs and more recently the acquisition of Envoy Therapeutics last year for $140 million.
Takeda has formed a New Frontier Science group to scout out the hottest research in academia and elsewhere and form collaborations with scientists behind those innovations. At the J.P. Morgan conference, Yamada said, he was attending many meetings with members of the biotech community.

Takeda Pharmaceutical Company Limited (武田薬品工業株式会社 Takeda Yakuhin Kōgyō Kabushiki-gaisha?) is the largest pharmaceutical company in Japan and Asia and a top 15 pharmaceutical company. The company has over 30,000 employees worldwide and achieved $16.2 billion USD in revenue during the 2012 fiscal year.[1] The company is focused on metabolic disorders, gastroenterology, neurology, inflammation, as well asoncology through its independent subsidiary, Millennium: The Takeda Oncology Company.[2] Its headquarters is located in Chuo-ku, Osaka, and it has an office in Nihonbashi, Chuo, Tokyo.[3][4] In January 2012, Fortune Magazine ranked the Takeda Oncology Company as one the 100 best companies to work for in the United States.
Takeda Pharmaceuticals was founded on June 12, 1781 and was incorporated on January 29, 1925.
Takeda’s Japanese logo
In 1977, Takeda first entered the U.S. pharmaceutical market by developing a joint venture with Abbott Laboratories called TAP Pharmaceuticals.[5]Through TAP Pharmaceuticals, Takeda and Abbott launched the blockbusters Lupron (leuprolide) in 1985 and Prevacid (lansoprazole) in 1995.
One of the firm’s mainstay drugs is Actos, a compound in the thiazolidinedione class of drugs used in the treatment of type 2 diabetes. Launched in 1999, Actos has become the best-selling diabetes drug in the world with $4 billion USD in sales during the 2008 fiscal year.[6]
In February 2005, Takeda announced its acquisition of San Diego, California-based Syrrx, a company specializing in high-throughput X-ray crystallography, for $270 million.[7]
In February 2008, Takeda acquired the Japanese operations of Amgen and rights to a dozen of the California biotechnology company’s pipeline candidates for the Japanese market.[8]
In March 2008, Takeda and Abbott Laboratories announced plans to conclude their 30-year old joint venture, TAP Pharmaceuticals, that had over $3 billion in sales in its final year. The split resulted in Abbott acquiring U.S. rights to Lupron and the drug’s support staff. On the other hand, Takeda received rights to Prevacid and TAP’s pipeline candidates. The move also increased Takeda’s headcount by 3,000 employees.[9]
In April 2008, Takeda announced that it was acquiring Millennium Pharmaceuticals of Cambridge, Massachusetts, a company specializing in cancerdrug research, for $8.8 billion. The acquisition brought in Velcade, a drug indicated for hematological malignancies, as well as a portfolio of pipeline candidates in the oncology, inflammation, and cardiovascular therapeutic areas. Millennium now operates as an independent subsidiary, serving as the global center of excellence in oncology under its new name: “Millennium: The Takeda Oncology Company.” [10]
In May 2008, the company licensed non-exclusively the RNAi technology platform developed by Alnylam Pharmaceuticals, creating a potentially long-term partnership between the companies.[11]
On May 19, 2011, Takeda Pharmaceutical and Nycomed announced that Takeda will acquire Nycomed for € 9.6 billion. The acquisition was completed by September 30, 2011.[12]
On April 11, 2012, Takeda Pharmaceutical and URL Pharma announced that Takeda will acquire URL Pharma for $800 million. The acquisition is expected to be completed within 60 days.
On 25 May 2012, Takeda announced the purchase of Brazilian pharmaceutical company Multilab by R$ 540 million.[13]

Takeda Midosuji Building, headquarters of Takeda Pharmaceutical Company, inChuo-ku, Osaka, Japan
Takeda operates two primary bases in Japan in Osaka and Tokyo. Its United States subsidiary is based in Deerfield, Illinois, and all Global Operations outside of Japan and U.S. are based in Opfikon (Zurich), Switzerland. The company maintains research & development sites in Osaka and Tsukuba, Japan; San Diego andSan Francisco, United States; Cambridge, United Kingdom; and Singapore.[14]
The company has manufacturing facilities in Japan, China, Indonesia, Italy, and Ireland.[15] Following the Nycomed acquisition, the Takeda manufacturing sites have been extended with facilities in Argentina,Austria,Belgium,Brazil,Denmark, Estonia,Germany,Mexico,Norway and Poland. Takeda has overseas marketing presences in the U.S., UK, France, Italy, Germany, Austria, Switzerland, Spain, China, Taiwan, Philippines, Thailand, Indonesia, and Singapore. It has recently[when?] announced its first foray into Canada, Portugal, Spain, Mexico, and Ireland.[15]

