
Home » PATENTS (Page 2)
Category Archives: PATENTS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ALCAFTADINE, WO 2017211246, NEW PATENT, SHENZHEN TARGETRX, INC.

Alcaftadine
NEW PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017211246&redirectedID=true
WO-2017211246, SHENZHEN TARGETRX, INC.
SUBSTITUTED FUSED IMIDAZOLE CYCLIC COMPOUND AND PHARMACEUTICAL COMPOSITION THEREOF
WANG, Yihan; (CN).
XING, Qingfeng; (CN)
Novel deuterated analogs of substituted fused imidazole cyclic compounds, particularly alcaftadine are histamine H1-receptor antagonists and mast cell stabilizers, useful for treating allergy and nasal congestion.
The present invention relates to a substituted fused imidazole cyclic compound and a composition containing said compound and application thereof. Specifically disclosed is the fused imidazole cyclic compound represented by formula (I), or a pharmaceutical composition of its crystalline form, pharmaceutically acceptable salt, prodrug, stereoisomer, hydrate, or solvate. The compound of the present invention may be used as a histamine H1-receptor antagonist and mast-cell stabilizer, and is capable of inhibiting mast-cell release of histamine and preventing histamine function, thereby reducing allergic reaction
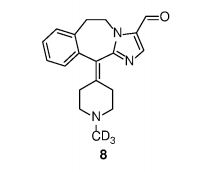

Example 1 Preparation of 6,11-dihydro -11- (1- (d3- methyl) piperidin-4-ylidene) -5H- imidazo [2,1-b] [3] benzazepine – 3- aldehyde (compound 8)
N-benzyloxycarbonylpiperidine-4-carboxylic acid (2.63 g, 10 mmol) was dissolved in 20 mL of dichloromethane, 6 mL of oxalyl chloride and 1 drop of DMF were added and the mixture was reacted at room temperature for 2 hours under nitrogen. The reaction mixture was concentrated to dryness under reduced pressure, dissolved in 20 mL of acetonitrile, and added with triethylamine (4.1 mL, 30 mmol) in an ice bath and stirred for 3 minutes. A solution of 1-phenethyl-1H-imidazole (2.06 g, 12 mmol) in 5 mL of acetonitrile was slowly added dropwise and the reaction was allowed to warm to room temperature overnight after the addition was completed. The reaction was completed, concentrated to dryness, 30 mL of ethyl acetate and 20 mL of water were added and the mixture was stirred for 5 minutes. The layers were separated and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate and concentrated to give 3.34 g of a colorless oil, benzyl-4- (1-phenethyl-1H-imidazole-2-formyl) piperidine-1-carboxylate (Compound 3) was obtained in a yield of 80%. ESI-MS: 418 [M ++ 1].
//////////////
IMIGLIPTIN, NEW PATENT, WO 2017211293, XUANZHU PHARMA CO., LTD.
(WO2017211293) CRYSTALLINE FORM OF SUCCINATE USED AS DIPEPTIDYL PEPTIDASE-4 INHIBITOR
WO-2017211293,
XUANZHU PHARMA CO., LTD. [CN/CN]; 2518, Tianchen Street, National High-Tech Development Zone Jinan, Shandong 250101 (CN)
SHU, Chutian; (CN)
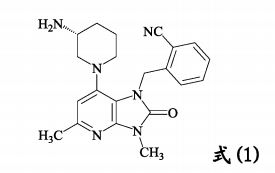
| The present invention relates to a crystalline form of a succinate used as a dipeptidyl peptidase-4 inhibitor, and a manufacturing method, pharmaceutical composition, and application thereof. The invention specifically relates to a dipeptidyl peptidase-4 inhibitor compound as represented by formula (1), a crystalline form of a succinate, wherein the succinate is an (R)-2-((7-(3-aminopiperidin-1-yl)-3,5-dimethyl-2-oxo-2,3-dihydro-1H-imidazo(4,5-b)pyridin-1-yl)methyl)benzonitrile, and a manufacturing method, pharmaceutical composition, and application thereof.
Example 1: Preparation of the succinate salt form I of the compound of formula (1) [0056]
[0057] The compound of formula (1) (44.6 g, 0.12 mol) was added to a 2 L round bottom flask and suspended in 1593 mL of acetonitrile. The mixture was heated to 80 ° C. and dissolved in free form. Immediately after the addition of 15.4 g A white solid precipitated, maintained at 80 ℃ for 1 hour and then cooled to room temperature, filtered and the filter cake was dried in vacuo at 40 ℃ for 10 hours, weighed 57.6g, yield 98.3%. The succinate salt Form I was tested by XRPD. [0058] Example 2: Preparation of the succinate salt form I of the compound of formula (1) II [0059] A quantity of succinate salt of the compound of formula (1) was weighed into glass vials in a total of 26 parts. A total of 26 vials of methanol, ethanol, isopropanol, isobutanol, 2-butanone, tetrahydrofuran, acetonitrile, methyl tert-butyl ether, acetone, water, toluene, Isopropyl acetate, n-propanol, isoamyl alcohol, butyl acetate, ethyl formate, 1,4-dioxane, n-butanol, pentane, heptane, cyclohexane, Ketone, xylene, isobutyl acetate, diethyl ether). After stirring, ultrasound and other means to make the sample fully dissolved. Subsequently, about 2 mL of liquid was removed from each bottle and filtered into 26 reagent tubes numbered 1-26. The resulting 26 filtrates were distributed in two 96-well plates. One or two of the above 1-13 solvents are sequentially added into the first 96-well plate, one or two of the above-mentioned 14-26 kinds of solvents are sequentially added into the second 96-well plate, Zha Kong sealing film sealed, placed in a fume hood, the natural environment to dry. Wherein Form I is obtained in the following mixed solvent, and Form I is also precipitated in the methyl isobutyl ketone in the remaining solution after plating. [0060] The solvent used to prepare succinate salt Form I was prepared [0061] [Table 0001]
[0062] Example 3: Preparation of the succinate salt form II of the compound of formula (1) [0063] Take 8 parts of the compound of formula (1), 200mg each, placed in a 10mL round bottom flask, add the solvent in the following table to each solvent, warmed until the solvent is refluxed, after dissolving it, add 69mg (1.1eq) succinic acid and cool to At room temperature, the solid precipitated and was filtered. The resulting solid was subjected to XRPD testing as succinate crystal form II. [0064] [Table 0002]
|
Enclomiphene citrate, New patent, WO 2017182097, F.I.S. – FABBRICA ITALIANA SINTETICI S.P.A

Enclomiphene citrate, New patent, WO 2017182097, F.I.S. – FABBRICA ITALIANA SINTETICI S.P.A
WO-2017182097
F.I.S. – FABBRICA ITALIANA SINTETICI S.P.A
CARUANA, Lorenzo; (IT).
PADOVAN, Pierluigi; (IT).
DAL SANTO, Claudio; (IT)
![]()
Enclomiphene citrate is an active pharmaceutical ingredient currently under evaluation in clinical phase III for the treatment of secondary hypergonadism. Moreover, it also could be potentially used for an adjuvant therapy in hypogonadal men with Type 2 diabetes.
Enclomiphene citrate of formula (I):

has chemical name of Ethanamine, 2-[4-[(1 )-2-chloro-1 ,2-diphenyl ethenyl]phenoxy]-/V,/V-diethyl-, 2-hydroxy-1 ,2,3-propanetricarboxylate (1 : 1 ); has CAS RN. 7599-79-3, and it is also named trans-Clomiphene monocitrate, E-Clomiphene citrate or Enclomiphene monocitrate.
Enclomiphene is component of Clomiphene, an active pharmaceutical ingredient, having chemical name Ethanamine, 2-[4-(2-chloro-1 ,2- diphenylethenyl)phenoxy]-N,N-diethyl, since Clomiphene is a mixture of the geometric isomers trans-Clomiphene (i.e. Enclomiphene) and cis- Clomiphene.
The US patent 3,848,030, in examples 31 and 32, discloses a process for the resolution of the geometric isomers of Clomiphene through the preparation of salts with racemic binaphthyl-phosphoric acid.
In the later publication Acta Cryst. (1976), B32, pag. 291 -293, the actual geometric isomery has been definitely established by single crystal X-Ray diffraction.
Finally, in the publication “Analytical profiles of drug substances and excipients”, vol. 25, (1998), pag. 85-121 , in particular at pag. 99, it is stated that prior to 1976 the cis stereochemistry was wrongly assigned to the trans-isomer of Clomiphene (E-Chlomiphene or Enclomiphene), and only after the above publication on Acta Cryst. the correct geometric isomery has been definitively assigned.
These observations in the prior art have been confirmed by our experimentation. In particular, repeating the experiment 31 of US patent 3,848,030, the trans-Clomiphene salt with racemic binaphthyl-phosphoric acid was isolated and not the salt with cis-Clomiphene as stated in said patent, as confirmed by 2D H-NMR analysis (NOESY experiment). Thus, Example 31 of US3,848,030, provides, at the end, Enclomiphene citrate, crystallized from a mixture of ethyl ether and ethanol, having a m.p. of 133-135°C. Example 32, instead provided Cis-Clomiphene citrate, crystallized from a mixture of ethyl ether and ethanol, having a m.p. of 120-126°C.
Thus, with the aim of preparing Enclomiphene citrate, whole experiment 31 of US3,848,030 has been reworked also carrying out the crystallization of the product form a mixture of ethyl ether and ethanol, hence providing a not crystalline solid with two DSC peaks respectively at 1 14°C and 188°C, although the starting material used for the reworking example was quite a pure substance (HPLC Analysis (A A%) is 98.95% of Enclomiphene), and having a substantially the same chemical purity of that used in the prior art experiment (m.p. of our Enclomiphene BPA salt was 218°C versus 220- 222°C of the prior art Enclomiphene BPA salt of Example 31 ).
The patent US2,914,563, in example 3, discloses a process for the preparation of trans-Clomiphene citrate, containing from 30% to 50% of cis-Clomiphene, as citrate, by reaction of 1 -ρ-(β- diethylaminoethoxy)phenyl]-1 ,2-diphenylethylene hydrochloride with N- chlorosuccinimmide in dry chloroform under reflux.
Khimiko-Farmatsevticheskii Zhurnal (1984), 18(1 1 ), 1318-24 English translation in the review Pharmaceutical Chemistry Journal November 1984, Volume 18, Issue 1 1 , pag. 758-764 (Title: Synthesis and biological study of the cis- and trans-isomers of Clomiphene citrate and some intermediates of its synthesis) discloses the trans-isomer of Clomiphene citrate, i.e. Enclomiphene citrate, characterized by:
1 H-NMR (MeOD) d 7.4-6.7 (m, 14H); 4.27 (t, 2H, -OCH2); 3.51 (t, 2H, CH2- N); 3.28 (q, 4H, 2xN-CH2)); 2.73 (2H); 2.78 (2H); 1.31 (t, 6H, 2xN-C-CHs)) Melting point: 138-139°C (98% purity by GLC);
IR spectrum, v cm-1 (suspension in mineral oil): 3640, 3430, 1720, 1710
(citrate), 1600-1555 (broad band, stilbene system); 750.
UV spectrum: λ max = 243 nm, ε 21 ,800 and λ max 300 nm, ε 1 1 ,400.
These prior art methods for the preparation of Enclomiphene citrate do not allow the preparation of Enclomiphene citrate having needle shaped crystal habit, indeed the crystallization by means of a mixture of ethyl ether and ethanol does not provide a crystalline solid having needle crystals.
Moreover, Enclomiphene citrate was described in literature with different melting points, in particular, 133-135°C and 138-139°C. Said solid forms of Enclomiphene citrate fail to comply with stabilities studies and furthermore show relatively poor solubility in water either in neutral or acid pH.
Furthermore, the prior art methods have the drawbacks related to the poor reproducibility of the process and of the solid form thus obtained.


EXPERIMENTAL SECTION
The starting material Clomiphene citrate can be prepared according to well-known prior art methods, or for example, as described in the example 1 of PCT/EP2015/074746 or can be purchased on the market.
[00190] Example 1 : Preparation of salt of Enclomiphene with racemic binaphthyl- phosphoric acid, starting from Clomiphene citrate.

Clomiphene citrate
[00191] A round bottom flask was charged 100 gr of Clomiphene Citrate (HPLC analysis (A/A%): 65.21 % Enclomiphene, 34.06% Z-Clomiphene) and 1000 mL of methanol. The suspension was stirred at 30°C up the complete
dissolution. Then a solution of racemic binaphthyl-phosphoric acid (abbreviated BPA) 30 gr (0.515 eq) in 30 ml_ of DMF was added. At the end of addition the mixture was stirred for 1 h at 30°C. The obtained suspension was filtered and the solid was washed with 100 ml_ of methanol.
[00192] 50.4 gr of Enclomiphene BPA salt (III) were obtained.
[00193] HPLC Analysis (A/A%): 97.04% Enchlomiphene, 2.5% Z-Clomiphene.
[00194] Example 1 b: Preparation of salt of Enclomiphene with racemic binaphthyl- phosphoric acid, starting from Clomiphene citrate.
[00195] A round bottom flask was charged 50 gr of Clomiphene Citrate and 500 ml_ of methanol. The suspension was heated at 40-45°C and stirred up to the complete dissolution. Then a solution of BPA 15 gr (0.515 eq) in 300 ml_ of methanol was added. At the end of addition the mixture was stirred for 1 h at 20°C. The obtained suspension was filtered and the solid was washed with 100 ml_ of methanol.
24.1 gr of Enclomiphene BPA salt were obtained.
HPLC Analysis (A/A%): 98.96% Enchlomiphene, 0.69% Z-Clomiphene.
[00196] Example 1 c: Preparation of salt of Enclomiphene with racemic binaphthyl- phosphoric acid, starting from Clomiphene citrate.
[00197] In a round bottom flask was charged 100 gr of Clomiphene Citrate and 1000 ml_ of methanol. The suspension was heated at 40-45°C and stirred up the complete dissolution. Then a solution of BPA 30 gr (0.515 eq) in 1000 ml_ of methanol was added. At the end of addition the mixture was stirred for 1 h at 20°C. the obtained suspension was filtered and the solid was wash with 100 ml_ of methanol.
47.9 gr of Enclomiphene BPA salt were obtained.
HPLC Analysis (A/A%): 98.81 % Enclomiphene, 0.79% Z-Clomiphene.
[00198] Example 1d: Preparation of salt of Enclomiphene with racemic binaphthyl- phosphoric, starting from Clomiphene citrate.
[00199] In a round bottom flask was charged 150 gr of Clomiphene citrate and 1500 mL of methanol. The suspension was heater at 40-45°C and stirred up the complete dissolution. Then a solution of BPA 45 gr (0.515 eq) in 900 mL of methanol was added. At the end of addition the mixture was
stirred for 1 h at 20°C. the obtained suspension was filtered and the solid was wash with 100 ml_ of methanol.
76.4 gr of E-Clomiphene BPA salt were obtained.
HPLC Analysis (A/A%): 98.82% Enchlomiphene, 0.80% Z-Clomiphene.
[00200] Example 2: Recrystallization of Enclomiphene BPA salt of formula (III) (the step A).

(Ill)
[00201] Into a proper 0.5 L reactor, equipped with propeller, temperature probes, condenser; Enclomiphene BPA salt (III) (50 g) and having Z-isomer of 1.64 % was suspended in DMF (2.1 L/Kg of Enclomiphene BPA (III)) and methanol (1.4 L/Kg of Enclomiphene BPA salt (III)). The suspension was heated to reflux (~ 76-79°C). Further DMF (0.1 L/Kg of Enclomiphene BPA (III)) might be required to improve the solubility of the starting material. Once the starting material was completely dissolved, methanol was added as anti-solvent (3.5 L/Kg of Enclomiphene BPA (III)). The temperature was decreased to 60°C and the mixture was stirred for 2 – 3 h. Then, the temperature was further decreased to 20 °C and filtered. The wet cake was washed twice with methanol (1.5 L/Kg of Enclomiphene BPA salt (III)). The product was dried under vacuum at 60 – 70 °C for 12 – 24 h. Time of drying could be prolonged until residual DMF is < 2500 ppm.
[00202] Analysis of quality of the final product of the above mentioned example and of the same product, obtained from repetition following the same process, it is shown in the following table:
Enclomiphene BPA (III) salt Enclomiphene BPA (III) salt rixx (Starting product) (finale product)
Z-isomer = 1.64 A/A% Z-isomer = 0.07 A/A%
Z-isomer = 0.79 A/A% Z- isomer = 0.03 A/A%
[00203] Example 3: Preparation of Enclomiphene citrate of formula (I), having needle shaped crystal habit, starting from Enclomiphene BPA salt formula (III).

