
Home » NDA (Page 5)
Category Archives: NDA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gilead Submits NDA to FDA for Sofosbuvir for the Treatment of Hepatitis C

Sofosbuvir
Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate

9 APRIL 2013
Gilead Sciences today announced that the company has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of sofosbuvir, a once-daily oral nucleotide analogue for the treatment of chronic hepatitis C virus (HCV) infection. The data submitted in this NDA support the use of sofosbuvir and ribavirin (RBV) as an all-oral therapy for patients with genotype 2 and 3 HCV infection, and for sofosbuvir in combination with RBV and pegylated interferon (peg-IFN) for treatment-naïve patients with genotype 1, 4, 5 and 6 HCV infection.
Chronic HCV infection affects up to four million Americans, particularly individuals born between 1946 and 1964. The disease is the leading cause of liver cancer and liver transplantation in the United States. Treatment for HCV currently includes 24-48 weeks of therapy with peg-IFN, which has to be injected and is associated with significant side effects, leaving some patients unable to complete therapy. If approved, sofosbuvir would shorten HCV therapy to 12 to 16 weeks, and depending on the genotype, would either eliminate or reduce the duration of peg-IFN injections.
“Current therapies are not suitable for large numbers of patients with HCV infection, and are challenging to take and tolerate,” said John C. Martin, PhD, Chairman and Chief Executive Officer of Gilead Sciences. “Sofosbuvir’s antiviral potency, safety profile and once-daily administration have the potential to improve cure rates by simplifying and shortening therapy for patients with this disease.”
The sofosbuvir NDA is supported primarily by data from four phase 3 studies, NEUTRINO, FISSION, POSITRON and FUSION, in which 12 or 16 weeks of sofosbuvir-based therapy was found to be superior or non-inferior to currently available treatment options or historical controls, based on the proportion of patients who had a sustained virologic response (HCV undetectable) 12 weeks after completing therapy (SVR12). Patients who achieve SVR12 are considered cured of HCV.
Gilead plans to file for regulatory approval of sofosbuvir in other geographies, including the European Union, in the second quarter of 2013. The European Medicines Agency (EMA) has accepted Gilead’s request for accelerated assessment for sofosbuvir, a designation that is granted to new medicines of major public health interest. Accelerated assessment could shorten the EMA’s review time of sofosbuvir by two months. Granting of accelerated assessment does not guarantee a positive opinion from the CHMP or approval by the European Commission.
Sofosbuvir (formerly PSI-7977 or GS-7977) is an experimental drug candidate for the treatment of hepatitis C.[1] It was discovered at Pharmasset and then acquired for development by Gilead Sciences. It is currently in Phase III clinical trials.[2]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-monophosphate.[3]
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) polymerase.[4] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[5] It has shown excellent clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[6]
- Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S. et al. (2010). “Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus”. Journal of Medicinal Chemistry 53 (19): 7202–7218. doi:10.1021/jm100863x. PMID 20845908. edit
- “PSI-7977″. Gilead Sciences.
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H. M. M.; Bao, D.; Chang, W.; Espiritu, C. et al. (2010). “Mechanism of Activation of PSI-7851 and Its Diastereoisomer PSI-7977″. Journal of Biological Chemistry 285 (45): 34337–34347. doi:10.1074/jbc.M110.161802. PMC 2966047. PMID 20801890. edit
- Alejandro Soza (November 11, 2012). “Sofosbuvir”. Hepaton.
- Tom Murphy (November 21, 2011). “Gilead Sciences to buy Pharmasset for $11 billion”. Bloomberg Businessweek.
- http://www.gilead.com/pr_1757156
- AASLD: PSI-7977 plus Ribavirin Can Cure Hepatitis C in 12 Weeks without Interferon. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Nucleotide Polymerase Inhibitor Sofosbuvir plus Ribavirin for Hepatitis C. Gane, E et al. New England Journal of Medicine 368:3444. January 3, 2013.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, L. HIVandHepatitis.com. 4 March 2013.
Bayer PAH drug Riociguat gets priority review at FDA

RIOCIQUAT
CAS NO 625115-55-1
Methyl N-[4,6-Diamino-2-[1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl]-5-pyrimidinyl]-N-methyl-carbaminate
9 APRIL2013
Bayer has been boosted by the news that regulators in the USA are fast-tracking the German group’s investigational pulmonary arterial hypertension riociguat.
The US Food and Drug Administration has granted priority review to the New Drug Application for riociguat, which Bayer filed in February on both sides of the Atlantic for PAH and a related condition, inoperable chronic thromboembolic pulmonary hypertension (CTEPH). The FDA bestows a priority review on medicines that offer major advances in care or that provide a treatment where no adequate therapy exists. The agency aims to complete its assessment within eight months from the submission of the NDA, rather than the standard 12 months.
Riociguat (BAY 63-2521) is a novel drug that is currently in clinical development by Bayer. It is a stimulator of soluble guanylate cyclase (sGC). At the moment Phase III clinical trialsinvestigate the use of riociguat as a new approach to treat two forms of pulmonary hypertension (PH): chronic thromboembolic pulmonary hypertension (CTEPH) andpulmonary arterial hypertension (PAH). Riociguat constitutes the first drug of a novel class of sGC stimulators
The submissions are based on two Phase III studies and riociguat, the first member of a novel class of compounds called stimulators of soluble guanylate cyclase (sGC), met its primary endpoint in both trials, a change in exercise capacity after 12- or 16 weeks respectively. The drug was generally well tolerated, with a good safety profile.

