
Home » PATENT (Page 7)
Category Archives: PATENT
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Regorafenib, SHILPA MEDICARE LIMITED, New patent, WO 2016005874
![]()
WO2016005874, PROCESS FOR THE PREPARATION OF REGORAFENIB AND ITS CRYSTALLINE FORMS
SHILPA MEDICARE LIMITED [IN/IN]; 10/80,Second Floor,Rajendra Gunj, Raichur, ರಾಯಚೂರು , karnataka 584102 (IN)
RAMPALLI, Sriram; (IN).
UPALLA, Lav Kumar; (IN).
RAMACHANDRULA, Krishna Kumar; (IN).
PUROHIT, Prashant; (IN).
AKSHAY KANT, Chaturvedi; (IN)
The present invention relates to a process for the preparation of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methylpyridine-2- carboxamide or Regorafenib (I): Formula (I). The present invention further relates to a process for the purification of 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2- carboxamide or Regorafenib (I) to provide highly pure material. The present invention further relates to a process for the preparation stable crystalline material of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]- N-methyl pyridine-2-carboxamide or Regorafenib (I) useful in the preparation of pharmaceutical compositions for the treatment of cancer.
4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide or Regorafenib is low molecular weight, orally available, inhibitor of multiple protein kinases, including kinases involved in tumour angiogenesis (VEGFR1, -2, -3, TIE2), oncogenesis (KIT, RET, RAF-1, BRAF, BRAFV600E), and the tumour microenvironment (PDGFR, FGFR). In preclinical studies regorafenib has demonstrated antitumour activity in a broad spectrum of tumour models including colorectal tumour models which is mediated both by its antiangiogenic and antiproliferative effects. Major human metabolites (M-2 and M-5) exhibited similar efficacies compared to Regorafenib both in vitro and in vivo models.
Regorafenib was approved by USFDA in 2012 and is marketed under the brand name Stivarga®, is an important chemotherapeutic agent useful for the treatment of adult patients with metastatic colorectal cancer (CRC) who have been previously treated with, or are not considered candidates for, available therapies. These include fluoropyrimidine-based chemotherapy, an anti-VEGF therapy and an anti-EGFR therapy.
Regorafenib is chemically known as 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide (I). Regorafenib is a white to slightly pink or slightly brownish solid substance with the empirical formula C2iHi5ClF4N403 and a molecular weight of 482.82. Regorafenib is practically insoluble in water, dilute alkaline solution, dilute acid solution, n-heptane, glycerine and toluene. It is slightly soluble in acetonitrile, dichloromethane, propylene glycol, methanol, 2-propanol, ethanol and ethyl acetate. It is sparingly soluble in acetone and soluble in PEG 400 (macrogol). Regorafenib is not hygroscopic.
Regorafenib is generically disclosed in US 7351834, and specifically disclosed in US 8637553. US ‘553 disclose a process for the preparation of Regorafenib starting from 3-fluoro-4-nitrophenol. The process is as demonstrated below:

The present inventors has repeated the above process and found the following disadvantages:
Unwanted reactions are observed during the formation of Regorafenib, due to the involvement of prolonged time in process.
> Incomplete reactions were observed with excessive impurity formations due to incomplete conversion.
Removal of impurities from final product
US 2010173953 disclose Regorafenib monohydrate and crystalline Form I of Regorafenib. This patent application further discloses that crystalline Form I of Regorafenib stated in this application is obtained as per the process disclosed in WO 2005009961 A2 (Equivalent to US ‘553). The compound obtained was having a melting point of 186-206° C.
This patent publication discloses a process for the preparation of Regorafenib monohydrate comprises dissolving Regorafenib Form I obtained as per WO ‘961 in acetone
and the solution is filtered, followed by addition of water until precipitation, which was filtered and dried at room temperature
US 2010/0113533 discloses crystalline Form II of Regorafenib, comprises dissolving Regorafenib Form I obtained as per WO ‘961 in ethyl acetate, the suspension was heated to 40-45°C, addition of isocyanate solution (isocyanate in ethyl acetate) and is cooled to room temperature to yield the crystals, which was filtered, washed with ethyl acetate and dried at room temperature.
US 2010/0063112 discloses Form III of Regorafenib, process comprises of heating
Regorafenib monohydrate at 100°C or 60 min, and further 15 min at 110°C, followed by cooling to room temperature.
As polymorphism has been given importance in the recent literatures owing to its relevance to the drugs having oral dosage forms due to its apparent relation to dose preparation/suitability in composition steps/ bioavailability and other pharmaceutical profiles, stable polymorphic form of a drug has often remained the clear choice in compositions due to various reasons of handling, mixing and further processing including bioavailability and stability.
Exploring new process for these stable polymorphic forms which are amenable to scale up for pharmaceutically active / useful compounds such as 4-[4-({[4-chloro-3-(trifluoro methyl)phenyl]carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2 -carboxamide or Regorafenib may thus provide an opportunity to improve the drug performance characteristics of such products.
Hence, inventors of the present application report a process for the preparation of a stable and usable form of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluoi phenoxy]-N-methylpyridine-2-carboxamide or Regorafenib, which may be industrially amenable and usable for preparing the corresponding pharmaceutical compositions. The present invention provides an improved process for the preparation of 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fiuorophenoxy]-N-methylpyridine-2-carboxamide or Regorafenib crystalline forms specifically for crystalline polymorphic forms Form I and Form III. Crystalline polymorphic forms of 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2 -carboxamide or Regorafenib obtained by the process of the present invention is non-hygroscopic and chemically stable and has good dissolution properties.
The process related impurities that appear in the impurity profile of the Regorafenib may be substantially removed by the process of the present invention resulting in the formation of highly pure material. The process of the present invention is as summarized below:

Example 1
Preparation of 4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide
4-Amino-3-fiuorophenol (l lg, 0.08 moles) and of 4-Chloro-N-methyl-2-pyridinecarboxamide (8.85 g, 0.05 moles) was added to a reaction flask containing N, N-dimethylacetamide (55 ml) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 110-115°C and then potassium tert-butoxide in tetrahydrofuran (60 ml, 0.06 moles) was added slowly over a period of 3 to 4hours. Distill off solvent at same temperature, cooled the reaction mass to 25-30°Cand water(110 ml) was added slowly over a period of 15min. and cooled the reaction mass to 0-5°C . Adjust the pH of the reaction mass in between 7 and 7.5 by using 10% aqueous hydrochloric acid (~7 ml). Stir the reaction mass for 30min at the same temperature. Filter the product, washed with water (22 mL) and Dried at 50-55 °C for 12hrs. The obtained crude material was added to the flask containing Ethyl acetate (55 mL).The reaction mass was heated to reflux to get a clear solution and stirred for 15min at reflux. Cooled to 0-5°C, stir for 2hrs at the same temperature. Filter the product, washed with Toluene (9 mL) and dried at 50-55°C for 3-5hrs.
Above recrystallized material was added to the reaction flask containing methylene dichloride (270 mL) at 25-30°C and stirred for 10-15 min. Activated carbon (1 g) and silica gel (4.4 g) was added to the reaction mass and stir for lh at the same temperature. Filter the reaction mass through hyflow bed and wash with methylene dichloride (18 mL).Distill off solvent still~l-2 volumes of methylene dichloride remains in the flask and then cooled to 25-30°C. Toluene (20 mL) was added and stirred for 30min at the same temperature. Filtered the product, washed with Toluene (9 mL) and dried at 50-55°C for 12h.
Yield: 9 gm
Chromatographic Purity (By HPLC): 98%
Example 2
Preparation of Regorafenib
4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide (4g, 0.01 moles) was added in to a reaction flask containing acetone (20 ml) at 25-30°C and stirred for 15 minutes. 4-chloro-3-trifluoromethylisocyanate (6.1g, 0.02 moles) was added slowly over a period of 5 to 10 minutes and stirred the reaction mixture 3 to 4 hours. Toluene (20 n L) was added to the reaction mass and stirred for 30 min at 25-30°C.The obtained reaction mass was filtered and washed with toluene (8 mL). Dried the material still constant weight appears to yield title product a crystalline material.
Yield: 5.5 gm
Chromatographic Purity (By HPLC): 97%
Example 3
Purification of Regorafenib using acetone and toluene mixture
4- [4-( { [4-chloro-3 -(trifluoromethyl)phenyl] carbamoyl } amino)-3 -fluorophenoxy] -N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (2 mL) and toluene (3 mL) at 25-30°C and stirred for 15 minutes.
The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes.
Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and suck dried for 15 min, followed by drying at 50-55°C for 10-12h to yield
Pure 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methyl pyridine-2-carboxamide (I) or Regorafenib.
Yield: 0.88gm
Chromatographic Purity (By HPLC): 99.3 %
Example 4
Purification of Regorafenib using acetone
4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3 -fluorophenoxy] -N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (5 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 0-5°C and stirred for 1 hour. Filter the material, washed with acetone (1 mL) and suck dried for 15 min. The obtained wet cake was added in to the reaction flask containing acetone (5 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50- 55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 0-5°C and stirred for 1 hour. Filter the material, washed with acetone (1 mL) and dried at 60-65°C for 12 h to yield Pure 4-[4-({[4-chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methyl pyridine -2-carboxamide (I) or Regorafenib.
Yield: 0.7 gm
Chromatographic Purity (By HPLC): 99.77%
Example 5
Double – Purification of Regorafenib using acetone and toluene mixture
4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] Carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide (I) or Regorafenib (1 g) was added slowly in to the reaction flask containing acetone (2 mL) and toluene (3 mL) at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and suck dried for 15 min. The obtained wet cake was added in to the reaction flask containing acetone (2 mL) and toluene (3 mL) mixture at 25-30°C and stirred for 15 minutes. The reaction mixture was heated to 50-55°C and stirred the reaction mixture for 30 minutes. Cooled the reaction mass to 25-30°C and stirred for 1 hour. Filter the material, washed with toluene (2 mL) and dry at 60-65°C for 12h.
Yield: 0.80gm
Chromatographic Purity (By HPLC): 99.79 %
Moisture content: 0.09%
Impurity-A: 0.03%
Impurity-B: Not detected
Impurity-C: 0.02%
Example 6
Preparation of Regorafenib Form I
4-(4-amino-3-fluorophenoxy) pyridine-2-carboxylic acid methyl amide (1.3 g, 0.004 moles) was added in to a reaction flask containing acetone (13 mL) at 25-30°C and stirred for 15 minutes.4-chloro-3-trifluoromethylisocyanate (6.6 g, 0.006 moles) wasadded slowly over a period of 15 to 20 minutes and stirred the reaction mixture 3 to 4 hours. The obtained reaction mass was filtered and washed with acetone. Dried the material still constant weight appears to yield title product a crystalline material.
Yield: 1.9 g
Chromatographic Purity (By HPLC): 98.4 %
XRPD was found to resemble similar to Fig-1.
![]()
Omprakash Inani – Chairman, Vishnukant C Bhutada – Managing Director, Namrata Bhutada – Non Executive Director, Ajeet Singh Karan – Independent Director, Carlton Felix Pereira – Independent Director, Pramod Kasat – Independent Director, Rajender Sunki Reddy – Independent Director, N P S Shinh – Independent Director,
 Mr. Omprakash Inani |
Mr. Omprakash Inani – CHAIRMAN
Mr. Omprakash Inani has more than 30 years of Business experience. He monitors business and functional aspects of the Company along with the operations of all the plants. Additionally, he is member of Audit and Remuneration committee of Shilpa Medicare Group of Companies. Currently he is also a council Member in “Academy of Medical Education, Dental College & V.L. College of Pharmacy”, “Taranath Shikshana Samsthe, Raichur” and a trustee in “Akhil Bhartiya Maheshwari Education Trust, Pune”. Mr. Omprakash Inani is also Managing Committee Member of “Karnataka State Cotton Assn., Hubli”. |
|
|
|
||
 Mr. Vishnukant C. Bhutada Mr. Vishnukant C. Bhutada |
Mr. Vishnukant C. Bhutada – MANAGING DIRECTOR
Mr. Vishnukant has vast and diverse Business experience of API and Intermediates and presently leads the core Business and functional teams which accelerate growth and performance by Innovating for Affordable solutions at Shilpa Medicare Group of Companies. He is the key decision maker with the teams for Shilpa Group for successful API and Generics formulation strategies. His untiring efforts have led the company to a leadership position in the Indian pharmaceutical domain and helped create a prominent presence for Oncology APIs globally. For his efforts on APIs Business, Mr. Vishnukant was awarded “Best Entrepreneur Award” by Late Dr Shankar Dayal Sharma – President of India in 1995. Subsequently, various state honours were conferred upon him -like -“Best Entrepreneur” from Karnataka State Govt. in 1996; “Excellence in Exports” from Vishweshwarayya Industrial Trade Centre, Bangalore 1996; and Export Excellence Award-2006” by FKCCI, Bangalore. Success has never stopped coming his way- as he was awarded “First runner up” at the Emerging India Awards London 2008 by CNBC TV18. Recently, his efforts in the Shilpa Group for environment sustainability, has led to “Best National Energy Conservation Award in Drugs & Pharmaceutical Sector for the year 2012” by Hon’ble President of India, Dr. Pranab Mukherjee. |
|
|
|
||
 Dr. Vimal Kumar Shrawat Dr. Vimal Kumar Shrawat |
Dr. Vimal Kumar Shrawat – CHIEF OPERATING OFFICER
Dr. Shrawat by qualification holds degrees of M.Sc (Organic Chemistry), Ph.D. (from Delhi University) and joined Shilpa Medicare in 2009. He has vast experience of more than 25 years of working in large pharma industries like Ranbaxy/ Dabur Pharma- presently known as Fresenius Kabi Oncology Ltd., spanning across activities of R&D, Pilot and Plant Productions, QA/QC, Administration, CRAMS, Project management etc. Presently, Dr. Shrawat is spearheading the entire Operations/ Control of Shilpa Medicare. His vision of team work and time bound approach always guides and motivates teams at all operational sites. His keen interest and consistent efforts for R&D has led him to become one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Pramod Kumar |
Dr. Pramod Kumar – MANAGING DIRECTOR(LOBA FEINCHEMIE GMBH AUSTRIA), SENIOR VICE-PRESIDENT (SHILPA MEDICARE LTD)
Dr. Pramod Kumar, who by qualification holds degrees of M.Pharm, Ph.D (Pharmaceutical chemistry) and a PGDBA, joined Shilpa Medicare in 1989. Since 2009 he is Managing Director of Loba FeinchemieGmBH, Austria and driving all R&D driven commercial processes. Dr. Pramod Kumar has more than 25 years of experience in Pharmaceutical industry, spanning across activities of production, QA/QC, administration, import/export, CRAMS etc. His efforts in CRAMS have led to the formation of Joint venture company RAICHEM MEDICARE Pvt LTD with Italian companies ICE SPA / P.C.A SPA. |
|
|
|
||
 Mr. Prashant Purohit |
Mr. Prashant Purohit – VICE-PRESIDENT-CRD
Mr. Prashant Purohit by qualification holds degrees of, M.Sc.(Organic Chemistry) and Diploma in Business Management and joined Shilpa Medicare in 1996. He is presently heading Chemical R&D wings of Shilpa Medicare Group. He has vast experience of handling CRAMS and Generics APIs R&D. His vast experience of nearly 35 years in R & D and production in Pharmaceutical Industry has consistently enriched the portfolio of Shilpa Medicare Group of Companies. He is one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Akshay Kant Chaturvedi |
Dr. Akshay Kant Chaturvedi – HEAD- CORPORATE IPM & LEGAL AFFAIRS
Dr. Akshay Kant by qualification holds degrees of M.Sc, Organic Chemistry (Univ. Gold Medalist), Ph.D. (Medicinal Chem), LL.B., M.B.A. and joined Shilpa Medicare in Jun 2012. Presently, Dr. Akshay is spearheading the entire IP portfolio management/ Legal Affairs of Contractual Business of Shilpa Medicare Group. His vision of innovative and creative thinking, team work and time bound approach always guide and motivate teams at all locations.His keen interest and consistent efforts for R&D has led him to become one of key contributor in large number of Patent/applications of Shilpa Medicare. |
|
|
|
||
 Dr. Seshachalam U. |
Dr. Seshachalam U. -ASSOCIATE VICEPRESIDENT- QUALITY AND RA
Dr. Seshachalam by qualification holds M.Sc (Chemistry) and Ph.D. (Chemistry) and joined Shilpa Medicare in 2008. He is presently heading Regulatory Affairs wings of Shilpa Medicare Group of Companies. He has vast experience of handling regulatory affairs related to Generics APIs. Being instrumental in Shilpa Medicare’s efforts to achieve recognition of different authorities, his key contribution in successful inspection and audit by various regulatory authorities is one of the core strength to the organization’s aims and objectives. |
|
|
|
||
 Mr. Sharath Reddy |
Mr. Sharath Reddy – VICE-PRESIDENT PROJECTS & OPERATIONS
Mr. Sharath Reddy by qualification holds M.Pharm from BITS Pilani and has overall experience of about 22 years predominately in the field of pharmaceuticals new projects and operations. His expertise of Oncology specialized equipment and Utilities designing has boosted organizations confidence to takeover new endeavors of upcoming projects with faster pace of time. His efforts have led to successfully executing Energy Saving projects of Shilpa Medicare Group of Companies and registration of the project under Clean Development Mechanism with UNFCC (Under Kyoto Protocol). |
|
|
|
||
 Mr. R K Somani |
Mr. R K Somani – VICE-PRESIDENT FORMULATION -BUSINESS DEVELOPMENT
Mr. R. K. Somani is a professional Chartered Accountant and holds a Diploma in Central Excise.He has overall business experience of more than 21 years predominately in the field of pharmaceuticals. Mr. Somani is one of the key drivers of Formulation business besides handling various key Contract Businesses of advanced oncology/ Non-Oncology APIs. He is known for successfully building formulations portfolio and spearheading the Generic sales operation. |
|
Shilpa Medicare Limited
1st Floor, 10/80,
Rajendra Gunj,
RAICHUR ರಾಯಚೂರು – 584 102.
Karnataka, India.
Telephone: +91-8532-236494
Fax: +91-8532-235876
Email: info@vbshilpa.com
RAICHUR, ರಾಯಚೂರು Karnataka, India