(II)
[00204] Into a proper 4 L reactor, equipped with propeller, temperature probes, condenser; Enclomiphene BPA salt of formula (III) (400 g, assay 99.8 wt% 0.528 mol, 1 equiv.) was suspended in methyl-tert-butyl ether (MTBE, 2 L), isopropanol (IPA, 0.5 L) and water (2 L). The mixture was stirred for 15 minutes, then 0.48 L of ammonia solution 30 wt% was added and the mixture was further stirred for one hour. The aqueous phase was separated and the organic layer was washed with a solution of ammonia solution 30 wt% (0.12 L) and water (0.6 L). The aqueous phase was separated and the organic layer was finally washed with water (0.6 L). The organic solution was evaporated to residue under vacuum at 60-65°C. The residue was dissolved in 1.36 L of absolute ethanol. The assay of the solution was determined at this stage through a potentiometric titration and results in 15.125 wt% as Enclomiphene of formula (II) (0.466 mol). Then 0.24 L of water were added and the solution was heated to 65°C. Meanwhile, citric acid monohydrate (100.8 g, 0.475 mol, 1.02 equiv.) was dissolved in absolute ethanol (1.7 L) and water (0.3 L), the solution was heated to 65°C. The solution of citric acid was dropped into the solution of Enclomiphene (II), while maintaining 65°C. The dosage takes place in 30- 40 minutes. The inner temperature was decreased very slowly to 60°C over 80 minutes, then it was further decrease to 55°C over 40 minutes. When the inner temperature was in the range 60-55°C (typically at 58°C), the crystallization mixture was seeded with Enclomiphene citrate needle- shaped and a white product began to precipitate. Once reached 55°C the temperature was further decreased to 30°C over 30 minutes, then to 0°C over 30 minutes. The slurry was stirred at 0°C for at least two hours, then it was filtered and the wet cake was washed with 0.4 L of absolute ethanol. The product was dried under vacuum at 65°C. At the end of drying, 269 g of Enclomiphene citrate of formula (I) as needle crystal were isolated, corresponding to 91.8% molar yield.
[00205] HPLC Analysis (A/A%): 99.79% Enchlomiphene, 0.04% Z-Clomiphene (i.e. Z-isomer).
[00206] Example 4: Preparation of Enclomiphene citrate of formula (I), having a needle shaped crystal habit, with a mixture of ethanol and water, wherein the amount of water is 15%.

(I)
[00207] Into a proper 1 L reactor, equipped with propeller, temperature probes, condenser; Enclomiphene of fomula (II) (15,0 g, assay 99.9 wt% 0.0369 mol, 1 equiv.) was dissolved in absolute ethanol (102 ml_, 6.8 mL/g of free base), then 18 ml_ (1.2 mL/g of free base) of water were added and the solution was heated to 65°C. Meanwhile, citric acid monohydrate (7.92 g, 0.0377 mol, 1.02 equiv.) was dissolved in absolute ethanol (127 ml_) and water (23 ml_), the solution was heated to 65°C. The solution of citric acid was dropped into the solution of Enclomiphene (II), while maintaining 65°C. The dosage takes place in 30-40 minutes. The inner temperature was decreased very slowly to 60°C over 80 minutes, then it was further decrease to 55°C over 40 minutes. When the inner temperature was in the range 60-55°C (typically at 58°C), the crystallization mixture was seeded with Enclomiphene citrate needle-shaped and a white product began to precipitate. Once reached 55°C the temperature was further decreased to 30°C over 30 minutes, then to 0°C over 30 minutes. The slurry was stirred at 0°C for at least two hours, then it was filtered and the wet cake was washed with 30 ml_ of absolute ethanol. The product was dried under
vacuum at 65°C. At the end of drying, 20.2 g of Enclomiphene citrate of formula (I) as needle crystal were isolated, corresponding to 91.4% molar yield.
[00208] HPLC Analysis (A/A%): 99.86% Enchlomiphene, 0.03% Z-Clomiphene.
[00209] Example 4a: Preparation of Enclomiphene citrate of formula (I), having a needle shaped crystal habit, with a mixture of isopropanol and water, wherein the amount of water is 15%.
[00210] Into a proper 1 L reactor, equipped with propeller, temperature probes, condenser; Enclomiphene of fomula (II) (40,0 g, assay 99.9 wt% 0.0985 mol, 1 equiv.) was dissolved in isopropanol (272 ml_, 6.8 mL/g of free base), then 48 ml_ (1.2 mL/g of free base) of water were added and the solution was heated to 65°C. Meanwhile, citric acid monohydrate (21.10 g, 0.100 mol, 1.02 equiv.) was dissolved in isopropanol (340 ml_, 8.5 mL/g of free base) and water (60 mL, 1.5 mL/g of free base), the solution was heated to 65°C. The solution of citric acid was dropped into the solution of Enclomiphene (II), while maintaining 65°C. The dosage takes place in 30- 40 minutes. The inner temperature was decreased very slowly to 60°C over 80 minutes, then it was further decrease to 55°C over 40 minutes. When the inner temperature was in the range 60-55°C (typically at 58°C), the crystallization mixture was seeded with Enclomiphene citrate needle- shaped and a white product began to precipitate. Once reached 55°C the temperature was further decreased to 30°C over 30 minutes, then to 0°C over 30 minutes. The slurry was stirred at 0°C for at least two hours, then it was filtered and the wet cake was washed with 30 mL of isopropanol. The product was dried under vacuum at 65°C. At the end of drying, 56.5 g of Enclomiphene citrate of formula (I) as needle crystal were isolated, corresponding to 95.9% molar yield.
[0021 1] Example 4b: Preparation of Enclomiphene citrate of formula (I), having a needle shaped crystal habit, with a mixture of n-propanol and water, wherein the amount of water is 15%.
[00212] Into a proper 0.5 L reactor, equipped with propeller, temperature probes, condenser; Enclomiphene of fomula (II) (9,0 g, assay 99.9 wt% 0.0985 mol, 1 equiv.) was dissolved in 7-propanol (61 mL, 6.8 mL/g of free base), then 1 1 ml_ (1.2 mL/g of free base) of water were added and the solution was heated to 65°C. Meanwhile, citric acid monohydrate (4.70 g, 0.0224 mol, 1.02 equiv.) was dissolved in 7-propanol (77 ml_, 8.5 mL/g of free base) and water (14 ml_, 1.5 mL/g of free base), the solution was heated to 65°C. The solution of citric acid was dropped into the solution of Enclomiphene (II), while maintaining 65°C. The dosage takes place in 30- 40 minutes. The inner temperature was decreased very slowly to 60°C over 80 minutes, then it was further decrease to 55°C over 40 minutes. When the inner temperature was in the range 60-55°C (typically at 58°C), the crystallization mixture was seeded with Enclomiphene citrate needle- shaped and a white product began to precipitate. Once reached 55°C the temperature was further decreased to 30°C over 30 minutes, then to 0°C over 30 minutes. The slurry was stirred at 0°C for at least two hours, then it was filtered and the wet cake was washed with 30 mL of 7-propanol I. The product was dried under vacuum at 65°C. At the end of drying, 1 1.7 g of Enclomiphene citrate of formula (I) as needle crystal were isolated, corresponding to 88.1 % molar yield
[00213] Example 4c: Preparation of Enclomiphene citrate of formula (I), having a needle shaped crystal habit, with a mixture of n-butanol and water, wherein the amount of water is 15%.
[00214] Into a proper 0.5 L reactor, equipped with propeller, temperature probes, condenser; Enclomiphene of fomula (II) (9,0 g, assay 99.9 wt% 0.0985 mol, 1 equiv.) was dissolved in 7-butanol (61 mL, 6.8 mL/g of free base), then 1 1 mL (1.2 mL/g of free base) of water were added and the solution was heated to 65°C. Meanwhile, citric acid monohydrate (4.70 g, 0.0224 mol, 1.02 equiv.) was dissolved in 7-butanol (77 mL, 8.5 mL/g of free base) and water (14 mL, 1.5 mL/g of free base), the solution was heated to 65°C. The solution of citric acid was dropped into the solution of Enclomiphene (II), while maintaining 65°C. The dosage takes place in 30- 40 minutes. The inner temperature was decreased very slowly to 60°C over 80 minutes, then it was further decrease to 55°C over 40 minutes. When the inner temperature was in the range 60-55°C (typically at 58°C), the crystallization mixture was seeded with Enclomiphene citrate needle- shaped and a white product began to precipitate. Once reached 55°C the temperature was further decreased to 30°C over 30 minutes, then to 0°C over 30 minutes. The slurry was stirred at 0°C for at least two hours, then it was filtered and the wet cake was washed with 30 ml_ of 7-butanol. The product was dried under vacuum at 65°C. At the end of drying, 1 1.6 g of Enclomiphene citrate of formula (I) as needle crystal were isolated, corresponding to 87.4% molar yield.
[00215] Example 4d: Preparation of Enclomiphene citrate of formula (I), having a needle shaped crystal habit, with a mixture of tert-butanol and water, wherein the amount of water is 15%.
[00216] Into a proper 0.5 L reactor, equipped with propeller, temperature probes, condenser; Enclomiphene of fomula (II) (9,0 g, assay 99.9 wt% 0.0985 mol, 1 equiv.) was dissolved in te T-butanol (61 ml_, 6.8 mL/g of free base), then 1 1 ml_ (1.2 mL/g of free base) of water were added and the solution was heated to 65°C. Meanwhile, citric acid monohydrate (4.70 g, 0.0224 mol, 1.02 equiv.) was dissolved in te T-butanol (77 ml_, 8.5 mL/g of free base) and water (14 mL, 1.5 mL/g of free base), the solution was heated to 65°C. The solution of citric acid was dropped into the solution of Enclomiphene (II), while maintaining 65°C. The dosage takes place in 30- 40 minutes. The inner temperature was decreased very slowly to 60°C over 80 minutes, then it was further decrease to 55°C over 40 minutes. When the inner temperature was in the range 60-55°C (typically at 58°C), the crystallization mixture was seeded with Enclomiphene citrate needle- shaped and a white product began to precipitate. Once reached 55°C the temperature was further decreased to 30°C over 30 minutes, then to 0°C over 30 minutes. The slurry was stirred at 0°C for at least two hours, then it was filtered and the wet cake was washed with 30 mL of te T-butanol. The product was dried under vacuum at 65°C. At the end of drying, 1 1.2 g of Enclomiphene citrate of formula (I) as needle crystal were isolated, corresponding to 84.4% molar yield.
[00217] Example 5: Preparation of Enclomiphene citrate of formula (I), having a needle shaped crystal habit. Preparation of the seed crystal.
[00218] Into a proper 1 L reactor, equipped with propeller, temperature probes, condenser; Enclomiphene of fomula (II) (15,0 g, assay 99.9 wt% 0.0369 mol, 1 equiv.) was dissolved in absolute ethanol (102 ml_, 6.8 mL/g of free base), then 18 ml_ (1.2 mL/g of free base) of water were added and the solution was heated to 65°C. Meanwhile, citric acid monohydrate (7.92 g,
0.0377 mol, 1.02 equiv.) was dissolved in absolute ethanol (127 ml_, 8.5 mL/g of free base) and water (23 mL 1.5 mL/g of free base), the solution was heated to 50°C. The solution of citric acid was dropped into the solution of Enclomiphene (II), while maintaining 50°C. The dosage takes place in 30-40 minutes. At the end of the dosage, the stirring was turned off and the mixture was allowed to cool down to room temperature without stirring. The product began to crystallize at 40-30°C. Once reached 20- 25°C the stirring was turned on and the temperature was further decreased to 0°C over 30 minutes. The slurry was stirred at 0°C for at least two hours, then it was filtered and the wet cake was washed with 30 mL of absolute ethanol. The product was dried under vacuum at 65°C. At the end of drying, 13.9 g of Enclomiphene citrate of formula (I) were isolated, corresponding to 62.3% molar yield
[00219] Example 6: Preparation of Enclomiphene citrate of formula (I), having a non-needle shaped crystals, with a mixture of acetone and water, wherein the amount of water is 15%.
Comparative example (see Fig. 8) and evidence example of the invention. Following the same process described in the example 4, substituting ethanol solvent with acetone solvent. Starting from 15,0 g of Enclomiphene of formula (II), following the above mentioned process, 22.3 g of Enclomiphene citrate of formula (I) were isolated, corresponding to 94.2% molar yield product. For the morphology of the crystal see fig. 8.
[00220] Indeed, the microscopy analysis provides a better further evidence of the crystal habit of Enclomiphene citrate (I) of the example 6 (see Fig.8) which has a form more different than/to Enclomiphene citrate (I) having a needle shaped crystal habit, obtained according to above described examples,
1. e. 4, 4a, 4b, 4c, 4d (see Fig. 5, 6 and 7).
[00221] HPLC Analysis (A/A%): 99.63% Enchlomiphene, 0.20% Z-Clomiphene.
[00222] Example 7: Analytical method to identify and quantify Z-Clomiphene of formula (IV) into Enclomiphene of formula (II) or Enclomiphene citrate of formula (I) or Enclomiphene BPA salt of formula (III) and for determining the chemical purity.
[00223] Chromatographic conditions:
Dim. Column: 250 mm x 4.6 mm , 5 pm
Stationaly phase: Butyl sylane (USP phase L26, Vydac 4C is suggested) Temp. Column: room temperature
Mobile Phase: Methanol / water / triethylamine 55 : 45 : 0.3 v/v
Adjust at pH 2.5 with phosphoric acid
Flow: 1.0 mL/min
Detector UV a 233 nm,
Injection Volume: 10 μΙ_
Sample diluent: mobile phase.
Applying the conditions described above the expected retention times are as indicated below:

/////////////////Enclomiphene citrate, New patent, WO 2017182097, F.I.S. – FABBRICA ITALIANA SINTETICI S.P.A

Enclomiphene
citrate); 7619-53-6 ((Z)-clomifene citrate)
 Chemical Structure of Clomifene
Chemical Structure of Clomifene
NOTE:
Clomifene may be separated into its Z-and E-isomers, zuclomifene and enclomifene
.
NEW PATENT, PONESIMOD, CRYSTAL PHARMATECH, WO 2017107972

NEW PATENT, PONESIMOD, CRYSTAL PHARMATECH, WO 2017107972
Novel crystalline forms I, II and III of ponesimod . Useful as a selective sphingosine-1-phosphate receptor-1 (S1P1) receptor agonist, for the treatment of psoriasis. Appears to be first filing from Crystal Pharmatech claiming ponesimod. Johnson & Johnson , following its acquisition of Actelion , is developing ponesimod (phase III clinical trial), a S1P1 agonist, for the treatment of autoimmune disorders.
| Applicants: | CRYSTAL PHARMATECH CO., LTD. [CN/CN]; B4-101, Biobay, 218 Xinghu Street, Suzhou Industrial Park Suzhou, Jiangsu 215123 (CN) |
| Inventors: | CHEN, Minhua; (CN). ZHANG, Yanfeng; (CN). LI, Jiaoyang; (CN). ZHANG, Xiaoyu; (CN) |
Most of the family members of the product case ( WO2005054215 ) of ponesimod expire in European countries until November, 2023 and in the US by December, 2024 with US154 extension.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017107972&redirectedID=true
Disclosed are crystalline forms 1, 2, and 3 of a selective S1P1 receptor agonist, namely Ponesimod, and a method for preparing the same. An X-ray powder diffraction pattern of the crystalline form 1 has characteristic peaks at 2 theta values of 18.1° ± 0.2°, 14.6° ± 0.2°, and 11.3° ± 0.2°. An X-ray powder diffraction pattern of the crystalline form 2 has characteristic peaks at 2 theta values of 3.8° ± 0.2°, 10.8° ± 0.2°, and 6.1° ± 0.2°. An X-ray powder diffraction pattern of the crystalline form 3 has characteristic peaks at 2 theta values of 12.2° ± 0.2°, 6.2° ± 0.2°, and 5.6° ± 0.2°. Compared with existing crystalline forms, the present invention has better stability and a greatly increased solubility, and is more suitable for development of a pharmaceutical preparation containing Ponesimod
Ponesimod (compound of formula I) is a selective S1P1 receptor antagonist developed by Actelion. The drug was used to treat moderate to severe chronic plaque psoriasis in the two medium-term trial was successful, and will carry out the treatment of psoriasis in 3 clinical trials.
The present invention discloses a process for the preparation of a compound of formula I, which is disclosed in patent CN 102177144B, which is an amorphous form prepared by the process of CN100567275C, and discloses a process for the preparation of a compound of formula I, crystalline form C, crystalline form III, Type II. The results show that the crystallinity of crystalline form III is poor and it is converted to crystalline form II at room temperature. The crystalline form II is difficult to repeat and prepare a certain amount of propionic acid. The thermodynamics stability of crystalline form A is inferior to that of crystal form C. In contrast, For the crystal form suitable for the development of the drug, the solubility of the crystalline form C is not ideal.
Example 1
Preparation of Ponesimod Form 1:
48.1 mg of Ponesimod was added to 0.40 mL of 1,4-dioxane and the filtrate was filtered. To the solution was stirred at room temperature, 1.20 mL of n-heptane was added dropwise to precipitate the crystals and stirred overnight. The supernatant was filtered off by centrifugation Liquid to obtain Ponesimod crystal form 1.