If approved, riociguat would be going up against Actelion’s Tracleer (bosentan) and Gilead Sciences/GlaxoSmithKline’s Letairis/Volibris (ambrisentan). Actelion, which has dominated the PAH market, has already filed its follow-up to Tracleer, Opsumit (macitentan).
Links
Medivir AB :New Drug Application has been filed with FDA for Simeprevir (TMC435) for combination treatment of adult patients with genotype 1 chronic hepatitis C

Simeprevir
03/28/2013| Medivir AB announced that a new drug application (NDA) has been filed with the U.S. Food and Drug Administration (FDA) seeking approval for simeprevir. The filing is based on phase III data in treatment-naïve and treatment-experienced patients with compensated liver disease.
The filing of a regulatory application in the US triggers a milestone payment of ?10m to Medivir.
Simeprevir is jointly developed by Medivir and Janssen Pharmaceuticals, Inc. (Janssen), and is an investigational NS3/4A protease inhibitor, administered as a 150 mg capsule once daily with pegylated interferon and ribavirin for the treatment of genotype 1 chronic hepatitis C in adult patients.
“The filing in the U.S. is a very important milestone for simeprevir, the hepatitis C patients and for Medivir as a company. In addition it triggers a ? 10m milestone payment to us, which strengthens our solid financial situation even more.” comments Maris Hartmanis, CEO of Medivir.
The regulatory submission for simeprevir is supported in part by data from three pivotal phase III studies: QUEST-1 and QUEST-2 in treatment-naïve patients and PROMISE in patients who have relapsed after prior interferon-based treatment. In each study, participants were treated with one 150 mg simeprevir capsule once daily for 12 weeks plus pegylated interferon and ribavirin for 24 or 48 weeks. Primary efficacy data from the phase III studies will be presented at different upcoming medical meetings.
About Simeprevir
Simeprevir, an investigational next generation NS3/4A protease inhibitor jointly developed by Janssen R&D Ireland and Medivir AB, is currently in late phase III studies as a once-daily capsule (150 mg) taken in combination with pegylated interferon and ribavirin for the treatment of genotypes 1 and 4 HCV.
Global phase III studies of simeprevir include QUEST-1 and QUEST-2 in treatment-naïve patients, PROMISE in patients who have relapsed after prior interferon-based treatment and ATTAIN in null-responder patients. In parallel to these trials, phase III studies for simeprevir are ongoing in treatment-naïve and treatment-experienced HIV-HCV co-infected patients, HCV genotype 4 patients and Japanese HCV genotype 1 patients. Janssen recently announced the submission of a new drug application for simeprevir in Japan for the treatment of genotype 1 hepatitis C.
Simeprevir is being studied in phase II interferon-free trials with and without ribavirin in combination with:
- Janssen’s non-nucleoside inhibitor TMC647055 and ritonavir in treatment-naïve genotype 1a and 1b HCV patients;
- Gilead Sciences, Inc.’s nucleotide inhibitor sofosbuvir (GS-7977) in treatment-naïve and previous null-responder genotype 1 HCV patients; and
- Bristol-Myers Squibb’s NS5A replication complex inhibitor daclatasvir (BMS-790052) in treatment-naive and previous null-responder genotype 1 HCV patients.
In addition, Janssen has a non-exclusive collaboration with Vertex Pharmaceuticals to evaluate in a phase II study the safety and efficacy of an all-oral regimen of simeprevir and Vertex’s investigational nucleotide analogue polymerase inhibitor VX-135 for the treatment of HCV. As a first step, Janssen Pharmaceutical Inc. will conduct a drug-drug interaction (DDI) study with simeprevir and VX-135.
We also recently announced plans to initiate a phase II trial of an investigational interferon-free regimen with simeprevir, TMC647055 and Idenix’s IDX719, a once-daily, pan-genotypic NS5A inhibitor, with and without ribavirin.
About Hepatitis C
Hepatitis C, a blood-borne infectious disease of the liver and a leading cause of chronic liver disease and liver transplants, is a rapidly evolving treatment area with a clear need for innovative treatments. Approximately 150 million people are infected with hepatitis C worldwide, and 350,000 people per year die from the disease.
About Medivir AB
Medivir is an emerging research-based pharmaceutical company focused on infectious diseases. Medivir has world class expertise in polymerase and protease drug targets and drug development which has resulted in a strong infectious disease R&D portfolio. The Company’s key pipeline asset is simeprevir, a novel protease inhibitor in late phase III clinical development for hepatitis C that is being developed in collaboration with Janssen R&D Ireland.
Simeprevir (formerly TMC435) is an experimental drug candidate for the treatment of hepatitis C. It is being developed by Medivir and Johnson & Johnson’s pharmaceutical division Janssen Pharmaceutica and is currently in Phase III clinical trials.[1]
Simeprevir is a hepatitis C virus protease inhibitor.[2]
Simeprevir is being tested in combination regimens with pegylated interferon alfa-2a and ribavirin,[3] and in interferon-free regimens with other direct-acting antiviral agents including daclatasvir[4] and sofosbuvir [5]
- “Medivir Announces That Simeprevir (TMC435) Data Will Be Presented at the Upcoming AASLD Meeting”. Yahoo News. October 1, 2012. Retrieved November 6, 2012.
- Lin, TI; Lenz, O; Fanning, G; Verbinnen, T; Delouvroy, F; Scholliers, A; Vermeiren, K; Rosenquist, A et al. (2009). “In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor”. Antimicrobial agents and chemotherapy 53 (4): 1377–85. doi:10.1128/AAC.01058-08. PMC 2663092. PMID 19171797.
- “Phase 3 Studies Show Simeprevir plus Interferon/Ribavirin Cures Most Patients in 24 Weeks”. hivandhepatitis.com. December 27, 2012.
- Medivir announces TMC435 in an expanded clinical collaboration. Medivir. 18 April 2012.
- [http://www.medivir.se/v4/en/ir_media/pressrelease.cfm Results from a phase IIa study evaluating Simeprevir and Sofosbuvir in prior null responder Hepatitis C patients have been presented at CROI. 6 March 2013.
FDA Approves New Drug Application (NDA) for Teva’s Quartette (levonorgestrel/ethinyl estradiol and ethinyl estradiol) Tablets for the Prevention of Pregnancy