Historical Stone Elephants in Malayabad, Raichur Taluk …

View of Raichur city and lake Aam Talab
///Regorafenib, SHILPA MEDICARE LIMITED, new patent, WO 2016005874, raichur, ರಾಯಚೂರು , karnataka, india
Lupin Ltd, Patent, Pitavastatin, WO2014203045
![]()
Lupin Ltd, Patent, Pitavastatin, WO2014203045
A NOVEL, GREEN AND COST EFFECTIVE PROCESS FOR SYNTHESIS OF TERT-BUTYL (3R,5S)-6-OXO-3,5-DIHYDROXY-3,5-O-ISOPROPYLIDENE-HEXANOATE
ROY, Bhairabnath; (IN).
SINGH, Girij, Pal; (IN).
LATHI, Piyush, Suresh; (IN).
AGRAWAL, Manoj, Kunjabihari; (IN).
MITRA, Rangan; (IN).
TRIVEDI, Anurag; (IN).
PISE, Vijay, Sadashiv; (IN).
RUPANWAR, Manoj; (IN)
The present invention describes an eco-friendly and cost effective process for the synthesis of teri-butyl (3R,5S)-6-oxo-3,5-dihydroxy-3,5-0-isopropylidene-hexanoate [I]
 PITAVASTATIN
PITAVASTATIN
![]()
TEXT
tert-b tyl (3R,5S)-6-oxo-3,5-dihydroxy-3,5-0-isopropylidene-hexanoate [I] [CAS No. 124752-23-4] is key intermediate for the preparation of statins such as Atorvastatin (Tetrahedron 63, 2007, 8124 -8134), Cerivastatin (Journal of Labeled Compounds and Radiopharmaceuticals, 49, 2006 311-319), Fluvastatin [WO2007125547; US 4739073], Pitavastatin [WO2007/132482; US2012/22102 Al, WO2010/77062 A2; WO2012/63254 Al ; EP 304063; Tetrahedron Letters, 1993, 34, 513 – 516; Bulletin of the Chemical Society of Japan, 1995, 68, 364 – 372] and Rosuvastatin [WO2007/125547 A2; WO2011/132172 Al ; EP 521471]. Statins are used for treatment of hypercholesterolemia, which reduces the LDL cholesterol levels by inhibiting activity of HMG-CoA reductase enzyme, which is involved in the synthesis of cholesterol in liver.

[I]
Compound [I] is generally obtained by various methods of oxidation of teri-butyl 2- ((4R,65)-6-(hydroxymethyl)-2,2-dimethyl-l,3-dioxan-4-yl)acetate [compound II] and are discussed in details hereinafter. In addition, various methods for synthesis of compound [II] are also elaborated below.

[II]
[II]
A) tert-butyl2-((4«,6.S)-6-(hydroxymethyl)-2,2-dimethyl-l,3-dioxan-4-yl)acetate
[compound II]
US patent Number 5278313 describes a process for synthesis of compound [II]
(Schemel). In the said process, (5)-methyl 4-chloro-3-hydroxybutanoate has been obtained in only 70% yield through whole cell enzymatic reduction of methyl 4-chloro-3- oxobutanoate, which has a necessity of special equipment such as fermenters as well as other microbial facilities such as sterile area, autoclaves, incubator for growing seed culture, etc.
(S)-mefhyl 4-chloro-3-hydroxybutanoate upon reaction with teri-butyl acetate in presence of LiHMDS or LDA at -78°C, yielded (S)-ieri-butyl 6-chloro-5-hydroxy-3- oxohexanoate, which was further transformed to corresponding diol through syn selective reduction in presence of methoxydiethyl borane/sodium borohydride at -78°C. The diol thus obtained was converted to compound [II] .
The overall yield for this process is low and required special equipment such as fermenters, etc and in addition to that, this process is not cost effective due to use of costly reagent such as methoxydiethyl borane.
Moreover, methoxydiethylborane is highly pyrophoric (Encyclopedia for organic synthesis, editor in chief L. Paquette; 2, 5304; Published by John and Wiley Sons;
Organic Process Research & Development 2006, 10, 1292-1295) and hence safety is a major concern.

Scheme 1
EP 1282719 B l (PCT application WO 01/85975 Al ) discloses a process for synthesis of compound ( R, 5S)-tert-bv y\ 3,5,6-trihydroxyhexanoate from (S)-tert-b tyl-5,6-dihydroxy-3-oxohexanoate through a) asymmetric hydrogenation in presence of a chiral catalyst e.g. di-mu-chlorobis-[(p-cymene)chlororuthenium(II)] along with an auxiliary such as (IS, 2S)-(+)-N- (4-toluenesulfonyl)-l ,2-diphenylethylenediamine as ligand, which gave desired product only in 70% diastereomeric excess (de); b) Whole cell enzymatic reduction of (S)-tert- butyl 5,6-dihydroxy-3-oxohexanoate to obtain compound (3R, 5S)-tert-bv y\ 3,5,6-trihydroxyhexanoate in 99% de (80% yield).
It is needless to mention that it has necessity of fermenter and other microbiological equipment (Scheme 2).
Moreover, conversion of (2>R,5S)-tert-bv y\ 6-acetoxy-3,5-dihydroxyhexanoate to tert-bv yl 2-((4R,65)-6-(acetoxymethyl)-2,2-dimethyl-l ,3-dioxan-4-yl)acetate was accomplished in only 25% yield and also required the flash chromatography for isolation of desired product.
Thus, overall yield for this process is poor and process is not operation friendly especially at large scale hence cannot be considered feasible for commercial manufacturing.

Scheme 2
EP1317440 Bl (PCT Application WO 02/06266 Al) has disclosed the process for synthesis of compound [II] from 6-chloro-2,4,6-trideoxy-D-erythro-hexose (Scheme 3) .
In the said patent application 6-chloro-2,4,6-trideoxy-D-erythro-hexose was converted to (4R, 65)-4-hydroxy-6-chloromethyl-tetrahydropyran-2one with excess of bromine in presence of potassium bicarbonate, which liberates environmentally undesired gas i.e. carbon dioxide.
Moreover, starting material i.e. 6-chloro-2,4,6-trideoxy-D-erythro-hexose is not commercially available and conversion efficiency of starting material at large scale towards (4R, 65)-4-hydroxy-6-chloromethyl-tetrahydropyran-2-one is only 67%.

Scheme 3
US Patent No. 6689591 B2 has demonstrated the whole cell enzymatic reduction of teri-butyl 6-chloro-3,5-dioxohexanoate to compound [II] (Scheme 4).
In the said process, whole cell enzymatic reduction is not specific; yield for desired product is only 34% and other partially reduced products are also obtained.
Hence, further purification is required for obtaining the desired compound. Thus, this process is not suitable for commercial scale.

Scheme 4
Tatsuya et al (Tetrahedron Letters; 34, 1993,513 – 516) has reported synthesis of compound [I] from derivative of L-tartatric acid (Scheme 5).
In the said process, tartaric acid di-i‘sopropyl ester is doubly protected by tert-butyldimethylsilyl group, which was reacted with dianion of teri-butyl acetoacetate to give β, δ-diketo ester compound.
β,δ-diketo ester was reacted with 2 equivalent of diisobutylaluminium hydride (which is a pyrophoric reagent) to afford -hydroxy,8-keto ester in only 60% yield.
This process is not industrially viable as overall yield is very low and also because of use of costly and pyrophoric reagents/chemicals.

Scheme 5
US7205418 (PCT application WO03/053950A1) has described the process for synthesis of compound [II] from (S)-ieri-butyl-3,4-epoxybutanoate (Scheme 6).
The overall yield for this process is very low and moreover, it required the diastereomeric separation of teri-butyl 2-(6-(iodomethyl)-2-oxo-l,3-dioxan-4-yl)acetate by flash chromatography.
Since overall requirement of title compound is very high, any operation involving flash chromatography will tend to render the process commercially unviable.

Scheme 6
Fengali et al (Tetrahedron: Asymmetry 17; 2006; 2907-2913) has reported the process for synthesis of compound [II] from racemic epichlorohydrin (Scheme 7).
In this process, racemic epichlorohydrin was converted to corresponding nitrile intermediate through reaction with sodium cyanide; nitrile intermediate thus obtained was further resolved through lipase catalyzed stereo-selective esterification to obtain (5)-4-(benzyloxy)-3-hydroxybutanenitrile and (R)-l-(benzyloxy)-3-cyanopropan-2-yl acetate;
separation of desired product i.e. (S)-4-(benzyloxy)-3-hydroxybutanenitrile having 98% de (40% yield) was done by column chromatography.
Needless to mention a commodity chemical like compound [I] cannot be manufactured by such a laboratory method, which involved number of steps.

Scheme 7
Bode et al (Organic letters, 2002, 4, 619-621) has reported diastereomer- specific hydrolysis of 1,3-diol-acetonides (Scheme 8).
In this publication, duration of the reaction for diastereomer- specific hydrolysis of 1,3, diol-acetonides is reported to be 4 h, however, in our hand it was observed that hardly any reaction took place in 4 h, which made it non-reproducible.
In addition to that, separation of desired product is achieved by flash chromatography and it is needless to mention that any process which involved flash chromatography would render the process to be commercially unviable.
Hence, additional innovation needs to be put in for making the process industrially viable.

Scheme 8
CN 101613341A has reported the process for synthesis of compound [II] (Scheme
9).
In the same patent application tert-b tyl (S)-6-chloro-5-hydroxy-3-oxohexanoate was synthesized through Blaise condensation of (5)-4-chloro-3-hydorxy-butanenitrile with zinc enolate of tert butyl bromo acetate.
In the literature, synthesis of tert-bv yl (S)-6-chloro-5-hydroxy-3-oxohexanoate was reported through Blaise condensation of silyl protected (5)-4-chloro-3-(trimethylsilyl)oxy-butanenitrile with zinc enolate of tert butyl bromo acetate, in good yield (Synthesis 2004, 16, 2629-2632). Thus, protection of hydroxy group in (5)-4-chloro-3-hydorxy-butanenitrile is imperative.
In the said Chinese patent application, in claim 7, it was mentioned that solvent used for conversion of tert-bv yl (5)-6-chloro-5-hydroxy-3-oxohexanoate to ( R,5S)-tert-butyl 6-chloro-3,5-dihydroxyhexanoate is anyone or mixture of more than one from tetrahydrofuran, ether, methanol, ethanol, n-propanol, /so-propanol and ethylene glycol.
However, in enablement the only example using mixture of solvent was that of THF-methanol (Experimental section, Example 4: The preparation of (R,5)-6-chloro-3,5- dihydroxyhexanoate) and same outcome was expected in other individual or mixture of solvents.
Claim 8 of CN 101613341A mentioned that reduction was carried out by any one or mixture of more than one reducing agents such as sodium borohydride, potassium borohydride, lithium aluminum hydride, diethylmethoxy borane, triethyl borane and tributyl borane.
It implies that either any one of the reducing agents or a mixture of the same can be employed. From reaction mechanism it is very much clear that diethylmethoxy borane, triethyl borane and tributyl borane form the six membered complex between optically active hydroxyl and carbonyl group, which gets reduced by sodium borohydride, signifying that individually diethylmethoxy borane, triethyl borane and tributyl borane are not reducing agents
Moreover, in claims 12 and 13 (Experimental section, Example 4: The preparation of (R,S)-6-chloro-3,5-dihydroxyhexanoate), it is mentioned that reduction should be carried out in temperature range -80 °C to -60 °C, implying that reaction would not work beyond this temperature range i.e. it would work in the temperature window of -80 °C to -60 °C only.
Summarizing, the teachings of the application are not workable.

Scheme 9
Wolberg et al (Angewandte Chemie International Edition, 2000, 4306) has reported that diastereomeric excess for syn selective reduction using mixture of diethyl methoxy borane/sodium borohydride of compound [VI] gave 93% de for compound [VIII], which required further re-crystallization to obtain compound [VIII] in 99% de and 70% yield.
Thus, all the reported methods for stereo-selective hydride reduction of compound [VI] were achieved through mixture of trialkyl borane or diethyl methoxy borane & sodium borohydride in THF, at -78°C. As mentioned earlier, trialkyl borane or diethyl methoxy borane are pyrophoric in nature; in addition to that anhydrous THF is costly and moreover, reaction required large dilution.
Hence, there is need for developing efficient, environment friendly, cost effective and green process for stereo-selective reduction compound [VI].
B) The process of Oxidation of compound [II] to compound [I] has been discussed in following literature processes.
1) Swern oxidation (US4970313; Tetrahedron Letters, 1990, 2545
Synthetic Communications, 2003, 2275 – 2284).
2) Parrkh-Doering oxidation (J. Am. Chem. Soc, 1967, 89, 5505-5507)
3) TEMPO/NaOCl oxidization (EP2351762)
4) Trichloroisocyanuric acid/ TEMPO (CN 101747313A)
5) Oxidation of compound [II] to compound [I] through IBX [CN101475558A].
It would be evident that most of the reported methods are not “green” and
environmentally benign; none of the reported methods use molecular oxygen as oxidizing agent in presence of metal catalyst/co-catalyst.
Example 18: Process for synthesis of tert-butyl 2-((4R,6S)-6-formyl-2,2-dimethyl-l,3-dioxan-4-yl)acetate [I]

A reactor was charged with 1.1 g of copper (I) chloride and 10 mL of acetonitrile. 2-2′ Bipyridyl (156 mg) and TEMPO (156 mg) were added to the reactor under oxygen environment at 25°C. A solution of (6-Hydroxymethyl-2,2-dimethyl-[l,3]dioxan-4-yl)-acetic acid tert-butyl ester 2.6 g in 26 mL DCM was added dropwise over a period of 10 min into it. The reaction mass was stirred at 40°C and progress of reaction was monitored on GLC, which shows that 90% conversion for desired product.
Example 19: Process for synthesis of tert-butyl 2-((4R,6S)-6-formyl-2,2-dimethyl-l,3-dioxan-4-yl)acetate [I]
A reactor was charged with 1.1 g of copper (I) chloride and 10 mL of dichlorome thane. 2-2′ Bipyridyl (156 mg) and TEMPO (156 mg) were added to the reactor under oxygen environment at 25°C. A solution of (6-Hydroxymethyl-2,2-dimethyl-[l,3]dioxan-4-yl)-acetic acid tert-butyl ester 2.6 g in 26 mL DCM was added dropwise over a period of 10 min into it. The reaction mass was stirred at 40°C and progress of reaction was monitored on GLC, which shows that 90% conversion for desired product.
AUTHORS