Follow “‘2014’ Suzhou International Elite Entrepreneurship Week” with interest Over 88 billion venture capital investment helps your pioneering dreams come true
Since 2009, there have been 1267 overseas high-level talent projects settled in Suzhou through International Elite Entrepreneurship Week and 54 talents have been introduced and fostered for the national “Thousand Talents Plan”. Among these 53 talents, Dr. Chen Minhua, the founder of Suzhou Crystal Pharmatech Co., Ltd., was deeply impressed by thoughtful services in Suzhou for innovative pioneering talents when he recalled the development in Suzhou. “Investment and financing services are placed with particular importance. Everything is thoroughly considered for fear that enterprise
In 2010, Chen Minhua quitted his job in a well-known pharmaceutical company in the United States and returned with his core 4-people R&D team. He founded Crystal Pharmatech Co., Ltd. in Suzhou Biobay through the Entrepreneurship Week. Till 2013, Crystal Pharmatech has made profits year by year. The yearly output value in 2013 reached 18 million Yuan, while the profits reached as high as 4 million Yuan. His clients involve half of top 20 pharmaceutical companies globally. Chen Minhua longs to fill the vacancy of drug crystals in China and take the lead in the international drug crystal research. Chen Minhua introduced that government service is an integral part to his growth. “Since it was settled down, Suzhou public sector organized several investment and financing activities and offered training and services in various aspects like the mode of financing, finance docking and enterprise strategic investment, which laid a solid foundation for Crystal Pharmatech’s capital expansion”, said by Chen Minhua.
To help high-level talents solve financial difficulty, Suzhou lays stress on the docking of science & technology and finance. The person in charge of the Municipal Science and Technology Bureau said that Suzhou guides and integrates social capital for equity investment of hi-tech enterprises at the start-up stage via the guiding funds set up by the government and follow-up investment, etc, thus evolving the venture capital investment cluster based on Shahu Equity Investment Center. After the national “Thousand Talents Plan” venture capital investment center was set up, pioneering talents and venture capital are further converging here. As of the end of 2013, there are 270 effective organizations engaged in various venture capital investment in Suzhou that manage the funds in excess of 88 billion Yuan. 30 million Yuan will be appropriated from the municipal science and technology fund budget for the newly established FOF of Angel Investment this year, so as to take avail of social capital for the development of small and medium-sized hi-tech enterprises.
Meanwhile, Suzhou sets up the special compensation fund against credit risks and offers “Kedaitong” with “low threshold and low interest rate”, so as to solve financial difficulty of small and medium-sized hi-tech enterprises and create favorable financing environment for the pioneering work of talents and corporate development. At present, the fund of credit risk pool has reached 500 million Yuan and “Kedaitong” loans of 8.52 billion Yuan have been granted for 1023 small and medium-sized hi-tech enterprises. Particularly, 120 pioneering enterprises that feature independent intellectual property, high content of technology and light assets were backed up with 1.314 billion Yuan, the special risk compensation fund of “Kedaitong”, thus vigorously supporting innovation and pioneering work of leading talents in the science and technology community in Suzhou.
Reporter Qian Yi
Quoted from Suzhou Daily on July 6, 2014
///////////
Imigliptin dihydrochloride, Xuanzhu Pharma Co Ltd, NEW PATENT, WO 2017107945
Imigliptin dihydrochloride, Xuanzhu Pharma Co Ltd, NEW PATENT, WO 2017107945
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017107945&redirectedID=true
| Applicants: | XUANZHU PHARMA CO.,LTD. [CN/CN]; 2518, Tianchen Street, National High-tech Development Zone Jinan, Shandong 250101 (CN) |
| Inventors: | SHU, Chutian; (CN). WANG, Zhenhua; (CN) |

The present invention relates to a crystalline form of benzoate of a dipeptidyl peptidase-IV inhibitor, a method for preparing the same, a pharmaceutical composition,and a use thereof. Specifically, the present invention relates to a crystalline form of benzoate of a compound used as a dipeptidyl peptidase-IV inhibitor and represented by formula (1), namely (R)-2-((7-(3-aminopiperidine-1-yl)-3,5-dimethyl-2-oxo-2,3-dihydro-1H-imidazo(4,5-b)pyridine-1-yl)methyl)benzonitrile, a method for preparing the same, a pharmaceutical composition, and a use thereof.
Novel crystalline form I of imigliptin dihydrochloride as dipeptidyl peptidase IV inhibitor (DPP-IV) for the treatment of and/or prevention of non-insulin dependent diabetes, hyperglycemia and hyperlipidemia. In June 2017, KBP Biosciences and Xuanzhu Pharma , subsidiaries of Sihuan Pharmaceutical , are developing an imigliptin dihydrochloride (phase II clinical trial), a DPP-IV inhibitor and a hypoglycemic agent,, for the treatment of type II diabetes. Follows on from WO2013007167 , claiming similar composition.
////////////////Imigliptin dihydrochloride, Xuanzhu Pharma Co Ltd, NEW PATENT, WO 2017107945
Phase II/ III Clinical Trials of Imigliptin Hydrochloride (KBP-3853) have been approved by CFDA; the Clinical Approval Numbers are 2016L05997 and 2016L06137.
WO 2016181414, IVACAFTOR, NEW PATENT, COUNCIL OF SCIENTIFIC & INDUSTRIAL RESEARCH
![]()


CSIR, Dr. D. Srinivasa Reddy
WO2016181414, PROCESS FOR THE SYNTHESIS OF IVACAFTOR AND RELATED COMPOUNDS
REDDY, Dumbala Srinivasa; (IN).
NATARAJAN, Vasudevan; (IN).
JACHAK, Gorakhnath Rajaram; (IN)
COUNCIL OF SCIENTIFIC & INDUSTRIAL RESEARCH [IN/IN]; Anusandhan Bhawan, Rafi Marg New Delhi 110001 (IN)
The present patent discloses a novel one pot two-step process for the synthesis of ivacaftor and related compounds of [Formula (I)], wherein R1, R2, R3, R4, R5, R6, R7 and Ar1are as described above; its tautomers or pharmaceutically acceptable salts thereof starting from indole acetic acid amides
See Eur J Org Chem, Nov 2015, for an article by the inventors, describing a process for preparing ivacaftor using 4-quinolone-3-carboxylic acid amides. The inventors appear to be based at National Chemical Laboratories of CSIR.
Ivacaftor, also known as N-(2,4-di-tert-butyl-5-hydroxyphenyl)-l,4-dihydro-4-oxoquinoline-3-carboxamide, having the following Formula (A):

Formula (A)
[003] Ivacaftor was approved by FDA and marketed by vertex pharma for the treatment of cystic fibrosis under the brand name KALYDECO® in the form of 150 mg oral tablets. Kalydeco® is indicated for the treatment of cystic fibrosis in patients age 6 years and older who have a G55ID mutation in the CFTR (cystic fibrosis transmembrane conductance regulator)gene.
[004] U.S. 20100267768 discloses a process for preparation of ivacaftor, which involves the coupling of 4-oxo-l,4-dihydro-3- quinoline carboxylic acid with hydroxyl protected phenol intermediate in the presence of propyl phosphonic anhydride (T3P®) followed by deprotection of hydroxyl protection group and optional crystallization with isopropyl acetate. The publication also discloses the use of highly expensive coupling reagent, propyl phosphonic anhydride; which in turn results to an increase in the manufacturing cost. The process disclosed is schematically represented as follows:

[005] Article titled “Discovery of N-(2,4-Di-te -butyl-5-hydroxyphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator” byHadida,S et. al in . Med. Chem., 2014, 57 (23), pp 9776-9795 reportsN-(2,4-di-teri-butyl-5-hydroxyphenyl)-4-oxo- 1 ,4-dihydroquinoline-3-carboxamide (VX-770, 48, ivacaftor), an investigational drug candidate approved by the FDA for the treatment of CF patients 6 years of age and older carrying the G551D mutation.

[006] WO 2014125506 A2 discloses a process for the preparation of ivacaftor in high yield and purity by using novel protected quinolone carboxylic acid compounds as intermediates.
[007] Article titled “Expeditious synthesis of ivacaftor” by Jingshan Shen et. al in Heterocycles, 2014, 89 (4), pp 1035 – 1040 reports an expeditious synthesis for ivacaftor featuring modified Leimgruber-Batcho procedure. The overall yield is 39% over six steps from commercially available 2-nitrobenzoyl chloride.
[008] U.S.2011/064811 discloses a process for preparation of ivacaftor, which involves condensation of 4-oxo-l,4-dihydro-3- quinolone carboxylic acid with 5- amino-2,4-di-(tert-butyl)phenol in the presence of HBTU followed by the formation of ethanol crystalate, which is then treated with diethyl ether to yield ivacaftor as a solid.

[010] U.S. 7,495,103 discloses modulators of ATP-binding cassette transporters such as ivacaftor and a process for the preparation of modulators of ATP-binding cassette transporters such as quinolone compounds. The process includes condensation of 4-oxo-l,4-dihydro-3 -quinolone carboxylic acid with aniline in presence of 2-(lH-7-azabenzotriazol-l-yl)-l,l,3,3-tetramethyluronium hexafluoro phosphate methanaminium (HATU) as shown:
![]()
[011] U.S. 2011/230519 discloses a process for preparation of 4-oxo-l,4-dihydro-3-quinoline carboxylic acid by reaction of aniline with diethylethoxymethylenemalonate at 100-110°C followed by cyclization in phenyl ether at temperature 228-232°C and then hydrolysis, as shown below:

[012] US 7,402,674 B2 discloses 7-Phenylamino-4-quinolone-3-carboxylic acid derivatives, process for their preparation and their use as medicaments.
[013] US 4,981,854 discloses l-aryl-4-quinolone-3 carboxylic acids, processes for their preparation and anti-bacterial agents and feed additives containing these compounds.
Article titled “Ozonolysis Applications in Drug Synthesis” by Van Ornum,S.G. ; Champeau,R.M.; Pariza,R. in Chem. Rev., 2006, 106 (7), pp 2990-3001 reports that ozonolysis for the synthesis of numerous interesting bioactive natural products and pharmaceutical agents.
[014] Article titled “Safe Execution of a Large-Scale Ozonolysis: Preparation of the Bisulfite Adduct of 2-Hydroxyindan-2-carbox-aldehyde and Its Utility in a Reductive Animation” by RaganJ.A. et. al. in Org. Proc. Res. Dev., 2003, 7 (2), pp 155-160 reports various routes to bisulfite adduct, the most efficient of which involved vinyl Grignard addition to 2-indanone followed by ozonolysis and workup with aqueous NaHS03 to effect reduction and bisulfite formation in a single pot. The utility of bisulfite adduct is as an aldehyde surrogate in a reductive amination reaction.

[015] The reported methods for the synthesis of ivacaftor suffered from several drawbacks such as harsh conditions, high temperature reactions and use of large excess of polyphosphoric acid and corrosive phosphoryl chloride etc. Furthermore, synthesis of ivacaftor requires use of high performance liquid chromatography (HPLC) techniques for the separation of ivacaftor and their analogues.
[016] Therefore, development of a simple and efficient synthetic route is in urgent need. Accordingly the present inventors developed environmentally benign, cost effective and short synthetic route for the synthesis of ivacaftor and their analogues.
Example 1:
Procedur A:

To a solution of indole acetic acid (500 mg, 2.85 mmol), aniline (2.85 mmol), HOBt (3.4 mmol) in acetonitrile (10 mL), EDC.HCl (3.4 mmol) followed by DIPEA (11.4 mmol) was added, and mixture was stirred for 16 h at ambient temperature. The
reaction mixture was evaporated to dryness, diluted with EtOAc (25 mL), washed with saturated aqueous NaHC03 solution (5 mL), H20 (5 mL), brine (5 mL), and dried over Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (silica gel 230-400 mesh, ethyl acetate – pet ether) to afford corresponding amide as a colorless solid.
[040] Example 2:
2-(lH-indol-3-yl)-N-phenylacetamide (1) :
Yield: 570 mg; 80%; 1H NMR (200MHz, DMSO-d6) δ = 10.95 (brs, 1 H), 10.14 (s, 1 H), 7.64 (d, J = 7.8 Hz, 3 H), 7.47 – 7.24 (m, 4 H), 7.21 – 6.92 (m, 3 H), 3.76 (s, 2H); MS: 273 (M+Na)+.
[041] Example 3:
5-(2-(lH-indol-3-yl)acetamido)-2,4-di-tert-butylphenyl methyl carbonate (2): Yield: 800 mg; 64%; 1H NMR (200 MHz, DMSO-d6) δ = 11.51 (brs, 1 H), 9.41 (s, 1 H), 8.12 (d, J = 7.6 Hz, 1 H), 7.96 – 7.78 (m, 3 H), 7.71 – 7.42 (m, 3 H), 4.34 (s, 3 H), 4.30 (s, 2 H), 1.79 (s, 9 H), 1.64 (s, 9 H); MS: 459 (M+Na)+.
[042] Example 4:
(S)-2-(lH-indol-3-yl)-N-(l-phenylethyl)acetamide (3):
Yield: 620 mg; 78%; 1H NMR (400MHz ,DMSO-d6)5 = 10.88 (brs, 1 H), 8.48 (d, J = 8.1 Hz, 1 H), 7.59 (d, J = 7.8 Hz, 1 H), 7.39 – 7.26 (m, 5 H), 7.25 – 7.16 (m, 2 H), 7.08 (t, J = 7.3 Hz, 1 H), 7.02 – 6.95 (m, 1 H), 4.96 (t, J = 7.3 Hz, 1 H), 3.59 (s, 2H), 1.38 (d, J = 7.1 Hz, 3 H).
[043] Example 5:
N-(4-Fluorophenyl)-2-(lH-indol-3-yl)acetamide (4):
1H NMR (400 MHz, DMSO-d6) : δ 10.93 (brs, 1H), 10.17 (s, 1H), 7.68 – 7.61 (m, 3H), 7.36 (d, J= 8.1 Hz, 1H), 7.27 (d, J= 2.0 Hz, 1H), 7.15 – 7.13 (m, 3H), 7.11 – 6.99 (m, 1H), 3.73 (s, 2H); 13C NMR (100 MHz, DMSO-d6) : δ 170.1, 159.5, 157.1, 136.6, 136.3, 127.7, 124.4, 121.5, 121.3, 121.2, 119.1, 118.9, 115.8, 115.6, 111.8, 108.9, 34.2; MS: 269 (M+H)+
[044] Example 6:
N-(4-Chlorophenyl)-2-(lH-indol-3-yl)acetamide (5):
1H NMR (200 MHz, DMSO-d6): 510.93 (brs, 1H),10.24 (s, 1H), 7.67 – 7.59 (m, 3H), 7.36 – 7.27 (m, 4H), 7.12 – 6.98 (m, 2H), 3.74 (s, 2H); 13CNMR (100 MHz, DMSO-d6): 5170.4, 138.9, 136.7, 129.1, 127.8, 127.1, 124.5, 121.6, 121.2, 119.2, 119.0, 115.7, 111.9, 108.9, 34.3; MS: 285 (M+H)+.
[045] Example 7:
2-(lH-Indol-3-yl)-N-(p-tolyl)acetamide (6) :
1H NMR (400 MHz, DMSO-d6): 510.91 (brs, 1H), 10.01 (s, 1H), 7.62 (d, J= 7.8 Hz, 1H), 7.50 (d, J= 8.6 Hz, 2H), 7.37 (d, J= 8.1 Hz, 1H), 7.29 – 7.26 (m, 1H), 7.10 – 7.07 (m, 3H), 7.01 – 6.99 (m, 1H), 3.71 (s, 2H), 2.23 (s, 3H); 13C NMR (100 MHz, DMSO-de): 5170.0, 137.4, 136.6, 132.4, 129.5, 127.7, 124.3, 121.4, 119.6, 119.2, 118.8, 111.8, 109.1, 34.2, 20.9; MS: 265 (M+H)+.
[046] Example 8:
N-(4-Ethylphenyl)-2-(lH-indol-3-yl)acetamide (7):
XH NMR (400 MHz, DMSO-d6): 510.91 (brs, 1H), 10.01 (s, 1H), 7.61 (s, 1H), 7.52 (d, J= 8.3 Hz, 2H), 7.36 (d, J= 8.1 Hz, 1H), 7.26 (s, 1H), 7.15 – 7.04 (m, 3H), 6.99 (s, 1H), 2.55 (t, J= 7.5 Hz, 2H), 1.15 (t, J= 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): 5169.9, 138.9, 137.6, 136.6, 128.3, 127.7, 124.3, 121.4, 119.6, 119.2, 118.8, 111.8, 109.1, 40.6, 40.4, 40.2, 40.0, 39.8, 39.6, 39.4, 34.2, 28.0, 16.2; MS: 279 (M+H)+.
[047] Example 9:
2-(lH-Indol-3-yl)-N-(4-propylphenyl)acetamide (8):
1H NMR (400 MHz, DMSO-d6): 58.48 (brs, 1H), 7.64 (d, J = 8.1 Hz, 1H), 7.50 – 7.42 (m, 2H), 7.33 – 7.15 (m, 6H), 7.07 (d, J= 8.3 Hz, 2H), 3.92 (s, 2H), 2.52 (t, J= 7.6 Hz, 2H), 1.65 – 1.53 (m, 2H), 0.91 (t, J= 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): 5169.7, 138.9, 136.5, 135.2, 128.8, 126.9, 124.0, 122.8, 120.4, 120.1, 118.7, 111.6, 108.7, 37.4, 34.5, 24.6, 13.7; MS: 315 (M+Na)+.
[048] Example 10:
2-(lH-Indol-3-yl)-N-(4-isopropylphenyl)acetamide (9) :
yield 79% ; 1H NMR (400 MHz, DMSO-d6): δ 10.91 (brs, 1H), 10.01 (s, 1H), 7.62 (d, = 7.8 Hz, 1H), 7.55 – 7.49 (m, = 8.6 Hz, 2H), 7.37 (d, = 8.1 Hz, 1H), 7.26 (d, = 2.0 Hz, 1H), 7.18 – 7.11 (m, = 8.6 Hz, 2H), 7.11 – 7.05 (m, 1H), 7.02 – 6.95 (m, 1H), 2.95 – 2.71 (m, 1H), 1.17 (d, = 6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6): δ 169.9, 143.5, 137.6, 136.6, 127.7, 126.8, 124.3, 121.4, 119.7, 119.2, 118.8, 111.8, 109.2, 24.4; MS: 315 (M+Na)+.
[049] Example 11:
2-(lH-indol-3-yl)-N-(4-(trifluoromethoxy)phenyl)acetamide (10):
Yield 85% ; 1H NMR (400 MHz, CDC13): δ 8.35 (brs., 1 H), 7.44 – 7.38 (m, 2 H), 7.27 – 7.21 (m, 3 H), 7.12 – 7.05 (m, 1H), 7.03 – 6.95 (m, 2H), 6.93 (d, = 8.6 Hz, 2H), 3.75 (s, 2H); 13C NMR (100 MHz, CDC13): δ 170.0, 145.3, 136.5, 136.2, 126.8, 124.1, 123.0, 121.6, 121.2, 120.5, 118.5, 111.7, 108.2, 34.4; MS: 335 (M+Na)+.
[050] Example 12:
N-(2-chloro-5-methoxyphenyl)-2-(lH-indol-3-yl)acetamide (11):
Yield 75% ; XH NMR (200 MHz, DMSO-d6): δ 10.98 (brs, 1H), 9.27 (s, 1H), 7.59 (d, = 7.8 Hz, 1H), 7.53 (d, = 2.9 Hz, 1H), 7.39 – 7.32 (m, 3H), 7.09 – 6.99 (m, 2H), 6.74 (dd, = 3.0, 8.8 Hz, 1H), 3.85 (s, 2H), 3.71 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ 170.4, 160.1, 141.1, 136.7, 130.0, 127.8, 124.4, 121.6, 119.2, 119.0, 111.9, 109.1, 105.4, 55.4, 34.4; MS: 315 (M+Na)+.
[051]Example 13:
N-(2-ethylphenyl)-2-(lH-indol-3-yl)acetamide (12):
Yield 78% ; 1H NMR (400 MHz, CDC13): δ 8.68 (brs, 1H), 7.95 (d, = 8.1 Hz, 1H), 7.67 (d, = 7.8 Hz, 1H), 7.48 – 7.44 (m, 2H), 7.29 – 7.23 (m, 1H), 7.22 – 7.20 (m, 3H), 7.05 (d, = 4.4 Hz, 2H), 2.00 (q, = 7.4 Hz, 2H), 0.67 (t, = 7.6 Hz, 3H); 13C NMR (100 MHz, CDC13): δ 169.9, 136.6, 135.0, 134.3, 128.7, 126.7, 125.1, 124.1, 123.0, 122.5, 120.4, 118.7, 111.6, 108.6, 34.4, 24.2, 13.6.
[052] Example 14:
N-(2-bromophenyl)-2-(lH-indol-3-yl)acetamide(13):
Yield 76%; 1H NMR (200 MHz, DMSO-d6): δ 11.00 (brs, 1H), 9.30 (s, 1H), 7.81 -7.77 (m, 1H), 7.63 – 7.56 (m, 2H), 7.41 – 7.35 (m, 3H), 7.11 – 7.05 (m, 3H), 3.85 (s, 2H);13C NMR (100 MHz, DMSO-d6): δ 169.9, 136.2, 132.5, 128.0, 127.2, 126.4, 125.5, 124.4, 121.2, 118.7, 118.5, 116.4, 111.4, 108.0, 33.2.
[053] Example 15:
N-benzyl-2-(lH-indol-3-yl)acetamide (14):
Yield 85%; 1H NMR (400 MHz, DMSO-d6): δ 10.89 (brs., 1H), 8.40 (t, = 5.7 Hz, 1H), 7.57 (d, = 7.8 Hz, 1H), 7.36 (d, = 8.1 Hz, 1H), 7.32 – 7.18 (m, 6H), 7.08 (t, = 7.5Hz, 1H), 7.03 – 6.90 (m, 1H), 4.28 (d, = 5.9Hz, 2H), 3.60 (s, 2H); 13C NMR (100 MHz, DMSO-de): δ 171.2, 140.1, 136.6, 128.7, 127.7, 127.2, 124.3, 121.4, 119.2, 118.7, 111.8, 109.3, 42.7, 33.2.
[054] Example 16:
2-(lH-indol-3-yl)-N-(4-methoxybenzyl)acetamide(15):
Yield 85% ; 1H NMR (400 MHz, DMSO-d6): δ 10.87 (brs, 1 H), 8.32 (t, = 5.6 Hz, 1 H), 7.55 (d, = 7.8 Hz, 1H), 7.35 (d, = 8.1 Hz, 1H), 7.22 – 7.13 (m, 3H), 7.11 – 7.05 (m, 1 H), 7.00 – 6.94 (m, 1H), 6.84 (d, = 8.6 Hz, 2H), 4.20 (d, = 6.1 Hz, 2H), 3.72 (s, 3H), 3.56 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 171.1, 158.6, 136.6, 132.0, 129.0, 127.7, 124.2, 121.4, 119.2, 118.7, 114.1, 111.8, 109.4, 55.5, 42.1, 33.2.
[055] Example 17:
N,N-dibenzyl-2-(lH-indol-3-yl)acetamide (16):
Yield 70% ; 1H NMR (400 MHz, DMSO-d6): δ 10.91 (brs, 1H), 7.50 (d, = 7.8 Hz, 1H), 7.37 – 7.34 (m, 3H), 7.30 (d, = 6.6 Hz, 1H), 7.25 – 7.19 (m, 3H), 7.17 (t, = 6.6 Hz, 5H), 7.16 (d, = 7.8 Hz, 1H), 7.00 – 6.97 (m, 1H), 4.59 (s, 2H), 4.50 (s, 2H), 3.86 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 171.7, 138.2, 136.6, 129.2, 128.8, 128.1, 127.8, 127.7, 127.5, 127.1, 124.2, 121.5, 119.2, 118.8, 111.8, 108.5, 50.7, 48.4, 31.2.
[056] Example 18:
2-(lH-indol-3-yl)-N-propylacetamide (17):
Yield 75% ; XH NMR (200 MHz, DMSO-d6): δ 10.86 (brs, 1H), 7.88 – 7.80 (m, 1H), 7.56 (d, = 7.6 Hz, 1H), 7.31 (d, = 7.8 Hz, 1H), 7.17 (d, = 2.3 Hz, 1H), 7.06 – 6.92 (m, 2H), 3.48 (s, 2H), 3.00 (q, J = 6.8 Hz, 2H), 1.39 (sxt, / = 7.2 Hz, 2H), 0.88 – 0.75 (t, = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 171.0, 136.6, 127.8, 124.2,
121.4, 119.2, 118.7, 111.8, 109.6, 39.4, 33.3, 22.9, 11.9.
[057] Example 19:
N-hexyl-2-(lH-indol-3-yl)acetamide (18) :
Yield 87% ; 1H NMR (400 MHz, DMSO-d6): δ 10.84 (brs, 1H), 7.83 (brs, 1H), 7.54 (d, = 7.8 Hz, 1H), 7.33 (d, = 8.1 Hz, 1H), 7.21 – 7.13 (m, 1H), 7.06 (t, = 7.6 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 3.47 (s, 2H), 3.03 (q, / = 6.8 Hz, 2H), 1.37 (t, = 6.5 Hz, 2H), 1.30 – 1.15 (m, 6H), 0.84 (t, = 6.7 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 170.9, 136.6, 127.7, 124.2, 121.3, 119.1, 118.7, 111.7, 109.5, 39.06, 33.2, 31.5, 29.6, 26.5, 22.5, 14.4.
[058] Example 20:
Methyl (2-(lH-indol-3-yl)acetyl)-L-alaninate (19):
Yield 79% ; 1H NMR (400 MHz, CDC13): δ 8.53 (brs, 1H), 7.60 (d, = 7.8 Hz, 1H), 7.41 (d, = 8.1 Hz, 1H), 7.25 – 7.23 (m, 1H), 7.19 – 7.14 (m, 2H), 6.27 (d, = 7.3 Hz, 1H), 4.63 (t, = 7.3 Hz, 1H), 3.78 (s, 2H), 3.68 (s, 3H), 1.31 (d, = 7.3 Hz, 3H); 13C NMR (100 MHz, CDC13): δ 173.4, 171.2, 136.4, 127.0, 123.8, 122.5, 119.9, 118.7,
111.5, 108.5, 52.4, 48.0, 33.3, 18.2.
[059] Example 21:
-(6-chloro-lH-indol-3-yl)-N-phenylacetamide(20):

To a solution of 6-Chloro indole 20a (300 mg, 1.98 mmol )in anhydrous THF, Oxalyl chloride (186 μΤ, 276 mg, 2.18 mmol) was added and the mixture stirred at room temperature. After 2 h, N,N-Diisopropylethylamine (758 μΤ, 562 mg, 4.35 mmol) was
introduced to the mixture, followed by the aniline (221.0 mg, 2.37 mmol). The temperature was raised to 45 °C, and heating continued for 18 h. The solvent was evaporated, and then mixture was diluted with EtOAC (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (10 – 20% EtOAc : Petroleum ether) to afford 20b (295 mg, 51% yield) as a yellow coloured solid. IR Omax(film): 3346, 3307,2853, 1724, 1678 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 12.40 (br. s., 1H), 10.68 (s, 1H), 8.79 (d, = 3.2 Hz, 1H), 8.25 (d, = 8.6 Hz, 1H), 7.85 (d, = 7.8 Hz, 2H), 7.62 (d, = 1.7 Hz, 1H), 7.41 – 7.30 (m, 3H), 7.19 – 7.13 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 182.5, 162.5, 140.0, 138.4, 137.4, 129.2, 128.5, 125.4, 124.8, 123.4, 122.9, 120.8, 113.0, 112.3; HRMS (ESI) Calculated for Ci6HnN2OCl[M+H]+: 299.0582, found 299.0580;
A solution of 20b (300 mg, 0.99 mmol) dissolved in MeOH (40 mL) was added to NaBH4 (45 mg, 1.23 mmol). The reaction was stirred for 4h and then added to saturated solution of Na2S04. The reaction mixture was further stirred for lh and then filtered through Celite.The filtrate obtained was concentrated in vacuo, and then mixture was diluted with EtOAc (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was forwarded for next step without further purification.In an N2 atmosphere, TMSC1 (1.272 mL, 9.9 mmol) in CH3CN (40 mL) was added to sodium iodide (1.488 mg, 9.9 mmol) and stirred for 2h. The reaction mixture was cooled to 0 °C and a solution of above crude alcohol (0.99 mmol) in CH3CN (10 mL) was then added drop wise over 30 min, followed by stirring for 3h. The reaction mixture was poured into NaOH (7g in 40 mL of water) and then extracted with ethyl acetate (15×2). The organic layer was washed with aq.Na2S203, dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel (EtOAc:Pet ether) to afford 20 as a off white solid (two steps 38 % ); IR Umax(film): 3273, 3084,2953, 2857, 1629, 1562 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 11.06 (br. s., 1H), 10.13 (br. s., 1H), 7.62 – 7.57 (m, 3H), 7.40 (s, 1H), 7.30 – 7.25 (m, 3H), 7.04 – 6.99 (m, 2H), 3.71 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 170.1,
139.7, 136.9, 129.2, 126.5, 126.3, 125.5, 123.7, 120.6, 119.6, 119.3, 111.5, 109.4, 34.0; HRMS (ESI):Calculated for Ci6Hi4N2OCl[M+H]+: 285.0789, found 285.0786.
[060] Example 22:
2-(5-chloro-lH-indol-3-yl)-N-phenylacetamide(21):