levonorgestrel

ethinyl estradiol
Quartette™ Represents the Next Generation of Extended Regimen Oral Contraceptives
JERUSALEM 30 MAR 2013
Teva Pharmaceutical Industries Ltd. today announced that the U.S. Food and Drug Administration (FDA) has approved Quartette™ (levonorgestrel/ethinyl estradiol and ethinyl estradiol) tablets for the prevention of pregnancy. Quartette™ represents the next generation of extended regimen oral contraceptives to be approved by the FDA, and was designed to minimize breakthrough bleeding (BTB) between scheduled periods. The approval of Quartette™ demonstrates Teva’s continued commitment to the development and production of an innovative range of pharmaceutical products that support the health of women around the world.
“Breakthrough bleeding can be experienced with any birth control pill, especially during the first few months, and is one of the reasons a large number of women discontinue extended regimens,” said Dr. James A. Simon, clinical professor of Obstetrics and Gynecology at the George Washington University School of Medicine. “The estrogen in Quartette™ increases at specific points and provides four short light periods a year. Breakthrough bleeding decreases over time, which might help encourage patient adherence.”
The approval was based on a development program that included results from Phase I, Phase II and Phase III clinical trials designed to evaluate the safety and efficacy of Quartette™. The Phase III clinical trial, which involved more than 3,000 women, found that Quartette™ was 97 percent effective at preventing pregnancy. Data further demonstrated that the most common adverse reactions (≥2%) in the Phase III clinical trial were headaches, heavy/irregular vaginal bleeding, nausea/vomiting, acne, dysmenorrhea, weight increased, mood changes, anxiety/panic attack, breast pain and migraines. The primary clinical trial that evaluated the efficacy of Quartette™ also assessed BTB. BTB and unscheduled spotting decreased over successive 91 day cycles.1
Quartette™ features a unique 91-day oral regimen, whereby the dose of estrogen increases at three distinct points over the first 84 days and the amount of progestin remains consistent; this is followed by seven days of 10 mcg of ethinyl estradiol.
“Teva is the leader in the pharmaceutical industry in the marketing and development of extended regimen oral contraceptives, and Quartette™ represents the next generation of these contraceptives. It is a uniquely differentiated product and is based on Teva’s research into when breakthrough bleeding is most likely to occur with these regimens,” said Jill DeSimone, senior vice president & general manager, Global Teva Women’s Health. “Quartette™ is the newest product in our global women’s health franchise and is an example of our dedication to providing a variety of contraceptive and family planning options that fit women’s lifestyles.”
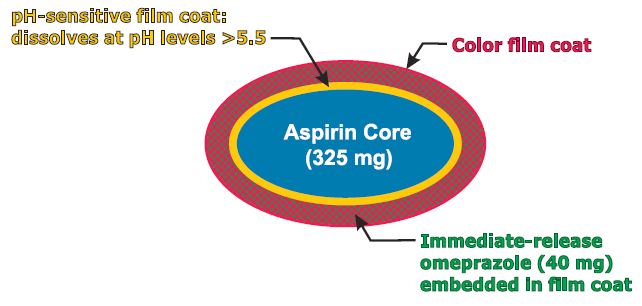
NDA PA32540, Pozen Submits NDA for Aspirin Alternatives
PA32540
PA-325/40 (EC-ASA 325 mg + IR omeprazole 40 mg)
enteric-coated aspirin (EC-ASA) 325 mg
MAR 27,2013
Pozen Inc. announced the submission of a new drug application to the U.S. Food and Drug Administration for the marketing approval of two potential cardiovascular drugs.
The move is a major step toward commercializing the drugs and means that the company thinks it has proven that the drugs are safe and effective.
Pozen’s PA32540 and PA8140 are both intended as alternatives to plain aspirin for the prevention of cardiovascular disease. Many people take aspirin to prevent heart problems, but long-term use of aspirin can cause ulcers.
Pozen’s drugs contain aspirin and the omeprazole, the active ingredient in heartburn drugs like Prilosec. The omeprazole is released as soon as the drug is taken and the aspirin is released over time.
Pozen is seeking approval for use in patients at risk of aspirin-induced ulcers.
Pozen CEO John Plachetka called the move “an important milestone for the drug” and said the company looks forward to completing a commercial deal with a partner “in the upcoming months.”
Pozen’s PA-325/40 is a combination pill containing enteric-coated aspirin 325 mg surrounded by a pH-sensitive film surrounded by an immediate-release omeprazole 40 mg.
Florbetaben (18F), FDA and EMA accept NDA and MAA for Piramal‘s Alzheimer’s imaging agent