///////
Lupin Ltd, New patent, Pitavastatin, WO 2016005919
Formula (1)
Lupin Ltd, New patent, Pitavastatin, WO 2016005919
MANE, Narendra, Dattatray; (IN).
NEHATE, Sagar, Purushottam; (IN).
GODBOLE, Himanshu, Madhav; (IN).
SINGH, Girij, Pal; (IN)
![]()
The present invention is directed to polymorphic forms of Pitavastatin sodium and processes for preparation of the same
Novel crystalline polymorphic forms (I and II) and an amorphous form of pitavastatin, useful for treating hyperlipidemia and mixed dyslipidemia.
Also claims a method for preparing the crystalline and amorphous forms of pitavastatin. In January 2016, Newport Premium™ reported that Lupin holds an active US DMF for pitavastatin calcium since July 2013.
Nissan Chemical Industries and licensee Kowa, with sub-licensees Sankyo, Eli Lilly, Esteve, JW Pharmaceutical, Recordati, Laboratorios Delta and Zydus-Cadila, have developed and launched pitavastatin.
WO2014203045, claiming a process for preparing an intermediate useful in the synthesis of statins (eg pitavastatin).
Pitavastatin is a cholesterol lowering agent of the class of HMG-CoA reductase inhibitor. The HMG-CoA reductase enzyme catalyzes the conversions of HMG- CoA to mevalonate. Inhibitors of HMG-CoA reductase are commonly referred to as “statins.” Statins are therapeutically effective drugs used for reducing low density lipoprotein (LDL) particle concentration in the blood stream of patients at risk for cardiovascular disease.
Pitavastatin is one of the synthetic statins which is chemically known as (3R, 5S, 6E)-7-[2-cyclopropyl-4-(4-fluorophenyl) quinoline-3-yl]-3, 5-dihydroxy-6- heptenoic acid represented by structural formula (1):
Formula (1)
Pitavastatin and its pharmaceutically acceptable salts are described in US 5,753,675 patent and US 5,856,336 patent, respectively.
Processes for the preparation of Pitavastatin are well documented in the literature. European patents, EP 0304063 and EP 1099694 and reports by Miyachi et al (Tetrahedron Letters
(1993) vol. 34, pages 8267-8270) and Takahashi et al (Bull. Chem. Soc. Japan (1995) Vol. 68, 2649-2656) describe processes for preparation of Pitavastatin.
US 5,872,130 patent discloses sodium salt of Pitavastatin. This patent, however, is silent about the solid state form of Pitavastatin Sodium.
It is generally known in the art that active pharmaceutical ingredients frequently do not exhibit the range of physical properties that makes them directly suitable for development. One of the approaches that is used to modify the characteristics of drug substances is to employ a salt form of the substance, since salts enable one to modify aqueous solubility, dissolution rate, solution pH, crystal form, hygroscopicity, chemical stability, melting point and even mechanical properties. The beneficial aspects of using salt forms of active pharmaceutical ingredients are well known and represent one of the means to increase the degree of solubility of otherwise intractable substances and to increase bioavailability.
Although the known salts of Pitavastatin like sodium, potassium, magnesium, calcium etc. and their polymorphic forms may address some of the deficiencies in terms of formulated product and its manufacturability. There remains a need for yet further improvement in these properties as well as improvements in other properties such as flowability, and solubility.
Polymorphism is a known phenomenon among pharmaceutical substances. It is commonly defined as the ability of any substance to exist in two or more crystalline phases that have a different arrangement and/or conformation of the molecules in the crystal lattice. Different polymorphic forms of the same pharmaceutically active moiety also differ in their physical properties such as melting point, solubility, chemical reactivity, etc. These properties may also appreciably influence pharmaceutical properties such as dissolution rate and bioavailability.
Further, the discovery of new polymorphic forms and solvates of an active pharmaceutical ingredient provides broader scope to a formulation scientist for formulation optimization, for example by providing a product with different properties, e.g., better processing or handling characteristics, improved dissolution profile, or improved shelf-life. For at least these reasons, there is a need for polymorphs of Pitavastatin salts such as Pitavastatin sodium.
New polymorphic forms and hydrates and/or solvates of a pharmaceutically acceptable salt of Pitavastatin can also provide an opportunity to improve the performance characteristics of a pharmaceutical product.
Therefore, there is a scope to prepare novel polymorphic forms of Pitavastatin sodium and hydrates and/or solvates.
Example-1: Preparation of Pitavastatin Sodium (Form-I)
A mixture of 40.0 gm Pitavastatin acid and 120 ml water was cooled to 15-20 °C temperature. Thereafter aqueous solution of sodium hydroxide (4.0 gm) in water (20 ml) was added to the reaction mixture. The reaction mixture was stirred for 30-45 min at 15-20 °C temperature. Ethyl acetate (80ml) was added into the reaction mixture at 15-20 °C temperature, stirred for 15-20 min and the layers were separated. The aqueous layer was filtered and acetonitrile (1200 ml) was gradually added to the aqueous layer under stirring till the precipitation was completed. The reaction mixture was cooled to 5-8 °C temperature and stirred for 2-3 hours at 5-8 °C temperature. The precipitated solid was filtered, washed with acetonitrile (40ml) and dried at 45-50 °C temperature under vacuum for 10-12 hours to afford the title compound (28.0 gm).
Yield (w/w): 0.70 (66.0%)
HPLC purity: 99.70 %
Example-2: Preparation of Pitavastatin Sodium (Form-II)
A mixture of 40.0 gm of Pitavastatin acid and 120 ml of water was cooled to 15-20°C temperature under stirring. Thereafter aqueous solution of sodium hydroxide (4.0 gm) in water (20 ml) was added to the reaction mixture. The reaction mixture was stirred for 30-45 min at 15-20 °C temperature. Ethyl acetate (80ml) was added to the reaction mixture at 15-20 °C temperature, stirred for 15-20 min and the layers were separated. The aqueous layer was filtered and acetonitrile (1200 ml) was gradually added to the aqueous layer under stirring till the precipitation was completed. The reaction mixture was cooled to 5-8 °C temperature and stirred for 2-3 hours at 5-8 °C temperature. The precipitated solid was filtered, washed with acetonitrile (40ml) and dried at 45-50 °C temperature under vacuum for 10-12 hours and kept in a petri dish at 25-30 °C and 60 ± 5 RH (relative humidity) for 18-24 hours to afford the title compound (31.6 gm).
Yield (w/w): 0.79 (65.8%)
HPLC purity: 99.70 %
Example-3: Preparation of Pitavastatin Sodium Amorphous
Pitavastatin sodium (3.0 gm) and ethanol (60 ml) were taken in a round bottomed flask at 25-30 °C temperature. The reaction mixture was filtered and the solvent was distilled off on rotatory evaporator under vacuum maintaining bath temperature at 45-50 °C temperature. Thereafter the reaction mixture was degassed under vacuum for 2-3 hours to afford the title compound (2.8gm).
HPLC purity: 99.70 %.
SEE……..https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016005919&redirectedID=true
/////////Lupin Ltd, New patent, Pitavastatin, WO 2016005919, statins, POLYMORPH
Dr Reddy’s Laboratories Ltd, New patent, WO 2016005960, Liraglutide

!e™A!a™Trp™leu™Va!~-Arg~~GIy-~Arg~~Gly~~OH
Formula (I)
LIRAGLUTIDE
![]()
Dr Reddy’s Laboratories Ltd, New patent, WO 2016005960, Liraglutide
Process for preparation of liraglutide
Kola, Lavanya; Ramasamy, Karthik; Thakur, Rajiv Vishnukant; Katkam, Srinivas; Komaravolu, Yagna Kiran Kumar; Nandivada, Giri Babu; Gandavadi, Sunil Kumar; Nariyam Munaswamy, Sekhar; Movva, Kishore Kumar
Improved process for preparing liraglutide, by solid phase synthesis, useful for treating type 2 diabetes.
It having been developed and launched by Novo Nordisk, under license from Scios and Massachusetts General Hospital.
Liraglutide, marketed under the brand name Victoza, is a long-acting glucagon like peptide agonist developed by Novo Nordisk for the treatment of type 2 diabetes.
Liraglutide is an injectable drug that reduces the level of sugar (glucose) in the blood. It is used for treating type 2 diabetes and is similar to exenatide (Byetta). Liraglutide belongs to a class of drugs called incretin mimetics because these drugs mimic the effects of incretins. Incretins, such as human-glucagon-like peptide-1 (GLP-1 ), are hormones that are produced and released into the blood by the intestine in response to food. GLP-1 increases the secretion of insulin from the pancreas, slows absorption of glucose from the gut, and reduces the action of glucagon. (Glucagon is a hormone that increases glucose production by the liver.)
All three of these actions reduce levels of glucose in the blood. In addition, GLP-1 reduces appetite. Liraglutide is a synthetic (man-made) hormone that resembles and acts like GLP-1 . In studies, Liraglutide treated patients achieved lower blood glucose levels and experienced weight loss.
Liraglutide, an analog of human GLP-1 acts as a GLP-1 receptor agonist. The peptide precursor of Liraglutide, produced by a process that includes expression of recombinant DNA in Saccharomyces cerevisiae, has been engineered to be 97% homologous to native human GLP-1 by substituting arginine for lysine at position 34. Liraglutide is made by attaching a C-16 fatty acid (palmitic acid) with a glutamic acid spacer on the remaining lysine residue at position 26 of the peptide precursor.
The molecular formula of Liraglutide is Ci72H265N4305i and the molecular weight is 3751 .2 Daltons. It is represented by the structure of formula (I)
!e™A!a™Trp™leu™Va!~-Arg~~GIy-~Arg~~Gly~~OH
Formula (I)
U.S. Patent No. 7572884 discloses a process for preparing Liraglutide by recombinant technology followed by acylation and removal of N-terminal extension.
U.S. Patent No. 7273921 and 6451974 discloses a process for acylation of Arg-34GLP-1 (7-37) to obtain Liraglutide.
U.S. Patent No. 8445433 discloses a solid phase synthesis of Liraglutide using a fragment approach.
International Application publication No. WO2013037266A1 discloses solid phase synthesis of Liraglutide, characterized in that comprises A) the presence of the activator system, solid phase carrier and by resin Fmoc protection N end obtained by coupling of glycine (Fmoc-Gly-OH) Fmoc-Gly-resin; B) by solid phase synthesis, prepared in accordance with the sequentially advantage Liraglutide principal chain N end of the coupling with Fmoc protected amino acid side chain protection and, wherein the lysine using Fmoc-Lys (Alloc)-OH; C) Alloc getting rid of the lysine side chain protecting group; D) by solid phase synthesis, the lysine side chain coupling Palmitoyl-Glu-OtBu; E) cracking, get rid of protecting group and resin to obtain crude Liraglutide ; F) purification, freeze-dried, to obtain Liraglutide.
Even though, the above mentioned prior art discloses diverse processes for the preparation of Liraglutide, they are often not amenable on commercial scale because of expensive amino acid derivatives such as pseudo prolines used in those processes.
Hence, there remains a need to provide simple, cost effective, scalable and robust processes for the preparation of Liraglutide involving commercially viable amino acid derivatives and reagents.

EXAMPLE 1 :
Stage I Preparation of Wang resin-Gly-Arg(pbf)-Gly-Arg(pbf)-Val-Leu-Trp(Boc)-Ala-lleu-Phe-Glu(Otbu)-Lys-{Glu(OH)-NH(palmitoyl)}-Ala-Ala-Gln(trt)-Gly-OH-Glu(Otbu)-Leu-Tyr(Otbu)-Ser(Otbu)-Ser(Otbu)-Val-Asp(Otbu)-Ser(Otbu)-Thr(Otbu)-Phe-Thr(Otbu)-Gly-Glu(Otbu)-Ala-Boc-His(trt)-OH.
Wang resin (50gm) is swelled in DCM (500ml) for 1 hr in a sintered flask. DCM was filtered using Vacuum. Fmoc-Glycine (44.6 gm, 150 mmol) was dissolved in dichloromethane (250 ml). 1 -(2-mesitylene sulfonyl)-3-nitro-1 H-1 ,2,4 triazole (44.4 gm, 150 mmol) and 1 -methyl imidazole (9 ml, 1 12 mmol) was then added. The reaction mixture was added to wang resin and stirred for 3hrs at about 25° C. The resin was washed with DCM and a second lot of Fmoc-Glycine (27 gm, 90 mmol) was dissolved in dichloromethane (250 ml). 1 -(2-mesitylene sulfonyl)-3-nitro-1 H-1 ,2,4 triazole (26.6 gm, 90 mmol) and 1 -methyl imidazole (5.3 ml, 90 mmol) was then added and stirred for 3hrs. The resin was washed with DCM and a sample of resin beads were checked for UV analysis. The capping was carried out using acetic anhydride (15 ml) DCM (120 ml) and pyridine (120 ml). The resin was washed with dichloromethane and DMF. The Fmoc protecting group was removed by treatment with 20% piperidine in DMF. The
resin was washed repeatedly with DMF. The next amino acid Fmoc-Arg(pbf)-OH (52 gm, 80 mmol) dissolved in 250 ml DMF was then added. The coupling was carried out by addition of HOBt (10.8gm, 80 mmol) and DIC (6.2ml, 80 mmol) in DMF. The completion of the coupling was confirmed by a ninhydrin test. After washing the resin, the Fmoc protecting group was removed with 20% piperidine in DMF. These steps were repeated each time with the respective amino acid according to the peptide sequence. After coupling 12th amino acid Fmoc-Lys (Alloc)-OH, deprotection of alloc group is carried out with palladium tetrakis and phenyl silane in DCM. The resin was washed repeatedly with DMF. The next amino acid H-Glu(OH)-NH(palmitoyl)-Otbu (9.9 gm, 0.023 moles) dissolved in 250 ml DMF was then added. The coupling was carried out by addition of HOBt (10.8gm, 80 mmol) and DIC (6.2ml, 80 mmol) in DMF. The completion of the coupling was confirmed by a ninhydrin test. After washing the resin, the Fmoc protecting group of Lys was removed with 20% piperidine in DMF. The next amino acid Fmoc-Ala-OH (52 gm, 80 mmol) dissolved in 250 ml DMF was then added. The coupling was carried out by addition of HOBt (10.8gm, 80 mmol) and DIC (6.2ml, 80 mmol) in DMF. The completion of the coupling was confirmed by a ninhydrin test. After washing the resin, the Fmoc protecting group was removed with 20% piperidine in DMF. These steps were repeated each time with the respective amino acid according to the peptide sequence. The resin was washed repeatedly with DMF, Methanol and MTBE and dried under vacuum.
Stage II: Cleavage of Liraglutide from resin along with global deprotection
45gms of resin obtained in stage I was treated with cleavage cocktail mixture of TFA (462.5ml), TIPS (12.5ml), Water (12.5ml), and Phenol (12.5 ml), stirred at 0°C for 30 min. and at 25°C for 3hrs at 200RPM. Then the reaction mixture was filtered, repeatedly wash the resin with TFA and the filtrate was concentrated on Rotary evaporator at 30°C. Pour the concentrated solution to MTBE (2L) at 4°C slowly and stir for 1 hr. The precipitate obtained is filtered and dried in a vacuum tray drier to afford 18 gm of Liraglutide crude with a purity of 27.5%.
Stage III: Purification of crude Liraglutide using RP HPLC.
The crude Liraglutide (4 gm) of purity around 27.5% is dissolved in 10 mM Tris buffer (120ml) of pH: 8.00 and 0.5 N NaOH is further added drop wise to the solution for making the crude solid completely dissolved. The solution is further passed through 0.2 micron filter. The Reverse phase C 18 – 150 Angstrom media (C18 silica media – 10 micron particle size) is equilibrated with 10mM Tris buffer of pH: 8.0 The crude solution is loaded onto the column and the gradient elution is performed as per the below tabular column against the mobile phase B (Acetonitrile).
Table 1 : Gradient program for pre purification

The desired fractions are collected in the gradient range of and the fractions (F1 , F2, F3, F4 and F5) whose purity > 80% are pooled. The pooled fractions are then subjected to further purification.
The Pooled fractions having purity >80% are then subjected to C18 RPHPLC silica media (5 micron particle size) for further purification. The pooled fractions – Feed is diluted with purified water in the ratio of 1 :2 (one part of pooled fraction to two parts of purified water) as a part of sample preparation before loading into the column. The media C18 is first equilibrated with 0.1 % TFA for 3 column volumes (1 CV = bed volume of media). After equilibration, the sample is loaded onto the column and the gradient
elution is performed as per the below tabular column against the mobile phase B (Acetonitrile).
Table 2: Gradient program for second purification

The desired fractions are collected in the gradient range of and the fraction whose purity > 96% are pooled together and lyophilized to afford 220mg of Liraglutide trifluoro acetate salt. The pooled fractions and their purity by HPLC are listed in the below table.