21a 21b 21
To a solution of 5-Chloro indole 21a (300 mg, 1.98 mmol )in anhydrous THF(20 mL), Oxalyl chloride (186 ^L, 276 mg, 2.18 mmol) was added and the mixture stirred at room temperature. After 2 h, N,N-diisopropylethylamine (758 μΕ, 562 mg, 4.35 mmol) was introduced to the mixture, followed by the aniline (221.0 mg, 2.37 mmol). The tempera ture was raised to 45 °C, and heating continued for 18 h. The solvent was evaporated, and then mixture was diluted with EtOAC (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (10 – 20% EtOAc : Petroleum ether) to afford (21b) (305 mg, 53% yield) as a yellow coloured solid. IR rjmax(film): 3346, 3307,2853, 1724, 1678 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 12.40 (br. s., 1H), 10.68 (s, 1H), 8.79 (d, = 3.2 Hz, 1H), 8.25 (d, = 8.6 Hz, 1H), 7.85 (d, = 7.8 Hz, 2H), 7.62 (d, = 1.7 Hz, 1H), 7.42 – 7.30 (m, 3H), 7.20 – 7.14 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 182.4, 162.4, 140.3, 138.4, 135.4, 129.2, 127.9, 124.8, 124.1, 120.8, 114.8, 112.0; HRMS (ESI) Calculated for Ci6HnN2OCl[M+H]+: 299.0582, found 299.0580; A solution of 21b (200 mg, 0.66 mmol) dissolved in MeOH (30 mL) was added to NaBH4 (30 mg, 0.82 mmol). The reaction was stirred for 4h and then added to saturated solution of Na2S04. The reaction mixture was further stirred for lh and then filtered through Celite. The filtrate obtained was concentrated in vacuo, and then mixture was diluted with EtOAc (15 mL), washed with brine and dried over anhydrous Na2S04. The crude material obtained after removal of solvent was forwarded for next step without further purification. In an N2 atmosphere, TMSC1 (848 mL, 6.6 mmol) in CH3CN (25 mL) was added to sodium iodide (992 mg, 6.6 mmol) and stirred for 2h. The reaction mixture was cooled to 0 °C and a solution of above crude alcohol(0.66 mmol) in CH3CN (5 mL) was then added dropwise over 30 min, followed by stirring for 3h. The reaction mixture was poured into NaOH (5g in 30 mL of water) and then extracted with ethyl acetate(15×2). The organic layer was washed with aq.Na2S203, dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel (EtOAc:Pet ether) to afford 22 as a off white solid (two steps 42 % ); IR Umax(film): 3273, 3084,2955, 2857, 1629, 1562 cm“1; 1H NMR (400 MHz, DMSO-d6): δ 11.13 (br. s., 1H), 10.11 (s, 1H), 7.67 (s, 1H), 7.60 (d, = 7.8 Hz, 2H), 7.39 – 7.27 (m, 4H), 7.13 – 7.02 (m, 2H), 3.16 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 169.9, 139.8, 135.0, 129.2, 128.9, 126.2, 123.6, 121.4, 119.6, 118.6, 113.4, 109.0, 34.0; HRMS (ESI) Calculated for Ci6H14N2OCl[M+H]+: 285.0789, found 285.0786.
[061] Example 23:
2-(l-benzyl-lH-indol-3-yl)-N-phenylacetamide (22):
Yield 79% ; 1H NMR (400 MHz, DMSO-d6): δ 7.67 (d, = 7.8 Hz, 1H), 7.54 (brs, 1H), 7.43 – 7.31 (m, 6H), 7.31 – 7.25 (m, 3H), 7.23 – 7.15 (m, 4H), 7.12 – 7.06 (m, 1H), 5.36 (s, 2H), 3.91 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 169.7, 137.7, 137.2, 137.0, 128.9, 128.9, 127.9, 127.6, 126.9, 124.3, 122.7, 120.2, 119.9, 119.0, 110.2, 107.9, 77.4, 77.1, 76.8, 50.1, 34.5.
[062] Example 24:
Procedure B:

2-(lH-indol-3-yl)-N-phenylacetamidel(100 mg; 0.4 mmol) was dissolved in DCM:MeOH(50 mL; 5: 1), then a stream of 03 was passed through the solution until a blue color developed (10 min). The 03 stream was continued for 4 min. Then surplus O3 was removed by passing a stream of 02 through the solution for 10 min or until the blue colorcompletely vanished. Afterwards pyridine (0.1 mL;1.2mmol) was added to the cold (- 78 °C) mixture. The mixture was allowed to warm to room temperature (1 h) and then Et3N (0.35 mL; 2.4 mmol) were added. After stirring at room temperature overnight the reaction mass was concentrated under reduced pressure to dryness, diluted with EtOAc (30 mL), washed with H20 (5 mL), brine (5 mL), and dried over Na2S04. The crude material obtained after removal of solvent was purified by column chromatography (silica gel 230-400 mesh, MeOH – DCM) to give desired quinolone carboxamide as colorless solid.
[063] Example 25:
4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide (23):
Yield: 65 mg; 62%; XH NMR (200MHz ,DMSO-d6) δ = 12.97 (brs, 1 H), 12.49 (s, 1 H), 8.89 (s, 1 H), 8.33 (d, J = 8.2 Hz, 1 H), 7.91 – 7.69 (m, 4 H), 7.62 – 7.50 (m, 1 H), 7.37 (t, J = 7.8 Hz, 2 H), 7.18 – 7.01 (m, 1 H); MS: 287 (M+Na)+.
[064] Example 26:
2,4-di-tert-butyl-5-(4-oxo-l,4-dihydroquinoline-3-carboxamido)phenyl methyl carbonate (24):
Yield: 35 mg; 34%; 1H NMR (400MHz ,DMSO-d6) δ = 12.96 (brs, 1 H), 12.08 (s, 1 H), 8.94 – 8.82 (m, 1 H), 8.44 – 8.28 (m, 1 H), 7.86 – 7.79 (m, 1 H), 7.78 – 7.73 (m, 1 H), 7.59 (s, 1 H), 7.53 (t, J = 7.5 Hz, 1 H), 7.39 (s, 1 H), 3.86 (s, 3 H), 1.46 (s, 9 H), 1.32 (s, 9 H).
[065] Example 27:
(S)-4-oxo-N-(l-phenylethyl)-l,4-dihydroquinoline-3-carboxamide (25):
Yield: 56 mg; 53%; 1H NMR (500MHz ,DMSO-d6) δ = 12.75 (brs, 1H), 10.54 (d, J = 7.6 Hz, 1H), 8.73 (brs, 1H), 8.28 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 7.9 Hz, 1H), 7.73 -7.68 (m, 1 H), 7.50 (t, J = 7.5 Hz, 1 H), 7.42 – 7.34 (m, 4 H), 7.29 – 7.23 (m, 1 H), 5.18 (t, J = 7.2 Hz, 1 H), 1.50 (d, J = 6.7 Hz, 3 H).
[066] Example 28:
Synthesis of ivacaftor (26):

To a solution of 2,4-di-tert-butyl-5-(4-oxo-l,4-dihydroquinoline-3-carboxamido)phenyl methyl carbonate 5 (30 mg, 0.06mmol) in MeOH (2 mL) was added NaOH (5.3 mg, 0.13mmol) dissolved in H20 (2 mL), and the reaction mixture was stirred at room temperature for 5h. Reaction mass was evaporated to one third of its volume (temperature not exceeding 40°C) and acidified with aq.2N HC1 to pH 2-3. The resulting precipitate was collected by suction filtration give desired compound 7 (19 mg, 76%) as off white solid H NMR (400MHz ,DMSO-d6) δ = 12.88 (d, J = 6.6 Hz, 1 H), 11.81 (s, 1 H), 9.20 (s, 1 H), 8.86 (d, J = 6.6 Hz, 1 H), 8.32 (d, J = 7.8 Hz, 1 H), 7.88 – 7.65 (m, 2 H), 7.51 (t, J = 7.5 Hz, 1 H), 7.16 (s, 1 H), 7.10 (s, 1 H), 1.38 (s,9H), 1.36 (s, 9H).
[067] Example 29:
N-(4-fluorophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (27):
Yield 56% ; 1H NMR (400 MHz, DMSO-d6): δ 12.96 (br. s., 1H), 12.50 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.3 Hz, 1H), 7.86 – 7.72 (m, 4H), 7.54 (t, = 7.3 Hz, 1H), 7.20 (t, = 8.8 Hz, 2H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.2, 159.7, 157.3, 144.6, 139.6, 135.7, 133.5, 126.4, 125.9, 125.8, 121.8, 119.7, 116.1, 115.9, 110.9.
[068] Example 30:
N-(4-chlorophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (28):
Yield 51% ; 1H NMR (400 MHz, DMSO-d6): δ 13.00 (brs., 1H), 12.59 (br. s., 1H), 8.89 (s, 1H), 8.34 (d, = 7.6 Hz, 1H), 7.83 – 7.76 (m, 4H), 7.56 (s, 1H), 7.42 (d, = 7.9 Hz, 2H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.4, 144.7, 139.6, 138.2, 133.5, 129.4, 127.4, 126.4, 125.9, 125.8, 121.6, 119.7, 110.8.
[069] Example 31:
4-oxo-N-(p-tolyl)-l,4-dihydroquinoline-3-carboxamide (29):
Yield 57% ; 1H NMR (400 MHz, DMSO-d6): δ 12.94 (brs., 1H), 12.40 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.8Hz, 1H), 7.82 – 7.80 (m, 1H), 7.76 – 7.7 (m, 1H), 7.63 (d, = 8.3 Hz, 2H), 7.53 (t, = 7.3 Hz, 1H), 7.17 (d, = 8.1 Hz, 2H), 2.29 (s, 3H); 13C NMR (100 MHz, DMSO-de): δ 176.8, 163.1, 144.5, 139.6, 136.8, 133.4, 132.8, 129.9, 126.4, 125.9, 125.7, 120.0, 119.6, 111.1, 20.9; HRMS (ESI):Calculated for Ci7H1502N2[M+H]+: 279.1128, found 279.1127.
[070] Example 32:
N-(4-ethylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (30):
Yield 51% ; 1H NMR (400 MHz, DMSO-d6): δ 12.95 (br. s., 1H), 12.40 (d, = 7.8 Hz, 1H), 8.87 (d, = 6.1 Hz, 1H), 8.33 (d, = 8.1 Hz, 1H), 7.81 – 7.76 (m, 2H), 7.66 – 7.62 (m, = 8.3 Hz, 2H), 7.53 (t, 7 = 7.5 Hz, 1H), 7.22 – 7.17 (m, = 8.3 Hz, 2H), 2.58 (q, = 7.6 Hz, 2H), 1.18 (t, = 7.6 Hz, 3H); 13C NMR (400 MHz, DMSO-d6): δ 181.5, 167.8, 149.3, 144.3, 144.0, 141.7, 138.2, 133.4, 131.1, 130.7, 130.5, 124.8, 124.4, 115.9, 32.8, 20.9.
[071] Example 33:
4-Oxo-N-(4-propylphenyl)-l,4-dihydroquinoline-3-carboxamide (31):
Yield 51%; 1H NMR (500 MHz, DMSO-d6): δ12.93 (brs, 1H), 12.40 (s, 1H), 8.87 (s, 1H), 8.36 – 8.29 (m, 1H), 7.86 – 7.78 (m, 1H), 7.75 (d, J= 7.9 Hz, 1H), 7.68 – 7.61 (m, J= 8.2 Hz, 2H), 7.54 (t, J= 7.6 Hz, 1H), 7.22 – 7.14 (m, J= 8.2 Hz, 2H), 2.55 – 2.51 (m, 2H), 1.64 – 1.53 (m, 2H), 0.90 (t, J= 7.3 Hz, 3H); 13C NMR (500 MHz, DMSO-d6): 176.8, 163.1, 144.5, 139.6, 137.6, 137.0, 133.5, 129.3, 126.4, 125.9, 125.7, 120.0, 119.7, 111.1, 37.2, 24.6, 14.1.
[072] Example 34:
N-(4-isopropylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide (32):
Yield 46% ; 1H NMR (500 MHz, DMSO-d6): δ 12.93 (br. s., 1H), 12.40 (br. s., 1H), 8.89 – 8.86 (m, 1H), 8.33(d, = 7.6 Hz, 1H), 7.81 – 7.50 (m, 5H), 7.25 – 7.21 (m, 2H), 2.90-2.83 (m, 1H), 1.22-1. l l(m, 6H); 13C NMR (100 MHz, DMSO-d6): δ 176.8, 163.1, 144.5, 143.9, 139.6, 137.1, 133.4, 127.2, 126.4, 125.9, 125.7, 120.1, 119.6, 111.1, 33.4, 24.4.
[073] Example 35:
4-oxo-N-(4-(trifluoromethoxy)phenyl)-l,4-dihydroquinoline-3-carboxamide(33):
Yield 57% ; 1H NMR (400 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.63 (s, 1H), 8.88 (d, = 4.9 Hz, 1H), 8.32 (d, = 7.8 Hz, 1H), 7.89 – 7.83 (m, = 8.8 Hz, 2H), 7.79 (d, = 7.6 Hz, 1H), 7.77 – 7.73 (m, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.40 – 7.34 (m, = 8.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 176.8, 163.5, 144.7, 144.0, 139.5, 138.5, 133.5, 126.3, 125.9, 125.8, 122.3, 121.4, 119.7, 110.7.
[074] Example 36:
N-(2-chloro-5-methoxyphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(34):
Yield 54% ; XH NMR (400 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.49 (s, 1H), 8.88 (s, 1H), 8.33 (d, = 7.8 Hz, 1H), 7.83 – 7.75 (m, 1H), 7.56-7.48 (m, 3H), 7.27 – 7.21 (m, 1H), 6.67 (d, = 7.8 Hz, 1H), 3.77 (s, 3H); 13C NMR (400 MHz, DMSO-d6): δ 176.8, 163.4, 160.2, 144.7, 140.4, 139.6, 133.5, 130.3, 126.4, 125.9, 125.8, 119.7, 112.3, 111.0, 109.5, 105.7, 55.5.
[075] Example 37:
N-(2-ethylphenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(35):
Yield 58% ; 1H NMR (400 MHz, DMSO-d6): δ 12.94 (br. s., 1H), 12.37 (s, 1H), 8.90 (s, 1H), 8.36 (dd, = 8.1, 1.4 Hz, 2H), 8.32 (dd, = 8.1, 1.4 Hz, 2H), 7.82 – 7.74 (m, 1H), 7.53- 7.19 (m, 3H), 7.15 – 7.06(m, 1H), 2.79 (q, = 7.3 Hz, 2H), 1.26 (t, = 7.5 Hz, 3H); 293 (M+H)+.
[076] Example 38:
N-(2-bromophenyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(36):
Yield 47% ; 1H NMR (200 MHz, DMSO-d6): δ 12.98 (br. s., 1H), 12.69 (s, 1H), 8.90 (d, = 5.9 Hz, 1H), 8.54 (dd, 7 = 1.4, 8.3 Hz, 1H), 8.34 (d, = 7.6 Hz, 1H), 7.86 – 7.67 (m, 3H), 7.57 – 7.49 (m, 1H), 7.40 (t, = 7.2 Hz, 1H), 7.10 – 7.05 (m, 1H); 13C NMR (100 MHz, DMSO-de): δ 176.7, 163.7, 145.0, 139.5, 137.7, 133.5, 133.1, 128.6, 126.4, 126.0, 125.8, 125.3, 122.9, 119.7, 113.4, 110.8.
[077] Example 39:
N-benzyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(37):
Yield 58% ; 1H NMR (400 MHz, CD3OD-d6): δ 8.82 (s, 1 H), 8.35 (d, = 8.1 Hz, 1 H), 7.79 – 7.77 (m, 1 H), 7.65 (d, = 8.3 Hz, 1 H), 7.52 (t, = 7.6 Hz, 1 H), 7.42 – 7.34 (m, 4 H), 7.31 – 7.26 (m, 1 H), 4.67 (s, 2 H); 13C NMR (400 MHz, DMSO-d6): δ 176.6, 165.0, 144.2, 140.0, 139.5, 133.2, 128.9, 128.7, 127.8, 127.3, 126.6, 125.9, 125.4, 119.5, 111.2, 42.6.
[078] ] Example 40:
N-(4-methoxybenzyl)-4-oxo-l,4-dihydroquinoline-3-carboxamide(38):
Yield 56% ; 1H NMR (400 MHz, DMSO-d6): δ 12.73 (br. s., 1H), 10.35 (t, = 5.3 Hz, 1H), 8.78 (d, = 6.1 Hz, 1H), 8.24 (d, = 8.1 Hz, 1H), 7.76 (d, = 7.1 Hz, 1H), 7.73 -7.68 (m, 1H), 7.48 (t, = 7.5 Hz, 1H), 7.28 (d, = 8.3 Hz, 2H), 6.91 (d, = 8.1 Hz, 2H), 4.49 (d, = 5.6 Hz, 2H), 3.74 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 176.6, 164.8, 158.8, 144.1, 139.5, 133.1, 131.9, 129.2, 126.6, 125.8, 125.4, 119.5, 114.3, 111.3, 55.5, 42.0.
[079] Example 41:
N,N-dibenzyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(39):
Yield 43% ; 1H NMR (400 MHz, DMSO-d6): δ 12.21 (br. s., 1H), 8.27 (d, = 4.9 Hz, 1H), 8.21 (d, = 7.6 Hz, 1H), 7.49 – 7.41 (m, 2H), 7.41 – 7.35 (m, 3H), 7.33 – 7.20 (m, 5H), 7.20 – 7.11 (m, 7 = 7.1 Hz, 2H), 4.59 (br. s., 2H), 4.42 (s, 2H).
[080] Example 42:
4-oxo-N-propyl-l,4-dihydroquinoline-3-carboxamide(40):
Yield 47% ;1H NMR (400 MHz, DMSO-d6): δ 12.7 (br.s., 1H)10.05 (t, = 5.5 Hz, 1H), 8.74 (s, 1H), 8.26 (d, = 8.1 Hz, 1H), 7.83 – 7.66 (m, 2H), 7.52 – 7.44 (m, 1H), 3.33 – 3.22 (m, 2H), 1.61 – 1.49 (m, 2H), 0.93 (t, = 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-de): δ 176.6, 164.8, 143.9, 139.5, 133.1, 126.6, 125.9, 125.3, 119.4, 111.4, 39.3, 23.1, 12.0
[081] Example 43:
N-hexyl-4-oxo-l,4-dihydroquinoline-3-carboxamide(41):
Yield 51% ;1H NMR (400 MHz, DMSO-d6): δ 12.68 (m, 1H), 10.02 (t, = 5.5 Hz, 1H), 8.73 (d, = 6.1 Hz, 1H), 8.27 – 8.25 (m, 1H), 7.77 – 7.67 (m, 2H), 7.47 (t, = 7.5 Hz, 1H), 3.33 – 3.29 (m, 2H), 1.56 – 1.45 (m, 2H), 1.34 – 1.25 (m, 6H), 0.88 – 0.82 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 176.6, 164.8, 143.9, 139.5, 133.1, 126.6, 125.9, 125.3, 119.4, 111.4, 38.7, 31.5, 29.8, 26.7, 22.5, 14.4.
[082] Example 44:
Methyl (4-oxo-l,4-dihydroquinoline-3-carbonyl)-L-alaninate(42):
Yield 38% ; 1H NMR (400 MHz, CD3OD): δ 8.74 (s, 1H), 8.47 – 8.29 (m, 1H), 7.86 -7.76 (m, 1H), 7.64 (d, = 8.3 Hz, 1H), 7.58 – 7.44 (m, 1H), 4.69 (d, = 7.3 Hz, 1H), 3.79 (s, 3H), 1.55 (d, = 7.3 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ 177.3, 173.3, 165.5, 143.6, 139.2, 132.9, 126.3, 125.4, 125.2, 118.5, 110.3, 51.5, 47.0, 17.0.
[083] Example 45:
7-chloro-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(43):
Yield 48% ; IR Omax(film): 2920, 2868, 1661, 1601 cm” 1; 1H NMR (400 MHz, DMSO-de): δ 12.91 (br. s., 1H), 12.30 (s, 1H), 8.90 (s, 1H), 8.29 (d, = 8.8 Hz, 1H), 7.80 -7.67 (m, 3H), 7.58 – 7.51 (m, 1H), 7.36 (t, = 7.7 Hz, 2H), 7.09 (t, = 7.3 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 176.3, 162.9, 145.4, 140.3, 139.2, 138.0, 129.5, 128.2, 126.1, 125.1, 123.9, 120.1, 118.8, 111.6.
[084] Example 46:
6-chloro-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(44):
Yield 52% ; 1H NMR (400 MHz, DMSO-d6): δ 13.05 (brs, 1H), 12.27 (s, 1H), 8.88 (s, 1H), 8.21 (d, = 2.2 Hz, 1H), 7.86 – 7.67 (m, 4H), 7.36 (t, = 7.8 Hz, 2H), 7.16 – 7.04 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 175.6, 162.9, 144.9, 139.1, 138.2, 133.5, 130.4, 129.5, 127.5, 124.9, 123.9, 122.0, 120.1, 111.4.
[085] Example 47:
l-benzyl-4-oxo-N-phenyl-l,4-dihydroquinoline-3-carboxamide(45)
Yield 55% ; 1H NMR (400 MHz, DMSO-d6): δ 12.30 (s, 1H), 9.05 (s, 1H), 8.60 (dd, = 1.7, 8.1 Hz, 1H), 7.82 (d, = 7.8 Hz, 2H), 7.69 – 7.62 (m, 1H), 7.55 – 7.45 (m, 2H), 7.43 – 7.34 (m, 5H), 7.24 – 7.18 (m, 2H), 7.17 – 7.10 (m, 1H), 5.53 (s, 2H); 13C NMR (100 MHz, DMSO-d6): δ 176.9, 162.9, 148.7, 139.3, 138.7, 134.1, 133.1, 129.4, 128.9, 128.7, 128.0, 127.4, 126.2, 125.5, 123.9, 120.5, 116.9, 112.3, 57.9; HRMS (ESI): Calculated for C23H1802N2Na [M+Na]+: 377.1260, found 377.1259; MS: 355 (M+H)+.
[086] Advantages of invention:
1. Cost-effective process for synthesis.
2. Carried out at environmentally benign conditions.
3. Short synthetic route.
4. Useful for making several related compounds of medicinal