Florbetaben (18F)
PHOTO CREDIT-KEGG
902143-01-5 cas no
(18F-AV-1/ZK; BAY-94-9172; 18F-BAY-94-9172; ZK-6013443)

Mr Ajay Piramal, Chairman, Piramal Healthcare
Imaging with amyloid-β PET can potentially aid the early and accurate diagnosis of Alzheimer’s disease. Florbetaben (¹⁸F) is a promising ¹⁸F-labelled amyloid-β-targeted PET tracer in clinical development. We aimed to assess the sensitivity and specificity of florbetaben (¹⁸F) PET in discriminating between patients with probable Alzheimer’s disease and elderly healthy controls.
METHODS:
We did a multicentre, open-label, non-randomised phase 2 study in 18 centres in Australia, Germany, Switzerland, and the USA. Imaging with florbetaben (¹⁸F) PET was done on patients with probable Alzheimer’s disease (age 55 years or older, mini-mental state examination [MMSE] score=18-26, clinical dementia rating [CDR]=0·5-2·0) and age-matched healthy controls (MMSE ≥ 28, CDR=0). Our primary objective was to establish the diagnostic efficacy of the scans in differentiating between patients with probable disease and age-matched healthy controls on the basis of neocortical tracer uptake pattern 90-110 min post-injection. PET images were assessed visually by three readers masked to the clinical diagnosis and all other clinical findings, and quantitatively by use of pre-established brain volumes of interest to obtain standard uptake value ratios (SUVRs), taking the cerebellar cortex as the reference region. This study is registered with ClinicalTrials.gov, number NCT00750282.
FINDINGS:
81 participants with probable Alzheimer’s disease and 69 healthy controls were assessed. Independent visual assessment of the PET scans showed a sensitivity of 80% (95% CI 71-89) and a specificity of 91% (84-98) for discriminating participants with Alzheimer’s disease from healthy controls. The SUVRs in all neocortical grey-matter regions in participants with Alzheimer’s disease were significantly higher (p < 0·0001) compared with the healthy controls, with the posterior cingulate being the best discriminator. Linear discriminant analysis of regional SUVRs yielded a sensitivity of 85% and a specificity of 91%. Regional SUVRs also correlated well with scores of cognitive impairment such as the MMSE and the word-list memory and word-list recall scores (r -0·27 to -0·33, p ≤ 0·021). APOE ɛ4 was more common in participants with positive PET images compared with those with negative scans (65%vs 22% [p=0·027
MAR 21 2013
Piramal Imaging SA, a division of Piramal Enterprises, today announced that the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have accepted its applications for review of the investigational PET amyloid imaging agent [18F] florbetaben. A New Drug Application (NDA) was submitted to the U.S. Food and Drug Administration (FDA) and a Marketing Authorization Application to the EMA for [18F] florbetabenuse in the visual detection of beta-amyloid in the brains of adultswith cognitive impairment who are being evaluated for Alzheimer’s disease and other causes of cognitive decline.[18F] florbetaben binds to beta-amyloid plaques in the human brain, a hallmark characteristic in Alzheimer’s disease.
Today, Alzheimer’s disease is usually diagnosed after a person with a cognitive impairment undergoes an extensive clinical examination which typically includes family and medical history, physical and neurological examinations, laboratory tests, and imaging procedures such as computed tomography (CT) and magnetic resonance imaging (MRI) scans. Still, a definitive diagnosis of Alzheimer’s disease can only be made after death where an autopsy can reveal the presence of beta-amyloid plaques and neurofibrillary tangles in the brain. However, post-mortem studies looking for accumulations of beta-amyloid in the brain have shown that 10 to 30 percent of diagnoses based on clinical examinations are incorrect. [18F] florbetaben is being studied to determine its potential ability to detect beta-amyloid plaquesin living subjects with cognitive impairment.
FLORBETABEN F18
Diagnostic radiopharmaceutical
1. Benzenamine, 4-[(1E)-2-[4-[2-[2-[2-(fluoro-18F)ethoxy]ethoxy]ethoxy]phenyl]
ethenyl]-N-methyl-
2. 4-{(1E)-2-(4-{2-[2-(2-[18F]fluoroethoxy)ethoxy]ethoxy}phenyl)eth- 1-en-1-yl}-N-methylaniline
C21H26[18F]NO3
358.5
Bayer Healthcare
UNII-TLA7312TOI
CAS REGISTRY NUMBER 902143-01-5
https://www.ama-assn.org/resources/doc/usan/florbetaben-f18.pdf

4-[(E)-2-(4-{2-[2-(2-fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline has been labeled with [F-18]fluoride and is claimed by patent application WO2006066104 and members of the corresponding patent family.