AT INDONESIA
Products
Some of the key products that Takeda produces on behalf of partners include:[16]
- Actos (pioglitazone) – Type 2 Diabetes
- Amitiza (lubiprostone) – Chronic idiopathic constipation
- Basen (voglibose) – Type 2 Diabetes
- Benet (risedronic acid) – Osteoporosis (Japan)
- Blopress (candesartan) – Hypertension
- Enbrel (etanercept) – Inflammatory diseases (Japan)
- Dexilant (dexlansoprazole) – Gastroesophageal reflux disease – name changed to Dexilant in U.S.
- Lupron/Leuplin (leuprorelin) – GnRH agonist for prostate cancer and endometriosis
- Prevacid/Takepron (lansoprazole) – Gastroesophageal reflux disease
- Rozerem (ramelteon) – Insomnia
- Uloric (febuxostat) – Gout
- Velcade (bortezomib) – Multiple myeloma and mantle cell lymphoma (Millennium Pharmaceuticals)
![]() AT UK
AT UK
References
- “Financial Results for Fiscal 2012” (PDF). Takeda Pharmaceutical Company Limited. May 9, 2013. Retrieved June 13, 2013.
- “Takeda Initiates Cardiovascular Outcomes Trial for Alogliptin, An Investigational Treatment for Type 2 Diabetes”. Newsblaze.com. 2009-08-28. Retrieved 2010-09-18.
- “FAQ.” Takeda Pharmaceutical Company. Retrieved on February 2, 2011. “Q : Where is Takeda located? A : The Head Office is located in Osaka, Japan, and the Tokyo Head Office is located in Tokyo, Japan.”
- “Overview.” Takeda Pharmaceutical Company. Retrieved on February 2, 2011. “Headquarters Head Office 1-1, Doshomachi 4-chome, Chuo-ku, Osaka 540-8645” and “Tokyo Head Office 12-10, Nihonbashi 2-chome, Chuo-ku, Tokyo 103-8668”
- “TAP Pharmaceutical Products, Inc.: Private Company Information – BusinessWeek”. Investing.businessweek.com. 2008-04-30. Retrieved 2010-09-18.
- Decker, Susan (2009-07-06). “Takeda Sues Torrent to Stop Generic Copy of Actos Diabetes Pill”. Bloomberg. Retrieved 2010-09-18.
- Somers, Terri (2005-02-08). “Japanese drug giant taking over Syrrx here | The San Diego Union-Tribune”. Signonsandiego.com. Retrieved 2010-09-18.
- “Takeda, Amgen in exclusive tie-up for Japanese market”. MarketWatch. 2008-02-04. Retrieved 2010-09-18.
- Marrazzo, Amanda (2008-05-15). “Featured Articles From The Chicago Tribune”. Archives.chicagotribune.com. Retrieved 2010-09-18.
- “MILLENNIUM: The Takeda Oncology Company | About Millennium | Our History”. Mlnm.com. Retrieved 2010-09-18.
- staff (2008-06-15). “Takeda Signs On as Alnylam’s Asian Partner for $150M Upfront”. Genetic Engineering & Biotechnology News (print) (Mary Ann Liebert, Inc.). p. 14.
- http://www.takeda.com/press/article_43116.html
- Hirschler, Ben (May 25, 2012). “Farmacêutica Takeda comprará Multilab por até R$ 540 mi”. Grupo Abril (in portuguese). Exame. Retrieved January 27, 2013.
- “Locations | Worldwide | Takeda Pharmaceutical Company Limited”. Takedaism.com. Retrieved 2010-09-18.
- “By Business | Worldwide | Takeda Pharmaceutical Company Limited”. Takedaism.com. Retrieved 2010-09-18.
- “Annual Reports | Investor Information | Takeda Pharmaceutical Company Limited”. Takeda.com. Retrieved 2010-09-18.
 |
|
| Native name | 武田薬品工業株式会社 |
|---|---|
| Type | Public KK |
| Traded as | |
| Industry | Pharmaceuticals |
| Founded | Doshomachi, Osaka, Japan (June 12, 1781) |
| Headquarters | 1-1, Doshomachi Yonchome,Chuo-ku, Osaka, Japan |
| Key people | Yasuchika Hasegawa (President & CEO) |
| Revenue | |
| Operating income | |
| Net income | |
| Total assets | |
| Total equity | |
| Employees | 30,481 (2012) |
| Website | takeda.com (Global website) |
References:
|
|