DR SRINIVASA REDDY recieving NASI – Reliance Industries Platinum Jubilee Award (2015) for Application Oriented Innovations in Physical Sciences.
MYSELF WITH HIM

From left to right: Dr. D. Srinivasa Reddy, Shri Y. S. Chowdary, Dr. Harsh Vardhan, Dr. Girish Sahni
- Dr D. Srinivasa Reddy receiving the prestigious “SHANTI SWARUP BHATNAGAR” award at the occasion of the 75th Foundation day of CSIR.

Shanti Swarup Bhatnagar awardees with the honorable Prime Minister of India

NCL PUNE

DSR Group
//////////WO-2016181414, WO 2016181414, IVACAFTOR, new patent, COUNCIL OF SCIENTIFIC & INDUSTRIAL RESEARCH, Anusandhan Bhawan, Rafi Marg New Delhi, INDIA, CSIR, Dr. D. Srinivasa Reddy
IPRAGLIFLOZIN, NEW PATENT, WO2016173551, China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry


WO 2016173551 China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016173551&redirectedID=true
MA, Shuai; (CN).
ZHOU, Weicheng; (CN)
WO2016173551, IPRAGLIFLOZIN PREPARATION METHOD
CHINA STATE INSTITUTE OF PHARMACEUTICAL INDUSTRY [CN/CN]; 4th Floor, Building 1, No.1111 Halley Road,pudong New Area Shanghai 201203 (CN).
SHANGHAI INSTITUTE OF PHARMACEUTICAL INDUSTRY [CN/CN]; No.1320,West Beijing Road,Jing’an District Shanghai 200040 (CN)

MACHINE TRANSLATED FROM CHINESE……
Claims
////////WO 2016173551, China State Institute of Pharmaceutical Industry; Shanghai Institute of Pharmaceutical Industry, IPRAGLIFLOZIN, NEW PATENT,
Sacubitril, WO 2016180275, New patent, SUZHOU PENGXU PHARMATECH CO., LTD

Sacubitril, WO 2016180275, New patent, SUZHOU PENGXU PHARMATECH CO., LTD
AHU-377 INTERMEDIATES AND METHOD FOR PREPARING AHU-377 AND AHU-377 INTERMEDIATES PATENT
WO2016180275, new patent, SUZHOU PENGXU PHARMATECH CO., LTD. [CN/CN]; 3rd Floor Building 7, 2358 Chang An Road, Wujiang Suzhou, Jiangsu 215200 (CN)
WANG, Peng; (CN).
LI, Pixu; (CN).
GU, Xiangyong; (CN)