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
The usefulness of this radiotracer for the detection of Αβ plaques have been reported in the literature (W. Zhang et al., Nuclear Medicine and Biology 32 (2005) 799-809; C. Rowe et al., Lancet Neurology 7 (2008) 1 -7).
The synthesis of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)- vinyl]-N-methylaniline has been described before:
a) W. Zhang et al., Nuclear Medicine and Biology 32 (2005) 799-809.

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
4 mg precursor 2a (2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate) in 0.2 mL
DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediate was deprotected with HCI and neutralized with
NaOH. The mixture was extracted with ethyl acetate. The solvent was dried and evaporated, the residue was dissolved in acetonitrile and purified by semi-preparative HPLC. 20% (decay corrected), 1 1 % (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N- methylaniline were obtained in 90 min.
WO2006066104
4 mg precursor 2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate in 0.2 mL DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediates was deprotected with HCI and neutralized with NaOH. The mixture was extracted with ethyl acetate. The solvent was dried and evaporated, the residue was dissolved in acetonitrile and purified by semi- preparative HPLC. 30% (decay corrected), 17% (not corrected for decay) 4- [(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N- methylaniline were obtained in 90 min. to yield N-Boc protected 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline. The unreacted perfluorinated precursor was removed using a fluorous phase cartridge.
Deprotection, final purification and formulation to obtain a product, suitable for injection into human is not disclosed. Furthermore, the usefulness (e.g. regarding unwanted F-19/F-18 exchange) of this approach at a higher radioactivity level is not demonstrated. Finally, this method would demand a two-pot setup (first reaction vessel: fluorination, followed by solid-phase- extraction, and deprotection in the second reaction vessel).
However, the focus of the present invention are compounds and methods for an improved “one-pot process” for the manufacturing of 4-[(E)-2-(4-{2-[2-(2- [F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline.
Very recently, further methods have been described:
d) US201001 13763
The mesylate precursor 2a was reacted with [F-18]fluoride species in a solvent mixture consisting of 100 μΙ_ acetonitrile and 500 μΙ_ tertiary alcohol. After fluorination for 10 min at 100-150 °C, the solvent was evaporated. After deprotection (1 N HCI, 5 min, 100-120 °C), the crude product was purified by HPLC (C18 silica, acetonitrile / 0.1 M ammonium formate).
e) H. Wang et al., Nuclear Medicine and Biology 38 (201 1 ) 121 -127
5 mg precursor 2a (2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate) in 0.5 ml_
DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediate was deprotected with HCI and neutralized with NaOH. The crude product was diluted with acetonitrile / 0.1 M ammonium dformate (6/4) and purified by semi-preparative HPLC. The product fraction was collected, diluted with water, passed through a C18 cartridge and eluted with ethanol, yielding 17% (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline within 50 min. In the paper, the conversion of an unprotected mesylate precursor (is described:
5 mg unprotected mesylate precursor (2-{2-[2-(4-{(E)-2-[4- (methylamino)phenyl]vinyl}phenoxy)ethoxy]-ethoxy}ethyl 4- methanesulfonate) in 0.5 ml_ DMSO were reacted with [F- 18]fluoride/kryptofix/potassium carbonate complex. The crude product was diluted with acetonitrile / 0.1 M ammonium formate (6/4) and purified by semi- preparative HPLC. The product fraction was collected, diluted with water, passed through a C18 cartridge and eluted with ethanol, yielding 23% (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F-
18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline within 30 min. Beside the purification by HPLC, a process based on solid-phase-extraction was investigated, but the purity was inferior to that with HPLC purification. So far, one-pot radiolabelings have been performed using a mesylate precursor. It is know, that for F-18 labeling of stilbenes, mesylates have advantages over corresponding tosylates by providing more clean reactions with less amount of by-products (W. Zhang et al. Journal of Medicinal Chemistry 48 (2005) 5980- 5988), whereas the purification starting from the tosylate precursor was tedious and time consuming resulting in a low yield.
In contrast to this teaching of the prior art, we found advantages of tosylate and further arylsulfonate precursors for 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline compared to the corresponding mesylate. Less non-radioactive by-products that eluted close to the retention time of 4-[(E)-2-(4-{2-[2-(2-[F-
18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline were found in the crude products if arylsulfonate precursors were used (Example 2 – Example 6) compared to the crude mixture that was obtained after conversion of the mesylate precursor (Example 1 ).
The favorable by-product profile after radiolabeling of tosylate precursor 2b (Figure 10) compared to the radiolabeling of mesylate precursor 2a (Figure 7) supported an improved cartridge based purification (Example 8, Example 9).
…………………
The term “F-18” means fluorine isotope 18F. The term”F-19″ means fluorine isotope 19F. EXAMPLES
Example 1 Radiolabeling of mesylate precursor 2a

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2a (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 1 ). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 21 % (corrected for decay).
Example 2 Synthesis and radiolabeling of tosylate precursor 2b