The pooled fractions with the purity of average 97% are subjected further to de solvation to remove the Acetonitrile content by Rota vapor. The final solution was filtered through 0.2 micron filter and lyophilized to get Liraglutide API.
EXAMPLE 2:
Stage I Preparation of Tentagel SPHB resin-Gly-Arg(pbf)-Gly-Arg(pbf)-Val-Leu-Trp(Boc)-Ala-lleu-Phe-Glu(Otbu)-Lys-{Glu(OH)-NH(palmitoyl)}-Ala-Ala-Gln(trt)-Gly-OH-Glu(Otbu)-Leu-Tyr(Otbu)-Ser(Otbu)-Ser(Otbu)-Val-Asp(Otbu)-Ser(Otbu)-Thr(Otbu)-Phe-Thr(Otbu)-Gly-Glu(Otbu)-Ala-Boc-His(trt)-OH using Fragment approach.
Fragments used are as follows
1 . Fmoc-Arg(pbf)-Gly-OH.
2. Fmoc-Leu-Ala-Arg(pbf)-OH.
3. Fmoc-lle-Ala-Trp(boc)-OH.
4. Fmoc-Glu(Otbu)-Phe-OH.
5. Fmoc-Glu(Otbu)-Phe-OH.
6. Fmoc-Lys-Glu-Palmitic acid.
7. Fmoc-Gly-Gln(trt)-Ala-Ala-OH.
8. Fmoc-Tyr(Otbu)-Leu-Glu(Otbu)-OH.
9. Fmoc-Val-Ser(Otbu)-Ser(Otbu)-OH.
10. Fmoc-Phe-Thr(Otbu)-Ser(Otbu)-Asp(Otbu)-OH
1 1 . Fmoc-Gly-Thr(Otbu)-OH.
12. Boc-His(Trt)-Ala-Glu(Otbu)-OH.
Tentagel SPHB resin (30gm) is swelled in DCM (300ml) for 1 hr in a sintered flask. DCM was filtered using Vacuum. Fmoc-Glycine (13.8 gm, 46.8 moles) was dissolved in dichloromethane (150 ml). 1 -(2-mesitylene sulfonyl)-3-nitro-1 H-1 ,2,4 triazole (13.8 gm, 46.8 moles) and 1 -methyl imidazole (2.4 ml, 29.25 moles) was then added. The resulting solution was added to tentagel resin and stirred for 2hrs at about 25° C. The resin was washed with DCM and a second lot of Fmoc-Glycine (13.8 gm, 46.8 moles) was dissolved in dichloromethane (150 ml). 1 -(2-mesitylene sulfonyl)-3-nitro-I H-1 ,2,4 triazole (13.8 gm, 46.8 moles) and 1 -methyl imidazole (2.4 ml, 29.25 moles) was then added and stirred for 2hrs. The resin was washed with DCM and a sample of resin beads were checked for UV analysis. The Fmoc protecting group was removed by treatment with 20% piperidine in DMF. The resin was washed repeatedly
with DMF. The next amino acid fragment 1 Fmoc-Gly-Arg(pbf)-OH (8.25 gm, 1 1 .7 moles) dissolved in 150 ml DMF was then added. The coupling was carried out by addition of HOBt (2.1 gm, 1 1 .7 moles) and DIC (2.5ml, 1 1 .7 moles) in DMF for 2hrs. The completion of the coupling was confirmed by a ninhydrin test. After washing the resin, the Fmoc protecting group was removed with 20% piperidine in DMF. These steps were repeated each time with the respective amino acid fragments according to the peptide sequence. The resin was washed repeatedly with DMF, Methanol and MTBE and dried under vacuum.
Stage II: Cleavage of Liraglutide from resin along with global deprotection
58gms of resin obtained from stage I was treated with cleavage cocktail mixture of TFA (555ml), TIPS (15ml), Water (15ml), and Phenol (15 ml) and stirred at 0°C for 30 min. at 25°C for 3hrs at 200RPM. Then filter the reaction mixture, repeatedly wash the resin with TFA and concentrate on Rotary evaporator at 30°C. Pour the concentrated solution to MTBE at 4°C slowly and stirred for 1 hr. The precipitate obtained was filtered and dried in a vacuum tray drier to afford 23.12 gm of crude Liraglutide with a purity of 36.89%.
Stage III: Purification of crude Liraglutide using RP HPLC.
The crude Liraglutide (4 gm) of purity around 27.5% is dissolved in 10 mM Tris buffer (120ml) of pH: 8.00 and 0.5 N NaOH is further added drop wise to the solution for making the crude solid completely dissolved. The solution is further passed through 0.2 micron filter. The Reverse phase C 18 – 150 Angstrom media (Irregular C18 silica media – 10 micron particle size) is equilibrated with 10mM Tris buffer of pH: 8.0 The crude solution is loaded onto the column and the gradient elution is performed as per the below tabular column against the mobile phase B (Acetonitrile).
Table 1 : Gradient program for pre purification

60 40 30
55 45 30
52 48 30
51 49 60
The desired fractions are collected in the gradient range of and the fractions (F1 , F2, F3, F4 and F5) whose purity > 80% are pooled. The pooled fractions then subjected to further purification.
The Pooled fractions having purity >80% are then subjected to C18 RPHPLC silica media (5 micron particle size) for further purification. The pooled fractions – Feed is diluted with purified water in the ratio of 1 :2 (one part of pooled fraction to two parts of purified water) as a part of sample preparation before loading into the column. The media C18 is first equilibrated with 0.1 % TFA for 3 column volumes (1 CV = bed volume of media). After equilibration, the sample is loaded onto the column and the gradient elution is performed as per the below tabular column against the mobile phase B (Acetonitrile).
Table 2: Gradient program for second purification

The desired fractions are collected in the gradient range and the fraction whose purity > 96% are pooled together and Lyophilized to afford 865 mg of Liraglutide trifluoro acetate salt. The pooled fractions and their purity by HPLC are listed in the below table.

The pooled fractions with the purity of average 97% are subjected further to de solvation to remove the Acetonitrile content by Rota vapor. The final solution was filtered through 0.2 micron filter and lyophilized to get Liraglutide API.

G.V. Prasad, chairman, Dr Reddy’s Laboratories.
REFERENCE
IN2014CH3453 INDIAN PATENT
WO 2016005960, CLICK FOR PATENT
//////
New patent, WO 2016001885, Dr Reddy’s Laboratories Ltd, Eliglustat hemitartarate
![]()
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills, Telangana, India Hyderabad 500034 (IN)
VELAGA, Dharma Jagannadha Rao; (IN).
PEDDY, Vishweshwar; (IN).
VYALA, Sunitha; (IN)
(WO2016001885) AMORPHOUS FORM OF ELIGLUSTAT HEMITARTARATE
Chemically Eliglustat is named N-[(1 R,2R)-2-(2,3-dihydro-1 ,4-benzodioxin-6-yl)-2-hydroxy-1 -(1 -pyrrolidinylmethyl)ethyl]-Octanamide(2R!3R)-2,3-dihydroxybutanedioate and the hemitartarate salt of eliglustat has the structural formula as shown in Formula I.

Formula I
Eliglustat hemitartrate (Genz-1 12638), currently under development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy. Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase, and is currently under development by Genzyme.
U.S. patent No. 7,196,205 discloses a process for the preparation of Eliglustat or a pharmaceutically acceptable salt thereof.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 discloses process for preparation of Eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of Eliglustat, (ii) a hemitartrate salt of Eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
It has been disclosed earlier that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailablity patterns compared to crystalline forms [Konne T., Chem pharm Bull., 38, 2003(1990)]. For some therapeutic indications one bioavailabihty pattern may be favoured over another. An amorphous form of Cefuroxime axetil is a good example for exhibiting higher bioavailability than the crystalline form.
Solid amorphous dispersions of drugs are known generally to improve the stability and solubility of drug products. However, such dispersions are generally unstable over time. Amorphous dispersions of drugs tend to convert to crystalline forms over time, which can lead to improper dosing due to differences of the solubility of crystalline drug material compared to amorphous drug material. The present invention, however, provides stable amorphous dispersions of eliglustat hemitartrate. Moreover, the present invention provides solid dispersions of eliglustat hemitartrate which may be reproduced easily and is amenable for processing into a dosage form.
There remains a need to provide solid state forms of eliglustat hemitartarate which are advantageous in a cost effective and environment friendly manner.

EXAMPLES
Example 1 : Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 14 mL of dichloromethane at 26°C and stirred for 15 min. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 45°C. After distillation the solid was dried under vacuum at 45°C.
Example 2: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 70 mL of ethanol and stirred for 15 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid was dried under vacuum at 48°C.
Example 3: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 20 mL of methanol and stirred for 15 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid was dried under vacuum at 48°C.
Example 4: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and PVP-K30.
500mg of eliglustat hemitartarate and 500mg of PVP-K30 was dissolved in 20 mL of methanol and stirred for 10 min at 25° – 30°C. The solution is filtered to remove the undissolved particles and the filtrate is distilled under reduced pressure at 48°C. After distillation the solid is dried under vacuum at 48°C.
Example 5: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and hydroxy propyl cellulose.
500mg of eliglustat hemitartarate and 500 mg of hydroxy propyl cellulose was dissolved in 30 ml of methanol and stirred for 10 min at 25° – 30°C. The solution is distilled under reduced pressure at 49°C. After distillation the solid is dried under vacuum at 49°C.
Example 6: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and hydroxy propyl methyl cellulose.
500mg of eliglustat hemitartarate and 500 mg of hydroxy propyl methyl cellulose was dissolved in 30 mL of methanol and stirred for 10 min at 25° – 30°C. The solution is distilled under reduced pressure at 48°C. After distillation the solid is dried under vacuum at 48°C.
Example 7 Preparation of amorphous form of eliglustat hemitartarate.
3g of eliglustat hemitartarate was dissolved in 75 mL of methanol and stirred at 25°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected for spray drying at inlet temperature of 70°C and outlet temperature of 42°C to afford the title compound.
Example 8: Preparation of amorphous form of eliglustat hemitartarate.
500mg of eliglustat hemitartarate was dissolved in 30 mL of isopropanol and stirred at 56°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected to complete distillation under reduced pressure and drying at about 56°C to afford the title compound.
Example 9: Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 40 mL of ethyl acetate and stirred at about 63°C. Then methanol (5 mL) is added at the same temperature to obtain clear solution which was filtered to remove the undissolved particles. Then additional quantity of methanol (5mL) is added to the filtrate and the filtrate was again filtered to remove particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 10: Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 40 mL of acetone and stirred at about 55°C followed by addition of methanol (15 mL). The mixture is stirred at 55°C for clear solution and filtered to remove the undissolved particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 11 : Preparation of amorphous form of eliglustat hemitartarate.
1 g of eliglustat hemitartarate was provided in 25 mL of isopropyl alcohol and 25 mL of ethanol. The mixture was stirred at about 58°C for dissolution and filtered to remove the undissolved particles. The obtained filtrate was subjected to complete distillation under reduced pressure and drying at about 57°C to afford the title compound.
Example 12 Preparation of amorphous form of eliglustat hemitartarate.
5g of eliglustat hemitartarate was provided in 300 mL of isopropyl alcohol and stirred at about 59°C for dissolution. The solution was filtered to remove the undissolved particles and the filtrate is subjected for spray drying at inlet temperature of 65°C and outlet temperature of 37°C to afford the title compound according to Fig. 6
Example 13: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Copovidone
500mg of eliglustat hemitartarate and 500mg of Copovidone were dissolved in 30 mL of methanol and stirred for clear solution, then filtered to make it particle free. The solvent from the filtrate was evaporated under reduced pressure at 45°C and obtained solid was subjected to drying at 45°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction.
Example 14: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Copovidone
2g of eliglustat hemitartarate and 2g of Copovidone were dissolved in 100 mL of methanol and stirred for clear solution, then filtered to make it particle free. The solvent from the filtrate was subjected to spray drying at inlet temperature of 70 at 45°C and outlet temperature of 42°C to afford the title compound. The resulting dispersion was found to be amorphous by X-ray powder diffraction.
Example 15: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate
2g of eliglustat hemitartarate was charged in 40 mL of methanol followed by addition of 2g of PVP K-30. The mixture was stirred for clear solution and filtered to make it particle free, the bed was washed with 20 mL of methanol. Then 2g of Syloid is added to the filtrate and filtrate is subjected to distillation under reduced pressure at about 57°C and obtained solid was subjected to drying at about 57°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 7a. The said dispersion is kept at 25°C under 40% relative humidity for 24 hours and PXRD was recorded and found to be amorphous according to Fig 7b.
Example 16: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate
2g of eliglustat hemitartarate was charged in 40 mL of methanol followed by addition of 2g of Copovidone. The mixture was stirred for clear solution and filtered to make it particle free, the bed was washed with 20 mL of methanol. Then 2g of Syloid is added to the filtrate and filtrate is subjected to distillation under reduced pressure at about 57°C and obtained solid was subjected to drying at about 57°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 8a. The said dispersion is kept at 25°C under 40% relative humidity for 24 hours and PXRD was recorded and found to be amorphous according to Fig. 8b and D90 of the resultant solid is about 437 microns.
Example 17: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Syloid
1 g of eliglustat hemitartarate was dissolved in 25 ml_ of methanol and filtered to make it particle free. Then 1 g of Syloid 244 FPNF was added to the filtrate and solvent from the filtrate was evaporated under reduced pressure at 56°C and obtained solid was subjected to drying at 56°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction according to Fig. 9 and D90 of the resultant solid is about 4 microns.
Example 18: Preparation of a solid dispersion comprising an amorphous form of eliglustat hemitartarate and Syloid
1 g of eliglustat hemitartarate was dissolved in 25 ml_ of methanol and filtered to make it particle free. Then 500mg of Syloid 244 FPNF was added to the filtrate and solvent from the filtrate was evaporated under reduced pressure at 56°C and obtained solid was subjected to drying at 56°C to afford the title solid. The resulting dispersion was found to be amorphous by X-ray powder diffraction.

PATENT
(WO2015059679) IMPROVED PROCESS FOR THE PREPARATION OF ELIGLUSTAT
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara Hills Hyderabad 500034 (IN)
JAVED, Iqbal; (IN).
DAHANUKAR, Vilas Hareshwar; (IN).
ORUGANTI, Srinivas; (IN).
KANDAGATLA, Bhaskar; (IN)
Eliglustat tartrate (Genz-1 12638) is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of gaucher disease and other lysosomal storage disorders, which is currently under development.
Eliglustat is chemically known as 1 R, 2R-Octanoic acid [2-(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1 -ylmethyl]-ethyl]-amide, having a structural formula I depicted here under.