Heart failure is a very high mortality syndrome, for patients with heart failure, so far no drug can significantly improve mortality and morbidity, and thus a new type of therapy is necessary. AHU-377 (CAS No. 149709-62-6) is an enkephalinase inhibitor, which is a prodrug ester groups can be lost through hydrolysis, converted to pharmaceutically active LBQ657, inhibit endorphin enzyme (NEP) the role of the main biological effects of NEP is to natriuretic peptides, bradykinin and other vasoactive peptide degradation failure. AHU-377 and angiotensin valsartan composition according to the molar ratio of 1 LCZ696. LCZ696 is an angiotensin receptor enkephalinase inhibitors, which can lower blood pressure, treat heart failure may become a new drug. Clinical data show, LCZ696 is more effective for the treatment of hypertension than valsartan alone.
Patents US 5,217,996 and US 5,354,892 reported the first synthesis of AHU-377, the synthetic route is as follows:
Reaction with unnatural D-tyrosine derivative as a substrate, more expensive, while the second step in the synthesis is necessary to use Pd-catalyzed Suzuki coupling reaction, whereby preparative route costs than the AHU-377 high.
Patent US 8,115,016 above routes also reported the departure from the pyroglutamate, through multi-step process for preparing a reaction AHU-377, which is more difficult methylation reaction, and the yield is not high. Patent US 8,580,974 also reported a carbonyl group of the a- introducing N, N- dimethyl enamine is converted to methyl, however, there are some problems in the route for constructing methyl chiral centers, are not suitable for scale-up synthesis route as follows:
About the latest AHU377 synthesis intermediates, Patent WO2014032627A1 reported using a Grignard reagent to react with epichlorohydrin, a quicker been important intermediates, synthetic route Compound AHU377 synthesized as follows:
However, the second step of the synthetic route use succinimide nitrogen atoms introduced by Mitsunobu reaction with hydrochloric acid hydrolysis to remove, then converted to Boc protected at the end of the synthesis process AHU377 Boc will have to take off protection, then any connection with succinic anhydride reaction product introduced into the structure of succinic acid portion, so that this method of atom economy and the economy of the steps are low.
Example 1
Synthesis of Compound 2
In inert atmosphere, a solution of three 500mL flask was added compound 1 (10g, 1eq), dissolved after 90mL THF, was added CuI (4.814g, 0.1eq), the system moves to the low temperature in the cooling bath to -20 ℃ when, biphenyl magnesium bromide dropwise addition, the internal temperature was controlled not higher than -10 ℃. Bi closed refrigeration drop, return to room temperature overnight. Completion of the reaction, the reaction solution was poured into saturated the NH 4 of Cl (10vol, 100 mL) was stirred at room temperature for 0.5h. Suction filtered, the filter cake was rinsed with a small amount of EA, and the filtrate was transferred to a separatory funnel carved, and the aqueous phase was extracted with EA (10vol × 2,100mL × 2) and the combined organic phases with saturated NaHC [theta] 3 , the NH 4 of Cl, each Brine 150mL (15vol) washed once, dried over anhydrous over MgSO 4 dried, suction filtered, and concentrated to give a white solid. Product obtained was purified by column 15.2g, yield 78%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.52 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.38-7.25 (m, 8H), 4.62-4.47 ( m, 2H), 4.09 (dd, J = 6.7,3.5Hz, 1H), 3.54 (dd, J = 9.5,3.5Hz, 1H), 3.43 (dd, J = 9.4 , 6.9Hz, 1H), 2.84 ( d, J = 6.6Hz, 2H), 2.38 (s, 1H).
Example 2
Synthesis of Compound 3
In an inert gas, at room temperature was added to the flask 500mL three Ph3P (18.54g, 2eq), 240mL DCM dissolution, butyryl diimide (of 6.44 g), compound 2 (15g), an ice-water bath cooling to 0 ℃ or so, was added dropwise DIAD (14mL) was complete, the reaction go to room temperature.Starting material the reaction was complete, the system was added to water (100 mL) quenched the reaction was stirred for 10min; liquid separation, the aqueous phase was extracted with DCM (100mL × 2), the combined organic phases with saturated Brine 100mL × 2), dried over anhydrous over MgSO 4 dried , filtration, spin dry to give a white solid; product was purified by column 15.4g, yield 82%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.56 (D, J = 7.4Hz, 2H), 7.49 (D, J = 8.0Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.37-7.30 (m, 3H), 7.27 ( d, J = 6.7Hz, 3H), 7.22 (d, J = 8.0Hz, 2H), 4.75 (s, 1H), 4.56 (d, J = 12.0Hz, 1H), 4.45 (d, J = 12.0Hz, 1H ), 4.06 (t, J = 9.6Hz, 1H), 3.70 (dd, J = 10.0,5.2Hz, 1H), 3.23 (dd, J = 13.8,10.3Hz, 1H) , 3.14-3.00 (m, 1H), 2.48 (d, J = 4.0Hz.4H).
Example 3
Synthesis of Compound 4
Protection of inert gas, at room temperature was added to the flask 1L three compound 3 (18.81g), 470mL EtOH was dissolved, was added Pd / C, replaced the H 2 three times, move heated on an oil bath at 60 ℃ reaction. Raw reaction was complete, the system was removed from the oil bath, the reaction solution was suction filtered through Celite and concentrated to give the crude product. It was purified by column pure 11.8g, a yield of 81.2%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.8Hz, 2H), 7.51 (D, J = 7.8Hz, 2H), 7.42 (T, J = 7.5Hz, 2H), 7.33 (T , J = 7.2Hz, 1H), 7.26 (d, J = 7.2Hz, 2H), 4.55 (d, J = 5.2Hz, 1H), 4.06-3.97 (m, 1H), 3.86 (dd, J = 12.0, 3.1Hz, 1H), 3.16 (dd , J = 8.1,2.9Hz, 2H), 2.58 (t, J = 7.0Hz, 4H), 1.26 (s, 2H).
Example 4
Synthesis of Compound 7
Protection of inert gas, at room temperature to a 25mL flask was added three Dess-Martin oxidant (767.7mg), 10mL DCM was dissolved, the system was cooled down to -10 deg.] C, was added 4 (500mg). Starting material the reaction was complete, to the system was added saturated NaHCO3 and Na2S2O3 each 5mL, quench the reaction stirred for 10min; aqueous phase was extracted with DCM (10mL × 3) and the combined organic phases with saturated NaHCO3, Brine 30mL each wash, dried over anhydrous MgSO4, filtration, spin dried to give the crude product used directly in the next reaction cast.
Example 5
Synthesis of Compound 8
Inert gas, at room temperature for three to 500mL flask 7 (497.5mg), 10mL DCM to dissolve an ice water bath to cool, added phosphorus ylide reagent (880.6mg), the system was removed from the ice water bath at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. Liquid separation, the aqueous phase was extracted with DCM (10mL × 2), organic phases were combined, washed with saturated Brine 20mL × 2, dried over anhydrous MgSO4, filtration, spin crude done. Product obtained was purified by column 563mg, 90% yield.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.60-7.53 (m, 2H), 7.51 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.33 (D, J = 7.3Hz, 1H), 7.23 (d , J = 8.1Hz, 2H), 7.13 (dd, J = 9.2,1.5Hz, 1H), 5.26 (td, J = 9.5,6.9Hz, 1H), 4.25-4.05 ( m, 2H), 3.40 (dd , J = 13.7,9.7Hz, 1H), 3.13 (dd, J = 13.8,6.7Hz, 1H), 2.53 (d, J = 2.2Hz, 4H), 1.85 (d, J = 1.4Hz, 3H), 1.30 ( t, J = 7.1Hz, 3H).
Example 6
Synthesis of Compound 9
Protection of inert gas, at room temperature to a 50mL flask was added three 8 (365mg, 1eq), 9mL of ethanol and stirred to dissolve, the system was replaced with hydrogen three times, was added Pd / C (25% w / w) at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. The reaction mixture was suction filtered through Celite and concentrated to give the crude product. Product was purified by column, yield 80.2%, purity 97.2%.
Example 7
Synthesis of Compound 10
Equipped with Compound 9 (100mg) acetic acid A reaction flask (9mL), hydrochloric acid (1mL). The reaction was heated oil bath at 80 deg.] C. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. After saturated NaHCO3 and extracted with EA and concentrated to give crude product. Product obtained was purified by column 90mg, yield 84%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.54 (m, 2H), 7.53-7.48 (m, 2H), 7.41 (dd, J = 10.5,4.9Hz, 2H), 7.31 (dd, J = 8.3 , 6.4Hz, 1H), 7.22 ( d, J = 8.2Hz, 2H), 5.93 (t, J = 9.7Hz, 1H), 4.34-4.00 (m, 3H), 2.91-2.71 (m, 2H), 2.68 -2.57 (m, 2H), 2.55 (ddd, J = 9.4,7.0,4.3Hz, 1H), 2.42 (dt, J = 13.3,6.8Hz, 2H), 1.97-1.74 (m, 1H), 1.64-1.46 (m, 1H), 1.23 ( td, J = 7.1,3.3Hz, 3H), 1.14 (dd, J = 7.1,3.9Hz, 3H)
Example 8
Synthesis of Compound 5
Example 8-1: The reaction flask was added compound 4 (1eq) was added water (2VOL), concentrated hydrochloric acid (2VOL), 110 ℃ reaction was heated in an oil bath overnight, complete conversion of starting material, the HPLC peak area 97%. 10% NaOH solution was added to adjust the pH to about 10, filtration products. Yield 85%.
Example 8-2: The reaction flask was added compound 4 (1eq) was added ethanol (5 vol), water (5 vol), potassium hydroxide (8 eq), was heated in an oil bath overnight at 110 ℃ reaction, complete conversion of the starting material, the HPLC peak area 99%. Water was added (5Vol), filtered to obtain the product. Yield 95%. Product was dissolved in toluene, was added ethanolic hydrochloric acid, the precipitated hydrochloride Compound 5.
NMR data for the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.31 (S, 3H), 7.70-7.61 (m, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.31 (m, 3H), 4.09 (the dq- , J = 42.6,7.1Hz, 1H), 3.62-3.51 (m, 1H), 3.50-3.41 (m, 1H), 3.11-3.00 (m, 1H), 2.95-2.84 (m, 1H), 1.30-1.10 (m, 1H).
EXAMPLE 9
Synthesis of Compound 6
To the reactor was added compound 5, was added absolute ethanol (3vol). Temperature of the outer set 30 ℃ heating, stirring was continued after the temperature reached 25 ℃ 20min. Was added 30% NaOH aqueous solution (1.1eq). External temperature 65 ℃ heating provided, after the internal temperature reached 60 deg.] C was slowly added (of Boc) 2 O (1.1 eq). Stirring 0.5h, reaction monitoring. After completion of the reaction, water was added slowly dropwise (8vol), turn off the heating and natural cooling. The system temperature was lowered to 25 deg.] C and continue stirring for 2h. Filter cake at 50 ℃ blast oven drying to obtain the product.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Synthesis of Intermediate Compound 6 to Compound 10, i.e., the AHU-377, a synthetic route in the background of the present invention, the cited patent application WO2014032627A1 loaded in detail, not in this repeat.
Example 10
Synthesis of Compound 2
Benzyl glycidyl ether preparation (50g) in THF (200mL) was added. Under inert gas protection, the biphenyl magnesium bromide (365mmol) was added to THF (1020mL) was added the reaction flask is placed in a low temperature bath -40 ℃ cooling. Cuprous iodide (O.leq) when the internal temperature dropped to -9 ℃. Continued to decrease the temperature of -23 ℃ dropwise addition of benzyl glycidyl ether in THF was added dropwise to control the internal temperature process of not higher than -15 deg.] C, 47 min when used, the addition was completed the cooling off the reaction was stirred overnight. The cooling system to -20 ℃ quenched with 1N HCl aqueous solution, <10 ℃ Go stirred 30min at room temperature. Liquid separation, the aqueous phase was extracted with THF, the combined THF phases. Respectively saturated ammonium chloride (250mL), saturated brine (250mL) washed. Rotary evaporation to remove THF, and water (200 mL) Continue rotary evaporation 1h, cool to precipitate a solid. Suction crude. Crude n-heptane was added 2Vol beating, suction filtration to obtain the product in a yield of 90 ~ 95%, HPLC peak area 94%. In another column purification was pure, columned yield 88.6%, HPLC 99.1%.
Example 11
Synthesis of Compound 3
Preparation Example 9, said compound taking the embodiment 2 (5g) added to the reaction flask, the reaction flask was added toluene (80mL), phthalimide (2.55 g of) and triphenylphosphine (5.35g of), the nitrogen was replaced protection. An ice-salt bath cooling to -5 deg.] C, was added dropwise DIAD (4.12g), dropwise addition was exothermic, the temperature was raised to 5 ℃. The reaction was continued 1h sampling HPLC test material substantially complete reaction. Join 12g silica spin column done to collect the product (including DIEA derivative).
Example 12
Synthesis of Compound 11
Compound 3 (3g) was added to the reaction flask embodiment taken in Preparation Example 10, was added ethanol (30 mL), with stirring. Was added hydrazine hydrate (2g) was heated in an oil bath reflux 1h, when supplemented with 20mL ethanol was stirred difficulties, the reaction was continued to 2.5h, HPLC showed the starting material the reaction was complete. Add EA / H2O 100mL each liquid separation, the EA phase was washed with water (100mL) and the combined organic phases were washed with water (100mL) and saturated brine (100mL) washed. Anhydrous magnesium sulfate and filtered spin column was done product 1.88g, yield 88%, HPLC 94%.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 7.64 (D, J = 7.2Hz, 2H), 7.57 (D, J = 8.1Hz, 2H), 7.45 (T, J = 7.6Hz, 2H), 7.39-7.32 ( m, 5H), 7.29 (d , J = 8.1Hz, 3H), 4.55-4.43 (m, 2H), 3.38-3.23 (m, 3H), 3.18-3.10 (m, 1H), 2.82-2.74 (m, 1H), 2.61-2.52 (m, 1H ).
Example 13
Synthesis of Compound 11
To the toluene solution of the compound 2 was added phthalimide (1.1 eq), triphenylphosphine (1.3 eq) with stirring. External bath set -10 ℃, to cool the system, the internal temperature dropped to 0 ~ 5 ℃, start dropping DIAD (1.3eq), control the internal temperature -5 ~ 5 ℃. Completion of the dropwise addition, the cooling bath was turned off outside the reaction was stirred at room temperature. The reaction was stirred for 1 to 4 hours. The reaction solution to give compound 3, administered directly in the next reaction. To the above reaction mixture was added hydrazine hydrate (6 eq), heated to 70 ~ 80 ℃, to complete the reaction, filtered hot, the filtrate. Aqueous sodium hydroxide solution (20vol 10%) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. Water was added (20vol) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. The toluene phase was added hydrochloric acid (20vol, 3N), stirred for 0.5h, to form a solid precipitate. Filtration and drying to obtain a product, i.e. compound 11, the hydrochloride salt, yield 60% in two steps.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.46 (S, 3H), 7.63 (dd, J = 16.4,7.7Hz, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.22 (m, 8H ), 4.56 (d, J = 12.1Hz, 1H), 4.48 (d, J = 12.1Hz, 1H), 3.58 (d, J = 7.9Hz, 2H), 3.47 (dd, J = 10.9,6.3Hz, 1H ), 3.11 (dd, J = 13.5,4.9Hz, 1H), 2.92 (dd, J = 13.4,9.1Hz, 1H).
Example 14
Synthesis of Compound 12
Weigh Compound 11 (1.38g) was added to the reaction flask. To the reaction flask plus DCM (14ml) and Et3N (462mg, 0.73ml). Weighed (of Boc) 2O (1.23 g of) was added to DCM (5ml) was dissolved. Room temperature (8 ℃), a solution (of Boc) 2 DCM solution O was added dropwise to the reaction, (2ml) rinsed with DCM. The reaction mixture was stirred at room temperature, detected by HPLC, the reaction ends 4h. Reaction mixture was washed (15ml) 3 times with Brine (15ml) The reaction solution was washed 1 times. Inorganic sulfate, concentrated and purified by column PE:EA = 15:1 give product 560mg, yield 30.8%, HPLC 99.92%.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.49 (D, J = 7.4Hz, 2H), 7.43 (T, J = 7.3Hz, 2H), 7.39-7.28 (m, 5H), 7.24 ( d, J = 9.0Hz, 3H), 5.00-4.80 (br, 1H), 4.51 (q, J = 11.8Hz, 2H), 4.08-3.85 (br, 1H), 3.43 ( d, J = 2.9Hz, 2H) , 3.02-2.77 (m, 2H), 1.42 (s, 9H).
Example 15
Synthesis of Compound 6
Weigh Compound 12 (250mg) and methanol (9ml) was added to the reaction flask. Added Pd / C (138mg, 1 / 4w / w, water content 55%). The H 2replaced 3 times, 50 ℃ stirred and heated. HPLC detection reaction, the reaction end 30h. Filtered off Pd / C, 40 ℃ concentrated under reduced pressure to remove methanol. PE:EA = 3:1 florisil column to give the product 196mg, 100% yield, 99.34% purity.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Method for preparing the AHU-377, characterized by comprising the steps of: (a) Compound (1) S- benzyl glycidyl ether and biphenyl Grignard reagent produced by the reaction of the compound (2) in an organic solvent; ( b) compound (2) with a succinimide or phthalimide Mitsunobu reaction occurs in an organic solvent to form a compound (3); (C) compound (3) in an organic solvent in the role of a catalyst under removal debenzylation protected form compound (4); (D) compound (4) with an oxidizing agent oxidation reaction occurs in an organic solvent to form a compound (7); (E) compound (7) with a phosphorus ylide reagent in an organic solvent to give the compound (8); (F.) compound (8) in an organic solvent in the selective catalytic hydrogenation of the compound (9); and (g) of the compound (9) in an organic solvent in the hydrolysis reaction of the amide compound occurs in the presence of an acid ( 10), i.e., AHU-377;
SUVEN LIFE SCIENCES LTD, WO 2016178064, POLYMORPH OF NINTEDANIB ETHANESULPHONATE, NEW PATENT

NINTEDANIB ETHANESULPHONATE
NEW PATENT
WO2016178064, CLICK FOR PATENT
POLYMORPH OF NINTEDANIB ETHANESULPHONATE, PROCESSES AND INTERMEDIATES THEREOF
SUVEN LIFE SCIENCES LIMITED [IN/IN]; 5th floor, Serene Chamber, Road No.5, Off Avenue No. 7, Banjara Hills, Telangana Hyderabad 500034 (IN)
ARAVA, Veera Reddy; (IN).
GOGIREDDY, Surendra Reddy; (IN).
JASTI, Venkateswarlu; (IN)
DR VEERA ARAVA REDDY

Vice President

Surendra Reddy Gogireddy, Sr.Research Associate

The present invention provides novel crystalline Form of Nintedanib and process for its preparation. The present invention also provides to a novel process for the preparation of Nintedanib. The present invention further provides to novel intermediates used in the preparation of Nintedanib and process for their preparation.
Nintedanib inhibits multiple receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (nRTKs).The chemical name of Nintedanib is lH-Indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl-(4-methyl-l-iperazinyl)acetyl]amino]phenyl]amino]phenylmethylene] -2-oxo-,methylester, (3Z)-, ethanesulfonate (1 : 1) and is structurally represented by compound of Formula I.
Formula I
Nintedanib is marketed in the United States under the trade name OFEV and is indicated for the treatment of Idiopathic Pulmonary Fibrosis (IPF).
Nintedanib was first described and claimed in U.S. Pat.No. 6,762, 180 and EP 1224 170. These patents disclose a process for the preparation of Nintedanib as depicted in scheme I given below:

U.S.Pat.No. 8,067,617 discloses a process for the preparation of Nintedanib intermediate Enolindole derivative), which is shown in the scheme-II given below:

Scheme-II
U.S.Pat. No. 7,119,093 discloses Nintedanib monoethanesulphonate in crystalline form characterised by X-ray powder diffraction pattern having 2Θ values at 7.70, 8.78, 9.47, 9.82, 11.59, 11.93, 13.15, 13.69, 14.17, 16.32, 16.72, 16.92, 17.43, 17.77, 18.58, 18.81, 19.03, 19.73, 19.87, 20.03, 20.61, 20.83, 21.26, 21.76, 22.05, 22.19, 22.57, 23.10, 23.81, 24.69, 24.78, 24.91, 25.42, 26.24, 26.91, 27.19, 27.61, 27.95, 28.71, 29.25.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, X-ray diffraction pattern, infrared absorption fingerprint and solid state NMR spectrum. One polymorph may give rise to thermal behaviour different from that of another polymorph. Thermal behaviour can be measured in the laboratory by such techniques as capillary melting point, thermo gravimetric analysis (“TGA”) and differential scanning calorimetry (“DSC”), which have been used to distinguish polymorphic forms.
The differences in the physical properties of different polymorphs results from the orientation and intermolecular interactions of adjacent molecules or complexes in the bulk solid. Accordingly, polymorphs are distinct solids sharing the same molecular Formula yet having distinct advantageous physical properties compared to other polymorphs of the same composition or complex. Hence there remains a need for polymorphic forms which have properties suitable for pharmaceutical processing on a commercial scale.
Considering the importance of Nintedanib, there exists a need to develop an alternate and improved process for the preparation of Nintedanib with better yield. Further, the process involved should be simple, convenient and cost-effective for large scale production. The inventors of the present invention during their continuous efforts also developed a novel high melting stable polymorphic form of Nintedanib ethanesulfonate.
EXAMPLES
Example 1: Process for the preparation of Nintedanib Monoethane Sulfonate:
Step-1: Preparation of methyl-3-(hydroxy(phenyl)methylene)-2-oxoindoline-6-carboxylate: To the suspension of methyl 2-oxoindoline-6-carboxylate (50 gm, 0.261 mol) in IPA (350 ml) was added slowly SMO-powder (33.8 gm, 0.626 mol) and stirred for about 15 min. Benzyl chloride (44 g, 0.313 mol) was added after completion of the reaction at a reaction temperature of -5 to -10°C for about 5hrs. The reaction mixture was quenched into ice-water (700 ml) and acidified with Cone. HC1 (2.0-2.5 ml). Filtered the reaction mixture, washed with water (2X100 ml) and dried the precipitate to obtain crude product which can be recrystallized from acetonitrile (28 ml) to obtain methyl-3-(hydroxy(phenyl)methylene)-2- oxoindoline-6-carboxylate pure crystalline solid (32 gm) (61%) (HPLC purity >97%). The filtrate was evaporated in vacuum to give unreacted methyl 2-oxoindoline-6-carboxylate. MR: 216-223°C; IR (KBr, cm“1): 3178, 1711, 1651; 1H-NMR (400 MHz, DMSO): δ 3.80 (s, 3H), 7.17 (s, 1H), 7.28-7.31 (m, 2H), 7.46-7.50 (m, 3H), 7.72 (d, 2H, J = 6.0 Hz), 9.52 (s, 1H), 11.53 (s, 1H); 13C-NMR (100 MHz, DMSO): δ 22.12, 52.41, 101.13, 111.13, 119.23, 123.06, 126.65, 127.06, 128.65, 129.21, 132.26, 134.47, 136.99, 166.58, 172.52 and 175.80; MS: m/z 294 [M]“1
Step-2: Preparation of methyl-3-(acetoxy(phenyl)methylene)-l-acetyl-2-oxoindoline-6-carboxylate (Acetyl derivative):
To the suspension of methyl-3-(hydroxy(phenyl)methylene)-2-oxoindoline-6-carboxylate (45 gm, 0.1512 mol) in acetic anhydride (300 ml) was added pyridine (4.5g) slowly (drop-wise) and stirred the reaction at temperature of 0-5°C for about30 min. After completion of the reaction raised the temperature of the reaction mass to 75-80°C and stirred for about lhr. Cooled the reaction mass and stirred for about 30 min at 25-28°C, filtered, washed with hexane (100ml) and dried the precipitate to obtain methyl-3-(acetoxy(phenyl)methylene)-l-acetyl-2-oxoindoline-6-carboxylate.
MR: 226-229°C; IR (KBr, cm“1): 3413, 1771, 1743, 1717, 1640; 1H-NMR (400 MHz, CDC13): δ 2.38 (s, 3H), 2.62 (s, 3H), 3.92 (s, 3H), 7.44 (m, 3H), 7.62 (d, 2H, J = 7.004 Hz), 7.68 (d, 1H, J = 8.12 Hz), 7.91 (d, 1H, J = 8.0 Hz), 8.90 (s, 1H); 13C-NMR (100 MHz, CDC13): δ 21.08, 21.38, 26.96, 52.25, 52.34, 115.17, 117.18, 121.33, 122.77, 125.82, 126.19, 126.56, 128.15, 128.87, 129.27, 129.34, 130.81, 130.90, 131.47, 131.82, 132.80, 138.55, 160.85, 165.95, 166.38, 166.42, 167.01, 170.67 and 170.76; MS: m/z 380 [M]+1.
Step-3: Preparation of methyl- l-acetyl-3-(((4-(2-chloro-N-methylacetamido)phenyl)amino) (phenyl)methylene)-2-oxoindoline-6-carboxylate) (Chloroacetyl derivative) :
Suspension of methyl-3-(acetoxy(phenyl)methylene)- l-acetyl-2-oxoindoline-6-carboxylate (Acetyl derivative) (49gm, 0.129mol) and N-(4-aminophenyl)-2-chloro-N-methylacetamide(25.66gm, 0.129 mol) in a mixture of methanol (350 ml) and DMF (88 ml) was heated to 60-65°C stirred for about 12hr at the same temperature. After completion of the reaction cooled the reaction mass to room temperature and stirred for about 30min. Filtered the reaction mixture, washed with methanol (2X50ml) and dried the precipitate to obtainmethyl-l-acetyl-3-(((4-(2-chloro-N-ethylacetamido)phenyl)amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate).
MR: 247-250°C; IR (KBr, cm“1): 3432, 1712, 1675, 1591; 1H-NMR (400 MHz, DMSO): δ 2.74 (s, 3H), 3.11 (s, 3H), 3.78(s, 3H), 3.87 (s, 2H), 5.75 (d, 1H, J = 8.08 Hz), 7.01 (d, 2H, J = 7.96 Hz), 7.22 (d, 2H, J = 6.08Hz), 7.36 (d, 1H, J = 8.48 Hz), 7.46 (d, 2H, J = 7.24 Hz), 7.54-7.64 (m, 3H), 8.74 (s, 1H0, 11.92 (s, 1H), 13C-NMR (100MHz, DMSO): δ 27.17, 37.76, 42.48, 52.40, 96.38, 116.17, 117.59, 124.80, 125.33, 125.68, 128.09, 129.00, 130.10, 131.35, 131.97, 134.05, 160.93, 165.63, 166.68, 168.49 and 171.28; MS: m/z 518 [M]+1 and 520 [M]+1.
Step-4: Preparation of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-lyl)acetamide) phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate (Nintedanib free base):
Suspension of methyl- l-acetyl-3-(((4-(2-chloro-N-methylacetamido)phenyl)amino) (phenyl)methylene)-2-oxoindoline-6-carboxylate)(40 gm, 0.077ml) and N-methylpiperidine (23.24 gm, 0.232 mol) in a mixture of DMF (160 ml) was heated to a reaction temperature of 45-50°C for about l-2hrs. The reaction mixture was quenched into ice-water (1.6 Lt) and stirred for about lhr at 15-20°C. Filtered the reaction mixture mass washed with water and dried the precipitate to obtain crystalline crude solid (36 gm). Purified with acetonitrile to obtain Nintedanib free base (34 gm) as a yellow crystals (93.74%) (HPLC purity: >98%). MR: 240-246°C; IR (KBr, cm“1): 3559, 3455, 2940, 2810, 1711, 1657; 1H-NMR (400 MHz, DMSO): δ 2.09 (s, 3H), 2.17 (s, 8H), 2.68 (s, 2H), 3.05 (s, 3H), 3.76 (s, 3H), 5.80 (d, 1H, J = 7.56 Hz), 6.86 (d, 2H, J = 6.72 Hz), 7.11 (d, 1H, J = 6.48 Hz), 7.17 (d, 2H, J = 7.68 Hz), 7.42-7.57 (m, 6H), 10.98 (s, 1H), 12.23 (s, 1H) ; 13C-NMR (100MHz, DMSO): δ 37.17, 46.18, 52.24, 52.79, 55.05, 59.68, 98.10, 109.94, 117.75, 121.96, 124.29, 124.52, 128.06, 128.90, 129.40, 129.92, 130.91, 132.50, 136.72, 140.66, 158.81, 166.84, 169.04 and 170.66; MS: m/z 540 [M]+1.
Step-5: Preparation of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-lyl)acetamide) phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate ethane sulfonate salt:
Suspension of (Z)-methyl-3-(((4-(N-methyl-2-(4-methylpiperazin-l-yl)acetamide)phenyl) amino)(phenyl)methylene)-2-oxoindoline-6-carboxylate (36 gm, 0.066 mol) in methanol (237 ml) and water (2.88 ml)was heated to 60-65°C and aq. ethane sulfonic acid was added to the reaction mixture. The resulting solution was cooled to 50°C, seeds diluted with isopropanol (237 ml) was added. The reaction mixture was cooled at 0°C for lhr. Filtered the precipitate, washed with mixture of methanol and isopropanol (50 ml), dried to obtain crude Nintedanib monoethane sulfonate (36.6 gm) and crystallized from methanol (5 Vol) to
obtained pure Nintedanib monoethane sulfonate salt as yellow crystals (33 gm) (80%) (HPLC purity >99%).
DSC: 298°C; IR (KBr, cm-1): 3321, 3273, 1710, 1652, 1615, 1515, 1435, 1378, 1289, 1209, 1161, 1087; 1H-NMR (400 MHz, DMSO): δ 1.08 (t, 3H, J = 7.31 Hz), 2.41-2.47 (q, 2H), 2.50-3.16 (broad m, 13H), 3.37 (s, 3H), 3.76 (s, 3H), 5.82 (d, 1H, J = 7.88Hz), 6.87 (d, 2H, J = 7.36 Hz), 7.14-7.20 (m, 3H), 7.49 (s, 1H), 7.49 (d, 2H, J = 6.68 Hz), 7.56-7.63 (m, 3H), 9.45 (s, 1H), 10.99 (s, 1H), 12.25 (s, 1H), 13C-NMR (100 MHz, DMSO): δ 37.15, 42.79, 45.65, 49.40, 52.26, 53.10, 58.04, 98.25, 110.01, 117.78, 121.97, 124.32, 124.59, 128.27, 128.90, 129.36, 130.00, 131.00, 132.52, 136.79, 137.93, 140.00, 158.66, 166.85, 168.47 and 170.65; MS: m/z 540[M]+1.
Example 2: Process for the preparation of Polymorph Form S of Nintedanib monoethanesulf onate :
Crude Nintedanib monoethane sulfonate was dissolved in methanol and heated to 60-64°C for about 15 min. After completion of the reaction cooled to room temperature for about lhr. Filtered the precipitate, washed with mixture of methanol (20ml) and isopropanol (30 ml) and dried to obtain pure crystalline solid (28 gm) (yield: 93.3%) with HPLC purity 99.72% and individual impurities 0.09%, 0.02% and 0.04%.
SUVEN, Chief executive and chairman Venkat Jasti



//////////WO2016178064, POLYMORPH, NINTEDANIB ETHANESULPHONATE, PROCESSES, INTERMEDIATES, suven, new patent
LUPIN LIMITED, WO 2016181313, NEW PATENT, SOFOSBUVIR
![]()
WO2016181313, A PROCESS FOR THE PREPARATION OF SOFOSBUVIR INTERMEDIATES & ITS POLYMORPH
| LUPIN LIMITED [IN/IN]; Kalpataru Inspire 3rd Floor, Off Western Express Highway Santacruz (East) Mumbai 400 055 (IN) |
SINGH, Girij, Pal; (IN).
SRIVASTAVA, Dhananjai; (IN).
MEHARE, Kishor, Gulabrao; (IN).
MALIK, Vineet; (IN).
DEOKAR, Sharad, Chandrabhan; (IN).
DANGE, Abhijeet, Avinash; (IN)


SUCCESS QUOTIENT: Lupin chairman DB Gupta (sitting) with managing director Kamal K Sharma (centre), directors Vinita Gupta (right) and Nilesh Gupta.
The present invention provides a novel process for preparation N-[(2,3,4,5,6- Pentafluorophenoxy)phenoxyphosphinyl]-L-alanine 1-methylethyl ester (formula 2) and resolving the formula 2 in the presence base to form N-[(S)-(2,3,4,5,6- Pentafluorophenoxy)phenoxyphosphinyl]-L-alanine 1-methylethyl ester (formula 2′).
Sofosbuvir is chemically named as (S)-isopropyl 2-((S)-(((2R,3R,4R,5R)-5-(2,4- dioxo3,4-dihydropyrimidin-l(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran- 2yl)methoxy)-(phenoxy)phosphorylamino)propanoate and is represented by the following chemical structure:

Formula 1
PCT publications WO2011123645 and WO2010135569 describes process for preparation of compound of formula 2′ by reacting isopropyl (chloro(phenoxy)phosphoryl)-L-alaninate and pentaflurophenol in the presence of base.

Formula 2′
Example-1:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium hydride
10.2g of sodium hydride was dissolved in 100 ml anhydrous THF. This solution was slowly added to a solution of pentafluorophenol (50g) in THF (100ml), Reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained solid was dried under vacuum at 45-50°C (yield=55g, confirmed by IR)
Example-2:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium methoxide
2,3,4,5, 6-pentafluorophenol (lOg) was dissolved in methanol (100ml), solution was cooled to 5-10°C. To this was added a solution of sodium methoxide in methanol. The reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained residue was striped with toluene. Obtained solid was dried under vacuum at 45-50°C (yield=l lg)
Example 3:
Preparation of sodium 2,3,4,5,6-pentaflurophenolate using sodium hydroxide
2,3,4,5, 6-pentafluorophenol (lOOg) dissolved in methanol (—ml), solution was cooled to 5-10°C. To this was added a solution of sodium hydroxide (— g) in methanol. The reaction mass was stirred for 60-120 min at 25-30°C. Reaction mass was distilled under reduced pressure, obtained residue was striped with dichloromethane. Obtained solid was dried under vacuum at 45-50°C (yield=— g)
Example 4:
Preparation of (2S)-isopropyl-2-((chloro(phenoxy)posphoryl)amino)propanoate:
phenyl phosphodichloridate (30.6g) was dissolved in dichloromethane , to this was added a solution of 1-alanine isopropyl ester free base (19.16g) in dichloromethane at-60°C under nitrogen. Solution of triethylamine (20.7ml) was added to above reaction mass. Reaction mass was stirredat -60°C for 30 min and then temperature was raised to 25 °C. Reaction mass was stirred at 20-25 °C for 60 min & filtered and washed with dichloromethane. Clear filtrate was distilled under reduced pressure obtained residue was stirred with diisopropyl ether & filtered. Clear filtrate was distilled under reduced pressure to get (2S)-isopropyl-2-((chloro(phenoxy)posphoryl)amino)propanoate compound.
Example 5:
Preparation of isopropyl ((perfluorophenoxy)(phenoxy)phosphoryl)-L-alaninate (formula 2):

(Formula 2)
Obtained (2S)-isopropyl-2-((chloro(phenoxy)phosphoryl)amino)propanoate (1.2 mol eq.) was dissolved in dichloromethane and cooled to 0-5°C under nitrogen atmosphere. To this was added solution of sodium 2,3,4,5,6-pentaflurophemolate (1 mol eq.) in tetrahydrofuran . Temperature of reaction mass was raised to 25°C and reaction mass was stirred for 3 hrs. After completion of reaction, reaction mass was distilled under reduced pressure & obtained residue was dissolved I ethyl acetate. Ethyl acetate layer was washed with water, dried over sodium sulfate & distilled off under reduced pressure. Diisopropyl ether was added to obtained residue and stirred for 60 min at 25 °C, obtained mass was filtered & washed with diisopropyl ether. Solid product was dried under vacuum at 40-45 °C .(yield=20g, enantiomer purity=93.45%)
Example 6:
Preparation of (S)-isopropyl 2-(((S)- (perfluorophenoxy)phenoxy)phosphoyl)amino)propanoate (Formula 2′):

Formula 2′
(2S)-isopropyl-2-((chloro(phenoxy)phosphoryl)amino)propanoate (1.2 mol eq.) was dissolved in tetrahydrofuran (3.5 volumes). The reaction mass was cooled to -10°C. Solution of sodium salt of pentafluorophenol (1 mol eq.) in tetrahydrofuran (3.5 volumes) was added dropwise to the reaction mass at -10°C. After completion of the reaction solvent was distilled off. Ethyl acetate and water were added to the reaction mass. Reaction mass was stirred, ethyl acetate layer was separated and washed with sodium bicarbonate solution and brine. Ethyl acetate layer was concentrated under reduced pressure. Reaction mass was stripped with n-hepatane to get crude product. Crude product was dissolved in Methyl tert-butyl ether and n-heptane (1 : 1 ratio). The pH of reaction mass was adjusted to pH 8 by using triethylamine. Reaction mass was stirred overnight. Solid mass was filtered and washed with a mixture of methyl tertiary-butyl ether: n-heptane (1 : 1). The obtained product was dissolved in ethyl-acetate and washed with water and 20% brine solution. Ethyl acetate layer was separated; solvent was distilled off under reduced pressure. Reaction mass was stripped with diisopropyl ether. Di-isopropyl ether was added to the reaction mass. Reaction mass was stirred at 45-50°C. Reaction mass was cooled to 5-10°C and stirred. The titled compound was isolated by filtration and washed with di-isopropyl ether. The titled compound was dried under reduced pressure at 40°C. Yield 66.81%.

Vinita Gupta, CEO, Lupin Pharmaceuticals Inc

Desh Bhandu Gupta- Founder and chairman of Lupin Limited
////////////LUPIN LIMITED, WO 2016181313, NEW PATENT, SOFOSBUVIR