CMC CENTRE
The Chemistry, Manufacturing and Controls (CMC) Center is a global organization responsible for overall R&D activities ranging from chemical information on development candidates to the processes leading to “manufacturing” of pharmaceutical products.
The main sites are located in Osaka and consist of the following laboratories: the Chemical Development Laboratories in charge of R&D for developing the manufacturing methods of active pharmaceutical ingredients and the manufacturing of drug substances for clinical samples; the Pharmaceutical Technology R&D Laboratories in charge of R&D for dosage forms, manufacturing and packaging, as well as manufacturing of clinical samples; and the Analytical Development Laboratories in charge of R&D for the development of analytical methods and stability studies of clinical samples. In addition, Hikari Bio-Manufacturing Technology Laboratories is located in Hikari (Yamaguchi) and this is where antibody drug substances are manufactured.
As for overseas sites, the Cambridge Biologics CMC Group (Massachusetts) and the Chicago Pharmaceutical Science Group (Illinois) are located in the USA, while the CMC Center Europe is mainly located in Roskilde, Denmark. All research and development activities at Takeda are promoted with the cooperation of these sites.
List of Publications of Takeda Research Laboratories
Trelagliptin succinate (SYR-472) for the treatment of type 2 diabetes.

Trelagliptin succinate (SYR-472)
2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2, 4-dioxopyrimidin-1-yl]methyl]-4-fluorobenzonitrile; butanedioic acid
2-[6-[3(R)-Aminopiperidin-1-yl]-3-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-ylmethyl]-4-fluorobenzonitrile
2- [ [6- [ (3R) -3-amino-l-piperidinyl] -3, 4-dihydro-3- methyl-2, 4-dioxo-l (2H) -pyrimidinyl]methyl] -4-fluorobenzonitrile
succinic acid salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Sponsor/Developer: Takeda Pharmaceuticals and Furiex Pharmaceuticals
Mechanism of action: DPP-4 inhibitor
865759-25-7 cas FREE BASE
1029877-94-8 succinate
- SYR 111472 succinate
- SYR 472
- Syr-472
- Syr111472 succinate
- Trelagliptin succinate
- UNII-4118932Z90
- clinical trials….http://clinicaltrials.gov/search/intervention=SYR+472
Trelagliptin-succinate M. Wt: 475.47
Trelagliptin-succinate Formula: C22H26FN5O6
SYR-472 is an oral dipeptidyl peptidase IV inhibitor originated by Takeda. It is in phase III clinical trials for the treatment of type 2 diabetes.
- Diabetes affects 25.8 million people of all ages, or roughly 8.3 percent of the U.S. population.
- The World Health Organization predicts that there will be 366 million people worldwide affected by diabetes by the year 2030.
- The advent of trelagliptin succinate, a unique once weekly medication for patients with type 2 Diabetes is now the focus of clinical trials and exciting research and development.
- Phase III clinical trials of trelagliptin succinate commenced in September 2011, and are estimated to be complete by the second half of 2013.

Indication (Phase): Japan—Once-weekly oral treatment for type 2 diabetes (Phase III; study expected to be completed in second half of 2013)

trelagliptin succinate
Compound I, A, TRELAGLIPTIN which has the formula:

is a DPP-IV inhibitor that is described in U.S. patent application Ser. No. 11/080,992 filed Mar. 15, 2005 (see Compound 34). Its dosing, administration and biological activities are described in U.S. patent application Ser. No. 11/531,671 filed Sep. 13, 2006. U.S. patent application Ser. No. 11/080,992 and Ser. No. 11/531,671 are incorporated herein by reference in their entirety.
Dipeptidyl peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) (referred herein as “DPP-IV”) is a type II membrane protein and a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (e.g., intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of human disease states, including, but are not limit to, diabetes, particularly type II diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (IFG), metabolic acidosis, ketosis, appetite regulation and obesity; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; AIDS; and cancers.
DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV.
Compound (A) or a salt thereof has been reported as an inhibitor of dipeptidyl peptidase (DPP-IV) , which is an enzyme that decomposes glucagon-like peptide-1 (GLP-1) , a hormone increasing insulin secretion (patent document 1) .
In addition, a method including administering 1 – 250 mg of compound (A) or a salt thereof to a patient once per week (patent documents 2, 3), crystal polymorphs of compound (A) (patent documents 4, 5) , and a preparation of compound (A)
(patent documents 6, 7) have also been reported. Compound (A) and a salt thereof are recommended for oral administration in view of the easiness of self-administration, and a tablet, particularly a tablet in the dosage form for administration once per week, is desired. [0006]
The dosage form of once per week is expected to improve drug compliance of patients, whereas it requires supply of compound (A) or a salt thereof to patients in a high dose as compared to, for example, the dosage form of once per day. Since a solid preparation containing compound (A) or a salt thereof in a high dose increases its size, it may conversely degrade the drug compliance for patients, particularly infants and elderly patients having difficulty in swallowing
……………………..
SYNTHESIS
Compound 34 IS TRELAGLIPTIN