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
4-Dimethylaminopyridine (26.7 mg) and triethylamine (225 μΙ_) were added to a solution of 1 .0 g terf-butyl {4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate (4) in dichloromethane (12 mL) at 0 °C. A solution of p- toluenesulfonyl chloride (417 mg) in dichloromethane (13.5 mL) was added at 0 °C. The resulting mixture was stirred at room temperature over night. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (silica, 0- 80% ethyl acetate in hexane). 850 mg 2b were obtained as colorless solid.
1 H NMR (300 MHz, CDCI3) δ ppm 1 .46 (s, 9 H), 2.43 (s, 3 H), 3.27 (s, 3 H), 3.59-3.73 (m, 6 H), 3.80- 3.86 (m, 2 H), 4.05-4.19 (m, 2 H), 6.88-7.05 (m, 4 H), 7.21 (d, J = 8.3 Hz, 2 H), 7.32 (d, J = 8.3 Hz, 2 H), 7.39-7-47 (m, 4 H), 7.80 (d, J = 8.3 Hz, 2 H). MS (ESIpos): m/z = 612 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2b (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 2). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 25% (corrected for decay).
Example 3 Synthesis and radiolabeling of 2c (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-bromobenzenesulfonate)

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline To a stirred solution of 100 mg (0,219 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104) in 2 mL THF was added a solution of 140 mg (0.548 mmol) 4-brombenzene sulfonylchlorid in 3 mL THF drop by drop. The reaction mixture was cooled to 0°C. 156.8 mg (1 .1 mmol) potassium trimethylsilanolat was added. The milky suspension was stirred at 0°C for 2 hours and at 80°C for another 2 hours. The reaction mixture was poured onto ice-cooled water. The aqueous solution was extracted with dichloromethane several times. The combined organic phases were dried with sodium sulphate and concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2c was obtained with 77 mg (0.1 14 mmol, 52.0 % yield).
1 H NMR (300 MHz, CDCI3) δ ppm 1 .39 (s, 10 H) 3.20 (s, 3 H) 3.50 – 3.57 (m, 2 H) 3.57 – 3.61 (m, 2 H) 3.61 – 3.66 (m, 2 H) 3.72 – 3.80 (m, 2 H) 4.02 – 4.10 (m, 2 H) 4.10 – 4.17 (m, 2 H) 6.79 – 6.85 (m, 2 H) 6.91 (d, J=8.10 Hz, 2 H) 7.10 – 7.17 (m, 2 H) 7.32 – 7.41 (m, 5 H) 7.57 – 7.65 (m, 2 H) 7.67 – 7.74 (m, 2 H)
MS (ESIpos): m/z = 676/678 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2c (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 3). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 43% (corrected for decay). Example 4 Synthesis and radiolabeling of 2d (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-(adamantan-1 -yl)benzenesulfonate)

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
To a stirred solution of 151 mg (0,330 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 4.03 mg (0,033 mmol) DMAP und 36.7 mg (363 mmol) triethylamine in 4 mL dichlormethane was added a solution of 97,4 mg (0,313 mmol) 4-(adamantan-1 -yl)benzene sulfonylchloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred at 0°C for 1 hour and over night at room temperature. 7.3 mg (0,072 mmol) triethylamin und 19.5 mg (0.062 mmol) 4- (adamantan-l -yl)benzenesulfonyl chloride were added to the reaction mixture. The reaction mixture was stirred at room temperature for 3 days. It was concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2d was obtained with 104 mg (0.142 mmol, 43.4% yield).
1 H NMR (300 MHz, CDCI3) δ ppm 1 .51 (s, 9 H), 1 .62 (s, 1 H), 1 .74 – 1 .91 (m, 6 H), 1 .94 (d, J=3.20 Hz, 6 H), 2.16 (br. s., 3 H), 3.31 (s, 3 H), 3.63 – 3.69 (m, 2 H), 3.69 – 3.73 (m, 2 H), 3.76 (dd, J=5.27, 4.52 Hz, 2 H), 3.89 (d, J=4.90 Hz, 2 H), 4.13 – 4.26 (m, 4 H), 6.95 (d, J=8.85 Hz, 2 H), 7.02 (d, J=8.29 Hz, 2 H), 7.25 (d, J=8.48 Hz, 2 H), 7.40 – 7.52 (m, 4 H), 7.55 (m, J=8.67 Hz, 2 H), 7.89 (m, J=8.67 Hz, 2 H)
MS (ESIpos): m/z = 732 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2d (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 4). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 27% (corrected for decay).
Example 5 Synthesis and radiolabeling of 2e (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-cyanobenzenesulfonate)


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
To a stirred solution of 151 mg (0.330 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 4.03 mg (0.033 mmol) DMAP und 36.7 mg (0.363 mmol) triethylamine in 4 mL dichlormethane was added a solution of 63.2 mg (0.313 mmol) 4-cyanobenzenesulfonyl chloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred over night and concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2e was obtained with 118 mg (0.190 mmol, 57.6 % yield).
1 H NMR (400 MHz, CDCI3) δ ppm 1 .47 (s, 9 H) 3.28 (s, 3 H) 3.58 – 3.63 (m, 2 H) 3.63 – 3.68 (m, 2 H) 3.70 – 3.75 (m, 2 H) 3.81 – 3.87 (m, 2 H) 4.1 1 – 4.18 (m, 2 H) 4.24 – 4.30 (m, 2 H) 6.91 (d, J=8.59 Hz, 2 H) 6.99 (dt, 2 H) 7.22 (d, J=8.34 Hz, 2 H) 7.39 – 7.50 (m, 4 H) 7.83 (m, J=8.59 Hz, 2 H) 7.98 – 8.1 1 (m, 2 H)
MS (ESIpos): m/z = 623 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2e (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 5). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 31 % (corrected for decay).
Example 6 Synthesis and radiolabeling of 2f (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
2-nitrobenzenesulfonate)