Formula I
Eliglustat hemitartrate (Genz-1 12638) development by Genzyme, is a glucocerebroside (glucosylceramide) synthase inhibitor for the treatment of Gaucher disease and other lysosomal storage disorders. Eliglustat hemitartrate is orally active with potent effects on the primary identified molecular target for type 1 Gaucher disease and other glycosphingolipidoses, appears likely to fulfill high expectations for clinical efficacy. Gaucher disease belongs to the class of lysosomal diseases known as glycosphingolipidoses, which result directly or indirectly from the accumulation of glycosphingolipids, many hundreds of which are derived from glucocerebroside. The first step in glycosphingolipid biosynthesis is the formation of glucocerebroside, the primary storage molecule in Gaucher disease, via glucocerebroside synthase (uridine diphosphate [UDP] – glucosylceramide glucosyl transferase). Eliglustat hemitartrate is based on improved inhibitors of glucocerebroside synthase.
U.S. patent No. 7,196,205 (herein described as US’205) discloses a process for the preparation of eliglustat or a pharmaceutically acceptable salt thereof. In this patent, eliglustat was synthesized via a seven-step process involving steps in that sequence: (i) coupling S-(+)-2-phenyl glycinol with phenyl bromoacetate followed by column chromatography for purification of the resulting intermediate, (ii) reacting the resulting (5S)-5-phenylmorpholin-2-one with 1 , 4-benzodioxan-6-carboxaldehyde to obtain a lactone, (iii) opening the lactone of the oxazolo-oxazinone cyclo adduct via reaction with pyrrolidine, (iv) hydrolyzing the oxazolidine ring, (v) reducing the amide to amine to obtain sphingosine like compound, (vi) reacting the resulting amine with octanoic acid and N-hydroxysuccinimide to obtain crude eliglustat, (vii) purifying the crude eliglustat by repeated isolation for four times from a mixture of ethyl acetate and n-heptane.
U.S. patent No. 6855830, 7265228, 7615573, 7763738, 8138353, U.S. patent application publication No. 2012/296088 disclose processes for preparation of eliglustat and intermediates thereof.
U.S. patent application publication No. 2013/137743 discloses (i) a hemitartrate salt of eliglustat, (ii) a hemitartrate salt of eliglustat, wherein at least 70% by weight of the salt is crystalline, (iii) a hemitartrate salt of Eliglustat, wherein at least 99% by weight of the salt is in a single crystalline form.
It is also an objective of the present application to provide an improved process for the preparation of eliglustat and a pharmaceutically acceptable salt thereof which is high yielding, simple, cost effective, environment friendly and commercially viable by avoiding repeated cumbersome and lengthy purification steps. It is a further objective of the present application to provide crystalline forms of eliglustat free base and its salts.
Example 6: Preparation of Eliglustat {(1 R, 2R)-Octanoic acid[2-(2′,3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-2-hydroxy-1 -pyrrolidin-1-ylmethyl-ethyl]-amide}.
(1 R, 2R)-2-Amino-1 -(2′, 3′-dihydro-benzo [1 , 4] dioxin-6′-yl)-3-pyrrolidin-1 -yl-propan-1 -ol (15g) obtained from above stage 5 was dissolved in dry dichloromethane (150ml) at room temperature under nitrogen atmosphere and cooled to 10-15° C. Octanoic acid N-hydroxy succinimide ester (13.0 g)was added to the above reaction mass at 10-15° C and stirred for 15 min. The reaction mixture was stirred at room temperature for 16h-18h. Progress of the reaction was monitored by thin layer chromatography. After completion of reaction, the reaction mixture was cooled to 15°C and diluted with 2M NaOH solution (100 ml_) and stirred for 20 min at 20 °C. The organic layer was separated and washed with 2M sodium hydroxide (3x90ml).The organic layer was dried over anhydrous sodium sulphate (30g) and concentrated under reduced pressure at a water bath temperature of 45°C to give the crude compound (20g).The crude is again dissolved in methyl tertiary butyl ether (25 ml_) and precipitated with Hexane (60ml). It is stirred for 10 min, filtered and dried under vacuum to afford Eliglustat as a white solid (16g). Yield: 74%, Mass (m/zj: 404.7 HPLC (% Area Method): 97.5 %, ELSD (% Area Method): 99.78%, Chiral HPLC (% Area Method): 99.78 %.
Example 7: Preparation of Eliglustat oxalate.
Eliglustat (5g) obtained from above stage 6 is dissolved in Ethyl acetate (5ml) at room temperature under nitrogen atmosphere. Oxalic acid (2.22g) dissolved in ethyl acetate (5ml) was added to the above solution at room temperature and stirred for 14h. White solid observed in the reaction mixture was filtered and dried under vacuum at room temperature for 1 h to afford Eliglustat oxalate as a white solid (4g). Yield: 65.46%, Mass (m/zj: 404.8 [M+H] +> HPLC (% Area Method): 95.52 %, Chiral HPLC (% Area Method): 99.86 %
G.V. Prasad, chairman, Dr Reddy’s Laboratories
//////////////New patent, WO 2016001885, Dr Reddy’s Laboratories Ltd, Eliglustat hemitartarate, WO 2015059679
NEW PATENT, WO2016001844, SUN PHARMACEUTICALS, AFATINIB DIMALEATE

AMORPHOUS FORM OF AFATINIB DIMALEATE
SUN PHARMACEUTICAL INDUSTRIES LIMITED
VERMA, Shyam Sunder; (IN).
SINGH, Shravan Kumar; (IN).
SINGH, Kaptan; (IN).
PRASAD, Mohan; (IN)
Afatinib dimaleate is a tyrosine kinase inhibitor, chemically designated as 2-butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(35)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-,(2£)-, (2Z)-2-butenedioate (1:2) having the structure depicted by Formula I.

Formula I
U.S. Patent Nos. RE43,431 and 6,251,912 provide processes for the preparation of afatinib dimaleate.
U.S. Patent No. 8,426,586 and PCT Publication Nos. WO 2012/121764 and WO
2013/052157 provide processes for the preparation of crystalline forms of afatinib and their salts.
Example: Preparation of an amorphous form of afatinib dimaleate
In a round bottom flask, a mixture of afatinib (3 g) and ethyl acetate (30 mL) was heated to about 65°C to obtain a turbid solution. In another round bottom flask, a mixture of maleic acid (1.6 g) and ethyl acetate (30 mL) was heated to about 50°C to obtain a clear solution. The maleic acid solution was added to the afatinib solution, and then the reaction mixture was heated at about 75°C to about 80°C. The reaction mixture was stirred at about 75°C to about 80°C for about 1 hour. The reaction mixture was cooled to about
20°C to obtain a sticky material. The sticky material was scratched with a spatula, and then the reaction mixture was further stirred at about 20°C to about 25°C for about 1 hour. The material obtained was filtered, and then washed with ethyl acetate (20 mL). The solid obtained was dried under vacuum at about 45°C to about 50°C for about 15 hours to obtain the amorphous form of afatinib dimaleate.
Yield: 2.5 g (56%)


Sun Pharma chief Dilip Shanghvi
///////
NEW PATENT, TICAGRELOR, DR. REDDY’S LABORATORIES LIMITED, WO 2016001851
![]()
DR. REDDY’S LABORATORIES LIMITED [IN/IN]; 8-2-337, Road No. 3, Banjara hills, Hyderabad, Telangana Hyderabad 500034 (IN)
DAHANUKAR, Vilas; (IN).
ELATI, Ravi Ram Chandrasekhar; (IN).
ORUGANTI, Srinivas; (IN).
RAPOLU, Rajesh Kumar; (IN).
KURELLA, Sreenivasulu; (IN)
![]()
The drug compound having the adopted name “ticagrelor” has chemical names: [1 S-(1 α,2α,3β(1 S*,2R*),5p)]-3-[7-[2-(3,4-difluorophenyl-cyclopropyl] amino]-5-(propylthio)-3H-1 ,2,3-triazolo[4,5-d]pyrimidin-3-yl)-5-(2-hydroxyethoxy)-cyclopentane-1 ,2-diol; or (1 S,2S,3fl,5S)-3-[7-{[(1 fl,2S)-2-(3,4-difluorophenyl) cyclopropyl]amino}-5-(propylthio)-3H 1 ,2,3]-triazolo[4,5-c/|pyrimidin-3-yl]-5-(2-hydroxyethoxy)cyclope ed by Formula I.

Formula I
Ticagrelor is the active ingredient in the commercially available BRILINTA® tablets for oral administration.
Ticagrelor and related compounds are disclosed in International Patent Application Publication Nos. WO 00/34283 and WO 99/05143 as pharmaceutically active Ρ2τ (which are now usually referred to as P2Y12) receptor antagonists. Such antagonists can be used, inter alia, as inhibitors of platelet activation, aggregation, or degranulation. International Patent Application Publication Nos. WO 01 /92263 and WO 2010/030224 A1 , WO 2012085665 A2, WO 2012138981 A2 and WO 2013037942 A1 disclose processes for preparing ticagrelor.
The processes for the preparation of traizolo [4,5-d] pyrimidine derivatives preferably Ticagrelor and related compounds, described in the above mentioned prior art suffer from disadvantages since the processes involve tedious and cumbersome procedures such as lengthy and multiple synthesis steps, reactions under pressure and high temperature, longer reaction times, tedious work up procedures and multiple crystallizations or isolation steps, column chromatographic purifications and thus resulting in low overall yields of the product. Ticagrelor obtained by the processes described in the prior art does not have satisfactory purity and unacceptable amounts of impurities are formed along with Ticagrelor at various stages of the processes that are difficult to purify and thus get carried forward in the subsequent steps thus affecting the purity of final compound. Thus, there remains a need to prepare compounds of Formula I of high purity and in good yield while overcoming the drawbacks presented by the previously described processes.

Formula V Formula V”
In a preferred embodiment, present application provide compounds of Formula IV with specific groups i.e. compounds of Formula IV and Formula IV”,

Formula IV Formula IV”
In a preferred embodiment, present application provides a compound of Formula II with specific groups i.e. compounds of Formula ΙΓ and Formula II”,

Formula ΙΓ Formula II”
In a preferred embodiment, present application provides a compound of Formula l la with specific grou

Formula lla

Formula VII’ Formula VII”
In a preferred embodiment, present application provides compounds of Formula Vila with specific groups i.e. compounds of Formula Vila’ and Formula “,