4-Fluoro-2-methylbenzonitrile (31).
A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32).
A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
Alternatively, 32 was made as follows.
4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AIBN (122 g) and heated to 75° C. A suspension of DBH (353 g) in DCE (500 mL) was added at 75° C. portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AIBN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10° C. over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20° C. over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (33).
A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows.
To a solution of 6-chloro-3-methyluracil (750 g) and N,N-diisopropylethylamine (998 mL) in NMP (3 L) was added (at <30° C. over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 60° C. for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5° C. for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2×2.25 L). The material was then dried in a vacuum oven at 40±5° C. for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
TFAsalt OF TRELAGLIPTIN
2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (34).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
FREE BASE NOF TRELAGLIPTIN
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 60° C. until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 60° C. and the mixture was allowed to cool to <25° C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2×3.6 L). The filtrate was concentrated at 45° C. under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
HCL salt OF TRELAGLIPTIN
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0° C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35° C. over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 50° C. (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
Succinate salt OF TRELAGLIPTIN

The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purified water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 30° C. over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 60° C. until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57° C. After stirring at 60° C. for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5° C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2×4.2 L). The material was then dried in a vacuum oven at 70±5° C. (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(−)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
Methanesulfonate salt
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75° C. and filtered through #3 Whatman filter paper. The solution was heated back to 75° C. and a 1M solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20° C./hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
…………………………
FORMULATION
COMPD A IS TRELAGLIPTIN
Examples (Comparative Example IA)
Succinate of compound (A) (26.6 mg) was weighed in a glass bottle and used as Comparative Example IA. (Comparative Example 2A)
The succinate of compound (A) and microcrystalline cellulose were uniformly mixed in a mortar at a ratio of 1:10, and the mixture (226.6 mg) was weighed in a glass bottle and used as Comparative Example 2A. (Comparative Example 3A)
The succinate of compound (A) and corn starch were uniformly mixed in a mortar at a ratio of 1:5, and the mixture (126.6 mg) was weighed in a glass bottle and used as Comparative Example 3A. (Example IA) Succinate of compound (A) , mannitol and corn starch according to the formulation of Table IA were uniformly mixed in a fluid bed granulator (LAB-I, POWREX CORPORATION) , and the mixture was granulated by spraying an aqueous solution of dissolved hypromellose 2910, and dried therein. The obtained granules were passed through a sieve -(16M) to give milled granules. To the milled granules were added croscarmellose sodium, microcrystalline cellulose and magnesium stearate, and they were mixed in a bag to give granules for tableting. The granules were punched by a rotary tableting machine (Correct 19K, Kikusui Seisakusho, Ltd.) with a 6.5 mmφ punch to give a plain tablet weighting 121 mg. On the other hand, titanium oxide, yellow ferric oxide and talc were dispersed in a hypromellose 2910 aqueous solution to prepare a film coating liquid. The aforementioned coating liquid was sprayed onto the above-mentioned plain tablet in a film coating machine (Hicoater HCP-75, Freund Corporation), to give 2500 film- coated tablets containing 3.125 mg of compound (A) (free form) per tablet. Table IA

………………………..
POLYMORPHS AND SYNTHESIS
FORM A
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared. For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days. The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE).
[0091] Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR.
[0092] Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1.
Table 1. Characteristic XRPD Peaks (CuKa) of Form A


Characterization Data of Form A of Compound I

8. Amorphous Form
[0137] The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis.
Table A. Approximate Solubilities of Compound I



Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool;CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation
…………………………
SYNTHESIS
EXAMPLES
1. Preparation of 2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro- 2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile and pharmaceutically acceptable salts