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- eth oxy} phe nyl )vi ny I] -N -methyla n i I i ne
To a stirred solution of 200 mg (0.437 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 5.34 mg (0.044 mmol) DMAP und 47.5 mg (0.470 mmol) triethylamine in 4 mL dichlormethane was added a solution of 92 mg (0,415 mmol) 2-nitrobenzenesulfonyl chloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred over night and concentrated in vacuum. The crude product was purified with ethyl acetate/hexane-gradient as mobile phase using silica gel. The desired product 2f was obtained with 77 mg (0.1 19 mmol, 59.5 % yield). 1 H NMR (400 MHz, CDCI3) δ ppm 1 .39 (s, 9 H) 3.20 (s, 3 H) 3.55 – 3.63 (m, 4 H) 3.59 (m, 4 H) 3.69 – 3.74 (m, 2 H) 3.75 – 3.80 (m, 2 H) 4.06 (dd, J=5.68, 3.92 Hz,
2 H) 4.32 – 4.37 (m, 2 H) 6.80 – 6.84 (m, 2 H) 6.84 – 6.98 (dt, 2 H) 7.14 (d, J=8.59 Hz, 2 H) 7.35 (d, J=3.03 Hz, 2 H) 7.37 (d, J=2.78 Hz, 2 H) 7.62 – 7.74 (m,
3 H) 8.06 – 8.1 1 (m, 1 H)
Morphine and oxycodone Dual-Opioid combination (MoxDuo)
SYDNEY and BEDMINSTER, N.J., March 14, 2013
QRxPharma Limited announced today the US Food and Drug Administration (FDA) has set 26 August 2013 as the Prescription Drug User Fee Act (PDUFA) date for action on the Company’s resubmitted MoxDuo New Drug Application (NDA).
“We are pleased that the FDA has formally accepted our resubmitted MoxDuo NDA” said Dr. John Holaday , Managing Director and Chief Executive Officer, QRxPharma. “We expect the Advisory Committee meeting to be scheduled between late June and late July and will update shareholders once formal notification has been received,” added Holaday.
The NDA is the basis for recommencing the regulatory approval process for MoxDuo for the treatment of moderate to severe acute pain, a $2.5 billion segment of the $8 billion spent annually on prescription opioids in the US. MoxDuo, an immediate release Dual Opioid® pain therapy, is a patented 3:2 fixed ratio combination of morphine

and
oxycodone

FDA Sets 26 August 2013 As New PDUFA Date For MoxDuo NDA.
NDA FDA-Nuvo reports FDA response to PENNSAID 2% , diclofenac sodium topical solution, 2% w/w

DICLOFENAC

PENNSAID 2%
7 MAR 2013
The US Food and Drug Administration (FDA) has issued a Complete Response Letter (CRL) to Nuvo Research’s US licensing partner, Mallinckrodt, following the review of Mallinckrodt’s New Drug Application (NDA) for diclofenac sodium topical solution, 2% w/w (PENNSAID 2%).
FDA in the letter mentioned that it requires Mallinckrodt’s complete pharmacokinetic study comparing PENNSAID 2% to original PENNSAID 1.5%.
FDA denied to review the similar pharmacokinetic studies submitted by Mallinckrodt with the NDA, as the reserve samples were not retained at the clinical site.
Pharmacokinetic studies are standard studies conducted during a drug development program to identify the total exposure or the amount of drug that reaches the blood stream after a patient receives both single and multiple doses of the product.
Mallinckrodt has suggested Nuvo that it expects to complete the study and submit the results to the FDA in the third quarter of 2013, and that it anticipates the FDA will provide a formal response to the filing within 6 months thereafter.
Nuvo’s Pain Group president Dr. Bradley Galer said with the new FDA’s letter the firm was disappointed that PENNSAID 2% will not be approved in this review cycle.
“We are pleased that the FDA has outlined a clear pathway to approval that we believe can be completed in a relatively short time frame,” Galer added.
“Upon approval, PENNSAID 2% will be the first and only topical NSAID in the U.S. featuring twice per day dosing and a metered dose pump bottle.”
NDA-US Marketing by Ranbaxy, Alembic has announced that it has received an NDA approval for extended release version of Pfizer’s anti depressant drug Pristiq, Desvenlafaxine Base

DESVENLAFAXINE
read at
5 march 2013
Alembic has announced that it has received an NDA approval for extended release version of Pfizer’s anti depressant drug Pristiq. Pristiq sell approximately $550m in the US. Alembic has outlicensed rights to Ranbaxy for marketing in the US. The company will start marketing the product immediately.
Alembic will manufacture and supply the drug to Ranbaxy for marketing in the US. Vadodara-based pharma player, Alembic Pharmaceuticals Limited has received the approval from the US Food and Drug Administration (USFDA) for a bioequivalent version of Pristiq by Pfizer.
NDA –Sefelsa (gabapentin) Extended Release Tablets – formerly Serada