Formula Vila’ Formula Vila”
G.V. Prasad, chairman, Dr Reddy’s Laboratories
EXAMPLES
EXAMPLE 1 : Preparation of 2-bromo-N,N-diphenylacetamide (FORMULA Vile).
A flask is charged with Ν,Ν-diphenyl amine (25 g) and dichloromethane (350 mL) under nitrogen atmosphere. The reaction mixture is cooled to 0°C followed by addition of solution of triethyl amine (20.7 mL) and bromoacetyl chloride (38.72 mL) in dichloromethane (181 mL). The mixture is cooled to room temperature and then stirred for about 16 hours. The completion of the reaction is monitored by TLC. The reaction mixture is diluted with dichloromethane (250 mL) and then washed with 0.5N aqueous hydrochloric acid solution (3×150 mL), brine (100 mL). The organic layer is separated and subjected to distillation under vacuum at 45°C. The obtained compound is recrystallized from hexane (250 mL) and methanol (100 mL) to afford the title compound.
EXAMPLE 2: Preparation of benzyl ((3aS,4R,6S,6aR)-6-(2-(diphenylamino)-2-oxoethoxy)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (FORMULA Vila).
A flask is charged with sodium hydride (2.85 mL, 60% dispersion in oil) and dimethyl formamide (10 mL) under nitrogen atmosphere. The reaction mixture is then cooled to -30°C followed by addition of a solution of benzyl
((3aS,4R,6S,6aR)-6-hydroxy-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (20 g) in dimethyl formamide (40 mL). The mixture is stirred at -30°C for about 45 minutes, then a solution of 2-bromo-N,N-diphenylacetamide (22.65 g) in dimethyl formamide (60 mL) is added at the same temperature. The reaction mixture is allowed to attain room temperature and stirred at the same for 3 hours and completion of the reaction is monitored by TLC. The reaction mixture is quenched with ice-cold water (200 mL) and extracted with ethyl acetate (3×150 mL). The organic layer is combined and washed with water (3×100 mL), brine (100 mL) and then organic layer is then subjected to complete distillation under vacuum at 45°C. The crude so obtained is treated with MTBE (150 mL) and stirred at room temperature for overnight followed by filtration of obtained solid to afford the title compound.
EXAMPLE 3: Preparation of 2-(((3aR,4S,6R,6aS)-6-amino-2,2-dimethyl tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula VII)
A flask is charged with benzyl ((3aS,4R,6S,6aR)-6-(2-(diphenylamino)-2-oxoethoxy)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (15 g), ethanol (300 mL), 10% Pd/C (1.5 g) and ammonium formate (5.49 g). The reaction mixture is stirred at 80°C for 8 hours and completion of the reaction is monitored by TLC. Then reaction mixture is cooled to room temperature and filtered through celite bed, bed is washed with ethyl acetate (100 mL). The filtrate is subjected to complete distillation under vacuum at 45°C. Then ethanol (120 mL), L-tartaric acid (4.88 g) is added to the crude compound and mixture is stirred for 4 hours at room temperature. To the mixture, MTBE (300 mL) is added at the same temperature. The solvent is distilled under vacuum at 35°C to afford the gummy solid. Then MTBE (100 mL) is added to the gummy solid and mixture is stirred for 10-12 hours. The solid obtained is filtered and washed with MTBE (50 mL). The solid obtained is dissolved in water and sodium bicarbonate solution (200 mL) is added, desired compound is extracted in ethyl acetate (100 mL). The solvent is subjected to distillation (upto 40%) followed by addition of hexane (150 mL) and ethyl acetate (20 mL). The mixture is stirred at -10°C, then solid is recovered followed by drying under vacuum at 40°C. The crude compound is purified by column chromatography using methanol and dichloromethane (5:95) to afford the title compound.
EXAMPLE 4: Preparation of 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2- (propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d]
[1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula V)
A flask is charged with 4,6-dichloro-2-(propylthio)pyrimidin-5-amine (6.5 g),
2- (((3aR,4S,6R,6aS)-6-amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (10.39 g), sodium bicarbonate (9.13 g) and water. The mixture is stirred at 95-100°C for 15-20 hours till completion of the reaction (as monitored by TLC). Then water (20 mL) and ethyl acetate (25 mL) are added at room temperature. The layers are separated and aqueous layer is extracted with ethyl acetate (20 mL). The organic layers are combined and washed with brine solution (2×25 mL). The organic layer is subjected to complete distillation under vacuum at 40-45°C. The obtained crude compound is purified by column chromatography using ethyl acetate and hexane (30:70) to afford the title compound.
EXAMPLE 5: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula IV)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2-(propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (6 g), acetic acid (30 mL) and water (6 mL). The mixture is cooled to 0 to -5°C followed by addition of addition of sodium nitrite solution (768 mg in 6 mL of water). The mixture is stirred for 1 hour at the same temperature and then mixture is allowed to attain room temperature, and further stirred for 1 hour. The completion of the reaction is monitored by TLC and then toluene (60 mL) is added. The layers are separated, organic layer is washed with saturated solution of potassium carbonate and subsequently organic layer is dried with sodium sulphate and used for next reaction.
EXAMPLE 6: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin- 3- yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula II)
A flask is charged with (1 R,2S)-2-(3,4-difluorophenyl)cyclopropan-1 -amine mandelate (3.25 g), diisopropylethyl amine (6.1 mL), toluene (60 mL) and stirred for 30 minutes at room temperature. Then slowly, toluene layer containing 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (60 mL) is added over a period of 10 minutes. The reaction mixture is stirred at room temperature for overnight and completion of the reaction is monitored by TLC. The reaction mixture is diluted with water (60 mL), layers are separated and aqueous layer is extracted with toluene (2×30 mL). The combined organic layers are washed with brine (60 mL) and then subjected to complete distillation under vacuum at 45°C to afford the crude compound. The crude compound is purified by column chromatography using ethyl acetate and hexane (80:20).
EXAMPLE 7: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)ethan-1 -ol (Formula lib)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (3 g), THF (90 mL) and mixture is cooled to 0°C. Then portion wise, Lithium aluminium hydride (940 mg) is added over a period of 10 minutes and mixture is stirred at 0°C for 1 hour. The reaction mixture is then stirred at room temperature for 5 hours and progress of the reaction is monitored by TLC. Then mixture is cooled to 0-5°C and quenched with ice cold water (100 mL) and then diluted with ethyl acetate (30 mL). The layers are separated and organic layer after drying is used for next step.
EXAMPLE 8: Preparation of Ticagrelor (Formula I)
A flask is charged with organic layer containing 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3] triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)ethan-1 -ol (1 10 mL) and 2% hydrochloric acid solution (75 mL). The reaction mixture is stirred at room temperature for 48 hours and progress of the reaction is monitored by TLC. Then the reaction mixture is diluted with ethyl acetate (50 mL), layers are separated. The organic layer is sequentially washed with water (50 mL), brine solution (50 mL) followed by complete distillation under vacuum at 45°C. The crude compound is dissolved in ethyl acetate (12 mL) and then hexane (50 mL) is added. The mixture is stirred for 2 hours followed by isolation of solid by filtration. The obtained solid is dissolved in ethyl acetate (12 mL) and treated with charcoal followed by filtration. The filtrate is subjected to complete distillation and obtained solid is purified by column chromatography using ethyl acetate:hexane (1 :1 ) and methanohdichloromethane (5:95) to afford the title compound.
EXAMPLE 9: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)ethan-1 -ol (Formula lib)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (100 mg), THF (3 mL) and cooled to 0-5°C followed by addition of Vitride (0.04 mL) at 0-5°C over a period of 5 minutes. The mixture is stirred at same temperature for 1 hour and progress of the reaction is monitored by TLC. Additional amount of Vitride (0.13 mL) is added to the mixture and stirred for additional 6 hours. After completion of reaction, reaction mixture is cooled to 0-5°C and quenched with saturated sodium potassium tartrate solution (10 mL) and extracted with ethyl acetate (20 mL). The organic layer is subjected to complete distillation under reduced pressure and obtained material is purified by column chromatography using ethyl acetate: hexane (1 :1 ) and methanohdichloromethane (5:95) to afford the title compound.
EXAMPLE 10: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)acetaldehyde (Formula lib’)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (100 mg), THF (5 mL) and mixture is cooled to 0°C. Then portion wise, Lithium aluminium hydride (15 mg) is added over a period of 1 0
minutes and mixture is stirred at 0°C for 1 hour. The progress of the reaction is monitored by TLC. After completion of the reaction, mixture is quenched with ice cold water (5 mL) and diluted with ethyl acetate (10 mL). The layers are separated and organic layer after drying is subjected to complete distillation followed by purification using preparative TLC using 40% ethyl acetate in hexane to afford the title compound.
EXAMPLE 11 : Preparation of 2-bromo-1 -morpholinoethan-1 -one
A flask is charged with bromoacetyl bromide (25 mL), dichloromethane (500 mL) and mixture is stirred under nitrogen atmosphere. The reaction mixture is cooled to -25°C followed by slow addition of morpholine (72.7 mL in 500 mL of DCM) at the same temperature over a period of 30 minutes. The reaction mixture is stirred at -25°C for 15 minutes, then allowed to attain room temperature at which it is further stirred for 4 hours. The completion of the reaction is monitored by TLC and reaction mixture is sequentially washed with water (2×250 mL) and brine solution (2×100 mL). The organic solvent is subjected to distillation to afford the title compound.
EXAMPLE 12: Preparation of benzyl ((3aS,4R,6S,6aR)-2,2-dimethyl-6-(2-morpholino-2-oxoethoxy)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (Formula Vila”)
A flask is charged with sodium hydride (60%, 4.29 g), DMF (90 mL) and cooled to -30°C. Then, benzyl ((3aS,4R,6S,6aR)-6-hydroxy-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate (30 g) is added to the reaction mixture at the same temperature over a period of 25 minutes and mixture is stirred at -30°C for 1 hour. Then 2-bromo-1 -morpholinoethan-1 -one (24.36 g) is added to the reaction mixture at -30°C over a period of 20 minutes and temperature is raised to room temperature. The mixture is stirred at RT for 1 hour. The progress of the reaction is monitored by TLC and after completion, the reaction mixture is quenched with ice cold water followed by extraction with ethyl acetate. The organic layer is separated and subjected to distillation to afford the title compound.
EXAMPLE 13: Preparation of 2-(((3aR,4S,6R,6aS)-6-amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (Formula VII”)
A flask is charged with benzyl ((3aS,4R,6S,6aR)-2,2-dimethyl-6-(2-morpholino-2-oxoethoxy)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)carbamate, ethanol (1 0 g), ammonium formate (4.35 g) and 10% Pd/C (1 g). The reaction mixture is heated to 80°C and then stirred for 2 hours. The progress of the reaction is monitored by TLC and after completion of the reaction, mixture is cooled to room temperature, filtered and washed with ethyl acetate (100 mL). The filtrate is distilled under reduced pressure and obtained compound is purified by column chromatography using methanol-DCM (5:95) to afford the title compound.
EXAMPLE 14: Preparation of 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2- (propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d]
[1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (Formula V”)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-amino-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (8 g), water (24 mL) and stirred for 10 minutes. Then sodium bicarbonate (8.9 g), 4,6-dichloro-2-(propylthio)pyrimidin-5-amine (6.3 g) and water (24 mL) is added and mixture is heated to 95-100°C at which point it is stirred for 15 hours. The progress of the reaction is monitored by TLC and on completion reaction mixture is cooled to room temperature followed by addition of water (24 mL) and ethyl acetate (40 mL). The layers are separated and aqueous layer is extracted with ethyl acetate (20 mL). The organic layers are combined, washed with brine solution (2×40 mL) and subjected to distillation under vacuum at 45°C to afford the title compound.
EXAMPLE 15: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3] dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (Formula IV”)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2-(propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d]
[1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (4 g), acetic acid (20 mL) and stirred under nitrogen atmosphere for 10 minutes. Then water (8 mL) is added and mixture is cooled to -5 to 0°C followed by slow addition of sodium nitrite (650 mg). The mixture is stirred at 0°C for 1 hour and progress of the reaction is monitored by TLC. After completion of the reaction, mixture is extracted with toluene (40 mL and 20 mL). The combined toluene layer is sequentially washed with potassium
carbonate solution (40 mL) and brine solution (2×20 mL) followed by distillation under vacuum to afford the desired compound.
EXAMPLE 16: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (Formula II”)
A flask is charged with (1 R,2S)-2-(3,4-difluorophenyl)cyclopropan-1 -amine mandelate (2.5 g) and toluene (20 mL) followed by drop-wise addition of diisopropylethylamine (4.7 mL), then mixture is stirred for 10 minutes at RT. Then toluene layer containing 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3] dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (4 g in 55 mL) is added to the above mixture and reaction mass is stirred for 15 hours at room temperature. The progress of the reaction is monitored by TLC followed by addition of water (20 mL) on completion of reaction. The layers are separated, aqueous layer is extracted with toluene (20 mL). The organic layers are combined, washed with brine solution (2×20 mL) and then subjected to distillation under vacuum at 45°C to afford the crude compound. The crude compound is purified by column chromatography using hexane to afford the title compound.
EXAMPLE 17: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)ethan-1 -ol
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-1 -morpholinoethan-1 -one (1 g) in tetrahydrofuran (20 mL) and stirred under nitrogen atmosphere followed by addition of vitride (1 .53 mL) over a period of 10 minutes. The reaction mixture is stirred for 1 hour at room temperature and progress of the reaction is monitored by TLC. On completion, the mixture is quenched with sodium potassium tartrate (5 mL). The mixture is extracted with ethyl acetate (10 mL), then layers are separated and organic layer is subjected to distillation under vacuum at 45°C. The obtained material is dissolved in THF (20 mL) and slowly lithium aluminiumhydride (0.1 17 g) is added to the mixture at 0-5°C. Then mixture is stirred at room temperature for 1 hour and progress of the reaction is monitored by TLC. On completion of reaction, it is quenched with ice-cold water (20 mL) and extracted with ethyl acetate (15 mL). The layers are separated and organic layer is used for next step.
EXAMPLE 18: Preparation of Ticagrelor
A flask is charged with organic layer containing 2-(((3aR,4S,6R,6aS)-6-(7-(((1 R,2S)-2-(3,4-difluorophenyl)cyclopropyl)amino)-5-(propylthio)-3H-[1 ,2,3] triazolo [4,5-d]pyrimidin-3-yl)-2,2-dimethyltetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy) ethan-1 -ol (30 mL) and 2% hydrochloric acid solution (30 mL). The reaction mixture is stirred at room temperature for overnight and progress of the reaction is monitored by TLC. Then the reaction mixture is diluted with ethyl acetate (20 mL), layers are separated. The organic layer is washed with brine solution (20 mL) followed by complete distillation under vacuum at 45°C. The crude compound is purified by column chromatography using ethyl acetate:hexane (7:10) and methanol :d ic h I oro methane (5:95) to afford the title compound.
EXAMPLE 19: Preparation of 2-(((3aR,4S,6R,6aS)-6-(7-chloro-5-(propylthio)-3H-[1 ,2,3]triazolo[4,5-d]pyrimidin-3-yl)tetrahydro-4H-cyclopenta[d][1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (Formula IV)
A flask is charged with 2-(((3aR,4S,6R,6aS)-6-((5-amino-6-chloro-2-(propylthio)pyrimidin-4-yl)amino)-2,2-dimethyltetrahydro-4H-cyclopenta[d]
[1 ,3]dioxol-4-yl)oxy)-N,N-diphenylacetamide (5 g) and acetonitrile (50 mL) for clear solution. To this, isoamyl nitrite (1 .5 g) is added over a period of 5 minutes. The reaction mixture is maintained at room temperature for 5 hours and completion of the reaction is monitored by TLC. Then water (50 mL) and toluene (50 mL) are added and layers are separated. The aqueous layer is extracted with toluene (50 mL) and total organic layers are combined, subjected to distillation under vacuum to afford the title compound.
Anji Reddy

Mr G.V. Prasad, CEO, Dr. Reddy’s Labs

G V Prasad and Mr K. Satish Reddy
///////////
NEW PATENT, TICAGRELOR, DR. REDDY’S LABORATORIES LIMITED, WO 2016001851
New Patent from Zydus Cadila, Canagliflozin, US 20160002275
CADILA HEALTHCARE LIMITED [IN]

(2S,3R,4R,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is also known as Canagliflozin, is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2) which is chemically represented as compound of Formula (I).
U.S. Pat. No. 7,943,788 B2 discloses canagliflozin and a process for its preparation.
U.S. Pat. No. 7,943,582 B2 (the ‘582 patent) discloses crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate and process for preparation thereof.
U.S. PG-Pub. No. 2011/0212905 discloses crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate and process for preparation thereof.
U.S. PG-Pub. Nos. 2009/0233874, 2010/099883 and 2008/0146515 discloses similar process for the preparation of canagliflozin substantially as same as shown in scheme-1 below.
International (PCT) Publication No. WO 2011/079772 discloses a process for the preparation of canagliflozin by reduction of keto group of acetyl protected compound followed by hydrolysis.
U.S. PG-Publication No. 2014/0128595 discloses a process for the preparation of canagliflozin from anhydroglucopyranose derivative substantially as same as shown in scheme-2 below.
The prior-art processes requires sequence of protection/deprotection of canagliflozin obtained in the course of the reactions and further purification or crystallization to obtain canagliflozin in reasonably pure form. This sequences of processes results in high amount of yield loss.
In view of the above prior art, there is provided a novel, efficient and convenient process for preparation of canagliflozin which is at least a useful alternative to the prior art as well as an efficient and convenient method for purification of canagliflozin without sequence of protection and deprotection.
Scheme-3.

Ahmedabad-based pharma giant Cadila Healthcare’s chairman and managing director, Pankaj Patel,
EXAMPLES
Example-1Preparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Va) (5 g) and 150 mL toluene at 25° C. 1.5 mL (1.6M) n-butyl lithium in hexane was added dropwise at room temperature and the solution was stirred for 30 minutes. This solution was cooled to −78° C. and added dropwise to a solution of 3,4,5-tris((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV) (6.4 g) in 100 mL toluene and the mixture was stirred for 3 hours. The reaction mixture was treated with 2.5 g methanesulfonic acid in 100 mL methanol and stirred for 1 hour. The reaction mass was warmed to 25° C. and then added to pre-cool saturated sodium bicarbonate solution and resulting mass was extracted with ethyl acetate. The extract was washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain compound of Formula (III).
Example-1APreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Va) (5 g) and 150 mL toluene at 25° C. 1.5 mL (1.6M) n-butyl lithium in hexane was added dropwise at room temperature and the solution was stirred for 30 minutes. This solution was cooled to −78° C. and added dropwise to a solution of 3,4,5-tris((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV) (6.4 g) in 100 mL toluene and the mixture was stirred for 3 hours. The reaction mixture was treated with 2.5 g methanesulfonic acid in 100 mL methanol and stirred for 1 hour. The reaction mixture warmed to room temperature and stirred for 8 hours. Saturated sodium bicarbonate solution was added to the reaction mixture and the separated aqueous layer was extracted with toluene. The organic layer was distilled to remove toluene and the residue was dissolved in 50 mL methylene dichloride, washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain residue. The residue was treated with 150 mL diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed withdiisopropyl ether to obtain compound of Formula (III).
Example-1BPreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 5 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 100 g 2-(5-iodo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Vb), 114.35 g 3,4,5-tris((trimethylsilyl)oxy)-6-(((tri-methylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV), 2 L toluene and 1 Ltetrahydrofuran at 30° C. The reaction mixture was cooled to −78° C. and 171.45 mL n-butyl lithium in hexane (1.6M) was added and the solution was stirred for 3 hours. The reaction mixture was treated with 94.16 g methanesulfonic acid in 1500 mL methanol and stirred for 1 hour. The reaction mixture warmed to 25° C. and stirred for 8 hours. The reaction mixture was cooled to 5° C. and saturated sodium bicarbonate solution was added to the reaction mixture and stirred for 30 min. The separated aqueous layer was extracted with toluene. The organic layer was distilled to remove toluene and the residue was dissolved in 300 mL methylene dichloride and 200 g silica gel of 60-120 mesh was added. The reaction mixture was stirred for 30 min at 30° C., washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain residue. The residue was treated with 1 L diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed with diisopropyl ether to obtain compound of Formula (III).
Example-2APreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triol (IIa1)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 10 g compound of Formula (III), 80 mL methylene dichloride and 4.3 g N-methylmorpholine at −5 to 5° C. 2.7 g trimethylsilyl chloride was added slowly and stirred for 1 hour. After confirming the reaction completion TLC, 30 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The organic layer was evaporated under reduced pressure to obtain compound of Formula (IIa).
Example-2BPreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triol (IIa1)
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 100 g compound of Formula (III) and 900 mL methanol at 30° C. and stirred for 1 hour. The reaction mixture was filtered to remove silica gel and washed with methanol. The filtrate was distilled under vacuum to remove methanol completely, 350 mL methylene dichloride and 42.63 g N-methylmorpholine were added to the residue and cooled to at −5 to 5° C., 34.34 g trimethylsilyl chloride was lot-wise added and stirred for 45 min. After confirming the reaction completion TLC, 300 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The separated organic layer was dried over sodium sulfate and filtered to obtain compound of Formula (IIa1).
Example-3APreparation of Canagliflozin of Formula (I)
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added solution of compound (IIa) prepared in example-2B and cooled to −70° C. 8 mL triethylsilane and 5.5 mL boron trifluoridediethyl etherate were added dropwise within 1 hour maintaining the reaction temperature between −70° C. The reaction was warmed to −30° C. and stirred for 30 min. The reaction mixture was then added to freshly preparedsodium bicarbonate solution at 5° C. and then allowed to warm to room temperature and stirred for 20 mints to adjust the pH of 7-8. The reaction mass was then slowly added to cold water. The resulting mass was extracted with ethyl acetate. The combined organic layers were washed with saturated bicarbonatesolution, dried over Na2SO4 and evaporated under reduced pressure to obtain canagliflozin having purity 86% by HPLC.
Example-3BPreparation of Canagliflozin of Formula (I)
In 2 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added the solution of compound (IIa) prepared in example-2B and cooled to −70° C. 67.38 g triethylsilane and 83.08 g boron trifluoridediethyl etherate were added dropwise within 1 hour maintaining the reaction temperature between −70° C. The reaction was warmed to −30° C. and stirred for 3 hours. The reaction mixture was then added to freshly prepared sodium bicarbonate solution at 5° C. and then allowed to warm to room temperature and stirred for 20 mints to adjust the pH of 7-8. The reaction mixture was then slowly added to cold water. The separated aqueous layer was extracted with 200 mL methylene dichloride. The combined organic layer was washed with 300 mL water and distilled completely to remove methylene dichloride. The resulting residue extracted with 500 mL ethyl acetate and stirred to obtain clear solution. The reaction mixture was treated with brine and saturated bicarbonate solution to separate the layers. The separated organic layer was dried over sodium sulfate, charcoalized and filtered. The filtrate is distilled to remove ethyl acetate completely under vacuum. The residue was dissolved in 300 mL methylene dichloride and 200 g silica gel of 60-120 mesh was added. The reaction mixture was stirred for 30 min at 30° C. and distilled completely under reduced pressure to obtain residue. The residue was treated with 500 L diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed with diisopropyl ether to obtain canagliflozin (I) having purity 87% by HPLC.
Example-4Preparation of (3R,4S,5R,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl)tris(oxy)tris(trimethylsilane) (IIb1)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 10 g compound of Formula (III), 100 mL methylene dichloride and 15 g N-methylmorpholine at 0 to 5° C. 12.7 g trimethylsilyl chloride was added slowly and stirred for 1 hour. After confirming the reaction completion by TLC, 300 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The organic layer was evaporated under reduced pressure to obtain compound of Formula (IIb1).
Example-5Preparation of Canagliflozin
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added 20 g compound (IIb1) prepared in example-4 and 100 mL methylene dichloride at −25° C. to −30° C. 11 mL triethylsilane and 7.8 mL boron trifluoridediethyl etherate was added drop wise within 1-2 hours maintaining the reaction temperature between −25° C. to −30° C. The reaction was stirred for 30 min and then allowed to warn to room temperature and stirred for 1.5-2 hours. The reaction mixture was then slowly added to cold water. The reaction mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated bicarbonate solution, dried over sodium sulfate and evaporated under reduced pressure to obtain canagliflozin having purity 86% by HPLC.
Example-6Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 85%) and 100 mLtoluene were stirred to obtain a clear solution. 10 g Polyvinylpyrrolidone was added to the solution and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in ethyl acetate and water mixture for 30 min. The separated ethyl acetate layer was evaporated to dryness to obtain pure canagliflozin. (7.1 g. Purity 96.55% by HPLC).
Example-7Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 87%) and 100 mLtoluene were stirred in in a round bottom flask to obtain a clear solution. 10 g β-cyclodextrin was added to the solution and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in ethyl acetate and water mixture for 30 min. The separated ethyl acetate layer was treated with activated carbon, filtered and evaporated to dryness to obtain pure canagliflozin. (7.9 g, Purity 98.93% by HPLC).
Example-8Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 87%) and 0.25 g activated carbon were stirred in 100 mL toluene for 15-20 min and filtered. 10 g β-cyclodextrin was added to the filtrate and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in isopropyl acetate and water mixture for 30 min. The separated isopropyl acetate layer was evaporated to dryness to obtain pure canagliflozin. (7.7 g, Purity 99.12% by HPLC).
Example-9Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g canagliflozin (purity 87%) and 100 mLtoluene were stirred to obtain a clear solution. 10 g hydroxy propyl methyl cellulose was added to the solution and stirred for 2-3 hour. The reaction mixture was filtered, washed with toluene. The solid was stirred in isopropyl acetate and water mixture for 30 min and dried to obtain pure canagliflozin. (Purity 97-98% by HPLC).
Example-10Purification of Canagliflozin
In 2 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 100 g canagliflozin (purity 87%) obtained in example-3B and 900 mL methanol were stirred for 45 min at 30° C. The reaction mixture was filtered to remove silica gel. The filtrate was distilled under vacuum completely below 45° C. 400 mL toluene was added and heated to 55° C. to obtain a clear solution. The reaction mixture was filtered and the filtrate was added 100 g β-cyclodextrine. The reaction mixture was heated at 75° C. for 30 min and cooled to 30° C. and further stirred for 30 min. 5 g canagliflozin β-cyclodextrin complex was added to the solution and further cooled to 5° C. The reaction mixture was stirred for 3 hours and filtered. The wet-cake was treated with 300 mL isopropyl acetate and heated at 75° C. for 30 min. The reaction mixture was cooled to 30° C. and stirred for 6 hours and further cooled to 5° C. and stirred for 3 hours. The reaction mixture was filtered and washed with isopropyl acetate and dried at 30° C. to obtain crystalline canagliflozin β-cyclodextrine complex having 40 g pure canagliflozin with 99% purity by HPLC.
Example-11Preparation of Amorphous Canagliflozin
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 100 g canagliflozin β-cyclodextrine (purity 98%) obtained in example-10 and 400 mL acetone were stirred for 30 min at 30° C. The reaction mixture was filtered to remove β-cyclodextrine. The filtrate was distilled under vacuum completely below 45° C. 400 mL acetone was added to the residue to get clear solution at 30° C. 5 g activated charcoal was added and stirred for 20 min. The reaction mixture was filtered and the filtrate was spray dried using JISL Mini spray drier LSD-48 keeping feed pump at 30 rpm, inlet temperature at 60° C., outlet temperature at 40° C. and 2 Kg/cm2 hot air supply. The product was collected from cyclone and is further dried at 40° C.±5° C. under vacuum for 12 hours to get 80 g of amorphous canagliflozin having 99.6% purity by HPLC.
WO2014195966
https://www.google.co.in/patents/WO2014195966A2?cl=un
Canagliflozin is inhibitor of sodium dependent glucose transporter inhibitor (SGLT) which is chemically represented as (25′,3i?,4/?,55,,6 ?)-2-{3-[5-[4-Fluoro-phenyl]-thiophen-2-ylmethyl]-4-methyl-phenyl}-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol having (I).