4-Fluoro-2-methylbenzonitrile (3)
[0166] A mixture of 2-bromo-5fluorotoluene ( 2) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was re fluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 3 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H). 2-Bromomethyl-4-fluorobenzonitrile (4)
[0167] A mixture of 4-fluoro-2-methylbenzonitrile (3) (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification.1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro- benzonitrile (6)
[0168] A mixture of crude 3-methyl-6-chlorouracil (5) (0.6 g, 3.8 mmol), 2- Bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO
(10 mL) was stirred at 60 C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=12, 8.4Hz, IH), 7.26 (d, J- 4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for Ci3H9ClFN3O2, 293.68; found 293.68.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, TFA salt (1) (TFA salt of Compound I)

[0169] 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4- fluoro-benzonitrile (5) (300 mg, 1.0 mmol), (i?)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 0C for 2 hrs. The final compound was obtained as a TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, HCl salt

[0170] The TFA salt of Compound I was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0 C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
General procedure for the preparation of salts of Compound I.
[0171] The benzonitrile product may be isolated as the free base if desired, but preferably, the product may be further converted to a corresponding acid addition salt. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of Compound I were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. [0172] The isolation and/or purification steps of the intermediate compounds in the above described process may optionally be avoided if the intermediates from the reaction mixture are obtained as relatively pure compounds and the by-products or impurities of the reaction mixture do not interfere with the subsequent reaction steps. Where feasible, one or more isolation steps may be eliminated to provide shorter processing times, and the elimination of further processing may also afford higher overall reaction yields.
…………………..
TABLET
2. Exemplary formulations comprising succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Provided are examples of tablet formulations that may be used to administer succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (Succinate salt of Compound I) according to the present invention. It is noted that the formulations provided herein may be varied as is known in the art.
The exemplary tablet formulations are as follows:
| 12.5 mg of Compound I (weight of free base form) per tablet | ||||
| Core Tablet Formulation | ||||
| (1) | 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4- | 17.0 | mg | |
| dioxo-3,4-dihydro-2H-pyrimidin-1- | ||||
| ylmethyl]-4-fluoro-benzonitrile (succinate salt) | ||||
| (2) | Lactose Monohydrate, NF, Ph, Eur | 224.6 | mg | |
| (FOREMOST 316 FAST FLO) | ||||
| (3) | Microcrystalline Cellulose, NF, Ph, Eur | 120.1 | mg | |
| (AVICEL PH 102) | ||||
| (4) | Croscarmellose Sodium, NF, Ph, Eur | 32.0 | mg | |
| (AC-DO-SOL) | ||||
| (5) | Colloidal Silicon Dioxide, NF, Ph, Eur | 3.2 | mg | |
| (CAB-O-SIL M-5P) | ||||
| (6) | Magnesium Stearate, NF, Ph, Eur | 3.2 | mg | |
| (MALLINCKRODT, Non-bovine Hyqual) | ||||
| TOTAL | 400.0 | mg | ||
| (per tablet) | ||||
…………..
US20080227798 AND US20120197018
POLYMORPHS AND SYNTHESIS
EXAMPLES Example 1 Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)

Compound I may be prepared by the follow synthetic route (Scheme 1)

A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)

Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)

Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)

Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)

Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile

The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent.
SUCCINATE SALT OF TRELAGLIPTIN
1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.
……………
patents
1. US 2013172377
2. WO 2011013639
3. WO 2009099172
4.WO 2009099171
5. WO 2008114807
6.WO 2008114800
7. WO 2008033851
8. WO 2007074884
9WO 2007035629
patent document 1: US2005/0261271
patent document 2: US2007/0060530
patent document 3: US2008/0287476
patent document 4: US2008/0227798
patent document 5: US2008/0280931
patent document 6: WO2008/114800
patent document 7: WO2011/013639
| US7906523 * | Oct 30, 2007 | Mar 15, 2011 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605 * | Nov 29, 2007 | Dec 27, 2011 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8188275 * | Oct 30, 2007 | May 29, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8222411 * | Sep 15, 2006 | Jul 17, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US20090275750 * | Sep 15, 2006 | Nov 5, 2009 | Jun Feng | Dipeptidyl peptidase inhibitors |
| WO2013183784A1 | Jun 4, 2013 | Dec 12, 2013 | Takeda Pharmaceutical Company Limited | Solid preparation |
| US20080227798 * | Nov 29, 2007 | Sep 18, 2008 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US20120197018 * | Feb 15, 2012 | Aug 2, 2012 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2007033265A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033266A2 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033350A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetes |
| EP1586571A1 * | Dec 21, 2004 | Oct 19, 2005 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
13 NMR TRELAGLIPTIN SUCCINATE

1H NMR TRELAGLIPTIN SUCCINATE