GABAPENTIN
Sefelsa (gabapentin) Extended Release Tablets – formerly Serada
Company: Depomed, Inc.
Treatment for: Menopausal Hot Flashes
Sefelsa (gabapentin) is an investigational extended-release formulation of gabapentin in development for the treatment of menopausal hot flashes.
- Depomed Provides Update on Sefelsa FDA Advisory Committee – March 4, 2013
- Depomed Announces Serada NDA Acceptance and FDA Advisory Committee Meeting – October 15, 2012
- Depomed Announces Submission Of Serada NDA – August 17, 2012
- Depomed Announces Intent to File NDA for Serada for Treatment of Menopausal Hot Flashes – April 18, 2012
NEWARK, Calif., March 4, 2013
Depomed, Inc., a specialty pharmaceutical company, announced today that the Reproductive Health Drugs Advisory Committee (RHDAC) of the U.S. Food and Drug Administration (FDA) voted 2-12 against approval for Sefelsa, Depomed’s investigational, oral, twice daily formulation of gabapentin, to treat moderate to severe vasomotor symptoms due to menopause. Sefelsa is the proposed trade name for the medication and was formerly referred to as Serada. Based on the outcome of committee meeting, the company will not have a conference call today as previously indicated.
“Depomed today is a product-focused, growth-oriented specialty pharmaceutical company with a growing franchise of treatments for pain and potentially other CNS indications. With revenues from two marketed products, Gralise and Zipsor, significant royalty income from our partnered products and technology, a strong balance sheet and potential to turn cash flow positive in the second half of this year, we believe that 2013 has the potential to be a landmark year in our company’s history,” said Jim Schoeneck, President and Chief Executive Officer of Depomed. “We recognize and appreciate the concerns that were raised by the members of the Advisory Committee. Based on today’s meeting we believe the hurdles for approval of a non-hormonal treatment for hot flashes remain high. Until we believe there is a positive direction for Sefelsa, we will cease all spending relating to the product candidate.”
Data presented at today’s Advisory Committee Meeting included results from the Phase 3 clinical program, which enrolled 1706 patients in three studies.
The FDA will consider the Advisory Committee recommendation in its review of the New Drug Application (NDA) for Sefelsa that Depomed submitted on July 31, 2012, though the FDA is not bound to follow it. The Prescription Drug User Fee Act (PDUFA) date for Sefelsa is May 31, 2013. The PDUFA date is the goal date for the FDA to complete its review of the NDA.
About Vasomotor Symptoms
Vasomotor symptoms include hot flashes and night sweats. A hot flash is a sudden flushing and sensation of heat caused by dilation of skin capillaries. Currently, the leading prescription drug product for the treatment of hot flashes associated with menopause is hormone replacement therapy (HRT). HRT involves the administration of the hormone estrogen, either alone or in combination with the hormone progestin.
About Depomed
Depomed, Inc. is a specialty pharmaceutical company with three approved and marketed products. Gralise® (gabapentin) is a once-daily treatment approved for the management of postherpetic neuralgia (PHN). Zipsor® (diclofenac potassium) Liquid Filled Capsules is a non-steroidal anti-inflammatory drug (NSAID) indicated for relief of mild to moderate acute pain in adults. Glumetza® (metformin hydrochloride extended release tablets) is approved for use in adults with type 2 diabetes and is commercialized by Santarus, Inc. in the United States. Depomed formulates its products and product candidates with its proven, proprietary Acuform® drug delivery technology, which is designed to improve existing oral medications, allowing for extended release of medications to the upper gastrointestinal tract when dosed with food. Additional information about Depomed may be found on its website, www.depomed.com.
Gabapentin (Neurontin) is a pharmaceutical drug, specifically a GABA analog. It was originally developed for the treatment of epilepsy, and currently is also used to relieve neuropathic pain. There are, however, concerns regarding the quality of the trials conducted for a number of conditions.[1]
Web Site: http://www.depomed.com
- Vedula, SS; Bero L; Scherer RW; Dickersin K (November 2009). “Outcome reporting in industry-sponsored trials of gabapentin for off-label use”. The New England Journal of Medicine 361 (20): 1963–71. doi:10.1056/NEJMsa0906126. PMID 19907043.
- “Gabapentin”. The American Society of Health-System Pharmacists. http://www.drugs.com/monograph/gabapentin.html. Retrieved 3 April 2011.
- Vedula, SS; Bero L; Scherer RW; Dickersin K (November 2009). “Outcome reporting in industry-sponsored trials of gabapentin for off-label use”. The New England Journal of Medicine 361 (20): 1963–71. doi:10.1056/NEJMsa0906126. PMID 19907043.
- Moore, RA; Wiffen PJ; Derry S; McQuay HJ (2011-03-16). Moore, R Andrew. ed. “Gabapentin for chronic neuropathic pain and fibromyalgia in adults”. Cochrane database of systematic reviews (Online) (3): CD007938. doi:10.1002/14651858.CD007938.pub2. PMID 21412914.
- Ho, KY; Gan TJ; Habib AS (2006-12-15). “Gabapentin and postoperative pain–a systematic review of randomized controlled trials”. Pain 126 (1–3): 91–101. doi:10.1016/j.pain.2006.06.018. PMID 16846695.