Formula (I)
U.S. Patent No, 7,943,788 B2 (the ‘788 patent) discloses canagliflozin or salts thereof and the process for its preparation.
U.S. Patent Nos. 7,943,582 B2 and 8,513,202 B2 discloses crystalline form of 1 -(P-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene hemihydrate and process for preparation thereof. The US ‘582 B2 and US ‘202 B2 further discloses that preparation of the crystalline form of hemi-hydrate canagliflozin typically involves dissolving in a good solvent (e.g. ketones or esters) crude or amorphous compound prepared in accordance with the procedures described in WO 2005/012326 pamphlet, and adding water and a poor solvent (e.g. alkanes or ethers) to the resulting solution, followed by filtration.
U.S. PG-Pub. No. 2013/0237487 Al (the US ‘487 Al) discloses amorphous dapagliflozin and amorphous canagliflozin. The US ‘487 Al also discloses 1:1 crystalline complex of canagliflozin with L-proline (Form CS1), ethanol solvate of a 1: 1 crystalline complex of canagliflozin with D-proline (Form CS2), 1 :1 crystalline complex of canagliflozin with L-phenylalanine (Form CS3), 1:1 crystalline complex of canagliflozin with D-proline (Form CS4).
The US ‘487 Al discloses preparation of amorphous canagliflozin by adding its heated toluene solution into n-heptane. After drying in vacuo the product was obtained as a white solid of with melting point of 54.7°C to 72.0°C. However, upon repetition of the said experiment, the obtained amorphous canagliflozin was having higher amount of residual solvents. Therefore, the amorphous canagliflozin obtained by process as disclosed in US ‘487 Al is not suitable for pharmaceutical preparations.
The US ‘487 Al further discloses that amorphous canagliflozin obtained by the above process is hygroscopic in nature which was confirmed by Dynamic vapor sorption (DVS) analysis. Further, it was observed that the amorphous form underwent a physical change between the sorption/desorption cycle, making the sorption/desorption behavior different between the two cycles. The physical change that occurred was determined to be a conversion or partial conversion from the amorphous state to a crystalline state. This change was supported by a change in the overall appearance of the sample as the humidity increased from 70% to 90% RH.
The canagliflozin assessment report EMA/718531/2013 published by EMEA discloses that Canagliflozin hemihydrate is a white to off-white powder^ practically insoluble in water and freely soluble in ethanol and non-hygroscopic. Polymorphism has been observed for canagliflozin and the manufactured Form I is a hemihydrate, and an unstable amorphous Form II. Form I is consistently produced by the proposed commercial synthesis process.
Therefore, it is evident from the prior art that the reported amorphous form of canagliflozin is unstable and hygroscopic as well as not suitable for pharmaceutical preparations due to higher amount of residual solvents above the ICH acceptable limits.
Hence, there is a need to provide a stable amorphous form of canagliflozin which is suitable for pharmaceutical preparations.
Crystalline solids normally require a significant amount of energy for dissolution due to their highly organized, lattice like structures. For example, the energy required for a drug molecule to escape from a crystal is more than from an amorphous or a non-crystalline form. It is known that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailability patterns compared to the crystalline form (Econno T., Chem. Pharm. Bull., 1990; 38: 2003-2007). For some therapeutic indications, one bioavailability pattern may be favoured over another.
An amorphous form of some of the drugs exhibit much higher bioavailability than the crystalline forms, which leads to the selection of the amorphous form as the final drug substance for pharmaceutical dosage from development. Additionally, the aqueous solubility of crystalline form is lower than its amorphous form in some of the drugs, which may resulted in the difference in their in vivo bioavailability. Therefore, it is desirable to have amorphous forms of drugs with high purity to meet the needs of regulatory agencies and also highly reproducible processes for their preparation.
In view of the above, it is therefore, desirable to provide canagliflozin amorphous form as well as an efficient, economical and eco-friendly process for the preparation of highly pure canagliflozin amorphous form.
Example-l:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 25.0 g of canagliflozin was dissolved in 250.0 mL of methanol mixture at 25°C to 30°C. The content was stirred for 30 minutes at 25°C to 30°C. To this, 1.0 g charcoal was added and stirred for 30 minutes at 25°C to 30°C. The content was filtered through Hyflo-supercel, and the Hyflo-supercel pad was washed with 50.0 mL methanol. The filtrate was concentrated under vacuum below 45°C followed by spray drying in JISL Mini spray drier LSD-48 under the below conditions. The product was collected from cyclone and is further dried at 55°C±5°C under vacuum for 16 hours to get 19.0 g of amorphous canagliflozin.

The spray-dried canagliflozin is amorphous in nature. The obtained product contains residual solvent well within ICH limit.
The obtained solid was amorphous canagliflozin as is shown by the X-ray diffraction pattern shown in FIG.1.
Example-2:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 25.0 g of canagliflozin was dissolved in 250.0 mL of acetone mixture at 25°C to 3O°C. The content was stirred for 30 minutes at 25°C to 30°C. To this, 1.0 g charcoal was added and stirred for 30 minutes at 25°C to 30°C. The content was filtered through Hyflo-supercel, and the Hyflo-supercel pad was washed with 50.0 mL acetone. The filtrate was concentrated under vacuum below 45°C followed by spray drying in JISL Mini spray drier LSD-48 under the below conditions. The product was collected from cyclone and is further dried at 55°C±5°C under vacuum for 16 hours to get 20.0 g of amorphous canagliflozin.

The spray-dried canagliflozin is amorphous in nature. The compound is having residual acetone less than 0.5% by GC.
The obtained solid was amorphous canagliflozin as is shown by the X-ray diffraction pattern shown in FIG.2.
Example-3:
Preparation of amorphous form of canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g of canagliflozin was dissolved in 125 mL methanol and heated to obtain clear solution at 65°C. The solution was distilled to remove methanol completely. The compound thus obtained was amorphous canagliflozin.
Example-4:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g of canagiiflozin was dissolved in 125 mL acetone and heated to obtain clear solution at 65°C. The solution was distilled to remove acetone completely. The compound thus obtained was amorphous canagiiflozin. The compound is having residual acetone less than 0.5% by GC.
Example 5:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottom flask equipped with mechanical stirrer, thermometer and an addition funnel, canagiiflozin (0.5 gm, 1.02 mmol), PVP K-30 (4 gm, 8 times) and 88% methanol in water (12.5ml, 25V) were heated to 65-70°C to get clear solution. The reaction mixture was stirred for 1 hour, concentrated under vacuum (1.5 mbar) at 65-70°C and degassed under vacuum (1.5 mbar) for 1 hour at 70°C to obtain the title compound in amorphous form.
Example 6:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottom flask equipped with mechanical stirrer, thermometer and an addition funnel, canagiiflozin (0.5 gm, 1.02 mmol), HPMC-AS (1 gm, 2 times) in 88% methanol in water (12.5 ml, 25V) were heated at 65 to 70°C to get clear solution. The reaction mixture was stirred for 2 hours, concentrated under vacuum (1.5 mbar) at 70°C and degassed under vacuum (1.5 mbar) for lhr at 70°C to obtain the title compound in amorphous form.
Example-7:
Preparation of canagliflozin-L-Proline crystalline complex
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel; 25.0 g of canagiiflozin, 6.06 g L-proline and 250 mL ethanol were heated to 75-80°C, stirred for 15 min and then cooled down to 25-30°C. The mass was filtered and washed with ethanol to obtain canagliflozin-L-proline crystalline complex.
Example-8:
Preparation of amorphous canagliflozin from canagliflozin-L-proline crystaUine complex
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of Canagliflozin-L-Proline Crystalline Complex and 250 mL of ethyl acetate were stirred to get a clear solution, washed with 2×150 mL of water and the organic layer was distilled. To the residue 100 mL of isopropyl acetate and 2.5 mL of water was added and heated to 75-80°C, stirred for 15 min and cooled down to 25-30°C. The mass filtered and washed with isopropyl acetate to obtain canagliflozin. The obtained canagliflozin was subjected to spray dyring under conditions of example-2 using acetone solvent to obtain amorphous canagliflozin. Purity > 99.5% by HPLC. The compound is having residual acetone less than 0.5% by GC.
The obtained solid was amorphous canagliflozin as shown by the X-ray diffraction pattern shown in FIG.2.
HPLC Purity of amorphous canagliflozin was measured by using following chromatographic conditions:
Equipment: Shimadzu LC2010C HPLC system equipped with a dual
wavelength UV-VIS detector or equivalent
Column: romasil C-8 (250mmx4.6 mm, 5 μπι) or equivalent
Flow rate: 1.5 mL/minute
Column oven temp.: 30°C
Wavelength: 210 nm
Injection Volume: 10 μΐ, .
Diluent: Mobile Phase A: Mobile Phase B (30:70)
Mobile Phase A: Buffer:Acetonitrile:Methanol (60:30: 10)
Mobile Phase B: Acetonitrile: Methanol (80:20)
Example-9:
Preparation of amorphous form of Canagliflozin as per Example-2 of US ‘487 Al In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to get clear solution. 100 mL of n-heptane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 65°C under vacuum for 72 hours. The residual n-heptane was 44000 ppm by GC after 72 hours drying.
Example-10:
Replacing toluene with ethyl acetate in above example-9
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to obtain clear solution. 100 mL of n-heptane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 65°C under vacuum for 72 hours. The residual n-heptane was -44000 ppm by GC after 72 hours drying.
Example-11:
Replacing n-heptane with cyclohexane in above example-9
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to obtain clear solution. 100 mL of cyclohexane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 55°C under vacuum for 72 hours. The residual cyclohexane was >5000 ppm by GC after 72 hours drying.
Example-12:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel; 25.0 g of canagliflozin and 250 mL of ethyl acetate were stirred to get clear solution and then ethyl acetate was removed under reduced pressure to obtain 20.0 g of amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 55°C under vacuum for 72 hours. The residual ethyl acetate was -8450 ppm by GC after 72 hours drying.
///////////////New Patent, Zydus Cadila, Canagliflozin, US 20160002275
WO 2015177807, New patent on AVANAFIL by Wanbury
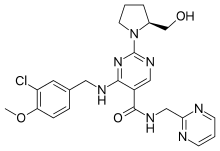

WO 2015177807
Suryakant Shivaji Pol; Nitin Sharadchandra Pradhan; Shashikant Balu Padwal; Vihar Raghunath Telange; Nitn Shankar Bondre
Wanbury ltd
![]()
The present invention relates to a novel compound of Formula (II), and its use in preparation of Avanafil, [Formula should be inserted here] wherein R is -OH, -CI or -OR1 and R1 is C1 to C3 alkyl group

It having been developed and launched by VIVUS and JW Pharmaceutical, under license from Mitsubishi Tanabe Pharma, and Auxilium Pharmaceuticals, for treating ED.
A process for preparation of Avanafil was first disclosed in US 6,797,709 (depicted in Scheme I), wherein 4-chloro-5-ethoxycarbonyl-2-methylthio-pyrimidine is coupled with 3-chloro-4-methoxybenzylamine in presence of triethylamine to provide compound of Formula (A), which on oxidization provides a sulfonyl compound of Formula (B). Said compound of Formula (B) is reacted with L-prolinol and exert compound of Formula (C). The resulting compound of Formula (C) undergoes column chromatographic purification and crystallization, while further subjected to hydrolysis to obtain compound of Formula (D). The compound of Formula (D) is coupled with 2-aminomethylpyrimidine to obtain Avanafil of Formula (I). The final product obtained is purified by column chromatography. The need to purify the intermediate compound of Formula (C) and final product, by column chromatography makes this process cumbersome, time consuming and unviable for large scale production thereby contributing to main disadvantages of the process.
Scheme I

Formula (A)
m-CPBA/chloroform

Formula (C) Formula (B)
NaOH/DMSO

Formula (D) Formula (I)
CN 103254179, discloses a process for preparation of Avanafi, wherein 3-chloro-4-methoxybenzylhalide is coupled with cytosine to result compound of Formula (E), later on condensation with L-prolinol yields 4-[(3-chloro-4-methoxy benzyl)amino-2-(2-hydroxymethyl)-l -pyrrolinyl]pyrimidine of Formula (F). The compound of Formula (F) is then condensed with N-(2-pyrimidylmethyl)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme II
Scheme II

Formula (F) Formula (I)
CN 103254180 describes an alternate process for preparation of Avanafil of Formula (I), wherein a substitution reaction on 6-amino-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester and 3-chloro-4-methoxybenzylchloride provides 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester of Formula (G) which on condensation with L-prolinoI generates 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid ethyl ester of Formula (H). The compound of Formula (H) is then hydrolysed and coupled with N-(2-pyrimidylmethyI)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme III
Scheme III

Formula (H) Formula (Γ)
In all the prior art discussed above, chiral compound L-prolinol is coupled in molecule in earlier steps of synthesis. This approach seems to be less feasible for large scale production; the insertion of L-prolinol in early stage may need to exert number of purifications for intermediates. Further the main shortcoming in such process is that the chirality of molecule is disturbed by inserting L-prolinol in early stages because there are number of operations in line in process to obtain the target compound.
CN 103483323, discloses a synthetic method for preparation of avanafil, wherein amidation of pyrimidine-5-carbonyl chlorides with 2-(aminomethyl)pyrimidine at temperature ranging from -10 to 5°C resulted an amide (intermediates A); which underwent condensation with 3-chloro-4-methoxybenzylamine at the temperature ranging from 0 -3°C to give 4-[(3-chloro-4-methoxybenzyl)amino]-5-
pyrimidinecarboxamides (intermediates B), which further on condensation with L-prolinol gave avanafil. The disadvantage of this process is the need to maintain the reaction temperature in range of – 10 to 5°C which adds up to cost of process and makes the process complicated. The process is depicted in Scheme IV.
Scheme IV

Intermediate (A)

wherein, R’ & R2 are independently, hydrogen, halogen, alkoxy, alkoxyalkyl, cyno group, amino group
Hence, to overcome shortcomings of prior art the inventors of present invention have skillfully designed a process with novel intermediate which concomitantly result Avanafil compound of Formula (I), substantially free from impurities. Further this invention encompass L-proline in last stage of molecule in order to avoid the number of purifications of intermediate which relent the economic significances by taking into account yield of each stage.
![]()
Object of the invention
1. The main object of the invention is to provide a novel compound of Formula
(ID-
2. Another object of present invention is to provide a process for preparation of a novel compound of Formula (II).
3. Yet another object of present invention is to provide a process for preparation of Avanafil of Formula (I), in high yield and purity using a novel compound of Formula (II).
4. Yet another object of the present invention to provide simple, economic and industrially scalable process for the preparation of Avanafil o Formula (I).
Summary of the invention
According to an aspect of present invention, there is provided a novel compound of Formula (II).

Formula (II)
wherein R is -OH, -CI or -OR and R is Q to C3 alkyl group
The invention will be specifically described below with reference to Examples but it should not be construed that the scope of the invention is limited thereto. Since the starting compound was produced by a modified method from that described in prior art, it will be described as Referential Example 1 to 3. Here synthesis routes of Referential Example 1 to 3 and Example 1 to 10 are illustrated below in Scheme (V).
Scheme (V)

Formula (I) Referential Examples
Referential Example 1 – Preparation of ethyl 4-[(3-chloro-4-methoxybenzyl)amino]-2-(methyl sulfanyl)pyrimidine-5-carboxylate
To 600ml of methylene dichloride was added l OOg of ethyl 4-chloro-2-(methylsulfanyl) pyrimidine-5-carboxylate and 91.2g of 3-chloro-4-methoxybenzylamine. The reaction mixture was stirred and 500ml of water, 48g of sodium carbonate and Ig of tetra-butylammonium bromide were added to it. The reaction mixture was then maintained overnight at 25-30°C. After completion of reaction, methylene dichloride layer was separated, washed with water and evaporated to obtain 145g of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate having 95% of HPLC purity.
Above reaction can also be carried out using ammonia or triethylamine in same reaction conditions and parameters, in place of sodium carbonate.
Referential Example 2 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid
To 600ml of methanol was added l OOg of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate (Referential Example 1) and an aqueous solution of sodium hydroxide (15g of NaOH in 140ml of water). The reaction mixture was heated to reflux temperature. After completion of reaction, the pH of mixture was adjusted to 1 -2 using concentrated hydrochloric acid followed by stirring the mixture for 1 hour at 10-15°C. The solid product obtained was filtered, washed sequentially with water and methanol, and dried overnight at 70-75°C to get 87g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid.
Referential Example 3 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III)
To a mixture of 400ml of toluene and 0.5ml of dimethyl formamide was added 50g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid (Referential Example 2) and 70g of thionyl chloride, and the reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain yellow solid mass. The solid mass thus obtained, was cooled to 15-20°C followed by addition of 1 75ml of methylene dichloride, 36. l g of 2-amino methyl pyrimidine mesylate and 35.55g of triaethylamine. The reaction mixture was stirred overnight at 25-30°C. After completion of reaction, methylene dichloride was distilled out to get residue. The residue was washed sequentially with 2.5% sodium carbonate solution and water. The residue was then treated with methanol to obtain 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) having HPLC purity of more than 95% (yield: 80%)
Referential Example 4 – Preparation of 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[(2S)-2-(hydroxymethyl)-l -pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide (Avanafil)
Step i)
To 200ml of dichloromethane was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyI)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting dichloromethane layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the dichloromethane layer obtained in step i), was added 2.57g of triethylamine followed by slow addition of 125ml solution of L-prolinol in dichloromethane (2.46g of L-prolinol in 125ml of dichlromethane). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water followed by evaporation of dichloromethane to obtain an oily mass. The oily mass thus obtained was treated with methanol to yield 8g of Avanafil.
Examples
Example 1 : Preparation of Compound of Formula (II) (wherein R is -OH)
Step i)
To 200ml of methylene dichloride was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting methylene dichloride layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the methylene dichloride layer comprising compound of Formula (IV) obtained in step i), was added 5g of triethylamine followed by slow addition of 125ml solution of L-proline in methylene dichloride (2.8g of L-proline in 125ml of methylene dichloride). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water and 5% sodium carbonate solution, followed by evaporation of methylene dichloride to obtain an oily mass. The oily mass obtained was stripped with 50ml acetone to yield 9g of compound of Formula (II) having HPLC purity 98%.
Example 2: Preparation of Compound of Formula (II) (wherein R is -OC2H5)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filtrate containing product. The ethanol in filtrate is completely distilled out to isolate 10.45g of esterified compound of Formula (II).
Example 3 : Preparation of Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of toluene and 0.5ml of dimethylformamide was added 50g of compound of Formula (II) obtained in example 1 , and 70g of thionyl chloride. The reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain 50.5g of oily carboxylic acid chloride compound of Formula (II).
Example 4: Preparation of Avanafil of Formula (I)
In an inert atmosphere, a solution of 30g of compound of Formula (II) obtained in example 1 or 2, in 150 ml of tetrahydrofuran was dropwise added to 180ml of suspension of 1.0M lithium aluminium hydride solution in tetrahydrofuran, The reaction mixture was refluxed for 5 hours. After completion of reaction, the mixture was cooled in ice-bath and saturated aqueous solution of sodium sulfate was added to decompose excess of lithium aluminium hydride. The mixture was then diluted with 200ml of methylene dichloride and thus formed organic layer was separated. The organic layer was washed with water (3 χ 100 ml), dried over MgS04 and concentrated to collect crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 22.8g of Avanafil of Formula (I) having HPLC purity of 99.20%.
Example 5 : Preparation of Avanafil of Formula (I)
To a mixture of 1.3g sodium borohydride, 1 ml methanesulfonic acid and 50ml ethanol was added l Og of compound of Formula (II) obtained in example 1 or 2, and the mixture was stirred at 25-30°C for 5 hours. After completion of reaction, 100ml water was added and the mixture was extracted with 1 00ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with ethyl acetate at 45- 50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 6 to Example 8
The procedure is carried out as in example 5 except for instead of methanesulfonic acid other reducing agents are used in combination with sodium borohydride. The results are given in Table I
Table I

Example 9: Preparation of Avanafil of Formula (I)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filterate containing product. To the fi Iterate was added 1.2g of sodium borohydride and 2.6g of lithium bromide, and the mixture was stirred for 5 hours. After complete conversion of ester to final product, l OOml water was added and the mixture was extracted with 100ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with 25ml ethyl acetate at 45-50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7.5g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 10: Preparation of Avanafil of Formula (I) from Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of tetrahydrofuran and 50g of carboxylic acid chloride compound of Formula (II) obtained in example 3, was added 12g sodium borohydride at 0-5°C. After completion of reaction, water was added to reaction mixture to decompose excess of sodium borohydride present. The reaction mixture was then concentrated and a solution of 30g of potassium hydroxide in 200 ml of water was added. The mixture was heated to 60-70°C and maintained for 15-18 hours. The mixture was then cooled to 25-30°C and 500 ml of methylene dichloride was added. The organic layer thus formed, was separated and evaporated to yield crude Avanafil
of Formula (I) which was then subjected to purification using methanol as solvent to obtain 40g of Avanafil of Formula (I) having HPLC purity of 99.01%.
![]()

| Mr. K. Chandran | ||
| Wholetime Director & Vice Chairman |
 Tarapur plant
Tarapur plant

, Director, Wanbury, and Mr Asok Shinkar, Director-Corporate Finance, at a press conference held in Mumbai on Monday. Paul Noronha")
MR K. CHANDRAN (left), Director, Wanbury, and Mr Asok Shinkar, Director-Corporate Finance, at a press conference held in Mumbai on Monday.
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
//////////////
Zydus Cadila Healthcare Ltd, WO 2015102017, lorcaserin












Processes for the preparation of lorcaserin
Zydus Cadila Healthcare Ltd
| Applicants: | CADILA HEALTHCARE LIMITED [IN/IN]; Zydus Tower, Satellite Cross Roads Ahmedabad – 380 015 Gujarat (IN) |
| Inventors: | DWIVEDI, Shriprakash Dhar; (IN). SHAH, Alpeshkumar Pravinchandra; (IN). GAJJAR, Samir Rameshbhai; (IN). KHERA, Brij; (IN) |

On 10 May 2012, after a new round of studies submitted by Arena, an FDA panel voted to recommend lorcaserin with certain restrictions and patient monitoring. The restrictions include patients with a BMI of over 30, or with a BMI over 27 and a comorbidity such as high blood pressure or type 2 diabetes.
On 27 June 2012, the FDA officially approved lorcaserin for use in the treatment of obesity for adults with a BMI equal to or greater than 30 or adults with a BMI of 27 or greater who “have at least one weight-related health condition, such as high blood pressure, type 2 diabetes, or high cholesterol
Useful for treating obesity.
The present invention relates to stable crystalline Form I of Iorcaserin hydrochloride of Formula (IA) and processes for its preparation. The invention also relates to processes for the preparation of lorcaserin and pharmaceutically acceptable salts, solvates and hydrates thereof.
Stable crystalline form I of lorcaserin hydrochloride and its process of preparation are claimed. Represents the first patenting from Cadila on lorcaserin, which was developed and launched by Arena Pharma and Eisai.
In July 2015, Newport Premium™ reported that Cadila is potentially interested in lorcaserin.
Lorcaserin hydrochloride is an agonist of the 5-HT2c receptor and shows effectiveness at reducing obesity in animal models and humans developed by Arena Pharmaceuticals. It is chemically represented as (R)-8-chloro-l -methyl -2,3,4,5-tetrahydro-lH-3-benzazepine hydrochloride having Formula (I) as depicted herein below.

(IA)
U.S. Patent No. 6,953,787 B2 discloses compound of Formula (I) and pharmaceutically acceptable salt, solvates or hydrates thereof and process for preparation thereof.
U.S. Patent No. 8,168,624 B2 discloses (R)-8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine hydrochloride hemihydrate and process for its preparation. The patent also discloses crystalline Form I, Form II and Form III of (R)-8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine hydrochloride. The crystalline Form
I and Form II are reported as anhydrous, non-solvated crystal forms. The crystalline Form III displays a dehydration feature calculated as a 3.7% weight loss which is consistent with the theoretical weight loss of 3.7% for a hemihydrate.
The patent discloses that anhydrous Form I and Form II readily converts to a hemihydrate, upon exposure to moisture. The dynamic vapor sorption (DVS) data for each of the three crystal forms reveals the hygroscopic nature of both Forms I and II, which readily adsorb moisture at relative humidity (RH) greater than about 40-60%. In addition, both Forms I and II were calculated to adsorb about 3.8% moisture between about 40 and about 80% RH which is consistent with conversion to the hemihydrate (Form III). X-ray powder diffraction (XRPD) carried out on both Forms I and II after the DVS cycle confirmed this conversion. In contrast, the DVS data in connection with Form III shows that it is substantially non-hygroscopic, adsorbing less than 0.5% water at 90% RH and the XRPD pattern showed no change in crystalline form after the DVS cycle.
International (PCT) Publication Nos. WO 2003/086306 Al, WO 2005/019179 Al, WO 2006/069363 Al, WO 2007/120517 Al, WO 2008/07011 1 Al and WO 2009/1 1 1004 Al disclose various synthetic approaches for the preparation of (R)-8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine, its related salts, enantiomers, crystalline forms and intermediates.
International (PCT) Publication No. WO 2006/071740 Al discloses combination of (R)-8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine with other agents. International (PCT) Publication No. WO 2012/030938 Al discloses various salts of lorcaserin with optically active acids.
U.S. PG-Pub No. US 2014/0187538 Al discloses amorphous lorcaserin hydrochloride and amorphous solid dispersion comprising lorcaserin hydrochloride and one or more pharmaceutically acceptable carriers and processes for their preparation.
International (PCT) Publication No. WO 2014/135545 Al discloses solid dispersion comprising amorphous lorcaserin hydrochloride and one or more pharmaceutically acceptable water soluble polymers.

Example-7: Preparation of crystalline Form I of lorcaserin hydrochloride. In a round bottom flask, 560g of methyl ethyl ketone and 40 ml water were taken and 100 g of 8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine was added and stirred for 10 minutes. The reaction mass heated to 55 to 60°C and 19.3 g of. L-(+)-tartaric acid was added slowly and stirred for one to two hours. The reaction mass was further stirred at 10-15°C for an hour and the product was filtered and washed with a mixture of methyl ethyl ketone and water. The wet cake and 150 ml methyl ethyl ketone were taken in another flask and heated to 75-80°C. 20-25 ml water was, added and stirred for an hour. Further, the reaction mass was stirred for an hour at 0-5°C. The product was filtered and washed with methyl ethyl ketone.
100 g tartrate salt of 8-chloro-l-methyl-2,3,4,5-tetrahydro-lH-3-benzazepine and 300 mL water were taken in another round bottom flask. 200 mL methylene dichloride was added and the reaction mass was cooled to 10-20°C. 17.2 g sodium hydroxide dissolved in 89 ml water was added into the reaction mass at 10-20°C. The reaction mass was stirred for an hour at 25-30°C and the layers were separated. The solvent was removed from the organic layer under vacuum and then 100 mL ethyl acetate was added into that and distilled out. Further, 100 mL ethyl acetate was added and stirred for 15 minutes. The reaction mass was filtered through a hyflow bed and the filtrate was treated with dry HC1 gas till a pH of 1.5 to 2.5 was obtained at 0-10°C and it was stirred for about 30 minutes to an hour. The product was then filtered and washed with ethyl acetate and then dried in a vacuum oven at 50°C to 55°C for 2 hours. The product was further dried at 90°C to 110°C for 20 hours to obtain crystalline Form I of lorcaserin hydrochloride. Yield: 87.5-98.6 %.
Example-8: Preparation of crystalline Form I of lorcaserin hydrochloride

In a round bottom flask, 2.20 g lorcaserin, 30 mL methylene chloride, 17.4 mL of 1M HCI in ether were added and the mixture was stirred for 5-15 minutes at room temperature. The solvent was removed under reduced pressure to give a white solid. This solid was again dissolved in 30 ml methylene chloride, 17.4 mL of 1M HCI solution and stirred for 5-15 minutes at room temperature. The solvent was removed under reduced pressure to give lorcaserin hydrochloride. The product was dried in a vacuum oven at 50°C to 55°C for 2 hours. The product was further dried at 90°C to 110°C for 20 hours to obtain crystalline Form I of lorcaserin hydrochloride.
Example-9: Preparation of crystalline Form I lorcaserin hydrochloride
50 g of lorcaserin hydrochloride hemihydrate and 50 g of hydroxypropylmethyl cellulose (HPMC) 3CPC were mixed in a blender at 25°C to 35°C. The mixture was mixed for 30 minutes and unloaded. The solid thus obtained was dried in a vacuum oven at 50°C to 55°C for 2 hours. The product was further dried at 90°C to 110°C for 20 hours to obtain crystalline Form I of lorcaserin hydrochloride.

Pankaj R. Patel (right), Chairman and Managing Director,
/////////
